User login
Lenalidomide best option for myeloma maintenance therapy
Lenalidomide may be the best maintenance treatment option for patients with newly diagnosed multiple myeloma, say the authors of a systematic review and meta-analysis.
Francesca M. Gay, MD, from the division of hematology at the University of Torino (Italy), and her coauthors wrote that despite the well-recognized importance of novel agent–based maintenance therapy for multiple myeloma, there is a lack of direct or indirect comparisons between the available regimens.
In a paper published online in JAMA Oncology, the researchers reported the results of the systematic review and meta-analysis of 11 prospective, phase 3 randomized, controlled trials of eight varieties of maintenance therapy, in 5,073 participants with newly diagnosed multiple myeloma.
Their analysis found that lenalidomide-based regimens showed the best progression-free survival rates (hazard ratio, 0.39 for lenalidomide plus prednisone; HR, 0.47 for lenalidomide alone), compared with placebo, and in 74% of the network meta-analysis simulations, they were the most effective options.
Four other maintenance treatment options - thalidomide-interferon, thalidomide-bortezomib, bortezomib-prednisone, and thalidomide alone – also showed progression-free survival gains – but interferon therapy failed to show any benefit.
However, for overall survival, lenalidomide alone was the best option, followed by thalidomide-bortezomib and bortezomib-prednisone. None of the other regimens considered showed benefits for overall survival.
“Long-term use of lenalidomide undoubtedly has advantages, owing to the lack of neuropathy, which is the main factor limiting the long-term use of both thalidomide and bortezomib,” the authors wrote.
When the authors restricted their analysis to trials conducted in the setting of autologous stem cell transplantation they found similar results, with lenalidomide-based regimens having the best progression-free and overall survival.
Patients with a good prognosis and standard-risk chromosomal abnormalities also did best with lenalidomide-based maintenance, while those with a poor prognosis – for example, with ISS stage III disease – benefited more from bortezomib-based maintenance. However patients with high-risk chromosomal abnormalities gained no advantage from any regimen, which the authors suggested may relate to small sample size, different cut-off points or their extremely poor prognosis.
The authors noted that their analysis did not take into account adverse events, drug discontinuations, or quality of life but focused solely on progression-free survival and overall survival.
“An increase in second primary malignant disease with prolonged lenalidomide therapy has been reported, but the survival benefit overcame the risk in all the trials,” they wrote.
They also commented that better treatment options are needed for patients with aggressive disease, and there are ongoing trials looking at second-generation proteasome inhibitors, immunomodulatory agents, and monoclonal antibodies for maintenance therapy.
Most authors declared research funding, advisory board positions, fees and honoraria from the pharmaceutical industry, including lenalidomide manufacturer Celgene.
SOURCE: Gay F et al. 2018 Aug 9. doi:10.1001/jamaoncol.2018.2961.
Lenalidomide may be the best maintenance treatment option for patients with newly diagnosed multiple myeloma, say the authors of a systematic review and meta-analysis.
Francesca M. Gay, MD, from the division of hematology at the University of Torino (Italy), and her coauthors wrote that despite the well-recognized importance of novel agent–based maintenance therapy for multiple myeloma, there is a lack of direct or indirect comparisons between the available regimens.
In a paper published online in JAMA Oncology, the researchers reported the results of the systematic review and meta-analysis of 11 prospective, phase 3 randomized, controlled trials of eight varieties of maintenance therapy, in 5,073 participants with newly diagnosed multiple myeloma.
Their analysis found that lenalidomide-based regimens showed the best progression-free survival rates (hazard ratio, 0.39 for lenalidomide plus prednisone; HR, 0.47 for lenalidomide alone), compared with placebo, and in 74% of the network meta-analysis simulations, they were the most effective options.
Four other maintenance treatment options - thalidomide-interferon, thalidomide-bortezomib, bortezomib-prednisone, and thalidomide alone – also showed progression-free survival gains – but interferon therapy failed to show any benefit.
However, for overall survival, lenalidomide alone was the best option, followed by thalidomide-bortezomib and bortezomib-prednisone. None of the other regimens considered showed benefits for overall survival.
“Long-term use of lenalidomide undoubtedly has advantages, owing to the lack of neuropathy, which is the main factor limiting the long-term use of both thalidomide and bortezomib,” the authors wrote.
When the authors restricted their analysis to trials conducted in the setting of autologous stem cell transplantation they found similar results, with lenalidomide-based regimens having the best progression-free and overall survival.
Patients with a good prognosis and standard-risk chromosomal abnormalities also did best with lenalidomide-based maintenance, while those with a poor prognosis – for example, with ISS stage III disease – benefited more from bortezomib-based maintenance. However patients with high-risk chromosomal abnormalities gained no advantage from any regimen, which the authors suggested may relate to small sample size, different cut-off points or their extremely poor prognosis.
The authors noted that their analysis did not take into account adverse events, drug discontinuations, or quality of life but focused solely on progression-free survival and overall survival.
“An increase in second primary malignant disease with prolonged lenalidomide therapy has been reported, but the survival benefit overcame the risk in all the trials,” they wrote.
They also commented that better treatment options are needed for patients with aggressive disease, and there are ongoing trials looking at second-generation proteasome inhibitors, immunomodulatory agents, and monoclonal antibodies for maintenance therapy.
Most authors declared research funding, advisory board positions, fees and honoraria from the pharmaceutical industry, including lenalidomide manufacturer Celgene.
SOURCE: Gay F et al. 2018 Aug 9. doi:10.1001/jamaoncol.2018.2961.
Lenalidomide may be the best maintenance treatment option for patients with newly diagnosed multiple myeloma, say the authors of a systematic review and meta-analysis.
Francesca M. Gay, MD, from the division of hematology at the University of Torino (Italy), and her coauthors wrote that despite the well-recognized importance of novel agent–based maintenance therapy for multiple myeloma, there is a lack of direct or indirect comparisons between the available regimens.
In a paper published online in JAMA Oncology, the researchers reported the results of the systematic review and meta-analysis of 11 prospective, phase 3 randomized, controlled trials of eight varieties of maintenance therapy, in 5,073 participants with newly diagnosed multiple myeloma.
Their analysis found that lenalidomide-based regimens showed the best progression-free survival rates (hazard ratio, 0.39 for lenalidomide plus prednisone; HR, 0.47 for lenalidomide alone), compared with placebo, and in 74% of the network meta-analysis simulations, they were the most effective options.
Four other maintenance treatment options - thalidomide-interferon, thalidomide-bortezomib, bortezomib-prednisone, and thalidomide alone – also showed progression-free survival gains – but interferon therapy failed to show any benefit.
However, for overall survival, lenalidomide alone was the best option, followed by thalidomide-bortezomib and bortezomib-prednisone. None of the other regimens considered showed benefits for overall survival.
“Long-term use of lenalidomide undoubtedly has advantages, owing to the lack of neuropathy, which is the main factor limiting the long-term use of both thalidomide and bortezomib,” the authors wrote.
When the authors restricted their analysis to trials conducted in the setting of autologous stem cell transplantation they found similar results, with lenalidomide-based regimens having the best progression-free and overall survival.
Patients with a good prognosis and standard-risk chromosomal abnormalities also did best with lenalidomide-based maintenance, while those with a poor prognosis – for example, with ISS stage III disease – benefited more from bortezomib-based maintenance. However patients with high-risk chromosomal abnormalities gained no advantage from any regimen, which the authors suggested may relate to small sample size, different cut-off points or their extremely poor prognosis.
The authors noted that their analysis did not take into account adverse events, drug discontinuations, or quality of life but focused solely on progression-free survival and overall survival.
“An increase in second primary malignant disease with prolonged lenalidomide therapy has been reported, but the survival benefit overcame the risk in all the trials,” they wrote.
They also commented that better treatment options are needed for patients with aggressive disease, and there are ongoing trials looking at second-generation proteasome inhibitors, immunomodulatory agents, and monoclonal antibodies for maintenance therapy.
Most authors declared research funding, advisory board positions, fees and honoraria from the pharmaceutical industry, including lenalidomide manufacturer Celgene.
SOURCE: Gay F et al. 2018 Aug 9. doi:10.1001/jamaoncol.2018.2961.
FROM JAMA ONCOLOGY
Key clinical point: Lenalidomide is the best option for maintenance therapy in multiple myeloma.
Major finding: Lenalidomide-based maintenance regimens show the best progression-free and overall survival in multiple myeloma.
Study details: Systematic review and network meta-analysis of 11 studies in 5073 participants with newly diagnosed multiple myeloma.
Disclosures: Most authors declared research funding, advisory board positions, fees and honoraria from the pharmaceutical industry, including lenalidomide manufacturer Celgene.
Source: Gay F et al. 2018 Aug 9. doi: 10.1001/jamaoncol.2018.2961.
Immunotherapies shape the treatment landscape for hematologic malignancies
The treatment landscape for hematologic malignancies is evolving faster than ever before, with a range of available therapeutic options that is now almost as diverse as this group of tumors. Immunotherapy in particular is front and center in the battle to control these diseases. Here, we describe the latest promising developments.
Exploiting T cells
The treatment landscape for hematologic malignancies is diverse, but one particular type of therapy has led the charge in improving patient outcomes. Several features of hematologic malignancies may make them particularly amenable to immunotherapy, including the fact that they are derived from corrupt immune cells and come into constant contact with other immune cells within the hematopoietic environment in which they reside. One of the oldest forms of immunotherapy, hematopoietic stem-cell transplantation (HSCT), remains the only curative option for many patients with hematologic malignancies.1,2
Given the central role of T lymphocytes in antitumor immunity, research efforts have focused on harnessing their activity for cancer treatment. One example of this is adoptive cellular therapy (ACT), in which T cells are collected from a patient, grown outside the body to increase their number and then reinfused back to the patient. Allogeneic HSCT, in which the stem cells are collected from a matching donor and transplanted into the patient, is a crude example of ACT. The graft-versus-tumor effect is driven by donor cells present in the transplant, but is limited by the development of graft-versus-host disease (GvHD), whereby the donor T cells attack healthy host tissue.
Other types of ACT have been developed in an effort to capitalize on the anti-tumor effects of the patients own T cells and thus avoid the potentially fatal complication of GvHD. Tumor-infiltrating lymphocyte (TIL) therapy was developed to exploit the presence of tumor-specific T cells in the tumor microenvironment. To date, the efficacy of TIL therapy has been predominantly limited to melanoma.1,3,4
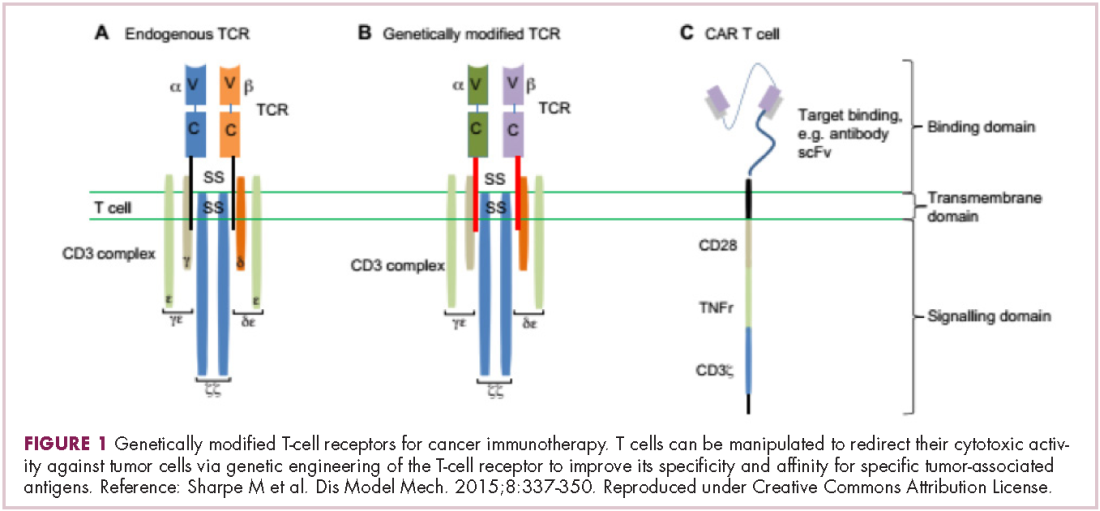
Most recently, there has been a substantial buzz around the idea of genetically engineering T cells before they are reintroduced into the patient, to increase their anti-tumor efficacy and minimize damage to healthy tissue. This is achieved either by manipulating the antigen binding portion of the T-cell receptor to alter its specificity (TCR T cells) or by generating artificial fusion receptors known as chimeric antigen receptors (CAR T cells; Figure 1). The former is limited by the need for the TCR to be genetically matched to the patient’s immune type, whereas the latter is more flexible in this regard and has proved most successful.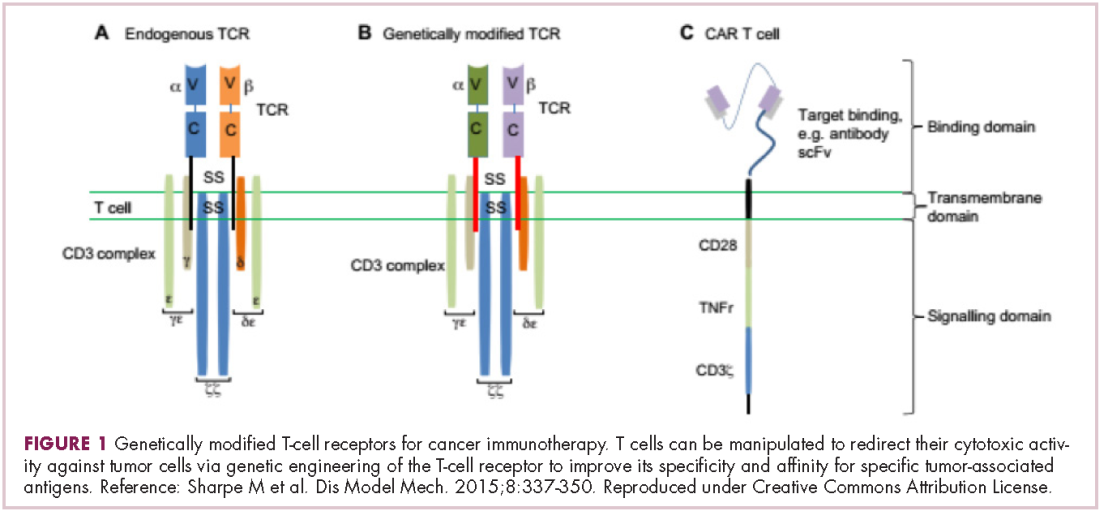
CARs are formed by fusing part of the single-chain variable fragment of a monoclonal antibody to part of the TCR and one or more costimulatory molecules. In this way, the T cell is guided to the tumor through antibody recognition of a particular tumor-associated antigen, whereupon its effector functions are activated by engagement of the TCR and costimulatory signal.5
Headlining advancements with CAR T cells
CAR T cells directed against the CD19 antigen, found on the surface of many hematologic malignancies, are the most clinically advanced in this rapidly evolving field (Table 1). Durable remissions have been demonstrated in patients with relapsed and refractory hematologic malignancies, including non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), and acute lymphoblastic lymphoma (ALL), with efficacy in both the pre- and posttransplant setting and in patients with chemotherapy-refractory disease.4,5
CTL019, a CD19-targeted CAR-T cell therapy, also known as tisagenlecleucel-T, has received breakthrough therapy designation from the US Food and Drug Administration (FDA) for the treatment of pediatric and adult patients with relapsed/refractory B-cell ALL and, more recently, for the treatment of adult patients with relapsed/refractory diffuse large B cell lymphoma.6
It is edging closer to FDA approval for the ALL indication, having been granted priority review in March on the basis of the phase 2 ELIANA trial, in which 50 patients received a single infusion of CTL019. Data presented at the American Society of Hematology annual meeting in December 2016 showed that 82% of patients achieved either complete remission (CR) or CR with incomplete blood count recovery (CRi) 3 months after treatment.7
Meanwhile, Kite Pharma has a rolling submission with the FDA for KTE-C19 (axicabtagene ciloleucel) for the treatment of patients with relapsed/refractory B-cell NHL who are ineligible for HSCT. In the ZUMA-1 trial, this therapy demonstrated an overall response rate (ORR) of 71%.8 Juno Therapeutics is developing several CAR T-cell therapies, including JCAR017, which elicited CR in 60% of patients with relapsed/refractory NHL.9
Target antigens other than CD19 are being explored, but these are mostly in the early stages of clinical development. While the focus has predominantly been on the treatment of lymphoma and leukemia, a presentation at the American Society for Clinical Oncology annual meeting in June reported the efficacy of a CAR-T cell therapy targeting the B-cell maturation antigen in patients with multiple myeloma. Results from 19 patients enrolled in an ongoing phase 1 trial in China showed that 14 had achieved stringent CR, 1 partial remission (PR) and 4 very good partial remission (VGPR).10
Antibodies evolve
Another type of immunotherapy that has revolutionized the treatment of hematologic malignancies is monoclonal antibodies (mAbs), targeting antigens on the surface of malignant B and T cells, in particular CD20. The approval of CD20-targeting mAb rituximab in 1997 was the first coup for the development of immunotherapy for the treatment of hematologic malignancies. It has become part of the standard treatment regimen for B-cell malignancies, including NHL and CLL, in combination with various types of chemotherapy.
Several other CD20-targeting antibodies have been developed (Table 2), some of which work in the same way as rituximab (eg, ofatumumab) and some that have a slightly different mechanism of action (eg, obinutuzumab).11 Both types of antibody have proved highly effective; ofatumumab is FDA approved for the treatment of advanced CLL and is being evaluated in phase 3 trials in other hematologic malignancies, while obinutuzumab has received regulatory approval for the first-line treatment of CLL, replacing the standard rituximab-containing regimen.12
The use of ofatumumab as maintenance therapy is supported by the results of the phase 3 PROLONG study in which 474 patients were randomly assigned to ofatumumab maintenance for 2 years or observation. Over a median follow-up of close to 20 months, ofatumumab-treated patients experienced improved progression-free survival (PFS; median PFS: 29.4 months vs 15.2 months; hazard ratio [HR], 0.50; P < .0001).13 Obinutuzumab’s new indication is based on data from the phase 3 GADOLIN trial, in which the obinutuzumab arm showed improved 3-year PFS compared with rituximab.14Until recently, multiple myeloma had proven relatively resistant to mAb therapy, but two new drug targets have dramatically altered the treatment landscape for this type of hematologic malignancy. CD2 subset 1 (CS1), also known as signaling lymphocytic activation molecule 7 (SLAMF7), and CD38 are glycoproteins expressed highly and nearly uniformly on the surface of multiple myeloma cells and only at low levels on other lymphoid and myeloid cells.15
Several antibodies directed at these targets are in clinical development, but daratumumab and elotuzumab, targeting CD38 and CS1, respectively, are both newly approved by the FDA for relapsed/refractory disease, daratumumab as monotherapy and elotuzumab in combination with lenalidomide and dexamethasone.
The indication for daratumumab was subsequently expanded to include its use in combination with lenalidomide plus dexamethasone or bortezomib plus dexamethasone. Support for this new indication came from 2 pivotal phase 3 trials. In the CASTOR trial, the combination of daratumumab with bortezomib–dexamethasone reduced the risk of disease progression or death by 61%, compared with bortezomib–dexamethasone alone, whereas daratumumab with lenalidomide–dexamethasone reduced the risk of disease progression or death by 63% in the POLLUX trial.16,17
Numerous clinical trials for both drugs are ongoing, including in the front-line setting in multiple myeloma, as well as trials in other types of B-cell malignancy, and several other CD38-targeting mAbs are also in development, including isatuximab, which has reached the phase 3 stage (NCT02990338).
Innovative design
Newer drug designs, which have sought to take mAb therapy to the next level, have also shown significant efficacy in hematologic malignancies. Antibody-drug conjugates (ADCs) combine the cytotoxic efficacy of chemotherapeutic agents with the specificity of a mAb targeting a tumor-specific antigen. This essentially creates a targeted payload that improves upon the efficacy of mAb monotherapy but mitigates some of the side effects of chemotherapy related to their indiscriminate killing of both cancerous and healthy cells.
The development of ADCs has been somewhat of a rollercoaster ride, with the approval and subsequent withdrawal of the first-in-class drug gemtuzumab ozogamicin in 2010, but the field was reinvigorated with the successful development of brentuximab vedotin, which targets the CD30 antigen and is approved for the treatment of multiple different hematologic malignancies, including, most recently, for posttransplant consolidation therapy in patients with Hodgkin lymphoma at high risk of relapse or progression.18
Brentuximab vedotin may soon be joined by another FDA-approved ADC, this one targeting CD22. Inotuzumab ozogamicin was recently granted priority review for the treatment of relapsed/refractory ALL. The FDA is reviewing data from the phase 3 INO-VATE study in which inotuzumab ozogamicin reduced the risk of disease progression or death by 55% compared with standard therapy, and a decision is expected by August.19 Other ADC targets being investigated in clinical trials include CD138, CD19, and CD33 (Table 3). Meanwhile, a meta-analysis of randomized trials suggested that the withdrawal of gemtuzumab ozogamicin may have been premature, indicating that it does improve long-term overall survival (OS) and reduces the risk of relapse.20
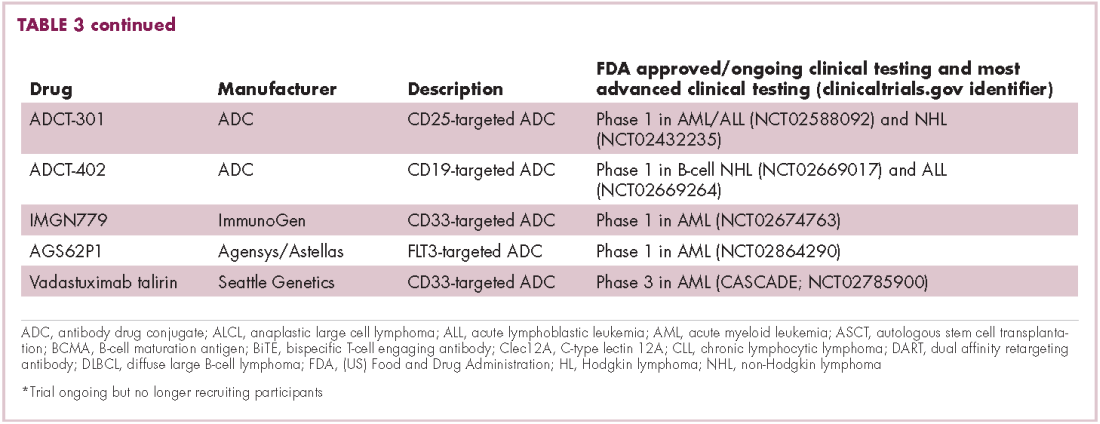
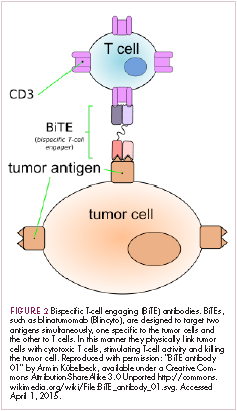
Bispecific antibodies that link natural killer (NK) cells to tumor cells, by targeting the NK-cell receptor CD16, known as BiKEs, are also in development in an attempt to harness the power of the innate immune response.
B-cell signaling a ripe target
Beyond immunotherapy, molecularly targeted drugs directed against key drivers of hematologic malignancies are also showing great promise. In particular, the B-cell receptor (BCR) signaling pathway, a central regulator of B-cell function, and its constituent kinases that are frequently dysregulated in B cell malignancies, has emerged as an exciting therapeutic avenue.
A variety of small molecule inhibitors targeting different nodes of the BCR pathway have been developed (Table 4), but the greatest success to date has been achieved with drugs targeting Bruton’s tyrosine kinase (BTK). Their clinical development culminated in the approval of ibrutinib for the treatment of patients with mantle cell lymphoma in 2013 and subsequently for patients with CLL, Waldenström macroglobulinemia, and most recently for patients with marginal zone lymphoma.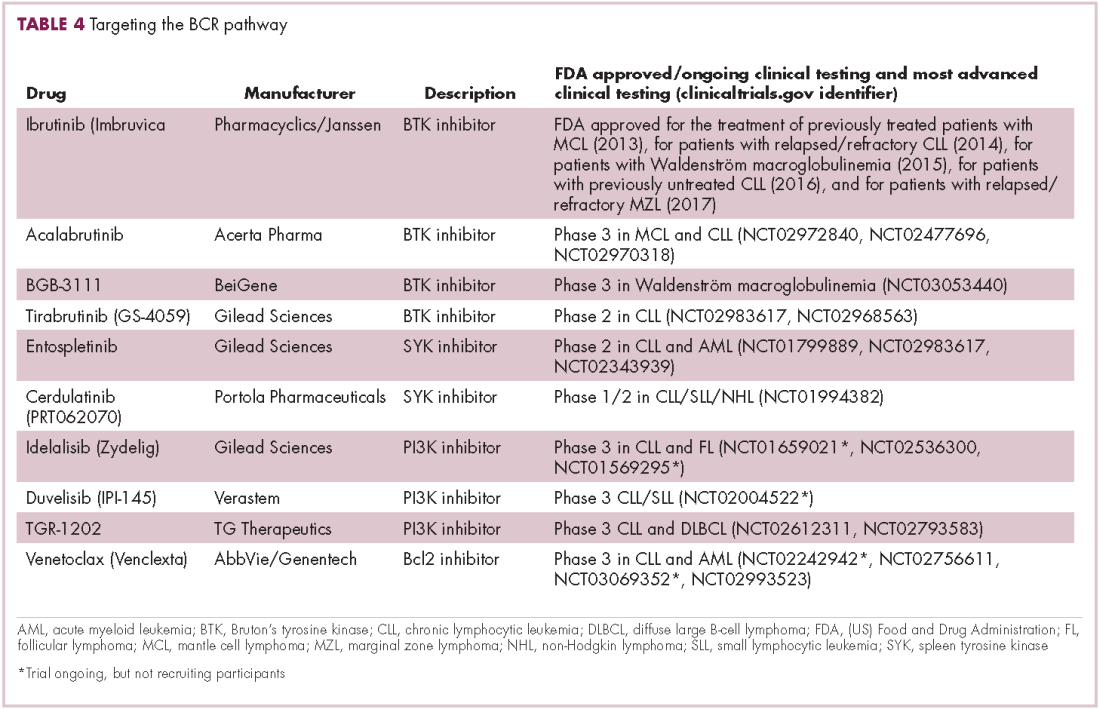
More than 100 clinical trials of ibrutinib are ongoing in an effort to further clarify its role in a variety of different disease settings. Furthermore, in an effort to address some of the toxicity concerns with ibrutinib, more specific BTK inhibitors are also being developed.
Other kinases that orchestrate the BCR pathway, including phosphatidylinositol-3-kinase (PI3K) and SYK, are also being targeted. The delta isoform of PI3K is expressed exclusively in hematopoietic cells and a number of PI3K delta inhibitors have been developed. Idelalisib received regulatory approval for the treatment of patients with CLL in combination with rituximab, and for patients with follicular lymphoma and small lymphocytic leukemia.
As with ibrutinib, a plethora of clinical trials are ongoing, however a major setback was suffered in the frontline setting when Gilead Sciences halted 6 clinical trials due to reports of increased rates of adverse events, including deaths.26 Meanwhile, SYK inhibitors have lagged behind somewhat in their development, but one such offering, entospletinib, is showing promise in patients with AML.27
Finally, there has been some success in targeting one of the downstream targets of the BCR signaling pathway, the Bcl2 protein that is involved in the regulation of apoptosis. Venetoclax was approved last year for the treatment of patients with relapsed/refractory CLL in patients who have a chromosome 17p deletion, based on the demonstration of impressive, durable responses.28
1. Bachireddy P, Burkhardt UE, Rajasagi M, Wu CJ. Haemato- logical malignancies: at the forefront of immunotherapeutic innovation. Nat Rev Cancer. 2015;15(4):201-215.
2. Im A, Pavletic SZ. Immunotherapy in hematologic malignancies: past, present, and future. J Hematol Oncol. 2017;10(1):94.
3. Gill S. Planes, trains, and automobiles: perspectives on CAR T cells and other cellular therapies for hematologic malignancies. Curr Hematol Malig Rep. 2016;11(4):318-325.
4. Ye B, Stary CM, Gao Q, et al. Genetically modified T-cell-based adoptive immunotherapy in hematological malignancies. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5237740/. Published January 2, 2017. Accessed July 22, 2017.
5. Sharpe M, Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
6. Novartis. Novartis personalized cell therapy CTL019 receives FDA breakthrough therapy designation. https://www.novartis.com/news/media-releases/novartis-personalized-cell-therapy-ctl019-receivesfda-breakthrough-therapy. Published July 7, 2014. Accessed June 19,
2017.
7. Novartis. Novartis presents results from first global registration trial of CTL019 in pediatric and young adult patients with r/r B-ALL. https://www.novartis.com/news/media-releases/novartis-presentsresults-first-global-registration-trial-ctl019-pediatric-and. Published December 4, 2016. Accessed June 19, 2017.
8. Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 Results of ZUMA1: a multicenter study of KTE-C19 Anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25(1):285-295.
9. Abramson JS, Palomba L, Gordon L. Transcend NHL 001: immunotherapy with the CD19-Directd CAR T-cell product JCAR017 results in high complete response rates in relapsed or refractory B-cell non-Hodgkin lymphoma. Paper presented at 58th American Society of Hematology Annual Meeting; December 3-6, 2016; San Diego, CA.
10. Fan F, Zhao W, Liu J, et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. J Clin Oncol. 2017;35(suppl;):Abstr LBA3001.
11. Okroj M, Osterborg A, Blom AM. Effector mechanisms of anti-CD20 monoclonal antibodies in B cell malignancies. Cancer Treat Rev. 2013;39(6):632-639.
12. Safdari Y, Ahmadzadeh V, Farajnia S. CD20-targeting in B-cell malignancies: novel prospects for antibodies and combination therapies. Invest New Drugs. 2016;34(4):497-512.
13. van Oers MH, Kuliczkowski K, Smolej L, et al. Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): an open-label, multicentre, randomised phase 3 study. Lancet Oncol. 2015;16(13):1370-1379.
14. Sehn LH, Chua N, Mayer J, et al. Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): a randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol. 2016;17(8):1081-1093.
15. Touzeau C, Moreau P, Dumontet C. Monoclonal antibody therapy in multiple myeloma. Leukemia. 2017;31(5):1039-1047.
16. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754-766.
17. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319-1331.
18. Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315-337.
19. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740-753.
20. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
21. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93(3):290-296.
22. Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836-847.
23. Koehrer S, Burger JA. B-cell receptor signaling in chronic lymphocytic leukemia and other B-cell malignancies. Clin Adv Hematol Oncol. 2016;14(1):55-65.
24. Seda V, Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol. 2015;94(3):193-205.
25. Bojarczuk K, Bobrowicz M, Dwojak M, et al. B-cell receptor signaling in the pathogenesis of lymphoid malignancies. Blood Cells Mol Dis. 2015;55(3):255-265.
26. Medscape Medical News. Gilead stops six trials adding idelalisib to other drugs. http://www.medscape.com/viewarticle/860372. Published March 14, 2016. Accessed June 19, 2017.
27. Sharman J, Di Paolo J. Targeting B-cell receptor signaling kinases in chronic lymphocytic leukemia: the promise of entospletinib. Ther Adv Hematol. 2016;7(3):157-170.
28. Food and Drug Administration. FDA approves new drug for chronic lymphocytic leukemia in patients with a specific chromosomal abnormality. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm495253.htm. Released April 11, 2016. Accessed June 19, 2017.
The treatment landscape for hematologic malignancies is evolving faster than ever before, with a range of available therapeutic options that is now almost as diverse as this group of tumors. Immunotherapy in particular is front and center in the battle to control these diseases. Here, we describe the latest promising developments.
Exploiting T cells
The treatment landscape for hematologic malignancies is diverse, but one particular type of therapy has led the charge in improving patient outcomes. Several features of hematologic malignancies may make them particularly amenable to immunotherapy, including the fact that they are derived from corrupt immune cells and come into constant contact with other immune cells within the hematopoietic environment in which they reside. One of the oldest forms of immunotherapy, hematopoietic stem-cell transplantation (HSCT), remains the only curative option for many patients with hematologic malignancies.1,2
Given the central role of T lymphocytes in antitumor immunity, research efforts have focused on harnessing their activity for cancer treatment. One example of this is adoptive cellular therapy (ACT), in which T cells are collected from a patient, grown outside the body to increase their number and then reinfused back to the patient. Allogeneic HSCT, in which the stem cells are collected from a matching donor and transplanted into the patient, is a crude example of ACT. The graft-versus-tumor effect is driven by donor cells present in the transplant, but is limited by the development of graft-versus-host disease (GvHD), whereby the donor T cells attack healthy host tissue.
Other types of ACT have been developed in an effort to capitalize on the anti-tumor effects of the patients own T cells and thus avoid the potentially fatal complication of GvHD. Tumor-infiltrating lymphocyte (TIL) therapy was developed to exploit the presence of tumor-specific T cells in the tumor microenvironment. To date, the efficacy of TIL therapy has been predominantly limited to melanoma.1,3,4
Most recently, there has been a substantial buzz around the idea of genetically engineering T cells before they are reintroduced into the patient, to increase their anti-tumor efficacy and minimize damage to healthy tissue. This is achieved either by manipulating the antigen binding portion of the T-cell receptor to alter its specificity (TCR T cells) or by generating artificial fusion receptors known as chimeric antigen receptors (CAR T cells; Figure 1). The former is limited by the need for the TCR to be genetically matched to the patient’s immune type, whereas the latter is more flexible in this regard and has proved most successful.
CARs are formed by fusing part of the single-chain variable fragment of a monoclonal antibody to part of the TCR and one or more costimulatory molecules. In this way, the T cell is guided to the tumor through antibody recognition of a particular tumor-associated antigen, whereupon its effector functions are activated by engagement of the TCR and costimulatory signal.5
Headlining advancements with CAR T cells
CAR T cells directed against the CD19 antigen, found on the surface of many hematologic malignancies, are the most clinically advanced in this rapidly evolving field (Table 1). Durable remissions have been demonstrated in patients with relapsed and refractory hematologic malignancies, including non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), and acute lymphoblastic lymphoma (ALL), with efficacy in both the pre- and posttransplant setting and in patients with chemotherapy-refractory disease.4,5
CTL019, a CD19-targeted CAR-T cell therapy, also known as tisagenlecleucel-T, has received breakthrough therapy designation from the US Food and Drug Administration (FDA) for the treatment of pediatric and adult patients with relapsed/refractory B-cell ALL and, more recently, for the treatment of adult patients with relapsed/refractory diffuse large B cell lymphoma.6
It is edging closer to FDA approval for the ALL indication, having been granted priority review in March on the basis of the phase 2 ELIANA trial, in which 50 patients received a single infusion of CTL019. Data presented at the American Society of Hematology annual meeting in December 2016 showed that 82% of patients achieved either complete remission (CR) or CR with incomplete blood count recovery (CRi) 3 months after treatment.7
Meanwhile, Kite Pharma has a rolling submission with the FDA for KTE-C19 (axicabtagene ciloleucel) for the treatment of patients with relapsed/refractory B-cell NHL who are ineligible for HSCT. In the ZUMA-1 trial, this therapy demonstrated an overall response rate (ORR) of 71%.8 Juno Therapeutics is developing several CAR T-cell therapies, including JCAR017, which elicited CR in 60% of patients with relapsed/refractory NHL.9
Target antigens other than CD19 are being explored, but these are mostly in the early stages of clinical development. While the focus has predominantly been on the treatment of lymphoma and leukemia, a presentation at the American Society for Clinical Oncology annual meeting in June reported the efficacy of a CAR-T cell therapy targeting the B-cell maturation antigen in patients with multiple myeloma. Results from 19 patients enrolled in an ongoing phase 1 trial in China showed that 14 had achieved stringent CR, 1 partial remission (PR) and 4 very good partial remission (VGPR).10
Antibodies evolve
Another type of immunotherapy that has revolutionized the treatment of hematologic malignancies is monoclonal antibodies (mAbs), targeting antigens on the surface of malignant B and T cells, in particular CD20. The approval of CD20-targeting mAb rituximab in 1997 was the first coup for the development of immunotherapy for the treatment of hematologic malignancies. It has become part of the standard treatment regimen for B-cell malignancies, including NHL and CLL, in combination with various types of chemotherapy.
Several other CD20-targeting antibodies have been developed (Table 2), some of which work in the same way as rituximab (eg, ofatumumab) and some that have a slightly different mechanism of action (eg, obinutuzumab).11 Both types of antibody have proved highly effective; ofatumumab is FDA approved for the treatment of advanced CLL and is being evaluated in phase 3 trials in other hematologic malignancies, while obinutuzumab has received regulatory approval for the first-line treatment of CLL, replacing the standard rituximab-containing regimen.12
The use of ofatumumab as maintenance therapy is supported by the results of the phase 3 PROLONG study in which 474 patients were randomly assigned to ofatumumab maintenance for 2 years or observation. Over a median follow-up of close to 20 months, ofatumumab-treated patients experienced improved progression-free survival (PFS; median PFS: 29.4 months vs 15.2 months; hazard ratio [HR], 0.50; P < .0001).13 Obinutuzumab’s new indication is based on data from the phase 3 GADOLIN trial, in which the obinutuzumab arm showed improved 3-year PFS compared with rituximab.14Until recently, multiple myeloma had proven relatively resistant to mAb therapy, but two new drug targets have dramatically altered the treatment landscape for this type of hematologic malignancy. CD2 subset 1 (CS1), also known as signaling lymphocytic activation molecule 7 (SLAMF7), and CD38 are glycoproteins expressed highly and nearly uniformly on the surface of multiple myeloma cells and only at low levels on other lymphoid and myeloid cells.15
Several antibodies directed at these targets are in clinical development, but daratumumab and elotuzumab, targeting CD38 and CS1, respectively, are both newly approved by the FDA for relapsed/refractory disease, daratumumab as monotherapy and elotuzumab in combination with lenalidomide and dexamethasone.
The indication for daratumumab was subsequently expanded to include its use in combination with lenalidomide plus dexamethasone or bortezomib plus dexamethasone. Support for this new indication came from 2 pivotal phase 3 trials. In the CASTOR trial, the combination of daratumumab with bortezomib–dexamethasone reduced the risk of disease progression or death by 61%, compared with bortezomib–dexamethasone alone, whereas daratumumab with lenalidomide–dexamethasone reduced the risk of disease progression or death by 63% in the POLLUX trial.16,17
Numerous clinical trials for both drugs are ongoing, including in the front-line setting in multiple myeloma, as well as trials in other types of B-cell malignancy, and several other CD38-targeting mAbs are also in development, including isatuximab, which has reached the phase 3 stage (NCT02990338).
Innovative design
Newer drug designs, which have sought to take mAb therapy to the next level, have also shown significant efficacy in hematologic malignancies. Antibody-drug conjugates (ADCs) combine the cytotoxic efficacy of chemotherapeutic agents with the specificity of a mAb targeting a tumor-specific antigen. This essentially creates a targeted payload that improves upon the efficacy of mAb monotherapy but mitigates some of the side effects of chemotherapy related to their indiscriminate killing of both cancerous and healthy cells.
The development of ADCs has been somewhat of a rollercoaster ride, with the approval and subsequent withdrawal of the first-in-class drug gemtuzumab ozogamicin in 2010, but the field was reinvigorated with the successful development of brentuximab vedotin, which targets the CD30 antigen and is approved for the treatment of multiple different hematologic malignancies, including, most recently, for posttransplant consolidation therapy in patients with Hodgkin lymphoma at high risk of relapse or progression.18
Brentuximab vedotin may soon be joined by another FDA-approved ADC, this one targeting CD22. Inotuzumab ozogamicin was recently granted priority review for the treatment of relapsed/refractory ALL. The FDA is reviewing data from the phase 3 INO-VATE study in which inotuzumab ozogamicin reduced the risk of disease progression or death by 55% compared with standard therapy, and a decision is expected by August.19 Other ADC targets being investigated in clinical trials include CD138, CD19, and CD33 (Table 3). Meanwhile, a meta-analysis of randomized trials suggested that the withdrawal of gemtuzumab ozogamicin may have been premature, indicating that it does improve long-term overall survival (OS) and reduces the risk of relapse.20
Bispecific antibodies that link natural killer (NK) cells to tumor cells, by targeting the NK-cell receptor CD16, known as BiKEs, are also in development in an attempt to harness the power of the innate immune response.
B-cell signaling a ripe target
Beyond immunotherapy, molecularly targeted drugs directed against key drivers of hematologic malignancies are also showing great promise. In particular, the B-cell receptor (BCR) signaling pathway, a central regulator of B-cell function, and its constituent kinases that are frequently dysregulated in B cell malignancies, has emerged as an exciting therapeutic avenue.
A variety of small molecule inhibitors targeting different nodes of the BCR pathway have been developed (Table 4), but the greatest success to date has been achieved with drugs targeting Bruton’s tyrosine kinase (BTK). Their clinical development culminated in the approval of ibrutinib for the treatment of patients with mantle cell lymphoma in 2013 and subsequently for patients with CLL, Waldenström macroglobulinemia, and most recently for patients with marginal zone lymphoma.
More than 100 clinical trials of ibrutinib are ongoing in an effort to further clarify its role in a variety of different disease settings. Furthermore, in an effort to address some of the toxicity concerns with ibrutinib, more specific BTK inhibitors are also being developed.
Other kinases that orchestrate the BCR pathway, including phosphatidylinositol-3-kinase (PI3K) and SYK, are also being targeted. The delta isoform of PI3K is expressed exclusively in hematopoietic cells and a number of PI3K delta inhibitors have been developed. Idelalisib received regulatory approval for the treatment of patients with CLL in combination with rituximab, and for patients with follicular lymphoma and small lymphocytic leukemia.
As with ibrutinib, a plethora of clinical trials are ongoing, however a major setback was suffered in the frontline setting when Gilead Sciences halted 6 clinical trials due to reports of increased rates of adverse events, including deaths.26 Meanwhile, SYK inhibitors have lagged behind somewhat in their development, but one such offering, entospletinib, is showing promise in patients with AML.27
Finally, there has been some success in targeting one of the downstream targets of the BCR signaling pathway, the Bcl2 protein that is involved in the regulation of apoptosis. Venetoclax was approved last year for the treatment of patients with relapsed/refractory CLL in patients who have a chromosome 17p deletion, based on the demonstration of impressive, durable responses.28
The treatment landscape for hematologic malignancies is evolving faster than ever before, with a range of available therapeutic options that is now almost as diverse as this group of tumors. Immunotherapy in particular is front and center in the battle to control these diseases. Here, we describe the latest promising developments.
Exploiting T cells
The treatment landscape for hematologic malignancies is diverse, but one particular type of therapy has led the charge in improving patient outcomes. Several features of hematologic malignancies may make them particularly amenable to immunotherapy, including the fact that they are derived from corrupt immune cells and come into constant contact with other immune cells within the hematopoietic environment in which they reside. One of the oldest forms of immunotherapy, hematopoietic stem-cell transplantation (HSCT), remains the only curative option for many patients with hematologic malignancies.1,2
Given the central role of T lymphocytes in antitumor immunity, research efforts have focused on harnessing their activity for cancer treatment. One example of this is adoptive cellular therapy (ACT), in which T cells are collected from a patient, grown outside the body to increase their number and then reinfused back to the patient. Allogeneic HSCT, in which the stem cells are collected from a matching donor and transplanted into the patient, is a crude example of ACT. The graft-versus-tumor effect is driven by donor cells present in the transplant, but is limited by the development of graft-versus-host disease (GvHD), whereby the donor T cells attack healthy host tissue.
Other types of ACT have been developed in an effort to capitalize on the anti-tumor effects of the patients own T cells and thus avoid the potentially fatal complication of GvHD. Tumor-infiltrating lymphocyte (TIL) therapy was developed to exploit the presence of tumor-specific T cells in the tumor microenvironment. To date, the efficacy of TIL therapy has been predominantly limited to melanoma.1,3,4
Most recently, there has been a substantial buzz around the idea of genetically engineering T cells before they are reintroduced into the patient, to increase their anti-tumor efficacy and minimize damage to healthy tissue. This is achieved either by manipulating the antigen binding portion of the T-cell receptor to alter its specificity (TCR T cells) or by generating artificial fusion receptors known as chimeric antigen receptors (CAR T cells; Figure 1). The former is limited by the need for the TCR to be genetically matched to the patient’s immune type, whereas the latter is more flexible in this regard and has proved most successful.
CARs are formed by fusing part of the single-chain variable fragment of a monoclonal antibody to part of the TCR and one or more costimulatory molecules. In this way, the T cell is guided to the tumor through antibody recognition of a particular tumor-associated antigen, whereupon its effector functions are activated by engagement of the TCR and costimulatory signal.5
Headlining advancements with CAR T cells
CAR T cells directed against the CD19 antigen, found on the surface of many hematologic malignancies, are the most clinically advanced in this rapidly evolving field (Table 1). Durable remissions have been demonstrated in patients with relapsed and refractory hematologic malignancies, including non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), and acute lymphoblastic lymphoma (ALL), with efficacy in both the pre- and posttransplant setting and in patients with chemotherapy-refractory disease.4,5
CTL019, a CD19-targeted CAR-T cell therapy, also known as tisagenlecleucel-T, has received breakthrough therapy designation from the US Food and Drug Administration (FDA) for the treatment of pediatric and adult patients with relapsed/refractory B-cell ALL and, more recently, for the treatment of adult patients with relapsed/refractory diffuse large B cell lymphoma.6
It is edging closer to FDA approval for the ALL indication, having been granted priority review in March on the basis of the phase 2 ELIANA trial, in which 50 patients received a single infusion of CTL019. Data presented at the American Society of Hematology annual meeting in December 2016 showed that 82% of patients achieved either complete remission (CR) or CR with incomplete blood count recovery (CRi) 3 months after treatment.7
Meanwhile, Kite Pharma has a rolling submission with the FDA for KTE-C19 (axicabtagene ciloleucel) for the treatment of patients with relapsed/refractory B-cell NHL who are ineligible for HSCT. In the ZUMA-1 trial, this therapy demonstrated an overall response rate (ORR) of 71%.8 Juno Therapeutics is developing several CAR T-cell therapies, including JCAR017, which elicited CR in 60% of patients with relapsed/refractory NHL.9
Target antigens other than CD19 are being explored, but these are mostly in the early stages of clinical development. While the focus has predominantly been on the treatment of lymphoma and leukemia, a presentation at the American Society for Clinical Oncology annual meeting in June reported the efficacy of a CAR-T cell therapy targeting the B-cell maturation antigen in patients with multiple myeloma. Results from 19 patients enrolled in an ongoing phase 1 trial in China showed that 14 had achieved stringent CR, 1 partial remission (PR) and 4 very good partial remission (VGPR).10
Antibodies evolve
Another type of immunotherapy that has revolutionized the treatment of hematologic malignancies is monoclonal antibodies (mAbs), targeting antigens on the surface of malignant B and T cells, in particular CD20. The approval of CD20-targeting mAb rituximab in 1997 was the first coup for the development of immunotherapy for the treatment of hematologic malignancies. It has become part of the standard treatment regimen for B-cell malignancies, including NHL and CLL, in combination with various types of chemotherapy.
Several other CD20-targeting antibodies have been developed (Table 2), some of which work in the same way as rituximab (eg, ofatumumab) and some that have a slightly different mechanism of action (eg, obinutuzumab).11 Both types of antibody have proved highly effective; ofatumumab is FDA approved for the treatment of advanced CLL and is being evaluated in phase 3 trials in other hematologic malignancies, while obinutuzumab has received regulatory approval for the first-line treatment of CLL, replacing the standard rituximab-containing regimen.12
The use of ofatumumab as maintenance therapy is supported by the results of the phase 3 PROLONG study in which 474 patients were randomly assigned to ofatumumab maintenance for 2 years or observation. Over a median follow-up of close to 20 months, ofatumumab-treated patients experienced improved progression-free survival (PFS; median PFS: 29.4 months vs 15.2 months; hazard ratio [HR], 0.50; P < .0001).13 Obinutuzumab’s new indication is based on data from the phase 3 GADOLIN trial, in which the obinutuzumab arm showed improved 3-year PFS compared with rituximab.14Until recently, multiple myeloma had proven relatively resistant to mAb therapy, but two new drug targets have dramatically altered the treatment landscape for this type of hematologic malignancy. CD2 subset 1 (CS1), also known as signaling lymphocytic activation molecule 7 (SLAMF7), and CD38 are glycoproteins expressed highly and nearly uniformly on the surface of multiple myeloma cells and only at low levels on other lymphoid and myeloid cells.15
Several antibodies directed at these targets are in clinical development, but daratumumab and elotuzumab, targeting CD38 and CS1, respectively, are both newly approved by the FDA for relapsed/refractory disease, daratumumab as monotherapy and elotuzumab in combination with lenalidomide and dexamethasone.
The indication for daratumumab was subsequently expanded to include its use in combination with lenalidomide plus dexamethasone or bortezomib plus dexamethasone. Support for this new indication came from 2 pivotal phase 3 trials. In the CASTOR trial, the combination of daratumumab with bortezomib–dexamethasone reduced the risk of disease progression or death by 61%, compared with bortezomib–dexamethasone alone, whereas daratumumab with lenalidomide–dexamethasone reduced the risk of disease progression or death by 63% in the POLLUX trial.16,17
Numerous clinical trials for both drugs are ongoing, including in the front-line setting in multiple myeloma, as well as trials in other types of B-cell malignancy, and several other CD38-targeting mAbs are also in development, including isatuximab, which has reached the phase 3 stage (NCT02990338).
Innovative design
Newer drug designs, which have sought to take mAb therapy to the next level, have also shown significant efficacy in hematologic malignancies. Antibody-drug conjugates (ADCs) combine the cytotoxic efficacy of chemotherapeutic agents with the specificity of a mAb targeting a tumor-specific antigen. This essentially creates a targeted payload that improves upon the efficacy of mAb monotherapy but mitigates some of the side effects of chemotherapy related to their indiscriminate killing of both cancerous and healthy cells.
The development of ADCs has been somewhat of a rollercoaster ride, with the approval and subsequent withdrawal of the first-in-class drug gemtuzumab ozogamicin in 2010, but the field was reinvigorated with the successful development of brentuximab vedotin, which targets the CD30 antigen and is approved for the treatment of multiple different hematologic malignancies, including, most recently, for posttransplant consolidation therapy in patients with Hodgkin lymphoma at high risk of relapse or progression.18
Brentuximab vedotin may soon be joined by another FDA-approved ADC, this one targeting CD22. Inotuzumab ozogamicin was recently granted priority review for the treatment of relapsed/refractory ALL. The FDA is reviewing data from the phase 3 INO-VATE study in which inotuzumab ozogamicin reduced the risk of disease progression or death by 55% compared with standard therapy, and a decision is expected by August.19 Other ADC targets being investigated in clinical trials include CD138, CD19, and CD33 (Table 3). Meanwhile, a meta-analysis of randomized trials suggested that the withdrawal of gemtuzumab ozogamicin may have been premature, indicating that it does improve long-term overall survival (OS) and reduces the risk of relapse.20
Bispecific antibodies that link natural killer (NK) cells to tumor cells, by targeting the NK-cell receptor CD16, known as BiKEs, are also in development in an attempt to harness the power of the innate immune response.
B-cell signaling a ripe target
Beyond immunotherapy, molecularly targeted drugs directed against key drivers of hematologic malignancies are also showing great promise. In particular, the B-cell receptor (BCR) signaling pathway, a central regulator of B-cell function, and its constituent kinases that are frequently dysregulated in B cell malignancies, has emerged as an exciting therapeutic avenue.
A variety of small molecule inhibitors targeting different nodes of the BCR pathway have been developed (Table 4), but the greatest success to date has been achieved with drugs targeting Bruton’s tyrosine kinase (BTK). Their clinical development culminated in the approval of ibrutinib for the treatment of patients with mantle cell lymphoma in 2013 and subsequently for patients with CLL, Waldenström macroglobulinemia, and most recently for patients with marginal zone lymphoma.
More than 100 clinical trials of ibrutinib are ongoing in an effort to further clarify its role in a variety of different disease settings. Furthermore, in an effort to address some of the toxicity concerns with ibrutinib, more specific BTK inhibitors are also being developed.
Other kinases that orchestrate the BCR pathway, including phosphatidylinositol-3-kinase (PI3K) and SYK, are also being targeted. The delta isoform of PI3K is expressed exclusively in hematopoietic cells and a number of PI3K delta inhibitors have been developed. Idelalisib received regulatory approval for the treatment of patients with CLL in combination with rituximab, and for patients with follicular lymphoma and small lymphocytic leukemia.
As with ibrutinib, a plethora of clinical trials are ongoing, however a major setback was suffered in the frontline setting when Gilead Sciences halted 6 clinical trials due to reports of increased rates of adverse events, including deaths.26 Meanwhile, SYK inhibitors have lagged behind somewhat in their development, but one such offering, entospletinib, is showing promise in patients with AML.27
Finally, there has been some success in targeting one of the downstream targets of the BCR signaling pathway, the Bcl2 protein that is involved in the regulation of apoptosis. Venetoclax was approved last year for the treatment of patients with relapsed/refractory CLL in patients who have a chromosome 17p deletion, based on the demonstration of impressive, durable responses.28
1. Bachireddy P, Burkhardt UE, Rajasagi M, Wu CJ. Haemato- logical malignancies: at the forefront of immunotherapeutic innovation. Nat Rev Cancer. 2015;15(4):201-215.
2. Im A, Pavletic SZ. Immunotherapy in hematologic malignancies: past, present, and future. J Hematol Oncol. 2017;10(1):94.
3. Gill S. Planes, trains, and automobiles: perspectives on CAR T cells and other cellular therapies for hematologic malignancies. Curr Hematol Malig Rep. 2016;11(4):318-325.
4. Ye B, Stary CM, Gao Q, et al. Genetically modified T-cell-based adoptive immunotherapy in hematological malignancies. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5237740/. Published January 2, 2017. Accessed July 22, 2017.
5. Sharpe M, Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
6. Novartis. Novartis personalized cell therapy CTL019 receives FDA breakthrough therapy designation. https://www.novartis.com/news/media-releases/novartis-personalized-cell-therapy-ctl019-receivesfda-breakthrough-therapy. Published July 7, 2014. Accessed June 19,
2017.
7. Novartis. Novartis presents results from first global registration trial of CTL019 in pediatric and young adult patients with r/r B-ALL. https://www.novartis.com/news/media-releases/novartis-presentsresults-first-global-registration-trial-ctl019-pediatric-and. Published December 4, 2016. Accessed June 19, 2017.
8. Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 Results of ZUMA1: a multicenter study of KTE-C19 Anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25(1):285-295.
9. Abramson JS, Palomba L, Gordon L. Transcend NHL 001: immunotherapy with the CD19-Directd CAR T-cell product JCAR017 results in high complete response rates in relapsed or refractory B-cell non-Hodgkin lymphoma. Paper presented at 58th American Society of Hematology Annual Meeting; December 3-6, 2016; San Diego, CA.
10. Fan F, Zhao W, Liu J, et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. J Clin Oncol. 2017;35(suppl;):Abstr LBA3001.
11. Okroj M, Osterborg A, Blom AM. Effector mechanisms of anti-CD20 monoclonal antibodies in B cell malignancies. Cancer Treat Rev. 2013;39(6):632-639.
12. Safdari Y, Ahmadzadeh V, Farajnia S. CD20-targeting in B-cell malignancies: novel prospects for antibodies and combination therapies. Invest New Drugs. 2016;34(4):497-512.
13. van Oers MH, Kuliczkowski K, Smolej L, et al. Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): an open-label, multicentre, randomised phase 3 study. Lancet Oncol. 2015;16(13):1370-1379.
14. Sehn LH, Chua N, Mayer J, et al. Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): a randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol. 2016;17(8):1081-1093.
15. Touzeau C, Moreau P, Dumontet C. Monoclonal antibody therapy in multiple myeloma. Leukemia. 2017;31(5):1039-1047.
16. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754-766.
17. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319-1331.
18. Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315-337.
19. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740-753.
20. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
21. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93(3):290-296.
22. Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836-847.
23. Koehrer S, Burger JA. B-cell receptor signaling in chronic lymphocytic leukemia and other B-cell malignancies. Clin Adv Hematol Oncol. 2016;14(1):55-65.
24. Seda V, Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol. 2015;94(3):193-205.
25. Bojarczuk K, Bobrowicz M, Dwojak M, et al. B-cell receptor signaling in the pathogenesis of lymphoid malignancies. Blood Cells Mol Dis. 2015;55(3):255-265.
26. Medscape Medical News. Gilead stops six trials adding idelalisib to other drugs. http://www.medscape.com/viewarticle/860372. Published March 14, 2016. Accessed June 19, 2017.
27. Sharman J, Di Paolo J. Targeting B-cell receptor signaling kinases in chronic lymphocytic leukemia: the promise of entospletinib. Ther Adv Hematol. 2016;7(3):157-170.
28. Food and Drug Administration. FDA approves new drug for chronic lymphocytic leukemia in patients with a specific chromosomal abnormality. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm495253.htm. Released April 11, 2016. Accessed June 19, 2017.
1. Bachireddy P, Burkhardt UE, Rajasagi M, Wu CJ. Haemato- logical malignancies: at the forefront of immunotherapeutic innovation. Nat Rev Cancer. 2015;15(4):201-215.
2. Im A, Pavletic SZ. Immunotherapy in hematologic malignancies: past, present, and future. J Hematol Oncol. 2017;10(1):94.
3. Gill S. Planes, trains, and automobiles: perspectives on CAR T cells and other cellular therapies for hematologic malignancies. Curr Hematol Malig Rep. 2016;11(4):318-325.
4. Ye B, Stary CM, Gao Q, et al. Genetically modified T-cell-based adoptive immunotherapy in hematological malignancies. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5237740/. Published January 2, 2017. Accessed July 22, 2017.
5. Sharpe M, Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
6. Novartis. Novartis personalized cell therapy CTL019 receives FDA breakthrough therapy designation. https://www.novartis.com/news/media-releases/novartis-personalized-cell-therapy-ctl019-receivesfda-breakthrough-therapy. Published July 7, 2014. Accessed June 19,
2017.
7. Novartis. Novartis presents results from first global registration trial of CTL019 in pediatric and young adult patients with r/r B-ALL. https://www.novartis.com/news/media-releases/novartis-presentsresults-first-global-registration-trial-ctl019-pediatric-and. Published December 4, 2016. Accessed June 19, 2017.
8. Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 Results of ZUMA1: a multicenter study of KTE-C19 Anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25(1):285-295.
9. Abramson JS, Palomba L, Gordon L. Transcend NHL 001: immunotherapy with the CD19-Directd CAR T-cell product JCAR017 results in high complete response rates in relapsed or refractory B-cell non-Hodgkin lymphoma. Paper presented at 58th American Society of Hematology Annual Meeting; December 3-6, 2016; San Diego, CA.
10. Fan F, Zhao W, Liu J, et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. J Clin Oncol. 2017;35(suppl;):Abstr LBA3001.
11. Okroj M, Osterborg A, Blom AM. Effector mechanisms of anti-CD20 monoclonal antibodies in B cell malignancies. Cancer Treat Rev. 2013;39(6):632-639.
12. Safdari Y, Ahmadzadeh V, Farajnia S. CD20-targeting in B-cell malignancies: novel prospects for antibodies and combination therapies. Invest New Drugs. 2016;34(4):497-512.
13. van Oers MH, Kuliczkowski K, Smolej L, et al. Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): an open-label, multicentre, randomised phase 3 study. Lancet Oncol. 2015;16(13):1370-1379.
14. Sehn LH, Chua N, Mayer J, et al. Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): a randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol. 2016;17(8):1081-1093.
15. Touzeau C, Moreau P, Dumontet C. Monoclonal antibody therapy in multiple myeloma. Leukemia. 2017;31(5):1039-1047.
16. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754-766.
17. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319-1331.
18. Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315-337.
19. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740-753.
20. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
21. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93(3):290-296.
22. Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836-847.
23. Koehrer S, Burger JA. B-cell receptor signaling in chronic lymphocytic leukemia and other B-cell malignancies. Clin Adv Hematol Oncol. 2016;14(1):55-65.
24. Seda V, Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol. 2015;94(3):193-205.
25. Bojarczuk K, Bobrowicz M, Dwojak M, et al. B-cell receptor signaling in the pathogenesis of lymphoid malignancies. Blood Cells Mol Dis. 2015;55(3):255-265.
26. Medscape Medical News. Gilead stops six trials adding idelalisib to other drugs. http://www.medscape.com/viewarticle/860372. Published March 14, 2016. Accessed June 19, 2017.
27. Sharman J, Di Paolo J. Targeting B-cell receptor signaling kinases in chronic lymphocytic leukemia: the promise of entospletinib. Ther Adv Hematol. 2016;7(3):157-170.
28. Food and Drug Administration. FDA approves new drug for chronic lymphocytic leukemia in patients with a specific chromosomal abnormality. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm495253.htm. Released April 11, 2016. Accessed June 19, 2017.
Study links communication, outcomes in cancer

Better communication between cancer patients and healthcare providers may provide tangible benefits, according to research published in JNCCN.
Cancer survivors who reported greater satisfaction in communicating with healthcare providers had better general health and mental health, fewer doctor visits, and reduced healthcare spending, when compared to patients who were less satisfied with communication.
“Our study suggests that when cancer care providers are more effective communicators, their patients are more likely to follow medical advice and medication protocols,” said study author Ashish Rai, PhD, of the American Cancer Society in Framingham, Massachusetts.
For this study, Dr Rai and his colleagues analyzed data from the Medical Expenditure Panel Survey (MEPS) from 2008 through 2014.
The researchers evaluated 4588 cancer survivors, dividing them into non-elderly and elderly groups. The non-elderly patients (n=2257) had a median age of 54 (range, 18-64), and the elderly patients (n=2331) had a median age of 75.
Communication satisfaction was measured by the Consumer Assessment of Healthcare Providers and Systems (CAHPS), in conjunction with the MEPS data.
Patients used a 4-point scale ranging from “never” to “always” to track whether their providers did the following:
- Listened carefully
- Explained things in a way that was easy to understand
- Showed respect for what the respondent had to say
- Spent enough time with the respondent.
A global satisfaction rating scale (0 to 10) was factored into a composite score and tracked across 12 months.
The researchers then assessed various patient outcomes.
Satisfaction and outcomes
Overall, patients who were the most satisfied with communication had the best outcomes with regard to general, physical, and mental health; fewer emergency department, hospital, and office visits; and reduced drug, out-of-pocket, and total healthcare expenditures.
However, the associations between communication satisfaction and outcomes were not always significant.
In an adjusted analysis, the elderly patients who were more satisfied with communication in year 1 had significantly better outcomes in year 2 for general health, mental health, and total healthcare expenditures.
| Elderly patients 65+ | |||
| Least satisfied
(tertile 1) |
Moderately satisfied (tertile 2) | Most satisfied
(tertile 3) |
|
| Excellent/very good general health | |||
| Unadjusted proportion | 23.6% | 31.8% | 45.8% |
| Predictive margin | 30.3
(95% CI 26.0–34.6) |
32.2
(95% CI 28.9–35.5) |
38.9
(95% CI 35.1–42.7) |
| P value | Reference | 0.466 | 0.007 |
| Highest quartile of mental health | |||
| Unadjusted proportion | 22.9% | 34.8% | 41.7% |
| Predictive margin | 27.1
(95% CI 22.1–32.1) |
35.5
(95% CI 31.5–39.5) |
37.0
(95% CI 32.7–41.4) |
| P value | Reference | 0.013 | 0.005 |
| Total healthcare expenditure | |||
| Unadjusted mean | $33,558 | $27,341 | $29,591 |
| Predictive margin | $34,071 ($29,011–$39,131) | $28,230 ($22,907–$33,553) | $26,995 ($22,568–$31,422) |
| P value | Reference | 0.301 | 0.049 |
For the non-elderly patients, those who were more satisfied with communication in year 1 had significantly better outcomes in year 2 for physician office visits and mental health.
| Non-elderly patients (18-64) | |||
| Least satisfied (tertile 1) | Moderately satisfied (tertile 2) | Most satisfied (tertile 3) | |
| Total physician office visits | |||
| Unadjusted mean | 7.96 | 6.96 | 5.85 |
| Predictive margin | 7.42
(95% CI 6.78–8.06) |
6.60
(95% CI 5.98–7.22) |
6.26
(95% CI 5.47–7.05) |
| P value | Reference | 0.211 | 0.038 |
| Highest quartile of mental health | |||
| Unadjusted proportion | 23.5% | 35.5% | 41.1% |
| Predictive margin | 29.7
(95% CI 25.3–34.1) |
36.0
(95% CI 31.3–40.7) |
34.0
(95% CI 29.5–38.4) |
| P value | Reference | 0.036 | 0.187 |
Baseline health and satisfaction
In both age groups, patients with better baseline health reported higher satisfaction with communication. Conversely, the more comorbidities patients had, the lower their satisfaction rating.
The researchers said this suggests that more complex circumstances negatively impacted patients’ perception of their communication, and the finding highlights the importance of coordinating care across a team of providers.
“The results of this study present an interesting challenge: those survivors most in need of good communication about complex medical issues may not be receiving the information they seek in a manner that they find helpful. That, in turn, results in higher healthcare utilization and expenditure,” said Crystal Denlinger, MD, of Fox Chase Cancer Center in Philadelphia, Pennsylvania, who was not involved in this study.
“This could be due to many factors, including time constraints, competing priorities, and increasingly complex cancer therapies. This study highlights the need for additional research into how to tailor the healthcare experience both during and after cancer treatment in order to communicate more effectively.”
Conclusions
“Communication needs vary from patient to patient,” Dr Rai noted. “While time constraints do pose a challenge, the amount of time spent is only one of the attributes of effective communication. By tailoring their communication strategy to a patient’s specific needs, providers may be able to communicate more effectively in the same amount of time.”
Dr Rai also pointed out the importance of delegating both clinical and communication duties as needed. Dr Rai and his colleagues also cited earlier research demonstrating better outcomes for patients who had the option of communicating with their provider electronically.1,2
Ultimately, the researchers concluded that effective provider communication can improve outcomes by streamlining care, alleviating anxiety, boosting mutual trust, and increasing treatment adherence.
1. Basch E, Deal AM, Dueck AC, et al. Overall survival results of a trial assessing patient-reported outcomes for symptom monitoring during routine cancer treatment. JAMA 2017;318:197–198.
2. Smith AB, Basch E. Role of patient-reported outcomes in postsurgical monitoring in oncology. J Oncol Pract 2017;13:535–538.
Better communication between cancer patients and healthcare providers may provide tangible benefits, according to research published in JNCCN.
Cancer survivors who reported greater satisfaction in communicating with healthcare providers had better general health and mental health, fewer doctor visits, and reduced healthcare spending, when compared to patients who were less satisfied with communication.
“Our study suggests that when cancer care providers are more effective communicators, their patients are more likely to follow medical advice and medication protocols,” said study author Ashish Rai, PhD, of the American Cancer Society in Framingham, Massachusetts.
For this study, Dr Rai and his colleagues analyzed data from the Medical Expenditure Panel Survey (MEPS) from 2008 through 2014.
The researchers evaluated 4588 cancer survivors, dividing them into non-elderly and elderly groups. The non-elderly patients (n=2257) had a median age of 54 (range, 18-64), and the elderly patients (n=2331) had a median age of 75.
Communication satisfaction was measured by the Consumer Assessment of Healthcare Providers and Systems (CAHPS), in conjunction with the MEPS data.
Patients used a 4-point scale ranging from “never” to “always” to track whether their providers did the following:
- Listened carefully
- Explained things in a way that was easy to understand
- Showed respect for what the respondent had to say
- Spent enough time with the respondent.
A global satisfaction rating scale (0 to 10) was factored into a composite score and tracked across 12 months.
The researchers then assessed various patient outcomes.
Satisfaction and outcomes
Overall, patients who were the most satisfied with communication had the best outcomes with regard to general, physical, and mental health; fewer emergency department, hospital, and office visits; and reduced drug, out-of-pocket, and total healthcare expenditures.
However, the associations between communication satisfaction and outcomes were not always significant.
In an adjusted analysis, the elderly patients who were more satisfied with communication in year 1 had significantly better outcomes in year 2 for general health, mental health, and total healthcare expenditures.
| Elderly patients 65+ | |||
| Least satisfied
(tertile 1) |
Moderately satisfied (tertile 2) | Most satisfied
(tertile 3) |
|
| Excellent/very good general health | |||
| Unadjusted proportion | 23.6% | 31.8% | 45.8% |
| Predictive margin | 30.3
(95% CI 26.0–34.6) |
32.2
(95% CI 28.9–35.5) |
38.9
(95% CI 35.1–42.7) |
| P value | Reference | 0.466 | 0.007 |
| Highest quartile of mental health | |||
| Unadjusted proportion | 22.9% | 34.8% | 41.7% |
| Predictive margin | 27.1
(95% CI 22.1–32.1) |
35.5
(95% CI 31.5–39.5) |
37.0
(95% CI 32.7–41.4) |
| P value | Reference | 0.013 | 0.005 |
| Total healthcare expenditure | |||
| Unadjusted mean | $33,558 | $27,341 | $29,591 |
| Predictive margin | $34,071 ($29,011–$39,131) | $28,230 ($22,907–$33,553) | $26,995 ($22,568–$31,422) |
| P value | Reference | 0.301 | 0.049 |
For the non-elderly patients, those who were more satisfied with communication in year 1 had significantly better outcomes in year 2 for physician office visits and mental health.
| Non-elderly patients (18-64) | |||
| Least satisfied (tertile 1) | Moderately satisfied (tertile 2) | Most satisfied (tertile 3) | |
| Total physician office visits | |||
| Unadjusted mean | 7.96 | 6.96 | 5.85 |
| Predictive margin | 7.42
(95% CI 6.78–8.06) |
6.60
(95% CI 5.98–7.22) |
6.26
(95% CI 5.47–7.05) |
| P value | Reference | 0.211 | 0.038 |
| Highest quartile of mental health | |||
| Unadjusted proportion | 23.5% | 35.5% | 41.1% |
| Predictive margin | 29.7
(95% CI 25.3–34.1) |
36.0
(95% CI 31.3–40.7) |
34.0
(95% CI 29.5–38.4) |
| P value | Reference | 0.036 | 0.187 |
Baseline health and satisfaction
In both age groups, patients with better baseline health reported higher satisfaction with communication. Conversely, the more comorbidities patients had, the lower their satisfaction rating.
The researchers said this suggests that more complex circumstances negatively impacted patients’ perception of their communication, and the finding highlights the importance of coordinating care across a team of providers.
“The results of this study present an interesting challenge: those survivors most in need of good communication about complex medical issues may not be receiving the information they seek in a manner that they find helpful. That, in turn, results in higher healthcare utilization and expenditure,” said Crystal Denlinger, MD, of Fox Chase Cancer Center in Philadelphia, Pennsylvania, who was not involved in this study.
“This could be due to many factors, including time constraints, competing priorities, and increasingly complex cancer therapies. This study highlights the need for additional research into how to tailor the healthcare experience both during and after cancer treatment in order to communicate more effectively.”
Conclusions
“Communication needs vary from patient to patient,” Dr Rai noted. “While time constraints do pose a challenge, the amount of time spent is only one of the attributes of effective communication. By tailoring their communication strategy to a patient’s specific needs, providers may be able to communicate more effectively in the same amount of time.”
Dr Rai also pointed out the importance of delegating both clinical and communication duties as needed. Dr Rai and his colleagues also cited earlier research demonstrating better outcomes for patients who had the option of communicating with their provider electronically.1,2
Ultimately, the researchers concluded that effective provider communication can improve outcomes by streamlining care, alleviating anxiety, boosting mutual trust, and increasing treatment adherence.
1. Basch E, Deal AM, Dueck AC, et al. Overall survival results of a trial assessing patient-reported outcomes for symptom monitoring during routine cancer treatment. JAMA 2017;318:197–198.
2. Smith AB, Basch E. Role of patient-reported outcomes in postsurgical monitoring in oncology. J Oncol Pract 2017;13:535–538.
Better communication between cancer patients and healthcare providers may provide tangible benefits, according to research published in JNCCN.
Cancer survivors who reported greater satisfaction in communicating with healthcare providers had better general health and mental health, fewer doctor visits, and reduced healthcare spending, when compared to patients who were less satisfied with communication.
“Our study suggests that when cancer care providers are more effective communicators, their patients are more likely to follow medical advice and medication protocols,” said study author Ashish Rai, PhD, of the American Cancer Society in Framingham, Massachusetts.
For this study, Dr Rai and his colleagues analyzed data from the Medical Expenditure Panel Survey (MEPS) from 2008 through 2014.
The researchers evaluated 4588 cancer survivors, dividing them into non-elderly and elderly groups. The non-elderly patients (n=2257) had a median age of 54 (range, 18-64), and the elderly patients (n=2331) had a median age of 75.
Communication satisfaction was measured by the Consumer Assessment of Healthcare Providers and Systems (CAHPS), in conjunction with the MEPS data.
Patients used a 4-point scale ranging from “never” to “always” to track whether their providers did the following:
- Listened carefully
- Explained things in a way that was easy to understand
- Showed respect for what the respondent had to say
- Spent enough time with the respondent.
A global satisfaction rating scale (0 to 10) was factored into a composite score and tracked across 12 months.
The researchers then assessed various patient outcomes.
Satisfaction and outcomes
Overall, patients who were the most satisfied with communication had the best outcomes with regard to general, physical, and mental health; fewer emergency department, hospital, and office visits; and reduced drug, out-of-pocket, and total healthcare expenditures.
However, the associations between communication satisfaction and outcomes were not always significant.
In an adjusted analysis, the elderly patients who were more satisfied with communication in year 1 had significantly better outcomes in year 2 for general health, mental health, and total healthcare expenditures.
| Elderly patients 65+ | |||
| Least satisfied
(tertile 1) |
Moderately satisfied (tertile 2) | Most satisfied
(tertile 3) |
|
| Excellent/very good general health | |||
| Unadjusted proportion | 23.6% | 31.8% | 45.8% |
| Predictive margin | 30.3
(95% CI 26.0–34.6) |
32.2
(95% CI 28.9–35.5) |
38.9
(95% CI 35.1–42.7) |
| P value | Reference | 0.466 | 0.007 |
| Highest quartile of mental health | |||
| Unadjusted proportion | 22.9% | 34.8% | 41.7% |
| Predictive margin | 27.1
(95% CI 22.1–32.1) |
35.5
(95% CI 31.5–39.5) |
37.0
(95% CI 32.7–41.4) |
| P value | Reference | 0.013 | 0.005 |
| Total healthcare expenditure | |||
| Unadjusted mean | $33,558 | $27,341 | $29,591 |
| Predictive margin | $34,071 ($29,011–$39,131) | $28,230 ($22,907–$33,553) | $26,995 ($22,568–$31,422) |
| P value | Reference | 0.301 | 0.049 |
For the non-elderly patients, those who were more satisfied with communication in year 1 had significantly better outcomes in year 2 for physician office visits and mental health.
| Non-elderly patients (18-64) | |||
| Least satisfied (tertile 1) | Moderately satisfied (tertile 2) | Most satisfied (tertile 3) | |
| Total physician office visits | |||
| Unadjusted mean | 7.96 | 6.96 | 5.85 |
| Predictive margin | 7.42
(95% CI 6.78–8.06) |
6.60
(95% CI 5.98–7.22) |
6.26
(95% CI 5.47–7.05) |
| P value | Reference | 0.211 | 0.038 |
| Highest quartile of mental health | |||
| Unadjusted proportion | 23.5% | 35.5% | 41.1% |
| Predictive margin | 29.7
(95% CI 25.3–34.1) |
36.0
(95% CI 31.3–40.7) |
34.0
(95% CI 29.5–38.4) |
| P value | Reference | 0.036 | 0.187 |
Baseline health and satisfaction
In both age groups, patients with better baseline health reported higher satisfaction with communication. Conversely, the more comorbidities patients had, the lower their satisfaction rating.
The researchers said this suggests that more complex circumstances negatively impacted patients’ perception of their communication, and the finding highlights the importance of coordinating care across a team of providers.
“The results of this study present an interesting challenge: those survivors most in need of good communication about complex medical issues may not be receiving the information they seek in a manner that they find helpful. That, in turn, results in higher healthcare utilization and expenditure,” said Crystal Denlinger, MD, of Fox Chase Cancer Center in Philadelphia, Pennsylvania, who was not involved in this study.
“This could be due to many factors, including time constraints, competing priorities, and increasingly complex cancer therapies. This study highlights the need for additional research into how to tailor the healthcare experience both during and after cancer treatment in order to communicate more effectively.”
Conclusions
“Communication needs vary from patient to patient,” Dr Rai noted. “While time constraints do pose a challenge, the amount of time spent is only one of the attributes of effective communication. By tailoring their communication strategy to a patient’s specific needs, providers may be able to communicate more effectively in the same amount of time.”
Dr Rai also pointed out the importance of delegating both clinical and communication duties as needed. Dr Rai and his colleagues also cited earlier research demonstrating better outcomes for patients who had the option of communicating with their provider electronically.1,2
Ultimately, the researchers concluded that effective provider communication can improve outcomes by streamlining care, alleviating anxiety, boosting mutual trust, and increasing treatment adherence.
1. Basch E, Deal AM, Dueck AC, et al. Overall survival results of a trial assessing patient-reported outcomes for symptom monitoring during routine cancer treatment. JAMA 2017;318:197–198.
2. Smith AB, Basch E. Role of patient-reported outcomes in postsurgical monitoring in oncology. J Oncol Pract 2017;13:535–538.
Drug under priority review for BPDCN

The US Food and Drug Administration(FDA) has accepted for priority review the biologics license application seeking approval for tagraxofusp (Elzonris, SL-401) to treat blastic plasmacytoid dendritic cell neoplasm (BPDCN).
The FDA expects to make a decision on this application by February 21, 2019.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency intends to take action on a priority review application within 6 months of receiving it rather than the standard 10 months.
About tagraxofusp
Tagraxofusp is a targeted therapy directed to CD123, a cell surface receptor expressed on a range of malignancies. The drug is being developed by Stemline Therapeutics, Inc.
In addition to priority review, tagraxofusp has breakthrough therapy designation and orphan drug designation from the FDA.
Tagraxofusp has produced favorable early results in a phase 2 trial of patients with BPDCN. Results from this trial were presented at the 23rd Congress of the European Hematology Association (EHA) in June.
Results were presented for 45 patients—32 with previously untreated BPDCN and 13 with relapsed/refractory BPDCN.
Three patients received tagraxofusp at 7 μg/kg/day on days 1 to 5 of a 21-day cycle, and the rest received the drug at 12 μg/kg on days 1 to 5 of a 21-day cycle.
Among patients who received the 12 μg/kg/day dose, the overall response rate was 83% (35/42). The overall response rate was 90% (26/29) in the previously untreated patients and 69% (9/13) in relapsed/refractory patients.
The composite complete response rate was 62% (n=26) overall, 72% (n=21) in previously untreated patients, and 38% (n=5) in relapsed/refractory patients.
Fourteen patients went on to stem cell transplant, 1 of whom had relapsed/refractory disease at baseline.
Overall survival results were only available for the 29 previously untreated patients who received tagraxofusp at 12 μg/kg/day. In this group, the median overall survival had not been reached at a median follow-up of 13.8 months (range, 0.2 to 37.4 months).
Safety results were presented for 114 patients who have received tagraxofusp at 12 μg/kg/day on all trials of the drug. These data include patients with diseases other than BPDCN, although adverse events (AEs) were similar regardless of disease.
Common treatment-related AEs (of any grade, occurring in at least 15% of patients) included hypoalbuminemia (49%), ALT increase (48%), AST increase (48%), thrombocytopenia (29%), nausea (27%), pyrexia (25%), chills (23%), fatigue (23%), weight increase (19%), hypotension (18%), peripheral edema (17%), and vomiting (15%).
Another common AE was capillary leak syndrome (CLS), which occurred in 20% of patients (n=23). Most cases of CLS were grade 1 or 2, but there were grade 3 (n=5) and 4 (n=2) cases, as well as a single case of grade 5 CLS that occurred in a BPDCN patient.
Researchers found they could manage the CLS with monitoring and protocol adjustments.
The US Food and Drug Administration(FDA) has accepted for priority review the biologics license application seeking approval for tagraxofusp (Elzonris, SL-401) to treat blastic plasmacytoid dendritic cell neoplasm (BPDCN).
The FDA expects to make a decision on this application by February 21, 2019.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency intends to take action on a priority review application within 6 months of receiving it rather than the standard 10 months.
About tagraxofusp
Tagraxofusp is a targeted therapy directed to CD123, a cell surface receptor expressed on a range of malignancies. The drug is being developed by Stemline Therapeutics, Inc.
In addition to priority review, tagraxofusp has breakthrough therapy designation and orphan drug designation from the FDA.
Tagraxofusp has produced favorable early results in a phase 2 trial of patients with BPDCN. Results from this trial were presented at the 23rd Congress of the European Hematology Association (EHA) in June.
Results were presented for 45 patients—32 with previously untreated BPDCN and 13 with relapsed/refractory BPDCN.
Three patients received tagraxofusp at 7 μg/kg/day on days 1 to 5 of a 21-day cycle, and the rest received the drug at 12 μg/kg on days 1 to 5 of a 21-day cycle.
Among patients who received the 12 μg/kg/day dose, the overall response rate was 83% (35/42). The overall response rate was 90% (26/29) in the previously untreated patients and 69% (9/13) in relapsed/refractory patients.
The composite complete response rate was 62% (n=26) overall, 72% (n=21) in previously untreated patients, and 38% (n=5) in relapsed/refractory patients.
Fourteen patients went on to stem cell transplant, 1 of whom had relapsed/refractory disease at baseline.
Overall survival results were only available for the 29 previously untreated patients who received tagraxofusp at 12 μg/kg/day. In this group, the median overall survival had not been reached at a median follow-up of 13.8 months (range, 0.2 to 37.4 months).
Safety results were presented for 114 patients who have received tagraxofusp at 12 μg/kg/day on all trials of the drug. These data include patients with diseases other than BPDCN, although adverse events (AEs) were similar regardless of disease.
Common treatment-related AEs (of any grade, occurring in at least 15% of patients) included hypoalbuminemia (49%), ALT increase (48%), AST increase (48%), thrombocytopenia (29%), nausea (27%), pyrexia (25%), chills (23%), fatigue (23%), weight increase (19%), hypotension (18%), peripheral edema (17%), and vomiting (15%).
Another common AE was capillary leak syndrome (CLS), which occurred in 20% of patients (n=23). Most cases of CLS were grade 1 or 2, but there were grade 3 (n=5) and 4 (n=2) cases, as well as a single case of grade 5 CLS that occurred in a BPDCN patient.
Researchers found they could manage the CLS with monitoring and protocol adjustments.
The US Food and Drug Administration(FDA) has accepted for priority review the biologics license application seeking approval for tagraxofusp (Elzonris, SL-401) to treat blastic plasmacytoid dendritic cell neoplasm (BPDCN).
The FDA expects to make a decision on this application by February 21, 2019.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency intends to take action on a priority review application within 6 months of receiving it rather than the standard 10 months.
About tagraxofusp
Tagraxofusp is a targeted therapy directed to CD123, a cell surface receptor expressed on a range of malignancies. The drug is being developed by Stemline Therapeutics, Inc.
In addition to priority review, tagraxofusp has breakthrough therapy designation and orphan drug designation from the FDA.
Tagraxofusp has produced favorable early results in a phase 2 trial of patients with BPDCN. Results from this trial were presented at the 23rd Congress of the European Hematology Association (EHA) in June.
Results were presented for 45 patients—32 with previously untreated BPDCN and 13 with relapsed/refractory BPDCN.
Three patients received tagraxofusp at 7 μg/kg/day on days 1 to 5 of a 21-day cycle, and the rest received the drug at 12 μg/kg on days 1 to 5 of a 21-day cycle.
Among patients who received the 12 μg/kg/day dose, the overall response rate was 83% (35/42). The overall response rate was 90% (26/29) in the previously untreated patients and 69% (9/13) in relapsed/refractory patients.
The composite complete response rate was 62% (n=26) overall, 72% (n=21) in previously untreated patients, and 38% (n=5) in relapsed/refractory patients.
Fourteen patients went on to stem cell transplant, 1 of whom had relapsed/refractory disease at baseline.
Overall survival results were only available for the 29 previously untreated patients who received tagraxofusp at 12 μg/kg/day. In this group, the median overall survival had not been reached at a median follow-up of 13.8 months (range, 0.2 to 37.4 months).
Safety results were presented for 114 patients who have received tagraxofusp at 12 μg/kg/day on all trials of the drug. These data include patients with diseases other than BPDCN, although adverse events (AEs) were similar regardless of disease.
Common treatment-related AEs (of any grade, occurring in at least 15% of patients) included hypoalbuminemia (49%), ALT increase (48%), AST increase (48%), thrombocytopenia (29%), nausea (27%), pyrexia (25%), chills (23%), fatigue (23%), weight increase (19%), hypotension (18%), peripheral edema (17%), and vomiting (15%).
Another common AE was capillary leak syndrome (CLS), which occurred in 20% of patients (n=23). Most cases of CLS were grade 1 or 2, but there were grade 3 (n=5) and 4 (n=2) cases, as well as a single case of grade 5 CLS that occurred in a BPDCN patient.
Researchers found they could manage the CLS with monitoring and protocol adjustments.
Auto-HSCT linked to higher AML, MDS risk

Patients undergoing autologous hematopoietic stem cell transplant (auto-HSCT) for lymphoma or myeloma have an increased risk of acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS), according to a retrospective study.
The study suggested these patients have 10 to 100 times the risk of AML or MDS as the general population.
The elevated risk also exceeds that of similar lymphoma and myeloma patients largely untreated with auto-HSCT.
Tomas Radivoyevitch, PhD, of the Cleveland Clinic Foundation in Ohio, and his colleagues reported these findings in Leukemia Research.
The investigators noted that exposure to DNA-damaging drugs and ionizing radiation—both used in auto-HSCT—is known to increase the risk of AML and MDS.
With this in mind, the team analyzed data on auto-HSCT recipients reported to the Center for International Blood and Marrow Transplant Research (CIBMTR).
Analyses were based on 9028 patients undergoing auto-HSCT from 1995 to 2010 for Hodgkin lymphoma (n=916), non-Hodgkin lymphoma (NHL, n=3546), or plasma cell myeloma (n=4566). Their median duration of follow-up was 90 months, 110 months, and 97 months, respectively.
Overall, 3.7% of the cohort developed AML or MDS after their transplant.
More aggressive transplant protocols increased the likelihood of this outcome. The risk of developing AML or MDS was higher for:
- Hodgkin lymphoma patients who received conditioning with total body radiation versus chemotherapy alone (hazard ratio [HR], 4.0)
- NHL patients who received conditioning with total body radiation (HR, 1.7) or with busulfan and melphalan or cyclophosphamide (HR, 1.8) versus the BEAM regimen (bischloroethylnitrosourea, etoposide, cytarabine, and melphalan)
- NHL or myeloma patients who received 3 or more lines of chemotherapy versus 1 line (HR, 1.9 for NHL and 1.8 for myeloma)
- NHL patients who underwent transplant in 2005 to 2010 versus 1995 to 1999 (HR, 2.1).
Patients reported to the Surveillance, Epidemiology and End Results database with the same lymphoma and myeloma diagnoses, few of whom underwent auto-HSCT, had risks of AML and MDS that were 5 to 10 times higher than the background level in the population.
However, the study auto-HSCT cohort had a risk of AML that was 10 to 50 times higher and a relative risk of MDS that was roughly 100 times higher than the background level.
“These increases may be related to exposure to high doses of DNA-damaging drugs given for [auto-HSCT], but this hypothesis can only be tested in a prospective study,” Dr Radivoyevitch and his coinvestigators wrote.
The reason for the greater elevation of MDS risk, compared with AML risk, is unknown.
“One possible explanation is that many cases of MDS evolve to AML, and that earlier diagnosis from increased post-transplant surveillance resulted in a deficiency of AML,” the investigators wrote. “A second is based on steeper MDS versus AML incidences versus age . . . and the possibility that transplantation recipient marrow ages (ie, marrow biological ages) are perhaps decades older than calendar ages.”
The study authors said they had no relevant conflicts of interest. The CIBMTR is supported by several US government agencies and numerous pharmaceutical companies.
Patients undergoing autologous hematopoietic stem cell transplant (auto-HSCT) for lymphoma or myeloma have an increased risk of acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS), according to a retrospective study.
The study suggested these patients have 10 to 100 times the risk of AML or MDS as the general population.
The elevated risk also exceeds that of similar lymphoma and myeloma patients largely untreated with auto-HSCT.
Tomas Radivoyevitch, PhD, of the Cleveland Clinic Foundation in Ohio, and his colleagues reported these findings in Leukemia Research.
The investigators noted that exposure to DNA-damaging drugs and ionizing radiation—both used in auto-HSCT—is known to increase the risk of AML and MDS.
With this in mind, the team analyzed data on auto-HSCT recipients reported to the Center for International Blood and Marrow Transplant Research (CIBMTR).
Analyses were based on 9028 patients undergoing auto-HSCT from 1995 to 2010 for Hodgkin lymphoma (n=916), non-Hodgkin lymphoma (NHL, n=3546), or plasma cell myeloma (n=4566). Their median duration of follow-up was 90 months, 110 months, and 97 months, respectively.
Overall, 3.7% of the cohort developed AML or MDS after their transplant.
More aggressive transplant protocols increased the likelihood of this outcome. The risk of developing AML or MDS was higher for:
- Hodgkin lymphoma patients who received conditioning with total body radiation versus chemotherapy alone (hazard ratio [HR], 4.0)
- NHL patients who received conditioning with total body radiation (HR, 1.7) or with busulfan and melphalan or cyclophosphamide (HR, 1.8) versus the BEAM regimen (bischloroethylnitrosourea, etoposide, cytarabine, and melphalan)
- NHL or myeloma patients who received 3 or more lines of chemotherapy versus 1 line (HR, 1.9 for NHL and 1.8 for myeloma)
- NHL patients who underwent transplant in 2005 to 2010 versus 1995 to 1999 (HR, 2.1).
Patients reported to the Surveillance, Epidemiology and End Results database with the same lymphoma and myeloma diagnoses, few of whom underwent auto-HSCT, had risks of AML and MDS that were 5 to 10 times higher than the background level in the population.
However, the study auto-HSCT cohort had a risk of AML that was 10 to 50 times higher and a relative risk of MDS that was roughly 100 times higher than the background level.
“These increases may be related to exposure to high doses of DNA-damaging drugs given for [auto-HSCT], but this hypothesis can only be tested in a prospective study,” Dr Radivoyevitch and his coinvestigators wrote.
The reason for the greater elevation of MDS risk, compared with AML risk, is unknown.
“One possible explanation is that many cases of MDS evolve to AML, and that earlier diagnosis from increased post-transplant surveillance resulted in a deficiency of AML,” the investigators wrote. “A second is based on steeper MDS versus AML incidences versus age . . . and the possibility that transplantation recipient marrow ages (ie, marrow biological ages) are perhaps decades older than calendar ages.”
The study authors said they had no relevant conflicts of interest. The CIBMTR is supported by several US government agencies and numerous pharmaceutical companies.
Patients undergoing autologous hematopoietic stem cell transplant (auto-HSCT) for lymphoma or myeloma have an increased risk of acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS), according to a retrospective study.
The study suggested these patients have 10 to 100 times the risk of AML or MDS as the general population.
The elevated risk also exceeds that of similar lymphoma and myeloma patients largely untreated with auto-HSCT.
Tomas Radivoyevitch, PhD, of the Cleveland Clinic Foundation in Ohio, and his colleagues reported these findings in Leukemia Research.
The investigators noted that exposure to DNA-damaging drugs and ionizing radiation—both used in auto-HSCT—is known to increase the risk of AML and MDS.
With this in mind, the team analyzed data on auto-HSCT recipients reported to the Center for International Blood and Marrow Transplant Research (CIBMTR).
Analyses were based on 9028 patients undergoing auto-HSCT from 1995 to 2010 for Hodgkin lymphoma (n=916), non-Hodgkin lymphoma (NHL, n=3546), or plasma cell myeloma (n=4566). Their median duration of follow-up was 90 months, 110 months, and 97 months, respectively.
Overall, 3.7% of the cohort developed AML or MDS after their transplant.
More aggressive transplant protocols increased the likelihood of this outcome. The risk of developing AML or MDS was higher for:
- Hodgkin lymphoma patients who received conditioning with total body radiation versus chemotherapy alone (hazard ratio [HR], 4.0)
- NHL patients who received conditioning with total body radiation (HR, 1.7) or with busulfan and melphalan or cyclophosphamide (HR, 1.8) versus the BEAM regimen (bischloroethylnitrosourea, etoposide, cytarabine, and melphalan)
- NHL or myeloma patients who received 3 or more lines of chemotherapy versus 1 line (HR, 1.9 for NHL and 1.8 for myeloma)
- NHL patients who underwent transplant in 2005 to 2010 versus 1995 to 1999 (HR, 2.1).
Patients reported to the Surveillance, Epidemiology and End Results database with the same lymphoma and myeloma diagnoses, few of whom underwent auto-HSCT, had risks of AML and MDS that were 5 to 10 times higher than the background level in the population.
However, the study auto-HSCT cohort had a risk of AML that was 10 to 50 times higher and a relative risk of MDS that was roughly 100 times higher than the background level.
“These increases may be related to exposure to high doses of DNA-damaging drugs given for [auto-HSCT], but this hypothesis can only be tested in a prospective study,” Dr Radivoyevitch and his coinvestigators wrote.
The reason for the greater elevation of MDS risk, compared with AML risk, is unknown.
“One possible explanation is that many cases of MDS evolve to AML, and that earlier diagnosis from increased post-transplant surveillance resulted in a deficiency of AML,” the investigators wrote. “A second is based on steeper MDS versus AML incidences versus age . . . and the possibility that transplantation recipient marrow ages (ie, marrow biological ages) are perhaps decades older than calendar ages.”
The study authors said they had no relevant conflicts of interest. The CIBMTR is supported by several US government agencies and numerous pharmaceutical companies.
Familial risk of myeloid malignancies

A large study has revealed “the strongest evidence yet” supporting genetic susceptibility to myeloid malignancies, according to a researcher.
The study showed that first-degree relatives of patients with myeloid malignancies had double the risk of developing a myeloid malignancy themselves, when compared to the general population.
The researchers observed significant risks for developing acute myeloid leukemia (AML), myelodysplastic syndromes (MDS), essential thrombocythemia (ET), and polycythemia vera (PV).
“Our study provides the strongest evidence yet for inherited risk for these diseases—evidence that has proved evasive before, in part, because these cancers are relatively uncommon, and our ability to characterize these diseases has, until recently, been limited,” said Amit Sud, MBChB, PhD, of The Institute of Cancer Research in London, UK.
Dr Sud and his colleagues described their research in a letter to Blood.
The researchers analyzed data from the Swedish Family-Cancer Database, which included 93,199 first-degree relatives of 35,037 patients with myeloid malignancies. The patients had been diagnosed between 1958 and 2015.
First-degree relatives of the patients had an increased risk of all myeloid malignancies, with a standardized incidence ratio (SIR) of 1.99 (95% CI 1.12-2.04).
For individual diseases, there was a significant association between family history and increased risk for:
- AML—SIR=1.53 (95% CI 1.21-2.17)
- ET—SIR=6.30 (95% CI 3.95-9.54)
- MDS—SIR=6.87 (95% CI 4.07-10.86)
- PV—SIR=7.66 (95% CI 5.74-10.02).
Dr Sud and his colleagues noted that the strongest familial relative risks tended to occur for the same disease, but there were significant associations between different myeloid malignancies as well.
Risk by age group
The researchers also looked at familial relative risk for the same disease by patients’ age at diagnosis and observed a significantly increased risk for younger cases for all myeloproliferative neoplasms (MPNs) combined, PV, and MDS.
The SIRs for MPNs were 6.46 (95% CI 5.12-8.04) for patients age 59 or younger and 4.15 (95% CI 3.38-5.04) for patients older than 59.
The SIRs for PV were 10.90 (95% CI 7.12-15.97) for patients age 59 or younger and 5.96 (95% CI 3.93-8.67) for patients older than 59.
The SIRs for MDS were 11.95 (95% CI 6.36-20.43) for patients age 68 or younger and 3.27 (95% CI 1.06-7.63) for patients older than 68.
Risk by number of relatives
Dr Sud and his colleagues also discovered that familial relative risks of all myeloid malignancies and MPNs were significantly associated with the number of first-degree relatives affected by myeloid malignancies or MPNs.
The SIRs for first-degree relatives with 2 or more affected relatives were 4.55 (95% CI 2.08-8.64) for all myeloid malignancies and 17.82 (95% CI 5.79-24.89) for MPNs.
The SIRs for first-degree relatives with 1 affected relative were 1.96 (95% CI 1.79-2.15) for all myeloid malignancies and 4.83 (95% CI 4.14-5.60) for MPNs.
The researchers said these results suggest inherited genetic changes increase the risk of myeloid malignancies, although environmental factors shared in families could also play a role.
“In the future, our findings could help identify people at higher risk than normal because of their family background who could be prioritized for medical help like screening to catch the disease earlier if it arises,” Dr Sud said.
This study was funded by German Cancer Aid, the Swedish Research Council, ALF funding from Region Skåne, DKFZ, and Bloodwise.
A large study has revealed “the strongest evidence yet” supporting genetic susceptibility to myeloid malignancies, according to a researcher.
The study showed that first-degree relatives of patients with myeloid malignancies had double the risk of developing a myeloid malignancy themselves, when compared to the general population.
The researchers observed significant risks for developing acute myeloid leukemia (AML), myelodysplastic syndromes (MDS), essential thrombocythemia (ET), and polycythemia vera (PV).
“Our study provides the strongest evidence yet for inherited risk for these diseases—evidence that has proved evasive before, in part, because these cancers are relatively uncommon, and our ability to characterize these diseases has, until recently, been limited,” said Amit Sud, MBChB, PhD, of The Institute of Cancer Research in London, UK.
Dr Sud and his colleagues described their research in a letter to Blood.
The researchers analyzed data from the Swedish Family-Cancer Database, which included 93,199 first-degree relatives of 35,037 patients with myeloid malignancies. The patients had been diagnosed between 1958 and 2015.
First-degree relatives of the patients had an increased risk of all myeloid malignancies, with a standardized incidence ratio (SIR) of 1.99 (95% CI 1.12-2.04).
For individual diseases, there was a significant association between family history and increased risk for:
- AML—SIR=1.53 (95% CI 1.21-2.17)
- ET—SIR=6.30 (95% CI 3.95-9.54)
- MDS—SIR=6.87 (95% CI 4.07-10.86)
- PV—SIR=7.66 (95% CI 5.74-10.02).
Dr Sud and his colleagues noted that the strongest familial relative risks tended to occur for the same disease, but there were significant associations between different myeloid malignancies as well.
Risk by age group
The researchers also looked at familial relative risk for the same disease by patients’ age at diagnosis and observed a significantly increased risk for younger cases for all myeloproliferative neoplasms (MPNs) combined, PV, and MDS.
The SIRs for MPNs were 6.46 (95% CI 5.12-8.04) for patients age 59 or younger and 4.15 (95% CI 3.38-5.04) for patients older than 59.
The SIRs for PV were 10.90 (95% CI 7.12-15.97) for patients age 59 or younger and 5.96 (95% CI 3.93-8.67) for patients older than 59.
The SIRs for MDS were 11.95 (95% CI 6.36-20.43) for patients age 68 or younger and 3.27 (95% CI 1.06-7.63) for patients older than 68.
Risk by number of relatives
Dr Sud and his colleagues also discovered that familial relative risks of all myeloid malignancies and MPNs were significantly associated with the number of first-degree relatives affected by myeloid malignancies or MPNs.
The SIRs for first-degree relatives with 2 or more affected relatives were 4.55 (95% CI 2.08-8.64) for all myeloid malignancies and 17.82 (95% CI 5.79-24.89) for MPNs.
The SIRs for first-degree relatives with 1 affected relative were 1.96 (95% CI 1.79-2.15) for all myeloid malignancies and 4.83 (95% CI 4.14-5.60) for MPNs.
The researchers said these results suggest inherited genetic changes increase the risk of myeloid malignancies, although environmental factors shared in families could also play a role.
“In the future, our findings could help identify people at higher risk than normal because of their family background who could be prioritized for medical help like screening to catch the disease earlier if it arises,” Dr Sud said.
This study was funded by German Cancer Aid, the Swedish Research Council, ALF funding from Region Skåne, DKFZ, and Bloodwise.
A large study has revealed “the strongest evidence yet” supporting genetic susceptibility to myeloid malignancies, according to a researcher.
The study showed that first-degree relatives of patients with myeloid malignancies had double the risk of developing a myeloid malignancy themselves, when compared to the general population.
The researchers observed significant risks for developing acute myeloid leukemia (AML), myelodysplastic syndromes (MDS), essential thrombocythemia (ET), and polycythemia vera (PV).
“Our study provides the strongest evidence yet for inherited risk for these diseases—evidence that has proved evasive before, in part, because these cancers are relatively uncommon, and our ability to characterize these diseases has, until recently, been limited,” said Amit Sud, MBChB, PhD, of The Institute of Cancer Research in London, UK.
Dr Sud and his colleagues described their research in a letter to Blood.
The researchers analyzed data from the Swedish Family-Cancer Database, which included 93,199 first-degree relatives of 35,037 patients with myeloid malignancies. The patients had been diagnosed between 1958 and 2015.
First-degree relatives of the patients had an increased risk of all myeloid malignancies, with a standardized incidence ratio (SIR) of 1.99 (95% CI 1.12-2.04).
For individual diseases, there was a significant association between family history and increased risk for:
- AML—SIR=1.53 (95% CI 1.21-2.17)
- ET—SIR=6.30 (95% CI 3.95-9.54)
- MDS—SIR=6.87 (95% CI 4.07-10.86)
- PV—SIR=7.66 (95% CI 5.74-10.02).
Dr Sud and his colleagues noted that the strongest familial relative risks tended to occur for the same disease, but there were significant associations between different myeloid malignancies as well.
Risk by age group
The researchers also looked at familial relative risk for the same disease by patients’ age at diagnosis and observed a significantly increased risk for younger cases for all myeloproliferative neoplasms (MPNs) combined, PV, and MDS.
The SIRs for MPNs were 6.46 (95% CI 5.12-8.04) for patients age 59 or younger and 4.15 (95% CI 3.38-5.04) for patients older than 59.
The SIRs for PV were 10.90 (95% CI 7.12-15.97) for patients age 59 or younger and 5.96 (95% CI 3.93-8.67) for patients older than 59.
The SIRs for MDS were 11.95 (95% CI 6.36-20.43) for patients age 68 or younger and 3.27 (95% CI 1.06-7.63) for patients older than 68.
Risk by number of relatives
Dr Sud and his colleagues also discovered that familial relative risks of all myeloid malignancies and MPNs were significantly associated with the number of first-degree relatives affected by myeloid malignancies or MPNs.
The SIRs for first-degree relatives with 2 or more affected relatives were 4.55 (95% CI 2.08-8.64) for all myeloid malignancies and 17.82 (95% CI 5.79-24.89) for MPNs.
The SIRs for first-degree relatives with 1 affected relative were 1.96 (95% CI 1.79-2.15) for all myeloid malignancies and 4.83 (95% CI 4.14-5.60) for MPNs.
The researchers said these results suggest inherited genetic changes increase the risk of myeloid malignancies, although environmental factors shared in families could also play a role.
“In the future, our findings could help identify people at higher risk than normal because of their family background who could be prioritized for medical help like screening to catch the disease earlier if it arises,” Dr Sud said.
This study was funded by German Cancer Aid, the Swedish Research Council, ALF funding from Region Skåne, DKFZ, and Bloodwise.
Group releases new CLL guidelines

Fludarabine, cyclophosphamide, and rituximab are recommended as initial therapy for fit patients with chronic lymphocytic leukemia (CLL) who do not have TP53 disruption, according to new guidelines from the British Society for Haematology.
The guidelines update the 2012 recommendations on CLL to include “significant” developments in treatment.
The new guidelines were published in the British Journal of Haematology.
Anna H. Schuh, MD, of the University of Oxford in the UK, and her coauthors noted that, while these guidelines apply to treatments available outside clinical trials, wherever possible, patients with CLL should be treated within the clinical trial setting.
While recommending fludarabine, cyclophosphamide, and rituximab as first-line therapy, the guideline authors acknowledged that the combination of bendamustine and rituximab is an acceptable alternative for patients who cannot take the triple therapy because of comorbidities such as advanced age, renal impairment, or issues with marrow capacity.
Similarly, less-fit patients can also be considered for chlorambucil-obinutuzumab or chlorambucil-ofatumumab combinations.
All patients diagnosed with CLL should be tested for TP53 deletions and mutations before each line of therapy, the guideline committee recommended.
TP53 disruption makes chemoimmunotherapy ineffective because of either a deletion of chromosome 17p or a mutation in the TP53 gene. However, there is compelling evidence for the efficacy of ibrutinib in these patients, or idelalisib and rituximab for those with cardiac disease or receiving vitamin K antagonists.
With respect to maintenance therapy, the guidelines noted that this was not routinely recommended in CLL as “it is unclear to what extent the progression-free survival benefit is offset by long-term toxicity.”
Patients who are refractory to chemoimmunotherapy, who have relapsed, or who cannot be retreated with chemoimmunotherapy should be treated with idelalisib plus rituximab or ibrutinib monotherapy, the guidelines suggested.
“Deciding whether ibrutinib or idelalisib with rituximab is most appropriate for an individual patient depends on a range of factors, including toxicity profile and convenience of delivery,” the authors wrote.
However, they noted that the value of adding bendamustine to either option was unclear as research had not shown significant, associated gains in median progression-free survival.
Allogeneic stem cell transplant should be considered as an option for patients who have failed chemotherapy, have a TP53 disruption and have not responded to B-cell receptor signaling pathway inhibitors such as ibrutinib, or have Richter’s transformation.
The guidelines also addressed the issue of autoimmune cytopenias, which occur in 5% to 10% of patients with CLL and can actually precede the diagnosis of CLL in about 9% of cases.
In patients where autoimmune cytopenia is the dominant clinical feature, they should be treated with corticosteroids, intravenous immunoglobulin, or rituximab. However, for patients where the cytopenia is triggered by CLL therapy, the guidelines recommended halting treatment and beginning immunosuppression.
The guideline development was supported by the British Society for Haematology. The UK CLL Forum, which was involved in development as well, is a registered charity that receives funding from a number of pharmaceutical companies.
Fludarabine, cyclophosphamide, and rituximab are recommended as initial therapy for fit patients with chronic lymphocytic leukemia (CLL) who do not have TP53 disruption, according to new guidelines from the British Society for Haematology.
The guidelines update the 2012 recommendations on CLL to include “significant” developments in treatment.
The new guidelines were published in the British Journal of Haematology.
Anna H. Schuh, MD, of the University of Oxford in the UK, and her coauthors noted that, while these guidelines apply to treatments available outside clinical trials, wherever possible, patients with CLL should be treated within the clinical trial setting.
While recommending fludarabine, cyclophosphamide, and rituximab as first-line therapy, the guideline authors acknowledged that the combination of bendamustine and rituximab is an acceptable alternative for patients who cannot take the triple therapy because of comorbidities such as advanced age, renal impairment, or issues with marrow capacity.
Similarly, less-fit patients can also be considered for chlorambucil-obinutuzumab or chlorambucil-ofatumumab combinations.
All patients diagnosed with CLL should be tested for TP53 deletions and mutations before each line of therapy, the guideline committee recommended.
TP53 disruption makes chemoimmunotherapy ineffective because of either a deletion of chromosome 17p or a mutation in the TP53 gene. However, there is compelling evidence for the efficacy of ibrutinib in these patients, or idelalisib and rituximab for those with cardiac disease or receiving vitamin K antagonists.
With respect to maintenance therapy, the guidelines noted that this was not routinely recommended in CLL as “it is unclear to what extent the progression-free survival benefit is offset by long-term toxicity.”
Patients who are refractory to chemoimmunotherapy, who have relapsed, or who cannot be retreated with chemoimmunotherapy should be treated with idelalisib plus rituximab or ibrutinib monotherapy, the guidelines suggested.
“Deciding whether ibrutinib or idelalisib with rituximab is most appropriate for an individual patient depends on a range of factors, including toxicity profile and convenience of delivery,” the authors wrote.
However, they noted that the value of adding bendamustine to either option was unclear as research had not shown significant, associated gains in median progression-free survival.
Allogeneic stem cell transplant should be considered as an option for patients who have failed chemotherapy, have a TP53 disruption and have not responded to B-cell receptor signaling pathway inhibitors such as ibrutinib, or have Richter’s transformation.
The guidelines also addressed the issue of autoimmune cytopenias, which occur in 5% to 10% of patients with CLL and can actually precede the diagnosis of CLL in about 9% of cases.
In patients where autoimmune cytopenia is the dominant clinical feature, they should be treated with corticosteroids, intravenous immunoglobulin, or rituximab. However, for patients where the cytopenia is triggered by CLL therapy, the guidelines recommended halting treatment and beginning immunosuppression.
The guideline development was supported by the British Society for Haematology. The UK CLL Forum, which was involved in development as well, is a registered charity that receives funding from a number of pharmaceutical companies.
Fludarabine, cyclophosphamide, and rituximab are recommended as initial therapy for fit patients with chronic lymphocytic leukemia (CLL) who do not have TP53 disruption, according to new guidelines from the British Society for Haematology.
The guidelines update the 2012 recommendations on CLL to include “significant” developments in treatment.
The new guidelines were published in the British Journal of Haematology.
Anna H. Schuh, MD, of the University of Oxford in the UK, and her coauthors noted that, while these guidelines apply to treatments available outside clinical trials, wherever possible, patients with CLL should be treated within the clinical trial setting.
While recommending fludarabine, cyclophosphamide, and rituximab as first-line therapy, the guideline authors acknowledged that the combination of bendamustine and rituximab is an acceptable alternative for patients who cannot take the triple therapy because of comorbidities such as advanced age, renal impairment, or issues with marrow capacity.
Similarly, less-fit patients can also be considered for chlorambucil-obinutuzumab or chlorambucil-ofatumumab combinations.
All patients diagnosed with CLL should be tested for TP53 deletions and mutations before each line of therapy, the guideline committee recommended.
TP53 disruption makes chemoimmunotherapy ineffective because of either a deletion of chromosome 17p or a mutation in the TP53 gene. However, there is compelling evidence for the efficacy of ibrutinib in these patients, or idelalisib and rituximab for those with cardiac disease or receiving vitamin K antagonists.
With respect to maintenance therapy, the guidelines noted that this was not routinely recommended in CLL as “it is unclear to what extent the progression-free survival benefit is offset by long-term toxicity.”
Patients who are refractory to chemoimmunotherapy, who have relapsed, or who cannot be retreated with chemoimmunotherapy should be treated with idelalisib plus rituximab or ibrutinib monotherapy, the guidelines suggested.
“Deciding whether ibrutinib or idelalisib with rituximab is most appropriate for an individual patient depends on a range of factors, including toxicity profile and convenience of delivery,” the authors wrote.
However, they noted that the value of adding bendamustine to either option was unclear as research had not shown significant, associated gains in median progression-free survival.
Allogeneic stem cell transplant should be considered as an option for patients who have failed chemotherapy, have a TP53 disruption and have not responded to B-cell receptor signaling pathway inhibitors such as ibrutinib, or have Richter’s transformation.
The guidelines also addressed the issue of autoimmune cytopenias, which occur in 5% to 10% of patients with CLL and can actually precede the diagnosis of CLL in about 9% of cases.
In patients where autoimmune cytopenia is the dominant clinical feature, they should be treated with corticosteroids, intravenous immunoglobulin, or rituximab. However, for patients where the cytopenia is triggered by CLL therapy, the guidelines recommended halting treatment and beginning immunosuppression.
The guideline development was supported by the British Society for Haematology. The UK CLL Forum, which was involved in development as well, is a registered charity that receives funding from a number of pharmaceutical companies.
Acute Leukemia of Ambiguous Lineage in Elderly Patients: A SEER-Medicare Database Analysis (FULL)
About Research in Context
In this article, the authors of recent scholarship have been asked to discuss the implications of their research on federal health care providers and specifically the veteran and active-duty service member patient populations. Because the article does not include new research and cannot be blinded, it has undergone an abbreviated peer review process. The original article can be found at Guru Murthy GS, Dhakal I, Lee JY, Mehta P. Acute leukemia of ambiguous lineage in elderly patients - analysis of survival using surveillance epidemiology and end results-Medicare database. Clin Lymphoma Myeloma Leuk. 2017;17(2):100-107.
Acute leukemia of ambiguous lineage (ALAL) is a rare disorder in adults, constituting about 3% to 5% of acute leukemia cases. Unlike acute myeloid leukemia (AML) or acute lymphoblastic leukemia (ALL), ALAL cannot be clearly differentiated into a single subtype based on immunophenotyping. The diagnostic criteria for accurately identifying ALAL has evolved over time. There is paucity of information regarding the outcomes and management of this rare leukemia especially in elderly patients, and it is unclear whether treatment improves survival in these patients.
We performed a retrospective analysis of the Surveillance, Epidemiology, and End Results (SEER)-Medicare linked database to describe the outcomes of ALAL in the elderly population in U.S.1 Patients included in the analysis were aged > 65 years, with a pathologically confirmed diagnosis of ALAL, diagnosed between 1992-2010, and on active follow-up. Information on patient demographics, treatment, chemotherapeutic agents used in treatment, and survival was obtained and analyzed using appropriate statistical methods. A total of 705 patients with a median age of 80 years were included. There was a higher proportion of males than females and a higher proportion of white patients compared with African Americans and other races. We found that the overall survival (OS) declined significantly with increasing age, and treatment with chemotherapy improved the survival. However, factors such as gender, race, or type of chemotherapy received (ALL based, AML based, or other regimens) did not significantly influence the survival.
Even in the current era, the optimal therapy for ALAL is not well established. Although options such as AML-based or ALL-based chemotherapy are available, the best chemotherapy regimen and its sequence is unknown as prior studies have demonstrated varying results.2-5 Among elderly patients, numerous factors such as performance status, comorbidities, and ability to tolerate therapy influence the treatment decision. In light of the poor prognosis in elderly patients, a question often arises in the clinician’s mind about whether chemotherapy would provide any benefit for the patient.
Our study results showed that chemotherapy likely improves survival in these patients. However, due to the smaller number of patients, caution is needed in interpreting the result that there was no significant difference between AML-directed or ALL-directed chemotherapy. Another factor highlighted in the study was that only about 21.5% of patients had been treated with chemotherapy. Due to the inherent nature of the database, we could not identify the factors that may have influenced treatment decisions in these patients. Additionally, patients with stem cell transplantation-related claims could not be included in the analysis due to noncontinuous Medicare coverage during the study period. Hence, the role of stem cell transplantation in these patients could not be determined.
Implications Among Veterans
Actual incidence of ALAL among veterans is not known. Whether the incidence of ALAL relates to exposures to chemicals or toxins during military training and service also is unknown. However, ALAL is likely to be at least as prevalent as it is in the nonveteran population and perhaps more so because of exposures and stresses during military training and service.
It is unclear whether veterans attending VA hospitals receive less or different treatment given the higher comorbidities. Finally, it also is not known whether the outcomes for veterans would be different with or without treatment.
Our findings suggest that treatment should be seriously considered in all patients (veterans or not) who are healthy enough to receive chemotherapy regardless of their age. More research is needed to determine the disease incidence and prevalence among veterans and to evaluate whether there are specific etiologic correlations between ALAL and military exposures, whether the natural history is similar to other populations, and to delineate responsiveness to treatment.
Conclusion
This study suggests a poor survival for elderly patients with ALAL in the U.S. Although treatment is associated with an improvement in survival, only 21.5% of patients have received therapy. The optimal choice of chemotherapy for this disease is still not known and warrants prospective studies.
Click here to read the digital edition.
1. Guru Murthy GS, Dhakal I, Lee JY, Mehta P. Acute leukemia of ambiguous lineage in elderly patients—analysis of survival using surveillance epidemiology and end results—Medicare database. Clin Lymphoma Myeloma Leuk. 2017;17(2):100-107.
2. Rubnitz JE, Onciu M, Pounds S, et al. Acute mixed lineage leukemia in children: the experience of St Jude Children’s Research Hospital. Blood. 2009;113(21):5083-5089.
3. Matutes E, Pickl WF, Van’t Veer M, et al. Mixed phenotype acute leukemia: clinical and laboratory features and out-come in 100 patients defined according to the WHO classification. Blood. 2011;117(11):3163-3171.
4. Wolach O, Stone RM. How I treat mixed-phenotype acute leukemia. Blood. 2015;125(16):2477-2485.
5. Lee JH, Min YH, Chung CW, et al; Korean Society of Hematology AML/MDS Working Party. Prognostic implications of the immunophenotype in biphenotypic acute leukemia. Leuk Lymphoma. 2008;49(4):700-709.
About Research in Context
In this article, the authors of recent scholarship have been asked to discuss the implications of their research on federal health care providers and specifically the veteran and active-duty service member patient populations. Because the article does not include new research and cannot be blinded, it has undergone an abbreviated peer review process. The original article can be found at Guru Murthy GS, Dhakal I, Lee JY, Mehta P. Acute leukemia of ambiguous lineage in elderly patients - analysis of survival using surveillance epidemiology and end results-Medicare database. Clin Lymphoma Myeloma Leuk. 2017;17(2):100-107.
Acute leukemia of ambiguous lineage (ALAL) is a rare disorder in adults, constituting about 3% to 5% of acute leukemia cases. Unlike acute myeloid leukemia (AML) or acute lymphoblastic leukemia (ALL), ALAL cannot be clearly differentiated into a single subtype based on immunophenotyping. The diagnostic criteria for accurately identifying ALAL has evolved over time. There is paucity of information regarding the outcomes and management of this rare leukemia especially in elderly patients, and it is unclear whether treatment improves survival in these patients.
We performed a retrospective analysis of the Surveillance, Epidemiology, and End Results (SEER)-Medicare linked database to describe the outcomes of ALAL in the elderly population in U.S.1 Patients included in the analysis were aged > 65 years, with a pathologically confirmed diagnosis of ALAL, diagnosed between 1992-2010, and on active follow-up. Information on patient demographics, treatment, chemotherapeutic agents used in treatment, and survival was obtained and analyzed using appropriate statistical methods. A total of 705 patients with a median age of 80 years were included. There was a higher proportion of males than females and a higher proportion of white patients compared with African Americans and other races. We found that the overall survival (OS) declined significantly with increasing age, and treatment with chemotherapy improved the survival. However, factors such as gender, race, or type of chemotherapy received (ALL based, AML based, or other regimens) did not significantly influence the survival.
Even in the current era, the optimal therapy for ALAL is not well established. Although options such as AML-based or ALL-based chemotherapy are available, the best chemotherapy regimen and its sequence is unknown as prior studies have demonstrated varying results.2-5 Among elderly patients, numerous factors such as performance status, comorbidities, and ability to tolerate therapy influence the treatment decision. In light of the poor prognosis in elderly patients, a question often arises in the clinician’s mind about whether chemotherapy would provide any benefit for the patient.
Our study results showed that chemotherapy likely improves survival in these patients. However, due to the smaller number of patients, caution is needed in interpreting the result that there was no significant difference between AML-directed or ALL-directed chemotherapy. Another factor highlighted in the study was that only about 21.5% of patients had been treated with chemotherapy. Due to the inherent nature of the database, we could not identify the factors that may have influenced treatment decisions in these patients. Additionally, patients with stem cell transplantation-related claims could not be included in the analysis due to noncontinuous Medicare coverage during the study period. Hence, the role of stem cell transplantation in these patients could not be determined.
Implications Among Veterans
Actual incidence of ALAL among veterans is not known. Whether the incidence of ALAL relates to exposures to chemicals or toxins during military training and service also is unknown. However, ALAL is likely to be at least as prevalent as it is in the nonveteran population and perhaps more so because of exposures and stresses during military training and service.
It is unclear whether veterans attending VA hospitals receive less or different treatment given the higher comorbidities. Finally, it also is not known whether the outcomes for veterans would be different with or without treatment.
Our findings suggest that treatment should be seriously considered in all patients (veterans or not) who are healthy enough to receive chemotherapy regardless of their age. More research is needed to determine the disease incidence and prevalence among veterans and to evaluate whether there are specific etiologic correlations between ALAL and military exposures, whether the natural history is similar to other populations, and to delineate responsiveness to treatment.
Conclusion
This study suggests a poor survival for elderly patients with ALAL in the U.S. Although treatment is associated with an improvement in survival, only 21.5% of patients have received therapy. The optimal choice of chemotherapy for this disease is still not known and warrants prospective studies.
Click here to read the digital edition.
About Research in Context
In this article, the authors of recent scholarship have been asked to discuss the implications of their research on federal health care providers and specifically the veteran and active-duty service member patient populations. Because the article does not include new research and cannot be blinded, it has undergone an abbreviated peer review process. The original article can be found at Guru Murthy GS, Dhakal I, Lee JY, Mehta P. Acute leukemia of ambiguous lineage in elderly patients - analysis of survival using surveillance epidemiology and end results-Medicare database. Clin Lymphoma Myeloma Leuk. 2017;17(2):100-107.
Acute leukemia of ambiguous lineage (ALAL) is a rare disorder in adults, constituting about 3% to 5% of acute leukemia cases. Unlike acute myeloid leukemia (AML) or acute lymphoblastic leukemia (ALL), ALAL cannot be clearly differentiated into a single subtype based on immunophenotyping. The diagnostic criteria for accurately identifying ALAL has evolved over time. There is paucity of information regarding the outcomes and management of this rare leukemia especially in elderly patients, and it is unclear whether treatment improves survival in these patients.
We performed a retrospective analysis of the Surveillance, Epidemiology, and End Results (SEER)-Medicare linked database to describe the outcomes of ALAL in the elderly population in U.S.1 Patients included in the analysis were aged > 65 years, with a pathologically confirmed diagnosis of ALAL, diagnosed between 1992-2010, and on active follow-up. Information on patient demographics, treatment, chemotherapeutic agents used in treatment, and survival was obtained and analyzed using appropriate statistical methods. A total of 705 patients with a median age of 80 years were included. There was a higher proportion of males than females and a higher proportion of white patients compared with African Americans and other races. We found that the overall survival (OS) declined significantly with increasing age, and treatment with chemotherapy improved the survival. However, factors such as gender, race, or type of chemotherapy received (ALL based, AML based, or other regimens) did not significantly influence the survival.
Even in the current era, the optimal therapy for ALAL is not well established. Although options such as AML-based or ALL-based chemotherapy are available, the best chemotherapy regimen and its sequence is unknown as prior studies have demonstrated varying results.2-5 Among elderly patients, numerous factors such as performance status, comorbidities, and ability to tolerate therapy influence the treatment decision. In light of the poor prognosis in elderly patients, a question often arises in the clinician’s mind about whether chemotherapy would provide any benefit for the patient.
Our study results showed that chemotherapy likely improves survival in these patients. However, due to the smaller number of patients, caution is needed in interpreting the result that there was no significant difference between AML-directed or ALL-directed chemotherapy. Another factor highlighted in the study was that only about 21.5% of patients had been treated with chemotherapy. Due to the inherent nature of the database, we could not identify the factors that may have influenced treatment decisions in these patients. Additionally, patients with stem cell transplantation-related claims could not be included in the analysis due to noncontinuous Medicare coverage during the study period. Hence, the role of stem cell transplantation in these patients could not be determined.
Implications Among Veterans
Actual incidence of ALAL among veterans is not known. Whether the incidence of ALAL relates to exposures to chemicals or toxins during military training and service also is unknown. However, ALAL is likely to be at least as prevalent as it is in the nonveteran population and perhaps more so because of exposures and stresses during military training and service.
It is unclear whether veterans attending VA hospitals receive less or different treatment given the higher comorbidities. Finally, it also is not known whether the outcomes for veterans would be different with or without treatment.
Our findings suggest that treatment should be seriously considered in all patients (veterans or not) who are healthy enough to receive chemotherapy regardless of their age. More research is needed to determine the disease incidence and prevalence among veterans and to evaluate whether there are specific etiologic correlations between ALAL and military exposures, whether the natural history is similar to other populations, and to delineate responsiveness to treatment.
Conclusion
This study suggests a poor survival for elderly patients with ALAL in the U.S. Although treatment is associated with an improvement in survival, only 21.5% of patients have received therapy. The optimal choice of chemotherapy for this disease is still not known and warrants prospective studies.
Click here to read the digital edition.
1. Guru Murthy GS, Dhakal I, Lee JY, Mehta P. Acute leukemia of ambiguous lineage in elderly patients—analysis of survival using surveillance epidemiology and end results—Medicare database. Clin Lymphoma Myeloma Leuk. 2017;17(2):100-107.
2. Rubnitz JE, Onciu M, Pounds S, et al. Acute mixed lineage leukemia in children: the experience of St Jude Children’s Research Hospital. Blood. 2009;113(21):5083-5089.
3. Matutes E, Pickl WF, Van’t Veer M, et al. Mixed phenotype acute leukemia: clinical and laboratory features and out-come in 100 patients defined according to the WHO classification. Blood. 2011;117(11):3163-3171.
4. Wolach O, Stone RM. How I treat mixed-phenotype acute leukemia. Blood. 2015;125(16):2477-2485.
5. Lee JH, Min YH, Chung CW, et al; Korean Society of Hematology AML/MDS Working Party. Prognostic implications of the immunophenotype in biphenotypic acute leukemia. Leuk Lymphoma. 2008;49(4):700-709.
1. Guru Murthy GS, Dhakal I, Lee JY, Mehta P. Acute leukemia of ambiguous lineage in elderly patients—analysis of survival using surveillance epidemiology and end results—Medicare database. Clin Lymphoma Myeloma Leuk. 2017;17(2):100-107.
2. Rubnitz JE, Onciu M, Pounds S, et al. Acute mixed lineage leukemia in children: the experience of St Jude Children’s Research Hospital. Blood. 2009;113(21):5083-5089.
3. Matutes E, Pickl WF, Van’t Veer M, et al. Mixed phenotype acute leukemia: clinical and laboratory features and out-come in 100 patients defined according to the WHO classification. Blood. 2011;117(11):3163-3171.
4. Wolach O, Stone RM. How I treat mixed-phenotype acute leukemia. Blood. 2015;125(16):2477-2485.
5. Lee JH, Min YH, Chung CW, et al; Korean Society of Hematology AML/MDS Working Party. Prognostic implications of the immunophenotype in biphenotypic acute leukemia. Leuk Lymphoma. 2008;49(4):700-709.
Ixazomib could improve treatment of AML

New research suggests the FOXM1 protein plays an important role in acute myeloid leukemia (AML) progression, and targeting FOXM1 could improve AML treatment.
With a retrospective study, researchers showed that overexpression of FOXM1 was associated with increased resistance to chemotherapy and inferior overall survival.
Subsequent preclinical research showed that ixazomib inhibits FOXM1, exhibits antileukemic activity, and sensitizes AML cells to chemotherapy.
Irum Khan, MD, of the University of Illinois in Chicago, and her colleagues reported these findings in JCI Insight.
Previous research showed that AML patients with NPM1 mutations have a higher rate of remission with chemotherapy, and the NPM1 protein affects the location and activity of FOXM1. NPM1 keeps FOXM1 in the nucleus where it can activate other cancer-promoting genes.
When the NPM1 gene is mutated, FOXM1 migrates out of the nucleus and into the cell’s cytoplasm, where it can’t interact with DNA. This may explain why AML patients with NPM1 mutations have a better response to chemotherapy and are less likely to relapse.
With the current research, Dr Khan and her colleagues further explored the role of FOXM1 in AML.
Retrospective analysis
The multicenter, retrospective study began with data from 111 adults with AML. They had intermediate-risk cytogenetics and a median age of 61.
Eighty-eight patients received induction with cytarabine and an anthracycline, and 80 achieved a complete remission with or without count recovery.
FOXM1 expression data were available for 74 of these patients. Fifty patients achieved remission with 1 cycle of induction, and 24 required more than 1 cycle.
“[Patients] with FOXM1 present in the nucleus of their cancer cells had worse treatment outcomes, higher rates of chemotherapy resistance, and lower survival rates compared to patients without FOXM1 present in the nucleus,” Dr Khan said.
The patients who failed their first line of induction had a more than 2-fold increase in the percentage of nuclei expressing FOXM1 in their bone marrow (P=0.004). And the average nuclear intensity of FOXM1 was significantly higher in the patients who failed their first line of induction (P=0.02).
The percentage of FOXM1-positive nuclei and the average nuclear intensity of FOXM1 both significantly predicted resistance to first-line chemotherapy. The odds ratio was 1.80 for a 10% increase in FOXM1-positive nuclei (P=0.005) and 2.5 for a 0.1 unit increase in nuclear intensity (P=0.02).
A multivariate analysis showed that the FOXM1 nuclear/cytoplasmic (N:C) ratio and nuclear FOXM1 intensity predicted inferior overall survival in a single institution. (Institutions were analyzed separately for survival). The hazard ratio was 4.7 for every 0.1 unit increase in N:C ratio (P=0.03) and 4.27 for every 0.1 unit increase in nuclear intensity (P=0.06).
Confirming the role of FOXM1
The researchers set out to confirm the role of FOXM1 via experiments in mice.
The team induced a FLT3-ITD-driven myeloproliferative neoplasm in a FOXM1-overexpressing transgenic mouse model.
These mice had more residual disease after treatment with cytarabine than control mice with normal levels of FOXM1.
“Our finding suggests that overexpression of FOXM1 directly induces chemoresistance, which matches what we saw in our analysis of patients’ FOXM1 levels and their treatment outcomes,” Dr Khan said.
Targeting FOXM1 with ixazomib
Next, the researchers showed they could produce a therapeutic response by inhibiting FOXM1 in AML. The team used ixazomib, which was shown to suppress FOXM1.
There was a 2-fold increase in apoptosis when AML patient cells were treated with ixazomib (compared to DMSO).
Ixazomib also exhibited antitumor activity in a xenograft model of AML (HL-60 cells) and reduced leukemic burden in an orthotopic model of AML (KG-1 cells).
Finally, the researchers found that ixazomib sensitized AML cells to chemotherapy. The team observed synergistic activity between ixazomib and cytarabine or 5-azacitidine.
“There is a real unmet need for new ways to get around the resistance to chemotherapy that patients who don’t have this beneficial [NPM1] mutation often face,” Dr Khan said.
“Drugs that suppress FOXM1 in combination with the standard treatment, such as ixazomib, should result in better outcomes, but clinical trials will ultimately be needed to prove this theory.”
This research was supported by grants from the National Institutes of Health and Takeda.
New research suggests the FOXM1 protein plays an important role in acute myeloid leukemia (AML) progression, and targeting FOXM1 could improve AML treatment.
With a retrospective study, researchers showed that overexpression of FOXM1 was associated with increased resistance to chemotherapy and inferior overall survival.
Subsequent preclinical research showed that ixazomib inhibits FOXM1, exhibits antileukemic activity, and sensitizes AML cells to chemotherapy.
Irum Khan, MD, of the University of Illinois in Chicago, and her colleagues reported these findings in JCI Insight.
Previous research showed that AML patients with NPM1 mutations have a higher rate of remission with chemotherapy, and the NPM1 protein affects the location and activity of FOXM1. NPM1 keeps FOXM1 in the nucleus where it can activate other cancer-promoting genes.
When the NPM1 gene is mutated, FOXM1 migrates out of the nucleus and into the cell’s cytoplasm, where it can’t interact with DNA. This may explain why AML patients with NPM1 mutations have a better response to chemotherapy and are less likely to relapse.
With the current research, Dr Khan and her colleagues further explored the role of FOXM1 in AML.
Retrospective analysis
The multicenter, retrospective study began with data from 111 adults with AML. They had intermediate-risk cytogenetics and a median age of 61.
Eighty-eight patients received induction with cytarabine and an anthracycline, and 80 achieved a complete remission with or without count recovery.
FOXM1 expression data were available for 74 of these patients. Fifty patients achieved remission with 1 cycle of induction, and 24 required more than 1 cycle.
“[Patients] with FOXM1 present in the nucleus of their cancer cells had worse treatment outcomes, higher rates of chemotherapy resistance, and lower survival rates compared to patients without FOXM1 present in the nucleus,” Dr Khan said.
The patients who failed their first line of induction had a more than 2-fold increase in the percentage of nuclei expressing FOXM1 in their bone marrow (P=0.004). And the average nuclear intensity of FOXM1 was significantly higher in the patients who failed their first line of induction (P=0.02).
The percentage of FOXM1-positive nuclei and the average nuclear intensity of FOXM1 both significantly predicted resistance to first-line chemotherapy. The odds ratio was 1.80 for a 10% increase in FOXM1-positive nuclei (P=0.005) and 2.5 for a 0.1 unit increase in nuclear intensity (P=0.02).
A multivariate analysis showed that the FOXM1 nuclear/cytoplasmic (N:C) ratio and nuclear FOXM1 intensity predicted inferior overall survival in a single institution. (Institutions were analyzed separately for survival). The hazard ratio was 4.7 for every 0.1 unit increase in N:C ratio (P=0.03) and 4.27 for every 0.1 unit increase in nuclear intensity (P=0.06).
Confirming the role of FOXM1
The researchers set out to confirm the role of FOXM1 via experiments in mice.
The team induced a FLT3-ITD-driven myeloproliferative neoplasm in a FOXM1-overexpressing transgenic mouse model.
These mice had more residual disease after treatment with cytarabine than control mice with normal levels of FOXM1.
“Our finding suggests that overexpression of FOXM1 directly induces chemoresistance, which matches what we saw in our analysis of patients’ FOXM1 levels and their treatment outcomes,” Dr Khan said.
Targeting FOXM1 with ixazomib
Next, the researchers showed they could produce a therapeutic response by inhibiting FOXM1 in AML. The team used ixazomib, which was shown to suppress FOXM1.
There was a 2-fold increase in apoptosis when AML patient cells were treated with ixazomib (compared to DMSO).
Ixazomib also exhibited antitumor activity in a xenograft model of AML (HL-60 cells) and reduced leukemic burden in an orthotopic model of AML (KG-1 cells).
Finally, the researchers found that ixazomib sensitized AML cells to chemotherapy. The team observed synergistic activity between ixazomib and cytarabine or 5-azacitidine.
“There is a real unmet need for new ways to get around the resistance to chemotherapy that patients who don’t have this beneficial [NPM1] mutation often face,” Dr Khan said.
“Drugs that suppress FOXM1 in combination with the standard treatment, such as ixazomib, should result in better outcomes, but clinical trials will ultimately be needed to prove this theory.”
This research was supported by grants from the National Institutes of Health and Takeda.
New research suggests the FOXM1 protein plays an important role in acute myeloid leukemia (AML) progression, and targeting FOXM1 could improve AML treatment.
With a retrospective study, researchers showed that overexpression of FOXM1 was associated with increased resistance to chemotherapy and inferior overall survival.
Subsequent preclinical research showed that ixazomib inhibits FOXM1, exhibits antileukemic activity, and sensitizes AML cells to chemotherapy.
Irum Khan, MD, of the University of Illinois in Chicago, and her colleagues reported these findings in JCI Insight.
Previous research showed that AML patients with NPM1 mutations have a higher rate of remission with chemotherapy, and the NPM1 protein affects the location and activity of FOXM1. NPM1 keeps FOXM1 in the nucleus where it can activate other cancer-promoting genes.
When the NPM1 gene is mutated, FOXM1 migrates out of the nucleus and into the cell’s cytoplasm, where it can’t interact with DNA. This may explain why AML patients with NPM1 mutations have a better response to chemotherapy and are less likely to relapse.
With the current research, Dr Khan and her colleagues further explored the role of FOXM1 in AML.
Retrospective analysis
The multicenter, retrospective study began with data from 111 adults with AML. They had intermediate-risk cytogenetics and a median age of 61.
Eighty-eight patients received induction with cytarabine and an anthracycline, and 80 achieved a complete remission with or without count recovery.
FOXM1 expression data were available for 74 of these patients. Fifty patients achieved remission with 1 cycle of induction, and 24 required more than 1 cycle.
“[Patients] with FOXM1 present in the nucleus of their cancer cells had worse treatment outcomes, higher rates of chemotherapy resistance, and lower survival rates compared to patients without FOXM1 present in the nucleus,” Dr Khan said.
The patients who failed their first line of induction had a more than 2-fold increase in the percentage of nuclei expressing FOXM1 in their bone marrow (P=0.004). And the average nuclear intensity of FOXM1 was significantly higher in the patients who failed their first line of induction (P=0.02).
The percentage of FOXM1-positive nuclei and the average nuclear intensity of FOXM1 both significantly predicted resistance to first-line chemotherapy. The odds ratio was 1.80 for a 10% increase in FOXM1-positive nuclei (P=0.005) and 2.5 for a 0.1 unit increase in nuclear intensity (P=0.02).
A multivariate analysis showed that the FOXM1 nuclear/cytoplasmic (N:C) ratio and nuclear FOXM1 intensity predicted inferior overall survival in a single institution. (Institutions were analyzed separately for survival). The hazard ratio was 4.7 for every 0.1 unit increase in N:C ratio (P=0.03) and 4.27 for every 0.1 unit increase in nuclear intensity (P=0.06).
Confirming the role of FOXM1
The researchers set out to confirm the role of FOXM1 via experiments in mice.
The team induced a FLT3-ITD-driven myeloproliferative neoplasm in a FOXM1-overexpressing transgenic mouse model.
These mice had more residual disease after treatment with cytarabine than control mice with normal levels of FOXM1.
“Our finding suggests that overexpression of FOXM1 directly induces chemoresistance, which matches what we saw in our analysis of patients’ FOXM1 levels and their treatment outcomes,” Dr Khan said.
Targeting FOXM1 with ixazomib
Next, the researchers showed they could produce a therapeutic response by inhibiting FOXM1 in AML. The team used ixazomib, which was shown to suppress FOXM1.
There was a 2-fold increase in apoptosis when AML patient cells were treated with ixazomib (compared to DMSO).
Ixazomib also exhibited antitumor activity in a xenograft model of AML (HL-60 cells) and reduced leukemic burden in an orthotopic model of AML (KG-1 cells).
Finally, the researchers found that ixazomib sensitized AML cells to chemotherapy. The team observed synergistic activity between ixazomib and cytarabine or 5-azacitidine.
“There is a real unmet need for new ways to get around the resistance to chemotherapy that patients who don’t have this beneficial [NPM1] mutation often face,” Dr Khan said.
“Drugs that suppress FOXM1 in combination with the standard treatment, such as ixazomib, should result in better outcomes, but clinical trials will ultimately be needed to prove this theory.”
This research was supported by grants from the National Institutes of Health and Takeda.
Team recommends melanoma screening in CLL

Patients with chronic lymphocytic leukemia (CLL) should be routinely monitored for melanoma, according to researchers.
A study of 470 CLL patients showed they have a significantly higher risk of invasive melanoma than the general population.
Most of the melanomas reported in this study were detected via routine surveillance, and most were discovered before they reached an advanced stage.
Clive Zent, MD, of Wilmot Cancer Institute at the University of Rochester Medical Center in Rochester, New York, and his colleagues described this study in Leukemia Research.
The researchers analyzed data on 470 CLL patients followed for 2849 person-years. Eighteen of these patients developed 22 melanomas. This included 14 cases of invasive melanoma in 13 patients.
The rate of invasive melanoma was significantly higher in this CLL cohort than the rate observed in the age- and sex-matched general population. The standardized incidence ratio was 6.32.
“We do not for sure know why CLL patients are more susceptible to melanoma, but the most likely cause is a suppressed immune system,” Dr Zent noted.
“Normally, in people with healthy immune systems, malignant skin cells might be detected and destroyed before they become a problem. But in CLL patients, failure of this control system increases the rate at which cancer cells can grow into tumors and also the likelihood that they will become invasive or spread to distant sites.”
Detection and management
Fifteen of the 22 melanomas (68.2%) in the CLL cohort were detected via surveillance in a dermatology clinic, and 2 (9.1%) were detected at the CLL/lymphoma clinic.
Three cases of melanoma (14.3%) were detected within the first year of a patient’s CLL diagnosis.
Seven melanomas (33.3%) were detected at pathologic stage 0, 8 (38.1%) at stage I, 2 (9.5%) at stage II, 3 (14.3%) at stage III, and 1 (4.8%) at stage IV. Detailed data were not available for the remaining case.
Melanomas were managed with wide local excision (n=19), sentinel node biopsies (n=6), Mohs surgery (n=1), drugs (n=2), palliative care (n=1), and comfort care (n=1).
The 4 patients who received drugs, palliative care, or comfort care had advanced melanoma.
The patient who received palliative care was still alive at 2.4 years of follow-up. The patient who received comfort care died of metastatic melanoma 1.4 years after diagnosis.
The third patient with advanced melanoma received 2 cycles of dacarbazine and palliative radiation to lung and brain metastases. This patient died 3.6 years after melanoma diagnosis.
The fourth patient received ipilimumab for the melanoma while also receiving ibrutinib to treat her CLL. When the ipilimumab failed, the patient proceeded to pembrolizumab and achieved a near-complete response within 3 months. Then, an intensely hypermetabolic abdominal node was detected and successfully treated with radiation.
The patient continued on pembrolizumab, and her melanoma was in sustained remission at last follow-up, after 23 cycles of pembrolizumab. Her CLL was still responding to ibrutinib at that point as well.
Based on these data, Dr Zent and his colleagues recommend routine melanoma screening for CLL patients. The team believes such surveillance might decrease morbidity and mortality in these patients, although more research is needed to confirm this theory.
Patients with chronic lymphocytic leukemia (CLL) should be routinely monitored for melanoma, according to researchers.
A study of 470 CLL patients showed they have a significantly higher risk of invasive melanoma than the general population.
Most of the melanomas reported in this study were detected via routine surveillance, and most were discovered before they reached an advanced stage.
Clive Zent, MD, of Wilmot Cancer Institute at the University of Rochester Medical Center in Rochester, New York, and his colleagues described this study in Leukemia Research.
The researchers analyzed data on 470 CLL patients followed for 2849 person-years. Eighteen of these patients developed 22 melanomas. This included 14 cases of invasive melanoma in 13 patients.
The rate of invasive melanoma was significantly higher in this CLL cohort than the rate observed in the age- and sex-matched general population. The standardized incidence ratio was 6.32.
“We do not for sure know why CLL patients are more susceptible to melanoma, but the most likely cause is a suppressed immune system,” Dr Zent noted.
“Normally, in people with healthy immune systems, malignant skin cells might be detected and destroyed before they become a problem. But in CLL patients, failure of this control system increases the rate at which cancer cells can grow into tumors and also the likelihood that they will become invasive or spread to distant sites.”
Detection and management
Fifteen of the 22 melanomas (68.2%) in the CLL cohort were detected via surveillance in a dermatology clinic, and 2 (9.1%) were detected at the CLL/lymphoma clinic.
Three cases of melanoma (14.3%) were detected within the first year of a patient’s CLL diagnosis.
Seven melanomas (33.3%) were detected at pathologic stage 0, 8 (38.1%) at stage I, 2 (9.5%) at stage II, 3 (14.3%) at stage III, and 1 (4.8%) at stage IV. Detailed data were not available for the remaining case.
Melanomas were managed with wide local excision (n=19), sentinel node biopsies (n=6), Mohs surgery (n=1), drugs (n=2), palliative care (n=1), and comfort care (n=1).
The 4 patients who received drugs, palliative care, or comfort care had advanced melanoma.
The patient who received palliative care was still alive at 2.4 years of follow-up. The patient who received comfort care died of metastatic melanoma 1.4 years after diagnosis.
The third patient with advanced melanoma received 2 cycles of dacarbazine and palliative radiation to lung and brain metastases. This patient died 3.6 years after melanoma diagnosis.
The fourth patient received ipilimumab for the melanoma while also receiving ibrutinib to treat her CLL. When the ipilimumab failed, the patient proceeded to pembrolizumab and achieved a near-complete response within 3 months. Then, an intensely hypermetabolic abdominal node was detected and successfully treated with radiation.
The patient continued on pembrolizumab, and her melanoma was in sustained remission at last follow-up, after 23 cycles of pembrolizumab. Her CLL was still responding to ibrutinib at that point as well.
Based on these data, Dr Zent and his colleagues recommend routine melanoma screening for CLL patients. The team believes such surveillance might decrease morbidity and mortality in these patients, although more research is needed to confirm this theory.
Patients with chronic lymphocytic leukemia (CLL) should be routinely monitored for melanoma, according to researchers.
A study of 470 CLL patients showed they have a significantly higher risk of invasive melanoma than the general population.
Most of the melanomas reported in this study were detected via routine surveillance, and most were discovered before they reached an advanced stage.
Clive Zent, MD, of Wilmot Cancer Institute at the University of Rochester Medical Center in Rochester, New York, and his colleagues described this study in Leukemia Research.
The researchers analyzed data on 470 CLL patients followed for 2849 person-years. Eighteen of these patients developed 22 melanomas. This included 14 cases of invasive melanoma in 13 patients.
The rate of invasive melanoma was significantly higher in this CLL cohort than the rate observed in the age- and sex-matched general population. The standardized incidence ratio was 6.32.
“We do not for sure know why CLL patients are more susceptible to melanoma, but the most likely cause is a suppressed immune system,” Dr Zent noted.
“Normally, in people with healthy immune systems, malignant skin cells might be detected and destroyed before they become a problem. But in CLL patients, failure of this control system increases the rate at which cancer cells can grow into tumors and also the likelihood that they will become invasive or spread to distant sites.”
Detection and management
Fifteen of the 22 melanomas (68.2%) in the CLL cohort were detected via surveillance in a dermatology clinic, and 2 (9.1%) were detected at the CLL/lymphoma clinic.
Three cases of melanoma (14.3%) were detected within the first year of a patient’s CLL diagnosis.
Seven melanomas (33.3%) were detected at pathologic stage 0, 8 (38.1%) at stage I, 2 (9.5%) at stage II, 3 (14.3%) at stage III, and 1 (4.8%) at stage IV. Detailed data were not available for the remaining case.
Melanomas were managed with wide local excision (n=19), sentinel node biopsies (n=6), Mohs surgery (n=1), drugs (n=2), palliative care (n=1), and comfort care (n=1).
The 4 patients who received drugs, palliative care, or comfort care had advanced melanoma.
The patient who received palliative care was still alive at 2.4 years of follow-up. The patient who received comfort care died of metastatic melanoma 1.4 years after diagnosis.
The third patient with advanced melanoma received 2 cycles of dacarbazine and palliative radiation to lung and brain metastases. This patient died 3.6 years after melanoma diagnosis.
The fourth patient received ipilimumab for the melanoma while also receiving ibrutinib to treat her CLL. When the ipilimumab failed, the patient proceeded to pembrolizumab and achieved a near-complete response within 3 months. Then, an intensely hypermetabolic abdominal node was detected and successfully treated with radiation.
The patient continued on pembrolizumab, and her melanoma was in sustained remission at last follow-up, after 23 cycles of pembrolizumab. Her CLL was still responding to ibrutinib at that point as well.
Based on these data, Dr Zent and his colleagues recommend routine melanoma screening for CLL patients. The team believes such surveillance might decrease morbidity and mortality in these patients, although more research is needed to confirm this theory.