User login
Oncologists are average in terms of happiness, survey suggests
When it comes to physician happiness both in and outside the workplace, oncologists are about average, according to Medscape’s 2020 Lifestyle, Happiness, and Burnout Report.
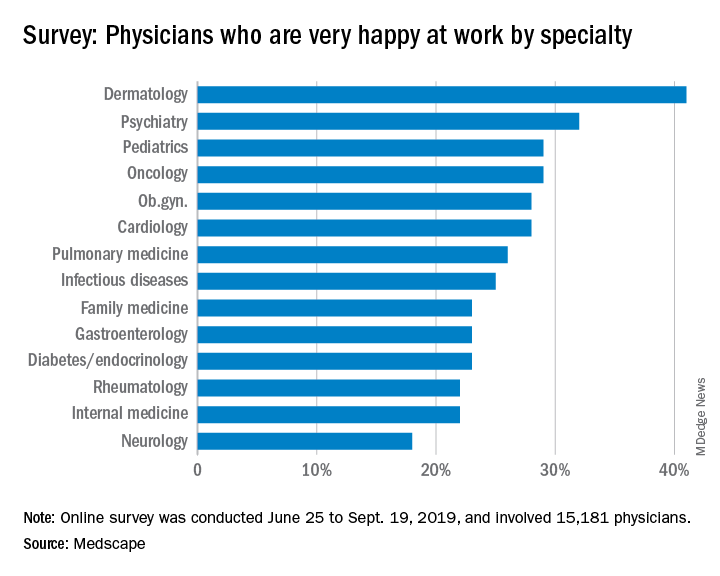
Oncologists landed in the middle of the pack among all physicians surveyed for happiness. Rheumatologists were most likely to report being very or extremely happy outside of work (60%) and neurologists were least likely to do so (44%), but about half of oncologists (51%) reported being very/extremely happy outside of work. For happiness at work, dermatologists topped the list (41%), neurologists came in last (18%), and oncologists remained in the middle (29%).
Oncologists were average when it came to burnout as well, matching the rate of overall physicians. Specifically, 32% of oncologists were burned out, 4% were depressed, and 9% were both burned out and depressed.
The most commonly reported factors contributing to burnout among oncologists were an overabundance of bureaucratic tasks (74%), spending too many hours at work (42%), and a lack of respect from colleagues in the workplace (36%).
Exercise was the most commonly reported way oncologists dealt with burnout (51%), followed by talking with family and friends (49%), and isolating themselves from others (38%). In addition, 57% of oncologists took 3-4 weeks’ vacation, compared with 44% of physicians overall; 29% of oncologists took less than 3 weeks’ vacation.
About 18% of oncologists said they had contemplated suicide, and 1% said they’d attempted it; 72% said they’d never had thoughts of suicide. Just under one-quarter of oncologists said they were currently seeking professional help or were planning to seek help for symptoms of depression and/or burnout.
“The survey results are concerning on several levels,” Maurie Markman, MD, of Cancer Treatment Centers of America, Philadelphia, said in an interview.
“First, the data suggest a considerable number of oncologists are simply burned out from the day-to-day bureaucracy (paperwork, etc.) of medical practice, which has absolutely nothing to do with the actual care delivered. This likely impacts the willingness to continue in this role. Second, one must be concerned for the future recruitment of physicians to become clinical oncologists. And finally, one must wonder about the impact of these concerning figures on the quality of care being provided to cancer patients.”
This survey was conducted from June 25 to Sept. 19, 2019, and involved 15,181 physicians. Oncologists made up 1% of the survey pool.
When it comes to physician happiness both in and outside the workplace, oncologists are about average, according to Medscape’s 2020 Lifestyle, Happiness, and Burnout Report.
Oncologists landed in the middle of the pack among all physicians surveyed for happiness. Rheumatologists were most likely to report being very or extremely happy outside of work (60%) and neurologists were least likely to do so (44%), but about half of oncologists (51%) reported being very/extremely happy outside of work. For happiness at work, dermatologists topped the list (41%), neurologists came in last (18%), and oncologists remained in the middle (29%).
Oncologists were average when it came to burnout as well, matching the rate of overall physicians. Specifically, 32% of oncologists were burned out, 4% were depressed, and 9% were both burned out and depressed.
The most commonly reported factors contributing to burnout among oncologists were an overabundance of bureaucratic tasks (74%), spending too many hours at work (42%), and a lack of respect from colleagues in the workplace (36%).
Exercise was the most commonly reported way oncologists dealt with burnout (51%), followed by talking with family and friends (49%), and isolating themselves from others (38%). In addition, 57% of oncologists took 3-4 weeks’ vacation, compared with 44% of physicians overall; 29% of oncologists took less than 3 weeks’ vacation.
About 18% of oncologists said they had contemplated suicide, and 1% said they’d attempted it; 72% said they’d never had thoughts of suicide. Just under one-quarter of oncologists said they were currently seeking professional help or were planning to seek help for symptoms of depression and/or burnout.
“The survey results are concerning on several levels,” Maurie Markman, MD, of Cancer Treatment Centers of America, Philadelphia, said in an interview.
“First, the data suggest a considerable number of oncologists are simply burned out from the day-to-day bureaucracy (paperwork, etc.) of medical practice, which has absolutely nothing to do with the actual care delivered. This likely impacts the willingness to continue in this role. Second, one must be concerned for the future recruitment of physicians to become clinical oncologists. And finally, one must wonder about the impact of these concerning figures on the quality of care being provided to cancer patients.”
This survey was conducted from June 25 to Sept. 19, 2019, and involved 15,181 physicians. Oncologists made up 1% of the survey pool.
When it comes to physician happiness both in and outside the workplace, oncologists are about average, according to Medscape’s 2020 Lifestyle, Happiness, and Burnout Report.
Oncologists landed in the middle of the pack among all physicians surveyed for happiness. Rheumatologists were most likely to report being very or extremely happy outside of work (60%) and neurologists were least likely to do so (44%), but about half of oncologists (51%) reported being very/extremely happy outside of work. For happiness at work, dermatologists topped the list (41%), neurologists came in last (18%), and oncologists remained in the middle (29%).
Oncologists were average when it came to burnout as well, matching the rate of overall physicians. Specifically, 32% of oncologists were burned out, 4% were depressed, and 9% were both burned out and depressed.
The most commonly reported factors contributing to burnout among oncologists were an overabundance of bureaucratic tasks (74%), spending too many hours at work (42%), and a lack of respect from colleagues in the workplace (36%).
Exercise was the most commonly reported way oncologists dealt with burnout (51%), followed by talking with family and friends (49%), and isolating themselves from others (38%). In addition, 57% of oncologists took 3-4 weeks’ vacation, compared with 44% of physicians overall; 29% of oncologists took less than 3 weeks’ vacation.
About 18% of oncologists said they had contemplated suicide, and 1% said they’d attempted it; 72% said they’d never had thoughts of suicide. Just under one-quarter of oncologists said they were currently seeking professional help or were planning to seek help for symptoms of depression and/or burnout.
“The survey results are concerning on several levels,” Maurie Markman, MD, of Cancer Treatment Centers of America, Philadelphia, said in an interview.
“First, the data suggest a considerable number of oncologists are simply burned out from the day-to-day bureaucracy (paperwork, etc.) of medical practice, which has absolutely nothing to do with the actual care delivered. This likely impacts the willingness to continue in this role. Second, one must be concerned for the future recruitment of physicians to become clinical oncologists. And finally, one must wonder about the impact of these concerning figures on the quality of care being provided to cancer patients.”
This survey was conducted from June 25 to Sept. 19, 2019, and involved 15,181 physicians. Oncologists made up 1% of the survey pool.
Gene therapy effective in hemophilia B patients with neutralizing antibodies
The gene therapy etranacogene dezaparvovec (AMT-061) continues to demonstrate safety and efficacy in patients with hemophilia B, according to a 1-year update of a phase 2b trial.

All three patients in this trial experienced sustained increases in factor IX (FIX) activity and were able to stop prophylaxis without suffering any bleeds. Adverse events related to treatment were mild and transient.
These favorable results are particularly noteworthy because all three patients had anti-AAV5 neutralizing antibodies at baseline, according to Steven W. Pipe, MD, of the University of Michigan, Ann Arbor. He noted that studies of etranacogene dezaparvovec and its predecessor, AMT-060, are the only studies that have not excluded hemophilia patients based on preexisting immunity.
Dr. Pipe presented the latest phase 2b results with etranacogene dezaparvovec at the annual congress of the European Association for Haemophilia and Allied Disorders.
Etranacogene dezaparvovec uses an AAV5 serotype with a transgene expression cassette that codes for the hyperactive Padua FIX variant, Dr. Pipe explained. Etranacogene dezaparvovec has a structure that is nearly identical to that of AMT-060, except for two nucleotide substitutions in the coding sequence for FIX.
AMT-060 enabled stable expression of FIX that has persisted for up to 4 years without any late-emergent safety signals (Blood 2019. 134 Supplement 1: 2059). Dr. Pipe said the “enhanced version” of AMT-060, etranacogene dezaparvovec, has produced even higher levels of FIX activity in the phase 2b study (NCT03489291).
The ongoing study enrolled three men with moderate to severe FIX deficiency at baseline. The patients were 43, 50, and 47 years of age, respectively. Two patients are HIV positive, and all had hepatitis C that resolved.
All three patients were receiving FIX prophylaxis and on-demand treatment at baseline. In the year prior to screening, patients had one, three, and five bleeds, respectively. All three patients had anti-AAV5 neutralizing antibodies.
Efficacy
Patients received a single dose of etranacogene dezaparvovec at 2 x 1013 genome copies/kg. All three patients achieved the primary endpoint, which was FIX activity of at least 5% at 6 weeks.
At 52 weeks, the mean FIX activity was 41%. Patients 1 and 3 have maintained FIX activity of 40% or greater, which is in the nonhemophilic range. Patient 2 has maintained FIX activity in the mild range. At 52 weeks, FIX activity levels were 50.2%, 40.8%, and 31.3%, respectively.
All patients remain free of prophylaxis and bleeds. Patient 3 received a single FIX infusion as a precaution in the perioperative setting. There was no evidence of a bleed in this patient.
Safety
Etranacogene dezaparvovec was generally well tolerated, Dr. Pipe said. One patient had two adverse events that were possibly related to etranacogene dezaparvovec. Both events – transient, self-limiting headache and slightly elevated C-reactive protein – resolved without intervention.
There was one serious adverse event, but it was considered unrelated to treatment. Patient 3 required hip surgery for preexisting avascular necrosis.
Dr. Pipe said there was no evidence of transaminitis. There were modest, transient elevations in liver enzymes, but this was not enough to trigger protocol-specified immunosuppression.
Specifically, one patient had ALT elevations at weeks 22 and 44, and one patient had AST elevations at weeks 2, 4, and 31. All of these resolved quickly without treatment or an impact on FIX activity, Dr. Pipe noted.
Next steps
This study is ongoing, and patients will be followed for 5 years. Dr. Pipe said the main focus of follow-up will be to determine if patients maintain durable expression of FIX.
A phase 3 trial of etranacogene dezaparvovec is ongoing as well. The trial, HOPE-B (NCT03569891), is fully enrolled, and dosing is planned for 55 patients.
“We’re looking forward to data analysis later this year,” Dr. Pipe said. “This will be the only phase 3 study, and really the only platform so far, that is not planning to exclude patients based on preexisting immunity.”
If all goes well in the phase 3 study, etranacogene dezaparvovec could be approved by the Food and Drug Administration very soon, Dr. Pipe added.
UniQure, the company developing etranacogene dezaparvovec, is planning to submit the biologics license application to the FDA next year. Etranacogene dezaparvovec was granted breakthrough designation from the FDA and is therefore eligible for priority review, so the gene therapy could be approved as early as 2021.
The phase 2b trial of etranacogene dezaparvovec is sponsored by uniQure. Dr. Pipe disclosed relationships with uniQure and other companies.
SOURCE: Pipe SW et al. EAHAD 2020, Abstract OR10.
The gene therapy etranacogene dezaparvovec (AMT-061) continues to demonstrate safety and efficacy in patients with hemophilia B, according to a 1-year update of a phase 2b trial.
All three patients in this trial experienced sustained increases in factor IX (FIX) activity and were able to stop prophylaxis without suffering any bleeds. Adverse events related to treatment were mild and transient.
These favorable results are particularly noteworthy because all three patients had anti-AAV5 neutralizing antibodies at baseline, according to Steven W. Pipe, MD, of the University of Michigan, Ann Arbor. He noted that studies of etranacogene dezaparvovec and its predecessor, AMT-060, are the only studies that have not excluded hemophilia patients based on preexisting immunity.
Dr. Pipe presented the latest phase 2b results with etranacogene dezaparvovec at the annual congress of the European Association for Haemophilia and Allied Disorders.
Etranacogene dezaparvovec uses an AAV5 serotype with a transgene expression cassette that codes for the hyperactive Padua FIX variant, Dr. Pipe explained. Etranacogene dezaparvovec has a structure that is nearly identical to that of AMT-060, except for two nucleotide substitutions in the coding sequence for FIX.
AMT-060 enabled stable expression of FIX that has persisted for up to 4 years without any late-emergent safety signals (Blood 2019. 134 Supplement 1: 2059). Dr. Pipe said the “enhanced version” of AMT-060, etranacogene dezaparvovec, has produced even higher levels of FIX activity in the phase 2b study (NCT03489291).
The ongoing study enrolled three men with moderate to severe FIX deficiency at baseline. The patients were 43, 50, and 47 years of age, respectively. Two patients are HIV positive, and all had hepatitis C that resolved.
All three patients were receiving FIX prophylaxis and on-demand treatment at baseline. In the year prior to screening, patients had one, three, and five bleeds, respectively. All three patients had anti-AAV5 neutralizing antibodies.
Efficacy
Patients received a single dose of etranacogene dezaparvovec at 2 x 1013 genome copies/kg. All three patients achieved the primary endpoint, which was FIX activity of at least 5% at 6 weeks.
At 52 weeks, the mean FIX activity was 41%. Patients 1 and 3 have maintained FIX activity of 40% or greater, which is in the nonhemophilic range. Patient 2 has maintained FIX activity in the mild range. At 52 weeks, FIX activity levels were 50.2%, 40.8%, and 31.3%, respectively.
All patients remain free of prophylaxis and bleeds. Patient 3 received a single FIX infusion as a precaution in the perioperative setting. There was no evidence of a bleed in this patient.
Safety
Etranacogene dezaparvovec was generally well tolerated, Dr. Pipe said. One patient had two adverse events that were possibly related to etranacogene dezaparvovec. Both events – transient, self-limiting headache and slightly elevated C-reactive protein – resolved without intervention.
There was one serious adverse event, but it was considered unrelated to treatment. Patient 3 required hip surgery for preexisting avascular necrosis.
Dr. Pipe said there was no evidence of transaminitis. There were modest, transient elevations in liver enzymes, but this was not enough to trigger protocol-specified immunosuppression.
Specifically, one patient had ALT elevations at weeks 22 and 44, and one patient had AST elevations at weeks 2, 4, and 31. All of these resolved quickly without treatment or an impact on FIX activity, Dr. Pipe noted.
Next steps
This study is ongoing, and patients will be followed for 5 years. Dr. Pipe said the main focus of follow-up will be to determine if patients maintain durable expression of FIX.
A phase 3 trial of etranacogene dezaparvovec is ongoing as well. The trial, HOPE-B (NCT03569891), is fully enrolled, and dosing is planned for 55 patients.
“We’re looking forward to data analysis later this year,” Dr. Pipe said. “This will be the only phase 3 study, and really the only platform so far, that is not planning to exclude patients based on preexisting immunity.”
If all goes well in the phase 3 study, etranacogene dezaparvovec could be approved by the Food and Drug Administration very soon, Dr. Pipe added.
UniQure, the company developing etranacogene dezaparvovec, is planning to submit the biologics license application to the FDA next year. Etranacogene dezaparvovec was granted breakthrough designation from the FDA and is therefore eligible for priority review, so the gene therapy could be approved as early as 2021.
The phase 2b trial of etranacogene dezaparvovec is sponsored by uniQure. Dr. Pipe disclosed relationships with uniQure and other companies.
SOURCE: Pipe SW et al. EAHAD 2020, Abstract OR10.
The gene therapy etranacogene dezaparvovec (AMT-061) continues to demonstrate safety and efficacy in patients with hemophilia B, according to a 1-year update of a phase 2b trial.
All three patients in this trial experienced sustained increases in factor IX (FIX) activity and were able to stop prophylaxis without suffering any bleeds. Adverse events related to treatment were mild and transient.
These favorable results are particularly noteworthy because all three patients had anti-AAV5 neutralizing antibodies at baseline, according to Steven W. Pipe, MD, of the University of Michigan, Ann Arbor. He noted that studies of etranacogene dezaparvovec and its predecessor, AMT-060, are the only studies that have not excluded hemophilia patients based on preexisting immunity.
Dr. Pipe presented the latest phase 2b results with etranacogene dezaparvovec at the annual congress of the European Association for Haemophilia and Allied Disorders.
Etranacogene dezaparvovec uses an AAV5 serotype with a transgene expression cassette that codes for the hyperactive Padua FIX variant, Dr. Pipe explained. Etranacogene dezaparvovec has a structure that is nearly identical to that of AMT-060, except for two nucleotide substitutions in the coding sequence for FIX.
AMT-060 enabled stable expression of FIX that has persisted for up to 4 years without any late-emergent safety signals (Blood 2019. 134 Supplement 1: 2059). Dr. Pipe said the “enhanced version” of AMT-060, etranacogene dezaparvovec, has produced even higher levels of FIX activity in the phase 2b study (NCT03489291).
The ongoing study enrolled three men with moderate to severe FIX deficiency at baseline. The patients were 43, 50, and 47 years of age, respectively. Two patients are HIV positive, and all had hepatitis C that resolved.
All three patients were receiving FIX prophylaxis and on-demand treatment at baseline. In the year prior to screening, patients had one, three, and five bleeds, respectively. All three patients had anti-AAV5 neutralizing antibodies.
Efficacy
Patients received a single dose of etranacogene dezaparvovec at 2 x 1013 genome copies/kg. All three patients achieved the primary endpoint, which was FIX activity of at least 5% at 6 weeks.
At 52 weeks, the mean FIX activity was 41%. Patients 1 and 3 have maintained FIX activity of 40% or greater, which is in the nonhemophilic range. Patient 2 has maintained FIX activity in the mild range. At 52 weeks, FIX activity levels were 50.2%, 40.8%, and 31.3%, respectively.
All patients remain free of prophylaxis and bleeds. Patient 3 received a single FIX infusion as a precaution in the perioperative setting. There was no evidence of a bleed in this patient.
Safety
Etranacogene dezaparvovec was generally well tolerated, Dr. Pipe said. One patient had two adverse events that were possibly related to etranacogene dezaparvovec. Both events – transient, self-limiting headache and slightly elevated C-reactive protein – resolved without intervention.
There was one serious adverse event, but it was considered unrelated to treatment. Patient 3 required hip surgery for preexisting avascular necrosis.
Dr. Pipe said there was no evidence of transaminitis. There were modest, transient elevations in liver enzymes, but this was not enough to trigger protocol-specified immunosuppression.
Specifically, one patient had ALT elevations at weeks 22 and 44, and one patient had AST elevations at weeks 2, 4, and 31. All of these resolved quickly without treatment or an impact on FIX activity, Dr. Pipe noted.
Next steps
This study is ongoing, and patients will be followed for 5 years. Dr. Pipe said the main focus of follow-up will be to determine if patients maintain durable expression of FIX.
A phase 3 trial of etranacogene dezaparvovec is ongoing as well. The trial, HOPE-B (NCT03569891), is fully enrolled, and dosing is planned for 55 patients.
“We’re looking forward to data analysis later this year,” Dr. Pipe said. “This will be the only phase 3 study, and really the only platform so far, that is not planning to exclude patients based on preexisting immunity.”
If all goes well in the phase 3 study, etranacogene dezaparvovec could be approved by the Food and Drug Administration very soon, Dr. Pipe added.
UniQure, the company developing etranacogene dezaparvovec, is planning to submit the biologics license application to the FDA next year. Etranacogene dezaparvovec was granted breakthrough designation from the FDA and is therefore eligible for priority review, so the gene therapy could be approved as early as 2021.
The phase 2b trial of etranacogene dezaparvovec is sponsored by uniQure. Dr. Pipe disclosed relationships with uniQure and other companies.
SOURCE: Pipe SW et al. EAHAD 2020, Abstract OR10.
REPORTING FROM EAHAD 2020
Genetic testing helps avoid false hemoglobinopathy diagnoses in newborns
Confirmatory genetic testing may be useful in the diagnosis of hemoglobinopathies for newborns with an abnormal hemoglobin (Hb) pattern, according to a recent study.
The findings suggest further research is needed to evaluate whether genetic testing programs for newborns could have diagnostic value in the clinical setting.
“We studied a consecutive cohort of newborns with an ‘FSA’ pattern (a suspected diagnosis of HbSbeta+) on the initial newborn screening test,” explained Lisa M. Shook of the University of Cincinnati and colleagues. The results were published in the International Journal of Neonatal Screening.
The retrospective study included a total of 1,151 newborns with an abnormal Hb pattern, 31 of which had an FSA pattern. The newborns were screened for hemoglobinopathies from 2015 to 2018. The findings of the initial newborn screening test (a suspected diagnosis of HbSbeta+) were compared with the diagnosis established using both protein-based and genetic confirmatory testing. Protein-based testing cannot accurately detect several hemoglobinopathies in newborns, especially when beta-thalassemia mutations are involved, according to the authors.
“During this study period, genetic testing was not universally applied in advance; it was used based on clinical suspicion,” the researchers wrote.
Among newborns with an FSA pattern, the mean gestational age was 38.7 weeks. In total, 17 newborns received genetic testing, and 30 had protein-based confirmatory testing.
“In this consecutive cohort of 31 newborns with a suspected diagnosis of HbSbeta+ based on initial newborn screening (an FSA pattern), none actually had HbSbeta+. All had the sickle cell trait (HbAS), instead; that is, we found that an initial FSA pattern was much more likely to indicate a final diagnosis of HbAS than HbSbeta+,” the authors wrote.
This meant that two-thirds of these newborns had a correct diagnosis of HbAS established at 2-4 weeks of age by protein-based confirmatory testing (and confirmed by genetic testing in a subset), but that the remaining one-third still had an incorrect, suspected diagnosis of HbSbeta+. This could lead to unnecessary treatment and testing of infants and incorrect, disease-focused counseling of parents and family members, according to the authors.
Two key limitations of the study were the small sample size and retrospective design.
“Based on this experience in which genetic testing was not universally applied, we now perform simultaneous protein-based and genetic testing as our standard clinical practice,” they concluded.
The study was funded by the National Institutes of Health and the Ohio Department of Health. The authors reported having no conflicts of interest.
SOURCE: Shook LM et al. Int J Neonatal Screen. 2020 Jan 31. doi: 10.3390/ijns6010007
Confirmatory genetic testing may be useful in the diagnosis of hemoglobinopathies for newborns with an abnormal hemoglobin (Hb) pattern, according to a recent study.
The findings suggest further research is needed to evaluate whether genetic testing programs for newborns could have diagnostic value in the clinical setting.
“We studied a consecutive cohort of newborns with an ‘FSA’ pattern (a suspected diagnosis of HbSbeta+) on the initial newborn screening test,” explained Lisa M. Shook of the University of Cincinnati and colleagues. The results were published in the International Journal of Neonatal Screening.
The retrospective study included a total of 1,151 newborns with an abnormal Hb pattern, 31 of which had an FSA pattern. The newborns were screened for hemoglobinopathies from 2015 to 2018. The findings of the initial newborn screening test (a suspected diagnosis of HbSbeta+) were compared with the diagnosis established using both protein-based and genetic confirmatory testing. Protein-based testing cannot accurately detect several hemoglobinopathies in newborns, especially when beta-thalassemia mutations are involved, according to the authors.
“During this study period, genetic testing was not universally applied in advance; it was used based on clinical suspicion,” the researchers wrote.
Among newborns with an FSA pattern, the mean gestational age was 38.7 weeks. In total, 17 newborns received genetic testing, and 30 had protein-based confirmatory testing.
“In this consecutive cohort of 31 newborns with a suspected diagnosis of HbSbeta+ based on initial newborn screening (an FSA pattern), none actually had HbSbeta+. All had the sickle cell trait (HbAS), instead; that is, we found that an initial FSA pattern was much more likely to indicate a final diagnosis of HbAS than HbSbeta+,” the authors wrote.
This meant that two-thirds of these newborns had a correct diagnosis of HbAS established at 2-4 weeks of age by protein-based confirmatory testing (and confirmed by genetic testing in a subset), but that the remaining one-third still had an incorrect, suspected diagnosis of HbSbeta+. This could lead to unnecessary treatment and testing of infants and incorrect, disease-focused counseling of parents and family members, according to the authors.
Two key limitations of the study were the small sample size and retrospective design.
“Based on this experience in which genetic testing was not universally applied, we now perform simultaneous protein-based and genetic testing as our standard clinical practice,” they concluded.
The study was funded by the National Institutes of Health and the Ohio Department of Health. The authors reported having no conflicts of interest.
SOURCE: Shook LM et al. Int J Neonatal Screen. 2020 Jan 31. doi: 10.3390/ijns6010007
Confirmatory genetic testing may be useful in the diagnosis of hemoglobinopathies for newborns with an abnormal hemoglobin (Hb) pattern, according to a recent study.
The findings suggest further research is needed to evaluate whether genetic testing programs for newborns could have diagnostic value in the clinical setting.
“We studied a consecutive cohort of newborns with an ‘FSA’ pattern (a suspected diagnosis of HbSbeta+) on the initial newborn screening test,” explained Lisa M. Shook of the University of Cincinnati and colleagues. The results were published in the International Journal of Neonatal Screening.
The retrospective study included a total of 1,151 newborns with an abnormal Hb pattern, 31 of which had an FSA pattern. The newborns were screened for hemoglobinopathies from 2015 to 2018. The findings of the initial newborn screening test (a suspected diagnosis of HbSbeta+) were compared with the diagnosis established using both protein-based and genetic confirmatory testing. Protein-based testing cannot accurately detect several hemoglobinopathies in newborns, especially when beta-thalassemia mutations are involved, according to the authors.
“During this study period, genetic testing was not universally applied in advance; it was used based on clinical suspicion,” the researchers wrote.
Among newborns with an FSA pattern, the mean gestational age was 38.7 weeks. In total, 17 newborns received genetic testing, and 30 had protein-based confirmatory testing.
“In this consecutive cohort of 31 newborns with a suspected diagnosis of HbSbeta+ based on initial newborn screening (an FSA pattern), none actually had HbSbeta+. All had the sickle cell trait (HbAS), instead; that is, we found that an initial FSA pattern was much more likely to indicate a final diagnosis of HbAS than HbSbeta+,” the authors wrote.
This meant that two-thirds of these newborns had a correct diagnosis of HbAS established at 2-4 weeks of age by protein-based confirmatory testing (and confirmed by genetic testing in a subset), but that the remaining one-third still had an incorrect, suspected diagnosis of HbSbeta+. This could lead to unnecessary treatment and testing of infants and incorrect, disease-focused counseling of parents and family members, according to the authors.
Two key limitations of the study were the small sample size and retrospective design.
“Based on this experience in which genetic testing was not universally applied, we now perform simultaneous protein-based and genetic testing as our standard clinical practice,” they concluded.
The study was funded by the National Institutes of Health and the Ohio Department of Health. The authors reported having no conflicts of interest.
SOURCE: Shook LM et al. Int J Neonatal Screen. 2020 Jan 31. doi: 10.3390/ijns6010007
FROM THE INTERNATIONAL JOURNAL OF NEONATAL SCREENING
Fear drives activity changes in hemophilia patients
Fear of negative events can drive changes in activity levels among patients with hemophilia A, results of the HemACTIVE study suggest.

Patients were more likely to adjust their level of physical activity due to fear of bleeding and joint damage rather than previously experienced bleeding or joint damage.
However, past experience was more likely than fear to make patients stop physical activities altogether.
Mark Skinner, of the Institute for Policy Advancement in Washington, D.C., and colleagues presented these findings in a poster from the annual congress of the European Association for Haemophilia and Allied Disorders.
Mr. Skinner, who is a hemophilia patient himself, said the goal of the HemACTIVE study is to better understand how hemophilia affects patients’ lives.
“We wanted to understand the limitations, challenges, and compromises of individuals living with hemophilia,” Mr. Skinner said. “What has motivated them or prevented them from living more full, active lives doing the kind of work, leisure, and social activities that those without hemophilia do? Is it treatment choice, is it satisfaction with treatment, is it fear?
“We wanted to do a comprehensive study that really looked at the intersection of treatment adherence and satisfaction, the emotional components that relate to those decisions, and the challenges and compromises so that we could better identify what we need to consider as patients think about either changing their therapy or changing their treatment regimen on existing therapy.”
Previous results from the HemACTIVE study showed that, although activity levels differed among hemophilia patients, all patients surveyed wanted greater activity levels, better protection from bleeding, better pain relief, and less-frequent infusions (EAHAD 2019, Abstract P084). In addition, patients who used factor VIII products with an extended half-life were more active and more likely to adhere to their prescribed treatment (ISTH 2019, Abstract PB0210).
The results reported at EAHAD 2020 focus on patients’ reasons for modifying physical activity. Patients and caregivers completed a screening phone interview, followed by a 25-minute, web-based questionnaire on patient activity.
There were 275 respondents – 194 patients with hemophilia A and 81 caregivers – from Canada, France, Germany, Italy, and the United States. Patients had severe (61%) or moderate (39%) hemophilia A, and most (67%) were receiving prophylaxis.
Most patients (70%) were “active” or “extremely/very active,” 77% of patients adjusted their activities because of their hemophilia, and nearly half of patients stopped activities because of their disease.
Fear drives adjustments in activity
Patients were sometimes more likely to adjust their activities based on fear of experiencing an event, as opposed to previously experiencing that event.
Specifically, 44% of patients adjusted their activities due to fear of joint damage, compared with 36% of patients who made adjustments because of past significant joint damage.
Similarly, 41% of patients adjusted activities due to fear of breakthrough bleeds, compared with 36% of patients who made adjustments because of past experience with bleeds and 25% who made adjustments because of significant past bleeds.
On the other hand, a similar percentage of patients adjusted activities because of past experience with pain (43%) and fear of pain (41%). And a similar percentage of patients adjusted activities because of existing joint damage restrictions (35%) and fear of joint deterioration (32%).
Past experience prompts discontinuation of activity
Overall, 47% of patients said anxiety was the most common emotional reason for stopping physical activities. However, patients were consistently more likely to stop activities because of past experience rather than fear or anxiety.
Specifically, 50% of patients stopped activities because of significant past joint damage, 46% stopped because of developing joint problems, and 38% stopped due to fear of joint damage.
More patients stopped activities because of significant past bleeds (41%) rather than fear of breakthrough bleeds (26%). More patients stopped activities because they developed chronic pain (38%) rather than fear of pain (less than 15%). And more patients stopped activities because of existing joint damage restrictions (62%) rather than fear of joint damage (34%).
Applying results to practice: Changing the conversation
Ideally, these findings would be used to promote individualized treatment of hemophilia driven by patients’ goals, Mr. Skinner said. By better understanding patients’ feelings and motivations, clinicians may devise more personalized treatment regimens that align with patients’ goals and improve their quality of life.
Rather than adjusting treatment based only on “hard metrics” such as bleeding events, “we need to take a more holistic approach to looking at outcomes that are more important to patients,” Mr. Skinner said. This type of approach is particularly important to Mr. Skinner as someone who has severe hemophilia A.
“Because hemophilia is a life-long disease, and you’re born with it, you make conscious or unconscious adaptations throughout your life,” he explained. “Your expectations or aspirations adjust to what you’ve been told you can or cannot do because of your hemophilia. The choices I made for my career, where I live, the type of vacations I go on, the type of sports I participate in have all been limited over the course of time, which has meant that I’ve made compromises. There are a lot of individuals with hemophilia who are making decisions that are not what their life goals are.
“What this research helps me understand is that we can change the conversation and build it around an individual patient and understand what their aspirations are. If a clinician understands what I’m wanting to achieve in life … we can build a treatment regime around helping me achieve those goals. That is known to improve adherence.
“The goal, really, is to have hemophilia as a secondary consideration. Instead of saying: ‘You have hemophilia, so these are the options available to you,’ you can say, ‘what is it that you would like to achieve, and then we’ll figure out how your treatment for hemophilia can be adjusted to help you achieve those goals.’ It may sound like a nuance, but it really is reversing the conversation. The goal setting first versus your disease comes first.”
The HemACTIVE study was supported by Bayer. Mr. Skinner disclosed relationships with Bayer and other pharmaceutical companies.
SOURCE: Skinner M et al. EAHAD 2020, Abstract P304.
Fear of negative events can drive changes in activity levels among patients with hemophilia A, results of the HemACTIVE study suggest.
Patients were more likely to adjust their level of physical activity due to fear of bleeding and joint damage rather than previously experienced bleeding or joint damage.
However, past experience was more likely than fear to make patients stop physical activities altogether.
Mark Skinner, of the Institute for Policy Advancement in Washington, D.C., and colleagues presented these findings in a poster from the annual congress of the European Association for Haemophilia and Allied Disorders.
Mr. Skinner, who is a hemophilia patient himself, said the goal of the HemACTIVE study is to better understand how hemophilia affects patients’ lives.
“We wanted to understand the limitations, challenges, and compromises of individuals living with hemophilia,” Mr. Skinner said. “What has motivated them or prevented them from living more full, active lives doing the kind of work, leisure, and social activities that those without hemophilia do? Is it treatment choice, is it satisfaction with treatment, is it fear?
“We wanted to do a comprehensive study that really looked at the intersection of treatment adherence and satisfaction, the emotional components that relate to those decisions, and the challenges and compromises so that we could better identify what we need to consider as patients think about either changing their therapy or changing their treatment regimen on existing therapy.”
Previous results from the HemACTIVE study showed that, although activity levels differed among hemophilia patients, all patients surveyed wanted greater activity levels, better protection from bleeding, better pain relief, and less-frequent infusions (EAHAD 2019, Abstract P084). In addition, patients who used factor VIII products with an extended half-life were more active and more likely to adhere to their prescribed treatment (ISTH 2019, Abstract PB0210).
The results reported at EAHAD 2020 focus on patients’ reasons for modifying physical activity. Patients and caregivers completed a screening phone interview, followed by a 25-minute, web-based questionnaire on patient activity.
There were 275 respondents – 194 patients with hemophilia A and 81 caregivers – from Canada, France, Germany, Italy, and the United States. Patients had severe (61%) or moderate (39%) hemophilia A, and most (67%) were receiving prophylaxis.
Most patients (70%) were “active” or “extremely/very active,” 77% of patients adjusted their activities because of their hemophilia, and nearly half of patients stopped activities because of their disease.
Fear drives adjustments in activity
Patients were sometimes more likely to adjust their activities based on fear of experiencing an event, as opposed to previously experiencing that event.
Specifically, 44% of patients adjusted their activities due to fear of joint damage, compared with 36% of patients who made adjustments because of past significant joint damage.
Similarly, 41% of patients adjusted activities due to fear of breakthrough bleeds, compared with 36% of patients who made adjustments because of past experience with bleeds and 25% who made adjustments because of significant past bleeds.
On the other hand, a similar percentage of patients adjusted activities because of past experience with pain (43%) and fear of pain (41%). And a similar percentage of patients adjusted activities because of existing joint damage restrictions (35%) and fear of joint deterioration (32%).
Past experience prompts discontinuation of activity
Overall, 47% of patients said anxiety was the most common emotional reason for stopping physical activities. However, patients were consistently more likely to stop activities because of past experience rather than fear or anxiety.
Specifically, 50% of patients stopped activities because of significant past joint damage, 46% stopped because of developing joint problems, and 38% stopped due to fear of joint damage.
More patients stopped activities because of significant past bleeds (41%) rather than fear of breakthrough bleeds (26%). More patients stopped activities because they developed chronic pain (38%) rather than fear of pain (less than 15%). And more patients stopped activities because of existing joint damage restrictions (62%) rather than fear of joint damage (34%).
Applying results to practice: Changing the conversation
Ideally, these findings would be used to promote individualized treatment of hemophilia driven by patients’ goals, Mr. Skinner said. By better understanding patients’ feelings and motivations, clinicians may devise more personalized treatment regimens that align with patients’ goals and improve their quality of life.
Rather than adjusting treatment based only on “hard metrics” such as bleeding events, “we need to take a more holistic approach to looking at outcomes that are more important to patients,” Mr. Skinner said. This type of approach is particularly important to Mr. Skinner as someone who has severe hemophilia A.
“Because hemophilia is a life-long disease, and you’re born with it, you make conscious or unconscious adaptations throughout your life,” he explained. “Your expectations or aspirations adjust to what you’ve been told you can or cannot do because of your hemophilia. The choices I made for my career, where I live, the type of vacations I go on, the type of sports I participate in have all been limited over the course of time, which has meant that I’ve made compromises. There are a lot of individuals with hemophilia who are making decisions that are not what their life goals are.
“What this research helps me understand is that we can change the conversation and build it around an individual patient and understand what their aspirations are. If a clinician understands what I’m wanting to achieve in life … we can build a treatment regime around helping me achieve those goals. That is known to improve adherence.
“The goal, really, is to have hemophilia as a secondary consideration. Instead of saying: ‘You have hemophilia, so these are the options available to you,’ you can say, ‘what is it that you would like to achieve, and then we’ll figure out how your treatment for hemophilia can be adjusted to help you achieve those goals.’ It may sound like a nuance, but it really is reversing the conversation. The goal setting first versus your disease comes first.”
The HemACTIVE study was supported by Bayer. Mr. Skinner disclosed relationships with Bayer and other pharmaceutical companies.
SOURCE: Skinner M et al. EAHAD 2020, Abstract P304.
Fear of negative events can drive changes in activity levels among patients with hemophilia A, results of the HemACTIVE study suggest.
Patients were more likely to adjust their level of physical activity due to fear of bleeding and joint damage rather than previously experienced bleeding or joint damage.
However, past experience was more likely than fear to make patients stop physical activities altogether.
Mark Skinner, of the Institute for Policy Advancement in Washington, D.C., and colleagues presented these findings in a poster from the annual congress of the European Association for Haemophilia and Allied Disorders.
Mr. Skinner, who is a hemophilia patient himself, said the goal of the HemACTIVE study is to better understand how hemophilia affects patients’ lives.
“We wanted to understand the limitations, challenges, and compromises of individuals living with hemophilia,” Mr. Skinner said. “What has motivated them or prevented them from living more full, active lives doing the kind of work, leisure, and social activities that those without hemophilia do? Is it treatment choice, is it satisfaction with treatment, is it fear?
“We wanted to do a comprehensive study that really looked at the intersection of treatment adherence and satisfaction, the emotional components that relate to those decisions, and the challenges and compromises so that we could better identify what we need to consider as patients think about either changing their therapy or changing their treatment regimen on existing therapy.”
Previous results from the HemACTIVE study showed that, although activity levels differed among hemophilia patients, all patients surveyed wanted greater activity levels, better protection from bleeding, better pain relief, and less-frequent infusions (EAHAD 2019, Abstract P084). In addition, patients who used factor VIII products with an extended half-life were more active and more likely to adhere to their prescribed treatment (ISTH 2019, Abstract PB0210).
The results reported at EAHAD 2020 focus on patients’ reasons for modifying physical activity. Patients and caregivers completed a screening phone interview, followed by a 25-minute, web-based questionnaire on patient activity.
There were 275 respondents – 194 patients with hemophilia A and 81 caregivers – from Canada, France, Germany, Italy, and the United States. Patients had severe (61%) or moderate (39%) hemophilia A, and most (67%) were receiving prophylaxis.
Most patients (70%) were “active” or “extremely/very active,” 77% of patients adjusted their activities because of their hemophilia, and nearly half of patients stopped activities because of their disease.
Fear drives adjustments in activity
Patients were sometimes more likely to adjust their activities based on fear of experiencing an event, as opposed to previously experiencing that event.
Specifically, 44% of patients adjusted their activities due to fear of joint damage, compared with 36% of patients who made adjustments because of past significant joint damage.
Similarly, 41% of patients adjusted activities due to fear of breakthrough bleeds, compared with 36% of patients who made adjustments because of past experience with bleeds and 25% who made adjustments because of significant past bleeds.
On the other hand, a similar percentage of patients adjusted activities because of past experience with pain (43%) and fear of pain (41%). And a similar percentage of patients adjusted activities because of existing joint damage restrictions (35%) and fear of joint deterioration (32%).
Past experience prompts discontinuation of activity
Overall, 47% of patients said anxiety was the most common emotional reason for stopping physical activities. However, patients were consistently more likely to stop activities because of past experience rather than fear or anxiety.
Specifically, 50% of patients stopped activities because of significant past joint damage, 46% stopped because of developing joint problems, and 38% stopped due to fear of joint damage.
More patients stopped activities because of significant past bleeds (41%) rather than fear of breakthrough bleeds (26%). More patients stopped activities because they developed chronic pain (38%) rather than fear of pain (less than 15%). And more patients stopped activities because of existing joint damage restrictions (62%) rather than fear of joint damage (34%).
Applying results to practice: Changing the conversation
Ideally, these findings would be used to promote individualized treatment of hemophilia driven by patients’ goals, Mr. Skinner said. By better understanding patients’ feelings and motivations, clinicians may devise more personalized treatment regimens that align with patients’ goals and improve their quality of life.
Rather than adjusting treatment based only on “hard metrics” such as bleeding events, “we need to take a more holistic approach to looking at outcomes that are more important to patients,” Mr. Skinner said. This type of approach is particularly important to Mr. Skinner as someone who has severe hemophilia A.
“Because hemophilia is a life-long disease, and you’re born with it, you make conscious or unconscious adaptations throughout your life,” he explained. “Your expectations or aspirations adjust to what you’ve been told you can or cannot do because of your hemophilia. The choices I made for my career, where I live, the type of vacations I go on, the type of sports I participate in have all been limited over the course of time, which has meant that I’ve made compromises. There are a lot of individuals with hemophilia who are making decisions that are not what their life goals are.
“What this research helps me understand is that we can change the conversation and build it around an individual patient and understand what their aspirations are. If a clinician understands what I’m wanting to achieve in life … we can build a treatment regime around helping me achieve those goals. That is known to improve adherence.
“The goal, really, is to have hemophilia as a secondary consideration. Instead of saying: ‘You have hemophilia, so these are the options available to you,’ you can say, ‘what is it that you would like to achieve, and then we’ll figure out how your treatment for hemophilia can be adjusted to help you achieve those goals.’ It may sound like a nuance, but it really is reversing the conversation. The goal setting first versus your disease comes first.”
The HemACTIVE study was supported by Bayer. Mr. Skinner disclosed relationships with Bayer and other pharmaceutical companies.
SOURCE: Skinner M et al. EAHAD 2020, Abstract P304.
REPORTING FROM EAHAD 2020
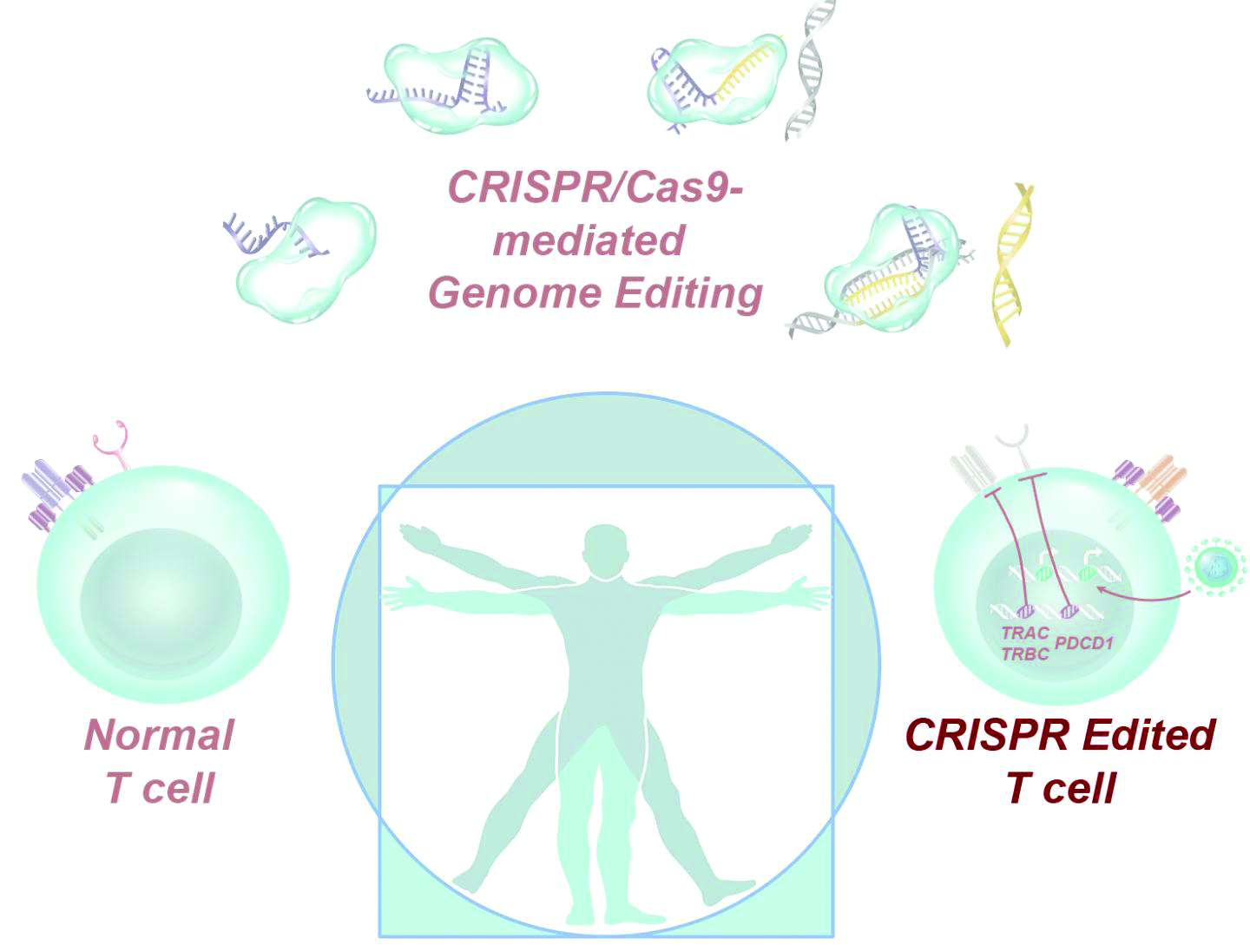
CRISPR-engineered T cells may be safe for cancer, but do they work?
according to a report in Science.
The results of no harm support this “promising” area of cancer immunotherapy, according to study investigator Edward A. Stadtmauer, MD, of the University of Pennsylvania in Philadelphia and colleagues.
However, there was no evidence of benefit in this trial. One patient transfused with CRISPR-engineered T cells has since died, and the other two have moved on to other treatments.
“The big question that remains unanswered by this study is whether gene-edited, engineered T cells are effective against advanced cancer,” Jennifer Hamilton, PhD, and Jennifer Doudna, PhD, both of the University of California, Berkeley, wrote in an accompanying editorial.
The study enrolled six patients with refractory cancer, and three of them received CRISPR-engineered T cells. Two patients had multiple myeloma, and one had metastatic sarcoma.
Dr. Stadtmauer and colleagues drew blood from the patients, isolated the T cells, and used CRISPR-Cas9 to modify the cells. The T cells were transfected with Cas9 protein complexed with single guide RNAs against TRAC and TRBC (genes encoding the T-cell receptor chains TCR-alpha and TCR-beta) as well as PDCD1 (a gene encoding programmed cell death protein 1). The T cells were then transduced with a lentiviral vector to express a transgenic NY-ESO-1 cancer-specific T-cell receptor.
The investigators expanded the cell lines and infused them back into the patients after administering lymphodepleting chemotherapy. The sarcoma patient initially had a 50% decrease in a large abdominal mass, but all three patients ultimately progressed.
The editorialists noted that gene disruption efficiencies in this study were “modest,” ranging from 15% to 45%, but the investigators used a protocol from 2016, when the study was given the go-ahead by the National Institutes of Health and the Food and Drug Administration. With current protocols, gene disruption efficiencies can exceed 90%, which means patients might do better in subsequent trials.
There was no more than mild toxicity in this trial, and most adverse events were attributed to the lymphodepleting chemotherapy.
There was concern about potential rejection of infused cells because of preexisting immune responses to Cas9, but it doesn’t seem “to be a barrier to the application of this promising technology,” the investigators said.
They noted that “the stable engraftment of our engineered T cells is remarkably different from previously reported trials ... where the half-life of the cells in blood was [about] 1 week. Biopsy specimens of bone marrow in the myeloma patients and tumor in the sarcoma patient demonstrated trafficking of the engineered T cells to the tumor in all three patients” beyond that point. The decay half-life of the transduced cells was 20.3 days, 121.8 days, and 293.5 days in these patients.
The editorialists said the details in the report are a model for other researchers to follow, but “as more gene-based therapies are demonstrated to be safe and effective, the barrier to clinical translation will become cell manufacturing and administration.”
This work was funded by the National Institutes of Health and others. Dr. Stadtmauer didn’t report any disclosures, but other investigators disclosed patent applications and commercialization efforts. Dr. Doudna disclosed that she is a cofounder or adviser for several companies developing gene-editing therapeutics.
SOURCE: Stadtmauer EA et al. Science. 2020 Feb 6. doi: 10.1126/science.aba7365.
according to a report in Science.
The results of no harm support this “promising” area of cancer immunotherapy, according to study investigator Edward A. Stadtmauer, MD, of the University of Pennsylvania in Philadelphia and colleagues.
However, there was no evidence of benefit in this trial. One patient transfused with CRISPR-engineered T cells has since died, and the other two have moved on to other treatments.
“The big question that remains unanswered by this study is whether gene-edited, engineered T cells are effective against advanced cancer,” Jennifer Hamilton, PhD, and Jennifer Doudna, PhD, both of the University of California, Berkeley, wrote in an accompanying editorial.
The study enrolled six patients with refractory cancer, and three of them received CRISPR-engineered T cells. Two patients had multiple myeloma, and one had metastatic sarcoma.
Dr. Stadtmauer and colleagues drew blood from the patients, isolated the T cells, and used CRISPR-Cas9 to modify the cells. The T cells were transfected with Cas9 protein complexed with single guide RNAs against TRAC and TRBC (genes encoding the T-cell receptor chains TCR-alpha and TCR-beta) as well as PDCD1 (a gene encoding programmed cell death protein 1). The T cells were then transduced with a lentiviral vector to express a transgenic NY-ESO-1 cancer-specific T-cell receptor.
The investigators expanded the cell lines and infused them back into the patients after administering lymphodepleting chemotherapy. The sarcoma patient initially had a 50% decrease in a large abdominal mass, but all three patients ultimately progressed.
The editorialists noted that gene disruption efficiencies in this study were “modest,” ranging from 15% to 45%, but the investigators used a protocol from 2016, when the study was given the go-ahead by the National Institutes of Health and the Food and Drug Administration. With current protocols, gene disruption efficiencies can exceed 90%, which means patients might do better in subsequent trials.
There was no more than mild toxicity in this trial, and most adverse events were attributed to the lymphodepleting chemotherapy.
There was concern about potential rejection of infused cells because of preexisting immune responses to Cas9, but it doesn’t seem “to be a barrier to the application of this promising technology,” the investigators said.
They noted that “the stable engraftment of our engineered T cells is remarkably different from previously reported trials ... where the half-life of the cells in blood was [about] 1 week. Biopsy specimens of bone marrow in the myeloma patients and tumor in the sarcoma patient demonstrated trafficking of the engineered T cells to the tumor in all three patients” beyond that point. The decay half-life of the transduced cells was 20.3 days, 121.8 days, and 293.5 days in these patients.
The editorialists said the details in the report are a model for other researchers to follow, but “as more gene-based therapies are demonstrated to be safe and effective, the barrier to clinical translation will become cell manufacturing and administration.”
This work was funded by the National Institutes of Health and others. Dr. Stadtmauer didn’t report any disclosures, but other investigators disclosed patent applications and commercialization efforts. Dr. Doudna disclosed that she is a cofounder or adviser for several companies developing gene-editing therapeutics.
SOURCE: Stadtmauer EA et al. Science. 2020 Feb 6. doi: 10.1126/science.aba7365.
according to a report in Science.
The results of no harm support this “promising” area of cancer immunotherapy, according to study investigator Edward A. Stadtmauer, MD, of the University of Pennsylvania in Philadelphia and colleagues.
However, there was no evidence of benefit in this trial. One patient transfused with CRISPR-engineered T cells has since died, and the other two have moved on to other treatments.
“The big question that remains unanswered by this study is whether gene-edited, engineered T cells are effective against advanced cancer,” Jennifer Hamilton, PhD, and Jennifer Doudna, PhD, both of the University of California, Berkeley, wrote in an accompanying editorial.
The study enrolled six patients with refractory cancer, and three of them received CRISPR-engineered T cells. Two patients had multiple myeloma, and one had metastatic sarcoma.
Dr. Stadtmauer and colleagues drew blood from the patients, isolated the T cells, and used CRISPR-Cas9 to modify the cells. The T cells were transfected with Cas9 protein complexed with single guide RNAs against TRAC and TRBC (genes encoding the T-cell receptor chains TCR-alpha and TCR-beta) as well as PDCD1 (a gene encoding programmed cell death protein 1). The T cells were then transduced with a lentiviral vector to express a transgenic NY-ESO-1 cancer-specific T-cell receptor.
The investigators expanded the cell lines and infused them back into the patients after administering lymphodepleting chemotherapy. The sarcoma patient initially had a 50% decrease in a large abdominal mass, but all three patients ultimately progressed.
The editorialists noted that gene disruption efficiencies in this study were “modest,” ranging from 15% to 45%, but the investigators used a protocol from 2016, when the study was given the go-ahead by the National Institutes of Health and the Food and Drug Administration. With current protocols, gene disruption efficiencies can exceed 90%, which means patients might do better in subsequent trials.
There was no more than mild toxicity in this trial, and most adverse events were attributed to the lymphodepleting chemotherapy.
There was concern about potential rejection of infused cells because of preexisting immune responses to Cas9, but it doesn’t seem “to be a barrier to the application of this promising technology,” the investigators said.
They noted that “the stable engraftment of our engineered T cells is remarkably different from previously reported trials ... where the half-life of the cells in blood was [about] 1 week. Biopsy specimens of bone marrow in the myeloma patients and tumor in the sarcoma patient demonstrated trafficking of the engineered T cells to the tumor in all three patients” beyond that point. The decay half-life of the transduced cells was 20.3 days, 121.8 days, and 293.5 days in these patients.
The editorialists said the details in the report are a model for other researchers to follow, but “as more gene-based therapies are demonstrated to be safe and effective, the barrier to clinical translation will become cell manufacturing and administration.”
This work was funded by the National Institutes of Health and others. Dr. Stadtmauer didn’t report any disclosures, but other investigators disclosed patent applications and commercialization efforts. Dr. Doudna disclosed that she is a cofounder or adviser for several companies developing gene-editing therapeutics.
SOURCE: Stadtmauer EA et al. Science. 2020 Feb 6. doi: 10.1126/science.aba7365.
FROM SCIENCE
Global project reveals cancer’s genomic playbook
A massive collaborative project spanning four continents and 744 research centers has revealed driver mutations in both protein-coding and noncoding regions of 38 cancer types.

The Pan-Cancer Analysis of Whole Genomes (PCAWG) is an integrative analysis of the whole-genome sequences from 2,658 donors across 38 common tumor types. The findings are expected to add exponentially to what’s currently known about the complex genetics of cancer, and they point to possible strategies for improving cancer prevention, diagnosis, and care.
Six articles summarizing the findings are presented in a series of papers in Nature, and 16 more appear in affiliated publications.
“It’s humbling that it was only 14 years ago that the genomics community sequenced its very first cancer exome, and it was able to identify mutations within the roughly 20,000 protein-coding genes in the human cell,” investigator Lincoln Stein, MD, PhD, of the Ontario Institute for Cancer Research in Toronto, said in a telephone briefing.
Exome sequencing, however, covers only protein-coding genomic regions, which constitute only about 1% of the entire genome, “so assembling an accurate portrait of the cancer genome using just the exome data is like trying to put together a 100,000-piece jigsaw puzzle when you’re missing 99% of the pieces and there’s no puzzle box with a completed picture to guide you,” Dr. Stein said.
Members of the PCAWG from centers in North America, Europe, Asia, and Australia screened 2,658 whole-cancer genomes and matched samples of noncancerous tissues from the same individuals, along with 1,188 transcriptomes cataloging the sequences and expression of RNA transcripts in a given tumor. The 6-year project netted more than 800 terabytes of genomic data, roughly equivalent to the digital holdings of the U.S. Library of Congress multiplied by 11.
The findings are summarized in papers focusing on cancer drivers, noncoding changes, mutational signatures, structural variants, cancer evolution over time, and RNA alterations.
Driver mutations
Investigators found that the average cancer genome contains four or five driver mutations located in both coding and noncoding regions. They also found, however, that in approximately 5% of cases no driver mutations could be identified.
A substantial proportion of tumors displayed “hallmarks of genomic catastrophes.” About 22% of tumors exhibited chromothripsis, a mutational process marked by hundreds or even thousands of clustered chromosomal rearrangements. About 18% showed chromoplexy, which is characterized by scattering and rearrangement of multiple strands of DNA from one or more chromosomes.
Analyzing driver point mutations and structural variants in noncoding regions, the investigators found the usual suspects – previously reported culprits – as well as novel candidates.
For example, they identified point mutations in the five prime region of the tumor suppressor gene TP53 and the three prime untranslated regions of NFKBIZ (a nuclear factor kappa B inhibitor) and TOB1 (an antiproliferative protein), focal deletion in BRD4 (a transcriptional and epigenetic regulator), and rearrangements in chromosomal loci in members of the AKR1C family of enzymes thought to play a role in disease progression.
In addition, investigators identified mutations in noncoding regions of TERT, a telomerase gene. These mutations result in ramped-up expression of telomerase, which in turn promotes uncontrollable division of tumor cells.
Mutational signatures
In a related line of research, PCAWG investigators identified new DNA mutational signatures ranging from single nucleotide polymorphisms to insertions and deletions, as well as to structural variants – rearrangements of large sections of the genome.
“The substantial size of our dataset, compared with previous analyses, enabled the discovery of new signatures, the separation of overlapping signatures, and the decomposition of signatures into components that may represent associated – but distinct – DNA damage, repair, and/or replication mechanisms. By estimating the contribution of each signature to the mutational catalogs of individual cancer genomes, we revealed associations of signatures to exogenous or endogenous exposures, as well as to defective DNA maintenance processes,” the investigators wrote.
They also acknowledged, however, that “many signatures are of unknown cause.”
Cancer evolution
One of the six main studies focused on the evolution of cancer over time. Instead of providing a “snapshot” of the genome as captured by sequencing tissue from a single biopsy, consortium investigators created full-length features of the “life history and evolution of mutational processes and driver mutation sequences.”
They found that early cancer development was marked by relatively few mutations in driver genes and by identifiable copy-number gains, including trisomy 7 in glioblastoma, and an abnormal mirroring of the arms (isochromosome) of chromosome 17 in medulloblastoma.
In 40% of the samples, however, there were significant changes in the mutational spectrum as the cancers grew, leading to a near quadrupling of driver genes and increased genomic instability in later-stage tumors.
“Copy-number alterations often occur in mitotic crises and lead to simultaneous gains of chromosomal segments,” the investigators wrote. “Timing analyses suggest that driver mutations often precede diagnosis by many years, if not decades. Together, these results determine the evolutionary trajectories of cancer and highlight opportunities for early cancer detection.”
Implications for cancer care
“When I used to treat patients with cancer, I was always completely amazed and puzzled by how two patients could have what looked like the same tumor. It would look the same under the microscope, have the same size, and the two patients would receive exactly the same treatment, but the two patients would have completely opposite outcomes; one would survive, and one would die. What this analysis … has done is really laid bare the reasons for that unpredictability in clinical outcomes,” Peter Campbell, MD, PhD, of the Wellcome Sanger Institute in Hinxton, England, said during the telebriefing.
“The most striking finding out of all of the suite of papers is just how different one person’s cancer genome is from another person’s. We see thousands of different combinations of mutations that can cause the cancer, and more than 80 different underlying processes generating the mutations in a cancer, and that leads to very different shapes and patterns in the genome that result,” he added.
On a positive note, the research shows that one or more driver mutations can be identified in about 95% of all cancer patients, and it elucidates the sequence of events leading to oncogenesis and tumor evolution, providing opportunities for earlier identification and potential interventions to prevent cancer, Dr. Campbell said.
The PCAWG was a collaborative multinational effort with multiple funding sources and many investigators.
SOURCE: Nature. 2020 Feb 5. https://www.nature.com/collections/pcawg/
A massive collaborative project spanning four continents and 744 research centers has revealed driver mutations in both protein-coding and noncoding regions of 38 cancer types.
The Pan-Cancer Analysis of Whole Genomes (PCAWG) is an integrative analysis of the whole-genome sequences from 2,658 donors across 38 common tumor types. The findings are expected to add exponentially to what’s currently known about the complex genetics of cancer, and they point to possible strategies for improving cancer prevention, diagnosis, and care.
Six articles summarizing the findings are presented in a series of papers in Nature, and 16 more appear in affiliated publications.
“It’s humbling that it was only 14 years ago that the genomics community sequenced its very first cancer exome, and it was able to identify mutations within the roughly 20,000 protein-coding genes in the human cell,” investigator Lincoln Stein, MD, PhD, of the Ontario Institute for Cancer Research in Toronto, said in a telephone briefing.
Exome sequencing, however, covers only protein-coding genomic regions, which constitute only about 1% of the entire genome, “so assembling an accurate portrait of the cancer genome using just the exome data is like trying to put together a 100,000-piece jigsaw puzzle when you’re missing 99% of the pieces and there’s no puzzle box with a completed picture to guide you,” Dr. Stein said.
Members of the PCAWG from centers in North America, Europe, Asia, and Australia screened 2,658 whole-cancer genomes and matched samples of noncancerous tissues from the same individuals, along with 1,188 transcriptomes cataloging the sequences and expression of RNA transcripts in a given tumor. The 6-year project netted more than 800 terabytes of genomic data, roughly equivalent to the digital holdings of the U.S. Library of Congress multiplied by 11.
The findings are summarized in papers focusing on cancer drivers, noncoding changes, mutational signatures, structural variants, cancer evolution over time, and RNA alterations.
Driver mutations
Investigators found that the average cancer genome contains four or five driver mutations located in both coding and noncoding regions. They also found, however, that in approximately 5% of cases no driver mutations could be identified.
A substantial proportion of tumors displayed “hallmarks of genomic catastrophes.” About 22% of tumors exhibited chromothripsis, a mutational process marked by hundreds or even thousands of clustered chromosomal rearrangements. About 18% showed chromoplexy, which is characterized by scattering and rearrangement of multiple strands of DNA from one or more chromosomes.
Analyzing driver point mutations and structural variants in noncoding regions, the investigators found the usual suspects – previously reported culprits – as well as novel candidates.
For example, they identified point mutations in the five prime region of the tumor suppressor gene TP53 and the three prime untranslated regions of NFKBIZ (a nuclear factor kappa B inhibitor) and TOB1 (an antiproliferative protein), focal deletion in BRD4 (a transcriptional and epigenetic regulator), and rearrangements in chromosomal loci in members of the AKR1C family of enzymes thought to play a role in disease progression.
In addition, investigators identified mutations in noncoding regions of TERT, a telomerase gene. These mutations result in ramped-up expression of telomerase, which in turn promotes uncontrollable division of tumor cells.
Mutational signatures
In a related line of research, PCAWG investigators identified new DNA mutational signatures ranging from single nucleotide polymorphisms to insertions and deletions, as well as to structural variants – rearrangements of large sections of the genome.
“The substantial size of our dataset, compared with previous analyses, enabled the discovery of new signatures, the separation of overlapping signatures, and the decomposition of signatures into components that may represent associated – but distinct – DNA damage, repair, and/or replication mechanisms. By estimating the contribution of each signature to the mutational catalogs of individual cancer genomes, we revealed associations of signatures to exogenous or endogenous exposures, as well as to defective DNA maintenance processes,” the investigators wrote.
They also acknowledged, however, that “many signatures are of unknown cause.”
Cancer evolution
One of the six main studies focused on the evolution of cancer over time. Instead of providing a “snapshot” of the genome as captured by sequencing tissue from a single biopsy, consortium investigators created full-length features of the “life history and evolution of mutational processes and driver mutation sequences.”
They found that early cancer development was marked by relatively few mutations in driver genes and by identifiable copy-number gains, including trisomy 7 in glioblastoma, and an abnormal mirroring of the arms (isochromosome) of chromosome 17 in medulloblastoma.
In 40% of the samples, however, there were significant changes in the mutational spectrum as the cancers grew, leading to a near quadrupling of driver genes and increased genomic instability in later-stage tumors.
“Copy-number alterations often occur in mitotic crises and lead to simultaneous gains of chromosomal segments,” the investigators wrote. “Timing analyses suggest that driver mutations often precede diagnosis by many years, if not decades. Together, these results determine the evolutionary trajectories of cancer and highlight opportunities for early cancer detection.”
Implications for cancer care
“When I used to treat patients with cancer, I was always completely amazed and puzzled by how two patients could have what looked like the same tumor. It would look the same under the microscope, have the same size, and the two patients would receive exactly the same treatment, but the two patients would have completely opposite outcomes; one would survive, and one would die. What this analysis … has done is really laid bare the reasons for that unpredictability in clinical outcomes,” Peter Campbell, MD, PhD, of the Wellcome Sanger Institute in Hinxton, England, said during the telebriefing.
“The most striking finding out of all of the suite of papers is just how different one person’s cancer genome is from another person’s. We see thousands of different combinations of mutations that can cause the cancer, and more than 80 different underlying processes generating the mutations in a cancer, and that leads to very different shapes and patterns in the genome that result,” he added.
On a positive note, the research shows that one or more driver mutations can be identified in about 95% of all cancer patients, and it elucidates the sequence of events leading to oncogenesis and tumor evolution, providing opportunities for earlier identification and potential interventions to prevent cancer, Dr. Campbell said.
The PCAWG was a collaborative multinational effort with multiple funding sources and many investigators.
SOURCE: Nature. 2020 Feb 5. https://www.nature.com/collections/pcawg/
A massive collaborative project spanning four continents and 744 research centers has revealed driver mutations in both protein-coding and noncoding regions of 38 cancer types.
The Pan-Cancer Analysis of Whole Genomes (PCAWG) is an integrative analysis of the whole-genome sequences from 2,658 donors across 38 common tumor types. The findings are expected to add exponentially to what’s currently known about the complex genetics of cancer, and they point to possible strategies for improving cancer prevention, diagnosis, and care.
Six articles summarizing the findings are presented in a series of papers in Nature, and 16 more appear in affiliated publications.
“It’s humbling that it was only 14 years ago that the genomics community sequenced its very first cancer exome, and it was able to identify mutations within the roughly 20,000 protein-coding genes in the human cell,” investigator Lincoln Stein, MD, PhD, of the Ontario Institute for Cancer Research in Toronto, said in a telephone briefing.
Exome sequencing, however, covers only protein-coding genomic regions, which constitute only about 1% of the entire genome, “so assembling an accurate portrait of the cancer genome using just the exome data is like trying to put together a 100,000-piece jigsaw puzzle when you’re missing 99% of the pieces and there’s no puzzle box with a completed picture to guide you,” Dr. Stein said.
Members of the PCAWG from centers in North America, Europe, Asia, and Australia screened 2,658 whole-cancer genomes and matched samples of noncancerous tissues from the same individuals, along with 1,188 transcriptomes cataloging the sequences and expression of RNA transcripts in a given tumor. The 6-year project netted more than 800 terabytes of genomic data, roughly equivalent to the digital holdings of the U.S. Library of Congress multiplied by 11.
The findings are summarized in papers focusing on cancer drivers, noncoding changes, mutational signatures, structural variants, cancer evolution over time, and RNA alterations.
Driver mutations
Investigators found that the average cancer genome contains four or five driver mutations located in both coding and noncoding regions. They also found, however, that in approximately 5% of cases no driver mutations could be identified.
A substantial proportion of tumors displayed “hallmarks of genomic catastrophes.” About 22% of tumors exhibited chromothripsis, a mutational process marked by hundreds or even thousands of clustered chromosomal rearrangements. About 18% showed chromoplexy, which is characterized by scattering and rearrangement of multiple strands of DNA from one or more chromosomes.
Analyzing driver point mutations and structural variants in noncoding regions, the investigators found the usual suspects – previously reported culprits – as well as novel candidates.
For example, they identified point mutations in the five prime region of the tumor suppressor gene TP53 and the three prime untranslated regions of NFKBIZ (a nuclear factor kappa B inhibitor) and TOB1 (an antiproliferative protein), focal deletion in BRD4 (a transcriptional and epigenetic regulator), and rearrangements in chromosomal loci in members of the AKR1C family of enzymes thought to play a role in disease progression.
In addition, investigators identified mutations in noncoding regions of TERT, a telomerase gene. These mutations result in ramped-up expression of telomerase, which in turn promotes uncontrollable division of tumor cells.
Mutational signatures
In a related line of research, PCAWG investigators identified new DNA mutational signatures ranging from single nucleotide polymorphisms to insertions and deletions, as well as to structural variants – rearrangements of large sections of the genome.
“The substantial size of our dataset, compared with previous analyses, enabled the discovery of new signatures, the separation of overlapping signatures, and the decomposition of signatures into components that may represent associated – but distinct – DNA damage, repair, and/or replication mechanisms. By estimating the contribution of each signature to the mutational catalogs of individual cancer genomes, we revealed associations of signatures to exogenous or endogenous exposures, as well as to defective DNA maintenance processes,” the investigators wrote.
They also acknowledged, however, that “many signatures are of unknown cause.”
Cancer evolution
One of the six main studies focused on the evolution of cancer over time. Instead of providing a “snapshot” of the genome as captured by sequencing tissue from a single biopsy, consortium investigators created full-length features of the “life history and evolution of mutational processes and driver mutation sequences.”
They found that early cancer development was marked by relatively few mutations in driver genes and by identifiable copy-number gains, including trisomy 7 in glioblastoma, and an abnormal mirroring of the arms (isochromosome) of chromosome 17 in medulloblastoma.
In 40% of the samples, however, there were significant changes in the mutational spectrum as the cancers grew, leading to a near quadrupling of driver genes and increased genomic instability in later-stage tumors.
“Copy-number alterations often occur in mitotic crises and lead to simultaneous gains of chromosomal segments,” the investigators wrote. “Timing analyses suggest that driver mutations often precede diagnosis by many years, if not decades. Together, these results determine the evolutionary trajectories of cancer and highlight opportunities for early cancer detection.”
Implications for cancer care
“When I used to treat patients with cancer, I was always completely amazed and puzzled by how two patients could have what looked like the same tumor. It would look the same under the microscope, have the same size, and the two patients would receive exactly the same treatment, but the two patients would have completely opposite outcomes; one would survive, and one would die. What this analysis … has done is really laid bare the reasons for that unpredictability in clinical outcomes,” Peter Campbell, MD, PhD, of the Wellcome Sanger Institute in Hinxton, England, said during the telebriefing.
“The most striking finding out of all of the suite of papers is just how different one person’s cancer genome is from another person’s. We see thousands of different combinations of mutations that can cause the cancer, and more than 80 different underlying processes generating the mutations in a cancer, and that leads to very different shapes and patterns in the genome that result,” he added.
On a positive note, the research shows that one or more driver mutations can be identified in about 95% of all cancer patients, and it elucidates the sequence of events leading to oncogenesis and tumor evolution, providing opportunities for earlier identification and potential interventions to prevent cancer, Dr. Campbell said.
The PCAWG was a collaborative multinational effort with multiple funding sources and many investigators.
SOURCE: Nature. 2020 Feb 5. https://www.nature.com/collections/pcawg/
FROM NATURE
U.S. cancer centers embroiled in Chinese research thefts
Academic cancer centers around the United States continue to get caught up in an ever-evolving investigation into researchers – American and Chinese – who did not disclose payments from or the work they did for Chinese institutions while simultaneously accepting taxpayer money through U.S. government grants.
The U.S. Federal Bureau of Investigation has been ferreting out researchers it says have acted illegally.
On Jan. 28, the agency arrested Charles Lieber, a chemist from Harvard University, Cambridge, Mass., and also unveiled charges against Zheng Zaosong, a cancer researcher who is in the United States on a Harvard-sponsored visa.
The FBI said Mr. Zheng, who worked at the Harvard-affiliated Beth Israel Deaconess Medical Center, Boston, tried to smuggle 21 vials of biological material and research to China. Mr. Zheng was arrested in December at Boston’s Logan Airport. He admitted he planned to conduct and publish research in China using the stolen samples, said the FBI.
“All of the individuals charged today were either directly or indirectly working for the Chinese government, at our country’s expense,” said the agent in charge of the FBI’s Boston office, Joseph R. Bonavolonta.
Sen. Charles Grassley (R-IA), who has been pushing for more government action against foreign theft of U.S. research, said in a statement, “I’m glad the FBI appears to be taking foreign threats to taxpayer-funded research seriously, but I fear that this case is only the tip of the iceberg.”
The FBI said it is investigating China-related cases in all 50 states.
Ross McKinney, MD, the chief scientific officer at the Association of American Medical Colleges (AAMC), said he is aware of some 200 investigations, not all of which are cancer related, at 70-75 institutions.
“It’s a very ubiquitous problem,” Dr. McKinney said in an interview.
He also pointed out that some 6,000 National Institutes of Health–funded principal investigators are of Asian background. “So that 200 is a pretty small proportion,” said Dr. McKinney.
The NIH warned some 10,000 institutions in August 2018 that it had uncovered Chinese manipulation of peer review and a lack of disclosure of work for Chinese institutions. It urged the institutions to report irregularities.
For universities, “the trouble is sorting out who is the violator from who is not,” said Dr. McKinney. He noted that they are not set up to investigate whether someone has a laboratory in China.
“The fact that the Chinese government exploited the fact that universities are typically fairly trusting is extremely disappointing,” he said.
Moffitt story still unfolding
The most serious allegations have been leveled against six former employees of the Moffitt Cancer Center and Research Institute in Tampa, Florida.
In December 2019, Moffitt announced that the six – including President and CEO Alan List, MD, and the center director, Thomas Sellers, PhD – had left Moffitt as a result of “violations of conflict of interest rules through their work in China.”
New details have emerged, thanks to a new investigative report from a committee of the Florida House of Representatives.
The report said that Sheng Wei, a naturalized U.S. citizen who had worked at Moffitt since 2008 – when Moffitt began its affiliation with the Tianjin Medical University Cancer Institute and Hospital – was instrumental in recruiting top executives into the Thousand Talents program, which Wei had joined in 2010, according to the report. These executives included Dr. List, Dr. Sellers, and also Daniel Sullivan, head of Moffitt’s clinical science program, and cancer biologist Pearlie Epling-Burnette, it noted.
Begun in 2008, China’s Thousand Talents Plan gave salaries, funding, laboratory space, and other incentives to researchers who promised to bring U.S.-gained knowledge and research to China.
All information about this program has been removed from the Internet, but the program may still be active, Dr. McKinney commented.
According to the report, Dr. List pledged to work for the Tianjin cancer center 9 months a year for $71,000 annually. He was appointed head of the hematology department ($85,300 a year) in 2016. He opened a bank account in China to receive that salary and other Thousand Talents payments, the report found. The report notes that the exact amount Dr. List was paid is still not known.
Initially, Dr. Sellers, who was the principal investigator for Moffitt’s National Cancer Institute core grant, said he had not been involved in the Thousand Talents program. He later admitted that he had pledged to work in China 2 months a year for the program and that he’d opened a Chinese bank account and had deposited at least $35,000 into the account, the report notes.
The others pledged to work for the Thousand Talents program and also opened bank accounts in China and received money in those accounts.
Another Moffitt employee, Howard McLeod, MD, had worked for Thousand Talents before he joined Moffitt but did not disclose his China work. Dr. McLeod also supervised and had a close relationship with another researcher, Yijing (Bob) He, MD, who was employed by Moffitt but who lived in China, unbeknownst to Moffitt. “Dr. He appears to have functioned as an agent of Dr. McLeod in China,” said the report.
The report concluded that “none of the Moffitt faculty who were Talents program participants properly or timely disclosed their Talents program involvement to Moffitt, and none disclosed the full extent of their Talents program activities prior to Moffitt’s internal investigation.”
No charges have been filed against any of the former Moffitt employees.
However, the Cancer Letter has reported that Dr. Sellers is claiming he was not involved in the program and that he is preparing to sue Moffitt.
AAMC’s Dr. McKinney notes that it is illegal for researchers to take U.S. government grant money and pledge a certain amount of time but not deliver on that commitment because they are working for someone else – in this case, China. They also lied about not having any other research support, which is also illegal, he said.
The researchers received Chinese money and deposited it in Chinese accounts, which was never reported to the U.S. Internal Revenue Service.
“One of the hallmarks of the Chinese recruitment program was that people were instructed to not tell their normal U.S. host institution and not tell any U.S. government agency about their relationship with China,” Dr. McKinney said. “It was creating a culture where dishonesty in this situation was norm,” he added.
The lack of honesty brings up bigger questions for the field, he said. “Once you start lying about one thing, do you lie about your science, too?”
Lack of oversight?
Dr. McKinney said the NIH, as well as universities and hospitals, had a long and trusting relationship with China and should not be blamed for falling prey to the Chinese government’s concerted effort to steal intellectual property.
But some government watchdog groups have chided the NIH for lax oversight. In February 2019, the federal Health & Human Services’ Office of Inspector General found that “NIH has not assessed the risks to national security when permitting data access to foreign [principal investigators].”
Federal investigators have said that Thousand Talents has been one of the biggest threats.
The U.S. Senate Permanent Subcommittee on Investigations reported in November 2019 that “the federal government’s grant-making agencies did little to prevent this from happening, nor did the FBI and other federal agencies develop a coordinated response to mitigate the threat.”
The NIH invests $31 billion a year in medical research through 50,000 competitive grants to more than 300,000 researchers, according to that report. Even after uncovering grant fraud and peer-review manipulation that benefited China, “significant gaps in NIH’s grant integrity process remain,” the report states. Site visits by the NIH’s Division of Grants Compliance and Oversight dropped from 28 in 2012 to just 3 in 2018, the report noted.
Widening dragnet
In April 2019, Science reported that the NIH identified five researchers at MD Anderson Cancer Center in Houston who had failed to disclose their ties to Chinese enterprises and who had failed to keep peer review confidential.
Two resigned before they could be fired, one was fired, another eventually left the institution, and the fifth was found to have not willfully engaged in subterfuge.
Just a month later, Emory University in Atlanta announced that it had fired a husband and wife research team. The neuroscientists were known for their studies of Huntington disease. Both were U.S. citizens and had worked at Emory for more than 2 decades, according to the Science report.
The Moffitt situation led to the Florida legislature’s investigation, and also prompted some soul searching. The Tampa Bay Times reported that U.S. Senator Rick Scott (R-FL) asked state universities to provide information on what they are doing to stop foreign influence. The University of Florida then acknowledged that four faculty members resigned or were terminated because of ties to a foreign recruitment program.
This article first appeared on Medscape.com.
Academic cancer centers around the United States continue to get caught up in an ever-evolving investigation into researchers – American and Chinese – who did not disclose payments from or the work they did for Chinese institutions while simultaneously accepting taxpayer money through U.S. government grants.
The U.S. Federal Bureau of Investigation has been ferreting out researchers it says have acted illegally.
On Jan. 28, the agency arrested Charles Lieber, a chemist from Harvard University, Cambridge, Mass., and also unveiled charges against Zheng Zaosong, a cancer researcher who is in the United States on a Harvard-sponsored visa.
The FBI said Mr. Zheng, who worked at the Harvard-affiliated Beth Israel Deaconess Medical Center, Boston, tried to smuggle 21 vials of biological material and research to China. Mr. Zheng was arrested in December at Boston’s Logan Airport. He admitted he planned to conduct and publish research in China using the stolen samples, said the FBI.
“All of the individuals charged today were either directly or indirectly working for the Chinese government, at our country’s expense,” said the agent in charge of the FBI’s Boston office, Joseph R. Bonavolonta.
Sen. Charles Grassley (R-IA), who has been pushing for more government action against foreign theft of U.S. research, said in a statement, “I’m glad the FBI appears to be taking foreign threats to taxpayer-funded research seriously, but I fear that this case is only the tip of the iceberg.”
The FBI said it is investigating China-related cases in all 50 states.
Ross McKinney, MD, the chief scientific officer at the Association of American Medical Colleges (AAMC), said he is aware of some 200 investigations, not all of which are cancer related, at 70-75 institutions.
“It’s a very ubiquitous problem,” Dr. McKinney said in an interview.
He also pointed out that some 6,000 National Institutes of Health–funded principal investigators are of Asian background. “So that 200 is a pretty small proportion,” said Dr. McKinney.
The NIH warned some 10,000 institutions in August 2018 that it had uncovered Chinese manipulation of peer review and a lack of disclosure of work for Chinese institutions. It urged the institutions to report irregularities.
For universities, “the trouble is sorting out who is the violator from who is not,” said Dr. McKinney. He noted that they are not set up to investigate whether someone has a laboratory in China.
“The fact that the Chinese government exploited the fact that universities are typically fairly trusting is extremely disappointing,” he said.
Moffitt story still unfolding
The most serious allegations have been leveled against six former employees of the Moffitt Cancer Center and Research Institute in Tampa, Florida.
In December 2019, Moffitt announced that the six – including President and CEO Alan List, MD, and the center director, Thomas Sellers, PhD – had left Moffitt as a result of “violations of conflict of interest rules through their work in China.”
New details have emerged, thanks to a new investigative report from a committee of the Florida House of Representatives.
The report said that Sheng Wei, a naturalized U.S. citizen who had worked at Moffitt since 2008 – when Moffitt began its affiliation with the Tianjin Medical University Cancer Institute and Hospital – was instrumental in recruiting top executives into the Thousand Talents program, which Wei had joined in 2010, according to the report. These executives included Dr. List, Dr. Sellers, and also Daniel Sullivan, head of Moffitt’s clinical science program, and cancer biologist Pearlie Epling-Burnette, it noted.
Begun in 2008, China’s Thousand Talents Plan gave salaries, funding, laboratory space, and other incentives to researchers who promised to bring U.S.-gained knowledge and research to China.
All information about this program has been removed from the Internet, but the program may still be active, Dr. McKinney commented.
According to the report, Dr. List pledged to work for the Tianjin cancer center 9 months a year for $71,000 annually. He was appointed head of the hematology department ($85,300 a year) in 2016. He opened a bank account in China to receive that salary and other Thousand Talents payments, the report found. The report notes that the exact amount Dr. List was paid is still not known.
Initially, Dr. Sellers, who was the principal investigator for Moffitt’s National Cancer Institute core grant, said he had not been involved in the Thousand Talents program. He later admitted that he had pledged to work in China 2 months a year for the program and that he’d opened a Chinese bank account and had deposited at least $35,000 into the account, the report notes.
The others pledged to work for the Thousand Talents program and also opened bank accounts in China and received money in those accounts.
Another Moffitt employee, Howard McLeod, MD, had worked for Thousand Talents before he joined Moffitt but did not disclose his China work. Dr. McLeod also supervised and had a close relationship with another researcher, Yijing (Bob) He, MD, who was employed by Moffitt but who lived in China, unbeknownst to Moffitt. “Dr. He appears to have functioned as an agent of Dr. McLeod in China,” said the report.
The report concluded that “none of the Moffitt faculty who were Talents program participants properly or timely disclosed their Talents program involvement to Moffitt, and none disclosed the full extent of their Talents program activities prior to Moffitt’s internal investigation.”
No charges have been filed against any of the former Moffitt employees.
However, the Cancer Letter has reported that Dr. Sellers is claiming he was not involved in the program and that he is preparing to sue Moffitt.
AAMC’s Dr. McKinney notes that it is illegal for researchers to take U.S. government grant money and pledge a certain amount of time but not deliver on that commitment because they are working for someone else – in this case, China. They also lied about not having any other research support, which is also illegal, he said.
The researchers received Chinese money and deposited it in Chinese accounts, which was never reported to the U.S. Internal Revenue Service.
“One of the hallmarks of the Chinese recruitment program was that people were instructed to not tell their normal U.S. host institution and not tell any U.S. government agency about their relationship with China,” Dr. McKinney said. “It was creating a culture where dishonesty in this situation was norm,” he added.
The lack of honesty brings up bigger questions for the field, he said. “Once you start lying about one thing, do you lie about your science, too?”
Lack of oversight?
Dr. McKinney said the NIH, as well as universities and hospitals, had a long and trusting relationship with China and should not be blamed for falling prey to the Chinese government’s concerted effort to steal intellectual property.
But some government watchdog groups have chided the NIH for lax oversight. In February 2019, the federal Health & Human Services’ Office of Inspector General found that “NIH has not assessed the risks to national security when permitting data access to foreign [principal investigators].”
Federal investigators have said that Thousand Talents has been one of the biggest threats.
The U.S. Senate Permanent Subcommittee on Investigations reported in November 2019 that “the federal government’s grant-making agencies did little to prevent this from happening, nor did the FBI and other federal agencies develop a coordinated response to mitigate the threat.”
The NIH invests $31 billion a year in medical research through 50,000 competitive grants to more than 300,000 researchers, according to that report. Even after uncovering grant fraud and peer-review manipulation that benefited China, “significant gaps in NIH’s grant integrity process remain,” the report states. Site visits by the NIH’s Division of Grants Compliance and Oversight dropped from 28 in 2012 to just 3 in 2018, the report noted.
Widening dragnet
In April 2019, Science reported that the NIH identified five researchers at MD Anderson Cancer Center in Houston who had failed to disclose their ties to Chinese enterprises and who had failed to keep peer review confidential.
Two resigned before they could be fired, one was fired, another eventually left the institution, and the fifth was found to have not willfully engaged in subterfuge.
Just a month later, Emory University in Atlanta announced that it had fired a husband and wife research team. The neuroscientists were known for their studies of Huntington disease. Both were U.S. citizens and had worked at Emory for more than 2 decades, according to the Science report.
The Moffitt situation led to the Florida legislature’s investigation, and also prompted some soul searching. The Tampa Bay Times reported that U.S. Senator Rick Scott (R-FL) asked state universities to provide information on what they are doing to stop foreign influence. The University of Florida then acknowledged that four faculty members resigned or were terminated because of ties to a foreign recruitment program.
This article first appeared on Medscape.com.
Academic cancer centers around the United States continue to get caught up in an ever-evolving investigation into researchers – American and Chinese – who did not disclose payments from or the work they did for Chinese institutions while simultaneously accepting taxpayer money through U.S. government grants.
The U.S. Federal Bureau of Investigation has been ferreting out researchers it says have acted illegally.
On Jan. 28, the agency arrested Charles Lieber, a chemist from Harvard University, Cambridge, Mass., and also unveiled charges against Zheng Zaosong, a cancer researcher who is in the United States on a Harvard-sponsored visa.
The FBI said Mr. Zheng, who worked at the Harvard-affiliated Beth Israel Deaconess Medical Center, Boston, tried to smuggle 21 vials of biological material and research to China. Mr. Zheng was arrested in December at Boston’s Logan Airport. He admitted he planned to conduct and publish research in China using the stolen samples, said the FBI.
“All of the individuals charged today were either directly or indirectly working for the Chinese government, at our country’s expense,” said the agent in charge of the FBI’s Boston office, Joseph R. Bonavolonta.
Sen. Charles Grassley (R-IA), who has been pushing for more government action against foreign theft of U.S. research, said in a statement, “I’m glad the FBI appears to be taking foreign threats to taxpayer-funded research seriously, but I fear that this case is only the tip of the iceberg.”
The FBI said it is investigating China-related cases in all 50 states.
Ross McKinney, MD, the chief scientific officer at the Association of American Medical Colleges (AAMC), said he is aware of some 200 investigations, not all of which are cancer related, at 70-75 institutions.
“It’s a very ubiquitous problem,” Dr. McKinney said in an interview.
He also pointed out that some 6,000 National Institutes of Health–funded principal investigators are of Asian background. “So that 200 is a pretty small proportion,” said Dr. McKinney.
The NIH warned some 10,000 institutions in August 2018 that it had uncovered Chinese manipulation of peer review and a lack of disclosure of work for Chinese institutions. It urged the institutions to report irregularities.
For universities, “the trouble is sorting out who is the violator from who is not,” said Dr. McKinney. He noted that they are not set up to investigate whether someone has a laboratory in China.
“The fact that the Chinese government exploited the fact that universities are typically fairly trusting is extremely disappointing,” he said.
Moffitt story still unfolding
The most serious allegations have been leveled against six former employees of the Moffitt Cancer Center and Research Institute in Tampa, Florida.
In December 2019, Moffitt announced that the six – including President and CEO Alan List, MD, and the center director, Thomas Sellers, PhD – had left Moffitt as a result of “violations of conflict of interest rules through their work in China.”
New details have emerged, thanks to a new investigative report from a committee of the Florida House of Representatives.
The report said that Sheng Wei, a naturalized U.S. citizen who had worked at Moffitt since 2008 – when Moffitt began its affiliation with the Tianjin Medical University Cancer Institute and Hospital – was instrumental in recruiting top executives into the Thousand Talents program, which Wei had joined in 2010, according to the report. These executives included Dr. List, Dr. Sellers, and also Daniel Sullivan, head of Moffitt’s clinical science program, and cancer biologist Pearlie Epling-Burnette, it noted.
Begun in 2008, China’s Thousand Talents Plan gave salaries, funding, laboratory space, and other incentives to researchers who promised to bring U.S.-gained knowledge and research to China.
All information about this program has been removed from the Internet, but the program may still be active, Dr. McKinney commented.
According to the report, Dr. List pledged to work for the Tianjin cancer center 9 months a year for $71,000 annually. He was appointed head of the hematology department ($85,300 a year) in 2016. He opened a bank account in China to receive that salary and other Thousand Talents payments, the report found. The report notes that the exact amount Dr. List was paid is still not known.
Initially, Dr. Sellers, who was the principal investigator for Moffitt’s National Cancer Institute core grant, said he had not been involved in the Thousand Talents program. He later admitted that he had pledged to work in China 2 months a year for the program and that he’d opened a Chinese bank account and had deposited at least $35,000 into the account, the report notes.
The others pledged to work for the Thousand Talents program and also opened bank accounts in China and received money in those accounts.
Another Moffitt employee, Howard McLeod, MD, had worked for Thousand Talents before he joined Moffitt but did not disclose his China work. Dr. McLeod also supervised and had a close relationship with another researcher, Yijing (Bob) He, MD, who was employed by Moffitt but who lived in China, unbeknownst to Moffitt. “Dr. He appears to have functioned as an agent of Dr. McLeod in China,” said the report.
The report concluded that “none of the Moffitt faculty who were Talents program participants properly or timely disclosed their Talents program involvement to Moffitt, and none disclosed the full extent of their Talents program activities prior to Moffitt’s internal investigation.”
No charges have been filed against any of the former Moffitt employees.
However, the Cancer Letter has reported that Dr. Sellers is claiming he was not involved in the program and that he is preparing to sue Moffitt.
AAMC’s Dr. McKinney notes that it is illegal for researchers to take U.S. government grant money and pledge a certain amount of time but not deliver on that commitment because they are working for someone else – in this case, China. They also lied about not having any other research support, which is also illegal, he said.
The researchers received Chinese money and deposited it in Chinese accounts, which was never reported to the U.S. Internal Revenue Service.
“One of the hallmarks of the Chinese recruitment program was that people were instructed to not tell their normal U.S. host institution and not tell any U.S. government agency about their relationship with China,” Dr. McKinney said. “It was creating a culture where dishonesty in this situation was norm,” he added.
The lack of honesty brings up bigger questions for the field, he said. “Once you start lying about one thing, do you lie about your science, too?”
Lack of oversight?
Dr. McKinney said the NIH, as well as universities and hospitals, had a long and trusting relationship with China and should not be blamed for falling prey to the Chinese government’s concerted effort to steal intellectual property.
But some government watchdog groups have chided the NIH for lax oversight. In February 2019, the federal Health & Human Services’ Office of Inspector General found that “NIH has not assessed the risks to national security when permitting data access to foreign [principal investigators].”
Federal investigators have said that Thousand Talents has been one of the biggest threats.
The U.S. Senate Permanent Subcommittee on Investigations reported in November 2019 that “the federal government’s grant-making agencies did little to prevent this from happening, nor did the FBI and other federal agencies develop a coordinated response to mitigate the threat.”
The NIH invests $31 billion a year in medical research through 50,000 competitive grants to more than 300,000 researchers, according to that report. Even after uncovering grant fraud and peer-review manipulation that benefited China, “significant gaps in NIH’s grant integrity process remain,” the report states. Site visits by the NIH’s Division of Grants Compliance and Oversight dropped from 28 in 2012 to just 3 in 2018, the report noted.
Widening dragnet
In April 2019, Science reported that the NIH identified five researchers at MD Anderson Cancer Center in Houston who had failed to disclose their ties to Chinese enterprises and who had failed to keep peer review confidential.
Two resigned before they could be fired, one was fired, another eventually left the institution, and the fifth was found to have not willfully engaged in subterfuge.
Just a month later, Emory University in Atlanta announced that it had fired a husband and wife research team. The neuroscientists were known for their studies of Huntington disease. Both were U.S. citizens and had worked at Emory for more than 2 decades, according to the Science report.
The Moffitt situation led to the Florida legislature’s investigation, and also prompted some soul searching. The Tampa Bay Times reported that U.S. Senator Rick Scott (R-FL) asked state universities to provide information on what they are doing to stop foreign influence. The University of Florida then acknowledged that four faculty members resigned or were terminated because of ties to a foreign recruitment program.
This article first appeared on Medscape.com.
Deferiprone noninferior to deferoxamine for iron overload in SCD, rare anemias
ORLANDO – The oral iron chelator deferiprone showed noninferiority to deferoxamine for treating iron overload in patients with sickle cell disease and other rare anemias in a randomized open-label trial.
The least squares mean change from baseline in liver iron concentration (LIC) – the primary study endpoint – was –4.04 mg/g dry weight (dw) in 152 patients randomized to receive deferiprone, and –4.45 mg/g dw in 76 who received deferoxamine, Janet L. Kwiatkowski, MD, of the Children’s Hospital of Philadelphia reported at the annual meeting of the American Society of Hematology.
The upper limit of the stringent 96.01% confidence interval used for the evaluation of noninferiority in the study was 1.57, thus the findings demonstrated noninferiority of deferiprone, Dr. Kwiatkowski said.
Deferiprone also showed noninferiority for the secondary endpoints of change in cardiac iron (about –0.02 ms on T2* MRI, log-transformed for both groups) and serum ferritin levels (–415 vs. –750 mcg/L for deferiprone vs. deferoxamine) at 12 months. The difference between the groups was not statistically significant for either endpoint.
Study participants, who had a mean age of 16.9 years, were aged 2 years and older with LIC between 7 and 30 mg/g dw. They were recruited from 33 sites in nine countries and randomized 2:1 to receive deferiprone or deferoxamine for up to 12 months; in patients with lower transfusional iron input and/or less severe iron load, deferiprone was dosed at 75 mg/kg daily and deferoxamine was dosed at 20 mg/kg for children and 40 mg/kg for adults. In those with higher iron input and/or more severe iron load, the deferiprone dose was 99 mg/kg daily and the deferoxamine doses were up to 40 mg/kg in children and up to 50 mg/kg for adults.
“Over the course of the treatment period, the dosage could be adjusted downward if there were side effects, or upward if there was no improvement in iron burden,” Dr Kwiatkowski said, adding that after 12 months, patients had the option of continuing on to a 2-year extension trial in which everyone received deferiprone.
No significant demographic differences were noted between the groups; 84% in both groups had sickle cell disease, and the remaining patients had other, rarer forms of transfusion-dependent anemia. Baseline iron burden was similar in the groups.
The rates of acceptable compliance over the course of the study were also similar at 69% and 79% in the deferiprone and deferoxamine arms, respectively, she noted.
No statistically significant difference between the groups was seen in the overall rate of adverse events, treatment-related AEs, serious AEs, or withdrawals from the study due to AEs. Agranulocytosis occurred in one deferiprone patient and zero deferoxamine patients, and mild or moderate neutropenia occurred in four patients and one patient in the groups, respectively.
All episodes resolved, no difference was seen in the rates of any of the serious AEs, and no unexpected serious adverse events occurred, she said.
Patients with sickle cell disease or other rare anemias whose care includes chronic blood transfusions require iron chelation to prevent iron overload. Currently, only deferoxamine and deferasirox are approved chelators in these patient populations, she said, noting that in 2011 deferiprone received accelerated Food and Drug Administration approval for the treatment of thalassemia.
The current study was conducted because of an FDA requirement for postmarket assessment of deferiprone’s efficacy and safety in patients with sickle cell disease and other anemias who develop transfusional iron overload. It was initiated prior to the approval of deferasirox for the first-line treatment of SCD, therefore it was compared only with deferoxamine, she explained.
Dr. Kwiatkowski reported research funding from Apopharma, bluebird bio, Novartis, and Terumo, and consultancy for Agios, bluebird bio, Celgene, and Imara.
ORLANDO – The oral iron chelator deferiprone showed noninferiority to deferoxamine for treating iron overload in patients with sickle cell disease and other rare anemias in a randomized open-label trial.
The least squares mean change from baseline in liver iron concentration (LIC) – the primary study endpoint – was –4.04 mg/g dry weight (dw) in 152 patients randomized to receive deferiprone, and –4.45 mg/g dw in 76 who received deferoxamine, Janet L. Kwiatkowski, MD, of the Children’s Hospital of Philadelphia reported at the annual meeting of the American Society of Hematology.
The upper limit of the stringent 96.01% confidence interval used for the evaluation of noninferiority in the study was 1.57, thus the findings demonstrated noninferiority of deferiprone, Dr. Kwiatkowski said.
Deferiprone also showed noninferiority for the secondary endpoints of change in cardiac iron (about –0.02 ms on T2* MRI, log-transformed for both groups) and serum ferritin levels (–415 vs. –750 mcg/L for deferiprone vs. deferoxamine) at 12 months. The difference between the groups was not statistically significant for either endpoint.
Study participants, who had a mean age of 16.9 years, were aged 2 years and older with LIC between 7 and 30 mg/g dw. They were recruited from 33 sites in nine countries and randomized 2:1 to receive deferiprone or deferoxamine for up to 12 months; in patients with lower transfusional iron input and/or less severe iron load, deferiprone was dosed at 75 mg/kg daily and deferoxamine was dosed at 20 mg/kg for children and 40 mg/kg for adults. In those with higher iron input and/or more severe iron load, the deferiprone dose was 99 mg/kg daily and the deferoxamine doses were up to 40 mg/kg in children and up to 50 mg/kg for adults.
“Over the course of the treatment period, the dosage could be adjusted downward if there were side effects, or upward if there was no improvement in iron burden,” Dr Kwiatkowski said, adding that after 12 months, patients had the option of continuing on to a 2-year extension trial in which everyone received deferiprone.
No significant demographic differences were noted between the groups; 84% in both groups had sickle cell disease, and the remaining patients had other, rarer forms of transfusion-dependent anemia. Baseline iron burden was similar in the groups.
The rates of acceptable compliance over the course of the study were also similar at 69% and 79% in the deferiprone and deferoxamine arms, respectively, she noted.
No statistically significant difference between the groups was seen in the overall rate of adverse events, treatment-related AEs, serious AEs, or withdrawals from the study due to AEs. Agranulocytosis occurred in one deferiprone patient and zero deferoxamine patients, and mild or moderate neutropenia occurred in four patients and one patient in the groups, respectively.
All episodes resolved, no difference was seen in the rates of any of the serious AEs, and no unexpected serious adverse events occurred, she said.
Patients with sickle cell disease or other rare anemias whose care includes chronic blood transfusions require iron chelation to prevent iron overload. Currently, only deferoxamine and deferasirox are approved chelators in these patient populations, she said, noting that in 2011 deferiprone received accelerated Food and Drug Administration approval for the treatment of thalassemia.
The current study was conducted because of an FDA requirement for postmarket assessment of deferiprone’s efficacy and safety in patients with sickle cell disease and other anemias who develop transfusional iron overload. It was initiated prior to the approval of deferasirox for the first-line treatment of SCD, therefore it was compared only with deferoxamine, she explained.
Dr. Kwiatkowski reported research funding from Apopharma, bluebird bio, Novartis, and Terumo, and consultancy for Agios, bluebird bio, Celgene, and Imara.
ORLANDO – The oral iron chelator deferiprone showed noninferiority to deferoxamine for treating iron overload in patients with sickle cell disease and other rare anemias in a randomized open-label trial.
The least squares mean change from baseline in liver iron concentration (LIC) – the primary study endpoint – was –4.04 mg/g dry weight (dw) in 152 patients randomized to receive deferiprone, and –4.45 mg/g dw in 76 who received deferoxamine, Janet L. Kwiatkowski, MD, of the Children’s Hospital of Philadelphia reported at the annual meeting of the American Society of Hematology.
The upper limit of the stringent 96.01% confidence interval used for the evaluation of noninferiority in the study was 1.57, thus the findings demonstrated noninferiority of deferiprone, Dr. Kwiatkowski said.
Deferiprone also showed noninferiority for the secondary endpoints of change in cardiac iron (about –0.02 ms on T2* MRI, log-transformed for both groups) and serum ferritin levels (–415 vs. –750 mcg/L for deferiprone vs. deferoxamine) at 12 months. The difference between the groups was not statistically significant for either endpoint.
Study participants, who had a mean age of 16.9 years, were aged 2 years and older with LIC between 7 and 30 mg/g dw. They were recruited from 33 sites in nine countries and randomized 2:1 to receive deferiprone or deferoxamine for up to 12 months; in patients with lower transfusional iron input and/or less severe iron load, deferiprone was dosed at 75 mg/kg daily and deferoxamine was dosed at 20 mg/kg for children and 40 mg/kg for adults. In those with higher iron input and/or more severe iron load, the deferiprone dose was 99 mg/kg daily and the deferoxamine doses were up to 40 mg/kg in children and up to 50 mg/kg for adults.
“Over the course of the treatment period, the dosage could be adjusted downward if there were side effects, or upward if there was no improvement in iron burden,” Dr Kwiatkowski said, adding that after 12 months, patients had the option of continuing on to a 2-year extension trial in which everyone received deferiprone.
No significant demographic differences were noted between the groups; 84% in both groups had sickle cell disease, and the remaining patients had other, rarer forms of transfusion-dependent anemia. Baseline iron burden was similar in the groups.
The rates of acceptable compliance over the course of the study were also similar at 69% and 79% in the deferiprone and deferoxamine arms, respectively, she noted.
No statistically significant difference between the groups was seen in the overall rate of adverse events, treatment-related AEs, serious AEs, or withdrawals from the study due to AEs. Agranulocytosis occurred in one deferiprone patient and zero deferoxamine patients, and mild or moderate neutropenia occurred in four patients and one patient in the groups, respectively.
All episodes resolved, no difference was seen in the rates of any of the serious AEs, and no unexpected serious adverse events occurred, she said.
Patients with sickle cell disease or other rare anemias whose care includes chronic blood transfusions require iron chelation to prevent iron overload. Currently, only deferoxamine and deferasirox are approved chelators in these patient populations, she said, noting that in 2011 deferiprone received accelerated Food and Drug Administration approval for the treatment of thalassemia.
The current study was conducted because of an FDA requirement for postmarket assessment of deferiprone’s efficacy and safety in patients with sickle cell disease and other anemias who develop transfusional iron overload. It was initiated prior to the approval of deferasirox for the first-line treatment of SCD, therefore it was compared only with deferoxamine, she explained.
Dr. Kwiatkowski reported research funding from Apopharma, bluebird bio, Novartis, and Terumo, and consultancy for Agios, bluebird bio, Celgene, and Imara.
REPORTING FROM ASH 2019
Novel mutations contribute to progression of venetoclax-treated CLL
Newly discovered gene mutations in the progression of venetoclax-treated relapsed chronic lymphocytic leukemia (CLL) may improve understanding of clinical resistance mechanisms underlying the disease, according to recent research.
“We investigated patients with progressive CLL on venetoclax harboring subclonal BCL2 Gly101Val mutations for the presence of additional acquired BCL2 resistance mutations,” wrote Piers Blombery, MBBS, of the University of Melbourne in Victoria, Australia, and his colleagues in Blood.
Among 67 patients with relapsed disease treated with the BCL2 inhibitor venetoclax, the researchers identified a total of 11 patients with co-occurring BCL2 Gly101Val mutations. Each patient was enrolled in an early phase clinical trial at an institution in Australia.
With respect to testing methods, next-generation sequencing (NGS) and hybridization-based target enrichment technologies were used to detect novel acquired mutations in the BCL2 coding region.
Among those harboring the Gly101Val mutation, additional BCL2 mutations were identified in 10 patients (91%), with a median of three mutations detected per patient (range, 1-7). Previously undescribed mutations included an in-frame insertion mutation (Arg107_Arg110dup), and other substitutions (Asp103/Val156) in the BCL2 gene.
“As with the Gly101Val, these observations support the specificity of these mutations for the context of venetoclax resistance,” they wrote.
The investigators further explained that the BCL2 Asp103Glu mutation could have particular significance in the context of venetoclax sensitivity because of selective targeting of the BCL2 gene.
In comparison to wild-type aspartic acid, the BCL2 Asp103Glu substitution was linked to an approximate 20-fold reduction in affinity for venetoclax, they reported.
“[Our findings] consolidate the paradigm emerging across hematological malignancies of multiple independent molecular mechanisms underpinning an ‘oligoclonal’ pattern of clinical relapse on targeted therapies,” they concluded.
Further studies are needed to fully characterize the relationship between acquired BCL2 mutations and venetoclax resistance.
The study was funded by the Snowdome Foundation, Vision Super and the Wilson Centre for Lymphoma Genomics, the Leukemia and Lymphoma Society, the National Health and Medical Research Council of Australia, and other grant funding sources provided to the study authors. The authors reported financial affiliations with AbbVie, Genentech, and the Walter and Eliza Hall Institute.
Newly discovered gene mutations in the progression of venetoclax-treated relapsed chronic lymphocytic leukemia (CLL) may improve understanding of clinical resistance mechanisms underlying the disease, according to recent research.
“We investigated patients with progressive CLL on venetoclax harboring subclonal BCL2 Gly101Val mutations for the presence of additional acquired BCL2 resistance mutations,” wrote Piers Blombery, MBBS, of the University of Melbourne in Victoria, Australia, and his colleagues in Blood.
Among 67 patients with relapsed disease treated with the BCL2 inhibitor venetoclax, the researchers identified a total of 11 patients with co-occurring BCL2 Gly101Val mutations. Each patient was enrolled in an early phase clinical trial at an institution in Australia.
With respect to testing methods, next-generation sequencing (NGS) and hybridization-based target enrichment technologies were used to detect novel acquired mutations in the BCL2 coding region.
Among those harboring the Gly101Val mutation, additional BCL2 mutations were identified in 10 patients (91%), with a median of three mutations detected per patient (range, 1-7). Previously undescribed mutations included an in-frame insertion mutation (Arg107_Arg110dup), and other substitutions (Asp103/Val156) in the BCL2 gene.
“As with the Gly101Val, these observations support the specificity of these mutations for the context of venetoclax resistance,” they wrote.
The investigators further explained that the BCL2 Asp103Glu mutation could have particular significance in the context of venetoclax sensitivity because of selective targeting of the BCL2 gene.
In comparison to wild-type aspartic acid, the BCL2 Asp103Glu substitution was linked to an approximate 20-fold reduction in affinity for venetoclax, they reported.
“[Our findings] consolidate the paradigm emerging across hematological malignancies of multiple independent molecular mechanisms underpinning an ‘oligoclonal’ pattern of clinical relapse on targeted therapies,” they concluded.
Further studies are needed to fully characterize the relationship between acquired BCL2 mutations and venetoclax resistance.
The study was funded by the Snowdome Foundation, Vision Super and the Wilson Centre for Lymphoma Genomics, the Leukemia and Lymphoma Society, the National Health and Medical Research Council of Australia, and other grant funding sources provided to the study authors. The authors reported financial affiliations with AbbVie, Genentech, and the Walter and Eliza Hall Institute.
Newly discovered gene mutations in the progression of venetoclax-treated relapsed chronic lymphocytic leukemia (CLL) may improve understanding of clinical resistance mechanisms underlying the disease, according to recent research.
“We investigated patients with progressive CLL on venetoclax harboring subclonal BCL2 Gly101Val mutations for the presence of additional acquired BCL2 resistance mutations,” wrote Piers Blombery, MBBS, of the University of Melbourne in Victoria, Australia, and his colleagues in Blood.
Among 67 patients with relapsed disease treated with the BCL2 inhibitor venetoclax, the researchers identified a total of 11 patients with co-occurring BCL2 Gly101Val mutations. Each patient was enrolled in an early phase clinical trial at an institution in Australia.
With respect to testing methods, next-generation sequencing (NGS) and hybridization-based target enrichment technologies were used to detect novel acquired mutations in the BCL2 coding region.
Among those harboring the Gly101Val mutation, additional BCL2 mutations were identified in 10 patients (91%), with a median of three mutations detected per patient (range, 1-7). Previously undescribed mutations included an in-frame insertion mutation (Arg107_Arg110dup), and other substitutions (Asp103/Val156) in the BCL2 gene.
“As with the Gly101Val, these observations support the specificity of these mutations for the context of venetoclax resistance,” they wrote.
The investigators further explained that the BCL2 Asp103Glu mutation could have particular significance in the context of venetoclax sensitivity because of selective targeting of the BCL2 gene.
In comparison to wild-type aspartic acid, the BCL2 Asp103Glu substitution was linked to an approximate 20-fold reduction in affinity for venetoclax, they reported.
“[Our findings] consolidate the paradigm emerging across hematological malignancies of multiple independent molecular mechanisms underpinning an ‘oligoclonal’ pattern of clinical relapse on targeted therapies,” they concluded.
Further studies are needed to fully characterize the relationship between acquired BCL2 mutations and venetoclax resistance.
The study was funded by the Snowdome Foundation, Vision Super and the Wilson Centre for Lymphoma Genomics, the Leukemia and Lymphoma Society, the National Health and Medical Research Council of Australia, and other grant funding sources provided to the study authors. The authors reported financial affiliations with AbbVie, Genentech, and the Walter and Eliza Hall Institute.
FROM BLOOD
ECHELON-1 update: A+AVD bests ABVD in Hodgkin lymphoma
Brentuximab vedotin plus doxorubicin, vinblastine, and dacarbazine (A+AVD) provides “robust, sustained efficacy” in patients with Hodgkin lymphoma, according to investigators.
In the ECHELON-1 trial, investigators compared A+AVD to doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) as frontline treatment for stage III or IV Hodgkin lymphoma. The 3-year progression-free survival (PFS) was superior in patients who received A+AVD, and this benefit was seen across most subgroups.
David J. Straus, MD, of Memorial Sloan Kettering Cancer Center in New York and his colleagues detailed these findings in Blood.
The phase 3 trial (NCT01712490) enrolled 1,334 patients with stage III or IV classical Hodgkin lymphoma. They were randomized to receive A+AVD (n = 664) or ABVD (n = 670). Baseline characteristics were similar between the treatment arms.
Positron emission tomography status after cycle 2 (PET2) was similar between the treatment arms as well. Most patients – 89% of the A+AVD arm and 86% of the ABVD arm – were PET2 negative. Treating physicians used PET2 status as a guide to potentially switch patients to an alternative regimen (radiotherapy or chemotherapy with or without transplant).
In a prior analysis, the study’s primary endpoint was modified PFS (time to progression, death, or noncomplete response after frontline therapy) per an independent review committee (N Engl J Med. 2018;378:331-44). The 2-year modified PFS rate was 82.1% in the A+AVD arm and 77.2% in the ABVD arm (hazard ratio, 0.77; P = .04).
PFS update
In the current analysis, the main exploratory endpoint was PFS per investigator. The 3-year PFS rate was significantly higher in the A+AVD arm than in the ABVD arm – 83.1% and 76.0%, respectively (HR, 0.704; P = .005).
The investigators observed a “consistent improvement in PFS” in the A+AVD arm, regardless of disease stage, International Prognostic score, Eastern Cooperative Oncology Group status, sex, or age. There was a significant improvement in PFS with A+AVD in PET2-negative patients and a trend toward improvement in PET2-positive patients. In the PET2-negative patients, the 3-year PFS was 85.8% in the A+AVD arm and 79.5% in the ABVD arm (HR, 0.69; P = .009). In PET2-positive patients, the 3-year PFS was 67.7% and 51.5%, respectively (HR, 0.59; P = .077).
“These data highlight that A+AVD provides a durable efficacy benefit, compared with ABVD, for frontline stage III/IV cHL [classical Hodgkin lymphoma], which is consistent across key subgroups regardless of patient status at PET2,” Dr. Straus and his colleagues wrote.
Safety update
In both treatment arms, peripheral neuropathy continued to improve or resolve with longer follow-up. Among patients who developed peripheral neuropathy, 78% in the A+AVD arm and 83% in the ABVD arm had improvement or resolution of the condition at 3 years.
Most patients had complete resolution of peripheral neuropathy; 62% in the A+AVD arm and 73% in the ABVD arm. The median time to complete resolution was 28 weeks (range, 0-167 weeks) after stopping A+AVD and 14 weeks (range, 0-188 weeks) after stopping ABVD.
The incidence of secondary malignancies was similar between the treatment arms. There were 14 secondary malignancies in the A+AVD arm (6 solid tumors, 8 hematologic malignancies) and 20 in the ABVD arm (9 solid tumors, 11 hematologic malignancies).
“A+AVD provided a sustained PFS benefit with a predictable and manageable safety profile,” Dr. Straus and colleagues wrote. “These data further support the advantages of A+AVD versus ABVD as frontline treatment of patients with advanced stage III or IV cHL [classical Hodgkin lymphoma].”
The ECHELON-1 trial was sponsored by Millennium Pharmaceuticals (a subsidiary of Takeda) and Seattle Genetics. The investigators disclosed relationships with Millennium, Takeda, Seattle Genetics, and a range of other companies.
SOURCE: Straus DJ et al. Blood. 2020 Jan 16. pii: blood.2019003127. doi: 10.1182/blood.2019003127.
Brentuximab vedotin plus doxorubicin, vinblastine, and dacarbazine (A+AVD) provides “robust, sustained efficacy” in patients with Hodgkin lymphoma, according to investigators.
In the ECHELON-1 trial, investigators compared A+AVD to doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) as frontline treatment for stage III or IV Hodgkin lymphoma. The 3-year progression-free survival (PFS) was superior in patients who received A+AVD, and this benefit was seen across most subgroups.
David J. Straus, MD, of Memorial Sloan Kettering Cancer Center in New York and his colleagues detailed these findings in Blood.
The phase 3 trial (NCT01712490) enrolled 1,334 patients with stage III or IV classical Hodgkin lymphoma. They were randomized to receive A+AVD (n = 664) or ABVD (n = 670). Baseline characteristics were similar between the treatment arms.
Positron emission tomography status after cycle 2 (PET2) was similar between the treatment arms as well. Most patients – 89% of the A+AVD arm and 86% of the ABVD arm – were PET2 negative. Treating physicians used PET2 status as a guide to potentially switch patients to an alternative regimen (radiotherapy or chemotherapy with or without transplant).
In a prior analysis, the study’s primary endpoint was modified PFS (time to progression, death, or noncomplete response after frontline therapy) per an independent review committee (N Engl J Med. 2018;378:331-44). The 2-year modified PFS rate was 82.1% in the A+AVD arm and 77.2% in the ABVD arm (hazard ratio, 0.77; P = .04).
PFS update
In the current analysis, the main exploratory endpoint was PFS per investigator. The 3-year PFS rate was significantly higher in the A+AVD arm than in the ABVD arm – 83.1% and 76.0%, respectively (HR, 0.704; P = .005).
The investigators observed a “consistent improvement in PFS” in the A+AVD arm, regardless of disease stage, International Prognostic score, Eastern Cooperative Oncology Group status, sex, or age. There was a significant improvement in PFS with A+AVD in PET2-negative patients and a trend toward improvement in PET2-positive patients. In the PET2-negative patients, the 3-year PFS was 85.8% in the A+AVD arm and 79.5% in the ABVD arm (HR, 0.69; P = .009). In PET2-positive patients, the 3-year PFS was 67.7% and 51.5%, respectively (HR, 0.59; P = .077).
“These data highlight that A+AVD provides a durable efficacy benefit, compared with ABVD, for frontline stage III/IV cHL [classical Hodgkin lymphoma], which is consistent across key subgroups regardless of patient status at PET2,” Dr. Straus and his colleagues wrote.
Safety update
In both treatment arms, peripheral neuropathy continued to improve or resolve with longer follow-up. Among patients who developed peripheral neuropathy, 78% in the A+AVD arm and 83% in the ABVD arm had improvement or resolution of the condition at 3 years.
Most patients had complete resolution of peripheral neuropathy; 62% in the A+AVD arm and 73% in the ABVD arm. The median time to complete resolution was 28 weeks (range, 0-167 weeks) after stopping A+AVD and 14 weeks (range, 0-188 weeks) after stopping ABVD.
The incidence of secondary malignancies was similar between the treatment arms. There were 14 secondary malignancies in the A+AVD arm (6 solid tumors, 8 hematologic malignancies) and 20 in the ABVD arm (9 solid tumors, 11 hematologic malignancies).
“A+AVD provided a sustained PFS benefit with a predictable and manageable safety profile,” Dr. Straus and colleagues wrote. “These data further support the advantages of A+AVD versus ABVD as frontline treatment of patients with advanced stage III or IV cHL [classical Hodgkin lymphoma].”
The ECHELON-1 trial was sponsored by Millennium Pharmaceuticals (a subsidiary of Takeda) and Seattle Genetics. The investigators disclosed relationships with Millennium, Takeda, Seattle Genetics, and a range of other companies.
SOURCE: Straus DJ et al. Blood. 2020 Jan 16. pii: blood.2019003127. doi: 10.1182/blood.2019003127.
Brentuximab vedotin plus doxorubicin, vinblastine, and dacarbazine (A+AVD) provides “robust, sustained efficacy” in patients with Hodgkin lymphoma, according to investigators.
In the ECHELON-1 trial, investigators compared A+AVD to doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) as frontline treatment for stage III or IV Hodgkin lymphoma. The 3-year progression-free survival (PFS) was superior in patients who received A+AVD, and this benefit was seen across most subgroups.
David J. Straus, MD, of Memorial Sloan Kettering Cancer Center in New York and his colleagues detailed these findings in Blood.
The phase 3 trial (NCT01712490) enrolled 1,334 patients with stage III or IV classical Hodgkin lymphoma. They were randomized to receive A+AVD (n = 664) or ABVD (n = 670). Baseline characteristics were similar between the treatment arms.
Positron emission tomography status after cycle 2 (PET2) was similar between the treatment arms as well. Most patients – 89% of the A+AVD arm and 86% of the ABVD arm – were PET2 negative. Treating physicians used PET2 status as a guide to potentially switch patients to an alternative regimen (radiotherapy or chemotherapy with or without transplant).
In a prior analysis, the study’s primary endpoint was modified PFS (time to progression, death, or noncomplete response after frontline therapy) per an independent review committee (N Engl J Med. 2018;378:331-44). The 2-year modified PFS rate was 82.1% in the A+AVD arm and 77.2% in the ABVD arm (hazard ratio, 0.77; P = .04).
PFS update
In the current analysis, the main exploratory endpoint was PFS per investigator. The 3-year PFS rate was significantly higher in the A+AVD arm than in the ABVD arm – 83.1% and 76.0%, respectively (HR, 0.704; P = .005).
The investigators observed a “consistent improvement in PFS” in the A+AVD arm, regardless of disease stage, International Prognostic score, Eastern Cooperative Oncology Group status, sex, or age. There was a significant improvement in PFS with A+AVD in PET2-negative patients and a trend toward improvement in PET2-positive patients. In the PET2-negative patients, the 3-year PFS was 85.8% in the A+AVD arm and 79.5% in the ABVD arm (HR, 0.69; P = .009). In PET2-positive patients, the 3-year PFS was 67.7% and 51.5%, respectively (HR, 0.59; P = .077).
“These data highlight that A+AVD provides a durable efficacy benefit, compared with ABVD, for frontline stage III/IV cHL [classical Hodgkin lymphoma], which is consistent across key subgroups regardless of patient status at PET2,” Dr. Straus and his colleagues wrote.
Safety update
In both treatment arms, peripheral neuropathy continued to improve or resolve with longer follow-up. Among patients who developed peripheral neuropathy, 78% in the A+AVD arm and 83% in the ABVD arm had improvement or resolution of the condition at 3 years.
Most patients had complete resolution of peripheral neuropathy; 62% in the A+AVD arm and 73% in the ABVD arm. The median time to complete resolution was 28 weeks (range, 0-167 weeks) after stopping A+AVD and 14 weeks (range, 0-188 weeks) after stopping ABVD.
The incidence of secondary malignancies was similar between the treatment arms. There were 14 secondary malignancies in the A+AVD arm (6 solid tumors, 8 hematologic malignancies) and 20 in the ABVD arm (9 solid tumors, 11 hematologic malignancies).
“A+AVD provided a sustained PFS benefit with a predictable and manageable safety profile,” Dr. Straus and colleagues wrote. “These data further support the advantages of A+AVD versus ABVD as frontline treatment of patients with advanced stage III or IV cHL [classical Hodgkin lymphoma].”
The ECHELON-1 trial was sponsored by Millennium Pharmaceuticals (a subsidiary of Takeda) and Seattle Genetics. The investigators disclosed relationships with Millennium, Takeda, Seattle Genetics, and a range of other companies.
SOURCE: Straus DJ et al. Blood. 2020 Jan 16. pii: blood.2019003127. doi: 10.1182/blood.2019003127.
FROM BLOOD