User login
MEDALIST: Erythroid maturation agent reduced transfusion burden in MDS
SAN DIEGO – A novel erythroid maturation agent significantly reduced transfusion burden versus placebo in patients with anemia caused by myelodysplastic syndromes (MDS) and ringed sideroblasts, results of a randomized, phase 3 trial demonstrate.
Luspatercept was “very well tolerated” and responses were durable, with approximately 40% of patients remaining transfusion free after 1 year of therapy, said Alan F. List, MD, of Moffitt Cancer Center, Tampa.
“Luspatercept is a potential new therapy that we think could be very effective in patients with lower-risk MDS with ringed sideroblasts who are red blood cell transfusion–dependent,” said Dr. List, senior author of the MEDALIST trial, said in a press conference at the annual meeting of the American Society of Hematology.
The first-in-class erythroid maturation agent is being developed as a treatment for anemia related to MDS and beta-thalassemia, Dr. List said.
In a separate randomized, placebo-controlled, phase 3 study presented at ASH 2018, Maria Domenica Cappellini, MD, of the University of Milan, reported that, in beta-thalassemia patients who were transfusion dependent, luspatercept treatment resulted in a statistically significant reductions in transfusion burden versus placebo, and was generally well tolerated.
Luspatercept is a soluble receptor chimera that binds to an array of ligands in the transforming growth factor–beta superfamily, which is known to be important in suppressing erythropoiesis in patients with MDS, Dr. List said.
The MDS study included patients with very low–, low-, or intermediate-risk disease and ringed sideroblasts who were RBC transfusion–dependent and were refractory to, unresponsive to, or ineligible for first-line treatment with an erythropoiesis-stimulating agent (ESA).
A total of 153 patients were randomly allocated to luspatercept 1.0 mg/kg, administered subcutaneously every 21 days for at least 24 weeks, while 76 were randomized to placebo every 21 days. The primary end point was the proportion of patients achieving RBC transfusion independence for at least 8 weeks during the first 24 weeks of treatment.
A total of 37.9% of luspatercept-treated patients achieved that primary endpoint, compared with 13.2% of placebo-treated patient (P less than .0001), Dr. List reported. The luspatercept-treated patients also had a 52.9% rate of erythroid response, compared with 11.8% in the placebo group (P less than .0001).
There were no differences in treatment-emergent adverse events, severe adverse events, or frequency of progression of acute myeloid leukemia. “This was a very clean drug and a very safe drug,” he said.
The decision to study luspatercept in patients with ringed sideroblasts was based on results of a large, phase 2 European study showing a higher response rate in that subset of MDS patients, according to Dr. List.
That study also included a small number of patients who had not previously received an ESA. Currently underway is a phase 3 trial looking at luspatercept in ESA-naive, lower-risk MDS patients with anemia who require RBC transfusions.
Luspatercept would be a useful therapy to have in clinic for patients with ring sideroblasts, who represent about 25% of patients overall, according to MDS expert David Steensma, MD, of Dana-Farber Cancer Institute and Harvard Medical School, Boston.
“It’s been 12 years since we had an FDA [Food and Drug Administration]-approved drug in MDS, and there have been seven in acute myeloid leukemia in the last year and a half, so it’s our turn, I think,” said Dr. Steensma, who moderated the press conference.
Dr. List reported research funding from Celgene.
SOURCE: List AF et al. ASH 2018, Abstract 1.
SAN DIEGO – A novel erythroid maturation agent significantly reduced transfusion burden versus placebo in patients with anemia caused by myelodysplastic syndromes (MDS) and ringed sideroblasts, results of a randomized, phase 3 trial demonstrate.
Luspatercept was “very well tolerated” and responses were durable, with approximately 40% of patients remaining transfusion free after 1 year of therapy, said Alan F. List, MD, of Moffitt Cancer Center, Tampa.
“Luspatercept is a potential new therapy that we think could be very effective in patients with lower-risk MDS with ringed sideroblasts who are red blood cell transfusion–dependent,” said Dr. List, senior author of the MEDALIST trial, said in a press conference at the annual meeting of the American Society of Hematology.
The first-in-class erythroid maturation agent is being developed as a treatment for anemia related to MDS and beta-thalassemia, Dr. List said.
In a separate randomized, placebo-controlled, phase 3 study presented at ASH 2018, Maria Domenica Cappellini, MD, of the University of Milan, reported that, in beta-thalassemia patients who were transfusion dependent, luspatercept treatment resulted in a statistically significant reductions in transfusion burden versus placebo, and was generally well tolerated.
Luspatercept is a soluble receptor chimera that binds to an array of ligands in the transforming growth factor–beta superfamily, which is known to be important in suppressing erythropoiesis in patients with MDS, Dr. List said.
The MDS study included patients with very low–, low-, or intermediate-risk disease and ringed sideroblasts who were RBC transfusion–dependent and were refractory to, unresponsive to, or ineligible for first-line treatment with an erythropoiesis-stimulating agent (ESA).
A total of 153 patients were randomly allocated to luspatercept 1.0 mg/kg, administered subcutaneously every 21 days for at least 24 weeks, while 76 were randomized to placebo every 21 days. The primary end point was the proportion of patients achieving RBC transfusion independence for at least 8 weeks during the first 24 weeks of treatment.
A total of 37.9% of luspatercept-treated patients achieved that primary endpoint, compared with 13.2% of placebo-treated patient (P less than .0001), Dr. List reported. The luspatercept-treated patients also had a 52.9% rate of erythroid response, compared with 11.8% in the placebo group (P less than .0001).
There were no differences in treatment-emergent adverse events, severe adverse events, or frequency of progression of acute myeloid leukemia. “This was a very clean drug and a very safe drug,” he said.
The decision to study luspatercept in patients with ringed sideroblasts was based on results of a large, phase 2 European study showing a higher response rate in that subset of MDS patients, according to Dr. List.
That study also included a small number of patients who had not previously received an ESA. Currently underway is a phase 3 trial looking at luspatercept in ESA-naive, lower-risk MDS patients with anemia who require RBC transfusions.
Luspatercept would be a useful therapy to have in clinic for patients with ring sideroblasts, who represent about 25% of patients overall, according to MDS expert David Steensma, MD, of Dana-Farber Cancer Institute and Harvard Medical School, Boston.
“It’s been 12 years since we had an FDA [Food and Drug Administration]-approved drug in MDS, and there have been seven in acute myeloid leukemia in the last year and a half, so it’s our turn, I think,” said Dr. Steensma, who moderated the press conference.
Dr. List reported research funding from Celgene.
SOURCE: List AF et al. ASH 2018, Abstract 1.
SAN DIEGO – A novel erythroid maturation agent significantly reduced transfusion burden versus placebo in patients with anemia caused by myelodysplastic syndromes (MDS) and ringed sideroblasts, results of a randomized, phase 3 trial demonstrate.
Luspatercept was “very well tolerated” and responses were durable, with approximately 40% of patients remaining transfusion free after 1 year of therapy, said Alan F. List, MD, of Moffitt Cancer Center, Tampa.
“Luspatercept is a potential new therapy that we think could be very effective in patients with lower-risk MDS with ringed sideroblasts who are red blood cell transfusion–dependent,” said Dr. List, senior author of the MEDALIST trial, said in a press conference at the annual meeting of the American Society of Hematology.
The first-in-class erythroid maturation agent is being developed as a treatment for anemia related to MDS and beta-thalassemia, Dr. List said.
In a separate randomized, placebo-controlled, phase 3 study presented at ASH 2018, Maria Domenica Cappellini, MD, of the University of Milan, reported that, in beta-thalassemia patients who were transfusion dependent, luspatercept treatment resulted in a statistically significant reductions in transfusion burden versus placebo, and was generally well tolerated.
Luspatercept is a soluble receptor chimera that binds to an array of ligands in the transforming growth factor–beta superfamily, which is known to be important in suppressing erythropoiesis in patients with MDS, Dr. List said.
The MDS study included patients with very low–, low-, or intermediate-risk disease and ringed sideroblasts who were RBC transfusion–dependent and were refractory to, unresponsive to, or ineligible for first-line treatment with an erythropoiesis-stimulating agent (ESA).
A total of 153 patients were randomly allocated to luspatercept 1.0 mg/kg, administered subcutaneously every 21 days for at least 24 weeks, while 76 were randomized to placebo every 21 days. The primary end point was the proportion of patients achieving RBC transfusion independence for at least 8 weeks during the first 24 weeks of treatment.
A total of 37.9% of luspatercept-treated patients achieved that primary endpoint, compared with 13.2% of placebo-treated patient (P less than .0001), Dr. List reported. The luspatercept-treated patients also had a 52.9% rate of erythroid response, compared with 11.8% in the placebo group (P less than .0001).
There were no differences in treatment-emergent adverse events, severe adverse events, or frequency of progression of acute myeloid leukemia. “This was a very clean drug and a very safe drug,” he said.
The decision to study luspatercept in patients with ringed sideroblasts was based on results of a large, phase 2 European study showing a higher response rate in that subset of MDS patients, according to Dr. List.
That study also included a small number of patients who had not previously received an ESA. Currently underway is a phase 3 trial looking at luspatercept in ESA-naive, lower-risk MDS patients with anemia who require RBC transfusions.
Luspatercept would be a useful therapy to have in clinic for patients with ring sideroblasts, who represent about 25% of patients overall, according to MDS expert David Steensma, MD, of Dana-Farber Cancer Institute and Harvard Medical School, Boston.
“It’s been 12 years since we had an FDA [Food and Drug Administration]-approved drug in MDS, and there have been seven in acute myeloid leukemia in the last year and a half, so it’s our turn, I think,” said Dr. Steensma, who moderated the press conference.
Dr. List reported research funding from Celgene.
SOURCE: List AF et al. ASH 2018, Abstract 1.
REPORTING FROM ASH 2018
Key clinical point: Luspatercept, a novel erythroid maturation agent, significantly reduced transfusion burden versus placebo in patients with anemia caused by myelodysplastic syndromes and ringed sideroblasts.
Major finding: The proportion of patients achieving RBC transfusion independence for at least 8 weeks during the first 24 weeks of treatment was 37.9% for luspatercept and 13.2% for placebo (P less than .0001).
Study details: A randomized, phase 3 trial including 220 lower-risk myelodysplastic syndromes patients with ringed sideroblasts who were RBC transfusion–dependent.
Disclosures: Dr. List reported research funding from Celgene.
Source: List AF et al. ASH 2018, abstract 1.
Stem cell transplant after CAR T cells may reduce B-ALL relapse risk
SAN DIEGO – A hematopoietic cell transplant following chimeric antigen receptor (CAR) T-cell therapy for B-cell acute lymphocytic leukemia (B-ALL) may reduce late relapse risk in certain patients, a retrospective analysis suggests.
Corinne Summers, MD, of Seattle Children’s Hospital, and her colleagues evaluated the potential benefits of allogeneic hematopoietic cell transplant (HCT) in 50 pediatric and young adult B-ALL patients who had sustained leukemic remission after receiving SCRI-CAR19v1, a CD19-specific CAR T-cell product.
Leukemia-free survival was significantly improved for patients with no history of HCT who received CD19 CAR T-cell therapy followed by consolidative HCT, Dr. Summers reported at the annual meeting of the American Society of Hematology.
However, the benefits of consolidative HCT are unclear for patients with a history of HCT, Dr. Summers said at the meeting, noting that larger studies are needed.
In her video interview at ASH 2018, Dr. Summers talked more about the challenges of late leukemic relapse and the potential role of HCT after CAR T-cell therapy.
Dr. Summers reported no disclosures related to her presentation.
SAN DIEGO – A hematopoietic cell transplant following chimeric antigen receptor (CAR) T-cell therapy for B-cell acute lymphocytic leukemia (B-ALL) may reduce late relapse risk in certain patients, a retrospective analysis suggests.
Corinne Summers, MD, of Seattle Children’s Hospital, and her colleagues evaluated the potential benefits of allogeneic hematopoietic cell transplant (HCT) in 50 pediatric and young adult B-ALL patients who had sustained leukemic remission after receiving SCRI-CAR19v1, a CD19-specific CAR T-cell product.
Leukemia-free survival was significantly improved for patients with no history of HCT who received CD19 CAR T-cell therapy followed by consolidative HCT, Dr. Summers reported at the annual meeting of the American Society of Hematology.
However, the benefits of consolidative HCT are unclear for patients with a history of HCT, Dr. Summers said at the meeting, noting that larger studies are needed.
In her video interview at ASH 2018, Dr. Summers talked more about the challenges of late leukemic relapse and the potential role of HCT after CAR T-cell therapy.
Dr. Summers reported no disclosures related to her presentation.
SAN DIEGO – A hematopoietic cell transplant following chimeric antigen receptor (CAR) T-cell therapy for B-cell acute lymphocytic leukemia (B-ALL) may reduce late relapse risk in certain patients, a retrospective analysis suggests.
Corinne Summers, MD, of Seattle Children’s Hospital, and her colleagues evaluated the potential benefits of allogeneic hematopoietic cell transplant (HCT) in 50 pediatric and young adult B-ALL patients who had sustained leukemic remission after receiving SCRI-CAR19v1, a CD19-specific CAR T-cell product.
Leukemia-free survival was significantly improved for patients with no history of HCT who received CD19 CAR T-cell therapy followed by consolidative HCT, Dr. Summers reported at the annual meeting of the American Society of Hematology.
However, the benefits of consolidative HCT are unclear for patients with a history of HCT, Dr. Summers said at the meeting, noting that larger studies are needed.
In her video interview at ASH 2018, Dr. Summers talked more about the challenges of late leukemic relapse and the potential role of HCT after CAR T-cell therapy.
Dr. Summers reported no disclosures related to her presentation.
REPORTING FROM ASH 2018
FDA warns of serious side effect of AML treatment
The (Idhifa).

Enasidenib is FDA approved to treat adults with relapsed or refractory acute myeloid leukemia (AML) and an IDH2 mutation. The drug is known to be associated with differentiation syndrome, and the drug’s prescribing information contains a boxed warning about this life-threatening condition.
However, the FDA has found that patients and health care providers are missing the signs and symptoms of differentiation syndrome, and some patients are not receiving the necessary treatment in time.
The FDA also is warning that differentiation syndrome has been observed in AML patients taking the IDH1 inhibitor ivosidenib (Tibsovo).
However, the agency has not provided many details on cases related to this drug, which is FDA approved to treat adults with relapsed or refractory AML who have an IDH1 mutation.The FDA says differentiation syndrome may occur anywhere from 10 days to 5 months after starting enasidenib.
The agency is advising health care providers to describe the symptoms of differentiation syndrome to patients, both when starting them on enasidenib and at follow-up visits.
Symptoms of differentiation syndrome include:
- Acute respiratory distress represented by dyspnea and/or hypoxia and a need for supplemental oxygen.
- Pulmonary infiltrates and pleural effusion.
- Fever.
- Lymphadenopathy.
- Bone pain.
- Peripheral edema with rapid weight gain.
- Pericardial effusion.
- Hepatic, renal, and multiorgan dysfunction.
The FDA notes that differentiation syndrome may be mistaken for cardiogenic pulmonary edema, pneumonia, or sepsis.If health care providers suspect differentiation syndrome, they should promptly administer oral or intravenous corticosteroids, such as dexamethasone at 10 mg every 12 hours, according to the FDA. Providers also should monitor hemodynamics until improvement and provide supportive care as necessary.
If patients continue to experience renal dysfunction or severe pulmonary symptoms that require intubation or ventilator support for more than 48 hours after starting corticosteroids, enasidenib should be stopped until signs and symptoms of differentiation syndrome are no longer severe.
Corticosteroids should be tapered only after the symptoms resolve completely, as differentiation syndrome may recur if treatment is stopped too soon. The FDA notes that in the clinical trial that supported approval of enasidenib at least 14% of patients experienced differentiation syndrome.
The manufacturer’s latest safety report includes five deaths (from May 1, 2018, to July 31, 2018) associated with differentiation syndrome in patients taking enasidenib.
Differentiation syndrome was listed as the only cause of death in two cases. In the other cases, patients also had hemorrhagic stroke, pneumonia and sepsis, and sepsis alone.
One patient received systemic corticosteroids promptly but may have died of sepsis during hospitalization. In another patient, differentiation syndrome was not diagnosed or treated promptly.
Treatment details are not available for the remaining three patients who died, according to the FDA.
The FDA has also performed a systematic analysis of differentiation syndrome in 293 patients treated with enasidenib (n = 214) or ivosidenib (n = 179).
With both drugs, the incidence of differentiation syndrome was 19%. The condition was fatal in two of the ivosidenib-treated patients (6%) and two of the enasidenib-treated patients (5%).
Additional results from this analysis are scheduled to be presented at the annual meeting of the American Society of Hematology (Abstract 288).
The (Idhifa).
Enasidenib is FDA approved to treat adults with relapsed or refractory acute myeloid leukemia (AML) and an IDH2 mutation. The drug is known to be associated with differentiation syndrome, and the drug’s prescribing information contains a boxed warning about this life-threatening condition.
However, the FDA has found that patients and health care providers are missing the signs and symptoms of differentiation syndrome, and some patients are not receiving the necessary treatment in time.
The FDA also is warning that differentiation syndrome has been observed in AML patients taking the IDH1 inhibitor ivosidenib (Tibsovo).
However, the agency has not provided many details on cases related to this drug, which is FDA approved to treat adults with relapsed or refractory AML who have an IDH1 mutation.The FDA says differentiation syndrome may occur anywhere from 10 days to 5 months after starting enasidenib.
The agency is advising health care providers to describe the symptoms of differentiation syndrome to patients, both when starting them on enasidenib and at follow-up visits.
Symptoms of differentiation syndrome include:
- Acute respiratory distress represented by dyspnea and/or hypoxia and a need for supplemental oxygen.
- Pulmonary infiltrates and pleural effusion.
- Fever.
- Lymphadenopathy.
- Bone pain.
- Peripheral edema with rapid weight gain.
- Pericardial effusion.
- Hepatic, renal, and multiorgan dysfunction.
The FDA notes that differentiation syndrome may be mistaken for cardiogenic pulmonary edema, pneumonia, or sepsis.If health care providers suspect differentiation syndrome, they should promptly administer oral or intravenous corticosteroids, such as dexamethasone at 10 mg every 12 hours, according to the FDA. Providers also should monitor hemodynamics until improvement and provide supportive care as necessary.
If patients continue to experience renal dysfunction or severe pulmonary symptoms that require intubation or ventilator support for more than 48 hours after starting corticosteroids, enasidenib should be stopped until signs and symptoms of differentiation syndrome are no longer severe.
Corticosteroids should be tapered only after the symptoms resolve completely, as differentiation syndrome may recur if treatment is stopped too soon. The FDA notes that in the clinical trial that supported approval of enasidenib at least 14% of patients experienced differentiation syndrome.
The manufacturer’s latest safety report includes five deaths (from May 1, 2018, to July 31, 2018) associated with differentiation syndrome in patients taking enasidenib.
Differentiation syndrome was listed as the only cause of death in two cases. In the other cases, patients also had hemorrhagic stroke, pneumonia and sepsis, and sepsis alone.
One patient received systemic corticosteroids promptly but may have died of sepsis during hospitalization. In another patient, differentiation syndrome was not diagnosed or treated promptly.
Treatment details are not available for the remaining three patients who died, according to the FDA.
The FDA has also performed a systematic analysis of differentiation syndrome in 293 patients treated with enasidenib (n = 214) or ivosidenib (n = 179).
With both drugs, the incidence of differentiation syndrome was 19%. The condition was fatal in two of the ivosidenib-treated patients (6%) and two of the enasidenib-treated patients (5%).
Additional results from this analysis are scheduled to be presented at the annual meeting of the American Society of Hematology (Abstract 288).
The (Idhifa).
Enasidenib is FDA approved to treat adults with relapsed or refractory acute myeloid leukemia (AML) and an IDH2 mutation. The drug is known to be associated with differentiation syndrome, and the drug’s prescribing information contains a boxed warning about this life-threatening condition.
However, the FDA has found that patients and health care providers are missing the signs and symptoms of differentiation syndrome, and some patients are not receiving the necessary treatment in time.
The FDA also is warning that differentiation syndrome has been observed in AML patients taking the IDH1 inhibitor ivosidenib (Tibsovo).
However, the agency has not provided many details on cases related to this drug, which is FDA approved to treat adults with relapsed or refractory AML who have an IDH1 mutation.The FDA says differentiation syndrome may occur anywhere from 10 days to 5 months after starting enasidenib.
The agency is advising health care providers to describe the symptoms of differentiation syndrome to patients, both when starting them on enasidenib and at follow-up visits.
Symptoms of differentiation syndrome include:
- Acute respiratory distress represented by dyspnea and/or hypoxia and a need for supplemental oxygen.
- Pulmonary infiltrates and pleural effusion.
- Fever.
- Lymphadenopathy.
- Bone pain.
- Peripheral edema with rapid weight gain.
- Pericardial effusion.
- Hepatic, renal, and multiorgan dysfunction.
The FDA notes that differentiation syndrome may be mistaken for cardiogenic pulmonary edema, pneumonia, or sepsis.If health care providers suspect differentiation syndrome, they should promptly administer oral or intravenous corticosteroids, such as dexamethasone at 10 mg every 12 hours, according to the FDA. Providers also should monitor hemodynamics until improvement and provide supportive care as necessary.
If patients continue to experience renal dysfunction or severe pulmonary symptoms that require intubation or ventilator support for more than 48 hours after starting corticosteroids, enasidenib should be stopped until signs and symptoms of differentiation syndrome are no longer severe.
Corticosteroids should be tapered only after the symptoms resolve completely, as differentiation syndrome may recur if treatment is stopped too soon. The FDA notes that in the clinical trial that supported approval of enasidenib at least 14% of patients experienced differentiation syndrome.
The manufacturer’s latest safety report includes five deaths (from May 1, 2018, to July 31, 2018) associated with differentiation syndrome in patients taking enasidenib.
Differentiation syndrome was listed as the only cause of death in two cases. In the other cases, patients also had hemorrhagic stroke, pneumonia and sepsis, and sepsis alone.
One patient received systemic corticosteroids promptly but may have died of sepsis during hospitalization. In another patient, differentiation syndrome was not diagnosed or treated promptly.
Treatment details are not available for the remaining three patients who died, according to the FDA.
The FDA has also performed a systematic analysis of differentiation syndrome in 293 patients treated with enasidenib (n = 214) or ivosidenib (n = 179).
With both drugs, the incidence of differentiation syndrome was 19%. The condition was fatal in two of the ivosidenib-treated patients (6%) and two of the enasidenib-treated patients (5%).
Additional results from this analysis are scheduled to be presented at the annual meeting of the American Society of Hematology (Abstract 288).
Serious side effect of AML treatment going unnoticed, FDA warns
The U.S. Food and Drug Administration (FDA) has released a safety communication warning that cases of differentiation syndrome are going unnoticed in patients treated with the IDH2 inhibitor enasidenib (Idhifa).
Enasidenib is FDA-approved to treat adults with relapsed or refractory acute myeloid leukemia (AML) and an IDH2 mutation.
The drug is known to be associated with differentiation syndrome, and the prescribing information contains a boxed warning about this life-threatening condition.
However, the FDA has found that patients and healthcare providers are missing the signs and symptoms of differentiation syndrome, and some patients are not receiving the necessary treatment in time.
The FDA is also warning that differentiation syndrome has been observed in AML patients taking the IDH1 inhibitor ivosidenib (Tibsovo).
However, the agency has not provided many details on cases related to this drug, which is FDA-approved to treat adults with relapsed or refractory AML who have an IDH1 mutation.
Recognizing differentiation syndrome
The FDA says differentiation syndrome may occur anywhere from 10 days to 5 months after starting enasidenib.
The agency is advising healthcare providers to describe the symptoms of differentiation syndrome to patients, both when starting them on enasidenib and at follow-up visits.
Symptoms of differentiation syndrome include:
- Acute respiratory distress represented by dyspnea and/or hypoxia and a need for supplemental oxygen
- Pulmonary infiltrates and pleural effusion
- Fever
- Lymphadenopathy
- Bone pain
- Peripheral edema with rapid weight gain
- Pericardial effusion
- Hepatic, renal, and multiorgan dysfunction.
The FDA notes that differentiation syndrome may be mistaken for cardiogenic pulmonary edema, pneumonia, or sepsis.
Treatment
If healthcare providers suspect differentiation syndrome, they should promptly administer oral or intravenous corticosteroids, such as dexamethasone at 10 mg every 12 hours, according to the FDA.
Providers should also monitor hemodynamics until improvement and provide supportive care as necessary.
If patients continue to experience renal dysfunction or severe pulmonary symptoms requiring intubation or ventilator support for more than 48 hours after starting corticosteroids, enasidenib should be stopped until signs and symptoms of differentiation syndrome are no longer severe.
Corticosteroids should be tapered only after the symptoms resolve completely, as differentiation syndrome may recur if treatment is stopped too soon.
Cases of differentiation syndrome
The FDA notes that, in the phase 1/2 trial that supported the U.S. approval of enasidenib, at least 14% of patients experienced differentiation syndrome.
The manufacturer’s latest safety report includes 5 deaths (from May 1, 2018, to July 31, 2018) associated with differentiation syndrome in patients taking enasidenib.
Differentiation syndrome was listed as the only cause of death in two cases. In the remaining three cases, patients also had hemorrhagic stroke, pneumonia and sepsis, and sepsis alone.
One patient received systemic corticosteroids promptly but may have died of sepsis during hospitalization. In another patient, differentiation syndrome was not diagnosed or treated promptly. Treatment details are not available for the remaining three patients, according to the FDA.
The FDA has also performed a systematic analysis of differentiation syndrome in 293 patients treated with enasidenib (n=214) or ivosidenib (n=179).
With both drugs, the incidence of differentiation syndrome was 19%. The condition was fatal in 6% (n=2) of ivosidenib-treated patients and 5% (n=2) of enasidenib-treated patients.
Additional results from this analysis are scheduled to be presented at the 2018 ASH Annual Meeting (abstract 288).
The U.S. Food and Drug Administration (FDA) has released a safety communication warning that cases of differentiation syndrome are going unnoticed in patients treated with the IDH2 inhibitor enasidenib (Idhifa).
Enasidenib is FDA-approved to treat adults with relapsed or refractory acute myeloid leukemia (AML) and an IDH2 mutation.
The drug is known to be associated with differentiation syndrome, and the prescribing information contains a boxed warning about this life-threatening condition.
However, the FDA has found that patients and healthcare providers are missing the signs and symptoms of differentiation syndrome, and some patients are not receiving the necessary treatment in time.
The FDA is also warning that differentiation syndrome has been observed in AML patients taking the IDH1 inhibitor ivosidenib (Tibsovo).
However, the agency has not provided many details on cases related to this drug, which is FDA-approved to treat adults with relapsed or refractory AML who have an IDH1 mutation.
Recognizing differentiation syndrome
The FDA says differentiation syndrome may occur anywhere from 10 days to 5 months after starting enasidenib.
The agency is advising healthcare providers to describe the symptoms of differentiation syndrome to patients, both when starting them on enasidenib and at follow-up visits.
Symptoms of differentiation syndrome include:
- Acute respiratory distress represented by dyspnea and/or hypoxia and a need for supplemental oxygen
- Pulmonary infiltrates and pleural effusion
- Fever
- Lymphadenopathy
- Bone pain
- Peripheral edema with rapid weight gain
- Pericardial effusion
- Hepatic, renal, and multiorgan dysfunction.
The FDA notes that differentiation syndrome may be mistaken for cardiogenic pulmonary edema, pneumonia, or sepsis.
Treatment
If healthcare providers suspect differentiation syndrome, they should promptly administer oral or intravenous corticosteroids, such as dexamethasone at 10 mg every 12 hours, according to the FDA.
Providers should also monitor hemodynamics until improvement and provide supportive care as necessary.
If patients continue to experience renal dysfunction or severe pulmonary symptoms requiring intubation or ventilator support for more than 48 hours after starting corticosteroids, enasidenib should be stopped until signs and symptoms of differentiation syndrome are no longer severe.
Corticosteroids should be tapered only after the symptoms resolve completely, as differentiation syndrome may recur if treatment is stopped too soon.
Cases of differentiation syndrome
The FDA notes that, in the phase 1/2 trial that supported the U.S. approval of enasidenib, at least 14% of patients experienced differentiation syndrome.
The manufacturer’s latest safety report includes 5 deaths (from May 1, 2018, to July 31, 2018) associated with differentiation syndrome in patients taking enasidenib.
Differentiation syndrome was listed as the only cause of death in two cases. In the remaining three cases, patients also had hemorrhagic stroke, pneumonia and sepsis, and sepsis alone.
One patient received systemic corticosteroids promptly but may have died of sepsis during hospitalization. In another patient, differentiation syndrome was not diagnosed or treated promptly. Treatment details are not available for the remaining three patients, according to the FDA.
The FDA has also performed a systematic analysis of differentiation syndrome in 293 patients treated with enasidenib (n=214) or ivosidenib (n=179).
With both drugs, the incidence of differentiation syndrome was 19%. The condition was fatal in 6% (n=2) of ivosidenib-treated patients and 5% (n=2) of enasidenib-treated patients.
Additional results from this analysis are scheduled to be presented at the 2018 ASH Annual Meeting (abstract 288).
The U.S. Food and Drug Administration (FDA) has released a safety communication warning that cases of differentiation syndrome are going unnoticed in patients treated with the IDH2 inhibitor enasidenib (Idhifa).
Enasidenib is FDA-approved to treat adults with relapsed or refractory acute myeloid leukemia (AML) and an IDH2 mutation.
The drug is known to be associated with differentiation syndrome, and the prescribing information contains a boxed warning about this life-threatening condition.
However, the FDA has found that patients and healthcare providers are missing the signs and symptoms of differentiation syndrome, and some patients are not receiving the necessary treatment in time.
The FDA is also warning that differentiation syndrome has been observed in AML patients taking the IDH1 inhibitor ivosidenib (Tibsovo).
However, the agency has not provided many details on cases related to this drug, which is FDA-approved to treat adults with relapsed or refractory AML who have an IDH1 mutation.
Recognizing differentiation syndrome
The FDA says differentiation syndrome may occur anywhere from 10 days to 5 months after starting enasidenib.
The agency is advising healthcare providers to describe the symptoms of differentiation syndrome to patients, both when starting them on enasidenib and at follow-up visits.
Symptoms of differentiation syndrome include:
- Acute respiratory distress represented by dyspnea and/or hypoxia and a need for supplemental oxygen
- Pulmonary infiltrates and pleural effusion
- Fever
- Lymphadenopathy
- Bone pain
- Peripheral edema with rapid weight gain
- Pericardial effusion
- Hepatic, renal, and multiorgan dysfunction.
The FDA notes that differentiation syndrome may be mistaken for cardiogenic pulmonary edema, pneumonia, or sepsis.
Treatment
If healthcare providers suspect differentiation syndrome, they should promptly administer oral or intravenous corticosteroids, such as dexamethasone at 10 mg every 12 hours, according to the FDA.
Providers should also monitor hemodynamics until improvement and provide supportive care as necessary.
If patients continue to experience renal dysfunction or severe pulmonary symptoms requiring intubation or ventilator support for more than 48 hours after starting corticosteroids, enasidenib should be stopped until signs and symptoms of differentiation syndrome are no longer severe.
Corticosteroids should be tapered only after the symptoms resolve completely, as differentiation syndrome may recur if treatment is stopped too soon.
Cases of differentiation syndrome
The FDA notes that, in the phase 1/2 trial that supported the U.S. approval of enasidenib, at least 14% of patients experienced differentiation syndrome.
The manufacturer’s latest safety report includes 5 deaths (from May 1, 2018, to July 31, 2018) associated with differentiation syndrome in patients taking enasidenib.
Differentiation syndrome was listed as the only cause of death in two cases. In the remaining three cases, patients also had hemorrhagic stroke, pneumonia and sepsis, and sepsis alone.
One patient received systemic corticosteroids promptly but may have died of sepsis during hospitalization. In another patient, differentiation syndrome was not diagnosed or treated promptly. Treatment details are not available for the remaining three patients, according to the FDA.
The FDA has also performed a systematic analysis of differentiation syndrome in 293 patients treated with enasidenib (n=214) or ivosidenib (n=179).
With both drugs, the incidence of differentiation syndrome was 19%. The condition was fatal in 6% (n=2) of ivosidenib-treated patients and 5% (n=2) of enasidenib-treated patients.
Additional results from this analysis are scheduled to be presented at the 2018 ASH Annual Meeting (abstract 288).
FDA approves gilteritinib for relapsed/refractory AML

The U.S. Food and Drug Administration (FDA) has approved gilteritinib (Xospata) for use in adults who have relapsed or refractory acute myeloid leukemia (AML) with a FLT3 mutation, as detected by an FDA-approved test.
The FDA also expanded the approved indication for the LeukoStrat CDx FLT3 Mutation Assay to include use with gilteritinib.
The LeukoStrat CDx FLT3 Mutation Assay, developed by Invivoscribe Technologies, Inc., is used to detect FLT3 mutations in patients with AML.
Gilteritinib, developed by Astellas Pharma, has demonstrated inhibitory activity against FLT3 internal tandem duplication (ITD) and FLT3 tyrosine kinase domain.
The FDA’s approval of gilteritinib was based on an interim analysis of the ADMIRAL trial (NCT02421939).
The trial enrolled adults with relapsed or refractory AML who had a FLT3 ITD, D835, or I836 mutation, according to the LeukoStrat CDx FLT3 Mutation Assay.
Patients received gilteritinib at 120 mg daily until they developed unacceptable toxicity or did not show a clinical benefit.
Efficacy results are available for 138 patients, with a median follow-up of 4.6 months (range, 2.8 to 15.8).
The complete response (CR) rate was 11.6% (16/138), the rate of CR with partial hematologic recovery (CRh) was 9.4% (13/138), and the rate of CR/CRh was 21% (29/138).
The median duration of CR/CRh was 4.6 months.
There were 106 patients who were transfusion-dependent at baseline, and 33 of these patients (31.1%) became transfusion-independent during the post-baseline period.
Seventeen of the 32 patients (53.1%) who were transfusion-independent at baseline remained transfusion-independent.
Safety results are available for 292 patients. The median duration of exposure to gilteritinib in this group was 3 months (range, 0.1 to 42.8).
The most common adverse events were myalgia/arthralgia (42%), transaminase increase (41%), fatigue/malaise (40%), fever (35%), noninfectious diarrhea (34%), dyspnea (34%), edema (34%), rash (30%), pneumonia (30%), nausea (27%), constipation (27%), stomatitis (26%), cough (25%), headache (21%), hypotension (21%), dizziness (20%), and vomiting (20%).
Eight percent of patients (n=22) discontinued gilteritinib due to adverse events. The most common were pneumonia (2%), sepsis (2%), and dyspnea (1%).
For more details on the ADMIRAL trial and gilteritinib, see the full prescribing information.
The U.S. Food and Drug Administration (FDA) has approved gilteritinib (Xospata) for use in adults who have relapsed or refractory acute myeloid leukemia (AML) with a FLT3 mutation, as detected by an FDA-approved test.
The FDA also expanded the approved indication for the LeukoStrat CDx FLT3 Mutation Assay to include use with gilteritinib.
The LeukoStrat CDx FLT3 Mutation Assay, developed by Invivoscribe Technologies, Inc., is used to detect FLT3 mutations in patients with AML.
Gilteritinib, developed by Astellas Pharma, has demonstrated inhibitory activity against FLT3 internal tandem duplication (ITD) and FLT3 tyrosine kinase domain.
The FDA’s approval of gilteritinib was based on an interim analysis of the ADMIRAL trial (NCT02421939).
The trial enrolled adults with relapsed or refractory AML who had a FLT3 ITD, D835, or I836 mutation, according to the LeukoStrat CDx FLT3 Mutation Assay.
Patients received gilteritinib at 120 mg daily until they developed unacceptable toxicity or did not show a clinical benefit.
Efficacy results are available for 138 patients, with a median follow-up of 4.6 months (range, 2.8 to 15.8).
The complete response (CR) rate was 11.6% (16/138), the rate of CR with partial hematologic recovery (CRh) was 9.4% (13/138), and the rate of CR/CRh was 21% (29/138).
The median duration of CR/CRh was 4.6 months.
There were 106 patients who were transfusion-dependent at baseline, and 33 of these patients (31.1%) became transfusion-independent during the post-baseline period.
Seventeen of the 32 patients (53.1%) who were transfusion-independent at baseline remained transfusion-independent.
Safety results are available for 292 patients. The median duration of exposure to gilteritinib in this group was 3 months (range, 0.1 to 42.8).
The most common adverse events were myalgia/arthralgia (42%), transaminase increase (41%), fatigue/malaise (40%), fever (35%), noninfectious diarrhea (34%), dyspnea (34%), edema (34%), rash (30%), pneumonia (30%), nausea (27%), constipation (27%), stomatitis (26%), cough (25%), headache (21%), hypotension (21%), dizziness (20%), and vomiting (20%).
Eight percent of patients (n=22) discontinued gilteritinib due to adverse events. The most common were pneumonia (2%), sepsis (2%), and dyspnea (1%).
For more details on the ADMIRAL trial and gilteritinib, see the full prescribing information.
The U.S. Food and Drug Administration (FDA) has approved gilteritinib (Xospata) for use in adults who have relapsed or refractory acute myeloid leukemia (AML) with a FLT3 mutation, as detected by an FDA-approved test.
The FDA also expanded the approved indication for the LeukoStrat CDx FLT3 Mutation Assay to include use with gilteritinib.
The LeukoStrat CDx FLT3 Mutation Assay, developed by Invivoscribe Technologies, Inc., is used to detect FLT3 mutations in patients with AML.
Gilteritinib, developed by Astellas Pharma, has demonstrated inhibitory activity against FLT3 internal tandem duplication (ITD) and FLT3 tyrosine kinase domain.
The FDA’s approval of gilteritinib was based on an interim analysis of the ADMIRAL trial (NCT02421939).
The trial enrolled adults with relapsed or refractory AML who had a FLT3 ITD, D835, or I836 mutation, according to the LeukoStrat CDx FLT3 Mutation Assay.
Patients received gilteritinib at 120 mg daily until they developed unacceptable toxicity or did not show a clinical benefit.
Efficacy results are available for 138 patients, with a median follow-up of 4.6 months (range, 2.8 to 15.8).
The complete response (CR) rate was 11.6% (16/138), the rate of CR with partial hematologic recovery (CRh) was 9.4% (13/138), and the rate of CR/CRh was 21% (29/138).
The median duration of CR/CRh was 4.6 months.
There were 106 patients who were transfusion-dependent at baseline, and 33 of these patients (31.1%) became transfusion-independent during the post-baseline period.
Seventeen of the 32 patients (53.1%) who were transfusion-independent at baseline remained transfusion-independent.
Safety results are available for 292 patients. The median duration of exposure to gilteritinib in this group was 3 months (range, 0.1 to 42.8).
The most common adverse events were myalgia/arthralgia (42%), transaminase increase (41%), fatigue/malaise (40%), fever (35%), noninfectious diarrhea (34%), dyspnea (34%), edema (34%), rash (30%), pneumonia (30%), nausea (27%), constipation (27%), stomatitis (26%), cough (25%), headache (21%), hypotension (21%), dizziness (20%), and vomiting (20%).
Eight percent of patients (n=22) discontinued gilteritinib due to adverse events. The most common were pneumonia (2%), sepsis (2%), and dyspnea (1%).
For more details on the ADMIRAL trial and gilteritinib, see the full prescribing information.
FDA approves gilteritinib for AML with FLT3 mutation
as detected by an FDA-approved test.
The FDA also expanded the approved indication for the LeukoStrat CDx FLT3 Mutation Assay to include use with gilteritinib. The LeukoStrat CDx FLT3 Mutation Assay, developed by Invivoscribe, is used to detect the FLT3 mutation in patients with AML.
Gilteritinib, developed by Astellas Pharma, has demonstrated inhibitory activity against FLT3 internal tandem duplication and FLT3 tyrosine kinase domain.
The FDA’s approval of gilteritinib was based on an interim analysis of the ADMIRAL trial, which enrolled adults with relapsed or refractory AML who had a FLT3 ITD, D835 or I836 mutation, according to the LeukoStrat CDx FLT3 Mutation Assay.
Patients received gilteritinib at 120 mg daily until they developed unacceptable toxicity or did not show a clinical benefit. Efficacy results are available for 138 patients, with a median follow-up of 4.6 months.
The complete response (CR) rate was 11.6% (16/138), the CR rate with partial hematologic recovery (CRh) was 9.4% (13/138), and the CR/CRh rate was 21% (29/138). The median duration of CR/CRh was 4.6 months.
There were 106 patients who were transfusion dependent at baseline, and 33 of these patients (31.1%) became transfusion independent during the post-baseline period.
Seventeen of the 32 patients (53.1%) who were transfusion independent at baseline remained transfusion independent.
Safety results are available for 292 patients. The median duration of exposure to gilteritinib in this group was 3 months.
The most common adverse events were myalgia/arthralgia, transaminase increase, fatigue/malaise, fever, noninfectious diarrhea, dyspnea, edema, rash, pneumonia, nausea, constipation, stomatitis, cough, headache, hypotension, dizziness, and vomiting.
A total of 8% of patients (n = 22) discontinued gilteritinib because of adverse events, the most common of which were pneumonia (2%), sepsis (2%), and dyspnea (1%).
as detected by an FDA-approved test.
The FDA also expanded the approved indication for the LeukoStrat CDx FLT3 Mutation Assay to include use with gilteritinib. The LeukoStrat CDx FLT3 Mutation Assay, developed by Invivoscribe, is used to detect the FLT3 mutation in patients with AML.
Gilteritinib, developed by Astellas Pharma, has demonstrated inhibitory activity against FLT3 internal tandem duplication and FLT3 tyrosine kinase domain.
The FDA’s approval of gilteritinib was based on an interim analysis of the ADMIRAL trial, which enrolled adults with relapsed or refractory AML who had a FLT3 ITD, D835 or I836 mutation, according to the LeukoStrat CDx FLT3 Mutation Assay.
Patients received gilteritinib at 120 mg daily until they developed unacceptable toxicity or did not show a clinical benefit. Efficacy results are available for 138 patients, with a median follow-up of 4.6 months.
The complete response (CR) rate was 11.6% (16/138), the CR rate with partial hematologic recovery (CRh) was 9.4% (13/138), and the CR/CRh rate was 21% (29/138). The median duration of CR/CRh was 4.6 months.
There were 106 patients who were transfusion dependent at baseline, and 33 of these patients (31.1%) became transfusion independent during the post-baseline period.
Seventeen of the 32 patients (53.1%) who were transfusion independent at baseline remained transfusion independent.
Safety results are available for 292 patients. The median duration of exposure to gilteritinib in this group was 3 months.
The most common adverse events were myalgia/arthralgia, transaminase increase, fatigue/malaise, fever, noninfectious diarrhea, dyspnea, edema, rash, pneumonia, nausea, constipation, stomatitis, cough, headache, hypotension, dizziness, and vomiting.
A total of 8% of patients (n = 22) discontinued gilteritinib because of adverse events, the most common of which were pneumonia (2%), sepsis (2%), and dyspnea (1%).
as detected by an FDA-approved test.
The FDA also expanded the approved indication for the LeukoStrat CDx FLT3 Mutation Assay to include use with gilteritinib. The LeukoStrat CDx FLT3 Mutation Assay, developed by Invivoscribe, is used to detect the FLT3 mutation in patients with AML.
Gilteritinib, developed by Astellas Pharma, has demonstrated inhibitory activity against FLT3 internal tandem duplication and FLT3 tyrosine kinase domain.
The FDA’s approval of gilteritinib was based on an interim analysis of the ADMIRAL trial, which enrolled adults with relapsed or refractory AML who had a FLT3 ITD, D835 or I836 mutation, according to the LeukoStrat CDx FLT3 Mutation Assay.
Patients received gilteritinib at 120 mg daily until they developed unacceptable toxicity or did not show a clinical benefit. Efficacy results are available for 138 patients, with a median follow-up of 4.6 months.
The complete response (CR) rate was 11.6% (16/138), the CR rate with partial hematologic recovery (CRh) was 9.4% (13/138), and the CR/CRh rate was 21% (29/138). The median duration of CR/CRh was 4.6 months.
There were 106 patients who were transfusion dependent at baseline, and 33 of these patients (31.1%) became transfusion independent during the post-baseline period.
Seventeen of the 32 patients (53.1%) who were transfusion independent at baseline remained transfusion independent.
Safety results are available for 292 patients. The median duration of exposure to gilteritinib in this group was 3 months.
The most common adverse events were myalgia/arthralgia, transaminase increase, fatigue/malaise, fever, noninfectious diarrhea, dyspnea, edema, rash, pneumonia, nausea, constipation, stomatitis, cough, headache, hypotension, dizziness, and vomiting.
A total of 8% of patients (n = 22) discontinued gilteritinib because of adverse events, the most common of which were pneumonia (2%), sepsis (2%), and dyspnea (1%).
Pegfilgrastim biosimilar approved by EC

The European Commission (EC) has granted marketing authorization for Sandoz’s pegfilgrastim product Ziextenzo®, a biosimilar of Amgen’s Neulasta.
Ziextenzo is approved for the same use as the reference medicine—to reduce the duration of neutropenia and the incidence of febrile neutropenia in adults receiving cytotoxic chemotherapy for malignancies except chronic myeloid leukemia and myelodysplastic syndromes.
The approval is valid in all countries of the European Union as well as Norway, Iceland, and Liechtenstein.
The EC’s approval was based on research suggesting Ziextenzo is comparable to Neulasta in terms of safety, efficacy, pharmacokinetics, and pharmacodynamics.1,2,3,4
1. Blackwell K. et al. Pooled analysis of two randomized, double-blind trials comparing proposed biosimilar LA-EP2006 with reference pegfilgrastim in breast cancer. Ann Oncol 28, 2272-2277 (2017).
2. Nakov R. et al. Proposed biosimilar pegfilgrastim LA-EP2006 shows similarity in pharmacokinetics and pharmacodynamics to reference pegfilgrastim in healthy subjects. 2017 San Antonio Breast Cancer Symposium, abstract P3-14-10.
3. Blackwell K. et al. A Comparison of Proposed Biosimilar LA-EP2006 and Reference Pegfilgrastim for the Prevention of Neutropenia in Patients With Early-Stage Breast Cancer Receiving Myelosuppressive Adjuvant or Neoadjuvant Chemotherapy: Pegfilgrastim Randomized Oncology (Supportive Care) Trial to Evaluate Comparative Treatment (PROTECT-2), a Phase III, Randomized, Double-Blind Trial. Oncologist 21, 789-794 (2016).
4. Harbeck N. et al. Randomized, double-blind study comparing proposed biosimilar LA-EP2006 with reference pegfilgrastim in breast cancer. Future Oncol 12, 1359-1367 (2016).
The European Commission (EC) has granted marketing authorization for Sandoz’s pegfilgrastim product Ziextenzo®, a biosimilar of Amgen’s Neulasta.
Ziextenzo is approved for the same use as the reference medicine—to reduce the duration of neutropenia and the incidence of febrile neutropenia in adults receiving cytotoxic chemotherapy for malignancies except chronic myeloid leukemia and myelodysplastic syndromes.
The approval is valid in all countries of the European Union as well as Norway, Iceland, and Liechtenstein.
The EC’s approval was based on research suggesting Ziextenzo is comparable to Neulasta in terms of safety, efficacy, pharmacokinetics, and pharmacodynamics.1,2,3,4
1. Blackwell K. et al. Pooled analysis of two randomized, double-blind trials comparing proposed biosimilar LA-EP2006 with reference pegfilgrastim in breast cancer. Ann Oncol 28, 2272-2277 (2017).
2. Nakov R. et al. Proposed biosimilar pegfilgrastim LA-EP2006 shows similarity in pharmacokinetics and pharmacodynamics to reference pegfilgrastim in healthy subjects. 2017 San Antonio Breast Cancer Symposium, abstract P3-14-10.
3. Blackwell K. et al. A Comparison of Proposed Biosimilar LA-EP2006 and Reference Pegfilgrastim for the Prevention of Neutropenia in Patients With Early-Stage Breast Cancer Receiving Myelosuppressive Adjuvant or Neoadjuvant Chemotherapy: Pegfilgrastim Randomized Oncology (Supportive Care) Trial to Evaluate Comparative Treatment (PROTECT-2), a Phase III, Randomized, Double-Blind Trial. Oncologist 21, 789-794 (2016).
4. Harbeck N. et al. Randomized, double-blind study comparing proposed biosimilar LA-EP2006 with reference pegfilgrastim in breast cancer. Future Oncol 12, 1359-1367 (2016).
The European Commission (EC) has granted marketing authorization for Sandoz’s pegfilgrastim product Ziextenzo®, a biosimilar of Amgen’s Neulasta.
Ziextenzo is approved for the same use as the reference medicine—to reduce the duration of neutropenia and the incidence of febrile neutropenia in adults receiving cytotoxic chemotherapy for malignancies except chronic myeloid leukemia and myelodysplastic syndromes.
The approval is valid in all countries of the European Union as well as Norway, Iceland, and Liechtenstein.
The EC’s approval was based on research suggesting Ziextenzo is comparable to Neulasta in terms of safety, efficacy, pharmacokinetics, and pharmacodynamics.1,2,3,4
1. Blackwell K. et al. Pooled analysis of two randomized, double-blind trials comparing proposed biosimilar LA-EP2006 with reference pegfilgrastim in breast cancer. Ann Oncol 28, 2272-2277 (2017).
2. Nakov R. et al. Proposed biosimilar pegfilgrastim LA-EP2006 shows similarity in pharmacokinetics and pharmacodynamics to reference pegfilgrastim in healthy subjects. 2017 San Antonio Breast Cancer Symposium, abstract P3-14-10.
3. Blackwell K. et al. A Comparison of Proposed Biosimilar LA-EP2006 and Reference Pegfilgrastim for the Prevention of Neutropenia in Patients With Early-Stage Breast Cancer Receiving Myelosuppressive Adjuvant or Neoadjuvant Chemotherapy: Pegfilgrastim Randomized Oncology (Supportive Care) Trial to Evaluate Comparative Treatment (PROTECT-2), a Phase III, Randomized, Double-Blind Trial. Oncologist 21, 789-794 (2016).
4. Harbeck N. et al. Randomized, double-blind study comparing proposed biosimilar LA-EP2006 with reference pegfilgrastim in breast cancer. Future Oncol 12, 1359-1367 (2016).
EC approves pegfilgrastim biosimilar

The European Commission (EC) has approved Mundipharma’s pegfilgrastim product Pelmeg, a biosimilar of Amgen’s Neulasta.
Pelmeg is approved for use in reducing the duration of neutropenia and the incidence of febrile neutropenia in adults who receive cytotoxic chemotherapy for malignancies, with the exceptions of chronic myeloid leukemia and myelodysplastic syndromes.
The approval is valid in all countries of the European Union as well as Norway, Iceland, and Liechtenstein.
The EC’s approval of Pelmeg was supported by research showing pharmacokinetic comparability between Pelmeg and Neulasta at a dose of 6 mg, pharmacodynamic comparability at doses of 6 mg and 3 mg, and no clinically meaningful differences in the safety and immunogenicity profiles of Pelmeg and Neulasta.1,2,3
1. Roth K. et al. Demonstration of pharmacokinetic and pharmacodynamic comparability in healthy volunteers for B12019, a proposed pegfilgrastim biosimilar. ECCO 2017, abstract 241.
2. Roth K. et al. Comparability of pharmacodynamics and immunogenicity of B12019, a proposed pegfilgrastim biosimilar to Neulasta®. ASH 2017, abstract 1002.
3. Roth K. et al. Pharmacokinetic and pharmacodynamic comparability of B12019, a proposed pegfilgrastim biosimilar. ESMO 2017, poster 1573.
The European Commission (EC) has approved Mundipharma’s pegfilgrastim product Pelmeg, a biosimilar of Amgen’s Neulasta.
Pelmeg is approved for use in reducing the duration of neutropenia and the incidence of febrile neutropenia in adults who receive cytotoxic chemotherapy for malignancies, with the exceptions of chronic myeloid leukemia and myelodysplastic syndromes.
The approval is valid in all countries of the European Union as well as Norway, Iceland, and Liechtenstein.
The EC’s approval of Pelmeg was supported by research showing pharmacokinetic comparability between Pelmeg and Neulasta at a dose of 6 mg, pharmacodynamic comparability at doses of 6 mg and 3 mg, and no clinically meaningful differences in the safety and immunogenicity profiles of Pelmeg and Neulasta.1,2,3
1. Roth K. et al. Demonstration of pharmacokinetic and pharmacodynamic comparability in healthy volunteers for B12019, a proposed pegfilgrastim biosimilar. ECCO 2017, abstract 241.
2. Roth K. et al. Comparability of pharmacodynamics and immunogenicity of B12019, a proposed pegfilgrastim biosimilar to Neulasta®. ASH 2017, abstract 1002.
3. Roth K. et al. Pharmacokinetic and pharmacodynamic comparability of B12019, a proposed pegfilgrastim biosimilar. ESMO 2017, poster 1573.
The European Commission (EC) has approved Mundipharma’s pegfilgrastim product Pelmeg, a biosimilar of Amgen’s Neulasta.
Pelmeg is approved for use in reducing the duration of neutropenia and the incidence of febrile neutropenia in adults who receive cytotoxic chemotherapy for malignancies, with the exceptions of chronic myeloid leukemia and myelodysplastic syndromes.
The approval is valid in all countries of the European Union as well as Norway, Iceland, and Liechtenstein.
The EC’s approval of Pelmeg was supported by research showing pharmacokinetic comparability between Pelmeg and Neulasta at a dose of 6 mg, pharmacodynamic comparability at doses of 6 mg and 3 mg, and no clinically meaningful differences in the safety and immunogenicity profiles of Pelmeg and Neulasta.1,2,3
1. Roth K. et al. Demonstration of pharmacokinetic and pharmacodynamic comparability in healthy volunteers for B12019, a proposed pegfilgrastim biosimilar. ECCO 2017, abstract 241.
2. Roth K. et al. Comparability of pharmacodynamics and immunogenicity of B12019, a proposed pegfilgrastim biosimilar to Neulasta®. ASH 2017, abstract 1002.
3. Roth K. et al. Pharmacokinetic and pharmacodynamic comparability of B12019, a proposed pegfilgrastim biosimilar. ESMO 2017, poster 1573.
FDA grants priority review to quizartinib
The U.S. Food and Drug Administration (FDA) has accepted for priority review a new drug application (NDA) for the FLT3 inhibitor quizartinib.
With this NDA, Daiichi Sankyo is seeking approval for quizartinib to treat adults with relapsed/refractory FLT3-ITD acute myeloid leukemia (AML).
The FDA grants priority review to applications for products that are expected to provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The FDA aims to take action on a priority review application within 6 months rather than the standard 10 months.
The FDA is expected to make a decision on the quizartinib NDA by May 25, 2019.
In addition to priority review, quizartinib has breakthrough therapy designation and fast track designation from the FDA.
Trial results
The NDA for quizartinib is supported by results from the phase 3 QuANTUM-R study. Topline results from this study were presented at the 23rd Congress of the European Hematology Association in June, and new analyses are set to be presented at the 2018 ASH Annual Meeting in December (abstract 563).
QuANTUM-R enrolled adults with FLT3-ITD AML (at least 3% FLT3-ITD allelic ratio) who had refractory disease or had relapsed within 6 months of their first complete response (CR).
Patients were randomized to receive once-daily treatment with quizartinib (n=245) or a salvage chemotherapy regimen (n=122)—low-dose cytarabine (LoDAC, n=29); combination mitoxantrone, etoposide, and cytarabine (MEC, n=40); or combination fludarabine, cytarabine, and idarubicin (FLAG-IDA, n=53).
Patients who responded to treatment could proceed to hematopoietic stem cell transplant (HSCT), and those in the quizartinib arm could resume quizartinib after HSCT.
In all, 241 patients received quizartinib, and 94 received salvage chemotherapy—LoDAC (n=22), MEC (n=25), and FLAG-IDA (n=47). Of the 28 patients in the chemotherapy group who were not treated, most withdrew consent.
Thirty-two percent of quizartinib-treated patients and 12% of the chemotherapy group went on to HSCT.
Efficacy
The median follow-up was 23.5 months. The efficacy results include all randomized patients.
The overall response rate was 69% in the quizartinib arm and 30% in the chemotherapy arm. The composite CR rate was 48% in the quizartinib arm and 27% in the chemotherapy arm. This includes:
- The CR rate (4% and 1%, respectively)
- The rate of CR with incomplete platelet recovery (4% and 0%, respectively)
- The rate of CR with incomplete hematologic recovery (40% and 26%, respectively).
The median event-free survival was 6.0 weeks in the quizartinib arm and 3.7 weeks in the chemotherapy arm (hazard ratio=0.90, P=0.1071).
The median overall survival was 6.2 months in the quizartinib arm and 4.7 months in the chemotherapy arm (hazard ratio=0.76, P=0.0177). The 1-year overall survival rate was 27% and 20%, respectively.
Safety
The safety results include only patients who received their assigned treatment.
Grade 3 or higher hematologic treatment-emergent adverse events occurring in at least 5% of patients (in the quizartinib and chemotherapy groups, respectively) included:
- Thrombocytopenia (35% and 34%)
- Anemia (30% and 29%)
- Neutropenia (32% and 25%)
- Febrile neutropenia (31% and 21%)
- Leukopenia (17% and 16%).
Grade 3 or higher non-hematologic treatment-emergent adverse events occurring in at least 5% of patients (in the quizartinib and chemotherapy groups, respectively) included:
- Sepsis/septic shock (16% and 18%)
- Hypokalemia (12% and 9%)
- Pneumonia (12% and 9%)
- Fatigue (8% and 1%)
- Dyspnea (5% for both)
- Hypophosphatemia (5% for both).
The U.S. Food and Drug Administration (FDA) has accepted for priority review a new drug application (NDA) for the FLT3 inhibitor quizartinib.
With this NDA, Daiichi Sankyo is seeking approval for quizartinib to treat adults with relapsed/refractory FLT3-ITD acute myeloid leukemia (AML).
The FDA grants priority review to applications for products that are expected to provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The FDA aims to take action on a priority review application within 6 months rather than the standard 10 months.
The FDA is expected to make a decision on the quizartinib NDA by May 25, 2019.
In addition to priority review, quizartinib has breakthrough therapy designation and fast track designation from the FDA.
Trial results
The NDA for quizartinib is supported by results from the phase 3 QuANTUM-R study. Topline results from this study were presented at the 23rd Congress of the European Hematology Association in June, and new analyses are set to be presented at the 2018 ASH Annual Meeting in December (abstract 563).
QuANTUM-R enrolled adults with FLT3-ITD AML (at least 3% FLT3-ITD allelic ratio) who had refractory disease or had relapsed within 6 months of their first complete response (CR).
Patients were randomized to receive once-daily treatment with quizartinib (n=245) or a salvage chemotherapy regimen (n=122)—low-dose cytarabine (LoDAC, n=29); combination mitoxantrone, etoposide, and cytarabine (MEC, n=40); or combination fludarabine, cytarabine, and idarubicin (FLAG-IDA, n=53).
Patients who responded to treatment could proceed to hematopoietic stem cell transplant (HSCT), and those in the quizartinib arm could resume quizartinib after HSCT.
In all, 241 patients received quizartinib, and 94 received salvage chemotherapy—LoDAC (n=22), MEC (n=25), and FLAG-IDA (n=47). Of the 28 patients in the chemotherapy group who were not treated, most withdrew consent.
Thirty-two percent of quizartinib-treated patients and 12% of the chemotherapy group went on to HSCT.
Efficacy
The median follow-up was 23.5 months. The efficacy results include all randomized patients.
The overall response rate was 69% in the quizartinib arm and 30% in the chemotherapy arm. The composite CR rate was 48% in the quizartinib arm and 27% in the chemotherapy arm. This includes:
- The CR rate (4% and 1%, respectively)
- The rate of CR with incomplete platelet recovery (4% and 0%, respectively)
- The rate of CR with incomplete hematologic recovery (40% and 26%, respectively).
The median event-free survival was 6.0 weeks in the quizartinib arm and 3.7 weeks in the chemotherapy arm (hazard ratio=0.90, P=0.1071).
The median overall survival was 6.2 months in the quizartinib arm and 4.7 months in the chemotherapy arm (hazard ratio=0.76, P=0.0177). The 1-year overall survival rate was 27% and 20%, respectively.
Safety
The safety results include only patients who received their assigned treatment.
Grade 3 or higher hematologic treatment-emergent adverse events occurring in at least 5% of patients (in the quizartinib and chemotherapy groups, respectively) included:
- Thrombocytopenia (35% and 34%)
- Anemia (30% and 29%)
- Neutropenia (32% and 25%)
- Febrile neutropenia (31% and 21%)
- Leukopenia (17% and 16%).
Grade 3 or higher non-hematologic treatment-emergent adverse events occurring in at least 5% of patients (in the quizartinib and chemotherapy groups, respectively) included:
- Sepsis/septic shock (16% and 18%)
- Hypokalemia (12% and 9%)
- Pneumonia (12% and 9%)
- Fatigue (8% and 1%)
- Dyspnea (5% for both)
- Hypophosphatemia (5% for both).
The U.S. Food and Drug Administration (FDA) has accepted for priority review a new drug application (NDA) for the FLT3 inhibitor quizartinib.
With this NDA, Daiichi Sankyo is seeking approval for quizartinib to treat adults with relapsed/refractory FLT3-ITD acute myeloid leukemia (AML).
The FDA grants priority review to applications for products that are expected to provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The FDA aims to take action on a priority review application within 6 months rather than the standard 10 months.
The FDA is expected to make a decision on the quizartinib NDA by May 25, 2019.
In addition to priority review, quizartinib has breakthrough therapy designation and fast track designation from the FDA.
Trial results
The NDA for quizartinib is supported by results from the phase 3 QuANTUM-R study. Topline results from this study were presented at the 23rd Congress of the European Hematology Association in June, and new analyses are set to be presented at the 2018 ASH Annual Meeting in December (abstract 563).
QuANTUM-R enrolled adults with FLT3-ITD AML (at least 3% FLT3-ITD allelic ratio) who had refractory disease or had relapsed within 6 months of their first complete response (CR).
Patients were randomized to receive once-daily treatment with quizartinib (n=245) or a salvage chemotherapy regimen (n=122)—low-dose cytarabine (LoDAC, n=29); combination mitoxantrone, etoposide, and cytarabine (MEC, n=40); or combination fludarabine, cytarabine, and idarubicin (FLAG-IDA, n=53).
Patients who responded to treatment could proceed to hematopoietic stem cell transplant (HSCT), and those in the quizartinib arm could resume quizartinib after HSCT.
In all, 241 patients received quizartinib, and 94 received salvage chemotherapy—LoDAC (n=22), MEC (n=25), and FLAG-IDA (n=47). Of the 28 patients in the chemotherapy group who were not treated, most withdrew consent.
Thirty-two percent of quizartinib-treated patients and 12% of the chemotherapy group went on to HSCT.
Efficacy
The median follow-up was 23.5 months. The efficacy results include all randomized patients.
The overall response rate was 69% in the quizartinib arm and 30% in the chemotherapy arm. The composite CR rate was 48% in the quizartinib arm and 27% in the chemotherapy arm. This includes:
- The CR rate (4% and 1%, respectively)
- The rate of CR with incomplete platelet recovery (4% and 0%, respectively)
- The rate of CR with incomplete hematologic recovery (40% and 26%, respectively).
The median event-free survival was 6.0 weeks in the quizartinib arm and 3.7 weeks in the chemotherapy arm (hazard ratio=0.90, P=0.1071).
The median overall survival was 6.2 months in the quizartinib arm and 4.7 months in the chemotherapy arm (hazard ratio=0.76, P=0.0177). The 1-year overall survival rate was 27% and 20%, respectively.
Safety
The safety results include only patients who received their assigned treatment.
Grade 3 or higher hematologic treatment-emergent adverse events occurring in at least 5% of patients (in the quizartinib and chemotherapy groups, respectively) included:
- Thrombocytopenia (35% and 34%)
- Anemia (30% and 29%)
- Neutropenia (32% and 25%)
- Febrile neutropenia (31% and 21%)
- Leukopenia (17% and 16%).
Grade 3 or higher non-hematologic treatment-emergent adverse events occurring in at least 5% of patients (in the quizartinib and chemotherapy groups, respectively) included:
- Sepsis/septic shock (16% and 18%)
- Hypokalemia (12% and 9%)
- Pneumonia (12% and 9%)
- Fatigue (8% and 1%)
- Dyspnea (5% for both)
- Hypophosphatemia (5% for both).
Immunotherapy may hold the key to defeating virally associated cancers
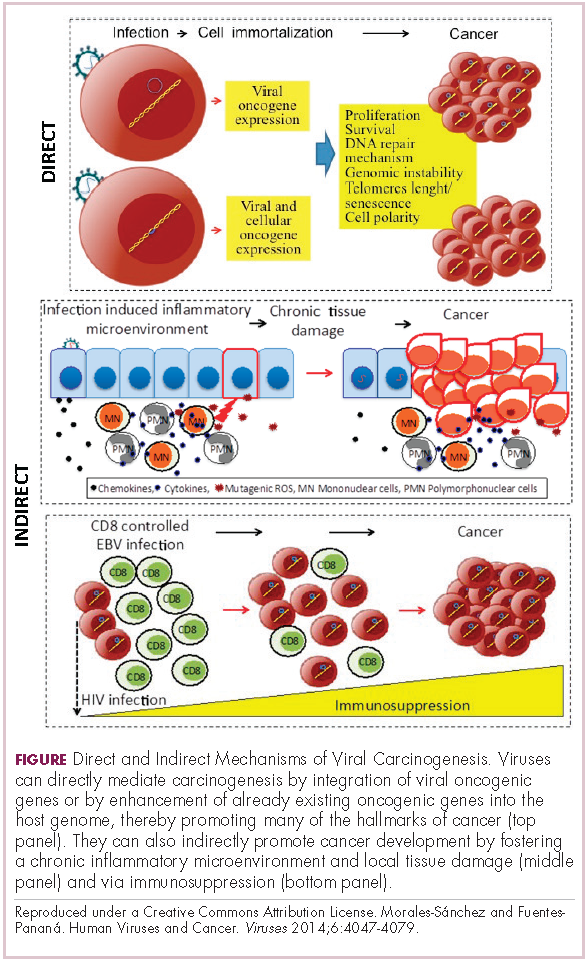
Infection with certain viruses has been causally linked to the development of cancer. In recent years, an improved understanding of the unique pathology and molecular underpinnings of these virally associated cancers has prompted the development of more personalized treatment strategies, with a particular focus on immunotherapy. Here, we describe some of the latest developments.
The link between viruses and cancer
Suspicions about a possible role of viral infections in the development of cancer were first aroused in the early 1900s. The seminal discovery is traced back to Peyton Rous, who showed that a malignant tumor growing in a chicken could be transferred to a healthy bird by injecting it with tumor extracts that contained no actual tumor cells.1
The infectious etiology of human cancer, however, remained controversial until many years later when the first cancer-causing virus, Epstein-Barr virus (EBV), was identified in cell cultures from patients with Burkitt lymphoma. Shortly afterward, the Rous sarcoma virus was unveiled as the oncogenic agent behind Rous’ observations.2Seven viruses have now been linked to the development of cancers and are thought to be responsible for around 12% of all cancer cases worldwide. The burden is likely to increase as technological advancements make it easier to establish a causal link between viruses and cancer development.3
In addition to making these links, researchers have also made significant headway in understanding how viruses cause cancer. Cancerous transformation of host cells occurs in only a minority of those who are infected with oncogenic viruses and often occurs in the setting of chronic infection.
Viruses can mediate carcinogenesis by direct and/or indirect mechanisms (Figure 1). Many of the hallmarks of cancer, the key attributes that drive the transformation from a normal cell to a malignant one, are compatible with the virus’s needs, such as needing to avoid cell death, increasing cell proliferation, and avoiding detection by the immune system.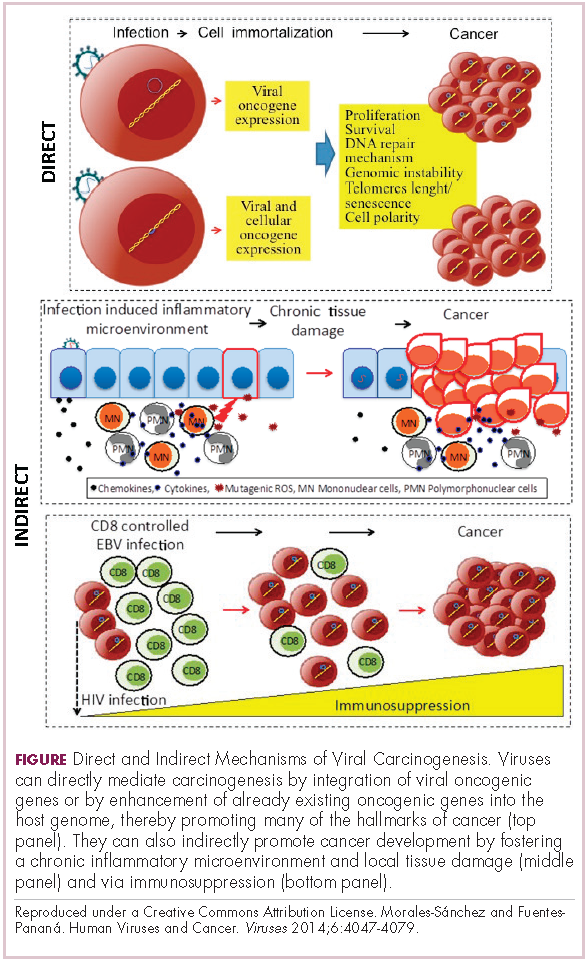
Viruses hijack the cellular machinery to meet those needs and they can do this either by producing viral proteins that have an oncogenic effect or by integrating their genetic material into the host cell genome. When the latter occurs, the process of integration can also cause damage to the DNA, which further increases the risk of cancer-promoting changes occurring in the host genome.
Viruses can indirectly contribute to carcinogenesis by fostering a microenvironment of chronic inflammation, causing oxidative stress and local tissue damage, and by suppressing the antitumor immune response.4,5
Screening and prevention efforts have helped to reduce the burden of several different virally associated cancers. However, for the substantial proportion of patients who are still affected by these cancers, there is a pressing need for new therapeutic options, particularly since genome sequencing studies have revealed that these cancers can often have distinct underlying molecular mechanisms.
Vaccines lead the charge in HPV-driven cancers
German virologist Harald zur Hausen received the Nobel Prize in 2008 for his discovery of the oncogenic role of human papillomaviruses (HPVs), a large family of more than 100 DNA viruses that infect the epithelial cells of the skin and mucous membranes. They are responsible for the largest number of virally associated cancer cases globally – around 5% (Table 1).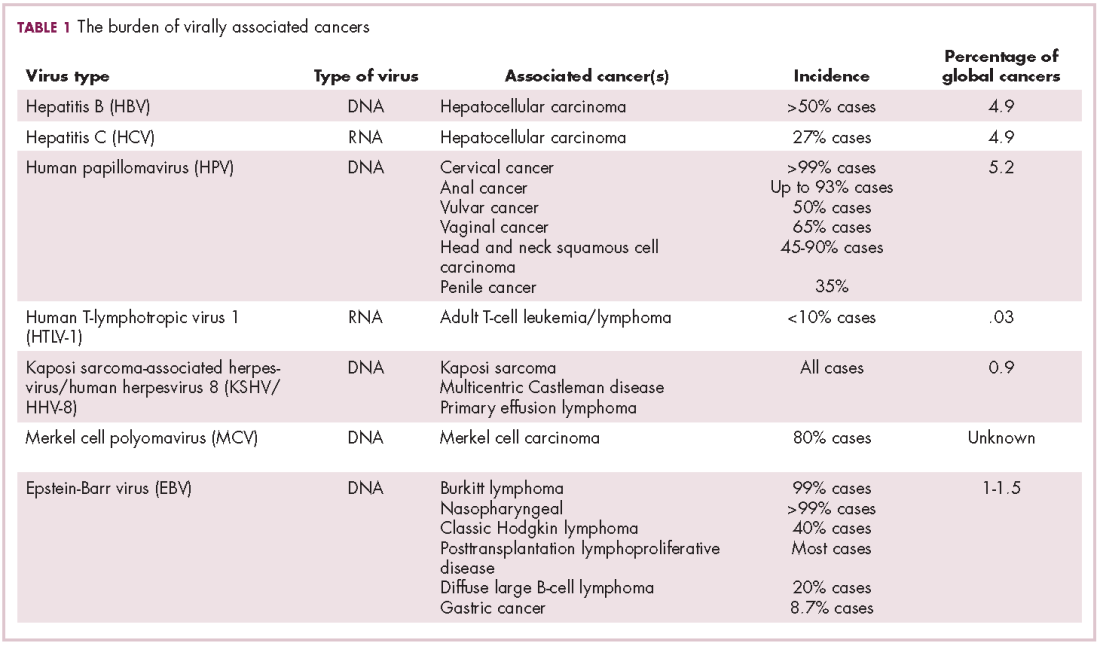
A number of different cancer types are linked to HPV infection, but it is best known as the cause of cervical cancer. The development of diagnostic blood tests and prophylactic vaccines for prevention and early intervention in HPV infection has helped to reduce the incidence of cervical cancer. Conversely, another type of HPV-associated cancer, head and neck squamous cell carcinoma (HNSCC), has seen increased incidence in recent years.
HPVs are categorized according to their oncogenic potential as high, intermediate, or low risk. The high-risk HPV16 and HPV18 strains are most commonly associated with cancer. They are thought to cause cancer predominantly through integration into the host genome. The HPV genome is composed of 8 genes encoding proteins that regulate viral replication and assembly. The E6 and E7 genes are the most highly oncogenic; as the HPV DNA is inserted into the host genome, the transcriptional regulator of E6/E7 is lost, leading to their increased expression. These genes have significant oncogenic potential because of their interaction with 2 tumor suppressor proteins, p53 and pRb.6,7
The largest investment in therapeutic development for HPV-positive cancers has been in the realm of immunotherapy in an effort to boost the anti-tumor immune response. In particular, there has been a focus on the development of therapeutic vaccines, designed to prime the anti-tumor immune response to recognize viral antigens. A variety of different types of vaccines are being developed, including live, attenuated and inactivated vaccines that are protein, DNA, or peptide based. Most developed to date target the E6/E7 proteins from the HPV16/18 strains (Table 2).8,9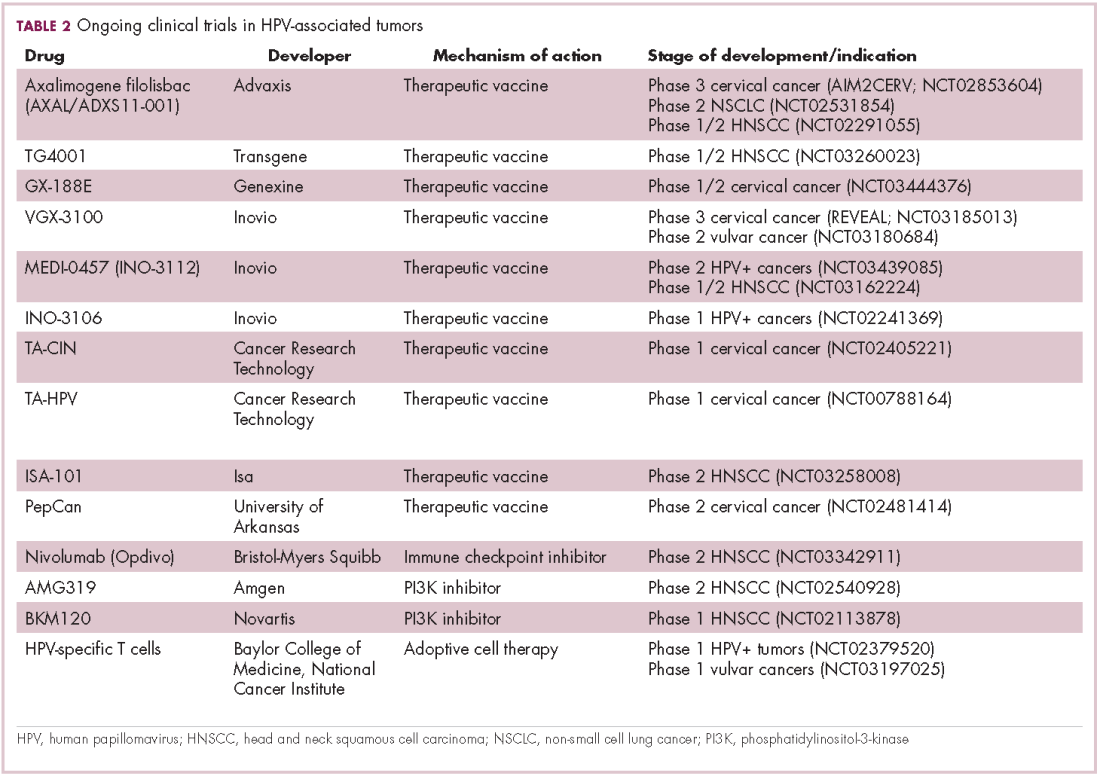
Other immunotherapies are also being evaluated, including immune checkpoint inhibitors, antibodies designed to target one of the principal mechanisms of immune evasion exploited by cancer cells. The combination of immune checkpoint inhibitors with vaccines is a particularly promising strategy in HPV-associated cancers. At the European Society for Medical Oncology Congress in 2017, the results of a phase 2 trial of nivolumab in combination with ISA-101 were presented.
Among 24 patients with HPV-positive tumors, the majority oropharyngeal cancers, the combination elicited an overall response rate (ORR) of 33%, including 2 complete responses (CRs). Most adverse events (AEs) were mild to moderate in severity and included fever, injection site reactions, fatigue and nausea.14
Hepatocellular carcinoma: a tale of two viruses
The hepatitis viruses are a group of 5 unrelated viruses that causes inflammation of the liver. Hepatitis B (HBV), a DNA virus, and hepatitis C (HCV), an RNA virus, are also oncoviruses; HBV in particular is one of the main causes of hepatocellular carcinoma (HCC), the most common type of liver cancer.
The highly inflammatory environment fostered by HBV and HCV infection causes liver damage that often leads to cirrhosis. Continued infection can drive permanent damage to the hepatocytes, leading to genetic and epigenetic damage and driving oncogenesis. As an RNA virus, HCV doesn’t integrate into the genome and no confirmed viral oncoproteins have been identified to date, therefore it mostly drives cancer through these indirect mechanisms, which is also reflected in the fact that HCV-associated HCC predominantly occurs against a backdrop of liver cirrhosis.
HBV does integrate into the host genome. Genome sequencing studies revealed hundreds of integration sites, but most commonly they disrupted host genes involved in telomere stability and cell cycle regulation, providing some insight into the mechanisms by which HBV-associated HCC develops. In addition, HBV produces several oncoproteins, including HBx, which disrupts gene transcription, cell signaling pathways, cell cycle progress, apoptosis and other cellular processes.15,16
Multitargeted tyrosine kinase inhibitors (TKIs) have been the focal point of therapeutic development in HCC. However, following the approval of sorafenib in 2008, there was a dearth of effective new treatment options despite substantial efforts and numerous phase 3 trials. More recently, immunotherapy has also come to the forefront, especially immune checkpoint inhibitors.
Last year marked the first new drug approvals in nearly a decade – the TKI regorafenib (Stivarga) and immune checkpoint inhibitor nivolumab (Opdivo), both in the second-line setting after failure of sorafenib. Treatment options in this setting may continue to expand, with the TKIs cabozantinib and lenvatinib and the immune checkpoint inhibitor pembrolizumab and the combination of durvalumab and tremelimumab hot on their heels.17-20 Many of these drugs are also being evaluated in the front-line setting in comparison with sorafenib (Table 3).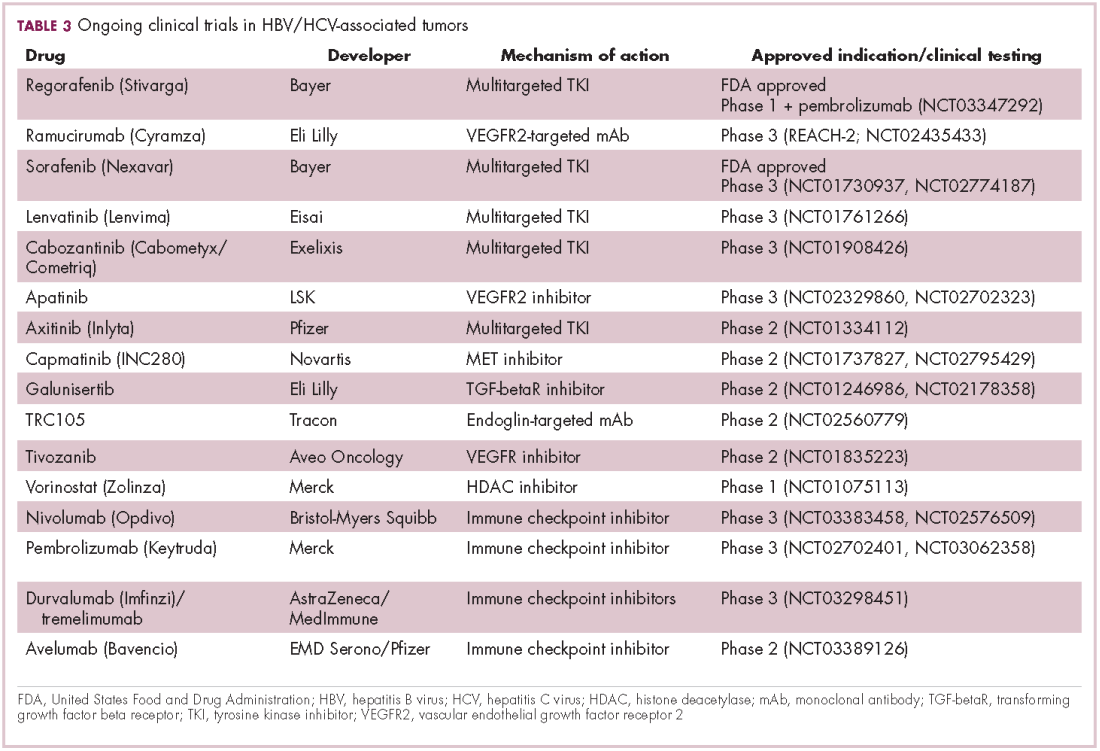
At the current time, the treatment strategy for patients with HCC is independent of etiology, however, there are significant ongoing efforts to try to tease out the implications of infection for treatment efficacy. A recent meta-analysis of patients treated with sorafenib in 3 randomized phase 3 trials (n = 3,526) suggested that it improved overall survival (OS) among patients who were HCV-positive, but HBV-negative.21
Studies of the vascular endothelial growth factor receptor 2-targeting monoclonal antibody ramucirumab, on the other hand, suggested that it may have a greater OS benefit in patients with HBV, while regorafenib seemed to have a comparable OS benefit in both subgroups.22-25 The immune checkpoint inhibitors studied thus far seem to elicit responses irrespective of infection status.
A phase 2 trial of the immune checkpoint inhibitor tremelimumab was conducted specifically in patients with advanced HCC and chronic HCV infection. The disease control rate (DCR) was 76.4%, with 17.6% partial response (PR) rate. There was also a significant drop in viral load, suggesting that tremelimumab may have antiviral effects.26,27,28
Adoptive cell therapy promising in EBV-positive cancers
More than 90% of the global population is infected with EBV, making it one of the most common human viruses. It is a member of the herpesvirus family that is probably best known as the cause of infectious mononucleosis. On rare occasions, however, EBV can cause tumor development, though our understanding of its exact pathogenic role in cancer is still incomplete.
EBV is a DNA virus that doesn’t tend to integrate into the host genome, but instead remains in the nucleus in the form of episomes and produces several oncoproteins, including latent membrane protein-1. It is associated with a range of different cancer types, including Burkitt lymphoma and other B-cell malignancies. It also infects epithelial cells and can cause nasopharyngeal carcinoma and gastric cancer, however, much less is known about the molecular underpinnings of these EBV-positive cancer types.26,27Gastric cancers actually comprise the largest group of EBV-associated tumors because of the global incidence of this cancer type. The Cancer Genome Atlas Research Network recently characterized gastric cancer on a molecular level and identified an EBV-positive subgroup as a distinct clinical entity with unique molecular characteristics.29
The focus of therapeutic development has again been on immunotherapy, however in this case the idea of collecting the patients T cells, engineering them to recognize EBV, and then reinfusing them into the patient – adoptive cell therapy – has gained the most traction (Table 4).
Two presentations at the American Society of Hematology annual meeting in 2017 detailed ongoing clinical trials of Atara Biotherapeutics’ ATA129 and Cell Medica’s CMD-003. ATA129 was associated with a high response rate and a low rate of serious AEs in patients with posttransplant lymphoproliferative disorder; ORR was 80% in 6 patients treated after hematopoietic stem cell transplantation, and 83% in 6 patients after solid organ transplant.30
CMD-003, meanwhile, demonstrated preliminary signs of activity and safety in patients with relapsed extranodal NK/T-cell lymphoma, according to early results from the phase 2 CITADEL trial. Among 6 evaluable patients, the ORR was 50% and the DCR was 67%.31
Newest oncovirus on the block
The most recently discovered cancer-associated virus is Merkel cell polyomavirus (MCV), a DNA virus that was identified in 2008. Like EBV, virtually the whole global adult population is infected with MCV. It is linked to the development of a highly aggressive and lethal, though rare, form of skin cancer – Merkel cell carcinoma.
MCV is found in around 80% of MCC cases and in fewer than 10% of melanomas and other skin cancers. Thus far, several direct mechanisms of oncogenesis have been described, including integration of MCV into the host genome and the production of viral oncogenes, though their precise function is as yet unclear.32-34
The American Cancer Society estimates that only 1500 cases of MCC are diagnosed each year in the United States.35 Its rarity makes it difficult to conduct clinical trials with sufficient power, yet some headway has still been made.
Around half of MCCs express the programmed cell death ligand 1 (PD-L1) on their surface, making them a logical candidate for immune checkpoint inhibition. In 2017, avelumab became the first FDA-approved drug for the treatment of MCC. Approval was based on the JAVELIN Merkel 200 study in which 88 patients received avelumab. After 1 year of follow-up the ORR was 31.8%, with a CR rate of 9%.36
Genome sequencing studies suggest that the mutational profile of MCV-positive tumors is quite different to those that are MCV-negative, which could have therapeutic implications. To date, these implications have not been delineated, given the challenge of small patient numbers, however an ongoing phase 1/2 trial is evaluating the combination of avelumab and radiation therapy or recombinant interferon beta, with or without MCV-specific cytotoxic T cells in patients with MCC and MCV infection.
The 2 other known cancer-causing viruses are human T-lymphotropic virus 1 (HTLV-1), a retrovirus associated with adult T-cell leukemia/lymphoma (ATL) and Kaposi sarcoma herpesvirus (KSHV). The latter is the causative agent of Kaposi sarcoma, often in combination with human immunodeficiency virus (HIV), a rare skin tumor that became renowned in the 1980s as an AIDS-defining illness.
The incidence of HTLV-1- and KSHV-positive tumors is substantially lower than the other virally associated cancers and, like MCC, this makes studying them and conducting clinical trials of novel therapeutic options a challenge. Nonetheless, several trials of targeted therapies and immunotherapies are underway.
1. Rous PA. Transmissible avain neoplasm. (Sarcoma of the common fowl). J Exp Med. 1910;12(5):696-705.
2. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet. 1964;1(7335):702-703.
3. Mesri Enrique A, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host & Microbe. 2014;15(3):266-282.
4. Santana-Davila R, Bhatia S, Chow LQ. Harnessing the immune system as a therapeutic tool in virus-associated cancers. JAMA Oncol. 2017;3(1):106-112.
5. Tashiro H, Brenner MK. Immunotherapy against cancer-related viruses. Cell Res. 2017;27(1):59-73.
6. Brianti P, De Flammineis E, Mercuri SR. Review of HPV-related diseases and cancers. New Microbiol. 2017;40(2):80-85.
7. Tulay P, Serakinci N. The route to HPV-associated neoplastic transformation: a review of the literature. Crit Rev Eukaryot Gene Expr. 2016;26(1):27-39.
8. Smola S. Immunopathogenesis of HPV-associated cancers and prospects for immunotherapy. Viruses. 2017;9(9).
9. Rosales R, Rosales C. Immune therapy for human papillomaviruses-related cancers. World Journal of Clinical Oncology. 2014;5(5):1002-1019.
10. Miles B, Safran HP, Monk BJ. Therapeutic options for treatment of human papillomavirus-associated cancers - novel immunologic vaccines: ADXS11-001. Gynecol Oncol Res Pract. 2017;4:10.
11. Miles BA, Monk BJ, Safran HP. Mechanistic insights into ADXS11-001 human papillomavirus-associated cancer immunotherapy. Gynecol Oncol Res Pract. 2017;4:9.
12. Huh W, Dizon D, Powell M, Landrum L, Leath C. A prospective phase II trial of the listeria-based human papillomavirus immunotherapy axalimogene filolisbac in second and third-line metastatic cervical cancer: A NRG oncology group trial. Paper presented at: Annual Meeting on Women's Cancer; March 12-15, 2017, 2017; National Harbor, MD.
13. Petit RG, Mehta A, Jain M, et al. ADXS11-001 immunotherapy targeting HPV-E7: final results from a Phase II study in Indian women with recurrent cervical cancer. Journal for Immunotherapy of Cancer. 2014;2(Suppl 3):P92-P92.
14. Glisson B, Massarelli E, William W, et al. Nivolumab and ISA 101 HPV vaccine in incurable HPV-16+ cancer. Ann Oncol. 2017;28(suppl_5):v403-v427.
15. Ding X-X, Zhu Q-G, Zhang S-M, et al. Precision medicine for hepatocellular carcinoma: driver mutations and targeted therapy. Oncotarget. 2017;8(33):55715-55730.
16. Ringehan M, McKeating JA, Protzer U. Viral hepatitis and liver cancer. Philosophical Transactions of the Royal Society B: Biological Sciences. 2017;372(1732):20160274.
17. Abou-Alfa G, Meyer T, Cheng AL, et al. Cabozantinib (C) versus placebo (P) in patients (pts) with advanced hepatocellular carcinoma (HCC) who have received prior sorafenib: results from the randomized phase III CELESTIAL trial. J Clin Oncol. 2017;36(Suppl 4S):abstr 207.
18. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018.
19. Zhu AX, Finn RS, Cattan S, et al. KEYNOTE-224: Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. J Clin Oncol. 2018;36(Suppl 4S):Abstr 209.
20. Kelley RK, Abou-Alfa GK, Bendell JC, et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. Journal of Clinical Oncology. 2017;35(15_suppl):4073-4073.
21. Jackson R, Psarelli E-E, Berhane S, Khan H, Johnson P. Impact of Viral Status on Survival in Patients Receiving Sorafenib for Advanced Hepatocellular Cancer: A Meta-Analysis of Randomized Phase III Trials. Journal of Clinical Oncology. 2017;35(6):622-628.
22. Kudo M. Molecular Targeted Agents for Hepatocellular Carcinoma: Current Status and Future Perspectives. Liver Cancer. 2017;6(2):101-112.
23. zur Hausen H, Meinhof W, Scheiber W, Bornkamm GW. Attempts to detect virus-secific DNA in human tumors. I. Nucleic acid hybridizations with complementary RNA of human wart virus. Int J Cancer. 1974;13(5):650-656.
24. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56-66.
25. Bruix J, Tak WY, Gasbarrini A, et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: multicentre, open-label, phase II safety study. Eur J Cancer. 2013;49(16):3412-3419.
26. Neparidze N, Lacy J. Malignancies associated with epstein-barr virus: pathobiology, clinical features, and evolving treatments. Clin Adv Hematol Oncol. 2014;12(6):358-371.
27. Ozoya OO, Sokol L, Dalia S. EBV-Related Malignancies, Outcomes and Novel Prevention Strategies. Infect Disord Drug Targets. 2016;16(1):4-21.
28. Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81-88.
29. The Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202.
30. Prockop S, Li A, Baiocchi R, et al. Efficacy and safety of ATA129, partially matched allogeneic third-party Epstein-Barr virus-targeted cytotoxic T lymphocytes in a multicenter study for post-transplant lymphoproliferative disorder. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
31. Kim W, Ardeshna K, Lin Y, et al. Autologous EBV-specific T cells (CMD-003): Early results from a multicenter, multinational Phase 2 trial for treatment of EBV-associated NK/T-cell lymphoma. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
32. Schadendorf D, Lebbé C, zur Hausen A, et al. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. European Journal of Cancer. 2017;71:53-69.
33. Spurgeon ME, Lambert PF. Merkel cell polyomavirus: a newly discovered human virus with oncogenic potential. Virology. 2013;435(1):118-130.
34. Tello TL, Coggshall K, Yom SS, Yu SS. Merkel cell carcinoma: An update and review: Current and future therapy. J Am Acad Dermatol. 2018;78(3):445-454.
35. American Cancer Society. Key Statistics for Merkel Cell Carcinoma. 2015; https://www.cancer.org/cancer/merkel-cell-skin-cancer/about/key-statistics.html#written_by. Accessed March 7th, 2017.
36. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. The Lancet Oncology.17(10):1374-1385.
Infection with certain viruses has been causally linked to the development of cancer. In recent years, an improved understanding of the unique pathology and molecular underpinnings of these virally associated cancers has prompted the development of more personalized treatment strategies, with a particular focus on immunotherapy. Here, we describe some of the latest developments.
The link between viruses and cancer
Suspicions about a possible role of viral infections in the development of cancer were first aroused in the early 1900s. The seminal discovery is traced back to Peyton Rous, who showed that a malignant tumor growing in a chicken could be transferred to a healthy bird by injecting it with tumor extracts that contained no actual tumor cells.1
The infectious etiology of human cancer, however, remained controversial until many years later when the first cancer-causing virus, Epstein-Barr virus (EBV), was identified in cell cultures from patients with Burkitt lymphoma. Shortly afterward, the Rous sarcoma virus was unveiled as the oncogenic agent behind Rous’ observations.2Seven viruses have now been linked to the development of cancers and are thought to be responsible for around 12% of all cancer cases worldwide. The burden is likely to increase as technological advancements make it easier to establish a causal link between viruses and cancer development.3
In addition to making these links, researchers have also made significant headway in understanding how viruses cause cancer. Cancerous transformation of host cells occurs in only a minority of those who are infected with oncogenic viruses and often occurs in the setting of chronic infection.
Viruses can mediate carcinogenesis by direct and/or indirect mechanisms (Figure 1). Many of the hallmarks of cancer, the key attributes that drive the transformation from a normal cell to a malignant one, are compatible with the virus’s needs, such as needing to avoid cell death, increasing cell proliferation, and avoiding detection by the immune system.
Viruses hijack the cellular machinery to meet those needs and they can do this either by producing viral proteins that have an oncogenic effect or by integrating their genetic material into the host cell genome. When the latter occurs, the process of integration can also cause damage to the DNA, which further increases the risk of cancer-promoting changes occurring in the host genome.
Viruses can indirectly contribute to carcinogenesis by fostering a microenvironment of chronic inflammation, causing oxidative stress and local tissue damage, and by suppressing the antitumor immune response.4,5
Screening and prevention efforts have helped to reduce the burden of several different virally associated cancers. However, for the substantial proportion of patients who are still affected by these cancers, there is a pressing need for new therapeutic options, particularly since genome sequencing studies have revealed that these cancers can often have distinct underlying molecular mechanisms.
Vaccines lead the charge in HPV-driven cancers
German virologist Harald zur Hausen received the Nobel Prize in 2008 for his discovery of the oncogenic role of human papillomaviruses (HPVs), a large family of more than 100 DNA viruses that infect the epithelial cells of the skin and mucous membranes. They are responsible for the largest number of virally associated cancer cases globally – around 5% (Table 1).
A number of different cancer types are linked to HPV infection, but it is best known as the cause of cervical cancer. The development of diagnostic blood tests and prophylactic vaccines for prevention and early intervention in HPV infection has helped to reduce the incidence of cervical cancer. Conversely, another type of HPV-associated cancer, head and neck squamous cell carcinoma (HNSCC), has seen increased incidence in recent years.
HPVs are categorized according to their oncogenic potential as high, intermediate, or low risk. The high-risk HPV16 and HPV18 strains are most commonly associated with cancer. They are thought to cause cancer predominantly through integration into the host genome. The HPV genome is composed of 8 genes encoding proteins that regulate viral replication and assembly. The E6 and E7 genes are the most highly oncogenic; as the HPV DNA is inserted into the host genome, the transcriptional regulator of E6/E7 is lost, leading to their increased expression. These genes have significant oncogenic potential because of their interaction with 2 tumor suppressor proteins, p53 and pRb.6,7
The largest investment in therapeutic development for HPV-positive cancers has been in the realm of immunotherapy in an effort to boost the anti-tumor immune response. In particular, there has been a focus on the development of therapeutic vaccines, designed to prime the anti-tumor immune response to recognize viral antigens. A variety of different types of vaccines are being developed, including live, attenuated and inactivated vaccines that are protein, DNA, or peptide based. Most developed to date target the E6/E7 proteins from the HPV16/18 strains (Table 2).8,9
Other immunotherapies are also being evaluated, including immune checkpoint inhibitors, antibodies designed to target one of the principal mechanisms of immune evasion exploited by cancer cells. The combination of immune checkpoint inhibitors with vaccines is a particularly promising strategy in HPV-associated cancers. At the European Society for Medical Oncology Congress in 2017, the results of a phase 2 trial of nivolumab in combination with ISA-101 were presented.
Among 24 patients with HPV-positive tumors, the majority oropharyngeal cancers, the combination elicited an overall response rate (ORR) of 33%, including 2 complete responses (CRs). Most adverse events (AEs) were mild to moderate in severity and included fever, injection site reactions, fatigue and nausea.14
Hepatocellular carcinoma: a tale of two viruses
The hepatitis viruses are a group of 5 unrelated viruses that causes inflammation of the liver. Hepatitis B (HBV), a DNA virus, and hepatitis C (HCV), an RNA virus, are also oncoviruses; HBV in particular is one of the main causes of hepatocellular carcinoma (HCC), the most common type of liver cancer.
The highly inflammatory environment fostered by HBV and HCV infection causes liver damage that often leads to cirrhosis. Continued infection can drive permanent damage to the hepatocytes, leading to genetic and epigenetic damage and driving oncogenesis. As an RNA virus, HCV doesn’t integrate into the genome and no confirmed viral oncoproteins have been identified to date, therefore it mostly drives cancer through these indirect mechanisms, which is also reflected in the fact that HCV-associated HCC predominantly occurs against a backdrop of liver cirrhosis.
HBV does integrate into the host genome. Genome sequencing studies revealed hundreds of integration sites, but most commonly they disrupted host genes involved in telomere stability and cell cycle regulation, providing some insight into the mechanisms by which HBV-associated HCC develops. In addition, HBV produces several oncoproteins, including HBx, which disrupts gene transcription, cell signaling pathways, cell cycle progress, apoptosis and other cellular processes.15,16
Multitargeted tyrosine kinase inhibitors (TKIs) have been the focal point of therapeutic development in HCC. However, following the approval of sorafenib in 2008, there was a dearth of effective new treatment options despite substantial efforts and numerous phase 3 trials. More recently, immunotherapy has also come to the forefront, especially immune checkpoint inhibitors.
Last year marked the first new drug approvals in nearly a decade – the TKI regorafenib (Stivarga) and immune checkpoint inhibitor nivolumab (Opdivo), both in the second-line setting after failure of sorafenib. Treatment options in this setting may continue to expand, with the TKIs cabozantinib and lenvatinib and the immune checkpoint inhibitor pembrolizumab and the combination of durvalumab and tremelimumab hot on their heels.17-20 Many of these drugs are also being evaluated in the front-line setting in comparison with sorafenib (Table 3).
At the current time, the treatment strategy for patients with HCC is independent of etiology, however, there are significant ongoing efforts to try to tease out the implications of infection for treatment efficacy. A recent meta-analysis of patients treated with sorafenib in 3 randomized phase 3 trials (n = 3,526) suggested that it improved overall survival (OS) among patients who were HCV-positive, but HBV-negative.21
Studies of the vascular endothelial growth factor receptor 2-targeting monoclonal antibody ramucirumab, on the other hand, suggested that it may have a greater OS benefit in patients with HBV, while regorafenib seemed to have a comparable OS benefit in both subgroups.22-25 The immune checkpoint inhibitors studied thus far seem to elicit responses irrespective of infection status.
A phase 2 trial of the immune checkpoint inhibitor tremelimumab was conducted specifically in patients with advanced HCC and chronic HCV infection. The disease control rate (DCR) was 76.4%, with 17.6% partial response (PR) rate. There was also a significant drop in viral load, suggesting that tremelimumab may have antiviral effects.26,27,28
Adoptive cell therapy promising in EBV-positive cancers
More than 90% of the global population is infected with EBV, making it one of the most common human viruses. It is a member of the herpesvirus family that is probably best known as the cause of infectious mononucleosis. On rare occasions, however, EBV can cause tumor development, though our understanding of its exact pathogenic role in cancer is still incomplete.
EBV is a DNA virus that doesn’t tend to integrate into the host genome, but instead remains in the nucleus in the form of episomes and produces several oncoproteins, including latent membrane protein-1. It is associated with a range of different cancer types, including Burkitt lymphoma and other B-cell malignancies. It also infects epithelial cells and can cause nasopharyngeal carcinoma and gastric cancer, however, much less is known about the molecular underpinnings of these EBV-positive cancer types.26,27Gastric cancers actually comprise the largest group of EBV-associated tumors because of the global incidence of this cancer type. The Cancer Genome Atlas Research Network recently characterized gastric cancer on a molecular level and identified an EBV-positive subgroup as a distinct clinical entity with unique molecular characteristics.29
The focus of therapeutic development has again been on immunotherapy, however in this case the idea of collecting the patients T cells, engineering them to recognize EBV, and then reinfusing them into the patient – adoptive cell therapy – has gained the most traction (Table 4).
Two presentations at the American Society of Hematology annual meeting in 2017 detailed ongoing clinical trials of Atara Biotherapeutics’ ATA129 and Cell Medica’s CMD-003. ATA129 was associated with a high response rate and a low rate of serious AEs in patients with posttransplant lymphoproliferative disorder; ORR was 80% in 6 patients treated after hematopoietic stem cell transplantation, and 83% in 6 patients after solid organ transplant.30
CMD-003, meanwhile, demonstrated preliminary signs of activity and safety in patients with relapsed extranodal NK/T-cell lymphoma, according to early results from the phase 2 CITADEL trial. Among 6 evaluable patients, the ORR was 50% and the DCR was 67%.31
Newest oncovirus on the block
The most recently discovered cancer-associated virus is Merkel cell polyomavirus (MCV), a DNA virus that was identified in 2008. Like EBV, virtually the whole global adult population is infected with MCV. It is linked to the development of a highly aggressive and lethal, though rare, form of skin cancer – Merkel cell carcinoma.
MCV is found in around 80% of MCC cases and in fewer than 10% of melanomas and other skin cancers. Thus far, several direct mechanisms of oncogenesis have been described, including integration of MCV into the host genome and the production of viral oncogenes, though their precise function is as yet unclear.32-34
The American Cancer Society estimates that only 1500 cases of MCC are diagnosed each year in the United States.35 Its rarity makes it difficult to conduct clinical trials with sufficient power, yet some headway has still been made.
Around half of MCCs express the programmed cell death ligand 1 (PD-L1) on their surface, making them a logical candidate for immune checkpoint inhibition. In 2017, avelumab became the first FDA-approved drug for the treatment of MCC. Approval was based on the JAVELIN Merkel 200 study in which 88 patients received avelumab. After 1 year of follow-up the ORR was 31.8%, with a CR rate of 9%.36
Genome sequencing studies suggest that the mutational profile of MCV-positive tumors is quite different to those that are MCV-negative, which could have therapeutic implications. To date, these implications have not been delineated, given the challenge of small patient numbers, however an ongoing phase 1/2 trial is evaluating the combination of avelumab and radiation therapy or recombinant interferon beta, with or without MCV-specific cytotoxic T cells in patients with MCC and MCV infection.
The 2 other known cancer-causing viruses are human T-lymphotropic virus 1 (HTLV-1), a retrovirus associated with adult T-cell leukemia/lymphoma (ATL) and Kaposi sarcoma herpesvirus (KSHV). The latter is the causative agent of Kaposi sarcoma, often in combination with human immunodeficiency virus (HIV), a rare skin tumor that became renowned in the 1980s as an AIDS-defining illness.
The incidence of HTLV-1- and KSHV-positive tumors is substantially lower than the other virally associated cancers and, like MCC, this makes studying them and conducting clinical trials of novel therapeutic options a challenge. Nonetheless, several trials of targeted therapies and immunotherapies are underway.
Infection with certain viruses has been causally linked to the development of cancer. In recent years, an improved understanding of the unique pathology and molecular underpinnings of these virally associated cancers has prompted the development of more personalized treatment strategies, with a particular focus on immunotherapy. Here, we describe some of the latest developments.
The link between viruses and cancer
Suspicions about a possible role of viral infections in the development of cancer were first aroused in the early 1900s. The seminal discovery is traced back to Peyton Rous, who showed that a malignant tumor growing in a chicken could be transferred to a healthy bird by injecting it with tumor extracts that contained no actual tumor cells.1
The infectious etiology of human cancer, however, remained controversial until many years later when the first cancer-causing virus, Epstein-Barr virus (EBV), was identified in cell cultures from patients with Burkitt lymphoma. Shortly afterward, the Rous sarcoma virus was unveiled as the oncogenic agent behind Rous’ observations.2Seven viruses have now been linked to the development of cancers and are thought to be responsible for around 12% of all cancer cases worldwide. The burden is likely to increase as technological advancements make it easier to establish a causal link between viruses and cancer development.3
In addition to making these links, researchers have also made significant headway in understanding how viruses cause cancer. Cancerous transformation of host cells occurs in only a minority of those who are infected with oncogenic viruses and often occurs in the setting of chronic infection.
Viruses can mediate carcinogenesis by direct and/or indirect mechanisms (Figure 1). Many of the hallmarks of cancer, the key attributes that drive the transformation from a normal cell to a malignant one, are compatible with the virus’s needs, such as needing to avoid cell death, increasing cell proliferation, and avoiding detection by the immune system.
Viruses hijack the cellular machinery to meet those needs and they can do this either by producing viral proteins that have an oncogenic effect or by integrating their genetic material into the host cell genome. When the latter occurs, the process of integration can also cause damage to the DNA, which further increases the risk of cancer-promoting changes occurring in the host genome.
Viruses can indirectly contribute to carcinogenesis by fostering a microenvironment of chronic inflammation, causing oxidative stress and local tissue damage, and by suppressing the antitumor immune response.4,5
Screening and prevention efforts have helped to reduce the burden of several different virally associated cancers. However, for the substantial proportion of patients who are still affected by these cancers, there is a pressing need for new therapeutic options, particularly since genome sequencing studies have revealed that these cancers can often have distinct underlying molecular mechanisms.
Vaccines lead the charge in HPV-driven cancers
German virologist Harald zur Hausen received the Nobel Prize in 2008 for his discovery of the oncogenic role of human papillomaviruses (HPVs), a large family of more than 100 DNA viruses that infect the epithelial cells of the skin and mucous membranes. They are responsible for the largest number of virally associated cancer cases globally – around 5% (Table 1).
A number of different cancer types are linked to HPV infection, but it is best known as the cause of cervical cancer. The development of diagnostic blood tests and prophylactic vaccines for prevention and early intervention in HPV infection has helped to reduce the incidence of cervical cancer. Conversely, another type of HPV-associated cancer, head and neck squamous cell carcinoma (HNSCC), has seen increased incidence in recent years.
HPVs are categorized according to their oncogenic potential as high, intermediate, or low risk. The high-risk HPV16 and HPV18 strains are most commonly associated with cancer. They are thought to cause cancer predominantly through integration into the host genome. The HPV genome is composed of 8 genes encoding proteins that regulate viral replication and assembly. The E6 and E7 genes are the most highly oncogenic; as the HPV DNA is inserted into the host genome, the transcriptional regulator of E6/E7 is lost, leading to their increased expression. These genes have significant oncogenic potential because of their interaction with 2 tumor suppressor proteins, p53 and pRb.6,7
The largest investment in therapeutic development for HPV-positive cancers has been in the realm of immunotherapy in an effort to boost the anti-tumor immune response. In particular, there has been a focus on the development of therapeutic vaccines, designed to prime the anti-tumor immune response to recognize viral antigens. A variety of different types of vaccines are being developed, including live, attenuated and inactivated vaccines that are protein, DNA, or peptide based. Most developed to date target the E6/E7 proteins from the HPV16/18 strains (Table 2).8,9
Other immunotherapies are also being evaluated, including immune checkpoint inhibitors, antibodies designed to target one of the principal mechanisms of immune evasion exploited by cancer cells. The combination of immune checkpoint inhibitors with vaccines is a particularly promising strategy in HPV-associated cancers. At the European Society for Medical Oncology Congress in 2017, the results of a phase 2 trial of nivolumab in combination with ISA-101 were presented.
Among 24 patients with HPV-positive tumors, the majority oropharyngeal cancers, the combination elicited an overall response rate (ORR) of 33%, including 2 complete responses (CRs). Most adverse events (AEs) were mild to moderate in severity and included fever, injection site reactions, fatigue and nausea.14
Hepatocellular carcinoma: a tale of two viruses
The hepatitis viruses are a group of 5 unrelated viruses that causes inflammation of the liver. Hepatitis B (HBV), a DNA virus, and hepatitis C (HCV), an RNA virus, are also oncoviruses; HBV in particular is one of the main causes of hepatocellular carcinoma (HCC), the most common type of liver cancer.
The highly inflammatory environment fostered by HBV and HCV infection causes liver damage that often leads to cirrhosis. Continued infection can drive permanent damage to the hepatocytes, leading to genetic and epigenetic damage and driving oncogenesis. As an RNA virus, HCV doesn’t integrate into the genome and no confirmed viral oncoproteins have been identified to date, therefore it mostly drives cancer through these indirect mechanisms, which is also reflected in the fact that HCV-associated HCC predominantly occurs against a backdrop of liver cirrhosis.
HBV does integrate into the host genome. Genome sequencing studies revealed hundreds of integration sites, but most commonly they disrupted host genes involved in telomere stability and cell cycle regulation, providing some insight into the mechanisms by which HBV-associated HCC develops. In addition, HBV produces several oncoproteins, including HBx, which disrupts gene transcription, cell signaling pathways, cell cycle progress, apoptosis and other cellular processes.15,16
Multitargeted tyrosine kinase inhibitors (TKIs) have been the focal point of therapeutic development in HCC. However, following the approval of sorafenib in 2008, there was a dearth of effective new treatment options despite substantial efforts and numerous phase 3 trials. More recently, immunotherapy has also come to the forefront, especially immune checkpoint inhibitors.
Last year marked the first new drug approvals in nearly a decade – the TKI regorafenib (Stivarga) and immune checkpoint inhibitor nivolumab (Opdivo), both in the second-line setting after failure of sorafenib. Treatment options in this setting may continue to expand, with the TKIs cabozantinib and lenvatinib and the immune checkpoint inhibitor pembrolizumab and the combination of durvalumab and tremelimumab hot on their heels.17-20 Many of these drugs are also being evaluated in the front-line setting in comparison with sorafenib (Table 3).
At the current time, the treatment strategy for patients with HCC is independent of etiology, however, there are significant ongoing efforts to try to tease out the implications of infection for treatment efficacy. A recent meta-analysis of patients treated with sorafenib in 3 randomized phase 3 trials (n = 3,526) suggested that it improved overall survival (OS) among patients who were HCV-positive, but HBV-negative.21
Studies of the vascular endothelial growth factor receptor 2-targeting monoclonal antibody ramucirumab, on the other hand, suggested that it may have a greater OS benefit in patients with HBV, while regorafenib seemed to have a comparable OS benefit in both subgroups.22-25 The immune checkpoint inhibitors studied thus far seem to elicit responses irrespective of infection status.
A phase 2 trial of the immune checkpoint inhibitor tremelimumab was conducted specifically in patients with advanced HCC and chronic HCV infection. The disease control rate (DCR) was 76.4%, with 17.6% partial response (PR) rate. There was also a significant drop in viral load, suggesting that tremelimumab may have antiviral effects.26,27,28
Adoptive cell therapy promising in EBV-positive cancers
More than 90% of the global population is infected with EBV, making it one of the most common human viruses. It is a member of the herpesvirus family that is probably best known as the cause of infectious mononucleosis. On rare occasions, however, EBV can cause tumor development, though our understanding of its exact pathogenic role in cancer is still incomplete.
EBV is a DNA virus that doesn’t tend to integrate into the host genome, but instead remains in the nucleus in the form of episomes and produces several oncoproteins, including latent membrane protein-1. It is associated with a range of different cancer types, including Burkitt lymphoma and other B-cell malignancies. It also infects epithelial cells and can cause nasopharyngeal carcinoma and gastric cancer, however, much less is known about the molecular underpinnings of these EBV-positive cancer types.26,27Gastric cancers actually comprise the largest group of EBV-associated tumors because of the global incidence of this cancer type. The Cancer Genome Atlas Research Network recently characterized gastric cancer on a molecular level and identified an EBV-positive subgroup as a distinct clinical entity with unique molecular characteristics.29
The focus of therapeutic development has again been on immunotherapy, however in this case the idea of collecting the patients T cells, engineering them to recognize EBV, and then reinfusing them into the patient – adoptive cell therapy – has gained the most traction (Table 4).
Two presentations at the American Society of Hematology annual meeting in 2017 detailed ongoing clinical trials of Atara Biotherapeutics’ ATA129 and Cell Medica’s CMD-003. ATA129 was associated with a high response rate and a low rate of serious AEs in patients with posttransplant lymphoproliferative disorder; ORR was 80% in 6 patients treated after hematopoietic stem cell transplantation, and 83% in 6 patients after solid organ transplant.30
CMD-003, meanwhile, demonstrated preliminary signs of activity and safety in patients with relapsed extranodal NK/T-cell lymphoma, according to early results from the phase 2 CITADEL trial. Among 6 evaluable patients, the ORR was 50% and the DCR was 67%.31
Newest oncovirus on the block
The most recently discovered cancer-associated virus is Merkel cell polyomavirus (MCV), a DNA virus that was identified in 2008. Like EBV, virtually the whole global adult population is infected with MCV. It is linked to the development of a highly aggressive and lethal, though rare, form of skin cancer – Merkel cell carcinoma.
MCV is found in around 80% of MCC cases and in fewer than 10% of melanomas and other skin cancers. Thus far, several direct mechanisms of oncogenesis have been described, including integration of MCV into the host genome and the production of viral oncogenes, though their precise function is as yet unclear.32-34
The American Cancer Society estimates that only 1500 cases of MCC are diagnosed each year in the United States.35 Its rarity makes it difficult to conduct clinical trials with sufficient power, yet some headway has still been made.
Around half of MCCs express the programmed cell death ligand 1 (PD-L1) on their surface, making them a logical candidate for immune checkpoint inhibition. In 2017, avelumab became the first FDA-approved drug for the treatment of MCC. Approval was based on the JAVELIN Merkel 200 study in which 88 patients received avelumab. After 1 year of follow-up the ORR was 31.8%, with a CR rate of 9%.36
Genome sequencing studies suggest that the mutational profile of MCV-positive tumors is quite different to those that are MCV-negative, which could have therapeutic implications. To date, these implications have not been delineated, given the challenge of small patient numbers, however an ongoing phase 1/2 trial is evaluating the combination of avelumab and radiation therapy or recombinant interferon beta, with or without MCV-specific cytotoxic T cells in patients with MCC and MCV infection.
The 2 other known cancer-causing viruses are human T-lymphotropic virus 1 (HTLV-1), a retrovirus associated with adult T-cell leukemia/lymphoma (ATL) and Kaposi sarcoma herpesvirus (KSHV). The latter is the causative agent of Kaposi sarcoma, often in combination with human immunodeficiency virus (HIV), a rare skin tumor that became renowned in the 1980s as an AIDS-defining illness.
The incidence of HTLV-1- and KSHV-positive tumors is substantially lower than the other virally associated cancers and, like MCC, this makes studying them and conducting clinical trials of novel therapeutic options a challenge. Nonetheless, several trials of targeted therapies and immunotherapies are underway.
1. Rous PA. Transmissible avain neoplasm. (Sarcoma of the common fowl). J Exp Med. 1910;12(5):696-705.
2. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet. 1964;1(7335):702-703.
3. Mesri Enrique A, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host & Microbe. 2014;15(3):266-282.
4. Santana-Davila R, Bhatia S, Chow LQ. Harnessing the immune system as a therapeutic tool in virus-associated cancers. JAMA Oncol. 2017;3(1):106-112.
5. Tashiro H, Brenner MK. Immunotherapy against cancer-related viruses. Cell Res. 2017;27(1):59-73.
6. Brianti P, De Flammineis E, Mercuri SR. Review of HPV-related diseases and cancers. New Microbiol. 2017;40(2):80-85.
7. Tulay P, Serakinci N. The route to HPV-associated neoplastic transformation: a review of the literature. Crit Rev Eukaryot Gene Expr. 2016;26(1):27-39.
8. Smola S. Immunopathogenesis of HPV-associated cancers and prospects for immunotherapy. Viruses. 2017;9(9).
9. Rosales R, Rosales C. Immune therapy for human papillomaviruses-related cancers. World Journal of Clinical Oncology. 2014;5(5):1002-1019.
10. Miles B, Safran HP, Monk BJ. Therapeutic options for treatment of human papillomavirus-associated cancers - novel immunologic vaccines: ADXS11-001. Gynecol Oncol Res Pract. 2017;4:10.
11. Miles BA, Monk BJ, Safran HP. Mechanistic insights into ADXS11-001 human papillomavirus-associated cancer immunotherapy. Gynecol Oncol Res Pract. 2017;4:9.
12. Huh W, Dizon D, Powell M, Landrum L, Leath C. A prospective phase II trial of the listeria-based human papillomavirus immunotherapy axalimogene filolisbac in second and third-line metastatic cervical cancer: A NRG oncology group trial. Paper presented at: Annual Meeting on Women's Cancer; March 12-15, 2017, 2017; National Harbor, MD.
13. Petit RG, Mehta A, Jain M, et al. ADXS11-001 immunotherapy targeting HPV-E7: final results from a Phase II study in Indian women with recurrent cervical cancer. Journal for Immunotherapy of Cancer. 2014;2(Suppl 3):P92-P92.
14. Glisson B, Massarelli E, William W, et al. Nivolumab and ISA 101 HPV vaccine in incurable HPV-16+ cancer. Ann Oncol. 2017;28(suppl_5):v403-v427.
15. Ding X-X, Zhu Q-G, Zhang S-M, et al. Precision medicine for hepatocellular carcinoma: driver mutations and targeted therapy. Oncotarget. 2017;8(33):55715-55730.
16. Ringehan M, McKeating JA, Protzer U. Viral hepatitis and liver cancer. Philosophical Transactions of the Royal Society B: Biological Sciences. 2017;372(1732):20160274.
17. Abou-Alfa G, Meyer T, Cheng AL, et al. Cabozantinib (C) versus placebo (P) in patients (pts) with advanced hepatocellular carcinoma (HCC) who have received prior sorafenib: results from the randomized phase III CELESTIAL trial. J Clin Oncol. 2017;36(Suppl 4S):abstr 207.
18. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018.
19. Zhu AX, Finn RS, Cattan S, et al. KEYNOTE-224: Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. J Clin Oncol. 2018;36(Suppl 4S):Abstr 209.
20. Kelley RK, Abou-Alfa GK, Bendell JC, et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. Journal of Clinical Oncology. 2017;35(15_suppl):4073-4073.
21. Jackson R, Psarelli E-E, Berhane S, Khan H, Johnson P. Impact of Viral Status on Survival in Patients Receiving Sorafenib for Advanced Hepatocellular Cancer: A Meta-Analysis of Randomized Phase III Trials. Journal of Clinical Oncology. 2017;35(6):622-628.
22. Kudo M. Molecular Targeted Agents for Hepatocellular Carcinoma: Current Status and Future Perspectives. Liver Cancer. 2017;6(2):101-112.
23. zur Hausen H, Meinhof W, Scheiber W, Bornkamm GW. Attempts to detect virus-secific DNA in human tumors. I. Nucleic acid hybridizations with complementary RNA of human wart virus. Int J Cancer. 1974;13(5):650-656.
24. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56-66.
25. Bruix J, Tak WY, Gasbarrini A, et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: multicentre, open-label, phase II safety study. Eur J Cancer. 2013;49(16):3412-3419.
26. Neparidze N, Lacy J. Malignancies associated with epstein-barr virus: pathobiology, clinical features, and evolving treatments. Clin Adv Hematol Oncol. 2014;12(6):358-371.
27. Ozoya OO, Sokol L, Dalia S. EBV-Related Malignancies, Outcomes and Novel Prevention Strategies. Infect Disord Drug Targets. 2016;16(1):4-21.
28. Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81-88.
29. The Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202.
30. Prockop S, Li A, Baiocchi R, et al. Efficacy and safety of ATA129, partially matched allogeneic third-party Epstein-Barr virus-targeted cytotoxic T lymphocytes in a multicenter study for post-transplant lymphoproliferative disorder. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
31. Kim W, Ardeshna K, Lin Y, et al. Autologous EBV-specific T cells (CMD-003): Early results from a multicenter, multinational Phase 2 trial for treatment of EBV-associated NK/T-cell lymphoma. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
32. Schadendorf D, Lebbé C, zur Hausen A, et al. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. European Journal of Cancer. 2017;71:53-69.
33. Spurgeon ME, Lambert PF. Merkel cell polyomavirus: a newly discovered human virus with oncogenic potential. Virology. 2013;435(1):118-130.
34. Tello TL, Coggshall K, Yom SS, Yu SS. Merkel cell carcinoma: An update and review: Current and future therapy. J Am Acad Dermatol. 2018;78(3):445-454.
35. American Cancer Society. Key Statistics for Merkel Cell Carcinoma. 2015; https://www.cancer.org/cancer/merkel-cell-skin-cancer/about/key-statistics.html#written_by. Accessed March 7th, 2017.
36. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. The Lancet Oncology.17(10):1374-1385.
1. Rous PA. Transmissible avain neoplasm. (Sarcoma of the common fowl). J Exp Med. 1910;12(5):696-705.
2. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet. 1964;1(7335):702-703.
3. Mesri Enrique A, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host & Microbe. 2014;15(3):266-282.
4. Santana-Davila R, Bhatia S, Chow LQ. Harnessing the immune system as a therapeutic tool in virus-associated cancers. JAMA Oncol. 2017;3(1):106-112.
5. Tashiro H, Brenner MK. Immunotherapy against cancer-related viruses. Cell Res. 2017;27(1):59-73.
6. Brianti P, De Flammineis E, Mercuri SR. Review of HPV-related diseases and cancers. New Microbiol. 2017;40(2):80-85.
7. Tulay P, Serakinci N. The route to HPV-associated neoplastic transformation: a review of the literature. Crit Rev Eukaryot Gene Expr. 2016;26(1):27-39.
8. Smola S. Immunopathogenesis of HPV-associated cancers and prospects for immunotherapy. Viruses. 2017;9(9).
9. Rosales R, Rosales C. Immune therapy for human papillomaviruses-related cancers. World Journal of Clinical Oncology. 2014;5(5):1002-1019.
10. Miles B, Safran HP, Monk BJ. Therapeutic options for treatment of human papillomavirus-associated cancers - novel immunologic vaccines: ADXS11-001. Gynecol Oncol Res Pract. 2017;4:10.
11. Miles BA, Monk BJ, Safran HP. Mechanistic insights into ADXS11-001 human papillomavirus-associated cancer immunotherapy. Gynecol Oncol Res Pract. 2017;4:9.
12. Huh W, Dizon D, Powell M, Landrum L, Leath C. A prospective phase II trial of the listeria-based human papillomavirus immunotherapy axalimogene filolisbac in second and third-line metastatic cervical cancer: A NRG oncology group trial. Paper presented at: Annual Meeting on Women's Cancer; March 12-15, 2017, 2017; National Harbor, MD.
13. Petit RG, Mehta A, Jain M, et al. ADXS11-001 immunotherapy targeting HPV-E7: final results from a Phase II study in Indian women with recurrent cervical cancer. Journal for Immunotherapy of Cancer. 2014;2(Suppl 3):P92-P92.
14. Glisson B, Massarelli E, William W, et al. Nivolumab and ISA 101 HPV vaccine in incurable HPV-16+ cancer. Ann Oncol. 2017;28(suppl_5):v403-v427.
15. Ding X-X, Zhu Q-G, Zhang S-M, et al. Precision medicine for hepatocellular carcinoma: driver mutations and targeted therapy. Oncotarget. 2017;8(33):55715-55730.
16. Ringehan M, McKeating JA, Protzer U. Viral hepatitis and liver cancer. Philosophical Transactions of the Royal Society B: Biological Sciences. 2017;372(1732):20160274.
17. Abou-Alfa G, Meyer T, Cheng AL, et al. Cabozantinib (C) versus placebo (P) in patients (pts) with advanced hepatocellular carcinoma (HCC) who have received prior sorafenib: results from the randomized phase III CELESTIAL trial. J Clin Oncol. 2017;36(Suppl 4S):abstr 207.
18. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018.
19. Zhu AX, Finn RS, Cattan S, et al. KEYNOTE-224: Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. J Clin Oncol. 2018;36(Suppl 4S):Abstr 209.
20. Kelley RK, Abou-Alfa GK, Bendell JC, et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. Journal of Clinical Oncology. 2017;35(15_suppl):4073-4073.
21. Jackson R, Psarelli E-E, Berhane S, Khan H, Johnson P. Impact of Viral Status on Survival in Patients Receiving Sorafenib for Advanced Hepatocellular Cancer: A Meta-Analysis of Randomized Phase III Trials. Journal of Clinical Oncology. 2017;35(6):622-628.
22. Kudo M. Molecular Targeted Agents for Hepatocellular Carcinoma: Current Status and Future Perspectives. Liver Cancer. 2017;6(2):101-112.
23. zur Hausen H, Meinhof W, Scheiber W, Bornkamm GW. Attempts to detect virus-secific DNA in human tumors. I. Nucleic acid hybridizations with complementary RNA of human wart virus. Int J Cancer. 1974;13(5):650-656.
24. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56-66.
25. Bruix J, Tak WY, Gasbarrini A, et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: multicentre, open-label, phase II safety study. Eur J Cancer. 2013;49(16):3412-3419.
26. Neparidze N, Lacy J. Malignancies associated with epstein-barr virus: pathobiology, clinical features, and evolving treatments. Clin Adv Hematol Oncol. 2014;12(6):358-371.
27. Ozoya OO, Sokol L, Dalia S. EBV-Related Malignancies, Outcomes and Novel Prevention Strategies. Infect Disord Drug Targets. 2016;16(1):4-21.
28. Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81-88.
29. The Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202.
30. Prockop S, Li A, Baiocchi R, et al. Efficacy and safety of ATA129, partially matched allogeneic third-party Epstein-Barr virus-targeted cytotoxic T lymphocytes in a multicenter study for post-transplant lymphoproliferative disorder. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
31. Kim W, Ardeshna K, Lin Y, et al. Autologous EBV-specific T cells (CMD-003): Early results from a multicenter, multinational Phase 2 trial for treatment of EBV-associated NK/T-cell lymphoma. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
32. Schadendorf D, Lebbé C, zur Hausen A, et al. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. European Journal of Cancer. 2017;71:53-69.
33. Spurgeon ME, Lambert PF. Merkel cell polyomavirus: a newly discovered human virus with oncogenic potential. Virology. 2013;435(1):118-130.
34. Tello TL, Coggshall K, Yom SS, Yu SS. Merkel cell carcinoma: An update and review: Current and future therapy. J Am Acad Dermatol. 2018;78(3):445-454.
35. American Cancer Society. Key Statistics for Merkel Cell Carcinoma. 2015; https://www.cancer.org/cancer/merkel-cell-skin-cancer/about/key-statistics.html#written_by. Accessed March 7th, 2017.
36. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. The Lancet Oncology.17(10):1374-1385.