User login
A ‘double-hit’ bone marrow rare co-occurrence of 2 different pathologies
Chronic myeloid leukemia and chronic lymphocytic leukemia are entirely different in terms of pathogenesis, presentation, diagnostic work-up, treatment, and prognosis: CML is a myeloproliferative condition, whereas CLL involves lymphoid population. Here we discuss a very rare case of co-occurrence of CML and CLL in the same patient.
Click on the PDF icon at the top of this introduction to read the full article.
Chronic myeloid leukemia and chronic lymphocytic leukemia are entirely different in terms of pathogenesis, presentation, diagnostic work-up, treatment, and prognosis: CML is a myeloproliferative condition, whereas CLL involves lymphoid population. Here we discuss a very rare case of co-occurrence of CML and CLL in the same patient.
Click on the PDF icon at the top of this introduction to read the full article.
Chronic myeloid leukemia and chronic lymphocytic leukemia are entirely different in terms of pathogenesis, presentation, diagnostic work-up, treatment, and prognosis: CML is a myeloproliferative condition, whereas CLL involves lymphoid population. Here we discuss a very rare case of co-occurrence of CML and CLL in the same patient.
Click on the PDF icon at the top of this introduction to read the full article.
Financial burden of blood cancers in the U.S.

An analysis of more than 2,000 U.S. patients with blood cancers revealed an average healthcare cost of almost $157,000 in the first year after diagnosis.
Costs were highest for acute leukemia patients—almost triple the average for all blood cancers.
Out-of-pocket (OOP) costs were initially highest for acute leukemia patients. However, over time, OOP costs became highest for patients with multiple myeloma.
These results are included in a report commissioned by the Leukemia & Lymphoma Society and prepared by the actuarial firm Milliman.
The report is based on data from the Truven Health MarketScan commercial claims databases.
The cost figures are drawn from data for 2,332 patients, ages 18 to 64, who were diagnosed with blood cancer in 2014 and followed through 2016. This includes the following:
- 1,468 patients with lymphoma
- 286 with chronic leukemia
- 282 with multiple myeloma
- 148 with acute leukemia
- 148 with bone marrow disorders (myelodysplastic syndromes).
The average allowed spending—the amount paid by the payer and patient combined—in the first 12 months after diagnosis was:
- $156,845 overall
- $463,414 for acute leukemia
- $213,879 for multiple myeloma
- $133,744 for bone marrow disorders
- $130,545 for lymphoma
- $88,913 for chronic leukemia.
Differences in OOP costs were smaller, although OOP spending was 32% higher for acute leukemia patients than the overall average.
Average OOP costs—which include coinsurance, copay, and deductible—in the first 12 months after diagnosis were:
- $3,877 overall
- $5,147 for acute leukemia
- $4,849 for multiple myeloma
- $3,695 for lymphoma
- $3,480 for chronic leukemia
- $3,336 for bone marrow disorders.
Although OOP costs were initially highest for acute leukemia patients, over time, costs for multiple myeloma patients became the highest.
The average OOP costs in the month of diagnosis were $1,637 for acute leukemia patients and $1,210 for multiple myeloma patients.
The total accumulated OOP costs 3 years after diagnosis were $8,797 for acute leukemia and $9,127 for multiple myeloma. For the other blood cancers, the average 3-year accumulated OOP costs were under $7,800.
The Leukemia & Lymphoma Society received support from Pfizer, Genentech, and Amgen for this work.
An analysis of more than 2,000 U.S. patients with blood cancers revealed an average healthcare cost of almost $157,000 in the first year after diagnosis.
Costs were highest for acute leukemia patients—almost triple the average for all blood cancers.
Out-of-pocket (OOP) costs were initially highest for acute leukemia patients. However, over time, OOP costs became highest for patients with multiple myeloma.
These results are included in a report commissioned by the Leukemia & Lymphoma Society and prepared by the actuarial firm Milliman.
The report is based on data from the Truven Health MarketScan commercial claims databases.
The cost figures are drawn from data for 2,332 patients, ages 18 to 64, who were diagnosed with blood cancer in 2014 and followed through 2016. This includes the following:
- 1,468 patients with lymphoma
- 286 with chronic leukemia
- 282 with multiple myeloma
- 148 with acute leukemia
- 148 with bone marrow disorders (myelodysplastic syndromes).
The average allowed spending—the amount paid by the payer and patient combined—in the first 12 months after diagnosis was:
- $156,845 overall
- $463,414 for acute leukemia
- $213,879 for multiple myeloma
- $133,744 for bone marrow disorders
- $130,545 for lymphoma
- $88,913 for chronic leukemia.
Differences in OOP costs were smaller, although OOP spending was 32% higher for acute leukemia patients than the overall average.
Average OOP costs—which include coinsurance, copay, and deductible—in the first 12 months after diagnosis were:
- $3,877 overall
- $5,147 for acute leukemia
- $4,849 for multiple myeloma
- $3,695 for lymphoma
- $3,480 for chronic leukemia
- $3,336 for bone marrow disorders.
Although OOP costs were initially highest for acute leukemia patients, over time, costs for multiple myeloma patients became the highest.
The average OOP costs in the month of diagnosis were $1,637 for acute leukemia patients and $1,210 for multiple myeloma patients.
The total accumulated OOP costs 3 years after diagnosis were $8,797 for acute leukemia and $9,127 for multiple myeloma. For the other blood cancers, the average 3-year accumulated OOP costs were under $7,800.
The Leukemia & Lymphoma Society received support from Pfizer, Genentech, and Amgen for this work.
An analysis of more than 2,000 U.S. patients with blood cancers revealed an average healthcare cost of almost $157,000 in the first year after diagnosis.
Costs were highest for acute leukemia patients—almost triple the average for all blood cancers.
Out-of-pocket (OOP) costs were initially highest for acute leukemia patients. However, over time, OOP costs became highest for patients with multiple myeloma.
These results are included in a report commissioned by the Leukemia & Lymphoma Society and prepared by the actuarial firm Milliman.
The report is based on data from the Truven Health MarketScan commercial claims databases.
The cost figures are drawn from data for 2,332 patients, ages 18 to 64, who were diagnosed with blood cancer in 2014 and followed through 2016. This includes the following:
- 1,468 patients with lymphoma
- 286 with chronic leukemia
- 282 with multiple myeloma
- 148 with acute leukemia
- 148 with bone marrow disorders (myelodysplastic syndromes).
The average allowed spending—the amount paid by the payer and patient combined—in the first 12 months after diagnosis was:
- $156,845 overall
- $463,414 for acute leukemia
- $213,879 for multiple myeloma
- $133,744 for bone marrow disorders
- $130,545 for lymphoma
- $88,913 for chronic leukemia.
Differences in OOP costs were smaller, although OOP spending was 32% higher for acute leukemia patients than the overall average.
Average OOP costs—which include coinsurance, copay, and deductible—in the first 12 months after diagnosis were:
- $3,877 overall
- $5,147 for acute leukemia
- $4,849 for multiple myeloma
- $3,695 for lymphoma
- $3,480 for chronic leukemia
- $3,336 for bone marrow disorders.
Although OOP costs were initially highest for acute leukemia patients, over time, costs for multiple myeloma patients became the highest.
The average OOP costs in the month of diagnosis were $1,637 for acute leukemia patients and $1,210 for multiple myeloma patients.
The total accumulated OOP costs 3 years after diagnosis were $8,797 for acute leukemia and $9,127 for multiple myeloma. For the other blood cancers, the average 3-year accumulated OOP costs were under $7,800.
The Leukemia & Lymphoma Society received support from Pfizer, Genentech, and Amgen for this work.
P-BCMA-101 gains FDA regenerative medicine designation
(MM), has received the regenerative medicine advanced therapy (RMAT) designation from the Food and Drug Administration.

P-BCMA-101 modifies patients’ T cells using a nonviral DNA modification system known as piggyBac. The modified T cells target cells expressing B-cell maturation antigen (BCMA), which is expressed on essentially all MM cells.
Early results from the phase 1 clinical trial of P-BCMA-101 were recently reported at the 2018 CAR-TCR Summit by Eric Ostertag, MD, PhD, chief executive officer of Poseida Therapeutics, the company developing P-BCMA-101.
Initial results of the trial (NCT03288493) included data on 11 patients with heavily pretreated MM. Patients were a median age of 60, and 73% were high risk. They had a median of six prior therapies.
Patients received conditioning treatment with fludarabine and cyclophosphamide for 3 days prior to receiving P-BCMA-101. They then received one of three doses of CAR T cells – 51×106 (n=3), 152×106 (n=7), or 430×106 (n=1).
The investigators observed no dose-limiting toxicities. Adverse events included neutropenia in eight patients and thrombocytopenia in five.
One patient may have had cytokine release syndrome, but the condition resolved without drug intervention. And investigators observed no neurotoxicity.
Seven of ten patients evaluable for response by International Myeloma Working Group criteria achieved at least a partial response, including very good partial responses and stringent complete response.
The eleventh patient has oligosecretory disease and was only evaluable by PET, which indicated a near-complete response.
Poseida expects to have additional data to report by the end of the year, according to Dr. Ostertag. The study is funded by the California Institute for Regenerative Medicine and Poseida Therapeutics.RMAT designation is intended to expedite development and review of regenerative medicines that are intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition.
Preliminary evidence must indicate that the therapy has the potential to address unmet medical needs for the disease or condition. RMAT designation includes all the benefits of fast track and breakthrough therapy designations, including early interactions with the FDA.
(MM), has received the regenerative medicine advanced therapy (RMAT) designation from the Food and Drug Administration.
P-BCMA-101 modifies patients’ T cells using a nonviral DNA modification system known as piggyBac. The modified T cells target cells expressing B-cell maturation antigen (BCMA), which is expressed on essentially all MM cells.
Early results from the phase 1 clinical trial of P-BCMA-101 were recently reported at the 2018 CAR-TCR Summit by Eric Ostertag, MD, PhD, chief executive officer of Poseida Therapeutics, the company developing P-BCMA-101.
Initial results of the trial (NCT03288493) included data on 11 patients with heavily pretreated MM. Patients were a median age of 60, and 73% were high risk. They had a median of six prior therapies.
Patients received conditioning treatment with fludarabine and cyclophosphamide for 3 days prior to receiving P-BCMA-101. They then received one of three doses of CAR T cells – 51×106 (n=3), 152×106 (n=7), or 430×106 (n=1).
The investigators observed no dose-limiting toxicities. Adverse events included neutropenia in eight patients and thrombocytopenia in five.
One patient may have had cytokine release syndrome, but the condition resolved without drug intervention. And investigators observed no neurotoxicity.
Seven of ten patients evaluable for response by International Myeloma Working Group criteria achieved at least a partial response, including very good partial responses and stringent complete response.
The eleventh patient has oligosecretory disease and was only evaluable by PET, which indicated a near-complete response.
Poseida expects to have additional data to report by the end of the year, according to Dr. Ostertag. The study is funded by the California Institute for Regenerative Medicine and Poseida Therapeutics.RMAT designation is intended to expedite development and review of regenerative medicines that are intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition.
Preliminary evidence must indicate that the therapy has the potential to address unmet medical needs for the disease or condition. RMAT designation includes all the benefits of fast track and breakthrough therapy designations, including early interactions with the FDA.
(MM), has received the regenerative medicine advanced therapy (RMAT) designation from the Food and Drug Administration.
P-BCMA-101 modifies patients’ T cells using a nonviral DNA modification system known as piggyBac. The modified T cells target cells expressing B-cell maturation antigen (BCMA), which is expressed on essentially all MM cells.
Early results from the phase 1 clinical trial of P-BCMA-101 were recently reported at the 2018 CAR-TCR Summit by Eric Ostertag, MD, PhD, chief executive officer of Poseida Therapeutics, the company developing P-BCMA-101.
Initial results of the trial (NCT03288493) included data on 11 patients with heavily pretreated MM. Patients were a median age of 60, and 73% were high risk. They had a median of six prior therapies.
Patients received conditioning treatment with fludarabine and cyclophosphamide for 3 days prior to receiving P-BCMA-101. They then received one of three doses of CAR T cells – 51×106 (n=3), 152×106 (n=7), or 430×106 (n=1).
The investigators observed no dose-limiting toxicities. Adverse events included neutropenia in eight patients and thrombocytopenia in five.
One patient may have had cytokine release syndrome, but the condition resolved without drug intervention. And investigators observed no neurotoxicity.
Seven of ten patients evaluable for response by International Myeloma Working Group criteria achieved at least a partial response, including very good partial responses and stringent complete response.
The eleventh patient has oligosecretory disease and was only evaluable by PET, which indicated a near-complete response.
Poseida expects to have additional data to report by the end of the year, according to Dr. Ostertag. The study is funded by the California Institute for Regenerative Medicine and Poseida Therapeutics.RMAT designation is intended to expedite development and review of regenerative medicines that are intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition.
Preliminary evidence must indicate that the therapy has the potential to address unmet medical needs for the disease or condition. RMAT designation includes all the benefits of fast track and breakthrough therapy designations, including early interactions with the FDA.
Aberrant RNA editing linked to aggressive myeloma
Overly zealous editing of messenger RNA in multiple myeloma cells appears to contribute to myeloma pathogenesis, and is prognostic of poor outcomes, investigators contend.

Over-expression of RNA editing enzymes in the adenosine deaminases acting on RNA (ADAR) family, specifically ADAR1, lead to hyperediting of the multiple myeloma (MM) transcriptome that in turn appears related to a drug-resistant disease phenotype and worse prognosis, reported Phaik Ju Teoh, PhD, of the Cancer Science Institute of Singapore, and colleagues.
The investigators implicate aberrant editing of adenosine to inosine (A-to-I) in malignant plasma cells, and its effects on NEIL1, a gene that encodes proteins involved in base excision repair of DNA, as important mechanisms in multiple myeloma pathogenesis.
“To the best of our knowledge, this is the first report of ADAR1-mediated hypereditome being an independent prognostic factor. The compromised integrity of MM transcriptome drives oncogenic phenotypes, likely contributing to the disease pathogenesis. Our current work, therefore, recognizes the clear biological and clinical importance of A-to-I editing at both the whole-transcriptome and gene-specific level (NEIL1) in MM,” they wrote in Blood.
A-to-I editing is the most prevalent form of RNA editing in humans, and aberrant editing mediated by ADAR1 has recently been linked to the development of several different cancer types, the investigators noted.
To see whether this process may also be involved in multiple myeloma, the investigators examined whole blood or bone marrow samples from healthy volunteers and patients with multiple myeloma.
They first looked at gene-expression profiling in the control and multiple myeloma samples and found that ADAR1 was overexpressed in the multiple myeloma cells, compared with nonmalignant plasma cells. Additionally, they saw that, at the protein level, ADAR1 was expressed at higher levels in patients with newly diagnosed or relapsed disease, compared with patients with smoldering myeloma or monoclonal gammopathy of undetermined significance.
They next determined that ADAR1 directly regulates hyperediting of the MM transcriptome, evidenced by the observation of a significant increase in A-to-G editing in the newly diagnosed and relapsed myeloma samples, compared with normal plasma cells. They confirmed this finding by observing the effects of ADAR1 levels on editing events across the transcriptome.
The authors followed this observation with experiments to see whether RNA editing by ADAR1 contributes to oncogenesis in myeloma cells. They silenced its expression and found that growth rate slowed and that ADAR1 wild-type protein introduced into cells promoted growth and proliferation.
“As the rescue with mutant ADAR1 is incomplete, we do not discount potential nonediting effects in ADAR1-induced oncogenesis in vivo. Nevertheless, taking into consideration the collective results from both the in vitro and in vivo studies, the RNA editing function of ADAR1 is important for its oncogenic effects in myeloma,” they wrote.
In the final steps, they identified NEIL1 as an important target for editing in multiple myeloma and observed that the editing compromised the ability of the proteins produced by the gene to accurately repair DNA damage.
“Further demonstrating its vital contribution to disease aggressiveness, patients with high ADAR1 expression showed less responsiveness toward standard and novel therapies. Therefore, our findings implied that a disturbed editome mediated by ADAR1 overexpression is both clinically and functionally crucial in our disease setting, and that ADAR1 confers oncogenic properties in myeloma in an editing-dependent manner,” they wrote.
The study was supported by the National Research Foundation Singapore, the Singapore Ministry of Education, and the National University of Singapore. The authors reported having no competing financial interests.
SOURCE: Teoh PJ et al. Blood. 2018;132(12):1304-17.
Overly zealous editing of messenger RNA in multiple myeloma cells appears to contribute to myeloma pathogenesis, and is prognostic of poor outcomes, investigators contend.
Over-expression of RNA editing enzymes in the adenosine deaminases acting on RNA (ADAR) family, specifically ADAR1, lead to hyperediting of the multiple myeloma (MM) transcriptome that in turn appears related to a drug-resistant disease phenotype and worse prognosis, reported Phaik Ju Teoh, PhD, of the Cancer Science Institute of Singapore, and colleagues.
The investigators implicate aberrant editing of adenosine to inosine (A-to-I) in malignant plasma cells, and its effects on NEIL1, a gene that encodes proteins involved in base excision repair of DNA, as important mechanisms in multiple myeloma pathogenesis.
“To the best of our knowledge, this is the first report of ADAR1-mediated hypereditome being an independent prognostic factor. The compromised integrity of MM transcriptome drives oncogenic phenotypes, likely contributing to the disease pathogenesis. Our current work, therefore, recognizes the clear biological and clinical importance of A-to-I editing at both the whole-transcriptome and gene-specific level (NEIL1) in MM,” they wrote in Blood.
A-to-I editing is the most prevalent form of RNA editing in humans, and aberrant editing mediated by ADAR1 has recently been linked to the development of several different cancer types, the investigators noted.
To see whether this process may also be involved in multiple myeloma, the investigators examined whole blood or bone marrow samples from healthy volunteers and patients with multiple myeloma.
They first looked at gene-expression profiling in the control and multiple myeloma samples and found that ADAR1 was overexpressed in the multiple myeloma cells, compared with nonmalignant plasma cells. Additionally, they saw that, at the protein level, ADAR1 was expressed at higher levels in patients with newly diagnosed or relapsed disease, compared with patients with smoldering myeloma or monoclonal gammopathy of undetermined significance.
They next determined that ADAR1 directly regulates hyperediting of the MM transcriptome, evidenced by the observation of a significant increase in A-to-G editing in the newly diagnosed and relapsed myeloma samples, compared with normal plasma cells. They confirmed this finding by observing the effects of ADAR1 levels on editing events across the transcriptome.
The authors followed this observation with experiments to see whether RNA editing by ADAR1 contributes to oncogenesis in myeloma cells. They silenced its expression and found that growth rate slowed and that ADAR1 wild-type protein introduced into cells promoted growth and proliferation.
“As the rescue with mutant ADAR1 is incomplete, we do not discount potential nonediting effects in ADAR1-induced oncogenesis in vivo. Nevertheless, taking into consideration the collective results from both the in vitro and in vivo studies, the RNA editing function of ADAR1 is important for its oncogenic effects in myeloma,” they wrote.
In the final steps, they identified NEIL1 as an important target for editing in multiple myeloma and observed that the editing compromised the ability of the proteins produced by the gene to accurately repair DNA damage.
“Further demonstrating its vital contribution to disease aggressiveness, patients with high ADAR1 expression showed less responsiveness toward standard and novel therapies. Therefore, our findings implied that a disturbed editome mediated by ADAR1 overexpression is both clinically and functionally crucial in our disease setting, and that ADAR1 confers oncogenic properties in myeloma in an editing-dependent manner,” they wrote.
The study was supported by the National Research Foundation Singapore, the Singapore Ministry of Education, and the National University of Singapore. The authors reported having no competing financial interests.
SOURCE: Teoh PJ et al. Blood. 2018;132(12):1304-17.
Overly zealous editing of messenger RNA in multiple myeloma cells appears to contribute to myeloma pathogenesis, and is prognostic of poor outcomes, investigators contend.
Over-expression of RNA editing enzymes in the adenosine deaminases acting on RNA (ADAR) family, specifically ADAR1, lead to hyperediting of the multiple myeloma (MM) transcriptome that in turn appears related to a drug-resistant disease phenotype and worse prognosis, reported Phaik Ju Teoh, PhD, of the Cancer Science Institute of Singapore, and colleagues.
The investigators implicate aberrant editing of adenosine to inosine (A-to-I) in malignant plasma cells, and its effects on NEIL1, a gene that encodes proteins involved in base excision repair of DNA, as important mechanisms in multiple myeloma pathogenesis.
“To the best of our knowledge, this is the first report of ADAR1-mediated hypereditome being an independent prognostic factor. The compromised integrity of MM transcriptome drives oncogenic phenotypes, likely contributing to the disease pathogenesis. Our current work, therefore, recognizes the clear biological and clinical importance of A-to-I editing at both the whole-transcriptome and gene-specific level (NEIL1) in MM,” they wrote in Blood.
A-to-I editing is the most prevalent form of RNA editing in humans, and aberrant editing mediated by ADAR1 has recently been linked to the development of several different cancer types, the investigators noted.
To see whether this process may also be involved in multiple myeloma, the investigators examined whole blood or bone marrow samples from healthy volunteers and patients with multiple myeloma.
They first looked at gene-expression profiling in the control and multiple myeloma samples and found that ADAR1 was overexpressed in the multiple myeloma cells, compared with nonmalignant plasma cells. Additionally, they saw that, at the protein level, ADAR1 was expressed at higher levels in patients with newly diagnosed or relapsed disease, compared with patients with smoldering myeloma or monoclonal gammopathy of undetermined significance.
They next determined that ADAR1 directly regulates hyperediting of the MM transcriptome, evidenced by the observation of a significant increase in A-to-G editing in the newly diagnosed and relapsed myeloma samples, compared with normal plasma cells. They confirmed this finding by observing the effects of ADAR1 levels on editing events across the transcriptome.
The authors followed this observation with experiments to see whether RNA editing by ADAR1 contributes to oncogenesis in myeloma cells. They silenced its expression and found that growth rate slowed and that ADAR1 wild-type protein introduced into cells promoted growth and proliferation.
“As the rescue with mutant ADAR1 is incomplete, we do not discount potential nonediting effects in ADAR1-induced oncogenesis in vivo. Nevertheless, taking into consideration the collective results from both the in vitro and in vivo studies, the RNA editing function of ADAR1 is important for its oncogenic effects in myeloma,” they wrote.
In the final steps, they identified NEIL1 as an important target for editing in multiple myeloma and observed that the editing compromised the ability of the proteins produced by the gene to accurately repair DNA damage.
“Further demonstrating its vital contribution to disease aggressiveness, patients with high ADAR1 expression showed less responsiveness toward standard and novel therapies. Therefore, our findings implied that a disturbed editome mediated by ADAR1 overexpression is both clinically and functionally crucial in our disease setting, and that ADAR1 confers oncogenic properties in myeloma in an editing-dependent manner,” they wrote.
The study was supported by the National Research Foundation Singapore, the Singapore Ministry of Education, and the National University of Singapore. The authors reported having no competing financial interests.
SOURCE: Teoh PJ et al. Blood. 2018;132(12):1304-17.
FROM BLOOD
Key clinical point:
Major finding: ADAR1-mediated editing of NEIL1 leads to a weakened DNA base excision repair mechanism.
Study details: Experimental series using plasma samples from healthy volunteers and patients with multiple myeloma.
Disclosures: The study was supported by the National Research Foundation Singapore, the Singapore Ministry of Education, and the National University of Singapore. The authors reported having no competing financial interests.
Source: Teoh PJ et al. Blood. 2018;132(12):1304-17.
FDA puts selinexor on fast track for DLBCL
The Food and Drug Administration has granted fast track designation to selinexor for the treatment of diffuse large B-cell lymphoma (DLBCL).
The designation is for selinexor to treat DLBCL patients who have received at least two prior therapies and who are not eligible for high-dose chemotherapy with stem cell rescue or chimeric antigen receptor (CAR) T-cell therapy.
Selinexor is being studied in the phase 2b SADAL trial (NCT02227251), which is enrolling patients with relapsed or refractory DLBCL who have received two to five prior therapies and are not eligible for stem cell transplant.
Top-line results from this trial are scheduled to be presented at the 2018 ASH Annual Meeting (Abstract 1677).
Selinexor is an oral selective inhibitor of nuclear export compound being developed by Karyopharm Therapeutics.
The company previously received fast track designation for selinexor to treat patients with penta-refractory multiple myeloma who have received at least three prior lines of therapy.
The FDA’s fast track program is designed to facilitate the development and expedite the review of products that are intended to treat serious conditions and have the potential to address unmet medical needs. Fast track designation provides developers with greater access to the FDA as well as eligibility for accelerated approval, priority review, and rolling review.
“Pending positive results from the phase 2b SADAL study, we plan to submit a second NDA [new drug application] to the FDA in the first half of 2019, with a request for accelerated approval, for oral selinexor as a potential new treatment for patients with relapsed or refractory DLBCL,” Sharon Shacham, PhD, founder, president, and chief scientific officer of Karyopharm, said in a statement.
In October, the FDA accepted an NDA for selinexor as a treatment for penta-refractory multiple myeloma. The agency granted the application priority review and set an action date of April 6, 2019.
The Food and Drug Administration has granted fast track designation to selinexor for the treatment of diffuse large B-cell lymphoma (DLBCL).
The designation is for selinexor to treat DLBCL patients who have received at least two prior therapies and who are not eligible for high-dose chemotherapy with stem cell rescue or chimeric antigen receptor (CAR) T-cell therapy.
Selinexor is being studied in the phase 2b SADAL trial (NCT02227251), which is enrolling patients with relapsed or refractory DLBCL who have received two to five prior therapies and are not eligible for stem cell transplant.
Top-line results from this trial are scheduled to be presented at the 2018 ASH Annual Meeting (Abstract 1677).
Selinexor is an oral selective inhibitor of nuclear export compound being developed by Karyopharm Therapeutics.
The company previously received fast track designation for selinexor to treat patients with penta-refractory multiple myeloma who have received at least three prior lines of therapy.
The FDA’s fast track program is designed to facilitate the development and expedite the review of products that are intended to treat serious conditions and have the potential to address unmet medical needs. Fast track designation provides developers with greater access to the FDA as well as eligibility for accelerated approval, priority review, and rolling review.
“Pending positive results from the phase 2b SADAL study, we plan to submit a second NDA [new drug application] to the FDA in the first half of 2019, with a request for accelerated approval, for oral selinexor as a potential new treatment for patients with relapsed or refractory DLBCL,” Sharon Shacham, PhD, founder, president, and chief scientific officer of Karyopharm, said in a statement.
In October, the FDA accepted an NDA for selinexor as a treatment for penta-refractory multiple myeloma. The agency granted the application priority review and set an action date of April 6, 2019.
The Food and Drug Administration has granted fast track designation to selinexor for the treatment of diffuse large B-cell lymphoma (DLBCL).
The designation is for selinexor to treat DLBCL patients who have received at least two prior therapies and who are not eligible for high-dose chemotherapy with stem cell rescue or chimeric antigen receptor (CAR) T-cell therapy.
Selinexor is being studied in the phase 2b SADAL trial (NCT02227251), which is enrolling patients with relapsed or refractory DLBCL who have received two to five prior therapies and are not eligible for stem cell transplant.
Top-line results from this trial are scheduled to be presented at the 2018 ASH Annual Meeting (Abstract 1677).
Selinexor is an oral selective inhibitor of nuclear export compound being developed by Karyopharm Therapeutics.
The company previously received fast track designation for selinexor to treat patients with penta-refractory multiple myeloma who have received at least three prior lines of therapy.
The FDA’s fast track program is designed to facilitate the development and expedite the review of products that are intended to treat serious conditions and have the potential to address unmet medical needs. Fast track designation provides developers with greater access to the FDA as well as eligibility for accelerated approval, priority review, and rolling review.
“Pending positive results from the phase 2b SADAL study, we plan to submit a second NDA [new drug application] to the FDA in the first half of 2019, with a request for accelerated approval, for oral selinexor as a potential new treatment for patients with relapsed or refractory DLBCL,” Sharon Shacham, PhD, founder, president, and chief scientific officer of Karyopharm, said in a statement.
In October, the FDA accepted an NDA for selinexor as a treatment for penta-refractory multiple myeloma. The agency granted the application priority review and set an action date of April 6, 2019.
Report details financial burden of blood cancers
with costs for acute leukemia almost tripling that amount, according to a new report from the Leukemia & Lymphoma Society (LLS).
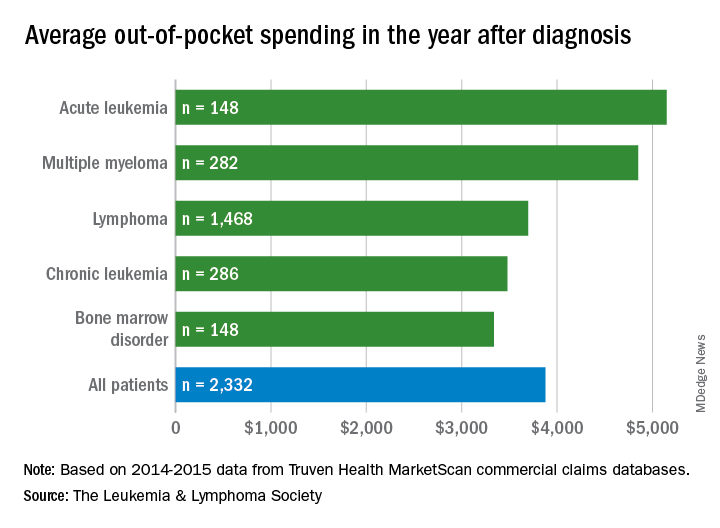
Total allowed cost – the average amount paid by the insurer and patient combined – for acute leukemia was more than $463,000 for the 12 months after initial diagnosis. Averages for the other four cancers included in the analysis came in at $214,000 for multiple myeloma, $134,000 for bone marrow disorders, $131,000 for lymphoma, and $89,000 for chronic leukemia, the LLS said.
The cost figures are drawn from claims data for 2,332 patients diagnosed in 2014.
Differences in out-of-pocket (OOP) costs were smaller, with the average for all patients at almost $3,900 in the year after diagnosis and acute leukemia coming in the highest at $5,100. Over time, however, OOP costs for multiple myeloma patients became the highest, totaling $9,100 for the 3 years after diagnosis, compared with $8,800 for acute leukemia and an average of less than $7,800 for the other blood cancers, the LLS said in the report, which was prepared by the actuarial firm Milliman.
OOP costs also varied by the type of plan. Patients in high-deductible plans averaged nearly $5,400 for the first year after diagnosis, compared with $3,300 for those with traditional insurance, the LLS noted. For acute leukemia, the OOP costs of high-deductible plans were more than twice as high as those of traditional plans.
The study was based on data for adults aged 18-64 years from the Truven Health MarketScan commercial claims databases for the years from 2013 to 2016. The LLS received support for the study from Pfizer, Genentech, and Amgen.
with costs for acute leukemia almost tripling that amount, according to a new report from the Leukemia & Lymphoma Society (LLS).
Total allowed cost – the average amount paid by the insurer and patient combined – for acute leukemia was more than $463,000 for the 12 months after initial diagnosis. Averages for the other four cancers included in the analysis came in at $214,000 for multiple myeloma, $134,000 for bone marrow disorders, $131,000 for lymphoma, and $89,000 for chronic leukemia, the LLS said.
The cost figures are drawn from claims data for 2,332 patients diagnosed in 2014.
Differences in out-of-pocket (OOP) costs were smaller, with the average for all patients at almost $3,900 in the year after diagnosis and acute leukemia coming in the highest at $5,100. Over time, however, OOP costs for multiple myeloma patients became the highest, totaling $9,100 for the 3 years after diagnosis, compared with $8,800 for acute leukemia and an average of less than $7,800 for the other blood cancers, the LLS said in the report, which was prepared by the actuarial firm Milliman.
OOP costs also varied by the type of plan. Patients in high-deductible plans averaged nearly $5,400 for the first year after diagnosis, compared with $3,300 for those with traditional insurance, the LLS noted. For acute leukemia, the OOP costs of high-deductible plans were more than twice as high as those of traditional plans.
The study was based on data for adults aged 18-64 years from the Truven Health MarketScan commercial claims databases for the years from 2013 to 2016. The LLS received support for the study from Pfizer, Genentech, and Amgen.
with costs for acute leukemia almost tripling that amount, according to a new report from the Leukemia & Lymphoma Society (LLS).
Total allowed cost – the average amount paid by the insurer and patient combined – for acute leukemia was more than $463,000 for the 12 months after initial diagnosis. Averages for the other four cancers included in the analysis came in at $214,000 for multiple myeloma, $134,000 for bone marrow disorders, $131,000 for lymphoma, and $89,000 for chronic leukemia, the LLS said.
The cost figures are drawn from claims data for 2,332 patients diagnosed in 2014.
Differences in out-of-pocket (OOP) costs were smaller, with the average for all patients at almost $3,900 in the year after diagnosis and acute leukemia coming in the highest at $5,100. Over time, however, OOP costs for multiple myeloma patients became the highest, totaling $9,100 for the 3 years after diagnosis, compared with $8,800 for acute leukemia and an average of less than $7,800 for the other blood cancers, the LLS said in the report, which was prepared by the actuarial firm Milliman.
OOP costs also varied by the type of plan. Patients in high-deductible plans averaged nearly $5,400 for the first year after diagnosis, compared with $3,300 for those with traditional insurance, the LLS noted. For acute leukemia, the OOP costs of high-deductible plans were more than twice as high as those of traditional plans.
The study was based on data for adults aged 18-64 years from the Truven Health MarketScan commercial claims databases for the years from 2013 to 2016. The LLS received support for the study from Pfizer, Genentech, and Amgen.
FDA clears portable hematology analyzer

The U.S. Food and Drug Administration (FDA) has granted 510(k) clearance for PixCell Medical’s HemoScreen™.
This portable hematology analyzer is used to perform a complete blood count at the point of care.
HemoScreen requires a single drop of blood and uses disposable cartridges that provide automatic sample preparation.
HemoScreen can analyze 20 standard complete blood count parameters and produces results within 5 minutes.
Study results suggested that HemoScreen provides results comparable to those of another hematology analyzer, Sysmex XE-2100. This study was published in the Journal of Clinical Pathology in 2016.
“The HemoScreen delivers lab-accurate results,” said Avishay Bransky, PhD, chief executive officer of PixCell Medical.
He added that HemoScreen “would be especially useful” in physicians’ offices, emergency rooms, intensive care units, oncology clinics, and remote locations.
HemoScreen makes use of a technology called viscoelastic focusing, which employs microfluidics and machine vision algorithms to analyze cells.
The U.S. Food and Drug Administration (FDA) has granted 510(k) clearance for PixCell Medical’s HemoScreen™.
This portable hematology analyzer is used to perform a complete blood count at the point of care.
HemoScreen requires a single drop of blood and uses disposable cartridges that provide automatic sample preparation.
HemoScreen can analyze 20 standard complete blood count parameters and produces results within 5 minutes.
Study results suggested that HemoScreen provides results comparable to those of another hematology analyzer, Sysmex XE-2100. This study was published in the Journal of Clinical Pathology in 2016.
“The HemoScreen delivers lab-accurate results,” said Avishay Bransky, PhD, chief executive officer of PixCell Medical.
He added that HemoScreen “would be especially useful” in physicians’ offices, emergency rooms, intensive care units, oncology clinics, and remote locations.
HemoScreen makes use of a technology called viscoelastic focusing, which employs microfluidics and machine vision algorithms to analyze cells.
The U.S. Food and Drug Administration (FDA) has granted 510(k) clearance for PixCell Medical’s HemoScreen™.
This portable hematology analyzer is used to perform a complete blood count at the point of care.
HemoScreen requires a single drop of blood and uses disposable cartridges that provide automatic sample preparation.
HemoScreen can analyze 20 standard complete blood count parameters and produces results within 5 minutes.
Study results suggested that HemoScreen provides results comparable to those of another hematology analyzer, Sysmex XE-2100. This study was published in the Journal of Clinical Pathology in 2016.
“The HemoScreen delivers lab-accurate results,” said Avishay Bransky, PhD, chief executive officer of PixCell Medical.
He added that HemoScreen “would be especially useful” in physicians’ offices, emergency rooms, intensive care units, oncology clinics, and remote locations.
HemoScreen makes use of a technology called viscoelastic focusing, which employs microfluidics and machine vision algorithms to analyze cells.
Selinexor on fast track for DLBCL

The U.S. Food and Drug Administration (FDA) has granted fast track designation to selinexor for the treatment of diffuse large B-cell lymphoma (DLBCL).
The designation is for selinexor to treat DLBCL patients who have received at least two prior therapies and are not eligible for high-dose chemotherapy with stem cell rescue or chimeric antigen receptor T-cell therapy.
Selinexor is being studied in the phase 2b SADAL trial (NCT02227251), which is enrolling patients with relapsed or refractory DLBCL who have received two to five prior therapies and are not eligible for stem cell transplant.
Top-line results from this trial are scheduled to be presented at the 2018 ASH Annual Meeting (abstract 1677).
Selinexor is an oral selective inhibitor of nuclear export (SINE) compound being developed by Karyopharm Therapeutics Inc.
The company previously received fast track designation for selinexor to treat patients with penta-refractory multiple myeloma who have received at least three prior lines of therapy.
The FDA says its fast track program is designed to facilitate the development and expedite the review of products that are intended to treat serious conditions and have the potential to address unmet medical needs.
Fast track designation provides developers with greater access to the FDA as well as eligibility for accelerated approval, priority review, and rolling review.
“The receipt of fast track designation from the FDA for selinexor in relapsed DLBCL underscores the great unmet medical need for this aggressive form of lymphoma,” said Sharon Shacham, PhD, founder, president, and chief scientific officer of Karyopharm.
“Pending positive results from the phase 2b SADAL study, we plan to submit a second NDA [new drug application] to the FDA in the first half of 2019, with a request for accelerated approval, for oral selinexor as a potential new treatment for patients with relapsed or refractory DLBCL.”
Last month, the FDA accepted a new drug application for selinexor as a treatment for penta-refractory multiple myeloma. The agency granted the application priority review and set an action date of April 6, 2019.
The U.S. Food and Drug Administration (FDA) has granted fast track designation to selinexor for the treatment of diffuse large B-cell lymphoma (DLBCL).
The designation is for selinexor to treat DLBCL patients who have received at least two prior therapies and are not eligible for high-dose chemotherapy with stem cell rescue or chimeric antigen receptor T-cell therapy.
Selinexor is being studied in the phase 2b SADAL trial (NCT02227251), which is enrolling patients with relapsed or refractory DLBCL who have received two to five prior therapies and are not eligible for stem cell transplant.
Top-line results from this trial are scheduled to be presented at the 2018 ASH Annual Meeting (abstract 1677).
Selinexor is an oral selective inhibitor of nuclear export (SINE) compound being developed by Karyopharm Therapeutics Inc.
The company previously received fast track designation for selinexor to treat patients with penta-refractory multiple myeloma who have received at least three prior lines of therapy.
The FDA says its fast track program is designed to facilitate the development and expedite the review of products that are intended to treat serious conditions and have the potential to address unmet medical needs.
Fast track designation provides developers with greater access to the FDA as well as eligibility for accelerated approval, priority review, and rolling review.
“The receipt of fast track designation from the FDA for selinexor in relapsed DLBCL underscores the great unmet medical need for this aggressive form of lymphoma,” said Sharon Shacham, PhD, founder, president, and chief scientific officer of Karyopharm.
“Pending positive results from the phase 2b SADAL study, we plan to submit a second NDA [new drug application] to the FDA in the first half of 2019, with a request for accelerated approval, for oral selinexor as a potential new treatment for patients with relapsed or refractory DLBCL.”
Last month, the FDA accepted a new drug application for selinexor as a treatment for penta-refractory multiple myeloma. The agency granted the application priority review and set an action date of April 6, 2019.
The U.S. Food and Drug Administration (FDA) has granted fast track designation to selinexor for the treatment of diffuse large B-cell lymphoma (DLBCL).
The designation is for selinexor to treat DLBCL patients who have received at least two prior therapies and are not eligible for high-dose chemotherapy with stem cell rescue or chimeric antigen receptor T-cell therapy.
Selinexor is being studied in the phase 2b SADAL trial (NCT02227251), which is enrolling patients with relapsed or refractory DLBCL who have received two to five prior therapies and are not eligible for stem cell transplant.
Top-line results from this trial are scheduled to be presented at the 2018 ASH Annual Meeting (abstract 1677).
Selinexor is an oral selective inhibitor of nuclear export (SINE) compound being developed by Karyopharm Therapeutics Inc.
The company previously received fast track designation for selinexor to treat patients with penta-refractory multiple myeloma who have received at least three prior lines of therapy.
The FDA says its fast track program is designed to facilitate the development and expedite the review of products that are intended to treat serious conditions and have the potential to address unmet medical needs.
Fast track designation provides developers with greater access to the FDA as well as eligibility for accelerated approval, priority review, and rolling review.
“The receipt of fast track designation from the FDA for selinexor in relapsed DLBCL underscores the great unmet medical need for this aggressive form of lymphoma,” said Sharon Shacham, PhD, founder, president, and chief scientific officer of Karyopharm.
“Pending positive results from the phase 2b SADAL study, we plan to submit a second NDA [new drug application] to the FDA in the first half of 2019, with a request for accelerated approval, for oral selinexor as a potential new treatment for patients with relapsed or refractory DLBCL.”
Last month, the FDA accepted a new drug application for selinexor as a treatment for penta-refractory multiple myeloma. The agency granted the application priority review and set an action date of April 6, 2019.
ICYMI: Elotuzumab reduces progression risk in lenalidomide-refractory multiple myeloma
Patients with multiple myeloma who did not respond to treatment with lenalidomide and a proteasome inhibitor had a significantly lower risk of progression or death when receiving elotuzumab plus pomalidomide and dexamethasone, compared with pomalidomide and dexamethasone alone (hazard ratio, 0.54; 95% confidence interval, 0.34-0.86; P = .008), according to results of a multicenter, randomized, open-label, phase 2 trial published in the New England Journal of Medicine 2018 Nov 7. doi: 10.1056/NEJMoa1805762.
Study results of ELOQUENT-3 were presented earlier this year at the Annual Congress of the European Hematology Association.
Patients with multiple myeloma who did not respond to treatment with lenalidomide and a proteasome inhibitor had a significantly lower risk of progression or death when receiving elotuzumab plus pomalidomide and dexamethasone, compared with pomalidomide and dexamethasone alone (hazard ratio, 0.54; 95% confidence interval, 0.34-0.86; P = .008), according to results of a multicenter, randomized, open-label, phase 2 trial published in the New England Journal of Medicine 2018 Nov 7. doi: 10.1056/NEJMoa1805762.
Study results of ELOQUENT-3 were presented earlier this year at the Annual Congress of the European Hematology Association.
Patients with multiple myeloma who did not respond to treatment with lenalidomide and a proteasome inhibitor had a significantly lower risk of progression or death when receiving elotuzumab plus pomalidomide and dexamethasone, compared with pomalidomide and dexamethasone alone (hazard ratio, 0.54; 95% confidence interval, 0.34-0.86; P = .008), according to results of a multicenter, randomized, open-label, phase 2 trial published in the New England Journal of Medicine 2018 Nov 7. doi: 10.1056/NEJMoa1805762.
Study results of ELOQUENT-3 were presented earlier this year at the Annual Congress of the European Hematology Association.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
FDA approves elotuzumab with pom/dex in refractory myeloma
The who have received at least two prior therapies, including lenalidomide and a proteasome inhibitor.
Elotuzumab is already approved in combination with lenalidomide and dexamethasone to treat adult myeloma patients who have received one to three prior therapies.
The FDA’s latest approval of elotuzumab is based on results from ELOQUENT-3. This phase 2 trial enrolled multiple myeloma patients who had refractory or relapsed disease and had received both lenalidomide and a proteasome inhibitor.
In the trial, patients were randomized to receive elotuzumab plus pomalidomide and dexamethasone (EPd, n = 60) or pomalidomide and dexamethasone (Pd, n = 57) in 28-day cycles until disease progression or unacceptable toxicity.
The overall response rate was 53.3% in the EPd arm and 26.3% in the Pd arm (P = .0029); the rate of complete response or stringent complete response was 8.3% and 1.8%, respectively.
Median progression-free survival was 10.25 months with EPd and 4.67 months with Pd (P = .0078).
Serious adverse events occurred in 22% of patients in the EPd arm and 15% in the Pd arm. The most frequent serious adverse events were pneumonia and respiratory tract infection.
Additional results from ELOQUENT-3 can be found in the full prescribing information for elotuzumab, which is available on the Empliciti website.
Bristol-Myers Squibb and AbbVie are codeveloping elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
The who have received at least two prior therapies, including lenalidomide and a proteasome inhibitor.
Elotuzumab is already approved in combination with lenalidomide and dexamethasone to treat adult myeloma patients who have received one to three prior therapies.
The FDA’s latest approval of elotuzumab is based on results from ELOQUENT-3. This phase 2 trial enrolled multiple myeloma patients who had refractory or relapsed disease and had received both lenalidomide and a proteasome inhibitor.
In the trial, patients were randomized to receive elotuzumab plus pomalidomide and dexamethasone (EPd, n = 60) or pomalidomide and dexamethasone (Pd, n = 57) in 28-day cycles until disease progression or unacceptable toxicity.
The overall response rate was 53.3% in the EPd arm and 26.3% in the Pd arm (P = .0029); the rate of complete response or stringent complete response was 8.3% and 1.8%, respectively.
Median progression-free survival was 10.25 months with EPd and 4.67 months with Pd (P = .0078).
Serious adverse events occurred in 22% of patients in the EPd arm and 15% in the Pd arm. The most frequent serious adverse events were pneumonia and respiratory tract infection.
Additional results from ELOQUENT-3 can be found in the full prescribing information for elotuzumab, which is available on the Empliciti website.
Bristol-Myers Squibb and AbbVie are codeveloping elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
The who have received at least two prior therapies, including lenalidomide and a proteasome inhibitor.
Elotuzumab is already approved in combination with lenalidomide and dexamethasone to treat adult myeloma patients who have received one to three prior therapies.
The FDA’s latest approval of elotuzumab is based on results from ELOQUENT-3. This phase 2 trial enrolled multiple myeloma patients who had refractory or relapsed disease and had received both lenalidomide and a proteasome inhibitor.
In the trial, patients were randomized to receive elotuzumab plus pomalidomide and dexamethasone (EPd, n = 60) or pomalidomide and dexamethasone (Pd, n = 57) in 28-day cycles until disease progression or unacceptable toxicity.
The overall response rate was 53.3% in the EPd arm and 26.3% in the Pd arm (P = .0029); the rate of complete response or stringent complete response was 8.3% and 1.8%, respectively.
Median progression-free survival was 10.25 months with EPd and 4.67 months with Pd (P = .0078).
Serious adverse events occurred in 22% of patients in the EPd arm and 15% in the Pd arm. The most frequent serious adverse events were pneumonia and respiratory tract infection.
Additional results from ELOQUENT-3 can be found in the full prescribing information for elotuzumab, which is available on the Empliciti website.
Bristol-Myers Squibb and AbbVie are codeveloping elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.