User login
Brentuximab vedotin plus nivolumab shows positive outcomes in PMBL
Combination brentuximab vedotin and nivolumab showed manageable safety and high activity in patients with relapsed/refractory primary mediastinal B-cell lymphoma (PMBL), according to results from a phase 2 trial.
“We evaluated whether the combination of nivolumab and [brentuximab vedotin] was safe and synergistically effective in patients with [relapsed/refractory] PMBL,” Pier Luigi Zinzani, MD, PhD, of the University of Bologna (Italy), and colleagues wrote in the Journal of Clinical Oncology.
The CheckMate 436 study is a multicenter, open-label, phase 1-2 study that included patients with relapsed/refractory disease who had previously received autologous stem cell transplantation (ASCT) or had two or more previous chemotherapy regimens for those ineligible for ASCT.
The phase 2 component evaluated the safety and efficacy of the two-drug combo in an expansion cohort of 30 patients. Study participants received intravenous brentuximab vedotin at 1.8 mg/kg and nivolumab at 240 mg every 3 weeks until cancer progression or intolerable adverse effects.
The primary outcomes were the investigator-evaluated objective response rate and safety. Secondary outcomes included progression-free survival, complete remission rate, overall duration of response, among other measures.
After analysis, the researchers reported that 53% of patients had grade 3 or 4 treatment-related toxicities following a median of five treatment cycles. The most common treatment-related toxicities were neutropenia (30%) and peripheral neuropathy (27%).
Five patients died during the study follow-up, four because of disease progression and one as a result of sepsis that was not considered related to treatment.
At a median follow-up of 11.1 months, the objective response rate was 73% in study participants, including 11 patients (37%) who achieved a complete response and 11 patients (37%) who had a partial response. An additional three patients had stable disease.
The median progression-free survival, duration of response, and overall survival were not reached in this study.
“The combination of nivolumab and [brentuximab vedotin] may be synergistic and is highly active in patients with [relapsed/refractory] PMBL, serving as a potential bridge to other consolidative therapies of curative intent,” the researchers wrote.
The study was funded by Bristol-Myers Squibb and Seattle Genetics. The authors reported financial affiliations with the study sponsors and several other companies.
SOURCE: Zinzani PL et al. J Clin Oncol. 2019 Aug 9. doi: 10.1200/JCO.19.01492.
Combination brentuximab vedotin and nivolumab showed manageable safety and high activity in patients with relapsed/refractory primary mediastinal B-cell lymphoma (PMBL), according to results from a phase 2 trial.
“We evaluated whether the combination of nivolumab and [brentuximab vedotin] was safe and synergistically effective in patients with [relapsed/refractory] PMBL,” Pier Luigi Zinzani, MD, PhD, of the University of Bologna (Italy), and colleagues wrote in the Journal of Clinical Oncology.
The CheckMate 436 study is a multicenter, open-label, phase 1-2 study that included patients with relapsed/refractory disease who had previously received autologous stem cell transplantation (ASCT) or had two or more previous chemotherapy regimens for those ineligible for ASCT.
The phase 2 component evaluated the safety and efficacy of the two-drug combo in an expansion cohort of 30 patients. Study participants received intravenous brentuximab vedotin at 1.8 mg/kg and nivolumab at 240 mg every 3 weeks until cancer progression or intolerable adverse effects.
The primary outcomes were the investigator-evaluated objective response rate and safety. Secondary outcomes included progression-free survival, complete remission rate, overall duration of response, among other measures.
After analysis, the researchers reported that 53% of patients had grade 3 or 4 treatment-related toxicities following a median of five treatment cycles. The most common treatment-related toxicities were neutropenia (30%) and peripheral neuropathy (27%).
Five patients died during the study follow-up, four because of disease progression and one as a result of sepsis that was not considered related to treatment.
At a median follow-up of 11.1 months, the objective response rate was 73% in study participants, including 11 patients (37%) who achieved a complete response and 11 patients (37%) who had a partial response. An additional three patients had stable disease.
The median progression-free survival, duration of response, and overall survival were not reached in this study.
“The combination of nivolumab and [brentuximab vedotin] may be synergistic and is highly active in patients with [relapsed/refractory] PMBL, serving as a potential bridge to other consolidative therapies of curative intent,” the researchers wrote.
The study was funded by Bristol-Myers Squibb and Seattle Genetics. The authors reported financial affiliations with the study sponsors and several other companies.
SOURCE: Zinzani PL et al. J Clin Oncol. 2019 Aug 9. doi: 10.1200/JCO.19.01492.
Combination brentuximab vedotin and nivolumab showed manageable safety and high activity in patients with relapsed/refractory primary mediastinal B-cell lymphoma (PMBL), according to results from a phase 2 trial.
“We evaluated whether the combination of nivolumab and [brentuximab vedotin] was safe and synergistically effective in patients with [relapsed/refractory] PMBL,” Pier Luigi Zinzani, MD, PhD, of the University of Bologna (Italy), and colleagues wrote in the Journal of Clinical Oncology.
The CheckMate 436 study is a multicenter, open-label, phase 1-2 study that included patients with relapsed/refractory disease who had previously received autologous stem cell transplantation (ASCT) or had two or more previous chemotherapy regimens for those ineligible for ASCT.
The phase 2 component evaluated the safety and efficacy of the two-drug combo in an expansion cohort of 30 patients. Study participants received intravenous brentuximab vedotin at 1.8 mg/kg and nivolumab at 240 mg every 3 weeks until cancer progression or intolerable adverse effects.
The primary outcomes were the investigator-evaluated objective response rate and safety. Secondary outcomes included progression-free survival, complete remission rate, overall duration of response, among other measures.
After analysis, the researchers reported that 53% of patients had grade 3 or 4 treatment-related toxicities following a median of five treatment cycles. The most common treatment-related toxicities were neutropenia (30%) and peripheral neuropathy (27%).
Five patients died during the study follow-up, four because of disease progression and one as a result of sepsis that was not considered related to treatment.
At a median follow-up of 11.1 months, the objective response rate was 73% in study participants, including 11 patients (37%) who achieved a complete response and 11 patients (37%) who had a partial response. An additional three patients had stable disease.
The median progression-free survival, duration of response, and overall survival were not reached in this study.
“The combination of nivolumab and [brentuximab vedotin] may be synergistic and is highly active in patients with [relapsed/refractory] PMBL, serving as a potential bridge to other consolidative therapies of curative intent,” the researchers wrote.
The study was funded by Bristol-Myers Squibb and Seattle Genetics. The authors reported financial affiliations with the study sponsors and several other companies.
SOURCE: Zinzani PL et al. J Clin Oncol. 2019 Aug 9. doi: 10.1200/JCO.19.01492.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Brentuximab vedotin plus nivolumab showed manageable safety and positive activity in patients with relapsed/refractory primary mediastinal B-cell lymphoma (PMBL).
Major finding: At 11.1 months, the objective response rate was 73% in study participants, including 37% of patients who achieved a complete response and 37% who had a partial response.
Study details: A phase 2 study of 30 patients with relapsed/refractory PMBL.
Disclosures: The study was funded by Bristol-Myers Squibb and Seattle Genetics. The authors reported financial affiliations with the study sponsors and several other companies.
Source: Zinzani PL et al. J Clin Oncol. 2019 Aug 9. doi: 10.1200/JCO.19.01492.
Ibrutinib/rituximab effective, safe as frontline treatment for older patients with MCL
LUGANO, SWITZERLAND – The chemotherapy-free combination of ibrutinib (Imbruvica) and rituximab is highly effective as frontline therapy for older, transplant-ineligible patients with nonblastoid mantle cell lymphoma, according to investigators.

In a phase 2 study of patients with a median age of 71 years, 38 of 41 patients (93%) had an objective response, and the regimen was both safe and easy to administer, reported Preetesh Jain, MD, PhD, of the University of Texas MD Anderson Cancer Center in Houston.
“The adverse event profile was generally favorable, with specific monitoring recommended for patients with cardiovascular comorbidities and a history of atrial fibrillation,” he said at the International Conference on Malignant Lymphoma.
The investigators enrolled 48 patients aged 65 years and older with previously untreated mantle cell lymphoma (MCL), of whom 41 were evaluable for the primary endpoints of overall response rate (ORR) and safety. The patients had good performance status and normal organ function, with the largest tumor size less than 10 cm. Patients with atrial fibrillation could participate, if the fibrillation was controlled. Patients with Ki-67 protein levels of 50% or greater and blastoid/pleomorphic histology were excluded.
Patients were treated with ibrutinib 560 mg orally daily for each 28-day cycle, with therapy continued until disease progression, or until therapy was stopped for any other reason. Patients also received intravenous rituximab 375 mg/m2 on days 1, 8, 15 and 22, plus or minus one day for cycle 1, on day 1 of cycles 3-8, and on day 1 of every other cycle for up to 2 years.
Of the 41 patients evaluable for response, 26 (64%) had a complete response (CR) and 12 (29%) had a partial response. Three additional patients had stable disease, for an objective response rate of 93%.
Of 34 patients with PET scans, all had negative scans. Of 37 patients evaluable for minimal residual disease (MRD) by flow cytometry, 21 (58%) were MRD negative.
Patients with low or intermediate Mantle Cell Lymphoma International Prognostic Index (MIPI) scores had a higher ORR (100% vs. 89% for patients with high MIPI scores), and patients with low Ki-67 levels had a higher response rate than that of patients with KI-67 of 30% or greater (80% vs. 87%).
Neither median 3-year progression-free survival nor median 3-year overall survival have been reached, with respective 3-year rates of 87% and 95%.
Four patients experienced disease progression at 4, 10, 13 and 33 months of treatment. Three of these patients had disease that had transformed to blastoid/pleomorphic variant, two had Ki-67 of 30% or greater, one had mutations in TP53, and one had FAT1 and SF3B1 mutations.
Two patients died after ibrutinib therapy, one who had discontinued therapy because of bleeding, and the other who died on treatment at 13 months from transformed disease. Both of these patients had high Ki-67 levels.
Grade 3 or 4 hematological adverse events were neutropenia in four patients, and thrombocytopenia in two patients. There were no cases of grade 3 or 4 anemia.
Grade 3 or 4 nonhematological adverse events were fatigue, myalgia, and atrial fibrillation in seven patients each, diarrhea in six patients, and petechiae/bleeding in three patients.
Patients will continue to be followed for late adverse events, secondary cancers, and relapses, and further studies on clonal evolution, mutation profiling, and MRD are ongoing and will be reported at a later date, Dr. Jain said.
The National Cancer Institute supported the study. Dr. Jain reported having no financial disclosures.
SOURCE: Jain P et al. 15-ICML. Abstract 011.
LUGANO, SWITZERLAND – The chemotherapy-free combination of ibrutinib (Imbruvica) and rituximab is highly effective as frontline therapy for older, transplant-ineligible patients with nonblastoid mantle cell lymphoma, according to investigators.
In a phase 2 study of patients with a median age of 71 years, 38 of 41 patients (93%) had an objective response, and the regimen was both safe and easy to administer, reported Preetesh Jain, MD, PhD, of the University of Texas MD Anderson Cancer Center in Houston.
“The adverse event profile was generally favorable, with specific monitoring recommended for patients with cardiovascular comorbidities and a history of atrial fibrillation,” he said at the International Conference on Malignant Lymphoma.
The investigators enrolled 48 patients aged 65 years and older with previously untreated mantle cell lymphoma (MCL), of whom 41 were evaluable for the primary endpoints of overall response rate (ORR) and safety. The patients had good performance status and normal organ function, with the largest tumor size less than 10 cm. Patients with atrial fibrillation could participate, if the fibrillation was controlled. Patients with Ki-67 protein levels of 50% or greater and blastoid/pleomorphic histology were excluded.
Patients were treated with ibrutinib 560 mg orally daily for each 28-day cycle, with therapy continued until disease progression, or until therapy was stopped for any other reason. Patients also received intravenous rituximab 375 mg/m2 on days 1, 8, 15 and 22, plus or minus one day for cycle 1, on day 1 of cycles 3-8, and on day 1 of every other cycle for up to 2 years.
Of the 41 patients evaluable for response, 26 (64%) had a complete response (CR) and 12 (29%) had a partial response. Three additional patients had stable disease, for an objective response rate of 93%.
Of 34 patients with PET scans, all had negative scans. Of 37 patients evaluable for minimal residual disease (MRD) by flow cytometry, 21 (58%) were MRD negative.
Patients with low or intermediate Mantle Cell Lymphoma International Prognostic Index (MIPI) scores had a higher ORR (100% vs. 89% for patients with high MIPI scores), and patients with low Ki-67 levels had a higher response rate than that of patients with KI-67 of 30% or greater (80% vs. 87%).
Neither median 3-year progression-free survival nor median 3-year overall survival have been reached, with respective 3-year rates of 87% and 95%.
Four patients experienced disease progression at 4, 10, 13 and 33 months of treatment. Three of these patients had disease that had transformed to blastoid/pleomorphic variant, two had Ki-67 of 30% or greater, one had mutations in TP53, and one had FAT1 and SF3B1 mutations.
Two patients died after ibrutinib therapy, one who had discontinued therapy because of bleeding, and the other who died on treatment at 13 months from transformed disease. Both of these patients had high Ki-67 levels.
Grade 3 or 4 hematological adverse events were neutropenia in four patients, and thrombocytopenia in two patients. There were no cases of grade 3 or 4 anemia.
Grade 3 or 4 nonhematological adverse events were fatigue, myalgia, and atrial fibrillation in seven patients each, diarrhea in six patients, and petechiae/bleeding in three patients.
Patients will continue to be followed for late adverse events, secondary cancers, and relapses, and further studies on clonal evolution, mutation profiling, and MRD are ongoing and will be reported at a later date, Dr. Jain said.
The National Cancer Institute supported the study. Dr. Jain reported having no financial disclosures.
SOURCE: Jain P et al. 15-ICML. Abstract 011.
LUGANO, SWITZERLAND – The chemotherapy-free combination of ibrutinib (Imbruvica) and rituximab is highly effective as frontline therapy for older, transplant-ineligible patients with nonblastoid mantle cell lymphoma, according to investigators.
In a phase 2 study of patients with a median age of 71 years, 38 of 41 patients (93%) had an objective response, and the regimen was both safe and easy to administer, reported Preetesh Jain, MD, PhD, of the University of Texas MD Anderson Cancer Center in Houston.
“The adverse event profile was generally favorable, with specific monitoring recommended for patients with cardiovascular comorbidities and a history of atrial fibrillation,” he said at the International Conference on Malignant Lymphoma.
The investigators enrolled 48 patients aged 65 years and older with previously untreated mantle cell lymphoma (MCL), of whom 41 were evaluable for the primary endpoints of overall response rate (ORR) and safety. The patients had good performance status and normal organ function, with the largest tumor size less than 10 cm. Patients with atrial fibrillation could participate, if the fibrillation was controlled. Patients with Ki-67 protein levels of 50% or greater and blastoid/pleomorphic histology were excluded.
Patients were treated with ibrutinib 560 mg orally daily for each 28-day cycle, with therapy continued until disease progression, or until therapy was stopped for any other reason. Patients also received intravenous rituximab 375 mg/m2 on days 1, 8, 15 and 22, plus or minus one day for cycle 1, on day 1 of cycles 3-8, and on day 1 of every other cycle for up to 2 years.
Of the 41 patients evaluable for response, 26 (64%) had a complete response (CR) and 12 (29%) had a partial response. Three additional patients had stable disease, for an objective response rate of 93%.
Of 34 patients with PET scans, all had negative scans. Of 37 patients evaluable for minimal residual disease (MRD) by flow cytometry, 21 (58%) were MRD negative.
Patients with low or intermediate Mantle Cell Lymphoma International Prognostic Index (MIPI) scores had a higher ORR (100% vs. 89% for patients with high MIPI scores), and patients with low Ki-67 levels had a higher response rate than that of patients with KI-67 of 30% or greater (80% vs. 87%).
Neither median 3-year progression-free survival nor median 3-year overall survival have been reached, with respective 3-year rates of 87% and 95%.
Four patients experienced disease progression at 4, 10, 13 and 33 months of treatment. Three of these patients had disease that had transformed to blastoid/pleomorphic variant, two had Ki-67 of 30% or greater, one had mutations in TP53, and one had FAT1 and SF3B1 mutations.
Two patients died after ibrutinib therapy, one who had discontinued therapy because of bleeding, and the other who died on treatment at 13 months from transformed disease. Both of these patients had high Ki-67 levels.
Grade 3 or 4 hematological adverse events were neutropenia in four patients, and thrombocytopenia in two patients. There were no cases of grade 3 or 4 anemia.
Grade 3 or 4 nonhematological adverse events were fatigue, myalgia, and atrial fibrillation in seven patients each, diarrhea in six patients, and petechiae/bleeding in three patients.
Patients will continue to be followed for late adverse events, secondary cancers, and relapses, and further studies on clonal evolution, mutation profiling, and MRD are ongoing and will be reported at a later date, Dr. Jain said.
The National Cancer Institute supported the study. Dr. Jain reported having no financial disclosures.
SOURCE: Jain P et al. 15-ICML. Abstract 011.
REPORTING FROM 15-ICML
HDAC/HMA combo shows ‘remarkable’ activity in PTCL
LUGANO, SWITZERLAND – A combination of 5-azacytidine and romidepsin showed promising activity in patients with peripheral T cell lymphomas, particularly angioimmunoblastic T-cell lymphoma (AITL) and primary cutaneous follicular helper T-cell lymphoma (PTCL-TFH), results of a phase 2 study showed.

Of 16 patients with AITL or PTCL-TFH, 11 (69%) had a clinical response to the 5-azacytidine (AZA)/romidepsin combination, including 8 (50%) with complete responses (CRs), and 3 with partial responses (PRs), reported Lorenzo Falchi, MD, of Columbia University Medical Center and New York Presbyterian Hospital, New York, and colleagues.
“We show that the combination of oral AZA/romidepsin is remarkably active in patients with T-cell lymphomas. Clearly more patients with other subtypes are needed to better evaluate this combination,” Dr. Falchi said at the International Conference on Malignant Lymphoma.
The combination is intended to target epigenetic changes in PTCLs, which often bear mutations in TET2, DNMT3A, and IDH2. These mutations create global hypermethylation and cause transcriptional silencing of tumor suppressor genes, Dr. Falchi said.
Both histone deacetylase inhibitors such as romidepsin, and hypomethylating agents such as AZA have been shown to have single-agent activity against PTCL, and as previously reported at the 2018 T-cell Lymphoma Forum, the combination produced a higher overall response rate (ORR) and prolonged progression-free survival (PFS) in patients with T-cell lymphomas.
Dr. Falchi presented the phase 2 results at 15-ICML. A total of 25 patients with newly diagnosed or relapsed/refractory PTCL were treated with AZA 300 mg daily on days 1-14 and romidepsin 14 mg/m2 on days 8, 15, and 22, every 35 days.
A total of 24 patients were evaluable for response. The ORR – the primary endpoint – was achieved in 14 patients (58%), and included 10 CRs and 4 PRs. Three additional patients had stable disease, and six patients experienced disease progression (response data for one patient was not complete at the time of the presentation).
In total, 11 of 16 patients with AITL/PTCL-TFH had responses, compared with 3 of 8 patients with other histologies.
A secondary analysis of 16 patients with information on mutational status showed that 10 of 12 patients with TET2 mutations (83%) had responses, including 8 CRs and 2 PRs. Two additional patients with TET2 mutations had disease progression. In contrast, among four patients without TET2 mutations, one had a CR, one a PR, and two had disease progression.
Of the 10 patients overall with CRs, 5 patients were receiving the combination in the first line, and 5 patients were receiving it for relapsed/refractory disease.
Median PFS among all patients was 8.7 months. The median overall survival has not been reached. Among patients with the AITL or PTCL-TFH subtypes, median PFS was 8.7 months, compared with 2.3 months for patients with other histologies.
The most frequent hematologic grade 3 or 4 adverse events were thrombocytopenia and neutropenia. The most frequent nonhematologic grade 3 or 4 events included lung infection and febrile neutropenia. Common grade 1 or 2 toxicities included anemia, diarrhea, fatigue, nausea, and vomiting. No patients discontinued therapy because of adverse events.
Dr. Falchi noted that a phase 1 trial evaluating the immune checkpoint inhibitor durvalumab (Imfinzi) with AZA or romidepsin alone or in combination, or pralatrexate and romidepsin, is currently recruiting.
Dr. Falchi reported having no financial disclosures. Other investigators reported funding from Celgene, which supported the study.
SOURCE: Falchi L et al. 15-ICML, Abstract 129.
LUGANO, SWITZERLAND – A combination of 5-azacytidine and romidepsin showed promising activity in patients with peripheral T cell lymphomas, particularly angioimmunoblastic T-cell lymphoma (AITL) and primary cutaneous follicular helper T-cell lymphoma (PTCL-TFH), results of a phase 2 study showed.
Of 16 patients with AITL or PTCL-TFH, 11 (69%) had a clinical response to the 5-azacytidine (AZA)/romidepsin combination, including 8 (50%) with complete responses (CRs), and 3 with partial responses (PRs), reported Lorenzo Falchi, MD, of Columbia University Medical Center and New York Presbyterian Hospital, New York, and colleagues.
“We show that the combination of oral AZA/romidepsin is remarkably active in patients with T-cell lymphomas. Clearly more patients with other subtypes are needed to better evaluate this combination,” Dr. Falchi said at the International Conference on Malignant Lymphoma.
The combination is intended to target epigenetic changes in PTCLs, which often bear mutations in TET2, DNMT3A, and IDH2. These mutations create global hypermethylation and cause transcriptional silencing of tumor suppressor genes, Dr. Falchi said.
Both histone deacetylase inhibitors such as romidepsin, and hypomethylating agents such as AZA have been shown to have single-agent activity against PTCL, and as previously reported at the 2018 T-cell Lymphoma Forum, the combination produced a higher overall response rate (ORR) and prolonged progression-free survival (PFS) in patients with T-cell lymphomas.
Dr. Falchi presented the phase 2 results at 15-ICML. A total of 25 patients with newly diagnosed or relapsed/refractory PTCL were treated with AZA 300 mg daily on days 1-14 and romidepsin 14 mg/m2 on days 8, 15, and 22, every 35 days.
A total of 24 patients were evaluable for response. The ORR – the primary endpoint – was achieved in 14 patients (58%), and included 10 CRs and 4 PRs. Three additional patients had stable disease, and six patients experienced disease progression (response data for one patient was not complete at the time of the presentation).
In total, 11 of 16 patients with AITL/PTCL-TFH had responses, compared with 3 of 8 patients with other histologies.
A secondary analysis of 16 patients with information on mutational status showed that 10 of 12 patients with TET2 mutations (83%) had responses, including 8 CRs and 2 PRs. Two additional patients with TET2 mutations had disease progression. In contrast, among four patients without TET2 mutations, one had a CR, one a PR, and two had disease progression.
Of the 10 patients overall with CRs, 5 patients were receiving the combination in the first line, and 5 patients were receiving it for relapsed/refractory disease.
Median PFS among all patients was 8.7 months. The median overall survival has not been reached. Among patients with the AITL or PTCL-TFH subtypes, median PFS was 8.7 months, compared with 2.3 months for patients with other histologies.
The most frequent hematologic grade 3 or 4 adverse events were thrombocytopenia and neutropenia. The most frequent nonhematologic grade 3 or 4 events included lung infection and febrile neutropenia. Common grade 1 or 2 toxicities included anemia, diarrhea, fatigue, nausea, and vomiting. No patients discontinued therapy because of adverse events.
Dr. Falchi noted that a phase 1 trial evaluating the immune checkpoint inhibitor durvalumab (Imfinzi) with AZA or romidepsin alone or in combination, or pralatrexate and romidepsin, is currently recruiting.
Dr. Falchi reported having no financial disclosures. Other investigators reported funding from Celgene, which supported the study.
SOURCE: Falchi L et al. 15-ICML, Abstract 129.
LUGANO, SWITZERLAND – A combination of 5-azacytidine and romidepsin showed promising activity in patients with peripheral T cell lymphomas, particularly angioimmunoblastic T-cell lymphoma (AITL) and primary cutaneous follicular helper T-cell lymphoma (PTCL-TFH), results of a phase 2 study showed.
Of 16 patients with AITL or PTCL-TFH, 11 (69%) had a clinical response to the 5-azacytidine (AZA)/romidepsin combination, including 8 (50%) with complete responses (CRs), and 3 with partial responses (PRs), reported Lorenzo Falchi, MD, of Columbia University Medical Center and New York Presbyterian Hospital, New York, and colleagues.
“We show that the combination of oral AZA/romidepsin is remarkably active in patients with T-cell lymphomas. Clearly more patients with other subtypes are needed to better evaluate this combination,” Dr. Falchi said at the International Conference on Malignant Lymphoma.
The combination is intended to target epigenetic changes in PTCLs, which often bear mutations in TET2, DNMT3A, and IDH2. These mutations create global hypermethylation and cause transcriptional silencing of tumor suppressor genes, Dr. Falchi said.
Both histone deacetylase inhibitors such as romidepsin, and hypomethylating agents such as AZA have been shown to have single-agent activity against PTCL, and as previously reported at the 2018 T-cell Lymphoma Forum, the combination produced a higher overall response rate (ORR) and prolonged progression-free survival (PFS) in patients with T-cell lymphomas.
Dr. Falchi presented the phase 2 results at 15-ICML. A total of 25 patients with newly diagnosed or relapsed/refractory PTCL were treated with AZA 300 mg daily on days 1-14 and romidepsin 14 mg/m2 on days 8, 15, and 22, every 35 days.
A total of 24 patients were evaluable for response. The ORR – the primary endpoint – was achieved in 14 patients (58%), and included 10 CRs and 4 PRs. Three additional patients had stable disease, and six patients experienced disease progression (response data for one patient was not complete at the time of the presentation).
In total, 11 of 16 patients with AITL/PTCL-TFH had responses, compared with 3 of 8 patients with other histologies.
A secondary analysis of 16 patients with information on mutational status showed that 10 of 12 patients with TET2 mutations (83%) had responses, including 8 CRs and 2 PRs. Two additional patients with TET2 mutations had disease progression. In contrast, among four patients without TET2 mutations, one had a CR, one a PR, and two had disease progression.
Of the 10 patients overall with CRs, 5 patients were receiving the combination in the first line, and 5 patients were receiving it for relapsed/refractory disease.
Median PFS among all patients was 8.7 months. The median overall survival has not been reached. Among patients with the AITL or PTCL-TFH subtypes, median PFS was 8.7 months, compared with 2.3 months for patients with other histologies.
The most frequent hematologic grade 3 or 4 adverse events were thrombocytopenia and neutropenia. The most frequent nonhematologic grade 3 or 4 events included lung infection and febrile neutropenia. Common grade 1 or 2 toxicities included anemia, diarrhea, fatigue, nausea, and vomiting. No patients discontinued therapy because of adverse events.
Dr. Falchi noted that a phase 1 trial evaluating the immune checkpoint inhibitor durvalumab (Imfinzi) with AZA or romidepsin alone or in combination, or pralatrexate and romidepsin, is currently recruiting.
Dr. Falchi reported having no financial disclosures. Other investigators reported funding from Celgene, which supported the study.
SOURCE: Falchi L et al. 15-ICML, Abstract 129.
REPORTING FROM 15-ICML
Cardiovascular complications most common with carfilzomib in relapsed myeloma
Cardiovascular (CV) adverse events were common in patients receiving proteasome inhibitor therapy for relapsed multiple myeloma, especially with carfilzomib-based therapy, according to results from the PROTECT study.
While prior studies have shown an increased risk for CV toxicities with proteasome inhibitor therapy, detailed descriptions of the events and risk factors have been lacking. “Furthermore, there is no validated protocol to help determine which patients are at highest risk of CV toxicity during therapy, nor is there management guidance for patients who experience a [CV adverse event],” wrote Robert F. Cornell, MD, of Vanderbilt University, Nashville, Tenn., and colleagues in the Journal of Clinical Oncology.
The PROTECT (Prospective Observation of Cardiac Safety with Proteasome Inhibitor) study was conducted at Vanderbilt University Medical Center and the University of Pennsylvania Abramson Cancer Center, Philadelphia, between September 2015 and March 2018.
Researchers followed 95 patients with relapsed multiple myeloma who were treated with either bortezomib or carfilzomib for a total duration of 18 months. A total of 65 patients received a carfilzomib-based therapy and 30 patients received a bortezomib-based therapy.
Study patients received a CV assessment at baseline and at the beginning of each treatment cycle for the initial six cycles of proteasome inhibitor therapy. Subsequently, patients were monitored for the development of CV adverse events. CV assessments included ECG, echocardiography, and measurement of other cardiac biomarkers, such as NTproBNP and troponin I or T.
CV toxicities were reported among 5 patients (16.7%) of patients treated with bortezomib and 33 patients (50.7%) treated with carfilzomib (P = .005).
In total, there were 64 CV adverse events reported, most of which were grade 2 or 3, and 56 of which occurred while on carfilzomib-based therapy. For carfilzomib, the most common complications were heart failure (23 cases), followed by grade 3 or 4 hypertension (13 cases). Cardiac chest pain, atrial fibrillation, and acute coronary syndrome were reported in fewer cases.
The researchers also found that elevated natriuretic peptides that occurred before starting carfilzomib therapy or within the first 3 weeks of carfilzomib therapy were associated with a substantially higher risk of CV adverse events.
Patients who have multiple CV risk factors, and especially patients with a history of CV complications and elevated baseline natriuretic peptides, should be referred for a comprehensive cardiac evaluation, the researchers advised. “Such patients are at highest risk of CV [adverse events] with carfilzomib-based therapy, and optimization of CV therapy seems to improve overall care, allow continuation of potentially lifesaving cancer treatment, and affect severity or development of CV [adverse events],” they wrote.
A key limitation of the study was the lack of standardized treatment regimens. As a result, there was a broad dosing range for carfilzomib, in comparison to bortezomib.
Some authors reported financial relationships with carfilzomib maker Amgen and bortezomib maker Takeda, as well as with other companies.
SOURCE: Cornell RF et al. J Clin Oncol. 2019 Jun 12. doi: 10.1200/JCO.19.00231.
Cardiovascular (CV) adverse events were common in patients receiving proteasome inhibitor therapy for relapsed multiple myeloma, especially with carfilzomib-based therapy, according to results from the PROTECT study.
While prior studies have shown an increased risk for CV toxicities with proteasome inhibitor therapy, detailed descriptions of the events and risk factors have been lacking. “Furthermore, there is no validated protocol to help determine which patients are at highest risk of CV toxicity during therapy, nor is there management guidance for patients who experience a [CV adverse event],” wrote Robert F. Cornell, MD, of Vanderbilt University, Nashville, Tenn., and colleagues in the Journal of Clinical Oncology.
The PROTECT (Prospective Observation of Cardiac Safety with Proteasome Inhibitor) study was conducted at Vanderbilt University Medical Center and the University of Pennsylvania Abramson Cancer Center, Philadelphia, between September 2015 and March 2018.
Researchers followed 95 patients with relapsed multiple myeloma who were treated with either bortezomib or carfilzomib for a total duration of 18 months. A total of 65 patients received a carfilzomib-based therapy and 30 patients received a bortezomib-based therapy.
Study patients received a CV assessment at baseline and at the beginning of each treatment cycle for the initial six cycles of proteasome inhibitor therapy. Subsequently, patients were monitored for the development of CV adverse events. CV assessments included ECG, echocardiography, and measurement of other cardiac biomarkers, such as NTproBNP and troponin I or T.
CV toxicities were reported among 5 patients (16.7%) of patients treated with bortezomib and 33 patients (50.7%) treated with carfilzomib (P = .005).
In total, there were 64 CV adverse events reported, most of which were grade 2 or 3, and 56 of which occurred while on carfilzomib-based therapy. For carfilzomib, the most common complications were heart failure (23 cases), followed by grade 3 or 4 hypertension (13 cases). Cardiac chest pain, atrial fibrillation, and acute coronary syndrome were reported in fewer cases.
The researchers also found that elevated natriuretic peptides that occurred before starting carfilzomib therapy or within the first 3 weeks of carfilzomib therapy were associated with a substantially higher risk of CV adverse events.
Patients who have multiple CV risk factors, and especially patients with a history of CV complications and elevated baseline natriuretic peptides, should be referred for a comprehensive cardiac evaluation, the researchers advised. “Such patients are at highest risk of CV [adverse events] with carfilzomib-based therapy, and optimization of CV therapy seems to improve overall care, allow continuation of potentially lifesaving cancer treatment, and affect severity or development of CV [adverse events],” they wrote.
A key limitation of the study was the lack of standardized treatment regimens. As a result, there was a broad dosing range for carfilzomib, in comparison to bortezomib.
Some authors reported financial relationships with carfilzomib maker Amgen and bortezomib maker Takeda, as well as with other companies.
SOURCE: Cornell RF et al. J Clin Oncol. 2019 Jun 12. doi: 10.1200/JCO.19.00231.
Cardiovascular (CV) adverse events were common in patients receiving proteasome inhibitor therapy for relapsed multiple myeloma, especially with carfilzomib-based therapy, according to results from the PROTECT study.
While prior studies have shown an increased risk for CV toxicities with proteasome inhibitor therapy, detailed descriptions of the events and risk factors have been lacking. “Furthermore, there is no validated protocol to help determine which patients are at highest risk of CV toxicity during therapy, nor is there management guidance for patients who experience a [CV adverse event],” wrote Robert F. Cornell, MD, of Vanderbilt University, Nashville, Tenn., and colleagues in the Journal of Clinical Oncology.
The PROTECT (Prospective Observation of Cardiac Safety with Proteasome Inhibitor) study was conducted at Vanderbilt University Medical Center and the University of Pennsylvania Abramson Cancer Center, Philadelphia, between September 2015 and March 2018.
Researchers followed 95 patients with relapsed multiple myeloma who were treated with either bortezomib or carfilzomib for a total duration of 18 months. A total of 65 patients received a carfilzomib-based therapy and 30 patients received a bortezomib-based therapy.
Study patients received a CV assessment at baseline and at the beginning of each treatment cycle for the initial six cycles of proteasome inhibitor therapy. Subsequently, patients were monitored for the development of CV adverse events. CV assessments included ECG, echocardiography, and measurement of other cardiac biomarkers, such as NTproBNP and troponin I or T.
CV toxicities were reported among 5 patients (16.7%) of patients treated with bortezomib and 33 patients (50.7%) treated with carfilzomib (P = .005).
In total, there were 64 CV adverse events reported, most of which were grade 2 or 3, and 56 of which occurred while on carfilzomib-based therapy. For carfilzomib, the most common complications were heart failure (23 cases), followed by grade 3 or 4 hypertension (13 cases). Cardiac chest pain, atrial fibrillation, and acute coronary syndrome were reported in fewer cases.
The researchers also found that elevated natriuretic peptides that occurred before starting carfilzomib therapy or within the first 3 weeks of carfilzomib therapy were associated with a substantially higher risk of CV adverse events.
Patients who have multiple CV risk factors, and especially patients with a history of CV complications and elevated baseline natriuretic peptides, should be referred for a comprehensive cardiac evaluation, the researchers advised. “Such patients are at highest risk of CV [adverse events] with carfilzomib-based therapy, and optimization of CV therapy seems to improve overall care, allow continuation of potentially lifesaving cancer treatment, and affect severity or development of CV [adverse events],” they wrote.
A key limitation of the study was the lack of standardized treatment regimens. As a result, there was a broad dosing range for carfilzomib, in comparison to bortezomib.
Some authors reported financial relationships with carfilzomib maker Amgen and bortezomib maker Takeda, as well as with other companies.
SOURCE: Cornell RF et al. J Clin Oncol. 2019 Jun 12. doi: 10.1200/JCO.19.00231.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Review of Radiologic Considerations in an Immunocompetent Patient With Primary Central Nervous System Lymphoma (FULL)
Central nervous system (CNS) lymphoma can be classified into 2 categories: primary CNS lymphoma (PCNSL), which includes disease limited to brain, eyes, spinal cord; and leptomeninges without coexisting or previous systemic lymphoma. Secondary CNS lymphoma (SCNSL) is essentially metastatic disease from a systemic primary site.1 The focus of this case presentation is PCNSL, with an emphasis on imaging characteristics and differential diagnosis.
The median age at diagnosis for PCNSL is 65 years, and the overall incidence has been decreasing since the mid-1990s, likely related to the increased use of highly-active antiretroviral therapy (HAART) in patients with AIDS.2,3 Although overall incidence has decreased, incidence in the elderly population has increased.4 Historically, PCNSL has been considered an AIDS-defining illness.5 These patients, among other immunocompromised patients, such as those on chronic immunosuppressive therapy, are at a higher risk for developing the malignancy.6
Clinical presentation varies because of the location of CNS involvement and may present with headache, mood or personality disturbances, or focal neurologic deficits. Seizures are less likely due to the tendency of PCNSL to spare gray matter. Initial workup generally includes a head computed tomography (CT) scan, as well as a contrast-enhanced magnetic resonance image (MRI), which may help direct clinicians to the appropriate diagnosis. However, there is significant overlap between the imaging characteristics of PCNSL and numerous other disease processes, including glioblastoma and demyelination. The imaging characteristics of PCNSL are considerably different depending on the patient’s immune status.7
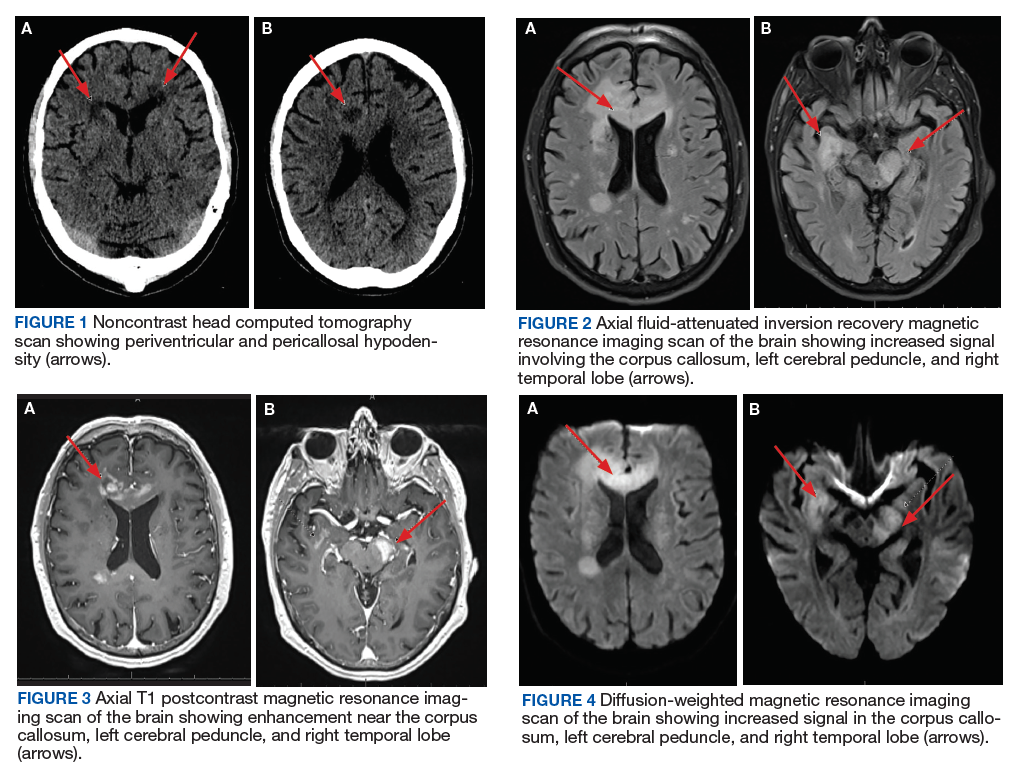
This case illustrates a rare presentation of PCNSL in an immunocompetent patient whose MRI characteristics were seemingly more consistent with those seen in patients with immunodeficiency. The main differential diagnoses and key imaging characteristics, which may help obtain accurate diagnosis, will be discussed.
Case Presentation
A 72-year-old male veteran presented with a 2-month history of subjective weakness in his upper and lower extremities progressing to multiple falls at home. He had no significant medical history other than a thymectomy at age 15 for an enlarged thymus, which per patient report, was benign. An initial laboratory test that included vitamin B12, folate, thyroid-stimulating hormone, complete blood cell count, and comprehensive metabolic panel, were unremarkable, with a white blood cell count of 8.5 K/uL. The initial neurologic evaluation did not show any focal neurologic deficits; however, during the initial hospital stay, the patient developed increasing lower extremity weakness on examination. A noncontrast CT head scan showed extensive nonspecific hypodensities within the periventricular white matter (Figure 1). A contrast-enhanced MRI showed enhancing lesions involving the corpus callosum, left cerebral peduncle, and right temporal lobe (Figures 2, 3, and 4). These lesions also exhibited significant restricted diffusion and a mild amount of surrounding vasogenic edema. The working diagnosis after the MRI included primary CNS lymphoma, multifocal glioblastoma, and tumefactive demyelinating disease. The patient was started on IV steroids and transferred for neurosurgical evaluation and biopsy at an outside hospital. The frontal lesion was biopsied, and the initial frozen section was consistent with lymphoma; a bone marrow biopsy was negative. The workup for immunodeficiency was unremarkable. Pathology revealed high-grade B-cell lymphoma, and the patient began a chemotherapy regimen.
Discussion
The workup of altered mental status, focal neurologic deficits, headaches, or other neurologic conditions often begins with a noncontrast CT scan. On CT, PCNSL generally appears isodense to hyperdense to gray matter, but appearance is variable. The often hyperdense appearance is attributable to the hypercellular nature of lymphoma. Many times, as in this case, CT may show only vague hypodensities, some of which may be associated with surrounding edema. This presentation is nonspecific and may be seen with advancing age due to changes of chronic microvascular ischemia as well as demyelination, other malignancies, and several other disease processes, both benign and malignant. After the initial CT scan, further workup requires evaluation with MRI. PCNSL exhibits restricted diffusion and variable signal intensity on T2-weighted imaging.
PCNSL is frequently centrally located within the periventricular white matter, often within the frontal lobe but can involve other lobes, the basal ganglia, brainstem, cerebellum, or less likely, the spinal canal.7 Contrary to primary CNS disease, secondary lymphoma within the CNS has been described classically as affecting a leptomeningeal (pia and arachnoid mater) distribution two-thirds of the time, with parenchymal involvement occurring in the other one-third of patients. A recent study by Malikova and colleagues found parenchymal involvement may be much more common than previously thought.1 Leptomeningeal spread of disease often involves the cranial nerves, subependymal regions, spinal cord, or spinal nerve roots. Dural involvement in primary or secondary lymphoma is rare.
PCNSL nearly always shows enhancement. Linear enhancement along perivascular spaces is highly characteristic of PCNSL. The typical appearance of PCNSL associated with immunodeficiency varies from that seen in an otherwise immunocompetent patient. Patients with immunodeficiency usually have multifocal involvement, central necrosis leading to a ring enhancement appearance, and have more propensity for spontaneous hemorrhage.7 Immunocompetent patients are less likely to present with multifocal disease and rarely show ring enhancement. Also, spontaneous hemorrhage is rare in immunocompetent patients. In our case, extensive multifocal involvement was present, whereas typically immunocompetent patients will present with a solitary homogeneously enhancing parenchymal mass.
The primary differential for PCNSL includes malignant glioma, tumefactive multiple sclerosis, metastatic disease, and in an immunocompromised patient, toxoplasmosis. The degree of associated vasogenic edema and mass effect is generally lower in PCNSL than that of malignant gliomas and metastasis. Also, PCNSL tends to spare the cerebral cortex.8
Classically, PCNSL, malignant gliomas, and demyelinating disease have been considered the main differential for lesions that cross midline and involve both cerebral hemispheres. Lymphoma generally exhibits more restricted diffusion than malignant gliomas and metastasis, attributable to the highly cellular nature of lymphoma.7 Tumefactive multiple sclerosis is associated with relatively minimal mass effect for lesion size and exhibits less restricted diffusion values when compared to high grade gliomas and PCNSL. One fairly specific finding for tumefactive demyelinating lesions is incomplete rim enhancement.9 Unfortunately, an MRI is not reliable in differentiating these entities, and biopsy is required for definitive diagnosis. Many advancing imaging modalities may help provide the correct diagnosis of PCNSL, including diffusion-weighted and apparent diffusion coefficient imaging, diffusion tensor imaging, MR spectroscopy and PET imaging.7
Conclusion
With the increasing use of HAART, the paradigm of PCNSL is shifting toward one predominantly affecting immunocompetent patients. PCNSL should be considered in any patient with multiple enhancing CNS lesions, regardless of immune status. Several key imaging characteristics may help differentiate PCNSL and other disease processes; however, at this time, biopsy is recommended for definitive diagnosis.
1. Malikova H, Burghardtova M, Koubska E, Mandys V, Kozak T, Weichet J. Secondary central nervous system lymphoma: spectrum of morphological MRI appearances. Neuropsychiatr Dis Treat. 2018;4:733-740.
2. Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-Oncol. 2012;14(suppl 5):v1-v49.
3. Diamond C, Taylor TH, Aboumrad T, Anton-Culver H. Changes in acquired immunodeficiency syndrome-related non-Hodgkin lymphoma in the era of highly active antiretroviral therapy: incidence, presentation, treatment, and survival. Cancer. 2006;106(1):128-135.
4. O’Neill BP, Decker PA, Tieu C, Cerhan JR. The changing incidence of primary central nervous system lymphoma is driven primarily by the changing incidence in young and middle-aged men and differs from time trends in systemic diffuse large B-cell non-Hodgkins lymphoma. Am J Hematol. 2013;88(12):997-1000.
5. [no authors listed]. 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. MMWR Recomm Rep. 1992;41(rr-17):1-19.
6. Maiuri F. Central nervous system lymphomas and immunodeficiency. Neurological Research. 1989;11(1):2-5.
7. Haldorsen IS, Espeland A, Larsson EM. Central nervous system lymphoma: characteristic findings on traditional and advanced imaging. AJNR Am J Neuroradiol. 2010;32(6):984-992.
8. Gómez Roselló E, Quiles Granado AM, Laguillo Sala G, Gutiérrez S. Primary central nervous system lymphoma in immunocompetent patients: spectrum of findings and differential characteristics. Radiología. 2018;60(4):280-289.
9. Mabray MC, Cohen BA, Villanueva-Meyer JE, et al. Performance of Apparent Diffusion Coefficient Values and Conventional MRI Features in Differentiating Tumefactive Demyelinating Lesions From Primary Brain Neoplasms. American Journal of Roentgenology. 2015;205(5):1075-1085.
Central nervous system (CNS) lymphoma can be classified into 2 categories: primary CNS lymphoma (PCNSL), which includes disease limited to brain, eyes, spinal cord; and leptomeninges without coexisting or previous systemic lymphoma. Secondary CNS lymphoma (SCNSL) is essentially metastatic disease from a systemic primary site.1 The focus of this case presentation is PCNSL, with an emphasis on imaging characteristics and differential diagnosis.
The median age at diagnosis for PCNSL is 65 years, and the overall incidence has been decreasing since the mid-1990s, likely related to the increased use of highly-active antiretroviral therapy (HAART) in patients with AIDS.2,3 Although overall incidence has decreased, incidence in the elderly population has increased.4 Historically, PCNSL has been considered an AIDS-defining illness.5 These patients, among other immunocompromised patients, such as those on chronic immunosuppressive therapy, are at a higher risk for developing the malignancy.6
Clinical presentation varies because of the location of CNS involvement and may present with headache, mood or personality disturbances, or focal neurologic deficits. Seizures are less likely due to the tendency of PCNSL to spare gray matter. Initial workup generally includes a head computed tomography (CT) scan, as well as a contrast-enhanced magnetic resonance image (MRI), which may help direct clinicians to the appropriate diagnosis. However, there is significant overlap between the imaging characteristics of PCNSL and numerous other disease processes, including glioblastoma and demyelination. The imaging characteristics of PCNSL are considerably different depending on the patient’s immune status.7
This case illustrates a rare presentation of PCNSL in an immunocompetent patient whose MRI characteristics were seemingly more consistent with those seen in patients with immunodeficiency. The main differential diagnoses and key imaging characteristics, which may help obtain accurate diagnosis, will be discussed.
Case Presentation
A 72-year-old male veteran presented with a 2-month history of subjective weakness in his upper and lower extremities progressing to multiple falls at home. He had no significant medical history other than a thymectomy at age 15 for an enlarged thymus, which per patient report, was benign. An initial laboratory test that included vitamin B12, folate, thyroid-stimulating hormone, complete blood cell count, and comprehensive metabolic panel, were unremarkable, with a white blood cell count of 8.5 K/uL. The initial neurologic evaluation did not show any focal neurologic deficits; however, during the initial hospital stay, the patient developed increasing lower extremity weakness on examination. A noncontrast CT head scan showed extensive nonspecific hypodensities within the periventricular white matter (Figure 1). A contrast-enhanced MRI showed enhancing lesions involving the corpus callosum, left cerebral peduncle, and right temporal lobe (Figures 2, 3, and 4). These lesions also exhibited significant restricted diffusion and a mild amount of surrounding vasogenic edema. The working diagnosis after the MRI included primary CNS lymphoma, multifocal glioblastoma, and tumefactive demyelinating disease. The patient was started on IV steroids and transferred for neurosurgical evaluation and biopsy at an outside hospital. The frontal lesion was biopsied, and the initial frozen section was consistent with lymphoma; a bone marrow biopsy was negative. The workup for immunodeficiency was unremarkable. Pathology revealed high-grade B-cell lymphoma, and the patient began a chemotherapy regimen.
Discussion
The workup of altered mental status, focal neurologic deficits, headaches, or other neurologic conditions often begins with a noncontrast CT scan. On CT, PCNSL generally appears isodense to hyperdense to gray matter, but appearance is variable. The often hyperdense appearance is attributable to the hypercellular nature of lymphoma. Many times, as in this case, CT may show only vague hypodensities, some of which may be associated with surrounding edema. This presentation is nonspecific and may be seen with advancing age due to changes of chronic microvascular ischemia as well as demyelination, other malignancies, and several other disease processes, both benign and malignant. After the initial CT scan, further workup requires evaluation with MRI. PCNSL exhibits restricted diffusion and variable signal intensity on T2-weighted imaging.
PCNSL is frequently centrally located within the periventricular white matter, often within the frontal lobe but can involve other lobes, the basal ganglia, brainstem, cerebellum, or less likely, the spinal canal.7 Contrary to primary CNS disease, secondary lymphoma within the CNS has been described classically as affecting a leptomeningeal (pia and arachnoid mater) distribution two-thirds of the time, with parenchymal involvement occurring in the other one-third of patients. A recent study by Malikova and colleagues found parenchymal involvement may be much more common than previously thought.1 Leptomeningeal spread of disease often involves the cranial nerves, subependymal regions, spinal cord, or spinal nerve roots. Dural involvement in primary or secondary lymphoma is rare.
PCNSL nearly always shows enhancement. Linear enhancement along perivascular spaces is highly characteristic of PCNSL. The typical appearance of PCNSL associated with immunodeficiency varies from that seen in an otherwise immunocompetent patient. Patients with immunodeficiency usually have multifocal involvement, central necrosis leading to a ring enhancement appearance, and have more propensity for spontaneous hemorrhage.7 Immunocompetent patients are less likely to present with multifocal disease and rarely show ring enhancement. Also, spontaneous hemorrhage is rare in immunocompetent patients. In our case, extensive multifocal involvement was present, whereas typically immunocompetent patients will present with a solitary homogeneously enhancing parenchymal mass.
The primary differential for PCNSL includes malignant glioma, tumefactive multiple sclerosis, metastatic disease, and in an immunocompromised patient, toxoplasmosis. The degree of associated vasogenic edema and mass effect is generally lower in PCNSL than that of malignant gliomas and metastasis. Also, PCNSL tends to spare the cerebral cortex.8
Classically, PCNSL, malignant gliomas, and demyelinating disease have been considered the main differential for lesions that cross midline and involve both cerebral hemispheres. Lymphoma generally exhibits more restricted diffusion than malignant gliomas and metastasis, attributable to the highly cellular nature of lymphoma.7 Tumefactive multiple sclerosis is associated with relatively minimal mass effect for lesion size and exhibits less restricted diffusion values when compared to high grade gliomas and PCNSL. One fairly specific finding for tumefactive demyelinating lesions is incomplete rim enhancement.9 Unfortunately, an MRI is not reliable in differentiating these entities, and biopsy is required for definitive diagnosis. Many advancing imaging modalities may help provide the correct diagnosis of PCNSL, including diffusion-weighted and apparent diffusion coefficient imaging, diffusion tensor imaging, MR spectroscopy and PET imaging.7
Conclusion
With the increasing use of HAART, the paradigm of PCNSL is shifting toward one predominantly affecting immunocompetent patients. PCNSL should be considered in any patient with multiple enhancing CNS lesions, regardless of immune status. Several key imaging characteristics may help differentiate PCNSL and other disease processes; however, at this time, biopsy is recommended for definitive diagnosis.
Central nervous system (CNS) lymphoma can be classified into 2 categories: primary CNS lymphoma (PCNSL), which includes disease limited to brain, eyes, spinal cord; and leptomeninges without coexisting or previous systemic lymphoma. Secondary CNS lymphoma (SCNSL) is essentially metastatic disease from a systemic primary site.1 The focus of this case presentation is PCNSL, with an emphasis on imaging characteristics and differential diagnosis.
The median age at diagnosis for PCNSL is 65 years, and the overall incidence has been decreasing since the mid-1990s, likely related to the increased use of highly-active antiretroviral therapy (HAART) in patients with AIDS.2,3 Although overall incidence has decreased, incidence in the elderly population has increased.4 Historically, PCNSL has been considered an AIDS-defining illness.5 These patients, among other immunocompromised patients, such as those on chronic immunosuppressive therapy, are at a higher risk for developing the malignancy.6
Clinical presentation varies because of the location of CNS involvement and may present with headache, mood or personality disturbances, or focal neurologic deficits. Seizures are less likely due to the tendency of PCNSL to spare gray matter. Initial workup generally includes a head computed tomography (CT) scan, as well as a contrast-enhanced magnetic resonance image (MRI), which may help direct clinicians to the appropriate diagnosis. However, there is significant overlap between the imaging characteristics of PCNSL and numerous other disease processes, including glioblastoma and demyelination. The imaging characteristics of PCNSL are considerably different depending on the patient’s immune status.7
This case illustrates a rare presentation of PCNSL in an immunocompetent patient whose MRI characteristics were seemingly more consistent with those seen in patients with immunodeficiency. The main differential diagnoses and key imaging characteristics, which may help obtain accurate diagnosis, will be discussed.
Case Presentation
A 72-year-old male veteran presented with a 2-month history of subjective weakness in his upper and lower extremities progressing to multiple falls at home. He had no significant medical history other than a thymectomy at age 15 for an enlarged thymus, which per patient report, was benign. An initial laboratory test that included vitamin B12, folate, thyroid-stimulating hormone, complete blood cell count, and comprehensive metabolic panel, were unremarkable, with a white blood cell count of 8.5 K/uL. The initial neurologic evaluation did not show any focal neurologic deficits; however, during the initial hospital stay, the patient developed increasing lower extremity weakness on examination. A noncontrast CT head scan showed extensive nonspecific hypodensities within the periventricular white matter (Figure 1). A contrast-enhanced MRI showed enhancing lesions involving the corpus callosum, left cerebral peduncle, and right temporal lobe (Figures 2, 3, and 4). These lesions also exhibited significant restricted diffusion and a mild amount of surrounding vasogenic edema. The working diagnosis after the MRI included primary CNS lymphoma, multifocal glioblastoma, and tumefactive demyelinating disease. The patient was started on IV steroids and transferred for neurosurgical evaluation and biopsy at an outside hospital. The frontal lesion was biopsied, and the initial frozen section was consistent with lymphoma; a bone marrow biopsy was negative. The workup for immunodeficiency was unremarkable. Pathology revealed high-grade B-cell lymphoma, and the patient began a chemotherapy regimen.
Discussion
The workup of altered mental status, focal neurologic deficits, headaches, or other neurologic conditions often begins with a noncontrast CT scan. On CT, PCNSL generally appears isodense to hyperdense to gray matter, but appearance is variable. The often hyperdense appearance is attributable to the hypercellular nature of lymphoma. Many times, as in this case, CT may show only vague hypodensities, some of which may be associated with surrounding edema. This presentation is nonspecific and may be seen with advancing age due to changes of chronic microvascular ischemia as well as demyelination, other malignancies, and several other disease processes, both benign and malignant. After the initial CT scan, further workup requires evaluation with MRI. PCNSL exhibits restricted diffusion and variable signal intensity on T2-weighted imaging.
PCNSL is frequently centrally located within the periventricular white matter, often within the frontal lobe but can involve other lobes, the basal ganglia, brainstem, cerebellum, or less likely, the spinal canal.7 Contrary to primary CNS disease, secondary lymphoma within the CNS has been described classically as affecting a leptomeningeal (pia and arachnoid mater) distribution two-thirds of the time, with parenchymal involvement occurring in the other one-third of patients. A recent study by Malikova and colleagues found parenchymal involvement may be much more common than previously thought.1 Leptomeningeal spread of disease often involves the cranial nerves, subependymal regions, spinal cord, or spinal nerve roots. Dural involvement in primary or secondary lymphoma is rare.
PCNSL nearly always shows enhancement. Linear enhancement along perivascular spaces is highly characteristic of PCNSL. The typical appearance of PCNSL associated with immunodeficiency varies from that seen in an otherwise immunocompetent patient. Patients with immunodeficiency usually have multifocal involvement, central necrosis leading to a ring enhancement appearance, and have more propensity for spontaneous hemorrhage.7 Immunocompetent patients are less likely to present with multifocal disease and rarely show ring enhancement. Also, spontaneous hemorrhage is rare in immunocompetent patients. In our case, extensive multifocal involvement was present, whereas typically immunocompetent patients will present with a solitary homogeneously enhancing parenchymal mass.
The primary differential for PCNSL includes malignant glioma, tumefactive multiple sclerosis, metastatic disease, and in an immunocompromised patient, toxoplasmosis. The degree of associated vasogenic edema and mass effect is generally lower in PCNSL than that of malignant gliomas and metastasis. Also, PCNSL tends to spare the cerebral cortex.8
Classically, PCNSL, malignant gliomas, and demyelinating disease have been considered the main differential for lesions that cross midline and involve both cerebral hemispheres. Lymphoma generally exhibits more restricted diffusion than malignant gliomas and metastasis, attributable to the highly cellular nature of lymphoma.7 Tumefactive multiple sclerosis is associated with relatively minimal mass effect for lesion size and exhibits less restricted diffusion values when compared to high grade gliomas and PCNSL. One fairly specific finding for tumefactive demyelinating lesions is incomplete rim enhancement.9 Unfortunately, an MRI is not reliable in differentiating these entities, and biopsy is required for definitive diagnosis. Many advancing imaging modalities may help provide the correct diagnosis of PCNSL, including diffusion-weighted and apparent diffusion coefficient imaging, diffusion tensor imaging, MR spectroscopy and PET imaging.7
Conclusion
With the increasing use of HAART, the paradigm of PCNSL is shifting toward one predominantly affecting immunocompetent patients. PCNSL should be considered in any patient with multiple enhancing CNS lesions, regardless of immune status. Several key imaging characteristics may help differentiate PCNSL and other disease processes; however, at this time, biopsy is recommended for definitive diagnosis.
1. Malikova H, Burghardtova M, Koubska E, Mandys V, Kozak T, Weichet J. Secondary central nervous system lymphoma: spectrum of morphological MRI appearances. Neuropsychiatr Dis Treat. 2018;4:733-740.
2. Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-Oncol. 2012;14(suppl 5):v1-v49.
3. Diamond C, Taylor TH, Aboumrad T, Anton-Culver H. Changes in acquired immunodeficiency syndrome-related non-Hodgkin lymphoma in the era of highly active antiretroviral therapy: incidence, presentation, treatment, and survival. Cancer. 2006;106(1):128-135.
4. O’Neill BP, Decker PA, Tieu C, Cerhan JR. The changing incidence of primary central nervous system lymphoma is driven primarily by the changing incidence in young and middle-aged men and differs from time trends in systemic diffuse large B-cell non-Hodgkins lymphoma. Am J Hematol. 2013;88(12):997-1000.
5. [no authors listed]. 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. MMWR Recomm Rep. 1992;41(rr-17):1-19.
6. Maiuri F. Central nervous system lymphomas and immunodeficiency. Neurological Research. 1989;11(1):2-5.
7. Haldorsen IS, Espeland A, Larsson EM. Central nervous system lymphoma: characteristic findings on traditional and advanced imaging. AJNR Am J Neuroradiol. 2010;32(6):984-992.
8. Gómez Roselló E, Quiles Granado AM, Laguillo Sala G, Gutiérrez S. Primary central nervous system lymphoma in immunocompetent patients: spectrum of findings and differential characteristics. Radiología. 2018;60(4):280-289.
9. Mabray MC, Cohen BA, Villanueva-Meyer JE, et al. Performance of Apparent Diffusion Coefficient Values and Conventional MRI Features in Differentiating Tumefactive Demyelinating Lesions From Primary Brain Neoplasms. American Journal of Roentgenology. 2015;205(5):1075-1085.
1. Malikova H, Burghardtova M, Koubska E, Mandys V, Kozak T, Weichet J. Secondary central nervous system lymphoma: spectrum of morphological MRI appearances. Neuropsychiatr Dis Treat. 2018;4:733-740.
2. Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-Oncol. 2012;14(suppl 5):v1-v49.
3. Diamond C, Taylor TH, Aboumrad T, Anton-Culver H. Changes in acquired immunodeficiency syndrome-related non-Hodgkin lymphoma in the era of highly active antiretroviral therapy: incidence, presentation, treatment, and survival. Cancer. 2006;106(1):128-135.
4. O’Neill BP, Decker PA, Tieu C, Cerhan JR. The changing incidence of primary central nervous system lymphoma is driven primarily by the changing incidence in young and middle-aged men and differs from time trends in systemic diffuse large B-cell non-Hodgkins lymphoma. Am J Hematol. 2013;88(12):997-1000.
5. [no authors listed]. 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. MMWR Recomm Rep. 1992;41(rr-17):1-19.
6. Maiuri F. Central nervous system lymphomas and immunodeficiency. Neurological Research. 1989;11(1):2-5.
7. Haldorsen IS, Espeland A, Larsson EM. Central nervous system lymphoma: characteristic findings on traditional and advanced imaging. AJNR Am J Neuroradiol. 2010;32(6):984-992.
8. Gómez Roselló E, Quiles Granado AM, Laguillo Sala G, Gutiérrez S. Primary central nervous system lymphoma in immunocompetent patients: spectrum of findings and differential characteristics. Radiología. 2018;60(4):280-289.
9. Mabray MC, Cohen BA, Villanueva-Meyer JE, et al. Performance of Apparent Diffusion Coefficient Values and Conventional MRI Features in Differentiating Tumefactive Demyelinating Lesions From Primary Brain Neoplasms. American Journal of Roentgenology. 2015;205(5):1075-1085.
Low-dose radiation therapy looks effective in hard-to-treat MCL
Low-dose radiation therapy – with or without concurrent chemotherapy – appears promising as a treatment for patients with relapsed or refractory mantle cell lymphoma (MCL) or at least a bridge to subsequent therapy, according to findings published in Blood Advances.

Matthew S. Ning, MD, of the department of radiation oncology at the University of Texas MD Anderson Cancer Center, Houston, and colleagues, said this is the first study to evaluate low-dose radiation therapy (LDRT) with chemotherapy as a treatment modality outside of palliative care for relapsed, multiple refractory MCL patients.
“Our findings indicate that LDRT imparts excellent [local control], minimal toxicity, and favorable outcomes in this setting,” the researchers said.
The study included 19 patients with a total of 98 sites of relapsed, refractory MCL who were treated from 2014 to 2018. The median follow-up was 51.3 months from initial diagnosis and 15.4 months from initial treatment with low-dose radiation therapy, given at a dose of 4 Gy.
These were hard-to-treat patients who had received multiple prior therapies since diagnosis, including carfilzomib, ibrutinib, bortezomib, anthracycline, and rituximab. In total, 8 of the patients had previously undergone autologous stem cell transplant and 11 were refractory to ibrutinib by the time of initial radiation therapy.
Median age of the patients was 69 years; 15 patients had classical histology and 4 had blastoid variant. Among the 98 tumor sites treated, the median tumor size was 2.8 cm.
In all, 14 patients received initial LDRT that was concurrent with chemotherapy. The remaining 5 patients had stopped chemotherapy prior to starting LDRT.
LDRT was given in 1-2 daily fractions via 3-dimensional conformal radiation therapy or electron beam.
Of the 98 tumor sites treated, complete response was achieved for 79 sites (81%) and the median time to complete response was 2.7 months after the start of LDRT. The researchers removed one patient who was an outlier with 27 tumor sites treated, and that dropped the complete response rate down to 76%. The overall response rate, which include an additional five sites with partial response, was 86%.
The researchers found links between complete response and soft tissue site versus non–soft tissue site (hazard ratio, 1.80; 1.12-2.90, P = .02). However, there were no associations between response and chemo-refractory status, ibrutinib-refractory status, prior chemotherapy courts, receipt of concurrent chemotherapy, tumor size, number of fractions, lesions treated per course, or blastoid variant.
The overall survival at 1 year after LDRT initiation was 90% and the 1-year progression-free survival was 55%. All five patients who died were refractory to ibrutinib.
The researchers reported finding no radiation therapy–related toxicities, even when patients received concurrent chemotherapy.
The use of LDRT has the potential to bridge refractory patients to subsequent therapies or to provide treatment breaks as patients recover from toxicities, the researchers said. However, they called for additional studies to confirm that this approach improves progression-free survival over chemotherapy alone.
The study was supported in part by a grant from the National Cancer Institute. The researchers reported having no competing financial interests.
SOURCE: Ning MS et al. Blood Adv. 2019. Jul 9;3(13):2035-9.
Low-dose radiation therapy – with or without concurrent chemotherapy – appears promising as a treatment for patients with relapsed or refractory mantle cell lymphoma (MCL) or at least a bridge to subsequent therapy, according to findings published in Blood Advances.
Matthew S. Ning, MD, of the department of radiation oncology at the University of Texas MD Anderson Cancer Center, Houston, and colleagues, said this is the first study to evaluate low-dose radiation therapy (LDRT) with chemotherapy as a treatment modality outside of palliative care for relapsed, multiple refractory MCL patients.
“Our findings indicate that LDRT imparts excellent [local control], minimal toxicity, and favorable outcomes in this setting,” the researchers said.
The study included 19 patients with a total of 98 sites of relapsed, refractory MCL who were treated from 2014 to 2018. The median follow-up was 51.3 months from initial diagnosis and 15.4 months from initial treatment with low-dose radiation therapy, given at a dose of 4 Gy.
These were hard-to-treat patients who had received multiple prior therapies since diagnosis, including carfilzomib, ibrutinib, bortezomib, anthracycline, and rituximab. In total, 8 of the patients had previously undergone autologous stem cell transplant and 11 were refractory to ibrutinib by the time of initial radiation therapy.
Median age of the patients was 69 years; 15 patients had classical histology and 4 had blastoid variant. Among the 98 tumor sites treated, the median tumor size was 2.8 cm.
In all, 14 patients received initial LDRT that was concurrent with chemotherapy. The remaining 5 patients had stopped chemotherapy prior to starting LDRT.
LDRT was given in 1-2 daily fractions via 3-dimensional conformal radiation therapy or electron beam.
Of the 98 tumor sites treated, complete response was achieved for 79 sites (81%) and the median time to complete response was 2.7 months after the start of LDRT. The researchers removed one patient who was an outlier with 27 tumor sites treated, and that dropped the complete response rate down to 76%. The overall response rate, which include an additional five sites with partial response, was 86%.
The researchers found links between complete response and soft tissue site versus non–soft tissue site (hazard ratio, 1.80; 1.12-2.90, P = .02). However, there were no associations between response and chemo-refractory status, ibrutinib-refractory status, prior chemotherapy courts, receipt of concurrent chemotherapy, tumor size, number of fractions, lesions treated per course, or blastoid variant.
The overall survival at 1 year after LDRT initiation was 90% and the 1-year progression-free survival was 55%. All five patients who died were refractory to ibrutinib.
The researchers reported finding no radiation therapy–related toxicities, even when patients received concurrent chemotherapy.
The use of LDRT has the potential to bridge refractory patients to subsequent therapies or to provide treatment breaks as patients recover from toxicities, the researchers said. However, they called for additional studies to confirm that this approach improves progression-free survival over chemotherapy alone.
The study was supported in part by a grant from the National Cancer Institute. The researchers reported having no competing financial interests.
SOURCE: Ning MS et al. Blood Adv. 2019. Jul 9;3(13):2035-9.
Low-dose radiation therapy – with or without concurrent chemotherapy – appears promising as a treatment for patients with relapsed or refractory mantle cell lymphoma (MCL) or at least a bridge to subsequent therapy, according to findings published in Blood Advances.
Matthew S. Ning, MD, of the department of radiation oncology at the University of Texas MD Anderson Cancer Center, Houston, and colleagues, said this is the first study to evaluate low-dose radiation therapy (LDRT) with chemotherapy as a treatment modality outside of palliative care for relapsed, multiple refractory MCL patients.
“Our findings indicate that LDRT imparts excellent [local control], minimal toxicity, and favorable outcomes in this setting,” the researchers said.
The study included 19 patients with a total of 98 sites of relapsed, refractory MCL who were treated from 2014 to 2018. The median follow-up was 51.3 months from initial diagnosis and 15.4 months from initial treatment with low-dose radiation therapy, given at a dose of 4 Gy.
These were hard-to-treat patients who had received multiple prior therapies since diagnosis, including carfilzomib, ibrutinib, bortezomib, anthracycline, and rituximab. In total, 8 of the patients had previously undergone autologous stem cell transplant and 11 were refractory to ibrutinib by the time of initial radiation therapy.
Median age of the patients was 69 years; 15 patients had classical histology and 4 had blastoid variant. Among the 98 tumor sites treated, the median tumor size was 2.8 cm.
In all, 14 patients received initial LDRT that was concurrent with chemotherapy. The remaining 5 patients had stopped chemotherapy prior to starting LDRT.
LDRT was given in 1-2 daily fractions via 3-dimensional conformal radiation therapy or electron beam.
Of the 98 tumor sites treated, complete response was achieved for 79 sites (81%) and the median time to complete response was 2.7 months after the start of LDRT. The researchers removed one patient who was an outlier with 27 tumor sites treated, and that dropped the complete response rate down to 76%. The overall response rate, which include an additional five sites with partial response, was 86%.
The researchers found links between complete response and soft tissue site versus non–soft tissue site (hazard ratio, 1.80; 1.12-2.90, P = .02). However, there were no associations between response and chemo-refractory status, ibrutinib-refractory status, prior chemotherapy courts, receipt of concurrent chemotherapy, tumor size, number of fractions, lesions treated per course, or blastoid variant.
The overall survival at 1 year after LDRT initiation was 90% and the 1-year progression-free survival was 55%. All five patients who died were refractory to ibrutinib.
The researchers reported finding no radiation therapy–related toxicities, even when patients received concurrent chemotherapy.
The use of LDRT has the potential to bridge refractory patients to subsequent therapies or to provide treatment breaks as patients recover from toxicities, the researchers said. However, they called for additional studies to confirm that this approach improves progression-free survival over chemotherapy alone.
The study was supported in part by a grant from the National Cancer Institute. The researchers reported having no competing financial interests.
SOURCE: Ning MS et al. Blood Adv. 2019. Jul 9;3(13):2035-9.
FROM BLOOD ADVANCES
Key clinical point:
Major finding: The overall survival was 90% at 1 year following the initiation of low-dose radiation therapy (4 Gy).
Study details: A study of 19 patients with relapsed, refractory mantle cell lymphoma who received low-dose radiation at doses of 4 Gy at 98 sites of disease.
Disclosures: The study was supported in part by a grant from the National Cancer Institute. The researchers reported having no competing financial interests.
Source: Ning MS et al. Blood Adv. 2019. Jul 9;3(13):2035-9.
TP53 double hit predicts aggressive myeloma
Relapsed multiple myeloma becomes increasingly aggressive and difficult to treat with each additional TP53 alteration, according to investigators.
Findings from the study help illuminate the mechanics of myeloma disease progression and demonstrate the value of clonal competition assays, reported lead author Umair Munawar of the University Hospital Würzburg (Germany) and colleagues.
“The implications of mono-allelic TP53 lesions for the clinical outcome remain controversial, but clonal selection and evolution is a common feature of myeloma progression, and patients with TP53 wild-type or mono-allelic inactivation may present a double hit on relapse,” the investigators wrote in Blood. “Here, we addressed the hypothesis that sequential acquisition of TP53 hits lead to a gain of proliferative fitness of [multiple myeloma] cancer cells, inducing the expansion and domination of the affected clones within the patient’s bone marrow.”
The investigators used sleeping beauty and CRISPR/Cas9 techniques to create double- and single-hit multiple myeloma cell lines that were stably transfected with fluorescent proteins. By observing coculture pairings of wild-type, single-hit, and double-hit cells, the investigators found a hierarchy of proliferation that depended on the number of TP53 alterations. For instance, when double-hit cells were cocultured with wild-type cells in a 1:3 ratio, it took 21 days for the double-hit cells to reach 50% of the total culture population. Similarly, single-hit cells outcompeted wild-type cells after 38 days, while double-hit cells took 35 days to overcome the single-hit population.
Further testing showed that comparatively smaller initial populations of TP53-aberrant cells required longer to outcompete larger wild-type populations, which could explain why deeper responses in the clinic are often followed by longer periods without disease progression, the investigators suggested.
A comparison of transcriptomes between wild-type cells and TP53 mutants revealed differences in about 900 genes, including 14 signaling pathways. Specifically, downregulation impacted antigen processing and presentation, chemokine signaling, and oxidative phosphorylation.
“These differences on the transcriptomic level well reflect the biology of ultra–high risk disease,” the investigators wrote, referring to increased glucose uptake on PET, resistance to immunotherapies, and extramedullary disease.
“[This study] underscores the power of clonal competition assays to decipher the effect of genomic lesions in tumors to better understand their impact on progression and disease relapse in [multiple myeloma],” the investigators concluded.
The study was funded by Deutsche Forschungsgemeinschaft, the CDW Stiftung, and the IZKF Würzburg. The investigators reported additional support from the CRIS foundation, the German Cancer Aid, and the University of Würzburg.
SOURCE: Munawar U et al. Blood. 2019 Jul 24. doi: 10.1182/blood.2019000080.
Relapsed multiple myeloma becomes increasingly aggressive and difficult to treat with each additional TP53 alteration, according to investigators.
Findings from the study help illuminate the mechanics of myeloma disease progression and demonstrate the value of clonal competition assays, reported lead author Umair Munawar of the University Hospital Würzburg (Germany) and colleagues.
“The implications of mono-allelic TP53 lesions for the clinical outcome remain controversial, but clonal selection and evolution is a common feature of myeloma progression, and patients with TP53 wild-type or mono-allelic inactivation may present a double hit on relapse,” the investigators wrote in Blood. “Here, we addressed the hypothesis that sequential acquisition of TP53 hits lead to a gain of proliferative fitness of [multiple myeloma] cancer cells, inducing the expansion and domination of the affected clones within the patient’s bone marrow.”
The investigators used sleeping beauty and CRISPR/Cas9 techniques to create double- and single-hit multiple myeloma cell lines that were stably transfected with fluorescent proteins. By observing coculture pairings of wild-type, single-hit, and double-hit cells, the investigators found a hierarchy of proliferation that depended on the number of TP53 alterations. For instance, when double-hit cells were cocultured with wild-type cells in a 1:3 ratio, it took 21 days for the double-hit cells to reach 50% of the total culture population. Similarly, single-hit cells outcompeted wild-type cells after 38 days, while double-hit cells took 35 days to overcome the single-hit population.
Further testing showed that comparatively smaller initial populations of TP53-aberrant cells required longer to outcompete larger wild-type populations, which could explain why deeper responses in the clinic are often followed by longer periods without disease progression, the investigators suggested.
A comparison of transcriptomes between wild-type cells and TP53 mutants revealed differences in about 900 genes, including 14 signaling pathways. Specifically, downregulation impacted antigen processing and presentation, chemokine signaling, and oxidative phosphorylation.
“These differences on the transcriptomic level well reflect the biology of ultra–high risk disease,” the investigators wrote, referring to increased glucose uptake on PET, resistance to immunotherapies, and extramedullary disease.
“[This study] underscores the power of clonal competition assays to decipher the effect of genomic lesions in tumors to better understand their impact on progression and disease relapse in [multiple myeloma],” the investigators concluded.
The study was funded by Deutsche Forschungsgemeinschaft, the CDW Stiftung, and the IZKF Würzburg. The investigators reported additional support from the CRIS foundation, the German Cancer Aid, and the University of Würzburg.
SOURCE: Munawar U et al. Blood. 2019 Jul 24. doi: 10.1182/blood.2019000080.
Relapsed multiple myeloma becomes increasingly aggressive and difficult to treat with each additional TP53 alteration, according to investigators.
Findings from the study help illuminate the mechanics of myeloma disease progression and demonstrate the value of clonal competition assays, reported lead author Umair Munawar of the University Hospital Würzburg (Germany) and colleagues.
“The implications of mono-allelic TP53 lesions for the clinical outcome remain controversial, but clonal selection and evolution is a common feature of myeloma progression, and patients with TP53 wild-type or mono-allelic inactivation may present a double hit on relapse,” the investigators wrote in Blood. “Here, we addressed the hypothesis that sequential acquisition of TP53 hits lead to a gain of proliferative fitness of [multiple myeloma] cancer cells, inducing the expansion and domination of the affected clones within the patient’s bone marrow.”
The investigators used sleeping beauty and CRISPR/Cas9 techniques to create double- and single-hit multiple myeloma cell lines that were stably transfected with fluorescent proteins. By observing coculture pairings of wild-type, single-hit, and double-hit cells, the investigators found a hierarchy of proliferation that depended on the number of TP53 alterations. For instance, when double-hit cells were cocultured with wild-type cells in a 1:3 ratio, it took 21 days for the double-hit cells to reach 50% of the total culture population. Similarly, single-hit cells outcompeted wild-type cells after 38 days, while double-hit cells took 35 days to overcome the single-hit population.
Further testing showed that comparatively smaller initial populations of TP53-aberrant cells required longer to outcompete larger wild-type populations, which could explain why deeper responses in the clinic are often followed by longer periods without disease progression, the investigators suggested.
A comparison of transcriptomes between wild-type cells and TP53 mutants revealed differences in about 900 genes, including 14 signaling pathways. Specifically, downregulation impacted antigen processing and presentation, chemokine signaling, and oxidative phosphorylation.
“These differences on the transcriptomic level well reflect the biology of ultra–high risk disease,” the investigators wrote, referring to increased glucose uptake on PET, resistance to immunotherapies, and extramedullary disease.
“[This study] underscores the power of clonal competition assays to decipher the effect of genomic lesions in tumors to better understand their impact on progression and disease relapse in [multiple myeloma],” the investigators concluded.
The study was funded by Deutsche Forschungsgemeinschaft, the CDW Stiftung, and the IZKF Würzburg. The investigators reported additional support from the CRIS foundation, the German Cancer Aid, and the University of Würzburg.
SOURCE: Munawar U et al. Blood. 2019 Jul 24. doi: 10.1182/blood.2019000080.
FROM BLOOD
Researchers combine genetic and clinical factors in new VTE risk score
MELBOURNE – A venous thromboembolism risk score that combines clinical risk factors, such as lymphoma type and stage, along with genetic variables, could offer a better way to predict venous thromboembolism in patients with lymphoma, according to new findings presented at the International Society on Thrombosis and Haemostasis congress.

Cristina Pascual, MD, of the Hospital Universitario Gregorio Marañon in Madrid presented data from a development and validation study of a clinical-genetic risk model for thrombosis in lymphoma in 208 patients with lymphoma, 31 of whom experienced a venous thromboembolic event.
While the relationship between cancer and increased thrombosis risk is well recognized, lymphoma patients are at particularly high risk, with an estimated thrombosis incidence of 5%-10%, Dr. Pascual said.
Currently, the Khorana score is the most validated risk score for thrombosis in patients with solid tumors, using factors such as tumor site, platelet and leukocyte count, hemoglobin levels, and body mass index. However, Dr. Pascual pointed out that just 10% of the validation cohort for the Khorana score were lymphoma patients, and it had previously been found to be not as useful for that population.
More recently, researchers had developed the ThroLy score for predicting thromboembolic events specifically in patients with lymphoma, incorporating clinical variables such as mediastinal involvement and extranodal localization.
Another group took a different approach by incorporating genetic risk factors for thrombosis to create Thrombo inCode-Oncology (TiC-Onco) for solid tumors. This assessment included four genetic variants known to increase the risk of thromboembolic events in cancer patients, as well as the clinical risk factors of body mass index, family history of thrombosis, primary tumor site, and tumor stage.
Dr. Pascual and colleagues developed a unique risk factor model that combined both the ThroLy and TiC-Onco elements.
In 208 patients with lymphoma who were not receiving anticoagulant treatment, researchers identified five clinical factors that were most predictive of venous thrombosis: a history of thrombosis, immobilization for more than 3 days, lymphoma type, Ann Arbor score for lymphoma stage, and mediastinal extension.
They combined these clinical risk factors with the genetic risk factors from the TiC-Onco score to develop the TiC-Onco–associated lymphoma score (TiC-Lympho).
When validated in the same group of patients, the TiC-Lympho score had a sensitivity of 93.55%, a specificity of 54.49%, positive predictive value of 26.36%, and negative predictive value of 97.94%.
The researchers also compared TiC-Lympho’s performance with that of the ThroLy and TiC-Onco models, and found it performed better on sensitivity and negative predictive value. The area under the curve for TiC-Lympho (0.783) was significantly higher than that seen with the other two risk models.
Session chair Kate Burbury, MBBS, of the Peter MacCallum Cancer Centre in Melbourne, raised the question of how the score – and particularly the genetic risk factor assessment – might be applied in the real-world clinical setting.
In an interview, Dr. Pascual said the findings represented preliminary data only, so the model was not ready to be applied to clinical practice yet. She also stressed that this was based on retrospective data, and needed to be further validated in other cohorts of lymphoma patients.
No conflicts of interest were reported.
SOURCE: Pascual C et al. 2019 ISTH Congress, Abstract OC 41.3.
MELBOURNE – A venous thromboembolism risk score that combines clinical risk factors, such as lymphoma type and stage, along with genetic variables, could offer a better way to predict venous thromboembolism in patients with lymphoma, according to new findings presented at the International Society on Thrombosis and Haemostasis congress.
Cristina Pascual, MD, of the Hospital Universitario Gregorio Marañon in Madrid presented data from a development and validation study of a clinical-genetic risk model for thrombosis in lymphoma in 208 patients with lymphoma, 31 of whom experienced a venous thromboembolic event.
While the relationship between cancer and increased thrombosis risk is well recognized, lymphoma patients are at particularly high risk, with an estimated thrombosis incidence of 5%-10%, Dr. Pascual said.
Currently, the Khorana score is the most validated risk score for thrombosis in patients with solid tumors, using factors such as tumor site, platelet and leukocyte count, hemoglobin levels, and body mass index. However, Dr. Pascual pointed out that just 10% of the validation cohort for the Khorana score were lymphoma patients, and it had previously been found to be not as useful for that population.
More recently, researchers had developed the ThroLy score for predicting thromboembolic events specifically in patients with lymphoma, incorporating clinical variables such as mediastinal involvement and extranodal localization.
Another group took a different approach by incorporating genetic risk factors for thrombosis to create Thrombo inCode-Oncology (TiC-Onco) for solid tumors. This assessment included four genetic variants known to increase the risk of thromboembolic events in cancer patients, as well as the clinical risk factors of body mass index, family history of thrombosis, primary tumor site, and tumor stage.
Dr. Pascual and colleagues developed a unique risk factor model that combined both the ThroLy and TiC-Onco elements.
In 208 patients with lymphoma who were not receiving anticoagulant treatment, researchers identified five clinical factors that were most predictive of venous thrombosis: a history of thrombosis, immobilization for more than 3 days, lymphoma type, Ann Arbor score for lymphoma stage, and mediastinal extension.
They combined these clinical risk factors with the genetic risk factors from the TiC-Onco score to develop the TiC-Onco–associated lymphoma score (TiC-Lympho).
When validated in the same group of patients, the TiC-Lympho score had a sensitivity of 93.55%, a specificity of 54.49%, positive predictive value of 26.36%, and negative predictive value of 97.94%.
The researchers also compared TiC-Lympho’s performance with that of the ThroLy and TiC-Onco models, and found it performed better on sensitivity and negative predictive value. The area under the curve for TiC-Lympho (0.783) was significantly higher than that seen with the other two risk models.
Session chair Kate Burbury, MBBS, of the Peter MacCallum Cancer Centre in Melbourne, raised the question of how the score – and particularly the genetic risk factor assessment – might be applied in the real-world clinical setting.
In an interview, Dr. Pascual said the findings represented preliminary data only, so the model was not ready to be applied to clinical practice yet. She also stressed that this was based on retrospective data, and needed to be further validated in other cohorts of lymphoma patients.
No conflicts of interest were reported.
SOURCE: Pascual C et al. 2019 ISTH Congress, Abstract OC 41.3.
MELBOURNE – A venous thromboembolism risk score that combines clinical risk factors, such as lymphoma type and stage, along with genetic variables, could offer a better way to predict venous thromboembolism in patients with lymphoma, according to new findings presented at the International Society on Thrombosis and Haemostasis congress.
Cristina Pascual, MD, of the Hospital Universitario Gregorio Marañon in Madrid presented data from a development and validation study of a clinical-genetic risk model for thrombosis in lymphoma in 208 patients with lymphoma, 31 of whom experienced a venous thromboembolic event.
While the relationship between cancer and increased thrombosis risk is well recognized, lymphoma patients are at particularly high risk, with an estimated thrombosis incidence of 5%-10%, Dr. Pascual said.
Currently, the Khorana score is the most validated risk score for thrombosis in patients with solid tumors, using factors such as tumor site, platelet and leukocyte count, hemoglobin levels, and body mass index. However, Dr. Pascual pointed out that just 10% of the validation cohort for the Khorana score were lymphoma patients, and it had previously been found to be not as useful for that population.
More recently, researchers had developed the ThroLy score for predicting thromboembolic events specifically in patients with lymphoma, incorporating clinical variables such as mediastinal involvement and extranodal localization.
Another group took a different approach by incorporating genetic risk factors for thrombosis to create Thrombo inCode-Oncology (TiC-Onco) for solid tumors. This assessment included four genetic variants known to increase the risk of thromboembolic events in cancer patients, as well as the clinical risk factors of body mass index, family history of thrombosis, primary tumor site, and tumor stage.
Dr. Pascual and colleagues developed a unique risk factor model that combined both the ThroLy and TiC-Onco elements.
In 208 patients with lymphoma who were not receiving anticoagulant treatment, researchers identified five clinical factors that were most predictive of venous thrombosis: a history of thrombosis, immobilization for more than 3 days, lymphoma type, Ann Arbor score for lymphoma stage, and mediastinal extension.
They combined these clinical risk factors with the genetic risk factors from the TiC-Onco score to develop the TiC-Onco–associated lymphoma score (TiC-Lympho).
When validated in the same group of patients, the TiC-Lympho score had a sensitivity of 93.55%, a specificity of 54.49%, positive predictive value of 26.36%, and negative predictive value of 97.94%.
The researchers also compared TiC-Lympho’s performance with that of the ThroLy and TiC-Onco models, and found it performed better on sensitivity and negative predictive value. The area under the curve for TiC-Lympho (0.783) was significantly higher than that seen with the other two risk models.
Session chair Kate Burbury, MBBS, of the Peter MacCallum Cancer Centre in Melbourne, raised the question of how the score – and particularly the genetic risk factor assessment – might be applied in the real-world clinical setting.
In an interview, Dr. Pascual said the findings represented preliminary data only, so the model was not ready to be applied to clinical practice yet. She also stressed that this was based on retrospective data, and needed to be further validated in other cohorts of lymphoma patients.
No conflicts of interest were reported.
SOURCE: Pascual C et al. 2019 ISTH Congress, Abstract OC 41.3.
REPORTING FROM 2019 ISTH CONGRESS
BRCA2 mutations linked to childhood NHL
Pediatric non-Hodgkin lymphomas should be added to the list of cancers associated with BRCA2 mutations, and survivors of childhood NHL should be considered for genetic counseling, investigators suggest.
Among 1,380 survivors of childhood lymphomas, those who were retrospectively found to be carriers of BRCA2 mutations had a fivefold higher risk for non-Hodgkin lymphoma than controls without cancer, reported Zhaoming Wang, PhD, and colleagues from St. Jude Children’s Research Hospital in Memphis.
“Genetic counseling and the option of BRCA2 genetic testing should be offered to survivors of pediatric or adolescent non–Hodgkin lymphoma, particularly those with a family history of BRCA2-associated cancers,” they wrote in JAMA Oncology.
The investigators had previously reported that BRCA2 was the third-most frequently mutated gene among 3006 survivors of childhood cancers, with the highest number of mutations seen in lymphoma survivors. In that study, 7 of 586 survivors of Hodgkin and non-Hodgkin lymphoma (1.2%) were found to carry BRCA2 mutations.
In the current study, the investigators performed germline whole-genome sequencing on samples from 815 survivors of childhood Hodgkin lymphoma and 748 survivors of non-Hodgkin lymphoma from the St. Jude Lifetime Cohort and Childhood Cancer Survivor studies and compared the data with those of controls without cancer from the Genome Aggregation Database.
They identified mutations in five Hodgkin lymphoma survivors (0.6%) and eight non-Hodgkin lymphoma survivors.
A comparison of cancer risk among lymphoma survivors and controls found that non-Hodgkin lymphoma survivors and BRCA2 carriers had an odds ratio for cancer of 5.0, compared with controls who were not BRCA2 carriers (P less than .001). Among Hodgkin lymphoma survivors the OR for carriers vs. controls was 2.1, but was not statistically significant.
Available family histories for seven of the eight non-Hodgkin lymphoma BRCA2 mutation carriers showed histories of BRCA2-linked cancers, including breast, prostate, and pancreas tumors and malignant melanoma.
“Survivors whose test results are positive for mutation should be offered surveillance for BRCA2-associated cancers, such as breast and ovarian, and counseled about cancer risk–reducing strategies. Currently, it remains unclear whether surveillance for non–Hodgkin lymphoma is associated with early detection of lymphomas or with other medical advantages,” the investigators wrote.
“This study was funded by a grant to St Jude Children’s Research Hospital from the American Lebanese Syrian Associated Charities and by grants to St Jude Children’s Research Hospital from the National Institutes of Health. The authors reported having no conflicts of interest.
SOURCE: Wang Z et al. JAMA Oncology. 2019 Jul 25. doi: 10.1001/jamaoncol.2019.2203.
Pediatric non-Hodgkin lymphomas should be added to the list of cancers associated with BRCA2 mutations, and survivors of childhood NHL should be considered for genetic counseling, investigators suggest.
Among 1,380 survivors of childhood lymphomas, those who were retrospectively found to be carriers of BRCA2 mutations had a fivefold higher risk for non-Hodgkin lymphoma than controls without cancer, reported Zhaoming Wang, PhD, and colleagues from St. Jude Children’s Research Hospital in Memphis.
“Genetic counseling and the option of BRCA2 genetic testing should be offered to survivors of pediatric or adolescent non–Hodgkin lymphoma, particularly those with a family history of BRCA2-associated cancers,” they wrote in JAMA Oncology.
The investigators had previously reported that BRCA2 was the third-most frequently mutated gene among 3006 survivors of childhood cancers, with the highest number of mutations seen in lymphoma survivors. In that study, 7 of 586 survivors of Hodgkin and non-Hodgkin lymphoma (1.2%) were found to carry BRCA2 mutations.
In the current study, the investigators performed germline whole-genome sequencing on samples from 815 survivors of childhood Hodgkin lymphoma and 748 survivors of non-Hodgkin lymphoma from the St. Jude Lifetime Cohort and Childhood Cancer Survivor studies and compared the data with those of controls without cancer from the Genome Aggregation Database.
They identified mutations in five Hodgkin lymphoma survivors (0.6%) and eight non-Hodgkin lymphoma survivors.
A comparison of cancer risk among lymphoma survivors and controls found that non-Hodgkin lymphoma survivors and BRCA2 carriers had an odds ratio for cancer of 5.0, compared with controls who were not BRCA2 carriers (P less than .001). Among Hodgkin lymphoma survivors the OR for carriers vs. controls was 2.1, but was not statistically significant.
Available family histories for seven of the eight non-Hodgkin lymphoma BRCA2 mutation carriers showed histories of BRCA2-linked cancers, including breast, prostate, and pancreas tumors and malignant melanoma.
“Survivors whose test results are positive for mutation should be offered surveillance for BRCA2-associated cancers, such as breast and ovarian, and counseled about cancer risk–reducing strategies. Currently, it remains unclear whether surveillance for non–Hodgkin lymphoma is associated with early detection of lymphomas or with other medical advantages,” the investigators wrote.
“This study was funded by a grant to St Jude Children’s Research Hospital from the American Lebanese Syrian Associated Charities and by grants to St Jude Children’s Research Hospital from the National Institutes of Health. The authors reported having no conflicts of interest.
SOURCE: Wang Z et al. JAMA Oncology. 2019 Jul 25. doi: 10.1001/jamaoncol.2019.2203.
Pediatric non-Hodgkin lymphomas should be added to the list of cancers associated with BRCA2 mutations, and survivors of childhood NHL should be considered for genetic counseling, investigators suggest.
Among 1,380 survivors of childhood lymphomas, those who were retrospectively found to be carriers of BRCA2 mutations had a fivefold higher risk for non-Hodgkin lymphoma than controls without cancer, reported Zhaoming Wang, PhD, and colleagues from St. Jude Children’s Research Hospital in Memphis.
“Genetic counseling and the option of BRCA2 genetic testing should be offered to survivors of pediatric or adolescent non–Hodgkin lymphoma, particularly those with a family history of BRCA2-associated cancers,” they wrote in JAMA Oncology.
The investigators had previously reported that BRCA2 was the third-most frequently mutated gene among 3006 survivors of childhood cancers, with the highest number of mutations seen in lymphoma survivors. In that study, 7 of 586 survivors of Hodgkin and non-Hodgkin lymphoma (1.2%) were found to carry BRCA2 mutations.
In the current study, the investigators performed germline whole-genome sequencing on samples from 815 survivors of childhood Hodgkin lymphoma and 748 survivors of non-Hodgkin lymphoma from the St. Jude Lifetime Cohort and Childhood Cancer Survivor studies and compared the data with those of controls without cancer from the Genome Aggregation Database.
They identified mutations in five Hodgkin lymphoma survivors (0.6%) and eight non-Hodgkin lymphoma survivors.
A comparison of cancer risk among lymphoma survivors and controls found that non-Hodgkin lymphoma survivors and BRCA2 carriers had an odds ratio for cancer of 5.0, compared with controls who were not BRCA2 carriers (P less than .001). Among Hodgkin lymphoma survivors the OR for carriers vs. controls was 2.1, but was not statistically significant.
Available family histories for seven of the eight non-Hodgkin lymphoma BRCA2 mutation carriers showed histories of BRCA2-linked cancers, including breast, prostate, and pancreas tumors and malignant melanoma.
“Survivors whose test results are positive for mutation should be offered surveillance for BRCA2-associated cancers, such as breast and ovarian, and counseled about cancer risk–reducing strategies. Currently, it remains unclear whether surveillance for non–Hodgkin lymphoma is associated with early detection of lymphomas or with other medical advantages,” the investigators wrote.
“This study was funded by a grant to St Jude Children’s Research Hospital from the American Lebanese Syrian Associated Charities and by grants to St Jude Children’s Research Hospital from the National Institutes of Health. The authors reported having no conflicts of interest.
SOURCE: Wang Z et al. JAMA Oncology. 2019 Jul 25. doi: 10.1001/jamaoncol.2019.2203.
FROM JAMA ONCOLOGY
Lymphoma risk prompts FDA recall of Allergan’s textured breast implants
The Food and Drug Administration requested on July 24 that Allergan pull six brands of textured breast implants and breast expanders from the U.S. market, an action the agency took because of new data that substantially increased the number of women who developed a rare cancer – anaplastic large-cell lymphoma – in association with receiving these textured breast devices.

This is the first product recall the FDA has made to address the issue of breast implant–associated anaplastic large-cell lymphoma (BIA-ALCL), a complication that first came to national attention with a 2011 FDA report that had tallied 60 identified BIA-ALCL cases worldwide. By the end of September 2018, the number of reported worldwide BIA-ALCL cases had jumped to 457 cases reported to the agency via medical device reporting. In July 2019, the FDA cited a total of 573 unique, global case reports for BIA-ALCL sent to the agency through July 6, including 33 episodes that led to death.
It was inclusion of these additional 116 cases since September 2018 and 24 additional deaths that led FDA researchers to conclude that “the risk of BIA-ALCL with Allergan BIOCELL textured implants is approximately six-times the risk of BIA-ALCL with textured implants from other manufacturers marketing in the U.S.,” according to a statement from the agency.
The FDA is not recommending that patients who received one of the six products covered by the recall have the material removed if symptoms have not appeared because of the potential risk from explantation.
The agency also stressed that its investigation of the risk posed by placement of other brands of textured breast implants is ongoing and that overall less than 5% of all breast implants performed in current U.S. practice involve the macrotextured implants of the type specified in the Allergan recall.
This U.S. recall follows similar actions taken in France (and the rest of the European Union), Canada, and Australia, and it contrasts with the agency’s prior decision in May 2019 not to start a recall or ban of textured implants following a advisory committee meeting that discussed BIA-ALCL.
The six products that Allergan agreed to recall from marketing at the FDA’s request are four textured breast implants (Natrelle Saline-Filled Breast Implants, Natrelle Silicone-Filled Breast Implants, Natrelle Inspira Silicone-Filled breast Implants, and Natrelle 410 Highly Cohesive Anatomically Shaped Silicone-Filled Breast Implants) and two tissue expanders used prior to a breast implant (Natrelle 133 Plus Tissue Expander and the Natrelle 133 Tissue Expander with Suture Tabs).

FDA officials said they are considering recommendations for changes to the labeling of breast implant products, including a possible boxed warning and beefed up patient information.
“The recall of these textured implants is a big deal in protecting women from the potential risks of developing, and dying from, this rare type of aggressive lymphoma,” Joshua Brody, MD, a medical oncologist and director of the lymphoma immunotherapy program at Mount Sinai Medical Center in New York said in a statement. “While case reports have suggested a potential link between some types of breast implants and this disease – anaplastic lymphoma – for over 20 years, it has taken time to gain sufficient evidence to suggest, and understand, the causality. Some types of implants induce inflammation, which can both increase the chance of developing cancer, and also help to ‘hide’ developing cancers from the immune system. By preventing further use of these implants, the FDA is helping women to protect themselves from the medically serious and emotionally exhausting effects of these risks.”
Dr. Brody reported having no relevant disclosures.
The Food and Drug Administration requested on July 24 that Allergan pull six brands of textured breast implants and breast expanders from the U.S. market, an action the agency took because of new data that substantially increased the number of women who developed a rare cancer – anaplastic large-cell lymphoma – in association with receiving these textured breast devices.
This is the first product recall the FDA has made to address the issue of breast implant–associated anaplastic large-cell lymphoma (BIA-ALCL), a complication that first came to national attention with a 2011 FDA report that had tallied 60 identified BIA-ALCL cases worldwide. By the end of September 2018, the number of reported worldwide BIA-ALCL cases had jumped to 457 cases reported to the agency via medical device reporting. In July 2019, the FDA cited a total of 573 unique, global case reports for BIA-ALCL sent to the agency through July 6, including 33 episodes that led to death.
It was inclusion of these additional 116 cases since September 2018 and 24 additional deaths that led FDA researchers to conclude that “the risk of BIA-ALCL with Allergan BIOCELL textured implants is approximately six-times the risk of BIA-ALCL with textured implants from other manufacturers marketing in the U.S.,” according to a statement from the agency.
The FDA is not recommending that patients who received one of the six products covered by the recall have the material removed if symptoms have not appeared because of the potential risk from explantation.
The agency also stressed that its investigation of the risk posed by placement of other brands of textured breast implants is ongoing and that overall less than 5% of all breast implants performed in current U.S. practice involve the macrotextured implants of the type specified in the Allergan recall.
This U.S. recall follows similar actions taken in France (and the rest of the European Union), Canada, and Australia, and it contrasts with the agency’s prior decision in May 2019 not to start a recall or ban of textured implants following a advisory committee meeting that discussed BIA-ALCL.
The six products that Allergan agreed to recall from marketing at the FDA’s request are four textured breast implants (Natrelle Saline-Filled Breast Implants, Natrelle Silicone-Filled Breast Implants, Natrelle Inspira Silicone-Filled breast Implants, and Natrelle 410 Highly Cohesive Anatomically Shaped Silicone-Filled Breast Implants) and two tissue expanders used prior to a breast implant (Natrelle 133 Plus Tissue Expander and the Natrelle 133 Tissue Expander with Suture Tabs).
FDA officials said they are considering recommendations for changes to the labeling of breast implant products, including a possible boxed warning and beefed up patient information.
“The recall of these textured implants is a big deal in protecting women from the potential risks of developing, and dying from, this rare type of aggressive lymphoma,” Joshua Brody, MD, a medical oncologist and director of the lymphoma immunotherapy program at Mount Sinai Medical Center in New York said in a statement. “While case reports have suggested a potential link between some types of breast implants and this disease – anaplastic lymphoma – for over 20 years, it has taken time to gain sufficient evidence to suggest, and understand, the causality. Some types of implants induce inflammation, which can both increase the chance of developing cancer, and also help to ‘hide’ developing cancers from the immune system. By preventing further use of these implants, the FDA is helping women to protect themselves from the medically serious and emotionally exhausting effects of these risks.”
Dr. Brody reported having no relevant disclosures.
The Food and Drug Administration requested on July 24 that Allergan pull six brands of textured breast implants and breast expanders from the U.S. market, an action the agency took because of new data that substantially increased the number of women who developed a rare cancer – anaplastic large-cell lymphoma – in association with receiving these textured breast devices.
This is the first product recall the FDA has made to address the issue of breast implant–associated anaplastic large-cell lymphoma (BIA-ALCL), a complication that first came to national attention with a 2011 FDA report that had tallied 60 identified BIA-ALCL cases worldwide. By the end of September 2018, the number of reported worldwide BIA-ALCL cases had jumped to 457 cases reported to the agency via medical device reporting. In July 2019, the FDA cited a total of 573 unique, global case reports for BIA-ALCL sent to the agency through July 6, including 33 episodes that led to death.
It was inclusion of these additional 116 cases since September 2018 and 24 additional deaths that led FDA researchers to conclude that “the risk of BIA-ALCL with Allergan BIOCELL textured implants is approximately six-times the risk of BIA-ALCL with textured implants from other manufacturers marketing in the U.S.,” according to a statement from the agency.
The FDA is not recommending that patients who received one of the six products covered by the recall have the material removed if symptoms have not appeared because of the potential risk from explantation.
The agency also stressed that its investigation of the risk posed by placement of other brands of textured breast implants is ongoing and that overall less than 5% of all breast implants performed in current U.S. practice involve the macrotextured implants of the type specified in the Allergan recall.
This U.S. recall follows similar actions taken in France (and the rest of the European Union), Canada, and Australia, and it contrasts with the agency’s prior decision in May 2019 not to start a recall or ban of textured implants following a advisory committee meeting that discussed BIA-ALCL.
The six products that Allergan agreed to recall from marketing at the FDA’s request are four textured breast implants (Natrelle Saline-Filled Breast Implants, Natrelle Silicone-Filled Breast Implants, Natrelle Inspira Silicone-Filled breast Implants, and Natrelle 410 Highly Cohesive Anatomically Shaped Silicone-Filled Breast Implants) and two tissue expanders used prior to a breast implant (Natrelle 133 Plus Tissue Expander and the Natrelle 133 Tissue Expander with Suture Tabs).
FDA officials said they are considering recommendations for changes to the labeling of breast implant products, including a possible boxed warning and beefed up patient information.
“The recall of these textured implants is a big deal in protecting women from the potential risks of developing, and dying from, this rare type of aggressive lymphoma,” Joshua Brody, MD, a medical oncologist and director of the lymphoma immunotherapy program at Mount Sinai Medical Center in New York said in a statement. “While case reports have suggested a potential link between some types of breast implants and this disease – anaplastic lymphoma – for over 20 years, it has taken time to gain sufficient evidence to suggest, and understand, the causality. Some types of implants induce inflammation, which can both increase the chance of developing cancer, and also help to ‘hide’ developing cancers from the immune system. By preventing further use of these implants, the FDA is helping women to protect themselves from the medically serious and emotionally exhausting effects of these risks.”
Dr. Brody reported having no relevant disclosures.