User login
CAR T-cell studies to be presented at ASH

Several studies set to be presented at the 2018 ASH Annual Meeting provide new insights regarding chimeric antigen receptor (CAR) T-cell therapies.
One study suggests ibrutinib may enhance CAR T-cell therapy in patients with chronic lymphocytic leukemia (CLL), and another suggests checkpoint inhibitors can augment CAR T-cell therapy in certain patients with B-cell acute lymphoblastic leukemia (ALL).
Two additional studies indicate that responses to tisagenlecleucel are durable in both ALL and diffuse large B-cell lymphoma (DLBCL).
A fifth study suggests hematopoietic stem cell transplant (HSCT) may reduce the risk of relapse after CAR T-cell therapy.
ASH Secretary Robert A. Brodsky, MD, of Johns Hopkins University in Baltimore, Maryland, discussed these studies during a media briefing ahead of the ASH Annual Meeting.
Ibrutinib
In the ibrutinib study (abstract 299), patients received the BTK inhibitor starting 2 weeks prior to leukapheresis and continued until 3 months after treatment with JCAR014.
Data suggest this strategy may improve responses and decrease the incidence of severe cytokine release syndrome in patients with relapsed or refractory CLL.
Responses occurred in 88% of patients who received ibrutinib and 56% of those who did not.
Grade 3-5 cytokine release syndrome occurred in 5 of 19 patients (26%) in the no-ibrutinib cohort and 0 of 17 patients in the ibrutinib cohort.
These findings are “early and preliminary but very exciting” Dr. Brodsky said.
Checkpoint inhibitors
Early results of the checkpoint inhibitor study (abstract 556) suggest that pembrolizumab or nivolumab may augment CD19-directed CAR T-cell therapy.
The 14 patients studied had early CAR T-cell loss, partial response, or no response to CAR T-cell therapy. Thirteen patients had B-cell ALL, and one had B lymphoblastic lymphoma.
CD19-directed CAR T-cell therapy consisted of tisagenlecleucel in four patients and CTL119 in 10. Thirteen patients received pembrolizumab, and one received nivolumab.
Three of six patients who had early B-cell recovery re-established B-cell aplasia with the addition of a checkpoint inhibitor. In two patients, B-cell aplasia persists with ongoing pembrolizumab.
Four patients who did not respond to or relapsed after their initial CAR T-cell therapy had a partial (n=2) or complete response (n=2) with the addition of pembrolizumab.
There were additional partial responses in the remaining four patients. However, one of these patients (with CD19-dim/negative disease) progressed.
“The idea was if you can give pembrolizumab, you can take the brakes off, and maybe you can reinitiate the immune attack,” Dr. Brodsky said.
“[This is a] very small [study with] preliminary data but very exciting that it is safe to give checkpoint inhibitors with CAR T cells, and it may be efficacious at getting the immune response back.”
Tisagenlecleucel follow-up
One of the two tisagenlecleucel updates (abstract 895) consists of data from the ELIANA trial, which includes pediatric and young adult patients with relapsed/refractory ALL.
The overall response rate was 82% (65/79). Of the 65 responders, 29 were still in response at follow-up.
The probability of relapse-free survival was 66% at 12 months and 18 months.
“These are some very fast-growing tumors, and these are refractory, resistant patients, so, as we get further and further out, it’s more encouraging to see that there are durable responses,” Dr. Brodsky said.
The other tisagenlecleucel update (abstract 1684) is from the JULIET trial, which includes adults with relapsed or refractory DLBCL (n=99).
The overall response rate was 54%. The probability of relapse-free survival was 66% at 6 months and 64% at both 12 months and 18 months.
HSCT consolidation
Dr. Brodsky also discussed long-term follow-up from a phase 1/2 trial of SCRI-CAR19v1, a CD19-specific CAR T-cell product, in patients with relapsed/refractory ALL (abstract 967).
Of the 50 evaluable patients, 17 had no history of HSCT prior to CAR T-cell therapy.
Three of the 17 patients did not proceed to HSCT after CAR T-cell therapy, and two of these patients relapsed. Of the 14 patients who did undergo HSCT after CAR T-cell therapy, two relapsed.
There were 33 patients with a prior history of HSCT, and 10 of them had another HSCT after CAR T-cell therapy. Five of them are still alive and in remission.
Of the 23 patients who did not undergo another HSCT, eight are still in remission.
“This study is very small, and it’s retrospective, but it suggests that bone marrow transplant is a good way to consolidate the remission after CAR T-cell therapy,” Dr. Brodsky said.
Several studies set to be presented at the 2018 ASH Annual Meeting provide new insights regarding chimeric antigen receptor (CAR) T-cell therapies.
One study suggests ibrutinib may enhance CAR T-cell therapy in patients with chronic lymphocytic leukemia (CLL), and another suggests checkpoint inhibitors can augment CAR T-cell therapy in certain patients with B-cell acute lymphoblastic leukemia (ALL).
Two additional studies indicate that responses to tisagenlecleucel are durable in both ALL and diffuse large B-cell lymphoma (DLBCL).
A fifth study suggests hematopoietic stem cell transplant (HSCT) may reduce the risk of relapse after CAR T-cell therapy.
ASH Secretary Robert A. Brodsky, MD, of Johns Hopkins University in Baltimore, Maryland, discussed these studies during a media briefing ahead of the ASH Annual Meeting.
Ibrutinib
In the ibrutinib study (abstract 299), patients received the BTK inhibitor starting 2 weeks prior to leukapheresis and continued until 3 months after treatment with JCAR014.
Data suggest this strategy may improve responses and decrease the incidence of severe cytokine release syndrome in patients with relapsed or refractory CLL.
Responses occurred in 88% of patients who received ibrutinib and 56% of those who did not.
Grade 3-5 cytokine release syndrome occurred in 5 of 19 patients (26%) in the no-ibrutinib cohort and 0 of 17 patients in the ibrutinib cohort.
These findings are “early and preliminary but very exciting” Dr. Brodsky said.
Checkpoint inhibitors
Early results of the checkpoint inhibitor study (abstract 556) suggest that pembrolizumab or nivolumab may augment CD19-directed CAR T-cell therapy.
The 14 patients studied had early CAR T-cell loss, partial response, or no response to CAR T-cell therapy. Thirteen patients had B-cell ALL, and one had B lymphoblastic lymphoma.
CD19-directed CAR T-cell therapy consisted of tisagenlecleucel in four patients and CTL119 in 10. Thirteen patients received pembrolizumab, and one received nivolumab.
Three of six patients who had early B-cell recovery re-established B-cell aplasia with the addition of a checkpoint inhibitor. In two patients, B-cell aplasia persists with ongoing pembrolizumab.
Four patients who did not respond to or relapsed after their initial CAR T-cell therapy had a partial (n=2) or complete response (n=2) with the addition of pembrolizumab.
There were additional partial responses in the remaining four patients. However, one of these patients (with CD19-dim/negative disease) progressed.
“The idea was if you can give pembrolizumab, you can take the brakes off, and maybe you can reinitiate the immune attack,” Dr. Brodsky said.
“[This is a] very small [study with] preliminary data but very exciting that it is safe to give checkpoint inhibitors with CAR T cells, and it may be efficacious at getting the immune response back.”
Tisagenlecleucel follow-up
One of the two tisagenlecleucel updates (abstract 895) consists of data from the ELIANA trial, which includes pediatric and young adult patients with relapsed/refractory ALL.
The overall response rate was 82% (65/79). Of the 65 responders, 29 were still in response at follow-up.
The probability of relapse-free survival was 66% at 12 months and 18 months.
“These are some very fast-growing tumors, and these are refractory, resistant patients, so, as we get further and further out, it’s more encouraging to see that there are durable responses,” Dr. Brodsky said.
The other tisagenlecleucel update (abstract 1684) is from the JULIET trial, which includes adults with relapsed or refractory DLBCL (n=99).
The overall response rate was 54%. The probability of relapse-free survival was 66% at 6 months and 64% at both 12 months and 18 months.
HSCT consolidation
Dr. Brodsky also discussed long-term follow-up from a phase 1/2 trial of SCRI-CAR19v1, a CD19-specific CAR T-cell product, in patients with relapsed/refractory ALL (abstract 967).
Of the 50 evaluable patients, 17 had no history of HSCT prior to CAR T-cell therapy.
Three of the 17 patients did not proceed to HSCT after CAR T-cell therapy, and two of these patients relapsed. Of the 14 patients who did undergo HSCT after CAR T-cell therapy, two relapsed.
There were 33 patients with a prior history of HSCT, and 10 of them had another HSCT after CAR T-cell therapy. Five of them are still alive and in remission.
Of the 23 patients who did not undergo another HSCT, eight are still in remission.
“This study is very small, and it’s retrospective, but it suggests that bone marrow transplant is a good way to consolidate the remission after CAR T-cell therapy,” Dr. Brodsky said.
Several studies set to be presented at the 2018 ASH Annual Meeting provide new insights regarding chimeric antigen receptor (CAR) T-cell therapies.
One study suggests ibrutinib may enhance CAR T-cell therapy in patients with chronic lymphocytic leukemia (CLL), and another suggests checkpoint inhibitors can augment CAR T-cell therapy in certain patients with B-cell acute lymphoblastic leukemia (ALL).
Two additional studies indicate that responses to tisagenlecleucel are durable in both ALL and diffuse large B-cell lymphoma (DLBCL).
A fifth study suggests hematopoietic stem cell transplant (HSCT) may reduce the risk of relapse after CAR T-cell therapy.
ASH Secretary Robert A. Brodsky, MD, of Johns Hopkins University in Baltimore, Maryland, discussed these studies during a media briefing ahead of the ASH Annual Meeting.
Ibrutinib
In the ibrutinib study (abstract 299), patients received the BTK inhibitor starting 2 weeks prior to leukapheresis and continued until 3 months after treatment with JCAR014.
Data suggest this strategy may improve responses and decrease the incidence of severe cytokine release syndrome in patients with relapsed or refractory CLL.
Responses occurred in 88% of patients who received ibrutinib and 56% of those who did not.
Grade 3-5 cytokine release syndrome occurred in 5 of 19 patients (26%) in the no-ibrutinib cohort and 0 of 17 patients in the ibrutinib cohort.
These findings are “early and preliminary but very exciting” Dr. Brodsky said.
Checkpoint inhibitors
Early results of the checkpoint inhibitor study (abstract 556) suggest that pembrolizumab or nivolumab may augment CD19-directed CAR T-cell therapy.
The 14 patients studied had early CAR T-cell loss, partial response, or no response to CAR T-cell therapy. Thirteen patients had B-cell ALL, and one had B lymphoblastic lymphoma.
CD19-directed CAR T-cell therapy consisted of tisagenlecleucel in four patients and CTL119 in 10. Thirteen patients received pembrolizumab, and one received nivolumab.
Three of six patients who had early B-cell recovery re-established B-cell aplasia with the addition of a checkpoint inhibitor. In two patients, B-cell aplasia persists with ongoing pembrolizumab.
Four patients who did not respond to or relapsed after their initial CAR T-cell therapy had a partial (n=2) or complete response (n=2) with the addition of pembrolizumab.
There were additional partial responses in the remaining four patients. However, one of these patients (with CD19-dim/negative disease) progressed.
“The idea was if you can give pembrolizumab, you can take the brakes off, and maybe you can reinitiate the immune attack,” Dr. Brodsky said.
“[This is a] very small [study with] preliminary data but very exciting that it is safe to give checkpoint inhibitors with CAR T cells, and it may be efficacious at getting the immune response back.”
Tisagenlecleucel follow-up
One of the two tisagenlecleucel updates (abstract 895) consists of data from the ELIANA trial, which includes pediatric and young adult patients with relapsed/refractory ALL.
The overall response rate was 82% (65/79). Of the 65 responders, 29 were still in response at follow-up.
The probability of relapse-free survival was 66% at 12 months and 18 months.
“These are some very fast-growing tumors, and these are refractory, resistant patients, so, as we get further and further out, it’s more encouraging to see that there are durable responses,” Dr. Brodsky said.
The other tisagenlecleucel update (abstract 1684) is from the JULIET trial, which includes adults with relapsed or refractory DLBCL (n=99).
The overall response rate was 54%. The probability of relapse-free survival was 66% at 6 months and 64% at both 12 months and 18 months.
HSCT consolidation
Dr. Brodsky also discussed long-term follow-up from a phase 1/2 trial of SCRI-CAR19v1, a CD19-specific CAR T-cell product, in patients with relapsed/refractory ALL (abstract 967).
Of the 50 evaluable patients, 17 had no history of HSCT prior to CAR T-cell therapy.
Three of the 17 patients did not proceed to HSCT after CAR T-cell therapy, and two of these patients relapsed. Of the 14 patients who did undergo HSCT after CAR T-cell therapy, two relapsed.
There were 33 patients with a prior history of HSCT, and 10 of them had another HSCT after CAR T-cell therapy. Five of them are still alive and in remission.
Of the 23 patients who did not undergo another HSCT, eight are still in remission.
“This study is very small, and it’s retrospective, but it suggests that bone marrow transplant is a good way to consolidate the remission after CAR T-cell therapy,” Dr. Brodsky said.
FDA approves glasdegib for AML
The U.S. Food and Drug Administration has approved the hedgehog pathway inhibitor glasdegib (Daurismo) for use in combination with low-dose cytarabine (LDAC) to treat adults with newly diagnosed acute myeloid leukemia who are aged 75 years and older or who are ineligible for intensive chemotherapy.

The prescribing information for glasdegib includes a boxed warning detailing the risk of embryo-fetal death or severe birth defects associated with the drug.
The FDA’s approval of glasdegib is based on results from the phase 2 BRIGHT AML 1003 trial (NCT01546038). This trial included 111 adults with newly diagnosed AML and 14 patients with other conditions. The patients were randomized to receive glasdegib (at 100 mg daily) in combination with LDAC (n = 84) or LDAC alone (n = 41).
The complete response rate among the AML patients was 18.2% (14/77) in the glasdegib-LDAC arm and 2.6% (1/38) in the LDAC-only arm. The median overall survival was 8.3 months in the glasdegib-LDAC arm and 4.3 months in the LDAC-only arm (hazard ratio, 0.46; P = .0002).
The most common adverse events in the first 90 days of treatment, occurring in at least 30% of patients in either arm (glasdegib-LDAC and LDAC alone, respectively, were anemia (43% and 42%), fatigue (36% and 32%), hemorrhage (36% and 42%), febrile neutropenia (31% and 22%), musculoskeletal pain (30% and 17%), edema (30% and 20%), and thrombocytopenia (30% and 27%).
The incidence of serious adverse events was 79% in the glasdegib arm, and the most common events were febrile neutropenia (29%), pneumonia (23%), hemorrhage (12%), anemia (7%) and sepsis (7%).
Additional data from this trial are included in the prescribing information for glasdegib.
Glasdegib is a product of Pfizer.
The U.S. Food and Drug Administration has approved the hedgehog pathway inhibitor glasdegib (Daurismo) for use in combination with low-dose cytarabine (LDAC) to treat adults with newly diagnosed acute myeloid leukemia who are aged 75 years and older or who are ineligible for intensive chemotherapy.
The prescribing information for glasdegib includes a boxed warning detailing the risk of embryo-fetal death or severe birth defects associated with the drug.
The FDA’s approval of glasdegib is based on results from the phase 2 BRIGHT AML 1003 trial (NCT01546038). This trial included 111 adults with newly diagnosed AML and 14 patients with other conditions. The patients were randomized to receive glasdegib (at 100 mg daily) in combination with LDAC (n = 84) or LDAC alone (n = 41).
The complete response rate among the AML patients was 18.2% (14/77) in the glasdegib-LDAC arm and 2.6% (1/38) in the LDAC-only arm. The median overall survival was 8.3 months in the glasdegib-LDAC arm and 4.3 months in the LDAC-only arm (hazard ratio, 0.46; P = .0002).
The most common adverse events in the first 90 days of treatment, occurring in at least 30% of patients in either arm (glasdegib-LDAC and LDAC alone, respectively, were anemia (43% and 42%), fatigue (36% and 32%), hemorrhage (36% and 42%), febrile neutropenia (31% and 22%), musculoskeletal pain (30% and 17%), edema (30% and 20%), and thrombocytopenia (30% and 27%).
The incidence of serious adverse events was 79% in the glasdegib arm, and the most common events were febrile neutropenia (29%), pneumonia (23%), hemorrhage (12%), anemia (7%) and sepsis (7%).
Additional data from this trial are included in the prescribing information for glasdegib.
Glasdegib is a product of Pfizer.
The U.S. Food and Drug Administration has approved the hedgehog pathway inhibitor glasdegib (Daurismo) for use in combination with low-dose cytarabine (LDAC) to treat adults with newly diagnosed acute myeloid leukemia who are aged 75 years and older or who are ineligible for intensive chemotherapy.
The prescribing information for glasdegib includes a boxed warning detailing the risk of embryo-fetal death or severe birth defects associated with the drug.
The FDA’s approval of glasdegib is based on results from the phase 2 BRIGHT AML 1003 trial (NCT01546038). This trial included 111 adults with newly diagnosed AML and 14 patients with other conditions. The patients were randomized to receive glasdegib (at 100 mg daily) in combination with LDAC (n = 84) or LDAC alone (n = 41).
The complete response rate among the AML patients was 18.2% (14/77) in the glasdegib-LDAC arm and 2.6% (1/38) in the LDAC-only arm. The median overall survival was 8.3 months in the glasdegib-LDAC arm and 4.3 months in the LDAC-only arm (hazard ratio, 0.46; P = .0002).
The most common adverse events in the first 90 days of treatment, occurring in at least 30% of patients in either arm (glasdegib-LDAC and LDAC alone, respectively, were anemia (43% and 42%), fatigue (36% and 32%), hemorrhage (36% and 42%), febrile neutropenia (31% and 22%), musculoskeletal pain (30% and 17%), edema (30% and 20%), and thrombocytopenia (30% and 27%).
The incidence of serious adverse events was 79% in the glasdegib arm, and the most common events were febrile neutropenia (29%), pneumonia (23%), hemorrhage (12%), anemia (7%) and sepsis (7%).
Additional data from this trial are included in the prescribing information for glasdegib.
Glasdegib is a product of Pfizer.
FDA approves venetoclax for AML

The U.S. Food and Drug Administration (FDA) has granted accelerated approval to venetoclax (Venclexta®) for use in acute myeloid leukemia (AML).
The BCL-2 inhibitor is now approved for use in combination with azacitidine, decitabine, or low-dose cytarabine to treat adults with newly diagnosed AML who are age 75 and older or who are ineligible for intensive chemotherapy.
The FDA grants accelerated approval based on a surrogate or intermediate endpoint that is reasonably likely to predict clinical benefit.
Therefore, continued approval of venetoclax in AML may be contingent upon verification of clinical benefit in confirmatory trials.
The approval is based on data from two studies—the phase 1 b M14-358 trial (NCT02203773) and the phase 1/2 M14-387 trial (NCT02287233).
M14-358 trial
In M14-358, newly diagnosed AML patients received venetoclax in combination with azacitidine (n=84) or decitabine (n=31). There were 67 patients in the azacitidine arm and 13 in the decitabine arm who were 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via a daily ramp-up to a final dose of 400 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
They received azacitidine at 75 mg/m2 on days 1-7 of each 28-day cycle or decitabine at 20 mg/m2 on days 1-5 of each cycle. Patients continued treatment until disease progression or unacceptable toxicity.
The median follow-up was 7.9 months for the azacitidine arm and 11 months for the decitabine arm.
The complete response (CR) rate was 37% (25/67) in the azacitidine arm and 54% (7/13) in the decitabine arm. The rates of CR with partial hematologic recovery were 24% (16/67) and 7.7% (1/13), respectively.
The most common adverse events (AEs)—occurring in at least 30% of patients in both arms—were nausea, diarrhea, constipation, neutropenia, thrombocytopenia, hemorrhage, peripheral edema, vomiting, fatigue, febrile neutropenia, rash, and anemia.
The incidence of serious AEs was 75% overall. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, pneumonia (excluding fungal), sepsis (excluding fungal), respiratory failure, and multiple organ dysfunction syndrome.
The incidence of fatal AEs was 1.5% within 30 days of treatment initiation.
M14-387 trial
The M14-387 trial included 82 AML patients who received venetoclax plus low-dose cytarabine. Patients were newly diagnosed with AML, but some had previous exposure to a hypomethylating agent for an antecedent hematologic disorder.
There were 61 patients who were 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via daily ramp-up to a final dose of 600 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
Cytarabine was given at 20 mg/m2 on days 1-10 of each 28-day cycle. Patients continued to receive treatment until disease progression or unacceptable toxicity.
At a median follow-up of 6.5 months, the CR rate was 21% (13/61), and the rate of CR with partial hematologic recovery was 21% (13/61).
The most common AEs (occurring in at least 30% of patients) were nausea, thrombocytopenia, hemorrhage, febrile neutropenia, neutropenia, diarrhea, fatigue, constipation, and dyspnea.
The incidence of serious AEs was 95%. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, sepsis (excluding fungal), hemorrhage, pneumonia (excluding fungal), and device-related infection.
The incidence of fatal AEs was 4.9% within 30 days of treatment initiation.
Additional details from the M14-358 and M14-387 trials are available in the prescribing information for venetoclax.
Venetoclax is being developed by AbbVie and Roche. It is jointly commercialized by AbbVie and Genentech, a member of the Roche Group, in the United States and by AbbVie elsewhere.
The U.S. Food and Drug Administration (FDA) has granted accelerated approval to venetoclax (Venclexta®) for use in acute myeloid leukemia (AML).
The BCL-2 inhibitor is now approved for use in combination with azacitidine, decitabine, or low-dose cytarabine to treat adults with newly diagnosed AML who are age 75 and older or who are ineligible for intensive chemotherapy.
The FDA grants accelerated approval based on a surrogate or intermediate endpoint that is reasonably likely to predict clinical benefit.
Therefore, continued approval of venetoclax in AML may be contingent upon verification of clinical benefit in confirmatory trials.
The approval is based on data from two studies—the phase 1 b M14-358 trial (NCT02203773) and the phase 1/2 M14-387 trial (NCT02287233).
M14-358 trial
In M14-358, newly diagnosed AML patients received venetoclax in combination with azacitidine (n=84) or decitabine (n=31). There were 67 patients in the azacitidine arm and 13 in the decitabine arm who were 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via a daily ramp-up to a final dose of 400 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
They received azacitidine at 75 mg/m2 on days 1-7 of each 28-day cycle or decitabine at 20 mg/m2 on days 1-5 of each cycle. Patients continued treatment until disease progression or unacceptable toxicity.
The median follow-up was 7.9 months for the azacitidine arm and 11 months for the decitabine arm.
The complete response (CR) rate was 37% (25/67) in the azacitidine arm and 54% (7/13) in the decitabine arm. The rates of CR with partial hematologic recovery were 24% (16/67) and 7.7% (1/13), respectively.
The most common adverse events (AEs)—occurring in at least 30% of patients in both arms—were nausea, diarrhea, constipation, neutropenia, thrombocytopenia, hemorrhage, peripheral edema, vomiting, fatigue, febrile neutropenia, rash, and anemia.
The incidence of serious AEs was 75% overall. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, pneumonia (excluding fungal), sepsis (excluding fungal), respiratory failure, and multiple organ dysfunction syndrome.
The incidence of fatal AEs was 1.5% within 30 days of treatment initiation.
M14-387 trial
The M14-387 trial included 82 AML patients who received venetoclax plus low-dose cytarabine. Patients were newly diagnosed with AML, but some had previous exposure to a hypomethylating agent for an antecedent hematologic disorder.
There were 61 patients who were 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via daily ramp-up to a final dose of 600 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
Cytarabine was given at 20 mg/m2 on days 1-10 of each 28-day cycle. Patients continued to receive treatment until disease progression or unacceptable toxicity.
At a median follow-up of 6.5 months, the CR rate was 21% (13/61), and the rate of CR with partial hematologic recovery was 21% (13/61).
The most common AEs (occurring in at least 30% of patients) were nausea, thrombocytopenia, hemorrhage, febrile neutropenia, neutropenia, diarrhea, fatigue, constipation, and dyspnea.
The incidence of serious AEs was 95%. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, sepsis (excluding fungal), hemorrhage, pneumonia (excluding fungal), and device-related infection.
The incidence of fatal AEs was 4.9% within 30 days of treatment initiation.
Additional details from the M14-358 and M14-387 trials are available in the prescribing information for venetoclax.
Venetoclax is being developed by AbbVie and Roche. It is jointly commercialized by AbbVie and Genentech, a member of the Roche Group, in the United States and by AbbVie elsewhere.
The U.S. Food and Drug Administration (FDA) has granted accelerated approval to venetoclax (Venclexta®) for use in acute myeloid leukemia (AML).
The BCL-2 inhibitor is now approved for use in combination with azacitidine, decitabine, or low-dose cytarabine to treat adults with newly diagnosed AML who are age 75 and older or who are ineligible for intensive chemotherapy.
The FDA grants accelerated approval based on a surrogate or intermediate endpoint that is reasonably likely to predict clinical benefit.
Therefore, continued approval of venetoclax in AML may be contingent upon verification of clinical benefit in confirmatory trials.
The approval is based on data from two studies—the phase 1 b M14-358 trial (NCT02203773) and the phase 1/2 M14-387 trial (NCT02287233).
M14-358 trial
In M14-358, newly diagnosed AML patients received venetoclax in combination with azacitidine (n=84) or decitabine (n=31). There were 67 patients in the azacitidine arm and 13 in the decitabine arm who were 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via a daily ramp-up to a final dose of 400 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
They received azacitidine at 75 mg/m2 on days 1-7 of each 28-day cycle or decitabine at 20 mg/m2 on days 1-5 of each cycle. Patients continued treatment until disease progression or unacceptable toxicity.
The median follow-up was 7.9 months for the azacitidine arm and 11 months for the decitabine arm.
The complete response (CR) rate was 37% (25/67) in the azacitidine arm and 54% (7/13) in the decitabine arm. The rates of CR with partial hematologic recovery were 24% (16/67) and 7.7% (1/13), respectively.
The most common adverse events (AEs)—occurring in at least 30% of patients in both arms—were nausea, diarrhea, constipation, neutropenia, thrombocytopenia, hemorrhage, peripheral edema, vomiting, fatigue, febrile neutropenia, rash, and anemia.
The incidence of serious AEs was 75% overall. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, pneumonia (excluding fungal), sepsis (excluding fungal), respiratory failure, and multiple organ dysfunction syndrome.
The incidence of fatal AEs was 1.5% within 30 days of treatment initiation.
M14-387 trial
The M14-387 trial included 82 AML patients who received venetoclax plus low-dose cytarabine. Patients were newly diagnosed with AML, but some had previous exposure to a hypomethylating agent for an antecedent hematologic disorder.
There were 61 patients who were 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via daily ramp-up to a final dose of 600 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
Cytarabine was given at 20 mg/m2 on days 1-10 of each 28-day cycle. Patients continued to receive treatment until disease progression or unacceptable toxicity.
At a median follow-up of 6.5 months, the CR rate was 21% (13/61), and the rate of CR with partial hematologic recovery was 21% (13/61).
The most common AEs (occurring in at least 30% of patients) were nausea, thrombocytopenia, hemorrhage, febrile neutropenia, neutropenia, diarrhea, fatigue, constipation, and dyspnea.
The incidence of serious AEs was 95%. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, sepsis (excluding fungal), hemorrhage, pneumonia (excluding fungal), and device-related infection.
The incidence of fatal AEs was 4.9% within 30 days of treatment initiation.
Additional details from the M14-358 and M14-387 trials are available in the prescribing information for venetoclax.
Venetoclax is being developed by AbbVie and Roche. It is jointly commercialized by AbbVie and Genentech, a member of the Roche Group, in the United States and by AbbVie elsewhere.
FDA approves glasdegib for AML

The U.S. Food and Drug Administration (FDA) has approved the hedgehog pathway inhibitor glasdegib (Daurismo™) to treat certain patients with acute myeloid leukemia (AML).
Glasdegib is approved for use in combination with low-dose cytarabine (LDAC) to treat adults with newly diagnosed AML who are age 75 and older or who are ineligible for intensive chemotherapy.
Glasdegib was approved under priority review and also received orphan drug designation from the FDA.
The prescribing information for glasdegib includes a boxed warning detailing the risk of embryo-fetal death or severe birth defects associated with the drug.
The FDA’s approval of glasdegib is based on results from the phase 2 BRIGHT AML 1003 trial (NCT01546038).
This trial included 111 adults with newly diagnosed AML and 14 patients with other conditions.
The patients were randomized to receive glasdegib (at 100 mg daily) in combination with LDAC (n=84) or LDAC alone (n=41).
The complete response rate among the AML patients was 18.2% (14/77) in the glasdegib arm and 2.6% (1/38) in the LDAC arm.
The median overall survival was 8.3 months in the glasdegib arm and 4.3 months in the LDAC arm (hazard ratio=0.46; P=0.0002).
The most common adverse events (AEs) in the first 90 days of treatment—occurring in at least 30% of patients in either arm (glasdegib-LDAC and LDAC alone, respectively)—were:
- Anemia (43% and 42%)
- Fatigue (36% and 32%)
- Hemorrhage (36% and 42%)
- Febrile neutropenia (31% and 22%)
- Musculoskeletal pain (30% and 17%)
- Edema (30% and 20%)
- Thrombocytopenia (30% and 27%).
The incidence of serious AEs was 79% in the glasdegib arm. The most common serious AEs occurring in at least 5% of patients in this arm were febrile neutropenia (29%), pneumonia (23%), hemorrhage (12%), anemia (7%), and sepsis (7%).
Additional data from this trial are included in the prescribing information for glasdegib. Glasdegib is a product of Pfizer.
The U.S. Food and Drug Administration (FDA) has approved the hedgehog pathway inhibitor glasdegib (Daurismo™) to treat certain patients with acute myeloid leukemia (AML).
Glasdegib is approved for use in combination with low-dose cytarabine (LDAC) to treat adults with newly diagnosed AML who are age 75 and older or who are ineligible for intensive chemotherapy.
Glasdegib was approved under priority review and also received orphan drug designation from the FDA.
The prescribing information for glasdegib includes a boxed warning detailing the risk of embryo-fetal death or severe birth defects associated with the drug.
The FDA’s approval of glasdegib is based on results from the phase 2 BRIGHT AML 1003 trial (NCT01546038).
This trial included 111 adults with newly diagnosed AML and 14 patients with other conditions.
The patients were randomized to receive glasdegib (at 100 mg daily) in combination with LDAC (n=84) or LDAC alone (n=41).
The complete response rate among the AML patients was 18.2% (14/77) in the glasdegib arm and 2.6% (1/38) in the LDAC arm.
The median overall survival was 8.3 months in the glasdegib arm and 4.3 months in the LDAC arm (hazard ratio=0.46; P=0.0002).
The most common adverse events (AEs) in the first 90 days of treatment—occurring in at least 30% of patients in either arm (glasdegib-LDAC and LDAC alone, respectively)—were:
- Anemia (43% and 42%)
- Fatigue (36% and 32%)
- Hemorrhage (36% and 42%)
- Febrile neutropenia (31% and 22%)
- Musculoskeletal pain (30% and 17%)
- Edema (30% and 20%)
- Thrombocytopenia (30% and 27%).
The incidence of serious AEs was 79% in the glasdegib arm. The most common serious AEs occurring in at least 5% of patients in this arm were febrile neutropenia (29%), pneumonia (23%), hemorrhage (12%), anemia (7%), and sepsis (7%).
Additional data from this trial are included in the prescribing information for glasdegib. Glasdegib is a product of Pfizer.
The U.S. Food and Drug Administration (FDA) has approved the hedgehog pathway inhibitor glasdegib (Daurismo™) to treat certain patients with acute myeloid leukemia (AML).
Glasdegib is approved for use in combination with low-dose cytarabine (LDAC) to treat adults with newly diagnosed AML who are age 75 and older or who are ineligible for intensive chemotherapy.
Glasdegib was approved under priority review and also received orphan drug designation from the FDA.
The prescribing information for glasdegib includes a boxed warning detailing the risk of embryo-fetal death or severe birth defects associated with the drug.
The FDA’s approval of glasdegib is based on results from the phase 2 BRIGHT AML 1003 trial (NCT01546038).
This trial included 111 adults with newly diagnosed AML and 14 patients with other conditions.
The patients were randomized to receive glasdegib (at 100 mg daily) in combination with LDAC (n=84) or LDAC alone (n=41).
The complete response rate among the AML patients was 18.2% (14/77) in the glasdegib arm and 2.6% (1/38) in the LDAC arm.
The median overall survival was 8.3 months in the glasdegib arm and 4.3 months in the LDAC arm (hazard ratio=0.46; P=0.0002).
The most common adverse events (AEs) in the first 90 days of treatment—occurring in at least 30% of patients in either arm (glasdegib-LDAC and LDAC alone, respectively)—were:
- Anemia (43% and 42%)
- Fatigue (36% and 32%)
- Hemorrhage (36% and 42%)
- Febrile neutropenia (31% and 22%)
- Musculoskeletal pain (30% and 17%)
- Edema (30% and 20%)
- Thrombocytopenia (30% and 27%).
The incidence of serious AEs was 79% in the glasdegib arm. The most common serious AEs occurring in at least 5% of patients in this arm were febrile neutropenia (29%), pneumonia (23%), hemorrhage (12%), anemia (7%), and sepsis (7%).
Additional data from this trial are included in the prescribing information for glasdegib. Glasdegib is a product of Pfizer.
Venetoclax gets accelerated approval in older AML patients
(AML).
The BCL-2 inhibitor is now approved for use in combination with azacitidine, decitabine, or low-dose cytarabine to treat newly diagnosed adults with AML who are aged 75 and older or who are ineligible for intensive chemotherapy.
The FDA grants accelerated approval based on a surrogate or intermediate endpoint that is reasonably likely to predict clinical benefit. Continued approval of venetoclax in AML may be contingent upon verification of clinical benefit in confirmatory trials.
The approval is based on data from two studies – the phase 1b M14-358 trial ( NCT02203773 ) and the phase 1/2 M14-387 trial ( NCT02287233 ).
In M14-358, newly diagnosed AML patients received venetoclax in combination with azacitidine (n=84) or decitabine (n=31). There were 67 patients in the azacitidine arm and 13 in the decitabine arm who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via a daily ramp-up to a final dose of 400 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
They received azacitidine at 75 mg/m 2 on days 1-7 of each 28-day cycle or decitabine at 20 mg/m 2 on days 1-5 of each cycle. Patients continued treatment until disease progression or unacceptable toxicity.
The median follow-up was 7.9 months for the azacitidine arm and 11 months for the decitabine arm.
The complete response (CR) rate was 37% (25/67) in the azacitidine arm and 54% (7/13) in the decitabine arm. The rates of CR with partial hematologic recovery were 24% (16/67) and 7.7% (1/13), respectively. The most common adverse events (AEs) – occurring in at least 30% of patients in both arms – were nausea, diarrhea, constipation, neutropenia, thrombocytopenia, hemorrhage, peripheral edema, vomiting, fatigue, febrile neutropenia, rash, and anemia. The incidence of serious AEs was 75% overall. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, pneumonia (excluding fungal), sepsis (excluding fungal), respiratory failure, and multiple organ dysfunction syndrome. The incidence of fatal AEs was 1.5% within 30 days of treatment initiation. The M14-387 trial included 82 AML patients who received venetoclax plus low-dose cytarabine. Patients were newly diagnosed with AML, but some had previous exposure to a hypomethylating agent for an antecedent hematologic disorder.
There were 61 patients who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via daily ramp-up to a final dose of 600 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
Cytarabine was given at 20 mg/m 2 on days 1-10 of each 28-day cycle. Patients continued to receive treatment until disease progression or unacceptable toxicity.
At a median follow-up of 6.5 months, the CR rate was 21% (13/61), and the rate of CR with partial hematologic recovery was 21% (13/61).
The most common AEs (occurring in at least 30% of patients) were nausea, thrombocytopenia, hemorrhage, febrile neutropenia, neutropenia, diarrhea, fatigue, constipation, and dyspnea. The incidence of serious AEs was 95%. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, sepsis (excluding fungal), hemorrhage, pneumonia (excluding fungal), and device-related infection. The incidence of fatal AEs was 4.9% within 30 days of treatment initiation.
Additional details from the M14-358 and M14-387 trials are available in the prescribing information for venetoclax.
Venetoclax is being developed by AbbVie and Roche.
(AML).
The BCL-2 inhibitor is now approved for use in combination with azacitidine, decitabine, or low-dose cytarabine to treat newly diagnosed adults with AML who are aged 75 and older or who are ineligible for intensive chemotherapy.
The FDA grants accelerated approval based on a surrogate or intermediate endpoint that is reasonably likely to predict clinical benefit. Continued approval of venetoclax in AML may be contingent upon verification of clinical benefit in confirmatory trials.
The approval is based on data from two studies – the phase 1b M14-358 trial ( NCT02203773 ) and the phase 1/2 M14-387 trial ( NCT02287233 ).
In M14-358, newly diagnosed AML patients received venetoclax in combination with azacitidine (n=84) or decitabine (n=31). There were 67 patients in the azacitidine arm and 13 in the decitabine arm who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via a daily ramp-up to a final dose of 400 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
They received azacitidine at 75 mg/m 2 on days 1-7 of each 28-day cycle or decitabine at 20 mg/m 2 on days 1-5 of each cycle. Patients continued treatment until disease progression or unacceptable toxicity.
The median follow-up was 7.9 months for the azacitidine arm and 11 months for the decitabine arm.
The complete response (CR) rate was 37% (25/67) in the azacitidine arm and 54% (7/13) in the decitabine arm. The rates of CR with partial hematologic recovery were 24% (16/67) and 7.7% (1/13), respectively. The most common adverse events (AEs) – occurring in at least 30% of patients in both arms – were nausea, diarrhea, constipation, neutropenia, thrombocytopenia, hemorrhage, peripheral edema, vomiting, fatigue, febrile neutropenia, rash, and anemia. The incidence of serious AEs was 75% overall. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, pneumonia (excluding fungal), sepsis (excluding fungal), respiratory failure, and multiple organ dysfunction syndrome. The incidence of fatal AEs was 1.5% within 30 days of treatment initiation. The M14-387 trial included 82 AML patients who received venetoclax plus low-dose cytarabine. Patients were newly diagnosed with AML, but some had previous exposure to a hypomethylating agent for an antecedent hematologic disorder.
There were 61 patients who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via daily ramp-up to a final dose of 600 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
Cytarabine was given at 20 mg/m 2 on days 1-10 of each 28-day cycle. Patients continued to receive treatment until disease progression or unacceptable toxicity.
At a median follow-up of 6.5 months, the CR rate was 21% (13/61), and the rate of CR with partial hematologic recovery was 21% (13/61).
The most common AEs (occurring in at least 30% of patients) were nausea, thrombocytopenia, hemorrhage, febrile neutropenia, neutropenia, diarrhea, fatigue, constipation, and dyspnea. The incidence of serious AEs was 95%. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, sepsis (excluding fungal), hemorrhage, pneumonia (excluding fungal), and device-related infection. The incidence of fatal AEs was 4.9% within 30 days of treatment initiation.
Additional details from the M14-358 and M14-387 trials are available in the prescribing information for venetoclax.
Venetoclax is being developed by AbbVie and Roche.
(AML).
The BCL-2 inhibitor is now approved for use in combination with azacitidine, decitabine, or low-dose cytarabine to treat newly diagnosed adults with AML who are aged 75 and older or who are ineligible for intensive chemotherapy.
The FDA grants accelerated approval based on a surrogate or intermediate endpoint that is reasonably likely to predict clinical benefit. Continued approval of venetoclax in AML may be contingent upon verification of clinical benefit in confirmatory trials.
The approval is based on data from two studies – the phase 1b M14-358 trial ( NCT02203773 ) and the phase 1/2 M14-387 trial ( NCT02287233 ).
In M14-358, newly diagnosed AML patients received venetoclax in combination with azacitidine (n=84) or decitabine (n=31). There were 67 patients in the azacitidine arm and 13 in the decitabine arm who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via a daily ramp-up to a final dose of 400 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
They received azacitidine at 75 mg/m 2 on days 1-7 of each 28-day cycle or decitabine at 20 mg/m 2 on days 1-5 of each cycle. Patients continued treatment until disease progression or unacceptable toxicity.
The median follow-up was 7.9 months for the azacitidine arm and 11 months for the decitabine arm.
The complete response (CR) rate was 37% (25/67) in the azacitidine arm and 54% (7/13) in the decitabine arm. The rates of CR with partial hematologic recovery were 24% (16/67) and 7.7% (1/13), respectively. The most common adverse events (AEs) – occurring in at least 30% of patients in both arms – were nausea, diarrhea, constipation, neutropenia, thrombocytopenia, hemorrhage, peripheral edema, vomiting, fatigue, febrile neutropenia, rash, and anemia. The incidence of serious AEs was 75% overall. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, pneumonia (excluding fungal), sepsis (excluding fungal), respiratory failure, and multiple organ dysfunction syndrome. The incidence of fatal AEs was 1.5% within 30 days of treatment initiation. The M14-387 trial included 82 AML patients who received venetoclax plus low-dose cytarabine. Patients were newly diagnosed with AML, but some had previous exposure to a hypomethylating agent for an antecedent hematologic disorder.
There were 61 patients who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via daily ramp-up to a final dose of 600 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
Cytarabine was given at 20 mg/m 2 on days 1-10 of each 28-day cycle. Patients continued to receive treatment until disease progression or unacceptable toxicity.
At a median follow-up of 6.5 months, the CR rate was 21% (13/61), and the rate of CR with partial hematologic recovery was 21% (13/61).
The most common AEs (occurring in at least 30% of patients) were nausea, thrombocytopenia, hemorrhage, febrile neutropenia, neutropenia, diarrhea, fatigue, constipation, and dyspnea. The incidence of serious AEs was 95%. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, sepsis (excluding fungal), hemorrhage, pneumonia (excluding fungal), and device-related infection. The incidence of fatal AEs was 4.9% within 30 days of treatment initiation.
Additional details from the M14-358 and M14-387 trials are available in the prescribing information for venetoclax.
Venetoclax is being developed by AbbVie and Roche.
Americans concerned about cost of cancer care

A recent survey suggests Americans are nearly as worried about the cost of a cancer diagnosis as they are about dying from cancer.
The cost of cancer care was a top concern even among people who had no prior experience with cancer.
At the same time, cancer patients/survivors admitted to delaying or forgoing care due to costs, and caregivers reported taking “dramatic” actions to pay for their loved one’s care.
These are findings from the American Society of Clinical Oncology (ASCO)’s second annual National Cancer Opinion Survey.
The survey was conducted online by The Harris Poll from July 10, 2018, to August 10, 2018. It included 4,887 U.S. adults age 18 and older—1,001 of whom have or had cancer.
Cost among top concerns
Death and pain/suffering were the top concerns related to a cancer diagnosis. Fifty-four percent of respondents said death would be one of their greatest concerns if they were diagnosed with cancer, and the same percentage rated pain/suffering a top concern.
Forty-four percent of respondents said paying for cancer treatment would be a top concern, and 45% said the same about the financial impact of a cancer diagnosis on their family. When combined, financial issues were a top concern for 57% of respondents.
Paying for treatment was a top concern for:
- 36% of respondents who had/have cancer
- 51% of caregivers
- 43% of people with no prior cancer experience.
The financial impact on family was a top concern for:
- 39% of respondents who had/have cancer
- 55% of caregivers
- 42% of people with no prior cancer experience.
Cutting costs
Sixty-one percent of caregivers surveyed said they or another relative have taken a “dramatic” step to help pay for their loved one’s care, including:
- Dipping into savings accounts (35%)
- Working extra hours (23%)
- Taking an early withdrawal from a retirement account or college fund (14%)
- Postponing retirement (14%)
- Taking out a second mortgage or other type of loan (13%)
- Taking an additional job (13%)
- Selling family heirlooms (9%).
Twenty percent of cancer patients/survivors said they have taken actions to reduce treatment costs, including:
- Delaying scans (7%)
- Skipping or delaying appointments (7%)
- Skipping doses of prescribed treatment (6%)
- Postponing or not filling prescriptions (5%)
- Refusing treatment (3%).
“Patients are right to be concerned about the financial impact of a cancer diagnosis on their families,” said Richard L. Schilsky, MD, ASCO’s chief medical officer.
“It’s clear that high treatment costs are taking a serious toll not only on patients, but also on the people who care for them. If a family member has been diagnosed with cancer, the sole focus should be helping them get well. Instead, Americans are worrying about affording treatment, and, in many cases, they’re making serious personal sacrifices to help pay for their loved ones’ care.”
A recent survey suggests Americans are nearly as worried about the cost of a cancer diagnosis as they are about dying from cancer.
The cost of cancer care was a top concern even among people who had no prior experience with cancer.
At the same time, cancer patients/survivors admitted to delaying or forgoing care due to costs, and caregivers reported taking “dramatic” actions to pay for their loved one’s care.
These are findings from the American Society of Clinical Oncology (ASCO)’s second annual National Cancer Opinion Survey.
The survey was conducted online by The Harris Poll from July 10, 2018, to August 10, 2018. It included 4,887 U.S. adults age 18 and older—1,001 of whom have or had cancer.
Cost among top concerns
Death and pain/suffering were the top concerns related to a cancer diagnosis. Fifty-four percent of respondents said death would be one of their greatest concerns if they were diagnosed with cancer, and the same percentage rated pain/suffering a top concern.
Forty-four percent of respondents said paying for cancer treatment would be a top concern, and 45% said the same about the financial impact of a cancer diagnosis on their family. When combined, financial issues were a top concern for 57% of respondents.
Paying for treatment was a top concern for:
- 36% of respondents who had/have cancer
- 51% of caregivers
- 43% of people with no prior cancer experience.
The financial impact on family was a top concern for:
- 39% of respondents who had/have cancer
- 55% of caregivers
- 42% of people with no prior cancer experience.
Cutting costs
Sixty-one percent of caregivers surveyed said they or another relative have taken a “dramatic” step to help pay for their loved one’s care, including:
- Dipping into savings accounts (35%)
- Working extra hours (23%)
- Taking an early withdrawal from a retirement account or college fund (14%)
- Postponing retirement (14%)
- Taking out a second mortgage or other type of loan (13%)
- Taking an additional job (13%)
- Selling family heirlooms (9%).
Twenty percent of cancer patients/survivors said they have taken actions to reduce treatment costs, including:
- Delaying scans (7%)
- Skipping or delaying appointments (7%)
- Skipping doses of prescribed treatment (6%)
- Postponing or not filling prescriptions (5%)
- Refusing treatment (3%).
“Patients are right to be concerned about the financial impact of a cancer diagnosis on their families,” said Richard L. Schilsky, MD, ASCO’s chief medical officer.
“It’s clear that high treatment costs are taking a serious toll not only on patients, but also on the people who care for them. If a family member has been diagnosed with cancer, the sole focus should be helping them get well. Instead, Americans are worrying about affording treatment, and, in many cases, they’re making serious personal sacrifices to help pay for their loved ones’ care.”
A recent survey suggests Americans are nearly as worried about the cost of a cancer diagnosis as they are about dying from cancer.
The cost of cancer care was a top concern even among people who had no prior experience with cancer.
At the same time, cancer patients/survivors admitted to delaying or forgoing care due to costs, and caregivers reported taking “dramatic” actions to pay for their loved one’s care.
These are findings from the American Society of Clinical Oncology (ASCO)’s second annual National Cancer Opinion Survey.
The survey was conducted online by The Harris Poll from July 10, 2018, to August 10, 2018. It included 4,887 U.S. adults age 18 and older—1,001 of whom have or had cancer.
Cost among top concerns
Death and pain/suffering were the top concerns related to a cancer diagnosis. Fifty-four percent of respondents said death would be one of their greatest concerns if they were diagnosed with cancer, and the same percentage rated pain/suffering a top concern.
Forty-four percent of respondents said paying for cancer treatment would be a top concern, and 45% said the same about the financial impact of a cancer diagnosis on their family. When combined, financial issues were a top concern for 57% of respondents.
Paying for treatment was a top concern for:
- 36% of respondents who had/have cancer
- 51% of caregivers
- 43% of people with no prior cancer experience.
The financial impact on family was a top concern for:
- 39% of respondents who had/have cancer
- 55% of caregivers
- 42% of people with no prior cancer experience.
Cutting costs
Sixty-one percent of caregivers surveyed said they or another relative have taken a “dramatic” step to help pay for their loved one’s care, including:
- Dipping into savings accounts (35%)
- Working extra hours (23%)
- Taking an early withdrawal from a retirement account or college fund (14%)
- Postponing retirement (14%)
- Taking out a second mortgage or other type of loan (13%)
- Taking an additional job (13%)
- Selling family heirlooms (9%).
Twenty percent of cancer patients/survivors said they have taken actions to reduce treatment costs, including:
- Delaying scans (7%)
- Skipping or delaying appointments (7%)
- Skipping doses of prescribed treatment (6%)
- Postponing or not filling prescriptions (5%)
- Refusing treatment (3%).
“Patients are right to be concerned about the financial impact of a cancer diagnosis on their families,” said Richard L. Schilsky, MD, ASCO’s chief medical officer.
“It’s clear that high treatment costs are taking a serious toll not only on patients, but also on the people who care for them. If a family member has been diagnosed with cancer, the sole focus should be helping them get well. Instead, Americans are worrying about affording treatment, and, in many cases, they’re making serious personal sacrifices to help pay for their loved ones’ care.”
CHMP backs blinatumomab for MRD

The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding marketing authorization for blinatumomab (Blincyto) to include treatment of minimal residual disease (MRD).
The CHMP has recommended approval for blinatumomab as monotherapy for adults with Philadelphia chromosome-negative, CD19-positive, B-cell precursor acute lymphoblastic leukemia (BCP-ALL) in first or second complete remission with MRD greater than or equal to 0.1%.
The CHMP had originally adopted a negative opinion on extending the use of blinatumomab to these patients. However, the committee re-examined its opinion and reversed that decision.
The CHMP has requested that Amgen, the company developing blinatumomab, provide results from ongoing studies to support the new approval.
The CHMP’s recommendations are reviewed by the European Commission (EC), which has the authority to approve medicines for use in the European Union, Norway, Iceland, and Liechtenstein.
The EC usually makes a decision within 67 days of CHMP recommendations.
Blinatumomab is already EC-approved as monotherapy for:
- Adults with Philadelphia chromosome-negative, CD19-positive, relapsed or refractory BCP-ALL.
- Pediatric patients age 1 year or older who have relapsed/refractory, Philadelphia chromosome-negative, CD19-positive BCP-ALL and have received at least two prior therapies or relapsed after allogeneic hematopoietic stem cell transplant.
CHMP’s reversal of opinion
In July, the CHMP recommended against approving blinatumomab to treat patients with MRD based on data from the BLAST trial. Results from this phase 2 trial were published in Blood in April.
The CHMP noted that, although blinatumomab produced MRD negativity in many patients in the BLAST trial, there is no strong evidence that this leads to improved survival.
Given the uncertainty, the CHMP was of the opinion that the benefits of blinatumomab do not outweigh its risks in MRD-positive BCP-ALL patients.
However, Amgen request a re-examination of the CHMP’s opinion, and the CHMP complied.
During the re-examination, the CHMP reviewed all the data and consulted a group of experts.
The experts concluded, and the CHMP agreed, that, although there is no strong evidence of improved survival, the available data indicate a good response to blinatumomab, with around 78% of patients becoming MRD-negative after treatment.
The CHMP also noted that MRD-positive patients have a high risk of relapse and few treatment options.
Therefore, the committee concluded that the benefits of blinatumomab outweigh its risks in this patient population.
The CHMP recommended granting the change to the marketing authorization but also requested that Amgen provide data from ongoing studies of blinatumomab in MRD-positive patients, once those data are available.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding marketing authorization for blinatumomab (Blincyto) to include treatment of minimal residual disease (MRD).
The CHMP has recommended approval for blinatumomab as monotherapy for adults with Philadelphia chromosome-negative, CD19-positive, B-cell precursor acute lymphoblastic leukemia (BCP-ALL) in first or second complete remission with MRD greater than or equal to 0.1%.
The CHMP had originally adopted a negative opinion on extending the use of blinatumomab to these patients. However, the committee re-examined its opinion and reversed that decision.
The CHMP has requested that Amgen, the company developing blinatumomab, provide results from ongoing studies to support the new approval.
The CHMP’s recommendations are reviewed by the European Commission (EC), which has the authority to approve medicines for use in the European Union, Norway, Iceland, and Liechtenstein.
The EC usually makes a decision within 67 days of CHMP recommendations.
Blinatumomab is already EC-approved as monotherapy for:
- Adults with Philadelphia chromosome-negative, CD19-positive, relapsed or refractory BCP-ALL.
- Pediatric patients age 1 year or older who have relapsed/refractory, Philadelphia chromosome-negative, CD19-positive BCP-ALL and have received at least two prior therapies or relapsed after allogeneic hematopoietic stem cell transplant.
CHMP’s reversal of opinion
In July, the CHMP recommended against approving blinatumomab to treat patients with MRD based on data from the BLAST trial. Results from this phase 2 trial were published in Blood in April.
The CHMP noted that, although blinatumomab produced MRD negativity in many patients in the BLAST trial, there is no strong evidence that this leads to improved survival.
Given the uncertainty, the CHMP was of the opinion that the benefits of blinatumomab do not outweigh its risks in MRD-positive BCP-ALL patients.
However, Amgen request a re-examination of the CHMP’s opinion, and the CHMP complied.
During the re-examination, the CHMP reviewed all the data and consulted a group of experts.
The experts concluded, and the CHMP agreed, that, although there is no strong evidence of improved survival, the available data indicate a good response to blinatumomab, with around 78% of patients becoming MRD-negative after treatment.
The CHMP also noted that MRD-positive patients have a high risk of relapse and few treatment options.
Therefore, the committee concluded that the benefits of blinatumomab outweigh its risks in this patient population.
The CHMP recommended granting the change to the marketing authorization but also requested that Amgen provide data from ongoing studies of blinatumomab in MRD-positive patients, once those data are available.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding marketing authorization for blinatumomab (Blincyto) to include treatment of minimal residual disease (MRD).
The CHMP has recommended approval for blinatumomab as monotherapy for adults with Philadelphia chromosome-negative, CD19-positive, B-cell precursor acute lymphoblastic leukemia (BCP-ALL) in first or second complete remission with MRD greater than or equal to 0.1%.
The CHMP had originally adopted a negative opinion on extending the use of blinatumomab to these patients. However, the committee re-examined its opinion and reversed that decision.
The CHMP has requested that Amgen, the company developing blinatumomab, provide results from ongoing studies to support the new approval.
The CHMP’s recommendations are reviewed by the European Commission (EC), which has the authority to approve medicines for use in the European Union, Norway, Iceland, and Liechtenstein.
The EC usually makes a decision within 67 days of CHMP recommendations.
Blinatumomab is already EC-approved as monotherapy for:
- Adults with Philadelphia chromosome-negative, CD19-positive, relapsed or refractory BCP-ALL.
- Pediatric patients age 1 year or older who have relapsed/refractory, Philadelphia chromosome-negative, CD19-positive BCP-ALL and have received at least two prior therapies or relapsed after allogeneic hematopoietic stem cell transplant.
CHMP’s reversal of opinion
In July, the CHMP recommended against approving blinatumomab to treat patients with MRD based on data from the BLAST trial. Results from this phase 2 trial were published in Blood in April.
The CHMP noted that, although blinatumomab produced MRD negativity in many patients in the BLAST trial, there is no strong evidence that this leads to improved survival.
Given the uncertainty, the CHMP was of the opinion that the benefits of blinatumomab do not outweigh its risks in MRD-positive BCP-ALL patients.
However, Amgen request a re-examination of the CHMP’s opinion, and the CHMP complied.
During the re-examination, the CHMP reviewed all the data and consulted a group of experts.
The experts concluded, and the CHMP agreed, that, although there is no strong evidence of improved survival, the available data indicate a good response to blinatumomab, with around 78% of patients becoming MRD-negative after treatment.
The CHMP also noted that MRD-positive patients have a high risk of relapse and few treatment options.
Therefore, the committee concluded that the benefits of blinatumomab outweigh its risks in this patient population.
The CHMP recommended granting the change to the marketing authorization but also requested that Amgen provide data from ongoing studies of blinatumomab in MRD-positive patients, once those data are available.
FDA approves generic drugs for APL
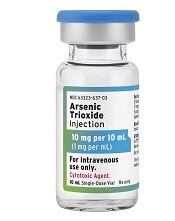
The U.S. Food and Drug Administration (FDA) has now approved three generic arsenic trioxide products for use in patients with acute promyelocytic leukemia (APL).
Two of the products—from Zydus Cadila and Amring Pharmaceuticals—were approved on November 13.
The third—from Fresenius Kabi—was approved in August and launched in the United States last month.
All three injectable arsenic trioxide products (1 mg/mL) are generic versions of Teva’s Trisenox.
Since 2000, Trisenox has been FDA-approved to induce remission and as consolidation therapy for patients with APL who are refractory to, or have relapsed after, retinoid and anthracycline chemotherapy, and whose APL is characterized by presence of the t(15;17) translocation or PML/RAR-alpha gene expression.
In January, the FDA approved Trisenox for use in combination with tretinoin to treat adults with newly diagnosed, low-risk APL with the t(15;17) translocation or PML/RAR-alpha gene expression.
The U.S. Food and Drug Administration (FDA) has now approved three generic arsenic trioxide products for use in patients with acute promyelocytic leukemia (APL).
Two of the products—from Zydus Cadila and Amring Pharmaceuticals—were approved on November 13.
The third—from Fresenius Kabi—was approved in August and launched in the United States last month.
All three injectable arsenic trioxide products (1 mg/mL) are generic versions of Teva’s Trisenox.
Since 2000, Trisenox has been FDA-approved to induce remission and as consolidation therapy for patients with APL who are refractory to, or have relapsed after, retinoid and anthracycline chemotherapy, and whose APL is characterized by presence of the t(15;17) translocation or PML/RAR-alpha gene expression.
In January, the FDA approved Trisenox for use in combination with tretinoin to treat adults with newly diagnosed, low-risk APL with the t(15;17) translocation or PML/RAR-alpha gene expression.
The U.S. Food and Drug Administration (FDA) has now approved three generic arsenic trioxide products for use in patients with acute promyelocytic leukemia (APL).
Two of the products—from Zydus Cadila and Amring Pharmaceuticals—were approved on November 13.
The third—from Fresenius Kabi—was approved in August and launched in the United States last month.
All three injectable arsenic trioxide products (1 mg/mL) are generic versions of Teva’s Trisenox.
Since 2000, Trisenox has been FDA-approved to induce remission and as consolidation therapy for patients with APL who are refractory to, or have relapsed after, retinoid and anthracycline chemotherapy, and whose APL is characterized by presence of the t(15;17) translocation or PML/RAR-alpha gene expression.
In January, the FDA approved Trisenox for use in combination with tretinoin to treat adults with newly diagnosed, low-risk APL with the t(15;17) translocation or PML/RAR-alpha gene expression.
Health Canada approves two tests for CML patients

Health Canada has approved the use of two tests designed to detect BCR-ABL transcripts in patients with chronic myeloid leukemia (CML)—the MRDx® BCR-ABL Test and the QuantideX qPCR BCR-ABL IS Kit.
MolecularMD’s MRDx® BCR-ABL Test is approved as an aid to monitor tyrosine kinase inhibitor (TKI) therapy in Philadelphia chromosome-positive CML patients.
This test is also approved to identify CML patients in the chronic phase being treated with nilotinib who, after sustaining a deep molecular response of MR4.5, may be eligible to stop treatment and be monitored for treatment-free remission.
Asuragen, Inc.’s QuantideX qPCR BCR-ABL IS Kit is approved for use in CML patients, regardless of whether they are taking a TKI or how their disease is being managed.
The kit is designed to detect and quantify major breakpoint (e13a2, e14a2) BCR-ABL1 fusion transcripts for use in monitoring molecular response.
Both the MRDx® BCR-ABL Test and the QuantideX qPCR BCR-ABL IS Kit are approved in the United States as well.
The QuantideX qPCR BCR-ABL IS Kit is also CE marked for clinical use in the European Union.
Health Canada has approved the use of two tests designed to detect BCR-ABL transcripts in patients with chronic myeloid leukemia (CML)—the MRDx® BCR-ABL Test and the QuantideX qPCR BCR-ABL IS Kit.
MolecularMD’s MRDx® BCR-ABL Test is approved as an aid to monitor tyrosine kinase inhibitor (TKI) therapy in Philadelphia chromosome-positive CML patients.
This test is also approved to identify CML patients in the chronic phase being treated with nilotinib who, after sustaining a deep molecular response of MR4.5, may be eligible to stop treatment and be monitored for treatment-free remission.
Asuragen, Inc.’s QuantideX qPCR BCR-ABL IS Kit is approved for use in CML patients, regardless of whether they are taking a TKI or how their disease is being managed.
The kit is designed to detect and quantify major breakpoint (e13a2, e14a2) BCR-ABL1 fusion transcripts for use in monitoring molecular response.
Both the MRDx® BCR-ABL Test and the QuantideX qPCR BCR-ABL IS Kit are approved in the United States as well.
The QuantideX qPCR BCR-ABL IS Kit is also CE marked for clinical use in the European Union.
Health Canada has approved the use of two tests designed to detect BCR-ABL transcripts in patients with chronic myeloid leukemia (CML)—the MRDx® BCR-ABL Test and the QuantideX qPCR BCR-ABL IS Kit.
MolecularMD’s MRDx® BCR-ABL Test is approved as an aid to monitor tyrosine kinase inhibitor (TKI) therapy in Philadelphia chromosome-positive CML patients.
This test is also approved to identify CML patients in the chronic phase being treated with nilotinib who, after sustaining a deep molecular response of MR4.5, may be eligible to stop treatment and be monitored for treatment-free remission.
Asuragen, Inc.’s QuantideX qPCR BCR-ABL IS Kit is approved for use in CML patients, regardless of whether they are taking a TKI or how their disease is being managed.
The kit is designed to detect and quantify major breakpoint (e13a2, e14a2) BCR-ABL1 fusion transcripts for use in monitoring molecular response.
Both the MRDx® BCR-ABL Test and the QuantideX qPCR BCR-ABL IS Kit are approved in the United States as well.
The QuantideX qPCR BCR-ABL IS Kit is also CE marked for clinical use in the European Union.
Azacitidine-nivolumab combo 'encouraging' in AML
The combination of azacitidine and nivolumab produced “encouraging” results in a phase 2 trial of patients with relapsed or refractory acute myeloid leukemia (AML), according to researchers.
The overall response rate was 33%, and the median overall survival (OS) was 6.3 months. However, the researchers identified factors associated with improved response and survival that could be used to select patients for this treatment.
A quarter of patients on this trial had immune-related adverse events (AEs) that were considered related to treatment, and two patients died of AEs that may have been treatment related.
Naval Daver, MD, of the University of Texas MD Anderson Cancer Center, Houston, and his colleagues reported these results in Cancer Discovery.
The trial included 70 patients with a median age of 70 years. More than half of the patients (56%) had de novo AML, and 44% had secondary AML. The median number of prior therapies was two; 64% of patients had received hypomethylating agents, 47% had received targeted therapies, and 19% had received allogeneic stem cell transplant (SCT).
For this trial, patients received azacitidine at 75 mg/m2 on days 1 to 7 and nivolumab at 3 mg/kg on days 1 and 14 of each cycle. The median number of cycles was three. Patients had a median time on study of 3.5 months and reasons for discontinuation included primary refractory disease, relapse after initial response, proceeding to SCT, patient preference, and death.
The most common treatment-related, nonhematologic AEs were constipation, diarrhea, pneumonitis, nausea, and lung infection. The rate of immune-related AEs was 25% (n = 18), with grade 2-4 immune-related AEs occurring in 16 patients (8 with grade 3-4); 14 responded to steroids and were safely rechallenged with nivolumab, according to the researchers.
Nine patients (13%) discontinued nivolumab (but continued with azacitidine) because of AEs. Two patients died of AEs that were considered possibly related to treatment. One death was caused by progressive pneumonia/pneumonitis, and one was caused by hemophagocytic lymphohistiocytosis.
The overall response rate was 33% (n = 23), with 4 patients achieving a complete response (CR) and 11 achieving a CR with incomplete count recovery (CRi). One patient had a partial response, and seven had hematologic improvement in one or more parameter maintained for more than 6 months. Six patients had stable disease lasting more than 6 months.
The researchers noted that the response rate was higher among patients who had not received prior treatment with hypomethylating agents. Additionally, a higher frequency of pretherapy CD3 and CD8 cells in the bone marrow or peripheral blood appeared to predict response.
“In particular, CD3 appeared to have a high sensitivity and specificity rate for predicting response, indicating it might serve as a reliable biomarker for selecting patients for this combination therapy,” Dr. Daver said in a statement.
At a median follow-up of 21.4 months, 81% of patients (n = 57) had died; 16 died on study treatment and 41 died after discontinuation. The median OS overall was 6.3 months, and the median event-free survival was 4.5 months.
The median OS was 16.1 months in patients with CR/CRi, partial response, hematologic improvement, or stable disease and 4.1 months in nonresponders (P less than .0001). This difference was still significant after the researchers censored the three patients who had gone on to SCT in CR/CRi (P less than .001).
The researchers also found that being in first salvage was associated with improved OS in a univariate analysis and in a comparison with historical controls.
Dr. Daver and his colleagues concluded that azacitidine and nivolumab “produced an encouraging response rate and overall survival” in patients with relapsed/refractory AML.
“We believe that implementation of clinical and immune biomarkers to select patients are likely to yield further improved outcomes with these types of therapies in AML,” Dr. Daver said.
This research was supported by Bristol-Myers Squibb, the University of Texas MD Anderson Cancer Center, and the Dick Clark Immunotherapy Research Fund. Individual researchers also reported financial relationships with Bristol-Myers Squibb.
SOURCE: Daver N et al. Cancer Discov. 2018 Nov 8. doi: 10.1158/2159-8290.CD-18-0774.
The combination of azacitidine and nivolumab produced “encouraging” results in a phase 2 trial of patients with relapsed or refractory acute myeloid leukemia (AML), according to researchers.
The overall response rate was 33%, and the median overall survival (OS) was 6.3 months. However, the researchers identified factors associated with improved response and survival that could be used to select patients for this treatment.
A quarter of patients on this trial had immune-related adverse events (AEs) that were considered related to treatment, and two patients died of AEs that may have been treatment related.
Naval Daver, MD, of the University of Texas MD Anderson Cancer Center, Houston, and his colleagues reported these results in Cancer Discovery.
The trial included 70 patients with a median age of 70 years. More than half of the patients (56%) had de novo AML, and 44% had secondary AML. The median number of prior therapies was two; 64% of patients had received hypomethylating agents, 47% had received targeted therapies, and 19% had received allogeneic stem cell transplant (SCT).
For this trial, patients received azacitidine at 75 mg/m2 on days 1 to 7 and nivolumab at 3 mg/kg on days 1 and 14 of each cycle. The median number of cycles was three. Patients had a median time on study of 3.5 months and reasons for discontinuation included primary refractory disease, relapse after initial response, proceeding to SCT, patient preference, and death.
The most common treatment-related, nonhematologic AEs were constipation, diarrhea, pneumonitis, nausea, and lung infection. The rate of immune-related AEs was 25% (n = 18), with grade 2-4 immune-related AEs occurring in 16 patients (8 with grade 3-4); 14 responded to steroids and were safely rechallenged with nivolumab, according to the researchers.
Nine patients (13%) discontinued nivolumab (but continued with azacitidine) because of AEs. Two patients died of AEs that were considered possibly related to treatment. One death was caused by progressive pneumonia/pneumonitis, and one was caused by hemophagocytic lymphohistiocytosis.
The overall response rate was 33% (n = 23), with 4 patients achieving a complete response (CR) and 11 achieving a CR with incomplete count recovery (CRi). One patient had a partial response, and seven had hematologic improvement in one or more parameter maintained for more than 6 months. Six patients had stable disease lasting more than 6 months.
The researchers noted that the response rate was higher among patients who had not received prior treatment with hypomethylating agents. Additionally, a higher frequency of pretherapy CD3 and CD8 cells in the bone marrow or peripheral blood appeared to predict response.
“In particular, CD3 appeared to have a high sensitivity and specificity rate for predicting response, indicating it might serve as a reliable biomarker for selecting patients for this combination therapy,” Dr. Daver said in a statement.
At a median follow-up of 21.4 months, 81% of patients (n = 57) had died; 16 died on study treatment and 41 died after discontinuation. The median OS overall was 6.3 months, and the median event-free survival was 4.5 months.
The median OS was 16.1 months in patients with CR/CRi, partial response, hematologic improvement, or stable disease and 4.1 months in nonresponders (P less than .0001). This difference was still significant after the researchers censored the three patients who had gone on to SCT in CR/CRi (P less than .001).
The researchers also found that being in first salvage was associated with improved OS in a univariate analysis and in a comparison with historical controls.
Dr. Daver and his colleagues concluded that azacitidine and nivolumab “produced an encouraging response rate and overall survival” in patients with relapsed/refractory AML.
“We believe that implementation of clinical and immune biomarkers to select patients are likely to yield further improved outcomes with these types of therapies in AML,” Dr. Daver said.
This research was supported by Bristol-Myers Squibb, the University of Texas MD Anderson Cancer Center, and the Dick Clark Immunotherapy Research Fund. Individual researchers also reported financial relationships with Bristol-Myers Squibb.
SOURCE: Daver N et al. Cancer Discov. 2018 Nov 8. doi: 10.1158/2159-8290.CD-18-0774.
The combination of azacitidine and nivolumab produced “encouraging” results in a phase 2 trial of patients with relapsed or refractory acute myeloid leukemia (AML), according to researchers.
The overall response rate was 33%, and the median overall survival (OS) was 6.3 months. However, the researchers identified factors associated with improved response and survival that could be used to select patients for this treatment.
A quarter of patients on this trial had immune-related adverse events (AEs) that were considered related to treatment, and two patients died of AEs that may have been treatment related.
Naval Daver, MD, of the University of Texas MD Anderson Cancer Center, Houston, and his colleagues reported these results in Cancer Discovery.
The trial included 70 patients with a median age of 70 years. More than half of the patients (56%) had de novo AML, and 44% had secondary AML. The median number of prior therapies was two; 64% of patients had received hypomethylating agents, 47% had received targeted therapies, and 19% had received allogeneic stem cell transplant (SCT).
For this trial, patients received azacitidine at 75 mg/m2 on days 1 to 7 and nivolumab at 3 mg/kg on days 1 and 14 of each cycle. The median number of cycles was three. Patients had a median time on study of 3.5 months and reasons for discontinuation included primary refractory disease, relapse after initial response, proceeding to SCT, patient preference, and death.
The most common treatment-related, nonhematologic AEs were constipation, diarrhea, pneumonitis, nausea, and lung infection. The rate of immune-related AEs was 25% (n = 18), with grade 2-4 immune-related AEs occurring in 16 patients (8 with grade 3-4); 14 responded to steroids and were safely rechallenged with nivolumab, according to the researchers.
Nine patients (13%) discontinued nivolumab (but continued with azacitidine) because of AEs. Two patients died of AEs that were considered possibly related to treatment. One death was caused by progressive pneumonia/pneumonitis, and one was caused by hemophagocytic lymphohistiocytosis.
The overall response rate was 33% (n = 23), with 4 patients achieving a complete response (CR) and 11 achieving a CR with incomplete count recovery (CRi). One patient had a partial response, and seven had hematologic improvement in one or more parameter maintained for more than 6 months. Six patients had stable disease lasting more than 6 months.
The researchers noted that the response rate was higher among patients who had not received prior treatment with hypomethylating agents. Additionally, a higher frequency of pretherapy CD3 and CD8 cells in the bone marrow or peripheral blood appeared to predict response.
“In particular, CD3 appeared to have a high sensitivity and specificity rate for predicting response, indicating it might serve as a reliable biomarker for selecting patients for this combination therapy,” Dr. Daver said in a statement.
At a median follow-up of 21.4 months, 81% of patients (n = 57) had died; 16 died on study treatment and 41 died after discontinuation. The median OS overall was 6.3 months, and the median event-free survival was 4.5 months.
The median OS was 16.1 months in patients with CR/CRi, partial response, hematologic improvement, or stable disease and 4.1 months in nonresponders (P less than .0001). This difference was still significant after the researchers censored the three patients who had gone on to SCT in CR/CRi (P less than .001).
The researchers also found that being in first salvage was associated with improved OS in a univariate analysis and in a comparison with historical controls.
Dr. Daver and his colleagues concluded that azacitidine and nivolumab “produced an encouraging response rate and overall survival” in patients with relapsed/refractory AML.
“We believe that implementation of clinical and immune biomarkers to select patients are likely to yield further improved outcomes with these types of therapies in AML,” Dr. Daver said.
This research was supported by Bristol-Myers Squibb, the University of Texas MD Anderson Cancer Center, and the Dick Clark Immunotherapy Research Fund. Individual researchers also reported financial relationships with Bristol-Myers Squibb.
SOURCE: Daver N et al. Cancer Discov. 2018 Nov 8. doi: 10.1158/2159-8290.CD-18-0774.
FROM CANCER DISCOVERY
Key clinical point:
Major finding: The overall response rate was 33%.
Study details: This phase 2 trial included 70 patients with relapsed/refractory acute myeloid leukemia.
Disclosures: The research was supported by Bristol-Myers Squibb, the University of Texas MD Anderson Cancer Center, and the Dick Clark Immunotherapy Research Fund. Researchers reported financial relationships with Bristol-Myers Squibb.
Source: Daver N et al. Cancer Discov. 2018 Nov 8. doi: 10.1158/2159-8290.CD-18-0774.