User login
Combo produces high response rate in CLL

Bendamustine followed by obinutuzumab and venetoclax produces a high overall response rate in chronic lymphocytic leukemia (CLL), according to research published in The Lancet Oncology.
In an ongoing, phase 2 study, researchers examined the outcomes of this treatment in 66 patients with CLL.
Patients underwent initial debulking with two cycles of bendamustine, received six cycles of obinutuzumab and venetoclax for induction, and could receive up to 24 months of maintenance with obinutuzumab and venetoclax.
Efficacy
Of the 63 patients included in the efficacy analysis, 34 (54%) were treatment-naïve, and 29 (46%) had relapsed or refractory disease.
At the end of induction, the overall response rate was 95% (60/63), with responses observed in 100% (34/34) of treatment-naive patients and 90% (26/29) of relapsed/refractory patients.
Five patients (8%) achieved complete remission (CR)—3 who were treatment-naïve and 2 with relapsed/refractory disease.
Twenty patients (32%) had a clinical CR or CR with incomplete bone marrow recovery—14 who were treatment-naïve and 6 with relapsed/refractory disease.
Thirty-five patients (56%) had a partial response—17 who were treatment-naïve and 18 with relapsed/refractory disease.
By 15 months, both progression-free and overall survival were 100% among treatment-naive patients.
In the relapsed/refractory patients, progression-free survival was 83%, and overall survival was 90%.
Researchers observed minimal residual disease negativity in the peripheral blood of 91% (31/34) of treatment-naive patients and 83% (24/29) of relapsed/refractory patients. (Most patients did not have data for MRD in the bone marrow.)
Study author Paula Cramer, MD, from the German CLL Study Group at University Hospital, Cologne, and her colleagues described the efficacy of the combination as “encouraging.”
Safety
Safety data were available for all 66 patients. Of the 677 AEs, 427 (63%) were deemed related to study treatment, and 69 of these were serious AEs. Twelve patients had related, serious AEs during debulking, and 23 patients had related, serious AEs during induction.
The most common serious AEs were infections and cytopenias. There were four infections during debulking and 18 infections in 11 patients during induction. The most common infections were pneumonia and sepsis.
There were two cases of neutropenia during debulking and six cases in five patients during induction. There were four cases of thrombocytopenia in three patients during induction.
Other common treatment-related, serious AEs were:
- Infusion-related reactions—six cases during induction
- Coronary artery disorder—one case during debulking and three during induction
- Tumor lysis syndrome —one case during debulking and two during induction
- Neoplasms—two squamous cell carcinomas and one malignant melanoma during induction
- Increased creatinine—two cases during debulking.
Five patients in the relapsed/refractory group died—three of sepsis related to study treatment and two from unrelated Richter’s transformation.
“With three deaths from sepsis in 66 enrolled patients, the treatment-related mortality seems high; however, in cases of low patient numbers, a few patients can have a substantial effect on the overall results,” the researchers wrote.
The study was funded by F Hoffmann-La Roche and AbbVie. Several authors reported research funding, grants, honoraria, and other support from the pharmaceutical industry, including from the study sponsors.
Bendamustine followed by obinutuzumab and venetoclax produces a high overall response rate in chronic lymphocytic leukemia (CLL), according to research published in The Lancet Oncology.
In an ongoing, phase 2 study, researchers examined the outcomes of this treatment in 66 patients with CLL.
Patients underwent initial debulking with two cycles of bendamustine, received six cycles of obinutuzumab and venetoclax for induction, and could receive up to 24 months of maintenance with obinutuzumab and venetoclax.
Efficacy
Of the 63 patients included in the efficacy analysis, 34 (54%) were treatment-naïve, and 29 (46%) had relapsed or refractory disease.
At the end of induction, the overall response rate was 95% (60/63), with responses observed in 100% (34/34) of treatment-naive patients and 90% (26/29) of relapsed/refractory patients.
Five patients (8%) achieved complete remission (CR)—3 who were treatment-naïve and 2 with relapsed/refractory disease.
Twenty patients (32%) had a clinical CR or CR with incomplete bone marrow recovery—14 who were treatment-naïve and 6 with relapsed/refractory disease.
Thirty-five patients (56%) had a partial response—17 who were treatment-naïve and 18 with relapsed/refractory disease.
By 15 months, both progression-free and overall survival were 100% among treatment-naive patients.
In the relapsed/refractory patients, progression-free survival was 83%, and overall survival was 90%.
Researchers observed minimal residual disease negativity in the peripheral blood of 91% (31/34) of treatment-naive patients and 83% (24/29) of relapsed/refractory patients. (Most patients did not have data for MRD in the bone marrow.)
Study author Paula Cramer, MD, from the German CLL Study Group at University Hospital, Cologne, and her colleagues described the efficacy of the combination as “encouraging.”
Safety
Safety data were available for all 66 patients. Of the 677 AEs, 427 (63%) were deemed related to study treatment, and 69 of these were serious AEs. Twelve patients had related, serious AEs during debulking, and 23 patients had related, serious AEs during induction.
The most common serious AEs were infections and cytopenias. There were four infections during debulking and 18 infections in 11 patients during induction. The most common infections were pneumonia and sepsis.
There were two cases of neutropenia during debulking and six cases in five patients during induction. There were four cases of thrombocytopenia in three patients during induction.
Other common treatment-related, serious AEs were:
- Infusion-related reactions—six cases during induction
- Coronary artery disorder—one case during debulking and three during induction
- Tumor lysis syndrome —one case during debulking and two during induction
- Neoplasms—two squamous cell carcinomas and one malignant melanoma during induction
- Increased creatinine—two cases during debulking.
Five patients in the relapsed/refractory group died—three of sepsis related to study treatment and two from unrelated Richter’s transformation.
“With three deaths from sepsis in 66 enrolled patients, the treatment-related mortality seems high; however, in cases of low patient numbers, a few patients can have a substantial effect on the overall results,” the researchers wrote.
The study was funded by F Hoffmann-La Roche and AbbVie. Several authors reported research funding, grants, honoraria, and other support from the pharmaceutical industry, including from the study sponsors.
Bendamustine followed by obinutuzumab and venetoclax produces a high overall response rate in chronic lymphocytic leukemia (CLL), according to research published in The Lancet Oncology.
In an ongoing, phase 2 study, researchers examined the outcomes of this treatment in 66 patients with CLL.
Patients underwent initial debulking with two cycles of bendamustine, received six cycles of obinutuzumab and venetoclax for induction, and could receive up to 24 months of maintenance with obinutuzumab and venetoclax.
Efficacy
Of the 63 patients included in the efficacy analysis, 34 (54%) were treatment-naïve, and 29 (46%) had relapsed or refractory disease.
At the end of induction, the overall response rate was 95% (60/63), with responses observed in 100% (34/34) of treatment-naive patients and 90% (26/29) of relapsed/refractory patients.
Five patients (8%) achieved complete remission (CR)—3 who were treatment-naïve and 2 with relapsed/refractory disease.
Twenty patients (32%) had a clinical CR or CR with incomplete bone marrow recovery—14 who were treatment-naïve and 6 with relapsed/refractory disease.
Thirty-five patients (56%) had a partial response—17 who were treatment-naïve and 18 with relapsed/refractory disease.
By 15 months, both progression-free and overall survival were 100% among treatment-naive patients.
In the relapsed/refractory patients, progression-free survival was 83%, and overall survival was 90%.
Researchers observed minimal residual disease negativity in the peripheral blood of 91% (31/34) of treatment-naive patients and 83% (24/29) of relapsed/refractory patients. (Most patients did not have data for MRD in the bone marrow.)
Study author Paula Cramer, MD, from the German CLL Study Group at University Hospital, Cologne, and her colleagues described the efficacy of the combination as “encouraging.”
Safety
Safety data were available for all 66 patients. Of the 677 AEs, 427 (63%) were deemed related to study treatment, and 69 of these were serious AEs. Twelve patients had related, serious AEs during debulking, and 23 patients had related, serious AEs during induction.
The most common serious AEs were infections and cytopenias. There were four infections during debulking and 18 infections in 11 patients during induction. The most common infections were pneumonia and sepsis.
There were two cases of neutropenia during debulking and six cases in five patients during induction. There were four cases of thrombocytopenia in three patients during induction.
Other common treatment-related, serious AEs were:
- Infusion-related reactions—six cases during induction
- Coronary artery disorder—one case during debulking and three during induction
- Tumor lysis syndrome —one case during debulking and two during induction
- Neoplasms—two squamous cell carcinomas and one malignant melanoma during induction
- Increased creatinine—two cases during debulking.
Five patients in the relapsed/refractory group died—three of sepsis related to study treatment and two from unrelated Richter’s transformation.
“With three deaths from sepsis in 66 enrolled patients, the treatment-related mortality seems high; however, in cases of low patient numbers, a few patients can have a substantial effect on the overall results,” the researchers wrote.
The study was funded by F Hoffmann-La Roche and AbbVie. Several authors reported research funding, grants, honoraria, and other support from the pharmaceutical industry, including from the study sponsors.
Combo treatment yields MRD-negative remissions in CLL
The combination of the anti-CD20 antibody obinutuzumab and venetoclax in chronic lymphocytic leukemia shows a high overall response rate and compares favorably with established therapies, according to a new report.
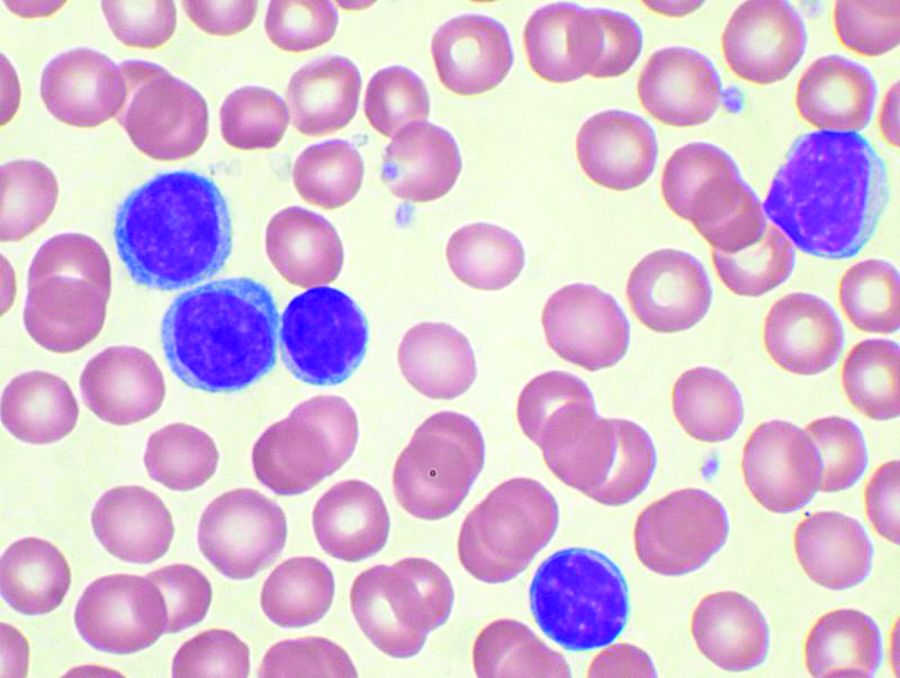
The ongoing, open-label, phase 2 study examined the outcomes of six induction cycles, followed by up to 24 months of maintenance treatment with obinutuzumab and venetoclax, in 66 patients with chronic lymphocytic leukemia (CLL). Of the 63 patients included in the efficacy analysis, 34 (54%) had treatment-naive and 29 (46%) had relapsed or refractory disease.
After an initial debulking with two cycles of bendamustine, followed by the obinutuzumab and venetoclax treatment, researchers observed an overall response rate of 95%. By the end of the induction phase, all the treatment-naive patients responded, as did 90% of the relapsed or refractory patients. Five patients had achieved complete remission and 55 patients had a partial response, the researchers reported in Lancet Oncology.
By 15 months, both progression-free and overall survival was 100% among treatment-naive patients, while progression-free survival was 83% and overall survival was 90% among the relapsed or refractory patients at this point.
Researchers observed minimal residual disease (MRD) negativity in the peripheral blood of 91% of treatment-naive patients and 83% of relapsed or refractory patients.
The combination of venetoclax and obinutuzumab was chosen based on earlier trial data, which suggested a synergy between venetoclax and the less-potent anti-CD20 antibody rituximab.
Paula Cramer, MD, from the German CLL Study Group at University Hospital, Cologne, and her coauthors described the efficacy of the venetoclax and obinutuzumab combination as “encouraging.”
“The combination of venetoclax and obinutuzumab yields fast responses with MRD-negative remissions in most patients,” they wrote. “Based on the experience with venetoclax combined with rituximab in another trial and with venetoclax and obinutuzumab in this and another study, these deep, MRD-negative remissions seem to last for a substantial time after treatment termination.”
Of the 677 adverse events, 427 (63%) were deemed to be related to the study treatment, and 69 of these were serious adverse events.
The most common of these were infections, experienced by four patients during the debulking with bendamustine, and 18 cases in 11 patients during the induction treatment. This included pneumonia, sepsis and cytomegalovirus infection, as well as neutropenia and thrombocytopenia.
Six patients also experienced infusion-related reactions, four had coronary artery disorder – one during debulking and three during induction – and there were three cases of neoplasms.
Five patients in the relapsed or refractory group died; three of sepsis related to study treatment, and two from unrelated Richter’s transformation.
“With three deaths from sepsis in 66 enrolled patients, the treatment-related mortality seems high; however, in cases of low patient numbers, a few patients can have a substantial effect on the overall results,” the researchers wrote.
The study was funded by F Hoffmann-La Roche and AbbVie. Several authors reported research funding, grants, honoraria and other support from the pharmaceutical industry, including from the study sponsors.
SOURCE: Cramer P et al. Lancet Oncol. 2018 Aug 13. doi: 10.1016/S1470-2045(18)30414-5.
The combination of the anti-CD20 antibody obinutuzumab and venetoclax in chronic lymphocytic leukemia shows a high overall response rate and compares favorably with established therapies, according to a new report.
The ongoing, open-label, phase 2 study examined the outcomes of six induction cycles, followed by up to 24 months of maintenance treatment with obinutuzumab and venetoclax, in 66 patients with chronic lymphocytic leukemia (CLL). Of the 63 patients included in the efficacy analysis, 34 (54%) had treatment-naive and 29 (46%) had relapsed or refractory disease.
After an initial debulking with two cycles of bendamustine, followed by the obinutuzumab and venetoclax treatment, researchers observed an overall response rate of 95%. By the end of the induction phase, all the treatment-naive patients responded, as did 90% of the relapsed or refractory patients. Five patients had achieved complete remission and 55 patients had a partial response, the researchers reported in Lancet Oncology.
By 15 months, both progression-free and overall survival was 100% among treatment-naive patients, while progression-free survival was 83% and overall survival was 90% among the relapsed or refractory patients at this point.
Researchers observed minimal residual disease (MRD) negativity in the peripheral blood of 91% of treatment-naive patients and 83% of relapsed or refractory patients.
The combination of venetoclax and obinutuzumab was chosen based on earlier trial data, which suggested a synergy between venetoclax and the less-potent anti-CD20 antibody rituximab.
Paula Cramer, MD, from the German CLL Study Group at University Hospital, Cologne, and her coauthors described the efficacy of the venetoclax and obinutuzumab combination as “encouraging.”
“The combination of venetoclax and obinutuzumab yields fast responses with MRD-negative remissions in most patients,” they wrote. “Based on the experience with venetoclax combined with rituximab in another trial and with venetoclax and obinutuzumab in this and another study, these deep, MRD-negative remissions seem to last for a substantial time after treatment termination.”
Of the 677 adverse events, 427 (63%) were deemed to be related to the study treatment, and 69 of these were serious adverse events.
The most common of these were infections, experienced by four patients during the debulking with bendamustine, and 18 cases in 11 patients during the induction treatment. This included pneumonia, sepsis and cytomegalovirus infection, as well as neutropenia and thrombocytopenia.
Six patients also experienced infusion-related reactions, four had coronary artery disorder – one during debulking and three during induction – and there were three cases of neoplasms.
Five patients in the relapsed or refractory group died; three of sepsis related to study treatment, and two from unrelated Richter’s transformation.
“With three deaths from sepsis in 66 enrolled patients, the treatment-related mortality seems high; however, in cases of low patient numbers, a few patients can have a substantial effect on the overall results,” the researchers wrote.
The study was funded by F Hoffmann-La Roche and AbbVie. Several authors reported research funding, grants, honoraria and other support from the pharmaceutical industry, including from the study sponsors.
SOURCE: Cramer P et al. Lancet Oncol. 2018 Aug 13. doi: 10.1016/S1470-2045(18)30414-5.
The combination of the anti-CD20 antibody obinutuzumab and venetoclax in chronic lymphocytic leukemia shows a high overall response rate and compares favorably with established therapies, according to a new report.
The ongoing, open-label, phase 2 study examined the outcomes of six induction cycles, followed by up to 24 months of maintenance treatment with obinutuzumab and venetoclax, in 66 patients with chronic lymphocytic leukemia (CLL). Of the 63 patients included in the efficacy analysis, 34 (54%) had treatment-naive and 29 (46%) had relapsed or refractory disease.
After an initial debulking with two cycles of bendamustine, followed by the obinutuzumab and venetoclax treatment, researchers observed an overall response rate of 95%. By the end of the induction phase, all the treatment-naive patients responded, as did 90% of the relapsed or refractory patients. Five patients had achieved complete remission and 55 patients had a partial response, the researchers reported in Lancet Oncology.
By 15 months, both progression-free and overall survival was 100% among treatment-naive patients, while progression-free survival was 83% and overall survival was 90% among the relapsed or refractory patients at this point.
Researchers observed minimal residual disease (MRD) negativity in the peripheral blood of 91% of treatment-naive patients and 83% of relapsed or refractory patients.
The combination of venetoclax and obinutuzumab was chosen based on earlier trial data, which suggested a synergy between venetoclax and the less-potent anti-CD20 antibody rituximab.
Paula Cramer, MD, from the German CLL Study Group at University Hospital, Cologne, and her coauthors described the efficacy of the venetoclax and obinutuzumab combination as “encouraging.”
“The combination of venetoclax and obinutuzumab yields fast responses with MRD-negative remissions in most patients,” they wrote. “Based on the experience with venetoclax combined with rituximab in another trial and with venetoclax and obinutuzumab in this and another study, these deep, MRD-negative remissions seem to last for a substantial time after treatment termination.”
Of the 677 adverse events, 427 (63%) were deemed to be related to the study treatment, and 69 of these were serious adverse events.
The most common of these were infections, experienced by four patients during the debulking with bendamustine, and 18 cases in 11 patients during the induction treatment. This included pneumonia, sepsis and cytomegalovirus infection, as well as neutropenia and thrombocytopenia.
Six patients also experienced infusion-related reactions, four had coronary artery disorder – one during debulking and three during induction – and there were three cases of neoplasms.
Five patients in the relapsed or refractory group died; three of sepsis related to study treatment, and two from unrelated Richter’s transformation.
“With three deaths from sepsis in 66 enrolled patients, the treatment-related mortality seems high; however, in cases of low patient numbers, a few patients can have a substantial effect on the overall results,” the researchers wrote.
The study was funded by F Hoffmann-La Roche and AbbVie. Several authors reported research funding, grants, honoraria and other support from the pharmaceutical industry, including from the study sponsors.
SOURCE: Cramer P et al. Lancet Oncol. 2018 Aug 13. doi: 10.1016/S1470-2045(18)30414-5.
FROM LANCET ONCOLOGY
Key clinical point:
Major finding: The overall response rate for obinutuzumab plus venetoclax in CLL was 95%.
Study details: An ongoing, phase 2, open-label trial in 66 patients with chronic lymphocytic leukemia.
Disclosures: The study was funded by F Hoffmann-La Roche and AbbVie. Several authors reported research funding, grants, honoraria, and other support from the pharmaceutical industry, including from the study sponsors.
Source: Cramer P et al. Lancet Oncol. 2018 Aug 13. doi: 10.1016/S1470-2045(18)30414-5.
AMP publishes report on DNA variants in CMNs

A new report addresses the clinical relevance of DNA variants in chronic myeloid neoplasms (CMNs).
The report is intended to aid clinical laboratory professionals with the management of most CMNs and the development of high-throughput pan-myeloid sequencing testing panels.
The authors list 34 genes they consider “critical” for sequencing tests to help standardize clinical practice and improve care of patients with CMNs.
The Association for Molecular Pathology (AMP) established a CMN Working Group to generate the report, which was published in The Journal of Molecular Diagnostics.
“The molecular pathology community has witnessed a recent explosion of scientific literature highlighting the clinical significance of small DNA variants in CMNs,” said Rebecca F. McClure, MD, a member of the AMP CMN Working Group and an associate professor at Health Sciences North/Horizon Santé-Nord in Sudbury, Ontario, Canada.
“AMP’s working group recognized a clear, unmet need for evidence-based recommendations to assist in the development of the high-quality pan-myeloid gene panels that provide relevant diagnostic and prognostic information and enable monitoring of clonal architecture.”
The increasing availability of targeted, high-throughput, next-generation sequencing panels has enabled scientists to explore the genetic heterogeneity and clinical relevance of the small DNA variants in CMNs.
However, the biological complexity and multiple forms of CMNs have led to variability in the genes included on the available panels that are used to make an accurate diagnosis, provide reliable prognostic information, and select an appropriate therapy based on DNA variant profiles present at various time points.
AMP established its CMN Working Group to review the published literature on CMNs, summarize key findings that support clinical utility, and define a set of critical gene inclusions for all high-throughput pan-myeloid sequencing testing panels.
The group proposed the following 34 genes as a minimum recommended testing list: ASXL1, BCOR, BCORL1, CALR, CBL, CEBPA, CSF3R, DNMT3A, ETV6, EZH2, FLT3, IDH1, IDH2, JAK2, KIT, KRAS, MPL, NF1, NPM1, NRAS, PHF6, PPM1D, PTPN11, RAD21, RUNX1, SETBP1, SF3B1, SMC3, SRSF2, STAG2, TET2, TP53, U2AF1, and ZRSR2.
“While the goal of the study was to distill the literature for molecular pathologists, in doing so, we also revealed recurrent mutational patterns of clonal evolution that will [help] hematologist/oncologists, researchers, and pathologists understand how to interpret the results of these panels as they reveal critical biology of the neoplasms,” said Annette S. Kim, MD, PhD, CMN Working Group Chair and an associate professor at Harvard Medical School and Brigham and Women’s Hospital in Boston, Massachusetts.
A new report addresses the clinical relevance of DNA variants in chronic myeloid neoplasms (CMNs).
The report is intended to aid clinical laboratory professionals with the management of most CMNs and the development of high-throughput pan-myeloid sequencing testing panels.
The authors list 34 genes they consider “critical” for sequencing tests to help standardize clinical practice and improve care of patients with CMNs.
The Association for Molecular Pathology (AMP) established a CMN Working Group to generate the report, which was published in The Journal of Molecular Diagnostics.
“The molecular pathology community has witnessed a recent explosion of scientific literature highlighting the clinical significance of small DNA variants in CMNs,” said Rebecca F. McClure, MD, a member of the AMP CMN Working Group and an associate professor at Health Sciences North/Horizon Santé-Nord in Sudbury, Ontario, Canada.
“AMP’s working group recognized a clear, unmet need for evidence-based recommendations to assist in the development of the high-quality pan-myeloid gene panels that provide relevant diagnostic and prognostic information and enable monitoring of clonal architecture.”
The increasing availability of targeted, high-throughput, next-generation sequencing panels has enabled scientists to explore the genetic heterogeneity and clinical relevance of the small DNA variants in CMNs.
However, the biological complexity and multiple forms of CMNs have led to variability in the genes included on the available panels that are used to make an accurate diagnosis, provide reliable prognostic information, and select an appropriate therapy based on DNA variant profiles present at various time points.
AMP established its CMN Working Group to review the published literature on CMNs, summarize key findings that support clinical utility, and define a set of critical gene inclusions for all high-throughput pan-myeloid sequencing testing panels.
The group proposed the following 34 genes as a minimum recommended testing list: ASXL1, BCOR, BCORL1, CALR, CBL, CEBPA, CSF3R, DNMT3A, ETV6, EZH2, FLT3, IDH1, IDH2, JAK2, KIT, KRAS, MPL, NF1, NPM1, NRAS, PHF6, PPM1D, PTPN11, RAD21, RUNX1, SETBP1, SF3B1, SMC3, SRSF2, STAG2, TET2, TP53, U2AF1, and ZRSR2.
“While the goal of the study was to distill the literature for molecular pathologists, in doing so, we also revealed recurrent mutational patterns of clonal evolution that will [help] hematologist/oncologists, researchers, and pathologists understand how to interpret the results of these panels as they reveal critical biology of the neoplasms,” said Annette S. Kim, MD, PhD, CMN Working Group Chair and an associate professor at Harvard Medical School and Brigham and Women’s Hospital in Boston, Massachusetts.
A new report addresses the clinical relevance of DNA variants in chronic myeloid neoplasms (CMNs).
The report is intended to aid clinical laboratory professionals with the management of most CMNs and the development of high-throughput pan-myeloid sequencing testing panels.
The authors list 34 genes they consider “critical” for sequencing tests to help standardize clinical practice and improve care of patients with CMNs.
The Association for Molecular Pathology (AMP) established a CMN Working Group to generate the report, which was published in The Journal of Molecular Diagnostics.
“The molecular pathology community has witnessed a recent explosion of scientific literature highlighting the clinical significance of small DNA variants in CMNs,” said Rebecca F. McClure, MD, a member of the AMP CMN Working Group and an associate professor at Health Sciences North/Horizon Santé-Nord in Sudbury, Ontario, Canada.
“AMP’s working group recognized a clear, unmet need for evidence-based recommendations to assist in the development of the high-quality pan-myeloid gene panels that provide relevant diagnostic and prognostic information and enable monitoring of clonal architecture.”
The increasing availability of targeted, high-throughput, next-generation sequencing panels has enabled scientists to explore the genetic heterogeneity and clinical relevance of the small DNA variants in CMNs.
However, the biological complexity and multiple forms of CMNs have led to variability in the genes included on the available panels that are used to make an accurate diagnosis, provide reliable prognostic information, and select an appropriate therapy based on DNA variant profiles present at various time points.
AMP established its CMN Working Group to review the published literature on CMNs, summarize key findings that support clinical utility, and define a set of critical gene inclusions for all high-throughput pan-myeloid sequencing testing panels.
The group proposed the following 34 genes as a minimum recommended testing list: ASXL1, BCOR, BCORL1, CALR, CBL, CEBPA, CSF3R, DNMT3A, ETV6, EZH2, FLT3, IDH1, IDH2, JAK2, KIT, KRAS, MPL, NF1, NPM1, NRAS, PHF6, PPM1D, PTPN11, RAD21, RUNX1, SETBP1, SF3B1, SMC3, SRSF2, STAG2, TET2, TP53, U2AF1, and ZRSR2.
“While the goal of the study was to distill the literature for molecular pathologists, in doing so, we also revealed recurrent mutational patterns of clonal evolution that will [help] hematologist/oncologists, researchers, and pathologists understand how to interpret the results of these panels as they reveal critical biology of the neoplasms,” said Annette S. Kim, MD, PhD, CMN Working Group Chair and an associate professor at Harvard Medical School and Brigham and Women’s Hospital in Boston, Massachusetts.
FDA grants orphan designation to DHODH inhibitor for AML

ASLAN003 is a small molecule inhibitor of the human dihydroorotate dehydrogenase (DHODH) enzyme. This second-generation DHODH inhibitor is being developed by Aslan Pharmaceuticals. The company is currently conducting a phase 2 trial (NCT03451084) of ASLAN003 in patients with newly diagnosed or relapsed/refractory AML. Aslan expects to report interim data from this trial in the second half of 2018.
Aslan has already completed a phase 1 trial (NCT01992367) of ASLAN003 in healthy volunteers. The results suggested that ASLAN003 has an “excellent” pharmacokinetic profile, according to Aslan, and the drug was considered well tolerated in the volunteers.
ASLAN003 has also demonstrated “potent” inhibition of DHODH, according to the drug sponsor. In fact, the company said the binding affinity of ASLAN003 to DHODH has proven to be up to two orders of magnitude stronger than first-generation DHODH inhibitors, such as leflunomide and teriflunomide, but it has less toxicity.
In addition, ASLAN003 has been shown to differentiate blast cells into granulocytes in AML cell lines that do not respond to all-trans retinoic acid. These results were published in Cell in 2016.
ASLAN003 is a small molecule inhibitor of the human dihydroorotate dehydrogenase (DHODH) enzyme. This second-generation DHODH inhibitor is being developed by Aslan Pharmaceuticals. The company is currently conducting a phase 2 trial (NCT03451084) of ASLAN003 in patients with newly diagnosed or relapsed/refractory AML. Aslan expects to report interim data from this trial in the second half of 2018.
Aslan has already completed a phase 1 trial (NCT01992367) of ASLAN003 in healthy volunteers. The results suggested that ASLAN003 has an “excellent” pharmacokinetic profile, according to Aslan, and the drug was considered well tolerated in the volunteers.
ASLAN003 has also demonstrated “potent” inhibition of DHODH, according to the drug sponsor. In fact, the company said the binding affinity of ASLAN003 to DHODH has proven to be up to two orders of magnitude stronger than first-generation DHODH inhibitors, such as leflunomide and teriflunomide, but it has less toxicity.
In addition, ASLAN003 has been shown to differentiate blast cells into granulocytes in AML cell lines that do not respond to all-trans retinoic acid. These results were published in Cell in 2016.
ASLAN003 is a small molecule inhibitor of the human dihydroorotate dehydrogenase (DHODH) enzyme. This second-generation DHODH inhibitor is being developed by Aslan Pharmaceuticals. The company is currently conducting a phase 2 trial (NCT03451084) of ASLAN003 in patients with newly diagnosed or relapsed/refractory AML. Aslan expects to report interim data from this trial in the second half of 2018.
Aslan has already completed a phase 1 trial (NCT01992367) of ASLAN003 in healthy volunteers. The results suggested that ASLAN003 has an “excellent” pharmacokinetic profile, according to Aslan, and the drug was considered well tolerated in the volunteers.
ASLAN003 has also demonstrated “potent” inhibition of DHODH, according to the drug sponsor. In fact, the company said the binding affinity of ASLAN003 to DHODH has proven to be up to two orders of magnitude stronger than first-generation DHODH inhibitors, such as leflunomide and teriflunomide, but it has less toxicity.
In addition, ASLAN003 has been shown to differentiate blast cells into granulocytes in AML cell lines that do not respond to all-trans retinoic acid. These results were published in Cell in 2016.
Role of SES in childhood cancer survival disparities

Socioeconomic status (SES) may explain some racial/ethnic disparities in childhood cancer survival, according to new research.
The study showed that whites had a significant survival advantage over blacks and Hispanics for several childhood cancers.
SES significantly mediated the association between race/ethnicity and survival for acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), neuroblastoma, and non-Hodgkin lymphoma (NHL).
Rebecca Kehm, PhD, of Columbia University in New York, New York, and her colleagues reported these findings in Cancer alongside a related editorial.
The researchers examined population-based cancer survival data from the Surveillance, Epidemiology, and End Results database.
The team collected information on 31,866 patients, ages 0 to 19, who were diagnosed with cancer between 2000 and 2011.
Survival differences by race/ethnicity
The researchers found that whites had a significant survival advantage over blacks for the cancers listed in the following table.
| Survival—black vs white | |||
| Cancer | Mortality hazard ratio | 95% confidence interval | P value |
| ALL | 1.43 | 1.15-1.77 | <0.01 |
| AML | 1.68 | 1.36-2.07 | <0.001 |
| Neuroblastoma | 1.38 | 1.08-1.75 | 0.01 |
| NHL | 1.53 | 1.14-2.07 | 0.01 |
| Hodgkin lymphoma | 1.66 | 1.06-2.60 | 0.03 |
| Astrocytoma | 1.95 | 1.57-2.43 | <0.001 |
| Non-astrocytoma CNS tumor | 1.53 | 1.25-1.88 | <0.001 |
| Non-rhabdomyosarcoma STS | 1.40 | 1.06-1.84 | 0.02 |
| Rhabdomyosarcoma | 1.44 | 1.10-1.88 | 0.01 |
In addition, whites had a significant survival advantage over Hispanics for the following cancers.
| Survival—Hispanic vs white | |||
| Cancer | Mortality hazard ratio | 95% confidence interval | P value |
| ALL | 1.63 | 1.43-1.86 | <0.001 |
| Neuroblastoma | 1.31 | 1.04-1.65 | 0.02 |
| NHL | 1.65 | 1.29-2.12 | <0.001 |
| Astrocytoma | 1.34 | 1.10-1.64 | <0.01 |
| Wilms tumor | 1.60 | 1.04-2.45 | 0.03 |
| Germ cell tumor | 1.63 | 1.19-2.24 | <0.01 |
Impact of SES
SES significantly mediated the association between race/ethnicity and survival for ALL, AML, neuroblastoma, and NHL but not for Hodgkin lymphoma or other cancers.
For black versus white patients, SES reduced the original association between race/ethnicity and survival by:
- 44% for ALL
- 28% for AML
- 49% for neuroblastoma
- 34% for NHL.
For Hispanics versus whites, SES reduced the original association between race/ethnicity and survival by:
- 31% for ALL
- 73% for AML
- 48% for neuroblastoma
- 28% for NHL.
“These findings provide insight for future intervention efforts aimed at closing the survival gap,” Dr Kehm said.
“For cancers in which socioeconomic status is a key factor in explaining racial and ethnic survival disparities, behavioral and supportive interventions that address social and economic barriers to effective care are warranted. However, for cancers in which survival is less influenced by socioeconomic status, more research is needed on underlying differences in tumor biology and drug processing.”
This research was supported by a grant from the National Institutes of Health, and the study’s authors made no disclosures.
Socioeconomic status (SES) may explain some racial/ethnic disparities in childhood cancer survival, according to new research.
The study showed that whites had a significant survival advantage over blacks and Hispanics for several childhood cancers.
SES significantly mediated the association between race/ethnicity and survival for acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), neuroblastoma, and non-Hodgkin lymphoma (NHL).
Rebecca Kehm, PhD, of Columbia University in New York, New York, and her colleagues reported these findings in Cancer alongside a related editorial.
The researchers examined population-based cancer survival data from the Surveillance, Epidemiology, and End Results database.
The team collected information on 31,866 patients, ages 0 to 19, who were diagnosed with cancer between 2000 and 2011.
Survival differences by race/ethnicity
The researchers found that whites had a significant survival advantage over blacks for the cancers listed in the following table.
| Survival—black vs white | |||
| Cancer | Mortality hazard ratio | 95% confidence interval | P value |
| ALL | 1.43 | 1.15-1.77 | <0.01 |
| AML | 1.68 | 1.36-2.07 | <0.001 |
| Neuroblastoma | 1.38 | 1.08-1.75 | 0.01 |
| NHL | 1.53 | 1.14-2.07 | 0.01 |
| Hodgkin lymphoma | 1.66 | 1.06-2.60 | 0.03 |
| Astrocytoma | 1.95 | 1.57-2.43 | <0.001 |
| Non-astrocytoma CNS tumor | 1.53 | 1.25-1.88 | <0.001 |
| Non-rhabdomyosarcoma STS | 1.40 | 1.06-1.84 | 0.02 |
| Rhabdomyosarcoma | 1.44 | 1.10-1.88 | 0.01 |
In addition, whites had a significant survival advantage over Hispanics for the following cancers.
| Survival—Hispanic vs white | |||
| Cancer | Mortality hazard ratio | 95% confidence interval | P value |
| ALL | 1.63 | 1.43-1.86 | <0.001 |
| Neuroblastoma | 1.31 | 1.04-1.65 | 0.02 |
| NHL | 1.65 | 1.29-2.12 | <0.001 |
| Astrocytoma | 1.34 | 1.10-1.64 | <0.01 |
| Wilms tumor | 1.60 | 1.04-2.45 | 0.03 |
| Germ cell tumor | 1.63 | 1.19-2.24 | <0.01 |
Impact of SES
SES significantly mediated the association between race/ethnicity and survival for ALL, AML, neuroblastoma, and NHL but not for Hodgkin lymphoma or other cancers.
For black versus white patients, SES reduced the original association between race/ethnicity and survival by:
- 44% for ALL
- 28% for AML
- 49% for neuroblastoma
- 34% for NHL.
For Hispanics versus whites, SES reduced the original association between race/ethnicity and survival by:
- 31% for ALL
- 73% for AML
- 48% for neuroblastoma
- 28% for NHL.
“These findings provide insight for future intervention efforts aimed at closing the survival gap,” Dr Kehm said.
“For cancers in which socioeconomic status is a key factor in explaining racial and ethnic survival disparities, behavioral and supportive interventions that address social and economic barriers to effective care are warranted. However, for cancers in which survival is less influenced by socioeconomic status, more research is needed on underlying differences in tumor biology and drug processing.”
This research was supported by a grant from the National Institutes of Health, and the study’s authors made no disclosures.
Socioeconomic status (SES) may explain some racial/ethnic disparities in childhood cancer survival, according to new research.
The study showed that whites had a significant survival advantage over blacks and Hispanics for several childhood cancers.
SES significantly mediated the association between race/ethnicity and survival for acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), neuroblastoma, and non-Hodgkin lymphoma (NHL).
Rebecca Kehm, PhD, of Columbia University in New York, New York, and her colleagues reported these findings in Cancer alongside a related editorial.
The researchers examined population-based cancer survival data from the Surveillance, Epidemiology, and End Results database.
The team collected information on 31,866 patients, ages 0 to 19, who were diagnosed with cancer between 2000 and 2011.
Survival differences by race/ethnicity
The researchers found that whites had a significant survival advantage over blacks for the cancers listed in the following table.
| Survival—black vs white | |||
| Cancer | Mortality hazard ratio | 95% confidence interval | P value |
| ALL | 1.43 | 1.15-1.77 | <0.01 |
| AML | 1.68 | 1.36-2.07 | <0.001 |
| Neuroblastoma | 1.38 | 1.08-1.75 | 0.01 |
| NHL | 1.53 | 1.14-2.07 | 0.01 |
| Hodgkin lymphoma | 1.66 | 1.06-2.60 | 0.03 |
| Astrocytoma | 1.95 | 1.57-2.43 | <0.001 |
| Non-astrocytoma CNS tumor | 1.53 | 1.25-1.88 | <0.001 |
| Non-rhabdomyosarcoma STS | 1.40 | 1.06-1.84 | 0.02 |
| Rhabdomyosarcoma | 1.44 | 1.10-1.88 | 0.01 |
In addition, whites had a significant survival advantage over Hispanics for the following cancers.
| Survival—Hispanic vs white | |||
| Cancer | Mortality hazard ratio | 95% confidence interval | P value |
| ALL | 1.63 | 1.43-1.86 | <0.001 |
| Neuroblastoma | 1.31 | 1.04-1.65 | 0.02 |
| NHL | 1.65 | 1.29-2.12 | <0.001 |
| Astrocytoma | 1.34 | 1.10-1.64 | <0.01 |
| Wilms tumor | 1.60 | 1.04-2.45 | 0.03 |
| Germ cell tumor | 1.63 | 1.19-2.24 | <0.01 |
Impact of SES
SES significantly mediated the association between race/ethnicity and survival for ALL, AML, neuroblastoma, and NHL but not for Hodgkin lymphoma or other cancers.
For black versus white patients, SES reduced the original association between race/ethnicity and survival by:
- 44% for ALL
- 28% for AML
- 49% for neuroblastoma
- 34% for NHL.
For Hispanics versus whites, SES reduced the original association between race/ethnicity and survival by:
- 31% for ALL
- 73% for AML
- 48% for neuroblastoma
- 28% for NHL.
“These findings provide insight for future intervention efforts aimed at closing the survival gap,” Dr Kehm said.
“For cancers in which socioeconomic status is a key factor in explaining racial and ethnic survival disparities, behavioral and supportive interventions that address social and economic barriers to effective care are warranted. However, for cancers in which survival is less influenced by socioeconomic status, more research is needed on underlying differences in tumor biology and drug processing.”
This research was supported by a grant from the National Institutes of Health, and the study’s authors made no disclosures.
Inhibitor receives orphan designation for AML

The US Food and Drug Administration (FDA) has granted orphan drug designation to ASLAN003 as a treatment for acute myeloid leukemia (AML).
ASLAN003 is a small molecule inhibitor of the human dihydroorotate dehydrogenase (DHODH) enzyme.
This second-generation DHODH inhibitor is being developed by ASLAN Pharmaceuticals.
The company is currently conducting a phase 2 trial (NCT03451084) of ASLAN003 in patients with newly diagnosed or relapsed/refractory AML.
The goals of this trial are to determine the optimum dose of ASLAN003 as monotherapy and assess the drug’s efficacy by overall complete remission rate. ASLAN expects to report interim data from this trial in the second half of this year.
ASLAN has already completed a phase 1 trial (NCT01992367) of ASLAN003 in healthy volunteers.
Results suggested that ASLAN003 has an “excellent” pharmacokinetic profile, according to ASLAN, and the drug was considered well tolerated in the volunteers.
ASLAN003 has also demonstrated “potent” inhibition of DHODH, according to ASLAN. In fact, the company said the binding affinity of ASLAN003 to DHODH has proven to be up to 2 orders of magnitude stronger than first-generation DHODH inhibitors such as leflunomide and teriflunomide.
ASLAN also said ASLAN003 should not confer the same toxicities as first-generation DHODH inhibitors and other novel AML therapies.
For example, leflunomide and teriflunomide, which may cause significant liver toxicity, take between 3 and 4 weeks to build to therapeutic levels and 2 years to be cleared completely after treatment is stopped.
ASLAN003, on the other hand, reaches full exposure in 24 hours and has a half-life of 18 hours.
Finally, ASLAN003 has been shown to differentiate blast cells into granulocytes in AML cell lines that do not respond to all-trans retinoic acid. These results were published in Cell in 2016.
Because of this research, ASLAN believes ASLAN003 may be effective in patients who do not respond to all-trans retinoic acid.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The US Food and Drug Administration (FDA) has granted orphan drug designation to ASLAN003 as a treatment for acute myeloid leukemia (AML).
ASLAN003 is a small molecule inhibitor of the human dihydroorotate dehydrogenase (DHODH) enzyme.
This second-generation DHODH inhibitor is being developed by ASLAN Pharmaceuticals.
The company is currently conducting a phase 2 trial (NCT03451084) of ASLAN003 in patients with newly diagnosed or relapsed/refractory AML.
The goals of this trial are to determine the optimum dose of ASLAN003 as monotherapy and assess the drug’s efficacy by overall complete remission rate. ASLAN expects to report interim data from this trial in the second half of this year.
ASLAN has already completed a phase 1 trial (NCT01992367) of ASLAN003 in healthy volunteers.
Results suggested that ASLAN003 has an “excellent” pharmacokinetic profile, according to ASLAN, and the drug was considered well tolerated in the volunteers.
ASLAN003 has also demonstrated “potent” inhibition of DHODH, according to ASLAN. In fact, the company said the binding affinity of ASLAN003 to DHODH has proven to be up to 2 orders of magnitude stronger than first-generation DHODH inhibitors such as leflunomide and teriflunomide.
ASLAN also said ASLAN003 should not confer the same toxicities as first-generation DHODH inhibitors and other novel AML therapies.
For example, leflunomide and teriflunomide, which may cause significant liver toxicity, take between 3 and 4 weeks to build to therapeutic levels and 2 years to be cleared completely after treatment is stopped.
ASLAN003, on the other hand, reaches full exposure in 24 hours and has a half-life of 18 hours.
Finally, ASLAN003 has been shown to differentiate blast cells into granulocytes in AML cell lines that do not respond to all-trans retinoic acid. These results were published in Cell in 2016.
Because of this research, ASLAN believes ASLAN003 may be effective in patients who do not respond to all-trans retinoic acid.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The US Food and Drug Administration (FDA) has granted orphan drug designation to ASLAN003 as a treatment for acute myeloid leukemia (AML).
ASLAN003 is a small molecule inhibitor of the human dihydroorotate dehydrogenase (DHODH) enzyme.
This second-generation DHODH inhibitor is being developed by ASLAN Pharmaceuticals.
The company is currently conducting a phase 2 trial (NCT03451084) of ASLAN003 in patients with newly diagnosed or relapsed/refractory AML.
The goals of this trial are to determine the optimum dose of ASLAN003 as monotherapy and assess the drug’s efficacy by overall complete remission rate. ASLAN expects to report interim data from this trial in the second half of this year.
ASLAN has already completed a phase 1 trial (NCT01992367) of ASLAN003 in healthy volunteers.
Results suggested that ASLAN003 has an “excellent” pharmacokinetic profile, according to ASLAN, and the drug was considered well tolerated in the volunteers.
ASLAN003 has also demonstrated “potent” inhibition of DHODH, according to ASLAN. In fact, the company said the binding affinity of ASLAN003 to DHODH has proven to be up to 2 orders of magnitude stronger than first-generation DHODH inhibitors such as leflunomide and teriflunomide.
ASLAN also said ASLAN003 should not confer the same toxicities as first-generation DHODH inhibitors and other novel AML therapies.
For example, leflunomide and teriflunomide, which may cause significant liver toxicity, take between 3 and 4 weeks to build to therapeutic levels and 2 years to be cleared completely after treatment is stopped.
ASLAN003, on the other hand, reaches full exposure in 24 hours and has a half-life of 18 hours.
Finally, ASLAN003 has been shown to differentiate blast cells into granulocytes in AML cell lines that do not respond to all-trans retinoic acid. These results were published in Cell in 2016.
Because of this research, ASLAN believes ASLAN003 may be effective in patients who do not respond to all-trans retinoic acid.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
FDA puts partial hold on trial of vascular agent for AML, MDS
The Food and Drug Administration has placed a partial clinical hold on a phase 1b/2 study of OXi4503, a vascular disrupting agent.
In this trial (NCT02576301), researchers are evaluating OXi4503 alone and in combination with cytarabine in patients with relapsed/refractory acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS).
The partial clinical hold applies to the 12.2 mg/m2 dose of OXi4503. The FDA is allowing the continued treatment and enrollment of patients using a dose of 9.76 mg/m2. Additional data on patients receiving OXi4503 at 9.76 mg/m2 must be evaluated before dosing at 12.2 mg/m2 can be resumed.
The partial clinical hold is a result of two potential dose-limiting toxicities (DLTs) observed at the 12.2-mg/m2 dose level: hypotension, which occurred shortly after initial treatment with OXi4503, and acute hypoxic respiratory failure, which occurred approximately 2 weeks after receiving OXi4503 and cytarabine.
Both events were deemed “possibly related” to OXi4503, and both patients recovered following treatment.
“Although it is disappointing that we are not currently continuing with the higher dose of OXi4503, we look forward to gathering more safety and efficacy data at the previous dose level, where we observed 2 complete remissions in the 4 patients that we treated,” William D. Schwieterman, MD, chief executive officer of Mateon Therapeutics Inc., the company developing OXi4503, said in a statement.
In preclinical research, OXi4503 demonstrated activity against AML, both when given alone and in combination with bevacizumab. These results were published in Blood in 2010.
In a phase 1 trial (NCT01085656), researchers evaluated OXi4503 in patients with relapsed or refractory AML or MDS. OXi4503 demonstrated preliminary evidence of disease response in heavily pretreated, refractory AML and advanced MDS.
The maximum tolerated dose of OXi4503 was not identified, but adverse events attributable to the drug included hypertension, bone pain, fever, anemia, thrombocytopenia, and coagulopathies.
Results from this study were presented at the 2013 annual meeting of the American Society of Hematology.
In 2015, Mateon Therapeutics initiated the phase 1b/2 study of OXi4503 (NCT02576301) that is now on partial clinical hold.
The phase 1 portion of this study was designed to assess the safety, pharmacokinetics, pharmacodynamics, and preliminary efficacy of single-agent OXi4503 in patients with relapsed/refractory AML and MDS. It is also aimed at determining the safety, pharmacokinetics, and pharmacodynamics of OXi4503 plus intermediate-dose cytarabine.
The goal of the phase 2 portion is to assess the preliminary efficacy of OXi4503 and cytarabine in patients with AML and MDS.
The Food and Drug Administration has placed a partial clinical hold on a phase 1b/2 study of OXi4503, a vascular disrupting agent.
In this trial (NCT02576301), researchers are evaluating OXi4503 alone and in combination with cytarabine in patients with relapsed/refractory acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS).
The partial clinical hold applies to the 12.2 mg/m2 dose of OXi4503. The FDA is allowing the continued treatment and enrollment of patients using a dose of 9.76 mg/m2. Additional data on patients receiving OXi4503 at 9.76 mg/m2 must be evaluated before dosing at 12.2 mg/m2 can be resumed.
The partial clinical hold is a result of two potential dose-limiting toxicities (DLTs) observed at the 12.2-mg/m2 dose level: hypotension, which occurred shortly after initial treatment with OXi4503, and acute hypoxic respiratory failure, which occurred approximately 2 weeks after receiving OXi4503 and cytarabine.
Both events were deemed “possibly related” to OXi4503, and both patients recovered following treatment.
“Although it is disappointing that we are not currently continuing with the higher dose of OXi4503, we look forward to gathering more safety and efficacy data at the previous dose level, where we observed 2 complete remissions in the 4 patients that we treated,” William D. Schwieterman, MD, chief executive officer of Mateon Therapeutics Inc., the company developing OXi4503, said in a statement.
In preclinical research, OXi4503 demonstrated activity against AML, both when given alone and in combination with bevacizumab. These results were published in Blood in 2010.
In a phase 1 trial (NCT01085656), researchers evaluated OXi4503 in patients with relapsed or refractory AML or MDS. OXi4503 demonstrated preliminary evidence of disease response in heavily pretreated, refractory AML and advanced MDS.
The maximum tolerated dose of OXi4503 was not identified, but adverse events attributable to the drug included hypertension, bone pain, fever, anemia, thrombocytopenia, and coagulopathies.
Results from this study were presented at the 2013 annual meeting of the American Society of Hematology.
In 2015, Mateon Therapeutics initiated the phase 1b/2 study of OXi4503 (NCT02576301) that is now on partial clinical hold.
The phase 1 portion of this study was designed to assess the safety, pharmacokinetics, pharmacodynamics, and preliminary efficacy of single-agent OXi4503 in patients with relapsed/refractory AML and MDS. It is also aimed at determining the safety, pharmacokinetics, and pharmacodynamics of OXi4503 plus intermediate-dose cytarabine.
The goal of the phase 2 portion is to assess the preliminary efficacy of OXi4503 and cytarabine in patients with AML and MDS.
The Food and Drug Administration has placed a partial clinical hold on a phase 1b/2 study of OXi4503, a vascular disrupting agent.
In this trial (NCT02576301), researchers are evaluating OXi4503 alone and in combination with cytarabine in patients with relapsed/refractory acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS).
The partial clinical hold applies to the 12.2 mg/m2 dose of OXi4503. The FDA is allowing the continued treatment and enrollment of patients using a dose of 9.76 mg/m2. Additional data on patients receiving OXi4503 at 9.76 mg/m2 must be evaluated before dosing at 12.2 mg/m2 can be resumed.
The partial clinical hold is a result of two potential dose-limiting toxicities (DLTs) observed at the 12.2-mg/m2 dose level: hypotension, which occurred shortly after initial treatment with OXi4503, and acute hypoxic respiratory failure, which occurred approximately 2 weeks after receiving OXi4503 and cytarabine.
Both events were deemed “possibly related” to OXi4503, and both patients recovered following treatment.
“Although it is disappointing that we are not currently continuing with the higher dose of OXi4503, we look forward to gathering more safety and efficacy data at the previous dose level, where we observed 2 complete remissions in the 4 patients that we treated,” William D. Schwieterman, MD, chief executive officer of Mateon Therapeutics Inc., the company developing OXi4503, said in a statement.
In preclinical research, OXi4503 demonstrated activity against AML, both when given alone and in combination with bevacizumab. These results were published in Blood in 2010.
In a phase 1 trial (NCT01085656), researchers evaluated OXi4503 in patients with relapsed or refractory AML or MDS. OXi4503 demonstrated preliminary evidence of disease response in heavily pretreated, refractory AML and advanced MDS.
The maximum tolerated dose of OXi4503 was not identified, but adverse events attributable to the drug included hypertension, bone pain, fever, anemia, thrombocytopenia, and coagulopathies.
Results from this study were presented at the 2013 annual meeting of the American Society of Hematology.
In 2015, Mateon Therapeutics initiated the phase 1b/2 study of OXi4503 (NCT02576301) that is now on partial clinical hold.
The phase 1 portion of this study was designed to assess the safety, pharmacokinetics, pharmacodynamics, and preliminary efficacy of single-agent OXi4503 in patients with relapsed/refractory AML and MDS. It is also aimed at determining the safety, pharmacokinetics, and pharmacodynamics of OXi4503 plus intermediate-dose cytarabine.
The goal of the phase 2 portion is to assess the preliminary efficacy of OXi4503 and cytarabine in patients with AML and MDS.
Access to care drives disparity between urban, rural cancer patients

New research suggests that better access to quality care may reduce disparities in survival between cancer patients living in rural areas of the US and those living in urban areas.
The study showed that urban and rural cancer patients had similar survival outcomes when they were enrolled in clinical trials.
These results, published in JAMA Network Open, cast new light on decades of research showing that cancer patients living in rural areas don’t live as long as urban cancer patients.
“These findings were a surprise, since we thought we might find the same disparities others had found,” said study author Joseph Unger, PhD, of Fred Hutchinson Cancer Research Center in Seattle, Washington.
“But clinical trials are a key difference here. In trials, patients are uniformly assessed, treated, and followed under a strict, guideline-driven protocol. This suggests that giving people with cancer access to uniform treatment strategies could help resolve the disparities in outcomes that we see between rural and urban patients.”
Dr Unger and his colleagues studied data on 36,995 patients who were enrolled in 44 phase 3 or phase 2/3 SWOG trials from 1986 through 2012. All 50 states were represented.
Patients had 17 different cancer types, including acute myeloid leukemia (AML), non-Hodgkin lymphoma (NHL), and multiple myeloma (MM).
Using US Department of Agriculture population classifications known as Rural-Urban Continuum Codes, the researchers categorized the patients as either rural or urban and analyzed their outcomes.
A minority of patients (19.4%, n=7184) were from rural locations. They were significantly more likely than urban patients to be 65 or older (P<0.001) and significantly less likely to be black (vs all other races; P<0.001).
However, there was no significant between-group difference in sex (P=0.53), and all major US geographic regions (West, Midwest, South, and Northeast) were represented.
Results
The researchers limited their analysis of survival to the first 5 years after trial enrollment to emphasize outcomes related to cancer and its treatment. They looked at overall survival (OS) as well as cancer-specific survival.
The team found no meaningful difference in OS or cancer-specific survival between rural and urban patients for 16 of the 17 cancer types.
The exception was estrogen receptor-negative, progesterone receptor-negative breast cancer. Rural patients with this cancer didn’t live as long as their urban counterparts. The hazard ratio (HR) was 1.27 (95% CI, 1.06-1.51; P=0.008) for OS and 1.26 (95% CI, 1.04-1.52; P=0.02) for cancer-specific survival.
The researchers believe this finding could be attributed to a few factors, including timely access to follow-up chemotherapy after patients’ first round of cancer treatment.
Although there were no significant survival differences for patients with hematologic malignancies, rural patients had slightly better OS if they had advanced indolent NHL or AML but slightly worse OS if they had MM or advanced aggressive NHL. The HRs were as follows:
- Advanced indolent NHL—HR=0.91 (95% CI, 0.64-1.29; P=0.60)
- AML—HR=0.94 (95% CI, 0.83-1.06; P=0.29)
- MM—HR=1.05 (95% CI, 0.93-1.18, P=0.46)
- Advanced aggressive NHL—HR=1.05 (95% CI, 0.87-1.27; P=0.60).
Rural patients had slightly better cancer-specific survival if they had advanced indolent NHL but slightly worse cancer-specific survival if they had AML, MM, or advanced aggressive NHL. The HRs were as follows:
- Advanced indolent NHL—HR=0.98 (95% CI, 0.66-1.45; P=0.90)
- AML—HR=1.01 (95% CI, 0.86-1.20; P=0.87)
- MM—HR=1.04 (95% CI, 0.90-1.20; P=0.60)
- Advanced aggressive NHL—HR=1.08 (95% CI, 0.87-1.34; P=0.50).
The researchers said these findings suggest it is access to care, and not other characteristics, that drive the survival disparities typically observed between urban and rural cancer patients.
“If people diagnosed with cancer, regardless of where they live, receive similar care and have similar outcomes, then a reasonable inference is that the best way to improve outcomes for rural patients is to improve their access to quality care,” Dr Unger said.
This research was supported by the National Cancer Institute and the HOPE Foundation. The researchers reported financial relationships with various pharmaceutical companies.
New research suggests that better access to quality care may reduce disparities in survival between cancer patients living in rural areas of the US and those living in urban areas.
The study showed that urban and rural cancer patients had similar survival outcomes when they were enrolled in clinical trials.
These results, published in JAMA Network Open, cast new light on decades of research showing that cancer patients living in rural areas don’t live as long as urban cancer patients.
“These findings were a surprise, since we thought we might find the same disparities others had found,” said study author Joseph Unger, PhD, of Fred Hutchinson Cancer Research Center in Seattle, Washington.
“But clinical trials are a key difference here. In trials, patients are uniformly assessed, treated, and followed under a strict, guideline-driven protocol. This suggests that giving people with cancer access to uniform treatment strategies could help resolve the disparities in outcomes that we see between rural and urban patients.”
Dr Unger and his colleagues studied data on 36,995 patients who were enrolled in 44 phase 3 or phase 2/3 SWOG trials from 1986 through 2012. All 50 states were represented.
Patients had 17 different cancer types, including acute myeloid leukemia (AML), non-Hodgkin lymphoma (NHL), and multiple myeloma (MM).
Using US Department of Agriculture population classifications known as Rural-Urban Continuum Codes, the researchers categorized the patients as either rural or urban and analyzed their outcomes.
A minority of patients (19.4%, n=7184) were from rural locations. They were significantly more likely than urban patients to be 65 or older (P<0.001) and significantly less likely to be black (vs all other races; P<0.001).
However, there was no significant between-group difference in sex (P=0.53), and all major US geographic regions (West, Midwest, South, and Northeast) were represented.
Results
The researchers limited their analysis of survival to the first 5 years after trial enrollment to emphasize outcomes related to cancer and its treatment. They looked at overall survival (OS) as well as cancer-specific survival.
The team found no meaningful difference in OS or cancer-specific survival between rural and urban patients for 16 of the 17 cancer types.
The exception was estrogen receptor-negative, progesterone receptor-negative breast cancer. Rural patients with this cancer didn’t live as long as their urban counterparts. The hazard ratio (HR) was 1.27 (95% CI, 1.06-1.51; P=0.008) for OS and 1.26 (95% CI, 1.04-1.52; P=0.02) for cancer-specific survival.
The researchers believe this finding could be attributed to a few factors, including timely access to follow-up chemotherapy after patients’ first round of cancer treatment.
Although there were no significant survival differences for patients with hematologic malignancies, rural patients had slightly better OS if they had advanced indolent NHL or AML but slightly worse OS if they had MM or advanced aggressive NHL. The HRs were as follows:
- Advanced indolent NHL—HR=0.91 (95% CI, 0.64-1.29; P=0.60)
- AML—HR=0.94 (95% CI, 0.83-1.06; P=0.29)
- MM—HR=1.05 (95% CI, 0.93-1.18, P=0.46)
- Advanced aggressive NHL—HR=1.05 (95% CI, 0.87-1.27; P=0.60).
Rural patients had slightly better cancer-specific survival if they had advanced indolent NHL but slightly worse cancer-specific survival if they had AML, MM, or advanced aggressive NHL. The HRs were as follows:
- Advanced indolent NHL—HR=0.98 (95% CI, 0.66-1.45; P=0.90)
- AML—HR=1.01 (95% CI, 0.86-1.20; P=0.87)
- MM—HR=1.04 (95% CI, 0.90-1.20; P=0.60)
- Advanced aggressive NHL—HR=1.08 (95% CI, 0.87-1.34; P=0.50).
The researchers said these findings suggest it is access to care, and not other characteristics, that drive the survival disparities typically observed between urban and rural cancer patients.
“If people diagnosed with cancer, regardless of where they live, receive similar care and have similar outcomes, then a reasonable inference is that the best way to improve outcomes for rural patients is to improve their access to quality care,” Dr Unger said.
This research was supported by the National Cancer Institute and the HOPE Foundation. The researchers reported financial relationships with various pharmaceutical companies.
New research suggests that better access to quality care may reduce disparities in survival between cancer patients living in rural areas of the US and those living in urban areas.
The study showed that urban and rural cancer patients had similar survival outcomes when they were enrolled in clinical trials.
These results, published in JAMA Network Open, cast new light on decades of research showing that cancer patients living in rural areas don’t live as long as urban cancer patients.
“These findings were a surprise, since we thought we might find the same disparities others had found,” said study author Joseph Unger, PhD, of Fred Hutchinson Cancer Research Center in Seattle, Washington.
“But clinical trials are a key difference here. In trials, patients are uniformly assessed, treated, and followed under a strict, guideline-driven protocol. This suggests that giving people with cancer access to uniform treatment strategies could help resolve the disparities in outcomes that we see between rural and urban patients.”
Dr Unger and his colleagues studied data on 36,995 patients who were enrolled in 44 phase 3 or phase 2/3 SWOG trials from 1986 through 2012. All 50 states were represented.
Patients had 17 different cancer types, including acute myeloid leukemia (AML), non-Hodgkin lymphoma (NHL), and multiple myeloma (MM).
Using US Department of Agriculture population classifications known as Rural-Urban Continuum Codes, the researchers categorized the patients as either rural or urban and analyzed their outcomes.
A minority of patients (19.4%, n=7184) were from rural locations. They were significantly more likely than urban patients to be 65 or older (P<0.001) and significantly less likely to be black (vs all other races; P<0.001).
However, there was no significant between-group difference in sex (P=0.53), and all major US geographic regions (West, Midwest, South, and Northeast) were represented.
Results
The researchers limited their analysis of survival to the first 5 years after trial enrollment to emphasize outcomes related to cancer and its treatment. They looked at overall survival (OS) as well as cancer-specific survival.
The team found no meaningful difference in OS or cancer-specific survival between rural and urban patients for 16 of the 17 cancer types.
The exception was estrogen receptor-negative, progesterone receptor-negative breast cancer. Rural patients with this cancer didn’t live as long as their urban counterparts. The hazard ratio (HR) was 1.27 (95% CI, 1.06-1.51; P=0.008) for OS and 1.26 (95% CI, 1.04-1.52; P=0.02) for cancer-specific survival.
The researchers believe this finding could be attributed to a few factors, including timely access to follow-up chemotherapy after patients’ first round of cancer treatment.
Although there were no significant survival differences for patients with hematologic malignancies, rural patients had slightly better OS if they had advanced indolent NHL or AML but slightly worse OS if they had MM or advanced aggressive NHL. The HRs were as follows:
- Advanced indolent NHL—HR=0.91 (95% CI, 0.64-1.29; P=0.60)
- AML—HR=0.94 (95% CI, 0.83-1.06; P=0.29)
- MM—HR=1.05 (95% CI, 0.93-1.18, P=0.46)
- Advanced aggressive NHL—HR=1.05 (95% CI, 0.87-1.27; P=0.60).
Rural patients had slightly better cancer-specific survival if they had advanced indolent NHL but slightly worse cancer-specific survival if they had AML, MM, or advanced aggressive NHL. The HRs were as follows:
- Advanced indolent NHL—HR=0.98 (95% CI, 0.66-1.45; P=0.90)
- AML—HR=1.01 (95% CI, 0.86-1.20; P=0.87)
- MM—HR=1.04 (95% CI, 0.90-1.20; P=0.60)
- Advanced aggressive NHL—HR=1.08 (95% CI, 0.87-1.34; P=0.50).
The researchers said these findings suggest it is access to care, and not other characteristics, that drive the survival disparities typically observed between urban and rural cancer patients.
“If people diagnosed with cancer, regardless of where they live, receive similar care and have similar outcomes, then a reasonable inference is that the best way to improve outcomes for rural patients is to improve their access to quality care,” Dr Unger said.
This research was supported by the National Cancer Institute and the HOPE Foundation. The researchers reported financial relationships with various pharmaceutical companies.
Partial hold placed on trial of drug for AML, MDS

The US Food and Drug Administration (FDA) has placed a partial clinical hold on a phase 1b/2 study of OXi4503, a vascular disrupting agent.
In this trial (NCT02576301), researchers are evaluating OXi4503, alone and in combination with cytarabine, in patients with relapsed/refractory acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS).
The partial clinical hold applies to the 12.2 mg/m2 dose of OXi4503.
The FDA is allowing the continued treatment and enrollment of patients using a dose of 9.76 mg/m2.
The agency said additional data on patients receiving OXi4503 at 9.76 mg/m2 must be evaluated before dosing at 12.2 mg/m2 can be resumed.
The partial clinical hold is a result of 2 potential dose-limiting toxicities (DLTs) observed at the 12.2 mg/m2 dose level.
One DLT was hypotension, which occurred shortly after initial treatment with OXi4503. The other DLT was acute hypoxic respiratory failure, which occurred approximately 2 weeks after receiving OXi4503 and cytarabine.
Both events were deemed “possibly related” to OXi4503, and both patients recovered following treatment.
The study protocol generally defines a DLT as any grade 3 serious adverse event where a relationship to OXi4503 cannot be ruled out.
“Although it is disappointing that we are not currently continuing with the higher dose of OXi4503, we look forward to gathering more safety and efficacy data at the previous dose level, where we observed 2 complete remissions in the 4 patients that we treated,” said William D. Schwieterman, MD, chief executive officer of Mateon Therapeutics, Inc., the company developing OXi4503.
About OXi4503
According to Mateon Therapeutics, OXi4503 has a dual mechanism of action that disrupts the shape of tumor bone marrow endothelial cells through reversible binding to tubulin at the colchicine binding site, downregulating intercellular adhesion molecules.
This alters the endothelial cell shape, releasing quiescent adherent tumor cells from bone marrow endothelial cells and activating the cell cycle, which makes the tumor cells vulnerable to chemotherapy.
OXi4503 also kills tumor cells directly via myeloperoxidase activation of an orthoquinone cytotoxic mediator.
In preclinical research, OXi4503 demonstrated activity against AML, both when given alone and in combination with bevacizumab. These results were published in Blood in 2010.
Clinical trials
In a phase 1 trial (NCT01085656), researchers evaluated OXi4503 in patients with relapsed or refractory AML or MDS. The goals were to determine the safety profile, maximum tolerated dose, and biologic activity of OXi4503.
The researchers said OXi4503 demonstrated preliminary evidence of disease response in heavily pre-treated, refractory AML and advanced MDS.
The maximum tolerated dose of OXi4503 was not identified, but adverse events attributable to the drug included hypertension, bone pain, fever, anemia, thrombocytopenia, and coagulopathies.
Results from this study were presented at the 2013 ASH Annual Meeting.
In 2015, Mateon Therapeutics initiated the phase 1b/2 study of OXi4503 (NCT02576301) that is now on partial clinical hold.
The phase 1 portion of this study was designed to assess the safety, pharmacokinetics, pharmacodynamics, and preliminary efficacy of single-agent OXi4503 in patients with relapsed/refractory AML and MDS.
The phase 1 portion was also intended to determine the safety, pharmacokinetics, and pharmacodynamics of OXi4503 plus intermediate-dose cytarabine.
The goal of the phase 2 portion is to assess the preliminary efficacy of OXi4503 and cytarabine in patients with AML and MDS.
The US Food and Drug Administration (FDA) has placed a partial clinical hold on a phase 1b/2 study of OXi4503, a vascular disrupting agent.
In this trial (NCT02576301), researchers are evaluating OXi4503, alone and in combination with cytarabine, in patients with relapsed/refractory acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS).
The partial clinical hold applies to the 12.2 mg/m2 dose of OXi4503.
The FDA is allowing the continued treatment and enrollment of patients using a dose of 9.76 mg/m2.
The agency said additional data on patients receiving OXi4503 at 9.76 mg/m2 must be evaluated before dosing at 12.2 mg/m2 can be resumed.
The partial clinical hold is a result of 2 potential dose-limiting toxicities (DLTs) observed at the 12.2 mg/m2 dose level.
One DLT was hypotension, which occurred shortly after initial treatment with OXi4503. The other DLT was acute hypoxic respiratory failure, which occurred approximately 2 weeks after receiving OXi4503 and cytarabine.
Both events were deemed “possibly related” to OXi4503, and both patients recovered following treatment.
The study protocol generally defines a DLT as any grade 3 serious adverse event where a relationship to OXi4503 cannot be ruled out.
“Although it is disappointing that we are not currently continuing with the higher dose of OXi4503, we look forward to gathering more safety and efficacy data at the previous dose level, where we observed 2 complete remissions in the 4 patients that we treated,” said William D. Schwieterman, MD, chief executive officer of Mateon Therapeutics, Inc., the company developing OXi4503.
About OXi4503
According to Mateon Therapeutics, OXi4503 has a dual mechanism of action that disrupts the shape of tumor bone marrow endothelial cells through reversible binding to tubulin at the colchicine binding site, downregulating intercellular adhesion molecules.
This alters the endothelial cell shape, releasing quiescent adherent tumor cells from bone marrow endothelial cells and activating the cell cycle, which makes the tumor cells vulnerable to chemotherapy.
OXi4503 also kills tumor cells directly via myeloperoxidase activation of an orthoquinone cytotoxic mediator.
In preclinical research, OXi4503 demonstrated activity against AML, both when given alone and in combination with bevacizumab. These results were published in Blood in 2010.
Clinical trials
In a phase 1 trial (NCT01085656), researchers evaluated OXi4503 in patients with relapsed or refractory AML or MDS. The goals were to determine the safety profile, maximum tolerated dose, and biologic activity of OXi4503.
The researchers said OXi4503 demonstrated preliminary evidence of disease response in heavily pre-treated, refractory AML and advanced MDS.
The maximum tolerated dose of OXi4503 was not identified, but adverse events attributable to the drug included hypertension, bone pain, fever, anemia, thrombocytopenia, and coagulopathies.
Results from this study were presented at the 2013 ASH Annual Meeting.
In 2015, Mateon Therapeutics initiated the phase 1b/2 study of OXi4503 (NCT02576301) that is now on partial clinical hold.
The phase 1 portion of this study was designed to assess the safety, pharmacokinetics, pharmacodynamics, and preliminary efficacy of single-agent OXi4503 in patients with relapsed/refractory AML and MDS.
The phase 1 portion was also intended to determine the safety, pharmacokinetics, and pharmacodynamics of OXi4503 plus intermediate-dose cytarabine.
The goal of the phase 2 portion is to assess the preliminary efficacy of OXi4503 and cytarabine in patients with AML and MDS.
The US Food and Drug Administration (FDA) has placed a partial clinical hold on a phase 1b/2 study of OXi4503, a vascular disrupting agent.
In this trial (NCT02576301), researchers are evaluating OXi4503, alone and in combination with cytarabine, in patients with relapsed/refractory acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS).
The partial clinical hold applies to the 12.2 mg/m2 dose of OXi4503.
The FDA is allowing the continued treatment and enrollment of patients using a dose of 9.76 mg/m2.
The agency said additional data on patients receiving OXi4503 at 9.76 mg/m2 must be evaluated before dosing at 12.2 mg/m2 can be resumed.
The partial clinical hold is a result of 2 potential dose-limiting toxicities (DLTs) observed at the 12.2 mg/m2 dose level.
One DLT was hypotension, which occurred shortly after initial treatment with OXi4503. The other DLT was acute hypoxic respiratory failure, which occurred approximately 2 weeks after receiving OXi4503 and cytarabine.
Both events were deemed “possibly related” to OXi4503, and both patients recovered following treatment.
The study protocol generally defines a DLT as any grade 3 serious adverse event where a relationship to OXi4503 cannot be ruled out.
“Although it is disappointing that we are not currently continuing with the higher dose of OXi4503, we look forward to gathering more safety and efficacy data at the previous dose level, where we observed 2 complete remissions in the 4 patients that we treated,” said William D. Schwieterman, MD, chief executive officer of Mateon Therapeutics, Inc., the company developing OXi4503.
About OXi4503
According to Mateon Therapeutics, OXi4503 has a dual mechanism of action that disrupts the shape of tumor bone marrow endothelial cells through reversible binding to tubulin at the colchicine binding site, downregulating intercellular adhesion molecules.
This alters the endothelial cell shape, releasing quiescent adherent tumor cells from bone marrow endothelial cells and activating the cell cycle, which makes the tumor cells vulnerable to chemotherapy.
OXi4503 also kills tumor cells directly via myeloperoxidase activation of an orthoquinone cytotoxic mediator.
In preclinical research, OXi4503 demonstrated activity against AML, both when given alone and in combination with bevacizumab. These results were published in Blood in 2010.
Clinical trials
In a phase 1 trial (NCT01085656), researchers evaluated OXi4503 in patients with relapsed or refractory AML or MDS. The goals were to determine the safety profile, maximum tolerated dose, and biologic activity of OXi4503.
The researchers said OXi4503 demonstrated preliminary evidence of disease response in heavily pre-treated, refractory AML and advanced MDS.
The maximum tolerated dose of OXi4503 was not identified, but adverse events attributable to the drug included hypertension, bone pain, fever, anemia, thrombocytopenia, and coagulopathies.
Results from this study were presented at the 2013 ASH Annual Meeting.
In 2015, Mateon Therapeutics initiated the phase 1b/2 study of OXi4503 (NCT02576301) that is now on partial clinical hold.
The phase 1 portion of this study was designed to assess the safety, pharmacokinetics, pharmacodynamics, and preliminary efficacy of single-agent OXi4503 in patients with relapsed/refractory AML and MDS.
The phase 1 portion was also intended to determine the safety, pharmacokinetics, and pharmacodynamics of OXi4503 plus intermediate-dose cytarabine.
The goal of the phase 2 portion is to assess the preliminary efficacy of OXi4503 and cytarabine in patients with AML and MDS.
First CAR T-cell therapy approvals bolster booming immunotherapy market
There were a number of landmark approvals by the US Food and Drug Administration (FDA) in 2017 for cancer therapies, among them, the approval of the first two chimeric antigen receptor (CAR) T-cell therapies for cancer: tisagenlecleucel (in August) and axicabtagene ciloluecel (in October).1 CAR T-cells are a type of adoptive cell therapy or immunotherapy, in which the patient’s own immune cells are genetically engineered to target a tumor-associated antigen, in this case CD19. In tisagenlecleucel, CD19 proteins on B cells are targeted in the treatment of B-cell precursor acute lymphoblastic leukemia. Axicabtagene ciloluecel, the second anti-CD19 CAR T-cell therapy, was approved for the treatment of refractory, aggressive B-cell non-Hodgkin lymphoma.
Tisagenlecleucel
Tisagenlecleucel was approved for the treatment of pediatric patients up to 25 years of age with B-cell precursor acute lymphoblastic leukemia (ALL) whose disease is refractory to treatment or who have relapsed after second-line therapy or beyond.2 Approval was based on the pivotal ELIANA trial, a single-arm, global phase 2 trial conducted at 25 centers worldwide during April 2015 through April 2017. Patients were eligible for enrollment if they had relapsed or refractory B-cell ALL and were at least 3 years of age at screening and no older than 21 years of age at diagnosis, had at least 5% lymphoblasts in the bone marrow at screening, had tumor expression of CD19, had adequate organ function, and a Karnofsky (adult) or Lansky (child) Performance Status of ≥50 (with the worst allowable score, 50, indicating a patient who requires considerable assistance and frequent medical care [Karnofsky] and lying around much of the day, but gets dressed; no active playing but participates in all quiet play and activities [Lansky]). Exclusion criteria included previous receipt of anti-CD19 therapy, concomitant genetic syndromes associated with bone marrow failure, previous malignancy, and/or active or latent hepatitis B or C virus (HBV/HCV) infection.
The overall remission rate (ORR) was evaluated in 75 patients who were given a single dose of tisagenlecleucel (a median weight-adjusted dose of 3.1 x 106 transduced viable T cells per kg of body weight) within 14 days of completing a lymphodepleting chemotherapy regimen. The confirmed ORR after at least 3 months of follow-up, as assessed by independent central review, was 81%, which included 60% of patients in complete remission (CR) and 21% in complete remission with incomplete hematologic recovery, all of whom were negative for minimal residual disease.
The most common adverse events (AEs) associated with tisagenlecleucel treatment were cytokine release syndrome (CRS), hypogammaglobulinemia, infection, pyrexia, decreased appetite, headache, encephalopathy, hypotension, bleeding episodes, tachycardia, nausea, diarrhea, vomiting, viral infectious disorders, hypoxia, fatigue, acute kidney injury, and delirium. AEs were of grade 3/4 severity in 84% of patients.3
To combat serious safety issues, including CRS and neurologic toxicities, the FDA approved tisagenlecleucel with a Risk Evaluation and Mitigation Strategy (REMS) that, in part, requires health care providers who administer the drug to be trained in their management. It also requires the facility where treatment is administered to have immediate, onsite access to the drug tocilizumab, which was approved in conjunction with tisagenlecleucel for the treatment of patients who experience CRS.
In addition to information about the REMS, the prescribing information details warnings and precautions relating to several other common toxicities. These include hypersensitivity reactions, serious infections, prolonged cytopenias, and hypogammaglobulinemia.
Patients should be monitored for signs and symptoms of infection and treated appropriately. Viral reactivation can occur after tisagenlecleucel treatment, so patients should be screened for HBV, HCV, and human immunodeficiency virus before collection of cells.
The administration of myeloid growth factors is not recommended during the first 3 weeks after infusion or until CRS has resolved. Immunoglobulin levels should be monitored after treatment and hypogammaglobulinemia managed using infection precautions, antibiotic prophylaxis, and immunoglobulin replacement according to standard guidelines.
Patients treated with tisagenlecleucel should also be monitored for life for secondary malignancies, should not be treated with live vaccines from 2 weeks before the start of lymphodepleting chemotherapy until immune recovery after tisagenlecleucel infusion, and should be aware of the potential for neurological events to impact their ability to drive and use dangerous machinery.4
Tisagenlecleucel is marketed as Kymriah by Novartis Pharmaceuticals. The recommended dose is 1 infusion of 0.2-5 x 106 CAR-positive viable T cells per kilogram of body weight intravenously (for patients ≤50kg) and 0.1-2.5 x 108 cells/kg (for patients >50kg), administered 2-14 days after lymphodepleting chemotherapy.
Axicabtagene ciloleucel
Axicabtagene ciloleucel was approved for the treatment of adult patients with certain types of relapsed or refractory large B-cell lymphoma, including diffuse large B-cell lymphoma (DLBCL), primary mediastinal B-cell lymphoma (PMBCL), high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma.5 It is not indicated for the treatment of patients with primary central nervous system lymphoma.
Approval followed positive results from the phase 2 single-arm, multicenter ZUMA-1 trial.6 Patients were included if they were aged 18 years of age and older, had histologically confirmed aggressive B-cell non-Hodgkin lymphoma that was chemotherapy refractory, had received adequate previous therapy, had at least 1 measurable lesion, had completed radiation or systemic therapy at least 2 weeks before, had resolved toxicities related to previous therapy, and had an Eastern Cooperative Oncology Group Performance Status of 0 (asymptomatic) or 1 (symptomatic), an absolute neutrophil count of ≥1000/µL, a platelet count of ≥50,000/µL, and adequate hepatic, renal and cardiac function. They were treated with a single infusion of axicabtagene ciloleucel after lymphodepleting chemotherapy.
Patients who had received previous CD19-targeted therapy, who had concomitant genetic syndromes associated with bone marrow failure, who had previous malignancy, and who had active or latent HBV/HCV infection were among those excluded from the study.
Patients were enrolled in 2 cohorts; those with DLBCL (n = 77) and those with PMBCL or transformed follicular lymphoma (n = 24). The primary endpoint was objective response rate, and after a primary analysis at a minimum of 6 months follow-up, the objective response rate was 82%, with a CR rate of 52%. Among patients who achieved CR, the median duration of response was not reached after a median follow-up of 7.9 months.
A subsequent updated analysis was performed when 108 patients had been followed for a minimum of 1 year. The objective response rate was 82%, and the CR rate was 58%, with some patients having CR in the absence of additional therapies as late as 15 months after treatment. At this updated analysis, 42% of patients continued to have a response, 40% of whom remained in CR.
The most common grade 3 or higher AEs included febrile neutropenia, fever, CRS, encephalopathy, infections, hypotension, and hypoxia. Serious AEs occurred in 52% of patients and included CRS, neurologic toxicity, prolonged cytopenias, and serious infections. Grade 3 or higher CRS or neurologic toxicities occurred in 13% and 28% of patients, respectively. Three patients died during treatment.
To mitigate the risk of CRS and neurologic toxicity, axicabtagene ciloleucel is approved with an REMS that requires appropriate certification and training before hospitals are cleared to administer the therapy.
Other warnings and precautions in the prescribing information relate to serious infections (monitor for signs and symptoms and treat appropriately), prolonged cytopenias (monitor blood counts), hypogammaglobulinemia (monitor immunoglobulin levels and manage appropriately), secondary malignancies (life-long monitoring), and the potential effects of neurologic events on a patient’s ability to drive and operate dangerous machinery (avoid for at least 8 weeks after infusion).7
Axicabtagene ciloleucel is marketed as Yescarta by Kite Pharma Inc. The recommended dose is a single intravenous infusion with a target of 2 x 106 CAR-positive viable T cells per kilogram of body weight, preceded by fludarabine and cyclophosphamide lymphodepleting chemotherapy.
1. Bosserman LD. Cancer care in 2017: the promise of more cures with the challenges of an unstable health care system. JCSO 2017;15(6):e283-e290.
2. FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome. FDA News Release. August 30, 2017. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/
ucm574154.htm. Accessed March 31, 2018.
3. Maude S.L, Laetsch T.W, Buechner S, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N Engl J Med. 2018;378:439-48.
4. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Novartis Pharmaceuticals Corporation, August, 2017. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.
com/files/kymriah.pdf. Accessed March 31, 2018.
5. FDA approves axicabtagene ciloleucel for large B-cell lymphoma. FDA News Release. October 18, 2017. https://www.fda.gov/Drugs/
InformationOnDrugs/ApprovedDrugs/ucm581296.htm. Accessed March 31, 2018.
6. Neelapu, S.S, Locke F.L, Bartlett, L.J, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377:2531-44.
7. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Kite Pharma Inc. October 2017. https://www.yescarta.com/wp-content/uploads/yescarta-pi.pdf. Accessed March 31, 2018.
There were a number of landmark approvals by the US Food and Drug Administration (FDA) in 2017 for cancer therapies, among them, the approval of the first two chimeric antigen receptor (CAR) T-cell therapies for cancer: tisagenlecleucel (in August) and axicabtagene ciloluecel (in October).1 CAR T-cells are a type of adoptive cell therapy or immunotherapy, in which the patient’s own immune cells are genetically engineered to target a tumor-associated antigen, in this case CD19. In tisagenlecleucel, CD19 proteins on B cells are targeted in the treatment of B-cell precursor acute lymphoblastic leukemia. Axicabtagene ciloluecel, the second anti-CD19 CAR T-cell therapy, was approved for the treatment of refractory, aggressive B-cell non-Hodgkin lymphoma.
Tisagenlecleucel
Tisagenlecleucel was approved for the treatment of pediatric patients up to 25 years of age with B-cell precursor acute lymphoblastic leukemia (ALL) whose disease is refractory to treatment or who have relapsed after second-line therapy or beyond.2 Approval was based on the pivotal ELIANA trial, a single-arm, global phase 2 trial conducted at 25 centers worldwide during April 2015 through April 2017. Patients were eligible for enrollment if they had relapsed or refractory B-cell ALL and were at least 3 years of age at screening and no older than 21 years of age at diagnosis, had at least 5% lymphoblasts in the bone marrow at screening, had tumor expression of CD19, had adequate organ function, and a Karnofsky (adult) or Lansky (child) Performance Status of ≥50 (with the worst allowable score, 50, indicating a patient who requires considerable assistance and frequent medical care [Karnofsky] and lying around much of the day, but gets dressed; no active playing but participates in all quiet play and activities [Lansky]). Exclusion criteria included previous receipt of anti-CD19 therapy, concomitant genetic syndromes associated with bone marrow failure, previous malignancy, and/or active or latent hepatitis B or C virus (HBV/HCV) infection.
The overall remission rate (ORR) was evaluated in 75 patients who were given a single dose of tisagenlecleucel (a median weight-adjusted dose of 3.1 x 106 transduced viable T cells per kg of body weight) within 14 days of completing a lymphodepleting chemotherapy regimen. The confirmed ORR after at least 3 months of follow-up, as assessed by independent central review, was 81%, which included 60% of patients in complete remission (CR) and 21% in complete remission with incomplete hematologic recovery, all of whom were negative for minimal residual disease.
The most common adverse events (AEs) associated with tisagenlecleucel treatment were cytokine release syndrome (CRS), hypogammaglobulinemia, infection, pyrexia, decreased appetite, headache, encephalopathy, hypotension, bleeding episodes, tachycardia, nausea, diarrhea, vomiting, viral infectious disorders, hypoxia, fatigue, acute kidney injury, and delirium. AEs were of grade 3/4 severity in 84% of patients.3
To combat serious safety issues, including CRS and neurologic toxicities, the FDA approved tisagenlecleucel with a Risk Evaluation and Mitigation Strategy (REMS) that, in part, requires health care providers who administer the drug to be trained in their management. It also requires the facility where treatment is administered to have immediate, onsite access to the drug tocilizumab, which was approved in conjunction with tisagenlecleucel for the treatment of patients who experience CRS.
In addition to information about the REMS, the prescribing information details warnings and precautions relating to several other common toxicities. These include hypersensitivity reactions, serious infections, prolonged cytopenias, and hypogammaglobulinemia.
Patients should be monitored for signs and symptoms of infection and treated appropriately. Viral reactivation can occur after tisagenlecleucel treatment, so patients should be screened for HBV, HCV, and human immunodeficiency virus before collection of cells.
The administration of myeloid growth factors is not recommended during the first 3 weeks after infusion or until CRS has resolved. Immunoglobulin levels should be monitored after treatment and hypogammaglobulinemia managed using infection precautions, antibiotic prophylaxis, and immunoglobulin replacement according to standard guidelines.
Patients treated with tisagenlecleucel should also be monitored for life for secondary malignancies, should not be treated with live vaccines from 2 weeks before the start of lymphodepleting chemotherapy until immune recovery after tisagenlecleucel infusion, and should be aware of the potential for neurological events to impact their ability to drive and use dangerous machinery.4
Tisagenlecleucel is marketed as Kymriah by Novartis Pharmaceuticals. The recommended dose is 1 infusion of 0.2-5 x 106 CAR-positive viable T cells per kilogram of body weight intravenously (for patients ≤50kg) and 0.1-2.5 x 108 cells/kg (for patients >50kg), administered 2-14 days after lymphodepleting chemotherapy.
Axicabtagene ciloleucel
Axicabtagene ciloleucel was approved for the treatment of adult patients with certain types of relapsed or refractory large B-cell lymphoma, including diffuse large B-cell lymphoma (DLBCL), primary mediastinal B-cell lymphoma (PMBCL), high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma.5 It is not indicated for the treatment of patients with primary central nervous system lymphoma.
Approval followed positive results from the phase 2 single-arm, multicenter ZUMA-1 trial.6 Patients were included if they were aged 18 years of age and older, had histologically confirmed aggressive B-cell non-Hodgkin lymphoma that was chemotherapy refractory, had received adequate previous therapy, had at least 1 measurable lesion, had completed radiation or systemic therapy at least 2 weeks before, had resolved toxicities related to previous therapy, and had an Eastern Cooperative Oncology Group Performance Status of 0 (asymptomatic) or 1 (symptomatic), an absolute neutrophil count of ≥1000/µL, a platelet count of ≥50,000/µL, and adequate hepatic, renal and cardiac function. They were treated with a single infusion of axicabtagene ciloleucel after lymphodepleting chemotherapy.
Patients who had received previous CD19-targeted therapy, who had concomitant genetic syndromes associated with bone marrow failure, who had previous malignancy, and who had active or latent HBV/HCV infection were among those excluded from the study.
Patients were enrolled in 2 cohorts; those with DLBCL (n = 77) and those with PMBCL or transformed follicular lymphoma (n = 24). The primary endpoint was objective response rate, and after a primary analysis at a minimum of 6 months follow-up, the objective response rate was 82%, with a CR rate of 52%. Among patients who achieved CR, the median duration of response was not reached after a median follow-up of 7.9 months.
A subsequent updated analysis was performed when 108 patients had been followed for a minimum of 1 year. The objective response rate was 82%, and the CR rate was 58%, with some patients having CR in the absence of additional therapies as late as 15 months after treatment. At this updated analysis, 42% of patients continued to have a response, 40% of whom remained in CR.
The most common grade 3 or higher AEs included febrile neutropenia, fever, CRS, encephalopathy, infections, hypotension, and hypoxia. Serious AEs occurred in 52% of patients and included CRS, neurologic toxicity, prolonged cytopenias, and serious infections. Grade 3 or higher CRS or neurologic toxicities occurred in 13% and 28% of patients, respectively. Three patients died during treatment.
To mitigate the risk of CRS and neurologic toxicity, axicabtagene ciloleucel is approved with an REMS that requires appropriate certification and training before hospitals are cleared to administer the therapy.
Other warnings and precautions in the prescribing information relate to serious infections (monitor for signs and symptoms and treat appropriately), prolonged cytopenias (monitor blood counts), hypogammaglobulinemia (monitor immunoglobulin levels and manage appropriately), secondary malignancies (life-long monitoring), and the potential effects of neurologic events on a patient’s ability to drive and operate dangerous machinery (avoid for at least 8 weeks after infusion).7
Axicabtagene ciloleucel is marketed as Yescarta by Kite Pharma Inc. The recommended dose is a single intravenous infusion with a target of 2 x 106 CAR-positive viable T cells per kilogram of body weight, preceded by fludarabine and cyclophosphamide lymphodepleting chemotherapy.
There were a number of landmark approvals by the US Food and Drug Administration (FDA) in 2017 for cancer therapies, among them, the approval of the first two chimeric antigen receptor (CAR) T-cell therapies for cancer: tisagenlecleucel (in August) and axicabtagene ciloluecel (in October).1 CAR T-cells are a type of adoptive cell therapy or immunotherapy, in which the patient’s own immune cells are genetically engineered to target a tumor-associated antigen, in this case CD19. In tisagenlecleucel, CD19 proteins on B cells are targeted in the treatment of B-cell precursor acute lymphoblastic leukemia. Axicabtagene ciloluecel, the second anti-CD19 CAR T-cell therapy, was approved for the treatment of refractory, aggressive B-cell non-Hodgkin lymphoma.
Tisagenlecleucel
Tisagenlecleucel was approved for the treatment of pediatric patients up to 25 years of age with B-cell precursor acute lymphoblastic leukemia (ALL) whose disease is refractory to treatment or who have relapsed after second-line therapy or beyond.2 Approval was based on the pivotal ELIANA trial, a single-arm, global phase 2 trial conducted at 25 centers worldwide during April 2015 through April 2017. Patients were eligible for enrollment if they had relapsed or refractory B-cell ALL and were at least 3 years of age at screening and no older than 21 years of age at diagnosis, had at least 5% lymphoblasts in the bone marrow at screening, had tumor expression of CD19, had adequate organ function, and a Karnofsky (adult) or Lansky (child) Performance Status of ≥50 (with the worst allowable score, 50, indicating a patient who requires considerable assistance and frequent medical care [Karnofsky] and lying around much of the day, but gets dressed; no active playing but participates in all quiet play and activities [Lansky]). Exclusion criteria included previous receipt of anti-CD19 therapy, concomitant genetic syndromes associated with bone marrow failure, previous malignancy, and/or active or latent hepatitis B or C virus (HBV/HCV) infection.
The overall remission rate (ORR) was evaluated in 75 patients who were given a single dose of tisagenlecleucel (a median weight-adjusted dose of 3.1 x 106 transduced viable T cells per kg of body weight) within 14 days of completing a lymphodepleting chemotherapy regimen. The confirmed ORR after at least 3 months of follow-up, as assessed by independent central review, was 81%, which included 60% of patients in complete remission (CR) and 21% in complete remission with incomplete hematologic recovery, all of whom were negative for minimal residual disease.
The most common adverse events (AEs) associated with tisagenlecleucel treatment were cytokine release syndrome (CRS), hypogammaglobulinemia, infection, pyrexia, decreased appetite, headache, encephalopathy, hypotension, bleeding episodes, tachycardia, nausea, diarrhea, vomiting, viral infectious disorders, hypoxia, fatigue, acute kidney injury, and delirium. AEs were of grade 3/4 severity in 84% of patients.3
To combat serious safety issues, including CRS and neurologic toxicities, the FDA approved tisagenlecleucel with a Risk Evaluation and Mitigation Strategy (REMS) that, in part, requires health care providers who administer the drug to be trained in their management. It also requires the facility where treatment is administered to have immediate, onsite access to the drug tocilizumab, which was approved in conjunction with tisagenlecleucel for the treatment of patients who experience CRS.
In addition to information about the REMS, the prescribing information details warnings and precautions relating to several other common toxicities. These include hypersensitivity reactions, serious infections, prolonged cytopenias, and hypogammaglobulinemia.
Patients should be monitored for signs and symptoms of infection and treated appropriately. Viral reactivation can occur after tisagenlecleucel treatment, so patients should be screened for HBV, HCV, and human immunodeficiency virus before collection of cells.
The administration of myeloid growth factors is not recommended during the first 3 weeks after infusion or until CRS has resolved. Immunoglobulin levels should be monitored after treatment and hypogammaglobulinemia managed using infection precautions, antibiotic prophylaxis, and immunoglobulin replacement according to standard guidelines.
Patients treated with tisagenlecleucel should also be monitored for life for secondary malignancies, should not be treated with live vaccines from 2 weeks before the start of lymphodepleting chemotherapy until immune recovery after tisagenlecleucel infusion, and should be aware of the potential for neurological events to impact their ability to drive and use dangerous machinery.4
Tisagenlecleucel is marketed as Kymriah by Novartis Pharmaceuticals. The recommended dose is 1 infusion of 0.2-5 x 106 CAR-positive viable T cells per kilogram of body weight intravenously (for patients ≤50kg) and 0.1-2.5 x 108 cells/kg (for patients >50kg), administered 2-14 days after lymphodepleting chemotherapy.
Axicabtagene ciloleucel
Axicabtagene ciloleucel was approved for the treatment of adult patients with certain types of relapsed or refractory large B-cell lymphoma, including diffuse large B-cell lymphoma (DLBCL), primary mediastinal B-cell lymphoma (PMBCL), high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma.5 It is not indicated for the treatment of patients with primary central nervous system lymphoma.
Approval followed positive results from the phase 2 single-arm, multicenter ZUMA-1 trial.6 Patients were included if they were aged 18 years of age and older, had histologically confirmed aggressive B-cell non-Hodgkin lymphoma that was chemotherapy refractory, had received adequate previous therapy, had at least 1 measurable lesion, had completed radiation or systemic therapy at least 2 weeks before, had resolved toxicities related to previous therapy, and had an Eastern Cooperative Oncology Group Performance Status of 0 (asymptomatic) or 1 (symptomatic), an absolute neutrophil count of ≥1000/µL, a platelet count of ≥50,000/µL, and adequate hepatic, renal and cardiac function. They were treated with a single infusion of axicabtagene ciloleucel after lymphodepleting chemotherapy.
Patients who had received previous CD19-targeted therapy, who had concomitant genetic syndromes associated with bone marrow failure, who had previous malignancy, and who had active or latent HBV/HCV infection were among those excluded from the study.
Patients were enrolled in 2 cohorts; those with DLBCL (n = 77) and those with PMBCL or transformed follicular lymphoma (n = 24). The primary endpoint was objective response rate, and after a primary analysis at a minimum of 6 months follow-up, the objective response rate was 82%, with a CR rate of 52%. Among patients who achieved CR, the median duration of response was not reached after a median follow-up of 7.9 months.
A subsequent updated analysis was performed when 108 patients had been followed for a minimum of 1 year. The objective response rate was 82%, and the CR rate was 58%, with some patients having CR in the absence of additional therapies as late as 15 months after treatment. At this updated analysis, 42% of patients continued to have a response, 40% of whom remained in CR.
The most common grade 3 or higher AEs included febrile neutropenia, fever, CRS, encephalopathy, infections, hypotension, and hypoxia. Serious AEs occurred in 52% of patients and included CRS, neurologic toxicity, prolonged cytopenias, and serious infections. Grade 3 or higher CRS or neurologic toxicities occurred in 13% and 28% of patients, respectively. Three patients died during treatment.
To mitigate the risk of CRS and neurologic toxicity, axicabtagene ciloleucel is approved with an REMS that requires appropriate certification and training before hospitals are cleared to administer the therapy.
Other warnings and precautions in the prescribing information relate to serious infections (monitor for signs and symptoms and treat appropriately), prolonged cytopenias (monitor blood counts), hypogammaglobulinemia (monitor immunoglobulin levels and manage appropriately), secondary malignancies (life-long monitoring), and the potential effects of neurologic events on a patient’s ability to drive and operate dangerous machinery (avoid for at least 8 weeks after infusion).7
Axicabtagene ciloleucel is marketed as Yescarta by Kite Pharma Inc. The recommended dose is a single intravenous infusion with a target of 2 x 106 CAR-positive viable T cells per kilogram of body weight, preceded by fludarabine and cyclophosphamide lymphodepleting chemotherapy.
1. Bosserman LD. Cancer care in 2017: the promise of more cures with the challenges of an unstable health care system. JCSO 2017;15(6):e283-e290.
2. FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome. FDA News Release. August 30, 2017. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/
ucm574154.htm. Accessed March 31, 2018.
3. Maude S.L, Laetsch T.W, Buechner S, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N Engl J Med. 2018;378:439-48.
4. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Novartis Pharmaceuticals Corporation, August, 2017. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.
com/files/kymriah.pdf. Accessed March 31, 2018.
5. FDA approves axicabtagene ciloleucel for large B-cell lymphoma. FDA News Release. October 18, 2017. https://www.fda.gov/Drugs/
InformationOnDrugs/ApprovedDrugs/ucm581296.htm. Accessed March 31, 2018.
6. Neelapu, S.S, Locke F.L, Bartlett, L.J, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377:2531-44.
7. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Kite Pharma Inc. October 2017. https://www.yescarta.com/wp-content/uploads/yescarta-pi.pdf. Accessed March 31, 2018.
1. Bosserman LD. Cancer care in 2017: the promise of more cures with the challenges of an unstable health care system. JCSO 2017;15(6):e283-e290.
2. FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome. FDA News Release. August 30, 2017. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/
ucm574154.htm. Accessed March 31, 2018.
3. Maude S.L, Laetsch T.W, Buechner S, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N Engl J Med. 2018;378:439-48.
4. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Novartis Pharmaceuticals Corporation, August, 2017. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.
com/files/kymriah.pdf. Accessed March 31, 2018.
5. FDA approves axicabtagene ciloleucel for large B-cell lymphoma. FDA News Release. October 18, 2017. https://www.fda.gov/Drugs/
InformationOnDrugs/ApprovedDrugs/ucm581296.htm. Accessed March 31, 2018.
6. Neelapu, S.S, Locke F.L, Bartlett, L.J, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377:2531-44.
7. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Kite Pharma Inc. October 2017. https://www.yescarta.com/wp-content/uploads/yescarta-pi.pdf. Accessed March 31, 2018.