User login
Cirrhosis 30-day readmissions down 40% with quality improvement initiative
Using checklists and electronic decision support in an inpatient liver unit, quality improvement (QI) care protocols reduced 30-day readmissions of patients with cirrhosis by 40%, due mostly to a drop in readmissions for hepatic encephalopathy (HE) according to a report published in the May issue of Clinical Gastroenterology and Hepatology.
For patients initially admitted for overt HE, the 30-day readmission rate was 26.0% (27 of 104), compared with 48.9% (66 of 135) before implementation of QI. The proportion of total readmissions due to HE after QI was 9.6% (14 of 146), compared with 40.7% (79 of 194) before QI. In addition, length of stay for HE patients was significantly reduced (–1.34 days; 95% confidence interval, –2.38 to –0.32; P = .01). There were no significant changes in 90-day mortality.
Source: American Gastroenterological Association
“Our study advances the current literature on QI for patients with cirrhosis by presenting an inexpensive, easy to implement, and generalizable approach,” wrote Dr. Elliot Tapper of Beth Israel Deaconess Medical Center, Boston, and his colleagues. Previous studies have addressed readmission interventions among patients with cirrhosis, but the protocols required costly infrastructure, expertise, and institutional commitments. The current study supports the value of standard checklists and education, according to the investigators, “showing that outcomes improve further when checklist items are hard-wired into the ordering system.” (Clin Gastroenterol Hepatol. 2016 Apr 7. doi: 10.1016/j.cgh.2015.08.041).
The QI initiative encompassed several aspects of care. All HE patients were designated to receive rifaximin, and their lactulose dosing was adjusted to mental status using the Richmond Agitation and Sedation Scale. For patients with spontaneous bacterial peritonitis (SBP), timely administration of the correct dose of antibiotics and albumin was promoted, as were prophylactic measures for all patients, such as variceal hemorrhage prophylaxis and subcutaneous heparin for the prevention of venous thrombosis.
The three-part program entailed a run-in phase for preliminary checklist troubleshooting, a hand-held checklist phase, including the HE protocol, SBP treatment, and prophylactic measures, and a final electronic phase in which checklist items were incorporated into the hospital’s electronic provider order entry system using mandatory preset doses and linked medications.
Individual protocol items were demonstrated to affect the readmission rate. Rifaximin use for HE patients rose from 78.1% to 96.3%, and use of rifaximin was associated with lower adjusted odds of 30-day readmission (OR, 0.39; 95% CI, 0.16-0.87; P = .02). The dose/frequency of lactulose for HE patients increased, and patients who had 6 or more cups of lactulose on the day of their readmission had significantly lower adjusted length of stay (–2.36 days; 95% CI, –3.40 to –1.31; P less than .0001). Patients taking SBP prophylaxis had lower readmission rates (OR, 0.51; 95% CI, 0.31-0.83; P = .007).
The prospective study from 2011 to 2013 evaluated patients with cirrhosis who were admitted to the liver unit of Beth Israel Deaconess Medical Center, Boston. Patients were diagnosed with cirrhosis caused by hepatitis C (44.9%), alcoholic liver disease (34%), hepatitis B (5.4%), and biliary cirrhosis (1.8%). In total, 824 unique patients were admitted 1,720 times; 485 (58.9%) were admitted once, 268 (32.5%) were admitted 2-4 times, and 71 (8.6%) were admitted 5 or more times. The median length of stay for all patients was 4.0 days (interquartile range, 2.0-8.0).
Dr. Tapper and his coauthors reported having no disclosures.
Using checklists and electronic decision support in an inpatient liver unit, quality improvement (QI) care protocols reduced 30-day readmissions of patients with cirrhosis by 40%, due mostly to a drop in readmissions for hepatic encephalopathy (HE) according to a report published in the May issue of Clinical Gastroenterology and Hepatology.
For patients initially admitted for overt HE, the 30-day readmission rate was 26.0% (27 of 104), compared with 48.9% (66 of 135) before implementation of QI. The proportion of total readmissions due to HE after QI was 9.6% (14 of 146), compared with 40.7% (79 of 194) before QI. In addition, length of stay for HE patients was significantly reduced (–1.34 days; 95% confidence interval, –2.38 to –0.32; P = .01). There were no significant changes in 90-day mortality.
Source: American Gastroenterological Association
“Our study advances the current literature on QI for patients with cirrhosis by presenting an inexpensive, easy to implement, and generalizable approach,” wrote Dr. Elliot Tapper of Beth Israel Deaconess Medical Center, Boston, and his colleagues. Previous studies have addressed readmission interventions among patients with cirrhosis, but the protocols required costly infrastructure, expertise, and institutional commitments. The current study supports the value of standard checklists and education, according to the investigators, “showing that outcomes improve further when checklist items are hard-wired into the ordering system.” (Clin Gastroenterol Hepatol. 2016 Apr 7. doi: 10.1016/j.cgh.2015.08.041).
The QI initiative encompassed several aspects of care. All HE patients were designated to receive rifaximin, and their lactulose dosing was adjusted to mental status using the Richmond Agitation and Sedation Scale. For patients with spontaneous bacterial peritonitis (SBP), timely administration of the correct dose of antibiotics and albumin was promoted, as were prophylactic measures for all patients, such as variceal hemorrhage prophylaxis and subcutaneous heparin for the prevention of venous thrombosis.
The three-part program entailed a run-in phase for preliminary checklist troubleshooting, a hand-held checklist phase, including the HE protocol, SBP treatment, and prophylactic measures, and a final electronic phase in which checklist items were incorporated into the hospital’s electronic provider order entry system using mandatory preset doses and linked medications.
Individual protocol items were demonstrated to affect the readmission rate. Rifaximin use for HE patients rose from 78.1% to 96.3%, and use of rifaximin was associated with lower adjusted odds of 30-day readmission (OR, 0.39; 95% CI, 0.16-0.87; P = .02). The dose/frequency of lactulose for HE patients increased, and patients who had 6 or more cups of lactulose on the day of their readmission had significantly lower adjusted length of stay (–2.36 days; 95% CI, –3.40 to –1.31; P less than .0001). Patients taking SBP prophylaxis had lower readmission rates (OR, 0.51; 95% CI, 0.31-0.83; P = .007).
The prospective study from 2011 to 2013 evaluated patients with cirrhosis who were admitted to the liver unit of Beth Israel Deaconess Medical Center, Boston. Patients were diagnosed with cirrhosis caused by hepatitis C (44.9%), alcoholic liver disease (34%), hepatitis B (5.4%), and biliary cirrhosis (1.8%). In total, 824 unique patients were admitted 1,720 times; 485 (58.9%) were admitted once, 268 (32.5%) were admitted 2-4 times, and 71 (8.6%) were admitted 5 or more times. The median length of stay for all patients was 4.0 days (interquartile range, 2.0-8.0).
Dr. Tapper and his coauthors reported having no disclosures.
Using checklists and electronic decision support in an inpatient liver unit, quality improvement (QI) care protocols reduced 30-day readmissions of patients with cirrhosis by 40%, due mostly to a drop in readmissions for hepatic encephalopathy (HE) according to a report published in the May issue of Clinical Gastroenterology and Hepatology.
For patients initially admitted for overt HE, the 30-day readmission rate was 26.0% (27 of 104), compared with 48.9% (66 of 135) before implementation of QI. The proportion of total readmissions due to HE after QI was 9.6% (14 of 146), compared with 40.7% (79 of 194) before QI. In addition, length of stay for HE patients was significantly reduced (–1.34 days; 95% confidence interval, –2.38 to –0.32; P = .01). There were no significant changes in 90-day mortality.
Source: American Gastroenterological Association
“Our study advances the current literature on QI for patients with cirrhosis by presenting an inexpensive, easy to implement, and generalizable approach,” wrote Dr. Elliot Tapper of Beth Israel Deaconess Medical Center, Boston, and his colleagues. Previous studies have addressed readmission interventions among patients with cirrhosis, but the protocols required costly infrastructure, expertise, and institutional commitments. The current study supports the value of standard checklists and education, according to the investigators, “showing that outcomes improve further when checklist items are hard-wired into the ordering system.” (Clin Gastroenterol Hepatol. 2016 Apr 7. doi: 10.1016/j.cgh.2015.08.041).
The QI initiative encompassed several aspects of care. All HE patients were designated to receive rifaximin, and their lactulose dosing was adjusted to mental status using the Richmond Agitation and Sedation Scale. For patients with spontaneous bacterial peritonitis (SBP), timely administration of the correct dose of antibiotics and albumin was promoted, as were prophylactic measures for all patients, such as variceal hemorrhage prophylaxis and subcutaneous heparin for the prevention of venous thrombosis.
The three-part program entailed a run-in phase for preliminary checklist troubleshooting, a hand-held checklist phase, including the HE protocol, SBP treatment, and prophylactic measures, and a final electronic phase in which checklist items were incorporated into the hospital’s electronic provider order entry system using mandatory preset doses and linked medications.
Individual protocol items were demonstrated to affect the readmission rate. Rifaximin use for HE patients rose from 78.1% to 96.3%, and use of rifaximin was associated with lower adjusted odds of 30-day readmission (OR, 0.39; 95% CI, 0.16-0.87; P = .02). The dose/frequency of lactulose for HE patients increased, and patients who had 6 or more cups of lactulose on the day of their readmission had significantly lower adjusted length of stay (–2.36 days; 95% CI, –3.40 to –1.31; P less than .0001). Patients taking SBP prophylaxis had lower readmission rates (OR, 0.51; 95% CI, 0.31-0.83; P = .007).
The prospective study from 2011 to 2013 evaluated patients with cirrhosis who were admitted to the liver unit of Beth Israel Deaconess Medical Center, Boston. Patients were diagnosed with cirrhosis caused by hepatitis C (44.9%), alcoholic liver disease (34%), hepatitis B (5.4%), and biliary cirrhosis (1.8%). In total, 824 unique patients were admitted 1,720 times; 485 (58.9%) were admitted once, 268 (32.5%) were admitted 2-4 times, and 71 (8.6%) were admitted 5 or more times. The median length of stay for all patients was 4.0 days (interquartile range, 2.0-8.0).
Dr. Tapper and his coauthors reported having no disclosures.
Key clinical point: Care protocols implemented by electronic decision support reduced 30-day readmissions of patients with cirrhosis by 40% in an inpatient liver unit.
Major finding: The drop was likely driven by fewer readmissions for hepatic encephalopathy (HE): the 30-day HE readmission rate was 26.0% (27 of 104), compared with 48.9% (66 of 135) before implementation of quality improvement.
Data sources: The prospective study evaluated 824 patients who were admitted 1,720 times to the liver unit of Beth Israel Deaconess Medical Center, Boston.
Disclosures: Dr. Tapper and his coauthors reported having no disclosures.

Sitagliptin does not reduce liver fat in NAFLD
BARCELONA – Contrary to expectation, sitagliptin did not reduce liver fat in patients with early diabetes and nonalcoholic fatty liver disease in a double-blind, randomized, placebo-controlled trial.
Changes in liver fat were carefully measured in the trial using the proton density fat fraction (PDFF), a well-validated liver fat biomarker using magnetic resonance imaging (MRI), but no statistical differences between baseline and posttreatment liver fat values were seen in either sitagliptin (18.1% and 16.9%, P = .28) or placebo-treated patients (16.6% and 14.0%, P = .07), or between the two groups (–1.3% difference, P = .41), according to Jeffrey Cui, a medical student at the University of California, San Diego.

In fact, the percentage decrease in liver fat was greater for placebo (13.9%) than for sitagliptin (8.4%), although this was not statistically significant (P = .585), Mr. Cui said at the meeting, sponsored by the European Association for the Study of the Liver.
In addition, there were no changes in liver stiffness, assessed via magnetic resonance elastography (MRE), nor improvements in the liver enzymes aspartate aminotransferase (AST) or alanine aminotransferase (ALT), low-density lipoprotein, or homeostatic model assessment, a measure of insulin resistance.
“Uncontrolled pilot studies have shown that sitagliptin, an oral antihyperglycemic agent that inhibits DPP-4 [dipeptidyl peptidase 4], can improve AST, ALT, and liver histology in NAFLD [nonalcoholic fatty liver disease] patients,” said Mr. Cui.
The aim was to investigate whether sitagliptin could have beneficial effects on the liver by performing a more robustly designed trial and measuring liver fat objectively via MRI-PDFF. The hypothesis was that a 100-mg/day dosage of the DDP4-inhibitor given for 24 weeks would result in a significant decrease in liver fat, compared with placebo, Mr. Cui explained.
Although the results found no benefit for the diabetes drug, the “trial provides a prototype for co-localized assessment of MRI-PDFF and MRE that helps to noninvasively assess treatment response in NAFLD clinical trials,” he said, adding that MRI-PDFF has been shown to correlate well with magnetic resonance spectroscopy, which is a reference standard to quantify liver fat.
A total of 50 patients with pre- or early diabetes (hemoglobin A1c 5.7%-8.0%) with documented hepatic steatosis, defined as an MRI-PDFF of 5% or more, and ALT above the upper limit of normal, were recruited. Half were randomized to sitagliptin and half to placebo, with no major differences in baseline characteristics between the two. The mean age was 52-54 years, and the mean body mass index was 32 mg/m2. Roughly half had a diagnosis of type 2 diabetes.
With treatment given only for 24 weeks, perhaps this was too short to show any effect on a parameter like fibrosis, it was noted during discussion. Mr. Cui said it was unlikely that there would be any benefit from longer treatment with sitagliptin as there was no signal seen in the other liver markers such as AST and ALT.
“It is always disappointing when initial research cannot be replicated in a more rigorous trial setting,” Dr. Frank Tacke of University Hospital Aachen, Germany, said in a press release issued by the EASL, “but we hope that the experiences shared of the imaging techniques employed in this study will help introduce novel, noninvasive imaging techniques into the clinical management of patients.”
Mr. Cui and Dr. Tacke had no conflicts of interest to disclose.
BARCELONA – Contrary to expectation, sitagliptin did not reduce liver fat in patients with early diabetes and nonalcoholic fatty liver disease in a double-blind, randomized, placebo-controlled trial.
Changes in liver fat were carefully measured in the trial using the proton density fat fraction (PDFF), a well-validated liver fat biomarker using magnetic resonance imaging (MRI), but no statistical differences between baseline and posttreatment liver fat values were seen in either sitagliptin (18.1% and 16.9%, P = .28) or placebo-treated patients (16.6% and 14.0%, P = .07), or between the two groups (–1.3% difference, P = .41), according to Jeffrey Cui, a medical student at the University of California, San Diego.
In fact, the percentage decrease in liver fat was greater for placebo (13.9%) than for sitagliptin (8.4%), although this was not statistically significant (P = .585), Mr. Cui said at the meeting, sponsored by the European Association for the Study of the Liver.
In addition, there were no changes in liver stiffness, assessed via magnetic resonance elastography (MRE), nor improvements in the liver enzymes aspartate aminotransferase (AST) or alanine aminotransferase (ALT), low-density lipoprotein, or homeostatic model assessment, a measure of insulin resistance.
“Uncontrolled pilot studies have shown that sitagliptin, an oral antihyperglycemic agent that inhibits DPP-4 [dipeptidyl peptidase 4], can improve AST, ALT, and liver histology in NAFLD [nonalcoholic fatty liver disease] patients,” said Mr. Cui.
The aim was to investigate whether sitagliptin could have beneficial effects on the liver by performing a more robustly designed trial and measuring liver fat objectively via MRI-PDFF. The hypothesis was that a 100-mg/day dosage of the DDP4-inhibitor given for 24 weeks would result in a significant decrease in liver fat, compared with placebo, Mr. Cui explained.
Although the results found no benefit for the diabetes drug, the “trial provides a prototype for co-localized assessment of MRI-PDFF and MRE that helps to noninvasively assess treatment response in NAFLD clinical trials,” he said, adding that MRI-PDFF has been shown to correlate well with magnetic resonance spectroscopy, which is a reference standard to quantify liver fat.
A total of 50 patients with pre- or early diabetes (hemoglobin A1c 5.7%-8.0%) with documented hepatic steatosis, defined as an MRI-PDFF of 5% or more, and ALT above the upper limit of normal, were recruited. Half were randomized to sitagliptin and half to placebo, with no major differences in baseline characteristics between the two. The mean age was 52-54 years, and the mean body mass index was 32 mg/m2. Roughly half had a diagnosis of type 2 diabetes.
With treatment given only for 24 weeks, perhaps this was too short to show any effect on a parameter like fibrosis, it was noted during discussion. Mr. Cui said it was unlikely that there would be any benefit from longer treatment with sitagliptin as there was no signal seen in the other liver markers such as AST and ALT.
“It is always disappointing when initial research cannot be replicated in a more rigorous trial setting,” Dr. Frank Tacke of University Hospital Aachen, Germany, said in a press release issued by the EASL, “but we hope that the experiences shared of the imaging techniques employed in this study will help introduce novel, noninvasive imaging techniques into the clinical management of patients.”
Mr. Cui and Dr. Tacke had no conflicts of interest to disclose.
BARCELONA – Contrary to expectation, sitagliptin did not reduce liver fat in patients with early diabetes and nonalcoholic fatty liver disease in a double-blind, randomized, placebo-controlled trial.
Changes in liver fat were carefully measured in the trial using the proton density fat fraction (PDFF), a well-validated liver fat biomarker using magnetic resonance imaging (MRI), but no statistical differences between baseline and posttreatment liver fat values were seen in either sitagliptin (18.1% and 16.9%, P = .28) or placebo-treated patients (16.6% and 14.0%, P = .07), or between the two groups (–1.3% difference, P = .41), according to Jeffrey Cui, a medical student at the University of California, San Diego.
In fact, the percentage decrease in liver fat was greater for placebo (13.9%) than for sitagliptin (8.4%), although this was not statistically significant (P = .585), Mr. Cui said at the meeting, sponsored by the European Association for the Study of the Liver.
In addition, there were no changes in liver stiffness, assessed via magnetic resonance elastography (MRE), nor improvements in the liver enzymes aspartate aminotransferase (AST) or alanine aminotransferase (ALT), low-density lipoprotein, or homeostatic model assessment, a measure of insulin resistance.
“Uncontrolled pilot studies have shown that sitagliptin, an oral antihyperglycemic agent that inhibits DPP-4 [dipeptidyl peptidase 4], can improve AST, ALT, and liver histology in NAFLD [nonalcoholic fatty liver disease] patients,” said Mr. Cui.
The aim was to investigate whether sitagliptin could have beneficial effects on the liver by performing a more robustly designed trial and measuring liver fat objectively via MRI-PDFF. The hypothesis was that a 100-mg/day dosage of the DDP4-inhibitor given for 24 weeks would result in a significant decrease in liver fat, compared with placebo, Mr. Cui explained.
Although the results found no benefit for the diabetes drug, the “trial provides a prototype for co-localized assessment of MRI-PDFF and MRE that helps to noninvasively assess treatment response in NAFLD clinical trials,” he said, adding that MRI-PDFF has been shown to correlate well with magnetic resonance spectroscopy, which is a reference standard to quantify liver fat.
A total of 50 patients with pre- or early diabetes (hemoglobin A1c 5.7%-8.0%) with documented hepatic steatosis, defined as an MRI-PDFF of 5% or more, and ALT above the upper limit of normal, were recruited. Half were randomized to sitagliptin and half to placebo, with no major differences in baseline characteristics between the two. The mean age was 52-54 years, and the mean body mass index was 32 mg/m2. Roughly half had a diagnosis of type 2 diabetes.
With treatment given only for 24 weeks, perhaps this was too short to show any effect on a parameter like fibrosis, it was noted during discussion. Mr. Cui said it was unlikely that there would be any benefit from longer treatment with sitagliptin as there was no signal seen in the other liver markers such as AST and ALT.
“It is always disappointing when initial research cannot be replicated in a more rigorous trial setting,” Dr. Frank Tacke of University Hospital Aachen, Germany, said in a press release issued by the EASL, “but we hope that the experiences shared of the imaging techniques employed in this study will help introduce novel, noninvasive imaging techniques into the clinical management of patients.”
Mr. Cui and Dr. Tacke had no conflicts of interest to disclose.
AT THE INTERNATIONAL LIVER CONGRESS 2016
Key clinical point: Sitagliptin did not reduce liver fat in patients with early diabetes and nonalcoholic fatty liver disease.
Major finding: The percentage decrease in liver fat was 8.4% for sitagliptin and 13.9% for placebo (P = .585).
Data source: Randomized, double-blind placebo-controlled trial of 50 patients.
Disclosures: Mr. Cui and Dr. Tacke had no conflicts of interest to disclose.

DAAs, HCV-positive livers could reduce transplant waiting list
BARCELONA – Using direct-acting antiviral therapy and livers from HCV-positive donors are separate approaches that could help reduce waiting lists and ensure that patients with HCV in most need get a liver transplant, according to data from two studies presented at the International Liver Congress.
In a retrospective European cohort study, 33% of HCV-positive patients with decompensated cirrhosis who were awaiting a transplant were no longer considered to be in urgent need and almost 20% could be removed from the list altogether 60 weeks after starting treatment with direct-acting antivirals (DAAs).
Dr. Luca Belli of Niguarda Hospital, Milan, who presented the findings at the meeting, cautioned that while the results were encouraging, it is not clear how long the clinical improvement will last. “It will be critical to assess the long-term risks of death, further redeterioration, and developments of hepatocellular carcinoma more specifically, as all these factors still need to be verified,” he said at the meeting, sponsored by the European Association for the Study of the Liver (EASL).
Meanwhile, data from a large analysis of all solid transplant recipients in the United States showed that using livers from HCV-positive donors in HCV-positive recipients was associated with long-term patient outcomes similar to outcomes of using livers from non–HCV-negative donors in HCV-positive recipients, with no difference in mortality or graft survival.

“Over the past 2 decades, the use of HCV-positive organs for liver transplantation has tripled in the United States,” said Maria Stepanova, Ph.D., a senior biostatistician for Inova Health System in Falls Church, Va., who presented her findings at the meeting.
“Despite this, the medium- to long-term outcomes of HCV-positive liver transplant recipients transplanted from HCV-positive donors were not affected by HCV-positivity of a donor,” added Dr. Stepanova, who also works at the Center for Outcomes Research in Liver Disease in Washington, D.C.
Dr. Zobair Younossi, chairman of the department of medicine at Inova Fairfax (Va.) Hospital, and coauthor of the study, said during a press briefing that this does not mean that livers from HCV-positive donor could be used in HCV-negative recipients. The reason for that is that it would be causing an acute infection regardless of whether or not antiviral treatment is available and “at this point the evidence is not there,” he said.
Dr. Laurent Castera of Hôpital Beaujon in Paris, and Dr. Tom Hemming Karlsen of Oslo University Hospital Rikshospitalet in Norway commented on the significance of these data in press releases issued by the EASL. Both experts, who were not involved in the studies, noted the findings could help take the strain off the liver transplant list in the future.
“Treating patients with direct-acting antiviral therapy could result in those with a more pressing need for a liver transplant receiving the donation they need, potentially reducing the number of deaths that occur on the waiting list,” Dr. Castera said.
“With the number of people waiting for a liver transplant expected to rise, the study results should give hope over the coming years for those on the waiting list,” Dr. Karlsen said. Referring to the U.S. study, he said the results “clearly demonstrate a greater opportunity for use of HCV-positive livers over the coming years due to their comparable outcomes with healthy livers.”
The European study presented by Dr. Belli involved 134 patients with HCV and decompensated cirrhosis but without hepatocellular carcinoma listed for liver transplant between February 2014 and February 2015 at 11 centers in Austria, Italy, and France. Of these patients, 103 had been treated with DAAs while on the transplant list; approximately half had been treated with a single DAA (sofosbuvir plus ribavirin for 24-48 weeks) and half with two DAAs (sofosbuvir with either daclatasvir or ledipasvir for 12-24 weeks).
The primary endpoint was the probability of being “inactivated” and then “delisted.” Inactivated meant that there was clinical improvement resulting in patients being put “on hold,” and “delisted” was defined as patients being taken off the transplant list after a variable period of inactivation.
The median age of patients in the study was 54 years and 68% were male. Just under 50% of patients had a low (less than 16) MELD score, around 37% had a MELD score of 16-20, and 13% had a MELD score of more than 20. Around 45% had Child-Pugh B and 55% had Child-Pugh C cirrhosis. Two-thirds had medically controlled and 26% had medically uncontrolled ascites, and 46% had medically controlled and 1% had medically uncontrolled hepatic encephalopathy.

Good virologic efficacy was seen with both single- and dual-agent DAA therapy, with rapid virologic responses of 61% and 67% and early virologic responses of 98% and 98%. Of the 52 patients given single-agent DAA treatment, 22 had a transplant and one patient had a posttransplant relapse. Of the remaining 30 patients on the transplant list, four relapsed. Nineteen of 51 patients given dual-DAA therapy were transplanted and there was one relapse, with no relapses in the 32 patients who remained on the transplant list.
After about 60 weeks of follow-up, one in three patients were “inactivated” and not considered in urgent need of a transplant, but inactivation occurred as early as 12 weeks after DAA therapy, Dr. Belli observed.
Inactivation was associated with a 3.3 decrease in MELD score, a 2-point reduction in Child-Turcotte-Pugh score, and a 0.5-g/dL increase in serum albumin after 24 weeks. There was also regression or improvement in ascites and hepatic encephalopathy in most patients. The median time of delisting was 48 weeks.
These results suggest that there could be a wider role for DAA therapy even in the sickest of HCV-positive patients who are urgently awaiting a liver transplant. Another approach to increase the number of people receiving a transplant is to use HCV-positive livers. The practice has been increasing over the years, but it is not known if the approach is safe and if the risks outweigh the benefits.
To investigate, Dr. Stepanova and associates obtained data from the Scientific Registry of Transplant Recipients (SRTR) on all liver transplants performed in the United States between 1995 and 2003 involving HCV-positive patients. A total of 37,317 records were found, of which 33,668 had data on the donors’ HCV status and on the recipients’ mortality status. Of these, 1,930 (5.7%) had received a liver from an HCV-positive donor. Dr. Stepanova noted that there had been an increase in the percentage of patients who had received an HCV-positive liver, from less than 3% in 1995 to more than 9% in 2013.
Compared with HCV-negative donors, HCV-positive donors were older, were more likely to have a history of drug abuse, and more likely to be non–heart beating at the time of procurement.
The HCV-positive liver recipients also tended to be older, to be of African-American ethnicity, and to have liver cancer but lower MELD scores than those who received an HCV-negative liver. “So these are patients that cannot wait for a long time so maybe elect to use an HCV-positive donor,” Dr. Younossi said.
Adjusted hazard ratios (aHR) showed no statistically significant difference in posttransplant survival or posttransplant graft loss between HCV-positive and HCV-negative livers; aHR were a respective 1.03 and 0.905, with tight 95% confidence intervals. However, a more recent year of transplant did appear to suggest a possible advantage of using an HCV-positive donor liver in an HCV-positive patient, with lower mortality (aHR = 0.978 per year, P less than .0001) and graft failure (aHR = 0.960 per year, P less than .0001) rates.
While the use of HCV-positive livers in HCV-positive recipients was felt to be “reasonably safe,” these findings “cannot be used in support of indiscriminate use of HCV-positive donors,” Dr. Stepanova observed. Further studies are needed to establish criteria on which to select donors that would provide patients with the best possible risk-to-benefit ratio, she said.
Further avenues for research would also be to see if HCV-positive livers could be given to HCV-negative patients and if genotype matters. It would also need to be seen if posttransplantation antiviral treatment would be necessary.
Dr. Belli has received research support from Gilead, AbbVie and BMS and acted as a consultant to Gilead. Dr. Stepanova did not have conflicts of interest to disclose. Dr. Younossi has acted as a consultant to BMS, AbbVie, Gilead, GlaxoSmithKline, and Intercept. Dr. Castera and Dr. Karlsen had no conflicts of interest to disclose.
BARCELONA – Using direct-acting antiviral therapy and livers from HCV-positive donors are separate approaches that could help reduce waiting lists and ensure that patients with HCV in most need get a liver transplant, according to data from two studies presented at the International Liver Congress.
In a retrospective European cohort study, 33% of HCV-positive patients with decompensated cirrhosis who were awaiting a transplant were no longer considered to be in urgent need and almost 20% could be removed from the list altogether 60 weeks after starting treatment with direct-acting antivirals (DAAs).
Dr. Luca Belli of Niguarda Hospital, Milan, who presented the findings at the meeting, cautioned that while the results were encouraging, it is not clear how long the clinical improvement will last. “It will be critical to assess the long-term risks of death, further redeterioration, and developments of hepatocellular carcinoma more specifically, as all these factors still need to be verified,” he said at the meeting, sponsored by the European Association for the Study of the Liver (EASL).
Meanwhile, data from a large analysis of all solid transplant recipients in the United States showed that using livers from HCV-positive donors in HCV-positive recipients was associated with long-term patient outcomes similar to outcomes of using livers from non–HCV-negative donors in HCV-positive recipients, with no difference in mortality or graft survival.
“Over the past 2 decades, the use of HCV-positive organs for liver transplantation has tripled in the United States,” said Maria Stepanova, Ph.D., a senior biostatistician for Inova Health System in Falls Church, Va., who presented her findings at the meeting.
“Despite this, the medium- to long-term outcomes of HCV-positive liver transplant recipients transplanted from HCV-positive donors were not affected by HCV-positivity of a donor,” added Dr. Stepanova, who also works at the Center for Outcomes Research in Liver Disease in Washington, D.C.
Dr. Zobair Younossi, chairman of the department of medicine at Inova Fairfax (Va.) Hospital, and coauthor of the study, said during a press briefing that this does not mean that livers from HCV-positive donor could be used in HCV-negative recipients. The reason for that is that it would be causing an acute infection regardless of whether or not antiviral treatment is available and “at this point the evidence is not there,” he said.
Dr. Laurent Castera of Hôpital Beaujon in Paris, and Dr. Tom Hemming Karlsen of Oslo University Hospital Rikshospitalet in Norway commented on the significance of these data in press releases issued by the EASL. Both experts, who were not involved in the studies, noted the findings could help take the strain off the liver transplant list in the future.
“Treating patients with direct-acting antiviral therapy could result in those with a more pressing need for a liver transplant receiving the donation they need, potentially reducing the number of deaths that occur on the waiting list,” Dr. Castera said.
“With the number of people waiting for a liver transplant expected to rise, the study results should give hope over the coming years for those on the waiting list,” Dr. Karlsen said. Referring to the U.S. study, he said the results “clearly demonstrate a greater opportunity for use of HCV-positive livers over the coming years due to their comparable outcomes with healthy livers.”
The European study presented by Dr. Belli involved 134 patients with HCV and decompensated cirrhosis but without hepatocellular carcinoma listed for liver transplant between February 2014 and February 2015 at 11 centers in Austria, Italy, and France. Of these patients, 103 had been treated with DAAs while on the transplant list; approximately half had been treated with a single DAA (sofosbuvir plus ribavirin for 24-48 weeks) and half with two DAAs (sofosbuvir with either daclatasvir or ledipasvir for 12-24 weeks).
The primary endpoint was the probability of being “inactivated” and then “delisted.” Inactivated meant that there was clinical improvement resulting in patients being put “on hold,” and “delisted” was defined as patients being taken off the transplant list after a variable period of inactivation.
The median age of patients in the study was 54 years and 68% were male. Just under 50% of patients had a low (less than 16) MELD score, around 37% had a MELD score of 16-20, and 13% had a MELD score of more than 20. Around 45% had Child-Pugh B and 55% had Child-Pugh C cirrhosis. Two-thirds had medically controlled and 26% had medically uncontrolled ascites, and 46% had medically controlled and 1% had medically uncontrolled hepatic encephalopathy.
Good virologic efficacy was seen with both single- and dual-agent DAA therapy, with rapid virologic responses of 61% and 67% and early virologic responses of 98% and 98%. Of the 52 patients given single-agent DAA treatment, 22 had a transplant and one patient had a posttransplant relapse. Of the remaining 30 patients on the transplant list, four relapsed. Nineteen of 51 patients given dual-DAA therapy were transplanted and there was one relapse, with no relapses in the 32 patients who remained on the transplant list.
After about 60 weeks of follow-up, one in three patients were “inactivated” and not considered in urgent need of a transplant, but inactivation occurred as early as 12 weeks after DAA therapy, Dr. Belli observed.
Inactivation was associated with a 3.3 decrease in MELD score, a 2-point reduction in Child-Turcotte-Pugh score, and a 0.5-g/dL increase in serum albumin after 24 weeks. There was also regression or improvement in ascites and hepatic encephalopathy in most patients. The median time of delisting was 48 weeks.
These results suggest that there could be a wider role for DAA therapy even in the sickest of HCV-positive patients who are urgently awaiting a liver transplant. Another approach to increase the number of people receiving a transplant is to use HCV-positive livers. The practice has been increasing over the years, but it is not known if the approach is safe and if the risks outweigh the benefits.
To investigate, Dr. Stepanova and associates obtained data from the Scientific Registry of Transplant Recipients (SRTR) on all liver transplants performed in the United States between 1995 and 2003 involving HCV-positive patients. A total of 37,317 records were found, of which 33,668 had data on the donors’ HCV status and on the recipients’ mortality status. Of these, 1,930 (5.7%) had received a liver from an HCV-positive donor. Dr. Stepanova noted that there had been an increase in the percentage of patients who had received an HCV-positive liver, from less than 3% in 1995 to more than 9% in 2013.
Compared with HCV-negative donors, HCV-positive donors were older, were more likely to have a history of drug abuse, and more likely to be non–heart beating at the time of procurement.
The HCV-positive liver recipients also tended to be older, to be of African-American ethnicity, and to have liver cancer but lower MELD scores than those who received an HCV-negative liver. “So these are patients that cannot wait for a long time so maybe elect to use an HCV-positive donor,” Dr. Younossi said.
Adjusted hazard ratios (aHR) showed no statistically significant difference in posttransplant survival or posttransplant graft loss between HCV-positive and HCV-negative livers; aHR were a respective 1.03 and 0.905, with tight 95% confidence intervals. However, a more recent year of transplant did appear to suggest a possible advantage of using an HCV-positive donor liver in an HCV-positive patient, with lower mortality (aHR = 0.978 per year, P less than .0001) and graft failure (aHR = 0.960 per year, P less than .0001) rates.
While the use of HCV-positive livers in HCV-positive recipients was felt to be “reasonably safe,” these findings “cannot be used in support of indiscriminate use of HCV-positive donors,” Dr. Stepanova observed. Further studies are needed to establish criteria on which to select donors that would provide patients with the best possible risk-to-benefit ratio, she said.
Further avenues for research would also be to see if HCV-positive livers could be given to HCV-negative patients and if genotype matters. It would also need to be seen if posttransplantation antiviral treatment would be necessary.
Dr. Belli has received research support from Gilead, AbbVie and BMS and acted as a consultant to Gilead. Dr. Stepanova did not have conflicts of interest to disclose. Dr. Younossi has acted as a consultant to BMS, AbbVie, Gilead, GlaxoSmithKline, and Intercept. Dr. Castera and Dr. Karlsen had no conflicts of interest to disclose.
BARCELONA – Using direct-acting antiviral therapy and livers from HCV-positive donors are separate approaches that could help reduce waiting lists and ensure that patients with HCV in most need get a liver transplant, according to data from two studies presented at the International Liver Congress.
In a retrospective European cohort study, 33% of HCV-positive patients with decompensated cirrhosis who were awaiting a transplant were no longer considered to be in urgent need and almost 20% could be removed from the list altogether 60 weeks after starting treatment with direct-acting antivirals (DAAs).
Dr. Luca Belli of Niguarda Hospital, Milan, who presented the findings at the meeting, cautioned that while the results were encouraging, it is not clear how long the clinical improvement will last. “It will be critical to assess the long-term risks of death, further redeterioration, and developments of hepatocellular carcinoma more specifically, as all these factors still need to be verified,” he said at the meeting, sponsored by the European Association for the Study of the Liver (EASL).
Meanwhile, data from a large analysis of all solid transplant recipients in the United States showed that using livers from HCV-positive donors in HCV-positive recipients was associated with long-term patient outcomes similar to outcomes of using livers from non–HCV-negative donors in HCV-positive recipients, with no difference in mortality or graft survival.
“Over the past 2 decades, the use of HCV-positive organs for liver transplantation has tripled in the United States,” said Maria Stepanova, Ph.D., a senior biostatistician for Inova Health System in Falls Church, Va., who presented her findings at the meeting.
“Despite this, the medium- to long-term outcomes of HCV-positive liver transplant recipients transplanted from HCV-positive donors were not affected by HCV-positivity of a donor,” added Dr. Stepanova, who also works at the Center for Outcomes Research in Liver Disease in Washington, D.C.
Dr. Zobair Younossi, chairman of the department of medicine at Inova Fairfax (Va.) Hospital, and coauthor of the study, said during a press briefing that this does not mean that livers from HCV-positive donor could be used in HCV-negative recipients. The reason for that is that it would be causing an acute infection regardless of whether or not antiviral treatment is available and “at this point the evidence is not there,” he said.
Dr. Laurent Castera of Hôpital Beaujon in Paris, and Dr. Tom Hemming Karlsen of Oslo University Hospital Rikshospitalet in Norway commented on the significance of these data in press releases issued by the EASL. Both experts, who were not involved in the studies, noted the findings could help take the strain off the liver transplant list in the future.
“Treating patients with direct-acting antiviral therapy could result in those with a more pressing need for a liver transplant receiving the donation they need, potentially reducing the number of deaths that occur on the waiting list,” Dr. Castera said.
“With the number of people waiting for a liver transplant expected to rise, the study results should give hope over the coming years for those on the waiting list,” Dr. Karlsen said. Referring to the U.S. study, he said the results “clearly demonstrate a greater opportunity for use of HCV-positive livers over the coming years due to their comparable outcomes with healthy livers.”
The European study presented by Dr. Belli involved 134 patients with HCV and decompensated cirrhosis but without hepatocellular carcinoma listed for liver transplant between February 2014 and February 2015 at 11 centers in Austria, Italy, and France. Of these patients, 103 had been treated with DAAs while on the transplant list; approximately half had been treated with a single DAA (sofosbuvir plus ribavirin for 24-48 weeks) and half with two DAAs (sofosbuvir with either daclatasvir or ledipasvir for 12-24 weeks).
The primary endpoint was the probability of being “inactivated” and then “delisted.” Inactivated meant that there was clinical improvement resulting in patients being put “on hold,” and “delisted” was defined as patients being taken off the transplant list after a variable period of inactivation.
The median age of patients in the study was 54 years and 68% were male. Just under 50% of patients had a low (less than 16) MELD score, around 37% had a MELD score of 16-20, and 13% had a MELD score of more than 20. Around 45% had Child-Pugh B and 55% had Child-Pugh C cirrhosis. Two-thirds had medically controlled and 26% had medically uncontrolled ascites, and 46% had medically controlled and 1% had medically uncontrolled hepatic encephalopathy.
Good virologic efficacy was seen with both single- and dual-agent DAA therapy, with rapid virologic responses of 61% and 67% and early virologic responses of 98% and 98%. Of the 52 patients given single-agent DAA treatment, 22 had a transplant and one patient had a posttransplant relapse. Of the remaining 30 patients on the transplant list, four relapsed. Nineteen of 51 patients given dual-DAA therapy were transplanted and there was one relapse, with no relapses in the 32 patients who remained on the transplant list.
After about 60 weeks of follow-up, one in three patients were “inactivated” and not considered in urgent need of a transplant, but inactivation occurred as early as 12 weeks after DAA therapy, Dr. Belli observed.
Inactivation was associated with a 3.3 decrease in MELD score, a 2-point reduction in Child-Turcotte-Pugh score, and a 0.5-g/dL increase in serum albumin after 24 weeks. There was also regression or improvement in ascites and hepatic encephalopathy in most patients. The median time of delisting was 48 weeks.
These results suggest that there could be a wider role for DAA therapy even in the sickest of HCV-positive patients who are urgently awaiting a liver transplant. Another approach to increase the number of people receiving a transplant is to use HCV-positive livers. The practice has been increasing over the years, but it is not known if the approach is safe and if the risks outweigh the benefits.
To investigate, Dr. Stepanova and associates obtained data from the Scientific Registry of Transplant Recipients (SRTR) on all liver transplants performed in the United States between 1995 and 2003 involving HCV-positive patients. A total of 37,317 records were found, of which 33,668 had data on the donors’ HCV status and on the recipients’ mortality status. Of these, 1,930 (5.7%) had received a liver from an HCV-positive donor. Dr. Stepanova noted that there had been an increase in the percentage of patients who had received an HCV-positive liver, from less than 3% in 1995 to more than 9% in 2013.
Compared with HCV-negative donors, HCV-positive donors were older, were more likely to have a history of drug abuse, and more likely to be non–heart beating at the time of procurement.
The HCV-positive liver recipients also tended to be older, to be of African-American ethnicity, and to have liver cancer but lower MELD scores than those who received an HCV-negative liver. “So these are patients that cannot wait for a long time so maybe elect to use an HCV-positive donor,” Dr. Younossi said.
Adjusted hazard ratios (aHR) showed no statistically significant difference in posttransplant survival or posttransplant graft loss between HCV-positive and HCV-negative livers; aHR were a respective 1.03 and 0.905, with tight 95% confidence intervals. However, a more recent year of transplant did appear to suggest a possible advantage of using an HCV-positive donor liver in an HCV-positive patient, with lower mortality (aHR = 0.978 per year, P less than .0001) and graft failure (aHR = 0.960 per year, P less than .0001) rates.
While the use of HCV-positive livers in HCV-positive recipients was felt to be “reasonably safe,” these findings “cannot be used in support of indiscriminate use of HCV-positive donors,” Dr. Stepanova observed. Further studies are needed to establish criteria on which to select donors that would provide patients with the best possible risk-to-benefit ratio, she said.
Further avenues for research would also be to see if HCV-positive livers could be given to HCV-negative patients and if genotype matters. It would also need to be seen if posttransplantation antiviral treatment would be necessary.
Dr. Belli has received research support from Gilead, AbbVie and BMS and acted as a consultant to Gilead. Dr. Stepanova did not have conflicts of interest to disclose. Dr. Younossi has acted as a consultant to BMS, AbbVie, Gilead, GlaxoSmithKline, and Intercept. Dr. Castera and Dr. Karlsen had no conflicts of interest to disclose.
AT THE INTERNATIONAL LIVER CONGRESS 2016
Key clinical point: Using direct-acting antiviral (DAA) therapy and HCV-positive livers could help reduce the strain on liver transplant lists.
Major finding: One in three HCV-positive patients treated with DAAs were considered nonurgent cases and one in five could be delisted. HCV-positive livers were associated with similar graft and host survival.
Data source: A retrospective European cohort study of 103 patients with decompensated cirrhosis who were treated with DAAs while awaiting liver transplant and an analysis of more than 33,000 HCV patients awaiting a liver transplant in the United States.
Disclosures: Dr. Belli has received research support from Gilead, AbbVie, and BMS and acted as a consultant to Gilead. Dr. Stepanova did not have conflicts of interest to disclose. Dr. Younossi has acted as a consultant to BMS, AbbVie, Gilead, GlaxoSmithKline, and Intercept. Dr. Castera and Dr. Karlsen had no conflicts of interest to disclose.

Hepatitis B test identifies active carriers
A one-time test combining hepatitis B virus (HBV) surface antigen and HBV DNA measurement accurately distinguished inactive carriers from active cases of chronic HBV, based on data from 1,529 patients published online in the journal Hepatology.
Correct identification of active chronic HBV patients vs. inactive cases “is a crucial step in identifying patients in need of less stringent follow-up, as well as patients who may benefit from additional follow-up and earlier initiation of antiviral therapy,” wrote Dr. Jessica Liu of the Genomics Research Center, Academia Sinica, in Taipei, Taiwan, and her colleagues (Hepatology. 2016. doi: 10.1002/hep.28552).
The investigators reviewed data from patients in the REVEAL-HBV cohort, which included individuals aged 30-65 years with chronic HBV who were recruited between 1991 and 1992. All patients were free of liver cirrhosis and were seronegative for anti–hepatitis C virus and hepatitis B surface antigen (HBsAg) at baseline, and were characterized as inactive carriers or active cases based on their serologic profiles at 18 months’ follow-up. Blood was collected at study entry and every 6-12 months.
At 18 months’ follow-up, the combined test distinguished inactive carriers from active cases with a sensitivity of 71% and a specificity of 85%. The positive and negative predictive values were 83% and 74%, respectively, and the diagnostic accuracy was 78%. In addition, the one-time combination measurement predicted cirrhosis, hepatocelluar carcinoma, and HBsAg seroclearance with areas under the receiver operating characteristic curves of 0.72, 0.79, and 0.78, respectively. The combination test also yielded information about the predictability of inactive carrier status, showing that inactive carriers had a significantly lower risk of cirrhosis and hepatocelluar carcinoma; adjusted hazard ratios were 0.43 and 0.13, respectively.
“To our knowledge, this is the first study to externally validate the usage of a one-time measurement of serum HBsAg less than 1,000 IU/mL and HBV DNA less than 2,000 IU/mL,” the researchers wrote.
The study was limited by a relatively short follow-up period, but the results suggest that “this single-point strategy may provide new and complementary information useful for simplifying or tailoring management of patients with chronic hepatitis B infection,” they said.
The study was supported by the Taiwan Department of Health, Bristol-Myers Squibb, Roche Diagnostics, and Academia Sinica. One of the coauthors is employed by Roche Diagnostics.
A one-time test combining hepatitis B virus (HBV) surface antigen and HBV DNA measurement accurately distinguished inactive carriers from active cases of chronic HBV, based on data from 1,529 patients published online in the journal Hepatology.
Correct identification of active chronic HBV patients vs. inactive cases “is a crucial step in identifying patients in need of less stringent follow-up, as well as patients who may benefit from additional follow-up and earlier initiation of antiviral therapy,” wrote Dr. Jessica Liu of the Genomics Research Center, Academia Sinica, in Taipei, Taiwan, and her colleagues (Hepatology. 2016. doi: 10.1002/hep.28552).
The investigators reviewed data from patients in the REVEAL-HBV cohort, which included individuals aged 30-65 years with chronic HBV who were recruited between 1991 and 1992. All patients were free of liver cirrhosis and were seronegative for anti–hepatitis C virus and hepatitis B surface antigen (HBsAg) at baseline, and were characterized as inactive carriers or active cases based on their serologic profiles at 18 months’ follow-up. Blood was collected at study entry and every 6-12 months.
At 18 months’ follow-up, the combined test distinguished inactive carriers from active cases with a sensitivity of 71% and a specificity of 85%. The positive and negative predictive values were 83% and 74%, respectively, and the diagnostic accuracy was 78%. In addition, the one-time combination measurement predicted cirrhosis, hepatocelluar carcinoma, and HBsAg seroclearance with areas under the receiver operating characteristic curves of 0.72, 0.79, and 0.78, respectively. The combination test also yielded information about the predictability of inactive carrier status, showing that inactive carriers had a significantly lower risk of cirrhosis and hepatocelluar carcinoma; adjusted hazard ratios were 0.43 and 0.13, respectively.
“To our knowledge, this is the first study to externally validate the usage of a one-time measurement of serum HBsAg less than 1,000 IU/mL and HBV DNA less than 2,000 IU/mL,” the researchers wrote.
The study was limited by a relatively short follow-up period, but the results suggest that “this single-point strategy may provide new and complementary information useful for simplifying or tailoring management of patients with chronic hepatitis B infection,” they said.
The study was supported by the Taiwan Department of Health, Bristol-Myers Squibb, Roche Diagnostics, and Academia Sinica. One of the coauthors is employed by Roche Diagnostics.
A one-time test combining hepatitis B virus (HBV) surface antigen and HBV DNA measurement accurately distinguished inactive carriers from active cases of chronic HBV, based on data from 1,529 patients published online in the journal Hepatology.
Correct identification of active chronic HBV patients vs. inactive cases “is a crucial step in identifying patients in need of less stringent follow-up, as well as patients who may benefit from additional follow-up and earlier initiation of antiviral therapy,” wrote Dr. Jessica Liu of the Genomics Research Center, Academia Sinica, in Taipei, Taiwan, and her colleagues (Hepatology. 2016. doi: 10.1002/hep.28552).
The investigators reviewed data from patients in the REVEAL-HBV cohort, which included individuals aged 30-65 years with chronic HBV who were recruited between 1991 and 1992. All patients were free of liver cirrhosis and were seronegative for anti–hepatitis C virus and hepatitis B surface antigen (HBsAg) at baseline, and were characterized as inactive carriers or active cases based on their serologic profiles at 18 months’ follow-up. Blood was collected at study entry and every 6-12 months.
At 18 months’ follow-up, the combined test distinguished inactive carriers from active cases with a sensitivity of 71% and a specificity of 85%. The positive and negative predictive values were 83% and 74%, respectively, and the diagnostic accuracy was 78%. In addition, the one-time combination measurement predicted cirrhosis, hepatocelluar carcinoma, and HBsAg seroclearance with areas under the receiver operating characteristic curves of 0.72, 0.79, and 0.78, respectively. The combination test also yielded information about the predictability of inactive carrier status, showing that inactive carriers had a significantly lower risk of cirrhosis and hepatocelluar carcinoma; adjusted hazard ratios were 0.43 and 0.13, respectively.
“To our knowledge, this is the first study to externally validate the usage of a one-time measurement of serum HBsAg less than 1,000 IU/mL and HBV DNA less than 2,000 IU/mL,” the researchers wrote.
The study was limited by a relatively short follow-up period, but the results suggest that “this single-point strategy may provide new and complementary information useful for simplifying or tailoring management of patients with chronic hepatitis B infection,” they said.
The study was supported by the Taiwan Department of Health, Bristol-Myers Squibb, Roche Diagnostics, and Academia Sinica. One of the coauthors is employed by Roche Diagnostics.
FROM HEPATOLOGY
Key clinical point: The data confirm the predictability of a one-time test for chronic hepatitis B progression that combines hepatitis B surface antigen and hepatitis B DNA measurement.
Major finding: The one-time combined test distinguished inactive carriers from active cases with a sensitivity of 71% and specificity of 85%.
Data source: A study of 1,529 adults aged 30-65 years with chronic hepatitis B.
Disclosures: The study was supported by the Taiwan Department of Health, Bristol-Myers Squibb, Roche Diagnostics, and Academia Sinica. One of the coauthors is employed by Roche Diagnostics.
Investigational HBV core inhibitor shows early clinical promise
BARCELONA – An investigational drug that targets the hepatitis B virus (HBV) core protein is a “totally new approach” to treating chronic HBV infection according to the results of an early-phase clinical study.
In an ongoing, international, phase I study presented during the late-breaker session at the International Liver Congress, 4 weeks of treatment with NVR 3-778 was well tolerated and did not result in any drug discontinuations in chronically infected patients. There were dose-related reductions in HBV DNA and early reductions in HBeAg were observed.

“NVR 3-778 is a first-in-class HBV core inhibitor,” study investigator Dr. Man-Fung Yuen, Queen Mary Hospital, Hong Kong, said at a press briefing held at the meeting, which is sponsored by the European Association for the Study of the Liver (EASL). “The HBV core protein has many functions and is responsible for the most important pathway of the virus’ replication – nucleocapsid formation,” he explained.
The rationale behind its development is that although there are effective treatments such as nucleoside and nucleotide analogs (NAs) and pegylated interferon (peg-IFN) that can suppress the activity of HBV for many years and thus slow down damage to the liver, it is unusual to clear the virus permanently. Furthermore, as most patients require potentially life-long treatment, issues such as long-term safety, patients’ adherence, resistance, and cost of therapy come into play.
Dr. Yuen observed that achieving durable responses in patients with chronic HBV is likely to require combinations of potential agents, probably with complementary modes of action.
Preclinical data have shown that NVR 3-778 induces the rapid assembly of nonfunctional capsids and that it inhibits viral replication in a humanized mouse hepatocyte cell model. It has also been shown to reduce HBV DNA to a comparable extent as entecavir when used alone in a mouse model, with undetectable levels reached when used in combination with peg-IFN alpha-2a.
Dr. Yuen reported the preliminary results from the second part of the phase I study in 64 adults with chronic HBV, noting that the first part of the study in healthy volunteers had been completed successfully.
The aim was to examine the safety and efficacy of NVR 3-778 alone and in combination with peg-IFN alpha-2a in previously untreated patients aged between 18 and 65 years who were HBsAg- and HbeAg-positive, had HBV DNA of 20,000 or more copies per IU/mL, with no cirrhosis of the liver, as determined by liver biopsy or imaging.
Patients were randomly assigned to six groups, with four groups receiving the investigational therapy alone at four increasing doses (100, 200, 400, once daily and 600 mg twice daily). There was a staged initiation of each escalating dose of NVR 3-778 used in these groups, with the 100 mg initiated first and if, after 2 weeks, there were no interim safety concerns, the next dose groups, 200 mg, then 400 mg, then 600 mg were started. The other two groups of patients received NVR 3-788 (600 mg) with peg-IFN alpha-2a (180 microg/week) or peg-IFN alpha-2a plus placebo. Treatment for all groups was for 28 days with 28 days of follow-up.
The mean age of patients in each group ranged from 32 to 48 years, with the older patients more likely to be treated with peg-IFN alpha-2a alone or with NVR 3-788. Most of the patients studied were Chinese and the mean baseline HBV DNA was 7.5-8.3 log10 IU/mL.
A total of 98 adverse events were seen in 40 of 64 patients treated, although there was not any trend for more effects with the investigational treatment versus placebo as doses escalated. There were 21 adverse events seen in 20 of 46 patients that were possibly or probably related to the study drug, of which most were grade 1 nausea. Grade 2 nausea occurred only once in a patient given the 100-mg dose. There was one serious adverse event, a grade 3 papulovesicular rash on the hands and feet that occurred in a patient in the 100-mg group, but this responded to therapy. This side effect started about 20 days after treatment and resolved over a period of 7 months.
Although generally mild, more adverse effects were seen in the patients treated with a combination of NVR 3-788 and peg-IFN alpha-2a, which Dr. Yuen noted was thought to be more likely a result of the IFN therapy.
There was not much difference in the mean change in HBV DNA from baseline for the 100-400-mg groups, but there was a 1.72-log10 IU/mL decrease achieved with the 600-mg twice-daily dose. The greatest reduction, however, was seen when NVR 3-788 was combined with peg-IFN alpha-2a, with a 1.97-log10 IU/mL change.
“Encouraging early reductions in HBV RNA and quantitative serum HBeAg were observed,” Dr. Yuen said. He noted that the longer-term safety and efficacy of the novel agent in combination with peg-IFN alpha-2a and first-line HBV NAs will now be further assessed in phase II studies.
“At the moment we do not have a cure for hepatitis B, only treatment,” said Dr. Frank Tacke of the University Hospital Aachen (Germany), who chaired the press briefing where Dr. Yuen first aired the findings. NVR 3-788 represents “a totally new approach to this disease that we hope will lead to a cure,” Dr. Tacke said.
In an EASL-issued press release, Dr. Tacke also noted: “The results from this study are certainly interesting and promising for the treatment of patients with hepatitis B. The medical community is always on the lookout for treatments which can cure this condition, as opposed to simply suppressing the replication of the virus. More research is needed to confirm whether NVR 3-778 could really change the treatment paradigm.”
Novira Therapeutics financed the study. Dr. Yuen has been a consultant, speaker, or both for Arrowhead, Bristol-Myers Squibb, GlaxoSmithKline, Gilead, Novartis, and Roche. Dr. Tacke did not have any relevant disclosures.
BARCELONA – An investigational drug that targets the hepatitis B virus (HBV) core protein is a “totally new approach” to treating chronic HBV infection according to the results of an early-phase clinical study.
In an ongoing, international, phase I study presented during the late-breaker session at the International Liver Congress, 4 weeks of treatment with NVR 3-778 was well tolerated and did not result in any drug discontinuations in chronically infected patients. There were dose-related reductions in HBV DNA and early reductions in HBeAg were observed.
“NVR 3-778 is a first-in-class HBV core inhibitor,” study investigator Dr. Man-Fung Yuen, Queen Mary Hospital, Hong Kong, said at a press briefing held at the meeting, which is sponsored by the European Association for the Study of the Liver (EASL). “The HBV core protein has many functions and is responsible for the most important pathway of the virus’ replication – nucleocapsid formation,” he explained.
The rationale behind its development is that although there are effective treatments such as nucleoside and nucleotide analogs (NAs) and pegylated interferon (peg-IFN) that can suppress the activity of HBV for many years and thus slow down damage to the liver, it is unusual to clear the virus permanently. Furthermore, as most patients require potentially life-long treatment, issues such as long-term safety, patients’ adherence, resistance, and cost of therapy come into play.
Dr. Yuen observed that achieving durable responses in patients with chronic HBV is likely to require combinations of potential agents, probably with complementary modes of action.
Preclinical data have shown that NVR 3-778 induces the rapid assembly of nonfunctional capsids and that it inhibits viral replication in a humanized mouse hepatocyte cell model. It has also been shown to reduce HBV DNA to a comparable extent as entecavir when used alone in a mouse model, with undetectable levels reached when used in combination with peg-IFN alpha-2a.
Dr. Yuen reported the preliminary results from the second part of the phase I study in 64 adults with chronic HBV, noting that the first part of the study in healthy volunteers had been completed successfully.
The aim was to examine the safety and efficacy of NVR 3-778 alone and in combination with peg-IFN alpha-2a in previously untreated patients aged between 18 and 65 years who were HBsAg- and HbeAg-positive, had HBV DNA of 20,000 or more copies per IU/mL, with no cirrhosis of the liver, as determined by liver biopsy or imaging.
Patients were randomly assigned to six groups, with four groups receiving the investigational therapy alone at four increasing doses (100, 200, 400, once daily and 600 mg twice daily). There was a staged initiation of each escalating dose of NVR 3-778 used in these groups, with the 100 mg initiated first and if, after 2 weeks, there were no interim safety concerns, the next dose groups, 200 mg, then 400 mg, then 600 mg were started. The other two groups of patients received NVR 3-788 (600 mg) with peg-IFN alpha-2a (180 microg/week) or peg-IFN alpha-2a plus placebo. Treatment for all groups was for 28 days with 28 days of follow-up.
The mean age of patients in each group ranged from 32 to 48 years, with the older patients more likely to be treated with peg-IFN alpha-2a alone or with NVR 3-788. Most of the patients studied were Chinese and the mean baseline HBV DNA was 7.5-8.3 log10 IU/mL.
A total of 98 adverse events were seen in 40 of 64 patients treated, although there was not any trend for more effects with the investigational treatment versus placebo as doses escalated. There were 21 adverse events seen in 20 of 46 patients that were possibly or probably related to the study drug, of which most were grade 1 nausea. Grade 2 nausea occurred only once in a patient given the 100-mg dose. There was one serious adverse event, a grade 3 papulovesicular rash on the hands and feet that occurred in a patient in the 100-mg group, but this responded to therapy. This side effect started about 20 days after treatment and resolved over a period of 7 months.
Although generally mild, more adverse effects were seen in the patients treated with a combination of NVR 3-788 and peg-IFN alpha-2a, which Dr. Yuen noted was thought to be more likely a result of the IFN therapy.
There was not much difference in the mean change in HBV DNA from baseline for the 100-400-mg groups, but there was a 1.72-log10 IU/mL decrease achieved with the 600-mg twice-daily dose. The greatest reduction, however, was seen when NVR 3-788 was combined with peg-IFN alpha-2a, with a 1.97-log10 IU/mL change.
“Encouraging early reductions in HBV RNA and quantitative serum HBeAg were observed,” Dr. Yuen said. He noted that the longer-term safety and efficacy of the novel agent in combination with peg-IFN alpha-2a and first-line HBV NAs will now be further assessed in phase II studies.
“At the moment we do not have a cure for hepatitis B, only treatment,” said Dr. Frank Tacke of the University Hospital Aachen (Germany), who chaired the press briefing where Dr. Yuen first aired the findings. NVR 3-788 represents “a totally new approach to this disease that we hope will lead to a cure,” Dr. Tacke said.
In an EASL-issued press release, Dr. Tacke also noted: “The results from this study are certainly interesting and promising for the treatment of patients with hepatitis B. The medical community is always on the lookout for treatments which can cure this condition, as opposed to simply suppressing the replication of the virus. More research is needed to confirm whether NVR 3-778 could really change the treatment paradigm.”
Novira Therapeutics financed the study. Dr. Yuen has been a consultant, speaker, or both for Arrowhead, Bristol-Myers Squibb, GlaxoSmithKline, Gilead, Novartis, and Roche. Dr. Tacke did not have any relevant disclosures.
BARCELONA – An investigational drug that targets the hepatitis B virus (HBV) core protein is a “totally new approach” to treating chronic HBV infection according to the results of an early-phase clinical study.
In an ongoing, international, phase I study presented during the late-breaker session at the International Liver Congress, 4 weeks of treatment with NVR 3-778 was well tolerated and did not result in any drug discontinuations in chronically infected patients. There were dose-related reductions in HBV DNA and early reductions in HBeAg were observed.
“NVR 3-778 is a first-in-class HBV core inhibitor,” study investigator Dr. Man-Fung Yuen, Queen Mary Hospital, Hong Kong, said at a press briefing held at the meeting, which is sponsored by the European Association for the Study of the Liver (EASL). “The HBV core protein has many functions and is responsible for the most important pathway of the virus’ replication – nucleocapsid formation,” he explained.
The rationale behind its development is that although there are effective treatments such as nucleoside and nucleotide analogs (NAs) and pegylated interferon (peg-IFN) that can suppress the activity of HBV for many years and thus slow down damage to the liver, it is unusual to clear the virus permanently. Furthermore, as most patients require potentially life-long treatment, issues such as long-term safety, patients’ adherence, resistance, and cost of therapy come into play.
Dr. Yuen observed that achieving durable responses in patients with chronic HBV is likely to require combinations of potential agents, probably with complementary modes of action.
Preclinical data have shown that NVR 3-778 induces the rapid assembly of nonfunctional capsids and that it inhibits viral replication in a humanized mouse hepatocyte cell model. It has also been shown to reduce HBV DNA to a comparable extent as entecavir when used alone in a mouse model, with undetectable levels reached when used in combination with peg-IFN alpha-2a.
Dr. Yuen reported the preliminary results from the second part of the phase I study in 64 adults with chronic HBV, noting that the first part of the study in healthy volunteers had been completed successfully.
The aim was to examine the safety and efficacy of NVR 3-778 alone and in combination with peg-IFN alpha-2a in previously untreated patients aged between 18 and 65 years who were HBsAg- and HbeAg-positive, had HBV DNA of 20,000 or more copies per IU/mL, with no cirrhosis of the liver, as determined by liver biopsy or imaging.
Patients were randomly assigned to six groups, with four groups receiving the investigational therapy alone at four increasing doses (100, 200, 400, once daily and 600 mg twice daily). There was a staged initiation of each escalating dose of NVR 3-778 used in these groups, with the 100 mg initiated first and if, after 2 weeks, there were no interim safety concerns, the next dose groups, 200 mg, then 400 mg, then 600 mg were started. The other two groups of patients received NVR 3-788 (600 mg) with peg-IFN alpha-2a (180 microg/week) or peg-IFN alpha-2a plus placebo. Treatment for all groups was for 28 days with 28 days of follow-up.
The mean age of patients in each group ranged from 32 to 48 years, with the older patients more likely to be treated with peg-IFN alpha-2a alone or with NVR 3-788. Most of the patients studied were Chinese and the mean baseline HBV DNA was 7.5-8.3 log10 IU/mL.
A total of 98 adverse events were seen in 40 of 64 patients treated, although there was not any trend for more effects with the investigational treatment versus placebo as doses escalated. There were 21 adverse events seen in 20 of 46 patients that were possibly or probably related to the study drug, of which most were grade 1 nausea. Grade 2 nausea occurred only once in a patient given the 100-mg dose. There was one serious adverse event, a grade 3 papulovesicular rash on the hands and feet that occurred in a patient in the 100-mg group, but this responded to therapy. This side effect started about 20 days after treatment and resolved over a period of 7 months.
Although generally mild, more adverse effects were seen in the patients treated with a combination of NVR 3-788 and peg-IFN alpha-2a, which Dr. Yuen noted was thought to be more likely a result of the IFN therapy.
There was not much difference in the mean change in HBV DNA from baseline for the 100-400-mg groups, but there was a 1.72-log10 IU/mL decrease achieved with the 600-mg twice-daily dose. The greatest reduction, however, was seen when NVR 3-788 was combined with peg-IFN alpha-2a, with a 1.97-log10 IU/mL change.
“Encouraging early reductions in HBV RNA and quantitative serum HBeAg were observed,” Dr. Yuen said. He noted that the longer-term safety and efficacy of the novel agent in combination with peg-IFN alpha-2a and first-line HBV NAs will now be further assessed in phase II studies.
“At the moment we do not have a cure for hepatitis B, only treatment,” said Dr. Frank Tacke of the University Hospital Aachen (Germany), who chaired the press briefing where Dr. Yuen first aired the findings. NVR 3-788 represents “a totally new approach to this disease that we hope will lead to a cure,” Dr. Tacke said.
In an EASL-issued press release, Dr. Tacke also noted: “The results from this study are certainly interesting and promising for the treatment of patients with hepatitis B. The medical community is always on the lookout for treatments which can cure this condition, as opposed to simply suppressing the replication of the virus. More research is needed to confirm whether NVR 3-778 could really change the treatment paradigm.”
Novira Therapeutics financed the study. Dr. Yuen has been a consultant, speaker, or both for Arrowhead, Bristol-Myers Squibb, GlaxoSmithKline, Gilead, Novartis, and Roche. Dr. Tacke did not have any relevant disclosures.
AT THE INTERNATIONAL LIVER CONGRESS 2016
Key clinical point: NVR 3-778 is a “first-in-class” HBV core inhibitor in early clinical development showing promising results.
Major finding: There were no safety concerns. HBV DNA decreased by a respective 1.72 and 1.97 log10 IU/mL from baseline with a 600-mg twice-daily dose alone or in combination with peg-IFN alpha-2a.
Data source: Phase Ib study of 64 patients with chronic HBV infection treated with escalating doses of NVR 3-788.
Disclosures: Novira Therapeutics financed the study. Dr. Yuen has been a consultant, speaker, or both for Arrowhead, Bristol-Myers Squibb, GlaxoSmithKline, Gilead, Novartis, and Roche. Dr. Tacke did not have any relevant disclosures.

Biliary inflammation reduced by IBD drug
BARCELONA – The monoclonal antibody vedolizumab may reduce biliary inflammation in patients with primary sclerosing cholangitis and comorbid inflammatory bowel disease, according to early, open-label study findings reported at the meeting sponsored by the European Association for the Study of the Liver.
Vedolizumab given to 27 patients with primary sclerosing cholangitis (PSC) and inflammatory bowel disease (IBD) resulted in a 50% reduction or normalization of serum alkaline phosphatase levels in 17 cases (63%).

The findings are important as there is currently no medical treatment approved by the Food and Drug Administration for PSC. Although ursodeoxycholic acid is commonly used, there is no proof that it actually works in PSC, and the only option for patients, who are aged 30-40 years at diagnosis, is to wait until the disease has progressed far enough to warrant liver transplantation, which usually happens within 10-15 years.
The findings provide proof of concept that aberrant gut immunity could be behind the development of PSC, said Dr. Bertus Eksteenof the Snyder Institute for Chronic Diseases at the University of Calgary (Alta.).
“Many years ago we proposed that aberrant gut immunity has a significant role to play in PSC,” Dr. Eksteen said. He explained that the gut adhesion molecules CCL25 and MAdCAM-1 are possibly to blame as these can be aberrantly expressed in the liver of patients with PSC. This results in the recruitment of T cells expressing CCR9 and alpha 4 beta 7, which move from the gut and home in on the liver.
Vedolizumab (Entyvio), which targets alpha 4 beta 7, is licensed in the United States and in Europe for the treatment of IBD. The interest in using this particular drug in patients with PSC also stems from the fact that between 70% and 85% of patients with PSC have ulcerative colitis or Crohn’s disease.
More than two-thirds of patients were aged between 20 and 40 years old, around 38% had mild and 38% had moderate fibrosis, and only two had received prior treatment with ursodeoxycholic acid. Most (n = 17) of those studied had ulcerative colitis.
Three intravenous doses of vedolizumab (300 mg) were given at week 0, 2, and 6. Dr. Eksteen noted that this was the induction phase and the maintenance dosing phase of the study was ongoing. Vedolizumab is being given every 8 weeks for maintenance, he noted during discussion.
There was no change in fibrosis but that is not surprising with a short induction treatment period. Reductions in alkaline phosphatase levels often preceded clinical IBD responses, Dr. Eksteen said.
The most common adverse events were tiredness within 24 hours in six patients and redness at the infusion site in two patients, but no serious adverse events were reported.
Whether vedolizumab would be beneficial in patients with PSC without comorbid IBD remains to be seen and specifically studied. Dr. Eksteen suggested that because many of the patients included in the study had inactive or mild to moderate IBD at baseline the results were encouraging that an effect may be seen regardless of IBD.
A grant from the Canadian Institutes of Health Research funded the study. Dr. Eksteen did not report having any relevant financial disclosures. His research is funded by grants from the American Liver Foundation, the Medical Research Council (U.K.), and the Bowel Disease Research Foundation (U.K.).
BARCELONA – The monoclonal antibody vedolizumab may reduce biliary inflammation in patients with primary sclerosing cholangitis and comorbid inflammatory bowel disease, according to early, open-label study findings reported at the meeting sponsored by the European Association for the Study of the Liver.
Vedolizumab given to 27 patients with primary sclerosing cholangitis (PSC) and inflammatory bowel disease (IBD) resulted in a 50% reduction or normalization of serum alkaline phosphatase levels in 17 cases (63%).
The findings are important as there is currently no medical treatment approved by the Food and Drug Administration for PSC. Although ursodeoxycholic acid is commonly used, there is no proof that it actually works in PSC, and the only option for patients, who are aged 30-40 years at diagnosis, is to wait until the disease has progressed far enough to warrant liver transplantation, which usually happens within 10-15 years.
The findings provide proof of concept that aberrant gut immunity could be behind the development of PSC, said Dr. Bertus Eksteenof the Snyder Institute for Chronic Diseases at the University of Calgary (Alta.).
“Many years ago we proposed that aberrant gut immunity has a significant role to play in PSC,” Dr. Eksteen said. He explained that the gut adhesion molecules CCL25 and MAdCAM-1 are possibly to blame as these can be aberrantly expressed in the liver of patients with PSC. This results in the recruitment of T cells expressing CCR9 and alpha 4 beta 7, which move from the gut and home in on the liver.
Vedolizumab (Entyvio), which targets alpha 4 beta 7, is licensed in the United States and in Europe for the treatment of IBD. The interest in using this particular drug in patients with PSC also stems from the fact that between 70% and 85% of patients with PSC have ulcerative colitis or Crohn’s disease.
More than two-thirds of patients were aged between 20 and 40 years old, around 38% had mild and 38% had moderate fibrosis, and only two had received prior treatment with ursodeoxycholic acid. Most (n = 17) of those studied had ulcerative colitis.
Three intravenous doses of vedolizumab (300 mg) were given at week 0, 2, and 6. Dr. Eksteen noted that this was the induction phase and the maintenance dosing phase of the study was ongoing. Vedolizumab is being given every 8 weeks for maintenance, he noted during discussion.
There was no change in fibrosis but that is not surprising with a short induction treatment period. Reductions in alkaline phosphatase levels often preceded clinical IBD responses, Dr. Eksteen said.
The most common adverse events were tiredness within 24 hours in six patients and redness at the infusion site in two patients, but no serious adverse events were reported.
Whether vedolizumab would be beneficial in patients with PSC without comorbid IBD remains to be seen and specifically studied. Dr. Eksteen suggested that because many of the patients included in the study had inactive or mild to moderate IBD at baseline the results were encouraging that an effect may be seen regardless of IBD.
A grant from the Canadian Institutes of Health Research funded the study. Dr. Eksteen did not report having any relevant financial disclosures. His research is funded by grants from the American Liver Foundation, the Medical Research Council (U.K.), and the Bowel Disease Research Foundation (U.K.).
BARCELONA – The monoclonal antibody vedolizumab may reduce biliary inflammation in patients with primary sclerosing cholangitis and comorbid inflammatory bowel disease, according to early, open-label study findings reported at the meeting sponsored by the European Association for the Study of the Liver.
Vedolizumab given to 27 patients with primary sclerosing cholangitis (PSC) and inflammatory bowel disease (IBD) resulted in a 50% reduction or normalization of serum alkaline phosphatase levels in 17 cases (63%).
The findings are important as there is currently no medical treatment approved by the Food and Drug Administration for PSC. Although ursodeoxycholic acid is commonly used, there is no proof that it actually works in PSC, and the only option for patients, who are aged 30-40 years at diagnosis, is to wait until the disease has progressed far enough to warrant liver transplantation, which usually happens within 10-15 years.
The findings provide proof of concept that aberrant gut immunity could be behind the development of PSC, said Dr. Bertus Eksteenof the Snyder Institute for Chronic Diseases at the University of Calgary (Alta.).
“Many years ago we proposed that aberrant gut immunity has a significant role to play in PSC,” Dr. Eksteen said. He explained that the gut adhesion molecules CCL25 and MAdCAM-1 are possibly to blame as these can be aberrantly expressed in the liver of patients with PSC. This results in the recruitment of T cells expressing CCR9 and alpha 4 beta 7, which move from the gut and home in on the liver.
Vedolizumab (Entyvio), which targets alpha 4 beta 7, is licensed in the United States and in Europe for the treatment of IBD. The interest in using this particular drug in patients with PSC also stems from the fact that between 70% and 85% of patients with PSC have ulcerative colitis or Crohn’s disease.
More than two-thirds of patients were aged between 20 and 40 years old, around 38% had mild and 38% had moderate fibrosis, and only two had received prior treatment with ursodeoxycholic acid. Most (n = 17) of those studied had ulcerative colitis.
Three intravenous doses of vedolizumab (300 mg) were given at week 0, 2, and 6. Dr. Eksteen noted that this was the induction phase and the maintenance dosing phase of the study was ongoing. Vedolizumab is being given every 8 weeks for maintenance, he noted during discussion.
There was no change in fibrosis but that is not surprising with a short induction treatment period. Reductions in alkaline phosphatase levels often preceded clinical IBD responses, Dr. Eksteen said.
The most common adverse events were tiredness within 24 hours in six patients and redness at the infusion site in two patients, but no serious adverse events were reported.
Whether vedolizumab would be beneficial in patients with PSC without comorbid IBD remains to be seen and specifically studied. Dr. Eksteen suggested that because many of the patients included in the study had inactive or mild to moderate IBD at baseline the results were encouraging that an effect may be seen regardless of IBD.
A grant from the Canadian Institutes of Health Research funded the study. Dr. Eksteen did not report having any relevant financial disclosures. His research is funded by grants from the American Liver Foundation, the Medical Research Council (U.K.), and the Bowel Disease Research Foundation (U.K.).
AT THE INTERNATIONAL LIVER CONGRESS 2016
Key clinical point: The findings support an underlying inflammatory cause for primary biliary sclerosis.
Major finding: Alkaline phosphatase levels were reduced or normalized by 50% in 63% of patients (17 of 27) taking vedolizumab.
Data source: An open-label, proof-of-concept study involving 27 patients aged 25-30 years with PSC and comorbid IBD.
Disclosures: A grant from the Canadian Institutes of Health Research funded the study. Dr. Eksteen did not report having any disclosures. His research is funded by grants from the American Liver Foundation, the Medical Research Council (U.K.), and the Bowel Disease Research Foundation (U.K.).

New hope for primary sclerosing cholangitis treatment
BARCELONA – A modified form of ursodeoxycholic acid (UDCA) could offer patients with primary sclerosing cholangitis the first real pharmacologic treatment option, it was reported at the International Liver Congress.
Phase II study findings showed that a 1,500-mg daily dose of norursodeoxycholic acid (norUDCA) significantly (P less than .0001) reduced the primary endpoint of serum alkaline phosphatase (ALP) by 26% versus baseline levels within 12 weeks of treatment.
Other doses of norUDCA that were tested in the multicenter, randomized, double-blind, dose-finding study also produced significant reductions in serum ALP: –17.3% with a 1,000-mg dose (P = .0003), and –12.3% (P = .0029) with a 500-mg dose. The 1,500-mg dose has been selected for the follow-on phase III trial.

While this investigational drug is still only a symptomatic therapy and not a cure, it brings a viable option for managing the devastating but rare liver disease that currently lacks any effective therapy other than liver transplantation.
Primary sclerosing cholangitis (PSC) is an orphan disease that affects 1-16 in 100,000 people and typically strikes at a relatively young age, at around 30-40 years, with a male predominance. Often asymptomatic at first, the chronic disease can lead to liver transplant within 13-21 years of a diagnosis, with around half of all patients needing a transplant in 10-15 years.
This is the first trial of norUDCA in patients, lead study author Dr. Michael Trauner of the Medical University Vienna pointed out during the late-breaker session at the meeting sponsored by the European Association for the Study of the Liver (EASL).
“The role of UDCA in the treatment of PSC is still under debate and discussed controversially in the current guidelines,” Dr. Trauner noted. At a press briefing earlier in the day he had observed that there was not really any good evidence that it really worked in PSC, although it was approved as a treatment for primary biliary cholangitis.
norUDCA is a derivative of UDCA that has had a side-chain shortened by removing an ethylene group, he explained, and the resulting molecule is resistant to conjugation with taurine and glycine, which is part of process known as cholehepatic shunting. The resulting effect is protection of the bile ducts through the generation of bicarbonate-rich bile flow. Preclinical studies in mice have shown that norUDCA has potent antiproliferative, antifibrotic, and anti-inflammatory effects that, if translated into humans, could mean that norUDCA could have benefits beyond just addressing cholestasis.
“At the moment there is no medical treatment for PSC,” EASL spokesperson Dr. Frank Tacke of the University Hospital Aachen (Germany) commented at the press briefing. “We are very excited about these data because it is a new hope for this type of patient.” He added: “The fact that we have nothing to offer at the moment that works as a medical treatment makes this study so particular.”
Of 222 patients who were screened for inclusion into the study at 45 centers in 12 European countries, 161 met the criteria and were randomized, with 159 actually receiving their allocated treatment. There were 40 patients in the placebo arm, and 39, 41, and 39 patients, respectively, in the 500-, 1,000-, and 1,500-mg norUDCA arms. The two patients that did not receive allocated treatment had withdrawn their consent.
As expected, around 60%-70% of the patients in each group were male. The mean age was around 41-44 years, around one-fifth had a new diagnosis of PSC, and more than half (50%-77%) had inflammatory bowel disease (IBD) at screening, which was predominantly ulcerative colitis, Dr. Trauner observed. Patients also had pronounced cholestasis at the start of the study, signified by mean serum ALP of 400-500 IU/L. The normal range is between 44 and 147 IU/L.
“norUDCA reduced ALP in a dose-dependent fashion,” Dr. Trauner said. He noted that looking at changes in ALP over time, it was evident that there was a rebound effect after the treatment was stopped. The percentage of patients reaching an ALP equal to or below 1.5-fold the upper limit of normal, which has been shown to be prognostically meaningful in the disease, was 12.5% for placebo and 12.8%, 41.5%, and 30.8% for the three ascending doses of norUDCA.
Changes in serum levels of other important liver enzymes – gamma-glutamyl transferase, alanine aminotransferase, aspartate aminotransferase – showed a similar pattern in terms of absolute changes from baseline over time, with rebound effects once treatment stopped.
There were a comparable number of adverse drug reactions – 28%, 23%, 32%, and 28%, respectively, in the placebo, 500-, 1,000-, and 1,500-mg norUDCA groups. Treatment-emergent adverse events occurred in 80%, 59%, 73%, and 67%. The most common of these were gastrointestinal effects such as abdominal pain (12.5% for placebo vs. 2.6%-9.8% in the norUDCA groups) and diarrhea (10% vs. 0-7.7%), fatigue (10% vs. 4.9%-12.8%), arthralgia (10% vs. 0-2.4%), back pain (10% vs. 0-7.7%), headache (7.5% vs. 2.4%-17.9%), nasopharyngitis (17.5% vs. 15.4%-22%), and pruritus (10% vs. 7.7%-15.4%).
“We don’t think pruritus is an issue with norUDCA,” Dr. Trauner said in response to a delegate’s query on the numerically higher rates of itching in the active treatment groups. “Of course, we paid much attention to this, but overall pruritus was extremely mild, it led to no discontinuation of the study drug.” There is an ongoing, similar-dose study in nonalcoholic fatty liver disease where so far 150 patients have been treated and only a single patient so far has reported this side effect.
Based on these “highly encouraging” results, “the planning of the phase III trial in PSC is already ongoing,” Dr. Trauner said. Asked what the primary endpoint may be, he acknowledged that change in serum ALP was not an accepted endpoint in PSC by itself and that the trial would most likely use a combined endpoint of serum ALP and assessment of fibrosis, perhaps noninvasively with FibroScan.
He also noted that quality of life had been assessed in the phase II trial but no significant differences were seen. It could be that, at 12 weeks, the trial was too short to assess this parameter, he suggested, and that would be perhaps something to also address in the phase III trial. There was no change in IBD disease activity, which was important, he said.
Dr. Heiner Wedemeyer of Hannover (Germany) Medical School and who was not involved in the trial also commented on the importance of the study at the press briefing. Dr. Wedemeyer said that he was “extremely excited” by these data. “This is the most awful liver disease that we have because it is so difficult to judge when to transplant the patient,” he noted. This is a very difficult group of patients to handle and “this is the first time in decades that there is some hope on the horizon. This is the first time we see something that may work.”
Dr. Falk Pharma, Frieburg, Germany, sponsored the trial. Dr. Trauner has received honoraria, research grants, and travel support from Dr. Falk Pharma, and is listed as co-inventor on a patent for the medical use of norUDCA. Dr. Trauner has also received honoraria, research grants, or travel support from Albireo, Genfit, Gilead, Intercept, Merck Sharp & Dohme, Novartis, Phenex, and Roche. Dr. Tacke and Dr. Wedemeyer had no relevant disclosures.
BARCELONA – A modified form of ursodeoxycholic acid (UDCA) could offer patients with primary sclerosing cholangitis the first real pharmacologic treatment option, it was reported at the International Liver Congress.
Phase II study findings showed that a 1,500-mg daily dose of norursodeoxycholic acid (norUDCA) significantly (P less than .0001) reduced the primary endpoint of serum alkaline phosphatase (ALP) by 26% versus baseline levels within 12 weeks of treatment.
Other doses of norUDCA that were tested in the multicenter, randomized, double-blind, dose-finding study also produced significant reductions in serum ALP: –17.3% with a 1,000-mg dose (P = .0003), and –12.3% (P = .0029) with a 500-mg dose. The 1,500-mg dose has been selected for the follow-on phase III trial.
While this investigational drug is still only a symptomatic therapy and not a cure, it brings a viable option for managing the devastating but rare liver disease that currently lacks any effective therapy other than liver transplantation.
Primary sclerosing cholangitis (PSC) is an orphan disease that affects 1-16 in 100,000 people and typically strikes at a relatively young age, at around 30-40 years, with a male predominance. Often asymptomatic at first, the chronic disease can lead to liver transplant within 13-21 years of a diagnosis, with around half of all patients needing a transplant in 10-15 years.
This is the first trial of norUDCA in patients, lead study author Dr. Michael Trauner of the Medical University Vienna pointed out during the late-breaker session at the meeting sponsored by the European Association for the Study of the Liver (EASL).
“The role of UDCA in the treatment of PSC is still under debate and discussed controversially in the current guidelines,” Dr. Trauner noted. At a press briefing earlier in the day he had observed that there was not really any good evidence that it really worked in PSC, although it was approved as a treatment for primary biliary cholangitis.
norUDCA is a derivative of UDCA that has had a side-chain shortened by removing an ethylene group, he explained, and the resulting molecule is resistant to conjugation with taurine and glycine, which is part of process known as cholehepatic shunting. The resulting effect is protection of the bile ducts through the generation of bicarbonate-rich bile flow. Preclinical studies in mice have shown that norUDCA has potent antiproliferative, antifibrotic, and anti-inflammatory effects that, if translated into humans, could mean that norUDCA could have benefits beyond just addressing cholestasis.
“At the moment there is no medical treatment for PSC,” EASL spokesperson Dr. Frank Tacke of the University Hospital Aachen (Germany) commented at the press briefing. “We are very excited about these data because it is a new hope for this type of patient.” He added: “The fact that we have nothing to offer at the moment that works as a medical treatment makes this study so particular.”
Of 222 patients who were screened for inclusion into the study at 45 centers in 12 European countries, 161 met the criteria and were randomized, with 159 actually receiving their allocated treatment. There were 40 patients in the placebo arm, and 39, 41, and 39 patients, respectively, in the 500-, 1,000-, and 1,500-mg norUDCA arms. The two patients that did not receive allocated treatment had withdrawn their consent.
As expected, around 60%-70% of the patients in each group were male. The mean age was around 41-44 years, around one-fifth had a new diagnosis of PSC, and more than half (50%-77%) had inflammatory bowel disease (IBD) at screening, which was predominantly ulcerative colitis, Dr. Trauner observed. Patients also had pronounced cholestasis at the start of the study, signified by mean serum ALP of 400-500 IU/L. The normal range is between 44 and 147 IU/L.
“norUDCA reduced ALP in a dose-dependent fashion,” Dr. Trauner said. He noted that looking at changes in ALP over time, it was evident that there was a rebound effect after the treatment was stopped. The percentage of patients reaching an ALP equal to or below 1.5-fold the upper limit of normal, which has been shown to be prognostically meaningful in the disease, was 12.5% for placebo and 12.8%, 41.5%, and 30.8% for the three ascending doses of norUDCA.
Changes in serum levels of other important liver enzymes – gamma-glutamyl transferase, alanine aminotransferase, aspartate aminotransferase – showed a similar pattern in terms of absolute changes from baseline over time, with rebound effects once treatment stopped.
There were a comparable number of adverse drug reactions – 28%, 23%, 32%, and 28%, respectively, in the placebo, 500-, 1,000-, and 1,500-mg norUDCA groups. Treatment-emergent adverse events occurred in 80%, 59%, 73%, and 67%. The most common of these were gastrointestinal effects such as abdominal pain (12.5% for placebo vs. 2.6%-9.8% in the norUDCA groups) and diarrhea (10% vs. 0-7.7%), fatigue (10% vs. 4.9%-12.8%), arthralgia (10% vs. 0-2.4%), back pain (10% vs. 0-7.7%), headache (7.5% vs. 2.4%-17.9%), nasopharyngitis (17.5% vs. 15.4%-22%), and pruritus (10% vs. 7.7%-15.4%).
“We don’t think pruritus is an issue with norUDCA,” Dr. Trauner said in response to a delegate’s query on the numerically higher rates of itching in the active treatment groups. “Of course, we paid much attention to this, but overall pruritus was extremely mild, it led to no discontinuation of the study drug.” There is an ongoing, similar-dose study in nonalcoholic fatty liver disease where so far 150 patients have been treated and only a single patient so far has reported this side effect.
Based on these “highly encouraging” results, “the planning of the phase III trial in PSC is already ongoing,” Dr. Trauner said. Asked what the primary endpoint may be, he acknowledged that change in serum ALP was not an accepted endpoint in PSC by itself and that the trial would most likely use a combined endpoint of serum ALP and assessment of fibrosis, perhaps noninvasively with FibroScan.
He also noted that quality of life had been assessed in the phase II trial but no significant differences were seen. It could be that, at 12 weeks, the trial was too short to assess this parameter, he suggested, and that would be perhaps something to also address in the phase III trial. There was no change in IBD disease activity, which was important, he said.
Dr. Heiner Wedemeyer of Hannover (Germany) Medical School and who was not involved in the trial also commented on the importance of the study at the press briefing. Dr. Wedemeyer said that he was “extremely excited” by these data. “This is the most awful liver disease that we have because it is so difficult to judge when to transplant the patient,” he noted. This is a very difficult group of patients to handle and “this is the first time in decades that there is some hope on the horizon. This is the first time we see something that may work.”
Dr. Falk Pharma, Frieburg, Germany, sponsored the trial. Dr. Trauner has received honoraria, research grants, and travel support from Dr. Falk Pharma, and is listed as co-inventor on a patent for the medical use of norUDCA. Dr. Trauner has also received honoraria, research grants, or travel support from Albireo, Genfit, Gilead, Intercept, Merck Sharp & Dohme, Novartis, Phenex, and Roche. Dr. Tacke and Dr. Wedemeyer had no relevant disclosures.
BARCELONA – A modified form of ursodeoxycholic acid (UDCA) could offer patients with primary sclerosing cholangitis the first real pharmacologic treatment option, it was reported at the International Liver Congress.
Phase II study findings showed that a 1,500-mg daily dose of norursodeoxycholic acid (norUDCA) significantly (P less than .0001) reduced the primary endpoint of serum alkaline phosphatase (ALP) by 26% versus baseline levels within 12 weeks of treatment.
Other doses of norUDCA that were tested in the multicenter, randomized, double-blind, dose-finding study also produced significant reductions in serum ALP: –17.3% with a 1,000-mg dose (P = .0003), and –12.3% (P = .0029) with a 500-mg dose. The 1,500-mg dose has been selected for the follow-on phase III trial.
While this investigational drug is still only a symptomatic therapy and not a cure, it brings a viable option for managing the devastating but rare liver disease that currently lacks any effective therapy other than liver transplantation.
Primary sclerosing cholangitis (PSC) is an orphan disease that affects 1-16 in 100,000 people and typically strikes at a relatively young age, at around 30-40 years, with a male predominance. Often asymptomatic at first, the chronic disease can lead to liver transplant within 13-21 years of a diagnosis, with around half of all patients needing a transplant in 10-15 years.
This is the first trial of norUDCA in patients, lead study author Dr. Michael Trauner of the Medical University Vienna pointed out during the late-breaker session at the meeting sponsored by the European Association for the Study of the Liver (EASL).
“The role of UDCA in the treatment of PSC is still under debate and discussed controversially in the current guidelines,” Dr. Trauner noted. At a press briefing earlier in the day he had observed that there was not really any good evidence that it really worked in PSC, although it was approved as a treatment for primary biliary cholangitis.
norUDCA is a derivative of UDCA that has had a side-chain shortened by removing an ethylene group, he explained, and the resulting molecule is resistant to conjugation with taurine and glycine, which is part of process known as cholehepatic shunting. The resulting effect is protection of the bile ducts through the generation of bicarbonate-rich bile flow. Preclinical studies in mice have shown that norUDCA has potent antiproliferative, antifibrotic, and anti-inflammatory effects that, if translated into humans, could mean that norUDCA could have benefits beyond just addressing cholestasis.
“At the moment there is no medical treatment for PSC,” EASL spokesperson Dr. Frank Tacke of the University Hospital Aachen (Germany) commented at the press briefing. “We are very excited about these data because it is a new hope for this type of patient.” He added: “The fact that we have nothing to offer at the moment that works as a medical treatment makes this study so particular.”
Of 222 patients who were screened for inclusion into the study at 45 centers in 12 European countries, 161 met the criteria and were randomized, with 159 actually receiving their allocated treatment. There were 40 patients in the placebo arm, and 39, 41, and 39 patients, respectively, in the 500-, 1,000-, and 1,500-mg norUDCA arms. The two patients that did not receive allocated treatment had withdrawn their consent.
As expected, around 60%-70% of the patients in each group were male. The mean age was around 41-44 years, around one-fifth had a new diagnosis of PSC, and more than half (50%-77%) had inflammatory bowel disease (IBD) at screening, which was predominantly ulcerative colitis, Dr. Trauner observed. Patients also had pronounced cholestasis at the start of the study, signified by mean serum ALP of 400-500 IU/L. The normal range is between 44 and 147 IU/L.
“norUDCA reduced ALP in a dose-dependent fashion,” Dr. Trauner said. He noted that looking at changes in ALP over time, it was evident that there was a rebound effect after the treatment was stopped. The percentage of patients reaching an ALP equal to or below 1.5-fold the upper limit of normal, which has been shown to be prognostically meaningful in the disease, was 12.5% for placebo and 12.8%, 41.5%, and 30.8% for the three ascending doses of norUDCA.
Changes in serum levels of other important liver enzymes – gamma-glutamyl transferase, alanine aminotransferase, aspartate aminotransferase – showed a similar pattern in terms of absolute changes from baseline over time, with rebound effects once treatment stopped.
There were a comparable number of adverse drug reactions – 28%, 23%, 32%, and 28%, respectively, in the placebo, 500-, 1,000-, and 1,500-mg norUDCA groups. Treatment-emergent adverse events occurred in 80%, 59%, 73%, and 67%. The most common of these were gastrointestinal effects such as abdominal pain (12.5% for placebo vs. 2.6%-9.8% in the norUDCA groups) and diarrhea (10% vs. 0-7.7%), fatigue (10% vs. 4.9%-12.8%), arthralgia (10% vs. 0-2.4%), back pain (10% vs. 0-7.7%), headache (7.5% vs. 2.4%-17.9%), nasopharyngitis (17.5% vs. 15.4%-22%), and pruritus (10% vs. 7.7%-15.4%).
“We don’t think pruritus is an issue with norUDCA,” Dr. Trauner said in response to a delegate’s query on the numerically higher rates of itching in the active treatment groups. “Of course, we paid much attention to this, but overall pruritus was extremely mild, it led to no discontinuation of the study drug.” There is an ongoing, similar-dose study in nonalcoholic fatty liver disease where so far 150 patients have been treated and only a single patient so far has reported this side effect.
Based on these “highly encouraging” results, “the planning of the phase III trial in PSC is already ongoing,” Dr. Trauner said. Asked what the primary endpoint may be, he acknowledged that change in serum ALP was not an accepted endpoint in PSC by itself and that the trial would most likely use a combined endpoint of serum ALP and assessment of fibrosis, perhaps noninvasively with FibroScan.
He also noted that quality of life had been assessed in the phase II trial but no significant differences were seen. It could be that, at 12 weeks, the trial was too short to assess this parameter, he suggested, and that would be perhaps something to also address in the phase III trial. There was no change in IBD disease activity, which was important, he said.
Dr. Heiner Wedemeyer of Hannover (Germany) Medical School and who was not involved in the trial also commented on the importance of the study at the press briefing. Dr. Wedemeyer said that he was “extremely excited” by these data. “This is the most awful liver disease that we have because it is so difficult to judge when to transplant the patient,” he noted. This is a very difficult group of patients to handle and “this is the first time in decades that there is some hope on the horizon. This is the first time we see something that may work.”
Dr. Falk Pharma, Frieburg, Germany, sponsored the trial. Dr. Trauner has received honoraria, research grants, and travel support from Dr. Falk Pharma, and is listed as co-inventor on a patent for the medical use of norUDCA. Dr. Trauner has also received honoraria, research grants, or travel support from Albireo, Genfit, Gilead, Intercept, Merck Sharp & Dohme, Novartis, Phenex, and Roche. Dr. Tacke and Dr. Wedemeyer had no relevant disclosures.
AT THE INTERNATIONAL LIVER CONGRESS 2016
Key clinical point: Norursodeoxycholic acid (norUDCA) is a promising investigational treatment for primary sclerosing cholangitis (PSC).
Major finding: Serum alkaline phosphatase (ALP) was reduced by 26% (P less than .0001 versus baseline) within 12 weeks of treatment with the highest dose of norUDCA.
Data source: Multicenter, randomized, double-blind, placebo-controlled phase II dosing study of norUDCA in 159 patients with chronic PSC.
Disclosures: Dr. Falk Pharma, Frieburg, Germany, sponsored the trial. Dr. Trauner has received honoraria, research grants, and travel support from Dr. Falk Pharma, and is listed as co-inventor on a patent for the medical use of norUDCA. Dr. Trauner has also received honoraria, research grants, or travel support from Albireo, Genfit, Gilead, Intercept, Merck Sharp & Dohme, Novartis, Phenex, and Roche.

PBC: Female predominance lower than expected
The female-to-male predominance of primary biliary cholangitis (PBC), formerly cirrhosis, is much lower than previously believed, according to data from Swedish national registers.
Looking at a number of diseases thought to have at least some basis in autoimmunity, researchers found that the relative risk of PBC for women, compared with men, was 4.1, suggesting that, although it remains a disease predominantly affecting women, its prevalence in men might actually be higher than previously thought.
Of the three other gastrointestinal conditions among the 32 autoimmune diseases examined, one showed a female predominance, one showed no difference, and one had a male predominance, reported Dr. Jianguang Ji and his associates at Lund (Sweden) University (J Autoimmune. 2016;69:102-6).
Celiac disease had a slight female predominance, with a relative risk for women of 1.8, compared with men. There was no difference for Crohn’s disease (RR, 1.1), and ulcerative colitis had a slight male predominance (RR, 0.9), the investigators reported.
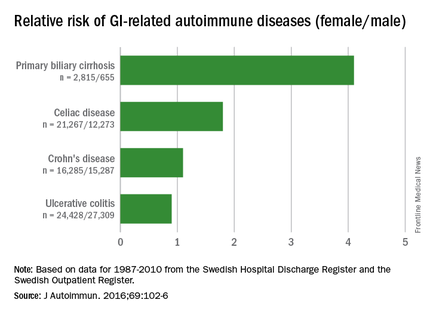
The situation was similar for the larger group of conditions: Eighteen showed a female predominance, 7 had no difference, and 7 showed a male predominance, they noted.
“Most review papers claim that the vast majority of autoimmune diseases have a striking female predominance, and most of them cited that more than 80% of cases affected are women. In this population-based study of 32 specific autoimmune diseases with more than 20 years of follow-up, we found that the incidence in women was only 60% higher as compared to that in men,” Dr. Ji and his associates wrote.
The investigators searched the Swedish Hospital Discharge Register and the Swedish Outpatient Register and found data on 403,757 individuals who had been diagnosed with one or more of the 32 autoimmune diseases between 1987 and 2010. About 62% of the subjects were female.
Dr. Ji and his associates had no conflicts of interest to report. The study was supported by the Swedish Research Council and Region Skåne.
The female-to-male predominance of primary biliary cholangitis (PBC), formerly cirrhosis, is much lower than previously believed, according to data from Swedish national registers.
Looking at a number of diseases thought to have at least some basis in autoimmunity, researchers found that the relative risk of PBC for women, compared with men, was 4.1, suggesting that, although it remains a disease predominantly affecting women, its prevalence in men might actually be higher than previously thought.
Of the three other gastrointestinal conditions among the 32 autoimmune diseases examined, one showed a female predominance, one showed no difference, and one had a male predominance, reported Dr. Jianguang Ji and his associates at Lund (Sweden) University (J Autoimmune. 2016;69:102-6).
Celiac disease had a slight female predominance, with a relative risk for women of 1.8, compared with men. There was no difference for Crohn’s disease (RR, 1.1), and ulcerative colitis had a slight male predominance (RR, 0.9), the investigators reported.
The situation was similar for the larger group of conditions: Eighteen showed a female predominance, 7 had no difference, and 7 showed a male predominance, they noted.
“Most review papers claim that the vast majority of autoimmune diseases have a striking female predominance, and most of them cited that more than 80% of cases affected are women. In this population-based study of 32 specific autoimmune diseases with more than 20 years of follow-up, we found that the incidence in women was only 60% higher as compared to that in men,” Dr. Ji and his associates wrote.
The investigators searched the Swedish Hospital Discharge Register and the Swedish Outpatient Register and found data on 403,757 individuals who had been diagnosed with one or more of the 32 autoimmune diseases between 1987 and 2010. About 62% of the subjects were female.
Dr. Ji and his associates had no conflicts of interest to report. The study was supported by the Swedish Research Council and Region Skåne.
The female-to-male predominance of primary biliary cholangitis (PBC), formerly cirrhosis, is much lower than previously believed, according to data from Swedish national registers.
Looking at a number of diseases thought to have at least some basis in autoimmunity, researchers found that the relative risk of PBC for women, compared with men, was 4.1, suggesting that, although it remains a disease predominantly affecting women, its prevalence in men might actually be higher than previously thought.
Of the three other gastrointestinal conditions among the 32 autoimmune diseases examined, one showed a female predominance, one showed no difference, and one had a male predominance, reported Dr. Jianguang Ji and his associates at Lund (Sweden) University (J Autoimmune. 2016;69:102-6).
Celiac disease had a slight female predominance, with a relative risk for women of 1.8, compared with men. There was no difference for Crohn’s disease (RR, 1.1), and ulcerative colitis had a slight male predominance (RR, 0.9), the investigators reported.
The situation was similar for the larger group of conditions: Eighteen showed a female predominance, 7 had no difference, and 7 showed a male predominance, they noted.
“Most review papers claim that the vast majority of autoimmune diseases have a striking female predominance, and most of them cited that more than 80% of cases affected are women. In this population-based study of 32 specific autoimmune diseases with more than 20 years of follow-up, we found that the incidence in women was only 60% higher as compared to that in men,” Dr. Ji and his associates wrote.
The investigators searched the Swedish Hospital Discharge Register and the Swedish Outpatient Register and found data on 403,757 individuals who had been diagnosed with one or more of the 32 autoimmune diseases between 1987 and 2010. About 62% of the subjects were female.
Dr. Ji and his associates had no conflicts of interest to report. The study was supported by the Swedish Research Council and Region Skåne.
FROM THE JOURNAL OF AUTOIMMUNITY
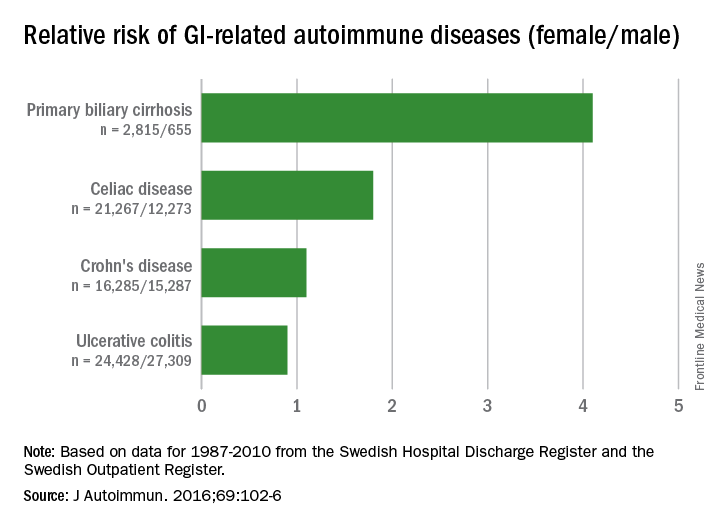
Report laid groundwork for ending the public health problem of viral hepatitis
The United States can end hepatitis B and C transmission and prevent related morbidity and mortality given enough time, resources, and will to surmount a variety of barriers, according to a new report from the National Academies of Sciences, Engineering, and Medicine.
A more feasible short-term goal is to control viral hepatitis by reducing the incidence and prevalence of hepatitis B (HBV) and C virus (HCV) infections, states the report, which is the first of a two-phase study. The second part, to be published in early 2017, will outline a strategy to achieve the goals.
“Disease elimination” once meant stopping all new infections in a population, but eliminating a public health problem “can be a less absolute goal,” Dr. Brian Strom, chancellor of Rutgers Biomedical and Health Sciences, wrote along with his associates. “The WHO has accepted a non-zero target in its work against viral hepatitis,” they added. “The organization’s provisional target is a 90% reduction in incidence, and a 65% reduction in mortality by 2030.”
Furthermore, those are global targets – countries should base their own strategies on their specific viral hepatitis disease burdens and epidemiologic features, Dr. Strom and his associates emphasized. For example, universal vaccination could eradicate HBV in the United States in two generations, but there is no cure for the 700,000-1.4 million Americans who are already infected. Mother-to-child HBV transmission is rare here, but better screening and surveillance could help prevent the 800-1,000 cases of vertical transmission that still occur annually.
More than a third of newborns in the United States do not receive the HBV vaccination on the day they are born, and 28% remain unvaccinated when they are 3 days old, “but childhood catch-up is possible,” the experts also wrote. Vaccinating adults is more complicated, but targeting high-risk groups such as prison inmates or patients at sexually transmitted disease clinics might work, they added. Vaccination is especially important, because chronically infected HBV patients need lifelong medical follow-up to prevent progression and death from cirrhosis and liver cancer, they emphasized.
The hepatitis C virus presents a different picture – there is no HCV vaccine, while the novel direct-acting antivirals can theoretically cure nearly all chronic infections. But in reality, high drug costs and the massive burden of undiagnosed HCV mean that only a small fraction of patients can be cured, the authors acknowledged. On an individual basis, the benefits of direct-acting antivirals outweigh the costs of treating genotype 1 HCV and associated morbidity, they add. But on a population level, “eliminating HCV infection would require near universal access to treatment, something that appears infeasible given the current pricing and policy environment.”
Several other barriers also impede progress against HBV and HCV. For example, only five states and two large cities receive enough funding for comprehensive viral hepatitis surveillance, Dr. Strom and his associates noted. If local and state public health departments cannot track infections, they cannot characterize their epidemics. In addition, viral hepatitis remains a source of stigma, adding to fear and avoidance of testing and undermining elimination efforts. Stigma, avoidance, and the fact that HBV and HCV are asymptomatic in the early stages all contribute to a large population of chronically infected patients who do not know their status.
Moreover, many state Medicaid programs will not cover foreigners until they have lived in the United States for 5 years, and the Affordable Care Act does not cover care for short-term residents and undocumented immigrants, the report stated. Newly infected HCV patients tend to have fewer resources, lower educational levels, and more often use injection drugs, making them harder to reach and screen through the health system. “Prisons are a promising venue in which to treat HCV, but treating HCV is expensive for the prison system. The cost of the direct-acting antivirals is high, and the staff time required to manage an inmate in treatment often far exceed the available resources,” the report stated.
The study was sponsored by the Centers for Disease Control and Prevention Office of Viral Hepatitis and the U.S. Department of Health and Human Services Office of Minority Health. The authors disclosed no conflicts of interest.

|
Dr. Sean Koppe |
The report offers a comprehensive view of the challenges and opportunities in regard to eliminating hepatitis B and C. As is well articulated in the report, we currently have excellent treatment for both hepatitis B and hepatitis C and a highly effective vaccine for eliminating transmission of hepatitis B. It is disappointing that we essentially already have the tools necessary to eliminate viral hepatitis as a top 10 cause of mortality worldwide, but currently lack the will and resources to accomplish the task.
I hope this report will serve to stimulate public health efforts to enhance screening and vaccination programs and also prod insurers and drug makers to make medications more affordable and accessible.
Dr. Sean Koppe is director of hepatology and medical director of transplantation, University of Illinois Hospital & Health Sciences System, Chicago. He has no conflicts of interest.
|
|
Dr. Sean Koppe |
The report offers a comprehensive view of the challenges and opportunities in regard to eliminating hepatitis B and C. As is well articulated in the report, we currently have excellent treatment for both hepatitis B and hepatitis C and a highly effective vaccine for eliminating transmission of hepatitis B. It is disappointing that we essentially already have the tools necessary to eliminate viral hepatitis as a top 10 cause of mortality worldwide, but currently lack the will and resources to accomplish the task.
I hope this report will serve to stimulate public health efforts to enhance screening and vaccination programs and also prod insurers and drug makers to make medications more affordable and accessible.
Dr. Sean Koppe is director of hepatology and medical director of transplantation, University of Illinois Hospital & Health Sciences System, Chicago. He has no conflicts of interest.
|
|
Dr. Sean Koppe |
The report offers a comprehensive view of the challenges and opportunities in regard to eliminating hepatitis B and C. As is well articulated in the report, we currently have excellent treatment for both hepatitis B and hepatitis C and a highly effective vaccine for eliminating transmission of hepatitis B. It is disappointing that we essentially already have the tools necessary to eliminate viral hepatitis as a top 10 cause of mortality worldwide, but currently lack the will and resources to accomplish the task.
I hope this report will serve to stimulate public health efforts to enhance screening and vaccination programs and also prod insurers and drug makers to make medications more affordable and accessible.
Dr. Sean Koppe is director of hepatology and medical director of transplantation, University of Illinois Hospital & Health Sciences System, Chicago. He has no conflicts of interest.
The United States can end hepatitis B and C transmission and prevent related morbidity and mortality given enough time, resources, and will to surmount a variety of barriers, according to a new report from the National Academies of Sciences, Engineering, and Medicine.
A more feasible short-term goal is to control viral hepatitis by reducing the incidence and prevalence of hepatitis B (HBV) and C virus (HCV) infections, states the report, which is the first of a two-phase study. The second part, to be published in early 2017, will outline a strategy to achieve the goals.
“Disease elimination” once meant stopping all new infections in a population, but eliminating a public health problem “can be a less absolute goal,” Dr. Brian Strom, chancellor of Rutgers Biomedical and Health Sciences, wrote along with his associates. “The WHO has accepted a non-zero target in its work against viral hepatitis,” they added. “The organization’s provisional target is a 90% reduction in incidence, and a 65% reduction in mortality by 2030.”
Furthermore, those are global targets – countries should base their own strategies on their specific viral hepatitis disease burdens and epidemiologic features, Dr. Strom and his associates emphasized. For example, universal vaccination could eradicate HBV in the United States in two generations, but there is no cure for the 700,000-1.4 million Americans who are already infected. Mother-to-child HBV transmission is rare here, but better screening and surveillance could help prevent the 800-1,000 cases of vertical transmission that still occur annually.
More than a third of newborns in the United States do not receive the HBV vaccination on the day they are born, and 28% remain unvaccinated when they are 3 days old, “but childhood catch-up is possible,” the experts also wrote. Vaccinating adults is more complicated, but targeting high-risk groups such as prison inmates or patients at sexually transmitted disease clinics might work, they added. Vaccination is especially important, because chronically infected HBV patients need lifelong medical follow-up to prevent progression and death from cirrhosis and liver cancer, they emphasized.
The hepatitis C virus presents a different picture – there is no HCV vaccine, while the novel direct-acting antivirals can theoretically cure nearly all chronic infections. But in reality, high drug costs and the massive burden of undiagnosed HCV mean that only a small fraction of patients can be cured, the authors acknowledged. On an individual basis, the benefits of direct-acting antivirals outweigh the costs of treating genotype 1 HCV and associated morbidity, they add. But on a population level, “eliminating HCV infection would require near universal access to treatment, something that appears infeasible given the current pricing and policy environment.”
Several other barriers also impede progress against HBV and HCV. For example, only five states and two large cities receive enough funding for comprehensive viral hepatitis surveillance, Dr. Strom and his associates noted. If local and state public health departments cannot track infections, they cannot characterize their epidemics. In addition, viral hepatitis remains a source of stigma, adding to fear and avoidance of testing and undermining elimination efforts. Stigma, avoidance, and the fact that HBV and HCV are asymptomatic in the early stages all contribute to a large population of chronically infected patients who do not know their status.
Moreover, many state Medicaid programs will not cover foreigners until they have lived in the United States for 5 years, and the Affordable Care Act does not cover care for short-term residents and undocumented immigrants, the report stated. Newly infected HCV patients tend to have fewer resources, lower educational levels, and more often use injection drugs, making them harder to reach and screen through the health system. “Prisons are a promising venue in which to treat HCV, but treating HCV is expensive for the prison system. The cost of the direct-acting antivirals is high, and the staff time required to manage an inmate in treatment often far exceed the available resources,” the report stated.
The study was sponsored by the Centers for Disease Control and Prevention Office of Viral Hepatitis and the U.S. Department of Health and Human Services Office of Minority Health. The authors disclosed no conflicts of interest.
The United States can end hepatitis B and C transmission and prevent related morbidity and mortality given enough time, resources, and will to surmount a variety of barriers, according to a new report from the National Academies of Sciences, Engineering, and Medicine.
A more feasible short-term goal is to control viral hepatitis by reducing the incidence and prevalence of hepatitis B (HBV) and C virus (HCV) infections, states the report, which is the first of a two-phase study. The second part, to be published in early 2017, will outline a strategy to achieve the goals.
“Disease elimination” once meant stopping all new infections in a population, but eliminating a public health problem “can be a less absolute goal,” Dr. Brian Strom, chancellor of Rutgers Biomedical and Health Sciences, wrote along with his associates. “The WHO has accepted a non-zero target in its work against viral hepatitis,” they added. “The organization’s provisional target is a 90% reduction in incidence, and a 65% reduction in mortality by 2030.”
Furthermore, those are global targets – countries should base their own strategies on their specific viral hepatitis disease burdens and epidemiologic features, Dr. Strom and his associates emphasized. For example, universal vaccination could eradicate HBV in the United States in two generations, but there is no cure for the 700,000-1.4 million Americans who are already infected. Mother-to-child HBV transmission is rare here, but better screening and surveillance could help prevent the 800-1,000 cases of vertical transmission that still occur annually.
More than a third of newborns in the United States do not receive the HBV vaccination on the day they are born, and 28% remain unvaccinated when they are 3 days old, “but childhood catch-up is possible,” the experts also wrote. Vaccinating adults is more complicated, but targeting high-risk groups such as prison inmates or patients at sexually transmitted disease clinics might work, they added. Vaccination is especially important, because chronically infected HBV patients need lifelong medical follow-up to prevent progression and death from cirrhosis and liver cancer, they emphasized.
The hepatitis C virus presents a different picture – there is no HCV vaccine, while the novel direct-acting antivirals can theoretically cure nearly all chronic infections. But in reality, high drug costs and the massive burden of undiagnosed HCV mean that only a small fraction of patients can be cured, the authors acknowledged. On an individual basis, the benefits of direct-acting antivirals outweigh the costs of treating genotype 1 HCV and associated morbidity, they add. But on a population level, “eliminating HCV infection would require near universal access to treatment, something that appears infeasible given the current pricing and policy environment.”
Several other barriers also impede progress against HBV and HCV. For example, only five states and two large cities receive enough funding for comprehensive viral hepatitis surveillance, Dr. Strom and his associates noted. If local and state public health departments cannot track infections, they cannot characterize their epidemics. In addition, viral hepatitis remains a source of stigma, adding to fear and avoidance of testing and undermining elimination efforts. Stigma, avoidance, and the fact that HBV and HCV are asymptomatic in the early stages all contribute to a large population of chronically infected patients who do not know their status.
Moreover, many state Medicaid programs will not cover foreigners until they have lived in the United States for 5 years, and the Affordable Care Act does not cover care for short-term residents and undocumented immigrants, the report stated. Newly infected HCV patients tend to have fewer resources, lower educational levels, and more often use injection drugs, making them harder to reach and screen through the health system. “Prisons are a promising venue in which to treat HCV, but treating HCV is expensive for the prison system. The cost of the direct-acting antivirals is high, and the staff time required to manage an inmate in treatment often far exceed the available resources,” the report stated.
The study was sponsored by the Centers for Disease Control and Prevention Office of Viral Hepatitis and the U.S. Department of Health and Human Services Office of Minority Health. The authors disclosed no conflicts of interest.
FROM THE NATIONAL ACADEMIES OF SCIENCES, ENGINEERING, AND MEDICINE

Hepatitis C virus transmission peaked in 1950
Iatrogenic factors, not high-risk behaviors, were the main cause of the exponential spread of hepatitis C virus infections in North America, according to a phylogenetic study published online in the Lancet Infectious Diseases.
The study shifted the epicenter of spread back by more than 15 years, to about 1950, when medical procedures were expanding after World War II at the same time that clinicians continued to reuse metal and glass syringes, said Dr. Jeffrey Joy of the British Columbia Centre for Excellence in HIV/AIDS, Vancouver, and his associates.
Exponential HCV transmission had mostly ended by 1965, negating “the idea that the epidemic among baby boomers and other demographic groups in North America is primarily due to injection drug use, unsafe tattooing, high-risk sex, and travel to high endemic areas during youth,” the researchers added.
The hepatitis C virus evolves rapidly, enabling researchers to characterize its spread based on the genetic divergence of infections. Dr. Joy and his associates studied 45,316 HCV genotype 1a sequences from the National Center for Biotechnology Information GenBank nucleotide database to carry out phylogenetic analyses of five HCV genes – E1, E2, NS2, NS4B, and NS5B (Lancet Infect Dis. 2016 Mar 30. doi: 10.1016/S1473-3099[16]00124-9).
The combined results for all gene regions suggested that the North American HCV epidemic expanded the most between 1940 and 1965, and that transmission rates peaked in 1950, when the oldest baby boomers were only 5 years old. Thus, nosocomial transmission rather than risky behaviors was the likely driver behind most infections, the researchers said.
“Our results could help to reduce stigma related to screening and diagnosis of hepatitis C virus, potentially increasing the numbers of providers offering screening, patients accepting testing, and if positive, presenting for care and treatment,” the authors noted.
The researchers also found that HCV transmission rates plateaued between 1965 and 1989, and dropped in the early 1990s, coinciding with increases in blood product testing and needle exchange programs aimed at preventing HIV infections. Furthermore, HCV infections rose slightly after 2000, mirroring epidemiologic reports of outbreaks among young injection drug users in nonurban areas and among men who have sex with men, the researchers said.
The Canadian Institutes of Health Research, Michael Smith Foundation for Health Research, and BC Centre for Excellence in HIV/AIDS funded the study. The investigators disclosed no competing interests.
Dr. Jeffrey Joy and colleagues offer evidence that the era of high rates of transmission and rapid expansion of hepatitis C virus infections was from 1940 to 1960, when reuse of glass and metal syringes was common.
The authors conclude that attributing the spread of hepatitis C virus to the medical establishment, rather than youthful behavior long outgrown by most baby boomers, could justify changing the message around screening of baby boomers. Baby boomers came of age when nosocomial transmission could have been rampant. For this additional reason, rather than just individual behavior, all Americans born between 1945 and 1965 should be tested, say the authors.
This change of message, while effective, could backfire. By accepting responsibility for the pre-1960 spread, the medical establishment might increase stigma for patients born after the uptake of disposable syringes and safer clinical practices. Also, screening by birth cohort might already be outlasting its usefulness in some populations. Lowering the cost of hepatitis C virus testing could lead to routine, universal testing becoming the norm, making birth cohort testing obsolete.
Last, the novel method used in this study could be replaced by an even newer, more precise technology; would our message need to change once more?
Here are the facts: Hepatitis C virus is common among Americans born between 1945 and 1965. A current Centers for Disease Control and Prevention fact sheet asserts that most people infected with hepatitis C virus are unaware of their diagnosis, despite aggressive campaigns since 2012 to find cases. Let’s just go ahead and test our baby boomers.
Dr. Anne C. Spaulding and Dr. Lesley S. Miller are at the Rollins School of Public Health, Emory University, Atlanta. They reported receiving grants and other funding from Gilead Sciences and Bristol-Myers Squibb. These comments are from their editorial (Lancet Infect Dis. 2016 Mar 30. doi: 10.1016/S1473-3099[16]30002-0).
Dr. Jeffrey Joy and colleagues offer evidence that the era of high rates of transmission and rapid expansion of hepatitis C virus infections was from 1940 to 1960, when reuse of glass and metal syringes was common.
The authors conclude that attributing the spread of hepatitis C virus to the medical establishment, rather than youthful behavior long outgrown by most baby boomers, could justify changing the message around screening of baby boomers. Baby boomers came of age when nosocomial transmission could have been rampant. For this additional reason, rather than just individual behavior, all Americans born between 1945 and 1965 should be tested, say the authors.
This change of message, while effective, could backfire. By accepting responsibility for the pre-1960 spread, the medical establishment might increase stigma for patients born after the uptake of disposable syringes and safer clinical practices. Also, screening by birth cohort might already be outlasting its usefulness in some populations. Lowering the cost of hepatitis C virus testing could lead to routine, universal testing becoming the norm, making birth cohort testing obsolete.
Last, the novel method used in this study could be replaced by an even newer, more precise technology; would our message need to change once more?
Here are the facts: Hepatitis C virus is common among Americans born between 1945 and 1965. A current Centers for Disease Control and Prevention fact sheet asserts that most people infected with hepatitis C virus are unaware of their diagnosis, despite aggressive campaigns since 2012 to find cases. Let’s just go ahead and test our baby boomers.
Dr. Anne C. Spaulding and Dr. Lesley S. Miller are at the Rollins School of Public Health, Emory University, Atlanta. They reported receiving grants and other funding from Gilead Sciences and Bristol-Myers Squibb. These comments are from their editorial (Lancet Infect Dis. 2016 Mar 30. doi: 10.1016/S1473-3099[16]30002-0).
Dr. Jeffrey Joy and colleagues offer evidence that the era of high rates of transmission and rapid expansion of hepatitis C virus infections was from 1940 to 1960, when reuse of glass and metal syringes was common.
The authors conclude that attributing the spread of hepatitis C virus to the medical establishment, rather than youthful behavior long outgrown by most baby boomers, could justify changing the message around screening of baby boomers. Baby boomers came of age when nosocomial transmission could have been rampant. For this additional reason, rather than just individual behavior, all Americans born between 1945 and 1965 should be tested, say the authors.
This change of message, while effective, could backfire. By accepting responsibility for the pre-1960 spread, the medical establishment might increase stigma for patients born after the uptake of disposable syringes and safer clinical practices. Also, screening by birth cohort might already be outlasting its usefulness in some populations. Lowering the cost of hepatitis C virus testing could lead to routine, universal testing becoming the norm, making birth cohort testing obsolete.
Last, the novel method used in this study could be replaced by an even newer, more precise technology; would our message need to change once more?
Here are the facts: Hepatitis C virus is common among Americans born between 1945 and 1965. A current Centers for Disease Control and Prevention fact sheet asserts that most people infected with hepatitis C virus are unaware of their diagnosis, despite aggressive campaigns since 2012 to find cases. Let’s just go ahead and test our baby boomers.
Dr. Anne C. Spaulding and Dr. Lesley S. Miller are at the Rollins School of Public Health, Emory University, Atlanta. They reported receiving grants and other funding from Gilead Sciences and Bristol-Myers Squibb. These comments are from their editorial (Lancet Infect Dis. 2016 Mar 30. doi: 10.1016/S1473-3099[16]30002-0).
Iatrogenic factors, not high-risk behaviors, were the main cause of the exponential spread of hepatitis C virus infections in North America, according to a phylogenetic study published online in the Lancet Infectious Diseases.
The study shifted the epicenter of spread back by more than 15 years, to about 1950, when medical procedures were expanding after World War II at the same time that clinicians continued to reuse metal and glass syringes, said Dr. Jeffrey Joy of the British Columbia Centre for Excellence in HIV/AIDS, Vancouver, and his associates.
Exponential HCV transmission had mostly ended by 1965, negating “the idea that the epidemic among baby boomers and other demographic groups in North America is primarily due to injection drug use, unsafe tattooing, high-risk sex, and travel to high endemic areas during youth,” the researchers added.
The hepatitis C virus evolves rapidly, enabling researchers to characterize its spread based on the genetic divergence of infections. Dr. Joy and his associates studied 45,316 HCV genotype 1a sequences from the National Center for Biotechnology Information GenBank nucleotide database to carry out phylogenetic analyses of five HCV genes – E1, E2, NS2, NS4B, and NS5B (Lancet Infect Dis. 2016 Mar 30. doi: 10.1016/S1473-3099[16]00124-9).
The combined results for all gene regions suggested that the North American HCV epidemic expanded the most between 1940 and 1965, and that transmission rates peaked in 1950, when the oldest baby boomers were only 5 years old. Thus, nosocomial transmission rather than risky behaviors was the likely driver behind most infections, the researchers said.
“Our results could help to reduce stigma related to screening and diagnosis of hepatitis C virus, potentially increasing the numbers of providers offering screening, patients accepting testing, and if positive, presenting for care and treatment,” the authors noted.
The researchers also found that HCV transmission rates plateaued between 1965 and 1989, and dropped in the early 1990s, coinciding with increases in blood product testing and needle exchange programs aimed at preventing HIV infections. Furthermore, HCV infections rose slightly after 2000, mirroring epidemiologic reports of outbreaks among young injection drug users in nonurban areas and among men who have sex with men, the researchers said.
The Canadian Institutes of Health Research, Michael Smith Foundation for Health Research, and BC Centre for Excellence in HIV/AIDS funded the study. The investigators disclosed no competing interests.
Iatrogenic factors, not high-risk behaviors, were the main cause of the exponential spread of hepatitis C virus infections in North America, according to a phylogenetic study published online in the Lancet Infectious Diseases.
The study shifted the epicenter of spread back by more than 15 years, to about 1950, when medical procedures were expanding after World War II at the same time that clinicians continued to reuse metal and glass syringes, said Dr. Jeffrey Joy of the British Columbia Centre for Excellence in HIV/AIDS, Vancouver, and his associates.
Exponential HCV transmission had mostly ended by 1965, negating “the idea that the epidemic among baby boomers and other demographic groups in North America is primarily due to injection drug use, unsafe tattooing, high-risk sex, and travel to high endemic areas during youth,” the researchers added.
The hepatitis C virus evolves rapidly, enabling researchers to characterize its spread based on the genetic divergence of infections. Dr. Joy and his associates studied 45,316 HCV genotype 1a sequences from the National Center for Biotechnology Information GenBank nucleotide database to carry out phylogenetic analyses of five HCV genes – E1, E2, NS2, NS4B, and NS5B (Lancet Infect Dis. 2016 Mar 30. doi: 10.1016/S1473-3099[16]00124-9).
The combined results for all gene regions suggested that the North American HCV epidemic expanded the most between 1940 and 1965, and that transmission rates peaked in 1950, when the oldest baby boomers were only 5 years old. Thus, nosocomial transmission rather than risky behaviors was the likely driver behind most infections, the researchers said.
“Our results could help to reduce stigma related to screening and diagnosis of hepatitis C virus, potentially increasing the numbers of providers offering screening, patients accepting testing, and if positive, presenting for care and treatment,” the authors noted.
The researchers also found that HCV transmission rates plateaued between 1965 and 1989, and dropped in the early 1990s, coinciding with increases in blood product testing and needle exchange programs aimed at preventing HIV infections. Furthermore, HCV infections rose slightly after 2000, mirroring epidemiologic reports of outbreaks among young injection drug users in nonurban areas and among men who have sex with men, the researchers said.
The Canadian Institutes of Health Research, Michael Smith Foundation for Health Research, and BC Centre for Excellence in HIV/AIDS funded the study. The investigators disclosed no competing interests.
FROM THE LANCET INFECTIOUS DISEASES
Key clinical point: Iatrogenic factors, rather than high-risk behaviors, were the most likely cause of the exponential spread of hepatitis C virus infections in North America.
Major finding: Transmission of HCV genotype 1a peaked in North America in 1950, when the oldest baby boomers were 5 years old.
Data source: A retrospective phylogenetic study of five HCV genotype 1a genes from 45,316 publicly available sequences.
Disclosures: The Canadian Institutes of Health Research, Michael Smith Foundation for Health Research, and BC Centre for Excellence in HIV/AIDS funded the study. The investigators disclosed no competing interests.