User login
Symptom burdens related to chemotherapy-induced anemia in stage IV cancer
Anemia is a common complication of cancer treatment as well as of cancer itself. Most cancer patients undergoing chemotherapy experience anemia sometime during their treatment course.1,2 Moderate to severe anemia is associated with an array of symptoms that are known to compromise the physical functioning and quality of life of cancer patients. Common anemia-related symptoms include fatigue, drowsiness, depression, dyspnea, tachycardia, and dizziness.1,3-7
Symptoms produced by cancer itself or the disease treatment (ie, side effects such as anemia) collectively compose a patient’s symptom burden.8 Although the occurrence of anemia-related fatigue has been described more systematically, other clinical presentations of chemotherapy-induced anemia (CIA) are not well characterized. Furthermore, the overall symptom burdens associated with different ranges of hemoglobin (Hb) concentrations have also not been well reported. Although various tools have been developed to facilitate the reporting of fatigue and other symptoms experienced by patients with CIA, such as the Functional Assessment of Cancer Therapy-Anemia (FACT-An) questionnaire and the MD Anderson Symptom Inventory (MDASI),9-11 these questionnaires have not been extensively used outside of the research context. As such, knowledge on symptom burdens associated with CIA in real-world patient populations remains lacking.
Given the common occurrence of CIA, management of CIA and associated symptoms plays an important role to patients’ quality of life during cancer treatment. Symptom control is often the main goal for patients with stage IV cancers, as treatment for disease is most likely palliative or noncurative. To facilitate supportive care planning, it is important to understand patient symptom burdens as chemotherapy progresses over cycles and Hb levels decline. We conducted a comprehensive medical record review study in patients diagnosed with stage IV non-Hodgkin lymphoma (NHL), breast cancer, and lung cancers at Kaiser Permanente Southern California (KPSC), a large community-based health care delivery system. The objective of this study was to report the occurrence of CIA-related symptoms throughout the course of chemotherapy and by Hb levels.
Methods
Study setting and population
KPSC is an integrated managed-care organization that provides comprehensive health services for 4 million racially, ethnically, and socioeconomically diverse members who broadly represent the population in Southern California.12 The organization maintains electronic records of health care received by its members, including physician record notes and clinical databases such as laboratory test results, diagnosis codes, medical procedures, medication dispenses, and disease registries. KPSC’s cancer registry is Surveillance, Epidemiology, and End Results, which is affiliated and routinely collects information on age, sex, race and/or ethnicity, cancer type, histology, and stage at diagnosis.
Patients who met the following inclusion criteria were included in this study: diagnosed with stage IV NHL, breast cancer, or lung cancer at age 18 years or older at KPSC between March 25, 2010 and December 31, 2012; initiated myelosuppressive chemotherapy at KPSC before June 30, 2013 (only the first chemotherapy course was included in this evaluation); and had at least 1 Hb measurement during the course of chemotherapy. Of those who met the inclusion criteria, patients who met the following criteria were excluded if they had less than 12 months KPSC membership before start of chemotherapy, missing information on cancer stage or chemotherapy regimen/agents, a diagnosis of myelodysplastic syndrome before chemotherapy initiation, a diagnosis of inherited anemia, an Hb concentration <10 g/L within 3 months before chemotherapy initiation, a transfusion within 2 weeks before chemotherapy initiation, radiation within 4 months before chemotherapy initiation, or bone marrow transplantation within 12 months before chemotherapy initiation or during the chemotherapy course. These exclusion criteria were applied to evaluate symptom burdens most likely related to CIA as opposed to other cancer treatment or pre-existing anemia.
CIA in this study was defined as moderate to severe anemia with Hb <10 g/dL after chemotherapy initiation. Based on this definition for CIA, all patients who developed CIA between the first chemotherapy administration to 60 days after the last dose of chemotherapy were included for the record review
Data collection
Data on anemia-related symptoms or signs and anemia-related comorbidities (Table 1) were collected by standardized review of physician record notes in the electronic medical records. A set of 24 anemia-related symptoms were identified based on the literature and clinical expertise and included abdominal pain, blurred vision/double vision/loss of vision, cold intolerance/coldness in hands or feet, depression/anxiety, diarrhea, dizziness/lightheadedness, dyspnea/shortness of breath/tachypnea, edema, fatigue, headache, heart failure, heat intolerance, hypotension, insomnia, leg pain, loss of appetite, nausea/vomiting, pale skin, palpitations/tachycardia, paralysis/ataxia/numbness or tingling in extremities, pectoral angina/chest pain, sweating/diaphoresis, syncope, and vertigo. Record review period was defined as 1 month before chemotherapy to 60 days after the last dose of chemotherapy in the first course. To understand the development of new symptoms during chemotherapy treatment, pre-existing symptoms documented within 1 month before chemotherapy initiation were recorded.
The data elements extracted included the date the symptom was documented, date the symptom started, symptom duration (when available), and any relevant comments regarding the symptom (ie, if dyspnea was at rest or on exertion, whether the symptom was a side effect caused by chemotherapy, or change in symptom severity). Ten percent of the records were reviewed independently by 2 abstractors to ensure quality control. Additional quality control measures included SAS algorithms (SAS Institute, Inc., Cary, North Carolina) to check reasonability and logical consistency in the abstracted data.
Patient demographic characteristics, cancer stage, additional selected comorbidities (Table 1), chemotherapy information, Hb test results, and anemia treatment, including erythrocyte stimulating agent (ESA) use and red blood cell transfusion, were collected using KPSC’s cancer registry and clinical databases. Anemia was defined by severity as grade 1 (10 g/dL to lower limit of normal, ie, 14 g/dL for men and 12 g/dL for women), grade 2 (8.0-9.9 g/dL), grade 3 (6.5-7.9 g/dL), and grade 4 (<6.5 g/dL) following the National Cancer Institute’s Common Terminology Criteria for Adverse Events.13
Statistical analysis
Distributions of demographic, cancer, and treatment characteristics were calculated by CIA status, overall and by cancer type. Differences between patients who did and did not develop CIA were assessed using chi-square test and Kruskal-Wallis test. For those who developed CIA, the distribution of the worst anemia grade was also calculated for each cycle of chemotherapy.
Next, the distributions for the following symptom categories were calculated in the 2 study samples defined by CIA status: pre-existing symptoms that occurred before chemotherapy, any symptoms during chemotherapy (ie, whether they started before chemotherapy), and incident symptoms during chemotherapy (ie, new symptoms that only started after chemotherapy). Specifically, the proportion of patients with each individual symptom and the distribution of the number of symptoms per patient were calculated. Differences in symptom distribution by CIA status were assessed using chi-square test.
The distribution of symptoms in each chemotherapy cycle was calculated up to 6 chemotherapy cycles (as >80% of the patients only had treatment up to 6 cycles) in the 2 study samples defined by CIA status. For this analysis, a symptom was “mapped” to a cycle if the date (or date range) of the symptom fell within the date range of that chemotherapy cycle. In patients who developed CIA, the distribution of symptoms was also calculated by anemia grade. This was again done on the chemotherapy cycle level. For each chemotherapy cycle, an anemia grade was assigned (no anemia or anemia grade 1, 2, 3, and 4) using the lowest Hb measurement in that cycle. Symptoms that occurred in a chemotherapy cycle were then “mapped” to the anemia grade of that cycle. Some patients had more than 1 anemia event of the same grade (eg, if a patient’s grade 2 anemia persist across cycles). For these patients, we randomly selected only 1 anemia event of the same grade from each patient to be included in this analysis. Patients could still contribute multiple events of different grades to this analysis. We calculated the mean number of symptoms per patient for each anemia grade (ie, 1-4) separately. Because of the small number of patients who developed grade 4 anemia (n = 11), they were combined with the grade 3 patients when the distributions of individual symptoms were evaluated.
All analyses were repeated stratified by gender. P values for differences between men and women were calculated using chi-square test or t test. All analyses were conducted using SAS version 9.3.
Results
A total of 402 stage IV NHL, breast, and lung cancer patients who developed CIA and 98 patients who did not develop CIA during the first course of chemotherapy were included (Figure 1).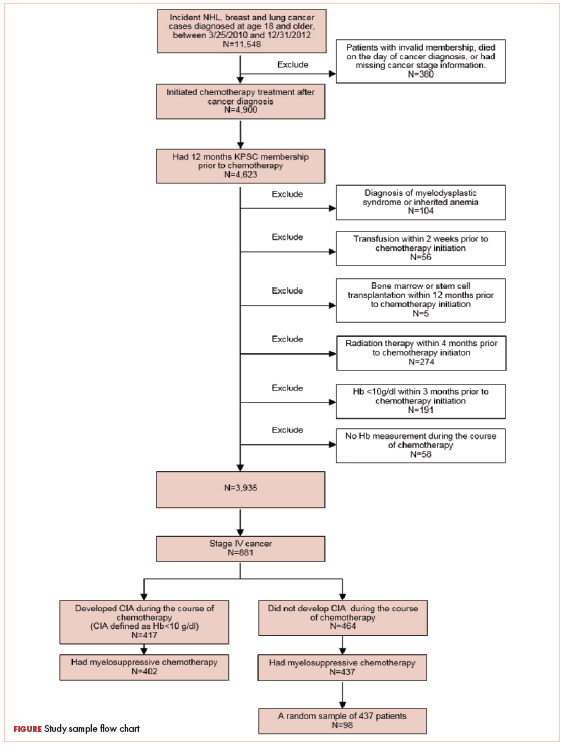
The distribution of cancer types in the study sample were similar across CIA status (Table 1). The mean age at diagnosis was 66 years in patients who developed CIA and 62 years in patients who did not develop CIA. Women accounted for half of the patients in both study samples (52% and 51%, respectively). Most of the study patients were of non-Hispanic white race/ethnicity. Chronic obstructive pulmonary disease/emphysema and gastroesophageal reflux disease were among the most common comorbidities examined in both study samples, while malnutrition and moderate to severe renal disease were also common in patients who developed CIA (Table 1).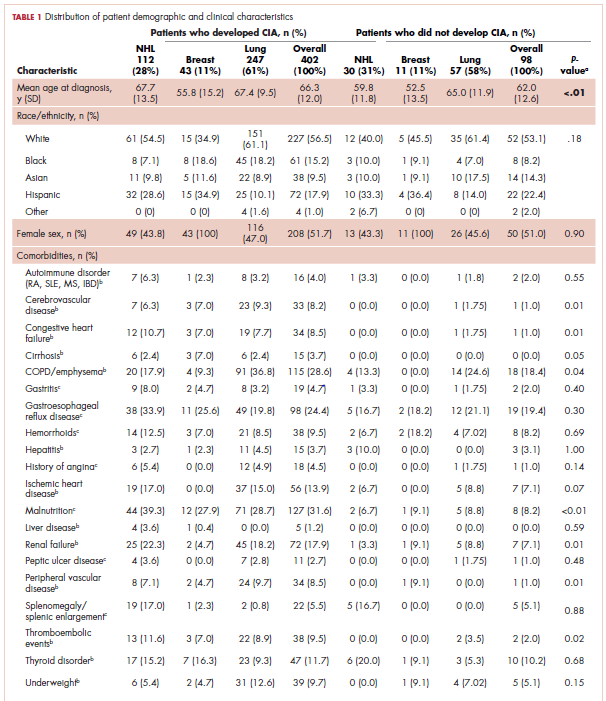
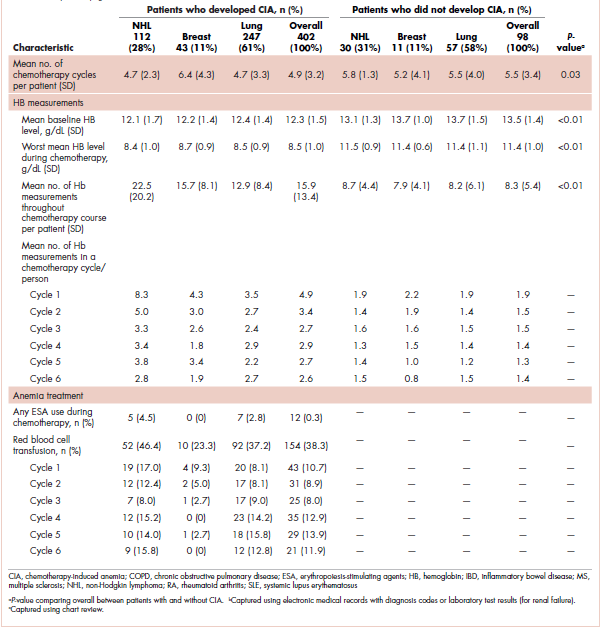
The mean Hb level before chemotherapy was lower for patients who developed CIA compared with patients who did not develop CIA (12.3 g/dL and 13.5 g/dL, respectively; Table 1). The mean lowest Hb level during chemotherapy was 8.5 g/dL for patients who developed CIA and 11.4 g/dL for patients without CIA (Table 1). The number of anemia events by grade in each chemotherapy cycle in patients who developed CIA is shown in Table 2. 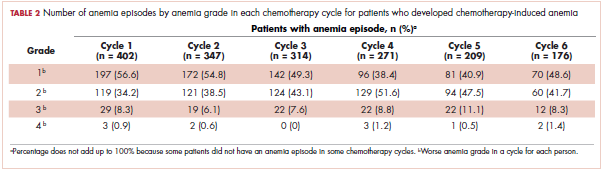
Table 3 shows the number and proportion of study patients with each of the symptoms documented before and after chemotherapy initiation for the 2 study samples. Patients who developed CIA had statistically significantly more pre-existing symptoms, incident symptoms, or any symptoms that occurred during chemotherapy compared with patients who did not develop CIA. 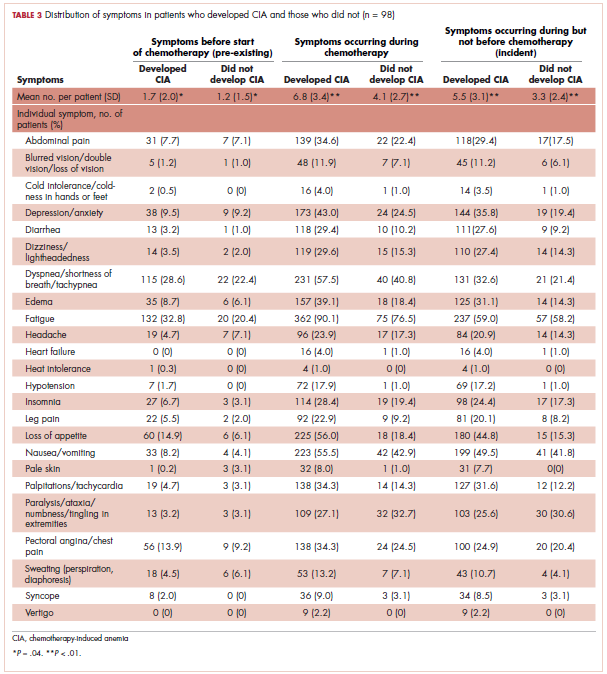
Table 4 shows the number and proportion of study patients with symptoms that occurred during each chemotherapy cycle. Again, fatigue is the predominant symptom documented throughout cycles for all patients. In patients who developed CIA, the proportion of patients experiencing the following symptoms was relatively stable across chemotherapy cycles: depression/anxiety, dizziness/lightheadedness, fatigue, pale skin, and sweating. The proportion of patients experiencing paralysis/ataxia/numbness/tingling in extremities increased over cycles. For headache, loss of appetite, hypotension, and nausea/vomiting, the proportion of patients with symptom documentation was highest in cycle 1, stabilizing in subsequent cycles (Table 4). In patients without CIA, the cycle-level prevalence of most of the symptoms did not increase over cycles, except for paralysis/ataxia/numbness or tingling in extremities. For insomnia, loss of appetite, and nausea/vomiting, the cycle-level prevalence dropped after the first cycle. There was no clear increasing trend of the mean number of symptoms per patient across chemotherapy cycles in both study samples (Table 4).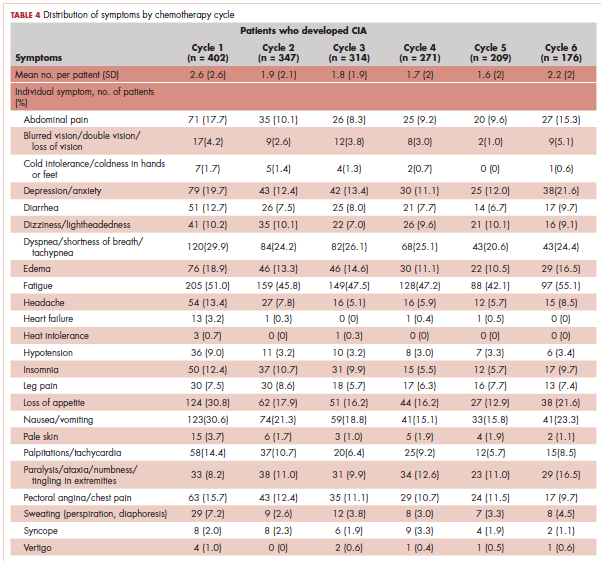
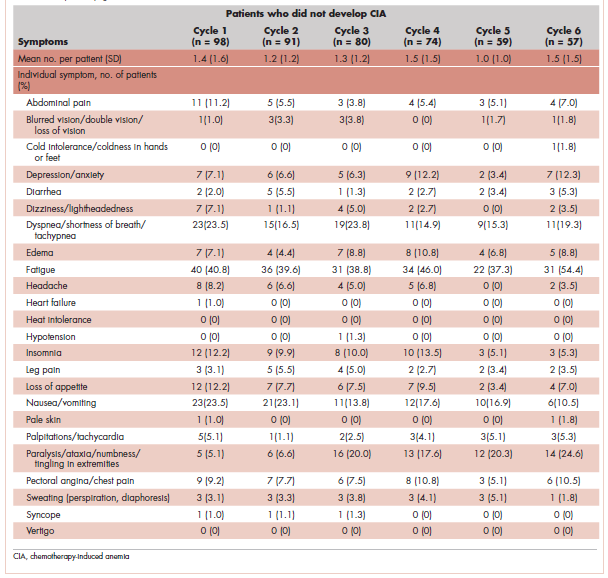
Table 5 shows the distribution of symptoms by anemia grade in patients who developed CIA. In general, the prevalence of symptoms increased with higher grades of anemia. The following symptoms especially have a clear increase in prevalence as the severity of anemia progressed: abdominal pain, depression, diarrhea, dizziness/lightheadedness, dyspnea, edema, fatigue, heart failure, headache, hypotension, insomnia, leg pain, loss of appetite, pale skin, palpitations, pectoral angina, and sweating. The mean number of symptoms per patient increased as CIA grade increased, from 3.6 (SD, 2.9) for grade 2 CIA to 5.4 (SD, 3.5) for grades 3 and 4 CIA (specifically, 5.3 [SD, 3.4] for grade 3 CIA and 6.4 [SD, 4.1] for grade 4 CIA; data not shown) (Table 5).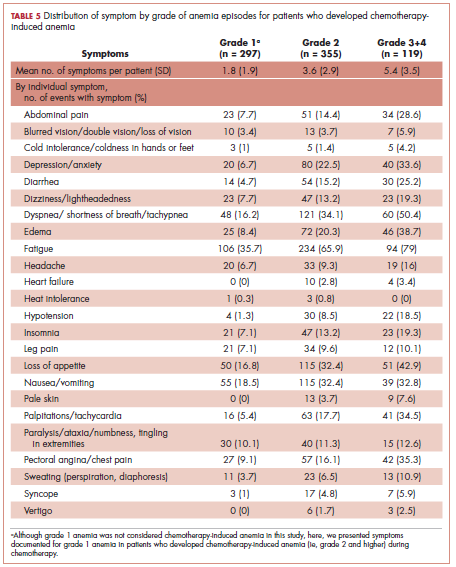
When stratified by gender, there are no material differences between men and women in most analyses. In men, the mean number of pre-existing symptoms was 1.7 (SD, 1.8) and 1.0 (SD, 1.2) for those with and without CIA, respectively (P = .02). The mean number of symptoms that occurred during chemotherapy was 7.0 (SD, 3.4) and 4.2 (SD, 2.4), respectively (P < .01). In women, the mean number of pre-existing symptoms was not statistically different in those with and without CIA (1.6 [SD, 2.2] and 1.3 [SD, 1.8], respectively; P = .46). However, like in men, the mean number of symptoms that occurred during chemotherapy was significantly more in those with CIA (6.5 [SD, 3.3] and 4.0 [SD, 2.9], respectively; P < .01). As in the overall analysis, there was no clear increasing trend of the number of symptoms per patients across chemotherapy cycles in both men and women, but the average number of symptoms increased as the CIA grade increased. For men, the mean number of symptoms per patient increased from 3.7 (SD, 3.0) for grade 2 CIA to 6.0 (SD, 3.5) for grades 3 and 4 CIA (data not shown). For women, the mean number of symptoms per patient increased from 3.6 (SD, 2.9) for grade 2 CIA to 4.7 (SD, 3.3) for grades 3 and 4 CIA (data not shown).
Discussion
In this study, we described the number and type of symptoms documented in the medical record notes among stage IV NHL, breast cancer, and lung cancer patients who did or did not develop CIA during chemotherapy.
Our findings on the prevalence of fatigue are in line with other studies in the literature. Maxwell reported that the prevalence of fatigue was 80% to 96% in cancer patients.17 Cella and colleagues found that using FACT-General questionnaire, 75% of cancer patients reported fatigue.11 The comparability of our estimate and those found in studies based on patient self-report offered some assurance of the validity of assessing symptom prevalence through physician record notes. In addition to fatigue, we described prevalence of 23 additional symptoms, most of which have not been extensively studied in the literature. Gabrilove and colleagues found that a substantial proportion of patients with CIA had moderate to severe score for lack of appetite (36%) and disturbed sleep (41%) using the MDASI.10 The prevalence of loss of appetite and insomnia was around 50% and 25%, respectively, in our study samples. A 2013 systematic review of 21 multinational studies reported the pooled prevalence of several nonfatigue symptoms in cancer patients including headache (23%), sleep disturbance/insomnia (49%), appetite changes (45%), nausea/vomiting (26%), diarrhea (15%), depression (34%), dyspnea (44%), dizziness (26%), numbness/tingling (42%), edema (14%), and sweating (28%).18 Our prevalence estimates in patients with CIA for most of these symptoms were higher, likely because Reilly and colleagues used source studies that included any cancer patients undergoing treatment and not just those with CIA. Our findings on the increased symptom burden in patients who experienced episodes of advanced anemia compared with patients with mild anemia were also consistent with the literature. To this end, several studies using MDASI or the FACT-An reported differential symptom burdens by Hb level based on patient self-report,10,11,19 including data on improvement in symptom burden and quality of life after anemia was amended with the use of ESA.20,21
We found that the number of pre-existing symptoms was significantly higher in patients who went on to develop CIA than in patients who did not develop CIA. Specifically, fatigue, loss of appetite, and pale skin before chemotherapy seemed to be significantly more common in patients who went on to develop CIA. This finding suggested that presentation of these symptoms before chemotherapy initiation may be a predictor for developing moderate or severe anemia during treatment. This is a novel hypothesis, as no studies have evaluated the relationship between pretreatment symptom and risk of CIA. However, our study was not designed to address this specific question. Additional investigation is needed to further shed light on whether the occurrence of anemia-related symptoms in nonanemic patients can be used to effectively risk-stratify patients for subsequent CIA.
Contrary to our expectation, the prevalence of most symptoms did not clearly increase as chemotherapy progressed. There are several possible explanations to this phenomenon, with the most likely being related to reporting of anemia-related symptoms. For example, patients might stop reporting the same symptom repeatedly or become adjusted to the new Hb levels, leading to less symptom manifestation. Clinicians may also be less likely to ask about symptoms in later treatment cycles and/or to document chronic symptoms. Several symptoms were rarely documented altogether, such as cold intolerance, heat intolerance, heart failure, and vertigo. Symptoms reported in earlier cycles could also be managed successfully. Another possible explanation is differential loss of follow-up. Patients who experienced severe adverse events or symptoms may terminate treatment prematurely. Thus, symptom burden found toward later cycles may not represent the true symptom burden should everyone who initiated the chemotherapy treatment complete all planned cycles.
Limitations
In addition to the limitations already discussed, there are several others that should be considered when interpreting our results. We did not have a consistent measure of symptom severity in the medical records. Duration of symptoms was also often poorly documented by physicians. Therefore, our results are not directly comparable with studies such as the MDASI that incorporate severity or duration in their prevalence measure.
Despite the potential limitations, our study has several important strengths.
Conclusions
Our data provide physicians a comprehensive picture of prevalence of various types of symptoms and how symptom burden evolves as chemotherapy cycle and anemia severity progress. High-grade CIA correlates with an increased symptom burden.
1. Barrett-Lee PJ, Ludwig H, Birgegård G, et al. Independent risk factors for anemia in cancer patients receiving chemotherapy: results from the European Cancer Anaemia Survey. Oncology. 2006;70(1):34-48.
2. Kitano T, Tada H, Nishimura T, et al. Prevalence and incidence of anemia in Japanese cancer patients receiving outpatient chemotherapy. Int J Hematol. 2007;86(1):37-41.
3. Birgegård G, Aapro MS, Bokemeyer C, et al. Cancer-related anemia: pathogenesis, prevalence and treatment. Oncology. 2005;68(Suppl 1):3-11.
4. Harper P, Littlewood T. Anaemia of cancer: impact on patient fatigue and long-term outcome. Oncology. 2005;69(Suppl 2):2-7.
5. Nieboer P, Buijs C, Rodenhuis S, et al. Fatigue and relating factors in high-risk breast cancer patients treated with adjuvant standard or high-dose chemotherapy: a longitudinal study. J Clin Oncol. 2005;23(33):8296-8304.
6. Bremberg ER, Brandberg Y, Hising C, Friesland S, Eksborg S. Anemia and quality of life including anemia-related symptoms in patients with solid tumors in clinical practice. Med Oncol. 2007;24(1):95-102.
7. Hofman M, Ryan JL, Figueroa-Moseley CD, Jean-Pierre P, Morrow GR. Cancer-related fatigue: the scale of the problem. Oncologist. 2007;12(Suppl 1):4-10.
8. Cleeland CS. Symptom burden: multiple symptoms and their impact as patient-reported outcomes. J Natl Cancer Inst Monogr. 2007(37):16-21.
9. Yellen SB, Cella DF, Webster K, Blendowski C, Kaplan E. Measuring fatigue and other anemia-related symptoms with the Functional Assessment of Cancer Therapy (FACT) measurement system. J Pain Symptom Manage. 1997;13(2):63-74.
10. Gabrilove JL, Perez EA, Tomita DK, Rossi G, Cleeland CS. Assessing symptom burden using the M. D. Anderson symptom inventory in patients with chemotherapy-induced anemia: results of a multicenter, open-label study (SURPASS) of patients treated with darbepoetin-alpha at a dose of 200 microg every 2 weeks. Cancer. 2007;110(7):1629-1640.
11. Cella D. The Functional Assessment of Cancer Therapy-Anemia (FACT-An) scale: a new tool for the assessment of outcomes in cancer anemia and fatigue. Semin Hematol. 1997;34(3 Suppl 2):13-19.
12. Koebnick C, Langer-Gould AM, Gould MK, et al. Sociodemographic characteristics of members of a large, integrated health care system: comparison with US Census Bureau data. Perm J. 2012;16(3):37-41.
13. Groopman JE, Itri LM. Chemotherapy-induced anemia in adults: incidence and treatment. J Natl Cancer Inst. 1999;91(19):1616-1634.
14. Gilreath JA, Stenehjem DD, Rodgers GM. Diagnosis and treatment of cancer-related anemia. Am J Hematol. 2014;89(2):203-212.
15. Rizzo JD, Somerfield MR, Hagerty KL, et al. Use of epoetin and darbepoetin in patients with cancer: 2007 American Society of Clinical Oncology/American Society of Hematology clinical practice guideline update. J Clin Oncol. 2008;26(1):132-149.
16. Bohlius J, Tonia T, Nüesch E, et al. Effects of erythropoiesis-stimulating agents on fatigue- and anaemia-related symptoms in cancer patients: systematic review and meta-analyses of published and unpublished data. Br J Cancer. 2014;111(1):33-45.
17. Maxwell MB. When the cancer patient becomes anemic. Cancer Nurs. 1984;7(4):321-326.
18. Reilly CM, Bruner DW, Mitchell SA, et al. A literature synthesis of symptom prevalence and severity in persons receiving active cancer treatment. Support Care Cancer. 2013;21(6):1525-1550.
19. Crawford J, Cella D, Cleeland CS, et al. Relationship between changes in hemoglobin level and quality of life during chemotherapy in anemic cancer patients receiving epoetin alfa therapy. Cancer. 2002;95(4):888-895.
20. Mouysset JL, Freier B, van den Bosch J, et al. Hemoglobin levels and quality of life in patients with symptomatic chemotherapy-induced anemia: the eAQUA study. Cancer Manag Res. 2016;8:1-10.
21. Vansteenkiste J, Pirker R, Massuti B, et al. Double-blind, placebo-controlled, randomized phase III trial of darbepoetin alfa in lung cancer patients receiving chemotherapy. J Natl Cancer Inst. 2002;94(16):1211-1220.
22. Kleinman L, Benjamin K, Viswanathan H, et al. The anemia impact measure (AIM): development and content validation of a patient-reported outcome measure of anemia symptoms and symptom impacts in cancer patients receiving chemotherapy. Qual Life Res. 2012;21(7):1255-1266.
Anemia is a common complication of cancer treatment as well as of cancer itself. Most cancer patients undergoing chemotherapy experience anemia sometime during their treatment course.1,2 Moderate to severe anemia is associated with an array of symptoms that are known to compromise the physical functioning and quality of life of cancer patients. Common anemia-related symptoms include fatigue, drowsiness, depression, dyspnea, tachycardia, and dizziness.1,3-7
Symptoms produced by cancer itself or the disease treatment (ie, side effects such as anemia) collectively compose a patient’s symptom burden.8 Although the occurrence of anemia-related fatigue has been described more systematically, other clinical presentations of chemotherapy-induced anemia (CIA) are not well characterized. Furthermore, the overall symptom burdens associated with different ranges of hemoglobin (Hb) concentrations have also not been well reported. Although various tools have been developed to facilitate the reporting of fatigue and other symptoms experienced by patients with CIA, such as the Functional Assessment of Cancer Therapy-Anemia (FACT-An) questionnaire and the MD Anderson Symptom Inventory (MDASI),9-11 these questionnaires have not been extensively used outside of the research context. As such, knowledge on symptom burdens associated with CIA in real-world patient populations remains lacking.
Given the common occurrence of CIA, management of CIA and associated symptoms plays an important role to patients’ quality of life during cancer treatment. Symptom control is often the main goal for patients with stage IV cancers, as treatment for disease is most likely palliative or noncurative. To facilitate supportive care planning, it is important to understand patient symptom burdens as chemotherapy progresses over cycles and Hb levels decline. We conducted a comprehensive medical record review study in patients diagnosed with stage IV non-Hodgkin lymphoma (NHL), breast cancer, and lung cancers at Kaiser Permanente Southern California (KPSC), a large community-based health care delivery system. The objective of this study was to report the occurrence of CIA-related symptoms throughout the course of chemotherapy and by Hb levels.
Methods
Study setting and population
KPSC is an integrated managed-care organization that provides comprehensive health services for 4 million racially, ethnically, and socioeconomically diverse members who broadly represent the population in Southern California.12 The organization maintains electronic records of health care received by its members, including physician record notes and clinical databases such as laboratory test results, diagnosis codes, medical procedures, medication dispenses, and disease registries. KPSC’s cancer registry is Surveillance, Epidemiology, and End Results, which is affiliated and routinely collects information on age, sex, race and/or ethnicity, cancer type, histology, and stage at diagnosis.
Patients who met the following inclusion criteria were included in this study: diagnosed with stage IV NHL, breast cancer, or lung cancer at age 18 years or older at KPSC between March 25, 2010 and December 31, 2012; initiated myelosuppressive chemotherapy at KPSC before June 30, 2013 (only the first chemotherapy course was included in this evaluation); and had at least 1 Hb measurement during the course of chemotherapy. Of those who met the inclusion criteria, patients who met the following criteria were excluded if they had less than 12 months KPSC membership before start of chemotherapy, missing information on cancer stage or chemotherapy regimen/agents, a diagnosis of myelodysplastic syndrome before chemotherapy initiation, a diagnosis of inherited anemia, an Hb concentration <10 g/L within 3 months before chemotherapy initiation, a transfusion within 2 weeks before chemotherapy initiation, radiation within 4 months before chemotherapy initiation, or bone marrow transplantation within 12 months before chemotherapy initiation or during the chemotherapy course. These exclusion criteria were applied to evaluate symptom burdens most likely related to CIA as opposed to other cancer treatment or pre-existing anemia.
CIA in this study was defined as moderate to severe anemia with Hb <10 g/dL after chemotherapy initiation. Based on this definition for CIA, all patients who developed CIA between the first chemotherapy administration to 60 days after the last dose of chemotherapy were included for the record review
Data collection
Data on anemia-related symptoms or signs and anemia-related comorbidities (Table 1) were collected by standardized review of physician record notes in the electronic medical records. A set of 24 anemia-related symptoms were identified based on the literature and clinical expertise and included abdominal pain, blurred vision/double vision/loss of vision, cold intolerance/coldness in hands or feet, depression/anxiety, diarrhea, dizziness/lightheadedness, dyspnea/shortness of breath/tachypnea, edema, fatigue, headache, heart failure, heat intolerance, hypotension, insomnia, leg pain, loss of appetite, nausea/vomiting, pale skin, palpitations/tachycardia, paralysis/ataxia/numbness or tingling in extremities, pectoral angina/chest pain, sweating/diaphoresis, syncope, and vertigo. Record review period was defined as 1 month before chemotherapy to 60 days after the last dose of chemotherapy in the first course. To understand the development of new symptoms during chemotherapy treatment, pre-existing symptoms documented within 1 month before chemotherapy initiation were recorded.
The data elements extracted included the date the symptom was documented, date the symptom started, symptom duration (when available), and any relevant comments regarding the symptom (ie, if dyspnea was at rest or on exertion, whether the symptom was a side effect caused by chemotherapy, or change in symptom severity). Ten percent of the records were reviewed independently by 2 abstractors to ensure quality control. Additional quality control measures included SAS algorithms (SAS Institute, Inc., Cary, North Carolina) to check reasonability and logical consistency in the abstracted data.
Patient demographic characteristics, cancer stage, additional selected comorbidities (Table 1), chemotherapy information, Hb test results, and anemia treatment, including erythrocyte stimulating agent (ESA) use and red blood cell transfusion, were collected using KPSC’s cancer registry and clinical databases. Anemia was defined by severity as grade 1 (10 g/dL to lower limit of normal, ie, 14 g/dL for men and 12 g/dL for women), grade 2 (8.0-9.9 g/dL), grade 3 (6.5-7.9 g/dL), and grade 4 (<6.5 g/dL) following the National Cancer Institute’s Common Terminology Criteria for Adverse Events.13
Statistical analysis
Distributions of demographic, cancer, and treatment characteristics were calculated by CIA status, overall and by cancer type. Differences between patients who did and did not develop CIA were assessed using chi-square test and Kruskal-Wallis test. For those who developed CIA, the distribution of the worst anemia grade was also calculated for each cycle of chemotherapy.
Next, the distributions for the following symptom categories were calculated in the 2 study samples defined by CIA status: pre-existing symptoms that occurred before chemotherapy, any symptoms during chemotherapy (ie, whether they started before chemotherapy), and incident symptoms during chemotherapy (ie, new symptoms that only started after chemotherapy). Specifically, the proportion of patients with each individual symptom and the distribution of the number of symptoms per patient were calculated. Differences in symptom distribution by CIA status were assessed using chi-square test.
The distribution of symptoms in each chemotherapy cycle was calculated up to 6 chemotherapy cycles (as >80% of the patients only had treatment up to 6 cycles) in the 2 study samples defined by CIA status. For this analysis, a symptom was “mapped” to a cycle if the date (or date range) of the symptom fell within the date range of that chemotherapy cycle. In patients who developed CIA, the distribution of symptoms was also calculated by anemia grade. This was again done on the chemotherapy cycle level. For each chemotherapy cycle, an anemia grade was assigned (no anemia or anemia grade 1, 2, 3, and 4) using the lowest Hb measurement in that cycle. Symptoms that occurred in a chemotherapy cycle were then “mapped” to the anemia grade of that cycle. Some patients had more than 1 anemia event of the same grade (eg, if a patient’s grade 2 anemia persist across cycles). For these patients, we randomly selected only 1 anemia event of the same grade from each patient to be included in this analysis. Patients could still contribute multiple events of different grades to this analysis. We calculated the mean number of symptoms per patient for each anemia grade (ie, 1-4) separately. Because of the small number of patients who developed grade 4 anemia (n = 11), they were combined with the grade 3 patients when the distributions of individual symptoms were evaluated.
All analyses were repeated stratified by gender. P values for differences between men and women were calculated using chi-square test or t test. All analyses were conducted using SAS version 9.3.
Results
A total of 402 stage IV NHL, breast, and lung cancer patients who developed CIA and 98 patients who did not develop CIA during the first course of chemotherapy were included (Figure 1).
The distribution of cancer types in the study sample were similar across CIA status (Table 1). The mean age at diagnosis was 66 years in patients who developed CIA and 62 years in patients who did not develop CIA. Women accounted for half of the patients in both study samples (52% and 51%, respectively). Most of the study patients were of non-Hispanic white race/ethnicity. Chronic obstructive pulmonary disease/emphysema and gastroesophageal reflux disease were among the most common comorbidities examined in both study samples, while malnutrition and moderate to severe renal disease were also common in patients who developed CIA (Table 1).
The mean Hb level before chemotherapy was lower for patients who developed CIA compared with patients who did not develop CIA (12.3 g/dL and 13.5 g/dL, respectively; Table 1). The mean lowest Hb level during chemotherapy was 8.5 g/dL for patients who developed CIA and 11.4 g/dL for patients without CIA (Table 1). The number of anemia events by grade in each chemotherapy cycle in patients who developed CIA is shown in Table 2.
Table 3 shows the number and proportion of study patients with each of the symptoms documented before and after chemotherapy initiation for the 2 study samples. Patients who developed CIA had statistically significantly more pre-existing symptoms, incident symptoms, or any symptoms that occurred during chemotherapy compared with patients who did not develop CIA.
Table 4 shows the number and proportion of study patients with symptoms that occurred during each chemotherapy cycle. Again, fatigue is the predominant symptom documented throughout cycles for all patients. In patients who developed CIA, the proportion of patients experiencing the following symptoms was relatively stable across chemotherapy cycles: depression/anxiety, dizziness/lightheadedness, fatigue, pale skin, and sweating. The proportion of patients experiencing paralysis/ataxia/numbness/tingling in extremities increased over cycles. For headache, loss of appetite, hypotension, and nausea/vomiting, the proportion of patients with symptom documentation was highest in cycle 1, stabilizing in subsequent cycles (Table 4). In patients without CIA, the cycle-level prevalence of most of the symptoms did not increase over cycles, except for paralysis/ataxia/numbness or tingling in extremities. For insomnia, loss of appetite, and nausea/vomiting, the cycle-level prevalence dropped after the first cycle. There was no clear increasing trend of the mean number of symptoms per patient across chemotherapy cycles in both study samples (Table 4).
Table 5 shows the distribution of symptoms by anemia grade in patients who developed CIA. In general, the prevalence of symptoms increased with higher grades of anemia. The following symptoms especially have a clear increase in prevalence as the severity of anemia progressed: abdominal pain, depression, diarrhea, dizziness/lightheadedness, dyspnea, edema, fatigue, heart failure, headache, hypotension, insomnia, leg pain, loss of appetite, pale skin, palpitations, pectoral angina, and sweating. The mean number of symptoms per patient increased as CIA grade increased, from 3.6 (SD, 2.9) for grade 2 CIA to 5.4 (SD, 3.5) for grades 3 and 4 CIA (specifically, 5.3 [SD, 3.4] for grade 3 CIA and 6.4 [SD, 4.1] for grade 4 CIA; data not shown) (Table 5).
When stratified by gender, there are no material differences between men and women in most analyses. In men, the mean number of pre-existing symptoms was 1.7 (SD, 1.8) and 1.0 (SD, 1.2) for those with and without CIA, respectively (P = .02). The mean number of symptoms that occurred during chemotherapy was 7.0 (SD, 3.4) and 4.2 (SD, 2.4), respectively (P < .01). In women, the mean number of pre-existing symptoms was not statistically different in those with and without CIA (1.6 [SD, 2.2] and 1.3 [SD, 1.8], respectively; P = .46). However, like in men, the mean number of symptoms that occurred during chemotherapy was significantly more in those with CIA (6.5 [SD, 3.3] and 4.0 [SD, 2.9], respectively; P < .01). As in the overall analysis, there was no clear increasing trend of the number of symptoms per patients across chemotherapy cycles in both men and women, but the average number of symptoms increased as the CIA grade increased. For men, the mean number of symptoms per patient increased from 3.7 (SD, 3.0) for grade 2 CIA to 6.0 (SD, 3.5) for grades 3 and 4 CIA (data not shown). For women, the mean number of symptoms per patient increased from 3.6 (SD, 2.9) for grade 2 CIA to 4.7 (SD, 3.3) for grades 3 and 4 CIA (data not shown).
Discussion
In this study, we described the number and type of symptoms documented in the medical record notes among stage IV NHL, breast cancer, and lung cancer patients who did or did not develop CIA during chemotherapy.
Our findings on the prevalence of fatigue are in line with other studies in the literature. Maxwell reported that the prevalence of fatigue was 80% to 96% in cancer patients.17 Cella and colleagues found that using FACT-General questionnaire, 75% of cancer patients reported fatigue.11 The comparability of our estimate and those found in studies based on patient self-report offered some assurance of the validity of assessing symptom prevalence through physician record notes. In addition to fatigue, we described prevalence of 23 additional symptoms, most of which have not been extensively studied in the literature. Gabrilove and colleagues found that a substantial proportion of patients with CIA had moderate to severe score for lack of appetite (36%) and disturbed sleep (41%) using the MDASI.10 The prevalence of loss of appetite and insomnia was around 50% and 25%, respectively, in our study samples. A 2013 systematic review of 21 multinational studies reported the pooled prevalence of several nonfatigue symptoms in cancer patients including headache (23%), sleep disturbance/insomnia (49%), appetite changes (45%), nausea/vomiting (26%), diarrhea (15%), depression (34%), dyspnea (44%), dizziness (26%), numbness/tingling (42%), edema (14%), and sweating (28%).18 Our prevalence estimates in patients with CIA for most of these symptoms were higher, likely because Reilly and colleagues used source studies that included any cancer patients undergoing treatment and not just those with CIA. Our findings on the increased symptom burden in patients who experienced episodes of advanced anemia compared with patients with mild anemia were also consistent with the literature. To this end, several studies using MDASI or the FACT-An reported differential symptom burdens by Hb level based on patient self-report,10,11,19 including data on improvement in symptom burden and quality of life after anemia was amended with the use of ESA.20,21
We found that the number of pre-existing symptoms was significantly higher in patients who went on to develop CIA than in patients who did not develop CIA. Specifically, fatigue, loss of appetite, and pale skin before chemotherapy seemed to be significantly more common in patients who went on to develop CIA. This finding suggested that presentation of these symptoms before chemotherapy initiation may be a predictor for developing moderate or severe anemia during treatment. This is a novel hypothesis, as no studies have evaluated the relationship between pretreatment symptom and risk of CIA. However, our study was not designed to address this specific question. Additional investigation is needed to further shed light on whether the occurrence of anemia-related symptoms in nonanemic patients can be used to effectively risk-stratify patients for subsequent CIA.
Contrary to our expectation, the prevalence of most symptoms did not clearly increase as chemotherapy progressed. There are several possible explanations to this phenomenon, with the most likely being related to reporting of anemia-related symptoms. For example, patients might stop reporting the same symptom repeatedly or become adjusted to the new Hb levels, leading to less symptom manifestation. Clinicians may also be less likely to ask about symptoms in later treatment cycles and/or to document chronic symptoms. Several symptoms were rarely documented altogether, such as cold intolerance, heat intolerance, heart failure, and vertigo. Symptoms reported in earlier cycles could also be managed successfully. Another possible explanation is differential loss of follow-up. Patients who experienced severe adverse events or symptoms may terminate treatment prematurely. Thus, symptom burden found toward later cycles may not represent the true symptom burden should everyone who initiated the chemotherapy treatment complete all planned cycles.
Limitations
In addition to the limitations already discussed, there are several others that should be considered when interpreting our results. We did not have a consistent measure of symptom severity in the medical records. Duration of symptoms was also often poorly documented by physicians. Therefore, our results are not directly comparable with studies such as the MDASI that incorporate severity or duration in their prevalence measure.
Despite the potential limitations, our study has several important strengths.
Conclusions
Our data provide physicians a comprehensive picture of prevalence of various types of symptoms and how symptom burden evolves as chemotherapy cycle and anemia severity progress. High-grade CIA correlates with an increased symptom burden.
Anemia is a common complication of cancer treatment as well as of cancer itself. Most cancer patients undergoing chemotherapy experience anemia sometime during their treatment course.1,2 Moderate to severe anemia is associated with an array of symptoms that are known to compromise the physical functioning and quality of life of cancer patients. Common anemia-related symptoms include fatigue, drowsiness, depression, dyspnea, tachycardia, and dizziness.1,3-7
Symptoms produced by cancer itself or the disease treatment (ie, side effects such as anemia) collectively compose a patient’s symptom burden.8 Although the occurrence of anemia-related fatigue has been described more systematically, other clinical presentations of chemotherapy-induced anemia (CIA) are not well characterized. Furthermore, the overall symptom burdens associated with different ranges of hemoglobin (Hb) concentrations have also not been well reported. Although various tools have been developed to facilitate the reporting of fatigue and other symptoms experienced by patients with CIA, such as the Functional Assessment of Cancer Therapy-Anemia (FACT-An) questionnaire and the MD Anderson Symptom Inventory (MDASI),9-11 these questionnaires have not been extensively used outside of the research context. As such, knowledge on symptom burdens associated with CIA in real-world patient populations remains lacking.
Given the common occurrence of CIA, management of CIA and associated symptoms plays an important role to patients’ quality of life during cancer treatment. Symptom control is often the main goal for patients with stage IV cancers, as treatment for disease is most likely palliative or noncurative. To facilitate supportive care planning, it is important to understand patient symptom burdens as chemotherapy progresses over cycles and Hb levels decline. We conducted a comprehensive medical record review study in patients diagnosed with stage IV non-Hodgkin lymphoma (NHL), breast cancer, and lung cancers at Kaiser Permanente Southern California (KPSC), a large community-based health care delivery system. The objective of this study was to report the occurrence of CIA-related symptoms throughout the course of chemotherapy and by Hb levels.
Methods
Study setting and population
KPSC is an integrated managed-care organization that provides comprehensive health services for 4 million racially, ethnically, and socioeconomically diverse members who broadly represent the population in Southern California.12 The organization maintains electronic records of health care received by its members, including physician record notes and clinical databases such as laboratory test results, diagnosis codes, medical procedures, medication dispenses, and disease registries. KPSC’s cancer registry is Surveillance, Epidemiology, and End Results, which is affiliated and routinely collects information on age, sex, race and/or ethnicity, cancer type, histology, and stage at diagnosis.
Patients who met the following inclusion criteria were included in this study: diagnosed with stage IV NHL, breast cancer, or lung cancer at age 18 years or older at KPSC between March 25, 2010 and December 31, 2012; initiated myelosuppressive chemotherapy at KPSC before June 30, 2013 (only the first chemotherapy course was included in this evaluation); and had at least 1 Hb measurement during the course of chemotherapy. Of those who met the inclusion criteria, patients who met the following criteria were excluded if they had less than 12 months KPSC membership before start of chemotherapy, missing information on cancer stage or chemotherapy regimen/agents, a diagnosis of myelodysplastic syndrome before chemotherapy initiation, a diagnosis of inherited anemia, an Hb concentration <10 g/L within 3 months before chemotherapy initiation, a transfusion within 2 weeks before chemotherapy initiation, radiation within 4 months before chemotherapy initiation, or bone marrow transplantation within 12 months before chemotherapy initiation or during the chemotherapy course. These exclusion criteria were applied to evaluate symptom burdens most likely related to CIA as opposed to other cancer treatment or pre-existing anemia.
CIA in this study was defined as moderate to severe anemia with Hb <10 g/dL after chemotherapy initiation. Based on this definition for CIA, all patients who developed CIA between the first chemotherapy administration to 60 days after the last dose of chemotherapy were included for the record review
Data collection
Data on anemia-related symptoms or signs and anemia-related comorbidities (Table 1) were collected by standardized review of physician record notes in the electronic medical records. A set of 24 anemia-related symptoms were identified based on the literature and clinical expertise and included abdominal pain, blurred vision/double vision/loss of vision, cold intolerance/coldness in hands or feet, depression/anxiety, diarrhea, dizziness/lightheadedness, dyspnea/shortness of breath/tachypnea, edema, fatigue, headache, heart failure, heat intolerance, hypotension, insomnia, leg pain, loss of appetite, nausea/vomiting, pale skin, palpitations/tachycardia, paralysis/ataxia/numbness or tingling in extremities, pectoral angina/chest pain, sweating/diaphoresis, syncope, and vertigo. Record review period was defined as 1 month before chemotherapy to 60 days after the last dose of chemotherapy in the first course. To understand the development of new symptoms during chemotherapy treatment, pre-existing symptoms documented within 1 month before chemotherapy initiation were recorded.
The data elements extracted included the date the symptom was documented, date the symptom started, symptom duration (when available), and any relevant comments regarding the symptom (ie, if dyspnea was at rest or on exertion, whether the symptom was a side effect caused by chemotherapy, or change in symptom severity). Ten percent of the records were reviewed independently by 2 abstractors to ensure quality control. Additional quality control measures included SAS algorithms (SAS Institute, Inc., Cary, North Carolina) to check reasonability and logical consistency in the abstracted data.
Patient demographic characteristics, cancer stage, additional selected comorbidities (Table 1), chemotherapy information, Hb test results, and anemia treatment, including erythrocyte stimulating agent (ESA) use and red blood cell transfusion, were collected using KPSC’s cancer registry and clinical databases. Anemia was defined by severity as grade 1 (10 g/dL to lower limit of normal, ie, 14 g/dL for men and 12 g/dL for women), grade 2 (8.0-9.9 g/dL), grade 3 (6.5-7.9 g/dL), and grade 4 (<6.5 g/dL) following the National Cancer Institute’s Common Terminology Criteria for Adverse Events.13
Statistical analysis
Distributions of demographic, cancer, and treatment characteristics were calculated by CIA status, overall and by cancer type. Differences between patients who did and did not develop CIA were assessed using chi-square test and Kruskal-Wallis test. For those who developed CIA, the distribution of the worst anemia grade was also calculated for each cycle of chemotherapy.
Next, the distributions for the following symptom categories were calculated in the 2 study samples defined by CIA status: pre-existing symptoms that occurred before chemotherapy, any symptoms during chemotherapy (ie, whether they started before chemotherapy), and incident symptoms during chemotherapy (ie, new symptoms that only started after chemotherapy). Specifically, the proportion of patients with each individual symptom and the distribution of the number of symptoms per patient were calculated. Differences in symptom distribution by CIA status were assessed using chi-square test.
The distribution of symptoms in each chemotherapy cycle was calculated up to 6 chemotherapy cycles (as >80% of the patients only had treatment up to 6 cycles) in the 2 study samples defined by CIA status. For this analysis, a symptom was “mapped” to a cycle if the date (or date range) of the symptom fell within the date range of that chemotherapy cycle. In patients who developed CIA, the distribution of symptoms was also calculated by anemia grade. This was again done on the chemotherapy cycle level. For each chemotherapy cycle, an anemia grade was assigned (no anemia or anemia grade 1, 2, 3, and 4) using the lowest Hb measurement in that cycle. Symptoms that occurred in a chemotherapy cycle were then “mapped” to the anemia grade of that cycle. Some patients had more than 1 anemia event of the same grade (eg, if a patient’s grade 2 anemia persist across cycles). For these patients, we randomly selected only 1 anemia event of the same grade from each patient to be included in this analysis. Patients could still contribute multiple events of different grades to this analysis. We calculated the mean number of symptoms per patient for each anemia grade (ie, 1-4) separately. Because of the small number of patients who developed grade 4 anemia (n = 11), they were combined with the grade 3 patients when the distributions of individual symptoms were evaluated.
All analyses were repeated stratified by gender. P values for differences between men and women were calculated using chi-square test or t test. All analyses were conducted using SAS version 9.3.
Results
A total of 402 stage IV NHL, breast, and lung cancer patients who developed CIA and 98 patients who did not develop CIA during the first course of chemotherapy were included (Figure 1).
The distribution of cancer types in the study sample were similar across CIA status (Table 1). The mean age at diagnosis was 66 years in patients who developed CIA and 62 years in patients who did not develop CIA. Women accounted for half of the patients in both study samples (52% and 51%, respectively). Most of the study patients were of non-Hispanic white race/ethnicity. Chronic obstructive pulmonary disease/emphysema and gastroesophageal reflux disease were among the most common comorbidities examined in both study samples, while malnutrition and moderate to severe renal disease were also common in patients who developed CIA (Table 1).
The mean Hb level before chemotherapy was lower for patients who developed CIA compared with patients who did not develop CIA (12.3 g/dL and 13.5 g/dL, respectively; Table 1). The mean lowest Hb level during chemotherapy was 8.5 g/dL for patients who developed CIA and 11.4 g/dL for patients without CIA (Table 1). The number of anemia events by grade in each chemotherapy cycle in patients who developed CIA is shown in Table 2.
Table 3 shows the number and proportion of study patients with each of the symptoms documented before and after chemotherapy initiation for the 2 study samples. Patients who developed CIA had statistically significantly more pre-existing symptoms, incident symptoms, or any symptoms that occurred during chemotherapy compared with patients who did not develop CIA.
Table 4 shows the number and proportion of study patients with symptoms that occurred during each chemotherapy cycle. Again, fatigue is the predominant symptom documented throughout cycles for all patients. In patients who developed CIA, the proportion of patients experiencing the following symptoms was relatively stable across chemotherapy cycles: depression/anxiety, dizziness/lightheadedness, fatigue, pale skin, and sweating. The proportion of patients experiencing paralysis/ataxia/numbness/tingling in extremities increased over cycles. For headache, loss of appetite, hypotension, and nausea/vomiting, the proportion of patients with symptom documentation was highest in cycle 1, stabilizing in subsequent cycles (Table 4). In patients without CIA, the cycle-level prevalence of most of the symptoms did not increase over cycles, except for paralysis/ataxia/numbness or tingling in extremities. For insomnia, loss of appetite, and nausea/vomiting, the cycle-level prevalence dropped after the first cycle. There was no clear increasing trend of the mean number of symptoms per patient across chemotherapy cycles in both study samples (Table 4).
Table 5 shows the distribution of symptoms by anemia grade in patients who developed CIA. In general, the prevalence of symptoms increased with higher grades of anemia. The following symptoms especially have a clear increase in prevalence as the severity of anemia progressed: abdominal pain, depression, diarrhea, dizziness/lightheadedness, dyspnea, edema, fatigue, heart failure, headache, hypotension, insomnia, leg pain, loss of appetite, pale skin, palpitations, pectoral angina, and sweating. The mean number of symptoms per patient increased as CIA grade increased, from 3.6 (SD, 2.9) for grade 2 CIA to 5.4 (SD, 3.5) for grades 3 and 4 CIA (specifically, 5.3 [SD, 3.4] for grade 3 CIA and 6.4 [SD, 4.1] for grade 4 CIA; data not shown) (Table 5).
When stratified by gender, there are no material differences between men and women in most analyses. In men, the mean number of pre-existing symptoms was 1.7 (SD, 1.8) and 1.0 (SD, 1.2) for those with and without CIA, respectively (P = .02). The mean number of symptoms that occurred during chemotherapy was 7.0 (SD, 3.4) and 4.2 (SD, 2.4), respectively (P < .01). In women, the mean number of pre-existing symptoms was not statistically different in those with and without CIA (1.6 [SD, 2.2] and 1.3 [SD, 1.8], respectively; P = .46). However, like in men, the mean number of symptoms that occurred during chemotherapy was significantly more in those with CIA (6.5 [SD, 3.3] and 4.0 [SD, 2.9], respectively; P < .01). As in the overall analysis, there was no clear increasing trend of the number of symptoms per patients across chemotherapy cycles in both men and women, but the average number of symptoms increased as the CIA grade increased. For men, the mean number of symptoms per patient increased from 3.7 (SD, 3.0) for grade 2 CIA to 6.0 (SD, 3.5) for grades 3 and 4 CIA (data not shown). For women, the mean number of symptoms per patient increased from 3.6 (SD, 2.9) for grade 2 CIA to 4.7 (SD, 3.3) for grades 3 and 4 CIA (data not shown).
Discussion
In this study, we described the number and type of symptoms documented in the medical record notes among stage IV NHL, breast cancer, and lung cancer patients who did or did not develop CIA during chemotherapy.
Our findings on the prevalence of fatigue are in line with other studies in the literature. Maxwell reported that the prevalence of fatigue was 80% to 96% in cancer patients.17 Cella and colleagues found that using FACT-General questionnaire, 75% of cancer patients reported fatigue.11 The comparability of our estimate and those found in studies based on patient self-report offered some assurance of the validity of assessing symptom prevalence through physician record notes. In addition to fatigue, we described prevalence of 23 additional symptoms, most of which have not been extensively studied in the literature. Gabrilove and colleagues found that a substantial proportion of patients with CIA had moderate to severe score for lack of appetite (36%) and disturbed sleep (41%) using the MDASI.10 The prevalence of loss of appetite and insomnia was around 50% and 25%, respectively, in our study samples. A 2013 systematic review of 21 multinational studies reported the pooled prevalence of several nonfatigue symptoms in cancer patients including headache (23%), sleep disturbance/insomnia (49%), appetite changes (45%), nausea/vomiting (26%), diarrhea (15%), depression (34%), dyspnea (44%), dizziness (26%), numbness/tingling (42%), edema (14%), and sweating (28%).18 Our prevalence estimates in patients with CIA for most of these symptoms were higher, likely because Reilly and colleagues used source studies that included any cancer patients undergoing treatment and not just those with CIA. Our findings on the increased symptom burden in patients who experienced episodes of advanced anemia compared with patients with mild anemia were also consistent with the literature. To this end, several studies using MDASI or the FACT-An reported differential symptom burdens by Hb level based on patient self-report,10,11,19 including data on improvement in symptom burden and quality of life after anemia was amended with the use of ESA.20,21
We found that the number of pre-existing symptoms was significantly higher in patients who went on to develop CIA than in patients who did not develop CIA. Specifically, fatigue, loss of appetite, and pale skin before chemotherapy seemed to be significantly more common in patients who went on to develop CIA. This finding suggested that presentation of these symptoms before chemotherapy initiation may be a predictor for developing moderate or severe anemia during treatment. This is a novel hypothesis, as no studies have evaluated the relationship between pretreatment symptom and risk of CIA. However, our study was not designed to address this specific question. Additional investigation is needed to further shed light on whether the occurrence of anemia-related symptoms in nonanemic patients can be used to effectively risk-stratify patients for subsequent CIA.
Contrary to our expectation, the prevalence of most symptoms did not clearly increase as chemotherapy progressed. There are several possible explanations to this phenomenon, with the most likely being related to reporting of anemia-related symptoms. For example, patients might stop reporting the same symptom repeatedly or become adjusted to the new Hb levels, leading to less symptom manifestation. Clinicians may also be less likely to ask about symptoms in later treatment cycles and/or to document chronic symptoms. Several symptoms were rarely documented altogether, such as cold intolerance, heat intolerance, heart failure, and vertigo. Symptoms reported in earlier cycles could also be managed successfully. Another possible explanation is differential loss of follow-up. Patients who experienced severe adverse events or symptoms may terminate treatment prematurely. Thus, symptom burden found toward later cycles may not represent the true symptom burden should everyone who initiated the chemotherapy treatment complete all planned cycles.
Limitations
In addition to the limitations already discussed, there are several others that should be considered when interpreting our results. We did not have a consistent measure of symptom severity in the medical records. Duration of symptoms was also often poorly documented by physicians. Therefore, our results are not directly comparable with studies such as the MDASI that incorporate severity or duration in their prevalence measure.
Despite the potential limitations, our study has several important strengths.
Conclusions
Our data provide physicians a comprehensive picture of prevalence of various types of symptoms and how symptom burden evolves as chemotherapy cycle and anemia severity progress. High-grade CIA correlates with an increased symptom burden.
1. Barrett-Lee PJ, Ludwig H, Birgegård G, et al. Independent risk factors for anemia in cancer patients receiving chemotherapy: results from the European Cancer Anaemia Survey. Oncology. 2006;70(1):34-48.
2. Kitano T, Tada H, Nishimura T, et al. Prevalence and incidence of anemia in Japanese cancer patients receiving outpatient chemotherapy. Int J Hematol. 2007;86(1):37-41.
3. Birgegård G, Aapro MS, Bokemeyer C, et al. Cancer-related anemia: pathogenesis, prevalence and treatment. Oncology. 2005;68(Suppl 1):3-11.
4. Harper P, Littlewood T. Anaemia of cancer: impact on patient fatigue and long-term outcome. Oncology. 2005;69(Suppl 2):2-7.
5. Nieboer P, Buijs C, Rodenhuis S, et al. Fatigue and relating factors in high-risk breast cancer patients treated with adjuvant standard or high-dose chemotherapy: a longitudinal study. J Clin Oncol. 2005;23(33):8296-8304.
6. Bremberg ER, Brandberg Y, Hising C, Friesland S, Eksborg S. Anemia and quality of life including anemia-related symptoms in patients with solid tumors in clinical practice. Med Oncol. 2007;24(1):95-102.
7. Hofman M, Ryan JL, Figueroa-Moseley CD, Jean-Pierre P, Morrow GR. Cancer-related fatigue: the scale of the problem. Oncologist. 2007;12(Suppl 1):4-10.
8. Cleeland CS. Symptom burden: multiple symptoms and their impact as patient-reported outcomes. J Natl Cancer Inst Monogr. 2007(37):16-21.
9. Yellen SB, Cella DF, Webster K, Blendowski C, Kaplan E. Measuring fatigue and other anemia-related symptoms with the Functional Assessment of Cancer Therapy (FACT) measurement system. J Pain Symptom Manage. 1997;13(2):63-74.
10. Gabrilove JL, Perez EA, Tomita DK, Rossi G, Cleeland CS. Assessing symptom burden using the M. D. Anderson symptom inventory in patients with chemotherapy-induced anemia: results of a multicenter, open-label study (SURPASS) of patients treated with darbepoetin-alpha at a dose of 200 microg every 2 weeks. Cancer. 2007;110(7):1629-1640.
11. Cella D. The Functional Assessment of Cancer Therapy-Anemia (FACT-An) scale: a new tool for the assessment of outcomes in cancer anemia and fatigue. Semin Hematol. 1997;34(3 Suppl 2):13-19.
12. Koebnick C, Langer-Gould AM, Gould MK, et al. Sociodemographic characteristics of members of a large, integrated health care system: comparison with US Census Bureau data. Perm J. 2012;16(3):37-41.
13. Groopman JE, Itri LM. Chemotherapy-induced anemia in adults: incidence and treatment. J Natl Cancer Inst. 1999;91(19):1616-1634.
14. Gilreath JA, Stenehjem DD, Rodgers GM. Diagnosis and treatment of cancer-related anemia. Am J Hematol. 2014;89(2):203-212.
15. Rizzo JD, Somerfield MR, Hagerty KL, et al. Use of epoetin and darbepoetin in patients with cancer: 2007 American Society of Clinical Oncology/American Society of Hematology clinical practice guideline update. J Clin Oncol. 2008;26(1):132-149.
16. Bohlius J, Tonia T, Nüesch E, et al. Effects of erythropoiesis-stimulating agents on fatigue- and anaemia-related symptoms in cancer patients: systematic review and meta-analyses of published and unpublished data. Br J Cancer. 2014;111(1):33-45.
17. Maxwell MB. When the cancer patient becomes anemic. Cancer Nurs. 1984;7(4):321-326.
18. Reilly CM, Bruner DW, Mitchell SA, et al. A literature synthesis of symptom prevalence and severity in persons receiving active cancer treatment. Support Care Cancer. 2013;21(6):1525-1550.
19. Crawford J, Cella D, Cleeland CS, et al. Relationship between changes in hemoglobin level and quality of life during chemotherapy in anemic cancer patients receiving epoetin alfa therapy. Cancer. 2002;95(4):888-895.
20. Mouysset JL, Freier B, van den Bosch J, et al. Hemoglobin levels and quality of life in patients with symptomatic chemotherapy-induced anemia: the eAQUA study. Cancer Manag Res. 2016;8:1-10.
21. Vansteenkiste J, Pirker R, Massuti B, et al. Double-blind, placebo-controlled, randomized phase III trial of darbepoetin alfa in lung cancer patients receiving chemotherapy. J Natl Cancer Inst. 2002;94(16):1211-1220.
22. Kleinman L, Benjamin K, Viswanathan H, et al. The anemia impact measure (AIM): development and content validation of a patient-reported outcome measure of anemia symptoms and symptom impacts in cancer patients receiving chemotherapy. Qual Life Res. 2012;21(7):1255-1266.
1. Barrett-Lee PJ, Ludwig H, Birgegård G, et al. Independent risk factors for anemia in cancer patients receiving chemotherapy: results from the European Cancer Anaemia Survey. Oncology. 2006;70(1):34-48.
2. Kitano T, Tada H, Nishimura T, et al. Prevalence and incidence of anemia in Japanese cancer patients receiving outpatient chemotherapy. Int J Hematol. 2007;86(1):37-41.
3. Birgegård G, Aapro MS, Bokemeyer C, et al. Cancer-related anemia: pathogenesis, prevalence and treatment. Oncology. 2005;68(Suppl 1):3-11.
4. Harper P, Littlewood T. Anaemia of cancer: impact on patient fatigue and long-term outcome. Oncology. 2005;69(Suppl 2):2-7.
5. Nieboer P, Buijs C, Rodenhuis S, et al. Fatigue and relating factors in high-risk breast cancer patients treated with adjuvant standard or high-dose chemotherapy: a longitudinal study. J Clin Oncol. 2005;23(33):8296-8304.
6. Bremberg ER, Brandberg Y, Hising C, Friesland S, Eksborg S. Anemia and quality of life including anemia-related symptoms in patients with solid tumors in clinical practice. Med Oncol. 2007;24(1):95-102.
7. Hofman M, Ryan JL, Figueroa-Moseley CD, Jean-Pierre P, Morrow GR. Cancer-related fatigue: the scale of the problem. Oncologist. 2007;12(Suppl 1):4-10.
8. Cleeland CS. Symptom burden: multiple symptoms and their impact as patient-reported outcomes. J Natl Cancer Inst Monogr. 2007(37):16-21.
9. Yellen SB, Cella DF, Webster K, Blendowski C, Kaplan E. Measuring fatigue and other anemia-related symptoms with the Functional Assessment of Cancer Therapy (FACT) measurement system. J Pain Symptom Manage. 1997;13(2):63-74.
10. Gabrilove JL, Perez EA, Tomita DK, Rossi G, Cleeland CS. Assessing symptom burden using the M. D. Anderson symptom inventory in patients with chemotherapy-induced anemia: results of a multicenter, open-label study (SURPASS) of patients treated with darbepoetin-alpha at a dose of 200 microg every 2 weeks. Cancer. 2007;110(7):1629-1640.
11. Cella D. The Functional Assessment of Cancer Therapy-Anemia (FACT-An) scale: a new tool for the assessment of outcomes in cancer anemia and fatigue. Semin Hematol. 1997;34(3 Suppl 2):13-19.
12. Koebnick C, Langer-Gould AM, Gould MK, et al. Sociodemographic characteristics of members of a large, integrated health care system: comparison with US Census Bureau data. Perm J. 2012;16(3):37-41.
13. Groopman JE, Itri LM. Chemotherapy-induced anemia in adults: incidence and treatment. J Natl Cancer Inst. 1999;91(19):1616-1634.
14. Gilreath JA, Stenehjem DD, Rodgers GM. Diagnosis and treatment of cancer-related anemia. Am J Hematol. 2014;89(2):203-212.
15. Rizzo JD, Somerfield MR, Hagerty KL, et al. Use of epoetin and darbepoetin in patients with cancer: 2007 American Society of Clinical Oncology/American Society of Hematology clinical practice guideline update. J Clin Oncol. 2008;26(1):132-149.
16. Bohlius J, Tonia T, Nüesch E, et al. Effects of erythropoiesis-stimulating agents on fatigue- and anaemia-related symptoms in cancer patients: systematic review and meta-analyses of published and unpublished data. Br J Cancer. 2014;111(1):33-45.
17. Maxwell MB. When the cancer patient becomes anemic. Cancer Nurs. 1984;7(4):321-326.
18. Reilly CM, Bruner DW, Mitchell SA, et al. A literature synthesis of symptom prevalence and severity in persons receiving active cancer treatment. Support Care Cancer. 2013;21(6):1525-1550.
19. Crawford J, Cella D, Cleeland CS, et al. Relationship between changes in hemoglobin level and quality of life during chemotherapy in anemic cancer patients receiving epoetin alfa therapy. Cancer. 2002;95(4):888-895.
20. Mouysset JL, Freier B, van den Bosch J, et al. Hemoglobin levels and quality of life in patients with symptomatic chemotherapy-induced anemia: the eAQUA study. Cancer Manag Res. 2016;8:1-10.
21. Vansteenkiste J, Pirker R, Massuti B, et al. Double-blind, placebo-controlled, randomized phase III trial of darbepoetin alfa in lung cancer patients receiving chemotherapy. J Natl Cancer Inst. 2002;94(16):1211-1220.
22. Kleinman L, Benjamin K, Viswanathan H, et al. The anemia impact measure (AIM): development and content validation of a patient-reported outcome measure of anemia symptoms and symptom impacts in cancer patients receiving chemotherapy. Qual Life Res. 2012;21(7):1255-1266.
LCAR-B38M CAR T therapy appears durable in myeloma
SAN DIEGO – The chimeric antigen receptor (CAR) T-cell therapy LCAR-B38M is in the race for approval in multiple myeloma following encouraging phase 1 results reported at the annual meeting of the American Society of Hematology.
In the LEGEND-2 phase 1/2 open study of 57 patients with advanced relapsed/refractory multiple myeloma treated with the investigational CAR T therapy, the overall response rate was 88% and the complete response rate was 74%. Among 42 patients who achieved complete response, 39 (68%) were negative for minimal residual disease (MRD).
With a median follow-up of 12 months, the median duration of response was 16 months and progression-free survival was 15 months. But in patients who achieved MRD-negative complete response, the median progression-free survival was extended to 24 months.
Pyrexia and cytokine release syndrome were reported in 90% or more of patients. Thrombocytopenia and leukopenia were reported in nearly half of patients.
The phase 1 study was conducted by researchers from the Second Affiliated Hospital of Xi’an Jiaotong University in Xi’an, China. The B-cell maturation antigen (BCMA)–directed CAR T-cell therapy is being jointly developed by Nanjing Legend Biotech and Janssen. A phase 2 study is currently being planned in China for LCAR-B38M. In parallel, Janssen and Legend are enrolling patients in a phase 1b/2 trial of the agent (also known as JNJ-68284528) in the United States.
The therapy joins a growing field of anti-BCMA CAR T-cell agents with promising initial trial results, including bb2121.
In a video interview at ASH, Sen Zhuang, MD, PhD, vice president of oncology clinical development at Janssen Research & Development, said this class of CAR T agents offers the potential for “very long remissions” and possibly even a “cure” for myeloma.
The LEGEND-2 study is sponsored by Nanjing Legend Biotech and two of the investigators reported employment with the company.
SAN DIEGO – The chimeric antigen receptor (CAR) T-cell therapy LCAR-B38M is in the race for approval in multiple myeloma following encouraging phase 1 results reported at the annual meeting of the American Society of Hematology.
In the LEGEND-2 phase 1/2 open study of 57 patients with advanced relapsed/refractory multiple myeloma treated with the investigational CAR T therapy, the overall response rate was 88% and the complete response rate was 74%. Among 42 patients who achieved complete response, 39 (68%) were negative for minimal residual disease (MRD).
With a median follow-up of 12 months, the median duration of response was 16 months and progression-free survival was 15 months. But in patients who achieved MRD-negative complete response, the median progression-free survival was extended to 24 months.
Pyrexia and cytokine release syndrome were reported in 90% or more of patients. Thrombocytopenia and leukopenia were reported in nearly half of patients.
The phase 1 study was conducted by researchers from the Second Affiliated Hospital of Xi’an Jiaotong University in Xi’an, China. The B-cell maturation antigen (BCMA)–directed CAR T-cell therapy is being jointly developed by Nanjing Legend Biotech and Janssen. A phase 2 study is currently being planned in China for LCAR-B38M. In parallel, Janssen and Legend are enrolling patients in a phase 1b/2 trial of the agent (also known as JNJ-68284528) in the United States.
The therapy joins a growing field of anti-BCMA CAR T-cell agents with promising initial trial results, including bb2121.
In a video interview at ASH, Sen Zhuang, MD, PhD, vice president of oncology clinical development at Janssen Research & Development, said this class of CAR T agents offers the potential for “very long remissions” and possibly even a “cure” for myeloma.
The LEGEND-2 study is sponsored by Nanjing Legend Biotech and two of the investigators reported employment with the company.
SAN DIEGO – The chimeric antigen receptor (CAR) T-cell therapy LCAR-B38M is in the race for approval in multiple myeloma following encouraging phase 1 results reported at the annual meeting of the American Society of Hematology.
In the LEGEND-2 phase 1/2 open study of 57 patients with advanced relapsed/refractory multiple myeloma treated with the investigational CAR T therapy, the overall response rate was 88% and the complete response rate was 74%. Among 42 patients who achieved complete response, 39 (68%) were negative for minimal residual disease (MRD).
With a median follow-up of 12 months, the median duration of response was 16 months and progression-free survival was 15 months. But in patients who achieved MRD-negative complete response, the median progression-free survival was extended to 24 months.
Pyrexia and cytokine release syndrome were reported in 90% or more of patients. Thrombocytopenia and leukopenia were reported in nearly half of patients.
The phase 1 study was conducted by researchers from the Second Affiliated Hospital of Xi’an Jiaotong University in Xi’an, China. The B-cell maturation antigen (BCMA)–directed CAR T-cell therapy is being jointly developed by Nanjing Legend Biotech and Janssen. A phase 2 study is currently being planned in China for LCAR-B38M. In parallel, Janssen and Legend are enrolling patients in a phase 1b/2 trial of the agent (also known as JNJ-68284528) in the United States.
The therapy joins a growing field of anti-BCMA CAR T-cell agents with promising initial trial results, including bb2121.
In a video interview at ASH, Sen Zhuang, MD, PhD, vice president of oncology clinical development at Janssen Research & Development, said this class of CAR T agents offers the potential for “very long remissions” and possibly even a “cure” for myeloma.
The LEGEND-2 study is sponsored by Nanjing Legend Biotech and two of the investigators reported employment with the company.
REPORTING FROM ASH 2018
Key clinical point:
Major finding: The complete response rate was 74% with median progression-free survival of 15 months.
Study details: A phase 1/2 study of 57 patients with advanced relapsed/refractory multiple myeloma.
Disclosures: The study is sponsored by Nanjing Legend Biotech. Two of the investigators reported employment with the company.

2018: A banner year for hematology drug approvals
SAN DIEGO – It was banner year for new hematology drug approvals, according to R. Angelo de Claro, MD, of the Food and Drug Administration.
, including 12 first-time approvals, 5 new biosimilars, and 15 new indications for previously approved drugs, Dr. de Claro, clinical team leader in the FDA’s division of hematology products in Silver Spring, Md., said during an overview of the approvals at the annual meeting of the American Society of Hematology.
These include six new approvals for first-line treatment, and eight for pediatric indications, he said.
Highlights were discussed at two ASH-FDA joint symposia at the meeting, including one focused on the malignant hematology approvals, and another on the nonmalignant hematology approvals. In a video interview, Dr. de Claro provides some additional insight into their importance and about what might lie ahead.
“I think what’s exciting is that you have drug development occurring in more common conditions such as chronic lymphocytic leukemia, as well as in rare conditions, including hairy cell leukemia – and the first-ever approval in hemophagocytic lymphohistiocytosis,” he said. “It’s been very busy at the FDA; stay tuned ... the year’s not done yet. There could be more coming and we certainly anticipate more applications in the future.”
Dr. de Claro is an FDA employee. He reported having no other relevant disclosures.
SAN DIEGO – It was banner year for new hematology drug approvals, according to R. Angelo de Claro, MD, of the Food and Drug Administration.
, including 12 first-time approvals, 5 new biosimilars, and 15 new indications for previously approved drugs, Dr. de Claro, clinical team leader in the FDA’s division of hematology products in Silver Spring, Md., said during an overview of the approvals at the annual meeting of the American Society of Hematology.
These include six new approvals for first-line treatment, and eight for pediatric indications, he said.
Highlights were discussed at two ASH-FDA joint symposia at the meeting, including one focused on the malignant hematology approvals, and another on the nonmalignant hematology approvals. In a video interview, Dr. de Claro provides some additional insight into their importance and about what might lie ahead.
“I think what’s exciting is that you have drug development occurring in more common conditions such as chronic lymphocytic leukemia, as well as in rare conditions, including hairy cell leukemia – and the first-ever approval in hemophagocytic lymphohistiocytosis,” he said. “It’s been very busy at the FDA; stay tuned ... the year’s not done yet. There could be more coming and we certainly anticipate more applications in the future.”
Dr. de Claro is an FDA employee. He reported having no other relevant disclosures.
SAN DIEGO – It was banner year for new hematology drug approvals, according to R. Angelo de Claro, MD, of the Food and Drug Administration.
, including 12 first-time approvals, 5 new biosimilars, and 15 new indications for previously approved drugs, Dr. de Claro, clinical team leader in the FDA’s division of hematology products in Silver Spring, Md., said during an overview of the approvals at the annual meeting of the American Society of Hematology.
These include six new approvals for first-line treatment, and eight for pediatric indications, he said.
Highlights were discussed at two ASH-FDA joint symposia at the meeting, including one focused on the malignant hematology approvals, and another on the nonmalignant hematology approvals. In a video interview, Dr. de Claro provides some additional insight into their importance and about what might lie ahead.
“I think what’s exciting is that you have drug development occurring in more common conditions such as chronic lymphocytic leukemia, as well as in rare conditions, including hairy cell leukemia – and the first-ever approval in hemophagocytic lymphohistiocytosis,” he said. “It’s been very busy at the FDA; stay tuned ... the year’s not done yet. There could be more coming and we certainly anticipate more applications in the future.”
Dr. de Claro is an FDA employee. He reported having no other relevant disclosures.
REPORTING FROM ASH 2018

Phase 3 study confirms biosimilarity of PF-05280586 with rituximab
SAN DIEGO – The potential rituximab biosimilar drug PF-05280586 showed efficacy, safety, immunogenicity, pharmacokinetics, and pharmacodynamics similar to those of rituximab at up to 26 weeks in a randomized phase 3 study of treatment-naive patients with CD20-positive low tumor burden follicular lymphoma (LTB-FL).

The primary endpoint of overall response rate at 26 weeks was 75.5% in 196 patients randomized to receive PF-05280586, and 70.7% in 198 patients who received a rituximab reference product sourced from the European Union (MabThera; rituximab‑EU), Jeff Sharman, MD, reported at the annual meeting of the American Society of Hematology.
“This resulted in a difference between the two arms of 4.66%,” said Dr. Sharman of Willamette Valley Cancer Institute and Research Center, Springfield, Ore.
The 95% confidence interval for this difference ... was entirely contained within the prespecified equivalence margin, he said.
“Depth of response was a key secondary endpoint, and rates of complete response were 29.3% and 30.4%, respectively,” he said, noting that rates of partial response, stable response, and progressive disease were also similar between the two study arms.
Estimated 1-year progression-free survival (PFS) rates were also highly similar at 76.4% and 81.2% in the PF-05280586 and rituximab-EU arms.
Rapid depletion in CD19-positive B-cell counts was observed in both groups after initial dosing, with recovery by week 39 and a sustained increase until the end of week 52.
Treatment-emergent adverse events (TEAEs) occurred in 78.6% vs. 72.1% of patients in the PF‑05280586 vs. rituximab‑EU arms, respectively, and the rates of serious adverse events and grade 3 events were similar in the groups, as were rates of infusion interruptions or infusion-related reactions (IRRs), Dr. Sharman said.
IRRs occurred in about 25% of patients in each arm, and most were grade 1 or 2. Grade 3 IRRs occurred in 2.6% vs. 0.5% of patients in the groups, respectively, and no grade 4 IRRs occurred.
Rates of anti-drug antibodies were also similar in the two groups, as were serum drug concentrations – regardless of anti-drug antibody status, he noted.
Study subjects were adults with a mean age of 60 years and histologically confirmed CD20-positive grade 1-3a follicular lymphoma with no prior rituximab or system therapy for B-cell non-Hodgkin lymphoma (NHL). They had Ann Arbor disease stages II (26.9%), III (44.2%) or IV (28.9%), ECOG performance status of 0-1, and at least 1 measurable disease lesion identifiable on imaging.
Risk level as assessed by the Follicular Lymphoma International Prognostic Index–2 was low in 28.4%, medium in 66%, and high in 5.6% of patients.
Treatment with each agent was given at intravenous doses of 375 mg/m2 weekly for 4 weeks at days 1, 8, 15, and 22.
PF-05280586 is being developed by Pfizer, and in this 52-week double-blind study – the largest study to date of the early use of the potential rituximab biosimilar in patients with previously untreated CD20-positive LTB-FL – the primary endpoint was met, demonstrating its therapeutic equivalence with rituximab-EU for overall response rate at week 26, Dr. Sharman said.
“These results therefore confirm the biosimilarity of PF-05280586 with rituximab-EU,” he concluded.
Of note, the reporting of these findings comes on the heels of the first Food and Drug Administration approval of a biosimilar rituximab product for the treatment of NHL; Celltrion’s product Truxima (formerly CT-P10), a biosimilar of Genentech’s Rituxan (rituximab), was approved Nov. 28 to treat adults with CD20-positive, B-cell NHL, either as a single agent or in combination with chemotherapy.
The PF-0528056 study was sponsored by Pfizer. Dr. Sharman has been a consultant for, and/or received research funding and honoraria from Acerta, Pharmacyclics (an AbbVie Company), Pfizer, TG Therapeutics, Abbvie, and Genentech.
SOURCE: Sharman J et al. ASH 2018: Abstract 394.
SAN DIEGO – The potential rituximab biosimilar drug PF-05280586 showed efficacy, safety, immunogenicity, pharmacokinetics, and pharmacodynamics similar to those of rituximab at up to 26 weeks in a randomized phase 3 study of treatment-naive patients with CD20-positive low tumor burden follicular lymphoma (LTB-FL).
The primary endpoint of overall response rate at 26 weeks was 75.5% in 196 patients randomized to receive PF-05280586, and 70.7% in 198 patients who received a rituximab reference product sourced from the European Union (MabThera; rituximab‑EU), Jeff Sharman, MD, reported at the annual meeting of the American Society of Hematology.
“This resulted in a difference between the two arms of 4.66%,” said Dr. Sharman of Willamette Valley Cancer Institute and Research Center, Springfield, Ore.
The 95% confidence interval for this difference ... was entirely contained within the prespecified equivalence margin, he said.
“Depth of response was a key secondary endpoint, and rates of complete response were 29.3% and 30.4%, respectively,” he said, noting that rates of partial response, stable response, and progressive disease were also similar between the two study arms.
Estimated 1-year progression-free survival (PFS) rates were also highly similar at 76.4% and 81.2% in the PF-05280586 and rituximab-EU arms.
Rapid depletion in CD19-positive B-cell counts was observed in both groups after initial dosing, with recovery by week 39 and a sustained increase until the end of week 52.
Treatment-emergent adverse events (TEAEs) occurred in 78.6% vs. 72.1% of patients in the PF‑05280586 vs. rituximab‑EU arms, respectively, and the rates of serious adverse events and grade 3 events were similar in the groups, as were rates of infusion interruptions or infusion-related reactions (IRRs), Dr. Sharman said.
IRRs occurred in about 25% of patients in each arm, and most were grade 1 or 2. Grade 3 IRRs occurred in 2.6% vs. 0.5% of patients in the groups, respectively, and no grade 4 IRRs occurred.
Rates of anti-drug antibodies were also similar in the two groups, as were serum drug concentrations – regardless of anti-drug antibody status, he noted.
Study subjects were adults with a mean age of 60 years and histologically confirmed CD20-positive grade 1-3a follicular lymphoma with no prior rituximab or system therapy for B-cell non-Hodgkin lymphoma (NHL). They had Ann Arbor disease stages II (26.9%), III (44.2%) or IV (28.9%), ECOG performance status of 0-1, and at least 1 measurable disease lesion identifiable on imaging.
Risk level as assessed by the Follicular Lymphoma International Prognostic Index–2 was low in 28.4%, medium in 66%, and high in 5.6% of patients.
Treatment with each agent was given at intravenous doses of 375 mg/m2 weekly for 4 weeks at days 1, 8, 15, and 22.
PF-05280586 is being developed by Pfizer, and in this 52-week double-blind study – the largest study to date of the early use of the potential rituximab biosimilar in patients with previously untreated CD20-positive LTB-FL – the primary endpoint was met, demonstrating its therapeutic equivalence with rituximab-EU for overall response rate at week 26, Dr. Sharman said.
“These results therefore confirm the biosimilarity of PF-05280586 with rituximab-EU,” he concluded.
Of note, the reporting of these findings comes on the heels of the first Food and Drug Administration approval of a biosimilar rituximab product for the treatment of NHL; Celltrion’s product Truxima (formerly CT-P10), a biosimilar of Genentech’s Rituxan (rituximab), was approved Nov. 28 to treat adults with CD20-positive, B-cell NHL, either as a single agent or in combination with chemotherapy.
The PF-0528056 study was sponsored by Pfizer. Dr. Sharman has been a consultant for, and/or received research funding and honoraria from Acerta, Pharmacyclics (an AbbVie Company), Pfizer, TG Therapeutics, Abbvie, and Genentech.
SOURCE: Sharman J et al. ASH 2018: Abstract 394.
SAN DIEGO – The potential rituximab biosimilar drug PF-05280586 showed efficacy, safety, immunogenicity, pharmacokinetics, and pharmacodynamics similar to those of rituximab at up to 26 weeks in a randomized phase 3 study of treatment-naive patients with CD20-positive low tumor burden follicular lymphoma (LTB-FL).
The primary endpoint of overall response rate at 26 weeks was 75.5% in 196 patients randomized to receive PF-05280586, and 70.7% in 198 patients who received a rituximab reference product sourced from the European Union (MabThera; rituximab‑EU), Jeff Sharman, MD, reported at the annual meeting of the American Society of Hematology.
“This resulted in a difference between the two arms of 4.66%,” said Dr. Sharman of Willamette Valley Cancer Institute and Research Center, Springfield, Ore.
The 95% confidence interval for this difference ... was entirely contained within the prespecified equivalence margin, he said.
“Depth of response was a key secondary endpoint, and rates of complete response were 29.3% and 30.4%, respectively,” he said, noting that rates of partial response, stable response, and progressive disease were also similar between the two study arms.
Estimated 1-year progression-free survival (PFS) rates were also highly similar at 76.4% and 81.2% in the PF-05280586 and rituximab-EU arms.
Rapid depletion in CD19-positive B-cell counts was observed in both groups after initial dosing, with recovery by week 39 and a sustained increase until the end of week 52.
Treatment-emergent adverse events (TEAEs) occurred in 78.6% vs. 72.1% of patients in the PF‑05280586 vs. rituximab‑EU arms, respectively, and the rates of serious adverse events and grade 3 events were similar in the groups, as were rates of infusion interruptions or infusion-related reactions (IRRs), Dr. Sharman said.
IRRs occurred in about 25% of patients in each arm, and most were grade 1 or 2. Grade 3 IRRs occurred in 2.6% vs. 0.5% of patients in the groups, respectively, and no grade 4 IRRs occurred.
Rates of anti-drug antibodies were also similar in the two groups, as were serum drug concentrations – regardless of anti-drug antibody status, he noted.
Study subjects were adults with a mean age of 60 years and histologically confirmed CD20-positive grade 1-3a follicular lymphoma with no prior rituximab or system therapy for B-cell non-Hodgkin lymphoma (NHL). They had Ann Arbor disease stages II (26.9%), III (44.2%) or IV (28.9%), ECOG performance status of 0-1, and at least 1 measurable disease lesion identifiable on imaging.
Risk level as assessed by the Follicular Lymphoma International Prognostic Index–2 was low in 28.4%, medium in 66%, and high in 5.6% of patients.
Treatment with each agent was given at intravenous doses of 375 mg/m2 weekly for 4 weeks at days 1, 8, 15, and 22.
PF-05280586 is being developed by Pfizer, and in this 52-week double-blind study – the largest study to date of the early use of the potential rituximab biosimilar in patients with previously untreated CD20-positive LTB-FL – the primary endpoint was met, demonstrating its therapeutic equivalence with rituximab-EU for overall response rate at week 26, Dr. Sharman said.
“These results therefore confirm the biosimilarity of PF-05280586 with rituximab-EU,” he concluded.
Of note, the reporting of these findings comes on the heels of the first Food and Drug Administration approval of a biosimilar rituximab product for the treatment of NHL; Celltrion’s product Truxima (formerly CT-P10), a biosimilar of Genentech’s Rituxan (rituximab), was approved Nov. 28 to treat adults with CD20-positive, B-cell NHL, either as a single agent or in combination with chemotherapy.
The PF-0528056 study was sponsored by Pfizer. Dr. Sharman has been a consultant for, and/or received research funding and honoraria from Acerta, Pharmacyclics (an AbbVie Company), Pfizer, TG Therapeutics, Abbvie, and Genentech.
SOURCE: Sharman J et al. ASH 2018: Abstract 394.
REPORTING FROM ASH 2018
Key clinical point: PF-05280586 shows biosimilarity to rituximab at up to 26 weeks.
Major finding: ORR at 26 weeks was 75.5% vs. 70.7% with PF-05280586 vs. rituximab, respectively.
Study details: A phase 3 study of 394 patients.
Disclosures: This study was sponsored by Pfizer. Dr. Sharman has been a consultant for, and/or received research funding and honoraria from Acerta, Pharmacyclics (an AbbVie Company), Pfizer, TG Therapeutics, Abbvie, and Genentech.
Source: Sharman J et al. ASH 2018: Abstract 394.
CLL resistance mechanism to venetoclax identified
SAN DIEGO – A recurrent mutation in BCL2, the therapeutic target of venetoclax (Venclexta), appears to be a major contributor to drug resistance in patients with chronic lymphocytic leukemia (CLL), investigators reported.
The mutation has been detected in some patients with CLL up to 2 years before resistance to venetoclax actually develops, said lead author Piers Blombery, MBBS, from the Peter MacCallum Cancer Center in Melbourne.
“We have identified the first acquired BCL2 mutation developed in patients clinically treated with venetoclax,” he said in a late-breaking oral abstract session at the annual meeting of the American Society of Hematology.
The mutation, which the investigators have labeled BCL2 Gly101Val, “is a recurrent and frequent mediator of resistance and may be detected years before clinical relapse occurs,” he added.
The paper was published online in Cancer Discovery (2018 Dec 4. doi: 10.1158/2159-8290.CD-18-1119) to coincide with the presentation at ASH.
Despite the demonstrated efficacy of venetoclax as continuous therapy in patients with relapsed or refractory CLL, the majority of patients experience disease progression, prompting the investigators to explore molecular mechanisms of secondary resistance.
To do this, they analyzed paired samples from 15 patients with CLL, enrolled in clinical trials of venetoclax, collected both before the start of venetoclax therapy and at the time of disease progression.
In seven of the patients, they identified a novel mutation that showed up at the time of progression, but was absent from the pre-venetoclax samples. The mutation first became detectable from about 19 to 42 months after the start of therapy and preceded clinical progression by as much as 25 months, the investigators found.
They pinned the mutation down to the BH3-binding groove on BCL2, the same molecular site targeted by venetoclax. They found that the mutation was not present in samples from 96 patients with venetoclax-naive CLL nor in any other B-cell malignancies. Searches for references to the mutation in both a cancer database (COSMIC) and a population database (gnomAD) came up empty.
In other experiments, they determined that cell lines overexpressing BCL2 Gly101Val are resistant to venetoclax, and that in the presence of venetoclax in vitro, BCL2 Gly101Val-expressing cells have a growth advantage, compared with wild type cells.
Additionally, they showed that the mutation results in impaired venetoclax binding in vitro.
“BCL2 Gly101Val is observed subclonally, implicating multiple mechanisms of venetoclax resistance in the same patient,” Dr. Blombery said.
In an interview, Dr. Blombery said that the identification of the resistance mutation is a strong rationale for using combination therapy to treat patients with relapsed or refractory CLL to help prevent or attenuate selection pressures that lead to resistance.
The investigators were supported by the Wilson Center for Lymphoma Genomics, Snowdome Foundation, National Health Medical Research Council, Leukemia and Lymphoma Society, Leukemia Foundation, Cancer Council of Victoria, and Australian Cancer Research Foundation. Dr. Blombery reported having no relevant disclosures.
SOURCE: Blombery P et al. ASH 2018, Abstract LBA-7.
SAN DIEGO – A recurrent mutation in BCL2, the therapeutic target of venetoclax (Venclexta), appears to be a major contributor to drug resistance in patients with chronic lymphocytic leukemia (CLL), investigators reported.
The mutation has been detected in some patients with CLL up to 2 years before resistance to venetoclax actually develops, said lead author Piers Blombery, MBBS, from the Peter MacCallum Cancer Center in Melbourne.
“We have identified the first acquired BCL2 mutation developed in patients clinically treated with venetoclax,” he said in a late-breaking oral abstract session at the annual meeting of the American Society of Hematology.
The mutation, which the investigators have labeled BCL2 Gly101Val, “is a recurrent and frequent mediator of resistance and may be detected years before clinical relapse occurs,” he added.
The paper was published online in Cancer Discovery (2018 Dec 4. doi: 10.1158/2159-8290.CD-18-1119) to coincide with the presentation at ASH.
Despite the demonstrated efficacy of venetoclax as continuous therapy in patients with relapsed or refractory CLL, the majority of patients experience disease progression, prompting the investigators to explore molecular mechanisms of secondary resistance.
To do this, they analyzed paired samples from 15 patients with CLL, enrolled in clinical trials of venetoclax, collected both before the start of venetoclax therapy and at the time of disease progression.
In seven of the patients, they identified a novel mutation that showed up at the time of progression, but was absent from the pre-venetoclax samples. The mutation first became detectable from about 19 to 42 months after the start of therapy and preceded clinical progression by as much as 25 months, the investigators found.
They pinned the mutation down to the BH3-binding groove on BCL2, the same molecular site targeted by venetoclax. They found that the mutation was not present in samples from 96 patients with venetoclax-naive CLL nor in any other B-cell malignancies. Searches for references to the mutation in both a cancer database (COSMIC) and a population database (gnomAD) came up empty.
In other experiments, they determined that cell lines overexpressing BCL2 Gly101Val are resistant to venetoclax, and that in the presence of venetoclax in vitro, BCL2 Gly101Val-expressing cells have a growth advantage, compared with wild type cells.
Additionally, they showed that the mutation results in impaired venetoclax binding in vitro.
“BCL2 Gly101Val is observed subclonally, implicating multiple mechanisms of venetoclax resistance in the same patient,” Dr. Blombery said.
In an interview, Dr. Blombery said that the identification of the resistance mutation is a strong rationale for using combination therapy to treat patients with relapsed or refractory CLL to help prevent or attenuate selection pressures that lead to resistance.
The investigators were supported by the Wilson Center for Lymphoma Genomics, Snowdome Foundation, National Health Medical Research Council, Leukemia and Lymphoma Society, Leukemia Foundation, Cancer Council of Victoria, and Australian Cancer Research Foundation. Dr. Blombery reported having no relevant disclosures.
SOURCE: Blombery P et al. ASH 2018, Abstract LBA-7.
SAN DIEGO – A recurrent mutation in BCL2, the therapeutic target of venetoclax (Venclexta), appears to be a major contributor to drug resistance in patients with chronic lymphocytic leukemia (CLL), investigators reported.
The mutation has been detected in some patients with CLL up to 2 years before resistance to venetoclax actually develops, said lead author Piers Blombery, MBBS, from the Peter MacCallum Cancer Center in Melbourne.
“We have identified the first acquired BCL2 mutation developed in patients clinically treated with venetoclax,” he said in a late-breaking oral abstract session at the annual meeting of the American Society of Hematology.
The mutation, which the investigators have labeled BCL2 Gly101Val, “is a recurrent and frequent mediator of resistance and may be detected years before clinical relapse occurs,” he added.
The paper was published online in Cancer Discovery (2018 Dec 4. doi: 10.1158/2159-8290.CD-18-1119) to coincide with the presentation at ASH.
Despite the demonstrated efficacy of venetoclax as continuous therapy in patients with relapsed or refractory CLL, the majority of patients experience disease progression, prompting the investigators to explore molecular mechanisms of secondary resistance.
To do this, they analyzed paired samples from 15 patients with CLL, enrolled in clinical trials of venetoclax, collected both before the start of venetoclax therapy and at the time of disease progression.
In seven of the patients, they identified a novel mutation that showed up at the time of progression, but was absent from the pre-venetoclax samples. The mutation first became detectable from about 19 to 42 months after the start of therapy and preceded clinical progression by as much as 25 months, the investigators found.
They pinned the mutation down to the BH3-binding groove on BCL2, the same molecular site targeted by venetoclax. They found that the mutation was not present in samples from 96 patients with venetoclax-naive CLL nor in any other B-cell malignancies. Searches for references to the mutation in both a cancer database (COSMIC) and a population database (gnomAD) came up empty.
In other experiments, they determined that cell lines overexpressing BCL2 Gly101Val are resistant to venetoclax, and that in the presence of venetoclax in vitro, BCL2 Gly101Val-expressing cells have a growth advantage, compared with wild type cells.
Additionally, they showed that the mutation results in impaired venetoclax binding in vitro.
“BCL2 Gly101Val is observed subclonally, implicating multiple mechanisms of venetoclax resistance in the same patient,” Dr. Blombery said.
In an interview, Dr. Blombery said that the identification of the resistance mutation is a strong rationale for using combination therapy to treat patients with relapsed or refractory CLL to help prevent or attenuate selection pressures that lead to resistance.
The investigators were supported by the Wilson Center for Lymphoma Genomics, Snowdome Foundation, National Health Medical Research Council, Leukemia and Lymphoma Society, Leukemia Foundation, Cancer Council of Victoria, and Australian Cancer Research Foundation. Dr. Blombery reported having no relevant disclosures.
SOURCE: Blombery P et al. ASH 2018, Abstract LBA-7.
REPORTING FROM ASH 2018
Key clinical point:
Major finding: The mutation was identified in samples from seven patients after venetoclax therapy, but not in any of the pretherapy samples.
Study details: Genetic analysis of CLL mutations in 15 patients enrolled in clinical trials of venetoclax.
Disclosures: The investigators were supported by the Wilson Center for Lymphoma Genomics, Snowdome Foundation, National Health Medical Research Council, Leukemia and Lymphoma Society, Leukemia Foundation, Cancer Council of Victoria, and Australian Cancer Research Foundation. Dr. Blombery reported having no relevant disclosures.
Source: Blombery P et al. ASH 2018, Abstract LBA-7.

In IDH-mutated AML, first-line IDH inhibitors plus chemo may boost remission
SAN DIEGO – In patients with newly diagnosed acute myeloid leukemia (AML) bearing IDH1 or IDH2 mutations, combinations of either ivosidenib (Tibsovo, for IDH1) or enasidenib (Idhifa, for IDH2) with standard induction and consolidation regimens are safe and well tolerated and are associated with encouraging remission rates, results of a phase 1 trial indicate.

In the open-label, phase 1 trial, ivosidenib plus chemotherapy was associated with elimination of minimal residual disease (MRD) by flow cytometry in 88% of treated patients and with IDH1-mutation clearance in 41% of patients.
Enasidenib plus chemotherapy was associated with elimination of MRD in 45% of patients and with IDH2-mutation clearance in 25% of patients, said Eytan M. Stein, MD, from Memorial Sloan Kettering Cancer Center in New York.
“The overall survival rates are robust, with greater than 75% 1-year survival in both ivosidenib- and enasidenib-treated patients,” he said at the annual meeting of the American Society of Hematology.
Both ivosidenib and enasidenib are approved in the United States for treatment of patients with relapsed or refractory AML bearing either IDH1 or IDH2 mutations, respectively. In this trial, the investigators explored the therapeutic potential of the IDH inhibitors in patients with previously untreated disease.
Dr. Stein and his colleagues investigated combining each of the agents with standard induction therapy with either daunorubicin 60 mg/m2 per day or idarubicin 12 mg/m2 per day for 3 days, plus cytarabine 200 mg/m2 per day for 7 days. Patients with IDH1 mutations received ivosidenib 500 mg once daily, and those with IDH2 mutations received enasidenib 100 mg once daily.
After induction, patients with a complete remission (CR), CR with incomplete recovery of hematologic counts (CRi), or CR with incomplete recovery of platelets (CRp) could receive up to four cycles of consolidation therapy while continuing the IDH inhibitor. Patients who completed consolidation or were ineligible for consolidation could continue on maintenance therapy with their assigned drug until the end of the study.
The drugs were discontinued in patients who went on to hematopoietic stem cell transplant.
The most frequent co-occurring baseline mutations were DNMT3A, NPM1, and NRAS for patients with IDH1 mutations, and DNMT3A, SRSF2, and ASXL1 for patients with IDH2 mutations.
A total of 60 patients were assigned to ivosidenib and chemotherapy and 93 were assigned to enasidenib/chemotherapy. The median patient age was about 63 years in each arm.
All patients in each arm received a least one induction dose and about 48% in each arm received at least some consolidation dosing. In all, 18% of patients in the ivosidenib arm and 19% in the enasidenib arm went on to maintenance.
Treatment discontinuations occurred in 55% of patients in the ivosidenib group and 84% in the enasidenib group. The primary reason for discontinuation included HSCT, adverse events, progressive disease, and death (one and four patients in the respective arms).
Adverse events of interest, regardless of attribution, included the IDH differentiation syndrome in two patients on ivosidenib and one on enasidenib, leukocytosis, QT interval prolongation, and increased blood bilirubin.
The 30-day and 60-day mortality rates were 5% and 8% in the ivosidenib arm and 5% and 9% in the enasidenib arm, respectively.
Best overall response rates (CR+CRi+CRp) among all patients were 80% in the ivosidenib arm and 72% in the enasidenib arm. In each trial arm, the response rates were higher among patients with de novo AML, compared with secondary AML.
Of 12 ivosidenib-treated patients who had IDH1-mutation clearance, 10 had clearance at the end of induction therapy, and 2 achieved clearance during or after consolidation. Of 15 ivosidenib-treated patients who became MRD negative, 12 had achieved it by the end of induction, and 3 became MRD negative during consolidation.
In the enasidenib arm, 15 patients had IDH2 mutation clearance (11 after induction, 4 during consolidation) and 9 became MRD negative (7 after induction and 2 during or after consolidation).
The probability of surviving to 1 year after the start of induction among ivosidenib-treated patients was 79%; the median overall survival had not been reached and was not estimable at the time of data cutoff. The probability of surviving to 1 year among patients in the enasidenib arm was 75%. In this group, too, median overall survival had not been reached.
The clinical benefit of adding either IDH inhibitor to induction, consolidation, and maintenance therapy for patients with newly diagnosed AML with IDH mutations will be further evaluated in a randomized, phase 3 trial, Dr. Stein said.
The study was funded by Agios Pharmaceuticals and Celgene. Dr. Stein reported consulting with those companies and others.
SOURCE: Stein EM et al. ASH 2018, Abstract 560.
SAN DIEGO – In patients with newly diagnosed acute myeloid leukemia (AML) bearing IDH1 or IDH2 mutations, combinations of either ivosidenib (Tibsovo, for IDH1) or enasidenib (Idhifa, for IDH2) with standard induction and consolidation regimens are safe and well tolerated and are associated with encouraging remission rates, results of a phase 1 trial indicate.
In the open-label, phase 1 trial, ivosidenib plus chemotherapy was associated with elimination of minimal residual disease (MRD) by flow cytometry in 88% of treated patients and with IDH1-mutation clearance in 41% of patients.
Enasidenib plus chemotherapy was associated with elimination of MRD in 45% of patients and with IDH2-mutation clearance in 25% of patients, said Eytan M. Stein, MD, from Memorial Sloan Kettering Cancer Center in New York.
“The overall survival rates are robust, with greater than 75% 1-year survival in both ivosidenib- and enasidenib-treated patients,” he said at the annual meeting of the American Society of Hematology.
Both ivosidenib and enasidenib are approved in the United States for treatment of patients with relapsed or refractory AML bearing either IDH1 or IDH2 mutations, respectively. In this trial, the investigators explored the therapeutic potential of the IDH inhibitors in patients with previously untreated disease.
Dr. Stein and his colleagues investigated combining each of the agents with standard induction therapy with either daunorubicin 60 mg/m2 per day or idarubicin 12 mg/m2 per day for 3 days, plus cytarabine 200 mg/m2 per day for 7 days. Patients with IDH1 mutations received ivosidenib 500 mg once daily, and those with IDH2 mutations received enasidenib 100 mg once daily.
After induction, patients with a complete remission (CR), CR with incomplete recovery of hematologic counts (CRi), or CR with incomplete recovery of platelets (CRp) could receive up to four cycles of consolidation therapy while continuing the IDH inhibitor. Patients who completed consolidation or were ineligible for consolidation could continue on maintenance therapy with their assigned drug until the end of the study.
The drugs were discontinued in patients who went on to hematopoietic stem cell transplant.
The most frequent co-occurring baseline mutations were DNMT3A, NPM1, and NRAS for patients with IDH1 mutations, and DNMT3A, SRSF2, and ASXL1 for patients with IDH2 mutations.
A total of 60 patients were assigned to ivosidenib and chemotherapy and 93 were assigned to enasidenib/chemotherapy. The median patient age was about 63 years in each arm.
All patients in each arm received a least one induction dose and about 48% in each arm received at least some consolidation dosing. In all, 18% of patients in the ivosidenib arm and 19% in the enasidenib arm went on to maintenance.
Treatment discontinuations occurred in 55% of patients in the ivosidenib group and 84% in the enasidenib group. The primary reason for discontinuation included HSCT, adverse events, progressive disease, and death (one and four patients in the respective arms).
Adverse events of interest, regardless of attribution, included the IDH differentiation syndrome in two patients on ivosidenib and one on enasidenib, leukocytosis, QT interval prolongation, and increased blood bilirubin.
The 30-day and 60-day mortality rates were 5% and 8% in the ivosidenib arm and 5% and 9% in the enasidenib arm, respectively.
Best overall response rates (CR+CRi+CRp) among all patients were 80% in the ivosidenib arm and 72% in the enasidenib arm. In each trial arm, the response rates were higher among patients with de novo AML, compared with secondary AML.
Of 12 ivosidenib-treated patients who had IDH1-mutation clearance, 10 had clearance at the end of induction therapy, and 2 achieved clearance during or after consolidation. Of 15 ivosidenib-treated patients who became MRD negative, 12 had achieved it by the end of induction, and 3 became MRD negative during consolidation.
In the enasidenib arm, 15 patients had IDH2 mutation clearance (11 after induction, 4 during consolidation) and 9 became MRD negative (7 after induction and 2 during or after consolidation).
The probability of surviving to 1 year after the start of induction among ivosidenib-treated patients was 79%; the median overall survival had not been reached and was not estimable at the time of data cutoff. The probability of surviving to 1 year among patients in the enasidenib arm was 75%. In this group, too, median overall survival had not been reached.
The clinical benefit of adding either IDH inhibitor to induction, consolidation, and maintenance therapy for patients with newly diagnosed AML with IDH mutations will be further evaluated in a randomized, phase 3 trial, Dr. Stein said.
The study was funded by Agios Pharmaceuticals and Celgene. Dr. Stein reported consulting with those companies and others.
SOURCE: Stein EM et al. ASH 2018, Abstract 560.
SAN DIEGO – In patients with newly diagnosed acute myeloid leukemia (AML) bearing IDH1 or IDH2 mutations, combinations of either ivosidenib (Tibsovo, for IDH1) or enasidenib (Idhifa, for IDH2) with standard induction and consolidation regimens are safe and well tolerated and are associated with encouraging remission rates, results of a phase 1 trial indicate.
In the open-label, phase 1 trial, ivosidenib plus chemotherapy was associated with elimination of minimal residual disease (MRD) by flow cytometry in 88% of treated patients and with IDH1-mutation clearance in 41% of patients.
Enasidenib plus chemotherapy was associated with elimination of MRD in 45% of patients and with IDH2-mutation clearance in 25% of patients, said Eytan M. Stein, MD, from Memorial Sloan Kettering Cancer Center in New York.
“The overall survival rates are robust, with greater than 75% 1-year survival in both ivosidenib- and enasidenib-treated patients,” he said at the annual meeting of the American Society of Hematology.
Both ivosidenib and enasidenib are approved in the United States for treatment of patients with relapsed or refractory AML bearing either IDH1 or IDH2 mutations, respectively. In this trial, the investigators explored the therapeutic potential of the IDH inhibitors in patients with previously untreated disease.
Dr. Stein and his colleagues investigated combining each of the agents with standard induction therapy with either daunorubicin 60 mg/m2 per day or idarubicin 12 mg/m2 per day for 3 days, plus cytarabine 200 mg/m2 per day for 7 days. Patients with IDH1 mutations received ivosidenib 500 mg once daily, and those with IDH2 mutations received enasidenib 100 mg once daily.
After induction, patients with a complete remission (CR), CR with incomplete recovery of hematologic counts (CRi), or CR with incomplete recovery of platelets (CRp) could receive up to four cycles of consolidation therapy while continuing the IDH inhibitor. Patients who completed consolidation or were ineligible for consolidation could continue on maintenance therapy with their assigned drug until the end of the study.
The drugs were discontinued in patients who went on to hematopoietic stem cell transplant.
The most frequent co-occurring baseline mutations were DNMT3A, NPM1, and NRAS for patients with IDH1 mutations, and DNMT3A, SRSF2, and ASXL1 for patients with IDH2 mutations.
A total of 60 patients were assigned to ivosidenib and chemotherapy and 93 were assigned to enasidenib/chemotherapy. The median patient age was about 63 years in each arm.
All patients in each arm received a least one induction dose and about 48% in each arm received at least some consolidation dosing. In all, 18% of patients in the ivosidenib arm and 19% in the enasidenib arm went on to maintenance.
Treatment discontinuations occurred in 55% of patients in the ivosidenib group and 84% in the enasidenib group. The primary reason for discontinuation included HSCT, adverse events, progressive disease, and death (one and four patients in the respective arms).
Adverse events of interest, regardless of attribution, included the IDH differentiation syndrome in two patients on ivosidenib and one on enasidenib, leukocytosis, QT interval prolongation, and increased blood bilirubin.
The 30-day and 60-day mortality rates were 5% and 8% in the ivosidenib arm and 5% and 9% in the enasidenib arm, respectively.
Best overall response rates (CR+CRi+CRp) among all patients were 80% in the ivosidenib arm and 72% in the enasidenib arm. In each trial arm, the response rates were higher among patients with de novo AML, compared with secondary AML.
Of 12 ivosidenib-treated patients who had IDH1-mutation clearance, 10 had clearance at the end of induction therapy, and 2 achieved clearance during or after consolidation. Of 15 ivosidenib-treated patients who became MRD negative, 12 had achieved it by the end of induction, and 3 became MRD negative during consolidation.
In the enasidenib arm, 15 patients had IDH2 mutation clearance (11 after induction, 4 during consolidation) and 9 became MRD negative (7 after induction and 2 during or after consolidation).
The probability of surviving to 1 year after the start of induction among ivosidenib-treated patients was 79%; the median overall survival had not been reached and was not estimable at the time of data cutoff. The probability of surviving to 1 year among patients in the enasidenib arm was 75%. In this group, too, median overall survival had not been reached.
The clinical benefit of adding either IDH inhibitor to induction, consolidation, and maintenance therapy for patients with newly diagnosed AML with IDH mutations will be further evaluated in a randomized, phase 3 trial, Dr. Stein said.
The study was funded by Agios Pharmaceuticals and Celgene. Dr. Stein reported consulting with those companies and others.
SOURCE: Stein EM et al. ASH 2018, Abstract 560.
REPORTING FROM ASH 2018
Key clinical point: The IDH1 inhibitor ivosidenib and IDH2 inhibitor enasidenib combined with induction and consolidation therapy showed promising efficacy against newly diagnosed acute myeloid leukemia.
Major finding: The 1-year survival rates were greater than 75% for patients with previously untreated acute myeloid leukemia who received either of the IDH-mutated inhibitors.
Study details: An open-label, prospective trial in 153 patients with newly diagnosed acute myeloid leukemia.
Disclosures: The study was funded by Agios Pharmaceuticals and Celgene. Dr. Stein reported consulting with those companies and others.
Source: Stein EM et al. ASH 2018, Abstract 560.
Shorter R-CHOP regimen noninferior in certain DLBCL patients

SAN DIEGO—A shortened regimen of four cycles of rituximab (R) plus cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) chemotherapy was noninferior in efficacy to the standard six cycles of R-CHOP in younger patients with favorable-risk diffuse large B-cell lymphoma (DLBCL), according to investigators of the FLYER trial.
In addition, the truncated regimen was associated with about a one-third reduction in non-hematologic adverse events.
Viola Poeschel, MD, of Saarland University Medical School in Homburg/Saar, Germany, reported results of this study on behalf of the German High-Grade Non-Hodgkin’s Lymphoma Study Group/German Lymphoma Alliance at the 2018 ASH Annual Meeting (abstract 781).
Among 588 evaluable patients younger than 60 with favorable-prognosis DLBCL, there were no significant differences in progression-free survival (PFS), event-free survival (EFS), or overall survival (OS) between patients who received four cycles of R-CHOP and those who received six cycles, Dr. Poeschel reported.
“Six cycles of R-CHOP led to a higher toxicity with respect to leukocytopenia and anemia, both of any grade and also of grades 3 to 4, compared to four cycles of R-CHOP,” she said.
The findings suggest that, for younger patients with favorable-prognosis DLBCL—defined as an age-adjusted International Prognostic Index score of 0 and low tumor burden (less than 7.5 cm)—four cycles of R-CHOP can be a new standard of care, Dr. Poeschel said.
The investigators were prompted to look at the question of a shorter R-CHOP regimen by results of the MInT trial, in which a subpopulation of favorable-prognosis DLBCL patients had a 3-year PFS rate of 89%.
The FLYER trial (NCT00278421) was designed as a non-inferiority study to see whether, in a similar group of patients, reducing the number of R-CHOP cycles could maintain efficacy while reducing toxicity.
At a median follow-up of 66 months, the PFS rate, the primary endpoint, was 94% in the six-cycle group and 96% for the four-cycle group.
“As the lower limit of the 95% confidence interval of our experimental arm was 94%, it is shown that it is definitely non-inferior to the standard arm, six cycles of R-CHOP,” Dr. Poeschel said.
Similarly, the rate of 3-year OS was 98% in the six-cycle group, compared with 99% in the four-cycle group, and the survival curves were virtually superimposable out to more than 10 years of follow-up.
Treatment with six cycles was associated with more frequent hematologic adverse events than four cycles. Leukopenia of any grade occurred in 237 and 171 patients, respectively. Grade 3-4 leukopenia occurred in 110 and 80 patients, respectively.
Any-grade anemia occurred in 172 patients assigned to six cycles and 107 assigned to four cycles. Rates of grade 3-4 anemia were similar between the groups, as were rates of thrombocytopenia of any grade or grade 3-4.
Non-hematologic adverse events of any grade or grade 3-4 that were more frequent with six cycles included parasthesia, nausea, infection, vomiting, and mucositis.
The total number of non-hematologic adverse events was reduced by about one-third.
“We are certainly always looking for ways to make treatments easier for our patients to reduce adverse effects, and, certainly, for this subgroup of patients, it appears that we can make their treatment shorter and have less burden but equivalent efficacy,” said David Steensma, MD, of the Dana-Farber Cancer Institute/Harvard Cancer Center in Boston, Massachusetts.
Drs. Steensma and Poeschel both cautioned that the results of this study pertain only to those patients with DLBCL who are younger and have favorable-prognosis disease.
“We can’t extend it to other subtypes of large-cell lymphoma, but that’s always a laudable goal, so I think this will immediately influence clinical practice,” Dr. Steensma said.
The study was supported by Deutsche Krebshilfe. Dr. Poeschel disclosed travel grants from Roche and Amgen. Dr. Steensma had no disclosures relevant to the study.
SAN DIEGO—A shortened regimen of four cycles of rituximab (R) plus cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) chemotherapy was noninferior in efficacy to the standard six cycles of R-CHOP in younger patients with favorable-risk diffuse large B-cell lymphoma (DLBCL), according to investigators of the FLYER trial.
In addition, the truncated regimen was associated with about a one-third reduction in non-hematologic adverse events.
Viola Poeschel, MD, of Saarland University Medical School in Homburg/Saar, Germany, reported results of this study on behalf of the German High-Grade Non-Hodgkin’s Lymphoma Study Group/German Lymphoma Alliance at the 2018 ASH Annual Meeting (abstract 781).
Among 588 evaluable patients younger than 60 with favorable-prognosis DLBCL, there were no significant differences in progression-free survival (PFS), event-free survival (EFS), or overall survival (OS) between patients who received four cycles of R-CHOP and those who received six cycles, Dr. Poeschel reported.
“Six cycles of R-CHOP led to a higher toxicity with respect to leukocytopenia and anemia, both of any grade and also of grades 3 to 4, compared to four cycles of R-CHOP,” she said.
The findings suggest that, for younger patients with favorable-prognosis DLBCL—defined as an age-adjusted International Prognostic Index score of 0 and low tumor burden (less than 7.5 cm)—four cycles of R-CHOP can be a new standard of care, Dr. Poeschel said.
The investigators were prompted to look at the question of a shorter R-CHOP regimen by results of the MInT trial, in which a subpopulation of favorable-prognosis DLBCL patients had a 3-year PFS rate of 89%.
The FLYER trial (NCT00278421) was designed as a non-inferiority study to see whether, in a similar group of patients, reducing the number of R-CHOP cycles could maintain efficacy while reducing toxicity.
At a median follow-up of 66 months, the PFS rate, the primary endpoint, was 94% in the six-cycle group and 96% for the four-cycle group.
“As the lower limit of the 95% confidence interval of our experimental arm was 94%, it is shown that it is definitely non-inferior to the standard arm, six cycles of R-CHOP,” Dr. Poeschel said.
Similarly, the rate of 3-year OS was 98% in the six-cycle group, compared with 99% in the four-cycle group, and the survival curves were virtually superimposable out to more than 10 years of follow-up.
Treatment with six cycles was associated with more frequent hematologic adverse events than four cycles. Leukopenia of any grade occurred in 237 and 171 patients, respectively. Grade 3-4 leukopenia occurred in 110 and 80 patients, respectively.
Any-grade anemia occurred in 172 patients assigned to six cycles and 107 assigned to four cycles. Rates of grade 3-4 anemia were similar between the groups, as were rates of thrombocytopenia of any grade or grade 3-4.
Non-hematologic adverse events of any grade or grade 3-4 that were more frequent with six cycles included parasthesia, nausea, infection, vomiting, and mucositis.
The total number of non-hematologic adverse events was reduced by about one-third.
“We are certainly always looking for ways to make treatments easier for our patients to reduce adverse effects, and, certainly, for this subgroup of patients, it appears that we can make their treatment shorter and have less burden but equivalent efficacy,” said David Steensma, MD, of the Dana-Farber Cancer Institute/Harvard Cancer Center in Boston, Massachusetts.
Drs. Steensma and Poeschel both cautioned that the results of this study pertain only to those patients with DLBCL who are younger and have favorable-prognosis disease.
“We can’t extend it to other subtypes of large-cell lymphoma, but that’s always a laudable goal, so I think this will immediately influence clinical practice,” Dr. Steensma said.
The study was supported by Deutsche Krebshilfe. Dr. Poeschel disclosed travel grants from Roche and Amgen. Dr. Steensma had no disclosures relevant to the study.
SAN DIEGO—A shortened regimen of four cycles of rituximab (R) plus cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) chemotherapy was noninferior in efficacy to the standard six cycles of R-CHOP in younger patients with favorable-risk diffuse large B-cell lymphoma (DLBCL), according to investigators of the FLYER trial.
In addition, the truncated regimen was associated with about a one-third reduction in non-hematologic adverse events.
Viola Poeschel, MD, of Saarland University Medical School in Homburg/Saar, Germany, reported results of this study on behalf of the German High-Grade Non-Hodgkin’s Lymphoma Study Group/German Lymphoma Alliance at the 2018 ASH Annual Meeting (abstract 781).
Among 588 evaluable patients younger than 60 with favorable-prognosis DLBCL, there were no significant differences in progression-free survival (PFS), event-free survival (EFS), or overall survival (OS) between patients who received four cycles of R-CHOP and those who received six cycles, Dr. Poeschel reported.
“Six cycles of R-CHOP led to a higher toxicity with respect to leukocytopenia and anemia, both of any grade and also of grades 3 to 4, compared to four cycles of R-CHOP,” she said.
The findings suggest that, for younger patients with favorable-prognosis DLBCL—defined as an age-adjusted International Prognostic Index score of 0 and low tumor burden (less than 7.5 cm)—four cycles of R-CHOP can be a new standard of care, Dr. Poeschel said.
The investigators were prompted to look at the question of a shorter R-CHOP regimen by results of the MInT trial, in which a subpopulation of favorable-prognosis DLBCL patients had a 3-year PFS rate of 89%.
The FLYER trial (NCT00278421) was designed as a non-inferiority study to see whether, in a similar group of patients, reducing the number of R-CHOP cycles could maintain efficacy while reducing toxicity.
At a median follow-up of 66 months, the PFS rate, the primary endpoint, was 94% in the six-cycle group and 96% for the four-cycle group.
“As the lower limit of the 95% confidence interval of our experimental arm was 94%, it is shown that it is definitely non-inferior to the standard arm, six cycles of R-CHOP,” Dr. Poeschel said.
Similarly, the rate of 3-year OS was 98% in the six-cycle group, compared with 99% in the four-cycle group, and the survival curves were virtually superimposable out to more than 10 years of follow-up.
Treatment with six cycles was associated with more frequent hematologic adverse events than four cycles. Leukopenia of any grade occurred in 237 and 171 patients, respectively. Grade 3-4 leukopenia occurred in 110 and 80 patients, respectively.
Any-grade anemia occurred in 172 patients assigned to six cycles and 107 assigned to four cycles. Rates of grade 3-4 anemia were similar between the groups, as were rates of thrombocytopenia of any grade or grade 3-4.
Non-hematologic adverse events of any grade or grade 3-4 that were more frequent with six cycles included parasthesia, nausea, infection, vomiting, and mucositis.
The total number of non-hematologic adverse events was reduced by about one-third.
“We are certainly always looking for ways to make treatments easier for our patients to reduce adverse effects, and, certainly, for this subgroup of patients, it appears that we can make their treatment shorter and have less burden but equivalent efficacy,” said David Steensma, MD, of the Dana-Farber Cancer Institute/Harvard Cancer Center in Boston, Massachusetts.
Drs. Steensma and Poeschel both cautioned that the results of this study pertain only to those patients with DLBCL who are younger and have favorable-prognosis disease.
“We can’t extend it to other subtypes of large-cell lymphoma, but that’s always a laudable goal, so I think this will immediately influence clinical practice,” Dr. Steensma said.
The study was supported by Deutsche Krebshilfe. Dr. Poeschel disclosed travel grants from Roche and Amgen. Dr. Steensma had no disclosures relevant to the study.

Regimen provides survival benefit in PTCL

SAN DIEGO—A newly approved treatment regimen provides a survival benefit over standard therapy for patients with CD30-positive peripheral T-cell lymphomas (PTCLs), according to a presentation at the 2018 ASH Annual Meeting.
In the ECHELON-2 trial, patients who received brentuximab vedotin (BV) plus cyclophosphamide, doxorubicin, and prednisone (CHP) had superior progression-free survival (PFS) and overall survival (OS) compared to patients who received standard treatment with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP).
These results supported the recent U.S. approval of BV in combination with CHP for adults with previously untreated, systemic anaplastic large-cell lymphoma or other CD30-expressing PTCLs, including angioimmunoblastic T-cell lymphoma and PTCL not otherwise specified.
“ECHELON-2 is the first prospective trial in peripheral T-cell lymphoma to show an overall survival benefit over CHOP,” said Steven M. Horwitz, MD, of Memorial Sloan Kettering Cancer Center in Basking Ridge, New Jersey.
Dr. Horwitz presented results from this trial at ASH as abstract 997. Results were simultaneously published in The Lancet.
Patients and treatment
ECHELON-2 (NCT01777152) enrolled 452 patients with previously untreated, CD30-positive PTCL. Subtypes included ALK-positive (n=98) or -negative (n=218) systemic anaplastic large-cell lymphoma, PTCL not otherwise specified (n=72), angioimmunoblastic T-cell lymphoma (n=54), enteropathy-associated T-cell lymphoma (n=7), and adult T-cell leukemia/lymphoma (n=3).
Patients were randomized to receive BV-CHP plus placebo (n=226) or CHOP plus placebo (n=226) every 3 weeks for six to eight cycles.
At baseline, the median age was 58 in both the BV-CHP arm (range, 18-85) and the CHOP arm (range, 18-83). The majority of patients were male—59% in the BV-CHP arm and 67% in the CHOP arm—and most patients had stage III/IV disease—81% and 80%, respectively.
Eighty-nine percent of patients in the BV-CHP arm and 81% in the CHOP arm completed six or more cycles of their assigned treatment.
Twenty-seven percent of patients in the BV-CHP arm and 19% in the CHOP arm received consolidation consisting of radiotherapy (6% and 3%, respectively) and/or stem cell transplant (22% and 17%).
Twenty-six percent of patients in the BV-CHP arm and 42% in the CHOP arm received systemic therapy for residual or progressive disease, and 4% of patients in each arm received palliative radiation.
Efficacy
The overall response rate was 83% in the BV-CHP arm and 72% in the CHOP arm (P=0.0032). The complete response rates were 68% and 56%, respectively (P=0.0066).
At a median follow-up of 36.2 months, the median PFS was 48.2 months in the BV-CHP arm and 20.8 months in the CHOP arm. The rate of death or progression was 42% in the BV-CHP arm and 55% in the CHOP arm (hazard ratio=0.71, P=0.011).
At a median follow-up of 42.1 months, the median OS was not reached in either treatment arm. The rate of death was 23% in the BV-CHP arm and 32% in the CHOP arm (hazard ratio=0.66, P=0.0244).
Dr. Horwitz noted that this study was not powered to determine differences in OS or PFS according to PTCL subtypes.
Safety
BV-CHP had a comparable safety profile to CHOP, Dr. Horwitz said.
The rate of adverse events (AEs) was 99% in the BV-CHP arm and 98% in the CHOP arm. Grade 3 or higher AEs occurred in 66% and 65% of patients, respectively. Serious AEs occurred in 39% and 38%, respectively.
Three percent of patients in the BV-CHP arm and 4% of those in the CHOP arm had fatal AEs.
The most common AEs of any grade occurring in at least 20% of patients (in the BV-CHP and CHOP arms, respectively) were:
- Nausea (46% and 38%)
- Peripheral sensory neuropathy (45% and 41%)
- Neutropenia (38% for both)
- Diarrhea (38% and 20%)
- Constipation (29% and 30%)
- Alopecia (26% and 25%)
- Pyrexia (26% and 19%)
- Vomiting (26% and 17%)
- Fatigue (24% and 20%)
- Anemia (21% and 16%).
This research was funded by Seattle Genetics Inc. and Millennium Pharmaceuticals Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
Dr. Horwitz disclosed relationships with Seattle Genetics, Aileron Therapeutics, Innate Pharma, Millennium/Takeda, Forty Seven, Corvus, Mundipharma, ADC Therapeutics, Trillium, Celgene, Portola, Infinity/Verastem, Spectrum, and Kyowa-Hakka-Kirin.
SAN DIEGO—A newly approved treatment regimen provides a survival benefit over standard therapy for patients with CD30-positive peripheral T-cell lymphomas (PTCLs), according to a presentation at the 2018 ASH Annual Meeting.
In the ECHELON-2 trial, patients who received brentuximab vedotin (BV) plus cyclophosphamide, doxorubicin, and prednisone (CHP) had superior progression-free survival (PFS) and overall survival (OS) compared to patients who received standard treatment with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP).
These results supported the recent U.S. approval of BV in combination with CHP for adults with previously untreated, systemic anaplastic large-cell lymphoma or other CD30-expressing PTCLs, including angioimmunoblastic T-cell lymphoma and PTCL not otherwise specified.
“ECHELON-2 is the first prospective trial in peripheral T-cell lymphoma to show an overall survival benefit over CHOP,” said Steven M. Horwitz, MD, of Memorial Sloan Kettering Cancer Center in Basking Ridge, New Jersey.
Dr. Horwitz presented results from this trial at ASH as abstract 997. Results were simultaneously published in The Lancet.
Patients and treatment
ECHELON-2 (NCT01777152) enrolled 452 patients with previously untreated, CD30-positive PTCL. Subtypes included ALK-positive (n=98) or -negative (n=218) systemic anaplastic large-cell lymphoma, PTCL not otherwise specified (n=72), angioimmunoblastic T-cell lymphoma (n=54), enteropathy-associated T-cell lymphoma (n=7), and adult T-cell leukemia/lymphoma (n=3).
Patients were randomized to receive BV-CHP plus placebo (n=226) or CHOP plus placebo (n=226) every 3 weeks for six to eight cycles.
At baseline, the median age was 58 in both the BV-CHP arm (range, 18-85) and the CHOP arm (range, 18-83). The majority of patients were male—59% in the BV-CHP arm and 67% in the CHOP arm—and most patients had stage III/IV disease—81% and 80%, respectively.
Eighty-nine percent of patients in the BV-CHP arm and 81% in the CHOP arm completed six or more cycles of their assigned treatment.
Twenty-seven percent of patients in the BV-CHP arm and 19% in the CHOP arm received consolidation consisting of radiotherapy (6% and 3%, respectively) and/or stem cell transplant (22% and 17%).
Twenty-six percent of patients in the BV-CHP arm and 42% in the CHOP arm received systemic therapy for residual or progressive disease, and 4% of patients in each arm received palliative radiation.
Efficacy
The overall response rate was 83% in the BV-CHP arm and 72% in the CHOP arm (P=0.0032). The complete response rates were 68% and 56%, respectively (P=0.0066).
At a median follow-up of 36.2 months, the median PFS was 48.2 months in the BV-CHP arm and 20.8 months in the CHOP arm. The rate of death or progression was 42% in the BV-CHP arm and 55% in the CHOP arm (hazard ratio=0.71, P=0.011).
At a median follow-up of 42.1 months, the median OS was not reached in either treatment arm. The rate of death was 23% in the BV-CHP arm and 32% in the CHOP arm (hazard ratio=0.66, P=0.0244).
Dr. Horwitz noted that this study was not powered to determine differences in OS or PFS according to PTCL subtypes.
Safety
BV-CHP had a comparable safety profile to CHOP, Dr. Horwitz said.
The rate of adverse events (AEs) was 99% in the BV-CHP arm and 98% in the CHOP arm. Grade 3 or higher AEs occurred in 66% and 65% of patients, respectively. Serious AEs occurred in 39% and 38%, respectively.
Three percent of patients in the BV-CHP arm and 4% of those in the CHOP arm had fatal AEs.
The most common AEs of any grade occurring in at least 20% of patients (in the BV-CHP and CHOP arms, respectively) were:
- Nausea (46% and 38%)
- Peripheral sensory neuropathy (45% and 41%)
- Neutropenia (38% for both)
- Diarrhea (38% and 20%)
- Constipation (29% and 30%)
- Alopecia (26% and 25%)
- Pyrexia (26% and 19%)
- Vomiting (26% and 17%)
- Fatigue (24% and 20%)
- Anemia (21% and 16%).
This research was funded by Seattle Genetics Inc. and Millennium Pharmaceuticals Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
Dr. Horwitz disclosed relationships with Seattle Genetics, Aileron Therapeutics, Innate Pharma, Millennium/Takeda, Forty Seven, Corvus, Mundipharma, ADC Therapeutics, Trillium, Celgene, Portola, Infinity/Verastem, Spectrum, and Kyowa-Hakka-Kirin.
SAN DIEGO—A newly approved treatment regimen provides a survival benefit over standard therapy for patients with CD30-positive peripheral T-cell lymphomas (PTCLs), according to a presentation at the 2018 ASH Annual Meeting.
In the ECHELON-2 trial, patients who received brentuximab vedotin (BV) plus cyclophosphamide, doxorubicin, and prednisone (CHP) had superior progression-free survival (PFS) and overall survival (OS) compared to patients who received standard treatment with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP).
These results supported the recent U.S. approval of BV in combination with CHP for adults with previously untreated, systemic anaplastic large-cell lymphoma or other CD30-expressing PTCLs, including angioimmunoblastic T-cell lymphoma and PTCL not otherwise specified.
“ECHELON-2 is the first prospective trial in peripheral T-cell lymphoma to show an overall survival benefit over CHOP,” said Steven M. Horwitz, MD, of Memorial Sloan Kettering Cancer Center in Basking Ridge, New Jersey.
Dr. Horwitz presented results from this trial at ASH as abstract 997. Results were simultaneously published in The Lancet.
Patients and treatment
ECHELON-2 (NCT01777152) enrolled 452 patients with previously untreated, CD30-positive PTCL. Subtypes included ALK-positive (n=98) or -negative (n=218) systemic anaplastic large-cell lymphoma, PTCL not otherwise specified (n=72), angioimmunoblastic T-cell lymphoma (n=54), enteropathy-associated T-cell lymphoma (n=7), and adult T-cell leukemia/lymphoma (n=3).
Patients were randomized to receive BV-CHP plus placebo (n=226) or CHOP plus placebo (n=226) every 3 weeks for six to eight cycles.
At baseline, the median age was 58 in both the BV-CHP arm (range, 18-85) and the CHOP arm (range, 18-83). The majority of patients were male—59% in the BV-CHP arm and 67% in the CHOP arm—and most patients had stage III/IV disease—81% and 80%, respectively.
Eighty-nine percent of patients in the BV-CHP arm and 81% in the CHOP arm completed six or more cycles of their assigned treatment.
Twenty-seven percent of patients in the BV-CHP arm and 19% in the CHOP arm received consolidation consisting of radiotherapy (6% and 3%, respectively) and/or stem cell transplant (22% and 17%).
Twenty-six percent of patients in the BV-CHP arm and 42% in the CHOP arm received systemic therapy for residual or progressive disease, and 4% of patients in each arm received palliative radiation.
Efficacy
The overall response rate was 83% in the BV-CHP arm and 72% in the CHOP arm (P=0.0032). The complete response rates were 68% and 56%, respectively (P=0.0066).
At a median follow-up of 36.2 months, the median PFS was 48.2 months in the BV-CHP arm and 20.8 months in the CHOP arm. The rate of death or progression was 42% in the BV-CHP arm and 55% in the CHOP arm (hazard ratio=0.71, P=0.011).
At a median follow-up of 42.1 months, the median OS was not reached in either treatment arm. The rate of death was 23% in the BV-CHP arm and 32% in the CHOP arm (hazard ratio=0.66, P=0.0244).
Dr. Horwitz noted that this study was not powered to determine differences in OS or PFS according to PTCL subtypes.
Safety
BV-CHP had a comparable safety profile to CHOP, Dr. Horwitz said.
The rate of adverse events (AEs) was 99% in the BV-CHP arm and 98% in the CHOP arm. Grade 3 or higher AEs occurred in 66% and 65% of patients, respectively. Serious AEs occurred in 39% and 38%, respectively.
Three percent of patients in the BV-CHP arm and 4% of those in the CHOP arm had fatal AEs.
The most common AEs of any grade occurring in at least 20% of patients (in the BV-CHP and CHOP arms, respectively) were:
- Nausea (46% and 38%)
- Peripheral sensory neuropathy (45% and 41%)
- Neutropenia (38% for both)
- Diarrhea (38% and 20%)
- Constipation (29% and 30%)
- Alopecia (26% and 25%)
- Pyrexia (26% and 19%)
- Vomiting (26% and 17%)
- Fatigue (24% and 20%)
- Anemia (21% and 16%).
This research was funded by Seattle Genetics Inc. and Millennium Pharmaceuticals Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
Dr. Horwitz disclosed relationships with Seattle Genetics, Aileron Therapeutics, Innate Pharma, Millennium/Takeda, Forty Seven, Corvus, Mundipharma, ADC Therapeutics, Trillium, Celgene, Portola, Infinity/Verastem, Spectrum, and Kyowa-Hakka-Kirin.
Update shows durable responses in rel/ref DLBCL

SAN DIEGO—An updated analysis of the JULIET trial showed that tisagenlecleucel produced a high rate of durable responses in adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL).
After a median follow-up of 19 months, two-thirds of adults with relapsed/refractory DLBCL who had early responses to the chimeric antigen receptor (CAR) T-cell therapy remained in remission with no evidence of minimal residual disease.
“Since the previous report, no new deaths have been reported due to any cause other than patient disease progression, no treatment-related mortality was seen throughout the study, and there were three early deaths, all related to lymphoma that progressed,” said study investigator Richard Thomas Maziarz, MD, of Oregon Health & Science University’s Knight Cancer Institute in Portland.
Dr. Maziarz and his colleagues reported the updated study results at the 2018 ASH Annual Meeting (abstract 1684). Results were published simultaneously in The New England Journal of Medicine. Data reported here are based on the ASH data.
JULIET then
In the phase 2, single-arm trial, investigators enrolled adults with DLBCL who had relapsed or were refractory after two or more prior lines of therapy and who were either ineligible for hematopoietic stem cell transplant (HSCT) or who experienced disease progression after HSCT.
Interim results of the study were previously reported at the 22nd Congress of the European Hematology Association in 2017.
At that meeting, Gilles Salles, MD, PhD, of the University of Lyon in France, presented results of an analysis of available efficacy data on 51 patients with at least 3 months of follow-up.
In this population, the best overall response rate (ORR) was 59%. Three-month ORR was 45%, consisting of 37% complete responses (CR) and 8% partial responses (PR).
Relapse-free survival at 6 months was 79%, and all patients who had responses at 3 months continued to have responses at the time of data cutoff.
JULIET now
The current analysis was completed after a median time from infusion to data cutoff of 19 months as of May 21, 2018. The analysis included 115 patients who received CAR T-cell infusions, 99 of whom were evaluable for efficacy.
As reported at ASH, the best ORR, the primary endpoint, was 54%, comprised of 40% CR and 13% PR.
Fifty-four percent of patients who had achieved PR converted to CR.
The response rates were consistent across all subgroups, regardless of age, sex, previous response status, International Prognostic Index score at enrollment, prior therapy, molecular subtype, and other factors.
Estimated relapse-free survival 12 months after documentation of an initial response was 64%.
The median duration of response had not been reached at the time of data cutoff, and the median overall survival had not been reached for patients with a CR.
Median overall survival in this heavily pretreated population as a whole (all patients who received CAR T-cell infusions) was 11.1 months and not reached for patients in CR.
Adverse events of special interest included grade 3 or 4 cytokine release syndrome (CRS) in 23% of patients, prolonged cytopenia in 34%, infections in 19%, neurologic events in 11%, febrile neutropenia in 15%, and tumor lysis syndrome in 2%.
There were no deaths attributable to the treatment, CRS, or to cerebral edema, a complication of CAR T-cell therapy that appears to be related to the costimulatory molecule used in various constructs.
The JULIET trial is supported by Novartis. Dr. Maziarz disclosed honoraria, consultancy fees, and/or research funding from Novartis, Incyte, Juno Therapeutics, and Kite Therapeutics as well as patents/royalties from Athersys, Inc.
SAN DIEGO—An updated analysis of the JULIET trial showed that tisagenlecleucel produced a high rate of durable responses in adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL).
After a median follow-up of 19 months, two-thirds of adults with relapsed/refractory DLBCL who had early responses to the chimeric antigen receptor (CAR) T-cell therapy remained in remission with no evidence of minimal residual disease.
“Since the previous report, no new deaths have been reported due to any cause other than patient disease progression, no treatment-related mortality was seen throughout the study, and there were three early deaths, all related to lymphoma that progressed,” said study investigator Richard Thomas Maziarz, MD, of Oregon Health & Science University’s Knight Cancer Institute in Portland.
Dr. Maziarz and his colleagues reported the updated study results at the 2018 ASH Annual Meeting (abstract 1684). Results were published simultaneously in The New England Journal of Medicine. Data reported here are based on the ASH data.
JULIET then
In the phase 2, single-arm trial, investigators enrolled adults with DLBCL who had relapsed or were refractory after two or more prior lines of therapy and who were either ineligible for hematopoietic stem cell transplant (HSCT) or who experienced disease progression after HSCT.
Interim results of the study were previously reported at the 22nd Congress of the European Hematology Association in 2017.
At that meeting, Gilles Salles, MD, PhD, of the University of Lyon in France, presented results of an analysis of available efficacy data on 51 patients with at least 3 months of follow-up.
In this population, the best overall response rate (ORR) was 59%. Three-month ORR was 45%, consisting of 37% complete responses (CR) and 8% partial responses (PR).
Relapse-free survival at 6 months was 79%, and all patients who had responses at 3 months continued to have responses at the time of data cutoff.
JULIET now
The current analysis was completed after a median time from infusion to data cutoff of 19 months as of May 21, 2018. The analysis included 115 patients who received CAR T-cell infusions, 99 of whom were evaluable for efficacy.
As reported at ASH, the best ORR, the primary endpoint, was 54%, comprised of 40% CR and 13% PR.
Fifty-four percent of patients who had achieved PR converted to CR.
The response rates were consistent across all subgroups, regardless of age, sex, previous response status, International Prognostic Index score at enrollment, prior therapy, molecular subtype, and other factors.
Estimated relapse-free survival 12 months after documentation of an initial response was 64%.
The median duration of response had not been reached at the time of data cutoff, and the median overall survival had not been reached for patients with a CR.
Median overall survival in this heavily pretreated population as a whole (all patients who received CAR T-cell infusions) was 11.1 months and not reached for patients in CR.
Adverse events of special interest included grade 3 or 4 cytokine release syndrome (CRS) in 23% of patients, prolonged cytopenia in 34%, infections in 19%, neurologic events in 11%, febrile neutropenia in 15%, and tumor lysis syndrome in 2%.
There were no deaths attributable to the treatment, CRS, or to cerebral edema, a complication of CAR T-cell therapy that appears to be related to the costimulatory molecule used in various constructs.
The JULIET trial is supported by Novartis. Dr. Maziarz disclosed honoraria, consultancy fees, and/or research funding from Novartis, Incyte, Juno Therapeutics, and Kite Therapeutics as well as patents/royalties from Athersys, Inc.
SAN DIEGO—An updated analysis of the JULIET trial showed that tisagenlecleucel produced a high rate of durable responses in adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL).
After a median follow-up of 19 months, two-thirds of adults with relapsed/refractory DLBCL who had early responses to the chimeric antigen receptor (CAR) T-cell therapy remained in remission with no evidence of minimal residual disease.
“Since the previous report, no new deaths have been reported due to any cause other than patient disease progression, no treatment-related mortality was seen throughout the study, and there were three early deaths, all related to lymphoma that progressed,” said study investigator Richard Thomas Maziarz, MD, of Oregon Health & Science University’s Knight Cancer Institute in Portland.
Dr. Maziarz and his colleagues reported the updated study results at the 2018 ASH Annual Meeting (abstract 1684). Results were published simultaneously in The New England Journal of Medicine. Data reported here are based on the ASH data.
JULIET then
In the phase 2, single-arm trial, investigators enrolled adults with DLBCL who had relapsed or were refractory after two or more prior lines of therapy and who were either ineligible for hematopoietic stem cell transplant (HSCT) or who experienced disease progression after HSCT.
Interim results of the study were previously reported at the 22nd Congress of the European Hematology Association in 2017.
At that meeting, Gilles Salles, MD, PhD, of the University of Lyon in France, presented results of an analysis of available efficacy data on 51 patients with at least 3 months of follow-up.
In this population, the best overall response rate (ORR) was 59%. Three-month ORR was 45%, consisting of 37% complete responses (CR) and 8% partial responses (PR).
Relapse-free survival at 6 months was 79%, and all patients who had responses at 3 months continued to have responses at the time of data cutoff.
JULIET now
The current analysis was completed after a median time from infusion to data cutoff of 19 months as of May 21, 2018. The analysis included 115 patients who received CAR T-cell infusions, 99 of whom were evaluable for efficacy.
As reported at ASH, the best ORR, the primary endpoint, was 54%, comprised of 40% CR and 13% PR.
Fifty-four percent of patients who had achieved PR converted to CR.
The response rates were consistent across all subgroups, regardless of age, sex, previous response status, International Prognostic Index score at enrollment, prior therapy, molecular subtype, and other factors.
Estimated relapse-free survival 12 months after documentation of an initial response was 64%.
The median duration of response had not been reached at the time of data cutoff, and the median overall survival had not been reached for patients with a CR.
Median overall survival in this heavily pretreated population as a whole (all patients who received CAR T-cell infusions) was 11.1 months and not reached for patients in CR.
Adverse events of special interest included grade 3 or 4 cytokine release syndrome (CRS) in 23% of patients, prolonged cytopenia in 34%, infections in 19%, neurologic events in 11%, febrile neutropenia in 15%, and tumor lysis syndrome in 2%.
There were no deaths attributable to the treatment, CRS, or to cerebral edema, a complication of CAR T-cell therapy that appears to be related to the costimulatory molecule used in various constructs.
The JULIET trial is supported by Novartis. Dr. Maziarz disclosed honoraria, consultancy fees, and/or research funding from Novartis, Incyte, Juno Therapeutics, and Kite Therapeutics as well as patents/royalties from Athersys, Inc.

TOURMALINE-MM3: Ixazomib improves PFS after myeloma transplant
SAN DIEGO – Ixazomib improved progression-free survival following autologous stem cell transplantation (ASCT) in patients with newly diagnosed multiple myeloma, according to results of a double-blind, randomized, placebo-controlled, phase 3 trial.

Treatment with the oral proteasome inhibitor for 24 months was well tolerated, had a low discontinuation rate, and improved progression-free survival by 39% versus placebo, according to Meletios A. Dimopoulos, MD, of the National and Kapodistrian University of Athens.
These findings suggest ixazomib (Ninlaro) represents a “new treatment option for maintenance after transplantation,” Dr. Dimopoulos said at the annual meeting of the American Society of Hematology.
The trial, known as TOURMALINE-MM3, is the first-ever randomized, double-blind, placebo-controlled study of a proteasome inhibitor used as maintenance after ASCT, according to Dr. Dimopoulos. Lenalidomide is approved in that setting, but 29% of patients who start the treatment discontinue because of treatment-related adverse events.
“Proteasome inhibitors have a different mechanism of action and may provide an alternative to lenalidomide,” Dr. Dimopoulos said at an oral abstract session. Ixazomib has a manageable toxicity profile and “convenient” weekly oral dosing, making it “well suited” for maintenance.
When asked by an attendee whether ixazomib would become “the standard of care” for younger patients with myeloma in this setting, Dr. Dimopoulos replied the results show that ixazomib “is an option for patients, especially for those where a physician may believe that a proteasome inhibitor may be indicated.”
However, when pressed by an attendee to comment on how ixazomib compares with lenalidomide for maintenance, Dr. Dimopoulos remarked that current maintenance studies are moving in the direction of combining therapies.
“I think that instead of saying, ‘is ixazomib better than lenalidomide?’ or vice versa, it is better to see how one may combine those drugs in subsets of patients, or even combine these drugs with other agents,” he said.
The TOURMALINE-MM3 study included 656 patients randomized posttransplantation to receive weekly ixazomib or placebo for up to 2 years.
The median progression-free survival was 26.5 months for ixazomib versus 21.3 months for placebo (P = .002; hazard ratio, 0.720; 95% confidence interval, 0.582-0.890). Median overall survival had not been reached in either ixazomib or placebo arms as of this report, with a median follow-up of 31 months.
The discontinuation rate was 7% for ixazomib versus 5% for placebo, according to the investigator. Moreover, ixazomib was associated with “low toxicity” and no difference in the rates of new primary malignancies, at 3% in both arms.
A manuscript describing results of the TOURMALINE-MM3 study is in press in the Lancet, with an expected online publication date of Dec. 10, Dr. Dimopoulos told attendees. Other studies are ongoing to evaluate ixazomib combinations and treatment to progression in this setting.
TOURMALINE-MM3 is sponsored by Takeda (Millennium), the maker of ixazomib. Dr. Dimopoulos reported honoraria and consultancy with Janssen, Takeda Pharmaceutical, Amgen, Bristol-Myers Squibb, and Celgene.
SOURCE: Dimopoulos MA et al. ASH 2018, Abstract 301.
SAN DIEGO – Ixazomib improved progression-free survival following autologous stem cell transplantation (ASCT) in patients with newly diagnosed multiple myeloma, according to results of a double-blind, randomized, placebo-controlled, phase 3 trial.
Treatment with the oral proteasome inhibitor for 24 months was well tolerated, had a low discontinuation rate, and improved progression-free survival by 39% versus placebo, according to Meletios A. Dimopoulos, MD, of the National and Kapodistrian University of Athens.
These findings suggest ixazomib (Ninlaro) represents a “new treatment option for maintenance after transplantation,” Dr. Dimopoulos said at the annual meeting of the American Society of Hematology.
The trial, known as TOURMALINE-MM3, is the first-ever randomized, double-blind, placebo-controlled study of a proteasome inhibitor used as maintenance after ASCT, according to Dr. Dimopoulos. Lenalidomide is approved in that setting, but 29% of patients who start the treatment discontinue because of treatment-related adverse events.
“Proteasome inhibitors have a different mechanism of action and may provide an alternative to lenalidomide,” Dr. Dimopoulos said at an oral abstract session. Ixazomib has a manageable toxicity profile and “convenient” weekly oral dosing, making it “well suited” for maintenance.
When asked by an attendee whether ixazomib would become “the standard of care” for younger patients with myeloma in this setting, Dr. Dimopoulos replied the results show that ixazomib “is an option for patients, especially for those where a physician may believe that a proteasome inhibitor may be indicated.”
However, when pressed by an attendee to comment on how ixazomib compares with lenalidomide for maintenance, Dr. Dimopoulos remarked that current maintenance studies are moving in the direction of combining therapies.
“I think that instead of saying, ‘is ixazomib better than lenalidomide?’ or vice versa, it is better to see how one may combine those drugs in subsets of patients, or even combine these drugs with other agents,” he said.
The TOURMALINE-MM3 study included 656 patients randomized posttransplantation to receive weekly ixazomib or placebo for up to 2 years.
The median progression-free survival was 26.5 months for ixazomib versus 21.3 months for placebo (P = .002; hazard ratio, 0.720; 95% confidence interval, 0.582-0.890). Median overall survival had not been reached in either ixazomib or placebo arms as of this report, with a median follow-up of 31 months.
The discontinuation rate was 7% for ixazomib versus 5% for placebo, according to the investigator. Moreover, ixazomib was associated with “low toxicity” and no difference in the rates of new primary malignancies, at 3% in both arms.
A manuscript describing results of the TOURMALINE-MM3 study is in press in the Lancet, with an expected online publication date of Dec. 10, Dr. Dimopoulos told attendees. Other studies are ongoing to evaluate ixazomib combinations and treatment to progression in this setting.
TOURMALINE-MM3 is sponsored by Takeda (Millennium), the maker of ixazomib. Dr. Dimopoulos reported honoraria and consultancy with Janssen, Takeda Pharmaceutical, Amgen, Bristol-Myers Squibb, and Celgene.
SOURCE: Dimopoulos MA et al. ASH 2018, Abstract 301.
SAN DIEGO – Ixazomib improved progression-free survival following autologous stem cell transplantation (ASCT) in patients with newly diagnosed multiple myeloma, according to results of a double-blind, randomized, placebo-controlled, phase 3 trial.
Treatment with the oral proteasome inhibitor for 24 months was well tolerated, had a low discontinuation rate, and improved progression-free survival by 39% versus placebo, according to Meletios A. Dimopoulos, MD, of the National and Kapodistrian University of Athens.
These findings suggest ixazomib (Ninlaro) represents a “new treatment option for maintenance after transplantation,” Dr. Dimopoulos said at the annual meeting of the American Society of Hematology.
The trial, known as TOURMALINE-MM3, is the first-ever randomized, double-blind, placebo-controlled study of a proteasome inhibitor used as maintenance after ASCT, according to Dr. Dimopoulos. Lenalidomide is approved in that setting, but 29% of patients who start the treatment discontinue because of treatment-related adverse events.
“Proteasome inhibitors have a different mechanism of action and may provide an alternative to lenalidomide,” Dr. Dimopoulos said at an oral abstract session. Ixazomib has a manageable toxicity profile and “convenient” weekly oral dosing, making it “well suited” for maintenance.
When asked by an attendee whether ixazomib would become “the standard of care” for younger patients with myeloma in this setting, Dr. Dimopoulos replied the results show that ixazomib “is an option for patients, especially for those where a physician may believe that a proteasome inhibitor may be indicated.”
However, when pressed by an attendee to comment on how ixazomib compares with lenalidomide for maintenance, Dr. Dimopoulos remarked that current maintenance studies are moving in the direction of combining therapies.
“I think that instead of saying, ‘is ixazomib better than lenalidomide?’ or vice versa, it is better to see how one may combine those drugs in subsets of patients, or even combine these drugs with other agents,” he said.
The TOURMALINE-MM3 study included 656 patients randomized posttransplantation to receive weekly ixazomib or placebo for up to 2 years.
The median progression-free survival was 26.5 months for ixazomib versus 21.3 months for placebo (P = .002; hazard ratio, 0.720; 95% confidence interval, 0.582-0.890). Median overall survival had not been reached in either ixazomib or placebo arms as of this report, with a median follow-up of 31 months.
The discontinuation rate was 7% for ixazomib versus 5% for placebo, according to the investigator. Moreover, ixazomib was associated with “low toxicity” and no difference in the rates of new primary malignancies, at 3% in both arms.
A manuscript describing results of the TOURMALINE-MM3 study is in press in the Lancet, with an expected online publication date of Dec. 10, Dr. Dimopoulos told attendees. Other studies are ongoing to evaluate ixazomib combinations and treatment to progression in this setting.
TOURMALINE-MM3 is sponsored by Takeda (Millennium), the maker of ixazomib. Dr. Dimopoulos reported honoraria and consultancy with Janssen, Takeda Pharmaceutical, Amgen, Bristol-Myers Squibb, and Celgene.
SOURCE: Dimopoulos MA et al. ASH 2018, Abstract 301.
REPORTING FROM ASH 2018
Key clinical point: The proteasome inhibitor ixazomib significantly improved progression-free survival following autologous stem cell transplantation in patients with newly diagnosed multiple myeloma.
Major finding: The median progression-free survival was 26.5 months for ixazomib, versus 21.3 months for placebo (P = .002; hazard ratio, 0.720; 95% confidence interval, 0.582-0.890).
Study details: TOURMALINE-MM3, a randomized, placebo-controlled trial, includes 656 patients with newly diagnosed myeloma who had undergone autologous stem cell transplantation.
Disclosures: TOURMALINE-MM3 is sponsored by Takeda (Millennium), the maker of ixazomib. Dr. Dimopoulos reported honoraria and consultancy with Janssen, Takeda Pharmaceutical, Amgen, Bristol-Myers Squibb, and Celgene.
Source: Dimopoulos MA et al. ASH 2018, Abstract 301.