User login
Antimalarial adherence is important for diabetes prevention in lupus
Adhering to antimalarial treatment offers some protection to patients with systemic lupus erythematosus (SLE) from developing type 2 diabetes mellitus (T2DM), according to new research.
Patients who took at least 90% of their prescribed antimalarial doses were 39% less likely to develop T2DM than patients who discontinued antimalarial therapy. Patients who took less than 90% of their prescribed doses but didn’t discontinue treatment were 22% less likely to develop T2DM.
“[O]ur study provides further support for the importance of adherence to antimalarials in SLE by demonstrating protective impacts on T2DM,” Shahrzad Salmasi, PhD, of the University of British Columbia, Vancouver, and colleagues wrote in Arthritis Care & Research.
Dr. Salmasi and colleagues conducted this retrospective study using administrative health data on patients in British Columbia. The researchers analyzed 1,498 patients with SLE. Their mean age was about 44 years, and 91% were women.
The researchers used data on prescription dates and days’ supply to establish antimalarial drug courses and gaps in treatment. A new treatment course occurred when a 90-day gap was exceeded between refills. The researchers calculated the proportion of days covered (PDC) – the total number of days with antimalarials divided by the length of the course – and separated patients into three categories:
- Adherent to treatment – PDC of 0.90 or greater
- Nonadherent – PDC greater than 0 but less than 0.90
- Discontinuer – PDC of 0
The patients had a mean of about 23 antimalarial prescriptions and a mean of about two courses. The mean course duration was 554 days.
At a median follow-up of 4.6 years, there were 140 incident cases of T2DM. The researchers calculated the risk of T2DM among adherent and nonadherent patients, comparing these groups with the discontinuers and adjusting for age, sex, comorbidities, and concomitant medications.
The adjusted hazard ratio for developing T2DM was 0.61 among adherent patients and 0.78 among nonadherent patients.
“This population-based study highlighted that taking less than 90% of the prescribed antimalarials compromises their effect in preventing T2DM in SLE patients,” Dr. Salmasi and colleagues wrote. “Our findings should be used to emphasize the importance of medication adherence in not only treating SLE but also preventing its complications.”
The researchers reported having no conflicts of interest.
SOURCE: Salmasi S et al. Arthritis Care Res. 2020 Jan 21. doi: 10.1002/acr.24147.
Adhering to antimalarial treatment offers some protection to patients with systemic lupus erythematosus (SLE) from developing type 2 diabetes mellitus (T2DM), according to new research.
Patients who took at least 90% of their prescribed antimalarial doses were 39% less likely to develop T2DM than patients who discontinued antimalarial therapy. Patients who took less than 90% of their prescribed doses but didn’t discontinue treatment were 22% less likely to develop T2DM.
“[O]ur study provides further support for the importance of adherence to antimalarials in SLE by demonstrating protective impacts on T2DM,” Shahrzad Salmasi, PhD, of the University of British Columbia, Vancouver, and colleagues wrote in Arthritis Care & Research.
Dr. Salmasi and colleagues conducted this retrospective study using administrative health data on patients in British Columbia. The researchers analyzed 1,498 patients with SLE. Their mean age was about 44 years, and 91% were women.
The researchers used data on prescription dates and days’ supply to establish antimalarial drug courses and gaps in treatment. A new treatment course occurred when a 90-day gap was exceeded between refills. The researchers calculated the proportion of days covered (PDC) – the total number of days with antimalarials divided by the length of the course – and separated patients into three categories:
- Adherent to treatment – PDC of 0.90 or greater
- Nonadherent – PDC greater than 0 but less than 0.90
- Discontinuer – PDC of 0
The patients had a mean of about 23 antimalarial prescriptions and a mean of about two courses. The mean course duration was 554 days.
At a median follow-up of 4.6 years, there were 140 incident cases of T2DM. The researchers calculated the risk of T2DM among adherent and nonadherent patients, comparing these groups with the discontinuers and adjusting for age, sex, comorbidities, and concomitant medications.
The adjusted hazard ratio for developing T2DM was 0.61 among adherent patients and 0.78 among nonadherent patients.
“This population-based study highlighted that taking less than 90% of the prescribed antimalarials compromises their effect in preventing T2DM in SLE patients,” Dr. Salmasi and colleagues wrote. “Our findings should be used to emphasize the importance of medication adherence in not only treating SLE but also preventing its complications.”
The researchers reported having no conflicts of interest.
SOURCE: Salmasi S et al. Arthritis Care Res. 2020 Jan 21. doi: 10.1002/acr.24147.
Adhering to antimalarial treatment offers some protection to patients with systemic lupus erythematosus (SLE) from developing type 2 diabetes mellitus (T2DM), according to new research.
Patients who took at least 90% of their prescribed antimalarial doses were 39% less likely to develop T2DM than patients who discontinued antimalarial therapy. Patients who took less than 90% of their prescribed doses but didn’t discontinue treatment were 22% less likely to develop T2DM.
“[O]ur study provides further support for the importance of adherence to antimalarials in SLE by demonstrating protective impacts on T2DM,” Shahrzad Salmasi, PhD, of the University of British Columbia, Vancouver, and colleagues wrote in Arthritis Care & Research.
Dr. Salmasi and colleagues conducted this retrospective study using administrative health data on patients in British Columbia. The researchers analyzed 1,498 patients with SLE. Their mean age was about 44 years, and 91% were women.
The researchers used data on prescription dates and days’ supply to establish antimalarial drug courses and gaps in treatment. A new treatment course occurred when a 90-day gap was exceeded between refills. The researchers calculated the proportion of days covered (PDC) – the total number of days with antimalarials divided by the length of the course – and separated patients into three categories:
- Adherent to treatment – PDC of 0.90 or greater
- Nonadherent – PDC greater than 0 but less than 0.90
- Discontinuer – PDC of 0
The patients had a mean of about 23 antimalarial prescriptions and a mean of about two courses. The mean course duration was 554 days.
At a median follow-up of 4.6 years, there were 140 incident cases of T2DM. The researchers calculated the risk of T2DM among adherent and nonadherent patients, comparing these groups with the discontinuers and adjusting for age, sex, comorbidities, and concomitant medications.
The adjusted hazard ratio for developing T2DM was 0.61 among adherent patients and 0.78 among nonadherent patients.
“This population-based study highlighted that taking less than 90% of the prescribed antimalarials compromises their effect in preventing T2DM in SLE patients,” Dr. Salmasi and colleagues wrote. “Our findings should be used to emphasize the importance of medication adherence in not only treating SLE but also preventing its complications.”
The researchers reported having no conflicts of interest.
SOURCE: Salmasi S et al. Arthritis Care Res. 2020 Jan 21. doi: 10.1002/acr.24147.
FROM ARTHRITIS CARE & RESEARCH
February 2020
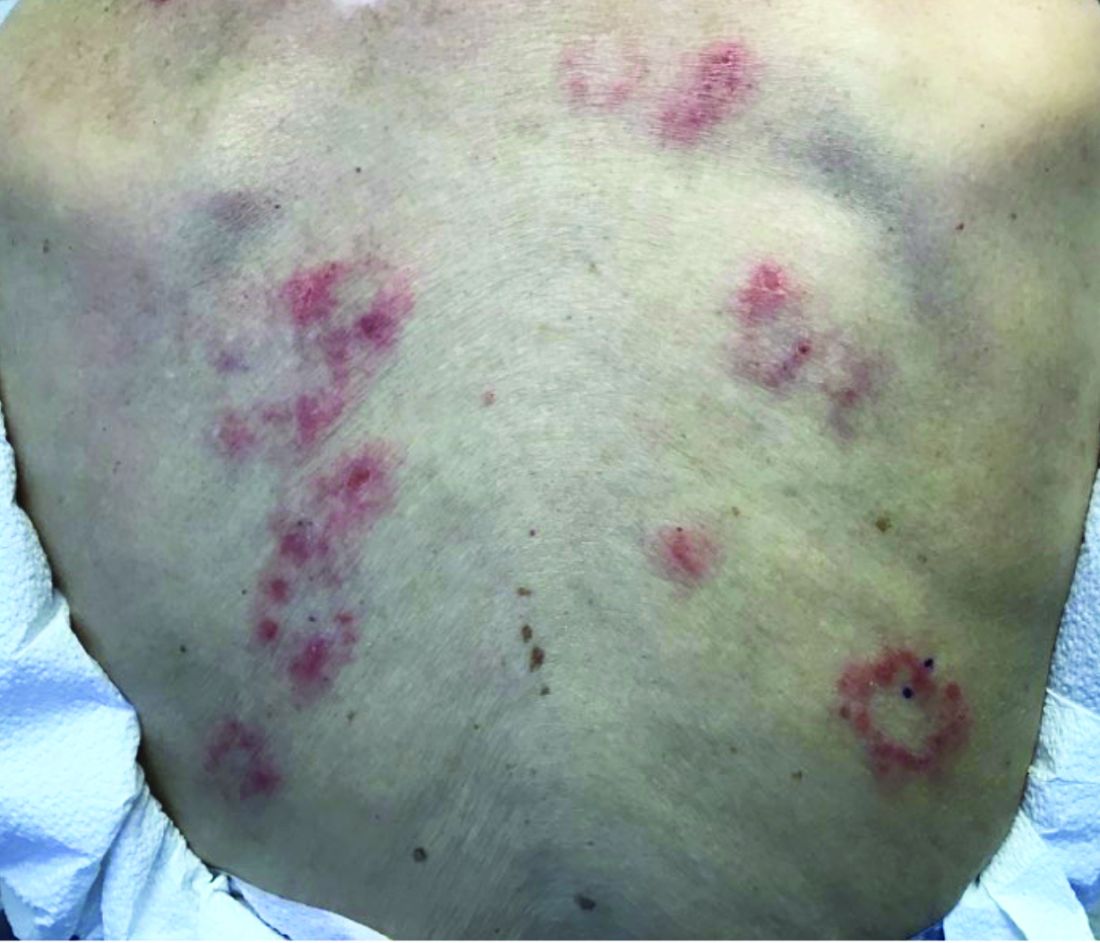
Subacute cutaneous lupus erythematosus
Subacute cutaneous lupus erythematosus (SCLE) is a type of cutaneous lupus erythematosus that may occur independently of or in combination with systemic lupus erythematosus. About 10%-15% of patients with SCLE will develop systemic lupus erythematosus. White females are more typically affected.
SCLE lesions often present as scaly, annular, or polycyclic scaly patches and plaques with central clearing. They may appear psoriasiform. They heal without atrophy or scarring but may leave dyspigmentation. Follicular plugging is absent. Lesions generally occur on sun exposed areas such as the neck, V of the chest, and upper extremities. Up to 75% of patients may exhibit associated symptoms such as photosensitivity, oral ulcers, and arthritis. Less than 20% of patients will develop internal disease, including nephritis and pulmonary disease. Symptoms of Sjögren’s syndrome and SCLE may overlap in some patients, and will portend higher risk for internal disease.
The differential diagnosis includes eczema, psoriasis, dermatophytosis, granuloma annulare, and erythema annulare centrifugum. Histology reveals epidermal atrophy and keratinocyte apoptosis, with a superficial and perivascular lymphohistiocytic infiltrate in the upper dermis. Interface changes at the dermal-epidermal junction can be seen. Direct immunofluorescence of lesional skin is positive in one-third of cases, often revealing granular deposits of IgG and IgM at the dermal-epidermal junction and around hair follicles (called the lupus-band test). Serology in SCLE may reveal a positive antinuclear antigen test, as well as positive Ro/SSA antigen. Other lupus serologies such as La/SSB, dsDNA, antihistone, and Sm antibodies may be positive, but are less commonly seen.

Several drugs may cause SCLE, such as hydrochlorothiazide, terbinafine, ACE inhibitors, NSAIDs, calcium-channel blockers, interferons, anticonvulsants, griseofulvin, penicillamine, spironolactone, tumor necrosis factor–alpha inhibitors, and statins. Discontinuing the offending medications may clear the lesions, but not always.
Treatment includes sunscreen and avoidance of sun exposure. Potent topical corticosteroids are helpful. If systemic treatment is indicated, antimalarials are first line.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
Subacute cutaneous lupus erythematosus
Subacute cutaneous lupus erythematosus (SCLE) is a type of cutaneous lupus erythematosus that may occur independently of or in combination with systemic lupus erythematosus. About 10%-15% of patients with SCLE will develop systemic lupus erythematosus. White females are more typically affected.
SCLE lesions often present as scaly, annular, or polycyclic scaly patches and plaques with central clearing. They may appear psoriasiform. They heal without atrophy or scarring but may leave dyspigmentation. Follicular plugging is absent. Lesions generally occur on sun exposed areas such as the neck, V of the chest, and upper extremities. Up to 75% of patients may exhibit associated symptoms such as photosensitivity, oral ulcers, and arthritis. Less than 20% of patients will develop internal disease, including nephritis and pulmonary disease. Symptoms of Sjögren’s syndrome and SCLE may overlap in some patients, and will portend higher risk for internal disease.
The differential diagnosis includes eczema, psoriasis, dermatophytosis, granuloma annulare, and erythema annulare centrifugum. Histology reveals epidermal atrophy and keratinocyte apoptosis, with a superficial and perivascular lymphohistiocytic infiltrate in the upper dermis. Interface changes at the dermal-epidermal junction can be seen. Direct immunofluorescence of lesional skin is positive in one-third of cases, often revealing granular deposits of IgG and IgM at the dermal-epidermal junction and around hair follicles (called the lupus-band test). Serology in SCLE may reveal a positive antinuclear antigen test, as well as positive Ro/SSA antigen. Other lupus serologies such as La/SSB, dsDNA, antihistone, and Sm antibodies may be positive, but are less commonly seen.
Several drugs may cause SCLE, such as hydrochlorothiazide, terbinafine, ACE inhibitors, NSAIDs, calcium-channel blockers, interferons, anticonvulsants, griseofulvin, penicillamine, spironolactone, tumor necrosis factor–alpha inhibitors, and statins. Discontinuing the offending medications may clear the lesions, but not always.
Treatment includes sunscreen and avoidance of sun exposure. Potent topical corticosteroids are helpful. If systemic treatment is indicated, antimalarials are first line.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
Subacute cutaneous lupus erythematosus
Subacute cutaneous lupus erythematosus (SCLE) is a type of cutaneous lupus erythematosus that may occur independently of or in combination with systemic lupus erythematosus. About 10%-15% of patients with SCLE will develop systemic lupus erythematosus. White females are more typically affected.
SCLE lesions often present as scaly, annular, or polycyclic scaly patches and plaques with central clearing. They may appear psoriasiform. They heal without atrophy or scarring but may leave dyspigmentation. Follicular plugging is absent. Lesions generally occur on sun exposed areas such as the neck, V of the chest, and upper extremities. Up to 75% of patients may exhibit associated symptoms such as photosensitivity, oral ulcers, and arthritis. Less than 20% of patients will develop internal disease, including nephritis and pulmonary disease. Symptoms of Sjögren’s syndrome and SCLE may overlap in some patients, and will portend higher risk for internal disease.
The differential diagnosis includes eczema, psoriasis, dermatophytosis, granuloma annulare, and erythema annulare centrifugum. Histology reveals epidermal atrophy and keratinocyte apoptosis, with a superficial and perivascular lymphohistiocytic infiltrate in the upper dermis. Interface changes at the dermal-epidermal junction can be seen. Direct immunofluorescence of lesional skin is positive in one-third of cases, often revealing granular deposits of IgG and IgM at the dermal-epidermal junction and around hair follicles (called the lupus-band test). Serology in SCLE may reveal a positive antinuclear antigen test, as well as positive Ro/SSA antigen. Other lupus serologies such as La/SSB, dsDNA, antihistone, and Sm antibodies may be positive, but are less commonly seen.
Several drugs may cause SCLE, such as hydrochlorothiazide, terbinafine, ACE inhibitors, NSAIDs, calcium-channel blockers, interferons, anticonvulsants, griseofulvin, penicillamine, spironolactone, tumor necrosis factor–alpha inhibitors, and statins. Discontinuing the offending medications may clear the lesions, but not always.
Treatment includes sunscreen and avoidance of sun exposure. Potent topical corticosteroids are helpful. If systemic treatment is indicated, antimalarials are first line.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
Big data breaks down Sjögren’s syndrome
The severity of Sjögren’s syndrome and its organ involvement has direct links to clinical characteristics that include age, gender, ethnicity, and geographical location, according to new research findings from a large, worldwide database of primary Sjögren’s syndrome patients.
Risk factors for more severe disease included male sex, earlier age at diagnosis, black/African-American (BAA) ethnicity, and living in more southerly countries, including below the equator. The authors hailed these factors as potentially helping to predict the presence of systemic disease in newly-diagnosed patients, as well as helping to determine the optimum follow-up strategy.
But not everyone agrees. In interviews, other experts suggested that the data were interesting and valuable, but were not ready to make reliable clinical predictions. “The predictive value of these data are limited when it comes to an individual patient sitting in front of you in the office,” said Tomas Mustelin, MD, professor of medicine at the University of Washington, Seattle.
“It would be an important clinical thing if you can figure out who’s at risk for systemic complications and who isn’t. This is a nice step in that direction,” said Hal Scofield, MD, professor of medicine and pathology at the University of Oklahoma, Oklahoma City, agreeing that the data were too preliminary to be broadly useful because there is too much overlap between different patient groups.
If there is one clinical message, it is that physicians should be looking beyond dry eyes and dry mouth in newly diagnosed Sjögren’s syndrome, according to Sarah Chung, MD, who is an acting instructor of rheumatology at the University of Washington. “It’s a reminder to investigate these extra-glandular manifestations, and a reminder that it is a heterogeneous presentation, so we have to keep an open mind and investigate thoroughly,” she said.
In the research published online in Rheumatology, first author Pilar Brito-Zerón, MD, PhD, of the University of Barcelona Hospital Clínic, and colleagues in the Sjögren Big Data Consortium used European League Against Rheumatism Sjögren’s syndrome disease activity index (ESSDAI) scores to examine phenotype and patient characteristics among 10,007 subjects drawn from the international consortium.

Overall, 93.5% of subjects were women, and the mean age at diagnosis was 53 (standard deviation [SD], 14.1 years). The mean total ESSDAI score was 6.1 (SD, 7.5).
Men had higher mean ESSDAI (8.1 vs. 6.0; P less than .001) and clinical ESSDAI (8.4 vs. 6.1; P less than .001) scores and were more likely to have a high disease activity state (22.5% vs. 11.7%; P less than .001). Domains that scored higher in men included lymphadenopathy (P less than .001), glandular (P less than .001), pulmonary (P = .001), peripheral nervous system (PNS) (P less than .001), and CNS (P less than .001).
The highest global ESSDAI scores were reported in patients younger than 35, but organ dominance differed by age group: The constitutional, lymphadenopathy, glandular, cutaneous, renal, hematologic, and biologic domains were highest in this age group, but pulmonary and peripheral nervous system were highest in patients over 65.
By ethnicity, the highest ESSDAI scores occurred in black/African-American patients (6.7), followed by white (6.5), Asian (5.4), and Hispanic (4.8; P less than .001). The organ-specific domains also tracked by ethnicity, with BAA patients experiencing the highest frequencies of activity in the lymphadenopathy, articular, neurologic, and biologic domains. White patients were more often affected in the glandular, cutaneous, and muscular domains, whereas Asians most often experienced the pulmonary, renal, and hematologic domains, and Hispanics the constitutional domain.
The survey included Europe, America, and Asia, and global ESSDAI scores were higher in the southern countries of Asia and Europe, and higher in countries below the equator in the Americas. The organ-by-organ activity scores had a differentiated pattern between northern and southern locales. Worldwide, the gradient of patients with moderate systemic activity (global ESSDAI score of 5 or higher) at diagnosis followed a north-south gradient.
The study had no specific funding, and the authors have no relevant financial disclosures. Dr. Mustelin, Dr. Scofield, and Dr. Chung have no relevant financial disclosures.
SOURCE: Brito-Zerón P et al. Rheumatology. 2019 Dec 24. doi: 10.1093/rheumatology/kez578.
The severity of Sjögren’s syndrome and its organ involvement has direct links to clinical characteristics that include age, gender, ethnicity, and geographical location, according to new research findings from a large, worldwide database of primary Sjögren’s syndrome patients.
Risk factors for more severe disease included male sex, earlier age at diagnosis, black/African-American (BAA) ethnicity, and living in more southerly countries, including below the equator. The authors hailed these factors as potentially helping to predict the presence of systemic disease in newly-diagnosed patients, as well as helping to determine the optimum follow-up strategy.
But not everyone agrees. In interviews, other experts suggested that the data were interesting and valuable, but were not ready to make reliable clinical predictions. “The predictive value of these data are limited when it comes to an individual patient sitting in front of you in the office,” said Tomas Mustelin, MD, professor of medicine at the University of Washington, Seattle.
“It would be an important clinical thing if you can figure out who’s at risk for systemic complications and who isn’t. This is a nice step in that direction,” said Hal Scofield, MD, professor of medicine and pathology at the University of Oklahoma, Oklahoma City, agreeing that the data were too preliminary to be broadly useful because there is too much overlap between different patient groups.
If there is one clinical message, it is that physicians should be looking beyond dry eyes and dry mouth in newly diagnosed Sjögren’s syndrome, according to Sarah Chung, MD, who is an acting instructor of rheumatology at the University of Washington. “It’s a reminder to investigate these extra-glandular manifestations, and a reminder that it is a heterogeneous presentation, so we have to keep an open mind and investigate thoroughly,” she said.
In the research published online in Rheumatology, first author Pilar Brito-Zerón, MD, PhD, of the University of Barcelona Hospital Clínic, and colleagues in the Sjögren Big Data Consortium used European League Against Rheumatism Sjögren’s syndrome disease activity index (ESSDAI) scores to examine phenotype and patient characteristics among 10,007 subjects drawn from the international consortium.
Overall, 93.5% of subjects were women, and the mean age at diagnosis was 53 (standard deviation [SD], 14.1 years). The mean total ESSDAI score was 6.1 (SD, 7.5).
Men had higher mean ESSDAI (8.1 vs. 6.0; P less than .001) and clinical ESSDAI (8.4 vs. 6.1; P less than .001) scores and were more likely to have a high disease activity state (22.5% vs. 11.7%; P less than .001). Domains that scored higher in men included lymphadenopathy (P less than .001), glandular (P less than .001), pulmonary (P = .001), peripheral nervous system (PNS) (P less than .001), and CNS (P less than .001).
The highest global ESSDAI scores were reported in patients younger than 35, but organ dominance differed by age group: The constitutional, lymphadenopathy, glandular, cutaneous, renal, hematologic, and biologic domains were highest in this age group, but pulmonary and peripheral nervous system were highest in patients over 65.
By ethnicity, the highest ESSDAI scores occurred in black/African-American patients (6.7), followed by white (6.5), Asian (5.4), and Hispanic (4.8; P less than .001). The organ-specific domains also tracked by ethnicity, with BAA patients experiencing the highest frequencies of activity in the lymphadenopathy, articular, neurologic, and biologic domains. White patients were more often affected in the glandular, cutaneous, and muscular domains, whereas Asians most often experienced the pulmonary, renal, and hematologic domains, and Hispanics the constitutional domain.
The survey included Europe, America, and Asia, and global ESSDAI scores were higher in the southern countries of Asia and Europe, and higher in countries below the equator in the Americas. The organ-by-organ activity scores had a differentiated pattern between northern and southern locales. Worldwide, the gradient of patients with moderate systemic activity (global ESSDAI score of 5 or higher) at diagnosis followed a north-south gradient.
The study had no specific funding, and the authors have no relevant financial disclosures. Dr. Mustelin, Dr. Scofield, and Dr. Chung have no relevant financial disclosures.
SOURCE: Brito-Zerón P et al. Rheumatology. 2019 Dec 24. doi: 10.1093/rheumatology/kez578.
The severity of Sjögren’s syndrome and its organ involvement has direct links to clinical characteristics that include age, gender, ethnicity, and geographical location, according to new research findings from a large, worldwide database of primary Sjögren’s syndrome patients.
Risk factors for more severe disease included male sex, earlier age at diagnosis, black/African-American (BAA) ethnicity, and living in more southerly countries, including below the equator. The authors hailed these factors as potentially helping to predict the presence of systemic disease in newly-diagnosed patients, as well as helping to determine the optimum follow-up strategy.
But not everyone agrees. In interviews, other experts suggested that the data were interesting and valuable, but were not ready to make reliable clinical predictions. “The predictive value of these data are limited when it comes to an individual patient sitting in front of you in the office,” said Tomas Mustelin, MD, professor of medicine at the University of Washington, Seattle.
“It would be an important clinical thing if you can figure out who’s at risk for systemic complications and who isn’t. This is a nice step in that direction,” said Hal Scofield, MD, professor of medicine and pathology at the University of Oklahoma, Oklahoma City, agreeing that the data were too preliminary to be broadly useful because there is too much overlap between different patient groups.
If there is one clinical message, it is that physicians should be looking beyond dry eyes and dry mouth in newly diagnosed Sjögren’s syndrome, according to Sarah Chung, MD, who is an acting instructor of rheumatology at the University of Washington. “It’s a reminder to investigate these extra-glandular manifestations, and a reminder that it is a heterogeneous presentation, so we have to keep an open mind and investigate thoroughly,” she said.
In the research published online in Rheumatology, first author Pilar Brito-Zerón, MD, PhD, of the University of Barcelona Hospital Clínic, and colleagues in the Sjögren Big Data Consortium used European League Against Rheumatism Sjögren’s syndrome disease activity index (ESSDAI) scores to examine phenotype and patient characteristics among 10,007 subjects drawn from the international consortium.
Overall, 93.5% of subjects were women, and the mean age at diagnosis was 53 (standard deviation [SD], 14.1 years). The mean total ESSDAI score was 6.1 (SD, 7.5).
Men had higher mean ESSDAI (8.1 vs. 6.0; P less than .001) and clinical ESSDAI (8.4 vs. 6.1; P less than .001) scores and were more likely to have a high disease activity state (22.5% vs. 11.7%; P less than .001). Domains that scored higher in men included lymphadenopathy (P less than .001), glandular (P less than .001), pulmonary (P = .001), peripheral nervous system (PNS) (P less than .001), and CNS (P less than .001).
The highest global ESSDAI scores were reported in patients younger than 35, but organ dominance differed by age group: The constitutional, lymphadenopathy, glandular, cutaneous, renal, hematologic, and biologic domains were highest in this age group, but pulmonary and peripheral nervous system were highest in patients over 65.
By ethnicity, the highest ESSDAI scores occurred in black/African-American patients (6.7), followed by white (6.5), Asian (5.4), and Hispanic (4.8; P less than .001). The organ-specific domains also tracked by ethnicity, with BAA patients experiencing the highest frequencies of activity in the lymphadenopathy, articular, neurologic, and biologic domains. White patients were more often affected in the glandular, cutaneous, and muscular domains, whereas Asians most often experienced the pulmonary, renal, and hematologic domains, and Hispanics the constitutional domain.
The survey included Europe, America, and Asia, and global ESSDAI scores were higher in the southern countries of Asia and Europe, and higher in countries below the equator in the Americas. The organ-by-organ activity scores had a differentiated pattern between northern and southern locales. Worldwide, the gradient of patients with moderate systemic activity (global ESSDAI score of 5 or higher) at diagnosis followed a north-south gradient.
The study had no specific funding, and the authors have no relevant financial disclosures. Dr. Mustelin, Dr. Scofield, and Dr. Chung have no relevant financial disclosures.
SOURCE: Brito-Zerón P et al. Rheumatology. 2019 Dec 24. doi: 10.1093/rheumatology/kez578.
FROM RHEUMATOLOGY
Half of SLE patients have incident neuropsychiatric events
Neuropsychiatric events occurred in just over half of all patients recently diagnosed with systemic lupus erythematosus and followed for an average of nearly 8 years in an international study of more than 1,800 patients.
Up to 30% of these neuropsychiatric (NP) events in up to 20% of the followed cohort were directly attributable to systemic lupus erythematosus (SLE) in a representative patient population, wrote John G. Hanly, MD, and associates in Annals of the Rheumatic Diseases. Their findings were consistent with prior reports, they added.
Another notable finding from follow-up of these 1,827 SLE patients was that among those without a history of SLE-related NP events at baseline, 74% remained free of NP events during the subsequent 10 years, wrote Dr. Hanly, professor of medicine and director of the lupus clinic at Dalhousie University, Halifax, N.S., and coauthors. Among patients free from SLE-associated NP events after 2 years, 84% remained event free during their remaining follow-up. SLE patients with a history of an NP event that subsequently resolved had a 72% rate of freedom from another NP event during 10 years of follow-up.
These findings came from patients recently diagnosed with SLE (within the preceding 15 months) and enrolled at any of 31 participating academic medical centers in North America, Europe, and Asia. The investigators considered preenrollment NP events to include those starting from 6 months prior to diagnosis of SLE until the time patients entered the study. They used case definitions for 19 SLE-associated NP events published by the American College of Rheumatology (Arthritis Rheum. 1999 Apr;42[4]:599-608). All enrolled patients underwent annual assessment for NP events, with follow-up continuing as long as 18 years.
The researchers identified NP events in 955 of the 1,827 enrolled patients, a 52% incidence, including 1,910 unique NP events that included episodes from each of the 19 NP event types, with 92% involving the central nervous system and 8% involving the peripheral nervous system. The percentage of NP events attributable to SLE ranged from 17% to 31%, and they occurred in 14%-21% of the studied patients, with the range reflecting various attribution models used in the analyses. Some patients remained in the same NP state, while others progressed through more than one state.
The study did not receive commercial funding. Dr. Hanly had no disclosures.
SOURCE: Hanly JG et al. Ann Rheum Dis. 2020 Jan 8. doi: 10.1136/annrheumdis-2019-216150.
Neuropsychiatric events occurred in just over half of all patients recently diagnosed with systemic lupus erythematosus and followed for an average of nearly 8 years in an international study of more than 1,800 patients.
Up to 30% of these neuropsychiatric (NP) events in up to 20% of the followed cohort were directly attributable to systemic lupus erythematosus (SLE) in a representative patient population, wrote John G. Hanly, MD, and associates in Annals of the Rheumatic Diseases. Their findings were consistent with prior reports, they added.
Another notable finding from follow-up of these 1,827 SLE patients was that among those without a history of SLE-related NP events at baseline, 74% remained free of NP events during the subsequent 10 years, wrote Dr. Hanly, professor of medicine and director of the lupus clinic at Dalhousie University, Halifax, N.S., and coauthors. Among patients free from SLE-associated NP events after 2 years, 84% remained event free during their remaining follow-up. SLE patients with a history of an NP event that subsequently resolved had a 72% rate of freedom from another NP event during 10 years of follow-up.
These findings came from patients recently diagnosed with SLE (within the preceding 15 months) and enrolled at any of 31 participating academic medical centers in North America, Europe, and Asia. The investigators considered preenrollment NP events to include those starting from 6 months prior to diagnosis of SLE until the time patients entered the study. They used case definitions for 19 SLE-associated NP events published by the American College of Rheumatology (Arthritis Rheum. 1999 Apr;42[4]:599-608). All enrolled patients underwent annual assessment for NP events, with follow-up continuing as long as 18 years.
The researchers identified NP events in 955 of the 1,827 enrolled patients, a 52% incidence, including 1,910 unique NP events that included episodes from each of the 19 NP event types, with 92% involving the central nervous system and 8% involving the peripheral nervous system. The percentage of NP events attributable to SLE ranged from 17% to 31%, and they occurred in 14%-21% of the studied patients, with the range reflecting various attribution models used in the analyses. Some patients remained in the same NP state, while others progressed through more than one state.
The study did not receive commercial funding. Dr. Hanly had no disclosures.
SOURCE: Hanly JG et al. Ann Rheum Dis. 2020 Jan 8. doi: 10.1136/annrheumdis-2019-216150.
Neuropsychiatric events occurred in just over half of all patients recently diagnosed with systemic lupus erythematosus and followed for an average of nearly 8 years in an international study of more than 1,800 patients.
Up to 30% of these neuropsychiatric (NP) events in up to 20% of the followed cohort were directly attributable to systemic lupus erythematosus (SLE) in a representative patient population, wrote John G. Hanly, MD, and associates in Annals of the Rheumatic Diseases. Their findings were consistent with prior reports, they added.
Another notable finding from follow-up of these 1,827 SLE patients was that among those without a history of SLE-related NP events at baseline, 74% remained free of NP events during the subsequent 10 years, wrote Dr. Hanly, professor of medicine and director of the lupus clinic at Dalhousie University, Halifax, N.S., and coauthors. Among patients free from SLE-associated NP events after 2 years, 84% remained event free during their remaining follow-up. SLE patients with a history of an NP event that subsequently resolved had a 72% rate of freedom from another NP event during 10 years of follow-up.
These findings came from patients recently diagnosed with SLE (within the preceding 15 months) and enrolled at any of 31 participating academic medical centers in North America, Europe, and Asia. The investigators considered preenrollment NP events to include those starting from 6 months prior to diagnosis of SLE until the time patients entered the study. They used case definitions for 19 SLE-associated NP events published by the American College of Rheumatology (Arthritis Rheum. 1999 Apr;42[4]:599-608). All enrolled patients underwent annual assessment for NP events, with follow-up continuing as long as 18 years.
The researchers identified NP events in 955 of the 1,827 enrolled patients, a 52% incidence, including 1,910 unique NP events that included episodes from each of the 19 NP event types, with 92% involving the central nervous system and 8% involving the peripheral nervous system. The percentage of NP events attributable to SLE ranged from 17% to 31%, and they occurred in 14%-21% of the studied patients, with the range reflecting various attribution models used in the analyses. Some patients remained in the same NP state, while others progressed through more than one state.
The study did not receive commercial funding. Dr. Hanly had no disclosures.
SOURCE: Hanly JG et al. Ann Rheum Dis. 2020 Jan 8. doi: 10.1136/annrheumdis-2019-216150.
FROM ANNALS OF THE RHEUMATIC DISEASES
Draft ACR Takayasu’s guidelines: Surgery is the last resort
ATLANTA – One of the goals in soon-to-be-published Takayasu’s arteritis guidelines from the American College of Rheumatology is to wean patients off high-dose steroids once they are in remission.

This recommendation is in opposition to another option – namely, switching these patients to low-dose glucocorticoids. The idea is to prevent long-term side effects, particularly in children. The guidelines also recommend against escalating immunotherapy for asymptomatic increases in inflammatory markers and generally recommend against surgery – stenting in most cases – unless there is threat to life, limb, or organ and also if limb pain is so severe it cramps quality of life and dose escalation doesn’t get the job done. If surgery is planned, patients should be on perioperative steroids if there’s active disease.
It’s draft guidance for now, but it’s probably what the final document will say when it’s published in 2020, according to a presentation at the annual meeting of the American College of Rheumatology by one of the authors, Anisha Dua, MD, an associate professor of rheumatology at Northwestern University, Chicago. She gave a sneak preview at the meeting.
In general, severe, active Takayasu’s calls for high-dose oral steroids in conjunction with a nonsteroid immunosuppressive, such as methotrexate, azathioprine, leflunomide, or mycophenolate mofetil. There’s evidence that dual therapy gives a more durable remission and also reduces the need for steroids.
When that approach doesn’t do the trick, the next step is a tumor necrosis factor (TNF) inhibitor. There’s evidence for infliximab, adalimumab, certolizumab, and etanercept. Dr. Dua noted, “We still can consider” tocilizumab, but it failed to meet its primary endpoint in a randomized trial, and evidence for other biologics is sparse or nonexistent. “TNF inhibitors are the first line” for refractory disease, Dr. Dua said.
The steroid taper comes after 6-12 months of remission. Given their toxicity, “our goal for steroids is zero,” especially in pediatric populations. Even in remission, patients should have a clinical assessment, including inflammatory markers, every 3-12 months.
A rise in C-reactive protein or erythrocyte sedimentation rate, with no new symptoms, might be a reason for more frequent monitoring, but it’s not a reason to escalate immunosuppression. That should be kept in reserve for new vascular lesions, rapid progression on an old one, or worsening of organ or limb ischemia.
“We recommend [escalation] over surgical intervention” because patients often develop collateral circulation that solves the problem; it also gives the disease time to quiet down should the patient eventually go into surgery. Immediate surgery is reserved for organ or life-threatening disease, Dr. Dua said.
“Takayasu’s is different from other vasculitides in the sense that patients often present with certain nonspecific constitutional symptoms,” and there’s not a lot of pathology or histology to work with, “so we do tend to rely on imaging a lot,” Dr. Dua said.
Angiography has fallen out of favor because it’s invasive and exposes patients to radiation, among other problems. The field has moved to noninvasive imaging such as color Doppler ultrasound, CT angiography, magnetic resonance angiography, and PET CT.
“We do recommend regularly scheduled, noninvasive imaging every 6-12 months, in addition to the routine clinical assessment,” except in children with inactive disease; the risk of sedation outweighs the imaging benefit, Dr. Dua said.
In patients with single-vessel cranial or cervical stenosis, without symptoms, “we recommend medical over surgical management because of the risk of surgery. Surgery can be considered for multivessel involvement,” she said.
She and her colleagues also recommend medical management for renal artery stenosis, including antihypertensives and immunotherapy escalation for active disease. Surgery is considered for refractory hypertension or worsening kidney function
Dr. Dua is a primary investigator and adviser for Chemocentryx and an adviser for Novartis and AbbVie.
ATLANTA – One of the goals in soon-to-be-published Takayasu’s arteritis guidelines from the American College of Rheumatology is to wean patients off high-dose steroids once they are in remission.
This recommendation is in opposition to another option – namely, switching these patients to low-dose glucocorticoids. The idea is to prevent long-term side effects, particularly in children. The guidelines also recommend against escalating immunotherapy for asymptomatic increases in inflammatory markers and generally recommend against surgery – stenting in most cases – unless there is threat to life, limb, or organ and also if limb pain is so severe it cramps quality of life and dose escalation doesn’t get the job done. If surgery is planned, patients should be on perioperative steroids if there’s active disease.
It’s draft guidance for now, but it’s probably what the final document will say when it’s published in 2020, according to a presentation at the annual meeting of the American College of Rheumatology by one of the authors, Anisha Dua, MD, an associate professor of rheumatology at Northwestern University, Chicago. She gave a sneak preview at the meeting.
In general, severe, active Takayasu’s calls for high-dose oral steroids in conjunction with a nonsteroid immunosuppressive, such as methotrexate, azathioprine, leflunomide, or mycophenolate mofetil. There’s evidence that dual therapy gives a more durable remission and also reduces the need for steroids.
When that approach doesn’t do the trick, the next step is a tumor necrosis factor (TNF) inhibitor. There’s evidence for infliximab, adalimumab, certolizumab, and etanercept. Dr. Dua noted, “We still can consider” tocilizumab, but it failed to meet its primary endpoint in a randomized trial, and evidence for other biologics is sparse or nonexistent. “TNF inhibitors are the first line” for refractory disease, Dr. Dua said.
The steroid taper comes after 6-12 months of remission. Given their toxicity, “our goal for steroids is zero,” especially in pediatric populations. Even in remission, patients should have a clinical assessment, including inflammatory markers, every 3-12 months.
A rise in C-reactive protein or erythrocyte sedimentation rate, with no new symptoms, might be a reason for more frequent monitoring, but it’s not a reason to escalate immunosuppression. That should be kept in reserve for new vascular lesions, rapid progression on an old one, or worsening of organ or limb ischemia.
“We recommend [escalation] over surgical intervention” because patients often develop collateral circulation that solves the problem; it also gives the disease time to quiet down should the patient eventually go into surgery. Immediate surgery is reserved for organ or life-threatening disease, Dr. Dua said.
“Takayasu’s is different from other vasculitides in the sense that patients often present with certain nonspecific constitutional symptoms,” and there’s not a lot of pathology or histology to work with, “so we do tend to rely on imaging a lot,” Dr. Dua said.
Angiography has fallen out of favor because it’s invasive and exposes patients to radiation, among other problems. The field has moved to noninvasive imaging such as color Doppler ultrasound, CT angiography, magnetic resonance angiography, and PET CT.
“We do recommend regularly scheduled, noninvasive imaging every 6-12 months, in addition to the routine clinical assessment,” except in children with inactive disease; the risk of sedation outweighs the imaging benefit, Dr. Dua said.
In patients with single-vessel cranial or cervical stenosis, without symptoms, “we recommend medical over surgical management because of the risk of surgery. Surgery can be considered for multivessel involvement,” she said.
She and her colleagues also recommend medical management for renal artery stenosis, including antihypertensives and immunotherapy escalation for active disease. Surgery is considered for refractory hypertension or worsening kidney function
Dr. Dua is a primary investigator and adviser for Chemocentryx and an adviser for Novartis and AbbVie.
ATLANTA – One of the goals in soon-to-be-published Takayasu’s arteritis guidelines from the American College of Rheumatology is to wean patients off high-dose steroids once they are in remission.
This recommendation is in opposition to another option – namely, switching these patients to low-dose glucocorticoids. The idea is to prevent long-term side effects, particularly in children. The guidelines also recommend against escalating immunotherapy for asymptomatic increases in inflammatory markers and generally recommend against surgery – stenting in most cases – unless there is threat to life, limb, or organ and also if limb pain is so severe it cramps quality of life and dose escalation doesn’t get the job done. If surgery is planned, patients should be on perioperative steroids if there’s active disease.
It’s draft guidance for now, but it’s probably what the final document will say when it’s published in 2020, according to a presentation at the annual meeting of the American College of Rheumatology by one of the authors, Anisha Dua, MD, an associate professor of rheumatology at Northwestern University, Chicago. She gave a sneak preview at the meeting.
In general, severe, active Takayasu’s calls for high-dose oral steroids in conjunction with a nonsteroid immunosuppressive, such as methotrexate, azathioprine, leflunomide, or mycophenolate mofetil. There’s evidence that dual therapy gives a more durable remission and also reduces the need for steroids.
When that approach doesn’t do the trick, the next step is a tumor necrosis factor (TNF) inhibitor. There’s evidence for infliximab, adalimumab, certolizumab, and etanercept. Dr. Dua noted, “We still can consider” tocilizumab, but it failed to meet its primary endpoint in a randomized trial, and evidence for other biologics is sparse or nonexistent. “TNF inhibitors are the first line” for refractory disease, Dr. Dua said.
The steroid taper comes after 6-12 months of remission. Given their toxicity, “our goal for steroids is zero,” especially in pediatric populations. Even in remission, patients should have a clinical assessment, including inflammatory markers, every 3-12 months.
A rise in C-reactive protein or erythrocyte sedimentation rate, with no new symptoms, might be a reason for more frequent monitoring, but it’s not a reason to escalate immunosuppression. That should be kept in reserve for new vascular lesions, rapid progression on an old one, or worsening of organ or limb ischemia.
“We recommend [escalation] over surgical intervention” because patients often develop collateral circulation that solves the problem; it also gives the disease time to quiet down should the patient eventually go into surgery. Immediate surgery is reserved for organ or life-threatening disease, Dr. Dua said.
“Takayasu’s is different from other vasculitides in the sense that patients often present with certain nonspecific constitutional symptoms,” and there’s not a lot of pathology or histology to work with, “so we do tend to rely on imaging a lot,” Dr. Dua said.
Angiography has fallen out of favor because it’s invasive and exposes patients to radiation, among other problems. The field has moved to noninvasive imaging such as color Doppler ultrasound, CT angiography, magnetic resonance angiography, and PET CT.
“We do recommend regularly scheduled, noninvasive imaging every 6-12 months, in addition to the routine clinical assessment,” except in children with inactive disease; the risk of sedation outweighs the imaging benefit, Dr. Dua said.
In patients with single-vessel cranial or cervical stenosis, without symptoms, “we recommend medical over surgical management because of the risk of surgery. Surgery can be considered for multivessel involvement,” she said.
She and her colleagues also recommend medical management for renal artery stenosis, including antihypertensives and immunotherapy escalation for active disease. Surgery is considered for refractory hypertension or worsening kidney function
Dr. Dua is a primary investigator and adviser for Chemocentryx and an adviser for Novartis and AbbVie.
REPORTING FROM ACR 2019
Methotrexate gives durable remission from idiopathic granulomatous mastitis
ATLANTA – Methotrexate, in combination with prednisone, might be emerging as the go-to option for idiopathic granulomatous mastitis, according to investigators from Oregon Health & Science University, Portland.

Idiopathic granulomatous mastitis (IGM) is an inflammatory disease in which granulomas form in breast tissue. It strikes mostly young to middle-aged women with painful, firm breast masses, sometimes with redness and drainage. Diagnosis is by biopsy with rule-out of known causes.
IGM does not respond to antibiotics. Prednisone and surgery have been the traditional approaches, but masses can recur after surgery, and a year or more of prednisone, with the weight gain and side effects, is problematic. As a result, cases are increasingly being referred to rheumatologists for other options, said lead investigator Sarah Ringsted, MD, a rheumatology fellow at the university.
A study she presented at the annual meeting of the American College of Rheumatology and previous work from others builds a case for methotrexate, which often seems to put the disease in remission and allows for shorter glucocorticoid courses. These days, “I offer this to patients as a great option. It’s really nice to have, instead of having women go on months and months of high-dose steroids, and I think we can save patients from unnecessary” surgery, Dr. Ringsted said.
Her usual regimen these days is methotrexate 15-20 mg/week for 12-18 months, with high-dose prednisone (greater than 20 mg/day) for the first 3 months, followed by a taper.
Dr. Ringsted and associates compared 23 women treated at the university during 2007-2018. Just 5 of the 12 women (42%) treated with high-dose prednisone alone went into remission and did not relapse over a mean follow-up of 27 months. Two out of three women who had both high-dose glucocorticoids and surgery achieved remission without relapse, as did all three women who received methotrexate and high-dose glucocorticoids (one also had surgery). Five other patients were treated with other options; just two had a durable remission.
The numbers are small, but they add to two previous reports. Among 19 women who had failed other treatments, 94% improved and 75% went into remission with 15 months of methotrexate in a review from Stanford (Calif.) University. An Iranian study of 17 patients treated with methotrexate, and also glucocorticoids in some, had a relapse rate of only 17.8%.
There were several cases of both inflammatory arthritis and erythema nodosum in the Oregon series, a higher incidence than what has been reported before for IGM. “It’s interesting because it makes me think of sarcoidosis. There have been cases of sarcoidosis causing mastitis, but mostly in patients with other features” of the disease. “It makes me wonder if any of these women will develop sarcoidosis later on; I think that’s an interesting question,” Dr. Ringsted said.
Women in the study were an average age of 32 years, and over half were Hispanic, which is associated with a higher risk for IGM. Almost all the women had been pregnant before and had breast fed in the previous 5 years. Cancer, tuberculosis, and fungal infections were among the things ruled out before mastitis was deemed idiopathic.
Women with IGM tend to be of childbearing age, and must be cautioned against the teratogenic effects of methotrexate, Dr. Ringsted noted.
There was no external funding, and the investigators didn’t report any disclosures.
SOURCE: Ringsted S et al. Arthritis Rheumatol. 2019;71(suppl 10), Abstract 386.
ATLANTA – Methotrexate, in combination with prednisone, might be emerging as the go-to option for idiopathic granulomatous mastitis, according to investigators from Oregon Health & Science University, Portland.
Idiopathic granulomatous mastitis (IGM) is an inflammatory disease in which granulomas form in breast tissue. It strikes mostly young to middle-aged women with painful, firm breast masses, sometimes with redness and drainage. Diagnosis is by biopsy with rule-out of known causes.
IGM does not respond to antibiotics. Prednisone and surgery have been the traditional approaches, but masses can recur after surgery, and a year or more of prednisone, with the weight gain and side effects, is problematic. As a result, cases are increasingly being referred to rheumatologists for other options, said lead investigator Sarah Ringsted, MD, a rheumatology fellow at the university.
A study she presented at the annual meeting of the American College of Rheumatology and previous work from others builds a case for methotrexate, which often seems to put the disease in remission and allows for shorter glucocorticoid courses. These days, “I offer this to patients as a great option. It’s really nice to have, instead of having women go on months and months of high-dose steroids, and I think we can save patients from unnecessary” surgery, Dr. Ringsted said.
Her usual regimen these days is methotrexate 15-20 mg/week for 12-18 months, with high-dose prednisone (greater than 20 mg/day) for the first 3 months, followed by a taper.
Dr. Ringsted and associates compared 23 women treated at the university during 2007-2018. Just 5 of the 12 women (42%) treated with high-dose prednisone alone went into remission and did not relapse over a mean follow-up of 27 months. Two out of three women who had both high-dose glucocorticoids and surgery achieved remission without relapse, as did all three women who received methotrexate and high-dose glucocorticoids (one also had surgery). Five other patients were treated with other options; just two had a durable remission.
The numbers are small, but they add to two previous reports. Among 19 women who had failed other treatments, 94% improved and 75% went into remission with 15 months of methotrexate in a review from Stanford (Calif.) University. An Iranian study of 17 patients treated with methotrexate, and also glucocorticoids in some, had a relapse rate of only 17.8%.
There were several cases of both inflammatory arthritis and erythema nodosum in the Oregon series, a higher incidence than what has been reported before for IGM. “It’s interesting because it makes me think of sarcoidosis. There have been cases of sarcoidosis causing mastitis, but mostly in patients with other features” of the disease. “It makes me wonder if any of these women will develop sarcoidosis later on; I think that’s an interesting question,” Dr. Ringsted said.
Women in the study were an average age of 32 years, and over half were Hispanic, which is associated with a higher risk for IGM. Almost all the women had been pregnant before and had breast fed in the previous 5 years. Cancer, tuberculosis, and fungal infections were among the things ruled out before mastitis was deemed idiopathic.
Women with IGM tend to be of childbearing age, and must be cautioned against the teratogenic effects of methotrexate, Dr. Ringsted noted.
There was no external funding, and the investigators didn’t report any disclosures.
SOURCE: Ringsted S et al. Arthritis Rheumatol. 2019;71(suppl 10), Abstract 386.
ATLANTA – Methotrexate, in combination with prednisone, might be emerging as the go-to option for idiopathic granulomatous mastitis, according to investigators from Oregon Health & Science University, Portland.
Idiopathic granulomatous mastitis (IGM) is an inflammatory disease in which granulomas form in breast tissue. It strikes mostly young to middle-aged women with painful, firm breast masses, sometimes with redness and drainage. Diagnosis is by biopsy with rule-out of known causes.
IGM does not respond to antibiotics. Prednisone and surgery have been the traditional approaches, but masses can recur after surgery, and a year or more of prednisone, with the weight gain and side effects, is problematic. As a result, cases are increasingly being referred to rheumatologists for other options, said lead investigator Sarah Ringsted, MD, a rheumatology fellow at the university.
A study she presented at the annual meeting of the American College of Rheumatology and previous work from others builds a case for methotrexate, which often seems to put the disease in remission and allows for shorter glucocorticoid courses. These days, “I offer this to patients as a great option. It’s really nice to have, instead of having women go on months and months of high-dose steroids, and I think we can save patients from unnecessary” surgery, Dr. Ringsted said.
Her usual regimen these days is methotrexate 15-20 mg/week for 12-18 months, with high-dose prednisone (greater than 20 mg/day) for the first 3 months, followed by a taper.
Dr. Ringsted and associates compared 23 women treated at the university during 2007-2018. Just 5 of the 12 women (42%) treated with high-dose prednisone alone went into remission and did not relapse over a mean follow-up of 27 months. Two out of three women who had both high-dose glucocorticoids and surgery achieved remission without relapse, as did all three women who received methotrexate and high-dose glucocorticoids (one also had surgery). Five other patients were treated with other options; just two had a durable remission.
The numbers are small, but they add to two previous reports. Among 19 women who had failed other treatments, 94% improved and 75% went into remission with 15 months of methotrexate in a review from Stanford (Calif.) University. An Iranian study of 17 patients treated with methotrexate, and also glucocorticoids in some, had a relapse rate of only 17.8%.
There were several cases of both inflammatory arthritis and erythema nodosum in the Oregon series, a higher incidence than what has been reported before for IGM. “It’s interesting because it makes me think of sarcoidosis. There have been cases of sarcoidosis causing mastitis, but mostly in patients with other features” of the disease. “It makes me wonder if any of these women will develop sarcoidosis later on; I think that’s an interesting question,” Dr. Ringsted said.
Women in the study were an average age of 32 years, and over half were Hispanic, which is associated with a higher risk for IGM. Almost all the women had been pregnant before and had breast fed in the previous 5 years. Cancer, tuberculosis, and fungal infections were among the things ruled out before mastitis was deemed idiopathic.
Women with IGM tend to be of childbearing age, and must be cautioned against the teratogenic effects of methotrexate, Dr. Ringsted noted.
There was no external funding, and the investigators didn’t report any disclosures.
SOURCE: Ringsted S et al. Arthritis Rheumatol. 2019;71(suppl 10), Abstract 386.
REPORTING FROM ACR 2019
Nearly 25% of U.S. adults take an obesogenic prescription drug
LAS VEGAS – based on national U.S. data collected during 2013-2016.

The Endocrine Society, the STOP Obesity Alliance, and other medical societies have recommended that clinicians try to minimize use of obesogenic drugs and focus on prescribing agents that are weight neutral or that trigger weight loss when those options are available and appropriate, and the new findings add further evidence that clinicians need to be more mindful of this issue, Craig M. Hales, MD, said at a meeting presented by the Obesity Society and the American Society for Metabolic and Bariatric Surgery.
Among the American adults interviewed for the survey, 40% of those on at least one prescription medication were on at least one drug that is considered obesogenic, said Dr. Hales, a medical epidemiologist at the Centers for Disease Control and Prevention in Hyattsville, Md.
According to practice guidelines published by the Endocrine Society, all drugs in the classes of glucocorticoids, beta-blockers, and antihistamines are obesogenic, as well as selected agents in the classes of antidepressant drugs, antipsychotics, antidepressants, antidiabetics, and contraceptives that are progestin only, said Dr. Hales (J Clin Endocrinol Metab. 2015 Feb;100[2]:342-62).
The data he reported came from the National Health and Nutrition Examination Survey (NHANES) run by the CDC during 2013-2016 that included 11,055 adults who were at least 20 years old. The findings showed that 23% of those adults had taken at least one drug that was considered obesogenic during the 30 days preceding the survey date. By comparison, 35% of the same adults had taken any type of prescription drug during the previous 30 days. That meant that overall, 40% of surveyed adults who had recently used any prescription medication had taken an obesogenic drug.
The 23% prevalence of recent obesogenic drug use was fairly stable at that level during several preceding NHANES surveys going back to 2001, suggesting that the increasing use of obesogenic drugs during the period since 2001 was not a factor in the recent increased prevalence of obesity among U.S. residents, added Dr. Hales.
The 2013-2016 analysis also showed a strong link between obesogenic drug use and increasing obesity severity. Among survey participants with a body mass index (BMI) in the normal range (18.5-24 kg/m2), 16% had recent use of an obesogenic drug. This prevalence increased to 22% among those who were overweight (BMI, 25-29 kg/m2), 29% among those with class 1 or 2 obesity (BMI, 30-39 kg/m2), and 33% among those with class 3 obesity (BMI, 40 kg/m2 or greater).
In contrast, recent use of prescription medications that do not contribute to obesity showed no significant relationship with BMI, with rates that ranged from 34% among those with a normal BMI, to 37% among those with class 3 obesity.
As an example of this relationship for a specific obesogenic drug class, the prevalence of beta-blocker use was about 7% among people with a normal BMI, about 10% among those who were overweight, about 14% among people with class 1 or 2 obesity, and about 17% among people with class 3 obesity, a statistically significant link suggesting that the relationship between use of obesogenic drugs and obesity is “bidirectional,” Dr. Hales said, in that increasing obesogenic drug use likely contributes to obesity, while simultaneously, the more obese people become, the more likely they are to take additional prescription drugs, particularly those that are obesogenic.
NHANES is run by the CDC and receives no commercial funding. The authors reported no conflicts of interest.
SOURCE: Hales CM et al. Obesity Week 2019, Abstract T-OR-2037.
LAS VEGAS – based on national U.S. data collected during 2013-2016.
The Endocrine Society, the STOP Obesity Alliance, and other medical societies have recommended that clinicians try to minimize use of obesogenic drugs and focus on prescribing agents that are weight neutral or that trigger weight loss when those options are available and appropriate, and the new findings add further evidence that clinicians need to be more mindful of this issue, Craig M. Hales, MD, said at a meeting presented by the Obesity Society and the American Society for Metabolic and Bariatric Surgery.
Among the American adults interviewed for the survey, 40% of those on at least one prescription medication were on at least one drug that is considered obesogenic, said Dr. Hales, a medical epidemiologist at the Centers for Disease Control and Prevention in Hyattsville, Md.
According to practice guidelines published by the Endocrine Society, all drugs in the classes of glucocorticoids, beta-blockers, and antihistamines are obesogenic, as well as selected agents in the classes of antidepressant drugs, antipsychotics, antidepressants, antidiabetics, and contraceptives that are progestin only, said Dr. Hales (J Clin Endocrinol Metab. 2015 Feb;100[2]:342-62).
The data he reported came from the National Health and Nutrition Examination Survey (NHANES) run by the CDC during 2013-2016 that included 11,055 adults who were at least 20 years old. The findings showed that 23% of those adults had taken at least one drug that was considered obesogenic during the 30 days preceding the survey date. By comparison, 35% of the same adults had taken any type of prescription drug during the previous 30 days. That meant that overall, 40% of surveyed adults who had recently used any prescription medication had taken an obesogenic drug.
The 23% prevalence of recent obesogenic drug use was fairly stable at that level during several preceding NHANES surveys going back to 2001, suggesting that the increasing use of obesogenic drugs during the period since 2001 was not a factor in the recent increased prevalence of obesity among U.S. residents, added Dr. Hales.
The 2013-2016 analysis also showed a strong link between obesogenic drug use and increasing obesity severity. Among survey participants with a body mass index (BMI) in the normal range (18.5-24 kg/m2), 16% had recent use of an obesogenic drug. This prevalence increased to 22% among those who were overweight (BMI, 25-29 kg/m2), 29% among those with class 1 or 2 obesity (BMI, 30-39 kg/m2), and 33% among those with class 3 obesity (BMI, 40 kg/m2 or greater).
In contrast, recent use of prescription medications that do not contribute to obesity showed no significant relationship with BMI, with rates that ranged from 34% among those with a normal BMI, to 37% among those with class 3 obesity.
As an example of this relationship for a specific obesogenic drug class, the prevalence of beta-blocker use was about 7% among people with a normal BMI, about 10% among those who were overweight, about 14% among people with class 1 or 2 obesity, and about 17% among people with class 3 obesity, a statistically significant link suggesting that the relationship between use of obesogenic drugs and obesity is “bidirectional,” Dr. Hales said, in that increasing obesogenic drug use likely contributes to obesity, while simultaneously, the more obese people become, the more likely they are to take additional prescription drugs, particularly those that are obesogenic.
NHANES is run by the CDC and receives no commercial funding. The authors reported no conflicts of interest.
SOURCE: Hales CM et al. Obesity Week 2019, Abstract T-OR-2037.
LAS VEGAS – based on national U.S. data collected during 2013-2016.
The Endocrine Society, the STOP Obesity Alliance, and other medical societies have recommended that clinicians try to minimize use of obesogenic drugs and focus on prescribing agents that are weight neutral or that trigger weight loss when those options are available and appropriate, and the new findings add further evidence that clinicians need to be more mindful of this issue, Craig M. Hales, MD, said at a meeting presented by the Obesity Society and the American Society for Metabolic and Bariatric Surgery.
Among the American adults interviewed for the survey, 40% of those on at least one prescription medication were on at least one drug that is considered obesogenic, said Dr. Hales, a medical epidemiologist at the Centers for Disease Control and Prevention in Hyattsville, Md.
According to practice guidelines published by the Endocrine Society, all drugs in the classes of glucocorticoids, beta-blockers, and antihistamines are obesogenic, as well as selected agents in the classes of antidepressant drugs, antipsychotics, antidepressants, antidiabetics, and contraceptives that are progestin only, said Dr. Hales (J Clin Endocrinol Metab. 2015 Feb;100[2]:342-62).
The data he reported came from the National Health and Nutrition Examination Survey (NHANES) run by the CDC during 2013-2016 that included 11,055 adults who were at least 20 years old. The findings showed that 23% of those adults had taken at least one drug that was considered obesogenic during the 30 days preceding the survey date. By comparison, 35% of the same adults had taken any type of prescription drug during the previous 30 days. That meant that overall, 40% of surveyed adults who had recently used any prescription medication had taken an obesogenic drug.
The 23% prevalence of recent obesogenic drug use was fairly stable at that level during several preceding NHANES surveys going back to 2001, suggesting that the increasing use of obesogenic drugs during the period since 2001 was not a factor in the recent increased prevalence of obesity among U.S. residents, added Dr. Hales.
The 2013-2016 analysis also showed a strong link between obesogenic drug use and increasing obesity severity. Among survey participants with a body mass index (BMI) in the normal range (18.5-24 kg/m2), 16% had recent use of an obesogenic drug. This prevalence increased to 22% among those who were overweight (BMI, 25-29 kg/m2), 29% among those with class 1 or 2 obesity (BMI, 30-39 kg/m2), and 33% among those with class 3 obesity (BMI, 40 kg/m2 or greater).
In contrast, recent use of prescription medications that do not contribute to obesity showed no significant relationship with BMI, with rates that ranged from 34% among those with a normal BMI, to 37% among those with class 3 obesity.
As an example of this relationship for a specific obesogenic drug class, the prevalence of beta-blocker use was about 7% among people with a normal BMI, about 10% among those who were overweight, about 14% among people with class 1 or 2 obesity, and about 17% among people with class 3 obesity, a statistically significant link suggesting that the relationship between use of obesogenic drugs and obesity is “bidirectional,” Dr. Hales said, in that increasing obesogenic drug use likely contributes to obesity, while simultaneously, the more obese people become, the more likely they are to take additional prescription drugs, particularly those that are obesogenic.
NHANES is run by the CDC and receives no commercial funding. The authors reported no conflicts of interest.
SOURCE: Hales CM et al. Obesity Week 2019, Abstract T-OR-2037.
REPORTING FROM OBESITY WEEK 2019
Hydroxychloroquine prevents congenital heart block recurrence in anti-Ro pregnancies
ATLANTA – Hydroxychloroquine (Plaquenil) 400 mg/day starting by pregnancy week 10 reduces recurrence of congenital heart block in infants born to women with anti-Ro antibodies, according to an open-label, prospective study presented at the annual meeting of the American College of Rheumatology.

Among antibody-positive women who had a previous pregnancy complicated by congenital heart block (CHB), the regimen reduced recurrence in a subsequent pregnancy from the expected historical rate of 18% to 7.4%, a more than 50% drop. “Given the potential benefit of hydroxychloroquine” (HCQ) and its relative safety during pregnancy, “testing all pregnancies for anti-Ro antibodies, regardless of maternal health, should be considered,” concluded investigators led by rheumatologist Peter Izmirly, MD, associate professor of medicine at New York (N.Y.) University.
About 40% of women with systemic lupus erythematosus and nearly 100% of women with Sjögren’s syndrome, as well as about 1% of women in the general population, have anti-Ro antibodies. They can be present in completely asymptomatic women, which is why the authors called for general screening. Indeed, half of the women in the trial had no or only mild, undifferentiated rheumatic symptoms. Often, “women who carry anti-Ro antibodies have no idea they have them” until they have a child with CHB and are tested, Dr. Izmirly said.
The antibodies cross the placenta and interfere with the normal development of the AV node; about 18% of infants die and most of the rest require lifelong pacing. The risk of CHB in antibody-positive women is about 2%, but once a child is born with the condition, the risk climbs to about 18% in subsequent pregnancies.
Years ago, Dr. Izmirly and his colleagues had a hunch that HCQ might help because it disrupts the toll-like receptor signaling involved in the disease process. A database review he led added weight to the idea, finding that among 257 anti-Ro positive pregnancies, the rate of CHB was 7.5% among the 40 women who happened to take HCQ, versus 21.2% among the 217 who did not. “We wanted to see if we could replicate that prospectively,” he said.
The Preventive Approach to Congenital Heart Block with Hydroxychloroquine (PATCH) trial enrolled 54 antibody positive women with a previous CHB pregnancy. They were started on 400 mg/day HCQ by gestation week 10.
There were four cases of second- or third-degree CHB among the women (7.4%, P = 0.02), all detected by fetal echocardiogram around week 20.
Nine of the women were treated with IVIG and/or dexamethasone for lupus flares or fetal heart issues other than advanced block, which confounded the results. To analyze the effect in a purely HCQ cohort, the team recruited an additional nine women not treated with any other medication during pregnancy, one of whose fetus developed third-degree heart block.
In total, 5 of 63 pregnancies (7.9%) resulted in advanced block. Among the 54 women exposed only to HCQ, the rate of second- or third-degree block was again 7.4% (4 of 54, P = .02). HCQ compliance, assessed by maternal blood levels above 200 ng/mL at least once, was 98%, and cord blood confirmed fetal exposure to HCQ.
Once detected, CHB was treated with dexamethasone or IVIG. One case progressed to cardiomyopathy, and the pregnancy was terminated. Another child required pacing after birth. Other children reverted to normal sinus rhythm but had intermittent second-degree block at age 2.
Overall, “the safety in this study was excellent,” said rheumatologist and senior investigator Jill Buyon, MD, director of the division of rheumatology at New York University.
The complications – nine births before 37 weeks, one infant small for gestational age – were not unexpected in a rheumatic population. “We were very nervous about Plaquenil cardiomyopathy” in the pregnancy that was terminated, but there was no evidence of it on histology.
The children will have ocular optical coherence tomography at age 5 to check for retinal toxicity; the 12 who have been tested so far show no obvious signs. Dr. Izmirly said he doesn’t expect to see any problems. “We are just being super cautious.”
The audience had questions about why the trial didn’t have a placebo arm. He explained that CHB is a rare event – one in 15,000 pregnancies – and it took 8 years just to adequately power the single-arm study; recruiting more than 100 additional women for a placebo-controlled trial wasn’t practical.
Also, “there was no way” women were going to be randomized to placebo when HCQ seemed so promising; 35% of the enrollees had already lost a child to CHB. “Everyone wanted the drug,” Dr. Izmirly said.
The majority of women were white, and about half met criteria for lupus and/or Sjögren’s. Anti-Ro levels remained above 1,000 EU throughout pregnancy. Women were excluded if they were taking high-dose prednisone or any dose of fluorinated corticosteroids at baseline.
The National Institutes of Health funded the work. The investigators had no relevant disclosures.
SOURCE: Izmirly P et al. Arthritis Rheumatol. 2019;71(suppl 10). Abstract 1761.
ATLANTA – Hydroxychloroquine (Plaquenil) 400 mg/day starting by pregnancy week 10 reduces recurrence of congenital heart block in infants born to women with anti-Ro antibodies, according to an open-label, prospective study presented at the annual meeting of the American College of Rheumatology.
Among antibody-positive women who had a previous pregnancy complicated by congenital heart block (CHB), the regimen reduced recurrence in a subsequent pregnancy from the expected historical rate of 18% to 7.4%, a more than 50% drop. “Given the potential benefit of hydroxychloroquine” (HCQ) and its relative safety during pregnancy, “testing all pregnancies for anti-Ro antibodies, regardless of maternal health, should be considered,” concluded investigators led by rheumatologist Peter Izmirly, MD, associate professor of medicine at New York (N.Y.) University.
About 40% of women with systemic lupus erythematosus and nearly 100% of women with Sjögren’s syndrome, as well as about 1% of women in the general population, have anti-Ro antibodies. They can be present in completely asymptomatic women, which is why the authors called for general screening. Indeed, half of the women in the trial had no or only mild, undifferentiated rheumatic symptoms. Often, “women who carry anti-Ro antibodies have no idea they have them” until they have a child with CHB and are tested, Dr. Izmirly said.
The antibodies cross the placenta and interfere with the normal development of the AV node; about 18% of infants die and most of the rest require lifelong pacing. The risk of CHB in antibody-positive women is about 2%, but once a child is born with the condition, the risk climbs to about 18% in subsequent pregnancies.
Years ago, Dr. Izmirly and his colleagues had a hunch that HCQ might help because it disrupts the toll-like receptor signaling involved in the disease process. A database review he led added weight to the idea, finding that among 257 anti-Ro positive pregnancies, the rate of CHB was 7.5% among the 40 women who happened to take HCQ, versus 21.2% among the 217 who did not. “We wanted to see if we could replicate that prospectively,” he said.
The Preventive Approach to Congenital Heart Block with Hydroxychloroquine (PATCH) trial enrolled 54 antibody positive women with a previous CHB pregnancy. They were started on 400 mg/day HCQ by gestation week 10.
There were four cases of second- or third-degree CHB among the women (7.4%, P = 0.02), all detected by fetal echocardiogram around week 20.
Nine of the women were treated with IVIG and/or dexamethasone for lupus flares or fetal heart issues other than advanced block, which confounded the results. To analyze the effect in a purely HCQ cohort, the team recruited an additional nine women not treated with any other medication during pregnancy, one of whose fetus developed third-degree heart block.
In total, 5 of 63 pregnancies (7.9%) resulted in advanced block. Among the 54 women exposed only to HCQ, the rate of second- or third-degree block was again 7.4% (4 of 54, P = .02). HCQ compliance, assessed by maternal blood levels above 200 ng/mL at least once, was 98%, and cord blood confirmed fetal exposure to HCQ.
Once detected, CHB was treated with dexamethasone or IVIG. One case progressed to cardiomyopathy, and the pregnancy was terminated. Another child required pacing after birth. Other children reverted to normal sinus rhythm but had intermittent second-degree block at age 2.
Overall, “the safety in this study was excellent,” said rheumatologist and senior investigator Jill Buyon, MD, director of the division of rheumatology at New York University.
The complications – nine births before 37 weeks, one infant small for gestational age – were not unexpected in a rheumatic population. “We were very nervous about Plaquenil cardiomyopathy” in the pregnancy that was terminated, but there was no evidence of it on histology.
The children will have ocular optical coherence tomography at age 5 to check for retinal toxicity; the 12 who have been tested so far show no obvious signs. Dr. Izmirly said he doesn’t expect to see any problems. “We are just being super cautious.”
The audience had questions about why the trial didn’t have a placebo arm. He explained that CHB is a rare event – one in 15,000 pregnancies – and it took 8 years just to adequately power the single-arm study; recruiting more than 100 additional women for a placebo-controlled trial wasn’t practical.
Also, “there was no way” women were going to be randomized to placebo when HCQ seemed so promising; 35% of the enrollees had already lost a child to CHB. “Everyone wanted the drug,” Dr. Izmirly said.
The majority of women were white, and about half met criteria for lupus and/or Sjögren’s. Anti-Ro levels remained above 1,000 EU throughout pregnancy. Women were excluded if they were taking high-dose prednisone or any dose of fluorinated corticosteroids at baseline.
The National Institutes of Health funded the work. The investigators had no relevant disclosures.
SOURCE: Izmirly P et al. Arthritis Rheumatol. 2019;71(suppl 10). Abstract 1761.
ATLANTA – Hydroxychloroquine (Plaquenil) 400 mg/day starting by pregnancy week 10 reduces recurrence of congenital heart block in infants born to women with anti-Ro antibodies, according to an open-label, prospective study presented at the annual meeting of the American College of Rheumatology.
Among antibody-positive women who had a previous pregnancy complicated by congenital heart block (CHB), the regimen reduced recurrence in a subsequent pregnancy from the expected historical rate of 18% to 7.4%, a more than 50% drop. “Given the potential benefit of hydroxychloroquine” (HCQ) and its relative safety during pregnancy, “testing all pregnancies for anti-Ro antibodies, regardless of maternal health, should be considered,” concluded investigators led by rheumatologist Peter Izmirly, MD, associate professor of medicine at New York (N.Y.) University.
About 40% of women with systemic lupus erythematosus and nearly 100% of women with Sjögren’s syndrome, as well as about 1% of women in the general population, have anti-Ro antibodies. They can be present in completely asymptomatic women, which is why the authors called for general screening. Indeed, half of the women in the trial had no or only mild, undifferentiated rheumatic symptoms. Often, “women who carry anti-Ro antibodies have no idea they have them” until they have a child with CHB and are tested, Dr. Izmirly said.
The antibodies cross the placenta and interfere with the normal development of the AV node; about 18% of infants die and most of the rest require lifelong pacing. The risk of CHB in antibody-positive women is about 2%, but once a child is born with the condition, the risk climbs to about 18% in subsequent pregnancies.
Years ago, Dr. Izmirly and his colleagues had a hunch that HCQ might help because it disrupts the toll-like receptor signaling involved in the disease process. A database review he led added weight to the idea, finding that among 257 anti-Ro positive pregnancies, the rate of CHB was 7.5% among the 40 women who happened to take HCQ, versus 21.2% among the 217 who did not. “We wanted to see if we could replicate that prospectively,” he said.
The Preventive Approach to Congenital Heart Block with Hydroxychloroquine (PATCH) trial enrolled 54 antibody positive women with a previous CHB pregnancy. They were started on 400 mg/day HCQ by gestation week 10.
There were four cases of second- or third-degree CHB among the women (7.4%, P = 0.02), all detected by fetal echocardiogram around week 20.
Nine of the women were treated with IVIG and/or dexamethasone for lupus flares or fetal heart issues other than advanced block, which confounded the results. To analyze the effect in a purely HCQ cohort, the team recruited an additional nine women not treated with any other medication during pregnancy, one of whose fetus developed third-degree heart block.
In total, 5 of 63 pregnancies (7.9%) resulted in advanced block. Among the 54 women exposed only to HCQ, the rate of second- or third-degree block was again 7.4% (4 of 54, P = .02). HCQ compliance, assessed by maternal blood levels above 200 ng/mL at least once, was 98%, and cord blood confirmed fetal exposure to HCQ.
Once detected, CHB was treated with dexamethasone or IVIG. One case progressed to cardiomyopathy, and the pregnancy was terminated. Another child required pacing after birth. Other children reverted to normal sinus rhythm but had intermittent second-degree block at age 2.
Overall, “the safety in this study was excellent,” said rheumatologist and senior investigator Jill Buyon, MD, director of the division of rheumatology at New York University.
The complications – nine births before 37 weeks, one infant small for gestational age – were not unexpected in a rheumatic population. “We were very nervous about Plaquenil cardiomyopathy” in the pregnancy that was terminated, but there was no evidence of it on histology.
The children will have ocular optical coherence tomography at age 5 to check for retinal toxicity; the 12 who have been tested so far show no obvious signs. Dr. Izmirly said he doesn’t expect to see any problems. “We are just being super cautious.”
The audience had questions about why the trial didn’t have a placebo arm. He explained that CHB is a rare event – one in 15,000 pregnancies – and it took 8 years just to adequately power the single-arm study; recruiting more than 100 additional women for a placebo-controlled trial wasn’t practical.
Also, “there was no way” women were going to be randomized to placebo when HCQ seemed so promising; 35% of the enrollees had already lost a child to CHB. “Everyone wanted the drug,” Dr. Izmirly said.
The majority of women were white, and about half met criteria for lupus and/or Sjögren’s. Anti-Ro levels remained above 1,000 EU throughout pregnancy. Women were excluded if they were taking high-dose prednisone or any dose of fluorinated corticosteroids at baseline.
The National Institutes of Health funded the work. The investigators had no relevant disclosures.
SOURCE: Izmirly P et al. Arthritis Rheumatol. 2019;71(suppl 10). Abstract 1761.
REPORTING FROM ACR 2019
ACR and EULAR release first classification criteria for IgG4-related disease
The American College of Rheumatology and the European League Against Rheumatism have released the first classification criteria for IgG4-related disease.
Although it was first recognized as a distinct disease in 2003, investigators have since learned that IgG4-related disease (IgG4-RD) is not particularly rare. Specialists across many different fields of medicine now treat IgG4-RD, which affects multiple organ systems, and the pancreas, kidneys, and orbits are most commonly affected by severe disease.
“IgG4-RD has proven to be a remarkable window into human immunology, and the insights investigators have made from studying this disease have already led to important discoveries in other rheumatic diseases, such as scleroderma,” John H. Stone, MD, professor of medicine at Harvard Medical School and director of clinical rheumatology at Massachusetts General Hospital, both in Boston, said in an interview.
To develop the classification criteria, 86 experts from five continents across various subspecialties including rheumatology, internal medicine, ophthalmology, pathology, gastroenterology, allergology, pulmonology, radiology, neurology, nephrology, and others met as a working group in 2016, achieving consensus on 79 criteria. They then narrowed down the number of items to 8 domains and 29 items within a set of inclusion and exclusion criteria for the draft classification criteria. For the final classification criteria, the working group applied weighting to each inclusion criteria item within a domain on a Likert scale (–5 to 5 range), removing items that were not significantly attributable to IgG4-RD classification (those with –2 to 2 scores).
The final IgG4-RD criteria are divided into three classification steps: entry criteria, exclusion criteria, and inclusion criteria. Patients who meet the entry criteria should have clinical or radiologic involvement of one or more organs consistent with IgG4-RD, such as the pancreas, salivary glands, bile ducts, orbits, kidney, lung, aorta, retroperitoneum, pachymeninges, or thyroid gland. Patients could alternatively meet the entry criteria by having “pathologic evidence of an inflammatory process accompanied by a lymphoplasmacytic infiltrate of uncertain etiology in one of these same organs,” the authors wrote.
If a patient meets the entry criteria, their case is examined against 32 clinical, serologic, radiologic, and pathologic items and specific disease inclusions. Any exclusion criteria present in a case means the patient does not meet the criteria for IgG4-RD classification.
The third step is to evaluate whether a patient meets inclusion criteria consisting of clinical findings, serologic results, radiology assessments, and pathology interpretations across eight domains: immunostaining, head and neck gland involvement, chest, pancreas and biliary tree, kidney, and the retroperitoneum. Each criterion has a weight, and if a patient has a score of 20 or higher, they meet the classification criteria for IgG4-RD.
“The final criteria set is easy to use and lends itself well to adaptation in an electronic format, which we have already instituted at my hospital,” said Dr. Stone, who is also director of the international panel of experts who created the criteria.
Two cohorts were used to validate the IgG4-RD classification criteria. In the first cohort, investigators used 771 patients (85% of the total cohort) in whom they were “confident” or “very confident” of a diagnosis of IgG4-RD or a mimicker to assess the test performance with a classification threshold of 20 points. The researchers found the criteria had a specificity of 99.2% (95% confidence interval, 97.2%-99.8%) and a sensitivity of 85.5% (95% CI, 81.9%-88.5%). The experts used a second validation cohort of 402 additional patients (83% of the total cohort) with suspected IgG4-RD or a mimicker using the same confident and very confident metric. The panel assembled this cohort because of minor definition changes in inclusion and exclusion criteria that had been made after the derivation set of patients had been collected, but the definitions of inclusion and exclusion criteria used in the two validation cohorts were exactly the same. Overall, the specificity of the criteria was 97.8% (95% CI, 93.7%-99.2%) and the sensitivity was 82.0% (95% CI, 77.0%-86.1%) for IgG4-RD classification in this second group.
Dr. Stone said that more investigations, including multicenter clinical trials, are being organized for patients with IgG4-RD, and these classification criteria will help to identify which patients to include in these studies.
“These rigorous ACR/EULAR classification criteria will help guide us through some of the most important challenges of studying this disease well,” Dr. Stone said. “I’m anticipating major advances in this field in the years to come, triggered in part by the strength of having sound classification criteria.”
The authors reported no relevant conflicts of interest.
SOURCE: Wallace ZS et al. Arthritis Rheumatol. 2019 Dec 2. doi: 10.1002/art.41120.
The American College of Rheumatology and the European League Against Rheumatism have released the first classification criteria for IgG4-related disease.
Although it was first recognized as a distinct disease in 2003, investigators have since learned that IgG4-related disease (IgG4-RD) is not particularly rare. Specialists across many different fields of medicine now treat IgG4-RD, which affects multiple organ systems, and the pancreas, kidneys, and orbits are most commonly affected by severe disease.
“IgG4-RD has proven to be a remarkable window into human immunology, and the insights investigators have made from studying this disease have already led to important discoveries in other rheumatic diseases, such as scleroderma,” John H. Stone, MD, professor of medicine at Harvard Medical School and director of clinical rheumatology at Massachusetts General Hospital, both in Boston, said in an interview.
To develop the classification criteria, 86 experts from five continents across various subspecialties including rheumatology, internal medicine, ophthalmology, pathology, gastroenterology, allergology, pulmonology, radiology, neurology, nephrology, and others met as a working group in 2016, achieving consensus on 79 criteria. They then narrowed down the number of items to 8 domains and 29 items within a set of inclusion and exclusion criteria for the draft classification criteria. For the final classification criteria, the working group applied weighting to each inclusion criteria item within a domain on a Likert scale (–5 to 5 range), removing items that were not significantly attributable to IgG4-RD classification (those with –2 to 2 scores).
The final IgG4-RD criteria are divided into three classification steps: entry criteria, exclusion criteria, and inclusion criteria. Patients who meet the entry criteria should have clinical or radiologic involvement of one or more organs consistent with IgG4-RD, such as the pancreas, salivary glands, bile ducts, orbits, kidney, lung, aorta, retroperitoneum, pachymeninges, or thyroid gland. Patients could alternatively meet the entry criteria by having “pathologic evidence of an inflammatory process accompanied by a lymphoplasmacytic infiltrate of uncertain etiology in one of these same organs,” the authors wrote.
If a patient meets the entry criteria, their case is examined against 32 clinical, serologic, radiologic, and pathologic items and specific disease inclusions. Any exclusion criteria present in a case means the patient does not meet the criteria for IgG4-RD classification.
The third step is to evaluate whether a patient meets inclusion criteria consisting of clinical findings, serologic results, radiology assessments, and pathology interpretations across eight domains: immunostaining, head and neck gland involvement, chest, pancreas and biliary tree, kidney, and the retroperitoneum. Each criterion has a weight, and if a patient has a score of 20 or higher, they meet the classification criteria for IgG4-RD.
“The final criteria set is easy to use and lends itself well to adaptation in an electronic format, which we have already instituted at my hospital,” said Dr. Stone, who is also director of the international panel of experts who created the criteria.
Two cohorts were used to validate the IgG4-RD classification criteria. In the first cohort, investigators used 771 patients (85% of the total cohort) in whom they were “confident” or “very confident” of a diagnosis of IgG4-RD or a mimicker to assess the test performance with a classification threshold of 20 points. The researchers found the criteria had a specificity of 99.2% (95% confidence interval, 97.2%-99.8%) and a sensitivity of 85.5% (95% CI, 81.9%-88.5%). The experts used a second validation cohort of 402 additional patients (83% of the total cohort) with suspected IgG4-RD or a mimicker using the same confident and very confident metric. The panel assembled this cohort because of minor definition changes in inclusion and exclusion criteria that had been made after the derivation set of patients had been collected, but the definitions of inclusion and exclusion criteria used in the two validation cohorts were exactly the same. Overall, the specificity of the criteria was 97.8% (95% CI, 93.7%-99.2%) and the sensitivity was 82.0% (95% CI, 77.0%-86.1%) for IgG4-RD classification in this second group.
Dr. Stone said that more investigations, including multicenter clinical trials, are being organized for patients with IgG4-RD, and these classification criteria will help to identify which patients to include in these studies.
“These rigorous ACR/EULAR classification criteria will help guide us through some of the most important challenges of studying this disease well,” Dr. Stone said. “I’m anticipating major advances in this field in the years to come, triggered in part by the strength of having sound classification criteria.”
The authors reported no relevant conflicts of interest.
SOURCE: Wallace ZS et al. Arthritis Rheumatol. 2019 Dec 2. doi: 10.1002/art.41120.
The American College of Rheumatology and the European League Against Rheumatism have released the first classification criteria for IgG4-related disease.
Although it was first recognized as a distinct disease in 2003, investigators have since learned that IgG4-related disease (IgG4-RD) is not particularly rare. Specialists across many different fields of medicine now treat IgG4-RD, which affects multiple organ systems, and the pancreas, kidneys, and orbits are most commonly affected by severe disease.
“IgG4-RD has proven to be a remarkable window into human immunology, and the insights investigators have made from studying this disease have already led to important discoveries in other rheumatic diseases, such as scleroderma,” John H. Stone, MD, professor of medicine at Harvard Medical School and director of clinical rheumatology at Massachusetts General Hospital, both in Boston, said in an interview.
To develop the classification criteria, 86 experts from five continents across various subspecialties including rheumatology, internal medicine, ophthalmology, pathology, gastroenterology, allergology, pulmonology, radiology, neurology, nephrology, and others met as a working group in 2016, achieving consensus on 79 criteria. They then narrowed down the number of items to 8 domains and 29 items within a set of inclusion and exclusion criteria for the draft classification criteria. For the final classification criteria, the working group applied weighting to each inclusion criteria item within a domain on a Likert scale (–5 to 5 range), removing items that were not significantly attributable to IgG4-RD classification (those with –2 to 2 scores).
The final IgG4-RD criteria are divided into three classification steps: entry criteria, exclusion criteria, and inclusion criteria. Patients who meet the entry criteria should have clinical or radiologic involvement of one or more organs consistent with IgG4-RD, such as the pancreas, salivary glands, bile ducts, orbits, kidney, lung, aorta, retroperitoneum, pachymeninges, or thyroid gland. Patients could alternatively meet the entry criteria by having “pathologic evidence of an inflammatory process accompanied by a lymphoplasmacytic infiltrate of uncertain etiology in one of these same organs,” the authors wrote.
If a patient meets the entry criteria, their case is examined against 32 clinical, serologic, radiologic, and pathologic items and specific disease inclusions. Any exclusion criteria present in a case means the patient does not meet the criteria for IgG4-RD classification.
The third step is to evaluate whether a patient meets inclusion criteria consisting of clinical findings, serologic results, radiology assessments, and pathology interpretations across eight domains: immunostaining, head and neck gland involvement, chest, pancreas and biliary tree, kidney, and the retroperitoneum. Each criterion has a weight, and if a patient has a score of 20 or higher, they meet the classification criteria for IgG4-RD.
“The final criteria set is easy to use and lends itself well to adaptation in an electronic format, which we have already instituted at my hospital,” said Dr. Stone, who is also director of the international panel of experts who created the criteria.
Two cohorts were used to validate the IgG4-RD classification criteria. In the first cohort, investigators used 771 patients (85% of the total cohort) in whom they were “confident” or “very confident” of a diagnosis of IgG4-RD or a mimicker to assess the test performance with a classification threshold of 20 points. The researchers found the criteria had a specificity of 99.2% (95% confidence interval, 97.2%-99.8%) and a sensitivity of 85.5% (95% CI, 81.9%-88.5%). The experts used a second validation cohort of 402 additional patients (83% of the total cohort) with suspected IgG4-RD or a mimicker using the same confident and very confident metric. The panel assembled this cohort because of minor definition changes in inclusion and exclusion criteria that had been made after the derivation set of patients had been collected, but the definitions of inclusion and exclusion criteria used in the two validation cohorts were exactly the same. Overall, the specificity of the criteria was 97.8% (95% CI, 93.7%-99.2%) and the sensitivity was 82.0% (95% CI, 77.0%-86.1%) for IgG4-RD classification in this second group.
Dr. Stone said that more investigations, including multicenter clinical trials, are being organized for patients with IgG4-RD, and these classification criteria will help to identify which patients to include in these studies.
“These rigorous ACR/EULAR classification criteria will help guide us through some of the most important challenges of studying this disease well,” Dr. Stone said. “I’m anticipating major advances in this field in the years to come, triggered in part by the strength of having sound classification criteria.”
The authors reported no relevant conflicts of interest.
SOURCE: Wallace ZS et al. Arthritis Rheumatol. 2019 Dec 2. doi: 10.1002/art.41120.
FROM ARTHRITIS & RHEUMATOLOGY
VEDOSS study describes predictors of progression to systemic sclerosis
ATLANTA – , according to recent results from the Very Early Diagnosis Of Systemic Sclerosis (VEDOSS) study presented at the annual meeting of the American College of Rheumatology.

“Our data show that thanks [to a] combination of the signs that characterize the various phases of the disease, patients can be diagnosed [with systemic sclerosis] in the very early stages,” first author Silvia Bellando-Randone, MD, PhD, assistant professor in the division of rheumatology at the University of Florence (Italy), said in her presentation.
Dr. Bellando-Randone and colleagues performed a longitudinal, observational study of 742 patients (mean 45.7 years old) at 42 centers in a cohort of mostly women (90%), nearly all of whom had had Raynaud’s phenomenon for longer than 36 months (97.5%). Patients were excluded if they had systemic sclerosis based on ACR 1980 classification criteria and/or ACR–European League Against Rheumatism 2013 criteria, had systemic sclerosis together with other connective-tissue diseases, or were unlikely to be present for three consecutive annual exams. Data collection began in March 2012 with follow-up of 5 years.
The researchers determined the positive predictive values (PPV) and negative predictive values (NPV) of clinical features, systemic sclerosis–specific antibodies, and nailfold video capillaroscopy (NVC) abnormalities on progression from Raynaud’s phenomenon to systemic sclerosis. Laboratory data collected at baseline included presence of antinuclear antibodies (ANA), anticentromere antibodies (ACA), anti-DNA topoisomerase I antibodies (anti-Scl-70), anti-U1RNP antibodies, anti-RNA polymerase III antibodies (ARA), N-terminal pro b-type natriuretic peptides (NT-proBNP), and C-reactive protein/erythrocyte sedimentation rate. Dr. Bellando-Randone and colleagues also collected clinical, pulmonary function, lung high-resolution CT, echocardiographic, and ECG data at baseline.
Predictions were based on these factors alone and in combination. Overall, 65% of patients had positive ANA. Other baseline characteristics present in patients that predicted systemic sclerosis included positive ACA/anti-Scl-70/ARA (32%), NVC abnormalities such as giant capillaries (25%), and puffy fingers (17%).
Using Kaplan-Meier analysis, the researchers found 7.4% of 401 patients who were ANA positive progressed to meet ACR-EULAR 2013 criteria, and the percentage of these patients increased to 29.3% at 3 years and 44.1% at 5 years. When the researchers considered disease-specific antibodies alone, 10.6% of 90 patients progressed from Raynaud’s phenomenon to systemic sclerosis within 1 year, 39.6% within 3 years, and 50.3% within 5 years. When the researchers analyzed disease-specific antibodies and NVC abnormalities together, 16% of 72 patients progressed to systemic sclerosis within 1 year, 61.7% within 3 years, and 77.4% within 5 years.
Puffy fingers also were a predictor of progression, and 14.4% of 69 patients with puffy fingers alone progressed from Raynaud’s phenomenon to systemic sclerosis at 1 year, 47.7% at 3 years, and 67.9% at 5 years. Considering puffy fingers and disease-specific antibodies together, 20% of 27 patients progressed at 1 year, 56.3% at 3 years, and 91.3% at 5 years. No patients with puffy fingers together and NVC abnormalities progressed to systemic sclerosis at 1 year, but 60.4% of 22 patients progressed at 3 years before plateauing at 5 years. For patients with NVC abnormalities alone, 7.1% progressed to systemic sclerosis from Raynaud’s phenomenon at 1 year, 39.4% at 3 years, and 52.7% at 5 years.
“Regarding capillaroscopy, we have to say that not all centers that participated were equally screened in capillaroscopy, and so we cannot assume the accuracy of this data,” she said.
Dr. Bellando-Randone noted that, apart from puffy fingers, disease-specific antibodies, and NVC abnormalities, patients were more likely to have a history of esophageal symptoms if they progressed to systemic sclerosis (37.3%), compared with patients who did not progress (23.6%; P = .003).
Puffy fingers alone were an independent predictor of systemic sclerosis (PPV, 78.9%; NPV, 45.1%) as well as in combination with disease-specific antibodies (PPV, 94.1%; NPV, 43.9%). The combination of disease-specific antibodies plus NVC abnormalities also independently predicted progression to systemic sclerosis (PPV, 82.2%; NPV, 50.4%). In a Cox multivariate analysis, disease-specific antibodies (relative risk, 5.4; 95% confidence interval, 3.7-7.9) and puffy fingers (RR, 3.0; 95% CI, 2.0-4.4) together were strongly predictive of progression from Raynaud’s phenomenon to systemic sclerosis (RR, 4.3; 95% CI, 2.6-7.3).
“This is really important for the risk stratification of patients [in] the very early stages of the disease, even if these data should be corroborated by larger data in larger studies in the future,” said Dr. Bellando-Randone.
The investigators reported having no conflicts of interest.
SOURCE: Bellando-Randone S et al. Arthritis Rheumatol. 2019;71(suppl 10). Abstract 2914.
ATLANTA – , according to recent results from the Very Early Diagnosis Of Systemic Sclerosis (VEDOSS) study presented at the annual meeting of the American College of Rheumatology.
“Our data show that thanks [to a] combination of the signs that characterize the various phases of the disease, patients can be diagnosed [with systemic sclerosis] in the very early stages,” first author Silvia Bellando-Randone, MD, PhD, assistant professor in the division of rheumatology at the University of Florence (Italy), said in her presentation.
Dr. Bellando-Randone and colleagues performed a longitudinal, observational study of 742 patients (mean 45.7 years old) at 42 centers in a cohort of mostly women (90%), nearly all of whom had had Raynaud’s phenomenon for longer than 36 months (97.5%). Patients were excluded if they had systemic sclerosis based on ACR 1980 classification criteria and/or ACR–European League Against Rheumatism 2013 criteria, had systemic sclerosis together with other connective-tissue diseases, or were unlikely to be present for three consecutive annual exams. Data collection began in March 2012 with follow-up of 5 years.
The researchers determined the positive predictive values (PPV) and negative predictive values (NPV) of clinical features, systemic sclerosis–specific antibodies, and nailfold video capillaroscopy (NVC) abnormalities on progression from Raynaud’s phenomenon to systemic sclerosis. Laboratory data collected at baseline included presence of antinuclear antibodies (ANA), anticentromere antibodies (ACA), anti-DNA topoisomerase I antibodies (anti-Scl-70), anti-U1RNP antibodies, anti-RNA polymerase III antibodies (ARA), N-terminal pro b-type natriuretic peptides (NT-proBNP), and C-reactive protein/erythrocyte sedimentation rate. Dr. Bellando-Randone and colleagues also collected clinical, pulmonary function, lung high-resolution CT, echocardiographic, and ECG data at baseline.
Predictions were based on these factors alone and in combination. Overall, 65% of patients had positive ANA. Other baseline characteristics present in patients that predicted systemic sclerosis included positive ACA/anti-Scl-70/ARA (32%), NVC abnormalities such as giant capillaries (25%), and puffy fingers (17%).
Using Kaplan-Meier analysis, the researchers found 7.4% of 401 patients who were ANA positive progressed to meet ACR-EULAR 2013 criteria, and the percentage of these patients increased to 29.3% at 3 years and 44.1% at 5 years. When the researchers considered disease-specific antibodies alone, 10.6% of 90 patients progressed from Raynaud’s phenomenon to systemic sclerosis within 1 year, 39.6% within 3 years, and 50.3% within 5 years. When the researchers analyzed disease-specific antibodies and NVC abnormalities together, 16% of 72 patients progressed to systemic sclerosis within 1 year, 61.7% within 3 years, and 77.4% within 5 years.
Puffy fingers also were a predictor of progression, and 14.4% of 69 patients with puffy fingers alone progressed from Raynaud’s phenomenon to systemic sclerosis at 1 year, 47.7% at 3 years, and 67.9% at 5 years. Considering puffy fingers and disease-specific antibodies together, 20% of 27 patients progressed at 1 year, 56.3% at 3 years, and 91.3% at 5 years. No patients with puffy fingers together and NVC abnormalities progressed to systemic sclerosis at 1 year, but 60.4% of 22 patients progressed at 3 years before plateauing at 5 years. For patients with NVC abnormalities alone, 7.1% progressed to systemic sclerosis from Raynaud’s phenomenon at 1 year, 39.4% at 3 years, and 52.7% at 5 years.
“Regarding capillaroscopy, we have to say that not all centers that participated were equally screened in capillaroscopy, and so we cannot assume the accuracy of this data,” she said.
Dr. Bellando-Randone noted that, apart from puffy fingers, disease-specific antibodies, and NVC abnormalities, patients were more likely to have a history of esophageal symptoms if they progressed to systemic sclerosis (37.3%), compared with patients who did not progress (23.6%; P = .003).
Puffy fingers alone were an independent predictor of systemic sclerosis (PPV, 78.9%; NPV, 45.1%) as well as in combination with disease-specific antibodies (PPV, 94.1%; NPV, 43.9%). The combination of disease-specific antibodies plus NVC abnormalities also independently predicted progression to systemic sclerosis (PPV, 82.2%; NPV, 50.4%). In a Cox multivariate analysis, disease-specific antibodies (relative risk, 5.4; 95% confidence interval, 3.7-7.9) and puffy fingers (RR, 3.0; 95% CI, 2.0-4.4) together were strongly predictive of progression from Raynaud’s phenomenon to systemic sclerosis (RR, 4.3; 95% CI, 2.6-7.3).
“This is really important for the risk stratification of patients [in] the very early stages of the disease, even if these data should be corroborated by larger data in larger studies in the future,” said Dr. Bellando-Randone.
The investigators reported having no conflicts of interest.
SOURCE: Bellando-Randone S et al. Arthritis Rheumatol. 2019;71(suppl 10). Abstract 2914.
ATLANTA – , according to recent results from the Very Early Diagnosis Of Systemic Sclerosis (VEDOSS) study presented at the annual meeting of the American College of Rheumatology.
“Our data show that thanks [to a] combination of the signs that characterize the various phases of the disease, patients can be diagnosed [with systemic sclerosis] in the very early stages,” first author Silvia Bellando-Randone, MD, PhD, assistant professor in the division of rheumatology at the University of Florence (Italy), said in her presentation.
Dr. Bellando-Randone and colleagues performed a longitudinal, observational study of 742 patients (mean 45.7 years old) at 42 centers in a cohort of mostly women (90%), nearly all of whom had had Raynaud’s phenomenon for longer than 36 months (97.5%). Patients were excluded if they had systemic sclerosis based on ACR 1980 classification criteria and/or ACR–European League Against Rheumatism 2013 criteria, had systemic sclerosis together with other connective-tissue diseases, or were unlikely to be present for three consecutive annual exams. Data collection began in March 2012 with follow-up of 5 years.
The researchers determined the positive predictive values (PPV) and negative predictive values (NPV) of clinical features, systemic sclerosis–specific antibodies, and nailfold video capillaroscopy (NVC) abnormalities on progression from Raynaud’s phenomenon to systemic sclerosis. Laboratory data collected at baseline included presence of antinuclear antibodies (ANA), anticentromere antibodies (ACA), anti-DNA topoisomerase I antibodies (anti-Scl-70), anti-U1RNP antibodies, anti-RNA polymerase III antibodies (ARA), N-terminal pro b-type natriuretic peptides (NT-proBNP), and C-reactive protein/erythrocyte sedimentation rate. Dr. Bellando-Randone and colleagues also collected clinical, pulmonary function, lung high-resolution CT, echocardiographic, and ECG data at baseline.
Predictions were based on these factors alone and in combination. Overall, 65% of patients had positive ANA. Other baseline characteristics present in patients that predicted systemic sclerosis included positive ACA/anti-Scl-70/ARA (32%), NVC abnormalities such as giant capillaries (25%), and puffy fingers (17%).
Using Kaplan-Meier analysis, the researchers found 7.4% of 401 patients who were ANA positive progressed to meet ACR-EULAR 2013 criteria, and the percentage of these patients increased to 29.3% at 3 years and 44.1% at 5 years. When the researchers considered disease-specific antibodies alone, 10.6% of 90 patients progressed from Raynaud’s phenomenon to systemic sclerosis within 1 year, 39.6% within 3 years, and 50.3% within 5 years. When the researchers analyzed disease-specific antibodies and NVC abnormalities together, 16% of 72 patients progressed to systemic sclerosis within 1 year, 61.7% within 3 years, and 77.4% within 5 years.
Puffy fingers also were a predictor of progression, and 14.4% of 69 patients with puffy fingers alone progressed from Raynaud’s phenomenon to systemic sclerosis at 1 year, 47.7% at 3 years, and 67.9% at 5 years. Considering puffy fingers and disease-specific antibodies together, 20% of 27 patients progressed at 1 year, 56.3% at 3 years, and 91.3% at 5 years. No patients with puffy fingers together and NVC abnormalities progressed to systemic sclerosis at 1 year, but 60.4% of 22 patients progressed at 3 years before plateauing at 5 years. For patients with NVC abnormalities alone, 7.1% progressed to systemic sclerosis from Raynaud’s phenomenon at 1 year, 39.4% at 3 years, and 52.7% at 5 years.
“Regarding capillaroscopy, we have to say that not all centers that participated were equally screened in capillaroscopy, and so we cannot assume the accuracy of this data,” she said.
Dr. Bellando-Randone noted that, apart from puffy fingers, disease-specific antibodies, and NVC abnormalities, patients were more likely to have a history of esophageal symptoms if they progressed to systemic sclerosis (37.3%), compared with patients who did not progress (23.6%; P = .003).
Puffy fingers alone were an independent predictor of systemic sclerosis (PPV, 78.9%; NPV, 45.1%) as well as in combination with disease-specific antibodies (PPV, 94.1%; NPV, 43.9%). The combination of disease-specific antibodies plus NVC abnormalities also independently predicted progression to systemic sclerosis (PPV, 82.2%; NPV, 50.4%). In a Cox multivariate analysis, disease-specific antibodies (relative risk, 5.4; 95% confidence interval, 3.7-7.9) and puffy fingers (RR, 3.0; 95% CI, 2.0-4.4) together were strongly predictive of progression from Raynaud’s phenomenon to systemic sclerosis (RR, 4.3; 95% CI, 2.6-7.3).
“This is really important for the risk stratification of patients [in] the very early stages of the disease, even if these data should be corroborated by larger data in larger studies in the future,” said Dr. Bellando-Randone.
The investigators reported having no conflicts of interest.
SOURCE: Bellando-Randone S et al. Arthritis Rheumatol. 2019;71(suppl 10). Abstract 2914.
REPORTING FROM ACR 2019