User login
Encouraging early results for CB-derived NK cells in MM

CHICAGO—Cord blood (CB) is a viable source of natural killer (NK) cells for adoptive cellular therapy for multiple myeloma (MM), according to a speaker at the 2018 ASCO Annual Meeting.
Ex-vivo expanded cord blood NK cells were well tolerated without significant graft-versus-host disease (GVHD) or cytokine release syndrome (CRS) in a phase 2 study.
Nina Shah, MD, of the University of California San Francisco, reported these results as abstract 8006.*
The phase 2 study (NCT01729091) included 33 patients with symptomatic MM who were appropriate candidates for autologous stem cell transplant (ASCT).
For each patient, investigators chose cord blood units with at least a 4/6 match at HLA-A, -B and –DR.
Prior to the autologous graft, patients received lenalidomide and melphalan. Lenalidomide was given based on preclinical data suggesting synergy between that immunomodulatory agent and NK cells, Dr Shah said.
Patients were a median age of 59 (range, 25 – 72), 36% had a history of progressive disease or relapse, and 73% had adverse cytogenetics/FISH, were ISS III, or had a history of progressive disease or relapse.
Results
Dr Shah observed that in a generally high-risk population, responses to treatment with cord blood NK cells in the setting of ASCT were “encouraging,” with 79% of patients achieving very good partial response (VGPR) or better.
Twenty-one patients (64%) achieved a complete response (CR) or near CR. And 61% achieved minimal residual disease (MRD) negativity by day 100.
Patients had an estimated 3-year progression-free survival of 52%.
Three patients died, all from disease progression, and 13 patients have progressed.
The investigators observed no infusional toxicities, no GVHD, no CRS, and no neurotoxicity.
One patient experienced graft failure and was rescued with an autologous back-up graft.
"We are able to detect the donor-derived NK cells up to 13 days after infusion,” Dr Shah said, “but I think a more sensitive analysis with flow chimerism will not only allow us to detect more patients, but also better interrogate them to truly understand the in vivo phenotype and activation status of these cells."
Dr Shah indicated she and her colleagues became interested in studying cord blood for NK cell therapy because it is a known source of hematopoietic cells that is immediately available, does not require donor manipulation, and has more flexibility in genetic matching.
Previously, Dr Shah and colleagues conducted a phase 1 study, in which 12 patients received cord blood NK cells up to a dose of 1 x 108 NK cells/kg. “This was determined to be adequate and safe to move on to the phase 2,” she said.
Despite encouraging results, more research needs to be done, according to Dr Shah. “I don't think this is the end-all, be-all for NK cell therapy.”
Some future directions include combination with antibody therapy, improving NK persistence in vivo using cytokine manipulation, and possibly engineering chimeric antigen receptor (CAR)-modified NK cells, Dr Shah observed.
It’s also possible that HLA match may not be needed: “If that is the case, we will truly have an off-the-shelf source of NK cells that we can apply more readily to various patients,” she said.
The study was supported by Celgene Corporation, Stading-Younger Cancer Research Foundation, and the MD Anderson High-Risk Multiple Myeloma Moonshot Project.
*Data in the presentation differ from the abstract.
CHICAGO—Cord blood (CB) is a viable source of natural killer (NK) cells for adoptive cellular therapy for multiple myeloma (MM), according to a speaker at the 2018 ASCO Annual Meeting.
Ex-vivo expanded cord blood NK cells were well tolerated without significant graft-versus-host disease (GVHD) or cytokine release syndrome (CRS) in a phase 2 study.
Nina Shah, MD, of the University of California San Francisco, reported these results as abstract 8006.*
The phase 2 study (NCT01729091) included 33 patients with symptomatic MM who were appropriate candidates for autologous stem cell transplant (ASCT).
For each patient, investigators chose cord blood units with at least a 4/6 match at HLA-A, -B and –DR.
Prior to the autologous graft, patients received lenalidomide and melphalan. Lenalidomide was given based on preclinical data suggesting synergy between that immunomodulatory agent and NK cells, Dr Shah said.
Patients were a median age of 59 (range, 25 – 72), 36% had a history of progressive disease or relapse, and 73% had adverse cytogenetics/FISH, were ISS III, or had a history of progressive disease or relapse.
Results
Dr Shah observed that in a generally high-risk population, responses to treatment with cord blood NK cells in the setting of ASCT were “encouraging,” with 79% of patients achieving very good partial response (VGPR) or better.
Twenty-one patients (64%) achieved a complete response (CR) or near CR. And 61% achieved minimal residual disease (MRD) negativity by day 100.
Patients had an estimated 3-year progression-free survival of 52%.
Three patients died, all from disease progression, and 13 patients have progressed.
The investigators observed no infusional toxicities, no GVHD, no CRS, and no neurotoxicity.
One patient experienced graft failure and was rescued with an autologous back-up graft.
"We are able to detect the donor-derived NK cells up to 13 days after infusion,” Dr Shah said, “but I think a more sensitive analysis with flow chimerism will not only allow us to detect more patients, but also better interrogate them to truly understand the in vivo phenotype and activation status of these cells."
Dr Shah indicated she and her colleagues became interested in studying cord blood for NK cell therapy because it is a known source of hematopoietic cells that is immediately available, does not require donor manipulation, and has more flexibility in genetic matching.
Previously, Dr Shah and colleagues conducted a phase 1 study, in which 12 patients received cord blood NK cells up to a dose of 1 x 108 NK cells/kg. “This was determined to be adequate and safe to move on to the phase 2,” she said.
Despite encouraging results, more research needs to be done, according to Dr Shah. “I don't think this is the end-all, be-all for NK cell therapy.”
Some future directions include combination with antibody therapy, improving NK persistence in vivo using cytokine manipulation, and possibly engineering chimeric antigen receptor (CAR)-modified NK cells, Dr Shah observed.
It’s also possible that HLA match may not be needed: “If that is the case, we will truly have an off-the-shelf source of NK cells that we can apply more readily to various patients,” she said.
The study was supported by Celgene Corporation, Stading-Younger Cancer Research Foundation, and the MD Anderson High-Risk Multiple Myeloma Moonshot Project.
*Data in the presentation differ from the abstract.
CHICAGO—Cord blood (CB) is a viable source of natural killer (NK) cells for adoptive cellular therapy for multiple myeloma (MM), according to a speaker at the 2018 ASCO Annual Meeting.
Ex-vivo expanded cord blood NK cells were well tolerated without significant graft-versus-host disease (GVHD) or cytokine release syndrome (CRS) in a phase 2 study.
Nina Shah, MD, of the University of California San Francisco, reported these results as abstract 8006.*
The phase 2 study (NCT01729091) included 33 patients with symptomatic MM who were appropriate candidates for autologous stem cell transplant (ASCT).
For each patient, investigators chose cord blood units with at least a 4/6 match at HLA-A, -B and –DR.
Prior to the autologous graft, patients received lenalidomide and melphalan. Lenalidomide was given based on preclinical data suggesting synergy between that immunomodulatory agent and NK cells, Dr Shah said.
Patients were a median age of 59 (range, 25 – 72), 36% had a history of progressive disease or relapse, and 73% had adverse cytogenetics/FISH, were ISS III, or had a history of progressive disease or relapse.
Results
Dr Shah observed that in a generally high-risk population, responses to treatment with cord blood NK cells in the setting of ASCT were “encouraging,” with 79% of patients achieving very good partial response (VGPR) or better.
Twenty-one patients (64%) achieved a complete response (CR) or near CR. And 61% achieved minimal residual disease (MRD) negativity by day 100.
Patients had an estimated 3-year progression-free survival of 52%.
Three patients died, all from disease progression, and 13 patients have progressed.
The investigators observed no infusional toxicities, no GVHD, no CRS, and no neurotoxicity.
One patient experienced graft failure and was rescued with an autologous back-up graft.
"We are able to detect the donor-derived NK cells up to 13 days after infusion,” Dr Shah said, “but I think a more sensitive analysis with flow chimerism will not only allow us to detect more patients, but also better interrogate them to truly understand the in vivo phenotype and activation status of these cells."
Dr Shah indicated she and her colleagues became interested in studying cord blood for NK cell therapy because it is a known source of hematopoietic cells that is immediately available, does not require donor manipulation, and has more flexibility in genetic matching.
Previously, Dr Shah and colleagues conducted a phase 1 study, in which 12 patients received cord blood NK cells up to a dose of 1 x 108 NK cells/kg. “This was determined to be adequate and safe to move on to the phase 2,” she said.
Despite encouraging results, more research needs to be done, according to Dr Shah. “I don't think this is the end-all, be-all for NK cell therapy.”
Some future directions include combination with antibody therapy, improving NK persistence in vivo using cytokine manipulation, and possibly engineering chimeric antigen receptor (CAR)-modified NK cells, Dr Shah observed.
It’s also possible that HLA match may not be needed: “If that is the case, we will truly have an off-the-shelf source of NK cells that we can apply more readily to various patients,” she said.
The study was supported by Celgene Corporation, Stading-Younger Cancer Research Foundation, and the MD Anderson High-Risk Multiple Myeloma Moonshot Project.
*Data in the presentation differ from the abstract.
CAR T-cell technology making headway in MM
Chimeric antigen receptor (CAR) T-cell technology has been successfully used in the treatment of hematologic malignancies such as leukemias and lymphomas. Now, this technology has come to the forefront in multiple myeloma (MM).
Researchers at the National Cancer Institute, led by James N. Kochenderfer, MD, generated CAR T cells expressing the B-cell maturation antigen (BCMA), which is uniquely found on MM cells.
In the first in humans study, 24 patients received CAR-BCMA T cells at doses ranging between 0.3 and 3 x 106 CAR+ T cells/kg. Sixteen patients were treated at the highest dose. These patients had a median of 9.5 lines of prior therapy and 63% were refractory to their last treatment.
Thirteen patients had responses that were either partial or better and the overall response rate was 81%. Median event-free survival was 31 weeks. At the time of the report, 6 patients still have ongoing responses.
Patient cases
The report highlights a case study of a patient who had a large abdominal mass that resolved on computed tomography imaging, with λ light chains decreasing dramatically and becoming undetectable after CAR T-cell infusion. Recovery of normal plasma cells was noticeable with λ chain increases, but the ratio of κ to λ ratio remained normal 6 months after CAR T-cell infusion.
Using immunohistochemistry staining of CS138 before and 2 months after CAR-BMCA infusion, the researchers found effective depletion in bone marrow plasma cells in patients who were evaluated.
In patients who responded to treatment, decline in serum BCMA was also observed. However, in a patient who later progressed, BCMA increases were seen, leading the researchers to suggest that BCMA may be a tumor marker for MM.
The researchers noted that peak levels of CAR T cells occurred between 7 and 14 days after infusion and highest levels were seen in patients who showed antimyeloma responses.
Toxicity
CAR T-cell technology is also associated with accompanying toxicities.
At the highest dose, cytokine release syndrome (CRS) was a substantial toxicity especially in 2 patients who had a significant MM burden: in one case 80% of bone marrow cells were MM plasma cells and in the other the MM burden was 90%.
Six of the 16 patients required vasopressor support for hypotension and 1 patient required mechanical ventilation. The researchers noted that CRS of grade 3/4 was seen in patients who had a higher MM plasma cell burden.
The researchers indicated there is room for improvement. “The importance of persistence of CAR T cells in treating MM requires additional study,” they stated.
The sometimes low or absence of BCMA expression on MM cells may prompt the search for other antigens. “Treatment outcomes varied substantially between patients, and much room for improvement remains in improving the durability of antimyeloma responses and in reducing toxicity,” they concluded.
The researchers reported their findings in the Journal of Clinical Oncology.
Chimeric antigen receptor (CAR) T-cell technology has been successfully used in the treatment of hematologic malignancies such as leukemias and lymphomas. Now, this technology has come to the forefront in multiple myeloma (MM).
Researchers at the National Cancer Institute, led by James N. Kochenderfer, MD, generated CAR T cells expressing the B-cell maturation antigen (BCMA), which is uniquely found on MM cells.
In the first in humans study, 24 patients received CAR-BCMA T cells at doses ranging between 0.3 and 3 x 106 CAR+ T cells/kg. Sixteen patients were treated at the highest dose. These patients had a median of 9.5 lines of prior therapy and 63% were refractory to their last treatment.
Thirteen patients had responses that were either partial or better and the overall response rate was 81%. Median event-free survival was 31 weeks. At the time of the report, 6 patients still have ongoing responses.
Patient cases
The report highlights a case study of a patient who had a large abdominal mass that resolved on computed tomography imaging, with λ light chains decreasing dramatically and becoming undetectable after CAR T-cell infusion. Recovery of normal plasma cells was noticeable with λ chain increases, but the ratio of κ to λ ratio remained normal 6 months after CAR T-cell infusion.
Using immunohistochemistry staining of CS138 before and 2 months after CAR-BMCA infusion, the researchers found effective depletion in bone marrow plasma cells in patients who were evaluated.
In patients who responded to treatment, decline in serum BCMA was also observed. However, in a patient who later progressed, BCMA increases were seen, leading the researchers to suggest that BCMA may be a tumor marker for MM.
The researchers noted that peak levels of CAR T cells occurred between 7 and 14 days after infusion and highest levels were seen in patients who showed antimyeloma responses.
Toxicity
CAR T-cell technology is also associated with accompanying toxicities.
At the highest dose, cytokine release syndrome (CRS) was a substantial toxicity especially in 2 patients who had a significant MM burden: in one case 80% of bone marrow cells were MM plasma cells and in the other the MM burden was 90%.
Six of the 16 patients required vasopressor support for hypotension and 1 patient required mechanical ventilation. The researchers noted that CRS of grade 3/4 was seen in patients who had a higher MM plasma cell burden.
The researchers indicated there is room for improvement. “The importance of persistence of CAR T cells in treating MM requires additional study,” they stated.
The sometimes low or absence of BCMA expression on MM cells may prompt the search for other antigens. “Treatment outcomes varied substantially between patients, and much room for improvement remains in improving the durability of antimyeloma responses and in reducing toxicity,” they concluded.
The researchers reported their findings in the Journal of Clinical Oncology.
Chimeric antigen receptor (CAR) T-cell technology has been successfully used in the treatment of hematologic malignancies such as leukemias and lymphomas. Now, this technology has come to the forefront in multiple myeloma (MM).
Researchers at the National Cancer Institute, led by James N. Kochenderfer, MD, generated CAR T cells expressing the B-cell maturation antigen (BCMA), which is uniquely found on MM cells.
In the first in humans study, 24 patients received CAR-BCMA T cells at doses ranging between 0.3 and 3 x 106 CAR+ T cells/kg. Sixteen patients were treated at the highest dose. These patients had a median of 9.5 lines of prior therapy and 63% were refractory to their last treatment.
Thirteen patients had responses that were either partial or better and the overall response rate was 81%. Median event-free survival was 31 weeks. At the time of the report, 6 patients still have ongoing responses.
Patient cases
The report highlights a case study of a patient who had a large abdominal mass that resolved on computed tomography imaging, with λ light chains decreasing dramatically and becoming undetectable after CAR T-cell infusion. Recovery of normal plasma cells was noticeable with λ chain increases, but the ratio of κ to λ ratio remained normal 6 months after CAR T-cell infusion.
Using immunohistochemistry staining of CS138 before and 2 months after CAR-BMCA infusion, the researchers found effective depletion in bone marrow plasma cells in patients who were evaluated.
In patients who responded to treatment, decline in serum BCMA was also observed. However, in a patient who later progressed, BCMA increases were seen, leading the researchers to suggest that BCMA may be a tumor marker for MM.
The researchers noted that peak levels of CAR T cells occurred between 7 and 14 days after infusion and highest levels were seen in patients who showed antimyeloma responses.
Toxicity
CAR T-cell technology is also associated with accompanying toxicities.
At the highest dose, cytokine release syndrome (CRS) was a substantial toxicity especially in 2 patients who had a significant MM burden: in one case 80% of bone marrow cells were MM plasma cells and in the other the MM burden was 90%.
Six of the 16 patients required vasopressor support for hypotension and 1 patient required mechanical ventilation. The researchers noted that CRS of grade 3/4 was seen in patients who had a higher MM plasma cell burden.
The researchers indicated there is room for improvement. “The importance of persistence of CAR T cells in treating MM requires additional study,” they stated.
The sometimes low or absence of BCMA expression on MM cells may prompt the search for other antigens. “Treatment outcomes varied substantially between patients, and much room for improvement remains in improving the durability of antimyeloma responses and in reducing toxicity,” they concluded.
The researchers reported their findings in the Journal of Clinical Oncology.
FDA approves first biosimilar pegfilgrastim

The US Food and Drug Association (FDA) has approved pegfilgrastim-jmdb (Fulphila™) as the first biosimilar to Neulasta®.
The agents reduce the risk of infection or the duration of febrile neutropenia in patients treated with immunosuppressive chemotherapy for non-myeloid hematologic malignancies.
The FDA approved Fulphila based on evidence that included extensive structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamic data, clinical immunogenicity data, and other clinical safety and effectiveness data.
The evidence demonstrated that Fulphila is biosimilar to Amgen’s Neulasta. The FDA, in its announcement, noted that Fulphila has been approved as a biosimilar and not as an interchangeable product.
A biosimilar is a biological product approved based on data showing it is highly similar to a biological product already approved by the FDA, termed the reference product.
A biosimilar has no clinically meaningful differences from the reference product in terms of safety, purity, and effectiveness.
Common side effects of Fulphila include bone pain and pain in extremities.
The FDA cautions that patients with a history of serious allergic reaction to human granulocyte colony-stimulating factors, such as pegfilgrastim or filgrastim products, should not take Fulphila.
Serious side effects from Fulphila include:
- rupture of the spleen
- acute respiratory distress syndrome
- serious allergic reactions including anaphylaxis
- glomerulonephritis
- leukocytosis
- capillary leak syndrome
- potential for tumor growth
Fatal sickle cell crises have also occurred with Fulphila use.
Fulphila is not indicated for the mobilization of peripheral blood progenitor cells for hematopoietic stem cell transplantation.
The FDA is planning to release a comprehensive new plan to advance policy efforts that promote biosimilar product development, according to FDA Commissioner Scott Gotlieb, MD.
“We want to make sure that the pathway for developing biosimilar versions of approved biologics is efficient and effective, so that patients benefit from competition to existing biologics once lawful intellectual property has lapsed on these products,” he said in the announcement.
The FDA granted approval of Fulphila to Mylan GmbH. Mylan is co-developing Fulphila with Biocon.
Last fall, the agency had issued a complete response letter saying it could not approve the proposed biosimilar pending an update to the application.
The complete response letter did not raise any questions on the biosimilarity of Fulphila (investigational drug product MYL-1401H), pharmacokinetic/pharmacodynamic data, clinical data, or immunogenicity, however.
Mylan anticipates launching Fulphila in the coming weeks.
The US Food and Drug Association (FDA) has approved pegfilgrastim-jmdb (Fulphila™) as the first biosimilar to Neulasta®.
The agents reduce the risk of infection or the duration of febrile neutropenia in patients treated with immunosuppressive chemotherapy for non-myeloid hematologic malignancies.
The FDA approved Fulphila based on evidence that included extensive structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamic data, clinical immunogenicity data, and other clinical safety and effectiveness data.
The evidence demonstrated that Fulphila is biosimilar to Amgen’s Neulasta. The FDA, in its announcement, noted that Fulphila has been approved as a biosimilar and not as an interchangeable product.
A biosimilar is a biological product approved based on data showing it is highly similar to a biological product already approved by the FDA, termed the reference product.
A biosimilar has no clinically meaningful differences from the reference product in terms of safety, purity, and effectiveness.
Common side effects of Fulphila include bone pain and pain in extremities.
The FDA cautions that patients with a history of serious allergic reaction to human granulocyte colony-stimulating factors, such as pegfilgrastim or filgrastim products, should not take Fulphila.
Serious side effects from Fulphila include:
- rupture of the spleen
- acute respiratory distress syndrome
- serious allergic reactions including anaphylaxis
- glomerulonephritis
- leukocytosis
- capillary leak syndrome
- potential for tumor growth
Fatal sickle cell crises have also occurred with Fulphila use.
Fulphila is not indicated for the mobilization of peripheral blood progenitor cells for hematopoietic stem cell transplantation.
The FDA is planning to release a comprehensive new plan to advance policy efforts that promote biosimilar product development, according to FDA Commissioner Scott Gotlieb, MD.
“We want to make sure that the pathway for developing biosimilar versions of approved biologics is efficient and effective, so that patients benefit from competition to existing biologics once lawful intellectual property has lapsed on these products,” he said in the announcement.
The FDA granted approval of Fulphila to Mylan GmbH. Mylan is co-developing Fulphila with Biocon.
Last fall, the agency had issued a complete response letter saying it could not approve the proposed biosimilar pending an update to the application.
The complete response letter did not raise any questions on the biosimilarity of Fulphila (investigational drug product MYL-1401H), pharmacokinetic/pharmacodynamic data, clinical data, or immunogenicity, however.
Mylan anticipates launching Fulphila in the coming weeks.
The US Food and Drug Association (FDA) has approved pegfilgrastim-jmdb (Fulphila™) as the first biosimilar to Neulasta®.
The agents reduce the risk of infection or the duration of febrile neutropenia in patients treated with immunosuppressive chemotherapy for non-myeloid hematologic malignancies.
The FDA approved Fulphila based on evidence that included extensive structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamic data, clinical immunogenicity data, and other clinical safety and effectiveness data.
The evidence demonstrated that Fulphila is biosimilar to Amgen’s Neulasta. The FDA, in its announcement, noted that Fulphila has been approved as a biosimilar and not as an interchangeable product.
A biosimilar is a biological product approved based on data showing it is highly similar to a biological product already approved by the FDA, termed the reference product.
A biosimilar has no clinically meaningful differences from the reference product in terms of safety, purity, and effectiveness.
Common side effects of Fulphila include bone pain and pain in extremities.
The FDA cautions that patients with a history of serious allergic reaction to human granulocyte colony-stimulating factors, such as pegfilgrastim or filgrastim products, should not take Fulphila.
Serious side effects from Fulphila include:
- rupture of the spleen
- acute respiratory distress syndrome
- serious allergic reactions including anaphylaxis
- glomerulonephritis
- leukocytosis
- capillary leak syndrome
- potential for tumor growth
Fatal sickle cell crises have also occurred with Fulphila use.
Fulphila is not indicated for the mobilization of peripheral blood progenitor cells for hematopoietic stem cell transplantation.
The FDA is planning to release a comprehensive new plan to advance policy efforts that promote biosimilar product development, according to FDA Commissioner Scott Gotlieb, MD.
“We want to make sure that the pathway for developing biosimilar versions of approved biologics is efficient and effective, so that patients benefit from competition to existing biologics once lawful intellectual property has lapsed on these products,” he said in the announcement.
The FDA granted approval of Fulphila to Mylan GmbH. Mylan is co-developing Fulphila with Biocon.
Last fall, the agency had issued a complete response letter saying it could not approve the proposed biosimilar pending an update to the application.
The complete response letter did not raise any questions on the biosimilarity of Fulphila (investigational drug product MYL-1401H), pharmacokinetic/pharmacodynamic data, clinical data, or immunogenicity, however.
Mylan anticipates launching Fulphila in the coming weeks.
When is denosumab an option in myeloma?
CHICAGO – , G. David Roodman, MD, PhD, reported at the annual meeting of the American Society of Clinical Oncology.
“We use denosumab in patients with compromised renal function,” said Dr. Roodman, director of the Division of Hematology-Oncology at Indiana University, Indianapolis, noting one such scenario. That use of denosumab echoes recently published ASCO guidelines on bone-modifying therapy.

The second scenario for denosumab use is in patients who aren’t tolerating bisphosphonates: “We switch them from zoledronic acid to pamidronate, and they still have terrible acute phase reactions,” Dr. Roodman said.
Dr. Roodman’s comments on use of denosumab were in response to an audience question about when he would use denosumab, given the considerable cost difference between the RANK ligand inhibitor and bisphosphonates.
The recent ASCO guidelines, of which Dr. Roodman is a coauthor, state that denosumab “is more expensive than zoledronic acid or pamidronate and must be considered in treatment decisions.”
Previously, ASCO guidelines recommended use of intravenous bisphosphonates for patients with myeloma and evidence of bone disease. Based on consideration of new evidence, the guideline authors eliminated the requirement for evidence of bone disease and added denosumab as an alternative treatment choice.
The addition of denosumab was based in part on results of a recent randomized phase 3 trial that comprised 1,718 myeloma patients who were treated with either denosumab or zoledronic acid.
The primary endpoint, time to first on-study skeletal-related event, was evaluated after 676 skeletal-related events had accrued on study. The investigators found no difference in time to first event (hazard ratio [HR], 0.98; 95% confidence interval, 0.85-1.14; P = 0.82).
Likewise, the secondary endpoint of overall survival showed no difference between arms (HR, 0.90; 95% CI, 0.70-1.16), though an exploratory analysis did suggest denosumab was superior on the endpoint of progression-free survival (HR, 0.82; 95% CI, 0.68-0.99).
The ASCO guidelines also recommend that clinicians consider less-frequent dosing in patients with responsive or stable disease. That recommendation is based on results of two studies of less-frequent dosing prompted by concerns over the risk of osteonecrosis of the jaw, an uncommon but potentially serious complication associated with bone-modifying agents.
Both studies suggested every-3-months dosing of zoledronic acid could be effective. However, Dr. Roodman noted that both studies had limitations that need to be considered, including small numbers of myeloma patients, limited duration of therapy studied, and a high dropout rate in the case of one study. Due to those limitations, “it’s very difficult to draw conclusions about this today,” Dr. Roodman said.
Dr. Roodman reported that he had a consulting or advisory role with Amgen.
CHICAGO – , G. David Roodman, MD, PhD, reported at the annual meeting of the American Society of Clinical Oncology.
“We use denosumab in patients with compromised renal function,” said Dr. Roodman, director of the Division of Hematology-Oncology at Indiana University, Indianapolis, noting one such scenario. That use of denosumab echoes recently published ASCO guidelines on bone-modifying therapy.
The second scenario for denosumab use is in patients who aren’t tolerating bisphosphonates: “We switch them from zoledronic acid to pamidronate, and they still have terrible acute phase reactions,” Dr. Roodman said.
Dr. Roodman’s comments on use of denosumab were in response to an audience question about when he would use denosumab, given the considerable cost difference between the RANK ligand inhibitor and bisphosphonates.
The recent ASCO guidelines, of which Dr. Roodman is a coauthor, state that denosumab “is more expensive than zoledronic acid or pamidronate and must be considered in treatment decisions.”
Previously, ASCO guidelines recommended use of intravenous bisphosphonates for patients with myeloma and evidence of bone disease. Based on consideration of new evidence, the guideline authors eliminated the requirement for evidence of bone disease and added denosumab as an alternative treatment choice.
The addition of denosumab was based in part on results of a recent randomized phase 3 trial that comprised 1,718 myeloma patients who were treated with either denosumab or zoledronic acid.
The primary endpoint, time to first on-study skeletal-related event, was evaluated after 676 skeletal-related events had accrued on study. The investigators found no difference in time to first event (hazard ratio [HR], 0.98; 95% confidence interval, 0.85-1.14; P = 0.82).
Likewise, the secondary endpoint of overall survival showed no difference between arms (HR, 0.90; 95% CI, 0.70-1.16), though an exploratory analysis did suggest denosumab was superior on the endpoint of progression-free survival (HR, 0.82; 95% CI, 0.68-0.99).
The ASCO guidelines also recommend that clinicians consider less-frequent dosing in patients with responsive or stable disease. That recommendation is based on results of two studies of less-frequent dosing prompted by concerns over the risk of osteonecrosis of the jaw, an uncommon but potentially serious complication associated with bone-modifying agents.
Both studies suggested every-3-months dosing of zoledronic acid could be effective. However, Dr. Roodman noted that both studies had limitations that need to be considered, including small numbers of myeloma patients, limited duration of therapy studied, and a high dropout rate in the case of one study. Due to those limitations, “it’s very difficult to draw conclusions about this today,” Dr. Roodman said.
Dr. Roodman reported that he had a consulting or advisory role with Amgen.
CHICAGO – , G. David Roodman, MD, PhD, reported at the annual meeting of the American Society of Clinical Oncology.
“We use denosumab in patients with compromised renal function,” said Dr. Roodman, director of the Division of Hematology-Oncology at Indiana University, Indianapolis, noting one such scenario. That use of denosumab echoes recently published ASCO guidelines on bone-modifying therapy.
The second scenario for denosumab use is in patients who aren’t tolerating bisphosphonates: “We switch them from zoledronic acid to pamidronate, and they still have terrible acute phase reactions,” Dr. Roodman said.
Dr. Roodman’s comments on use of denosumab were in response to an audience question about when he would use denosumab, given the considerable cost difference between the RANK ligand inhibitor and bisphosphonates.
The recent ASCO guidelines, of which Dr. Roodman is a coauthor, state that denosumab “is more expensive than zoledronic acid or pamidronate and must be considered in treatment decisions.”
Previously, ASCO guidelines recommended use of intravenous bisphosphonates for patients with myeloma and evidence of bone disease. Based on consideration of new evidence, the guideline authors eliminated the requirement for evidence of bone disease and added denosumab as an alternative treatment choice.
The addition of denosumab was based in part on results of a recent randomized phase 3 trial that comprised 1,718 myeloma patients who were treated with either denosumab or zoledronic acid.
The primary endpoint, time to first on-study skeletal-related event, was evaluated after 676 skeletal-related events had accrued on study. The investigators found no difference in time to first event (hazard ratio [HR], 0.98; 95% confidence interval, 0.85-1.14; P = 0.82).
Likewise, the secondary endpoint of overall survival showed no difference between arms (HR, 0.90; 95% CI, 0.70-1.16), though an exploratory analysis did suggest denosumab was superior on the endpoint of progression-free survival (HR, 0.82; 95% CI, 0.68-0.99).
The ASCO guidelines also recommend that clinicians consider less-frequent dosing in patients with responsive or stable disease. That recommendation is based on results of two studies of less-frequent dosing prompted by concerns over the risk of osteonecrosis of the jaw, an uncommon but potentially serious complication associated with bone-modifying agents.
Both studies suggested every-3-months dosing of zoledronic acid could be effective. However, Dr. Roodman noted that both studies had limitations that need to be considered, including small numbers of myeloma patients, limited duration of therapy studied, and a high dropout rate in the case of one study. Due to those limitations, “it’s very difficult to draw conclusions about this today,” Dr. Roodman said.
Dr. Roodman reported that he had a consulting or advisory role with Amgen.
EXPERT ANALYSIS FROM ASCO 2018
Over 1100 new meds, vaccines being developed to treat cancer

Currently, 1,120 new medicines and vaccines are being developed to treat cancer, according to a new report of the Pharmaceutical Research and Manufacturers of America (PhRMA).
And all of them, the organization states, are in clinical trials or awaiting review by the US Food and Drug Administration (FDA).
Leading the way are treatments for solid tumors, with 397 in development. Treatments for blood cancers are not far behind, with nearly 340 medicines in development: 137 for leukemias, 135 for lymphomas, and 62 for multiple myeloma.
Immuno-oncology and personalized medicine have a hand in this increase.
In the last year, according to PhRMA’s "Medicines in Development for Cancer 2018 Report," 47 new immune-oncology treatments have been added to the development pipeline, including CAR-T therapies and checkpoint inhibitors.
This brings the total to 295 immuno-oncology medicines and vaccines in the development pipeline this year.
The report also states that about 85% of these medicines in the oncology pipeline are first-in-class.
And PhRMA attributes the approximately 73% of survival gains in cancer to the new medicines.
Despite the bright picture, PhRMA acknowledges the financial burden and medical care challenges patients encounter.
It addresses them in a new chart pack, "Cancer Medicines: Value in Context," which puts cancer costs in perspective and offers solutions for improving the current system in the United States.
The association reports the top medical financial concerns of patients to be diagnostic tests or scans (53%), prescription medicines (43%), physician office visits (39%), outpatient treatments-including radiation (37%), and surgery (36%).
Spending on cancer medicines represents about 1% of overall healthcare spending, according to the organization, with cancer medications accounting for $49.8 billion of the $3.49 trillion healthcare spending in the United States.
Cancer medicines represent about 20% of spending on cancer, PhrMA notes, and some insurance plans place treatments for certain high-cost conditions on the highest drug formulary cost-sharing tier.
And patients with the highest copay were 5 times more likely to abandon treatment than the lowest copay group, PhRMA points out.
“No patient should struggle to afford their needed treatments,” PhRMA stated in a release, “and it is important that we address patient access challenges.”
Currently, 1,120 new medicines and vaccines are being developed to treat cancer, according to a new report of the Pharmaceutical Research and Manufacturers of America (PhRMA).
And all of them, the organization states, are in clinical trials or awaiting review by the US Food and Drug Administration (FDA).
Leading the way are treatments for solid tumors, with 397 in development. Treatments for blood cancers are not far behind, with nearly 340 medicines in development: 137 for leukemias, 135 for lymphomas, and 62 for multiple myeloma.
Immuno-oncology and personalized medicine have a hand in this increase.
In the last year, according to PhRMA’s "Medicines in Development for Cancer 2018 Report," 47 new immune-oncology treatments have been added to the development pipeline, including CAR-T therapies and checkpoint inhibitors.
This brings the total to 295 immuno-oncology medicines and vaccines in the development pipeline this year.
The report also states that about 85% of these medicines in the oncology pipeline are first-in-class.
And PhRMA attributes the approximately 73% of survival gains in cancer to the new medicines.
Despite the bright picture, PhRMA acknowledges the financial burden and medical care challenges patients encounter.
It addresses them in a new chart pack, "Cancer Medicines: Value in Context," which puts cancer costs in perspective and offers solutions for improving the current system in the United States.
The association reports the top medical financial concerns of patients to be diagnostic tests or scans (53%), prescription medicines (43%), physician office visits (39%), outpatient treatments-including radiation (37%), and surgery (36%).
Spending on cancer medicines represents about 1% of overall healthcare spending, according to the organization, with cancer medications accounting for $49.8 billion of the $3.49 trillion healthcare spending in the United States.
Cancer medicines represent about 20% of spending on cancer, PhrMA notes, and some insurance plans place treatments for certain high-cost conditions on the highest drug formulary cost-sharing tier.
And patients with the highest copay were 5 times more likely to abandon treatment than the lowest copay group, PhRMA points out.
“No patient should struggle to afford their needed treatments,” PhRMA stated in a release, “and it is important that we address patient access challenges.”
Currently, 1,120 new medicines and vaccines are being developed to treat cancer, according to a new report of the Pharmaceutical Research and Manufacturers of America (PhRMA).
And all of them, the organization states, are in clinical trials or awaiting review by the US Food and Drug Administration (FDA).
Leading the way are treatments for solid tumors, with 397 in development. Treatments for blood cancers are not far behind, with nearly 340 medicines in development: 137 for leukemias, 135 for lymphomas, and 62 for multiple myeloma.
Immuno-oncology and personalized medicine have a hand in this increase.
In the last year, according to PhRMA’s "Medicines in Development for Cancer 2018 Report," 47 new immune-oncology treatments have been added to the development pipeline, including CAR-T therapies and checkpoint inhibitors.
This brings the total to 295 immuno-oncology medicines and vaccines in the development pipeline this year.
The report also states that about 85% of these medicines in the oncology pipeline are first-in-class.
And PhRMA attributes the approximately 73% of survival gains in cancer to the new medicines.
Despite the bright picture, PhRMA acknowledges the financial burden and medical care challenges patients encounter.
It addresses them in a new chart pack, "Cancer Medicines: Value in Context," which puts cancer costs in perspective and offers solutions for improving the current system in the United States.
The association reports the top medical financial concerns of patients to be diagnostic tests or scans (53%), prescription medicines (43%), physician office visits (39%), outpatient treatments-including radiation (37%), and surgery (36%).
Spending on cancer medicines represents about 1% of overall healthcare spending, according to the organization, with cancer medications accounting for $49.8 billion of the $3.49 trillion healthcare spending in the United States.
Cancer medicines represent about 20% of spending on cancer, PhrMA notes, and some insurance plans place treatments for certain high-cost conditions on the highest drug formulary cost-sharing tier.
And patients with the highest copay were 5 times more likely to abandon treatment than the lowest copay group, PhRMA points out.
“No patient should struggle to afford their needed treatments,” PhRMA stated in a release, “and it is important that we address patient access challenges.”
Ruxolitinib overcame lenalidomide resistance in myeloma
based on phase I trial results presented at the annual meeting of the American Society of Clinical Oncology (ASCO).
The clinical trial is the first to demonstrate the activity of a JAK inhibitor in the treatment of myeloma patients, according to investigator James R. Berenson, MD, medical and scientific director for the Institute for Myeloma & Bone Cancer Research (IMBCR), West Hollywood, Calif.

“These promising results have led to the expansion of the current clinical trial and provide the basis for exploration of this and other JAK inhibitor-containing combinations for treating patients with myeloma and other malignant diseases,” he added.
The dose-escalation study enrolled 28 patients with relapsed/refractory multiple myeloma who had previous treatment with lenalidomide/steroids and a proteasome inhibitor. Subjects received ruxolitinib twice daily continuously, lenalidomide daily on days 1-21 of a 28-day cycle, and methylprednisolone orally every other day. A traditional 3+3 dose escalation design was used to enroll subjects in four cohorts. In DL0, patients received ruxolitinib 5 mg, lenalidomide 5 mg, and methylprednisolone 40 mg. In DL+1 and +2, both doses of lenalidomide and methylprednisolone remained unchanged, and ruxolitinib was escalated to 10 and 15 mg, respectively. In DL+3, lenalidomide was escalated to 10 mg with methylprednisolone unchanged and ruxolitinib at 15 mg. A total of 19 patients were treated at the highest dose level, which was ruxolitinib 15 mg twice daily on days 1-28, lenalidomide 10 mg daily on days 1-21, and methylprednisolone 40 mg every other day.
The overall response rate was 39% (10 of 26 evaluable patients), Dr. Berenson reported. The clinical benefit rate was 50% (13 of 26 patients), with a median duration of response of 5.6 months in that group.
There were no dose-limiting toxicities. Grade 3 toxicities reported included thrombocytopenia in 11% (3 patients, gastrointestinal bleeding in 11% (3 patients), and anemia in 7% (2 patients).
These results are encouraging, though challenges remain, said Craig Hofmeister, MD, MPH, of Winship Cancer Institute, Emory University, Atlanta.
“Clearly in these patients who are 100% lenalidomide refractory, the overall response rate of anything greater than or close to 20, 30, 40% is very appealing,” Dr. Hofmeister said in an ASCO presentation discussing the results of this trial.
The usual rationale for JAK inhibition is targeting of the bone microenvironment, but the microenvironment is a formidable opponent, Dr. Hofmeister said in his presentation.
“That’s an uphill battle,” he said. “There is an upcoming carfilzomib and ruxolitinib trial in multiple myeloma moving forward, and I’d be excited to see” the results.
The study (NCT03110822) was sponsored by Oncotherapeutics in collaboration with Incyte, the maker of ruxolitinib (Jakafi). Dr. Berenson, the presenting author, had disclosures related to Incyte, as well as Amgen, Bristol-Myers Squibb, Celgene, Janssen, Takeda, and OncoTracker.
SOURCE: Berenson JR, et al. J Clin Oncol 36, 2018 (suppl; abstr 8005).
based on phase I trial results presented at the annual meeting of the American Society of Clinical Oncology (ASCO).
The clinical trial is the first to demonstrate the activity of a JAK inhibitor in the treatment of myeloma patients, according to investigator James R. Berenson, MD, medical and scientific director for the Institute for Myeloma & Bone Cancer Research (IMBCR), West Hollywood, Calif.
“These promising results have led to the expansion of the current clinical trial and provide the basis for exploration of this and other JAK inhibitor-containing combinations for treating patients with myeloma and other malignant diseases,” he added.
The dose-escalation study enrolled 28 patients with relapsed/refractory multiple myeloma who had previous treatment with lenalidomide/steroids and a proteasome inhibitor. Subjects received ruxolitinib twice daily continuously, lenalidomide daily on days 1-21 of a 28-day cycle, and methylprednisolone orally every other day. A traditional 3+3 dose escalation design was used to enroll subjects in four cohorts. In DL0, patients received ruxolitinib 5 mg, lenalidomide 5 mg, and methylprednisolone 40 mg. In DL+1 and +2, both doses of lenalidomide and methylprednisolone remained unchanged, and ruxolitinib was escalated to 10 and 15 mg, respectively. In DL+3, lenalidomide was escalated to 10 mg with methylprednisolone unchanged and ruxolitinib at 15 mg. A total of 19 patients were treated at the highest dose level, which was ruxolitinib 15 mg twice daily on days 1-28, lenalidomide 10 mg daily on days 1-21, and methylprednisolone 40 mg every other day.
The overall response rate was 39% (10 of 26 evaluable patients), Dr. Berenson reported. The clinical benefit rate was 50% (13 of 26 patients), with a median duration of response of 5.6 months in that group.
There were no dose-limiting toxicities. Grade 3 toxicities reported included thrombocytopenia in 11% (3 patients, gastrointestinal bleeding in 11% (3 patients), and anemia in 7% (2 patients).
These results are encouraging, though challenges remain, said Craig Hofmeister, MD, MPH, of Winship Cancer Institute, Emory University, Atlanta.
“Clearly in these patients who are 100% lenalidomide refractory, the overall response rate of anything greater than or close to 20, 30, 40% is very appealing,” Dr. Hofmeister said in an ASCO presentation discussing the results of this trial.
The usual rationale for JAK inhibition is targeting of the bone microenvironment, but the microenvironment is a formidable opponent, Dr. Hofmeister said in his presentation.
“That’s an uphill battle,” he said. “There is an upcoming carfilzomib and ruxolitinib trial in multiple myeloma moving forward, and I’d be excited to see” the results.
The study (NCT03110822) was sponsored by Oncotherapeutics in collaboration with Incyte, the maker of ruxolitinib (Jakafi). Dr. Berenson, the presenting author, had disclosures related to Incyte, as well as Amgen, Bristol-Myers Squibb, Celgene, Janssen, Takeda, and OncoTracker.
SOURCE: Berenson JR, et al. J Clin Oncol 36, 2018 (suppl; abstr 8005).
based on phase I trial results presented at the annual meeting of the American Society of Clinical Oncology (ASCO).
The clinical trial is the first to demonstrate the activity of a JAK inhibitor in the treatment of myeloma patients, according to investigator James R. Berenson, MD, medical and scientific director for the Institute for Myeloma & Bone Cancer Research (IMBCR), West Hollywood, Calif.
“These promising results have led to the expansion of the current clinical trial and provide the basis for exploration of this and other JAK inhibitor-containing combinations for treating patients with myeloma and other malignant diseases,” he added.
The dose-escalation study enrolled 28 patients with relapsed/refractory multiple myeloma who had previous treatment with lenalidomide/steroids and a proteasome inhibitor. Subjects received ruxolitinib twice daily continuously, lenalidomide daily on days 1-21 of a 28-day cycle, and methylprednisolone orally every other day. A traditional 3+3 dose escalation design was used to enroll subjects in four cohorts. In DL0, patients received ruxolitinib 5 mg, lenalidomide 5 mg, and methylprednisolone 40 mg. In DL+1 and +2, both doses of lenalidomide and methylprednisolone remained unchanged, and ruxolitinib was escalated to 10 and 15 mg, respectively. In DL+3, lenalidomide was escalated to 10 mg with methylprednisolone unchanged and ruxolitinib at 15 mg. A total of 19 patients were treated at the highest dose level, which was ruxolitinib 15 mg twice daily on days 1-28, lenalidomide 10 mg daily on days 1-21, and methylprednisolone 40 mg every other day.
The overall response rate was 39% (10 of 26 evaluable patients), Dr. Berenson reported. The clinical benefit rate was 50% (13 of 26 patients), with a median duration of response of 5.6 months in that group.
There were no dose-limiting toxicities. Grade 3 toxicities reported included thrombocytopenia in 11% (3 patients, gastrointestinal bleeding in 11% (3 patients), and anemia in 7% (2 patients).
These results are encouraging, though challenges remain, said Craig Hofmeister, MD, MPH, of Winship Cancer Institute, Emory University, Atlanta.
“Clearly in these patients who are 100% lenalidomide refractory, the overall response rate of anything greater than or close to 20, 30, 40% is very appealing,” Dr. Hofmeister said in an ASCO presentation discussing the results of this trial.
The usual rationale for JAK inhibition is targeting of the bone microenvironment, but the microenvironment is a formidable opponent, Dr. Hofmeister said in his presentation.
“That’s an uphill battle,” he said. “There is an upcoming carfilzomib and ruxolitinib trial in multiple myeloma moving forward, and I’d be excited to see” the results.
The study (NCT03110822) was sponsored by Oncotherapeutics in collaboration with Incyte, the maker of ruxolitinib (Jakafi). Dr. Berenson, the presenting author, had disclosures related to Incyte, as well as Amgen, Bristol-Myers Squibb, Celgene, Janssen, Takeda, and OncoTracker.
SOURCE: Berenson JR, et al. J Clin Oncol 36, 2018 (suppl; abstr 8005).
REPORTING FROM ASCO 2018
Key clinical point: The JAK 1/2 inhibitor ruxolitinib, in combination with lenalidomide and methylprednisolone, overcame resistance to lenalidomide in about half of heavily pre-treated patients with relapsed/refractory multiple myeloma in a phase I trial.
Major finding: The overall response rate was 39%, and the clinical benefit rate was 50% (13 of 26 patients).
Study details: A phase 1 study including 28 patients with relapsed/refractory multiple myeloma who had previous treatment with lenalidomide/steroids and a proteasome inhibitor.
Disclosures: Dr. Berenson, the presenting author, had disclosures related to Amgen, Bristol-Myers Squibb, Celgene, Incyte, Janssen, Takeda, and OncoTracker.
Source: Berenson JR, et al. J Clin Oncol 36, 2018 (suppl; abstr 8005).
Liquid biopsies may be the future in managing MM patients
Scientists from the Dana-Farber Cancer Institute and the Broad Institute in Boston, Massachusetts, have shown that in multiple myeloma (MM), liquid biopsies provide similar genetic information as tumor biopsies.
Unlike tumor biopsies, liquid biopsies are noninvasive and, in the future, may offer a cost-effective way of following disease state, disease progression, and response to treatment.
The scientists used cell-free DNA (cfDNA) from 107 patients, circulating tumor cells (CTCs) from 56 patients, and compared their genomic profiling with bone marrow biopsies from 9 patients with MM.
The researchers used a two-step approach using the 2 kinds of liquid biopsies—CTCs and cfDNA. They first used “ultra-low pass” whole genome sequencing, which was a rapid and cost-effective method to identify blood samples with tumor DNA of at least 5%-10% (tumor fraction).
These samples were then subject to whole-exome sequencing (WES), which analyzes protein-coding sequence of the genome.
The researchers found genetic alterations in MM such as copy number alterations (1p, 13q deletions or 11q gain) and somatic single nucleotide variations (SSNVs), among others, which they observed in matched cfDNA, CTCs, and tumor DNA (tDNA) from tumor biopsies.
The liquid biopsies also captured the clonal heterogeneity characteristic of MM—99% of clonal mutations present in tDNA were also seen in cfDNA or CTCs, and 94% of mutations present in cfDNA or CTCs were seen in tDNA.
In addition, CTCs or cfDNA samples with higher tumor purity uncovered more mutations than were seen in tDNA.
“[The] combination of CTCs and cfDNA was able to detect almost all clonal mutations identified in the bone marrow biopsy sample and defined other subclones that were not identified in the bone marrow,” the scientists noted.
This increased sensitivity was “potentially due to the limitation of sampling site of the bone marrow,” they pointed out.
The scientists also showed that the tumor fraction in cfDNA and enriched CTCs correlated with disease progression from MGUS (monoclonal gammopathy of undetermined significance) and SMM (smoldering MM), to overt MM.
The scientists also evaluated liquid biopsies to determine response to treatment.
For example, a patient who responded to carfilzomib, lenalidomide, and dexamethasone, had a decreased cfDNA tumor fraction—from 22% to 2%.
Another patient who progressed on daratumumab over a period of 2 months showed an increase in tumor fraction—from 11% to 46%.
CTCs and cfDNA captured the mutational landscape of MM and provided a non-invasive profiling of tumor evolution. The liquid biopsy approach may be used as a novel biomarker for disease progression and response to therapy, the scientists suggest.
"Our discovery that cfDNA and CTC analyses agree with each other at the comprehensive level is an important finding,” said co-senior author Irene Ghobrial, MD, “because this means that routine genetic profiling of patient tumors from blood would be feasible."
"Our ultimate goal is to eventually use all the samples to monitor disease progression," added Jihye Park, PhD, researcher in the Ghobrial lab and co-first author on the paper.
Currently, bone marrow biopsies are the gold standard for diagnosing and monitoring the progression of MM.
However, due to their invasive nature and associated costs, they are not done at every visit. Liquid biopsies are an attractive way forward.
Nevertheless, before they can be used in the clinical management of patients with MM, these results need to be confirmed in larger prospective studies.
The investigators reported their findings in Nature Communications.
Scientists from the Dana-Farber Cancer Institute and the Broad Institute in Boston, Massachusetts, have shown that in multiple myeloma (MM), liquid biopsies provide similar genetic information as tumor biopsies.
Unlike tumor biopsies, liquid biopsies are noninvasive and, in the future, may offer a cost-effective way of following disease state, disease progression, and response to treatment.
The scientists used cell-free DNA (cfDNA) from 107 patients, circulating tumor cells (CTCs) from 56 patients, and compared their genomic profiling with bone marrow biopsies from 9 patients with MM.
The researchers used a two-step approach using the 2 kinds of liquid biopsies—CTCs and cfDNA. They first used “ultra-low pass” whole genome sequencing, which was a rapid and cost-effective method to identify blood samples with tumor DNA of at least 5%-10% (tumor fraction).
These samples were then subject to whole-exome sequencing (WES), which analyzes protein-coding sequence of the genome.
The researchers found genetic alterations in MM such as copy number alterations (1p, 13q deletions or 11q gain) and somatic single nucleotide variations (SSNVs), among others, which they observed in matched cfDNA, CTCs, and tumor DNA (tDNA) from tumor biopsies.
The liquid biopsies also captured the clonal heterogeneity characteristic of MM—99% of clonal mutations present in tDNA were also seen in cfDNA or CTCs, and 94% of mutations present in cfDNA or CTCs were seen in tDNA.
In addition, CTCs or cfDNA samples with higher tumor purity uncovered more mutations than were seen in tDNA.
“[The] combination of CTCs and cfDNA was able to detect almost all clonal mutations identified in the bone marrow biopsy sample and defined other subclones that were not identified in the bone marrow,” the scientists noted.
This increased sensitivity was “potentially due to the limitation of sampling site of the bone marrow,” they pointed out.
The scientists also showed that the tumor fraction in cfDNA and enriched CTCs correlated with disease progression from MGUS (monoclonal gammopathy of undetermined significance) and SMM (smoldering MM), to overt MM.
The scientists also evaluated liquid biopsies to determine response to treatment.
For example, a patient who responded to carfilzomib, lenalidomide, and dexamethasone, had a decreased cfDNA tumor fraction—from 22% to 2%.
Another patient who progressed on daratumumab over a period of 2 months showed an increase in tumor fraction—from 11% to 46%.
CTCs and cfDNA captured the mutational landscape of MM and provided a non-invasive profiling of tumor evolution. The liquid biopsy approach may be used as a novel biomarker for disease progression and response to therapy, the scientists suggest.
"Our discovery that cfDNA and CTC analyses agree with each other at the comprehensive level is an important finding,” said co-senior author Irene Ghobrial, MD, “because this means that routine genetic profiling of patient tumors from blood would be feasible."
"Our ultimate goal is to eventually use all the samples to monitor disease progression," added Jihye Park, PhD, researcher in the Ghobrial lab and co-first author on the paper.
Currently, bone marrow biopsies are the gold standard for diagnosing and monitoring the progression of MM.
However, due to their invasive nature and associated costs, they are not done at every visit. Liquid biopsies are an attractive way forward.
Nevertheless, before they can be used in the clinical management of patients with MM, these results need to be confirmed in larger prospective studies.
The investigators reported their findings in Nature Communications.
Scientists from the Dana-Farber Cancer Institute and the Broad Institute in Boston, Massachusetts, have shown that in multiple myeloma (MM), liquid biopsies provide similar genetic information as tumor biopsies.
Unlike tumor biopsies, liquid biopsies are noninvasive and, in the future, may offer a cost-effective way of following disease state, disease progression, and response to treatment.
The scientists used cell-free DNA (cfDNA) from 107 patients, circulating tumor cells (CTCs) from 56 patients, and compared their genomic profiling with bone marrow biopsies from 9 patients with MM.
The researchers used a two-step approach using the 2 kinds of liquid biopsies—CTCs and cfDNA. They first used “ultra-low pass” whole genome sequencing, which was a rapid and cost-effective method to identify blood samples with tumor DNA of at least 5%-10% (tumor fraction).
These samples were then subject to whole-exome sequencing (WES), which analyzes protein-coding sequence of the genome.
The researchers found genetic alterations in MM such as copy number alterations (1p, 13q deletions or 11q gain) and somatic single nucleotide variations (SSNVs), among others, which they observed in matched cfDNA, CTCs, and tumor DNA (tDNA) from tumor biopsies.
The liquid biopsies also captured the clonal heterogeneity characteristic of MM—99% of clonal mutations present in tDNA were also seen in cfDNA or CTCs, and 94% of mutations present in cfDNA or CTCs were seen in tDNA.
In addition, CTCs or cfDNA samples with higher tumor purity uncovered more mutations than were seen in tDNA.
“[The] combination of CTCs and cfDNA was able to detect almost all clonal mutations identified in the bone marrow biopsy sample and defined other subclones that were not identified in the bone marrow,” the scientists noted.
This increased sensitivity was “potentially due to the limitation of sampling site of the bone marrow,” they pointed out.
The scientists also showed that the tumor fraction in cfDNA and enriched CTCs correlated with disease progression from MGUS (monoclonal gammopathy of undetermined significance) and SMM (smoldering MM), to overt MM.
The scientists also evaluated liquid biopsies to determine response to treatment.
For example, a patient who responded to carfilzomib, lenalidomide, and dexamethasone, had a decreased cfDNA tumor fraction—from 22% to 2%.
Another patient who progressed on daratumumab over a period of 2 months showed an increase in tumor fraction—from 11% to 46%.
CTCs and cfDNA captured the mutational landscape of MM and provided a non-invasive profiling of tumor evolution. The liquid biopsy approach may be used as a novel biomarker for disease progression and response to therapy, the scientists suggest.
"Our discovery that cfDNA and CTC analyses agree with each other at the comprehensive level is an important finding,” said co-senior author Irene Ghobrial, MD, “because this means that routine genetic profiling of patient tumors from blood would be feasible."
"Our ultimate goal is to eventually use all the samples to monitor disease progression," added Jihye Park, PhD, researcher in the Ghobrial lab and co-first author on the paper.
Currently, bone marrow biopsies are the gold standard for diagnosing and monitoring the progression of MM.
However, due to their invasive nature and associated costs, they are not done at every visit. Liquid biopsies are an attractive way forward.
Nevertheless, before they can be used in the clinical management of patients with MM, these results need to be confirmed in larger prospective studies.
The investigators reported their findings in Nature Communications.
CAR T therapy to enter early testing in multiple myeloma
Janssen Biotech is launching a phase 1b/2 trial of an .
The trial, which was cleared by the Food and Drug Administration to begin in the second half of 2018, will evaluate the safety and efficacy of LCAR-B38M (JNJ-68284528). The CAR T therapy targets B-cell Maturation Antigen and expresses a CAR protein that is identical to a product that was developed by Legend Biotech and evaluated in a first-in-human clinical study in China.
The drug is being developed as part of a collaboration between Legend Biotech and Janssen Biotech.
Janssen Biotech is launching a phase 1b/2 trial of an .
The trial, which was cleared by the Food and Drug Administration to begin in the second half of 2018, will evaluate the safety and efficacy of LCAR-B38M (JNJ-68284528). The CAR T therapy targets B-cell Maturation Antigen and expresses a CAR protein that is identical to a product that was developed by Legend Biotech and evaluated in a first-in-human clinical study in China.
The drug is being developed as part of a collaboration between Legend Biotech and Janssen Biotech.
Janssen Biotech is launching a phase 1b/2 trial of an .
The trial, which was cleared by the Food and Drug Administration to begin in the second half of 2018, will evaluate the safety and efficacy of LCAR-B38M (JNJ-68284528). The CAR T therapy targets B-cell Maturation Antigen and expresses a CAR protein that is identical to a product that was developed by Legend Biotech and evaluated in a first-in-human clinical study in China.
The drug is being developed as part of a collaboration between Legend Biotech and Janssen Biotech.
Study of daratumamb with anti-PD-1 antibody in MM discontinued
Janssen is discontinuing the phase 1 MMY2036 study of daratumumab in combination with the anti PD-1 antibody JNJ-63723283 in patients with multiple myeloma (MM).
Janssen made the decision based on a Data Monitoring Committee review of Genmab’s phase 1b/2 study (LUC2001) of daratumumab plus the anti-PD-L1 antibody atezolizumab in non-small cell lung cancer (NSCLC).
Based on the DMC findings, Janssen also decided to discontinue its daratumumab-PD-1 combination study.
Janssen has an exclusive worldwide license from Genmab to develop, manufacture, and commercialize daratumumab.
In the planned review, the DMC determined there was no observed benefit within the daratumumab plus atezolizumab arm compared to the atezolizumab monotherapy arm. The DMC recommended termination of the NSCLC study.
The DMC also noted an increase in mortality-related events in the combination arm.
Janssen has informed health authorities about these events and has contacted its partner companies conducting daratumumab and anti-PD-1 combination studies to discuss ceasing enrollment and dosing of the combination while the data is being further investigated.
MMY2036 study (NCT03357952)
The randomized, multicenter, multiphase study was expected to enroll up to 386 patients with relapsed or refractory MM who had received at least 3 prior lines of therapy including a proteasome inhibitor (PI) and an immunomodulatory (IMiD) agent. Refractory patients had to be double refractory to both a PI and an IMiD.
The trial was to be conducted in 3 parts. Part 1 was to assess the safety of the combination of JNJ-63723283 and daratumumab. Part 2 was intended to compare the overall response rate in patients treated with the combination compared to those treated with daratumumab alone. And Part 3 was to compare progression-free survival between the 2 arms.
Daratumumab dose was planned to be 16 mg/kg weekly for 8 weeks, then once every other week for 16 weeks; then once every 4 weeks.
JNJ-63723283 dose was planned to be 240 milligrams IV fixed dose during week 1 on cycle 1 (28 days) day 2, cycle 1 day 15, then every 2 weeks thereafter.
The study was started in November 2017 and planned to be completed in December 2019.
In a news release, Genmab’s chief executive officer, Jan van de Winkel, PhD, expressed disappointment that the studies will be discontinued. He said Genmab “fully supports Janssen’s decision as patient safety is paramount in drug development. We look forward to gaining a better understanding of the data upon further analysis.”
Janssen is discontinuing the phase 1 MMY2036 study of daratumumab in combination with the anti PD-1 antibody JNJ-63723283 in patients with multiple myeloma (MM).
Janssen made the decision based on a Data Monitoring Committee review of Genmab’s phase 1b/2 study (LUC2001) of daratumumab plus the anti-PD-L1 antibody atezolizumab in non-small cell lung cancer (NSCLC).
Based on the DMC findings, Janssen also decided to discontinue its daratumumab-PD-1 combination study.
Janssen has an exclusive worldwide license from Genmab to develop, manufacture, and commercialize daratumumab.
In the planned review, the DMC determined there was no observed benefit within the daratumumab plus atezolizumab arm compared to the atezolizumab monotherapy arm. The DMC recommended termination of the NSCLC study.
The DMC also noted an increase in mortality-related events in the combination arm.
Janssen has informed health authorities about these events and has contacted its partner companies conducting daratumumab and anti-PD-1 combination studies to discuss ceasing enrollment and dosing of the combination while the data is being further investigated.
MMY2036 study (NCT03357952)
The randomized, multicenter, multiphase study was expected to enroll up to 386 patients with relapsed or refractory MM who had received at least 3 prior lines of therapy including a proteasome inhibitor (PI) and an immunomodulatory (IMiD) agent. Refractory patients had to be double refractory to both a PI and an IMiD.
The trial was to be conducted in 3 parts. Part 1 was to assess the safety of the combination of JNJ-63723283 and daratumumab. Part 2 was intended to compare the overall response rate in patients treated with the combination compared to those treated with daratumumab alone. And Part 3 was to compare progression-free survival between the 2 arms.
Daratumumab dose was planned to be 16 mg/kg weekly for 8 weeks, then once every other week for 16 weeks; then once every 4 weeks.
JNJ-63723283 dose was planned to be 240 milligrams IV fixed dose during week 1 on cycle 1 (28 days) day 2, cycle 1 day 15, then every 2 weeks thereafter.
The study was started in November 2017 and planned to be completed in December 2019.
In a news release, Genmab’s chief executive officer, Jan van de Winkel, PhD, expressed disappointment that the studies will be discontinued. He said Genmab “fully supports Janssen’s decision as patient safety is paramount in drug development. We look forward to gaining a better understanding of the data upon further analysis.”
Janssen is discontinuing the phase 1 MMY2036 study of daratumumab in combination with the anti PD-1 antibody JNJ-63723283 in patients with multiple myeloma (MM).
Janssen made the decision based on a Data Monitoring Committee review of Genmab’s phase 1b/2 study (LUC2001) of daratumumab plus the anti-PD-L1 antibody atezolizumab in non-small cell lung cancer (NSCLC).
Based on the DMC findings, Janssen also decided to discontinue its daratumumab-PD-1 combination study.
Janssen has an exclusive worldwide license from Genmab to develop, manufacture, and commercialize daratumumab.
In the planned review, the DMC determined there was no observed benefit within the daratumumab plus atezolizumab arm compared to the atezolizumab monotherapy arm. The DMC recommended termination of the NSCLC study.
The DMC also noted an increase in mortality-related events in the combination arm.
Janssen has informed health authorities about these events and has contacted its partner companies conducting daratumumab and anti-PD-1 combination studies to discuss ceasing enrollment and dosing of the combination while the data is being further investigated.
MMY2036 study (NCT03357952)
The randomized, multicenter, multiphase study was expected to enroll up to 386 patients with relapsed or refractory MM who had received at least 3 prior lines of therapy including a proteasome inhibitor (PI) and an immunomodulatory (IMiD) agent. Refractory patients had to be double refractory to both a PI and an IMiD.
The trial was to be conducted in 3 parts. Part 1 was to assess the safety of the combination of JNJ-63723283 and daratumumab. Part 2 was intended to compare the overall response rate in patients treated with the combination compared to those treated with daratumumab alone. And Part 3 was to compare progression-free survival between the 2 arms.
Daratumumab dose was planned to be 16 mg/kg weekly for 8 weeks, then once every other week for 16 weeks; then once every 4 weeks.
JNJ-63723283 dose was planned to be 240 milligrams IV fixed dose during week 1 on cycle 1 (28 days) day 2, cycle 1 day 15, then every 2 weeks thereafter.
The study was started in November 2017 and planned to be completed in December 2019.
In a news release, Genmab’s chief executive officer, Jan van de Winkel, PhD, expressed disappointment that the studies will be discontinued. He said Genmab “fully supports Janssen’s decision as patient safety is paramount in drug development. We look forward to gaining a better understanding of the data upon further analysis.”
Multiple myeloma rates rising fastest in East Asia
The global incidence of multiple myeloma rose by 126% from 1990 to 2016, with the largest regional increases occurring in East Asia and tropical Latin America, according to data from the Global Burden of Disease 2016 study.
East Asia (China, North Korea, and Taiwan) saw incident cases of multiple myeloma jump by 262% – up to 1.0 per 100,000 population – from 1990 to 2016, which was the largest increase among any of the 21 global regions; tropical Latin America’s 256% rise took its age-standardized incidence rate to 1.8 per 100,000. Worldwide, incidence of multiple myeloma was 2.1 cases per 100,000 in 2016, Andrew J. Cowan, MD, and his associates reported in JAMA Oncology.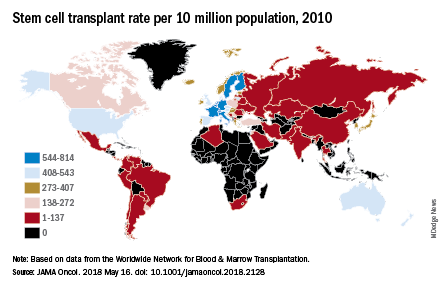
They also looked at treatment availability, with data on stem cell transplants for 2010 coming from the Worldwide Network for Blood & Marrow Transplantation (Lancet Haematol. 2015 Mar;2[3]:e91-100). The countries with the highest rates for all indications that year were Israel (814 per 10 million population), Italy (671), Germany (665), Sweden (625), and the Netherlands (614).
“Some regions of the world lack access to stem cell transplantation entirely, particularly sub-Saharan Africa (with the exception of South Africa),” wrote Dr. Cowan of the University of Washington, Seattle, and his associates.
The approval status of lenalidomide (Revlimid) and bortezomib (Velcade) in 2016 was used as a surrogate for availability of drug treatment: Lenalidomide had been approved in 73 countries out of 195 countries and territories and bortezomib in 103 countries. “On a global level, there are marked discrepancies in the availability of effective therapies. In addition to ensuring universal access to health care … it is imperative to at least ensure access to highly effective medications,” they wrote.
Dr. Cowan reported that he has received research funding from Janssen and AbbVie.
SOURCE: Cowan AJ et al. JAMA Oncol. 2018 May 16. doi: 10.1001/jamaoncol.2018.2128.
The global incidence of multiple myeloma rose by 126% from 1990 to 2016, with the largest regional increases occurring in East Asia and tropical Latin America, according to data from the Global Burden of Disease 2016 study.
East Asia (China, North Korea, and Taiwan) saw incident cases of multiple myeloma jump by 262% – up to 1.0 per 100,000 population – from 1990 to 2016, which was the largest increase among any of the 21 global regions; tropical Latin America’s 256% rise took its age-standardized incidence rate to 1.8 per 100,000. Worldwide, incidence of multiple myeloma was 2.1 cases per 100,000 in 2016, Andrew J. Cowan, MD, and his associates reported in JAMA Oncology.
They also looked at treatment availability, with data on stem cell transplants for 2010 coming from the Worldwide Network for Blood & Marrow Transplantation (Lancet Haematol. 2015 Mar;2[3]:e91-100). The countries with the highest rates for all indications that year were Israel (814 per 10 million population), Italy (671), Germany (665), Sweden (625), and the Netherlands (614).
“Some regions of the world lack access to stem cell transplantation entirely, particularly sub-Saharan Africa (with the exception of South Africa),” wrote Dr. Cowan of the University of Washington, Seattle, and his associates.
The approval status of lenalidomide (Revlimid) and bortezomib (Velcade) in 2016 was used as a surrogate for availability of drug treatment: Lenalidomide had been approved in 73 countries out of 195 countries and territories and bortezomib in 103 countries. “On a global level, there are marked discrepancies in the availability of effective therapies. In addition to ensuring universal access to health care … it is imperative to at least ensure access to highly effective medications,” they wrote.
Dr. Cowan reported that he has received research funding from Janssen and AbbVie.
SOURCE: Cowan AJ et al. JAMA Oncol. 2018 May 16. doi: 10.1001/jamaoncol.2018.2128.
The global incidence of multiple myeloma rose by 126% from 1990 to 2016, with the largest regional increases occurring in East Asia and tropical Latin America, according to data from the Global Burden of Disease 2016 study.
East Asia (China, North Korea, and Taiwan) saw incident cases of multiple myeloma jump by 262% – up to 1.0 per 100,000 population – from 1990 to 2016, which was the largest increase among any of the 21 global regions; tropical Latin America’s 256% rise took its age-standardized incidence rate to 1.8 per 100,000. Worldwide, incidence of multiple myeloma was 2.1 cases per 100,000 in 2016, Andrew J. Cowan, MD, and his associates reported in JAMA Oncology.
They also looked at treatment availability, with data on stem cell transplants for 2010 coming from the Worldwide Network for Blood & Marrow Transplantation (Lancet Haematol. 2015 Mar;2[3]:e91-100). The countries with the highest rates for all indications that year were Israel (814 per 10 million population), Italy (671), Germany (665), Sweden (625), and the Netherlands (614).
“Some regions of the world lack access to stem cell transplantation entirely, particularly sub-Saharan Africa (with the exception of South Africa),” wrote Dr. Cowan of the University of Washington, Seattle, and his associates.
The approval status of lenalidomide (Revlimid) and bortezomib (Velcade) in 2016 was used as a surrogate for availability of drug treatment: Lenalidomide had been approved in 73 countries out of 195 countries and territories and bortezomib in 103 countries. “On a global level, there are marked discrepancies in the availability of effective therapies. In addition to ensuring universal access to health care … it is imperative to at least ensure access to highly effective medications,” they wrote.
Dr. Cowan reported that he has received research funding from Janssen and AbbVie.
SOURCE: Cowan AJ et al. JAMA Oncol. 2018 May 16. doi: 10.1001/jamaoncol.2018.2128.
FROM JAMA ONCOLOGY