User login
Intermittent fasting fights ALL, not AML, in mice

Photo by Steve Berger
Intermittent fasting inhibits the development and progression of acute lymphoblastic leukemia (ALL), according to preclinical research published in Nature Medicine.
Fasting had an inhibitory effect in mouse models of T-cell and B-cell ALL but not acute myeloid leukemia (AML).
“This study using mouse models indicates that the effects of fasting on blood cancers are type-dependent and provides a platform for identifying new targets for leukemia treatments,” said study author Chengcheng “Alec” Zhang, PhD, of UT Southwestern Medical Center in Dallas, Texas.
“We also identified a mechanism responsible for the differing response to the fasting treatment.”
For this study, Dr Zhang and his colleagues created mouse models of acute leukemia—N-Myc B-ALL, activated Notch1 T-ALL, MLL-AF9 AML, and AML driven by the AML1-Eto9a oncogene—and tested the effects of various dietary restriction plans.
The team used green or yellow florescent proteins to mark and trace the leukemia cells so they could determine if the cells’ levels rose or fell in response to the fasting treatment.
“Strikingly, we found that, in models of ALL, a regimen consisting of 6 cycles of 1 day of fasting followed by 1 day of feeding completely inhibited cancer development,” Dr Zhang said.
At the end of 7 weeks, fasted mice with B-ALL had virtually no detectible cancerous cells—an average of 0.48%—compared to an average of 67.68% of cells found to be cancerous in the test areas of the non-fasted B-ALL mice.
Dr Zhang noted that, compared to B-ALL mice that ate normally, the mice on alternate-day fasting had dramatic reductions in the percentage of ALL cells in the bone marrow and spleen, as well as reduced numbers of white blood cells.
In addition, the spleens and lymph nodes in the fasted mice with B-ALL were similar in size to those of normal mice.
“Although initially cancerous, the few fluorescent cells that remained in the fasted mice after 7 weeks appeared to behave like normal cells,” Dr Zhang said. “Mice in the [B-ALL] model group that ate normally died within 59 days, while 75% of the fasted mice survived more than 120 days without signs of leukemia.”
Dr Zhang and his colleagues said they observed similar results in the T-ALL model but not the AML models. There was no decrease in leukemia cells among fasted mice with AML. And fasting actually shortened survival time in these mice.
Identifying the mechanism
Fasting is known to reduce the level of leptin, a cell signaling molecule created by fat tissue. In addition, previous studies have shown weakened activity by leptin receptors in humans with ALL. For those reasons, the researchers studied both leptin levels and leptin receptors in the mouse models.
The team found that mice with ALL showed reduced leptin receptor activity that increased with intermittent fasting.
“We found that fasting decreased the levels of leptin circulating in the bloodstream as well as decreased the leptin levels in the bone marrow,” Dr Zhang said. “These effects became more pronounced with repeated cycles of fasting. After fasting, the rate at which the leptin levels recovered seemed to correspond to the rate at which the cancerous ALL cells were cleared from the blood.”
The researchers also found that AML was associated with higher levels of leptin receptors that were unaffected by fasting, which could help explain why the fasting treatment was ineffective against this type of leukemia.
It also suggests a mechanism—the leptin receptor pathway—by which fasting exerts its effects in ALL, Dr Zhang said.
“It will be important to determine whether ALL cells can become resistant to the effects of fasting,” he noted. “It also will be interesting to investigate whether we can find alternative ways that mimic fasting to block ALL development.”
Given that this study did not involve drug treatment, researchers are discussing with clinicians whether the tested regimen might be able to move forward quickly to clinical trials. ![]()
Photo by Steve Berger
Intermittent fasting inhibits the development and progression of acute lymphoblastic leukemia (ALL), according to preclinical research published in Nature Medicine.
Fasting had an inhibitory effect in mouse models of T-cell and B-cell ALL but not acute myeloid leukemia (AML).
“This study using mouse models indicates that the effects of fasting on blood cancers are type-dependent and provides a platform for identifying new targets for leukemia treatments,” said study author Chengcheng “Alec” Zhang, PhD, of UT Southwestern Medical Center in Dallas, Texas.
“We also identified a mechanism responsible for the differing response to the fasting treatment.”
For this study, Dr Zhang and his colleagues created mouse models of acute leukemia—N-Myc B-ALL, activated Notch1 T-ALL, MLL-AF9 AML, and AML driven by the AML1-Eto9a oncogene—and tested the effects of various dietary restriction plans.
The team used green or yellow florescent proteins to mark and trace the leukemia cells so they could determine if the cells’ levels rose or fell in response to the fasting treatment.
“Strikingly, we found that, in models of ALL, a regimen consisting of 6 cycles of 1 day of fasting followed by 1 day of feeding completely inhibited cancer development,” Dr Zhang said.
At the end of 7 weeks, fasted mice with B-ALL had virtually no detectible cancerous cells—an average of 0.48%—compared to an average of 67.68% of cells found to be cancerous in the test areas of the non-fasted B-ALL mice.
Dr Zhang noted that, compared to B-ALL mice that ate normally, the mice on alternate-day fasting had dramatic reductions in the percentage of ALL cells in the bone marrow and spleen, as well as reduced numbers of white blood cells.
In addition, the spleens and lymph nodes in the fasted mice with B-ALL were similar in size to those of normal mice.
“Although initially cancerous, the few fluorescent cells that remained in the fasted mice after 7 weeks appeared to behave like normal cells,” Dr Zhang said. “Mice in the [B-ALL] model group that ate normally died within 59 days, while 75% of the fasted mice survived more than 120 days without signs of leukemia.”
Dr Zhang and his colleagues said they observed similar results in the T-ALL model but not the AML models. There was no decrease in leukemia cells among fasted mice with AML. And fasting actually shortened survival time in these mice.
Identifying the mechanism
Fasting is known to reduce the level of leptin, a cell signaling molecule created by fat tissue. In addition, previous studies have shown weakened activity by leptin receptors in humans with ALL. For those reasons, the researchers studied both leptin levels and leptin receptors in the mouse models.
The team found that mice with ALL showed reduced leptin receptor activity that increased with intermittent fasting.
“We found that fasting decreased the levels of leptin circulating in the bloodstream as well as decreased the leptin levels in the bone marrow,” Dr Zhang said. “These effects became more pronounced with repeated cycles of fasting. After fasting, the rate at which the leptin levels recovered seemed to correspond to the rate at which the cancerous ALL cells were cleared from the blood.”
The researchers also found that AML was associated with higher levels of leptin receptors that were unaffected by fasting, which could help explain why the fasting treatment was ineffective against this type of leukemia.
It also suggests a mechanism—the leptin receptor pathway—by which fasting exerts its effects in ALL, Dr Zhang said.
“It will be important to determine whether ALL cells can become resistant to the effects of fasting,” he noted. “It also will be interesting to investigate whether we can find alternative ways that mimic fasting to block ALL development.”
Given that this study did not involve drug treatment, researchers are discussing with clinicians whether the tested regimen might be able to move forward quickly to clinical trials. ![]()
Photo by Steve Berger
Intermittent fasting inhibits the development and progression of acute lymphoblastic leukemia (ALL), according to preclinical research published in Nature Medicine.
Fasting had an inhibitory effect in mouse models of T-cell and B-cell ALL but not acute myeloid leukemia (AML).
“This study using mouse models indicates that the effects of fasting on blood cancers are type-dependent and provides a platform for identifying new targets for leukemia treatments,” said study author Chengcheng “Alec” Zhang, PhD, of UT Southwestern Medical Center in Dallas, Texas.
“We also identified a mechanism responsible for the differing response to the fasting treatment.”
For this study, Dr Zhang and his colleagues created mouse models of acute leukemia—N-Myc B-ALL, activated Notch1 T-ALL, MLL-AF9 AML, and AML driven by the AML1-Eto9a oncogene—and tested the effects of various dietary restriction plans.
The team used green or yellow florescent proteins to mark and trace the leukemia cells so they could determine if the cells’ levels rose or fell in response to the fasting treatment.
“Strikingly, we found that, in models of ALL, a regimen consisting of 6 cycles of 1 day of fasting followed by 1 day of feeding completely inhibited cancer development,” Dr Zhang said.
At the end of 7 weeks, fasted mice with B-ALL had virtually no detectible cancerous cells—an average of 0.48%—compared to an average of 67.68% of cells found to be cancerous in the test areas of the non-fasted B-ALL mice.
Dr Zhang noted that, compared to B-ALL mice that ate normally, the mice on alternate-day fasting had dramatic reductions in the percentage of ALL cells in the bone marrow and spleen, as well as reduced numbers of white blood cells.
In addition, the spleens and lymph nodes in the fasted mice with B-ALL were similar in size to those of normal mice.
“Although initially cancerous, the few fluorescent cells that remained in the fasted mice after 7 weeks appeared to behave like normal cells,” Dr Zhang said. “Mice in the [B-ALL] model group that ate normally died within 59 days, while 75% of the fasted mice survived more than 120 days without signs of leukemia.”
Dr Zhang and his colleagues said they observed similar results in the T-ALL model but not the AML models. There was no decrease in leukemia cells among fasted mice with AML. And fasting actually shortened survival time in these mice.
Identifying the mechanism
Fasting is known to reduce the level of leptin, a cell signaling molecule created by fat tissue. In addition, previous studies have shown weakened activity by leptin receptors in humans with ALL. For those reasons, the researchers studied both leptin levels and leptin receptors in the mouse models.
The team found that mice with ALL showed reduced leptin receptor activity that increased with intermittent fasting.
“We found that fasting decreased the levels of leptin circulating in the bloodstream as well as decreased the leptin levels in the bone marrow,” Dr Zhang said. “These effects became more pronounced with repeated cycles of fasting. After fasting, the rate at which the leptin levels recovered seemed to correspond to the rate at which the cancerous ALL cells were cleared from the blood.”
The researchers also found that AML was associated with higher levels of leptin receptors that were unaffected by fasting, which could help explain why the fasting treatment was ineffective against this type of leukemia.
It also suggests a mechanism—the leptin receptor pathway—by which fasting exerts its effects in ALL, Dr Zhang said.
“It will be important to determine whether ALL cells can become resistant to the effects of fasting,” he noted. “It also will be interesting to investigate whether we can find alternative ways that mimic fasting to block ALL development.”
Given that this study did not involve drug treatment, researchers are discussing with clinicians whether the tested regimen might be able to move forward quickly to clinical trials. ![]()
Ruxolitinib may prevent CRS after CAR T-cell therapy
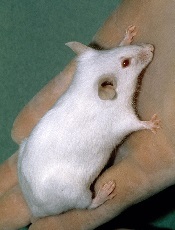
Photo courtesy of NCI
SAN DIEGO—A novel xenograft model of acute myeloid leukemia (AML) demonstrated that the JAK/STAT inhibitor ruxolitinib can prevent severe cytokine release syndrome (CRS) without impairing the anti-tumor effect of chimeric antigen receptor (CAR) T cells, according to research presented at the 2016 ASH Annual Meeting.
Almost all patients responding to CART-cell therapy develop CRS, and up to 60% develop severe CRS.
The research team believes the mouse model and findings with ruxolitinib will provide an important platform for studying CRS prevention and treatment.
At ASH, Saad Kenderian, MD, of the Mayo Clinic in Rochester, Minnesota, explained that CRS produces very high levels of the inflammatory protein IL-6.
Treatment with ruxolitinib in clinical studies has reduced human inflammatory cytokines. Therefore, it made sense to the investigators to study ruxolitinib as a means to prevent CRS after CAR T-cell therapy.
Tocilizumab has been used to treat grade 3 and 4 CRS, but physicians are concerned that earlier introduction during the course of CRS may impair CAR T-cell function.
At present, no relevant preclinical model for CRS after CAR T-cell therapy exists, “which is limiting the development of CRS preventative modalities that could, in turn, enhance the feasibility of CAR T-cell therapy,” Dr Kenderian said.
And so the investigators decided to create an animal model.
Dr Kenderian described the work at the meeting as abstract 652.
Mouse model for human CRS
Using NSG-S mice (non-obese diabetic, SCID ɣ -/- mice additionally transgenic for human stem cell factor, IL-3, and GM-CSF), investigators injected them with blasts from AML patients. After 3 to 4 weeks, investigators treated the mice with 1 x 106 CD123-directed CAR T cells.
Dr Kenderian noted this dose of CART123 was 10 times higher than doses previously used in primary AML xenograft models.
The mice became weak, emaciated, developed hunched bodies, became withdrawn, had poor motor responses, and died in 7 to 10 days. The illness started within 1 week of CAR T-cell injection and correlated with significant expansion of T cells in the peripheral blood of these mice.
The team studied the serum from these mice 7 days after CART123 injection. They found extreme elevation of human IL-6, interferon-γ, tumor necrosis factor-α, and other inflammatory cytokines. This response resembled human CRS after CAR T-cell therapy.
Ruxolitinib treatment
The investigators first studied ruxolitinib activity in vitro with CART123 cells and found that ruxolitinib did not impair CAR T-cell effector functions.
“And also, ruxolitinib was not directly toxic to CAR T cells,” Dr Kenderian added.
But ruxolitinib did slow CAR T-cell proliferation in vitro.
They next tested ruxolitinib and CART123 in the mouse model.
Once the mice experienced high-burden disease, investigators treated them with CART123. That same day, investigators began treating the mice with ruxolitinib for 1 week. The mice were randomized to 30, 60, 90 mg/kg, or vehicle twice a day.
Twenty-nine days after AML injection, the mice treated with CART123 plus 90 mg or 60 mg of ruxolitinib experienced less weight loss than those treated with CART123 plus 30 mg of ruxolitinib or CART123-only.
“And more importantly, all mice had eradication of their disease,” Dr Kenderian said.
Mice treated with CART123 plus 90 mg, 60 mg, or 30 mg of ruxolitinib or CART123 alone had fewer AML blasts at day 28 than mice treated with 60 mg of ruxolitinib alone.
The investigators then analyzed the effect of ruxolitinib on the anti-tumor effect of CART123 and found that ruxolitinib did not impair it.
The attenuation of inflammatory cytokines translated to a survival advantage for mice treated with CART123 and ruxolitinib.
The investigators believe the addition of ruxolitinib to CAR T-cell therapy is a modality that should be investigated in patients at high-risk of developing CRS.
Dr Kenderian disclosed patents, royalties, and research funding from Novartis. ![]()
Photo courtesy of NCI
SAN DIEGO—A novel xenograft model of acute myeloid leukemia (AML) demonstrated that the JAK/STAT inhibitor ruxolitinib can prevent severe cytokine release syndrome (CRS) without impairing the anti-tumor effect of chimeric antigen receptor (CAR) T cells, according to research presented at the 2016 ASH Annual Meeting.
Almost all patients responding to CART-cell therapy develop CRS, and up to 60% develop severe CRS.
The research team believes the mouse model and findings with ruxolitinib will provide an important platform for studying CRS prevention and treatment.
At ASH, Saad Kenderian, MD, of the Mayo Clinic in Rochester, Minnesota, explained that CRS produces very high levels of the inflammatory protein IL-6.
Treatment with ruxolitinib in clinical studies has reduced human inflammatory cytokines. Therefore, it made sense to the investigators to study ruxolitinib as a means to prevent CRS after CAR T-cell therapy.
Tocilizumab has been used to treat grade 3 and 4 CRS, but physicians are concerned that earlier introduction during the course of CRS may impair CAR T-cell function.
At present, no relevant preclinical model for CRS after CAR T-cell therapy exists, “which is limiting the development of CRS preventative modalities that could, in turn, enhance the feasibility of CAR T-cell therapy,” Dr Kenderian said.
And so the investigators decided to create an animal model.
Dr Kenderian described the work at the meeting as abstract 652.
Mouse model for human CRS
Using NSG-S mice (non-obese diabetic, SCID ɣ -/- mice additionally transgenic for human stem cell factor, IL-3, and GM-CSF), investigators injected them with blasts from AML patients. After 3 to 4 weeks, investigators treated the mice with 1 x 106 CD123-directed CAR T cells.
Dr Kenderian noted this dose of CART123 was 10 times higher than doses previously used in primary AML xenograft models.
The mice became weak, emaciated, developed hunched bodies, became withdrawn, had poor motor responses, and died in 7 to 10 days. The illness started within 1 week of CAR T-cell injection and correlated with significant expansion of T cells in the peripheral blood of these mice.
The team studied the serum from these mice 7 days after CART123 injection. They found extreme elevation of human IL-6, interferon-γ, tumor necrosis factor-α, and other inflammatory cytokines. This response resembled human CRS after CAR T-cell therapy.
Ruxolitinib treatment
The investigators first studied ruxolitinib activity in vitro with CART123 cells and found that ruxolitinib did not impair CAR T-cell effector functions.
“And also, ruxolitinib was not directly toxic to CAR T cells,” Dr Kenderian added.
But ruxolitinib did slow CAR T-cell proliferation in vitro.
They next tested ruxolitinib and CART123 in the mouse model.
Once the mice experienced high-burden disease, investigators treated them with CART123. That same day, investigators began treating the mice with ruxolitinib for 1 week. The mice were randomized to 30, 60, 90 mg/kg, or vehicle twice a day.
Twenty-nine days after AML injection, the mice treated with CART123 plus 90 mg or 60 mg of ruxolitinib experienced less weight loss than those treated with CART123 plus 30 mg of ruxolitinib or CART123-only.
“And more importantly, all mice had eradication of their disease,” Dr Kenderian said.
Mice treated with CART123 plus 90 mg, 60 mg, or 30 mg of ruxolitinib or CART123 alone had fewer AML blasts at day 28 than mice treated with 60 mg of ruxolitinib alone.
The investigators then analyzed the effect of ruxolitinib on the anti-tumor effect of CART123 and found that ruxolitinib did not impair it.
The attenuation of inflammatory cytokines translated to a survival advantage for mice treated with CART123 and ruxolitinib.
The investigators believe the addition of ruxolitinib to CAR T-cell therapy is a modality that should be investigated in patients at high-risk of developing CRS.
Dr Kenderian disclosed patents, royalties, and research funding from Novartis. ![]()
Photo courtesy of NCI
SAN DIEGO—A novel xenograft model of acute myeloid leukemia (AML) demonstrated that the JAK/STAT inhibitor ruxolitinib can prevent severe cytokine release syndrome (CRS) without impairing the anti-tumor effect of chimeric antigen receptor (CAR) T cells, according to research presented at the 2016 ASH Annual Meeting.
Almost all patients responding to CART-cell therapy develop CRS, and up to 60% develop severe CRS.
The research team believes the mouse model and findings with ruxolitinib will provide an important platform for studying CRS prevention and treatment.
At ASH, Saad Kenderian, MD, of the Mayo Clinic in Rochester, Minnesota, explained that CRS produces very high levels of the inflammatory protein IL-6.
Treatment with ruxolitinib in clinical studies has reduced human inflammatory cytokines. Therefore, it made sense to the investigators to study ruxolitinib as a means to prevent CRS after CAR T-cell therapy.
Tocilizumab has been used to treat grade 3 and 4 CRS, but physicians are concerned that earlier introduction during the course of CRS may impair CAR T-cell function.
At present, no relevant preclinical model for CRS after CAR T-cell therapy exists, “which is limiting the development of CRS preventative modalities that could, in turn, enhance the feasibility of CAR T-cell therapy,” Dr Kenderian said.
And so the investigators decided to create an animal model.
Dr Kenderian described the work at the meeting as abstract 652.
Mouse model for human CRS
Using NSG-S mice (non-obese diabetic, SCID ɣ -/- mice additionally transgenic for human stem cell factor, IL-3, and GM-CSF), investigators injected them with blasts from AML patients. After 3 to 4 weeks, investigators treated the mice with 1 x 106 CD123-directed CAR T cells.
Dr Kenderian noted this dose of CART123 was 10 times higher than doses previously used in primary AML xenograft models.
The mice became weak, emaciated, developed hunched bodies, became withdrawn, had poor motor responses, and died in 7 to 10 days. The illness started within 1 week of CAR T-cell injection and correlated with significant expansion of T cells in the peripheral blood of these mice.
The team studied the serum from these mice 7 days after CART123 injection. They found extreme elevation of human IL-6, interferon-γ, tumor necrosis factor-α, and other inflammatory cytokines. This response resembled human CRS after CAR T-cell therapy.
Ruxolitinib treatment
The investigators first studied ruxolitinib activity in vitro with CART123 cells and found that ruxolitinib did not impair CAR T-cell effector functions.
“And also, ruxolitinib was not directly toxic to CAR T cells,” Dr Kenderian added.
But ruxolitinib did slow CAR T-cell proliferation in vitro.
They next tested ruxolitinib and CART123 in the mouse model.
Once the mice experienced high-burden disease, investigators treated them with CART123. That same day, investigators began treating the mice with ruxolitinib for 1 week. The mice were randomized to 30, 60, 90 mg/kg, or vehicle twice a day.
Twenty-nine days after AML injection, the mice treated with CART123 plus 90 mg or 60 mg of ruxolitinib experienced less weight loss than those treated with CART123 plus 30 mg of ruxolitinib or CART123-only.
“And more importantly, all mice had eradication of their disease,” Dr Kenderian said.
Mice treated with CART123 plus 90 mg, 60 mg, or 30 mg of ruxolitinib or CART123 alone had fewer AML blasts at day 28 than mice treated with 60 mg of ruxolitinib alone.
The investigators then analyzed the effect of ruxolitinib on the anti-tumor effect of CART123 and found that ruxolitinib did not impair it.
The attenuation of inflammatory cytokines translated to a survival advantage for mice treated with CART123 and ruxolitinib.
The investigators believe the addition of ruxolitinib to CAR T-cell therapy is a modality that should be investigated in patients at high-risk of developing CRS.
Dr Kenderian disclosed patents, royalties, and research funding from Novartis. ![]()
MDS patients with mutated IDH2 benefit from enasidenib

Photo courtesy of ASH
SAN DIEGO—Daily treatment with enasidenib monotherapy in patients with mutated IDH2-positive myelodysplastic syndromes (MDS) induced responses in the majority of patients treated, according to a presentation at the 2016 ASH Annual Meeting.
The study was a portion of a larger phase 1/2 trial of the agent in patients with acute myeloid leukemia (AML) and other hematologic malignancies, so the subset was relatively small, numbering 17 patients.
Nevertheless, enasidenib was well tolerated and induced responses in these predominantly higher-risk patients.
Enasidenib (AG-221/CC-9007) is a selective, oral, potent inhibitor of mutant IDH2 (mIDH2), which produces 2-HG and thus alters DNA methylation and blocks cellular differentiation of hematopoietic progenitor cells.
Approximately 15% of AML patients and 5% of MDS patients have mIDH2. So investigators undertook the study to evaluate the safety and efficacy of enasidenib monotherapy in these diseases.
Eytan Stein, MD, of Memorial Sloan Kettering Cancer Center in New York, New York, presented the analysis of enasidenib in mIDH2-positive MDS patients as abstract 343.*
Study design
MDS patients were allowed to enroll during the dose-escalation and expansion phase of the study, Dr Stein explained.
Patients had to be 18 or older and have an advanced hematologic malignancy with mutated IDH2—relapsed or refractory AML, relapsed or refractory MDS, untreated AML, or other hematologic malignancy with mIDH2.
MDS patients could not be candidates for other therapies, had to be IPSS-R high risk, and had to have relapsed or refractory RAEB-1/RAEB-2 disease.
Investigators also performed co-molecular profiling using next-generation sequencing with a FoundationOne® Heme Panel.
All patients received daily oral enasidenib at 100 mg daily in 28-day cycles.
Patient characteristics
The study accrued a total of 239 patients—176 with relapsed or refractory AML, 37 with untreated AML, 9 with another hematologic malignancy, and 17 with MDS.
The median age of the MDS patients was 67 (range, 45-78), and 71% were male. All had the IDH2 mutation, 88% had R140 mutations, and 12% had R172.
Thirteen patients (76%) had an ECOG performance status of 0-1, and 4 (24%) had a performance status of 2.
A little more than a third (35%) of patients had 2 or more prior anti-cancer regimens.
Two patients (12%) received prior lenalidomide therapy, 8 (47%) received other treatments, including sorafenib (n=2) and vosaroxin, epoetin alfa, pracinostat, cytarabine plus clofarabine, ruxolitinib, and rigosertib (n=1 each). Four patients (24%) were untreated.
“I want to make the point,” Dr Stein said, “that, of those patients, three quarters of them, 76% [n=13], had received a prior hypomethylating agent, really understanding that this is a very poor-risk group of patients that we are studying here.”
About half of patients (47%) had intermediate-2/high IPSS risk status, good MDS cytogenetic risk, and high/very high IPSS-R risk status.
Adverse events
Grade 3-4 treatment-emergent adverse events (AEs) occurring in 2 or more patients were hyperbilirubinemia (n=5), pneumonia (n=4), thrombocytopenia (n=4), anemia (n=3), hypokalemia (n=3), dyspnea (n=2), and tumor lysis syndrome (n=2).
“As I’ve said in a number of meetings where I’ve talked about IDH2 inhibitors, and specifically enasidenib, the hyperbilirubinemia that is seen with this drug is an indirect hyperbilirubinemia,” Dr Stein said.
“A known off-target effect of this drug is inhibition of the UGT1a1 enzyme, which conjugates bilirubin, and this indirect hyperbilirubinemia, which is typically relatively mild [and] does not appear to have any clinical sequelae.”
Investigators considered 9 of the AEs reported for 6 patients to be drug-related.
Four serious enasidenib-related AEs included tumor lysis syndrome (n=2), increased blood bilirubin (n=1), and transaminitis (n=1).
There were no treatment-related deaths.
Response and survival
Ten of 17 patients (59%) achieved an overall response, defined as complete response (CR) plus partial response, plus marrow CR, plus hematologic improvement (HI).
One patient achieved CR, 1 had a partial response, 3 had marrow CR, and 5 had HI.
Dr Stein noted that, of the 13 patients who had received prior hypomethylating agent therapy, 7 (54%) had a response with enasidenib.
Of the patients who attained HI, 2 had trilineage and 2 had bilineage improvement.
The median time to response was 21 days (range, 10-87), and the median number of treatment cycles was 3.0.
Patients remained on study for up to 24 months. Four patients in hematologic remission are still on study, and 3 patients proceeded to stem cell or bone marrow transplant, Dr Stein said.
Two patients discontinued because of investigator decision, and 8 discontinued therapy due to death or disease progression.
The limitation of the study regarding overall survival, Dr Stein said, is that the number is very small.
At a median follow-up of 7.5 months, the median overall survival was not reached.
“So I’m not arguing that this is the end word of this,” he said. “This is going to be dynamic as more patients come on these studies. But I think it’s a nice indication that this treatment appears to be well-tolerated and doing good for a large subset of patients.”
Co-occurring mutations
The investigators also analyzed co-occurring mutations in 13 MDS patients.
The small patient population prevented the investigators from making definitive conclusions regarding potential correlations between response and co-mutations.
Nevertheless, Dr Stein said the analysis revealed something “very, very intriguing.”
He noted that 7 patients had ASXL1 mutations, and “those are typically patients who are bad actors.”
“Five of those 7 patients had a response,” Dr Stein said. “And of those 5, 3 of them had received a prior hypomethylating agent. I think it’s at least food for thought that you can salvage a patient who has failed a hypomethylating agent, with poor risk disease. I think this is very, very exciting.”
Celgene Corporation and its collaborator, Agios Pharmaceuticals, sponsored the trial. ![]()
*Information in the abstract differs from the presentation.
Photo courtesy of ASH
SAN DIEGO—Daily treatment with enasidenib monotherapy in patients with mutated IDH2-positive myelodysplastic syndromes (MDS) induced responses in the majority of patients treated, according to a presentation at the 2016 ASH Annual Meeting.
The study was a portion of a larger phase 1/2 trial of the agent in patients with acute myeloid leukemia (AML) and other hematologic malignancies, so the subset was relatively small, numbering 17 patients.
Nevertheless, enasidenib was well tolerated and induced responses in these predominantly higher-risk patients.
Enasidenib (AG-221/CC-9007) is a selective, oral, potent inhibitor of mutant IDH2 (mIDH2), which produces 2-HG and thus alters DNA methylation and blocks cellular differentiation of hematopoietic progenitor cells.
Approximately 15% of AML patients and 5% of MDS patients have mIDH2. So investigators undertook the study to evaluate the safety and efficacy of enasidenib monotherapy in these diseases.
Eytan Stein, MD, of Memorial Sloan Kettering Cancer Center in New York, New York, presented the analysis of enasidenib in mIDH2-positive MDS patients as abstract 343.*
Study design
MDS patients were allowed to enroll during the dose-escalation and expansion phase of the study, Dr Stein explained.
Patients had to be 18 or older and have an advanced hematologic malignancy with mutated IDH2—relapsed or refractory AML, relapsed or refractory MDS, untreated AML, or other hematologic malignancy with mIDH2.
MDS patients could not be candidates for other therapies, had to be IPSS-R high risk, and had to have relapsed or refractory RAEB-1/RAEB-2 disease.
Investigators also performed co-molecular profiling using next-generation sequencing with a FoundationOne® Heme Panel.
All patients received daily oral enasidenib at 100 mg daily in 28-day cycles.
Patient characteristics
The study accrued a total of 239 patients—176 with relapsed or refractory AML, 37 with untreated AML, 9 with another hematologic malignancy, and 17 with MDS.
The median age of the MDS patients was 67 (range, 45-78), and 71% were male. All had the IDH2 mutation, 88% had R140 mutations, and 12% had R172.
Thirteen patients (76%) had an ECOG performance status of 0-1, and 4 (24%) had a performance status of 2.
A little more than a third (35%) of patients had 2 or more prior anti-cancer regimens.
Two patients (12%) received prior lenalidomide therapy, 8 (47%) received other treatments, including sorafenib (n=2) and vosaroxin, epoetin alfa, pracinostat, cytarabine plus clofarabine, ruxolitinib, and rigosertib (n=1 each). Four patients (24%) were untreated.
“I want to make the point,” Dr Stein said, “that, of those patients, three quarters of them, 76% [n=13], had received a prior hypomethylating agent, really understanding that this is a very poor-risk group of patients that we are studying here.”
About half of patients (47%) had intermediate-2/high IPSS risk status, good MDS cytogenetic risk, and high/very high IPSS-R risk status.
Adverse events
Grade 3-4 treatment-emergent adverse events (AEs) occurring in 2 or more patients were hyperbilirubinemia (n=5), pneumonia (n=4), thrombocytopenia (n=4), anemia (n=3), hypokalemia (n=3), dyspnea (n=2), and tumor lysis syndrome (n=2).
“As I’ve said in a number of meetings where I’ve talked about IDH2 inhibitors, and specifically enasidenib, the hyperbilirubinemia that is seen with this drug is an indirect hyperbilirubinemia,” Dr Stein said.
“A known off-target effect of this drug is inhibition of the UGT1a1 enzyme, which conjugates bilirubin, and this indirect hyperbilirubinemia, which is typically relatively mild [and] does not appear to have any clinical sequelae.”
Investigators considered 9 of the AEs reported for 6 patients to be drug-related.
Four serious enasidenib-related AEs included tumor lysis syndrome (n=2), increased blood bilirubin (n=1), and transaminitis (n=1).
There were no treatment-related deaths.
Response and survival
Ten of 17 patients (59%) achieved an overall response, defined as complete response (CR) plus partial response, plus marrow CR, plus hematologic improvement (HI).
One patient achieved CR, 1 had a partial response, 3 had marrow CR, and 5 had HI.
Dr Stein noted that, of the 13 patients who had received prior hypomethylating agent therapy, 7 (54%) had a response with enasidenib.
Of the patients who attained HI, 2 had trilineage and 2 had bilineage improvement.
The median time to response was 21 days (range, 10-87), and the median number of treatment cycles was 3.0.
Patients remained on study for up to 24 months. Four patients in hematologic remission are still on study, and 3 patients proceeded to stem cell or bone marrow transplant, Dr Stein said.
Two patients discontinued because of investigator decision, and 8 discontinued therapy due to death or disease progression.
The limitation of the study regarding overall survival, Dr Stein said, is that the number is very small.
At a median follow-up of 7.5 months, the median overall survival was not reached.
“So I’m not arguing that this is the end word of this,” he said. “This is going to be dynamic as more patients come on these studies. But I think it’s a nice indication that this treatment appears to be well-tolerated and doing good for a large subset of patients.”
Co-occurring mutations
The investigators also analyzed co-occurring mutations in 13 MDS patients.
The small patient population prevented the investigators from making definitive conclusions regarding potential correlations between response and co-mutations.
Nevertheless, Dr Stein said the analysis revealed something “very, very intriguing.”
He noted that 7 patients had ASXL1 mutations, and “those are typically patients who are bad actors.”
“Five of those 7 patients had a response,” Dr Stein said. “And of those 5, 3 of them had received a prior hypomethylating agent. I think it’s at least food for thought that you can salvage a patient who has failed a hypomethylating agent, with poor risk disease. I think this is very, very exciting.”
Celgene Corporation and its collaborator, Agios Pharmaceuticals, sponsored the trial. ![]()
*Information in the abstract differs from the presentation.
Photo courtesy of ASH
SAN DIEGO—Daily treatment with enasidenib monotherapy in patients with mutated IDH2-positive myelodysplastic syndromes (MDS) induced responses in the majority of patients treated, according to a presentation at the 2016 ASH Annual Meeting.
The study was a portion of a larger phase 1/2 trial of the agent in patients with acute myeloid leukemia (AML) and other hematologic malignancies, so the subset was relatively small, numbering 17 patients.
Nevertheless, enasidenib was well tolerated and induced responses in these predominantly higher-risk patients.
Enasidenib (AG-221/CC-9007) is a selective, oral, potent inhibitor of mutant IDH2 (mIDH2), which produces 2-HG and thus alters DNA methylation and blocks cellular differentiation of hematopoietic progenitor cells.
Approximately 15% of AML patients and 5% of MDS patients have mIDH2. So investigators undertook the study to evaluate the safety and efficacy of enasidenib monotherapy in these diseases.
Eytan Stein, MD, of Memorial Sloan Kettering Cancer Center in New York, New York, presented the analysis of enasidenib in mIDH2-positive MDS patients as abstract 343.*
Study design
MDS patients were allowed to enroll during the dose-escalation and expansion phase of the study, Dr Stein explained.
Patients had to be 18 or older and have an advanced hematologic malignancy with mutated IDH2—relapsed or refractory AML, relapsed or refractory MDS, untreated AML, or other hematologic malignancy with mIDH2.
MDS patients could not be candidates for other therapies, had to be IPSS-R high risk, and had to have relapsed or refractory RAEB-1/RAEB-2 disease.
Investigators also performed co-molecular profiling using next-generation sequencing with a FoundationOne® Heme Panel.
All patients received daily oral enasidenib at 100 mg daily in 28-day cycles.
Patient characteristics
The study accrued a total of 239 patients—176 with relapsed or refractory AML, 37 with untreated AML, 9 with another hematologic malignancy, and 17 with MDS.
The median age of the MDS patients was 67 (range, 45-78), and 71% were male. All had the IDH2 mutation, 88% had R140 mutations, and 12% had R172.
Thirteen patients (76%) had an ECOG performance status of 0-1, and 4 (24%) had a performance status of 2.
A little more than a third (35%) of patients had 2 or more prior anti-cancer regimens.
Two patients (12%) received prior lenalidomide therapy, 8 (47%) received other treatments, including sorafenib (n=2) and vosaroxin, epoetin alfa, pracinostat, cytarabine plus clofarabine, ruxolitinib, and rigosertib (n=1 each). Four patients (24%) were untreated.
“I want to make the point,” Dr Stein said, “that, of those patients, three quarters of them, 76% [n=13], had received a prior hypomethylating agent, really understanding that this is a very poor-risk group of patients that we are studying here.”
About half of patients (47%) had intermediate-2/high IPSS risk status, good MDS cytogenetic risk, and high/very high IPSS-R risk status.
Adverse events
Grade 3-4 treatment-emergent adverse events (AEs) occurring in 2 or more patients were hyperbilirubinemia (n=5), pneumonia (n=4), thrombocytopenia (n=4), anemia (n=3), hypokalemia (n=3), dyspnea (n=2), and tumor lysis syndrome (n=2).
“As I’ve said in a number of meetings where I’ve talked about IDH2 inhibitors, and specifically enasidenib, the hyperbilirubinemia that is seen with this drug is an indirect hyperbilirubinemia,” Dr Stein said.
“A known off-target effect of this drug is inhibition of the UGT1a1 enzyme, which conjugates bilirubin, and this indirect hyperbilirubinemia, which is typically relatively mild [and] does not appear to have any clinical sequelae.”
Investigators considered 9 of the AEs reported for 6 patients to be drug-related.
Four serious enasidenib-related AEs included tumor lysis syndrome (n=2), increased blood bilirubin (n=1), and transaminitis (n=1).
There were no treatment-related deaths.
Response and survival
Ten of 17 patients (59%) achieved an overall response, defined as complete response (CR) plus partial response, plus marrow CR, plus hematologic improvement (HI).
One patient achieved CR, 1 had a partial response, 3 had marrow CR, and 5 had HI.
Dr Stein noted that, of the 13 patients who had received prior hypomethylating agent therapy, 7 (54%) had a response with enasidenib.
Of the patients who attained HI, 2 had trilineage and 2 had bilineage improvement.
The median time to response was 21 days (range, 10-87), and the median number of treatment cycles was 3.0.
Patients remained on study for up to 24 months. Four patients in hematologic remission are still on study, and 3 patients proceeded to stem cell or bone marrow transplant, Dr Stein said.
Two patients discontinued because of investigator decision, and 8 discontinued therapy due to death or disease progression.
The limitation of the study regarding overall survival, Dr Stein said, is that the number is very small.
At a median follow-up of 7.5 months, the median overall survival was not reached.
“So I’m not arguing that this is the end word of this,” he said. “This is going to be dynamic as more patients come on these studies. But I think it’s a nice indication that this treatment appears to be well-tolerated and doing good for a large subset of patients.”
Co-occurring mutations
The investigators also analyzed co-occurring mutations in 13 MDS patients.
The small patient population prevented the investigators from making definitive conclusions regarding potential correlations between response and co-mutations.
Nevertheless, Dr Stein said the analysis revealed something “very, very intriguing.”
He noted that 7 patients had ASXL1 mutations, and “those are typically patients who are bad actors.”
“Five of those 7 patients had a response,” Dr Stein said. “And of those 5, 3 of them had received a prior hypomethylating agent. I think it’s at least food for thought that you can salvage a patient who has failed a hypomethylating agent, with poor risk disease. I think this is very, very exciting.”
Celgene Corporation and its collaborator, Agios Pharmaceuticals, sponsored the trial. ![]()
*Information in the abstract differs from the presentation.
Combo improves ORR, PFS in relapsed/refractory CLL

Image by Mary Ann Thompson
Adding an anti-CD37 molecule to treatment with bendamustine can improve outcomes in patients with relapsed/refractory chronic lymphocytic leukemia (CLL), according to research published in the British Journal of Haematology.
In a phase 2 trial, researchers found that combining the anti-CD37 molecule otlertuzumab with bendamustine significantly improved overall response rates (ORRs) and progression-free survival (PFS) when compared to bendamustine alone.
“We’re very encouraged by the phase 2 data, which demonstrated a significant increase in median progression-free survival, from approximately 10 to 16 months in patients receiving combination otlertuzumab/bendamustine therapy,” said Marvin L. White, president and chief executive officer of Aptevo Therapeutics Inc, the company developing otlertuzumab.
“These data, coupled with additional results from ongoing studies of otlertuzumab used in combination with current CLL therapies, should help position otlertuzumab for a potential partnership to advance into phase 3 clinical development.”
The phase 2 trial was sponsored by Emergent Product Development Seattle LLC. Aptevo Therapeutics is a spin-off of Emergent Biosolutions.
About otlertuzumab
Otlertuzumab (formerly TRU-016) is a humanized, monospecific ADAPTIR™ molecule that targets CD37.
Aptevo Therapeutics says the company is applying its ADAPTIR technology to develop immuno-oncology candidates that focus on redirected T-cell cytotoxicity. ADAPTIR technology can be used to generate immunotherapeutics with unique mechanisms of action, including targeted cytokine delivery, targeting 2 cell surface receptors, or neutralization of multiple soluble proteins.
According to Aptevo, otlertuzumab mediates death of CD37-expressing cells through various mechanisms, including direct cell death, antibody-dependent cell-mediated cytotoxicity, and phagocytosis. Otlertuzumab is being investigated as part of combination therapies for the treatment of CLL.
Study design
This phase 2 study enrolled 65 patients with relapsed/refractory CLL—32 who received a combination of otlertuzumab and bendamustine and 33 who received bendamustine alone.
Patients in the combination arm received otlertuzumab at 20 mg/kg weekly by intravenous infusion for two 28-day cycles, then every 14 days for four 28-day cycles.
Patients in both arms received intravenous bendamustine at 70 mg/m2 on days 1 and 2 of each cycle for up to six 28-day cycles. Dosing was adjusted according to neutrophil and platelet counts.
The study’s primary endpoint was ORR (per IWCLL criteria), and secondary endpoints included PFS and safety.
Patient characteristics
The researchers said the treatment arms were generally well balanced. However, patients in the combination arm were older, had more prior treatment regimens, a longer time from diagnosis, and more bulky disease. More patients in the control arm were Rai stage III or IV.
Two patients in the combination arm and 5 in the control arm had 17p deletion. Four patients in the combination arm and 6 in the control arm had TP53 mutations.
There were 5 patients in the combination arm and 3 in the control arm who were refractory to their prior treatment.
In both arms, patients received a median of 6 cycles of study treatment. Bendamustine exposure was similar between the arms—a median of 143 days. The median treatment duration for otlertuzumab was 156 days.
Seven patients (22%) in the combination arm and 12 (36%) in the control arm discontinued treatment early.
In the combination arm, 3 patients discontinued due to adverse events (AEs), 3 due to disease progression, and 1 patient withdrew to have a stem cell transplant.
In the control arm, 7 patients discontinued due to AEs, 3 due to progression, 1 withdrew for an unspecified reason, and 1 patient died of acute heart failure.
Response and survival
The ORR was 69% in the combination arm and 39% in the control arm (P=0.025).
In the combination arm, 3 patients (9%) had a complete response (CR), 1 patient had a CR with incomplete marrow recovery, and 19 (59%) had a partial response.
In the control arm, 1 patient (3%) had a CR and 12 (36%) had a partial response.
The median PFS was 15.9 months in the combination arm and 10.2 months in the control arm (P=0.0192).
The median overall survival was not reached in either arm. After 2 years of follow-up, there were no deaths in the combination arm and 3 deaths in the control arm.
Safety
Ninety-one percent of patients in the combination arm and 100% of those in the control arm experienced an AE. Serious AEs occurred in 31% and 45%, respectively.
Severe neutropenia was more frequent in the combination arm than the control arm (56% vs 39%), as was severe thrombocytopenia (19% vs 15%).
However, there were fewer grade 3/4 infections in the combination arm than the control arm (13% vs 27%). And 2 patients in the control arm had febrile neutropenia, but there were no cases in the combination arm.
“These latest data show the combination of otlertuzumab and bendamustine is well tolerated and significantly increases the response rate and PFS in patients with relapsed or refractory CLL,” said Scott Stromatt, MD, study director and chief medical officer for Aptevo.
“Consequently, we are now exploring the utility of otlertuzumab in combination with additional CLL therapies to evaluate clinical benefit in distinct CLL patient subgroups.” ![]()
Image by Mary Ann Thompson
Adding an anti-CD37 molecule to treatment with bendamustine can improve outcomes in patients with relapsed/refractory chronic lymphocytic leukemia (CLL), according to research published in the British Journal of Haematology.
In a phase 2 trial, researchers found that combining the anti-CD37 molecule otlertuzumab with bendamustine significantly improved overall response rates (ORRs) and progression-free survival (PFS) when compared to bendamustine alone.
“We’re very encouraged by the phase 2 data, which demonstrated a significant increase in median progression-free survival, from approximately 10 to 16 months in patients receiving combination otlertuzumab/bendamustine therapy,” said Marvin L. White, president and chief executive officer of Aptevo Therapeutics Inc, the company developing otlertuzumab.
“These data, coupled with additional results from ongoing studies of otlertuzumab used in combination with current CLL therapies, should help position otlertuzumab for a potential partnership to advance into phase 3 clinical development.”
The phase 2 trial was sponsored by Emergent Product Development Seattle LLC. Aptevo Therapeutics is a spin-off of Emergent Biosolutions.
About otlertuzumab
Otlertuzumab (formerly TRU-016) is a humanized, monospecific ADAPTIR™ molecule that targets CD37.
Aptevo Therapeutics says the company is applying its ADAPTIR technology to develop immuno-oncology candidates that focus on redirected T-cell cytotoxicity. ADAPTIR technology can be used to generate immunotherapeutics with unique mechanisms of action, including targeted cytokine delivery, targeting 2 cell surface receptors, or neutralization of multiple soluble proteins.
According to Aptevo, otlertuzumab mediates death of CD37-expressing cells through various mechanisms, including direct cell death, antibody-dependent cell-mediated cytotoxicity, and phagocytosis. Otlertuzumab is being investigated as part of combination therapies for the treatment of CLL.
Study design
This phase 2 study enrolled 65 patients with relapsed/refractory CLL—32 who received a combination of otlertuzumab and bendamustine and 33 who received bendamustine alone.
Patients in the combination arm received otlertuzumab at 20 mg/kg weekly by intravenous infusion for two 28-day cycles, then every 14 days for four 28-day cycles.
Patients in both arms received intravenous bendamustine at 70 mg/m2 on days 1 and 2 of each cycle for up to six 28-day cycles. Dosing was adjusted according to neutrophil and platelet counts.
The study’s primary endpoint was ORR (per IWCLL criteria), and secondary endpoints included PFS and safety.
Patient characteristics
The researchers said the treatment arms were generally well balanced. However, patients in the combination arm were older, had more prior treatment regimens, a longer time from diagnosis, and more bulky disease. More patients in the control arm were Rai stage III or IV.
Two patients in the combination arm and 5 in the control arm had 17p deletion. Four patients in the combination arm and 6 in the control arm had TP53 mutations.
There were 5 patients in the combination arm and 3 in the control arm who were refractory to their prior treatment.
In both arms, patients received a median of 6 cycles of study treatment. Bendamustine exposure was similar between the arms—a median of 143 days. The median treatment duration for otlertuzumab was 156 days.
Seven patients (22%) in the combination arm and 12 (36%) in the control arm discontinued treatment early.
In the combination arm, 3 patients discontinued due to adverse events (AEs), 3 due to disease progression, and 1 patient withdrew to have a stem cell transplant.
In the control arm, 7 patients discontinued due to AEs, 3 due to progression, 1 withdrew for an unspecified reason, and 1 patient died of acute heart failure.
Response and survival
The ORR was 69% in the combination arm and 39% in the control arm (P=0.025).
In the combination arm, 3 patients (9%) had a complete response (CR), 1 patient had a CR with incomplete marrow recovery, and 19 (59%) had a partial response.
In the control arm, 1 patient (3%) had a CR and 12 (36%) had a partial response.
The median PFS was 15.9 months in the combination arm and 10.2 months in the control arm (P=0.0192).
The median overall survival was not reached in either arm. After 2 years of follow-up, there were no deaths in the combination arm and 3 deaths in the control arm.
Safety
Ninety-one percent of patients in the combination arm and 100% of those in the control arm experienced an AE. Serious AEs occurred in 31% and 45%, respectively.
Severe neutropenia was more frequent in the combination arm than the control arm (56% vs 39%), as was severe thrombocytopenia (19% vs 15%).
However, there were fewer grade 3/4 infections in the combination arm than the control arm (13% vs 27%). And 2 patients in the control arm had febrile neutropenia, but there were no cases in the combination arm.
“These latest data show the combination of otlertuzumab and bendamustine is well tolerated and significantly increases the response rate and PFS in patients with relapsed or refractory CLL,” said Scott Stromatt, MD, study director and chief medical officer for Aptevo.
“Consequently, we are now exploring the utility of otlertuzumab in combination with additional CLL therapies to evaluate clinical benefit in distinct CLL patient subgroups.” ![]()
Image by Mary Ann Thompson
Adding an anti-CD37 molecule to treatment with bendamustine can improve outcomes in patients with relapsed/refractory chronic lymphocytic leukemia (CLL), according to research published in the British Journal of Haematology.
In a phase 2 trial, researchers found that combining the anti-CD37 molecule otlertuzumab with bendamustine significantly improved overall response rates (ORRs) and progression-free survival (PFS) when compared to bendamustine alone.
“We’re very encouraged by the phase 2 data, which demonstrated a significant increase in median progression-free survival, from approximately 10 to 16 months in patients receiving combination otlertuzumab/bendamustine therapy,” said Marvin L. White, president and chief executive officer of Aptevo Therapeutics Inc, the company developing otlertuzumab.
“These data, coupled with additional results from ongoing studies of otlertuzumab used in combination with current CLL therapies, should help position otlertuzumab for a potential partnership to advance into phase 3 clinical development.”
The phase 2 trial was sponsored by Emergent Product Development Seattle LLC. Aptevo Therapeutics is a spin-off of Emergent Biosolutions.
About otlertuzumab
Otlertuzumab (formerly TRU-016) is a humanized, monospecific ADAPTIR™ molecule that targets CD37.
Aptevo Therapeutics says the company is applying its ADAPTIR technology to develop immuno-oncology candidates that focus on redirected T-cell cytotoxicity. ADAPTIR technology can be used to generate immunotherapeutics with unique mechanisms of action, including targeted cytokine delivery, targeting 2 cell surface receptors, or neutralization of multiple soluble proteins.
According to Aptevo, otlertuzumab mediates death of CD37-expressing cells through various mechanisms, including direct cell death, antibody-dependent cell-mediated cytotoxicity, and phagocytosis. Otlertuzumab is being investigated as part of combination therapies for the treatment of CLL.
Study design
This phase 2 study enrolled 65 patients with relapsed/refractory CLL—32 who received a combination of otlertuzumab and bendamustine and 33 who received bendamustine alone.
Patients in the combination arm received otlertuzumab at 20 mg/kg weekly by intravenous infusion for two 28-day cycles, then every 14 days for four 28-day cycles.
Patients in both arms received intravenous bendamustine at 70 mg/m2 on days 1 and 2 of each cycle for up to six 28-day cycles. Dosing was adjusted according to neutrophil and platelet counts.
The study’s primary endpoint was ORR (per IWCLL criteria), and secondary endpoints included PFS and safety.
Patient characteristics
The researchers said the treatment arms were generally well balanced. However, patients in the combination arm were older, had more prior treatment regimens, a longer time from diagnosis, and more bulky disease. More patients in the control arm were Rai stage III or IV.
Two patients in the combination arm and 5 in the control arm had 17p deletion. Four patients in the combination arm and 6 in the control arm had TP53 mutations.
There were 5 patients in the combination arm and 3 in the control arm who were refractory to their prior treatment.
In both arms, patients received a median of 6 cycles of study treatment. Bendamustine exposure was similar between the arms—a median of 143 days. The median treatment duration for otlertuzumab was 156 days.
Seven patients (22%) in the combination arm and 12 (36%) in the control arm discontinued treatment early.
In the combination arm, 3 patients discontinued due to adverse events (AEs), 3 due to disease progression, and 1 patient withdrew to have a stem cell transplant.
In the control arm, 7 patients discontinued due to AEs, 3 due to progression, 1 withdrew for an unspecified reason, and 1 patient died of acute heart failure.
Response and survival
The ORR was 69% in the combination arm and 39% in the control arm (P=0.025).
In the combination arm, 3 patients (9%) had a complete response (CR), 1 patient had a CR with incomplete marrow recovery, and 19 (59%) had a partial response.
In the control arm, 1 patient (3%) had a CR and 12 (36%) had a partial response.
The median PFS was 15.9 months in the combination arm and 10.2 months in the control arm (P=0.0192).
The median overall survival was not reached in either arm. After 2 years of follow-up, there were no deaths in the combination arm and 3 deaths in the control arm.
Safety
Ninety-one percent of patients in the combination arm and 100% of those in the control arm experienced an AE. Serious AEs occurred in 31% and 45%, respectively.
Severe neutropenia was more frequent in the combination arm than the control arm (56% vs 39%), as was severe thrombocytopenia (19% vs 15%).
However, there were fewer grade 3/4 infections in the combination arm than the control arm (13% vs 27%). And 2 patients in the control arm had febrile neutropenia, but there were no cases in the combination arm.
“These latest data show the combination of otlertuzumab and bendamustine is well tolerated and significantly increases the response rate and PFS in patients with relapsed or refractory CLL,” said Scott Stromatt, MD, study director and chief medical officer for Aptevo.
“Consequently, we are now exploring the utility of otlertuzumab in combination with additional CLL therapies to evaluate clinical benefit in distinct CLL patient subgroups.” ![]()
CHMP recommends authorization of rituximab biosimilar

Photo by Linda Bartlett
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended that the rituximab biosimilar Truxima receive marketing authorization for the treatment of several conditions.
The CHMP said studies have shown that Truxima is comparable to the reference product, Mabthera, which was authorized for use in the European Union in 1998.
The active substance in both products is the anti-CD20 monoclonal antibody rituximab.
Truxima is being developed by Celltrion Healthcare Hungary Kft.
The CHMP’s recommendation for Truxima will be reviewed by the European Commission, which normally issues its decision on a product within 67 days of the time the CHMP adopts its opinion.
If the European Commission grants marketing authorization for Truxima, it will be available as a 500 mg concentrate for solution for infusion.
The CHMP has recommended marketing authorization for Truxima in the treatment of non-Hodgkin lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, and granulomatosis with polyangiitis and microscopic polyangiitis.
The full indications are as follows.
Non-Hodgkin lymphoma
Truxima is indicated for use in combination with chemotherapy to treat previously untreated patients with stage III-IV follicular lymphoma.
Truxima maintenance therapy is indicated for the treatment of follicular lymphoma patients responding to induction therapy.
Truxima monotherapy is indicated for treatment of patients with stage III-IV follicular lymphoma who are chemo-resistant or are in their second or subsequent relapse after chemotherapy.
Truxima is indicated for use in combination with CHOP (cyclophosphamide, doxorubicin, vincristine, prednisolone) for the treatment of patients with CD20-positive diffuse large B-cell lymphoma.
Chronic lymphocytic leukemia
Truxima in combination with chemotherapy is indicated for the treatment of patients with previously untreated and relapsed/refractory chronic lymphocytic leukemia.
The CHMP noted that only limited data are available on efficacy and safety for patients previously treated with monoclonal antibodies, including Truxima, or patients refractory to previous Truxima plus chemotherapy.
Rheumatoid arthritis
Truxima in combination with methotrexate is indicated for the treatment of adults with severe active rheumatoid arthritis who have had an inadequate response or cannot tolerate other disease-modifying anti-rheumatic drugs, including one or more tumor necrosis factor inhibitor therapies.
The CHMP noted that Truxima has been shown to reduce the rate of progression of joint damage as measured by X-ray and to improve physical function, when given in combination with methotrexate.
Granulomatosis with polyangiitis and microscopic polyangiitis
Truxima in combination with glucocorticoids is indicated for the induction of remission in adults with severe, active granulomatosis with polyangiitis and microscopic polyangiitis.
The CHMP proposed that Truxima be administered under the close supervision of an experienced healthcare professional and in an environment where full resuscitation facilities are immediately available. ![]()
Photo by Linda Bartlett
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended that the rituximab biosimilar Truxima receive marketing authorization for the treatment of several conditions.
The CHMP said studies have shown that Truxima is comparable to the reference product, Mabthera, which was authorized for use in the European Union in 1998.
The active substance in both products is the anti-CD20 monoclonal antibody rituximab.
Truxima is being developed by Celltrion Healthcare Hungary Kft.
The CHMP’s recommendation for Truxima will be reviewed by the European Commission, which normally issues its decision on a product within 67 days of the time the CHMP adopts its opinion.
If the European Commission grants marketing authorization for Truxima, it will be available as a 500 mg concentrate for solution for infusion.
The CHMP has recommended marketing authorization for Truxima in the treatment of non-Hodgkin lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, and granulomatosis with polyangiitis and microscopic polyangiitis.
The full indications are as follows.
Non-Hodgkin lymphoma
Truxima is indicated for use in combination with chemotherapy to treat previously untreated patients with stage III-IV follicular lymphoma.
Truxima maintenance therapy is indicated for the treatment of follicular lymphoma patients responding to induction therapy.
Truxima monotherapy is indicated for treatment of patients with stage III-IV follicular lymphoma who are chemo-resistant or are in their second or subsequent relapse after chemotherapy.
Truxima is indicated for use in combination with CHOP (cyclophosphamide, doxorubicin, vincristine, prednisolone) for the treatment of patients with CD20-positive diffuse large B-cell lymphoma.
Chronic lymphocytic leukemia
Truxima in combination with chemotherapy is indicated for the treatment of patients with previously untreated and relapsed/refractory chronic lymphocytic leukemia.
The CHMP noted that only limited data are available on efficacy and safety for patients previously treated with monoclonal antibodies, including Truxima, or patients refractory to previous Truxima plus chemotherapy.
Rheumatoid arthritis
Truxima in combination with methotrexate is indicated for the treatment of adults with severe active rheumatoid arthritis who have had an inadequate response or cannot tolerate other disease-modifying anti-rheumatic drugs, including one or more tumor necrosis factor inhibitor therapies.
The CHMP noted that Truxima has been shown to reduce the rate of progression of joint damage as measured by X-ray and to improve physical function, when given in combination with methotrexate.
Granulomatosis with polyangiitis and microscopic polyangiitis
Truxima in combination with glucocorticoids is indicated for the induction of remission in adults with severe, active granulomatosis with polyangiitis and microscopic polyangiitis.
The CHMP proposed that Truxima be administered under the close supervision of an experienced healthcare professional and in an environment where full resuscitation facilities are immediately available. ![]()
Photo by Linda Bartlett
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended that the rituximab biosimilar Truxima receive marketing authorization for the treatment of several conditions.
The CHMP said studies have shown that Truxima is comparable to the reference product, Mabthera, which was authorized for use in the European Union in 1998.
The active substance in both products is the anti-CD20 monoclonal antibody rituximab.
Truxima is being developed by Celltrion Healthcare Hungary Kft.
The CHMP’s recommendation for Truxima will be reviewed by the European Commission, which normally issues its decision on a product within 67 days of the time the CHMP adopts its opinion.
If the European Commission grants marketing authorization for Truxima, it will be available as a 500 mg concentrate for solution for infusion.
The CHMP has recommended marketing authorization for Truxima in the treatment of non-Hodgkin lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, and granulomatosis with polyangiitis and microscopic polyangiitis.
The full indications are as follows.
Non-Hodgkin lymphoma
Truxima is indicated for use in combination with chemotherapy to treat previously untreated patients with stage III-IV follicular lymphoma.
Truxima maintenance therapy is indicated for the treatment of follicular lymphoma patients responding to induction therapy.
Truxima monotherapy is indicated for treatment of patients with stage III-IV follicular lymphoma who are chemo-resistant or are in their second or subsequent relapse after chemotherapy.
Truxima is indicated for use in combination with CHOP (cyclophosphamide, doxorubicin, vincristine, prednisolone) for the treatment of patients with CD20-positive diffuse large B-cell lymphoma.
Chronic lymphocytic leukemia
Truxima in combination with chemotherapy is indicated for the treatment of patients with previously untreated and relapsed/refractory chronic lymphocytic leukemia.
The CHMP noted that only limited data are available on efficacy and safety for patients previously treated with monoclonal antibodies, including Truxima, or patients refractory to previous Truxima plus chemotherapy.
Rheumatoid arthritis
Truxima in combination with methotrexate is indicated for the treatment of adults with severe active rheumatoid arthritis who have had an inadequate response or cannot tolerate other disease-modifying anti-rheumatic drugs, including one or more tumor necrosis factor inhibitor therapies.
The CHMP noted that Truxima has been shown to reduce the rate of progression of joint damage as measured by X-ray and to improve physical function, when given in combination with methotrexate.
Granulomatosis with polyangiitis and microscopic polyangiitis
Truxima in combination with glucocorticoids is indicated for the induction of remission in adults with severe, active granulomatosis with polyangiitis and microscopic polyangiitis.
The CHMP proposed that Truxima be administered under the close supervision of an experienced healthcare professional and in an environment where full resuscitation facilities are immediately available. ![]()
Congenital CMV linked to increased risk of ALL

Photo by Vera Kratochvil
Newborns with congenital cytomegalovirus (CMV) infection may have an increased risk of developing acute lymphoblastic leukemia (ALL), according to a study published in Blood.
The data also suggest the risk may be particularly high among Hispanic children.
Researchers said this is the first study to suggest that congenital CMV infection is a risk factor for childhood ALL and is more prominent in Hispanic children.
To conduct this study, the researchers first identified all known infections present in the bone marrow of 127 children diagnosed with ALL and 38 children diagnosed with acute myeloid leukemia (AML).
The team found CMV infection was prevalent in children with ALL but rare in those with AML.
Next, the researchers looked for CMV in newborn blood samples from 268 children who went on to develop ALL. The team compared the samples with samples from 270 healthy children.
“Our goal in tracking CMV back from the time of diagnosis to the womb was to establish that this infection occurred well before initiation of disease,” said lead study author Stephen Francis, PhD, of the University of Nevada and University of California, San Francisco.
He and his colleagues found that children who went on to develop ALL were nearly 4 times more likely than control subjects to be CMV-positive at birth. The odds ratio was 3.71 (P=0.0016).
The odds ratio was 5.9 in Hispanic children and 2.1 in non-Hispanic whites. The researchers said this finding is particularly interesting because of the high rate of ALL observed in Hispanics.
“If it’s true that in utero CMV is one of the initiating events in the development of childhood leukemia, then control of the virus has the potential to be a prevention target,” Dr Francis said. “That’s the real take-home message.”
While this research is in the early stages, the researchers hope these results will inspire more studies that will validate these findings and lead to the development of a CMV vaccine.
“This is the first step, but if we do end up finding a causal link to the most common childhood cancer, we hope that will light a fire in terms of stopping mother-to-child transmission of CMV,” Dr Francis said. ![]()
Photo by Vera Kratochvil
Newborns with congenital cytomegalovirus (CMV) infection may have an increased risk of developing acute lymphoblastic leukemia (ALL), according to a study published in Blood.
The data also suggest the risk may be particularly high among Hispanic children.
Researchers said this is the first study to suggest that congenital CMV infection is a risk factor for childhood ALL and is more prominent in Hispanic children.
To conduct this study, the researchers first identified all known infections present in the bone marrow of 127 children diagnosed with ALL and 38 children diagnosed with acute myeloid leukemia (AML).
The team found CMV infection was prevalent in children with ALL but rare in those with AML.
Next, the researchers looked for CMV in newborn blood samples from 268 children who went on to develop ALL. The team compared the samples with samples from 270 healthy children.
“Our goal in tracking CMV back from the time of diagnosis to the womb was to establish that this infection occurred well before initiation of disease,” said lead study author Stephen Francis, PhD, of the University of Nevada and University of California, San Francisco.
He and his colleagues found that children who went on to develop ALL were nearly 4 times more likely than control subjects to be CMV-positive at birth. The odds ratio was 3.71 (P=0.0016).
The odds ratio was 5.9 in Hispanic children and 2.1 in non-Hispanic whites. The researchers said this finding is particularly interesting because of the high rate of ALL observed in Hispanics.
“If it’s true that in utero CMV is one of the initiating events in the development of childhood leukemia, then control of the virus has the potential to be a prevention target,” Dr Francis said. “That’s the real take-home message.”
While this research is in the early stages, the researchers hope these results will inspire more studies that will validate these findings and lead to the development of a CMV vaccine.
“This is the first step, but if we do end up finding a causal link to the most common childhood cancer, we hope that will light a fire in terms of stopping mother-to-child transmission of CMV,” Dr Francis said. ![]()
Photo by Vera Kratochvil
Newborns with congenital cytomegalovirus (CMV) infection may have an increased risk of developing acute lymphoblastic leukemia (ALL), according to a study published in Blood.
The data also suggest the risk may be particularly high among Hispanic children.
Researchers said this is the first study to suggest that congenital CMV infection is a risk factor for childhood ALL and is more prominent in Hispanic children.
To conduct this study, the researchers first identified all known infections present in the bone marrow of 127 children diagnosed with ALL and 38 children diagnosed with acute myeloid leukemia (AML).
The team found CMV infection was prevalent in children with ALL but rare in those with AML.
Next, the researchers looked for CMV in newborn blood samples from 268 children who went on to develop ALL. The team compared the samples with samples from 270 healthy children.
“Our goal in tracking CMV back from the time of diagnosis to the womb was to establish that this infection occurred well before initiation of disease,” said lead study author Stephen Francis, PhD, of the University of Nevada and University of California, San Francisco.
He and his colleagues found that children who went on to develop ALL were nearly 4 times more likely than control subjects to be CMV-positive at birth. The odds ratio was 3.71 (P=0.0016).
The odds ratio was 5.9 in Hispanic children and 2.1 in non-Hispanic whites. The researchers said this finding is particularly interesting because of the high rate of ALL observed in Hispanics.
“If it’s true that in utero CMV is one of the initiating events in the development of childhood leukemia, then control of the virus has the potential to be a prevention target,” Dr Francis said. “That’s the real take-home message.”
While this research is in the early stages, the researchers hope these results will inspire more studies that will validate these findings and lead to the development of a CMV vaccine.
“This is the first step, but if we do end up finding a causal link to the most common childhood cancer, we hope that will light a fire in terms of stopping mother-to-child transmission of CMV,” Dr Francis said. ![]()
Study reveals CML patients likely to benefit from HSCT long-term
![]()
Photo by Chad McNeeley
SAN DIEGO—Researchers believe they have identified patients with chronic myeloid leukemia (CML) who are likely to derive long-term benefit from allogeneic hematopoietic stem cell transplant (allo-HSCT).
The researchers found that CML patients have a low risk of long-term morbidity if they undergo HSCT before the age of 45, are conditioned with busulfan and cyclophosphamide (Bu/Cy), and receive a graft from a matched, related donor (MRD).
Jessica Wu, of the University of Alabama at Birmingham, presented these findings at the 2016 ASH Annual Meeting (abstract 823*).
Wu noted that allogeneic HSCT is potentially curative for CML, but this method of treatment has been on the decline since the introduction of tyrosine kinase inhibitors (TKIs). And today, few CML patients undergo allo-HSCT.
She said that although TKIs can induce remission in CML patients, the drugs also fail to eradicate leukemia, can produce side effects that impact patients’ quality of life, and come with a significant financial burden (estimated at $92,000 to $138,000 per patient per year).
With this in mind, Wu and her colleagues set out to determine if certain CML patients might benefit from allo-HSCT long-term. The team also wanted to quantify overall and cause-specific late mortality after allo-HSCT and the long-term burden of severe/life-threatening chronic health conditions after allo-HSCT.
Patient population
The researchers studied 637 CML patients treated with allo-HSCT between 1981 and 2010 at City of Hope in Duarte, California, or the University of Minnesota in Minneapolis/Saint Paul. The patients had to have survived at least 2 years post-transplant.
About 60% of patients were male, and 67% were non-Hispanic white. Their median age at HSCT was 36.4 years, and 65% received an MRD graft. Nineteen percent of patients were transplanted in 1980-1989, 52% were transplanted in 1990-1999, and 29% were transplanted in 2000-2010.
Fifty-eight percent of patients received Cy/total body irradiation (TBI), 18% received Bu/Cy, and 3% received reduced-intensity conditioning (RIC).
Sixty-one percent of patients had chronic graft-vs-host disease (cGVHD), and 32% had high-risk disease at the time of HSCT.
Survival
The patients were followed for a median of 16.7 years. Thirty percent (n=192) died after surviving at least 2 years post-HSCT.
The median time to death was 8.3 years (range, 2-29.5), and the median age at death was 49.2 (range, 7.8-69.8). At 20 years from HSCT, the overall survival was 68.6%.
HSCT recipients had a 4.4-fold increased risk of death compared with the age-, sex-, and race-matched general population.
“Non-relapse mortality was the major contributor to late mortality, with infection, second malignancies, and cGVHD being the most common causes of death,” Wu said.
Non-relapse mortality was 20%, and relapse-related mortality was 4%. Eight percent of patients died of infection, 6.3% died of cGVHD, and 3.7% died of second malignancies.
Health outcomes
Patients who were still alive at the time of the study were asked to complete the BMTSS-2 health questionnaire, which was used to examine the risk of grade 3/4 chronic health conditions.
A total of 288 patients completed the questionnaire, as did a sibling comparison group of 404 individuals.
Among the patients, the median age at allo-HSCT was 37.5 (range, 3.6-71.4), and the median duration of follow-up was 13.9 years (range, 2-34.6).
Sixty-two percent of patients received an MRD graft, and 38% had a matched, unrelated donor. Eighty-three percent of patients had TBI-based conditioning, 16% received Bu/Cy, and 2.7% received RIC.
The prevalence of grade 3/4 chronic health conditions was significantly higher among patients than among siblings—38% and 24%, respectively (P<0.0001).
The odds ratio (OR)—adjusted for age, sex, race, and socioeconomic status—was 2.7 (P<0.0001).
The cumulative incidence of any grade 3/4 condition at 20 years after HSCT was 47.2% among patients. Common conditions were diabetes (14.9%), second malignancies (12.6%), and coronary artery disease (10%).
The researchers found the risk of grade 3/4 morbidity was significantly higher for the following patient groups:
- Those age 45 and older (hazard ratio [HR]=3.3, P<0.0001)
- Patients with a matched, unrelated donor (HR=3.0, P<0.0001)
- Those who received peripheral blood or cord blood grafts as opposed to bone marrow (HR=2.7, P=0.006).
(This analysis was adjusted for race/ethnicity, sex, education, household income, insurance, cGVHD, and conditioning regimen).
Lower risk
To identify subpopulations with a reduced risk of long-term morbidity, the researchers calculated the risk in various CML patient groups compared to siblings.
The overall OR for CML patients compared with siblings was 2.7 (P<0.0001).
The OR for patients in first chronic phase who underwent HSCT before the age of 45 and had an MRD was 1.5 (P=0.1).
The OR for CML patients in first chronic phase who underwent HSCT before the age of 45, had an MRD, and received Bu/Cy conditioning was 0.8 (P=0.7).
“[W]e found that patients who received a matched, related donor transplant under the age of 45, with busulfan/cyclophosphamide, carried the same burden of morbidity as the sibling cohort,” Wu said. “These findings could help inform decisions regarding therapeutic options for the management of CML.”
Wu noted that the limited sample size in this study prevented the researchers from examining outcomes with RIC. And a lack of data at analysis prevented them from examining pre-HSCT and post-HSCT management of CML, the interval between diagnosis and HSCT, and the life-long economic burden of allo-HSCT.
However, she said data collection is ongoing, and the researchers hope to address some of these limitations.![]()
*Information presented at the meeting differs from the abstract.
![]()
Photo by Chad McNeeley
SAN DIEGO—Researchers believe they have identified patients with chronic myeloid leukemia (CML) who are likely to derive long-term benefit from allogeneic hematopoietic stem cell transplant (allo-HSCT).
The researchers found that CML patients have a low risk of long-term morbidity if they undergo HSCT before the age of 45, are conditioned with busulfan and cyclophosphamide (Bu/Cy), and receive a graft from a matched, related donor (MRD).
Jessica Wu, of the University of Alabama at Birmingham, presented these findings at the 2016 ASH Annual Meeting (abstract 823*).
Wu noted that allogeneic HSCT is potentially curative for CML, but this method of treatment has been on the decline since the introduction of tyrosine kinase inhibitors (TKIs). And today, few CML patients undergo allo-HSCT.
She said that although TKIs can induce remission in CML patients, the drugs also fail to eradicate leukemia, can produce side effects that impact patients’ quality of life, and come with a significant financial burden (estimated at $92,000 to $138,000 per patient per year).
With this in mind, Wu and her colleagues set out to determine if certain CML patients might benefit from allo-HSCT long-term. The team also wanted to quantify overall and cause-specific late mortality after allo-HSCT and the long-term burden of severe/life-threatening chronic health conditions after allo-HSCT.
Patient population
The researchers studied 637 CML patients treated with allo-HSCT between 1981 and 2010 at City of Hope in Duarte, California, or the University of Minnesota in Minneapolis/Saint Paul. The patients had to have survived at least 2 years post-transplant.
About 60% of patients were male, and 67% were non-Hispanic white. Their median age at HSCT was 36.4 years, and 65% received an MRD graft. Nineteen percent of patients were transplanted in 1980-1989, 52% were transplanted in 1990-1999, and 29% were transplanted in 2000-2010.
Fifty-eight percent of patients received Cy/total body irradiation (TBI), 18% received Bu/Cy, and 3% received reduced-intensity conditioning (RIC).
Sixty-one percent of patients had chronic graft-vs-host disease (cGVHD), and 32% had high-risk disease at the time of HSCT.
Survival
The patients were followed for a median of 16.7 years. Thirty percent (n=192) died after surviving at least 2 years post-HSCT.
The median time to death was 8.3 years (range, 2-29.5), and the median age at death was 49.2 (range, 7.8-69.8). At 20 years from HSCT, the overall survival was 68.6%.
HSCT recipients had a 4.4-fold increased risk of death compared with the age-, sex-, and race-matched general population.
“Non-relapse mortality was the major contributor to late mortality, with infection, second malignancies, and cGVHD being the most common causes of death,” Wu said.
Non-relapse mortality was 20%, and relapse-related mortality was 4%. Eight percent of patients died of infection, 6.3% died of cGVHD, and 3.7% died of second malignancies.
Health outcomes
Patients who were still alive at the time of the study were asked to complete the BMTSS-2 health questionnaire, which was used to examine the risk of grade 3/4 chronic health conditions.
A total of 288 patients completed the questionnaire, as did a sibling comparison group of 404 individuals.
Among the patients, the median age at allo-HSCT was 37.5 (range, 3.6-71.4), and the median duration of follow-up was 13.9 years (range, 2-34.6).
Sixty-two percent of patients received an MRD graft, and 38% had a matched, unrelated donor. Eighty-three percent of patients had TBI-based conditioning, 16% received Bu/Cy, and 2.7% received RIC.
The prevalence of grade 3/4 chronic health conditions was significantly higher among patients than among siblings—38% and 24%, respectively (P<0.0001).
The odds ratio (OR)—adjusted for age, sex, race, and socioeconomic status—was 2.7 (P<0.0001).
The cumulative incidence of any grade 3/4 condition at 20 years after HSCT was 47.2% among patients. Common conditions were diabetes (14.9%), second malignancies (12.6%), and coronary artery disease (10%).
The researchers found the risk of grade 3/4 morbidity was significantly higher for the following patient groups:
- Those age 45 and older (hazard ratio [HR]=3.3, P<0.0001)
- Patients with a matched, unrelated donor (HR=3.0, P<0.0001)
- Those who received peripheral blood or cord blood grafts as opposed to bone marrow (HR=2.7, P=0.006).
(This analysis was adjusted for race/ethnicity, sex, education, household income, insurance, cGVHD, and conditioning regimen).
Lower risk
To identify subpopulations with a reduced risk of long-term morbidity, the researchers calculated the risk in various CML patient groups compared to siblings.
The overall OR for CML patients compared with siblings was 2.7 (P<0.0001).
The OR for patients in first chronic phase who underwent HSCT before the age of 45 and had an MRD was 1.5 (P=0.1).
The OR for CML patients in first chronic phase who underwent HSCT before the age of 45, had an MRD, and received Bu/Cy conditioning was 0.8 (P=0.7).
“[W]e found that patients who received a matched, related donor transplant under the age of 45, with busulfan/cyclophosphamide, carried the same burden of morbidity as the sibling cohort,” Wu said. “These findings could help inform decisions regarding therapeutic options for the management of CML.”
Wu noted that the limited sample size in this study prevented the researchers from examining outcomes with RIC. And a lack of data at analysis prevented them from examining pre-HSCT and post-HSCT management of CML, the interval between diagnosis and HSCT, and the life-long economic burden of allo-HSCT.
However, she said data collection is ongoing, and the researchers hope to address some of these limitations.![]()
*Information presented at the meeting differs from the abstract.
![]()
Photo by Chad McNeeley
SAN DIEGO—Researchers believe they have identified patients with chronic myeloid leukemia (CML) who are likely to derive long-term benefit from allogeneic hematopoietic stem cell transplant (allo-HSCT).
The researchers found that CML patients have a low risk of long-term morbidity if they undergo HSCT before the age of 45, are conditioned with busulfan and cyclophosphamide (Bu/Cy), and receive a graft from a matched, related donor (MRD).
Jessica Wu, of the University of Alabama at Birmingham, presented these findings at the 2016 ASH Annual Meeting (abstract 823*).
Wu noted that allogeneic HSCT is potentially curative for CML, but this method of treatment has been on the decline since the introduction of tyrosine kinase inhibitors (TKIs). And today, few CML patients undergo allo-HSCT.
She said that although TKIs can induce remission in CML patients, the drugs also fail to eradicate leukemia, can produce side effects that impact patients’ quality of life, and come with a significant financial burden (estimated at $92,000 to $138,000 per patient per year).
With this in mind, Wu and her colleagues set out to determine if certain CML patients might benefit from allo-HSCT long-term. The team also wanted to quantify overall and cause-specific late mortality after allo-HSCT and the long-term burden of severe/life-threatening chronic health conditions after allo-HSCT.
Patient population
The researchers studied 637 CML patients treated with allo-HSCT between 1981 and 2010 at City of Hope in Duarte, California, or the University of Minnesota in Minneapolis/Saint Paul. The patients had to have survived at least 2 years post-transplant.
About 60% of patients were male, and 67% were non-Hispanic white. Their median age at HSCT was 36.4 years, and 65% received an MRD graft. Nineteen percent of patients were transplanted in 1980-1989, 52% were transplanted in 1990-1999, and 29% were transplanted in 2000-2010.
Fifty-eight percent of patients received Cy/total body irradiation (TBI), 18% received Bu/Cy, and 3% received reduced-intensity conditioning (RIC).
Sixty-one percent of patients had chronic graft-vs-host disease (cGVHD), and 32% had high-risk disease at the time of HSCT.
Survival
The patients were followed for a median of 16.7 years. Thirty percent (n=192) died after surviving at least 2 years post-HSCT.
The median time to death was 8.3 years (range, 2-29.5), and the median age at death was 49.2 (range, 7.8-69.8). At 20 years from HSCT, the overall survival was 68.6%.
HSCT recipients had a 4.4-fold increased risk of death compared with the age-, sex-, and race-matched general population.
“Non-relapse mortality was the major contributor to late mortality, with infection, second malignancies, and cGVHD being the most common causes of death,” Wu said.
Non-relapse mortality was 20%, and relapse-related mortality was 4%. Eight percent of patients died of infection, 6.3% died of cGVHD, and 3.7% died of second malignancies.
Health outcomes
Patients who were still alive at the time of the study were asked to complete the BMTSS-2 health questionnaire, which was used to examine the risk of grade 3/4 chronic health conditions.
A total of 288 patients completed the questionnaire, as did a sibling comparison group of 404 individuals.
Among the patients, the median age at allo-HSCT was 37.5 (range, 3.6-71.4), and the median duration of follow-up was 13.9 years (range, 2-34.6).
Sixty-two percent of patients received an MRD graft, and 38% had a matched, unrelated donor. Eighty-three percent of patients had TBI-based conditioning, 16% received Bu/Cy, and 2.7% received RIC.
The prevalence of grade 3/4 chronic health conditions was significantly higher among patients than among siblings—38% and 24%, respectively (P<0.0001).
The odds ratio (OR)—adjusted for age, sex, race, and socioeconomic status—was 2.7 (P<0.0001).
The cumulative incidence of any grade 3/4 condition at 20 years after HSCT was 47.2% among patients. Common conditions were diabetes (14.9%), second malignancies (12.6%), and coronary artery disease (10%).
The researchers found the risk of grade 3/4 morbidity was significantly higher for the following patient groups:
- Those age 45 and older (hazard ratio [HR]=3.3, P<0.0001)
- Patients with a matched, unrelated donor (HR=3.0, P<0.0001)
- Those who received peripheral blood or cord blood grafts as opposed to bone marrow (HR=2.7, P=0.006).
(This analysis was adjusted for race/ethnicity, sex, education, household income, insurance, cGVHD, and conditioning regimen).
Lower risk
To identify subpopulations with a reduced risk of long-term morbidity, the researchers calculated the risk in various CML patient groups compared to siblings.
The overall OR for CML patients compared with siblings was 2.7 (P<0.0001).
The OR for patients in first chronic phase who underwent HSCT before the age of 45 and had an MRD was 1.5 (P=0.1).
The OR for CML patients in first chronic phase who underwent HSCT before the age of 45, had an MRD, and received Bu/Cy conditioning was 0.8 (P=0.7).
“[W]e found that patients who received a matched, related donor transplant under the age of 45, with busulfan/cyclophosphamide, carried the same burden of morbidity as the sibling cohort,” Wu said. “These findings could help inform decisions regarding therapeutic options for the management of CML.”
Wu noted that the limited sample size in this study prevented the researchers from examining outcomes with RIC. And a lack of data at analysis prevented them from examining pre-HSCT and post-HSCT management of CML, the interval between diagnosis and HSCT, and the life-long economic burden of allo-HSCT.
However, she said data collection is ongoing, and the researchers hope to address some of these limitations.
*Information presented at the meeting differs from the abstract.
Platform could optimize treatment of ALL, other diseases

A digital health technology platform may prove useful for optimizing treatment of acute lymphoblastic leukemia (ALL), according to research published in SLAS Technology.
The platform is based on phenotypic personalized medicine (PPM), in which a patient’s response to treatment can be visually represented in the shape of a parabola.
The graph plots the drug dose along the horizontal axis and the patient’s response on the vertical axis.
Researchers said PPM has the ability to accurately identify a person’s optimal drug and dose combinations throughout an entire course of treatment.
“Phenotypic personalized medicine is like turbocharged artificial intelligence,” said study author Dean Ho, PhD, of the University of California, Los Angeles.
“It personalizes combination therapy to optimize efficacy and safety. The ability for our technology to continuously pinpoint the proper dosages of multiple drugs from such a large pool of possible combinations overcomes a challenge that is substantially more difficult than finding a needle in a haystack.”
In this study, Dr Ho and his colleagues used PPM to (retrospectively) individualize drug ratios/dosages in 2 pediatric patients with standard-risk ALL.
The researchers looked at the patients’ records to examine the administration of 4-drug maintenance regimens (dexamethasone, vincristine, mercaptopurine, and methotrexate).
The team said the drug doses served as the inputs, and maintaining absolute neutrophil counts and platelet counts within target ranges served as the outputs for optimization.
Using PPM, the researchers generated individualized 3-dimensional maps to determine the optimal drug ratios.
The team found their technology-suggested drug dosages were as much as 40% lower than clinical chemotherapy dosages, but they still maintained target neutrophil/platelet levels.
The parabolas showed that markedly different dosages of each drug were required to maintain normal cell counts for each patient.
The researchers said their results demonstrate a clear need to personalize ALL treatment and will serve as a foundation for a pending clinical trial to optimize multidrug chemotherapy.
“PPM has the ability to personalize combination therapy for a wide spectrum of diseases, making it a broadly applicable technology,” said study author Chih-Ming Ho, PhD, of the University of California, Los Angeles.
“The fact that we don’t need any information pertaining to a disease’s biological process in order to optimize and personalize treatment is a revolutionary advance. We’re at the interface of digital health and cancer treatment.”
The research team is planning to recruit patients for a prospective trial within the next year. The technology is approved for additional infectious disease and oncology studies.
A digital health technology platform may prove useful for optimizing treatment of acute lymphoblastic leukemia (ALL), according to research published in SLAS Technology.
The platform is based on phenotypic personalized medicine (PPM), in which a patient’s response to treatment can be visually represented in the shape of a parabola.
The graph plots the drug dose along the horizontal axis and the patient’s response on the vertical axis.
Researchers said PPM has the ability to accurately identify a person’s optimal drug and dose combinations throughout an entire course of treatment.
“Phenotypic personalized medicine is like turbocharged artificial intelligence,” said study author Dean Ho, PhD, of the University of California, Los Angeles.
“It personalizes combination therapy to optimize efficacy and safety. The ability for our technology to continuously pinpoint the proper dosages of multiple drugs from such a large pool of possible combinations overcomes a challenge that is substantially more difficult than finding a needle in a haystack.”
In this study, Dr Ho and his colleagues used PPM to (retrospectively) individualize drug ratios/dosages in 2 pediatric patients with standard-risk ALL.
The researchers looked at the patients’ records to examine the administration of 4-drug maintenance regimens (dexamethasone, vincristine, mercaptopurine, and methotrexate).
The team said the drug doses served as the inputs, and maintaining absolute neutrophil counts and platelet counts within target ranges served as the outputs for optimization.
Using PPM, the researchers generated individualized 3-dimensional maps to determine the optimal drug ratios.
The team found their technology-suggested drug dosages were as much as 40% lower than clinical chemotherapy dosages, but they still maintained target neutrophil/platelet levels.
The parabolas showed that markedly different dosages of each drug were required to maintain normal cell counts for each patient.
The researchers said their results demonstrate a clear need to personalize ALL treatment and will serve as a foundation for a pending clinical trial to optimize multidrug chemotherapy.
“PPM has the ability to personalize combination therapy for a wide spectrum of diseases, making it a broadly applicable technology,” said study author Chih-Ming Ho, PhD, of the University of California, Los Angeles.
“The fact that we don’t need any information pertaining to a disease’s biological process in order to optimize and personalize treatment is a revolutionary advance. We’re at the interface of digital health and cancer treatment.”
The research team is planning to recruit patients for a prospective trial within the next year. The technology is approved for additional infectious disease and oncology studies.
A digital health technology platform may prove useful for optimizing treatment of acute lymphoblastic leukemia (ALL), according to research published in SLAS Technology.
The platform is based on phenotypic personalized medicine (PPM), in which a patient’s response to treatment can be visually represented in the shape of a parabola.
The graph plots the drug dose along the horizontal axis and the patient’s response on the vertical axis.
Researchers said PPM has the ability to accurately identify a person’s optimal drug and dose combinations throughout an entire course of treatment.
“Phenotypic personalized medicine is like turbocharged artificial intelligence,” said study author Dean Ho, PhD, of the University of California, Los Angeles.
“It personalizes combination therapy to optimize efficacy and safety. The ability for our technology to continuously pinpoint the proper dosages of multiple drugs from such a large pool of possible combinations overcomes a challenge that is substantially more difficult than finding a needle in a haystack.”
In this study, Dr Ho and his colleagues used PPM to (retrospectively) individualize drug ratios/dosages in 2 pediatric patients with standard-risk ALL.
The researchers looked at the patients’ records to examine the administration of 4-drug maintenance regimens (dexamethasone, vincristine, mercaptopurine, and methotrexate).
The team said the drug doses served as the inputs, and maintaining absolute neutrophil counts and platelet counts within target ranges served as the outputs for optimization.
Using PPM, the researchers generated individualized 3-dimensional maps to determine the optimal drug ratios.
The team found their technology-suggested drug dosages were as much as 40% lower than clinical chemotherapy dosages, but they still maintained target neutrophil/platelet levels.
The parabolas showed that markedly different dosages of each drug were required to maintain normal cell counts for each patient.
The researchers said their results demonstrate a clear need to personalize ALL treatment and will serve as a foundation for a pending clinical trial to optimize multidrug chemotherapy.
“PPM has the ability to personalize combination therapy for a wide spectrum of diseases, making it a broadly applicable technology,” said study author Chih-Ming Ho, PhD, of the University of California, Los Angeles.
“The fact that we don’t need any information pertaining to a disease’s biological process in order to optimize and personalize treatment is a revolutionary advance. We’re at the interface of digital health and cancer treatment.”
The research team is planning to recruit patients for a prospective trial within the next year. The technology is approved for additional infectious disease and oncology studies.
Novel trial aims to BEAT AML
SAN DIEGO – A multi-arm clinical trial aims to transform the treatment of acute myeloid leukemia, a deadly blood cancer whose standard of care has remained essentially unchanged for 4 decades.
Launched in October 2016, the multicenter BEAT AML Master Trial is based on a simple but radical goal – turn around genomic tests of bone marrow biopsies within 7 days to allow targeted therapy, said lead investigator Brian Druker, MD, of Oregon Health and Science University Knight Cancer Institute in Portland.

Speaking at a press conference at the annual meeting of the American Society of Hematology, he emphasized that rapid, accurate genomic testing is the only way to prescribe targeted agents for AML in time for them to help patients. “It really is about matching the right patient with the right drug,” he said. He also spoke about AML in a video interview at the conference.
That is a major departure from the current approach to treating AML, in which patients receive standard chemotherapy regimens that are toxic and largely ineffective. “Patients themselves call this barbaric therapy,” said John Byrd, MD, who is co-leading the trial on behalf of the Ohio State University Wexner Medical Center in Columbus. “In this trial, we’re going to move away from toxic therapy that is not potentially curative to give more targeted medicine instead.”
In addition to Dr. Druker’s and Dr. Byrd’s centers, Memorial Sloan Kettering Cancer Center, New York, and Dana-Farber Cancer Institute and Massachusetts General Hospital, both in Boston, are onboard for the study. The lead investigators hope to add another six centers to the study group and to have 10 arms of the study underway by mid-2017.
Older patients with AML find chemotherapy especially hard to tolerate and typically respond poorly. Accordingly, the trial will enroll those aged 60 years and up regardless of their genomic profile, the researchers said. Patients lacking targetable markers will be offered investigational therapies showing broad activity in AML.
Another complexity of AML is that any patient can have a variety of mutations, including some affecting only a small subset of leukemia cells, Dr. Byrd noted. Targeting those mutations cannot eradicate disease, but past trials did not rank or choose therapies based on mutation prevalence. Thus, this trial is the first to ask “which mutation is in all of the cells, which gives you the opportunity to get rid of all the disease,” he emphasized. Again, patients – not individual markers or agents – are the priority.
The study also is meant to be nimble – arms can be quickly opened or closed if bench or clinical data are promising or lackluster. This design does not preclude FDA approvals, said Louis J. DeGennaro, PhD, of the Leukemia and Lymphoma Society, which is sponsoring the trial. “We have worked closely with FDA to design a unique protocol that we believe will change the paradigm of AML treatment and future clinical trials,” he added. “This is an unprecedented collaboration.”
Dr. Druker agreed. “If we do this correctly, we can potentially see large effects, and that can become the impetus for rapid FDA approval of these drugs for the right patients,” he said. “That one of the things this trial is designed to do.”
Dr. DeGennaro is president and chief executive officer of the Leukemia and Lymphoma Society, which is sponsoring the BEAT AML Master trial. Dr. Druker disclosed ties to a number of pharmaceutical companies. Dr. Byrd had no relevant disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN DIEGO – A multi-arm clinical trial aims to transform the treatment of acute myeloid leukemia, a deadly blood cancer whose standard of care has remained essentially unchanged for 4 decades.
Launched in October 2016, the multicenter BEAT AML Master Trial is based on a simple but radical goal – turn around genomic tests of bone marrow biopsies within 7 days to allow targeted therapy, said lead investigator Brian Druker, MD, of Oregon Health and Science University Knight Cancer Institute in Portland.
Speaking at a press conference at the annual meeting of the American Society of Hematology, he emphasized that rapid, accurate genomic testing is the only way to prescribe targeted agents for AML in time for them to help patients. “It really is about matching the right patient with the right drug,” he said. He also spoke about AML in a video interview at the conference.
That is a major departure from the current approach to treating AML, in which patients receive standard chemotherapy regimens that are toxic and largely ineffective. “Patients themselves call this barbaric therapy,” said John Byrd, MD, who is co-leading the trial on behalf of the Ohio State University Wexner Medical Center in Columbus. “In this trial, we’re going to move away from toxic therapy that is not potentially curative to give more targeted medicine instead.”
In addition to Dr. Druker’s and Dr. Byrd’s centers, Memorial Sloan Kettering Cancer Center, New York, and Dana-Farber Cancer Institute and Massachusetts General Hospital, both in Boston, are onboard for the study. The lead investigators hope to add another six centers to the study group and to have 10 arms of the study underway by mid-2017.
Older patients with AML find chemotherapy especially hard to tolerate and typically respond poorly. Accordingly, the trial will enroll those aged 60 years and up regardless of their genomic profile, the researchers said. Patients lacking targetable markers will be offered investigational therapies showing broad activity in AML.
Another complexity of AML is that any patient can have a variety of mutations, including some affecting only a small subset of leukemia cells, Dr. Byrd noted. Targeting those mutations cannot eradicate disease, but past trials did not rank or choose therapies based on mutation prevalence. Thus, this trial is the first to ask “which mutation is in all of the cells, which gives you the opportunity to get rid of all the disease,” he emphasized. Again, patients – not individual markers or agents – are the priority.
The study also is meant to be nimble – arms can be quickly opened or closed if bench or clinical data are promising or lackluster. This design does not preclude FDA approvals, said Louis J. DeGennaro, PhD, of the Leukemia and Lymphoma Society, which is sponsoring the trial. “We have worked closely with FDA to design a unique protocol that we believe will change the paradigm of AML treatment and future clinical trials,” he added. “This is an unprecedented collaboration.”
Dr. Druker agreed. “If we do this correctly, we can potentially see large effects, and that can become the impetus for rapid FDA approval of these drugs for the right patients,” he said. “That one of the things this trial is designed to do.”
Dr. DeGennaro is president and chief executive officer of the Leukemia and Lymphoma Society, which is sponsoring the BEAT AML Master trial. Dr. Druker disclosed ties to a number of pharmaceutical companies. Dr. Byrd had no relevant disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN DIEGO – A multi-arm clinical trial aims to transform the treatment of acute myeloid leukemia, a deadly blood cancer whose standard of care has remained essentially unchanged for 4 decades.
Launched in October 2016, the multicenter BEAT AML Master Trial is based on a simple but radical goal – turn around genomic tests of bone marrow biopsies within 7 days to allow targeted therapy, said lead investigator Brian Druker, MD, of Oregon Health and Science University Knight Cancer Institute in Portland.
Speaking at a press conference at the annual meeting of the American Society of Hematology, he emphasized that rapid, accurate genomic testing is the only way to prescribe targeted agents for AML in time for them to help patients. “It really is about matching the right patient with the right drug,” he said. He also spoke about AML in a video interview at the conference.
That is a major departure from the current approach to treating AML, in which patients receive standard chemotherapy regimens that are toxic and largely ineffective. “Patients themselves call this barbaric therapy,” said John Byrd, MD, who is co-leading the trial on behalf of the Ohio State University Wexner Medical Center in Columbus. “In this trial, we’re going to move away from toxic therapy that is not potentially curative to give more targeted medicine instead.”
In addition to Dr. Druker’s and Dr. Byrd’s centers, Memorial Sloan Kettering Cancer Center, New York, and Dana-Farber Cancer Institute and Massachusetts General Hospital, both in Boston, are onboard for the study. The lead investigators hope to add another six centers to the study group and to have 10 arms of the study underway by mid-2017.
Older patients with AML find chemotherapy especially hard to tolerate and typically respond poorly. Accordingly, the trial will enroll those aged 60 years and up regardless of their genomic profile, the researchers said. Patients lacking targetable markers will be offered investigational therapies showing broad activity in AML.
Another complexity of AML is that any patient can have a variety of mutations, including some affecting only a small subset of leukemia cells, Dr. Byrd noted. Targeting those mutations cannot eradicate disease, but past trials did not rank or choose therapies based on mutation prevalence. Thus, this trial is the first to ask “which mutation is in all of the cells, which gives you the opportunity to get rid of all the disease,” he emphasized. Again, patients – not individual markers or agents – are the priority.
The study also is meant to be nimble – arms can be quickly opened or closed if bench or clinical data are promising or lackluster. This design does not preclude FDA approvals, said Louis J. DeGennaro, PhD, of the Leukemia and Lymphoma Society, which is sponsoring the trial. “We have worked closely with FDA to design a unique protocol that we believe will change the paradigm of AML treatment and future clinical trials,” he added. “This is an unprecedented collaboration.”
Dr. Druker agreed. “If we do this correctly, we can potentially see large effects, and that can become the impetus for rapid FDA approval of these drugs for the right patients,” he said. “That one of the things this trial is designed to do.”
Dr. DeGennaro is president and chief executive officer of the Leukemia and Lymphoma Society, which is sponsoring the BEAT AML Master trial. Dr. Druker disclosed ties to a number of pharmaceutical companies. Dr. Byrd had no relevant disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT ASH 2016

Lenalidomide improves PFS after 1st and 2nd line CLL therapy
SAN DIEGO – Lenalidomide, a mainstay of maintenance therapy for multiple myeloma, is now making inroads into maintenance therapy following first- and second-line therapy for chronic lymphocytic leukemia (CLL), investigators reported in two phase III studies.
Among patients with previously untreated CLL who were at high risk for early disease progression following standard chemotherapy, lenalidomide (Revlimid) maintenance therapy was associated with an 80% reduction in the relative risk for disease progression compared with placebo, reported Anna Fink, MD, of the University of Cologne, Germany, and her colleagues in the German CLL Study Group.
Similarly, lenalidomide maintenance significantly improved progression-free survival (PFS) compared with placebo among patients with CLL who had at least partial responses to second-line therapy, reported Anna Schuh, MD, of the University of Oxford, England, and her colleagues in the CONTINUUM trial.
“Lenalidomide maintenance therapy significantly improved progression-free survival, from just about 9 months to almost 40 months when given to patients with CLL who responded to second-line therapy,” Dr. Schuh said at the American Society of Hematology annual meeting.
Both studies were unblinded because of the superiority of lenalidomide after prespecified analyses, on the recommendation of the respective data safety monitoring boards (DSMBs).
The PFS advantage with lenalidomide seen in each study did not translate into differences in overall survival in either study, however.
Maintenance after first-line therapy
In the CLLM1 trial, Dr. Fink and her colleagues enrolled physically fit, previously untreated patients with CLL and delivered chemoimmunotherapy at the investigator’s choice: either FCR (fludarabine, cyclophosphamide, and rituximab), FR (fludarabine and rituximab), FC (fludarabine and cyclophosphamide), or BR (bendamustine and rituximab).
Patients who had at least a partial response after a minimum of four cycles were identified as being at high risk for progression if they had minimal residual disease (MRD) levels of at least 10-2 cells, or MRD levels from 10-4 to less than 10-2 combined with either an unmutated IGHV gene status, del(17p) or TP53 mutation at baseline.
Of 468 screened patients, 89 were deemed to have high risk disease, and these patients were randomly assigned on a 2:1 basis to maintenance with lenalidomide given 5 mg orally for the first cycle and escalated to a target dose of 15 mg by the seventh cycle, or to placebo.
Additional dose escalations could be performed based on MRD assessments every 6 months, with the drug continued until progression or unacceptable toxicity. Patients also were assigned to daily low-dose aspirin or to an anticoagulation agent depending on their individual risk for thromboembolic events.
The study was stopped and unblinded after a planned interim analysis showed that the difference in PFS met the stopping boundary for efficacy.
Ultimately, 56 patients assigned to lenalidomide received study treatment, as did 29 assigned to placebo.
At a median follow-up of 17.5 months, the median PFS according to independent review was not reached for lenalidomide, vs. 13.3 months for placebo. Lenalidomide was associated with a hazard ratio (HR) for progression of 0.148 (P less than .00001), and a relative risk reduction of 80%.
Lenalidomide was also significantly better for PFS in analysis by MRD at baseline, with a PFS of 19.4 months for placebo vs. not reached among patients with less than 10-2 but more than 10-4 cells (HR, 0.125) and 3.7 vs. 32.3 months, respectively, for patients with MRD greater than 10-2 (HR 0.165).
There were three deaths (two in patients on placebo), and at the last analysis there was no difference in overall survival. In all, 42.9% of patients on lenalidomide discontinued because of adverse events, compared with 72.4% of those on placebo.
Maintenance after second-line therapy
In CONTINUUM, patients with at least a partial response after two prior lines of therapy and an Eastern Cooperative Oncology Group performance score of 0-2 were enrolled and randomized to receive either lenalidomide (160 patients) at a starting dose of 2.5 mg/day for the first 28-day cycle, 5 mg/day from cycle 2, and, if well tolerated, up to 10 mg/day from cycle 7 on, or to placebo (154 patients).
This study, as noted before, was also unblinded at the time of the primary analysis as recommended by the DSMB, after a prespecified number of events had occurred.
At a median follow-up of 31.5 months, the median PFS, a co-primary endpoint with OS, was 33.9 months in the lenalidomide arm, compared with 9.2 months in the placebo arm, translating into a HR for lenalidomide of 0.40 (P less than .001).
The lenalidomide advantage also was seen in a subgroup analysis by age, prior response to chemotherapy, and number of factors for poor prognosis. Of note, among patients older than age 70, the PFS with lenalidomide was 52.5 months, compared with 7.3 months for placebo (HR 0.34, P = .005).
In a second PFS analysis conducted after 71 months of follow-up, lenalidomide remained superior, with a median PFS of 57.5 months vs. 32.7 months in the placebo arm. As noted, there was no difference in overall survival in this study.
Grade 3 or greater adverse events occurring more frequently with lenalidomide were neutropenia, thrombocytopenia, diarrhea, pneumonia, fatigue, hypokalemia, pulmonary embolism, and sepsis. There was no difference in the incidence of second primary malignancies, however.
CLLM1 was sponsored by the German CLL Study Group with support from Celgene. CONTINUUM was supported by Celgene. Dr. Fink disclosed research funding from the company, and travel grants and honoraria from others. Dr. Schuh disclosed honoraria from and consulting with Celgene and other companies.
SAN DIEGO – Lenalidomide, a mainstay of maintenance therapy for multiple myeloma, is now making inroads into maintenance therapy following first- and second-line therapy for chronic lymphocytic leukemia (CLL), investigators reported in two phase III studies.
Among patients with previously untreated CLL who were at high risk for early disease progression following standard chemotherapy, lenalidomide (Revlimid) maintenance therapy was associated with an 80% reduction in the relative risk for disease progression compared with placebo, reported Anna Fink, MD, of the University of Cologne, Germany, and her colleagues in the German CLL Study Group.
Similarly, lenalidomide maintenance significantly improved progression-free survival (PFS) compared with placebo among patients with CLL who had at least partial responses to second-line therapy, reported Anna Schuh, MD, of the University of Oxford, England, and her colleagues in the CONTINUUM trial.
“Lenalidomide maintenance therapy significantly improved progression-free survival, from just about 9 months to almost 40 months when given to patients with CLL who responded to second-line therapy,” Dr. Schuh said at the American Society of Hematology annual meeting.
Both studies were unblinded because of the superiority of lenalidomide after prespecified analyses, on the recommendation of the respective data safety monitoring boards (DSMBs).
The PFS advantage with lenalidomide seen in each study did not translate into differences in overall survival in either study, however.
Maintenance after first-line therapy
In the CLLM1 trial, Dr. Fink and her colleagues enrolled physically fit, previously untreated patients with CLL and delivered chemoimmunotherapy at the investigator’s choice: either FCR (fludarabine, cyclophosphamide, and rituximab), FR (fludarabine and rituximab), FC (fludarabine and cyclophosphamide), or BR (bendamustine and rituximab).
Patients who had at least a partial response after a minimum of four cycles were identified as being at high risk for progression if they had minimal residual disease (MRD) levels of at least 10-2 cells, or MRD levels from 10-4 to less than 10-2 combined with either an unmutated IGHV gene status, del(17p) or TP53 mutation at baseline.
Of 468 screened patients, 89 were deemed to have high risk disease, and these patients were randomly assigned on a 2:1 basis to maintenance with lenalidomide given 5 mg orally for the first cycle and escalated to a target dose of 15 mg by the seventh cycle, or to placebo.
Additional dose escalations could be performed based on MRD assessments every 6 months, with the drug continued until progression or unacceptable toxicity. Patients also were assigned to daily low-dose aspirin or to an anticoagulation agent depending on their individual risk for thromboembolic events.
The study was stopped and unblinded after a planned interim analysis showed that the difference in PFS met the stopping boundary for efficacy.
Ultimately, 56 patients assigned to lenalidomide received study treatment, as did 29 assigned to placebo.
At a median follow-up of 17.5 months, the median PFS according to independent review was not reached for lenalidomide, vs. 13.3 months for placebo. Lenalidomide was associated with a hazard ratio (HR) for progression of 0.148 (P less than .00001), and a relative risk reduction of 80%.
Lenalidomide was also significantly better for PFS in analysis by MRD at baseline, with a PFS of 19.4 months for placebo vs. not reached among patients with less than 10-2 but more than 10-4 cells (HR, 0.125) and 3.7 vs. 32.3 months, respectively, for patients with MRD greater than 10-2 (HR 0.165).
There were three deaths (two in patients on placebo), and at the last analysis there was no difference in overall survival. In all, 42.9% of patients on lenalidomide discontinued because of adverse events, compared with 72.4% of those on placebo.
Maintenance after second-line therapy
In CONTINUUM, patients with at least a partial response after two prior lines of therapy and an Eastern Cooperative Oncology Group performance score of 0-2 were enrolled and randomized to receive either lenalidomide (160 patients) at a starting dose of 2.5 mg/day for the first 28-day cycle, 5 mg/day from cycle 2, and, if well tolerated, up to 10 mg/day from cycle 7 on, or to placebo (154 patients).
This study, as noted before, was also unblinded at the time of the primary analysis as recommended by the DSMB, after a prespecified number of events had occurred.
At a median follow-up of 31.5 months, the median PFS, a co-primary endpoint with OS, was 33.9 months in the lenalidomide arm, compared with 9.2 months in the placebo arm, translating into a HR for lenalidomide of 0.40 (P less than .001).
The lenalidomide advantage also was seen in a subgroup analysis by age, prior response to chemotherapy, and number of factors for poor prognosis. Of note, among patients older than age 70, the PFS with lenalidomide was 52.5 months, compared with 7.3 months for placebo (HR 0.34, P = .005).
In a second PFS analysis conducted after 71 months of follow-up, lenalidomide remained superior, with a median PFS of 57.5 months vs. 32.7 months in the placebo arm. As noted, there was no difference in overall survival in this study.
Grade 3 or greater adverse events occurring more frequently with lenalidomide were neutropenia, thrombocytopenia, diarrhea, pneumonia, fatigue, hypokalemia, pulmonary embolism, and sepsis. There was no difference in the incidence of second primary malignancies, however.
CLLM1 was sponsored by the German CLL Study Group with support from Celgene. CONTINUUM was supported by Celgene. Dr. Fink disclosed research funding from the company, and travel grants and honoraria from others. Dr. Schuh disclosed honoraria from and consulting with Celgene and other companies.
SAN DIEGO – Lenalidomide, a mainstay of maintenance therapy for multiple myeloma, is now making inroads into maintenance therapy following first- and second-line therapy for chronic lymphocytic leukemia (CLL), investigators reported in two phase III studies.
Among patients with previously untreated CLL who were at high risk for early disease progression following standard chemotherapy, lenalidomide (Revlimid) maintenance therapy was associated with an 80% reduction in the relative risk for disease progression compared with placebo, reported Anna Fink, MD, of the University of Cologne, Germany, and her colleagues in the German CLL Study Group.
Similarly, lenalidomide maintenance significantly improved progression-free survival (PFS) compared with placebo among patients with CLL who had at least partial responses to second-line therapy, reported Anna Schuh, MD, of the University of Oxford, England, and her colleagues in the CONTINUUM trial.
“Lenalidomide maintenance therapy significantly improved progression-free survival, from just about 9 months to almost 40 months when given to patients with CLL who responded to second-line therapy,” Dr. Schuh said at the American Society of Hematology annual meeting.
Both studies were unblinded because of the superiority of lenalidomide after prespecified analyses, on the recommendation of the respective data safety monitoring boards (DSMBs).
The PFS advantage with lenalidomide seen in each study did not translate into differences in overall survival in either study, however.
Maintenance after first-line therapy
In the CLLM1 trial, Dr. Fink and her colleagues enrolled physically fit, previously untreated patients with CLL and delivered chemoimmunotherapy at the investigator’s choice: either FCR (fludarabine, cyclophosphamide, and rituximab), FR (fludarabine and rituximab), FC (fludarabine and cyclophosphamide), or BR (bendamustine and rituximab).
Patients who had at least a partial response after a minimum of four cycles were identified as being at high risk for progression if they had minimal residual disease (MRD) levels of at least 10-2 cells, or MRD levels from 10-4 to less than 10-2 combined with either an unmutated IGHV gene status, del(17p) or TP53 mutation at baseline.
Of 468 screened patients, 89 were deemed to have high risk disease, and these patients were randomly assigned on a 2:1 basis to maintenance with lenalidomide given 5 mg orally for the first cycle and escalated to a target dose of 15 mg by the seventh cycle, or to placebo.
Additional dose escalations could be performed based on MRD assessments every 6 months, with the drug continued until progression or unacceptable toxicity. Patients also were assigned to daily low-dose aspirin or to an anticoagulation agent depending on their individual risk for thromboembolic events.
The study was stopped and unblinded after a planned interim analysis showed that the difference in PFS met the stopping boundary for efficacy.
Ultimately, 56 patients assigned to lenalidomide received study treatment, as did 29 assigned to placebo.
At a median follow-up of 17.5 months, the median PFS according to independent review was not reached for lenalidomide, vs. 13.3 months for placebo. Lenalidomide was associated with a hazard ratio (HR) for progression of 0.148 (P less than .00001), and a relative risk reduction of 80%.
Lenalidomide was also significantly better for PFS in analysis by MRD at baseline, with a PFS of 19.4 months for placebo vs. not reached among patients with less than 10-2 but more than 10-4 cells (HR, 0.125) and 3.7 vs. 32.3 months, respectively, for patients with MRD greater than 10-2 (HR 0.165).
There were three deaths (two in patients on placebo), and at the last analysis there was no difference in overall survival. In all, 42.9% of patients on lenalidomide discontinued because of adverse events, compared with 72.4% of those on placebo.
Maintenance after second-line therapy
In CONTINUUM, patients with at least a partial response after two prior lines of therapy and an Eastern Cooperative Oncology Group performance score of 0-2 were enrolled and randomized to receive either lenalidomide (160 patients) at a starting dose of 2.5 mg/day for the first 28-day cycle, 5 mg/day from cycle 2, and, if well tolerated, up to 10 mg/day from cycle 7 on, or to placebo (154 patients).
This study, as noted before, was also unblinded at the time of the primary analysis as recommended by the DSMB, after a prespecified number of events had occurred.
At a median follow-up of 31.5 months, the median PFS, a co-primary endpoint with OS, was 33.9 months in the lenalidomide arm, compared with 9.2 months in the placebo arm, translating into a HR for lenalidomide of 0.40 (P less than .001).
The lenalidomide advantage also was seen in a subgroup analysis by age, prior response to chemotherapy, and number of factors for poor prognosis. Of note, among patients older than age 70, the PFS with lenalidomide was 52.5 months, compared with 7.3 months for placebo (HR 0.34, P = .005).
In a second PFS analysis conducted after 71 months of follow-up, lenalidomide remained superior, with a median PFS of 57.5 months vs. 32.7 months in the placebo arm. As noted, there was no difference in overall survival in this study.
Grade 3 or greater adverse events occurring more frequently with lenalidomide were neutropenia, thrombocytopenia, diarrhea, pneumonia, fatigue, hypokalemia, pulmonary embolism, and sepsis. There was no difference in the incidence of second primary malignancies, however.
CLLM1 was sponsored by the German CLL Study Group with support from Celgene. CONTINUUM was supported by Celgene. Dr. Fink disclosed research funding from the company, and travel grants and honoraria from others. Dr. Schuh disclosed honoraria from and consulting with Celgene and other companies.
AT ASH 2016
Key clinical point: Lenalidomide (Revlimid) maintenance improves progression free survival following first- and second-line therapies for chronic lymphocytic leukemia.
Major finding: Median PFS vs. placebo was not reached vs. 13.3 months in CLLM1, and 33.9 vs. 9.2 months in CONTINUUM.
Data source: Phase III randomized studies of lenalidomide maintenance following first-line therapy (CLLM1) and second-line therapy (CONTINUUM).
Disclosures: CLLM1 was sponsored by the German CLL Study Group with support from Celgene. CONTINUUM was supported by Celgene. Dr. Fink disclosed research funding from Celgene, and travel grants and honoraria from other drug companies. Dr. Schuh disclosed honoraria from and consulting with Celgene and other companies.