User login
AGA: Biopsy normal gastric mucosa for H. pylori during esophagogastroduodenoscopy
Obtain gastric biopsies to check for Helicobacter pylori infection in patients undergoing routine esophagogastroduodenoscopy for dyspepsia, even if their mucosa appears normal and they’re immunocompetent, the American Gastroenterological Association advises in a new guideline for upper gastrointestinal biopsy to evaluate dyspepsia in the adult patient in the absence of visible mucosal lesions, which was published in the October issue of Gastroenterology.
The group suggests taking five biopsy specimens from the gastric body and antrum using the updated Sydney System; placing the samples in the same jar; and relying on routine staining to make the call. “Experienced GI pathologists can determine the anatomic location of biopsy specimens sent from the stomach, which obviates the need for” – and cost of – “separating specimens into multiple jars.” They can also identify “virtually all cases of H. pylori .... Therefore, routine use of ancillary special staining” needlessly adds cost, said the guideline authors, led by Dr. Yu-Xiao Yang of the University of Pennsylvania in Philadelphia (Gastroenterology. 2015 Aug 14. doi: 10.1053/j.gastro.2015.07.039).
The group also counsels against routine biopsies of normal-appearing esophagus and gastroesophageal junctions in patients undergoing esophagogastroduodenoscopy (EGD) for dyspepsia, regardless of their immune status.
It does recommend biopsies of normal-appearing duodenum in immunocompromised patients to check for opportunistic infections and graft-versus-host disease (GVHD) after bone marrow transplant, but counsels against routine duodenum biopsies in immunocompetent patients undergoing EGD solely for dyspepsia if signs and symptoms of celiac disease are absent. Again, routine use of special staining isn’t necessary. “Studies using [hematoxylin and eosin] counting methods show similar results as those obtained using CD3 [T-cell marker] stains,” the authors wrote.
The purpose of the guideline is to establish evidence-based standards for biopsies of normal-appearing mucosa in the upper gastrointestinal tract. No such standards existed until now, so “there [is] likely wide practice variation in whether or not such biopsies of normal-appearing mucosa are obtained,” they said.
The advice is based on a thorough review of the medical literature, and a grading of its strength. The guideline is meant for adult patients undergoing EGD solely for dyspepsia, and assumes that endoscopic biopsy has negligible complications.
The esophageal biopsy recommendations are strong, meaning that “most individuals should receive the recommended course of action.” The gastric biopsy advice is a mix of both strong and conditional recommendations, while the duodenal biopsy recommendations are conditional. Evidence for each recommendation ranged from very low to moderate.
Regarding gastric biopsy, H. pylori can lurk in normal-looking mucosa, and evidence supports detection and treatment for both symptom relief and cancer risk reduction. In the immunocompromised, gastric biopsies can also help detect cytomegalovirus infection.
The five-biopsy Sydney System gathers specimens from the lesser and greater curve of the gastric antrum, lesser curvature of the corpus, middle portion of the greater curvature of the corpus, and the incisura angularis. The thorough approach probably detects H. pylori missed by a three-biopsy method, with little added cost.
Esophageal biopsies of normal mucosa aren’t necessary, the authors said, because “although a number of microscopic changes in the esophageal mucosa can be seen in patients with” gastroesophageal reflux disease, they aren’t much help in distinguishing it from heartburn or even healthy controls. For immune compromised patients, GVHD is more likely to show up at other sites, such as the duodenum.
Although the group counsels against routine duodenum biopsies in immunocompetent patients, it did note that “if the suspicion of celiac disease is high, biopsies of the normal-appearing duodenum may be of value even if serologies ... are negative.”
Funding source and disclosures for the work are available from AGA.
Obtain gastric biopsies to check for Helicobacter pylori infection in patients undergoing routine esophagogastroduodenoscopy for dyspepsia, even if their mucosa appears normal and they’re immunocompetent, the American Gastroenterological Association advises in a new guideline for upper gastrointestinal biopsy to evaluate dyspepsia in the adult patient in the absence of visible mucosal lesions, which was published in the October issue of Gastroenterology.
The group suggests taking five biopsy specimens from the gastric body and antrum using the updated Sydney System; placing the samples in the same jar; and relying on routine staining to make the call. “Experienced GI pathologists can determine the anatomic location of biopsy specimens sent from the stomach, which obviates the need for” – and cost of – “separating specimens into multiple jars.” They can also identify “virtually all cases of H. pylori .... Therefore, routine use of ancillary special staining” needlessly adds cost, said the guideline authors, led by Dr. Yu-Xiao Yang of the University of Pennsylvania in Philadelphia (Gastroenterology. 2015 Aug 14. doi: 10.1053/j.gastro.2015.07.039).
The group also counsels against routine biopsies of normal-appearing esophagus and gastroesophageal junctions in patients undergoing esophagogastroduodenoscopy (EGD) for dyspepsia, regardless of their immune status.
It does recommend biopsies of normal-appearing duodenum in immunocompromised patients to check for opportunistic infections and graft-versus-host disease (GVHD) after bone marrow transplant, but counsels against routine duodenum biopsies in immunocompetent patients undergoing EGD solely for dyspepsia if signs and symptoms of celiac disease are absent. Again, routine use of special staining isn’t necessary. “Studies using [hematoxylin and eosin] counting methods show similar results as those obtained using CD3 [T-cell marker] stains,” the authors wrote.
The purpose of the guideline is to establish evidence-based standards for biopsies of normal-appearing mucosa in the upper gastrointestinal tract. No such standards existed until now, so “there [is] likely wide practice variation in whether or not such biopsies of normal-appearing mucosa are obtained,” they said.
The advice is based on a thorough review of the medical literature, and a grading of its strength. The guideline is meant for adult patients undergoing EGD solely for dyspepsia, and assumes that endoscopic biopsy has negligible complications.
The esophageal biopsy recommendations are strong, meaning that “most individuals should receive the recommended course of action.” The gastric biopsy advice is a mix of both strong and conditional recommendations, while the duodenal biopsy recommendations are conditional. Evidence for each recommendation ranged from very low to moderate.
Regarding gastric biopsy, H. pylori can lurk in normal-looking mucosa, and evidence supports detection and treatment for both symptom relief and cancer risk reduction. In the immunocompromised, gastric biopsies can also help detect cytomegalovirus infection.
The five-biopsy Sydney System gathers specimens from the lesser and greater curve of the gastric antrum, lesser curvature of the corpus, middle portion of the greater curvature of the corpus, and the incisura angularis. The thorough approach probably detects H. pylori missed by a three-biopsy method, with little added cost.
Esophageal biopsies of normal mucosa aren’t necessary, the authors said, because “although a number of microscopic changes in the esophageal mucosa can be seen in patients with” gastroesophageal reflux disease, they aren’t much help in distinguishing it from heartburn or even healthy controls. For immune compromised patients, GVHD is more likely to show up at other sites, such as the duodenum.
Although the group counsels against routine duodenum biopsies in immunocompetent patients, it did note that “if the suspicion of celiac disease is high, biopsies of the normal-appearing duodenum may be of value even if serologies ... are negative.”
Funding source and disclosures for the work are available from AGA.
Obtain gastric biopsies to check for Helicobacter pylori infection in patients undergoing routine esophagogastroduodenoscopy for dyspepsia, even if their mucosa appears normal and they’re immunocompetent, the American Gastroenterological Association advises in a new guideline for upper gastrointestinal biopsy to evaluate dyspepsia in the adult patient in the absence of visible mucosal lesions, which was published in the October issue of Gastroenterology.
The group suggests taking five biopsy specimens from the gastric body and antrum using the updated Sydney System; placing the samples in the same jar; and relying on routine staining to make the call. “Experienced GI pathologists can determine the anatomic location of biopsy specimens sent from the stomach, which obviates the need for” – and cost of – “separating specimens into multiple jars.” They can also identify “virtually all cases of H. pylori .... Therefore, routine use of ancillary special staining” needlessly adds cost, said the guideline authors, led by Dr. Yu-Xiao Yang of the University of Pennsylvania in Philadelphia (Gastroenterology. 2015 Aug 14. doi: 10.1053/j.gastro.2015.07.039).
The group also counsels against routine biopsies of normal-appearing esophagus and gastroesophageal junctions in patients undergoing esophagogastroduodenoscopy (EGD) for dyspepsia, regardless of their immune status.
It does recommend biopsies of normal-appearing duodenum in immunocompromised patients to check for opportunistic infections and graft-versus-host disease (GVHD) after bone marrow transplant, but counsels against routine duodenum biopsies in immunocompetent patients undergoing EGD solely for dyspepsia if signs and symptoms of celiac disease are absent. Again, routine use of special staining isn’t necessary. “Studies using [hematoxylin and eosin] counting methods show similar results as those obtained using CD3 [T-cell marker] stains,” the authors wrote.
The purpose of the guideline is to establish evidence-based standards for biopsies of normal-appearing mucosa in the upper gastrointestinal tract. No such standards existed until now, so “there [is] likely wide practice variation in whether or not such biopsies of normal-appearing mucosa are obtained,” they said.
The advice is based on a thorough review of the medical literature, and a grading of its strength. The guideline is meant for adult patients undergoing EGD solely for dyspepsia, and assumes that endoscopic biopsy has negligible complications.
The esophageal biopsy recommendations are strong, meaning that “most individuals should receive the recommended course of action.” The gastric biopsy advice is a mix of both strong and conditional recommendations, while the duodenal biopsy recommendations are conditional. Evidence for each recommendation ranged from very low to moderate.
Regarding gastric biopsy, H. pylori can lurk in normal-looking mucosa, and evidence supports detection and treatment for both symptom relief and cancer risk reduction. In the immunocompromised, gastric biopsies can also help detect cytomegalovirus infection.
The five-biopsy Sydney System gathers specimens from the lesser and greater curve of the gastric antrum, lesser curvature of the corpus, middle portion of the greater curvature of the corpus, and the incisura angularis. The thorough approach probably detects H. pylori missed by a three-biopsy method, with little added cost.
Esophageal biopsies of normal mucosa aren’t necessary, the authors said, because “although a number of microscopic changes in the esophageal mucosa can be seen in patients with” gastroesophageal reflux disease, they aren’t much help in distinguishing it from heartburn or even healthy controls. For immune compromised patients, GVHD is more likely to show up at other sites, such as the duodenum.
Although the group counsels against routine duodenum biopsies in immunocompetent patients, it did note that “if the suspicion of celiac disease is high, biopsies of the normal-appearing duodenum may be of value even if serologies ... are negative.”
Funding source and disclosures for the work are available from AGA.
FROM GASTROENTEROLOGY
Guidelines ineffective for reprocessing of endoscopes
Reprocessing guidelines currently in place for clinical gastrointestinal endoscopes need to be reevaluated and updated as soon as possible, according to a study published in the American Journal of Infection Control (doi: 10.1016/j.ajic.2015.03.003).
Procedures that follow current guidelines do not fully decontaminate the colonoscopes and gastroscopes after they’re used for procedures, leaving behind microbe levels that are well above the accepted benchmark standards.
“Previous studies show that cleaning endoscope channels is laborious and time consuming, and technicians often skip steps,” wrote the study’s authors – led by Cori L. Ofstead, MSPH, of Ofstead & Associates, St. Paul, Minn. “This is concerning, given that the procedure-related risk of endoscopy may be higher than previously thought [and] without objective verification, clinicians can unknowingly use contaminated endoscopes for procedures.”
The study, conducted at the Mayo Clinic in Rochester, Minn., examined the use of 15 endoscopes involved in 60 separate procedures over the period of Nov. 4-8, 2013, to determine how each endoscope was treated after an operation and whether these cleaning procedures followed guidelines and effectively decontaminated the devices.
“Endoscope testing was performed in a dedicated room adjacent to the procedure room, which allowed for rapid sampling and testing [and] barrier separation [minimized] potential for environmental cross-contamination.” the authors explained.
They maintained aseptic environmental conditions while gathering data, using disinfectant wipes on surfaces, using disposable absorbent pads, and restricting room access.
The researchers wore gloves, impervious gowns, face masks with splash protection, hair nets, and shoe covers; the gloves were changed between each sample collection, and gowns were changed between endoscope collections.
Microbial cultures and rapid indicator tests were conducted for ATP, protein, hemoglobin, and carbohydrate residue. Viable microbe samples were collected from 92% of bedside-cleaned endoscopes, 46% of manually cleaned endoscopes, 64% of “high-level disinfected” endoscopes, and 9% of stored endoscopes. Results from rapid indicator tests showed that some form of contamination was present on 100% of bedside-cleaned endoscopes, 92% of manually cleaned endoscopes, 73% of high-level disinfected endoscopes, and 82% of stored endoscopes which had viable microbes.
“Current guidelines rely on visual inspection to verify cleaning [but] visible contamination was never present, potentially because blood and feces may be difficult to discern from endoscopes’ black exteriors, and microscopic organisms cannot be seen by the naked eye,” the researchers wrote.
Consequently, Ms. Ofstead and colleagues called for a reexamination of current guidelines to move away from the eyeball test, saying that the results found here should pave the way for future studies to devise methods of more effectively cleaning and disinfecting endoscopes in a shorter amount of time.
“In the meantime, methods to ensure effectiveness of reprocessing practices are needed, including the potential use of routine monitoring with rapid indicators and microbiologic cultures,” they suggested.
Ms. Ofstead is president and CEO of Ofstead & Associates, which has received research funding and speaking honoraria related to endoscopic procedures from 3M, Advanced Sterilization Products, Medivators, Invendo Medical, Boston Scientific, and Steris. Three coauthors are employed by Ofstead & Associates, but have no other disclosures to report. The remaining coauthors had no disclosures to report. Funding was provided by 3M, Mayo Clinic, and Ofstead & Associates.
Reprocessing guidelines currently in place for clinical gastrointestinal endoscopes need to be reevaluated and updated as soon as possible, according to a study published in the American Journal of Infection Control (doi: 10.1016/j.ajic.2015.03.003).
Procedures that follow current guidelines do not fully decontaminate the colonoscopes and gastroscopes after they’re used for procedures, leaving behind microbe levels that are well above the accepted benchmark standards.
“Previous studies show that cleaning endoscope channels is laborious and time consuming, and technicians often skip steps,” wrote the study’s authors – led by Cori L. Ofstead, MSPH, of Ofstead & Associates, St. Paul, Minn. “This is concerning, given that the procedure-related risk of endoscopy may be higher than previously thought [and] without objective verification, clinicians can unknowingly use contaminated endoscopes for procedures.”
The study, conducted at the Mayo Clinic in Rochester, Minn., examined the use of 15 endoscopes involved in 60 separate procedures over the period of Nov. 4-8, 2013, to determine how each endoscope was treated after an operation and whether these cleaning procedures followed guidelines and effectively decontaminated the devices.
“Endoscope testing was performed in a dedicated room adjacent to the procedure room, which allowed for rapid sampling and testing [and] barrier separation [minimized] potential for environmental cross-contamination.” the authors explained.
They maintained aseptic environmental conditions while gathering data, using disinfectant wipes on surfaces, using disposable absorbent pads, and restricting room access.
The researchers wore gloves, impervious gowns, face masks with splash protection, hair nets, and shoe covers; the gloves were changed between each sample collection, and gowns were changed between endoscope collections.
Microbial cultures and rapid indicator tests were conducted for ATP, protein, hemoglobin, and carbohydrate residue. Viable microbe samples were collected from 92% of bedside-cleaned endoscopes, 46% of manually cleaned endoscopes, 64% of “high-level disinfected” endoscopes, and 9% of stored endoscopes. Results from rapid indicator tests showed that some form of contamination was present on 100% of bedside-cleaned endoscopes, 92% of manually cleaned endoscopes, 73% of high-level disinfected endoscopes, and 82% of stored endoscopes which had viable microbes.
“Current guidelines rely on visual inspection to verify cleaning [but] visible contamination was never present, potentially because blood and feces may be difficult to discern from endoscopes’ black exteriors, and microscopic organisms cannot be seen by the naked eye,” the researchers wrote.
Consequently, Ms. Ofstead and colleagues called for a reexamination of current guidelines to move away from the eyeball test, saying that the results found here should pave the way for future studies to devise methods of more effectively cleaning and disinfecting endoscopes in a shorter amount of time.
“In the meantime, methods to ensure effectiveness of reprocessing practices are needed, including the potential use of routine monitoring with rapid indicators and microbiologic cultures,” they suggested.
Ms. Ofstead is president and CEO of Ofstead & Associates, which has received research funding and speaking honoraria related to endoscopic procedures from 3M, Advanced Sterilization Products, Medivators, Invendo Medical, Boston Scientific, and Steris. Three coauthors are employed by Ofstead & Associates, but have no other disclosures to report. The remaining coauthors had no disclosures to report. Funding was provided by 3M, Mayo Clinic, and Ofstead & Associates.
Reprocessing guidelines currently in place for clinical gastrointestinal endoscopes need to be reevaluated and updated as soon as possible, according to a study published in the American Journal of Infection Control (doi: 10.1016/j.ajic.2015.03.003).
Procedures that follow current guidelines do not fully decontaminate the colonoscopes and gastroscopes after they’re used for procedures, leaving behind microbe levels that are well above the accepted benchmark standards.
“Previous studies show that cleaning endoscope channels is laborious and time consuming, and technicians often skip steps,” wrote the study’s authors – led by Cori L. Ofstead, MSPH, of Ofstead & Associates, St. Paul, Minn. “This is concerning, given that the procedure-related risk of endoscopy may be higher than previously thought [and] without objective verification, clinicians can unknowingly use contaminated endoscopes for procedures.”
The study, conducted at the Mayo Clinic in Rochester, Minn., examined the use of 15 endoscopes involved in 60 separate procedures over the period of Nov. 4-8, 2013, to determine how each endoscope was treated after an operation and whether these cleaning procedures followed guidelines and effectively decontaminated the devices.
“Endoscope testing was performed in a dedicated room adjacent to the procedure room, which allowed for rapid sampling and testing [and] barrier separation [minimized] potential for environmental cross-contamination.” the authors explained.
They maintained aseptic environmental conditions while gathering data, using disinfectant wipes on surfaces, using disposable absorbent pads, and restricting room access.
The researchers wore gloves, impervious gowns, face masks with splash protection, hair nets, and shoe covers; the gloves were changed between each sample collection, and gowns were changed between endoscope collections.
Microbial cultures and rapid indicator tests were conducted for ATP, protein, hemoglobin, and carbohydrate residue. Viable microbe samples were collected from 92% of bedside-cleaned endoscopes, 46% of manually cleaned endoscopes, 64% of “high-level disinfected” endoscopes, and 9% of stored endoscopes. Results from rapid indicator tests showed that some form of contamination was present on 100% of bedside-cleaned endoscopes, 92% of manually cleaned endoscopes, 73% of high-level disinfected endoscopes, and 82% of stored endoscopes which had viable microbes.
“Current guidelines rely on visual inspection to verify cleaning [but] visible contamination was never present, potentially because blood and feces may be difficult to discern from endoscopes’ black exteriors, and microscopic organisms cannot be seen by the naked eye,” the researchers wrote.
Consequently, Ms. Ofstead and colleagues called for a reexamination of current guidelines to move away from the eyeball test, saying that the results found here should pave the way for future studies to devise methods of more effectively cleaning and disinfecting endoscopes in a shorter amount of time.
“In the meantime, methods to ensure effectiveness of reprocessing practices are needed, including the potential use of routine monitoring with rapid indicators and microbiologic cultures,” they suggested.
Ms. Ofstead is president and CEO of Ofstead & Associates, which has received research funding and speaking honoraria related to endoscopic procedures from 3M, Advanced Sterilization Products, Medivators, Invendo Medical, Boston Scientific, and Steris. Three coauthors are employed by Ofstead & Associates, but have no other disclosures to report. The remaining coauthors had no disclosures to report. Funding was provided by 3M, Mayo Clinic, and Ofstead & Associates.
FDA approves ReShape intragastric balloon device
The first intragastric balloon–based device designed to help obese people lose weight has been approved by the Food and Drug Administration, providing a treatment option that is less invasive than bariatric surgery and gastric banding.
The FDA approved the ReShape Integrated Dual Balloon System on July 28, for “weight reduction when used in conjunction with diet and exercise, in obese patients with a body mass index (BMI) of 30-40 kg/m2 and one or more obesity-related comorbid conditions,” in adults who have not been able to lose weight with diet and exercise alone, according to the agency’s approval letter. Laparoscopic gastric banding is indicated for patients with a BMI of at least 40 kg/m2 (or at least 30 kg/m2 in people with one or more obesity-related comorbidities) and bariatric surgery is usually recommended for patients with a BMI of at least 40 kg/m2 (or at least 35 kg/m2 in people with at least one obesity-related comorbidity).

The ReShape device is made up of two attached balloons that are placed in the stomach through a minimally invasive endoscopic procedure, where they are filled with about 2 cups of saline and methylene blue dye, under mild sedation; the balloons are sealed with mineral oil and left in place for up to 6 months. If a balloon ruptures, the dye appears in the urine. When it is time to remove the balloons, they are deflated then removed using another endoscopic procedure.
The device was evaluated in a pivotal study at eight U.S. sites of over 300 mostly female obese patients whose mean age was about 44 years; their mean weight was about 209-213 pounds, and their mean BMI was about 35 kg/m2; 187 received the device and 139 had the endoscopy only. All participants were on a medically managed diet and exercise program. At 6 months, those in the device group had lost a mean of about 24% of their weight, vs. a mean of about 11% among controls, a statistically significant difference (P = .0041). Those who had lost weight at 6 months “maintained 60% of this weight loss through 48 weeks of follow-up,” according to the FDA.
After placement of the device, common adverse events were vomiting, nausea, and abdominal pain, but most symptoms resolved within 30 days, according to the FDA. The development of gastric ulcerations is described as the “most worrisome” device-related risk, but “there were no unanticipated adverse device effects, no deaths, no intestinal obstructions, and no gastric perforations” in the study.
Among the 265 patients who received the device (those initially enrolled in the pivotal trial plus 78 who were in the control group and opted to receive the device after the first 6 months), 20 (7.5%) experienced severe adverse events; vomiting was the most common, in 4.5%. Serious events included gastric ulcers in two patients (0.8%) at 19 and at 97 days after the device was placed; in both cases, the device was removed. Almost 15% of those who received the device had to have it removed because of an adverse event. The rate of gastric ulcers after a minor change was made to the device was 10%; and the rate of balloon deflations without migration was 6%.
The FDA summary of the approval refers to the “marginal benefit of weight loss” among those in the treatment group, compared with controls, but adds that the decision to approve the device “is based in part on the limited options available to patients with mild to moderate obesity who have failed other means for conservative weight loss.”
While the effectiveness of the device is better than what would be expected with diet and exercise or pharmacologic therapy,” it is “substantially less than what would be expected with gastric banding or other surgical interventions.” The list of contraindications includes previous gastrointestinal surgery “with sequelae,” such as an obstruction or adhesions; previous bariatric surgery; any GI inflammatory disease, severe coagulopathy; and women who are pregnant or breastfeeding.
“The company plans to make the ReShape procedure available to patients first in select markets, as physicians and allied health professionals are trained in the procedure and support program to optimize patient outcome,” according to the company’s statement announcing approval.
The ReShape device has been available in Europe since 2007.
Information posted by the FDA, including labeling for professionals, is available at www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfTopic/pma/pma.cfm?num=P140012.
The AGA Center for GI Innovation and Technology is committed to supporting the development of new devices and their introduction to the market in a safe and efficient manner. The center has been working with the FDA for the past several years to help the agency determine how to assess obesity devices.
“It is gratifying to see that the FDA continues to facilitate technologies to address significant public health concerns,” said chair of the center, Dr. Michael L. Kochman, FACP, AGAF. “The AGA Center for GI Innovation and Technology’s obesity-focused meeting, held in conjunction with the FDA, and the annual AGA Tech Summits have helped inform the discussion surrounding the new and novel devices in the obesity and metabolism space.”
The first intragastric balloon–based device designed to help obese people lose weight has been approved by the Food and Drug Administration, providing a treatment option that is less invasive than bariatric surgery and gastric banding.
The FDA approved the ReShape Integrated Dual Balloon System on July 28, for “weight reduction when used in conjunction with diet and exercise, in obese patients with a body mass index (BMI) of 30-40 kg/m2 and one or more obesity-related comorbid conditions,” in adults who have not been able to lose weight with diet and exercise alone, according to the agency’s approval letter. Laparoscopic gastric banding is indicated for patients with a BMI of at least 40 kg/m2 (or at least 30 kg/m2 in people with one or more obesity-related comorbidities) and bariatric surgery is usually recommended for patients with a BMI of at least 40 kg/m2 (or at least 35 kg/m2 in people with at least one obesity-related comorbidity).
The ReShape device is made up of two attached balloons that are placed in the stomach through a minimally invasive endoscopic procedure, where they are filled with about 2 cups of saline and methylene blue dye, under mild sedation; the balloons are sealed with mineral oil and left in place for up to 6 months. If a balloon ruptures, the dye appears in the urine. When it is time to remove the balloons, they are deflated then removed using another endoscopic procedure.
The device was evaluated in a pivotal study at eight U.S. sites of over 300 mostly female obese patients whose mean age was about 44 years; their mean weight was about 209-213 pounds, and their mean BMI was about 35 kg/m2; 187 received the device and 139 had the endoscopy only. All participants were on a medically managed diet and exercise program. At 6 months, those in the device group had lost a mean of about 24% of their weight, vs. a mean of about 11% among controls, a statistically significant difference (P = .0041). Those who had lost weight at 6 months “maintained 60% of this weight loss through 48 weeks of follow-up,” according to the FDA.
After placement of the device, common adverse events were vomiting, nausea, and abdominal pain, but most symptoms resolved within 30 days, according to the FDA. The development of gastric ulcerations is described as the “most worrisome” device-related risk, but “there were no unanticipated adverse device effects, no deaths, no intestinal obstructions, and no gastric perforations” in the study.
Among the 265 patients who received the device (those initially enrolled in the pivotal trial plus 78 who were in the control group and opted to receive the device after the first 6 months), 20 (7.5%) experienced severe adverse events; vomiting was the most common, in 4.5%. Serious events included gastric ulcers in two patients (0.8%) at 19 and at 97 days after the device was placed; in both cases, the device was removed. Almost 15% of those who received the device had to have it removed because of an adverse event. The rate of gastric ulcers after a minor change was made to the device was 10%; and the rate of balloon deflations without migration was 6%.
The FDA summary of the approval refers to the “marginal benefit of weight loss” among those in the treatment group, compared with controls, but adds that the decision to approve the device “is based in part on the limited options available to patients with mild to moderate obesity who have failed other means for conservative weight loss.”
While the effectiveness of the device is better than what would be expected with diet and exercise or pharmacologic therapy,” it is “substantially less than what would be expected with gastric banding or other surgical interventions.” The list of contraindications includes previous gastrointestinal surgery “with sequelae,” such as an obstruction or adhesions; previous bariatric surgery; any GI inflammatory disease, severe coagulopathy; and women who are pregnant or breastfeeding.
“The company plans to make the ReShape procedure available to patients first in select markets, as physicians and allied health professionals are trained in the procedure and support program to optimize patient outcome,” according to the company’s statement announcing approval.
The ReShape device has been available in Europe since 2007.
Information posted by the FDA, including labeling for professionals, is available at www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfTopic/pma/pma.cfm?num=P140012.
The AGA Center for GI Innovation and Technology is committed to supporting the development of new devices and their introduction to the market in a safe and efficient manner. The center has been working with the FDA for the past several years to help the agency determine how to assess obesity devices.
“It is gratifying to see that the FDA continues to facilitate technologies to address significant public health concerns,” said chair of the center, Dr. Michael L. Kochman, FACP, AGAF. “The AGA Center for GI Innovation and Technology’s obesity-focused meeting, held in conjunction with the FDA, and the annual AGA Tech Summits have helped inform the discussion surrounding the new and novel devices in the obesity and metabolism space.”
The first intragastric balloon–based device designed to help obese people lose weight has been approved by the Food and Drug Administration, providing a treatment option that is less invasive than bariatric surgery and gastric banding.
The FDA approved the ReShape Integrated Dual Balloon System on July 28, for “weight reduction when used in conjunction with diet and exercise, in obese patients with a body mass index (BMI) of 30-40 kg/m2 and one or more obesity-related comorbid conditions,” in adults who have not been able to lose weight with diet and exercise alone, according to the agency’s approval letter. Laparoscopic gastric banding is indicated for patients with a BMI of at least 40 kg/m2 (or at least 30 kg/m2 in people with one or more obesity-related comorbidities) and bariatric surgery is usually recommended for patients with a BMI of at least 40 kg/m2 (or at least 35 kg/m2 in people with at least one obesity-related comorbidity).
The ReShape device is made up of two attached balloons that are placed in the stomach through a minimally invasive endoscopic procedure, where they are filled with about 2 cups of saline and methylene blue dye, under mild sedation; the balloons are sealed with mineral oil and left in place for up to 6 months. If a balloon ruptures, the dye appears in the urine. When it is time to remove the balloons, they are deflated then removed using another endoscopic procedure.
The device was evaluated in a pivotal study at eight U.S. sites of over 300 mostly female obese patients whose mean age was about 44 years; their mean weight was about 209-213 pounds, and their mean BMI was about 35 kg/m2; 187 received the device and 139 had the endoscopy only. All participants were on a medically managed diet and exercise program. At 6 months, those in the device group had lost a mean of about 24% of their weight, vs. a mean of about 11% among controls, a statistically significant difference (P = .0041). Those who had lost weight at 6 months “maintained 60% of this weight loss through 48 weeks of follow-up,” according to the FDA.
After placement of the device, common adverse events were vomiting, nausea, and abdominal pain, but most symptoms resolved within 30 days, according to the FDA. The development of gastric ulcerations is described as the “most worrisome” device-related risk, but “there were no unanticipated adverse device effects, no deaths, no intestinal obstructions, and no gastric perforations” in the study.
Among the 265 patients who received the device (those initially enrolled in the pivotal trial plus 78 who were in the control group and opted to receive the device after the first 6 months), 20 (7.5%) experienced severe adverse events; vomiting was the most common, in 4.5%. Serious events included gastric ulcers in two patients (0.8%) at 19 and at 97 days after the device was placed; in both cases, the device was removed. Almost 15% of those who received the device had to have it removed because of an adverse event. The rate of gastric ulcers after a minor change was made to the device was 10%; and the rate of balloon deflations without migration was 6%.
The FDA summary of the approval refers to the “marginal benefit of weight loss” among those in the treatment group, compared with controls, but adds that the decision to approve the device “is based in part on the limited options available to patients with mild to moderate obesity who have failed other means for conservative weight loss.”
While the effectiveness of the device is better than what would be expected with diet and exercise or pharmacologic therapy,” it is “substantially less than what would be expected with gastric banding or other surgical interventions.” The list of contraindications includes previous gastrointestinal surgery “with sequelae,” such as an obstruction or adhesions; previous bariatric surgery; any GI inflammatory disease, severe coagulopathy; and women who are pregnant or breastfeeding.
“The company plans to make the ReShape procedure available to patients first in select markets, as physicians and allied health professionals are trained in the procedure and support program to optimize patient outcome,” according to the company’s statement announcing approval.
The ReShape device has been available in Europe since 2007.
Information posted by the FDA, including labeling for professionals, is available at www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfTopic/pma/pma.cfm?num=P140012.
The AGA Center for GI Innovation and Technology is committed to supporting the development of new devices and their introduction to the market in a safe and efficient manner. The center has been working with the FDA for the past several years to help the agency determine how to assess obesity devices.
“It is gratifying to see that the FDA continues to facilitate technologies to address significant public health concerns,” said chair of the center, Dr. Michael L. Kochman, FACP, AGAF. “The AGA Center for GI Innovation and Technology’s obesity-focused meeting, held in conjunction with the FDA, and the annual AGA Tech Summits have helped inform the discussion surrounding the new and novel devices in the obesity and metabolism space.”

FDA: Extra steps for cleaner scopes
Addressing recent outbreaks of serious, sometimes fatal infections associated with duodenoscopes, the Food and Drug Administration has issued recommendations for taking extra steps to improve the reprocessing of these devices, which “may further help reduce the risk of infection transmission,” the agency said in an August 4 statement.
The extra measures are microbiological culturing, ethylene oxide sterilization, the use of a liquid chemical sterilant processing system, and repeated high-level disinfection. “Hospitals and health care facilities that utilize duodenoscopes can, in addition to meticulously following manufacturer reprocessing instructions, take one or more of these additional steps to further reduce the risk of infection and increase the safety of these medical devices,” according to the FDA’s statement announcing the measures.

The statement acknowledges that some health care facilities will not be able to implement one or more of these steps, so “it is critical that staff responsible for reprocessing duodenoscopes have the manufacturer’s instructions readily available to promote strict adherence to the reprocessing instructions in the device labeling, understand the importance of their role in reprocessing the device, and maintain proficiency in performing these reprocessing tasks.”
And while it is not possible to completely eliminate the risk of transmitting infections, “the benefits of these devices continue to outweigh the risks in appropriately selected patients,” the statement adds. These recommendations and the statement regarding the risk-benefit profile of these devices, used to perform endoscopic retrograde cholangiopancreatography, reflect comments of panelists at a meeting of the FDA’s s gastroenterology and urology devices panel on May 14 and 15, in which AGA participated, to address recent concerns about duodenoscopes and several outbreaks in U.S. hospitals of serious infections related to the devices.
The FDA’s statement is available here.
Any infections possibly related to duodenoscopes should be reported to the manufacturer and the FDA’s MedWatch program at 800-332-1088.
Addressing recent outbreaks of serious, sometimes fatal infections associated with duodenoscopes, the Food and Drug Administration has issued recommendations for taking extra steps to improve the reprocessing of these devices, which “may further help reduce the risk of infection transmission,” the agency said in an August 4 statement.
The extra measures are microbiological culturing, ethylene oxide sterilization, the use of a liquid chemical sterilant processing system, and repeated high-level disinfection. “Hospitals and health care facilities that utilize duodenoscopes can, in addition to meticulously following manufacturer reprocessing instructions, take one or more of these additional steps to further reduce the risk of infection and increase the safety of these medical devices,” according to the FDA’s statement announcing the measures.
The statement acknowledges that some health care facilities will not be able to implement one or more of these steps, so “it is critical that staff responsible for reprocessing duodenoscopes have the manufacturer’s instructions readily available to promote strict adherence to the reprocessing instructions in the device labeling, understand the importance of their role in reprocessing the device, and maintain proficiency in performing these reprocessing tasks.”
And while it is not possible to completely eliminate the risk of transmitting infections, “the benefits of these devices continue to outweigh the risks in appropriately selected patients,” the statement adds. These recommendations and the statement regarding the risk-benefit profile of these devices, used to perform endoscopic retrograde cholangiopancreatography, reflect comments of panelists at a meeting of the FDA’s s gastroenterology and urology devices panel on May 14 and 15, in which AGA participated, to address recent concerns about duodenoscopes and several outbreaks in U.S. hospitals of serious infections related to the devices.
The FDA’s statement is available here.
Any infections possibly related to duodenoscopes should be reported to the manufacturer and the FDA’s MedWatch program at 800-332-1088.
Addressing recent outbreaks of serious, sometimes fatal infections associated with duodenoscopes, the Food and Drug Administration has issued recommendations for taking extra steps to improve the reprocessing of these devices, which “may further help reduce the risk of infection transmission,” the agency said in an August 4 statement.
The extra measures are microbiological culturing, ethylene oxide sterilization, the use of a liquid chemical sterilant processing system, and repeated high-level disinfection. “Hospitals and health care facilities that utilize duodenoscopes can, in addition to meticulously following manufacturer reprocessing instructions, take one or more of these additional steps to further reduce the risk of infection and increase the safety of these medical devices,” according to the FDA’s statement announcing the measures.
The statement acknowledges that some health care facilities will not be able to implement one or more of these steps, so “it is critical that staff responsible for reprocessing duodenoscopes have the manufacturer’s instructions readily available to promote strict adherence to the reprocessing instructions in the device labeling, understand the importance of their role in reprocessing the device, and maintain proficiency in performing these reprocessing tasks.”
And while it is not possible to completely eliminate the risk of transmitting infections, “the benefits of these devices continue to outweigh the risks in appropriately selected patients,” the statement adds. These recommendations and the statement regarding the risk-benefit profile of these devices, used to perform endoscopic retrograde cholangiopancreatography, reflect comments of panelists at a meeting of the FDA’s s gastroenterology and urology devices panel on May 14 and 15, in which AGA participated, to address recent concerns about duodenoscopes and several outbreaks in U.S. hospitals of serious infections related to the devices.
The FDA’s statement is available here.
Any infections possibly related to duodenoscopes should be reported to the manufacturer and the FDA’s MedWatch program at 800-332-1088.

Orbera intragastric balloon approved for weight loss in obese adults
Another endoscopically delivered intragastric balloon indicated as a weight loss aid in obese adults has been approved by the Food and Drug Administration.
The Orbera intragastric balloon has been approved as a treatment for weight loss, in obese adults, with a body mass index between 30 and 40 kg/m2, the manufacturer, Apollo Endosurgery, announced on Aug. 6. It is intended for obese adults who are considering invasive surgery or for whom invasive surgery is not appropriate, when diet and exercise or pharmaceutical interventions have not worked, the statement said.

During a 20- to 30-minute procedure, the deflated Orbera silicone balloon is placed in the stomach via an endoscopic procedure under a mild sedative, where it is then filled with saline until it is about the size of a grapefruit, according to the company. The patient usually can go home on the same day; the balloon is deflated and removed 6 months later. The company will provide patients with an individualized weight-loss program for patients for 1 year, starting from the time of balloon placement.
The approval of this device follows the approval of the ReShape intragastric balloon for obese adults, for up to 6 months, announced by the FDA on July 28. ReShape was the first such device to be approved in the United States.
The results of the pivotal U.S. 12-month multicenter trial of the Orbera balloon in more than 250 obese adults with a BMI of 30-40 kg/m2 were reported at the Digestive Disease Week meeting in May, by Dr. Barham K. Abu Dayyeh of the Mayo Clinic in Rochester, Minn. For more than 2 years, patients were randomized to a 12-month behavioral modification program, with or without endoscopic placement of the balloon, which was removed at 6 months. Eighteen patients withdrew before treatment; 215 patients were evaluable at 6 months, 206 at 9 months, and 191 at 12 months.
At 6 months, the mean percent total body weight loss was about 10% in the balloon group, vs. 4% in the control group, a significant difference (P less than .001). In addition, the total body weight loss was significantly higher in the balloon group at 3, 6, 9, and 12 months, and the mean percent of excess weight loss at 6 months was better in the balloon group than in the control group (about 40% vs. 13%; P less than .001), he said at the meeting. The majority of excess weight loss achieved at 6 months was also maintained at 12 months.
Serious adverse events were reported in 7% of controls and almost 10% of the balloon group, which included eight early removals for intolerance, one gastric outlet obstruction, one laryngospasm during placement, one case of severe abdominal cramping, and one case of severe dehydration. Early device removals occurred in 22% of patients, 15 for symptoms and 13 at subject request, Dr. Abu Dayyeh said. There were no deaths during the study.
The Orbera balloon has been available in more than 80 countries, according to the manufacturer.
More information is available on the FDA website.
Another endoscopically delivered intragastric balloon indicated as a weight loss aid in obese adults has been approved by the Food and Drug Administration.
The Orbera intragastric balloon has been approved as a treatment for weight loss, in obese adults, with a body mass index between 30 and 40 kg/m2, the manufacturer, Apollo Endosurgery, announced on Aug. 6. It is intended for obese adults who are considering invasive surgery or for whom invasive surgery is not appropriate, when diet and exercise or pharmaceutical interventions have not worked, the statement said.
During a 20- to 30-minute procedure, the deflated Orbera silicone balloon is placed in the stomach via an endoscopic procedure under a mild sedative, where it is then filled with saline until it is about the size of a grapefruit, according to the company. The patient usually can go home on the same day; the balloon is deflated and removed 6 months later. The company will provide patients with an individualized weight-loss program for patients for 1 year, starting from the time of balloon placement.
The approval of this device follows the approval of the ReShape intragastric balloon for obese adults, for up to 6 months, announced by the FDA on July 28. ReShape was the first such device to be approved in the United States.
The results of the pivotal U.S. 12-month multicenter trial of the Orbera balloon in more than 250 obese adults with a BMI of 30-40 kg/m2 were reported at the Digestive Disease Week meeting in May, by Dr. Barham K. Abu Dayyeh of the Mayo Clinic in Rochester, Minn. For more than 2 years, patients were randomized to a 12-month behavioral modification program, with or without endoscopic placement of the balloon, which was removed at 6 months. Eighteen patients withdrew before treatment; 215 patients were evaluable at 6 months, 206 at 9 months, and 191 at 12 months.
At 6 months, the mean percent total body weight loss was about 10% in the balloon group, vs. 4% in the control group, a significant difference (P less than .001). In addition, the total body weight loss was significantly higher in the balloon group at 3, 6, 9, and 12 months, and the mean percent of excess weight loss at 6 months was better in the balloon group than in the control group (about 40% vs. 13%; P less than .001), he said at the meeting. The majority of excess weight loss achieved at 6 months was also maintained at 12 months.
Serious adverse events were reported in 7% of controls and almost 10% of the balloon group, which included eight early removals for intolerance, one gastric outlet obstruction, one laryngospasm during placement, one case of severe abdominal cramping, and one case of severe dehydration. Early device removals occurred in 22% of patients, 15 for symptoms and 13 at subject request, Dr. Abu Dayyeh said. There were no deaths during the study.
The Orbera balloon has been available in more than 80 countries, according to the manufacturer.
More information is available on the FDA website.
Another endoscopically delivered intragastric balloon indicated as a weight loss aid in obese adults has been approved by the Food and Drug Administration.
The Orbera intragastric balloon has been approved as a treatment for weight loss, in obese adults, with a body mass index between 30 and 40 kg/m2, the manufacturer, Apollo Endosurgery, announced on Aug. 6. It is intended for obese adults who are considering invasive surgery or for whom invasive surgery is not appropriate, when diet and exercise or pharmaceutical interventions have not worked, the statement said.
During a 20- to 30-minute procedure, the deflated Orbera silicone balloon is placed in the stomach via an endoscopic procedure under a mild sedative, where it is then filled with saline until it is about the size of a grapefruit, according to the company. The patient usually can go home on the same day; the balloon is deflated and removed 6 months later. The company will provide patients with an individualized weight-loss program for patients for 1 year, starting from the time of balloon placement.
The approval of this device follows the approval of the ReShape intragastric balloon for obese adults, for up to 6 months, announced by the FDA on July 28. ReShape was the first such device to be approved in the United States.
The results of the pivotal U.S. 12-month multicenter trial of the Orbera balloon in more than 250 obese adults with a BMI of 30-40 kg/m2 were reported at the Digestive Disease Week meeting in May, by Dr. Barham K. Abu Dayyeh of the Mayo Clinic in Rochester, Minn. For more than 2 years, patients were randomized to a 12-month behavioral modification program, with or without endoscopic placement of the balloon, which was removed at 6 months. Eighteen patients withdrew before treatment; 215 patients were evaluable at 6 months, 206 at 9 months, and 191 at 12 months.
At 6 months, the mean percent total body weight loss was about 10% in the balloon group, vs. 4% in the control group, a significant difference (P less than .001). In addition, the total body weight loss was significantly higher in the balloon group at 3, 6, 9, and 12 months, and the mean percent of excess weight loss at 6 months was better in the balloon group than in the control group (about 40% vs. 13%; P less than .001), he said at the meeting. The majority of excess weight loss achieved at 6 months was also maintained at 12 months.
Serious adverse events were reported in 7% of controls and almost 10% of the balloon group, which included eight early removals for intolerance, one gastric outlet obstruction, one laryngospasm during placement, one case of severe abdominal cramping, and one case of severe dehydration. Early device removals occurred in 22% of patients, 15 for symptoms and 13 at subject request, Dr. Abu Dayyeh said. There were no deaths during the study.
The Orbera balloon has been available in more than 80 countries, according to the manufacturer.
More information is available on the FDA website.

Pentoxifylline beat placebo in acute pancreatitis trial
Patients with acute pancreatitis who received pentoxifylline had fewer ICU admissions and shorter ICU and hospital stays than placebo-treated controls, according to a small, randomized double-blind trial reported in Gastroenterology.
“We showed that a single-institution drug trial for acute pancreatitis is feasible and that pentoxifylline is safe, cheap, and might have efficacy,” wrote Dr. Santhi Vege and his associates at the Mayo Clinic in Rochester, Minn. “This sets the stage for a larger trial of this drug in all patients with acute pancreatitis, to realize the goal of finding an effective drug that can be given within 24 hours of diagnosis in any setting.”
Tumor necrosis factor–alpha is a key culprit in severe acute pancreatitis, including pancreatic and peripancreatic necrosis, systemic inflammatory response syndrome, and persistent organ failure, the researchers noted. Pentoxifylline is a nonselective phosphodiesterase inhibitor that has been found safe and effective in other TNF-alpha–mediated diseases such as acute alcoholic hepatitis, but few studies in humans have evaluated the drug for acute pancreatitis, they said (Gastroenterology 2015 June 22 [doi:10.1053/j.gastro.2015.04.019]). For their study, the investigators randomized 28 patients with predicted severe acute pancreatitis to either placebo or 400 mg pentoxifylline given orally at enrollment and then three times a day for 72 hours. Both groups also received standard of care treatments such as antibiotics and fluid therapy, and had comparable baseline characteristics including age, sex, body mass index, Acute Physiology and Chronic Health Evaluation scores, systemic inflammatory response syndrome scores, and inflammatory marker levels, the researchers said.
Significantly fewer patients who received pentoxifylline needed to stay in the hospital for more than 4 days (14% vs. 57% for the placebo group; P = .046), and the maximum length of ICU stay was 0 days for the intervention group, compared with 13 days for the control group (P = .03), the investigators reported. Analyses of several other outcome measures also favored pentoxifylline over placebo, but did not reach statistical significance in the small study, including the need for ICU transfer (0% for pentoxifylline patients vs. 28% of the placebo group; P = .098) and the median length of hospitalization (for pentoxifylline: 3 days, range 1-5 days; for placebo: 5 days; range 1-30 days; P = .06).
The treatment and control groups did not significantly differ in terms of levels of inflammatory markers, including circulating TNF-alpha, said the investigators. Differences in levels of TNF-alpha, interleukin-6, IL-8, and C-reactive protein “may be significant if the sample size is larger,” they added.
The exact mechanism by which pentoxifylline affects acute pancreatitis is unclear, but the production of pancreatic TNF-alpha peaks about 24-36 hours into an episode of the disease, so patients might benefit from receiving pentoxifylline sooner than the 72-hour window dictated by the study protocol, said Dr. Vege and his associates. “Initiating drug therapy within a few hours is challenging, although a 24-hour cutoff time may be feasible in appropriate settings,” they wrote.
A scholarly opportunity award from the Mayo Clinic helped fund the work. The investigators reported having no relevant financial conflicts of interest.
The study of acute pancreatitis (AP) is economically and scientifically essential because acute pancreatitis is the most common reason for hospitalization among patients with GI diseases, consumes considerable resources, and is treated primarily with supportive measures. The pilot study by Dr. Vege and his colleagues reports that pentoxifylline treatment is safe for patients with severe acute pancreatitis and is associated with a promising reduction in ICU utilization and duration in patients requiring a hospital stay >4 days.

|
| Dr. Matthew J. DiMagno |
This study is not only provocative but also raises the hypothesis-generating question of how pentoxifylline might exert a salutary effect without reducing blood tumor necrosis factor–alpha levels (or IL-6, IL-8, or C-reactive protein levels). The authors ascribe this discordance to the timing of administering pentoxifylline and to potential TNF-alpha independent effects. Biologically, pancreatic TNF-alpha levels increase within the first 30-60 minutes of onset of acute pancreatitis (Am. J. Surg. 1998;175:76-83). In experimental AP, pentoxifylline ameliorates severity, but data are conflicting about whether prophylactic or delayed (Surgery 1996;120:515-21) antagonism of TNF-alpha signaling is more protective. Clinically relevant data suggest that prophylactic administration of pentoxifylline does not prevent postendoscopic retrograde cholangiopancreatography pancreatitis (Gastrointest. Endosc. 2007;66:513-8), but nonprophylactic administration of pentoxifylline improves short-term survival in alcoholic hepatitis without significantly reducing blood TNF-alpha levels (Gastroenterology 2000;119:1637-48). Hence, pentoxifylline appears to ameliorate AP and alcoholic hepatitis through TNF-alpha independent signaling, conceivably by targeting the microcirculation, as described for patients with claudication (Angiology 1994;45:339-45).
Future studies might test this hypothesis by determining whether pentoxifylline blunts increases in deleterious vascular factors (for example, angiopoietin-2) [Am. J. Gastroenterol. 2010;105:2287-92; J. Am. Coll. Surg. 2014;218:26-32; Am. J. Gastroenterol. 2011;106:1859-61]) and reduces vascular complications that correlate with the need for ICU care and more severe AP.
Dr. Matthew J. DiMagno is in the division of gastroenterology and hepatology, department of internal medicine, University of Michigan, Ann Arbor. He serves as chair of the American Gastroenterological Association Institute Council Section on Pancreatic Disorders. He declared no relevant financial conflicts of interest.
The study of acute pancreatitis (AP) is economically and scientifically essential because acute pancreatitis is the most common reason for hospitalization among patients with GI diseases, consumes considerable resources, and is treated primarily with supportive measures. The pilot study by Dr. Vege and his colleagues reports that pentoxifylline treatment is safe for patients with severe acute pancreatitis and is associated with a promising reduction in ICU utilization and duration in patients requiring a hospital stay >4 days.
|
|
| Dr. Matthew J. DiMagno |
This study is not only provocative but also raises the hypothesis-generating question of how pentoxifylline might exert a salutary effect without reducing blood tumor necrosis factor–alpha levels (or IL-6, IL-8, or C-reactive protein levels). The authors ascribe this discordance to the timing of administering pentoxifylline and to potential TNF-alpha independent effects. Biologically, pancreatic TNF-alpha levels increase within the first 30-60 minutes of onset of acute pancreatitis (Am. J. Surg. 1998;175:76-83). In experimental AP, pentoxifylline ameliorates severity, but data are conflicting about whether prophylactic or delayed (Surgery 1996;120:515-21) antagonism of TNF-alpha signaling is more protective. Clinically relevant data suggest that prophylactic administration of pentoxifylline does not prevent postendoscopic retrograde cholangiopancreatography pancreatitis (Gastrointest. Endosc. 2007;66:513-8), but nonprophylactic administration of pentoxifylline improves short-term survival in alcoholic hepatitis without significantly reducing blood TNF-alpha levels (Gastroenterology 2000;119:1637-48). Hence, pentoxifylline appears to ameliorate AP and alcoholic hepatitis through TNF-alpha independent signaling, conceivably by targeting the microcirculation, as described for patients with claudication (Angiology 1994;45:339-45).
Future studies might test this hypothesis by determining whether pentoxifylline blunts increases in deleterious vascular factors (for example, angiopoietin-2) [Am. J. Gastroenterol. 2010;105:2287-92; J. Am. Coll. Surg. 2014;218:26-32; Am. J. Gastroenterol. 2011;106:1859-61]) and reduces vascular complications that correlate with the need for ICU care and more severe AP.
Dr. Matthew J. DiMagno is in the division of gastroenterology and hepatology, department of internal medicine, University of Michigan, Ann Arbor. He serves as chair of the American Gastroenterological Association Institute Council Section on Pancreatic Disorders. He declared no relevant financial conflicts of interest.
The study of acute pancreatitis (AP) is economically and scientifically essential because acute pancreatitis is the most common reason for hospitalization among patients with GI diseases, consumes considerable resources, and is treated primarily with supportive measures. The pilot study by Dr. Vege and his colleagues reports that pentoxifylline treatment is safe for patients with severe acute pancreatitis and is associated with a promising reduction in ICU utilization and duration in patients requiring a hospital stay >4 days.
|
|
| Dr. Matthew J. DiMagno |
This study is not only provocative but also raises the hypothesis-generating question of how pentoxifylline might exert a salutary effect without reducing blood tumor necrosis factor–alpha levels (or IL-6, IL-8, or C-reactive protein levels). The authors ascribe this discordance to the timing of administering pentoxifylline and to potential TNF-alpha independent effects. Biologically, pancreatic TNF-alpha levels increase within the first 30-60 minutes of onset of acute pancreatitis (Am. J. Surg. 1998;175:76-83). In experimental AP, pentoxifylline ameliorates severity, but data are conflicting about whether prophylactic or delayed (Surgery 1996;120:515-21) antagonism of TNF-alpha signaling is more protective. Clinically relevant data suggest that prophylactic administration of pentoxifylline does not prevent postendoscopic retrograde cholangiopancreatography pancreatitis (Gastrointest. Endosc. 2007;66:513-8), but nonprophylactic administration of pentoxifylline improves short-term survival in alcoholic hepatitis without significantly reducing blood TNF-alpha levels (Gastroenterology 2000;119:1637-48). Hence, pentoxifylline appears to ameliorate AP and alcoholic hepatitis through TNF-alpha independent signaling, conceivably by targeting the microcirculation, as described for patients with claudication (Angiology 1994;45:339-45).
Future studies might test this hypothesis by determining whether pentoxifylline blunts increases in deleterious vascular factors (for example, angiopoietin-2) [Am. J. Gastroenterol. 2010;105:2287-92; J. Am. Coll. Surg. 2014;218:26-32; Am. J. Gastroenterol. 2011;106:1859-61]) and reduces vascular complications that correlate with the need for ICU care and more severe AP.
Dr. Matthew J. DiMagno is in the division of gastroenterology and hepatology, department of internal medicine, University of Michigan, Ann Arbor. He serves as chair of the American Gastroenterological Association Institute Council Section on Pancreatic Disorders. He declared no relevant financial conflicts of interest.
Patients with acute pancreatitis who received pentoxifylline had fewer ICU admissions and shorter ICU and hospital stays than placebo-treated controls, according to a small, randomized double-blind trial reported in Gastroenterology.
“We showed that a single-institution drug trial for acute pancreatitis is feasible and that pentoxifylline is safe, cheap, and might have efficacy,” wrote Dr. Santhi Vege and his associates at the Mayo Clinic in Rochester, Minn. “This sets the stage for a larger trial of this drug in all patients with acute pancreatitis, to realize the goal of finding an effective drug that can be given within 24 hours of diagnosis in any setting.”
Tumor necrosis factor–alpha is a key culprit in severe acute pancreatitis, including pancreatic and peripancreatic necrosis, systemic inflammatory response syndrome, and persistent organ failure, the researchers noted. Pentoxifylline is a nonselective phosphodiesterase inhibitor that has been found safe and effective in other TNF-alpha–mediated diseases such as acute alcoholic hepatitis, but few studies in humans have evaluated the drug for acute pancreatitis, they said (Gastroenterology 2015 June 22 [doi:10.1053/j.gastro.2015.04.019]). For their study, the investigators randomized 28 patients with predicted severe acute pancreatitis to either placebo or 400 mg pentoxifylline given orally at enrollment and then three times a day for 72 hours. Both groups also received standard of care treatments such as antibiotics and fluid therapy, and had comparable baseline characteristics including age, sex, body mass index, Acute Physiology and Chronic Health Evaluation scores, systemic inflammatory response syndrome scores, and inflammatory marker levels, the researchers said.
Significantly fewer patients who received pentoxifylline needed to stay in the hospital for more than 4 days (14% vs. 57% for the placebo group; P = .046), and the maximum length of ICU stay was 0 days for the intervention group, compared with 13 days for the control group (P = .03), the investigators reported. Analyses of several other outcome measures also favored pentoxifylline over placebo, but did not reach statistical significance in the small study, including the need for ICU transfer (0% for pentoxifylline patients vs. 28% of the placebo group; P = .098) and the median length of hospitalization (for pentoxifylline: 3 days, range 1-5 days; for placebo: 5 days; range 1-30 days; P = .06).
The treatment and control groups did not significantly differ in terms of levels of inflammatory markers, including circulating TNF-alpha, said the investigators. Differences in levels of TNF-alpha, interleukin-6, IL-8, and C-reactive protein “may be significant if the sample size is larger,” they added.
The exact mechanism by which pentoxifylline affects acute pancreatitis is unclear, but the production of pancreatic TNF-alpha peaks about 24-36 hours into an episode of the disease, so patients might benefit from receiving pentoxifylline sooner than the 72-hour window dictated by the study protocol, said Dr. Vege and his associates. “Initiating drug therapy within a few hours is challenging, although a 24-hour cutoff time may be feasible in appropriate settings,” they wrote.
A scholarly opportunity award from the Mayo Clinic helped fund the work. The investigators reported having no relevant financial conflicts of interest.
Patients with acute pancreatitis who received pentoxifylline had fewer ICU admissions and shorter ICU and hospital stays than placebo-treated controls, according to a small, randomized double-blind trial reported in Gastroenterology.
“We showed that a single-institution drug trial for acute pancreatitis is feasible and that pentoxifylline is safe, cheap, and might have efficacy,” wrote Dr. Santhi Vege and his associates at the Mayo Clinic in Rochester, Minn. “This sets the stage for a larger trial of this drug in all patients with acute pancreatitis, to realize the goal of finding an effective drug that can be given within 24 hours of diagnosis in any setting.”
Tumor necrosis factor–alpha is a key culprit in severe acute pancreatitis, including pancreatic and peripancreatic necrosis, systemic inflammatory response syndrome, and persistent organ failure, the researchers noted. Pentoxifylline is a nonselective phosphodiesterase inhibitor that has been found safe and effective in other TNF-alpha–mediated diseases such as acute alcoholic hepatitis, but few studies in humans have evaluated the drug for acute pancreatitis, they said (Gastroenterology 2015 June 22 [doi:10.1053/j.gastro.2015.04.019]). For their study, the investigators randomized 28 patients with predicted severe acute pancreatitis to either placebo or 400 mg pentoxifylline given orally at enrollment and then three times a day for 72 hours. Both groups also received standard of care treatments such as antibiotics and fluid therapy, and had comparable baseline characteristics including age, sex, body mass index, Acute Physiology and Chronic Health Evaluation scores, systemic inflammatory response syndrome scores, and inflammatory marker levels, the researchers said.
Significantly fewer patients who received pentoxifylline needed to stay in the hospital for more than 4 days (14% vs. 57% for the placebo group; P = .046), and the maximum length of ICU stay was 0 days for the intervention group, compared with 13 days for the control group (P = .03), the investigators reported. Analyses of several other outcome measures also favored pentoxifylline over placebo, but did not reach statistical significance in the small study, including the need for ICU transfer (0% for pentoxifylline patients vs. 28% of the placebo group; P = .098) and the median length of hospitalization (for pentoxifylline: 3 days, range 1-5 days; for placebo: 5 days; range 1-30 days; P = .06).
The treatment and control groups did not significantly differ in terms of levels of inflammatory markers, including circulating TNF-alpha, said the investigators. Differences in levels of TNF-alpha, interleukin-6, IL-8, and C-reactive protein “may be significant if the sample size is larger,” they added.
The exact mechanism by which pentoxifylline affects acute pancreatitis is unclear, but the production of pancreatic TNF-alpha peaks about 24-36 hours into an episode of the disease, so patients might benefit from receiving pentoxifylline sooner than the 72-hour window dictated by the study protocol, said Dr. Vege and his associates. “Initiating drug therapy within a few hours is challenging, although a 24-hour cutoff time may be feasible in appropriate settings,” they wrote.
A scholarly opportunity award from the Mayo Clinic helped fund the work. The investigators reported having no relevant financial conflicts of interest.
FROM GASTROENTEROLOGY
Key clinical point: Pentoxifylline topped placebo for several outcome measures among patients with severe acute pancreatitis.
Major finding: Significantly fewer patients who received pentoxifylline needed to stay in the hospital for more than 4 days (14% vs. 57% for the placebo group; P = .046).
Data source: A single-center, randomized placebo-controlled trial of 28 patients with predicted severe acute pancreatitis.
Disclosures: A scholarly opportunity award from the Mayo Clinic supported the work. The investigators reported having no relevant financial conflicts of interest.

Lifestyle changes, surgical weight loss benefit NAFLD
Losing weight through lifestyle changes can significantly improve several measures of nonalcoholic fatty liver disease, particularly if patients lose at least 10% of their body weight, and bariatric surgery is a valid alternative if diet and exercise fail, according to two studies reported in the August issue of Gastroenterology (2015 Apr. 9 [doi:10.1053/j.gastro.2015.04.005]).
Global rates of nonalcoholic fatty liver disease (NAFLD) are up because of the “parallel epidemics” of obesity and type 2 diabetes mellitus, said Dr. Eduardo Vilar-Gomez of the National Institute of Hepatology in Havana, Cuba, who authored the study of lifestyle changes. There are no approved therapies for the more aggressive form of NAFLD, steatohepatitis, he and his associates said. In past studies, patients who lost about 7%-10% of their body weight substantially improved their NAFLD activity score and had reductions in steatosis, lobular inflammation, and ballooning, but the results did not extend to fibrosis, and prospective studies of the effect of lifestyle changes on histology are lacking, the researchers added .
To fill the gap, they followed 293 adults with histologically confirmed nonalcoholic steatohepatitis who completed a 52-week lifestyle intervention program that included keeping a food diary, restricting saturated fats to less than 10% of total intake, and walking at least 200 minutes a week. Patients had not received hypolipidemic treatment in the preceding 3 months and were not allowed to take insulin sensitizers or vitamin E, both of which are potentially beneficial for nonalcoholic steatohepatitis, the investigators said.
At the end of the yearlong program, steatohepatitis had resolved in 25% of patients, 47% had lower NAFLD activity scores, and 19% had regression of fibrosis, the researchers reported. Although only 30% of participants lost at least 5% of their body weight, weight loss correlated positively with resolution of steatohepatitis and with 2-point reductions in histologic activity scores (P <.001). Among patients who lost at least 10% of their body weight, 90% had resolution of steatohepatitis, 45% had regression of fibrosis, and all had improved histologic activity scores, even if they had negative risk factors such as female sex, a baseline body mass index of at least 35 kg/m2, and a baseline fasting glucose level of at least 5.5 mmol/L, the investigators added. “Our findings support the current recommendation for weight loss using lifestyle modification as the first step in the management of patients with nonalcoholic steatohepatitis,” they wrote.
But what if lifestyle changes fail? The impact of bariatric surgery on nonalcoholic steatohepatitis has not been well studied, said Dr. Guillaume Lassailly at CHRU Lille (France). He and his associates therefore followed 109 morbidly obese patients (BMI ≥40 kg/m2) with biopsy-confirmed nonalcoholic steatohepatitis who underwent bariatric surgery at a single tertiary hospital (Gastroenterology 2015 Apr. 25 [doi:10.1053/j.gastro.2015.04.014]) .
One year after surgery, 85% of patients had achieved disease resolution (95% confidence interval, 75.8%-92.2%), the investigators reported. Stratifying patients by baseline Brunt scores showed that those with milder presurgical disease were more likely to have complete resolution than were patients whose disease was severe (94% vs. 70%; P <.05), the researchers added. Histologic analyses supported the findings, revealing steatosis in 60% of presurgical tissue samples, compared with 10% of samples taken a year after surgery. In addition, average NAFLD disease scores dropped from 5 to 1 (P <.001), hepatocyte ballooning decreased in 84% of samples, lobular inflammation decreased in 67%, and Metavir fibrosis scores dropped in one-third of specimens.
Notably, BMI scores for patients with persistent postsurgical disease dropped by an average of only 9.1, compared with 12.3 for patients whose disease resolved (P = .005), said the researchers. Gastric bypass surgery achieved greater weight loss and improvements in disease status, compared with laparoscopic banding, they added. “The encouraging results of the present study suggest that bariatric surgery should be tested in multicenter, randomized controlled trials in morbidly or severely obese patients with nonalcoholic steatohepatitis who did not respond to lifestyle therapy,” they wrote.
The Cuban National Institute of Gastroenterology and Ministry of Health partially funded the study by Dr. Vilar-Gomez and his associates. The French Ministry of Health and the Conseil Regional Nord-Pas de Calais supported the work by Dr. Lassailly and his colleagues. All investigators declared having no relevant financial conflicts of interest.
Losing weight through lifestyle changes can significantly improve several measures of nonalcoholic fatty liver disease, particularly if patients lose at least 10% of their body weight, and bariatric surgery is a valid alternative if diet and exercise fail, according to two studies reported in the August issue of Gastroenterology (2015 Apr. 9 [doi:10.1053/j.gastro.2015.04.005]).
Global rates of nonalcoholic fatty liver disease (NAFLD) are up because of the “parallel epidemics” of obesity and type 2 diabetes mellitus, said Dr. Eduardo Vilar-Gomez of the National Institute of Hepatology in Havana, Cuba, who authored the study of lifestyle changes. There are no approved therapies for the more aggressive form of NAFLD, steatohepatitis, he and his associates said. In past studies, patients who lost about 7%-10% of their body weight substantially improved their NAFLD activity score and had reductions in steatosis, lobular inflammation, and ballooning, but the results did not extend to fibrosis, and prospective studies of the effect of lifestyle changes on histology are lacking, the researchers added .
To fill the gap, they followed 293 adults with histologically confirmed nonalcoholic steatohepatitis who completed a 52-week lifestyle intervention program that included keeping a food diary, restricting saturated fats to less than 10% of total intake, and walking at least 200 minutes a week. Patients had not received hypolipidemic treatment in the preceding 3 months and were not allowed to take insulin sensitizers or vitamin E, both of which are potentially beneficial for nonalcoholic steatohepatitis, the investigators said.
At the end of the yearlong program, steatohepatitis had resolved in 25% of patients, 47% had lower NAFLD activity scores, and 19% had regression of fibrosis, the researchers reported. Although only 30% of participants lost at least 5% of their body weight, weight loss correlated positively with resolution of steatohepatitis and with 2-point reductions in histologic activity scores (P <.001). Among patients who lost at least 10% of their body weight, 90% had resolution of steatohepatitis, 45% had regression of fibrosis, and all had improved histologic activity scores, even if they had negative risk factors such as female sex, a baseline body mass index of at least 35 kg/m2, and a baseline fasting glucose level of at least 5.5 mmol/L, the investigators added. “Our findings support the current recommendation for weight loss using lifestyle modification as the first step in the management of patients with nonalcoholic steatohepatitis,” they wrote.
But what if lifestyle changes fail? The impact of bariatric surgery on nonalcoholic steatohepatitis has not been well studied, said Dr. Guillaume Lassailly at CHRU Lille (France). He and his associates therefore followed 109 morbidly obese patients (BMI ≥40 kg/m2) with biopsy-confirmed nonalcoholic steatohepatitis who underwent bariatric surgery at a single tertiary hospital (Gastroenterology 2015 Apr. 25 [doi:10.1053/j.gastro.2015.04.014]) .
One year after surgery, 85% of patients had achieved disease resolution (95% confidence interval, 75.8%-92.2%), the investigators reported. Stratifying patients by baseline Brunt scores showed that those with milder presurgical disease were more likely to have complete resolution than were patients whose disease was severe (94% vs. 70%; P <.05), the researchers added. Histologic analyses supported the findings, revealing steatosis in 60% of presurgical tissue samples, compared with 10% of samples taken a year after surgery. In addition, average NAFLD disease scores dropped from 5 to 1 (P <.001), hepatocyte ballooning decreased in 84% of samples, lobular inflammation decreased in 67%, and Metavir fibrosis scores dropped in one-third of specimens.
Notably, BMI scores for patients with persistent postsurgical disease dropped by an average of only 9.1, compared with 12.3 for patients whose disease resolved (P = .005), said the researchers. Gastric bypass surgery achieved greater weight loss and improvements in disease status, compared with laparoscopic banding, they added. “The encouraging results of the present study suggest that bariatric surgery should be tested in multicenter, randomized controlled trials in morbidly or severely obese patients with nonalcoholic steatohepatitis who did not respond to lifestyle therapy,” they wrote.
The Cuban National Institute of Gastroenterology and Ministry of Health partially funded the study by Dr. Vilar-Gomez and his associates. The French Ministry of Health and the Conseil Regional Nord-Pas de Calais supported the work by Dr. Lassailly and his colleagues. All investigators declared having no relevant financial conflicts of interest.
Losing weight through lifestyle changes can significantly improve several measures of nonalcoholic fatty liver disease, particularly if patients lose at least 10% of their body weight, and bariatric surgery is a valid alternative if diet and exercise fail, according to two studies reported in the August issue of Gastroenterology (2015 Apr. 9 [doi:10.1053/j.gastro.2015.04.005]).
Global rates of nonalcoholic fatty liver disease (NAFLD) are up because of the “parallel epidemics” of obesity and type 2 diabetes mellitus, said Dr. Eduardo Vilar-Gomez of the National Institute of Hepatology in Havana, Cuba, who authored the study of lifestyle changes. There are no approved therapies for the more aggressive form of NAFLD, steatohepatitis, he and his associates said. In past studies, patients who lost about 7%-10% of their body weight substantially improved their NAFLD activity score and had reductions in steatosis, lobular inflammation, and ballooning, but the results did not extend to fibrosis, and prospective studies of the effect of lifestyle changes on histology are lacking, the researchers added .
To fill the gap, they followed 293 adults with histologically confirmed nonalcoholic steatohepatitis who completed a 52-week lifestyle intervention program that included keeping a food diary, restricting saturated fats to less than 10% of total intake, and walking at least 200 minutes a week. Patients had not received hypolipidemic treatment in the preceding 3 months and were not allowed to take insulin sensitizers or vitamin E, both of which are potentially beneficial for nonalcoholic steatohepatitis, the investigators said.
At the end of the yearlong program, steatohepatitis had resolved in 25% of patients, 47% had lower NAFLD activity scores, and 19% had regression of fibrosis, the researchers reported. Although only 30% of participants lost at least 5% of their body weight, weight loss correlated positively with resolution of steatohepatitis and with 2-point reductions in histologic activity scores (P <.001). Among patients who lost at least 10% of their body weight, 90% had resolution of steatohepatitis, 45% had regression of fibrosis, and all had improved histologic activity scores, even if they had negative risk factors such as female sex, a baseline body mass index of at least 35 kg/m2, and a baseline fasting glucose level of at least 5.5 mmol/L, the investigators added. “Our findings support the current recommendation for weight loss using lifestyle modification as the first step in the management of patients with nonalcoholic steatohepatitis,” they wrote.
But what if lifestyle changes fail? The impact of bariatric surgery on nonalcoholic steatohepatitis has not been well studied, said Dr. Guillaume Lassailly at CHRU Lille (France). He and his associates therefore followed 109 morbidly obese patients (BMI ≥40 kg/m2) with biopsy-confirmed nonalcoholic steatohepatitis who underwent bariatric surgery at a single tertiary hospital (Gastroenterology 2015 Apr. 25 [doi:10.1053/j.gastro.2015.04.014]) .
One year after surgery, 85% of patients had achieved disease resolution (95% confidence interval, 75.8%-92.2%), the investigators reported. Stratifying patients by baseline Brunt scores showed that those with milder presurgical disease were more likely to have complete resolution than were patients whose disease was severe (94% vs. 70%; P <.05), the researchers added. Histologic analyses supported the findings, revealing steatosis in 60% of presurgical tissue samples, compared with 10% of samples taken a year after surgery. In addition, average NAFLD disease scores dropped from 5 to 1 (P <.001), hepatocyte ballooning decreased in 84% of samples, lobular inflammation decreased in 67%, and Metavir fibrosis scores dropped in one-third of specimens.
Notably, BMI scores for patients with persistent postsurgical disease dropped by an average of only 9.1, compared with 12.3 for patients whose disease resolved (P = .005), said the researchers. Gastric bypass surgery achieved greater weight loss and improvements in disease status, compared with laparoscopic banding, they added. “The encouraging results of the present study suggest that bariatric surgery should be tested in multicenter, randomized controlled trials in morbidly or severely obese patients with nonalcoholic steatohepatitis who did not respond to lifestyle therapy,” they wrote.
The Cuban National Institute of Gastroenterology and Ministry of Health partially funded the study by Dr. Vilar-Gomez and his associates. The French Ministry of Health and the Conseil Regional Nord-Pas de Calais supported the work by Dr. Lassailly and his colleagues. All investigators declared having no relevant financial conflicts of interest.
FROM GASTROENTEROLOGY
Key clinical point: Losing weight through lifestyle changes or bariatric surgery can significantly improve several measures of nonalcoholic steatohepatitis.
Major finding: A yearlong diet and exercise program led to resolution of nonalcoholic steatohepatitis in 25% of patients, while bariatric surgery achieved that outcome for 85% of patients in a separate study.
Data source: Two prospective uncontrolled cohort studies of 402 total adults with nonalcoholic steatohepatitis (the more severe form of nonalcoholic fatty liver disease).
Disclosures: The Cuban National Institute of Gastroenterology and Ministry of Health partially funded the study by Dr. Vilar-Gomez and his associates. The French Ministry of Health and the Conseil Regional Nord-Pas de Calais supported the work by Dr. Lassailly and his colleagues. All investigators declared having no relevant financial conflicts of interest.
Guideline adherence reduces biliary pancreatitis
WASHINGTON – Performed when recommended, cholecystecomy significantly decreases the risk of near-term rehospitalization for acute biliary pancreatitis, results of a retrospective study indicate.
Among more than 23,000 patients with mild to moderate acute biliary pancreatitis, less than 2% of those who underwent cholecystectomy within 30 days, as recommended under American Gastroenterological Association (AGA) guidelines, were rehospitalized for pancreatitis within 6 months.
In contrast, nearly 17% of patients who had cholecystectomy after 1 month or never had it were back in the hospital within half a year, said Dr. Ayesha Kamal, a postdoctoral research fellow at the Johns Hopkins Hospital in Baltimore.
“Cholecystectomy prevents future hospitalization for biliary pancreatitis,” she said at the annual Digestive Disease Week.
The study, based on claims data, also showed that adherence to AGA guidelines is fairly high, on the order of 75%, she said.
Acute pancreatitis is one of the most common gastrointestinal diseases in the United States, accounting for about 300,000 hospitalizations in 2009, at a total cost of about $2.6 billion.
Gallstone disease is the most common cause of acute pancreatitis, responsible for an estimated 40% of all cases, she said.
Guidelines from the AGA and other organizations recommend cholecystectomy either during the same hospitalization for acute biliary pancreatitis, or within 4 weeks.
To see whether clinicians were adhering to the AGA guidelines and whether the guideline-recommended timing of cholecystectomy made a difference, Dr. Kamal and colleagues analyzed data from the MarketScan Commercial Claims & Encounters database, which includes individual-level clinical utilization data for both inpatient and outpatient visits paid for by employer-sponsored health plans.
They looked at data both on patients who were treated in accordance with guidelines (first hospitalization for mild to moderate acute biliary pancreatitis, with cholecystectomy performed either on the day of hospitalization or within 30 days), and outside of the guidelines (no cholecystectomy, or cholecystectomy performed later than 30 days after the index hospitalization).
They assessed recurrences within 30 days of follow-up by International Classification of Diseases, 9th Revision (ICD-9) codes for acute pancreatitis and gallstone disease.
Combing through 8.8 million adult inpatient encounters for acute biliary pancreatitis, they excluded those patients with a diagnosis of severe or chronic pancreatitis, alcohol abuse, less than 30 days of follow-up, deaths during hospitalization, discharge to hospice, and those with a length of stay longer than 30 days.
This left them with a final cohort of 23,515 patients with mild to moderate acute biliary pancreatitis.
They found that 61% of patients had cholecystectomy during their initial hospitalizations, and an additional 14% had the surgery during a subsequent hospitalization within 30 days. Of the remaining patients, 7% had cholecystectomies after 30 days, and 18% never had one.
Among patients treated under the guidelines, 1.3% who had their gallbladders removed during the initial hospitalization had a pancreatitis recurrence within 6 months, and 0.2% had a recurrence more than 6 months later.
In contrast, 36.7% of patients who had a cholecystectomy more than a month after their first hospitalization for pancreatitis had a recurrence within 6 months, and 4.5% had a recurrence after 6 months.
Among patients who never underwent cholecystectomy, the respective recurrence rates were 5.4% and 1.1%.
“One in six patients who did not receive a cholecystectomy within 30 days will be hospitalized again within 6 months,” Dr. Kamal said.
She acknowledged that the study was limited by the authors’ inability to confirm acute biliary pancreatitis with chart review, and by the limitations of the database, which is confined to adults younger than 65 with employer-sponsored medical plans.
In the question and answer portion of the presentation, Dr. Nirav Thosani, a gastroenterologist at Memorial Hermann Hospital in Houston, noted that ICD-9 codes do not distinguish between different types of pancreatitis.
“It might be possible that those patients who never had cholecystectomy never had acute biliary pancreatitis, or some other reason for acute pancreatitis, and that’s the reason for the rehospitalization,” he said.
Dr. Kamal replied that they tried to control for other causes of pancreatitis by including ICD-9 codes for gallstone disease and by excluding patients with diagnoses of alcohol abuse.
WASHINGTON – Performed when recommended, cholecystecomy significantly decreases the risk of near-term rehospitalization for acute biliary pancreatitis, results of a retrospective study indicate.
Among more than 23,000 patients with mild to moderate acute biliary pancreatitis, less than 2% of those who underwent cholecystectomy within 30 days, as recommended under American Gastroenterological Association (AGA) guidelines, were rehospitalized for pancreatitis within 6 months.
In contrast, nearly 17% of patients who had cholecystectomy after 1 month or never had it were back in the hospital within half a year, said Dr. Ayesha Kamal, a postdoctoral research fellow at the Johns Hopkins Hospital in Baltimore.
“Cholecystectomy prevents future hospitalization for biliary pancreatitis,” she said at the annual Digestive Disease Week.
The study, based on claims data, also showed that adherence to AGA guidelines is fairly high, on the order of 75%, she said.
Acute pancreatitis is one of the most common gastrointestinal diseases in the United States, accounting for about 300,000 hospitalizations in 2009, at a total cost of about $2.6 billion.
Gallstone disease is the most common cause of acute pancreatitis, responsible for an estimated 40% of all cases, she said.
Guidelines from the AGA and other organizations recommend cholecystectomy either during the same hospitalization for acute biliary pancreatitis, or within 4 weeks.
To see whether clinicians were adhering to the AGA guidelines and whether the guideline-recommended timing of cholecystectomy made a difference, Dr. Kamal and colleagues analyzed data from the MarketScan Commercial Claims & Encounters database, which includes individual-level clinical utilization data for both inpatient and outpatient visits paid for by employer-sponsored health plans.
They looked at data both on patients who were treated in accordance with guidelines (first hospitalization for mild to moderate acute biliary pancreatitis, with cholecystectomy performed either on the day of hospitalization or within 30 days), and outside of the guidelines (no cholecystectomy, or cholecystectomy performed later than 30 days after the index hospitalization).
They assessed recurrences within 30 days of follow-up by International Classification of Diseases, 9th Revision (ICD-9) codes for acute pancreatitis and gallstone disease.
Combing through 8.8 million adult inpatient encounters for acute biliary pancreatitis, they excluded those patients with a diagnosis of severe or chronic pancreatitis, alcohol abuse, less than 30 days of follow-up, deaths during hospitalization, discharge to hospice, and those with a length of stay longer than 30 days.
This left them with a final cohort of 23,515 patients with mild to moderate acute biliary pancreatitis.
They found that 61% of patients had cholecystectomy during their initial hospitalizations, and an additional 14% had the surgery during a subsequent hospitalization within 30 days. Of the remaining patients, 7% had cholecystectomies after 30 days, and 18% never had one.
Among patients treated under the guidelines, 1.3% who had their gallbladders removed during the initial hospitalization had a pancreatitis recurrence within 6 months, and 0.2% had a recurrence more than 6 months later.
In contrast, 36.7% of patients who had a cholecystectomy more than a month after their first hospitalization for pancreatitis had a recurrence within 6 months, and 4.5% had a recurrence after 6 months.
Among patients who never underwent cholecystectomy, the respective recurrence rates were 5.4% and 1.1%.
“One in six patients who did not receive a cholecystectomy within 30 days will be hospitalized again within 6 months,” Dr. Kamal said.
She acknowledged that the study was limited by the authors’ inability to confirm acute biliary pancreatitis with chart review, and by the limitations of the database, which is confined to adults younger than 65 with employer-sponsored medical plans.
In the question and answer portion of the presentation, Dr. Nirav Thosani, a gastroenterologist at Memorial Hermann Hospital in Houston, noted that ICD-9 codes do not distinguish between different types of pancreatitis.
“It might be possible that those patients who never had cholecystectomy never had acute biliary pancreatitis, or some other reason for acute pancreatitis, and that’s the reason for the rehospitalization,” he said.
Dr. Kamal replied that they tried to control for other causes of pancreatitis by including ICD-9 codes for gallstone disease and by excluding patients with diagnoses of alcohol abuse.
WASHINGTON – Performed when recommended, cholecystecomy significantly decreases the risk of near-term rehospitalization for acute biliary pancreatitis, results of a retrospective study indicate.
Among more than 23,000 patients with mild to moderate acute biliary pancreatitis, less than 2% of those who underwent cholecystectomy within 30 days, as recommended under American Gastroenterological Association (AGA) guidelines, were rehospitalized for pancreatitis within 6 months.
In contrast, nearly 17% of patients who had cholecystectomy after 1 month or never had it were back in the hospital within half a year, said Dr. Ayesha Kamal, a postdoctoral research fellow at the Johns Hopkins Hospital in Baltimore.
“Cholecystectomy prevents future hospitalization for biliary pancreatitis,” she said at the annual Digestive Disease Week.
The study, based on claims data, also showed that adherence to AGA guidelines is fairly high, on the order of 75%, she said.
Acute pancreatitis is one of the most common gastrointestinal diseases in the United States, accounting for about 300,000 hospitalizations in 2009, at a total cost of about $2.6 billion.
Gallstone disease is the most common cause of acute pancreatitis, responsible for an estimated 40% of all cases, she said.
Guidelines from the AGA and other organizations recommend cholecystectomy either during the same hospitalization for acute biliary pancreatitis, or within 4 weeks.
To see whether clinicians were adhering to the AGA guidelines and whether the guideline-recommended timing of cholecystectomy made a difference, Dr. Kamal and colleagues analyzed data from the MarketScan Commercial Claims & Encounters database, which includes individual-level clinical utilization data for both inpatient and outpatient visits paid for by employer-sponsored health plans.
They looked at data both on patients who were treated in accordance with guidelines (first hospitalization for mild to moderate acute biliary pancreatitis, with cholecystectomy performed either on the day of hospitalization or within 30 days), and outside of the guidelines (no cholecystectomy, or cholecystectomy performed later than 30 days after the index hospitalization).
They assessed recurrences within 30 days of follow-up by International Classification of Diseases, 9th Revision (ICD-9) codes for acute pancreatitis and gallstone disease.
Combing through 8.8 million adult inpatient encounters for acute biliary pancreatitis, they excluded those patients with a diagnosis of severe or chronic pancreatitis, alcohol abuse, less than 30 days of follow-up, deaths during hospitalization, discharge to hospice, and those with a length of stay longer than 30 days.
This left them with a final cohort of 23,515 patients with mild to moderate acute biliary pancreatitis.
They found that 61% of patients had cholecystectomy during their initial hospitalizations, and an additional 14% had the surgery during a subsequent hospitalization within 30 days. Of the remaining patients, 7% had cholecystectomies after 30 days, and 18% never had one.
Among patients treated under the guidelines, 1.3% who had their gallbladders removed during the initial hospitalization had a pancreatitis recurrence within 6 months, and 0.2% had a recurrence more than 6 months later.
In contrast, 36.7% of patients who had a cholecystectomy more than a month after their first hospitalization for pancreatitis had a recurrence within 6 months, and 4.5% had a recurrence after 6 months.
Among patients who never underwent cholecystectomy, the respective recurrence rates were 5.4% and 1.1%.
“One in six patients who did not receive a cholecystectomy within 30 days will be hospitalized again within 6 months,” Dr. Kamal said.
She acknowledged that the study was limited by the authors’ inability to confirm acute biliary pancreatitis with chart review, and by the limitations of the database, which is confined to adults younger than 65 with employer-sponsored medical plans.
In the question and answer portion of the presentation, Dr. Nirav Thosani, a gastroenterologist at Memorial Hermann Hospital in Houston, noted that ICD-9 codes do not distinguish between different types of pancreatitis.
“It might be possible that those patients who never had cholecystectomy never had acute biliary pancreatitis, or some other reason for acute pancreatitis, and that’s the reason for the rehospitalization,” he said.
Dr. Kamal replied that they tried to control for other causes of pancreatitis by including ICD-9 codes for gallstone disease and by excluding patients with diagnoses of alcohol abuse.
AT DDW® 2015
Key clinical point: Cholecystectomy within 30 days of acute biliary pancreatitis protects against recurrence.
Major finding: Among patients treated under AGA guidelines, only 1.3% who had their gallbladders removed during the initial hospitalization had a pancreatitis recurrence within 6 months, and 0.2% had a recurrence more than 6 months later.
Data source: Retrospective cohort study of 23,515 patients with acute biliary pancreatitis in claims database.
Disclosures: The study funding source was not disclosed. Dr. Kamal and Dr. Thosani reported having no relevant disclosures.
Weight in America: Abnormal is the new normal
Three-quarters of men and two-thirds of women aged 25 years or older in the United States are either overweight or obese, according to a study published online June 22 in JAMA Internal Medicine.
During 2007-2012, about 40% of men were overweight and 35% were obese. For women, almost 30% were overweight and 37% were obese, Lin Yang, Ph.D., and Dr. Graham A. Colditz of Washington University, St. Louis, reported in a research letter (JAMA Intern. Med. 2015 June 22 [doi:10.1001/jamainternmed.2015.2405]).
Weight-status distributions across racial groups show that non-Hispanic whites were generally less obese than non-Hispanic blacks and Mexican Americans – an effect that was more pronounced among women. About 34% of white women were obese, compared with 57% of black women, with over 17% occupying the heaviest of three obesity levels, according to the investigators’ analysis of the most recent data from the National Health and Nutrition Examination Survey.
Weight groups in the study were defined by body mass index: underweight (< 18.5 kg/m2), normal weight (18.5-24.9), overweight (25.0-29.9), obesity class 1 (30.0-34.9), obesity class 2 (35.0-39.9), and obesity class 3 (≥ 40).
Compared with an earlier study reporting data for 1988-1994, “the greatest increase in the proportion of patients in the obesity class 3 category was among non-Hispanic black women,” Dr. Yang and Dr. Colditz wrote. Health and policy decision-makers should consider “population-based strategies helping to reduce modifiable risk factors such as physical environment interventions, enhancing primary care efforts to prevent and treat obesity, and altering societal norms of behavior,” they added.
The study was funded by the Washington University Transdiscliplinary Research on Energetics and Cancer Center, the Foundation for Barnes-Jewish Hospital, and the Breast Cancer Research Foundation. The investigators did not report any conflicts.
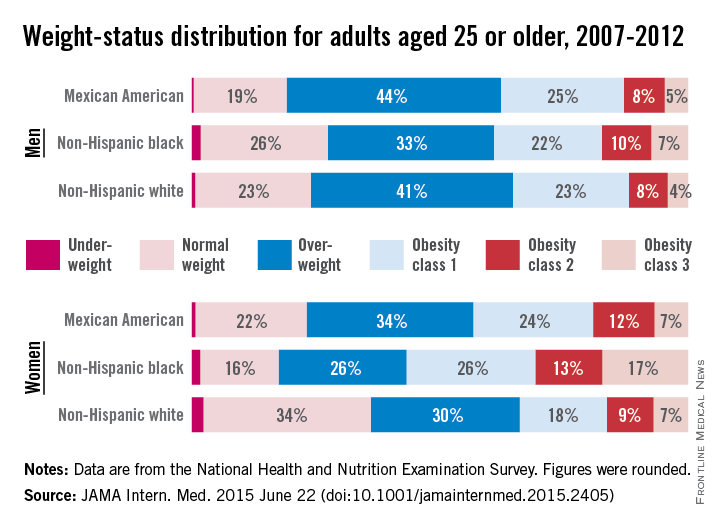
Three-quarters of men and two-thirds of women aged 25 years or older in the United States are either overweight or obese, according to a study published online June 22 in JAMA Internal Medicine.
During 2007-2012, about 40% of men were overweight and 35% were obese. For women, almost 30% were overweight and 37% were obese, Lin Yang, Ph.D., and Dr. Graham A. Colditz of Washington University, St. Louis, reported in a research letter (JAMA Intern. Med. 2015 June 22 [doi:10.1001/jamainternmed.2015.2405]).
Weight-status distributions across racial groups show that non-Hispanic whites were generally less obese than non-Hispanic blacks and Mexican Americans – an effect that was more pronounced among women. About 34% of white women were obese, compared with 57% of black women, with over 17% occupying the heaviest of three obesity levels, according to the investigators’ analysis of the most recent data from the National Health and Nutrition Examination Survey.
Weight groups in the study were defined by body mass index: underweight (< 18.5 kg/m2), normal weight (18.5-24.9), overweight (25.0-29.9), obesity class 1 (30.0-34.9), obesity class 2 (35.0-39.9), and obesity class 3 (≥ 40).
Compared with an earlier study reporting data for 1988-1994, “the greatest increase in the proportion of patients in the obesity class 3 category was among non-Hispanic black women,” Dr. Yang and Dr. Colditz wrote. Health and policy decision-makers should consider “population-based strategies helping to reduce modifiable risk factors such as physical environment interventions, enhancing primary care efforts to prevent and treat obesity, and altering societal norms of behavior,” they added.
The study was funded by the Washington University Transdiscliplinary Research on Energetics and Cancer Center, the Foundation for Barnes-Jewish Hospital, and the Breast Cancer Research Foundation. The investigators did not report any conflicts.
Three-quarters of men and two-thirds of women aged 25 years or older in the United States are either overweight or obese, according to a study published online June 22 in JAMA Internal Medicine.
During 2007-2012, about 40% of men were overweight and 35% were obese. For women, almost 30% were overweight and 37% were obese, Lin Yang, Ph.D., and Dr. Graham A. Colditz of Washington University, St. Louis, reported in a research letter (JAMA Intern. Med. 2015 June 22 [doi:10.1001/jamainternmed.2015.2405]).
Weight-status distributions across racial groups show that non-Hispanic whites were generally less obese than non-Hispanic blacks and Mexican Americans – an effect that was more pronounced among women. About 34% of white women were obese, compared with 57% of black women, with over 17% occupying the heaviest of three obesity levels, according to the investigators’ analysis of the most recent data from the National Health and Nutrition Examination Survey.
Weight groups in the study were defined by body mass index: underweight (< 18.5 kg/m2), normal weight (18.5-24.9), overweight (25.0-29.9), obesity class 1 (30.0-34.9), obesity class 2 (35.0-39.9), and obesity class 3 (≥ 40).
Compared with an earlier study reporting data for 1988-1994, “the greatest increase in the proportion of patients in the obesity class 3 category was among non-Hispanic black women,” Dr. Yang and Dr. Colditz wrote. Health and policy decision-makers should consider “population-based strategies helping to reduce modifiable risk factors such as physical environment interventions, enhancing primary care efforts to prevent and treat obesity, and altering societal norms of behavior,” they added.
The study was funded by the Washington University Transdiscliplinary Research on Energetics and Cancer Center, the Foundation for Barnes-Jewish Hospital, and the Breast Cancer Research Foundation. The investigators did not report any conflicts.
FROM JAMA INTERNAL MEDICINE
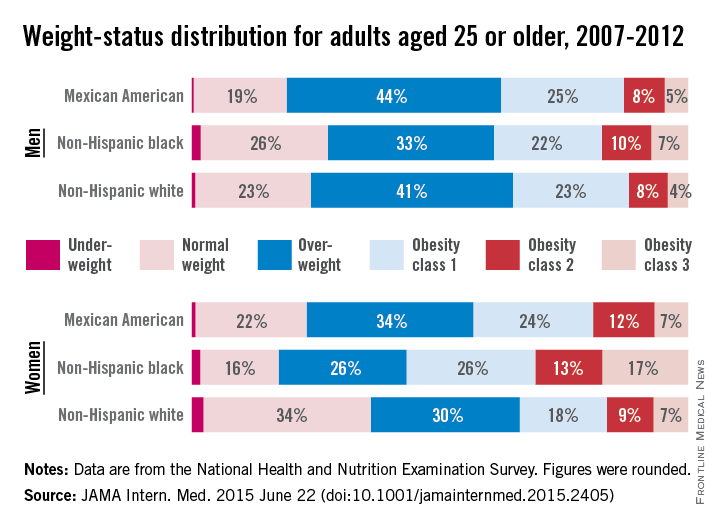
DDW: Early ERCP reduces LOS, costs of acute pancreatitis without cholangitis
WASHINGTON – When it’s given early in the course of treatment for patients who have acute pancreatitis and biliary obstruction without cholangitis, endoscopic retrograde cholangiopancreatography (ECRP) is associated with shorter lengths of stay, reductions in infectious complications, and substantially lower costs.
In more than 10,000 hospitalizations related to acute pancreatitis with choledocholithiasis/biliary obstruction with cholangitis, ECRP performed on the day of admission or the next day was associated with a significantly lower risk for septicemia than ECRP performed after the first full day of hospitalization, and hospitalization costs were nearly $20,000 lower when the procedure was done early in the course of care, reported Dr. Raxitkimar Jinjuvadia from the Henry Ford Hospital in Detroit.

“Even though the inpatient mortality did not differ among the early and late groups, early ERCP has significant other benefits in reducing health care resources utilization,” he said at the annual Digestive Disease Week.
The study’s use of a large, nationally representative hospital sample supports and adds weight to current guidelines on the use of ERCP from the American Society for Gastrointestinal Endoscopy (ASGE).
“We suggest that early ERCP should be encouraged for patients with acute biliary pancreatitis with biliary obstruction and without cholangitis,” he stated.
The investigators took a retrospective stroll through data from the National Inpatient Sample (NIS) database for the year 2011. From among approximately 8 million hospitalizations in about 1000 hospitals in the United States, they identified patients who presented with acute pancreatitis, choledocholithiasis, biliary obstruction, and cholangitis, as determined by International Classification of Disease, 9th Revision (ICD-9) codes.
The cohort included 10,364 hospitalizations related to acute pancreatitis with choledocholithiasis/biliary obstruction without cholangitis. The mean age of patients was 57.2 years, 66.4% were white and 62.2% were female. In all, 58.9% of patients underwent ERCP at some point during their hospitalizations, with 48.6% receiving it early (day 0 or day 1).
Patients who had ERCP had a significantly lower rate of inpatient deaths compared with those who did not have the procedure (0.5% vs, 1.7%, P < .001), but the timing of the procedure, the primary endpoint, did not make a difference in in-hospital mortality.
However, when the authors looked at secondary outcomes, they found that early ERCP was associated with significantly lower rates of septicemia (4.0% vs 7.2%, P < .001), shorter mean length of stay (5.2 vs. 8.0 days, P < .001) and lower costs ($52,400 vs $71,736, P <. 001).
In multivariable modeling adjusted for age, sex, race and Elixhauser comorbidities, independent risk factors for in-hospital mortality were age (adjusted odds ratio [aOR], 1.04, 95% confidence interval [CI] 1.03-1.06), Elixhauser comorbidities (aOR 1.17, 95% CI, 1.06-1.29), ERCP during hospitalization (aOR 0.30, 95% CI 0.19-0.47), and septicemia (aOR 13.5, 95% CI, 8.75-20.75).
As noted before, early ERCP was not an independent risk factor for inpatient mortality.
Dr. Jinjuvadia said that the study was limited by potential biases related to ICD-9 coding; the lack of lab values, imaging, or treatment data; and the fact that the database does not include data from Veterans Affairs hospitals.
WASHINGTON – When it’s given early in the course of treatment for patients who have acute pancreatitis and biliary obstruction without cholangitis, endoscopic retrograde cholangiopancreatography (ECRP) is associated with shorter lengths of stay, reductions in infectious complications, and substantially lower costs.
In more than 10,000 hospitalizations related to acute pancreatitis with choledocholithiasis/biliary obstruction with cholangitis, ECRP performed on the day of admission or the next day was associated with a significantly lower risk for septicemia than ECRP performed after the first full day of hospitalization, and hospitalization costs were nearly $20,000 lower when the procedure was done early in the course of care, reported Dr. Raxitkimar Jinjuvadia from the Henry Ford Hospital in Detroit.
“Even though the inpatient mortality did not differ among the early and late groups, early ERCP has significant other benefits in reducing health care resources utilization,” he said at the annual Digestive Disease Week.
The study’s use of a large, nationally representative hospital sample supports and adds weight to current guidelines on the use of ERCP from the American Society for Gastrointestinal Endoscopy (ASGE).
“We suggest that early ERCP should be encouraged for patients with acute biliary pancreatitis with biliary obstruction and without cholangitis,” he stated.
The investigators took a retrospective stroll through data from the National Inpatient Sample (NIS) database for the year 2011. From among approximately 8 million hospitalizations in about 1000 hospitals in the United States, they identified patients who presented with acute pancreatitis, choledocholithiasis, biliary obstruction, and cholangitis, as determined by International Classification of Disease, 9th Revision (ICD-9) codes.
The cohort included 10,364 hospitalizations related to acute pancreatitis with choledocholithiasis/biliary obstruction without cholangitis. The mean age of patients was 57.2 years, 66.4% were white and 62.2% were female. In all, 58.9% of patients underwent ERCP at some point during their hospitalizations, with 48.6% receiving it early (day 0 or day 1).
Patients who had ERCP had a significantly lower rate of inpatient deaths compared with those who did not have the procedure (0.5% vs, 1.7%, P < .001), but the timing of the procedure, the primary endpoint, did not make a difference in in-hospital mortality.
However, when the authors looked at secondary outcomes, they found that early ERCP was associated with significantly lower rates of septicemia (4.0% vs 7.2%, P < .001), shorter mean length of stay (5.2 vs. 8.0 days, P < .001) and lower costs ($52,400 vs $71,736, P <. 001).
In multivariable modeling adjusted for age, sex, race and Elixhauser comorbidities, independent risk factors for in-hospital mortality were age (adjusted odds ratio [aOR], 1.04, 95% confidence interval [CI] 1.03-1.06), Elixhauser comorbidities (aOR 1.17, 95% CI, 1.06-1.29), ERCP during hospitalization (aOR 0.30, 95% CI 0.19-0.47), and septicemia (aOR 13.5, 95% CI, 8.75-20.75).
As noted before, early ERCP was not an independent risk factor for inpatient mortality.
Dr. Jinjuvadia said that the study was limited by potential biases related to ICD-9 coding; the lack of lab values, imaging, or treatment data; and the fact that the database does not include data from Veterans Affairs hospitals.
WASHINGTON – When it’s given early in the course of treatment for patients who have acute pancreatitis and biliary obstruction without cholangitis, endoscopic retrograde cholangiopancreatography (ECRP) is associated with shorter lengths of stay, reductions in infectious complications, and substantially lower costs.
In more than 10,000 hospitalizations related to acute pancreatitis with choledocholithiasis/biliary obstruction with cholangitis, ECRP performed on the day of admission or the next day was associated with a significantly lower risk for septicemia than ECRP performed after the first full day of hospitalization, and hospitalization costs were nearly $20,000 lower when the procedure was done early in the course of care, reported Dr. Raxitkimar Jinjuvadia from the Henry Ford Hospital in Detroit.
“Even though the inpatient mortality did not differ among the early and late groups, early ERCP has significant other benefits in reducing health care resources utilization,” he said at the annual Digestive Disease Week.
The study’s use of a large, nationally representative hospital sample supports and adds weight to current guidelines on the use of ERCP from the American Society for Gastrointestinal Endoscopy (ASGE).
“We suggest that early ERCP should be encouraged for patients with acute biliary pancreatitis with biliary obstruction and without cholangitis,” he stated.
The investigators took a retrospective stroll through data from the National Inpatient Sample (NIS) database for the year 2011. From among approximately 8 million hospitalizations in about 1000 hospitals in the United States, they identified patients who presented with acute pancreatitis, choledocholithiasis, biliary obstruction, and cholangitis, as determined by International Classification of Disease, 9th Revision (ICD-9) codes.
The cohort included 10,364 hospitalizations related to acute pancreatitis with choledocholithiasis/biliary obstruction without cholangitis. The mean age of patients was 57.2 years, 66.4% were white and 62.2% were female. In all, 58.9% of patients underwent ERCP at some point during their hospitalizations, with 48.6% receiving it early (day 0 or day 1).
Patients who had ERCP had a significantly lower rate of inpatient deaths compared with those who did not have the procedure (0.5% vs, 1.7%, P < .001), but the timing of the procedure, the primary endpoint, did not make a difference in in-hospital mortality.
However, when the authors looked at secondary outcomes, they found that early ERCP was associated with significantly lower rates of septicemia (4.0% vs 7.2%, P < .001), shorter mean length of stay (5.2 vs. 8.0 days, P < .001) and lower costs ($52,400 vs $71,736, P <. 001).
In multivariable modeling adjusted for age, sex, race and Elixhauser comorbidities, independent risk factors for in-hospital mortality were age (adjusted odds ratio [aOR], 1.04, 95% confidence interval [CI] 1.03-1.06), Elixhauser comorbidities (aOR 1.17, 95% CI, 1.06-1.29), ERCP during hospitalization (aOR 0.30, 95% CI 0.19-0.47), and septicemia (aOR 13.5, 95% CI, 8.75-20.75).
As noted before, early ERCP was not an independent risk factor for inpatient mortality.
Dr. Jinjuvadia said that the study was limited by potential biases related to ICD-9 coding; the lack of lab values, imaging, or treatment data; and the fact that the database does not include data from Veterans Affairs hospitals.
AT DDW® 2015
Key clinical point: ECRP early in the course of hospitalization reduced adverse events, length of stay, and costs.
Major finding: For patients with acute pancreatitis with biliary obstruction without cholangitis, early ERCP was associated with a 4.0% rate of septicemia, compared with 7.2% for ERCP after the first day.
Data source: Retrospective review of inpatient data on 10,364 hospitalizations.
Disclosures: The study funding source was not disclosed. Dr. Jinjuvadia reported having no relevant disclosures.
