User login
Late-breaking abstracts highlight treatment advances in CLL, myeloma, and more
.
In a preplanned interim analysis of data from 389 patients in the randomized phase III Murano trial, venetoclax and rituximab therapy proved “superior to the standard of care and well tolerated, and a major advance in the management of [relapsed/refractory] CLL,” ASH President Kenneth C. Anderson, MD said during a premeeting preview session for the media.
In Murano, venetoclax plus rituximab bettered bendamustine plus rituximab in progression-free survival, overall survival, overall and complete response rates, and number of patients achieving minimal residual disease (MRD) negativity, said Dr. Anderson, who is also director of the Lebow Institute for Myeloma Therapeutics and Jerome Lipper Myeloma Center at Dana-Farber Cancer Institute, Boston.
The results were consistent in all risk subsets, including patients who had high-risk disease by virtue of chromosome 17p deletion, according Dr. Anderson.
In another late-breaking randomized phase III study, known as ALCYONE, adding the CD38-targeting monoclonal antibody daratumumab to standard therapy with bortezomib, melphalan, and prednisone (VMP) resulted in a “doubling” of progression-free survival in patients who had newly diagnosed multiple myeloma and were ineligible for transplantation, he reported.
In the trial of more than 700 patients, daratumumab plus VMP as initial treatment for nontransplant patients was well tolerated and improved outcomes, including overall response rate and the percent of patients who achieved MRD negative status.
“As we saw in CLL, so it’s true in this abstract in myeloma: this is a very major advance,” Dr. Anderson said.
Also during the preview session, ASH Secretary Robert A. Brodsky, MD, discussed the randomized, phase III HERCULES study results, which showed that patients with acquired thrombotic thrombocytopenic purpura (TTP) may benefit when caplacizumab is added to standard therapy. Caplacizumab targets the A1 domain of von Willebrand factor, which inhibits interaction between ultra-large von Willebrand factor and platelets.
In the trial, 145 patients were randomized to receive either plasma exchange alone or plasma exchange and caplacizumab.
Preliminary results suggest “this was a very positive trial” with a primary endpoint of time to platelet response that “greatly favored the caplacizumab arm,” said Dr. Brodsky, professor of medicine and oncology and director of the division of hematology at Johns Hopkins University, Baltimore. “Even the secondary composite endpoint of death, recurrence, and/or major thromboembolic events was much improved with caplacizumab, so this is a very positive trial and potentially a game-changing drug for the management of TTP, which can be very challenging.”
Dr. Brodsky also discussed the Hokusai VTE-Cancer Study, a randomized, open-label, blinded outcome assessment trial that showed the oral factor Xa inhibitor edoxaban was noninferior to subcutaneous dalteparin for the prevention of cancer-associated venous thromboembolism.
With more than 1,000 patients enrolled in 114 centers, the Hokusai VTE-Cancer Study had a primary outcome of the composite of the first recurrent VTE or major bleeding event during follow-up. The primary outcome occurred in 12.8% of patients in the edoxaban group, compared with 13.5% of patients in the dalteparin group (P = .0056 for noninferiority), according to the preliminary published results.
The key question addressed by the trial is whether a newer oral anticoagulant, edoxaban, can substitute for the older, subcutaneously administered low-molecular-weight heparin, dalteparin. The results “confirmed that a newer oral anticoagulant is at least as good and as safe as the low molecular weight heparin,” allowing patients the convenience of an oral therapy, Dr. Brodsky noted.
This year’s late-breaking abstracts at ASH are:
LBA-1 Results of the Randomized, Double-Blind, Placebo-Controlled, Phase III Hercules Study of Caplacizumab in Patients with Acquired Thrombotic Thrombocytopenic Purpura.
LBA-2 Venetoclax Plus Rituximab Is Superior to Bendamustine Plus Rituximab in Patients with Relapsed/ Refractory Chronic Lymphocytic Leukemia - Results from Pre-Planned Interim Analysis of the Randomized Phase III Murano Study.
LBA-3 Mutations in SRP54 Gene Cause Severe Primary Neutropenia As Well As Shwachman-Diamond-like Syndrome.
LBA-4 Phase III Randomized Study of Daratumumab Plus Bortezomib, Melphalan, and Prednisone (D-VMP) Versus Bortezomib, Melphalan, and Prednisone (VMP) in Newly Diagnosed Multiple Myeloma (NDMM) Patients (Pts) Ineligible for Transplant (ALCYONE).
LBA-5 Prospective Molecular MRD Detection By NGS: A Powerful Independent Predictor for Relapse and Survival in Adults with Newly Diagnosed AML.
LBA-6 A Randomized, Open-Label, Blinded Outcome Assessment Trial Evaluating the Efficacy and Safety of LMWH/Edoxaban Versus Dalteparin for Venous Thromboembolism Associated with Cancer: Hokusai VTE-Cancer Study
.
In a preplanned interim analysis of data from 389 patients in the randomized phase III Murano trial, venetoclax and rituximab therapy proved “superior to the standard of care and well tolerated, and a major advance in the management of [relapsed/refractory] CLL,” ASH President Kenneth C. Anderson, MD said during a premeeting preview session for the media.
In Murano, venetoclax plus rituximab bettered bendamustine plus rituximab in progression-free survival, overall survival, overall and complete response rates, and number of patients achieving minimal residual disease (MRD) negativity, said Dr. Anderson, who is also director of the Lebow Institute for Myeloma Therapeutics and Jerome Lipper Myeloma Center at Dana-Farber Cancer Institute, Boston.
The results were consistent in all risk subsets, including patients who had high-risk disease by virtue of chromosome 17p deletion, according Dr. Anderson.
In another late-breaking randomized phase III study, known as ALCYONE, adding the CD38-targeting monoclonal antibody daratumumab to standard therapy with bortezomib, melphalan, and prednisone (VMP) resulted in a “doubling” of progression-free survival in patients who had newly diagnosed multiple myeloma and were ineligible for transplantation, he reported.
In the trial of more than 700 patients, daratumumab plus VMP as initial treatment for nontransplant patients was well tolerated and improved outcomes, including overall response rate and the percent of patients who achieved MRD negative status.
“As we saw in CLL, so it’s true in this abstract in myeloma: this is a very major advance,” Dr. Anderson said.
Also during the preview session, ASH Secretary Robert A. Brodsky, MD, discussed the randomized, phase III HERCULES study results, which showed that patients with acquired thrombotic thrombocytopenic purpura (TTP) may benefit when caplacizumab is added to standard therapy. Caplacizumab targets the A1 domain of von Willebrand factor, which inhibits interaction between ultra-large von Willebrand factor and platelets.
In the trial, 145 patients were randomized to receive either plasma exchange alone or plasma exchange and caplacizumab.
Preliminary results suggest “this was a very positive trial” with a primary endpoint of time to platelet response that “greatly favored the caplacizumab arm,” said Dr. Brodsky, professor of medicine and oncology and director of the division of hematology at Johns Hopkins University, Baltimore. “Even the secondary composite endpoint of death, recurrence, and/or major thromboembolic events was much improved with caplacizumab, so this is a very positive trial and potentially a game-changing drug for the management of TTP, which can be very challenging.”
Dr. Brodsky also discussed the Hokusai VTE-Cancer Study, a randomized, open-label, blinded outcome assessment trial that showed the oral factor Xa inhibitor edoxaban was noninferior to subcutaneous dalteparin for the prevention of cancer-associated venous thromboembolism.
With more than 1,000 patients enrolled in 114 centers, the Hokusai VTE-Cancer Study had a primary outcome of the composite of the first recurrent VTE or major bleeding event during follow-up. The primary outcome occurred in 12.8% of patients in the edoxaban group, compared with 13.5% of patients in the dalteparin group (P = .0056 for noninferiority), according to the preliminary published results.
The key question addressed by the trial is whether a newer oral anticoagulant, edoxaban, can substitute for the older, subcutaneously administered low-molecular-weight heparin, dalteparin. The results “confirmed that a newer oral anticoagulant is at least as good and as safe as the low molecular weight heparin,” allowing patients the convenience of an oral therapy, Dr. Brodsky noted.
This year’s late-breaking abstracts at ASH are:
LBA-1 Results of the Randomized, Double-Blind, Placebo-Controlled, Phase III Hercules Study of Caplacizumab in Patients with Acquired Thrombotic Thrombocytopenic Purpura.
LBA-2 Venetoclax Plus Rituximab Is Superior to Bendamustine Plus Rituximab in Patients with Relapsed/ Refractory Chronic Lymphocytic Leukemia - Results from Pre-Planned Interim Analysis of the Randomized Phase III Murano Study.
LBA-3 Mutations in SRP54 Gene Cause Severe Primary Neutropenia As Well As Shwachman-Diamond-like Syndrome.
LBA-4 Phase III Randomized Study of Daratumumab Plus Bortezomib, Melphalan, and Prednisone (D-VMP) Versus Bortezomib, Melphalan, and Prednisone (VMP) in Newly Diagnosed Multiple Myeloma (NDMM) Patients (Pts) Ineligible for Transplant (ALCYONE).
LBA-5 Prospective Molecular MRD Detection By NGS: A Powerful Independent Predictor for Relapse and Survival in Adults with Newly Diagnosed AML.
LBA-6 A Randomized, Open-Label, Blinded Outcome Assessment Trial Evaluating the Efficacy and Safety of LMWH/Edoxaban Versus Dalteparin for Venous Thromboembolism Associated with Cancer: Hokusai VTE-Cancer Study
.
In a preplanned interim analysis of data from 389 patients in the randomized phase III Murano trial, venetoclax and rituximab therapy proved “superior to the standard of care and well tolerated, and a major advance in the management of [relapsed/refractory] CLL,” ASH President Kenneth C. Anderson, MD said during a premeeting preview session for the media.
In Murano, venetoclax plus rituximab bettered bendamustine plus rituximab in progression-free survival, overall survival, overall and complete response rates, and number of patients achieving minimal residual disease (MRD) negativity, said Dr. Anderson, who is also director of the Lebow Institute for Myeloma Therapeutics and Jerome Lipper Myeloma Center at Dana-Farber Cancer Institute, Boston.
The results were consistent in all risk subsets, including patients who had high-risk disease by virtue of chromosome 17p deletion, according Dr. Anderson.
In another late-breaking randomized phase III study, known as ALCYONE, adding the CD38-targeting monoclonal antibody daratumumab to standard therapy with bortezomib, melphalan, and prednisone (VMP) resulted in a “doubling” of progression-free survival in patients who had newly diagnosed multiple myeloma and were ineligible for transplantation, he reported.
In the trial of more than 700 patients, daratumumab plus VMP as initial treatment for nontransplant patients was well tolerated and improved outcomes, including overall response rate and the percent of patients who achieved MRD negative status.
“As we saw in CLL, so it’s true in this abstract in myeloma: this is a very major advance,” Dr. Anderson said.
Also during the preview session, ASH Secretary Robert A. Brodsky, MD, discussed the randomized, phase III HERCULES study results, which showed that patients with acquired thrombotic thrombocytopenic purpura (TTP) may benefit when caplacizumab is added to standard therapy. Caplacizumab targets the A1 domain of von Willebrand factor, which inhibits interaction between ultra-large von Willebrand factor and platelets.
In the trial, 145 patients were randomized to receive either plasma exchange alone or plasma exchange and caplacizumab.
Preliminary results suggest “this was a very positive trial” with a primary endpoint of time to platelet response that “greatly favored the caplacizumab arm,” said Dr. Brodsky, professor of medicine and oncology and director of the division of hematology at Johns Hopkins University, Baltimore. “Even the secondary composite endpoint of death, recurrence, and/or major thromboembolic events was much improved with caplacizumab, so this is a very positive trial and potentially a game-changing drug for the management of TTP, which can be very challenging.”
Dr. Brodsky also discussed the Hokusai VTE-Cancer Study, a randomized, open-label, blinded outcome assessment trial that showed the oral factor Xa inhibitor edoxaban was noninferior to subcutaneous dalteparin for the prevention of cancer-associated venous thromboembolism.
With more than 1,000 patients enrolled in 114 centers, the Hokusai VTE-Cancer Study had a primary outcome of the composite of the first recurrent VTE or major bleeding event during follow-up. The primary outcome occurred in 12.8% of patients in the edoxaban group, compared with 13.5% of patients in the dalteparin group (P = .0056 for noninferiority), according to the preliminary published results.
The key question addressed by the trial is whether a newer oral anticoagulant, edoxaban, can substitute for the older, subcutaneously administered low-molecular-weight heparin, dalteparin. The results “confirmed that a newer oral anticoagulant is at least as good and as safe as the low molecular weight heparin,” allowing patients the convenience of an oral therapy, Dr. Brodsky noted.
This year’s late-breaking abstracts at ASH are:
LBA-1 Results of the Randomized, Double-Blind, Placebo-Controlled, Phase III Hercules Study of Caplacizumab in Patients with Acquired Thrombotic Thrombocytopenic Purpura.
LBA-2 Venetoclax Plus Rituximab Is Superior to Bendamustine Plus Rituximab in Patients with Relapsed/ Refractory Chronic Lymphocytic Leukemia - Results from Pre-Planned Interim Analysis of the Randomized Phase III Murano Study.
LBA-3 Mutations in SRP54 Gene Cause Severe Primary Neutropenia As Well As Shwachman-Diamond-like Syndrome.
LBA-4 Phase III Randomized Study of Daratumumab Plus Bortezomib, Melphalan, and Prednisone (D-VMP) Versus Bortezomib, Melphalan, and Prednisone (VMP) in Newly Diagnosed Multiple Myeloma (NDMM) Patients (Pts) Ineligible for Transplant (ALCYONE).
LBA-5 Prospective Molecular MRD Detection By NGS: A Powerful Independent Predictor for Relapse and Survival in Adults with Newly Diagnosed AML.
LBA-6 A Randomized, Open-Label, Blinded Outcome Assessment Trial Evaluating the Efficacy and Safety of LMWH/Edoxaban Versus Dalteparin for Venous Thromboembolism Associated with Cancer: Hokusai VTE-Cancer Study
FROM ASH 2017
Obesity-Related Cancer Is on the Rise
Overweight and obesity are associated with an increased risk of 13 types of cancer—and those cancers account for about 40% of all cancers diagnosed in 2014, according to the CDC.
The 13 cancers are meningioma, adenocarcinoma of the esophagus, multiple myeloma, kidney, uterine, ovarian, thyroid, breast, liver, gallbladder, upper stomach, pancreas, and colorectal. About 630,000 people were diagnosed with 1 of those cancers in 2014; 2 in 3 cancers were in adults aged 50 to 74 years. In 2013-2014, about 2 of every 3 American adults were overweight or obese.
Related: The Impact of Obesity on Simvastatin for Lowering LDL-C Among Veterans
Overall, the rate of new cancer cases has been on the decline since the 1990s, the report says, but increases in overweight- and obesity-related cancers “are likely slowing this progress.” Obesity-related cancers (not including colorectal cancer, which declined by 23%) increased by 7% between 2005 and 2014, while the rates of nonobesity–related cancers declined 13%.
Health care providers can help, the CDC says, by counseling patients on healthy weight and its role in cancer prevention, referring obese patients to intensive management programs and connecting patients and their families to community services that give them easier access to healthful food. The National Comprehensive Cancer Control Program (https://www.cdc.gov/cancer/ncccp/index.htm) funds cancer coalitions across the U.S., which include strategies to prevent and control overweight and obesity.
Related: The Impact of Obesity on the Recovery of Patients With Cancer
“When people ask me if there’s a cure for cancer, I say, ‘Yes, good health is the best prescription for preventing chronic diseases, including cancer,” said Lisa Richardson, MD, director of CDC’s Division of Cancer Prevention and Control. That means, she says, giving people the information they need to make healthy choices where they live, work, learn, and play.
Overweight and obesity are associated with an increased risk of 13 types of cancer—and those cancers account for about 40% of all cancers diagnosed in 2014, according to the CDC.
The 13 cancers are meningioma, adenocarcinoma of the esophagus, multiple myeloma, kidney, uterine, ovarian, thyroid, breast, liver, gallbladder, upper stomach, pancreas, and colorectal. About 630,000 people were diagnosed with 1 of those cancers in 2014; 2 in 3 cancers were in adults aged 50 to 74 years. In 2013-2014, about 2 of every 3 American adults were overweight or obese.
Related: The Impact of Obesity on Simvastatin for Lowering LDL-C Among Veterans
Overall, the rate of new cancer cases has been on the decline since the 1990s, the report says, but increases in overweight- and obesity-related cancers “are likely slowing this progress.” Obesity-related cancers (not including colorectal cancer, which declined by 23%) increased by 7% between 2005 and 2014, while the rates of nonobesity–related cancers declined 13%.
Health care providers can help, the CDC says, by counseling patients on healthy weight and its role in cancer prevention, referring obese patients to intensive management programs and connecting patients and their families to community services that give them easier access to healthful food. The National Comprehensive Cancer Control Program (https://www.cdc.gov/cancer/ncccp/index.htm) funds cancer coalitions across the U.S., which include strategies to prevent and control overweight and obesity.
Related: The Impact of Obesity on the Recovery of Patients With Cancer
“When people ask me if there’s a cure for cancer, I say, ‘Yes, good health is the best prescription for preventing chronic diseases, including cancer,” said Lisa Richardson, MD, director of CDC’s Division of Cancer Prevention and Control. That means, she says, giving people the information they need to make healthy choices where they live, work, learn, and play.
Overweight and obesity are associated with an increased risk of 13 types of cancer—and those cancers account for about 40% of all cancers diagnosed in 2014, according to the CDC.
The 13 cancers are meningioma, adenocarcinoma of the esophagus, multiple myeloma, kidney, uterine, ovarian, thyroid, breast, liver, gallbladder, upper stomach, pancreas, and colorectal. About 630,000 people were diagnosed with 1 of those cancers in 2014; 2 in 3 cancers were in adults aged 50 to 74 years. In 2013-2014, about 2 of every 3 American adults were overweight or obese.
Related: The Impact of Obesity on Simvastatin for Lowering LDL-C Among Veterans
Overall, the rate of new cancer cases has been on the decline since the 1990s, the report says, but increases in overweight- and obesity-related cancers “are likely slowing this progress.” Obesity-related cancers (not including colorectal cancer, which declined by 23%) increased by 7% between 2005 and 2014, while the rates of nonobesity–related cancers declined 13%.
Health care providers can help, the CDC says, by counseling patients on healthy weight and its role in cancer prevention, referring obese patients to intensive management programs and connecting patients and their families to community services that give them easier access to healthful food. The National Comprehensive Cancer Control Program (https://www.cdc.gov/cancer/ncccp/index.htm) funds cancer coalitions across the U.S., which include strategies to prevent and control overweight and obesity.
Related: The Impact of Obesity on the Recovery of Patients With Cancer
“When people ask me if there’s a cure for cancer, I say, ‘Yes, good health is the best prescription for preventing chronic diseases, including cancer,” said Lisa Richardson, MD, director of CDC’s Division of Cancer Prevention and Control. That means, she says, giving people the information they need to make healthy choices where they live, work, learn, and play.
Eye Hemorrhage Signals Myeloid Leukemia
A 40-year-old man suddenly began to lose vision in his left eye. The retinal exam was normal for the right eye. But the left showed isolated subinternal limited membrane hemorrhage at the fovea along with a white-centered hemorrhage above the fovea.
The patient had no history of trauma or Valsalva retinopathy. His blood pressure was normal as was his blood glucose. However, when bloodwork showed a high total count, increased platelet count, and the peripheral smear indicated myeloid hyperplasia, clinicians at LV Prasad Eye Institute in Hyderabad, India, diagnosed the patient with underlying chronic myeloid leukemia (CML).
A physical examination revealed a palpable spleenomegaly—underscoring the fact, the clinicians note, that when an ophthalmologic finding suggests a systemic disease, a general physical examination will reveal more clinical clues. The patient was referred to an oncologist and started on imatinib for CML.
White-centered or pale-centered hemorrhages are believed to represent an accumulation of leukemic cells or platelet fibrin aggregates, the clinicians say. Blood dyscrasias, such as anemias, leukemia, multiple myeloma, and other platelet disorders may present with similar features. Such hemorrhages are known to resolve spontaneously when the patient is treated for the underlying condition, and the hematologic status improves, the clinicians say. This patient’s hemorrhage gradually resolved over the next month, and his visual acuity improved to 20/20.
Ocular manifestations as a presenting sign of leukemia, especially chronic, are rare, the clinicians say. They note that retinal hemorrhages are one of the “most striking findings” in leukemia, and because they can be directly observed, they provide a “subtle but important clue toward an otherwise asymptomatic disease.” If diagnosed early and treated promptly, patients with CML have a good survival rate.
Source:
Tyagi M, Agarwal K, Paulose RM, Rani PK. BMJ Case Rep. 2017;2017: pii: bcr-2017-21974.
doi: 10.1136/bcr-2017-219741.
A 40-year-old man suddenly began to lose vision in his left eye. The retinal exam was normal for the right eye. But the left showed isolated subinternal limited membrane hemorrhage at the fovea along with a white-centered hemorrhage above the fovea.
The patient had no history of trauma or Valsalva retinopathy. His blood pressure was normal as was his blood glucose. However, when bloodwork showed a high total count, increased platelet count, and the peripheral smear indicated myeloid hyperplasia, clinicians at LV Prasad Eye Institute in Hyderabad, India, diagnosed the patient with underlying chronic myeloid leukemia (CML).
A physical examination revealed a palpable spleenomegaly—underscoring the fact, the clinicians note, that when an ophthalmologic finding suggests a systemic disease, a general physical examination will reveal more clinical clues. The patient was referred to an oncologist and started on imatinib for CML.
White-centered or pale-centered hemorrhages are believed to represent an accumulation of leukemic cells or platelet fibrin aggregates, the clinicians say. Blood dyscrasias, such as anemias, leukemia, multiple myeloma, and other platelet disorders may present with similar features. Such hemorrhages are known to resolve spontaneously when the patient is treated for the underlying condition, and the hematologic status improves, the clinicians say. This patient’s hemorrhage gradually resolved over the next month, and his visual acuity improved to 20/20.
Ocular manifestations as a presenting sign of leukemia, especially chronic, are rare, the clinicians say. They note that retinal hemorrhages are one of the “most striking findings” in leukemia, and because they can be directly observed, they provide a “subtle but important clue toward an otherwise asymptomatic disease.” If diagnosed early and treated promptly, patients with CML have a good survival rate.
Source:
Tyagi M, Agarwal K, Paulose RM, Rani PK. BMJ Case Rep. 2017;2017: pii: bcr-2017-21974.
doi: 10.1136/bcr-2017-219741.
A 40-year-old man suddenly began to lose vision in his left eye. The retinal exam was normal for the right eye. But the left showed isolated subinternal limited membrane hemorrhage at the fovea along with a white-centered hemorrhage above the fovea.
The patient had no history of trauma or Valsalva retinopathy. His blood pressure was normal as was his blood glucose. However, when bloodwork showed a high total count, increased platelet count, and the peripheral smear indicated myeloid hyperplasia, clinicians at LV Prasad Eye Institute in Hyderabad, India, diagnosed the patient with underlying chronic myeloid leukemia (CML).
A physical examination revealed a palpable spleenomegaly—underscoring the fact, the clinicians note, that when an ophthalmologic finding suggests a systemic disease, a general physical examination will reveal more clinical clues. The patient was referred to an oncologist and started on imatinib for CML.
White-centered or pale-centered hemorrhages are believed to represent an accumulation of leukemic cells or platelet fibrin aggregates, the clinicians say. Blood dyscrasias, such as anemias, leukemia, multiple myeloma, and other platelet disorders may present with similar features. Such hemorrhages are known to resolve spontaneously when the patient is treated for the underlying condition, and the hematologic status improves, the clinicians say. This patient’s hemorrhage gradually resolved over the next month, and his visual acuity improved to 20/20.
Ocular manifestations as a presenting sign of leukemia, especially chronic, are rare, the clinicians say. They note that retinal hemorrhages are one of the “most striking findings” in leukemia, and because they can be directly observed, they provide a “subtle but important clue toward an otherwise asymptomatic disease.” If diagnosed early and treated promptly, patients with CML have a good survival rate.
Source:
Tyagi M, Agarwal K, Paulose RM, Rani PK. BMJ Case Rep. 2017;2017: pii: bcr-2017-21974.
doi: 10.1136/bcr-2017-219741.
Rare type of MCL mimics Castleman disease
A rare type of mantle cell lymphoma (MCL) has features that are similar to those of Castleman disease, according to a recent report based on three patient cases.
Lymph node biopsies for these patients initially indicated histologic features consistent with those of plasma cell (PC)-type Castleman disease, reported Takuro Igawa, MD, PhD, of Okayama (Japan) University Graduate School of Medicine, Dentistry, and Pharmaceutical Sciences, and his coauthors. Further work-up, including flow cytometric analysis and cyclin D1 immunostaining, showed features consistent with those of MCL.
“This rare type of MCL can mimic Castleman disease in the clinical setting and upon histological examination,” Dr. Igawa and his colleagues wrote (Pathol Res Pract. 2017 Sep 18. pii: S0344-0338[17]30684-2. doi: 10.1016/j.prp.2017.09.015). “These confusing characteristics can make the diagnosis challenging, and careful flow cytometric analysis is recommended when a histopathological diagnosis is made.”
The patients in the study, all male, were 51, 74, and 81 years of age. Each presented with systemic lymphadenopathy, along with abnormal laboratory findings that according to the investigators are not usually associated with B-cell lymphomas such as MCL, including anemia, polyclonal IgG hypergammaglobulinemia, and elevated levels of C-reactive protein.
In lymph node biopsy specimens, the MCL component was “masked by histological features of PC-type Castleman disease” such as interfollicular plasmacytosis and atrophic germinal centers, the researchers wrote.
However, further pathologic investigations revealed features that were “essential to distinguish these 3 cases of MCL from PC-type Castleman disease,” they added.
In particular, an abnormal B-cell population was found using flow cytometric analysis, while subsequent cyclin D1 immunostaining in all three cases showed abnormal B-cells primarily in the mantle zone that were positive for CD20 and CD5, both typically expressed by MCL, along with SOX11, which is an “excellent diagnostic marker for MCL, including atypical MCL,” the investigators wrote.
These case reports also provide some evidence that interleukin-6 (IL-6), which is thought to be a driver of Castleman disease, might also be implicated in the pathogenesis of this rare MCL variant. the researchers found that all three cases had positive IL-6 staining in the interfollicular areas.
If further studies confirm the role of IL-6 in this rare setting, “specific treatments other than chemotherapy could potentially be used for patients with MCL with features of Castleman disease, such as an anti-IL-6 receptor antibody (tocilizumab), which is already used for patients with Castleman disease,” they said.
A rare type of mantle cell lymphoma (MCL) has features that are similar to those of Castleman disease, according to a recent report based on three patient cases.
Lymph node biopsies for these patients initially indicated histologic features consistent with those of plasma cell (PC)-type Castleman disease, reported Takuro Igawa, MD, PhD, of Okayama (Japan) University Graduate School of Medicine, Dentistry, and Pharmaceutical Sciences, and his coauthors. Further work-up, including flow cytometric analysis and cyclin D1 immunostaining, showed features consistent with those of MCL.
“This rare type of MCL can mimic Castleman disease in the clinical setting and upon histological examination,” Dr. Igawa and his colleagues wrote (Pathol Res Pract. 2017 Sep 18. pii: S0344-0338[17]30684-2. doi: 10.1016/j.prp.2017.09.015). “These confusing characteristics can make the diagnosis challenging, and careful flow cytometric analysis is recommended when a histopathological diagnosis is made.”
The patients in the study, all male, were 51, 74, and 81 years of age. Each presented with systemic lymphadenopathy, along with abnormal laboratory findings that according to the investigators are not usually associated with B-cell lymphomas such as MCL, including anemia, polyclonal IgG hypergammaglobulinemia, and elevated levels of C-reactive protein.
In lymph node biopsy specimens, the MCL component was “masked by histological features of PC-type Castleman disease” such as interfollicular plasmacytosis and atrophic germinal centers, the researchers wrote.
However, further pathologic investigations revealed features that were “essential to distinguish these 3 cases of MCL from PC-type Castleman disease,” they added.
In particular, an abnormal B-cell population was found using flow cytometric analysis, while subsequent cyclin D1 immunostaining in all three cases showed abnormal B-cells primarily in the mantle zone that were positive for CD20 and CD5, both typically expressed by MCL, along with SOX11, which is an “excellent diagnostic marker for MCL, including atypical MCL,” the investigators wrote.
These case reports also provide some evidence that interleukin-6 (IL-6), which is thought to be a driver of Castleman disease, might also be implicated in the pathogenesis of this rare MCL variant. the researchers found that all three cases had positive IL-6 staining in the interfollicular areas.
If further studies confirm the role of IL-6 in this rare setting, “specific treatments other than chemotherapy could potentially be used for patients with MCL with features of Castleman disease, such as an anti-IL-6 receptor antibody (tocilizumab), which is already used for patients with Castleman disease,” they said.
A rare type of mantle cell lymphoma (MCL) has features that are similar to those of Castleman disease, according to a recent report based on three patient cases.
Lymph node biopsies for these patients initially indicated histologic features consistent with those of plasma cell (PC)-type Castleman disease, reported Takuro Igawa, MD, PhD, of Okayama (Japan) University Graduate School of Medicine, Dentistry, and Pharmaceutical Sciences, and his coauthors. Further work-up, including flow cytometric analysis and cyclin D1 immunostaining, showed features consistent with those of MCL.
“This rare type of MCL can mimic Castleman disease in the clinical setting and upon histological examination,” Dr. Igawa and his colleagues wrote (Pathol Res Pract. 2017 Sep 18. pii: S0344-0338[17]30684-2. doi: 10.1016/j.prp.2017.09.015). “These confusing characteristics can make the diagnosis challenging, and careful flow cytometric analysis is recommended when a histopathological diagnosis is made.”
The patients in the study, all male, were 51, 74, and 81 years of age. Each presented with systemic lymphadenopathy, along with abnormal laboratory findings that according to the investigators are not usually associated with B-cell lymphomas such as MCL, including anemia, polyclonal IgG hypergammaglobulinemia, and elevated levels of C-reactive protein.
In lymph node biopsy specimens, the MCL component was “masked by histological features of PC-type Castleman disease” such as interfollicular plasmacytosis and atrophic germinal centers, the researchers wrote.
However, further pathologic investigations revealed features that were “essential to distinguish these 3 cases of MCL from PC-type Castleman disease,” they added.
In particular, an abnormal B-cell population was found using flow cytometric analysis, while subsequent cyclin D1 immunostaining in all three cases showed abnormal B-cells primarily in the mantle zone that were positive for CD20 and CD5, both typically expressed by MCL, along with SOX11, which is an “excellent diagnostic marker for MCL, including atypical MCL,” the investigators wrote.
These case reports also provide some evidence that interleukin-6 (IL-6), which is thought to be a driver of Castleman disease, might also be implicated in the pathogenesis of this rare MCL variant. the researchers found that all three cases had positive IL-6 staining in the interfollicular areas.
If further studies confirm the role of IL-6 in this rare setting, “specific treatments other than chemotherapy could potentially be used for patients with MCL with features of Castleman disease, such as an anti-IL-6 receptor antibody (tocilizumab), which is already used for patients with Castleman disease,” they said.
FROM PATHOLOGY – RESEARCH AND PRACTICE
Key clinical point:
Major finding: Lymph node biopsy revealed histologic features consistent with plasma cell (PC)-type Castleman disease, but cyclin D1 immunostaining and flow cytometric analysis showed features consistent with a diagnosis of MCL.
Data source: A report on three patient cases of MCL with features of PC-type Castleman disease retrieved from surgical pathology consultation files.
Disclosures: The authors reported no conflicts of interest.
FDA Approves Treatment for Chronic GVHD
A treatment for cancer is finding a new purpose in treating another life-threatening condition. The FDA expanded approval of Ibrutinib for treatment of adults with chronic graft versus host disease (cGVHD) after ≥ 1 treatments have failed. Ibrutinib was previously approved for certain indications in treating chronic lymphocytic leukemia, Waldenström macroglobulinemia, and marginal zone lymphoma.
An estimated 30% to 70% of patients who receive hematopoietic stem cell transplantation for blood or bone marrow cancer develop cGVHD.
Ibrutinib , a kinase inhibitor, was tested in a single-arm trial of 42 patients with cGVHD. Most had mouth ulcers and skin rashes; > 50% had ≥ 2 organs affected. Their symptoms had persisted despite standard treatment with corticosteroids. In the study, cGVHD symptoms improved in 67%. For nearly half (48%), the improvements lasted for 5 months or longer.
Common adverse effects include fatigue, bruising, diarrhea, and thrombocytopenia. Serious adverse effects include severe bleeding, infections, and cytopenia.
A treatment for cancer is finding a new purpose in treating another life-threatening condition. The FDA expanded approval of Ibrutinib for treatment of adults with chronic graft versus host disease (cGVHD) after ≥ 1 treatments have failed. Ibrutinib was previously approved for certain indications in treating chronic lymphocytic leukemia, Waldenström macroglobulinemia, and marginal zone lymphoma.
An estimated 30% to 70% of patients who receive hematopoietic stem cell transplantation for blood or bone marrow cancer develop cGVHD.
Ibrutinib , a kinase inhibitor, was tested in a single-arm trial of 42 patients with cGVHD. Most had mouth ulcers and skin rashes; > 50% had ≥ 2 organs affected. Their symptoms had persisted despite standard treatment with corticosteroids. In the study, cGVHD symptoms improved in 67%. For nearly half (48%), the improvements lasted for 5 months or longer.
Common adverse effects include fatigue, bruising, diarrhea, and thrombocytopenia. Serious adverse effects include severe bleeding, infections, and cytopenia.
A treatment for cancer is finding a new purpose in treating another life-threatening condition. The FDA expanded approval of Ibrutinib for treatment of adults with chronic graft versus host disease (cGVHD) after ≥ 1 treatments have failed. Ibrutinib was previously approved for certain indications in treating chronic lymphocytic leukemia, Waldenström macroglobulinemia, and marginal zone lymphoma.
An estimated 30% to 70% of patients who receive hematopoietic stem cell transplantation for blood or bone marrow cancer develop cGVHD.
Ibrutinib , a kinase inhibitor, was tested in a single-arm trial of 42 patients with cGVHD. Most had mouth ulcers and skin rashes; > 50% had ≥ 2 organs affected. Their symptoms had persisted despite standard treatment with corticosteroids. In the study, cGVHD symptoms improved in 67%. For nearly half (48%), the improvements lasted for 5 months or longer.
Common adverse effects include fatigue, bruising, diarrhea, and thrombocytopenia. Serious adverse effects include severe bleeding, infections, and cytopenia.
Do Post-Transplant Tests Show Recurring Multiple Myeloma?
After stem cell therapy, profiles may show a pattern of antibodies that can look very much like the “M spike”—the signature of the monoclonal antibody produced by multiple myeloma (MM). But that pattern, called an oligoclonal band, can be misleading.
“Oligoclonal bands should mostly be recognized as a response to treatment and not be mistaken as a recurrence of the original tumor,” says Dr. Gurmukh Singh, vice chair of clinical affairs for the Department of Pathology at the Medical College of Georgia at Augusta University.
He and his research team analyzed data from 251 patients with MM, 159 of whom had received autologous stem cell transplants. The researchers performed tests using serum protein electrophoresis/serum immunofixation electrophoresis and serum free light chain assay. Each patient had at least 3 tests, with at least 2 following the transplant.
The researchers found the incidence of oligoclonal patterns was dramatically higher in patients who had a stem cell transplant, compared with patients who had chemotherapy alone (57.9% vs 8.8%). Moreover, only 5 of the 159 patients who received a transplant had an oligoclonal pattern before treatment, but 92 had 1 afterward. More than half the oligoclonal patterns developed within the first year following transplant. The earliest pattern was detected at 2 months and a few as long as 5 years later.
The key to assessing response, Singh says, is to see where the spike appears: that is, where the monoclonal spike is at diagnosis compared with any new spikes that appear in oligoclonal bands after stem cell treatment. “If the original peak was at location A, [and] now the peak is location B, that allows us to determine that it is not the same abnormal, malignant antibody.”
The finding that 58% of patients had the oligoclonal pattern after transplant is likely an underestimate due to irregular schedule of testing, the researchers say. They add that their findings highlight the need for higher resolution electrophoretic methods to obviate the need for using mass spectrometry for clinical samples. Their results “cast more doubt on the clinical usefulness and medical necessity of the serum free light chain assay.”
Source:
Baker T. Results after stem cell transplant can confuse patients and doctors about cancer’s status. Jagwire News. https://jagwire.augusta.edu/archives/46434. Published August 2017. Accessed September 20, 2017.
Singh G. J Clin Med Res. 2017;9(8):671-679.
doi: 10.14740/jocmr3049w.
After stem cell therapy, profiles may show a pattern of antibodies that can look very much like the “M spike”—the signature of the monoclonal antibody produced by multiple myeloma (MM). But that pattern, called an oligoclonal band, can be misleading.
“Oligoclonal bands should mostly be recognized as a response to treatment and not be mistaken as a recurrence of the original tumor,” says Dr. Gurmukh Singh, vice chair of clinical affairs for the Department of Pathology at the Medical College of Georgia at Augusta University.
He and his research team analyzed data from 251 patients with MM, 159 of whom had received autologous stem cell transplants. The researchers performed tests using serum protein electrophoresis/serum immunofixation electrophoresis and serum free light chain assay. Each patient had at least 3 tests, with at least 2 following the transplant.
The researchers found the incidence of oligoclonal patterns was dramatically higher in patients who had a stem cell transplant, compared with patients who had chemotherapy alone (57.9% vs 8.8%). Moreover, only 5 of the 159 patients who received a transplant had an oligoclonal pattern before treatment, but 92 had 1 afterward. More than half the oligoclonal patterns developed within the first year following transplant. The earliest pattern was detected at 2 months and a few as long as 5 years later.
The key to assessing response, Singh says, is to see where the spike appears: that is, where the monoclonal spike is at diagnosis compared with any new spikes that appear in oligoclonal bands after stem cell treatment. “If the original peak was at location A, [and] now the peak is location B, that allows us to determine that it is not the same abnormal, malignant antibody.”
The finding that 58% of patients had the oligoclonal pattern after transplant is likely an underestimate due to irregular schedule of testing, the researchers say. They add that their findings highlight the need for higher resolution electrophoretic methods to obviate the need for using mass spectrometry for clinical samples. Their results “cast more doubt on the clinical usefulness and medical necessity of the serum free light chain assay.”
Source:
Baker T. Results after stem cell transplant can confuse patients and doctors about cancer’s status. Jagwire News. https://jagwire.augusta.edu/archives/46434. Published August 2017. Accessed September 20, 2017.
Singh G. J Clin Med Res. 2017;9(8):671-679.
doi: 10.14740/jocmr3049w.
After stem cell therapy, profiles may show a pattern of antibodies that can look very much like the “M spike”—the signature of the monoclonal antibody produced by multiple myeloma (MM). But that pattern, called an oligoclonal band, can be misleading.
“Oligoclonal bands should mostly be recognized as a response to treatment and not be mistaken as a recurrence of the original tumor,” says Dr. Gurmukh Singh, vice chair of clinical affairs for the Department of Pathology at the Medical College of Georgia at Augusta University.
He and his research team analyzed data from 251 patients with MM, 159 of whom had received autologous stem cell transplants. The researchers performed tests using serum protein electrophoresis/serum immunofixation electrophoresis and serum free light chain assay. Each patient had at least 3 tests, with at least 2 following the transplant.
The researchers found the incidence of oligoclonal patterns was dramatically higher in patients who had a stem cell transplant, compared with patients who had chemotherapy alone (57.9% vs 8.8%). Moreover, only 5 of the 159 patients who received a transplant had an oligoclonal pattern before treatment, but 92 had 1 afterward. More than half the oligoclonal patterns developed within the first year following transplant. The earliest pattern was detected at 2 months and a few as long as 5 years later.
The key to assessing response, Singh says, is to see where the spike appears: that is, where the monoclonal spike is at diagnosis compared with any new spikes that appear in oligoclonal bands after stem cell treatment. “If the original peak was at location A, [and] now the peak is location B, that allows us to determine that it is not the same abnormal, malignant antibody.”
The finding that 58% of patients had the oligoclonal pattern after transplant is likely an underestimate due to irregular schedule of testing, the researchers say. They add that their findings highlight the need for higher resolution electrophoretic methods to obviate the need for using mass spectrometry for clinical samples. Their results “cast more doubt on the clinical usefulness and medical necessity of the serum free light chain assay.”
Source:
Baker T. Results after stem cell transplant can confuse patients and doctors about cancer’s status. Jagwire News. https://jagwire.augusta.edu/archives/46434. Published August 2017. Accessed September 20, 2017.
Singh G. J Clin Med Res. 2017;9(8):671-679.
doi: 10.14740/jocmr3049w.
FDA Approves New Leukemia Treatments
The FDA has approved Besponsa (inotuzumab ozogamicin) for adults with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL), a rapidly progressing cancer affecting about 6,000 people each year. About 1 in 4 patients affected will die of the disease.
Inotuzumab ozogamicin is a targeted therapy “thought to work” by binding to B-cell ALL cancer cells that express the CD22 antigen, blocking the growth of cancerous cells. In a study of 326 patients with relapsed or refractory B-cell ALL who had received 1 or 2 prior treatments, 36% of 218 evaluated patients experienced complete remission for a median 8 months. Of the patients who received alternative chemotherapy, 17% experienced complete remission for a median 5 months.
A second drug, Vyxeos ( daunorubicin and cytarabine) liposome injection, is approved for adults with 2 types of acute myeloid leukemia (AML): newly diagnosed therapy-related AML (t-AML) or AML with myelodysplasia-related changes (AML-MRC).
An estimated 8% to 10% of patients with AML develop t-AML as a complication of chemotherapy or radiation. AML-MRC is characterized by a history of certain blood disorders and other significant mutations within cancer cells. Patients with either disease have a low life expectancy. Vyxeos is a fixed-combination of daunorubicin and cytarabine. It’s the first approved treatment specifically for these patients, says Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence.
In a study of 309 patients with newly diagnosed t-AML or AML-MRC, those in the Vyxeos group lived longer: median survival, 9.56 months vs. 5.95 months in the patients who received separate treatments with daunorubicin and cytarabine.The third drug, Idhifa (enasidenib), is approved for adults with relapsed or refractory AML who have a mutation in the IDH2 gene. Idhifa is an isocitrate dehydrogenase-2 inhibitor that blocks several enzymes that promote cell growth.
The drug was studied in a single-arm trial of 199 patients. With a minimum of 6 months of treatment, 19% of patients experienced complete remission for a median of 8.2 months; 4% experienced complete remission with partial hematologic recovery for a median 9.6 months. Of the 157 patients who required blood or platelet transfusions due to AML at the start of the study, 34% no longer did after treatment with Idhifa.
Idhifa is approved for use with a companion diagnostic, the RealTime IDH2 Assay, which is used to detect mutations in the IDH2 gene in blood or bone marrow.
Source:
FDA approves new treatment for adults with relapsed or refractory acute lymphoblastic leukemia [news release]. Silver Spring, MD: U.S. Food & Drug Administration; August 17,2017. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm572131.htm. Accessed August 31, 2017.
FDA approves new targeted treatment for relapsed or refractory acute myeloid leukemia [news release]. Silver Spring, MD: U.S. Food & Drug Administration; August 1, 2017. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm569421.htm. Accessed August 31, 2017.
FDA approves first treatment for certain types of poor-prognosis acute myeloid leukemia [news release]. Silver Spring, MD: U.S. Food & Drug Administration; August 3, 2017. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm569883.htm. Accessed August 31, 2017.
The FDA has approved Besponsa (inotuzumab ozogamicin) for adults with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL), a rapidly progressing cancer affecting about 6,000 people each year. About 1 in 4 patients affected will die of the disease.
Inotuzumab ozogamicin is a targeted therapy “thought to work” by binding to B-cell ALL cancer cells that express the CD22 antigen, blocking the growth of cancerous cells. In a study of 326 patients with relapsed or refractory B-cell ALL who had received 1 or 2 prior treatments, 36% of 218 evaluated patients experienced complete remission for a median 8 months. Of the patients who received alternative chemotherapy, 17% experienced complete remission for a median 5 months.
A second drug, Vyxeos ( daunorubicin and cytarabine) liposome injection, is approved for adults with 2 types of acute myeloid leukemia (AML): newly diagnosed therapy-related AML (t-AML) or AML with myelodysplasia-related changes (AML-MRC).
An estimated 8% to 10% of patients with AML develop t-AML as a complication of chemotherapy or radiation. AML-MRC is characterized by a history of certain blood disorders and other significant mutations within cancer cells. Patients with either disease have a low life expectancy. Vyxeos is a fixed-combination of daunorubicin and cytarabine. It’s the first approved treatment specifically for these patients, says Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence.
In a study of 309 patients with newly diagnosed t-AML or AML-MRC, those in the Vyxeos group lived longer: median survival, 9.56 months vs. 5.95 months in the patients who received separate treatments with daunorubicin and cytarabine.The third drug, Idhifa (enasidenib), is approved for adults with relapsed or refractory AML who have a mutation in the IDH2 gene. Idhifa is an isocitrate dehydrogenase-2 inhibitor that blocks several enzymes that promote cell growth.
The drug was studied in a single-arm trial of 199 patients. With a minimum of 6 months of treatment, 19% of patients experienced complete remission for a median of 8.2 months; 4% experienced complete remission with partial hematologic recovery for a median 9.6 months. Of the 157 patients who required blood or platelet transfusions due to AML at the start of the study, 34% no longer did after treatment with Idhifa.
Idhifa is approved for use with a companion diagnostic, the RealTime IDH2 Assay, which is used to detect mutations in the IDH2 gene in blood or bone marrow.
Source:
FDA approves new treatment for adults with relapsed or refractory acute lymphoblastic leukemia [news release]. Silver Spring, MD: U.S. Food & Drug Administration; August 17,2017. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm572131.htm. Accessed August 31, 2017.
FDA approves new targeted treatment for relapsed or refractory acute myeloid leukemia [news release]. Silver Spring, MD: U.S. Food & Drug Administration; August 1, 2017. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm569421.htm. Accessed August 31, 2017.
FDA approves first treatment for certain types of poor-prognosis acute myeloid leukemia [news release]. Silver Spring, MD: U.S. Food & Drug Administration; August 3, 2017. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm569883.htm. Accessed August 31, 2017.
The FDA has approved Besponsa (inotuzumab ozogamicin) for adults with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL), a rapidly progressing cancer affecting about 6,000 people each year. About 1 in 4 patients affected will die of the disease.
Inotuzumab ozogamicin is a targeted therapy “thought to work” by binding to B-cell ALL cancer cells that express the CD22 antigen, blocking the growth of cancerous cells. In a study of 326 patients with relapsed or refractory B-cell ALL who had received 1 or 2 prior treatments, 36% of 218 evaluated patients experienced complete remission for a median 8 months. Of the patients who received alternative chemotherapy, 17% experienced complete remission for a median 5 months.
A second drug, Vyxeos ( daunorubicin and cytarabine) liposome injection, is approved for adults with 2 types of acute myeloid leukemia (AML): newly diagnosed therapy-related AML (t-AML) or AML with myelodysplasia-related changes (AML-MRC).
An estimated 8% to 10% of patients with AML develop t-AML as a complication of chemotherapy or radiation. AML-MRC is characterized by a history of certain blood disorders and other significant mutations within cancer cells. Patients with either disease have a low life expectancy. Vyxeos is a fixed-combination of daunorubicin and cytarabine. It’s the first approved treatment specifically for these patients, says Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence.
In a study of 309 patients with newly diagnosed t-AML or AML-MRC, those in the Vyxeos group lived longer: median survival, 9.56 months vs. 5.95 months in the patients who received separate treatments with daunorubicin and cytarabine.The third drug, Idhifa (enasidenib), is approved for adults with relapsed or refractory AML who have a mutation in the IDH2 gene. Idhifa is an isocitrate dehydrogenase-2 inhibitor that blocks several enzymes that promote cell growth.
The drug was studied in a single-arm trial of 199 patients. With a minimum of 6 months of treatment, 19% of patients experienced complete remission for a median of 8.2 months; 4% experienced complete remission with partial hematologic recovery for a median 9.6 months. Of the 157 patients who required blood or platelet transfusions due to AML at the start of the study, 34% no longer did after treatment with Idhifa.
Idhifa is approved for use with a companion diagnostic, the RealTime IDH2 Assay, which is used to detect mutations in the IDH2 gene in blood or bone marrow.
Source:
FDA approves new treatment for adults with relapsed or refractory acute lymphoblastic leukemia [news release]. Silver Spring, MD: U.S. Food & Drug Administration; August 17,2017. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm572131.htm. Accessed August 31, 2017.
FDA approves new targeted treatment for relapsed or refractory acute myeloid leukemia [news release]. Silver Spring, MD: U.S. Food & Drug Administration; August 1, 2017. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm569421.htm. Accessed August 31, 2017.
FDA approves first treatment for certain types of poor-prognosis acute myeloid leukemia [news release]. Silver Spring, MD: U.S. Food & Drug Administration; August 3, 2017. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm569883.htm. Accessed August 31, 2017.
FDA approves first gene therapy – tisagenlecleucel for ALL
The U.S. Food and Drug Administration has approved tisagenlecleucel (Kymriah), a first-of-its-kind chimeric antigen receptor T-cell (CAR T) therapy, for the treatment of children and young adults up to age 25 years with B-cell precursor acute lymphoblastic leukemia (ALL) that is refractory or in second or later relapse.

Tisagenlecleucel will carry a boxed warning regarding the CRS risk. Additionally, due to the CRS risk and risk of neurological events, the approval requires a risk evaluation and mitigation strategy (REMS), which includes elements to assure safe use, according to an FDA press release.
Special certification will be required for hospitals and associated clinics that dispense tisagenlecleucel. As part of certification, staff will be trained in the prescribing, dispensing, or administering of the therapy, and to recognize and manage CRS and neurological events.
Novartis, the maker of tisagenlecleucel, will be required to conduct postmarketing observational study.

Indeed, FDA commissioner Scott Gottlieb, MD, said the approval marks the entry to a “new frontier in medical innovation.”
“New technologies such as gene and cell therapies hold out the potential to transform medicine and create an inflection point in our ability to treat and even cure many intractable illnesses,” he said in the press statement.
Tisagenlecleucel is a genetically modified autologous T-cell immunotherapy involving customized treatment created using a patient’s own T cells. The T cells are genetically modified to include a chimeric antigen receptor that directs the T cells to target and kill leukemia cells with CD19 surface antigen, and are then infused back into the patient.
In a phase 2 clinical trial, the overall remission rate with tisagenlecleucel therapy was 83% in 63 children and young adults with relapsed/refractory B-cell precursor ALL for whom at least two prior lines of therapy had failed; the therapy was granted Fast Track, Priority Review, and Breakthrough Therapy designations.
“Kymriah is a first-of-its-kind treatment approach that fills an important unmet need for children and young adults with this serious disease,” Peter Marks, MD, director of the FDA’s Center for Biologics Evaluation and Research said in the press statement.
“Not only does Kymriah provide these patients with a new treatment option where very limited options existed, but a treatment option that shows promising remission and survival rates in clinical trials.”
At its meeting in July, the FDA ODAC agreed nearly unanimously that the risk mitigation plan put forward by Novartis, including planned 15-year follow-up and other mitigation measures, would be adequate for detecting serious consequences of CAR T-cell therapy.
This article was updated August 30, 2017.
The U.S. Food and Drug Administration has approved tisagenlecleucel (Kymriah), a first-of-its-kind chimeric antigen receptor T-cell (CAR T) therapy, for the treatment of children and young adults up to age 25 years with B-cell precursor acute lymphoblastic leukemia (ALL) that is refractory or in second or later relapse.
Tisagenlecleucel will carry a boxed warning regarding the CRS risk. Additionally, due to the CRS risk and risk of neurological events, the approval requires a risk evaluation and mitigation strategy (REMS), which includes elements to assure safe use, according to an FDA press release.
Special certification will be required for hospitals and associated clinics that dispense tisagenlecleucel. As part of certification, staff will be trained in the prescribing, dispensing, or administering of the therapy, and to recognize and manage CRS and neurological events.
Novartis, the maker of tisagenlecleucel, will be required to conduct postmarketing observational study.
Indeed, FDA commissioner Scott Gottlieb, MD, said the approval marks the entry to a “new frontier in medical innovation.”
“New technologies such as gene and cell therapies hold out the potential to transform medicine and create an inflection point in our ability to treat and even cure many intractable illnesses,” he said in the press statement.
Tisagenlecleucel is a genetically modified autologous T-cell immunotherapy involving customized treatment created using a patient’s own T cells. The T cells are genetically modified to include a chimeric antigen receptor that directs the T cells to target and kill leukemia cells with CD19 surface antigen, and are then infused back into the patient.
In a phase 2 clinical trial, the overall remission rate with tisagenlecleucel therapy was 83% in 63 children and young adults with relapsed/refractory B-cell precursor ALL for whom at least two prior lines of therapy had failed; the therapy was granted Fast Track, Priority Review, and Breakthrough Therapy designations.
“Kymriah is a first-of-its-kind treatment approach that fills an important unmet need for children and young adults with this serious disease,” Peter Marks, MD, director of the FDA’s Center for Biologics Evaluation and Research said in the press statement.
“Not only does Kymriah provide these patients with a new treatment option where very limited options existed, but a treatment option that shows promising remission and survival rates in clinical trials.”
At its meeting in July, the FDA ODAC agreed nearly unanimously that the risk mitigation plan put forward by Novartis, including planned 15-year follow-up and other mitigation measures, would be adequate for detecting serious consequences of CAR T-cell therapy.
This article was updated August 30, 2017.
The U.S. Food and Drug Administration has approved tisagenlecleucel (Kymriah), a first-of-its-kind chimeric antigen receptor T-cell (CAR T) therapy, for the treatment of children and young adults up to age 25 years with B-cell precursor acute lymphoblastic leukemia (ALL) that is refractory or in second or later relapse.
Tisagenlecleucel will carry a boxed warning regarding the CRS risk. Additionally, due to the CRS risk and risk of neurological events, the approval requires a risk evaluation and mitigation strategy (REMS), which includes elements to assure safe use, according to an FDA press release.
Special certification will be required for hospitals and associated clinics that dispense tisagenlecleucel. As part of certification, staff will be trained in the prescribing, dispensing, or administering of the therapy, and to recognize and manage CRS and neurological events.
Novartis, the maker of tisagenlecleucel, will be required to conduct postmarketing observational study.
Indeed, FDA commissioner Scott Gottlieb, MD, said the approval marks the entry to a “new frontier in medical innovation.”
“New technologies such as gene and cell therapies hold out the potential to transform medicine and create an inflection point in our ability to treat and even cure many intractable illnesses,” he said in the press statement.
Tisagenlecleucel is a genetically modified autologous T-cell immunotherapy involving customized treatment created using a patient’s own T cells. The T cells are genetically modified to include a chimeric antigen receptor that directs the T cells to target and kill leukemia cells with CD19 surface antigen, and are then infused back into the patient.
In a phase 2 clinical trial, the overall remission rate with tisagenlecleucel therapy was 83% in 63 children and young adults with relapsed/refractory B-cell precursor ALL for whom at least two prior lines of therapy had failed; the therapy was granted Fast Track, Priority Review, and Breakthrough Therapy designations.
“Kymriah is a first-of-its-kind treatment approach that fills an important unmet need for children and young adults with this serious disease,” Peter Marks, MD, director of the FDA’s Center for Biologics Evaluation and Research said in the press statement.
“Not only does Kymriah provide these patients with a new treatment option where very limited options existed, but a treatment option that shows promising remission and survival rates in clinical trials.”
At its meeting in July, the FDA ODAC agreed nearly unanimously that the risk mitigation plan put forward by Novartis, including planned 15-year follow-up and other mitigation measures, would be adequate for detecting serious consequences of CAR T-cell therapy.
This article was updated August 30, 2017.
The Challenges of Precision Medicine and New Advances in Molecular Diagnostic Testing in Hematolymphoid Malignancies: Impact on the VHA (FULL)
In January 2015, President Obama introduced the Precision Medicine Initiative, a program set up to identify new biomedical discoveries for the development of a personalized knowledge base of disease entities and individualized treatments. Advances in precision medicine typically involve the use of targeted therapies tailored to individual genetic characteristics identified with molecular testing. The goals are to improve survival and reduce adverse effects. With an initial budget of $215 million, this initiative presented a unique opportunity to combine efforts in genomic discovery, bioinformatic analysis, and health information technology to move toward data-driven, evidence-based precision medicine.1
The VHA is the largest comprehensive health care system in the U. S. and has more than 1,700 care sites serving nearly 9 million veterans each year. The budget for this single-payer system is proposed by the President and approved by Congress. As the VHA must treat a diverse and aging veteran population in an environment of rising costs and budget constraints, limited resources must be monitored and appropriated for the most cost-effective health care delivery. Precision medicine offers a model in which physicians can select the most appropriate diagnostic tests in defined clinical settings to direct clinical care. It supports the testing needed to subdivide each disease category into distinct subcategories. Nevertheless, the need for fiscal responsibility in a capitated health care system recommends testing in cases in which it can change therapy or prognosis rather than for purely academic reasons.
Pathology and Laboratory Medicine Service
Given limited resources and an increasing number of requests for advanced molecular testing, the VA Pathology and Laboratory Medicine Service (P&LMS) formed the Molecular Genetics Pathology Workgroup (MGPW) in September 2013. The charter listed the tasks of the MGPW to “provide recommendations on how to effectively use molecular genetics tests, promote increased quality and availability of testing within the VHA, encourage internal referral testing, provide an organizational structure for Molecular Genetics Testing Consortia, and create a P&LMS policy for molecular genetic testing in general, specifically addressing the issues surrounding laboratory developed testing.” The MGPW has 4 subcommittees: molecular oncology, pharmacogenetics, hematopathology molecular genetics (HMG), and genetic medicine. Since its inception, the HMG subcommittee has had several objectives:
- Standardize the molecular testing nomenclature for and develop practice guidelines for acute myeloid leukemia (AML), myeloproliferative neoplasms (MPN), myelodysplastic syndrome (MDS), chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma, lymphoma, and plasma cell neoplasms;
- Develop standardized reporting guidelines for current VA molecular laboratories;
- Identify new tests as they are being reported in the literature and collaborate with hematology and oncology services to evaluate the clinical utility of these tests for VA patients;
- Network current VA molecular laboratories, perform fact-finding for these laboratories, and compile test menus; and
- Assess for the formation of VA-wide interfacility consultation services for hematopathology so that all VA facilities, regardless of their complexity, will be able to access the expertise of hematopathology-trained pathologists (Appendix).
The HMG subcommittee met monthly and discussed various diagnostic entities in hematopathology. For hematolymphoid malignancies, it was generally agreed that the traditional laboratory tools of morphology, flow cytometry, and immunohistochemistry (IHC) are standard in initial assessment and often in diagnosis. As the clinical molecular and cytogenetic assays of karyotype, fluorescence in situ hybridization (FISH), advanced DNA sequencing, microarray, and highly sensitive polymerase chain reaction (PCR) analysis affect diagnosis, subclassification, minimal residual disease (MRD) monitoring, prognosis, and therapy selection, their use is marked by a high degree of variability. As a result, standardization is needed. As each laboratory develops and reports ancillary testing, the variable reporting formats may generate postanalytic errors.
A detailed description of all molecular methodologies is beyond the scope of this article. For practicing pathologists, challenges remain in overall cost and reimbursement, extensive and time-consuming data analysis, and in some cases, interpretation differences.
Myeloid Neoplasms
Myeloid malignancies were divided into AML, MPN, and MDS. Next-generation sequencing (NGS) information for these malignancies was used to identify various contributory functional categories, including cell signaling (FLT3, KIT, JAK2, MPL, KRAS/NRAS, PTPN11, NF1, CSF3R); transcription (CEBPA, RUNX1, GATA1/GATA2, PHF6, ETV6); splicing (SF3B1, SRSF2, ZRSR2, U2AF1); epigenetics (DNMT3A, TET2, IDH1/IDH2, ASXL1, EZH2, SUZ12, KDM6A); cohesin complex (STAG2, SMC1A, SMC3, RAD21); and cell cycle (TP53, NPM1).2
Acute Myeloid Leukemia
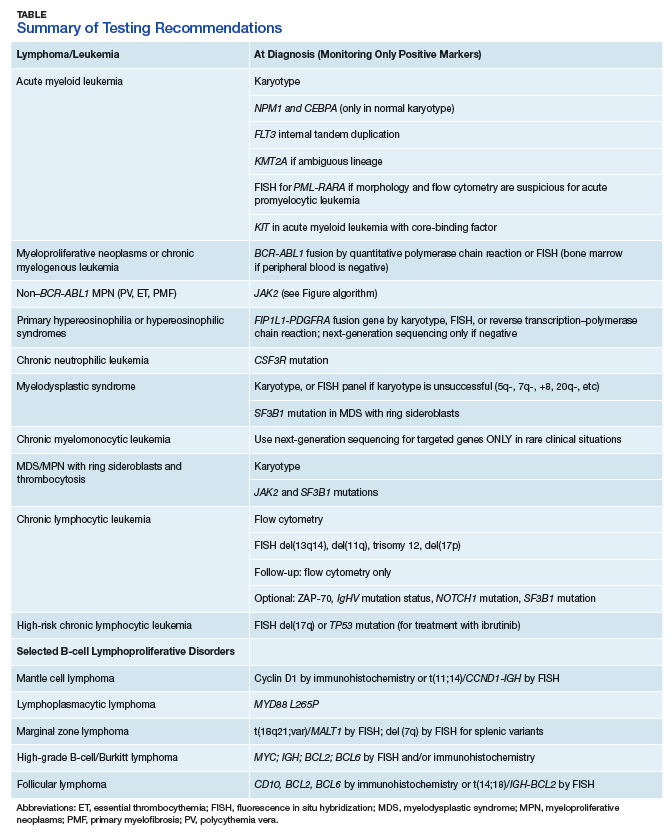
The HMG subcommittee reviewed the literature on prognostically significant genes in myeloid leukemias. Karyotype abnormalities, such as t(8;21) and inv(16), collectively known as the core-binding factor (CBF) leukemias, t(15;17), t(11q23) (KMT2A/MLL), and so forth, are recurrent lesions in AML. Included in the minimum set of genes recommended by the National Comprehensive Cancer Network (NCCN) for AML prognosis evaluation are nucleolar protein nucleophosmin (NPM1), CCAAT/enhancer-binding protein
Some of the chromosomal translocations, such as inv(16)/t(16;16) in AML and t(15;17) in acute promyelocytic leukemia, can be monitored with FISH or reverse transcription–PCR (RT-PCR) analysis. As NPM1 mutations tend to be seen in recurrence, they can be used as molecular markers for MRD. Other mutations that provide important prognostic information in AML include:
- Activating insertions/duplications in the FLT3 receptor tyrosine kinase, which can be detected with PCR sizing assays;
- Mutations in the KIT receptor tyrosine kinase, which can be detected with DNA sequencing or more limited hotspot PCR;
- Mutations in the DNA methyltransferase, DNMT3A, a poor prognostic indicator seen in 22% of cases of AML, also detected with gene sequencing or more limited hotspot PCR; and
- Another set of genes, TET2, IDH1, IDH2, KRAS, NRAS, EZH2, and ASXL1, is mutated in MPN as well as AML and MDS, making a common molecular panel with next-generation sequencing useful in diagnosing and risk-stratifying all myeloid neoplasms.
The HMG subcommittee agreed that, for de novo AML, chromosomal karyotype is the standard of care, necessary in detecting known cytogenetic abnormalities as well as a wide range of lesions that might indicate a diagnosis of AML with myelodysplasia-related changes at time of diagnosis. In addition, molecular analysis of FLT3 is useful in determining prognosis, and CEBPA (biallelic) and NPM1 mutations are good prognostic factors in normal-karyotype AML. KMT2A (MLL) rearrangements should be tested with FISH if the lineage is ambiguous. The PML-RARA fusion gene also should be tested with FISH if morphologic and flow cytometry results suggest acute promyelocytic leukemia (Table). At this time, testing for TP53, DNMT3A, RAS, and other such mutations is not recommended because it is not cost-effective for the VA.
Myeloproliferative Neoplasms
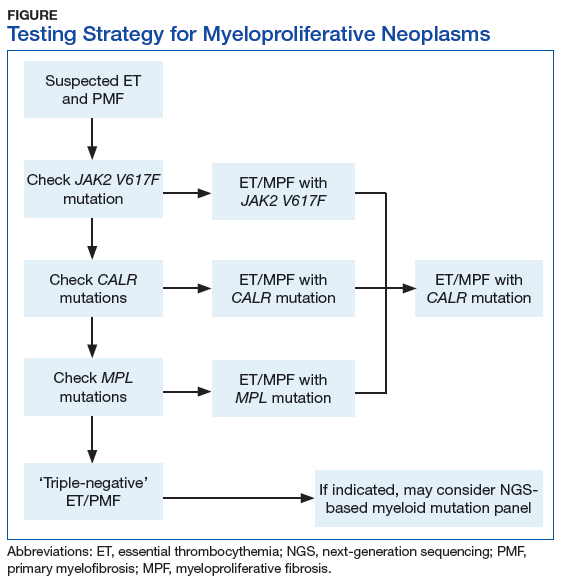
Myeloproliferative neoplasms are clonal hematopoietic stem cell disorders characterized by proliferation of at least 1 myeloid lineage: granulocytic, erythroid, or megakaryocytic. Myeloproliferative neoplasms show a range of recurrent chromosomal translocations, such as BCR-ABL1 fusion in chronic myelogenous leukemia (CML) that can be detected with RT-PCR analysis as well as FISH. In CML, BCR-ABL1 fusion transcript levels detected by a quantitative PCR (qPCR) method are now used to monitor the course of CML therapy with tyrosine kinase inhibitors (TKIs) and to trigger a treatment change in drug-resistant cases. Given the importance of qPCR in clinical management, significant progress has been made in standardizing both the PCR protocol and the reference materials used to calibrate the BCR-ABL1 PCR assay. BCR-ABL1–negative MPN, including polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF), are most commonly associated with mutations in the tyrosine kinase JAK2. Mutations in CALR and MPL are seen in a subset of patients with ET and PMF as well, whereas PV is essentially exclusively a disease of JAK2 mutations.
Chronic myelogenous leukemia is the prototypical MPN. To establish the initial diagnosis, FISH and/or qPCR for BCR-ABL1 fusion should be used. If CML is confirmed, the sample can be reflexed to qPCR BCR-ABL1 on the initial peripheral blood and/or bone marrow sample(s) to establish the patient’s baseline. In addition, a bone marrow sample (aspirate) should be used for a complete karyotype and for morphologic confirmation of disease phase.
For follow-up assessment of CML patients’ response to TKI treatment, qPCR for BCR-ABL1 should be tested with a peripheral blood sample or a bone marrow sample every 3 months.4 A peripheral blood sample is more commonly used because it is conveniently obtained. Early molecular response as indicated by a BCR-ABL1 transcript ratio of < 10% on the International Scale at 3 months, has a strong prognostic value.5 Major molecular response as indicated by a BCR-ABL1 transcript ratio of < 0.1% on the International Scale at 12 to 18 months is also highly prognostic.5
After the peripheral blood sample becomes negative for BCR-ABL1 by qPCR, testing bone marrow samples may be considered. If important treatment response benchmarks are not achieved, or response is lost with rising BCR-ABL1 levels (TKI resistance), ABL1 kinase domain mutation analysis as well as repeat FISH (to assess for copy number multiplication) should be performed to guide further management. Patients with the ABL1 T315I mutation are resistant to all first-line TKIs but may respond to later third-generation TKIs.6
BCR-ABL1–negative MPNs include PV, ET, and PMF. Bone marrow morphology remains the cornerstone of ET and PMF diagnosis. The discovery of JAK2, CALR, and MPL mutations has contributed to how these disorders are diagnosed.7-12 Besides providing the clonality proof that is crucial for diagnosis, the molecular markers influence the prognosis. The JAK2 (p.V617F) or less common JAK2 exon 12 mutations, which are detected in more than 95% of PV cases, are used as molecular markers to confirm diagnosis.7 Further, the JAK2 (p.V617F), CALR (exon 9), and MPL (exon 10) mutations are detected in ET (~60%, 25%, and 3%-5%, respectively) and PMF (~55%, 30%, and 5%, respectively).12 If ET or PMF is suspected clinically, first JAK2 (p.V617F) mutation analysis should be performed, then CALR mutation analysis, and finally MPL mutation analysis. Although novel gain-of-function JAK2 and MPL mutations were recently discovered in triple-negative ET (negative for canonical mutations in JAK2, CALR, and MPL) and PMF by whole exome sequencing,13 clinical testing is not readily available. Besides its utility in the initial diagnosis of ET and PMF, the JAK2 or CALR mutation assay also may be considered for bone marrow transplantation follow-up (Table).14
Despite the continuing debate on the classification of eosinophilic myeloid disorders, the discovery of the FIP1L1-PDGFRA fusion represents a major milestone in the understanding of these disorders.15,16 Unlike PDGFRB (5q33) and FGFR1 (8p11) rearrangements, which can be detected with routine chromosomal analysis (cytogenetics), the cryptic FIP1L1-PDGFRA fusion must be detected with FISH (for CHIC2 deletion) or RT-PCR analysis. It should be pointed out that, as most eosinophilia is reactive or secondary, molecular testing for FIP1L1-PDGFRA fusion is indicated only when primary hypereosinophilia or hypereosinophilic syndrome (HES) is suspected. This is particularly the case in the following hypereosinophilia accompanying conditions: CML-like morphology, but BCR-ABL1–negative; chronic myelomonocytic leukemia (CMML)–like morphology with a normal karyotype; and new onset of cardiac damage or dysfunction.17
Primary eosinophilic myeloid disorders with PDGFRA or PDGFRB rearrangements can be treated with TKIs (eg, imatinib). Next-generation sequencing may be considered in cases of presumed HES when there is no identifiable karyotypic or FISH abnormality. Recent studies have found that cases of HES with somatic mutations indicating clonality had adverse clinical outcomes similar to those of cases of chronic eosinophilic leukemia.18
The discovery of CSF3R mutations offers a new molecular marker for the diagnosis of chronic neutrophilic leukemia (CNL), an MPN.19 The CSF3R (p.T618I) mutation or another activating CSF3R mutation is now used as a diagnostic criterion for CNL. Identification of specific CSF3R mutations may have therapeutic implications as well. The test should be ordered only for patients with clinical and morphologic findings suggestive of CNL; reactive neutrophilic leukocytosis (eg, infection, inflammation) should be ruled out before the test is ordered.
Myelodysplastic Syndrome
Myelodysplastic syndrome is a group of clonal bone marrow disorders characterized by ineffective hematopoiesis, manifested by morphologic dysplasia in ≥ 1 hematopoietic lineages and peripheral cytopenias (hemoglobin level, < 10 g/dL; platelet count, < 100×103/µL; absolute neutrophil count, < 1.8×103/µL). Diagnosis and classification of MDS depend mainly on the degree of morphologic dysplasia and blast percentages, as determined by examining well-prepared cellular bone marrow aspirate smears and/or biopsy touch preparations and peripheral blood smears.
Conventional karyotyping is an essential part of the diagnostic workup for all presumptive cases of MDS and is of both diagnostic and prognostic importance.20 About 60% of MDS cases have recurrent cytogenetic abnormalities, which can be detected with conventional karyotyping. If a high-quality cytogenetic analysis cannot be performed (eg, the bone marrow sample is inadequate), or if quick turnaround is required, an alternative FISH panel may be used to detect some of the common MDS-associated chromosomal abnormalities (eg, 5q deletion, 7q deletion/monosomy 7, +8, 20q deletion).21 Sequencing with FISH also can be useful for assessing MRD by detecting a previously identified chromosomal abnormality.
Targeted sequencing of a limited number of genes can detect mutations in the vast majority of patients with MDS. The most commonly mutated genes in MDS are SF3B1, TET2, SRSF2, ASXL1, DNMT3A, RUNX1, U2AF1, TP53, and EZH2. Mutations in SRSF2 cause RNA splicing abnormalities. In addition, mutations in TP53, EZH2, RUNX1, and ASXL1 are associated with poor prognosis,22,23 whereas mutations in SF3B1 confer better event-free survival.24 Despite these developments, the HMG subcommittee agreed that NGS-based mutation panels are not cost-effective for the VA population at this time and should not be included in a MDS workup. Only in rare situations and when clinically indicated (to change disease classification or patient management) should evaluation for specific gene mutations be considered—for instance, the SF3B1 mutation for patients with probable MDS with ring sideroblasts, if ring sideroblasts are < 15%.25
Myelodysplastic/Myeloproliferative Neoplasms
Myelodysplastic/myeloproliferative neoplasms are a group of myeloid neoplasms with clinical, laboratory, and morphologic features that overlap both MDS and MPN. In MDS/MPN, the karyotype is often normal or shows abnormalities in common with MDS.
In cases of unexplained monocytosis for which there is clinical concern for CMML, morphologic evaluation and conventional chromosomal karyotyping should be performed after other secondary causes and known myeloproliferative and myelodysplastic entities have been excluded. If concomitant hypereosinophilia is present and the karyotype is normal, FISH or PCR-based assay should be performed to rule out FIP1L1-PDGFRA rearrangements. BCR-ABL1, PDGFRB, FGFR1, and t(8;9)/PCM1-JAK2 rearrangements typically are detected with high-quality cytogenetic analysis and thus do not require targeted molecular assays. Although certain gene mutations (eg, SRSF2, TET2, ASXL1, CBL) are commonly detected in CMML, the HMG subcommittee does not recommend sequencing-based mutation panels, as there is insufficient information for testing for prognostic or treatment stratification.
If MDS/MPN with ring sideroblasts and thrombocytosis is suspected on the basis of the clinical and morphologic criteria, molecular tests for the JAK2 (p.V617F) and SF3B1 mutations may be considered in an effort to help confirm the diagnosis.
Atypical CML is a rare MDS/MPN subtype that is now better characterized molecularly with SETBP1 and/or ETNK1 mutations, which are detectable in up to a third of cases. If clinical suspicion is high, sequencing may be diagnostically helpful.
Lymphoid Neoplasms
Chronic Lymphocytic Leukemia
In CLL, recurrent chromosomal abnormalities (eg, deletions of 13q, trisomy 12, deletions of 11q, deletions of 17p) have clear prognostic value and can be detected with FISH. Other prognostic information, such as somatic mutation of immunoglobulin heavy chain variable (IgHV) genes, TP53 mutations, SF3B1, and NOTCH1 mutation, are mostly derived from PCR-based assays. The discovery of recurrently mutated genes in CLL has increased with the use of highly sensitive sequencing methods constructing a more detailed landscape of CLL at genetic, epigenetic, and cellular levels. A recent literature review summarizes the vast heterogeneity of CLL with recurrent pathogenetic findings in MYD88, SF3B1, TP53, ATM, and NOTCH1 signaling pathways.26 The treatment of CLL is rapidly evolving, and many clinical trials are proposing a change from the “watch and wait” paradigm to treatment upon initial presentation based on molecular findings. Additional testing based on new treatment options from current clinical trials will be recommended.
Flow cytometry and morphology are standard for CLL diagnosis. The HMG subcommittee recommends FISH for del(13q14), del(11q), trisomy 12, and del(17p) at time of diagnosis or immediately before therapy initiation. Zeta-chain (
Other B-Cell Lymphoproliferative Disorders
Unlike the common molecular changes in CLL, in other mature B-cell lymphomas, chromosomal translocations that juxtapose a variety of different oncogenes next to an Ig gene enhancer usually are—and those that switch regions less commonly are—important initiating events that can be detected with PCR, DNA sequencing, or FISH. In follicular lymphoma (FL), Burkitt lymphoma, marginal zone lymphoma (MZL), and mantle cell lymphoma (MCL), these oncogenes driven by an Ig gene enhancer typically include BCL2, MYC, MALT1, and CCND1 (cyclin D1), respectively. Molecular variants of these lymphomas that lack these classical translocations often activate homologous genes (eg, cyclin D3/CCND3 is activated in variants of MCL).
Morphology, flow cytometry, and IHC are routinely used for diagnosis. In inconclusive cases, Ig gene rearrangement by PCR may be used. The Table summarizes common molecular changes in B-cell lymphomas.
Mantle cell lymphoma. MCL is a non-Hodgkin lymphoma subtype characterized by t(11;14) (q13;q32) translocations that in the majority of cases lead to overexpression of cyclin D1 (BCL1). Recent molecular profiling has identified an MCL variant that is cyclin D1–negative but SOX11-positive and may have a more aggressive clinical course.30 SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive MCL.
Lymphoplasmacytic lymphoma. Lymphoplasmacytic lymphoma (LPL), MZL, and CLL/small lymphocytic lymphoma are well-defined clinicopathologic entities. However, distinguishing LPL from MZL and atypical cases of CLL can sometimes be difficult because of overlapping clinical and morphologic features. Recent studies have identified a recurrent L265P mutation in the MYD88 gene in 90% to 95% of LPL cases with IgM paraprotein and in 40% to 50% of the rare non-IgM LPL cases. In contrast, the mutation is much less frequently present in MZL and other low-grade B-cell neoplasms (2%-7%).31 Therefore, testing for this abnormality can be a diagnostic aid in these difficult-to-classify cases. In addition, from a therapeutic perspective, presence or absence of MYD88 mutation may prove more significant than presence of a specific paraprotein or histopathologic features. Ibrutinib has shown efficacy in LPL and demonstrates improved response rates in patients with MYD88 mutation compared with that of their mutation-negative counterparts.32 Several MYD88 inhibitors are in clinical trials. This again indicates the need to more accurately identify and subclassify these non-IgM LPL cases to ensure appropriate molecular evaluation.
Hairy cell leukemia. Flow cytometry and morphology are usually sufficient for a hairy cell leukemia (HCL) diagnosis. However, rare cases are difficult to distinguish variant HCL from other mimics. The BRAF V600E mutation recently was described as a disease-defining molecular marker for HCL—present in nearly all HCL cases but virtually absent in HCL mimics. Therefore, detection of the BRAF mutation by IHC stain with specific antibody or PCR analysis is highly sensitive and specific for the diagnosis of HCL.33
Diffuse large B-cell lymphoma. Recent molecular analysis has created various risk stratification schemata for diffuse large B-cell lymphoma (DLBCL). The HGM subcommittee agrees that well-preserved morphology, IHC, flow cytometry, and FISH-specific markers (BCL2, BCL6, cMYC) are sufficient for diagnostic, prognostic, and therapeutic purposes. Although a wide range of genes have been implicated in the pathogenesis of DLBCL, sequencing and gene expression profiling are not cost-effective at this time and do not add benefit to patient treatment.
The MYD88 L265P mutation has been identified in DLBCL, particularly the activated B-cell-like type and primary central nervous system lymphoma (PCNSL), and may have implications for ibrutinib therapy. PCNSL commonly manifests aggressive clinical behavior and has a poor prognosis. It has been proposed that the MYD88 mutation can be used as a genetic hallmark for PCNSL to distinguish CNS involvement by systemic DLBCL from PCNSL.34
Plasma cell neoplasms. Flow cytometry is acceptable for the diagnosis of plasma cell neoplasms and for residual disease follow-up. Chromosomal karyotype or FISH for IGH/CCND1, IGH/MMSET, and IGH/CMAF dual fusion probes is recommended in conjunction with morphology, IHC, and flow cytometry. In plasma cell myeloma, several genetic mutations can be detected with NGS, including mutations in NRAS, KRAS, TP53, BCL7A, DIS3, and FAM46C.35 Less commonly, BRAF mutations, previously described in melanoma and several other solid tumors, can be detected with DNA sequencing in 4% of multiple myeloma cases, which may prove promising for targeted therapy with BRAF inhibitors. However, current therapeutic decisions are based on genetic and clinical factors, and sequence-based assays are not recommended at this time.
Follicular lymphoma. Cytology, histology, and IHC typically are sufficient for diagnosing FL. In difficult-to-diagnose cases and in cases with scant material, additional tests may help with diagnosis. Eighty to ninety percent of FL cases have t(14;18)(q32;q21), which places the BCL2 gene transcription under the control of the IGH promoter. In addition, about 10% of FL cases have 3q27 aberrancies at the BCL6 gene.36-38 More recently, cases of FL with bulky inguinal disease negative for IGH-BCL2 and BCL6 translocations were found to have 1p36 deletions. These 1p36-deleted FLs typically have a diffuse pattern and a good prognosis.39 For t(14;18), 3q27, or 1p36, FISH is a sensitive means for detecting these translocations, as is PCR for IGH-BCL2.40 There are reports that t(14;18) can be detected in a substantial fraction of otherwise healthy donors at levels and rates that depend on the type of detection test used.41-43 In addition, between one-fourth and one-third of de novo DLBCLs show t(14;18), and about one-third show BCL6 abnormalities at 3q27. Therefore, these genetic changes are not specific for FL and should not be used to subtype a lymphoma as follicular in origin.
Use of IGH-BCL2 as a marker for MRD is still controversial. Some studies have found that a postinduction and posttransplantation IGH-BCL2-positive finding by PCR predicted relapse.44,45 However, others studies have not found significance to postinduction IGH-BCL2 positivity.46 The NCCN guidelines recommend testing for IGH-BCL2 or BCL6 translocations or 1p36 deletion only if this testing is needed for diagnosis. The guidelines do not recommend using these genetic assays in follow-up biopsies, as the importance of treating early relapse has not been definitively demonstrated.
Therefore, if a lymphoma has morphologic, histologic, and IHC findings consistent with FL, then cytogenetic, FISH, or PCR testing is not needed for diagnosis but may be used as confirmation. Follow-up molecular and cytogenetic testing should be avoided if the original cytogenetic abnormality is unknown. That is, IGH-BCL2 FISH should be performed in follow-up samples only if the original lymphoma is known to contain the translocation. As follow-up genetic testing is of disputed clinical significance even in cases in which the original molecular change is known, the NCCN recommendations for therapy are no different. The HMG subcommittee does not recommend molecular or cytogenetic testing in FL beyond what is required for initial diagnosis.
T-Cell Lymphomas
Mature T-Cell Lymphoma and Leukemia
For mature T-cell lymphoma (TCL) and leukemia, the clinical and morphologic criteria have a very important role in the initial workup. However, IHC immunophenotyping is crucial for definitive diagnosis and subclassification. Flow cytometry is routinely used in diagnosing diseases such as T-cell prolymphocytic leukemia (TPLL), T-cell large granular lymphocytic (LGL) leukemia, and Sézary syndrome. T-cell clonality studies, preferably with BIOMED-II–validated primers against targets such as T-cell receptor
Significant advances in TCL classification have led to revisions and the inclusion of new provisional entities in the 2016 World Health Organization classification of lymphoid neoplasms.47 Many of these changes originated in studies of gene expression profiling and the genetic landscape of T-cell neoplasms. Even though subsets of peripheral TCL not otherwise specified (PTCL-NOS) have been recognized on the basis of phenotypic and molecular abnormalities with possible clinical implications, in most cases molecular testing is not part of routine practice. Typically, only a few cytogenetic abnormalities and genetic mutations are used in the evaluation of TCL and T-cell leukemia.
A group of T-cell lymphoproliferative disorders with expression of T follicular helper cell markers can be identified with IHC. These disorders include angioimmunoblastic TCL; follicular TCL, a new entity that is a PTCL-NOS subset; and primary cutaneous CD4-positive small/medium T-cell lymphoproliferative disorder. The neoplastic cells should express at least 2 or 3 T follicular helper cell–related antigens, including CD279/PD1, CD10, BCL6, CXCL13, ICOS, SAP, and CCR5; the most commonly used are PD1, BCL6, and CD10. Recurrent fusion of ITK-SYK translocation t(5;9) or CTLA4-CD28 is also common in follicular TCL. Although recurrent mutation is found in these entities, conventional karyotyping or IHC should be sufficient for diagnosis.
Cutaneous γ -Δ T-Cell Lymphoma
Among cutaneous TCLs, primary cutaneous
Peripheral T-Cell Lymphoma
Gene expression profiling analysis of PTCLs has identified at least 3 subtypes characterized by overexpression of GATA3, TBX21, and cytotoxic genes and expression of the corresponding proteins with IHC.47 These subtypes are associated with different clinical behavior and therapy responses. The GATA3 subtype has an inferior prognosis and shows a high level of T helper type 2 cytokines, which can be identified with IHC. As IHC-stained GATA3 has been available as a marker of urothelial carcinoma at most IHC laboratories, GATA3 IHC staining also may be considered in the evaluation of PTCLs.
Many monoclonal antibody therapies are being used as primary or secondary regimens in the treatment of TCL. Clinical trials are working to establish their efficacy. If treatment with a monoclonal antibody is being considered, it is appropriate to conduct IHC to demonstrate the presence of the target antigen and at follow-up, to demonstrate the efficacy of treatment. These therapies include alemtuzumab, which targets CD52, and brentuximab, which targets CD30.
T-Cell Large Granular Lymphocytic Leukemia
T-cell LGL leukemia is a complex diagnosis that requires persistent clonal expansion of LGLs and clinically peripheral blood cytopenia. In many cases, the diagnosis is difficult to establish, as benign large granular lymphocytosis with clonal T cells may occur in conjunction with viral infections or autoimmune disorders. Somatic mutations in the STAT3 (signal transducer and activator of transcription 3) gene are found in 40% of patients with T-cell LGL leukemia.49 More recently, somatic mutations in the STAT5B gene were identified in 2% of T-cell LGL leukemia subsets. The clinical course of T-cell LGL leukemia in patients with the STAT5B mutation is aggressive and fatal, clearly different from the relatively favorable course of typical T-cell LGL leukemia.50 The HMG subcommittee recommends considering a STAT3 and STAT5B mutation study for selected cases in which it is difficult to distinguish true T-cell LGL leukemia from its reactive expansions.
T-Cell Prolymphocytic Leukemia
T-cell prolymphocytic leukemia (T-PLL) is a rare, aggressive disease and is most commonly associated with a prolymphocytic morphology and expression of CD4. However, since a specific immunophenotypic profile of T-PLL has not been identified, flow cytometry is not adequate in isolation for definitive classification as T-PLL.51 A diagnosis of T-PLL often requires cytogenetics or a FISH study to confirm a suspected case. Most TPLL cases harbor characteristic chromosomal abnormalities involving 14q11.2 (TCR
Anaplastic Large Cell Lymphoma
The World Health Organization recognizes 3 distinct types of anaplastic large cell lymphoma (ALCL): systemic anaplastic lymphoma kinase (ALK)–positive ALCL, systemic ALK-negative ALCL, and primary cutaneous ALCL. Systemic ALK-positive ALCLs consistently have ALK gene rearrangements and favorable outcomes. The most common translocation is the t(2;5) rearrangement of NPM1 and ALK, though other ALK partners are also possible. In contrast, systemic ALK-negative ALCLs lack ALK gene rearrangements and as a whole have outcomes inferior to those of systemic ALK-positive ALCLs. However, studies have found systemic ALK-negative ALCL to be a genetically and clinically heterogeneous entity.54 About 30% of cases have rearrangements of the DUSP22-IRF4 locus on 6p25.3 (DUSP22 rearrangement), and these cases have favorable outcomes similar to those of systemic ALK-positive ALCL.55 Only 8% of patients have TP63 rearrangements and very poor outcomes. The remaining cases lack ALK, DUSP22, and TP63 rearrangements and have intermediate outcomes. The HMG subcommittee recommends considering DUSP22 rearrangement by FISH in the evaluation of systemic ALK-negative ALCL.
Conclusion
The pathologic diagnosis, classification, and risk stratification of lymphoma and leukemia require an approach that integrates morphology, flow cytometry, cytogenetics, and molecular pathology. Rapidly evolving molecular techniques currently allow for detailed description of the molecular defects in lymphoma and leukemia, including driver mutations, amplification/deletion events, and clonal evolution. Unfortunately, the technical ability to catalogue the molecular defects in lymphoma and leukemia, often at great expense, is outpacing the ability to use this detailed information in treating patients with hematologic malignancies. The challenge, then, is to identify best practices for the diagnosis and classification of lymphoma and leukemia in VHA hospitals that incorporate the most useful molecular tests without wasting financial resources.
In this report, the HMG subcommittee of the MGPW has presented its recommendations for molecular testing in AML, MPN, MDS, and lymphomas in the context of standard morphologic and immunophenotypic approaches to hematopathology diagnosis and classification. Adoption of these recommendations by VHA hospitals and clinics should help ensure that all VA patients with hematologic malignancies benefit from the latest advances in precision medicine.
Within the vast and comprehensive national VHA health care system are multiple centers of expertise in hematopathology. In addition, multiple VA clinical molecular diagnostic laboratories are performing state-of-the-art testing. The HMG subcommittee proposes that, to make best use of these expert resources, the VHA should establish an interfacility hematopathology consultation service. This service would allow any VA pathologist to consult a board-certified hematopathologist regarding use of ancillary molecular genetic testing in the diagnosis of hematologic malignancy.
In addition, the HMG subcommittee recommends consolidating VA molecular diagnostic reference laboratories and having them perform molecular testing for other VA hospitals rather than using commercial reference laboratories, where testing standards are not uniform and results may be difficult to interpret. Several well-established VA clinical laboratories with technical expertise and informatics support are already performing selected molecular diagnostic testing. These laboratories’ resources should be expanded, where practical, to cost-effectively provide VA expertise to all veterans and to improve access to appropriate molecular diagnostic testing.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of
Click here to read the digital edition.
1. Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372(9):793-795.
2. Matynia AP, Szankasi P, Shen W, Kelley TW. Molecular genetic biomarkers in myeloid malignancies. Arch Pathol Lab Med. 2015;139(5):594-601.
3. Wang ML, Bailey NG. Acute myeloid leukemia genetics: risk stratification and implications for therapy. Arch Pathol Lab Med. 2015;139(10):1215-1223.
4. Marum JE, Branford S. Current developments in molecular monitoring in chronic myeloid leukemia. Ther Adv Hematol. 2016;7(5):237-251.
5. Hughes TP, Saglio G, Kantarjian HM, et al. Early molecular response predicts outcomes in patients with chronic myeloid leukemia in chronic phase treated with frontline nilotinib or imatinib. Blood. 2014;123(9):1353-1360.
6. Pallera A, Altman JK, Berman E, et al. Guidelines insights: chronic myeloid leukemia, version 1.2017. J Natl Compr Canc Netw. 2016;14:1505-1512.
7. James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144-1148.
8. Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779-1790.
9. Beer PA, Campbell PJ, Scott LM, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood. 2008;112(1):141-149.
10. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Characteristics and clinical correlates of MPL 515W>L/K mutation in essential thrombocythemia. Blood. 2008;112(3):844-847.
11. Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391-2405.
12. Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379-2390.
13. Harrison CN, Vannucchi AM. Closing the gap: genetic landscape of MPN. Blood. 2016;127(3):276-278.
14. Tefferi A, Barbui T. Essential thrombocythemia and polycythemia vera: focus on clinical practice. Mayo Clin Proc. 2015;90(9):1283-1293.
15. Cools J, DeAngelo DJ, Gotlib J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348(13):1201-1214.
16. Valent P, Klion AD, Horny HP, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol. 2012;130(3):607-612.e609.
17. Bain B, Billiland D, Horny H, Verdiman J. Myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1. In: Swerdlow S, Campo E, Harris N, et al, eds. WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues. Vol 2. Lyon, France: IRAC Press; 2008:68-73.
18. Wang SA, Tam W, Tsai AG, et al. Targeted next-generation sequencing identifies a subset of idiopathic hypereosinophilic syndrome with features similar to chronic eosinophilic leukemia, not otherwise specified. Mod Pathol. 2016;29(8):854-864.
19. Maxson JE, Gotlib J, Pollyea DA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med. 2013;368(19):1781-1790.
20. Schanz J, Tüchler H, Solé F, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol. 2012;30(8):820-829.
21. Marshall D, Roboz GJ. Standardizing the initial evaluation for myelodysplastic syndromes. Curr Hematol Malig Rep. 2013;8(4):361-369.
22. Damm F, Chesnais V, Nagata Y, et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood. 2013;122(18):3169-3177.
23. Malcovati L, Papaemmanuil E, Ambaglio I, et al. Driver somatic mutations identify distinct disease entities within myeloid neoplasms with myelodysplasia. Blood. 2014;124(9):1513-1521.
24. Papaemmanuil E, Cazzola M, Boultwood J, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365(15):1384-1395.
25. Patnaik MM, Hanson CA, Sulai NH, et al. Prognostic irrelevance of ring sideroblast percentage in World Health Organization-defined myelodysplastic syndromes without excess blasts. Blood. 2012;119(24):5674-5677.
26. Guièze R, Wu CJ. Genomic and epigenomic heterogeneity in chronic lymphocytic leukemia. Blood. 2015;126(4):445-453.
27. Jeromin S, Weissmann S, Haferlach C, et al. SF3B1 mutations correlated to cytogenetics and mutations in NOTCH1, FBXW7, MYD88, XPO1 and TP53 in 1160 untreated CLL patients. Leukemia. 2014;28(1):108-117.
28. Del Giudice I, Rossi D, Chiaretti S, et al. NOTCH1 mutations in +12 chronic lymphocytic leukemia (CLL) confer an unfavorable prognosis, induce a distinctive transcriptional profiling and refine the intermediate prognosis of +12 CLL. Haematologica. 2012;97(3):437-441.
29. Weissmann S, Roller A, Jeromin S, et al. Prognostic impact and landscape of NOTCH1 mutations in chronic lymphocytic leukemia (CLL): a study on 852 patients. Leukemia. 2013;27(12):2393-2396.
30. Vegliante MC, Palomero J, Pérez-Galán P, et al. SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive mantle cell lymphoma. Blood. 2013;121(12):2175-2185.
31. King RL, Gonsalves WI, Ansell SM, et al. Lymphoplasmacytic lymphoma with a non-IgM paraprotein shows clinical and pathologic heterogeneity and may harbor MYD88 L265P mutations. Am J Clin Pathol. 2016;145(6):843-851.
32. Insuasti-Beltran G, Gale JM, Wilson CS, Foucar K, Czuchlewski DR. Significance of MYD88 L265P mutation status in the subclassification of low-grade B-cell lymphoma/leukemia. Arch Pathol Lab Med. 2015;139(8):1035-1041.
33. Wang XJ, Kim A, Li S. Immunohistochemical analysis using a BRAF V600E mutation specific antibody is highly sensitive and specific for the diagnosis of hairy cell leukemia. Int J Clin Exp Pathol. 2014;7(7):4323-4328.
34. Nakamura T, Tateishi K, Niwa T, et al. Recurrent mutations of CD79B and MYD88 are the hallmark of primary central nervous system lymphomas. Neuropathol Appl Neurobiol. 2016;42(3):279-290.
35. Chapman MA, Lawrence MS, Keats JJ, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471(7339):467-472.
36. Bosga-Bouwer AG, van Imhoff GW, Boonstra R, et al. Follicular lymphoma grade 3B includes 3 cytogenetically defined subgroups with primary t(14;18), 3q27, or other translocations: t(14;18) and 3q27 are mutually exclusive. Blood. 2003;101(3):1149-1154.
37. Gu K, Fu K, Jain S, et al. t(14;18)-negative follicular lymphomas are associated with a high frequency of BCL6 rearrangement at the alternative breakpoint region. Mod Pathol. 2009;22(9):1251-1257.
38. Katzenberger T, Ott G, Klein T, Kalla J, Müller-Hermelink HK, Ott MM. Cytogenetic alterations affecting BCL6 are predominantly found in follicular lymphomas grade 3B with a diffuse large B-cell component. Am J Pathol. 2004;165(2):481-490.
39. Katzenberger T, Kalla J, Leich E, et al. A distinctive subtype of t(14;18)-negative nodal follicular non-Hodgkin lymphoma characterized by a predominantly diffuse growth pattern and deletions in the chromosomal region 1p36. Blood. 2009;113(5):1053-1061.
40. Belaud-Rotureau MA, Parrens M, Carrere N, et al. Interphase fluorescence in situ hybridization is more sensitive than BIOMED-2 polymerase chain reaction protocol in detecting IGH-BCL2 rearrangement in both fixed and frozen lymph node with follicular lymphoma. Hum Pathol. 2007;38(2):365-372.
41. Limpens J, Stad R, Vos C, et al. Lymphoma-associated translocation t(14;18) in blood B cells of normal individuals. Blood. 1995;85(9):2528-2536.
42. Schmitt C, Balogh B, Grundt A, et al. The bcl-2/IgH rearrangement in a population of 204 healthy individuals: occurrence, age and gender distribution, breakpoints, and detection method validity. Leuk Res. 2006;30(6):745-750.
43. Summers KE, Goff LK, Wilson AG, Gupta RK, Lister TA, Fitzgibbon J. Frequency of the Bcl-2/IgH rearrangement in normal individuals: implications for the monitoring of disease in patients with follicular lymphoma. J Clin Oncol. 2001;19(2):420-424.
44. Galimberti S, Luminari S, Ciabatti E, et al. Minimal residual disease after conventional treatment significantly impacts on progression-free survival of patients with follicular lymphoma: the FIL FOLL05 trial. Clin Cancer Res. 2014;20(24):6398-6405.
45. Ladetto M, Lobetti-Bodoni C, Mantoan B, et al; Fondazione Italiana Linfomi. Persistence of minimal residual disease in bone marrow predicts outcome in follicular lymphomas treated with a rituximab-intensive program. Blood. 2013;122(23):3759-3766.
46. van Oers MHJ, Tönnissen E, Van Glabbeke M, et al. BCL-2/IgH polymerase chain reaction status at the end of induction treatment is not predictive for progression-free survival in relapsed/resistant follicular lymphoma: results of a prospective randomized EORTC 20981 phase III intergroup study. J Clin Oncol. 2010;28(13):2246-2252.
47. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375-2390.
48. Dewar R, Andea AA, Guitart J, Arber DA, Weiss LM. Best practices in diagnostic immunohistochemistry: workup of cutaneous lymphoid lesions in the diagnosis of primary cutaneous lymphoma. Arch Pathol Lab Med. 2015;139(3):338-350.
49. Koskela HLM, Eldfors S, Ellonen P, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012;366(20):1905-1913.
50. Rajala HL, Eldfors S, Kuusanmäki H, et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood. 2013;121(22):4541-4550.
51. Chen X, Cherian S. Immunophenotypic characterization of T-cell prolymphocytic leukemia. Am J Clin Pathol. 2013;140(5):727-735.
52. Delgado P, Starshak P, Rao N, Tirado C. A comprehensive update on molecular and cytogenetic abnormalities in T-cell prolymphocytic leukemia (T-PLL). J Assoc Genet Technol. 2012;38(4):193-198.
53. Stengel A, Kern W, Zenger M, et al. Genetic characterization of T-PLL reveals two major biologic subgroups and JAK3 mutations as prognostic marker. Genes Chromosomes Cancer. 2016;55(1):82-94.
54. Thompson MA, Stumph J, Henrickson SE, et al. Differential gene expression in anaplastic lymphoma kinase-positive and anaplastic lymphoma kinase-negative anaplastic large cell lymphomas. Hum Pathol. 2005;36(5):494-504.
55. King R, Dao L, McPhail E, et al. Morphologic features of ALK-negative anaplastic large cell lymphomas with DUSP22 rearrangements. Am J Surg Pathol. 2016;40(1):36-43.
In January 2015, President Obama introduced the Precision Medicine Initiative, a program set up to identify new biomedical discoveries for the development of a personalized knowledge base of disease entities and individualized treatments. Advances in precision medicine typically involve the use of targeted therapies tailored to individual genetic characteristics identified with molecular testing. The goals are to improve survival and reduce adverse effects. With an initial budget of $215 million, this initiative presented a unique opportunity to combine efforts in genomic discovery, bioinformatic analysis, and health information technology to move toward data-driven, evidence-based precision medicine.1
The VHA is the largest comprehensive health care system in the U. S. and has more than 1,700 care sites serving nearly 9 million veterans each year. The budget for this single-payer system is proposed by the President and approved by Congress. As the VHA must treat a diverse and aging veteran population in an environment of rising costs and budget constraints, limited resources must be monitored and appropriated for the most cost-effective health care delivery. Precision medicine offers a model in which physicians can select the most appropriate diagnostic tests in defined clinical settings to direct clinical care. It supports the testing needed to subdivide each disease category into distinct subcategories. Nevertheless, the need for fiscal responsibility in a capitated health care system recommends testing in cases in which it can change therapy or prognosis rather than for purely academic reasons.
Pathology and Laboratory Medicine Service
Given limited resources and an increasing number of requests for advanced molecular testing, the VA Pathology and Laboratory Medicine Service (P&LMS) formed the Molecular Genetics Pathology Workgroup (MGPW) in September 2013. The charter listed the tasks of the MGPW to “provide recommendations on how to effectively use molecular genetics tests, promote increased quality and availability of testing within the VHA, encourage internal referral testing, provide an organizational structure for Molecular Genetics Testing Consortia, and create a P&LMS policy for molecular genetic testing in general, specifically addressing the issues surrounding laboratory developed testing.” The MGPW has 4 subcommittees: molecular oncology, pharmacogenetics, hematopathology molecular genetics (HMG), and genetic medicine. Since its inception, the HMG subcommittee has had several objectives:
- Standardize the molecular testing nomenclature for and develop practice guidelines for acute myeloid leukemia (AML), myeloproliferative neoplasms (MPN), myelodysplastic syndrome (MDS), chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma, lymphoma, and plasma cell neoplasms;
- Develop standardized reporting guidelines for current VA molecular laboratories;
- Identify new tests as they are being reported in the literature and collaborate with hematology and oncology services to evaluate the clinical utility of these tests for VA patients;
- Network current VA molecular laboratories, perform fact-finding for these laboratories, and compile test menus; and
- Assess for the formation of VA-wide interfacility consultation services for hematopathology so that all VA facilities, regardless of their complexity, will be able to access the expertise of hematopathology-trained pathologists (Appendix).
The HMG subcommittee met monthly and discussed various diagnostic entities in hematopathology. For hematolymphoid malignancies, it was generally agreed that the traditional laboratory tools of morphology, flow cytometry, and immunohistochemistry (IHC) are standard in initial assessment and often in diagnosis. As the clinical molecular and cytogenetic assays of karyotype, fluorescence in situ hybridization (FISH), advanced DNA sequencing, microarray, and highly sensitive polymerase chain reaction (PCR) analysis affect diagnosis, subclassification, minimal residual disease (MRD) monitoring, prognosis, and therapy selection, their use is marked by a high degree of variability. As a result, standardization is needed. As each laboratory develops and reports ancillary testing, the variable reporting formats may generate postanalytic errors.
A detailed description of all molecular methodologies is beyond the scope of this article. For practicing pathologists, challenges remain in overall cost and reimbursement, extensive and time-consuming data analysis, and in some cases, interpretation differences.
Myeloid Neoplasms
Myeloid malignancies were divided into AML, MPN, and MDS. Next-generation sequencing (NGS) information for these malignancies was used to identify various contributory functional categories, including cell signaling (FLT3, KIT, JAK2, MPL, KRAS/NRAS, PTPN11, NF1, CSF3R); transcription (CEBPA, RUNX1, GATA1/GATA2, PHF6, ETV6); splicing (SF3B1, SRSF2, ZRSR2, U2AF1); epigenetics (DNMT3A, TET2, IDH1/IDH2, ASXL1, EZH2, SUZ12, KDM6A); cohesin complex (STAG2, SMC1A, SMC3, RAD21); and cell cycle (TP53, NPM1).2
Acute Myeloid Leukemia
The HMG subcommittee reviewed the literature on prognostically significant genes in myeloid leukemias. Karyotype abnormalities, such as t(8;21) and inv(16), collectively known as the core-binding factor (CBF) leukemias, t(15;17), t(11q23) (KMT2A/MLL), and so forth, are recurrent lesions in AML. Included in the minimum set of genes recommended by the National Comprehensive Cancer Network (NCCN) for AML prognosis evaluation are nucleolar protein nucleophosmin (NPM1), CCAAT/enhancer-binding protein
Some of the chromosomal translocations, such as inv(16)/t(16;16) in AML and t(15;17) in acute promyelocytic leukemia, can be monitored with FISH or reverse transcription–PCR (RT-PCR) analysis. As NPM1 mutations tend to be seen in recurrence, they can be used as molecular markers for MRD. Other mutations that provide important prognostic information in AML include:
- Activating insertions/duplications in the FLT3 receptor tyrosine kinase, which can be detected with PCR sizing assays;
- Mutations in the KIT receptor tyrosine kinase, which can be detected with DNA sequencing or more limited hotspot PCR;
- Mutations in the DNA methyltransferase, DNMT3A, a poor prognostic indicator seen in 22% of cases of AML, also detected with gene sequencing or more limited hotspot PCR; and
- Another set of genes, TET2, IDH1, IDH2, KRAS, NRAS, EZH2, and ASXL1, is mutated in MPN as well as AML and MDS, making a common molecular panel with next-generation sequencing useful in diagnosing and risk-stratifying all myeloid neoplasms.
The HMG subcommittee agreed that, for de novo AML, chromosomal karyotype is the standard of care, necessary in detecting known cytogenetic abnormalities as well as a wide range of lesions that might indicate a diagnosis of AML with myelodysplasia-related changes at time of diagnosis. In addition, molecular analysis of FLT3 is useful in determining prognosis, and CEBPA (biallelic) and NPM1 mutations are good prognostic factors in normal-karyotype AML. KMT2A (MLL) rearrangements should be tested with FISH if the lineage is ambiguous. The PML-RARA fusion gene also should be tested with FISH if morphologic and flow cytometry results suggest acute promyelocytic leukemia (Table). At this time, testing for TP53, DNMT3A, RAS, and other such mutations is not recommended because it is not cost-effective for the VA.
Myeloproliferative Neoplasms
Myeloproliferative neoplasms are clonal hematopoietic stem cell disorders characterized by proliferation of at least 1 myeloid lineage: granulocytic, erythroid, or megakaryocytic. Myeloproliferative neoplasms show a range of recurrent chromosomal translocations, such as BCR-ABL1 fusion in chronic myelogenous leukemia (CML) that can be detected with RT-PCR analysis as well as FISH. In CML, BCR-ABL1 fusion transcript levels detected by a quantitative PCR (qPCR) method are now used to monitor the course of CML therapy with tyrosine kinase inhibitors (TKIs) and to trigger a treatment change in drug-resistant cases. Given the importance of qPCR in clinical management, significant progress has been made in standardizing both the PCR protocol and the reference materials used to calibrate the BCR-ABL1 PCR assay. BCR-ABL1–negative MPN, including polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF), are most commonly associated with mutations in the tyrosine kinase JAK2. Mutations in CALR and MPL are seen in a subset of patients with ET and PMF as well, whereas PV is essentially exclusively a disease of JAK2 mutations.
Chronic myelogenous leukemia is the prototypical MPN. To establish the initial diagnosis, FISH and/or qPCR for BCR-ABL1 fusion should be used. If CML is confirmed, the sample can be reflexed to qPCR BCR-ABL1 on the initial peripheral blood and/or bone marrow sample(s) to establish the patient’s baseline. In addition, a bone marrow sample (aspirate) should be used for a complete karyotype and for morphologic confirmation of disease phase.
For follow-up assessment of CML patients’ response to TKI treatment, qPCR for BCR-ABL1 should be tested with a peripheral blood sample or a bone marrow sample every 3 months.4 A peripheral blood sample is more commonly used because it is conveniently obtained. Early molecular response as indicated by a BCR-ABL1 transcript ratio of < 10% on the International Scale at 3 months, has a strong prognostic value.5 Major molecular response as indicated by a BCR-ABL1 transcript ratio of < 0.1% on the International Scale at 12 to 18 months is also highly prognostic.5
After the peripheral blood sample becomes negative for BCR-ABL1 by qPCR, testing bone marrow samples may be considered. If important treatment response benchmarks are not achieved, or response is lost with rising BCR-ABL1 levels (TKI resistance), ABL1 kinase domain mutation analysis as well as repeat FISH (to assess for copy number multiplication) should be performed to guide further management. Patients with the ABL1 T315I mutation are resistant to all first-line TKIs but may respond to later third-generation TKIs.6
BCR-ABL1–negative MPNs include PV, ET, and PMF. Bone marrow morphology remains the cornerstone of ET and PMF diagnosis. The discovery of JAK2, CALR, and MPL mutations has contributed to how these disorders are diagnosed.7-12 Besides providing the clonality proof that is crucial for diagnosis, the molecular markers influence the prognosis. The JAK2 (p.V617F) or less common JAK2 exon 12 mutations, which are detected in more than 95% of PV cases, are used as molecular markers to confirm diagnosis.7 Further, the JAK2 (p.V617F), CALR (exon 9), and MPL (exon 10) mutations are detected in ET (~60%, 25%, and 3%-5%, respectively) and PMF (~55%, 30%, and 5%, respectively).12 If ET or PMF is suspected clinically, first JAK2 (p.V617F) mutation analysis should be performed, then CALR mutation analysis, and finally MPL mutation analysis. Although novel gain-of-function JAK2 and MPL mutations were recently discovered in triple-negative ET (negative for canonical mutations in JAK2, CALR, and MPL) and PMF by whole exome sequencing,13 clinical testing is not readily available. Besides its utility in the initial diagnosis of ET and PMF, the JAK2 or CALR mutation assay also may be considered for bone marrow transplantation follow-up (Table).14
Despite the continuing debate on the classification of eosinophilic myeloid disorders, the discovery of the FIP1L1-PDGFRA fusion represents a major milestone in the understanding of these disorders.15,16 Unlike PDGFRB (5q33) and FGFR1 (8p11) rearrangements, which can be detected with routine chromosomal analysis (cytogenetics), the cryptic FIP1L1-PDGFRA fusion must be detected with FISH (for CHIC2 deletion) or RT-PCR analysis. It should be pointed out that, as most eosinophilia is reactive or secondary, molecular testing for FIP1L1-PDGFRA fusion is indicated only when primary hypereosinophilia or hypereosinophilic syndrome (HES) is suspected. This is particularly the case in the following hypereosinophilia accompanying conditions: CML-like morphology, but BCR-ABL1–negative; chronic myelomonocytic leukemia (CMML)–like morphology with a normal karyotype; and new onset of cardiac damage or dysfunction.17
Primary eosinophilic myeloid disorders with PDGFRA or PDGFRB rearrangements can be treated with TKIs (eg, imatinib). Next-generation sequencing may be considered in cases of presumed HES when there is no identifiable karyotypic or FISH abnormality. Recent studies have found that cases of HES with somatic mutations indicating clonality had adverse clinical outcomes similar to those of cases of chronic eosinophilic leukemia.18
The discovery of CSF3R mutations offers a new molecular marker for the diagnosis of chronic neutrophilic leukemia (CNL), an MPN.19 The CSF3R (p.T618I) mutation or another activating CSF3R mutation is now used as a diagnostic criterion for CNL. Identification of specific CSF3R mutations may have therapeutic implications as well. The test should be ordered only for patients with clinical and morphologic findings suggestive of CNL; reactive neutrophilic leukocytosis (eg, infection, inflammation) should be ruled out before the test is ordered.
Myelodysplastic Syndrome
Myelodysplastic syndrome is a group of clonal bone marrow disorders characterized by ineffective hematopoiesis, manifested by morphologic dysplasia in ≥ 1 hematopoietic lineages and peripheral cytopenias (hemoglobin level, < 10 g/dL; platelet count, < 100×103/µL; absolute neutrophil count, < 1.8×103/µL). Diagnosis and classification of MDS depend mainly on the degree of morphologic dysplasia and blast percentages, as determined by examining well-prepared cellular bone marrow aspirate smears and/or biopsy touch preparations and peripheral blood smears.
Conventional karyotyping is an essential part of the diagnostic workup for all presumptive cases of MDS and is of both diagnostic and prognostic importance.20 About 60% of MDS cases have recurrent cytogenetic abnormalities, which can be detected with conventional karyotyping. If a high-quality cytogenetic analysis cannot be performed (eg, the bone marrow sample is inadequate), or if quick turnaround is required, an alternative FISH panel may be used to detect some of the common MDS-associated chromosomal abnormalities (eg, 5q deletion, 7q deletion/monosomy 7, +8, 20q deletion).21 Sequencing with FISH also can be useful for assessing MRD by detecting a previously identified chromosomal abnormality.
Targeted sequencing of a limited number of genes can detect mutations in the vast majority of patients with MDS. The most commonly mutated genes in MDS are SF3B1, TET2, SRSF2, ASXL1, DNMT3A, RUNX1, U2AF1, TP53, and EZH2. Mutations in SRSF2 cause RNA splicing abnormalities. In addition, mutations in TP53, EZH2, RUNX1, and ASXL1 are associated with poor prognosis,22,23 whereas mutations in SF3B1 confer better event-free survival.24 Despite these developments, the HMG subcommittee agreed that NGS-based mutation panels are not cost-effective for the VA population at this time and should not be included in a MDS workup. Only in rare situations and when clinically indicated (to change disease classification or patient management) should evaluation for specific gene mutations be considered—for instance, the SF3B1 mutation for patients with probable MDS with ring sideroblasts, if ring sideroblasts are < 15%.25
Myelodysplastic/Myeloproliferative Neoplasms
Myelodysplastic/myeloproliferative neoplasms are a group of myeloid neoplasms with clinical, laboratory, and morphologic features that overlap both MDS and MPN. In MDS/MPN, the karyotype is often normal or shows abnormalities in common with MDS.
In cases of unexplained monocytosis for which there is clinical concern for CMML, morphologic evaluation and conventional chromosomal karyotyping should be performed after other secondary causes and known myeloproliferative and myelodysplastic entities have been excluded. If concomitant hypereosinophilia is present and the karyotype is normal, FISH or PCR-based assay should be performed to rule out FIP1L1-PDGFRA rearrangements. BCR-ABL1, PDGFRB, FGFR1, and t(8;9)/PCM1-JAK2 rearrangements typically are detected with high-quality cytogenetic analysis and thus do not require targeted molecular assays. Although certain gene mutations (eg, SRSF2, TET2, ASXL1, CBL) are commonly detected in CMML, the HMG subcommittee does not recommend sequencing-based mutation panels, as there is insufficient information for testing for prognostic or treatment stratification.
If MDS/MPN with ring sideroblasts and thrombocytosis is suspected on the basis of the clinical and morphologic criteria, molecular tests for the JAK2 (p.V617F) and SF3B1 mutations may be considered in an effort to help confirm the diagnosis.
Atypical CML is a rare MDS/MPN subtype that is now better characterized molecularly with SETBP1 and/or ETNK1 mutations, which are detectable in up to a third of cases. If clinical suspicion is high, sequencing may be diagnostically helpful.
Lymphoid Neoplasms
Chronic Lymphocytic Leukemia
In CLL, recurrent chromosomal abnormalities (eg, deletions of 13q, trisomy 12, deletions of 11q, deletions of 17p) have clear prognostic value and can be detected with FISH. Other prognostic information, such as somatic mutation of immunoglobulin heavy chain variable (IgHV) genes, TP53 mutations, SF3B1, and NOTCH1 mutation, are mostly derived from PCR-based assays. The discovery of recurrently mutated genes in CLL has increased with the use of highly sensitive sequencing methods constructing a more detailed landscape of CLL at genetic, epigenetic, and cellular levels. A recent literature review summarizes the vast heterogeneity of CLL with recurrent pathogenetic findings in MYD88, SF3B1, TP53, ATM, and NOTCH1 signaling pathways.26 The treatment of CLL is rapidly evolving, and many clinical trials are proposing a change from the “watch and wait” paradigm to treatment upon initial presentation based on molecular findings. Additional testing based on new treatment options from current clinical trials will be recommended.
Flow cytometry and morphology are standard for CLL diagnosis. The HMG subcommittee recommends FISH for del(13q14), del(11q), trisomy 12, and del(17p) at time of diagnosis or immediately before therapy initiation. Zeta-chain (
Other B-Cell Lymphoproliferative Disorders
Unlike the common molecular changes in CLL, in other mature B-cell lymphomas, chromosomal translocations that juxtapose a variety of different oncogenes next to an Ig gene enhancer usually are—and those that switch regions less commonly are—important initiating events that can be detected with PCR, DNA sequencing, or FISH. In follicular lymphoma (FL), Burkitt lymphoma, marginal zone lymphoma (MZL), and mantle cell lymphoma (MCL), these oncogenes driven by an Ig gene enhancer typically include BCL2, MYC, MALT1, and CCND1 (cyclin D1), respectively. Molecular variants of these lymphomas that lack these classical translocations often activate homologous genes (eg, cyclin D3/CCND3 is activated in variants of MCL).
Morphology, flow cytometry, and IHC are routinely used for diagnosis. In inconclusive cases, Ig gene rearrangement by PCR may be used. The Table summarizes common molecular changes in B-cell lymphomas.
Mantle cell lymphoma. MCL is a non-Hodgkin lymphoma subtype characterized by t(11;14) (q13;q32) translocations that in the majority of cases lead to overexpression of cyclin D1 (BCL1). Recent molecular profiling has identified an MCL variant that is cyclin D1–negative but SOX11-positive and may have a more aggressive clinical course.30 SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive MCL.
Lymphoplasmacytic lymphoma. Lymphoplasmacytic lymphoma (LPL), MZL, and CLL/small lymphocytic lymphoma are well-defined clinicopathologic entities. However, distinguishing LPL from MZL and atypical cases of CLL can sometimes be difficult because of overlapping clinical and morphologic features. Recent studies have identified a recurrent L265P mutation in the MYD88 gene in 90% to 95% of LPL cases with IgM paraprotein and in 40% to 50% of the rare non-IgM LPL cases. In contrast, the mutation is much less frequently present in MZL and other low-grade B-cell neoplasms (2%-7%).31 Therefore, testing for this abnormality can be a diagnostic aid in these difficult-to-classify cases. In addition, from a therapeutic perspective, presence or absence of MYD88 mutation may prove more significant than presence of a specific paraprotein or histopathologic features. Ibrutinib has shown efficacy in LPL and demonstrates improved response rates in patients with MYD88 mutation compared with that of their mutation-negative counterparts.32 Several MYD88 inhibitors are in clinical trials. This again indicates the need to more accurately identify and subclassify these non-IgM LPL cases to ensure appropriate molecular evaluation.
Hairy cell leukemia. Flow cytometry and morphology are usually sufficient for a hairy cell leukemia (HCL) diagnosis. However, rare cases are difficult to distinguish variant HCL from other mimics. The BRAF V600E mutation recently was described as a disease-defining molecular marker for HCL—present in nearly all HCL cases but virtually absent in HCL mimics. Therefore, detection of the BRAF mutation by IHC stain with specific antibody or PCR analysis is highly sensitive and specific for the diagnosis of HCL.33
Diffuse large B-cell lymphoma. Recent molecular analysis has created various risk stratification schemata for diffuse large B-cell lymphoma (DLBCL). The HGM subcommittee agrees that well-preserved morphology, IHC, flow cytometry, and FISH-specific markers (BCL2, BCL6, cMYC) are sufficient for diagnostic, prognostic, and therapeutic purposes. Although a wide range of genes have been implicated in the pathogenesis of DLBCL, sequencing and gene expression profiling are not cost-effective at this time and do not add benefit to patient treatment.
The MYD88 L265P mutation has been identified in DLBCL, particularly the activated B-cell-like type and primary central nervous system lymphoma (PCNSL), and may have implications for ibrutinib therapy. PCNSL commonly manifests aggressive clinical behavior and has a poor prognosis. It has been proposed that the MYD88 mutation can be used as a genetic hallmark for PCNSL to distinguish CNS involvement by systemic DLBCL from PCNSL.34
Plasma cell neoplasms. Flow cytometry is acceptable for the diagnosis of plasma cell neoplasms and for residual disease follow-up. Chromosomal karyotype or FISH for IGH/CCND1, IGH/MMSET, and IGH/CMAF dual fusion probes is recommended in conjunction with morphology, IHC, and flow cytometry. In plasma cell myeloma, several genetic mutations can be detected with NGS, including mutations in NRAS, KRAS, TP53, BCL7A, DIS3, and FAM46C.35 Less commonly, BRAF mutations, previously described in melanoma and several other solid tumors, can be detected with DNA sequencing in 4% of multiple myeloma cases, which may prove promising for targeted therapy with BRAF inhibitors. However, current therapeutic decisions are based on genetic and clinical factors, and sequence-based assays are not recommended at this time.
Follicular lymphoma. Cytology, histology, and IHC typically are sufficient for diagnosing FL. In difficult-to-diagnose cases and in cases with scant material, additional tests may help with diagnosis. Eighty to ninety percent of FL cases have t(14;18)(q32;q21), which places the BCL2 gene transcription under the control of the IGH promoter. In addition, about 10% of FL cases have 3q27 aberrancies at the BCL6 gene.36-38 More recently, cases of FL with bulky inguinal disease negative for IGH-BCL2 and BCL6 translocations were found to have 1p36 deletions. These 1p36-deleted FLs typically have a diffuse pattern and a good prognosis.39 For t(14;18), 3q27, or 1p36, FISH is a sensitive means for detecting these translocations, as is PCR for IGH-BCL2.40 There are reports that t(14;18) can be detected in a substantial fraction of otherwise healthy donors at levels and rates that depend on the type of detection test used.41-43 In addition, between one-fourth and one-third of de novo DLBCLs show t(14;18), and about one-third show BCL6 abnormalities at 3q27. Therefore, these genetic changes are not specific for FL and should not be used to subtype a lymphoma as follicular in origin.
Use of IGH-BCL2 as a marker for MRD is still controversial. Some studies have found that a postinduction and posttransplantation IGH-BCL2-positive finding by PCR predicted relapse.44,45 However, others studies have not found significance to postinduction IGH-BCL2 positivity.46 The NCCN guidelines recommend testing for IGH-BCL2 or BCL6 translocations or 1p36 deletion only if this testing is needed for diagnosis. The guidelines do not recommend using these genetic assays in follow-up biopsies, as the importance of treating early relapse has not been definitively demonstrated.
Therefore, if a lymphoma has morphologic, histologic, and IHC findings consistent with FL, then cytogenetic, FISH, or PCR testing is not needed for diagnosis but may be used as confirmation. Follow-up molecular and cytogenetic testing should be avoided if the original cytogenetic abnormality is unknown. That is, IGH-BCL2 FISH should be performed in follow-up samples only if the original lymphoma is known to contain the translocation. As follow-up genetic testing is of disputed clinical significance even in cases in which the original molecular change is known, the NCCN recommendations for therapy are no different. The HMG subcommittee does not recommend molecular or cytogenetic testing in FL beyond what is required for initial diagnosis.
T-Cell Lymphomas
Mature T-Cell Lymphoma and Leukemia
For mature T-cell lymphoma (TCL) and leukemia, the clinical and morphologic criteria have a very important role in the initial workup. However, IHC immunophenotyping is crucial for definitive diagnosis and subclassification. Flow cytometry is routinely used in diagnosing diseases such as T-cell prolymphocytic leukemia (TPLL), T-cell large granular lymphocytic (LGL) leukemia, and Sézary syndrome. T-cell clonality studies, preferably with BIOMED-II–validated primers against targets such as T-cell receptor
Significant advances in TCL classification have led to revisions and the inclusion of new provisional entities in the 2016 World Health Organization classification of lymphoid neoplasms.47 Many of these changes originated in studies of gene expression profiling and the genetic landscape of T-cell neoplasms. Even though subsets of peripheral TCL not otherwise specified (PTCL-NOS) have been recognized on the basis of phenotypic and molecular abnormalities with possible clinical implications, in most cases molecular testing is not part of routine practice. Typically, only a few cytogenetic abnormalities and genetic mutations are used in the evaluation of TCL and T-cell leukemia.
A group of T-cell lymphoproliferative disorders with expression of T follicular helper cell markers can be identified with IHC. These disorders include angioimmunoblastic TCL; follicular TCL, a new entity that is a PTCL-NOS subset; and primary cutaneous CD4-positive small/medium T-cell lymphoproliferative disorder. The neoplastic cells should express at least 2 or 3 T follicular helper cell–related antigens, including CD279/PD1, CD10, BCL6, CXCL13, ICOS, SAP, and CCR5; the most commonly used are PD1, BCL6, and CD10. Recurrent fusion of ITK-SYK translocation t(5;9) or CTLA4-CD28 is also common in follicular TCL. Although recurrent mutation is found in these entities, conventional karyotyping or IHC should be sufficient for diagnosis.
Cutaneous γ -Δ T-Cell Lymphoma
Among cutaneous TCLs, primary cutaneous
Peripheral T-Cell Lymphoma
Gene expression profiling analysis of PTCLs has identified at least 3 subtypes characterized by overexpression of GATA3, TBX21, and cytotoxic genes and expression of the corresponding proteins with IHC.47 These subtypes are associated with different clinical behavior and therapy responses. The GATA3 subtype has an inferior prognosis and shows a high level of T helper type 2 cytokines, which can be identified with IHC. As IHC-stained GATA3 has been available as a marker of urothelial carcinoma at most IHC laboratories, GATA3 IHC staining also may be considered in the evaluation of PTCLs.
Many monoclonal antibody therapies are being used as primary or secondary regimens in the treatment of TCL. Clinical trials are working to establish their efficacy. If treatment with a monoclonal antibody is being considered, it is appropriate to conduct IHC to demonstrate the presence of the target antigen and at follow-up, to demonstrate the efficacy of treatment. These therapies include alemtuzumab, which targets CD52, and brentuximab, which targets CD30.
T-Cell Large Granular Lymphocytic Leukemia
T-cell LGL leukemia is a complex diagnosis that requires persistent clonal expansion of LGLs and clinically peripheral blood cytopenia. In many cases, the diagnosis is difficult to establish, as benign large granular lymphocytosis with clonal T cells may occur in conjunction with viral infections or autoimmune disorders. Somatic mutations in the STAT3 (signal transducer and activator of transcription 3) gene are found in 40% of patients with T-cell LGL leukemia.49 More recently, somatic mutations in the STAT5B gene were identified in 2% of T-cell LGL leukemia subsets. The clinical course of T-cell LGL leukemia in patients with the STAT5B mutation is aggressive and fatal, clearly different from the relatively favorable course of typical T-cell LGL leukemia.50 The HMG subcommittee recommends considering a STAT3 and STAT5B mutation study for selected cases in which it is difficult to distinguish true T-cell LGL leukemia from its reactive expansions.
T-Cell Prolymphocytic Leukemia
T-cell prolymphocytic leukemia (T-PLL) is a rare, aggressive disease and is most commonly associated with a prolymphocytic morphology and expression of CD4. However, since a specific immunophenotypic profile of T-PLL has not been identified, flow cytometry is not adequate in isolation for definitive classification as T-PLL.51 A diagnosis of T-PLL often requires cytogenetics or a FISH study to confirm a suspected case. Most TPLL cases harbor characteristic chromosomal abnormalities involving 14q11.2 (TCR
Anaplastic Large Cell Lymphoma
The World Health Organization recognizes 3 distinct types of anaplastic large cell lymphoma (ALCL): systemic anaplastic lymphoma kinase (ALK)–positive ALCL, systemic ALK-negative ALCL, and primary cutaneous ALCL. Systemic ALK-positive ALCLs consistently have ALK gene rearrangements and favorable outcomes. The most common translocation is the t(2;5) rearrangement of NPM1 and ALK, though other ALK partners are also possible. In contrast, systemic ALK-negative ALCLs lack ALK gene rearrangements and as a whole have outcomes inferior to those of systemic ALK-positive ALCLs. However, studies have found systemic ALK-negative ALCL to be a genetically and clinically heterogeneous entity.54 About 30% of cases have rearrangements of the DUSP22-IRF4 locus on 6p25.3 (DUSP22 rearrangement), and these cases have favorable outcomes similar to those of systemic ALK-positive ALCL.55 Only 8% of patients have TP63 rearrangements and very poor outcomes. The remaining cases lack ALK, DUSP22, and TP63 rearrangements and have intermediate outcomes. The HMG subcommittee recommends considering DUSP22 rearrangement by FISH in the evaluation of systemic ALK-negative ALCL.
Conclusion
The pathologic diagnosis, classification, and risk stratification of lymphoma and leukemia require an approach that integrates morphology, flow cytometry, cytogenetics, and molecular pathology. Rapidly evolving molecular techniques currently allow for detailed description of the molecular defects in lymphoma and leukemia, including driver mutations, amplification/deletion events, and clonal evolution. Unfortunately, the technical ability to catalogue the molecular defects in lymphoma and leukemia, often at great expense, is outpacing the ability to use this detailed information in treating patients with hematologic malignancies. The challenge, then, is to identify best practices for the diagnosis and classification of lymphoma and leukemia in VHA hospitals that incorporate the most useful molecular tests without wasting financial resources.
In this report, the HMG subcommittee of the MGPW has presented its recommendations for molecular testing in AML, MPN, MDS, and lymphomas in the context of standard morphologic and immunophenotypic approaches to hematopathology diagnosis and classification. Adoption of these recommendations by VHA hospitals and clinics should help ensure that all VA patients with hematologic malignancies benefit from the latest advances in precision medicine.
Within the vast and comprehensive national VHA health care system are multiple centers of expertise in hematopathology. In addition, multiple VA clinical molecular diagnostic laboratories are performing state-of-the-art testing. The HMG subcommittee proposes that, to make best use of these expert resources, the VHA should establish an interfacility hematopathology consultation service. This service would allow any VA pathologist to consult a board-certified hematopathologist regarding use of ancillary molecular genetic testing in the diagnosis of hematologic malignancy.
In addition, the HMG subcommittee recommends consolidating VA molecular diagnostic reference laboratories and having them perform molecular testing for other VA hospitals rather than using commercial reference laboratories, where testing standards are not uniform and results may be difficult to interpret. Several well-established VA clinical laboratories with technical expertise and informatics support are already performing selected molecular diagnostic testing. These laboratories’ resources should be expanded, where practical, to cost-effectively provide VA expertise to all veterans and to improve access to appropriate molecular diagnostic testing.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of
Click here to read the digital edition.
In January 2015, President Obama introduced the Precision Medicine Initiative, a program set up to identify new biomedical discoveries for the development of a personalized knowledge base of disease entities and individualized treatments. Advances in precision medicine typically involve the use of targeted therapies tailored to individual genetic characteristics identified with molecular testing. The goals are to improve survival and reduce adverse effects. With an initial budget of $215 million, this initiative presented a unique opportunity to combine efforts in genomic discovery, bioinformatic analysis, and health information technology to move toward data-driven, evidence-based precision medicine.1
The VHA is the largest comprehensive health care system in the U. S. and has more than 1,700 care sites serving nearly 9 million veterans each year. The budget for this single-payer system is proposed by the President and approved by Congress. As the VHA must treat a diverse and aging veteran population in an environment of rising costs and budget constraints, limited resources must be monitored and appropriated for the most cost-effective health care delivery. Precision medicine offers a model in which physicians can select the most appropriate diagnostic tests in defined clinical settings to direct clinical care. It supports the testing needed to subdivide each disease category into distinct subcategories. Nevertheless, the need for fiscal responsibility in a capitated health care system recommends testing in cases in which it can change therapy or prognosis rather than for purely academic reasons.
Pathology and Laboratory Medicine Service
Given limited resources and an increasing number of requests for advanced molecular testing, the VA Pathology and Laboratory Medicine Service (P&LMS) formed the Molecular Genetics Pathology Workgroup (MGPW) in September 2013. The charter listed the tasks of the MGPW to “provide recommendations on how to effectively use molecular genetics tests, promote increased quality and availability of testing within the VHA, encourage internal referral testing, provide an organizational structure for Molecular Genetics Testing Consortia, and create a P&LMS policy for molecular genetic testing in general, specifically addressing the issues surrounding laboratory developed testing.” The MGPW has 4 subcommittees: molecular oncology, pharmacogenetics, hematopathology molecular genetics (HMG), and genetic medicine. Since its inception, the HMG subcommittee has had several objectives:
- Standardize the molecular testing nomenclature for and develop practice guidelines for acute myeloid leukemia (AML), myeloproliferative neoplasms (MPN), myelodysplastic syndrome (MDS), chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma, lymphoma, and plasma cell neoplasms;
- Develop standardized reporting guidelines for current VA molecular laboratories;
- Identify new tests as they are being reported in the literature and collaborate with hematology and oncology services to evaluate the clinical utility of these tests for VA patients;
- Network current VA molecular laboratories, perform fact-finding for these laboratories, and compile test menus; and
- Assess for the formation of VA-wide interfacility consultation services for hematopathology so that all VA facilities, regardless of their complexity, will be able to access the expertise of hematopathology-trained pathologists (Appendix).
The HMG subcommittee met monthly and discussed various diagnostic entities in hematopathology. For hematolymphoid malignancies, it was generally agreed that the traditional laboratory tools of morphology, flow cytometry, and immunohistochemistry (IHC) are standard in initial assessment and often in diagnosis. As the clinical molecular and cytogenetic assays of karyotype, fluorescence in situ hybridization (FISH), advanced DNA sequencing, microarray, and highly sensitive polymerase chain reaction (PCR) analysis affect diagnosis, subclassification, minimal residual disease (MRD) monitoring, prognosis, and therapy selection, their use is marked by a high degree of variability. As a result, standardization is needed. As each laboratory develops and reports ancillary testing, the variable reporting formats may generate postanalytic errors.
A detailed description of all molecular methodologies is beyond the scope of this article. For practicing pathologists, challenges remain in overall cost and reimbursement, extensive and time-consuming data analysis, and in some cases, interpretation differences.
Myeloid Neoplasms
Myeloid malignancies were divided into AML, MPN, and MDS. Next-generation sequencing (NGS) information for these malignancies was used to identify various contributory functional categories, including cell signaling (FLT3, KIT, JAK2, MPL, KRAS/NRAS, PTPN11, NF1, CSF3R); transcription (CEBPA, RUNX1, GATA1/GATA2, PHF6, ETV6); splicing (SF3B1, SRSF2, ZRSR2, U2AF1); epigenetics (DNMT3A, TET2, IDH1/IDH2, ASXL1, EZH2, SUZ12, KDM6A); cohesin complex (STAG2, SMC1A, SMC3, RAD21); and cell cycle (TP53, NPM1).2
Acute Myeloid Leukemia
The HMG subcommittee reviewed the literature on prognostically significant genes in myeloid leukemias. Karyotype abnormalities, such as t(8;21) and inv(16), collectively known as the core-binding factor (CBF) leukemias, t(15;17), t(11q23) (KMT2A/MLL), and so forth, are recurrent lesions in AML. Included in the minimum set of genes recommended by the National Comprehensive Cancer Network (NCCN) for AML prognosis evaluation are nucleolar protein nucleophosmin (NPM1), CCAAT/enhancer-binding protein
Some of the chromosomal translocations, such as inv(16)/t(16;16) in AML and t(15;17) in acute promyelocytic leukemia, can be monitored with FISH or reverse transcription–PCR (RT-PCR) analysis. As NPM1 mutations tend to be seen in recurrence, they can be used as molecular markers for MRD. Other mutations that provide important prognostic information in AML include:
- Activating insertions/duplications in the FLT3 receptor tyrosine kinase, which can be detected with PCR sizing assays;
- Mutations in the KIT receptor tyrosine kinase, which can be detected with DNA sequencing or more limited hotspot PCR;
- Mutations in the DNA methyltransferase, DNMT3A, a poor prognostic indicator seen in 22% of cases of AML, also detected with gene sequencing or more limited hotspot PCR; and
- Another set of genes, TET2, IDH1, IDH2, KRAS, NRAS, EZH2, and ASXL1, is mutated in MPN as well as AML and MDS, making a common molecular panel with next-generation sequencing useful in diagnosing and risk-stratifying all myeloid neoplasms.
The HMG subcommittee agreed that, for de novo AML, chromosomal karyotype is the standard of care, necessary in detecting known cytogenetic abnormalities as well as a wide range of lesions that might indicate a diagnosis of AML with myelodysplasia-related changes at time of diagnosis. In addition, molecular analysis of FLT3 is useful in determining prognosis, and CEBPA (biallelic) and NPM1 mutations are good prognostic factors in normal-karyotype AML. KMT2A (MLL) rearrangements should be tested with FISH if the lineage is ambiguous. The PML-RARA fusion gene also should be tested with FISH if morphologic and flow cytometry results suggest acute promyelocytic leukemia (Table). At this time, testing for TP53, DNMT3A, RAS, and other such mutations is not recommended because it is not cost-effective for the VA.
Myeloproliferative Neoplasms
Myeloproliferative neoplasms are clonal hematopoietic stem cell disorders characterized by proliferation of at least 1 myeloid lineage: granulocytic, erythroid, or megakaryocytic. Myeloproliferative neoplasms show a range of recurrent chromosomal translocations, such as BCR-ABL1 fusion in chronic myelogenous leukemia (CML) that can be detected with RT-PCR analysis as well as FISH. In CML, BCR-ABL1 fusion transcript levels detected by a quantitative PCR (qPCR) method are now used to monitor the course of CML therapy with tyrosine kinase inhibitors (TKIs) and to trigger a treatment change in drug-resistant cases. Given the importance of qPCR in clinical management, significant progress has been made in standardizing both the PCR protocol and the reference materials used to calibrate the BCR-ABL1 PCR assay. BCR-ABL1–negative MPN, including polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF), are most commonly associated with mutations in the tyrosine kinase JAK2. Mutations in CALR and MPL are seen in a subset of patients with ET and PMF as well, whereas PV is essentially exclusively a disease of JAK2 mutations.
Chronic myelogenous leukemia is the prototypical MPN. To establish the initial diagnosis, FISH and/or qPCR for BCR-ABL1 fusion should be used. If CML is confirmed, the sample can be reflexed to qPCR BCR-ABL1 on the initial peripheral blood and/or bone marrow sample(s) to establish the patient’s baseline. In addition, a bone marrow sample (aspirate) should be used for a complete karyotype and for morphologic confirmation of disease phase.
For follow-up assessment of CML patients’ response to TKI treatment, qPCR for BCR-ABL1 should be tested with a peripheral blood sample or a bone marrow sample every 3 months.4 A peripheral blood sample is more commonly used because it is conveniently obtained. Early molecular response as indicated by a BCR-ABL1 transcript ratio of < 10% on the International Scale at 3 months, has a strong prognostic value.5 Major molecular response as indicated by a BCR-ABL1 transcript ratio of < 0.1% on the International Scale at 12 to 18 months is also highly prognostic.5
After the peripheral blood sample becomes negative for BCR-ABL1 by qPCR, testing bone marrow samples may be considered. If important treatment response benchmarks are not achieved, or response is lost with rising BCR-ABL1 levels (TKI resistance), ABL1 kinase domain mutation analysis as well as repeat FISH (to assess for copy number multiplication) should be performed to guide further management. Patients with the ABL1 T315I mutation are resistant to all first-line TKIs but may respond to later third-generation TKIs.6
BCR-ABL1–negative MPNs include PV, ET, and PMF. Bone marrow morphology remains the cornerstone of ET and PMF diagnosis. The discovery of JAK2, CALR, and MPL mutations has contributed to how these disorders are diagnosed.7-12 Besides providing the clonality proof that is crucial for diagnosis, the molecular markers influence the prognosis. The JAK2 (p.V617F) or less common JAK2 exon 12 mutations, which are detected in more than 95% of PV cases, are used as molecular markers to confirm diagnosis.7 Further, the JAK2 (p.V617F), CALR (exon 9), and MPL (exon 10) mutations are detected in ET (~60%, 25%, and 3%-5%, respectively) and PMF (~55%, 30%, and 5%, respectively).12 If ET or PMF is suspected clinically, first JAK2 (p.V617F) mutation analysis should be performed, then CALR mutation analysis, and finally MPL mutation analysis. Although novel gain-of-function JAK2 and MPL mutations were recently discovered in triple-negative ET (negative for canonical mutations in JAK2, CALR, and MPL) and PMF by whole exome sequencing,13 clinical testing is not readily available. Besides its utility in the initial diagnosis of ET and PMF, the JAK2 or CALR mutation assay also may be considered for bone marrow transplantation follow-up (Table).14
Despite the continuing debate on the classification of eosinophilic myeloid disorders, the discovery of the FIP1L1-PDGFRA fusion represents a major milestone in the understanding of these disorders.15,16 Unlike PDGFRB (5q33) and FGFR1 (8p11) rearrangements, which can be detected with routine chromosomal analysis (cytogenetics), the cryptic FIP1L1-PDGFRA fusion must be detected with FISH (for CHIC2 deletion) or RT-PCR analysis. It should be pointed out that, as most eosinophilia is reactive or secondary, molecular testing for FIP1L1-PDGFRA fusion is indicated only when primary hypereosinophilia or hypereosinophilic syndrome (HES) is suspected. This is particularly the case in the following hypereosinophilia accompanying conditions: CML-like morphology, but BCR-ABL1–negative; chronic myelomonocytic leukemia (CMML)–like morphology with a normal karyotype; and new onset of cardiac damage or dysfunction.17
Primary eosinophilic myeloid disorders with PDGFRA or PDGFRB rearrangements can be treated with TKIs (eg, imatinib). Next-generation sequencing may be considered in cases of presumed HES when there is no identifiable karyotypic or FISH abnormality. Recent studies have found that cases of HES with somatic mutations indicating clonality had adverse clinical outcomes similar to those of cases of chronic eosinophilic leukemia.18
The discovery of CSF3R mutations offers a new molecular marker for the diagnosis of chronic neutrophilic leukemia (CNL), an MPN.19 The CSF3R (p.T618I) mutation or another activating CSF3R mutation is now used as a diagnostic criterion for CNL. Identification of specific CSF3R mutations may have therapeutic implications as well. The test should be ordered only for patients with clinical and morphologic findings suggestive of CNL; reactive neutrophilic leukocytosis (eg, infection, inflammation) should be ruled out before the test is ordered.
Myelodysplastic Syndrome
Myelodysplastic syndrome is a group of clonal bone marrow disorders characterized by ineffective hematopoiesis, manifested by morphologic dysplasia in ≥ 1 hematopoietic lineages and peripheral cytopenias (hemoglobin level, < 10 g/dL; platelet count, < 100×103/µL; absolute neutrophil count, < 1.8×103/µL). Diagnosis and classification of MDS depend mainly on the degree of morphologic dysplasia and blast percentages, as determined by examining well-prepared cellular bone marrow aspirate smears and/or biopsy touch preparations and peripheral blood smears.
Conventional karyotyping is an essential part of the diagnostic workup for all presumptive cases of MDS and is of both diagnostic and prognostic importance.20 About 60% of MDS cases have recurrent cytogenetic abnormalities, which can be detected with conventional karyotyping. If a high-quality cytogenetic analysis cannot be performed (eg, the bone marrow sample is inadequate), or if quick turnaround is required, an alternative FISH panel may be used to detect some of the common MDS-associated chromosomal abnormalities (eg, 5q deletion, 7q deletion/monosomy 7, +8, 20q deletion).21 Sequencing with FISH also can be useful for assessing MRD by detecting a previously identified chromosomal abnormality.
Targeted sequencing of a limited number of genes can detect mutations in the vast majority of patients with MDS. The most commonly mutated genes in MDS are SF3B1, TET2, SRSF2, ASXL1, DNMT3A, RUNX1, U2AF1, TP53, and EZH2. Mutations in SRSF2 cause RNA splicing abnormalities. In addition, mutations in TP53, EZH2, RUNX1, and ASXL1 are associated with poor prognosis,22,23 whereas mutations in SF3B1 confer better event-free survival.24 Despite these developments, the HMG subcommittee agreed that NGS-based mutation panels are not cost-effective for the VA population at this time and should not be included in a MDS workup. Only in rare situations and when clinically indicated (to change disease classification or patient management) should evaluation for specific gene mutations be considered—for instance, the SF3B1 mutation for patients with probable MDS with ring sideroblasts, if ring sideroblasts are < 15%.25
Myelodysplastic/Myeloproliferative Neoplasms
Myelodysplastic/myeloproliferative neoplasms are a group of myeloid neoplasms with clinical, laboratory, and morphologic features that overlap both MDS and MPN. In MDS/MPN, the karyotype is often normal or shows abnormalities in common with MDS.
In cases of unexplained monocytosis for which there is clinical concern for CMML, morphologic evaluation and conventional chromosomal karyotyping should be performed after other secondary causes and known myeloproliferative and myelodysplastic entities have been excluded. If concomitant hypereosinophilia is present and the karyotype is normal, FISH or PCR-based assay should be performed to rule out FIP1L1-PDGFRA rearrangements. BCR-ABL1, PDGFRB, FGFR1, and t(8;9)/PCM1-JAK2 rearrangements typically are detected with high-quality cytogenetic analysis and thus do not require targeted molecular assays. Although certain gene mutations (eg, SRSF2, TET2, ASXL1, CBL) are commonly detected in CMML, the HMG subcommittee does not recommend sequencing-based mutation panels, as there is insufficient information for testing for prognostic or treatment stratification.
If MDS/MPN with ring sideroblasts and thrombocytosis is suspected on the basis of the clinical and morphologic criteria, molecular tests for the JAK2 (p.V617F) and SF3B1 mutations may be considered in an effort to help confirm the diagnosis.
Atypical CML is a rare MDS/MPN subtype that is now better characterized molecularly with SETBP1 and/or ETNK1 mutations, which are detectable in up to a third of cases. If clinical suspicion is high, sequencing may be diagnostically helpful.
Lymphoid Neoplasms
Chronic Lymphocytic Leukemia
In CLL, recurrent chromosomal abnormalities (eg, deletions of 13q, trisomy 12, deletions of 11q, deletions of 17p) have clear prognostic value and can be detected with FISH. Other prognostic information, such as somatic mutation of immunoglobulin heavy chain variable (IgHV) genes, TP53 mutations, SF3B1, and NOTCH1 mutation, are mostly derived from PCR-based assays. The discovery of recurrently mutated genes in CLL has increased with the use of highly sensitive sequencing methods constructing a more detailed landscape of CLL at genetic, epigenetic, and cellular levels. A recent literature review summarizes the vast heterogeneity of CLL with recurrent pathogenetic findings in MYD88, SF3B1, TP53, ATM, and NOTCH1 signaling pathways.26 The treatment of CLL is rapidly evolving, and many clinical trials are proposing a change from the “watch and wait” paradigm to treatment upon initial presentation based on molecular findings. Additional testing based on new treatment options from current clinical trials will be recommended.
Flow cytometry and morphology are standard for CLL diagnosis. The HMG subcommittee recommends FISH for del(13q14), del(11q), trisomy 12, and del(17p) at time of diagnosis or immediately before therapy initiation. Zeta-chain (
Other B-Cell Lymphoproliferative Disorders
Unlike the common molecular changes in CLL, in other mature B-cell lymphomas, chromosomal translocations that juxtapose a variety of different oncogenes next to an Ig gene enhancer usually are—and those that switch regions less commonly are—important initiating events that can be detected with PCR, DNA sequencing, or FISH. In follicular lymphoma (FL), Burkitt lymphoma, marginal zone lymphoma (MZL), and mantle cell lymphoma (MCL), these oncogenes driven by an Ig gene enhancer typically include BCL2, MYC, MALT1, and CCND1 (cyclin D1), respectively. Molecular variants of these lymphomas that lack these classical translocations often activate homologous genes (eg, cyclin D3/CCND3 is activated in variants of MCL).
Morphology, flow cytometry, and IHC are routinely used for diagnosis. In inconclusive cases, Ig gene rearrangement by PCR may be used. The Table summarizes common molecular changes in B-cell lymphomas.
Mantle cell lymphoma. MCL is a non-Hodgkin lymphoma subtype characterized by t(11;14) (q13;q32) translocations that in the majority of cases lead to overexpression of cyclin D1 (BCL1). Recent molecular profiling has identified an MCL variant that is cyclin D1–negative but SOX11-positive and may have a more aggressive clinical course.30 SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive MCL.
Lymphoplasmacytic lymphoma. Lymphoplasmacytic lymphoma (LPL), MZL, and CLL/small lymphocytic lymphoma are well-defined clinicopathologic entities. However, distinguishing LPL from MZL and atypical cases of CLL can sometimes be difficult because of overlapping clinical and morphologic features. Recent studies have identified a recurrent L265P mutation in the MYD88 gene in 90% to 95% of LPL cases with IgM paraprotein and in 40% to 50% of the rare non-IgM LPL cases. In contrast, the mutation is much less frequently present in MZL and other low-grade B-cell neoplasms (2%-7%).31 Therefore, testing for this abnormality can be a diagnostic aid in these difficult-to-classify cases. In addition, from a therapeutic perspective, presence or absence of MYD88 mutation may prove more significant than presence of a specific paraprotein or histopathologic features. Ibrutinib has shown efficacy in LPL and demonstrates improved response rates in patients with MYD88 mutation compared with that of their mutation-negative counterparts.32 Several MYD88 inhibitors are in clinical trials. This again indicates the need to more accurately identify and subclassify these non-IgM LPL cases to ensure appropriate molecular evaluation.
Hairy cell leukemia. Flow cytometry and morphology are usually sufficient for a hairy cell leukemia (HCL) diagnosis. However, rare cases are difficult to distinguish variant HCL from other mimics. The BRAF V600E mutation recently was described as a disease-defining molecular marker for HCL—present in nearly all HCL cases but virtually absent in HCL mimics. Therefore, detection of the BRAF mutation by IHC stain with specific antibody or PCR analysis is highly sensitive and specific for the diagnosis of HCL.33
Diffuse large B-cell lymphoma. Recent molecular analysis has created various risk stratification schemata for diffuse large B-cell lymphoma (DLBCL). The HGM subcommittee agrees that well-preserved morphology, IHC, flow cytometry, and FISH-specific markers (BCL2, BCL6, cMYC) are sufficient for diagnostic, prognostic, and therapeutic purposes. Although a wide range of genes have been implicated in the pathogenesis of DLBCL, sequencing and gene expression profiling are not cost-effective at this time and do not add benefit to patient treatment.
The MYD88 L265P mutation has been identified in DLBCL, particularly the activated B-cell-like type and primary central nervous system lymphoma (PCNSL), and may have implications for ibrutinib therapy. PCNSL commonly manifests aggressive clinical behavior and has a poor prognosis. It has been proposed that the MYD88 mutation can be used as a genetic hallmark for PCNSL to distinguish CNS involvement by systemic DLBCL from PCNSL.34
Plasma cell neoplasms. Flow cytometry is acceptable for the diagnosis of plasma cell neoplasms and for residual disease follow-up. Chromosomal karyotype or FISH for IGH/CCND1, IGH/MMSET, and IGH/CMAF dual fusion probes is recommended in conjunction with morphology, IHC, and flow cytometry. In plasma cell myeloma, several genetic mutations can be detected with NGS, including mutations in NRAS, KRAS, TP53, BCL7A, DIS3, and FAM46C.35 Less commonly, BRAF mutations, previously described in melanoma and several other solid tumors, can be detected with DNA sequencing in 4% of multiple myeloma cases, which may prove promising for targeted therapy with BRAF inhibitors. However, current therapeutic decisions are based on genetic and clinical factors, and sequence-based assays are not recommended at this time.
Follicular lymphoma. Cytology, histology, and IHC typically are sufficient for diagnosing FL. In difficult-to-diagnose cases and in cases with scant material, additional tests may help with diagnosis. Eighty to ninety percent of FL cases have t(14;18)(q32;q21), which places the BCL2 gene transcription under the control of the IGH promoter. In addition, about 10% of FL cases have 3q27 aberrancies at the BCL6 gene.36-38 More recently, cases of FL with bulky inguinal disease negative for IGH-BCL2 and BCL6 translocations were found to have 1p36 deletions. These 1p36-deleted FLs typically have a diffuse pattern and a good prognosis.39 For t(14;18), 3q27, or 1p36, FISH is a sensitive means for detecting these translocations, as is PCR for IGH-BCL2.40 There are reports that t(14;18) can be detected in a substantial fraction of otherwise healthy donors at levels and rates that depend on the type of detection test used.41-43 In addition, between one-fourth and one-third of de novo DLBCLs show t(14;18), and about one-third show BCL6 abnormalities at 3q27. Therefore, these genetic changes are not specific for FL and should not be used to subtype a lymphoma as follicular in origin.
Use of IGH-BCL2 as a marker for MRD is still controversial. Some studies have found that a postinduction and posttransplantation IGH-BCL2-positive finding by PCR predicted relapse.44,45 However, others studies have not found significance to postinduction IGH-BCL2 positivity.46 The NCCN guidelines recommend testing for IGH-BCL2 or BCL6 translocations or 1p36 deletion only if this testing is needed for diagnosis. The guidelines do not recommend using these genetic assays in follow-up biopsies, as the importance of treating early relapse has not been definitively demonstrated.
Therefore, if a lymphoma has morphologic, histologic, and IHC findings consistent with FL, then cytogenetic, FISH, or PCR testing is not needed for diagnosis but may be used as confirmation. Follow-up molecular and cytogenetic testing should be avoided if the original cytogenetic abnormality is unknown. That is, IGH-BCL2 FISH should be performed in follow-up samples only if the original lymphoma is known to contain the translocation. As follow-up genetic testing is of disputed clinical significance even in cases in which the original molecular change is known, the NCCN recommendations for therapy are no different. The HMG subcommittee does not recommend molecular or cytogenetic testing in FL beyond what is required for initial diagnosis.
T-Cell Lymphomas
Mature T-Cell Lymphoma and Leukemia
For mature T-cell lymphoma (TCL) and leukemia, the clinical and morphologic criteria have a very important role in the initial workup. However, IHC immunophenotyping is crucial for definitive diagnosis and subclassification. Flow cytometry is routinely used in diagnosing diseases such as T-cell prolymphocytic leukemia (TPLL), T-cell large granular lymphocytic (LGL) leukemia, and Sézary syndrome. T-cell clonality studies, preferably with BIOMED-II–validated primers against targets such as T-cell receptor
Significant advances in TCL classification have led to revisions and the inclusion of new provisional entities in the 2016 World Health Organization classification of lymphoid neoplasms.47 Many of these changes originated in studies of gene expression profiling and the genetic landscape of T-cell neoplasms. Even though subsets of peripheral TCL not otherwise specified (PTCL-NOS) have been recognized on the basis of phenotypic and molecular abnormalities with possible clinical implications, in most cases molecular testing is not part of routine practice. Typically, only a few cytogenetic abnormalities and genetic mutations are used in the evaluation of TCL and T-cell leukemia.
A group of T-cell lymphoproliferative disorders with expression of T follicular helper cell markers can be identified with IHC. These disorders include angioimmunoblastic TCL; follicular TCL, a new entity that is a PTCL-NOS subset; and primary cutaneous CD4-positive small/medium T-cell lymphoproliferative disorder. The neoplastic cells should express at least 2 or 3 T follicular helper cell–related antigens, including CD279/PD1, CD10, BCL6, CXCL13, ICOS, SAP, and CCR5; the most commonly used are PD1, BCL6, and CD10. Recurrent fusion of ITK-SYK translocation t(5;9) or CTLA4-CD28 is also common in follicular TCL. Although recurrent mutation is found in these entities, conventional karyotyping or IHC should be sufficient for diagnosis.
Cutaneous γ -Δ T-Cell Lymphoma
Among cutaneous TCLs, primary cutaneous
Peripheral T-Cell Lymphoma
Gene expression profiling analysis of PTCLs has identified at least 3 subtypes characterized by overexpression of GATA3, TBX21, and cytotoxic genes and expression of the corresponding proteins with IHC.47 These subtypes are associated with different clinical behavior and therapy responses. The GATA3 subtype has an inferior prognosis and shows a high level of T helper type 2 cytokines, which can be identified with IHC. As IHC-stained GATA3 has been available as a marker of urothelial carcinoma at most IHC laboratories, GATA3 IHC staining also may be considered in the evaluation of PTCLs.
Many monoclonal antibody therapies are being used as primary or secondary regimens in the treatment of TCL. Clinical trials are working to establish their efficacy. If treatment with a monoclonal antibody is being considered, it is appropriate to conduct IHC to demonstrate the presence of the target antigen and at follow-up, to demonstrate the efficacy of treatment. These therapies include alemtuzumab, which targets CD52, and brentuximab, which targets CD30.
T-Cell Large Granular Lymphocytic Leukemia
T-cell LGL leukemia is a complex diagnosis that requires persistent clonal expansion of LGLs and clinically peripheral blood cytopenia. In many cases, the diagnosis is difficult to establish, as benign large granular lymphocytosis with clonal T cells may occur in conjunction with viral infections or autoimmune disorders. Somatic mutations in the STAT3 (signal transducer and activator of transcription 3) gene are found in 40% of patients with T-cell LGL leukemia.49 More recently, somatic mutations in the STAT5B gene were identified in 2% of T-cell LGL leukemia subsets. The clinical course of T-cell LGL leukemia in patients with the STAT5B mutation is aggressive and fatal, clearly different from the relatively favorable course of typical T-cell LGL leukemia.50 The HMG subcommittee recommends considering a STAT3 and STAT5B mutation study for selected cases in which it is difficult to distinguish true T-cell LGL leukemia from its reactive expansions.
T-Cell Prolymphocytic Leukemia
T-cell prolymphocytic leukemia (T-PLL) is a rare, aggressive disease and is most commonly associated with a prolymphocytic morphology and expression of CD4. However, since a specific immunophenotypic profile of T-PLL has not been identified, flow cytometry is not adequate in isolation for definitive classification as T-PLL.51 A diagnosis of T-PLL often requires cytogenetics or a FISH study to confirm a suspected case. Most TPLL cases harbor characteristic chromosomal abnormalities involving 14q11.2 (TCR
Anaplastic Large Cell Lymphoma
The World Health Organization recognizes 3 distinct types of anaplastic large cell lymphoma (ALCL): systemic anaplastic lymphoma kinase (ALK)–positive ALCL, systemic ALK-negative ALCL, and primary cutaneous ALCL. Systemic ALK-positive ALCLs consistently have ALK gene rearrangements and favorable outcomes. The most common translocation is the t(2;5) rearrangement of NPM1 and ALK, though other ALK partners are also possible. In contrast, systemic ALK-negative ALCLs lack ALK gene rearrangements and as a whole have outcomes inferior to those of systemic ALK-positive ALCLs. However, studies have found systemic ALK-negative ALCL to be a genetically and clinically heterogeneous entity.54 About 30% of cases have rearrangements of the DUSP22-IRF4 locus on 6p25.3 (DUSP22 rearrangement), and these cases have favorable outcomes similar to those of systemic ALK-positive ALCL.55 Only 8% of patients have TP63 rearrangements and very poor outcomes. The remaining cases lack ALK, DUSP22, and TP63 rearrangements and have intermediate outcomes. The HMG subcommittee recommends considering DUSP22 rearrangement by FISH in the evaluation of systemic ALK-negative ALCL.
Conclusion
The pathologic diagnosis, classification, and risk stratification of lymphoma and leukemia require an approach that integrates morphology, flow cytometry, cytogenetics, and molecular pathology. Rapidly evolving molecular techniques currently allow for detailed description of the molecular defects in lymphoma and leukemia, including driver mutations, amplification/deletion events, and clonal evolution. Unfortunately, the technical ability to catalogue the molecular defects in lymphoma and leukemia, often at great expense, is outpacing the ability to use this detailed information in treating patients with hematologic malignancies. The challenge, then, is to identify best practices for the diagnosis and classification of lymphoma and leukemia in VHA hospitals that incorporate the most useful molecular tests without wasting financial resources.
In this report, the HMG subcommittee of the MGPW has presented its recommendations for molecular testing in AML, MPN, MDS, and lymphomas in the context of standard morphologic and immunophenotypic approaches to hematopathology diagnosis and classification. Adoption of these recommendations by VHA hospitals and clinics should help ensure that all VA patients with hematologic malignancies benefit from the latest advances in precision medicine.
Within the vast and comprehensive national VHA health care system are multiple centers of expertise in hematopathology. In addition, multiple VA clinical molecular diagnostic laboratories are performing state-of-the-art testing. The HMG subcommittee proposes that, to make best use of these expert resources, the VHA should establish an interfacility hematopathology consultation service. This service would allow any VA pathologist to consult a board-certified hematopathologist regarding use of ancillary molecular genetic testing in the diagnosis of hematologic malignancy.
In addition, the HMG subcommittee recommends consolidating VA molecular diagnostic reference laboratories and having them perform molecular testing for other VA hospitals rather than using commercial reference laboratories, where testing standards are not uniform and results may be difficult to interpret. Several well-established VA clinical laboratories with technical expertise and informatics support are already performing selected molecular diagnostic testing. These laboratories’ resources should be expanded, where practical, to cost-effectively provide VA expertise to all veterans and to improve access to appropriate molecular diagnostic testing.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of
Click here to read the digital edition.
1. Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372(9):793-795.
2. Matynia AP, Szankasi P, Shen W, Kelley TW. Molecular genetic biomarkers in myeloid malignancies. Arch Pathol Lab Med. 2015;139(5):594-601.
3. Wang ML, Bailey NG. Acute myeloid leukemia genetics: risk stratification and implications for therapy. Arch Pathol Lab Med. 2015;139(10):1215-1223.
4. Marum JE, Branford S. Current developments in molecular monitoring in chronic myeloid leukemia. Ther Adv Hematol. 2016;7(5):237-251.
5. Hughes TP, Saglio G, Kantarjian HM, et al. Early molecular response predicts outcomes in patients with chronic myeloid leukemia in chronic phase treated with frontline nilotinib or imatinib. Blood. 2014;123(9):1353-1360.
6. Pallera A, Altman JK, Berman E, et al. Guidelines insights: chronic myeloid leukemia, version 1.2017. J Natl Compr Canc Netw. 2016;14:1505-1512.
7. James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144-1148.
8. Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779-1790.
9. Beer PA, Campbell PJ, Scott LM, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood. 2008;112(1):141-149.
10. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Characteristics and clinical correlates of MPL 515W>L/K mutation in essential thrombocythemia. Blood. 2008;112(3):844-847.
11. Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391-2405.
12. Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379-2390.
13. Harrison CN, Vannucchi AM. Closing the gap: genetic landscape of MPN. Blood. 2016;127(3):276-278.
14. Tefferi A, Barbui T. Essential thrombocythemia and polycythemia vera: focus on clinical practice. Mayo Clin Proc. 2015;90(9):1283-1293.
15. Cools J, DeAngelo DJ, Gotlib J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348(13):1201-1214.
16. Valent P, Klion AD, Horny HP, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol. 2012;130(3):607-612.e609.
17. Bain B, Billiland D, Horny H, Verdiman J. Myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1. In: Swerdlow S, Campo E, Harris N, et al, eds. WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues. Vol 2. Lyon, France: IRAC Press; 2008:68-73.
18. Wang SA, Tam W, Tsai AG, et al. Targeted next-generation sequencing identifies a subset of idiopathic hypereosinophilic syndrome with features similar to chronic eosinophilic leukemia, not otherwise specified. Mod Pathol. 2016;29(8):854-864.
19. Maxson JE, Gotlib J, Pollyea DA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med. 2013;368(19):1781-1790.
20. Schanz J, Tüchler H, Solé F, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol. 2012;30(8):820-829.
21. Marshall D, Roboz GJ. Standardizing the initial evaluation for myelodysplastic syndromes. Curr Hematol Malig Rep. 2013;8(4):361-369.
22. Damm F, Chesnais V, Nagata Y, et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood. 2013;122(18):3169-3177.
23. Malcovati L, Papaemmanuil E, Ambaglio I, et al. Driver somatic mutations identify distinct disease entities within myeloid neoplasms with myelodysplasia. Blood. 2014;124(9):1513-1521.
24. Papaemmanuil E, Cazzola M, Boultwood J, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365(15):1384-1395.
25. Patnaik MM, Hanson CA, Sulai NH, et al. Prognostic irrelevance of ring sideroblast percentage in World Health Organization-defined myelodysplastic syndromes without excess blasts. Blood. 2012;119(24):5674-5677.
26. Guièze R, Wu CJ. Genomic and epigenomic heterogeneity in chronic lymphocytic leukemia. Blood. 2015;126(4):445-453.
27. Jeromin S, Weissmann S, Haferlach C, et al. SF3B1 mutations correlated to cytogenetics and mutations in NOTCH1, FBXW7, MYD88, XPO1 and TP53 in 1160 untreated CLL patients. Leukemia. 2014;28(1):108-117.
28. Del Giudice I, Rossi D, Chiaretti S, et al. NOTCH1 mutations in +12 chronic lymphocytic leukemia (CLL) confer an unfavorable prognosis, induce a distinctive transcriptional profiling and refine the intermediate prognosis of +12 CLL. Haematologica. 2012;97(3):437-441.
29. Weissmann S, Roller A, Jeromin S, et al. Prognostic impact and landscape of NOTCH1 mutations in chronic lymphocytic leukemia (CLL): a study on 852 patients. Leukemia. 2013;27(12):2393-2396.
30. Vegliante MC, Palomero J, Pérez-Galán P, et al. SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive mantle cell lymphoma. Blood. 2013;121(12):2175-2185.
31. King RL, Gonsalves WI, Ansell SM, et al. Lymphoplasmacytic lymphoma with a non-IgM paraprotein shows clinical and pathologic heterogeneity and may harbor MYD88 L265P mutations. Am J Clin Pathol. 2016;145(6):843-851.
32. Insuasti-Beltran G, Gale JM, Wilson CS, Foucar K, Czuchlewski DR. Significance of MYD88 L265P mutation status in the subclassification of low-grade B-cell lymphoma/leukemia. Arch Pathol Lab Med. 2015;139(8):1035-1041.
33. Wang XJ, Kim A, Li S. Immunohistochemical analysis using a BRAF V600E mutation specific antibody is highly sensitive and specific for the diagnosis of hairy cell leukemia. Int J Clin Exp Pathol. 2014;7(7):4323-4328.
34. Nakamura T, Tateishi K, Niwa T, et al. Recurrent mutations of CD79B and MYD88 are the hallmark of primary central nervous system lymphomas. Neuropathol Appl Neurobiol. 2016;42(3):279-290.
35. Chapman MA, Lawrence MS, Keats JJ, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471(7339):467-472.
36. Bosga-Bouwer AG, van Imhoff GW, Boonstra R, et al. Follicular lymphoma grade 3B includes 3 cytogenetically defined subgroups with primary t(14;18), 3q27, or other translocations: t(14;18) and 3q27 are mutually exclusive. Blood. 2003;101(3):1149-1154.
37. Gu K, Fu K, Jain S, et al. t(14;18)-negative follicular lymphomas are associated with a high frequency of BCL6 rearrangement at the alternative breakpoint region. Mod Pathol. 2009;22(9):1251-1257.
38. Katzenberger T, Ott G, Klein T, Kalla J, Müller-Hermelink HK, Ott MM. Cytogenetic alterations affecting BCL6 are predominantly found in follicular lymphomas grade 3B with a diffuse large B-cell component. Am J Pathol. 2004;165(2):481-490.
39. Katzenberger T, Kalla J, Leich E, et al. A distinctive subtype of t(14;18)-negative nodal follicular non-Hodgkin lymphoma characterized by a predominantly diffuse growth pattern and deletions in the chromosomal region 1p36. Blood. 2009;113(5):1053-1061.
40. Belaud-Rotureau MA, Parrens M, Carrere N, et al. Interphase fluorescence in situ hybridization is more sensitive than BIOMED-2 polymerase chain reaction protocol in detecting IGH-BCL2 rearrangement in both fixed and frozen lymph node with follicular lymphoma. Hum Pathol. 2007;38(2):365-372.
41. Limpens J, Stad R, Vos C, et al. Lymphoma-associated translocation t(14;18) in blood B cells of normal individuals. Blood. 1995;85(9):2528-2536.
42. Schmitt C, Balogh B, Grundt A, et al. The bcl-2/IgH rearrangement in a population of 204 healthy individuals: occurrence, age and gender distribution, breakpoints, and detection method validity. Leuk Res. 2006;30(6):745-750.
43. Summers KE, Goff LK, Wilson AG, Gupta RK, Lister TA, Fitzgibbon J. Frequency of the Bcl-2/IgH rearrangement in normal individuals: implications for the monitoring of disease in patients with follicular lymphoma. J Clin Oncol. 2001;19(2):420-424.
44. Galimberti S, Luminari S, Ciabatti E, et al. Minimal residual disease after conventional treatment significantly impacts on progression-free survival of patients with follicular lymphoma: the FIL FOLL05 trial. Clin Cancer Res. 2014;20(24):6398-6405.
45. Ladetto M, Lobetti-Bodoni C, Mantoan B, et al; Fondazione Italiana Linfomi. Persistence of minimal residual disease in bone marrow predicts outcome in follicular lymphomas treated with a rituximab-intensive program. Blood. 2013;122(23):3759-3766.
46. van Oers MHJ, Tönnissen E, Van Glabbeke M, et al. BCL-2/IgH polymerase chain reaction status at the end of induction treatment is not predictive for progression-free survival in relapsed/resistant follicular lymphoma: results of a prospective randomized EORTC 20981 phase III intergroup study. J Clin Oncol. 2010;28(13):2246-2252.
47. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375-2390.
48. Dewar R, Andea AA, Guitart J, Arber DA, Weiss LM. Best practices in diagnostic immunohistochemistry: workup of cutaneous lymphoid lesions in the diagnosis of primary cutaneous lymphoma. Arch Pathol Lab Med. 2015;139(3):338-350.
49. Koskela HLM, Eldfors S, Ellonen P, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012;366(20):1905-1913.
50. Rajala HL, Eldfors S, Kuusanmäki H, et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood. 2013;121(22):4541-4550.
51. Chen X, Cherian S. Immunophenotypic characterization of T-cell prolymphocytic leukemia. Am J Clin Pathol. 2013;140(5):727-735.
52. Delgado P, Starshak P, Rao N, Tirado C. A comprehensive update on molecular and cytogenetic abnormalities in T-cell prolymphocytic leukemia (T-PLL). J Assoc Genet Technol. 2012;38(4):193-198.
53. Stengel A, Kern W, Zenger M, et al. Genetic characterization of T-PLL reveals two major biologic subgroups and JAK3 mutations as prognostic marker. Genes Chromosomes Cancer. 2016;55(1):82-94.
54. Thompson MA, Stumph J, Henrickson SE, et al. Differential gene expression in anaplastic lymphoma kinase-positive and anaplastic lymphoma kinase-negative anaplastic large cell lymphomas. Hum Pathol. 2005;36(5):494-504.
55. King R, Dao L, McPhail E, et al. Morphologic features of ALK-negative anaplastic large cell lymphomas with DUSP22 rearrangements. Am J Surg Pathol. 2016;40(1):36-43.
1. Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372(9):793-795.
2. Matynia AP, Szankasi P, Shen W, Kelley TW. Molecular genetic biomarkers in myeloid malignancies. Arch Pathol Lab Med. 2015;139(5):594-601.
3. Wang ML, Bailey NG. Acute myeloid leukemia genetics: risk stratification and implications for therapy. Arch Pathol Lab Med. 2015;139(10):1215-1223.
4. Marum JE, Branford S. Current developments in molecular monitoring in chronic myeloid leukemia. Ther Adv Hematol. 2016;7(5):237-251.
5. Hughes TP, Saglio G, Kantarjian HM, et al. Early molecular response predicts outcomes in patients with chronic myeloid leukemia in chronic phase treated with frontline nilotinib or imatinib. Blood. 2014;123(9):1353-1360.
6. Pallera A, Altman JK, Berman E, et al. Guidelines insights: chronic myeloid leukemia, version 1.2017. J Natl Compr Canc Netw. 2016;14:1505-1512.
7. James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144-1148.
8. Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779-1790.
9. Beer PA, Campbell PJ, Scott LM, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood. 2008;112(1):141-149.
10. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Characteristics and clinical correlates of MPL 515W>L/K mutation in essential thrombocythemia. Blood. 2008;112(3):844-847.
11. Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391-2405.
12. Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379-2390.
13. Harrison CN, Vannucchi AM. Closing the gap: genetic landscape of MPN. Blood. 2016;127(3):276-278.
14. Tefferi A, Barbui T. Essential thrombocythemia and polycythemia vera: focus on clinical practice. Mayo Clin Proc. 2015;90(9):1283-1293.
15. Cools J, DeAngelo DJ, Gotlib J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348(13):1201-1214.
16. Valent P, Klion AD, Horny HP, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol. 2012;130(3):607-612.e609.
17. Bain B, Billiland D, Horny H, Verdiman J. Myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1. In: Swerdlow S, Campo E, Harris N, et al, eds. WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues. Vol 2. Lyon, France: IRAC Press; 2008:68-73.
18. Wang SA, Tam W, Tsai AG, et al. Targeted next-generation sequencing identifies a subset of idiopathic hypereosinophilic syndrome with features similar to chronic eosinophilic leukemia, not otherwise specified. Mod Pathol. 2016;29(8):854-864.
19. Maxson JE, Gotlib J, Pollyea DA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med. 2013;368(19):1781-1790.
20. Schanz J, Tüchler H, Solé F, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol. 2012;30(8):820-829.
21. Marshall D, Roboz GJ. Standardizing the initial evaluation for myelodysplastic syndromes. Curr Hematol Malig Rep. 2013;8(4):361-369.
22. Damm F, Chesnais V, Nagata Y, et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood. 2013;122(18):3169-3177.
23. Malcovati L, Papaemmanuil E, Ambaglio I, et al. Driver somatic mutations identify distinct disease entities within myeloid neoplasms with myelodysplasia. Blood. 2014;124(9):1513-1521.
24. Papaemmanuil E, Cazzola M, Boultwood J, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365(15):1384-1395.
25. Patnaik MM, Hanson CA, Sulai NH, et al. Prognostic irrelevance of ring sideroblast percentage in World Health Organization-defined myelodysplastic syndromes without excess blasts. Blood. 2012;119(24):5674-5677.
26. Guièze R, Wu CJ. Genomic and epigenomic heterogeneity in chronic lymphocytic leukemia. Blood. 2015;126(4):445-453.
27. Jeromin S, Weissmann S, Haferlach C, et al. SF3B1 mutations correlated to cytogenetics and mutations in NOTCH1, FBXW7, MYD88, XPO1 and TP53 in 1160 untreated CLL patients. Leukemia. 2014;28(1):108-117.
28. Del Giudice I, Rossi D, Chiaretti S, et al. NOTCH1 mutations in +12 chronic lymphocytic leukemia (CLL) confer an unfavorable prognosis, induce a distinctive transcriptional profiling and refine the intermediate prognosis of +12 CLL. Haematologica. 2012;97(3):437-441.
29. Weissmann S, Roller A, Jeromin S, et al. Prognostic impact and landscape of NOTCH1 mutations in chronic lymphocytic leukemia (CLL): a study on 852 patients. Leukemia. 2013;27(12):2393-2396.
30. Vegliante MC, Palomero J, Pérez-Galán P, et al. SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive mantle cell lymphoma. Blood. 2013;121(12):2175-2185.
31. King RL, Gonsalves WI, Ansell SM, et al. Lymphoplasmacytic lymphoma with a non-IgM paraprotein shows clinical and pathologic heterogeneity and may harbor MYD88 L265P mutations. Am J Clin Pathol. 2016;145(6):843-851.
32. Insuasti-Beltran G, Gale JM, Wilson CS, Foucar K, Czuchlewski DR. Significance of MYD88 L265P mutation status in the subclassification of low-grade B-cell lymphoma/leukemia. Arch Pathol Lab Med. 2015;139(8):1035-1041.
33. Wang XJ, Kim A, Li S. Immunohistochemical analysis using a BRAF V600E mutation specific antibody is highly sensitive and specific for the diagnosis of hairy cell leukemia. Int J Clin Exp Pathol. 2014;7(7):4323-4328.
34. Nakamura T, Tateishi K, Niwa T, et al. Recurrent mutations of CD79B and MYD88 are the hallmark of primary central nervous system lymphomas. Neuropathol Appl Neurobiol. 2016;42(3):279-290.
35. Chapman MA, Lawrence MS, Keats JJ, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471(7339):467-472.
36. Bosga-Bouwer AG, van Imhoff GW, Boonstra R, et al. Follicular lymphoma grade 3B includes 3 cytogenetically defined subgroups with primary t(14;18), 3q27, or other translocations: t(14;18) and 3q27 are mutually exclusive. Blood. 2003;101(3):1149-1154.
37. Gu K, Fu K, Jain S, et al. t(14;18)-negative follicular lymphomas are associated with a high frequency of BCL6 rearrangement at the alternative breakpoint region. Mod Pathol. 2009;22(9):1251-1257.
38. Katzenberger T, Ott G, Klein T, Kalla J, Müller-Hermelink HK, Ott MM. Cytogenetic alterations affecting BCL6 are predominantly found in follicular lymphomas grade 3B with a diffuse large B-cell component. Am J Pathol. 2004;165(2):481-490.
39. Katzenberger T, Kalla J, Leich E, et al. A distinctive subtype of t(14;18)-negative nodal follicular non-Hodgkin lymphoma characterized by a predominantly diffuse growth pattern and deletions in the chromosomal region 1p36. Blood. 2009;113(5):1053-1061.
40. Belaud-Rotureau MA, Parrens M, Carrere N, et al. Interphase fluorescence in situ hybridization is more sensitive than BIOMED-2 polymerase chain reaction protocol in detecting IGH-BCL2 rearrangement in both fixed and frozen lymph node with follicular lymphoma. Hum Pathol. 2007;38(2):365-372.
41. Limpens J, Stad R, Vos C, et al. Lymphoma-associated translocation t(14;18) in blood B cells of normal individuals. Blood. 1995;85(9):2528-2536.
42. Schmitt C, Balogh B, Grundt A, et al. The bcl-2/IgH rearrangement in a population of 204 healthy individuals: occurrence, age and gender distribution, breakpoints, and detection method validity. Leuk Res. 2006;30(6):745-750.
43. Summers KE, Goff LK, Wilson AG, Gupta RK, Lister TA, Fitzgibbon J. Frequency of the Bcl-2/IgH rearrangement in normal individuals: implications for the monitoring of disease in patients with follicular lymphoma. J Clin Oncol. 2001;19(2):420-424.
44. Galimberti S, Luminari S, Ciabatti E, et al. Minimal residual disease after conventional treatment significantly impacts on progression-free survival of patients with follicular lymphoma: the FIL FOLL05 trial. Clin Cancer Res. 2014;20(24):6398-6405.
45. Ladetto M, Lobetti-Bodoni C, Mantoan B, et al; Fondazione Italiana Linfomi. Persistence of minimal residual disease in bone marrow predicts outcome in follicular lymphomas treated with a rituximab-intensive program. Blood. 2013;122(23):3759-3766.
46. van Oers MHJ, Tönnissen E, Van Glabbeke M, et al. BCL-2/IgH polymerase chain reaction status at the end of induction treatment is not predictive for progression-free survival in relapsed/resistant follicular lymphoma: results of a prospective randomized EORTC 20981 phase III intergroup study. J Clin Oncol. 2010;28(13):2246-2252.
47. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375-2390.
48. Dewar R, Andea AA, Guitart J, Arber DA, Weiss LM. Best practices in diagnostic immunohistochemistry: workup of cutaneous lymphoid lesions in the diagnosis of primary cutaneous lymphoma. Arch Pathol Lab Med. 2015;139(3):338-350.
49. Koskela HLM, Eldfors S, Ellonen P, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012;366(20):1905-1913.
50. Rajala HL, Eldfors S, Kuusanmäki H, et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood. 2013;121(22):4541-4550.
51. Chen X, Cherian S. Immunophenotypic characterization of T-cell prolymphocytic leukemia. Am J Clin Pathol. 2013;140(5):727-735.
52. Delgado P, Starshak P, Rao N, Tirado C. A comprehensive update on molecular and cytogenetic abnormalities in T-cell prolymphocytic leukemia (T-PLL). J Assoc Genet Technol. 2012;38(4):193-198.
53. Stengel A, Kern W, Zenger M, et al. Genetic characterization of T-PLL reveals two major biologic subgroups and JAK3 mutations as prognostic marker. Genes Chromosomes Cancer. 2016;55(1):82-94.
54. Thompson MA, Stumph J, Henrickson SE, et al. Differential gene expression in anaplastic lymphoma kinase-positive and anaplastic lymphoma kinase-negative anaplastic large cell lymphomas. Hum Pathol. 2005;36(5):494-504.
55. King R, Dao L, McPhail E, et al. Morphologic features of ALK-negative anaplastic large cell lymphomas with DUSP22 rearrangements. Am J Surg Pathol. 2016;40(1):36-43.
Which Acute Myeloid Leukemia Patients are Good Immunotherapy Candidates?
Some patients with acute myeloid leukemia (AML) may have trouble with immunotherapy following chemotherapy. Researchers from the National Heart, Lung and Blood Institute may have found a reason why.
Related: Novel Treatment Shows Promise for Acute Lymphoblastic Leukemia
The researchers wanted to perform a “deep assessment” of the state of the adaptive immune system in AML patients in remission after chemotherapy. They used these patients’ response to seasonal influenza vaccination as a surrogate for the robustness of the immune system. The researchers say their approach was unique in that they established a comprehensive picture of the adaptive “immunome” by simultaneously examining the genetic, phenotypic, and functional consequences of chemotherapy.
Their assessment revealed a “dramatic impact” in the B-cell compartment, which appeared slower to recover than the T-cell compartment. Of 10 patients in the study, only 2 generated protective titers in response to vaccination. Most had abnormal frequencies of transitional and memory B-cells. The researchers say the inability of AML patients to produce protective antibody titers in response to influenza vaccination is likely due to multiple B-cell abnormalities.
Related: Six Open Clinical Trials That Are Expanding Our Understanding of Immunotherapies
The researchers “strikingly” found similar patterns of immune dysfunction across all the patients in the study. When they ranked patients based on time elapsed since chemotherapy, the degree of dysfunction was shown to be less in patients who had the most time elapsed form their chemotherapy treatment.
The researchers conclude the “consistent finding” of a reduction of memory B-cells in all the AML patients suggests that humoral immunity reconstitution is “a very long process.” They add that a better understanding of the changes in adaptive immune cell subsets after chemotherapy will be useful in designing immunotherapies that can work with existing immune capacity.
Source:
Goswami M, Prince G, Biancotto A, et al. J Transl Med. 2017;15:155.
doi: 10.1186/s12967-017-1252-2
Some patients with acute myeloid leukemia (AML) may have trouble with immunotherapy following chemotherapy. Researchers from the National Heart, Lung and Blood Institute may have found a reason why.
Related: Novel Treatment Shows Promise for Acute Lymphoblastic Leukemia
The researchers wanted to perform a “deep assessment” of the state of the adaptive immune system in AML patients in remission after chemotherapy. They used these patients’ response to seasonal influenza vaccination as a surrogate for the robustness of the immune system. The researchers say their approach was unique in that they established a comprehensive picture of the adaptive “immunome” by simultaneously examining the genetic, phenotypic, and functional consequences of chemotherapy.
Their assessment revealed a “dramatic impact” in the B-cell compartment, which appeared slower to recover than the T-cell compartment. Of 10 patients in the study, only 2 generated protective titers in response to vaccination. Most had abnormal frequencies of transitional and memory B-cells. The researchers say the inability of AML patients to produce protective antibody titers in response to influenza vaccination is likely due to multiple B-cell abnormalities.
Related: Six Open Clinical Trials That Are Expanding Our Understanding of Immunotherapies
The researchers “strikingly” found similar patterns of immune dysfunction across all the patients in the study. When they ranked patients based on time elapsed since chemotherapy, the degree of dysfunction was shown to be less in patients who had the most time elapsed form their chemotherapy treatment.
The researchers conclude the “consistent finding” of a reduction of memory B-cells in all the AML patients suggests that humoral immunity reconstitution is “a very long process.” They add that a better understanding of the changes in adaptive immune cell subsets after chemotherapy will be useful in designing immunotherapies that can work with existing immune capacity.
Source:
Goswami M, Prince G, Biancotto A, et al. J Transl Med. 2017;15:155.
doi: 10.1186/s12967-017-1252-2
Some patients with acute myeloid leukemia (AML) may have trouble with immunotherapy following chemotherapy. Researchers from the National Heart, Lung and Blood Institute may have found a reason why.
Related: Novel Treatment Shows Promise for Acute Lymphoblastic Leukemia
The researchers wanted to perform a “deep assessment” of the state of the adaptive immune system in AML patients in remission after chemotherapy. They used these patients’ response to seasonal influenza vaccination as a surrogate for the robustness of the immune system. The researchers say their approach was unique in that they established a comprehensive picture of the adaptive “immunome” by simultaneously examining the genetic, phenotypic, and functional consequences of chemotherapy.
Their assessment revealed a “dramatic impact” in the B-cell compartment, which appeared slower to recover than the T-cell compartment. Of 10 patients in the study, only 2 generated protective titers in response to vaccination. Most had abnormal frequencies of transitional and memory B-cells. The researchers say the inability of AML patients to produce protective antibody titers in response to influenza vaccination is likely due to multiple B-cell abnormalities.
Related: Six Open Clinical Trials That Are Expanding Our Understanding of Immunotherapies
The researchers “strikingly” found similar patterns of immune dysfunction across all the patients in the study. When they ranked patients based on time elapsed since chemotherapy, the degree of dysfunction was shown to be less in patients who had the most time elapsed form their chemotherapy treatment.
The researchers conclude the “consistent finding” of a reduction of memory B-cells in all the AML patients suggests that humoral immunity reconstitution is “a very long process.” They add that a better understanding of the changes in adaptive immune cell subsets after chemotherapy will be useful in designing immunotherapies that can work with existing immune capacity.
Source:
Goswami M, Prince G, Biancotto A, et al. J Transl Med. 2017;15:155.
doi: 10.1186/s12967-017-1252-2