User login
Parity laws don’t lower oral cancer drug costs for everyone

US state laws intended to ensure fair prices for oral cancer drugs have had a mixed impact on patients’ pocketbooks, according to a study published in JAMA Oncology.
A total of 43 states and Washington, DC, have enacted parity laws, which require that patients pay no more for an oral cancer treatment than they would for an infusion of the same treatment.
Researchers analyzed the impact of these laws and observed modest improvements in costs for some patients.
However, patients who were already paying the most for their medications saw their monthly costs go up.
“Although parity laws appear to help reduce out-of-pocket spending for some patients, they may not fully address affordability for patients needing cancer drugs,” said study author Stacie B. Dusetzina, PhD, of the University of North Carolina at Chapel Hill.
“We need to consider ways to address drug pricing directly and to improve benefit design to make sure that all patients can access prescribed drugs.”
To gauge the impact of parity laws on treatment costs, Dr Dusetzina and her colleagues analyzed health claims data for 63,780 adults from 3 large, nationwide insurance companies before and after the laws were enacted, from 2008 to 2012.
The team compared the cost of filling an oral cancer drug prescription for patients with health insurance plans that were covered by the state laws (fully insured) and patients whose plans were not (self-funded). All patients lived in 1 of 16 states that had passed parity laws at the time of the study.
About half of patients (51.4%) had fully insured plans, and the other half (48.6%) had self-funded plans.
For the entire cohort, the use of oral cancer drugs increased from 18% in the months before parity laws were passed to 22% in the months after (adjusted difference-in-differences risk ratio [aDDRR], 1.04; 95% confidence interval [CI], 0.96-1.13; P=0.34).
The proportion of prescription fills for oral drugs without copayment increased from 15.0% to 53.0% for patients with fully insured plans and from 12.3% to 18.0% in patients with self-funded plans (aDDRR, 2.36; 95% CI, 2.00-2.79; P<0.001).
“From our results, it looks like many plans decided they would just set the co-pay for oral drugs to $0,” Dr Dusetzina said. “Instead of $30 per month, those fills were now $0.”
The proportion of prescription fills with out-of-pocket cost of more than $100 per month increased from 8.4% to 11.1% for patients with fully insured plans but decreased from 12.0% to 11.7% for those with self-funded plans (aDDRR, 1.36; 95% CI, 1.11-1.68; P=0.004).
Patients paying the most for their oral cancer drug prescriptions experienced increases in their monthly out-of-pocket costs after parity laws were passed.
For patients whose costs were more expensive than 95% of other patients, their out-of-pocket costs increased an estimated $143.25 per month. Those paying more than 90% of what other patients paid saw their costs increase by $37.19 per month.
“One of the biggest problems with parity laws as they are written is that they don’t address the prices of these medications, which can be very high,” Dr Dusetzina noted.
“Parity can be reached as long as the coverage is the same for both oral and infused cancer therapies. Because we’re now seeing more people insured by plans with high deductibles or plans that require them to pay a percentage of their drug costs, parity may not reduce spending for some patients.”
However, Dr Dusetzina and her colleagues did find that patients who paid the least for their oral cancer treatments saw their estimated monthly out-of-pocket spending decrease.
Patients in the 25th percentile saw an estimated decrease of $19.44 per month, those in the 50th percentile saw a $32.13 decrease, and patients in the 75th percentile saw a decrease of $10.83.
On the other hand, the researchers also found that average 6-month healthcare costs—including what was paid by insurance companies and patients—did not change significantly as a result of parity laws.
The aDDRR was 0.96 (95% CI, 0.90-1.02; P=0.09) for all cancer treatments and 1.06 (95% CI, 0.93-1.20; P=0.40) for oral cancer drugs.
“One of the key arguments against passing parity, both for states that haven’t passed it and for legislation at the federal level, has been that it may increase costs to health plans,” Dr Dusetzina said. “But we didn’t find evidence of that.” ![]()
US state laws intended to ensure fair prices for oral cancer drugs have had a mixed impact on patients’ pocketbooks, according to a study published in JAMA Oncology.
A total of 43 states and Washington, DC, have enacted parity laws, which require that patients pay no more for an oral cancer treatment than they would for an infusion of the same treatment.
Researchers analyzed the impact of these laws and observed modest improvements in costs for some patients.
However, patients who were already paying the most for their medications saw their monthly costs go up.
“Although parity laws appear to help reduce out-of-pocket spending for some patients, they may not fully address affordability for patients needing cancer drugs,” said study author Stacie B. Dusetzina, PhD, of the University of North Carolina at Chapel Hill.
“We need to consider ways to address drug pricing directly and to improve benefit design to make sure that all patients can access prescribed drugs.”
To gauge the impact of parity laws on treatment costs, Dr Dusetzina and her colleagues analyzed health claims data for 63,780 adults from 3 large, nationwide insurance companies before and after the laws were enacted, from 2008 to 2012.
The team compared the cost of filling an oral cancer drug prescription for patients with health insurance plans that were covered by the state laws (fully insured) and patients whose plans were not (self-funded). All patients lived in 1 of 16 states that had passed parity laws at the time of the study.
About half of patients (51.4%) had fully insured plans, and the other half (48.6%) had self-funded plans.
For the entire cohort, the use of oral cancer drugs increased from 18% in the months before parity laws were passed to 22% in the months after (adjusted difference-in-differences risk ratio [aDDRR], 1.04; 95% confidence interval [CI], 0.96-1.13; P=0.34).
The proportion of prescription fills for oral drugs without copayment increased from 15.0% to 53.0% for patients with fully insured plans and from 12.3% to 18.0% in patients with self-funded plans (aDDRR, 2.36; 95% CI, 2.00-2.79; P<0.001).
“From our results, it looks like many plans decided they would just set the co-pay for oral drugs to $0,” Dr Dusetzina said. “Instead of $30 per month, those fills were now $0.”
The proportion of prescription fills with out-of-pocket cost of more than $100 per month increased from 8.4% to 11.1% for patients with fully insured plans but decreased from 12.0% to 11.7% for those with self-funded plans (aDDRR, 1.36; 95% CI, 1.11-1.68; P=0.004).
Patients paying the most for their oral cancer drug prescriptions experienced increases in their monthly out-of-pocket costs after parity laws were passed.
For patients whose costs were more expensive than 95% of other patients, their out-of-pocket costs increased an estimated $143.25 per month. Those paying more than 90% of what other patients paid saw their costs increase by $37.19 per month.
“One of the biggest problems with parity laws as they are written is that they don’t address the prices of these medications, which can be very high,” Dr Dusetzina noted.
“Parity can be reached as long as the coverage is the same for both oral and infused cancer therapies. Because we’re now seeing more people insured by plans with high deductibles or plans that require them to pay a percentage of their drug costs, parity may not reduce spending for some patients.”
However, Dr Dusetzina and her colleagues did find that patients who paid the least for their oral cancer treatments saw their estimated monthly out-of-pocket spending decrease.
Patients in the 25th percentile saw an estimated decrease of $19.44 per month, those in the 50th percentile saw a $32.13 decrease, and patients in the 75th percentile saw a decrease of $10.83.
On the other hand, the researchers also found that average 6-month healthcare costs—including what was paid by insurance companies and patients—did not change significantly as a result of parity laws.
The aDDRR was 0.96 (95% CI, 0.90-1.02; P=0.09) for all cancer treatments and 1.06 (95% CI, 0.93-1.20; P=0.40) for oral cancer drugs.
“One of the key arguments against passing parity, both for states that haven’t passed it and for legislation at the federal level, has been that it may increase costs to health plans,” Dr Dusetzina said. “But we didn’t find evidence of that.” ![]()
US state laws intended to ensure fair prices for oral cancer drugs have had a mixed impact on patients’ pocketbooks, according to a study published in JAMA Oncology.
A total of 43 states and Washington, DC, have enacted parity laws, which require that patients pay no more for an oral cancer treatment than they would for an infusion of the same treatment.
Researchers analyzed the impact of these laws and observed modest improvements in costs for some patients.
However, patients who were already paying the most for their medications saw their monthly costs go up.
“Although parity laws appear to help reduce out-of-pocket spending for some patients, they may not fully address affordability for patients needing cancer drugs,” said study author Stacie B. Dusetzina, PhD, of the University of North Carolina at Chapel Hill.
“We need to consider ways to address drug pricing directly and to improve benefit design to make sure that all patients can access prescribed drugs.”
To gauge the impact of parity laws on treatment costs, Dr Dusetzina and her colleagues analyzed health claims data for 63,780 adults from 3 large, nationwide insurance companies before and after the laws were enacted, from 2008 to 2012.
The team compared the cost of filling an oral cancer drug prescription for patients with health insurance plans that were covered by the state laws (fully insured) and patients whose plans were not (self-funded). All patients lived in 1 of 16 states that had passed parity laws at the time of the study.
About half of patients (51.4%) had fully insured plans, and the other half (48.6%) had self-funded plans.
For the entire cohort, the use of oral cancer drugs increased from 18% in the months before parity laws were passed to 22% in the months after (adjusted difference-in-differences risk ratio [aDDRR], 1.04; 95% confidence interval [CI], 0.96-1.13; P=0.34).
The proportion of prescription fills for oral drugs without copayment increased from 15.0% to 53.0% for patients with fully insured plans and from 12.3% to 18.0% in patients with self-funded plans (aDDRR, 2.36; 95% CI, 2.00-2.79; P<0.001).
“From our results, it looks like many plans decided they would just set the co-pay for oral drugs to $0,” Dr Dusetzina said. “Instead of $30 per month, those fills were now $0.”
The proportion of prescription fills with out-of-pocket cost of more than $100 per month increased from 8.4% to 11.1% for patients with fully insured plans but decreased from 12.0% to 11.7% for those with self-funded plans (aDDRR, 1.36; 95% CI, 1.11-1.68; P=0.004).
Patients paying the most for their oral cancer drug prescriptions experienced increases in their monthly out-of-pocket costs after parity laws were passed.
For patients whose costs were more expensive than 95% of other patients, their out-of-pocket costs increased an estimated $143.25 per month. Those paying more than 90% of what other patients paid saw their costs increase by $37.19 per month.
“One of the biggest problems with parity laws as they are written is that they don’t address the prices of these medications, which can be very high,” Dr Dusetzina noted.
“Parity can be reached as long as the coverage is the same for both oral and infused cancer therapies. Because we’re now seeing more people insured by plans with high deductibles or plans that require them to pay a percentage of their drug costs, parity may not reduce spending for some patients.”
However, Dr Dusetzina and her colleagues did find that patients who paid the least for their oral cancer treatments saw their estimated monthly out-of-pocket spending decrease.
Patients in the 25th percentile saw an estimated decrease of $19.44 per month, those in the 50th percentile saw a $32.13 decrease, and patients in the 75th percentile saw a decrease of $10.83.
On the other hand, the researchers also found that average 6-month healthcare costs—including what was paid by insurance companies and patients—did not change significantly as a result of parity laws.
The aDDRR was 0.96 (95% CI, 0.90-1.02; P=0.09) for all cancer treatments and 1.06 (95% CI, 0.93-1.20; P=0.40) for oral cancer drugs.
“One of the key arguments against passing parity, both for states that haven’t passed it and for legislation at the federal level, has been that it may increase costs to health plans,” Dr Dusetzina said. “But we didn’t find evidence of that.” ![]()
FDA approves IV formulation of aprepitant for CINV

The US Food and Drug Administration (FDA) has approved use of an intravenous (IV) formulation of aprepitant (CINVANTI™) to prevent chemotherapy-induced nausea and vomiting (CINV).
CINVANTI is intended to be used in combination with other antiemetic agents to prevent acute and delayed nausea and vomiting associated with initial and repeat courses of highly emetogenic chemotherapy (HEC) and moderately emetogenic chemotherapy (MEC).
CINVANTI is to be used in combination with a 5-HT3 receptor antagonist and dexamethasone.
The full prescribing information is available at www.cinvanti.com.
The US commercial launch of CINVANTI is planned for January 2018.
CINVANTI is the first IV formulation to directly deliver aprepitant, a substance P/neurokinin-1 (NK1) receptor antagonist.
Aprepitant is also the active ingredient in EMEND® capsules, which were approved by the FDA in 2003. EMEND IV®, which was approved in 2008, contains aprepitant’s prodrug, fosaprepitant.
Heron Therapeutics, Inc., developed CINVANTI in an attempt to provide an IV formulation of aprepitant that has the same efficacy as IV fosaprepitant but does not pose the risk of adverse events (AEs) related to polysorbate 80.
“Aprepitant has long been the standard in the NK1 class, and it remains the only single-agent NK1 with proven efficacy in preventing CINV in both the acute and delayed phases in HEC and MEC,” said Rudolph M. Navari, MD, PhD, of the University of Alabama Birmingham School of Medicine.
“Because CINVANTI is a novel, polysorbate 80-free, IV formulation of aprepitant, it enables physicians to provide patients with standard-of-care efficacy without the potential risk of polysorbate 80-related adverse events, such as infusion-site reactions.”
The FDA approved CINVANTI based on data demonstrating the bioequivalence of CINVANTI to EMEND IV.
A phase 1, randomized, 2-way cross-over study comparing the drugs enrolled 100 healthy subjects. The subjects received CINVANTI at 130 mg or EMEND IV at 150 mg, given over 30 minutes on day 1 of periods 1 and 2.
The researchers said 90% confidence intervals for CINVANTI AUC0-t (area under the time-concentration curve from time 0 to the last measurable concentration), AUC0-inf (area under the time-concentration curve from time 0 extrapolated to infinity), and C12h (plasma concentration at 12 hours) “were well within bioequivalence bounds,” which was 80% to 125%.
The team also found the incidence of treatment-emergent AEs was lower with CINVANTI than EMEND IV—21% and 28%, respectively. The same was true for related treatment-emergent AEs—15% and 28%, respectively.
These data were presented at the Hematology/Oncology Pharmacy Association Annual Conference in March/April and the Multinational Association of Supportive Care in Cancer (MASCC)/International Society of Oral Oncology (ISOO) Annual Meeting in June. ![]()
The US Food and Drug Administration (FDA) has approved use of an intravenous (IV) formulation of aprepitant (CINVANTI™) to prevent chemotherapy-induced nausea and vomiting (CINV).
CINVANTI is intended to be used in combination with other antiemetic agents to prevent acute and delayed nausea and vomiting associated with initial and repeat courses of highly emetogenic chemotherapy (HEC) and moderately emetogenic chemotherapy (MEC).
CINVANTI is to be used in combination with a 5-HT3 receptor antagonist and dexamethasone.
The full prescribing information is available at www.cinvanti.com.
The US commercial launch of CINVANTI is planned for January 2018.
CINVANTI is the first IV formulation to directly deliver aprepitant, a substance P/neurokinin-1 (NK1) receptor antagonist.
Aprepitant is also the active ingredient in EMEND® capsules, which were approved by the FDA in 2003. EMEND IV®, which was approved in 2008, contains aprepitant’s prodrug, fosaprepitant.
Heron Therapeutics, Inc., developed CINVANTI in an attempt to provide an IV formulation of aprepitant that has the same efficacy as IV fosaprepitant but does not pose the risk of adverse events (AEs) related to polysorbate 80.
“Aprepitant has long been the standard in the NK1 class, and it remains the only single-agent NK1 with proven efficacy in preventing CINV in both the acute and delayed phases in HEC and MEC,” said Rudolph M. Navari, MD, PhD, of the University of Alabama Birmingham School of Medicine.
“Because CINVANTI is a novel, polysorbate 80-free, IV formulation of aprepitant, it enables physicians to provide patients with standard-of-care efficacy without the potential risk of polysorbate 80-related adverse events, such as infusion-site reactions.”
The FDA approved CINVANTI based on data demonstrating the bioequivalence of CINVANTI to EMEND IV.
A phase 1, randomized, 2-way cross-over study comparing the drugs enrolled 100 healthy subjects. The subjects received CINVANTI at 130 mg or EMEND IV at 150 mg, given over 30 minutes on day 1 of periods 1 and 2.
The researchers said 90% confidence intervals for CINVANTI AUC0-t (area under the time-concentration curve from time 0 to the last measurable concentration), AUC0-inf (area under the time-concentration curve from time 0 extrapolated to infinity), and C12h (plasma concentration at 12 hours) “were well within bioequivalence bounds,” which was 80% to 125%.
The team also found the incidence of treatment-emergent AEs was lower with CINVANTI than EMEND IV—21% and 28%, respectively. The same was true for related treatment-emergent AEs—15% and 28%, respectively.
These data were presented at the Hematology/Oncology Pharmacy Association Annual Conference in March/April and the Multinational Association of Supportive Care in Cancer (MASCC)/International Society of Oral Oncology (ISOO) Annual Meeting in June. ![]()
The US Food and Drug Administration (FDA) has approved use of an intravenous (IV) formulation of aprepitant (CINVANTI™) to prevent chemotherapy-induced nausea and vomiting (CINV).
CINVANTI is intended to be used in combination with other antiemetic agents to prevent acute and delayed nausea and vomiting associated with initial and repeat courses of highly emetogenic chemotherapy (HEC) and moderately emetogenic chemotherapy (MEC).
CINVANTI is to be used in combination with a 5-HT3 receptor antagonist and dexamethasone.
The full prescribing information is available at www.cinvanti.com.
The US commercial launch of CINVANTI is planned for January 2018.
CINVANTI is the first IV formulation to directly deliver aprepitant, a substance P/neurokinin-1 (NK1) receptor antagonist.
Aprepitant is also the active ingredient in EMEND® capsules, which were approved by the FDA in 2003. EMEND IV®, which was approved in 2008, contains aprepitant’s prodrug, fosaprepitant.
Heron Therapeutics, Inc., developed CINVANTI in an attempt to provide an IV formulation of aprepitant that has the same efficacy as IV fosaprepitant but does not pose the risk of adverse events (AEs) related to polysorbate 80.
“Aprepitant has long been the standard in the NK1 class, and it remains the only single-agent NK1 with proven efficacy in preventing CINV in both the acute and delayed phases in HEC and MEC,” said Rudolph M. Navari, MD, PhD, of the University of Alabama Birmingham School of Medicine.
“Because CINVANTI is a novel, polysorbate 80-free, IV formulation of aprepitant, it enables physicians to provide patients with standard-of-care efficacy without the potential risk of polysorbate 80-related adverse events, such as infusion-site reactions.”
The FDA approved CINVANTI based on data demonstrating the bioequivalence of CINVANTI to EMEND IV.
A phase 1, randomized, 2-way cross-over study comparing the drugs enrolled 100 healthy subjects. The subjects received CINVANTI at 130 mg or EMEND IV at 150 mg, given over 30 minutes on day 1 of periods 1 and 2.
The researchers said 90% confidence intervals for CINVANTI AUC0-t (area under the time-concentration curve from time 0 to the last measurable concentration), AUC0-inf (area under the time-concentration curve from time 0 extrapolated to infinity), and C12h (plasma concentration at 12 hours) “were well within bioequivalence bounds,” which was 80% to 125%.
The team also found the incidence of treatment-emergent AEs was lower with CINVANTI than EMEND IV—21% and 28%, respectively. The same was true for related treatment-emergent AEs—15% and 28%, respectively.
These data were presented at the Hematology/Oncology Pharmacy Association Annual Conference in March/April and the Multinational Association of Supportive Care in Cancer (MASCC)/International Society of Oral Oncology (ISOO) Annual Meeting in June. ![]()
Is MRD ready for prime time in multiple myeloma?

NEW YORK, NY—Speakers faced off over the issue of minimal residual disease (MRD) testing in multiple myeloma (MM) at Lymphoma & Myeloma 2017.
Ola Landgren, MD, PhD, of Weill Cornell Medicine in New York, New York, said, “it’s really a necessary and logical step forward to look at MRD.”
On the other hand, Paul Richardson, MD, of Dana-Farber Cancer Institute in Boston, Massachusetts, took the clinicians’ perspective and suggested that, at this point, “we’re not yet ready to apply it to everyday practice.”
“[P]atients who have a complete response (CR) and are MRD negative have longer progression-free survival (PFS),” Dr Landgren pointed out, “and there are indications that their overall survival (OS) is better than in those patients who are just CR and MRD positive.”
“My position on this is that MRD testing is absolutely ready for prime time in the research and regulatory arena,” Dr Richardson contended. “The question for me, as a clinician, in my clinic, is ‘Do I apply it to everyday practice?’ And I would simply suggest to you, at this point, we’re not ready for that.”
Yes—MRD is ready for prime time
Dr Landgren based his argument on 2 meta-analyses published in 2016 and 2017 that outline the importance of MRD status in newly diagnosed MM patients.
The first analysis (Landgren et al 2016) showed that MRD negativity was associated with better PFS (hazard ratio [HR]=0.35] and OS (HR=0.48) than MRD positivity.
“So using more simple language,” Dr Landgren said, “this means that MRD negativity reduces the risk of progression by 65%, and it also reduces the risk of dying by 52%.”
The second analysis (Munshi et al 2017) also associated MRD-negative status with superior survival outcomes for both PFS (HR=0.41) and OS (HR=0.57).
As further confirmation of the importance of MRD status, the International Myeloma Working Group last year published response definitions that include MRD negativity at a sensitivity of 1 in 105 cells or higher as the deepest level of treatment response in MM.
Dr Landgren drew on additional studies to support routine MRD testing in patient care.
The IFM Study Group found that, in newly diagnosed patients treated with lenalidomide, bortezomib, and dexamethasone followed by 1 year of lenalidomide maintenance, patients who received a subsequent transplant achieved superior outcomes compared to non-transplanted patients, in terms of CR (58% vs 46%) and 3-year PFS (61% vs 48%).
However, in patients who were MRD negative in both arms, the PFS rates were very similar, Dr Landgren said. And in terms of 3-year OS, there was no difference, at 88% in both arms.
The experience with daratumumab in relapsed/refractory patients exhibited a similar pattern.
The phase 3 POLLUX trial first showed that adding daratumumab to lenalidomide and dexamethasone was superior to lenalidomide and dexamethasone only, with a PFS at 18 months of 78% and 52%, respectively. This amounted to a 63% reduction in the risk of disease progression.
Investigators then took one more step forward, Dr Landgren said, and looked at MRD.
At a sensitivity of 10-5, almost 25% of patients on the 3-drug regimen were MRD negative, “which is kind of amazing,” Dr Landgren said. “This is a very big step forward.”
“If you break down the results by MRD status, which is not the primary endpoint of the study, you see very similar patterns for PFS for MRD negative patients in each of the 2 arms,” he continued.
This raises the question of whether attaining MRD negativity is more important than the treatment modality.
MRD negativity has implications for speeding drug approvals, developing more sensitive assays, and future treatment management, Dr Landgren said.
No—MRD is not ready for prime time
Dr Richardson acknowledged that MRD assessment is important. However, he pointed out a number of caveats regarding how MRD assessment would be applied in clinical practice to support his position.
“I’d simply suggest to you that, in day-to-day practice, the definition [of MRD] is somewhat fluid,” he said. “And it varies, obviously, between diseases and technology used.”
For most malignancies, Dr Richardson said, 109 to 1010 malignant cells are undetectable with conventional methods. These may or may not lead to a full clinical relapse within months or even years.
Using a sensitive technique to determine the presence of MRD could permit analysis of treatments that induce a greater depth of response or identify patients at risk of early relapse who need further treatment.
Dr Richardson enumerated hematologic malignancies that utilize MRD as secondary endpoints—acute lymphoblastic leukemia, acute myeloid leukemia, acute promyelocytic leukemia, chronic lymphocytic leukemia, follicular lymphoma, and mantle cell lymphoma.
In chronic myeloid leukemia, MRD is used as a primary endpoint that dictates practice.
“And I would applaud the field in that area because, obviously, molecular response accepted as an endpoint by FDA for second-generation TKIs has been a bedrock of that approval process, and it now applies in clinical practice,” Dr Richardson said.
“Obviously, that’s where we’d like to be, but I’d suggest to you, just again, with a certain amount of moderation and a certain amount of caution, that we may not be quite there yet.”
Dr Richardson suggested that MRD assessment in MM is less advanced than in leukemia and lymphoma.
“[W]e are currently at the point where MRD assessments are clearly secondary endpoints, an important research tool,” he said.
Some “remarkable combination therapies,” he added, have abrogated some of the “extraordinary genetic complexity” in MM.
“The critical point here, though, is that, while we’re more successful in terms of these triplets and quadruplets and now with the introduction of monoclonal antibodies and similar approaches, we’re able to throw a bigger net around the disease,” Dr Richardson said.
“We’re not able to eradicate it completely, and cure remains, in myeloma, frankly, evasive. And I think that’s a critical point.”
Dr Richardson reviewed various strategies for molecular response monitoring, from flow cytometry to polymerase chain reaction and next-generation sequencing, noting that there is variance in applicability and sensitivity.
For example, the limits of detection among 91 labs ranged from 0.10% to 0.001%.
Dr Richardson returned to the “very robust” meta-analysis by Munshi and colleagues discussed by Dr Landgren.
While the authors’ analysis demonstrated that MRD is predictive of both longer PFS and OS, they concluded that the evidence supported MRD as an endpoint and research tool in clinical trials.
“So I would humbly suggest perhaps it’s not ready for clinical prime time yet,” Dr Richardson said.
He also referred to the IFM Study Group trial described by Dr Landgren, calling it a “critical forward effort.”
“[W]hat’s so interesting is that there was no difference in overall survival,” Dr Richardson said. “Now, that’s a very important point as we soberly look at these data and judge what they mean for each patient.”
And so Dr Richardson stood by his assessment that MRD is not yet a standard of care but may be one day. ![]()
NEW YORK, NY—Speakers faced off over the issue of minimal residual disease (MRD) testing in multiple myeloma (MM) at Lymphoma & Myeloma 2017.
Ola Landgren, MD, PhD, of Weill Cornell Medicine in New York, New York, said, “it’s really a necessary and logical step forward to look at MRD.”
On the other hand, Paul Richardson, MD, of Dana-Farber Cancer Institute in Boston, Massachusetts, took the clinicians’ perspective and suggested that, at this point, “we’re not yet ready to apply it to everyday practice.”
“[P]atients who have a complete response (CR) and are MRD negative have longer progression-free survival (PFS),” Dr Landgren pointed out, “and there are indications that their overall survival (OS) is better than in those patients who are just CR and MRD positive.”
“My position on this is that MRD testing is absolutely ready for prime time in the research and regulatory arena,” Dr Richardson contended. “The question for me, as a clinician, in my clinic, is ‘Do I apply it to everyday practice?’ And I would simply suggest to you, at this point, we’re not ready for that.”
Yes—MRD is ready for prime time
Dr Landgren based his argument on 2 meta-analyses published in 2016 and 2017 that outline the importance of MRD status in newly diagnosed MM patients.
The first analysis (Landgren et al 2016) showed that MRD negativity was associated with better PFS (hazard ratio [HR]=0.35] and OS (HR=0.48) than MRD positivity.
“So using more simple language,” Dr Landgren said, “this means that MRD negativity reduces the risk of progression by 65%, and it also reduces the risk of dying by 52%.”
The second analysis (Munshi et al 2017) also associated MRD-negative status with superior survival outcomes for both PFS (HR=0.41) and OS (HR=0.57).
As further confirmation of the importance of MRD status, the International Myeloma Working Group last year published response definitions that include MRD negativity at a sensitivity of 1 in 105 cells or higher as the deepest level of treatment response in MM.
Dr Landgren drew on additional studies to support routine MRD testing in patient care.
The IFM Study Group found that, in newly diagnosed patients treated with lenalidomide, bortezomib, and dexamethasone followed by 1 year of lenalidomide maintenance, patients who received a subsequent transplant achieved superior outcomes compared to non-transplanted patients, in terms of CR (58% vs 46%) and 3-year PFS (61% vs 48%).
However, in patients who were MRD negative in both arms, the PFS rates were very similar, Dr Landgren said. And in terms of 3-year OS, there was no difference, at 88% in both arms.
The experience with daratumumab in relapsed/refractory patients exhibited a similar pattern.
The phase 3 POLLUX trial first showed that adding daratumumab to lenalidomide and dexamethasone was superior to lenalidomide and dexamethasone only, with a PFS at 18 months of 78% and 52%, respectively. This amounted to a 63% reduction in the risk of disease progression.
Investigators then took one more step forward, Dr Landgren said, and looked at MRD.
At a sensitivity of 10-5, almost 25% of patients on the 3-drug regimen were MRD negative, “which is kind of amazing,” Dr Landgren said. “This is a very big step forward.”
“If you break down the results by MRD status, which is not the primary endpoint of the study, you see very similar patterns for PFS for MRD negative patients in each of the 2 arms,” he continued.
This raises the question of whether attaining MRD negativity is more important than the treatment modality.
MRD negativity has implications for speeding drug approvals, developing more sensitive assays, and future treatment management, Dr Landgren said.
No—MRD is not ready for prime time
Dr Richardson acknowledged that MRD assessment is important. However, he pointed out a number of caveats regarding how MRD assessment would be applied in clinical practice to support his position.
“I’d simply suggest to you that, in day-to-day practice, the definition [of MRD] is somewhat fluid,” he said. “And it varies, obviously, between diseases and technology used.”
For most malignancies, Dr Richardson said, 109 to 1010 malignant cells are undetectable with conventional methods. These may or may not lead to a full clinical relapse within months or even years.
Using a sensitive technique to determine the presence of MRD could permit analysis of treatments that induce a greater depth of response or identify patients at risk of early relapse who need further treatment.
Dr Richardson enumerated hematologic malignancies that utilize MRD as secondary endpoints—acute lymphoblastic leukemia, acute myeloid leukemia, acute promyelocytic leukemia, chronic lymphocytic leukemia, follicular lymphoma, and mantle cell lymphoma.
In chronic myeloid leukemia, MRD is used as a primary endpoint that dictates practice.
“And I would applaud the field in that area because, obviously, molecular response accepted as an endpoint by FDA for second-generation TKIs has been a bedrock of that approval process, and it now applies in clinical practice,” Dr Richardson said.
“Obviously, that’s where we’d like to be, but I’d suggest to you, just again, with a certain amount of moderation and a certain amount of caution, that we may not be quite there yet.”
Dr Richardson suggested that MRD assessment in MM is less advanced than in leukemia and lymphoma.
“[W]e are currently at the point where MRD assessments are clearly secondary endpoints, an important research tool,” he said.
Some “remarkable combination therapies,” he added, have abrogated some of the “extraordinary genetic complexity” in MM.
“The critical point here, though, is that, while we’re more successful in terms of these triplets and quadruplets and now with the introduction of monoclonal antibodies and similar approaches, we’re able to throw a bigger net around the disease,” Dr Richardson said.
“We’re not able to eradicate it completely, and cure remains, in myeloma, frankly, evasive. And I think that’s a critical point.”
Dr Richardson reviewed various strategies for molecular response monitoring, from flow cytometry to polymerase chain reaction and next-generation sequencing, noting that there is variance in applicability and sensitivity.
For example, the limits of detection among 91 labs ranged from 0.10% to 0.001%.
Dr Richardson returned to the “very robust” meta-analysis by Munshi and colleagues discussed by Dr Landgren.
While the authors’ analysis demonstrated that MRD is predictive of both longer PFS and OS, they concluded that the evidence supported MRD as an endpoint and research tool in clinical trials.
“So I would humbly suggest perhaps it’s not ready for clinical prime time yet,” Dr Richardson said.
He also referred to the IFM Study Group trial described by Dr Landgren, calling it a “critical forward effort.”
“[W]hat’s so interesting is that there was no difference in overall survival,” Dr Richardson said. “Now, that’s a very important point as we soberly look at these data and judge what they mean for each patient.”
And so Dr Richardson stood by his assessment that MRD is not yet a standard of care but may be one day. ![]()
NEW YORK, NY—Speakers faced off over the issue of minimal residual disease (MRD) testing in multiple myeloma (MM) at Lymphoma & Myeloma 2017.
Ola Landgren, MD, PhD, of Weill Cornell Medicine in New York, New York, said, “it’s really a necessary and logical step forward to look at MRD.”
On the other hand, Paul Richardson, MD, of Dana-Farber Cancer Institute in Boston, Massachusetts, took the clinicians’ perspective and suggested that, at this point, “we’re not yet ready to apply it to everyday practice.”
“[P]atients who have a complete response (CR) and are MRD negative have longer progression-free survival (PFS),” Dr Landgren pointed out, “and there are indications that their overall survival (OS) is better than in those patients who are just CR and MRD positive.”
“My position on this is that MRD testing is absolutely ready for prime time in the research and regulatory arena,” Dr Richardson contended. “The question for me, as a clinician, in my clinic, is ‘Do I apply it to everyday practice?’ And I would simply suggest to you, at this point, we’re not ready for that.”
Yes—MRD is ready for prime time
Dr Landgren based his argument on 2 meta-analyses published in 2016 and 2017 that outline the importance of MRD status in newly diagnosed MM patients.
The first analysis (Landgren et al 2016) showed that MRD negativity was associated with better PFS (hazard ratio [HR]=0.35] and OS (HR=0.48) than MRD positivity.
“So using more simple language,” Dr Landgren said, “this means that MRD negativity reduces the risk of progression by 65%, and it also reduces the risk of dying by 52%.”
The second analysis (Munshi et al 2017) also associated MRD-negative status with superior survival outcomes for both PFS (HR=0.41) and OS (HR=0.57).
As further confirmation of the importance of MRD status, the International Myeloma Working Group last year published response definitions that include MRD negativity at a sensitivity of 1 in 105 cells or higher as the deepest level of treatment response in MM.
Dr Landgren drew on additional studies to support routine MRD testing in patient care.
The IFM Study Group found that, in newly diagnosed patients treated with lenalidomide, bortezomib, and dexamethasone followed by 1 year of lenalidomide maintenance, patients who received a subsequent transplant achieved superior outcomes compared to non-transplanted patients, in terms of CR (58% vs 46%) and 3-year PFS (61% vs 48%).
However, in patients who were MRD negative in both arms, the PFS rates were very similar, Dr Landgren said. And in terms of 3-year OS, there was no difference, at 88% in both arms.
The experience with daratumumab in relapsed/refractory patients exhibited a similar pattern.
The phase 3 POLLUX trial first showed that adding daratumumab to lenalidomide and dexamethasone was superior to lenalidomide and dexamethasone only, with a PFS at 18 months of 78% and 52%, respectively. This amounted to a 63% reduction in the risk of disease progression.
Investigators then took one more step forward, Dr Landgren said, and looked at MRD.
At a sensitivity of 10-5, almost 25% of patients on the 3-drug regimen were MRD negative, “which is kind of amazing,” Dr Landgren said. “This is a very big step forward.”
“If you break down the results by MRD status, which is not the primary endpoint of the study, you see very similar patterns for PFS for MRD negative patients in each of the 2 arms,” he continued.
This raises the question of whether attaining MRD negativity is more important than the treatment modality.
MRD negativity has implications for speeding drug approvals, developing more sensitive assays, and future treatment management, Dr Landgren said.
No—MRD is not ready for prime time
Dr Richardson acknowledged that MRD assessment is important. However, he pointed out a number of caveats regarding how MRD assessment would be applied in clinical practice to support his position.
“I’d simply suggest to you that, in day-to-day practice, the definition [of MRD] is somewhat fluid,” he said. “And it varies, obviously, between diseases and technology used.”
For most malignancies, Dr Richardson said, 109 to 1010 malignant cells are undetectable with conventional methods. These may or may not lead to a full clinical relapse within months or even years.
Using a sensitive technique to determine the presence of MRD could permit analysis of treatments that induce a greater depth of response or identify patients at risk of early relapse who need further treatment.
Dr Richardson enumerated hematologic malignancies that utilize MRD as secondary endpoints—acute lymphoblastic leukemia, acute myeloid leukemia, acute promyelocytic leukemia, chronic lymphocytic leukemia, follicular lymphoma, and mantle cell lymphoma.
In chronic myeloid leukemia, MRD is used as a primary endpoint that dictates practice.
“And I would applaud the field in that area because, obviously, molecular response accepted as an endpoint by FDA for second-generation TKIs has been a bedrock of that approval process, and it now applies in clinical practice,” Dr Richardson said.
“Obviously, that’s where we’d like to be, but I’d suggest to you, just again, with a certain amount of moderation and a certain amount of caution, that we may not be quite there yet.”
Dr Richardson suggested that MRD assessment in MM is less advanced than in leukemia and lymphoma.
“[W]e are currently at the point where MRD assessments are clearly secondary endpoints, an important research tool,” he said.
Some “remarkable combination therapies,” he added, have abrogated some of the “extraordinary genetic complexity” in MM.
“The critical point here, though, is that, while we’re more successful in terms of these triplets and quadruplets and now with the introduction of monoclonal antibodies and similar approaches, we’re able to throw a bigger net around the disease,” Dr Richardson said.
“We’re not able to eradicate it completely, and cure remains, in myeloma, frankly, evasive. And I think that’s a critical point.”
Dr Richardson reviewed various strategies for molecular response monitoring, from flow cytometry to polymerase chain reaction and next-generation sequencing, noting that there is variance in applicability and sensitivity.
For example, the limits of detection among 91 labs ranged from 0.10% to 0.001%.
Dr Richardson returned to the “very robust” meta-analysis by Munshi and colleagues discussed by Dr Landgren.
While the authors’ analysis demonstrated that MRD is predictive of both longer PFS and OS, they concluded that the evidence supported MRD as an endpoint and research tool in clinical trials.
“So I would humbly suggest perhaps it’s not ready for clinical prime time yet,” Dr Richardson said.
He also referred to the IFM Study Group trial described by Dr Landgren, calling it a “critical forward effort.”
“[W]hat’s so interesting is that there was no difference in overall survival,” Dr Richardson said. “Now, that’s a very important point as we soberly look at these data and judge what they mean for each patient.”
And so Dr Richardson stood by his assessment that MRD is not yet a standard of care but may be one day. ![]()
Cancer drug costs increasing despite competition

Cancer drug costs in the US increase substantially after launch, regardless of competition, according to a study published in the Journal of Clinical Oncology.*
Researchers studied 24 cancer drugs approved over the last 20 years and found a mean cumulative cost increase of about 37%, or 19% when adjusted for inflation.
Among drugs approved to treat hematologic malignancies, the greatest inflation-adjusted price increases were for arsenic trioxide (57%), nelarabine (55%), and rituximab (49%).
The lowest inflation-adjusted price increases were for ofatumumab (8%), clofarabine (8%), and liposomal vincristine (18%).
For this study, Daniel A. Goldstein, MD, of Emory University in Atlanta, Georgia, and his colleagues measured the monthly price trajectories of 24 cancer drugs approved by the US Food and Drug Administration. This included 10 drugs approved to treat hematologic malignancies between 1997 and 2011.
To account for discounts and rebates, the researchers used the average sales prices published by the Centers for Medicare and Medicaid Services and adjusted to general and health-related inflation rates. For each drug, the researchers calculated the cumulative and annual drug cost changes.
Results
The mean follow-up was 8 years. The mean cumulative cost increase for all 24 drugs was +36.5% (95% CI, 24.7% to 48.3%).
The general inflation-adjusted increase was +19.1% (95% CI, 11.0% to 27.2%), and the health-related inflation-adjusted increase was +8.4% (95% CI, 1.4% to 15.4%).
Only 1 of the 24 drugs studied had a price decrease over time. That drug is ziv-aflibercept, which was approved to treat metastatic colorectal cancer in 2012.
Ziv-aflibercept was launched with an annual price exceeding $110,000. After public outcry, the drug’s manufacturer, Sanofi, cut the price in half. By the end of the study’s follow-up period in 2017, the cost of ziv-aflibercept had decreased 13% (inflation-adjusted decrease of 15%, health-related inflation-adjusted decrease of 20%).
Cost changes for the drugs approved to treat hematologic malignancies are listed in the following table.
| Drug (indication, approval date, years of follow-up) | Mean monthly cost at launch | Mean annual cost change (SD) | Cumulative cost change | General and health-related inflation-adjusted change, respectively |
| Arsenic trioxide (APL, 2000, 12) | $11,455 | +6% (4) | +95% | +57%, +39% |
| Bendamustine (CLL, NHL, 2008, 8) | $6924 | +5% (5) | +50% | +32%, +21% |
| Bortezomib (MM, MCL, 2003, 12) | $5490 | +4% (3) | +63% | +31%, +16% |
| Brentuximab (lymphoma, 2011, 4) | $19,482 | +8% (0.1) | +35% | +29%, +22% |
| Clofarabine (ALL, 2004, 11) | $56,486 | +3% (3) | +31% | +8%, -4% |
| Liposomal vincristine (ALL, 2012, 3) | $34,602 | +8% (0.5) | +21% | +18%, +14% |
| Nelarabine (ALL, lymphoma, 2005, 10) | $18,513 | +6% (2) | +83% | +55%, +39% |
| Ofatumumab (CLL, 2009, 6) | $4538 | +3% (2) | +17% | +8%, -0.5% |
| Pralatrexate (lymphoma, 2009, 6) | $31,684 | +6% (4) | +43% | +31%, +21% |
| Rituximab (NHL, CLL, 1997, 12) | $4111 | +5% (0.5) | +85% | +49%, +32% |
Abbreviations: ALL, acute lymphoblastic leukemia; APL, acute promyelocytic leukemia; CLL, chronic lymphocytic leukemia; MCL, mantle cell lymphoma; MM, multiple myeloma; NHL, non-Hodgkin lymphoma; SD, standard deviation.
The researchers noted that there was a steady increase in drug costs over the study period, regardless of whether a drug was granted a new supplemental indication, the drug had a new off-label indication, or a competitor drug was approved.
The only variable that was significantly associated with price change was the amount of time that had elapsed from a drug’s launch.
This association was significant in models in which the researchers used prices adjusted to inflation (P=0.002) and health-related inflation (P=0.023). However, it was not significant when the researchers used the actual drug price (P=0.085). ![]()
*Data in the abstract differ from data in the body of the JCO paper. This article includes data from the body of the JCO paper.
Cancer drug costs in the US increase substantially after launch, regardless of competition, according to a study published in the Journal of Clinical Oncology.*
Researchers studied 24 cancer drugs approved over the last 20 years and found a mean cumulative cost increase of about 37%, or 19% when adjusted for inflation.
Among drugs approved to treat hematologic malignancies, the greatest inflation-adjusted price increases were for arsenic trioxide (57%), nelarabine (55%), and rituximab (49%).
The lowest inflation-adjusted price increases were for ofatumumab (8%), clofarabine (8%), and liposomal vincristine (18%).
For this study, Daniel A. Goldstein, MD, of Emory University in Atlanta, Georgia, and his colleagues measured the monthly price trajectories of 24 cancer drugs approved by the US Food and Drug Administration. This included 10 drugs approved to treat hematologic malignancies between 1997 and 2011.
To account for discounts and rebates, the researchers used the average sales prices published by the Centers for Medicare and Medicaid Services and adjusted to general and health-related inflation rates. For each drug, the researchers calculated the cumulative and annual drug cost changes.
Results
The mean follow-up was 8 years. The mean cumulative cost increase for all 24 drugs was +36.5% (95% CI, 24.7% to 48.3%).
The general inflation-adjusted increase was +19.1% (95% CI, 11.0% to 27.2%), and the health-related inflation-adjusted increase was +8.4% (95% CI, 1.4% to 15.4%).
Only 1 of the 24 drugs studied had a price decrease over time. That drug is ziv-aflibercept, which was approved to treat metastatic colorectal cancer in 2012.
Ziv-aflibercept was launched with an annual price exceeding $110,000. After public outcry, the drug’s manufacturer, Sanofi, cut the price in half. By the end of the study’s follow-up period in 2017, the cost of ziv-aflibercept had decreased 13% (inflation-adjusted decrease of 15%, health-related inflation-adjusted decrease of 20%).
Cost changes for the drugs approved to treat hematologic malignancies are listed in the following table.
| Drug (indication, approval date, years of follow-up) | Mean monthly cost at launch | Mean annual cost change (SD) | Cumulative cost change | General and health-related inflation-adjusted change, respectively |
| Arsenic trioxide (APL, 2000, 12) | $11,455 | +6% (4) | +95% | +57%, +39% |
| Bendamustine (CLL, NHL, 2008, 8) | $6924 | +5% (5) | +50% | +32%, +21% |
| Bortezomib (MM, MCL, 2003, 12) | $5490 | +4% (3) | +63% | +31%, +16% |
| Brentuximab (lymphoma, 2011, 4) | $19,482 | +8% (0.1) | +35% | +29%, +22% |
| Clofarabine (ALL, 2004, 11) | $56,486 | +3% (3) | +31% | +8%, -4% |
| Liposomal vincristine (ALL, 2012, 3) | $34,602 | +8% (0.5) | +21% | +18%, +14% |
| Nelarabine (ALL, lymphoma, 2005, 10) | $18,513 | +6% (2) | +83% | +55%, +39% |
| Ofatumumab (CLL, 2009, 6) | $4538 | +3% (2) | +17% | +8%, -0.5% |
| Pralatrexate (lymphoma, 2009, 6) | $31,684 | +6% (4) | +43% | +31%, +21% |
| Rituximab (NHL, CLL, 1997, 12) | $4111 | +5% (0.5) | +85% | +49%, +32% |
Abbreviations: ALL, acute lymphoblastic leukemia; APL, acute promyelocytic leukemia; CLL, chronic lymphocytic leukemia; MCL, mantle cell lymphoma; MM, multiple myeloma; NHL, non-Hodgkin lymphoma; SD, standard deviation.
The researchers noted that there was a steady increase in drug costs over the study period, regardless of whether a drug was granted a new supplemental indication, the drug had a new off-label indication, or a competitor drug was approved.
The only variable that was significantly associated with price change was the amount of time that had elapsed from a drug’s launch.
This association was significant in models in which the researchers used prices adjusted to inflation (P=0.002) and health-related inflation (P=0.023). However, it was not significant when the researchers used the actual drug price (P=0.085). ![]()
*Data in the abstract differ from data in the body of the JCO paper. This article includes data from the body of the JCO paper.
Cancer drug costs in the US increase substantially after launch, regardless of competition, according to a study published in the Journal of Clinical Oncology.*
Researchers studied 24 cancer drugs approved over the last 20 years and found a mean cumulative cost increase of about 37%, or 19% when adjusted for inflation.
Among drugs approved to treat hematologic malignancies, the greatest inflation-adjusted price increases were for arsenic trioxide (57%), nelarabine (55%), and rituximab (49%).
The lowest inflation-adjusted price increases were for ofatumumab (8%), clofarabine (8%), and liposomal vincristine (18%).
For this study, Daniel A. Goldstein, MD, of Emory University in Atlanta, Georgia, and his colleagues measured the monthly price trajectories of 24 cancer drugs approved by the US Food and Drug Administration. This included 10 drugs approved to treat hematologic malignancies between 1997 and 2011.
To account for discounts and rebates, the researchers used the average sales prices published by the Centers for Medicare and Medicaid Services and adjusted to general and health-related inflation rates. For each drug, the researchers calculated the cumulative and annual drug cost changes.
Results
The mean follow-up was 8 years. The mean cumulative cost increase for all 24 drugs was +36.5% (95% CI, 24.7% to 48.3%).
The general inflation-adjusted increase was +19.1% (95% CI, 11.0% to 27.2%), and the health-related inflation-adjusted increase was +8.4% (95% CI, 1.4% to 15.4%).
Only 1 of the 24 drugs studied had a price decrease over time. That drug is ziv-aflibercept, which was approved to treat metastatic colorectal cancer in 2012.
Ziv-aflibercept was launched with an annual price exceeding $110,000. After public outcry, the drug’s manufacturer, Sanofi, cut the price in half. By the end of the study’s follow-up period in 2017, the cost of ziv-aflibercept had decreased 13% (inflation-adjusted decrease of 15%, health-related inflation-adjusted decrease of 20%).
Cost changes for the drugs approved to treat hematologic malignancies are listed in the following table.
| Drug (indication, approval date, years of follow-up) | Mean monthly cost at launch | Mean annual cost change (SD) | Cumulative cost change | General and health-related inflation-adjusted change, respectively |
| Arsenic trioxide (APL, 2000, 12) | $11,455 | +6% (4) | +95% | +57%, +39% |
| Bendamustine (CLL, NHL, 2008, 8) | $6924 | +5% (5) | +50% | +32%, +21% |
| Bortezomib (MM, MCL, 2003, 12) | $5490 | +4% (3) | +63% | +31%, +16% |
| Brentuximab (lymphoma, 2011, 4) | $19,482 | +8% (0.1) | +35% | +29%, +22% |
| Clofarabine (ALL, 2004, 11) | $56,486 | +3% (3) | +31% | +8%, -4% |
| Liposomal vincristine (ALL, 2012, 3) | $34,602 | +8% (0.5) | +21% | +18%, +14% |
| Nelarabine (ALL, lymphoma, 2005, 10) | $18,513 | +6% (2) | +83% | +55%, +39% |
| Ofatumumab (CLL, 2009, 6) | $4538 | +3% (2) | +17% | +8%, -0.5% |
| Pralatrexate (lymphoma, 2009, 6) | $31,684 | +6% (4) | +43% | +31%, +21% |
| Rituximab (NHL, CLL, 1997, 12) | $4111 | +5% (0.5) | +85% | +49%, +32% |
Abbreviations: ALL, acute lymphoblastic leukemia; APL, acute promyelocytic leukemia; CLL, chronic lymphocytic leukemia; MCL, mantle cell lymphoma; MM, multiple myeloma; NHL, non-Hodgkin lymphoma; SD, standard deviation.
The researchers noted that there was a steady increase in drug costs over the study period, regardless of whether a drug was granted a new supplemental indication, the drug had a new off-label indication, or a competitor drug was approved.
The only variable that was significantly associated with price change was the amount of time that had elapsed from a drug’s launch.
This association was significant in models in which the researchers used prices adjusted to inflation (P=0.002) and health-related inflation (P=0.023). However, it was not significant when the researchers used the actual drug price (P=0.085). ![]()
*Data in the abstract differ from data in the body of the JCO paper. This article includes data from the body of the JCO paper.
FDA grants product breakthrough designation for MM

The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to GSK2857916, an anti-B-cell maturation antigen (BCMA) monoclonal antibody conjugated to the cytotoxic agent monomethyl auristatin-F via a non-cleavable linker.
The designation is for GSK2857916 as monotherapy for patients with multiple myeloma (MM) who have failed at least 3 prior lines of therapy, including an anti-CD38 antibody, and who are refractory to a proteasome inhibitor and an immunomodulatory agent.
The designation is based on results from a phase 1, dose-escalation and expansion study in patients with relapsed/refractory MM, irrespective of BCMA expression (NCT02064387).
Data from this ongoing trial are scheduled to be presented December 11 in an oral presentation at the 59th Annual Meeting of the American Society of Hematology (ASH) in Atlanta, Georgia.
GSK2857916 has also received orphan drug designation from the FDA and the European Medicines Agency (EMA) as well as PRIME designation from the EMA.
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions. The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Orphan and PRIME designations
The FDA grants orphan designation to therapies intended to treat conditions that affect fewer than 200,000 people in the US. The designation qualifies a drug’s sponsor for various development incentives of the Orphan Drug Act, including tax credits for qualified clinical testing and 7 years of market exclusivity.
In Europe, sponsors who obtain orphan designation for a potential new medicine benefit from a range of incentives, including protocol assistance, access to the centralized procedure, 10 years of market exclusivity, and fee reductions.
The EMA grants PRIME designation to enhance support for the development of medicines that target an unmet medical need. The designation is based on enhanced interaction between sponsor companies and the EMA to optimize development plans and speed up evaluation so these medicines can reach patients earlier. ![]()
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to GSK2857916, an anti-B-cell maturation antigen (BCMA) monoclonal antibody conjugated to the cytotoxic agent monomethyl auristatin-F via a non-cleavable linker.
The designation is for GSK2857916 as monotherapy for patients with multiple myeloma (MM) who have failed at least 3 prior lines of therapy, including an anti-CD38 antibody, and who are refractory to a proteasome inhibitor and an immunomodulatory agent.
The designation is based on results from a phase 1, dose-escalation and expansion study in patients with relapsed/refractory MM, irrespective of BCMA expression (NCT02064387).
Data from this ongoing trial are scheduled to be presented December 11 in an oral presentation at the 59th Annual Meeting of the American Society of Hematology (ASH) in Atlanta, Georgia.
GSK2857916 has also received orphan drug designation from the FDA and the European Medicines Agency (EMA) as well as PRIME designation from the EMA.
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions. The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Orphan and PRIME designations
The FDA grants orphan designation to therapies intended to treat conditions that affect fewer than 200,000 people in the US. The designation qualifies a drug’s sponsor for various development incentives of the Orphan Drug Act, including tax credits for qualified clinical testing and 7 years of market exclusivity.
In Europe, sponsors who obtain orphan designation for a potential new medicine benefit from a range of incentives, including protocol assistance, access to the centralized procedure, 10 years of market exclusivity, and fee reductions.
The EMA grants PRIME designation to enhance support for the development of medicines that target an unmet medical need. The designation is based on enhanced interaction between sponsor companies and the EMA to optimize development plans and speed up evaluation so these medicines can reach patients earlier. ![]()
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to GSK2857916, an anti-B-cell maturation antigen (BCMA) monoclonal antibody conjugated to the cytotoxic agent monomethyl auristatin-F via a non-cleavable linker.
The designation is for GSK2857916 as monotherapy for patients with multiple myeloma (MM) who have failed at least 3 prior lines of therapy, including an anti-CD38 antibody, and who are refractory to a proteasome inhibitor and an immunomodulatory agent.
The designation is based on results from a phase 1, dose-escalation and expansion study in patients with relapsed/refractory MM, irrespective of BCMA expression (NCT02064387).
Data from this ongoing trial are scheduled to be presented December 11 in an oral presentation at the 59th Annual Meeting of the American Society of Hematology (ASH) in Atlanta, Georgia.
GSK2857916 has also received orphan drug designation from the FDA and the European Medicines Agency (EMA) as well as PRIME designation from the EMA.
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions. The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Orphan and PRIME designations
The FDA grants orphan designation to therapies intended to treat conditions that affect fewer than 200,000 people in the US. The designation qualifies a drug’s sponsor for various development incentives of the Orphan Drug Act, including tax credits for qualified clinical testing and 7 years of market exclusivity.
In Europe, sponsors who obtain orphan designation for a potential new medicine benefit from a range of incentives, including protocol assistance, access to the centralized procedure, 10 years of market exclusivity, and fee reductions.
The EMA grants PRIME designation to enhance support for the development of medicines that target an unmet medical need. The designation is based on enhanced interaction between sponsor companies and the EMA to optimize development plans and speed up evaluation so these medicines can reach patients earlier. ![]()
Cancer patients prefer computer-free interactions

SAN DIEGO—A new study suggests patients with advanced cancer may prefer doctors who do not use a computer while communicating with them.
Most of the 120 patients studied said they preferred face-to-face consultations in which a doctor used a notepad rather than a computer.
Doctors who did not use a computer were perceived as more compassionate, communicative, and professional.
These findings were presented at the 2017 Palliative and Supportive Care in Oncology Symposium (abstract 26*).
“To our knowledge, this is the only study that compares exam room interactions between people with advanced cancer and their physicians, with or without a computer present,” said study investigator Ali Haider, MD, of the University of Texas MD Anderson Cancer Center in Houston.
For this study, Dr Haider and his colleagues enrolled 120 patients with localized, recurrent, or metastatic disease. The patients’ median ECOG performance status was 2.
All patients were English speakers, they had a median age of 58 (range, 44-66), and 55% were female. Sixty-seven percent of patients were white, 18% were Hispanic, 13% were African American, and 2% were “other.” Forty-one percent of patients had completed college.
According to the Edmonton Symptom Assessment System, patients’ median pain score was 5 (range, 2-7), and their median fatigue score was 4 (range, 3-7). According to the Hospital Anxiety and Depression Scale, patients’ median anxiety score was 6 (range, 4-8), and their median depression score was 6 (range, 4-9).
The intervention
The investigators randomly assigned patients to watch different videos showing doctor-patient interactions with and without computer use. The team had filmed 4 short videos that featured actors playing the parts of doctor and patient.
All study participants were blinded to the hypothesis of the study. The actors were carefully scripted and used the same gestures, expressions, and other nonverbal communication in each video to minimize bias.
Video 1 involved Doctor A in a face-to-face consultation with just a notepad in hand, and Video 2 involved Doctor A in a consultation using a computer.
Video 3 involved Doctor B in a face-to-face consultation with just a notepad in hand, and Video 4 involved Doctor B in a consultation using a computer.
Doctors A and B looked similar, which was intended to minimize bias.
After viewing their first video, patients completed a validated questionnaire rating the doctor’s communication skills, professionalism, and compassion.
Subsequently, each group was assigned to a video topic (face-to-face or computer) they had not viewed previously featuring the doctor they had not viewed in the first video.
A follow-up questionnaire was given after this round of viewing, and the patients were also asked to rate their overall physician preference.
Results
After the first round of viewing, the patients gave better ratings to doctors (A or B) in the face-to-face videos than in the computer videos. Face-to-face doctors were rated significantly higher for compassion (P=0.0003), communication skills (P=0.0012), and professionalism (P=0.0001).
After patients had watched both videos, doctors in the face-to-face videos still had better scores for compassion, communication, and professionalism (P<0.001 for all).
Most patients (72%) said they preferred the face-to-face consultation, while 8% said they preferred the computer consultation, and 20% said they had no preference.
Dr Haider said a possible explanation for these findings is that patients with serious chronic illnesses might value undivided attention from their physicians, and patients might perceive providers using computers as more distracted or multitasking during visits.
“We know that having a good rapport with patients can be extremely beneficial for their health,” Dr Haider said. “Patients with advanced disease need the cues that come with direct interaction to help them along with their care.”
However, Dr Haider also noted that additional research is needed to confirm these results. And he said perceptions might be different in a younger population with higher computer literacy. ![]()
*Data in the abstract differ from the presentation.
SAN DIEGO—A new study suggests patients with advanced cancer may prefer doctors who do not use a computer while communicating with them.
Most of the 120 patients studied said they preferred face-to-face consultations in which a doctor used a notepad rather than a computer.
Doctors who did not use a computer were perceived as more compassionate, communicative, and professional.
These findings were presented at the 2017 Palliative and Supportive Care in Oncology Symposium (abstract 26*).
“To our knowledge, this is the only study that compares exam room interactions between people with advanced cancer and their physicians, with or without a computer present,” said study investigator Ali Haider, MD, of the University of Texas MD Anderson Cancer Center in Houston.
For this study, Dr Haider and his colleagues enrolled 120 patients with localized, recurrent, or metastatic disease. The patients’ median ECOG performance status was 2.
All patients were English speakers, they had a median age of 58 (range, 44-66), and 55% were female. Sixty-seven percent of patients were white, 18% were Hispanic, 13% were African American, and 2% were “other.” Forty-one percent of patients had completed college.
According to the Edmonton Symptom Assessment System, patients’ median pain score was 5 (range, 2-7), and their median fatigue score was 4 (range, 3-7). According to the Hospital Anxiety and Depression Scale, patients’ median anxiety score was 6 (range, 4-8), and their median depression score was 6 (range, 4-9).
The intervention
The investigators randomly assigned patients to watch different videos showing doctor-patient interactions with and without computer use. The team had filmed 4 short videos that featured actors playing the parts of doctor and patient.
All study participants were blinded to the hypothesis of the study. The actors were carefully scripted and used the same gestures, expressions, and other nonverbal communication in each video to minimize bias.
Video 1 involved Doctor A in a face-to-face consultation with just a notepad in hand, and Video 2 involved Doctor A in a consultation using a computer.
Video 3 involved Doctor B in a face-to-face consultation with just a notepad in hand, and Video 4 involved Doctor B in a consultation using a computer.
Doctors A and B looked similar, which was intended to minimize bias.
After viewing their first video, patients completed a validated questionnaire rating the doctor’s communication skills, professionalism, and compassion.
Subsequently, each group was assigned to a video topic (face-to-face or computer) they had not viewed previously featuring the doctor they had not viewed in the first video.
A follow-up questionnaire was given after this round of viewing, and the patients were also asked to rate their overall physician preference.
Results
After the first round of viewing, the patients gave better ratings to doctors (A or B) in the face-to-face videos than in the computer videos. Face-to-face doctors were rated significantly higher for compassion (P=0.0003), communication skills (P=0.0012), and professionalism (P=0.0001).
After patients had watched both videos, doctors in the face-to-face videos still had better scores for compassion, communication, and professionalism (P<0.001 for all).
Most patients (72%) said they preferred the face-to-face consultation, while 8% said they preferred the computer consultation, and 20% said they had no preference.
Dr Haider said a possible explanation for these findings is that patients with serious chronic illnesses might value undivided attention from their physicians, and patients might perceive providers using computers as more distracted or multitasking during visits.
“We know that having a good rapport with patients can be extremely beneficial for their health,” Dr Haider said. “Patients with advanced disease need the cues that come with direct interaction to help them along with their care.”
However, Dr Haider also noted that additional research is needed to confirm these results. And he said perceptions might be different in a younger population with higher computer literacy. ![]()
*Data in the abstract differ from the presentation.
SAN DIEGO—A new study suggests patients with advanced cancer may prefer doctors who do not use a computer while communicating with them.
Most of the 120 patients studied said they preferred face-to-face consultations in which a doctor used a notepad rather than a computer.
Doctors who did not use a computer were perceived as more compassionate, communicative, and professional.
These findings were presented at the 2017 Palliative and Supportive Care in Oncology Symposium (abstract 26*).
“To our knowledge, this is the only study that compares exam room interactions between people with advanced cancer and their physicians, with or without a computer present,” said study investigator Ali Haider, MD, of the University of Texas MD Anderson Cancer Center in Houston.
For this study, Dr Haider and his colleagues enrolled 120 patients with localized, recurrent, or metastatic disease. The patients’ median ECOG performance status was 2.
All patients were English speakers, they had a median age of 58 (range, 44-66), and 55% were female. Sixty-seven percent of patients were white, 18% were Hispanic, 13% were African American, and 2% were “other.” Forty-one percent of patients had completed college.
According to the Edmonton Symptom Assessment System, patients’ median pain score was 5 (range, 2-7), and their median fatigue score was 4 (range, 3-7). According to the Hospital Anxiety and Depression Scale, patients’ median anxiety score was 6 (range, 4-8), and their median depression score was 6 (range, 4-9).
The intervention
The investigators randomly assigned patients to watch different videos showing doctor-patient interactions with and without computer use. The team had filmed 4 short videos that featured actors playing the parts of doctor and patient.
All study participants were blinded to the hypothesis of the study. The actors were carefully scripted and used the same gestures, expressions, and other nonverbal communication in each video to minimize bias.
Video 1 involved Doctor A in a face-to-face consultation with just a notepad in hand, and Video 2 involved Doctor A in a consultation using a computer.
Video 3 involved Doctor B in a face-to-face consultation with just a notepad in hand, and Video 4 involved Doctor B in a consultation using a computer.
Doctors A and B looked similar, which was intended to minimize bias.
After viewing their first video, patients completed a validated questionnaire rating the doctor’s communication skills, professionalism, and compassion.
Subsequently, each group was assigned to a video topic (face-to-face or computer) they had not viewed previously featuring the doctor they had not viewed in the first video.
A follow-up questionnaire was given after this round of viewing, and the patients were also asked to rate their overall physician preference.
Results
After the first round of viewing, the patients gave better ratings to doctors (A or B) in the face-to-face videos than in the computer videos. Face-to-face doctors were rated significantly higher for compassion (P=0.0003), communication skills (P=0.0012), and professionalism (P=0.0001).
After patients had watched both videos, doctors in the face-to-face videos still had better scores for compassion, communication, and professionalism (P<0.001 for all).
Most patients (72%) said they preferred the face-to-face consultation, while 8% said they preferred the computer consultation, and 20% said they had no preference.
Dr Haider said a possible explanation for these findings is that patients with serious chronic illnesses might value undivided attention from their physicians, and patients might perceive providers using computers as more distracted or multitasking during visits.
“We know that having a good rapport with patients can be extremely beneficial for their health,” Dr Haider said. “Patients with advanced disease need the cues that come with direct interaction to help them along with their care.”
However, Dr Haider also noted that additional research is needed to confirm these results. And he said perceptions might be different in a younger population with higher computer literacy. ![]()
*Data in the abstract differ from the presentation.
BCMA emerging as a promising target in MM
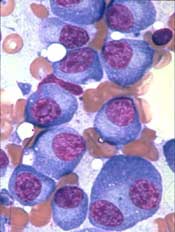
NEW YORK, NY—The B-cell maturation antigen (BCMA) is emerging as a promising target in multiple myeloma (MM), according to Adam D. Cohen, MD, of the University of Pennsylvania in Philadelphia.
BCMA is highly expressed on MM cells and, with its 2 ligands, is responsible for maintaining normal plasma cell homeostasis.
“And it’s really not expressed on any other normal tissues of the body,” Dr Cohen said.
“Importantly, BCMA is not just sitting on the cell surface as a target but actually promotes myeloma pathogenesis.”
Dr Cohen reviewed the progress being made using BCMA as a target in MM, paying particular attention to chimeric antigen receptor (CAR) T cells. He presented the update at Lymphoma & Myeloma 2017.
NCI BCMA-specific CARs
The first CAR to specifically target BCMA in MM was developed at the National Cancer Institute (NCI). It consisted of a murine single-chain variable fragment (scFv), CD3/CD28 signaling domains, and a gamma-retroviral vector.
Investigators conducted the first-in-human trial of this CAR T-cell therapy in 12 relapsed/refractory MM patients.
All patients received a lymphodepleting conditioning regimen of cyclophosphamide and fludarabine and a single infusion of 1 of 4 doses of the CAR T-cell therapy.
At higher dose levels, the BCMA-CAR produced objective responses “even in these highly refractory patients,” Dr Cohen said. “Some responses lasted 4 to 6 months.”
Patients who had the greatest degree of expansion of CAR T cells were the ones who had the best responses.
The BCMA-CAR is associated with the same toxicities as the CD19-directed CAR T-cell therapies now approved in acute lymphoblastic leukemia and non-Hodgkin lymphoma—cytokine release syndrome (CRS) and neurotoxicity.
The NCI study (NCT02215967) is ongoing.
Penn BCMA-specific CAR
A different BCMA CAR is being investigated at the University of Pennsylvania. It is a fully human CAR that consists of a human scFv, CD3/4-1BB costimulatory domains, and a lentiviral vector.
Investigators designed the first-in-human trial* (NCT02546167) with 3 different cohorts.
Patients in cohort 1 received 5 x 108 CAR T cells without any lymphodepleting chemotherapy.
The remaining patients received cyclophosphamide, followed by 5 x 107 CAR T cells in cohort 2 and 5 x 108 CAR T cells in cohort 3.
Dr Cohen reviewed current data from cohort 1, which included 9 patients. They were a median age of 57 (range, 44-70), and 67% were male. They were heavily pretreated with a median of 9 prior lines of therapy (range, 4-11).
All had high-risk cytogenetics, 67% had deletion 17p or TP53 mutation, and they had a median of 80% bone marrow plasma cells (range, 15%-95%).
“Despite this,” Dr Cohen said, “we were able to generate, successfully, CAR T cells from all patients, although 1 patient did require a second apheresis and manufacturing attempt.”
Four of the 9 patients achieved very good partial responses, and an additional 2 patients had minimal responses.
One patient had a stringent CR (sCR) for close to 2 years without having any intervening therapy.
“[The sCR] shows the potential for this [therapy] to create a durable remission in a patient without any other therapy,” Dr Cohen said. “And this patient still has circulating CAR cells detectable.”
Most of the other patients did not have as durable a response. Responses lasted a median of 3 to 5 months before the patients relapsed.
Dr Cohen noted that the Penn data confirm the NCI experience showing proof of principle.
“You can target BCMA with these cells and get objective responses that can lead to a durable one in a subset of patients,” he added.
Dr Cohen is scheduled to report the preliminary data on the cyclophosphamide cohorts (cohorts 2 and 3) at ASH 2017. Those cohorts, he said, are showing a bit more persistence and increased frequency of responses.
Other BCMA-specific CAR trials
Another BCMA CAR, bb2121, consists of a human anti-BCMA scFv, a lentiviral vector, and a CD3/4-1BB costimulatory domain.
In the first-in-human trial of bb2121* (NCT02658929), all 21 patients received cyclophosphamide and fludarabine conditioning first.
There were “very impressive response rates in this study,” Dr Cohen said.
Once patients were dosed at the higher levels of 150 million cells or greater, every patient responded.
“Many responses are still durable,” he pointed out, “some approaching a year.”
LCAR-B38M is structurally different from the other BCMA-specific CARs. It has 2 binding sites for BCMA.
Thirty-five patients enrolled on the trial of LCAR-B38M (NCT03090659), and 19 were evaluable for response.
Patients had a median of 3 or 4 lines of prior therapy. All received cyclophosphamide alone as conditioning.
All 19 evaluable patients responded, and 14 (74%) achieved an sCR.
Dr Cohen noted the differences among the 4 trials: costimulatory domains varied, the Penn study did not require a pre-existing level of BCMA on the MM cells, other studies excluded patients who didn’t meet a certain threshold level of BCMA expression, median lines of prior therapy differed, and conditioning regimens differed as well.
All trials, however, presented data on fewer than 20 patients.
“And so I think it’s a little too early to compare head-to-head between all of these, but they all are really demonstrating promising results so far,” he observed.
Toxicities
“Unfortunately, there’s no free lunch with CAR T cells,” Dr Cohen stated, “and these extraordinary responses do come at the cost of several somewhat unique toxicities that can be serious.”
Toxicities have included:
- Tumor lysis syndrome, which is expected and manageable
- B-cell aplasia, which is not as much of an issue with BCMA as with CD19-directed CAR Ts, since most B cells don’t express BCMA
- Hypogammaglobulinemia, which can be mitigated with IVIG
- CRS, which can be alleviated with tocilizumab
- Neurotoxicity and encephalopathy, which do not occur as frequently as CRS.
“These, I think, are things we still obviously need to learn more about and try to mitigate before this [BCMA-directed CAR therapy] is expanded and brought earlier to patients,” Dr Cohen said.
“The other issues with CAR T cells are logistical. Limited access (the procedure is available at only a few centers); manufacturing can take 2 to 4 weeks, during which time it is difficult to maintain disease control; and the cost is significant.”
“We are really in the early days of CAR cells for myeloma, but, certainly, this appears very bright in terms of its future.” ![]()
* Data from the presentation differ from the abstract.
NEW YORK, NY—The B-cell maturation antigen (BCMA) is emerging as a promising target in multiple myeloma (MM), according to Adam D. Cohen, MD, of the University of Pennsylvania in Philadelphia.
BCMA is highly expressed on MM cells and, with its 2 ligands, is responsible for maintaining normal plasma cell homeostasis.
“And it’s really not expressed on any other normal tissues of the body,” Dr Cohen said.
“Importantly, BCMA is not just sitting on the cell surface as a target but actually promotes myeloma pathogenesis.”
Dr Cohen reviewed the progress being made using BCMA as a target in MM, paying particular attention to chimeric antigen receptor (CAR) T cells. He presented the update at Lymphoma & Myeloma 2017.
NCI BCMA-specific CARs
The first CAR to specifically target BCMA in MM was developed at the National Cancer Institute (NCI). It consisted of a murine single-chain variable fragment (scFv), CD3/CD28 signaling domains, and a gamma-retroviral vector.
Investigators conducted the first-in-human trial of this CAR T-cell therapy in 12 relapsed/refractory MM patients.
All patients received a lymphodepleting conditioning regimen of cyclophosphamide and fludarabine and a single infusion of 1 of 4 doses of the CAR T-cell therapy.
At higher dose levels, the BCMA-CAR produced objective responses “even in these highly refractory patients,” Dr Cohen said. “Some responses lasted 4 to 6 months.”
Patients who had the greatest degree of expansion of CAR T cells were the ones who had the best responses.
The BCMA-CAR is associated with the same toxicities as the CD19-directed CAR T-cell therapies now approved in acute lymphoblastic leukemia and non-Hodgkin lymphoma—cytokine release syndrome (CRS) and neurotoxicity.
The NCI study (NCT02215967) is ongoing.
Penn BCMA-specific CAR
A different BCMA CAR is being investigated at the University of Pennsylvania. It is a fully human CAR that consists of a human scFv, CD3/4-1BB costimulatory domains, and a lentiviral vector.
Investigators designed the first-in-human trial* (NCT02546167) with 3 different cohorts.
Patients in cohort 1 received 5 x 108 CAR T cells without any lymphodepleting chemotherapy.
The remaining patients received cyclophosphamide, followed by 5 x 107 CAR T cells in cohort 2 and 5 x 108 CAR T cells in cohort 3.
Dr Cohen reviewed current data from cohort 1, which included 9 patients. They were a median age of 57 (range, 44-70), and 67% were male. They were heavily pretreated with a median of 9 prior lines of therapy (range, 4-11).
All had high-risk cytogenetics, 67% had deletion 17p or TP53 mutation, and they had a median of 80% bone marrow plasma cells (range, 15%-95%).
“Despite this,” Dr Cohen said, “we were able to generate, successfully, CAR T cells from all patients, although 1 patient did require a second apheresis and manufacturing attempt.”
Four of the 9 patients achieved very good partial responses, and an additional 2 patients had minimal responses.
One patient had a stringent CR (sCR) for close to 2 years without having any intervening therapy.
“[The sCR] shows the potential for this [therapy] to create a durable remission in a patient without any other therapy,” Dr Cohen said. “And this patient still has circulating CAR cells detectable.”
Most of the other patients did not have as durable a response. Responses lasted a median of 3 to 5 months before the patients relapsed.
Dr Cohen noted that the Penn data confirm the NCI experience showing proof of principle.
“You can target BCMA with these cells and get objective responses that can lead to a durable one in a subset of patients,” he added.
Dr Cohen is scheduled to report the preliminary data on the cyclophosphamide cohorts (cohorts 2 and 3) at ASH 2017. Those cohorts, he said, are showing a bit more persistence and increased frequency of responses.
Other BCMA-specific CAR trials
Another BCMA CAR, bb2121, consists of a human anti-BCMA scFv, a lentiviral vector, and a CD3/4-1BB costimulatory domain.
In the first-in-human trial of bb2121* (NCT02658929), all 21 patients received cyclophosphamide and fludarabine conditioning first.
There were “very impressive response rates in this study,” Dr Cohen said.
Once patients were dosed at the higher levels of 150 million cells or greater, every patient responded.
“Many responses are still durable,” he pointed out, “some approaching a year.”
LCAR-B38M is structurally different from the other BCMA-specific CARs. It has 2 binding sites for BCMA.
Thirty-five patients enrolled on the trial of LCAR-B38M (NCT03090659), and 19 were evaluable for response.
Patients had a median of 3 or 4 lines of prior therapy. All received cyclophosphamide alone as conditioning.
All 19 evaluable patients responded, and 14 (74%) achieved an sCR.
Dr Cohen noted the differences among the 4 trials: costimulatory domains varied, the Penn study did not require a pre-existing level of BCMA on the MM cells, other studies excluded patients who didn’t meet a certain threshold level of BCMA expression, median lines of prior therapy differed, and conditioning regimens differed as well.
All trials, however, presented data on fewer than 20 patients.
“And so I think it’s a little too early to compare head-to-head between all of these, but they all are really demonstrating promising results so far,” he observed.
Toxicities
“Unfortunately, there’s no free lunch with CAR T cells,” Dr Cohen stated, “and these extraordinary responses do come at the cost of several somewhat unique toxicities that can be serious.”
Toxicities have included:
- Tumor lysis syndrome, which is expected and manageable
- B-cell aplasia, which is not as much of an issue with BCMA as with CD19-directed CAR Ts, since most B cells don’t express BCMA
- Hypogammaglobulinemia, which can be mitigated with IVIG
- CRS, which can be alleviated with tocilizumab
- Neurotoxicity and encephalopathy, which do not occur as frequently as CRS.
“These, I think, are things we still obviously need to learn more about and try to mitigate before this [BCMA-directed CAR therapy] is expanded and brought earlier to patients,” Dr Cohen said.
“The other issues with CAR T cells are logistical. Limited access (the procedure is available at only a few centers); manufacturing can take 2 to 4 weeks, during which time it is difficult to maintain disease control; and the cost is significant.”
“We are really in the early days of CAR cells for myeloma, but, certainly, this appears very bright in terms of its future.” ![]()
* Data from the presentation differ from the abstract.
NEW YORK, NY—The B-cell maturation antigen (BCMA) is emerging as a promising target in multiple myeloma (MM), according to Adam D. Cohen, MD, of the University of Pennsylvania in Philadelphia.
BCMA is highly expressed on MM cells and, with its 2 ligands, is responsible for maintaining normal plasma cell homeostasis.
“And it’s really not expressed on any other normal tissues of the body,” Dr Cohen said.
“Importantly, BCMA is not just sitting on the cell surface as a target but actually promotes myeloma pathogenesis.”
Dr Cohen reviewed the progress being made using BCMA as a target in MM, paying particular attention to chimeric antigen receptor (CAR) T cells. He presented the update at Lymphoma & Myeloma 2017.
NCI BCMA-specific CARs
The first CAR to specifically target BCMA in MM was developed at the National Cancer Institute (NCI). It consisted of a murine single-chain variable fragment (scFv), CD3/CD28 signaling domains, and a gamma-retroviral vector.
Investigators conducted the first-in-human trial of this CAR T-cell therapy in 12 relapsed/refractory MM patients.
All patients received a lymphodepleting conditioning regimen of cyclophosphamide and fludarabine and a single infusion of 1 of 4 doses of the CAR T-cell therapy.
At higher dose levels, the BCMA-CAR produced objective responses “even in these highly refractory patients,” Dr Cohen said. “Some responses lasted 4 to 6 months.”
Patients who had the greatest degree of expansion of CAR T cells were the ones who had the best responses.
The BCMA-CAR is associated with the same toxicities as the CD19-directed CAR T-cell therapies now approved in acute lymphoblastic leukemia and non-Hodgkin lymphoma—cytokine release syndrome (CRS) and neurotoxicity.
The NCI study (NCT02215967) is ongoing.
Penn BCMA-specific CAR
A different BCMA CAR is being investigated at the University of Pennsylvania. It is a fully human CAR that consists of a human scFv, CD3/4-1BB costimulatory domains, and a lentiviral vector.
Investigators designed the first-in-human trial* (NCT02546167) with 3 different cohorts.
Patients in cohort 1 received 5 x 108 CAR T cells without any lymphodepleting chemotherapy.
The remaining patients received cyclophosphamide, followed by 5 x 107 CAR T cells in cohort 2 and 5 x 108 CAR T cells in cohort 3.
Dr Cohen reviewed current data from cohort 1, which included 9 patients. They were a median age of 57 (range, 44-70), and 67% were male. They were heavily pretreated with a median of 9 prior lines of therapy (range, 4-11).
All had high-risk cytogenetics, 67% had deletion 17p or TP53 mutation, and they had a median of 80% bone marrow plasma cells (range, 15%-95%).
“Despite this,” Dr Cohen said, “we were able to generate, successfully, CAR T cells from all patients, although 1 patient did require a second apheresis and manufacturing attempt.”
Four of the 9 patients achieved very good partial responses, and an additional 2 patients had minimal responses.
One patient had a stringent CR (sCR) for close to 2 years without having any intervening therapy.
“[The sCR] shows the potential for this [therapy] to create a durable remission in a patient without any other therapy,” Dr Cohen said. “And this patient still has circulating CAR cells detectable.”
Most of the other patients did not have as durable a response. Responses lasted a median of 3 to 5 months before the patients relapsed.
Dr Cohen noted that the Penn data confirm the NCI experience showing proof of principle.
“You can target BCMA with these cells and get objective responses that can lead to a durable one in a subset of patients,” he added.
Dr Cohen is scheduled to report the preliminary data on the cyclophosphamide cohorts (cohorts 2 and 3) at ASH 2017. Those cohorts, he said, are showing a bit more persistence and increased frequency of responses.
Other BCMA-specific CAR trials
Another BCMA CAR, bb2121, consists of a human anti-BCMA scFv, a lentiviral vector, and a CD3/4-1BB costimulatory domain.
In the first-in-human trial of bb2121* (NCT02658929), all 21 patients received cyclophosphamide and fludarabine conditioning first.
There were “very impressive response rates in this study,” Dr Cohen said.
Once patients were dosed at the higher levels of 150 million cells or greater, every patient responded.
“Many responses are still durable,” he pointed out, “some approaching a year.”
LCAR-B38M is structurally different from the other BCMA-specific CARs. It has 2 binding sites for BCMA.
Thirty-five patients enrolled on the trial of LCAR-B38M (NCT03090659), and 19 were evaluable for response.
Patients had a median of 3 or 4 lines of prior therapy. All received cyclophosphamide alone as conditioning.
All 19 evaluable patients responded, and 14 (74%) achieved an sCR.
Dr Cohen noted the differences among the 4 trials: costimulatory domains varied, the Penn study did not require a pre-existing level of BCMA on the MM cells, other studies excluded patients who didn’t meet a certain threshold level of BCMA expression, median lines of prior therapy differed, and conditioning regimens differed as well.
All trials, however, presented data on fewer than 20 patients.
“And so I think it’s a little too early to compare head-to-head between all of these, but they all are really demonstrating promising results so far,” he observed.
Toxicities
“Unfortunately, there’s no free lunch with CAR T cells,” Dr Cohen stated, “and these extraordinary responses do come at the cost of several somewhat unique toxicities that can be serious.”
Toxicities have included:
- Tumor lysis syndrome, which is expected and manageable
- B-cell aplasia, which is not as much of an issue with BCMA as with CD19-directed CAR Ts, since most B cells don’t express BCMA
- Hypogammaglobulinemia, which can be mitigated with IVIG
- CRS, which can be alleviated with tocilizumab
- Neurotoxicity and encephalopathy, which do not occur as frequently as CRS.
“These, I think, are things we still obviously need to learn more about and try to mitigate before this [BCMA-directed CAR therapy] is expanded and brought earlier to patients,” Dr Cohen said.
“The other issues with CAR T cells are logistical. Limited access (the procedure is available at only a few centers); manufacturing can take 2 to 4 weeks, during which time it is difficult to maintain disease control; and the cost is significant.”
“We are really in the early days of CAR cells for myeloma, but, certainly, this appears very bright in terms of its future.”
* Data from the presentation differ from the abstract.
Speaker advises caution in adding mAbs upfront in MM

NEW YORK, NY—Despite the attraction of incorporating monoclonal antibodies (mAbs) into upfront therapy for multiple myeloma (MM), a speaker at Lymphoma & Myeloma 2017 suggested mAbs are “not quite ready” for this use.
MAbs, particularly daratumumab, have shown single-agent activity in refractory MM and have been feasibly added to proteasome inhibitors and immunomodulatory drugs.
MAbs may even have the potential to enhance induction and shorten the time to minimal residual disease negativity.
“So the tendency is to simply add them to frontline therapy,” said the speaker, Joseph Mikhael, MD, from Mayo Clinic Arizona in Scottsdale.
However, he noted that there is little long-term experience with these agents.
“I’m going to suggest to you that they’re not quite ready [for upfront use] but will likely be ready in the future,” Dr Mikhael said. “We’ve had such a revolution in myeloma the last decade that it’s just easy for us to say, ‘Oh, throw it in there, just like we did, frankly, with rituximab in the lymphoma days. We added it to CVP, we added it to CHOP, we added it to bendamustine. It didn’t matter what we added it to, it just upgraded the response.”
“And, sometimes, I think we have the same approach with daratumumab or elotuzumab or some of the other mAbs that we have. I think we just have to do so cautiously.”
At present, the combination of a proteasome inhibitor and an immunomodulatory drug are the standard of care upfront in transplant-eligible and -ineligible MM patients.
Daratumumab plus KRd
Dr Mikhael described the experience of daratumumab added to carfilzomib, lenalidomide, and dexamethasone (KRd) in patients with newly diagnosed MM in the MMY1001 study.
Twenty-two patients were enrolled on the study, 91% achieved a very good partial response (VGPR) or better, and 43% achieved a complete response (CR). The depth of response improved with the duration of treatment.
Eight patients (36%) discontinued treatment.
Dr Mikhael emphasized that the preliminary data included very small numbers.
“There is a little bit of a yellow flag that pops up here,” he added, “when I see that 36% discontinued treatment, even in small numbers.”
The safety profile was consistent with previous reports for daratumumab or KRd.
The most common hematologic grade 3-4 treatment-emergent adverse events (AEs) occurring in 30% or more of patients were lymphopenia (64%), thrombocytopenia (9%), anemia (9%), leukopenia (9%), and neutropenia (14%).
Diarrhea (14%), cough (5%), fatigue (5%), insomnia (5%), and increased ALT (9%) were the most common grade 3-4 nonhematologic treatment-emergent AEs occurring in 30% or more of patients.
The treatment had no adverse impact on stem cell collection.
Elotuzumab plus VRd
Turning to elotuzumab in combination with bortezomib, lenalidomide, and dexamethasone (VRd), Dr Mikhael reviewed the phase 2a study (NCT02375555) presented at ASCO 2017 (abstract 8002).
Forty-one patients were enrolled on the study.
The overall response rate after 4 cycles was 100%, with 24% achieving a CR, 47% achieving a VGPR, and 29% a partial response.
Fatigue (60%), neuropathy (55%), musculoskeletal/joint pain (55%), infection (50%), back/neck pain (48%), diarrhea (45%), edema (38%), constipation (38%), cough (35%), mood alteration (35%), rash (35%), and insomnia (30%) occurred in 30% or more of patients.
“So again, not shocking,” Dr Mikhael said, “there was fatigue, there was neuropathy, and there were infections in 50% of patients.”
Grade 4 or greater AEs included thrombocytopenia, hyperglycemia, sepsis, cardiac arrest, and respiratory failure.
“However, here, [we have] maybe not even a yellow flag but a red flag of caution that there were 2 patients who died,” Dr Mikhael noted.
One patient died on study due to respiratory failure and sepsis that arose during cycle 2.
The other patient died more than 30 days after discontinuing study therapy due to febrile neutropenia and hypotension related to sepsis, followed by renal failure.
“Again, in a study that has such small numbers, I don’t want to overstate the case . . ., we don’t want to overreact, but whenever there is death involved, obviously, we have to be particularly cautious,” Dr Mikhael said.
Put into the context of 3 other VRd studies, he noted, the response rate with elotuzumab and VRd is relatively similar but not as good as the phase 3 study of VRd, which was a much larger study of 350 patients.
The situation with daratumumab and KRd is similar to elotuzumab, Dr Mikhael pointed out.
The initial response rates are impressive, but, when compared to other studies, “71% VGPR is good, only after 4 cycles, but we know that, in other studies, after a few more cycles, it was significantly higher.”
Cost
Dr Mikhael also considered cost in his assessment of daratumumab and elotuzumab integrated into frontline regimens.
Adding elotuzumab to VRd would almost double the cost of 12 weeks of therapy. And adding daratumumab to KRd would increase the cost even more.
“These costs are real,” Dr Mikhael said, “and, ultimately, if it’s the best thing for our patients, that’s what we are going to do. But until we have that convincing evidence, I think it’s critical to keep that in perspective. I would suggest that VRd, in many respects, is the standard of care for most patients.”
In terms of adding a mAb upfront, he said, “I don’t think we’re there yet. Do I think, in time, we will be? Quite likely, but I don’t think we are there yet.”
NEW YORK, NY—Despite the attraction of incorporating monoclonal antibodies (mAbs) into upfront therapy for multiple myeloma (MM), a speaker at Lymphoma & Myeloma 2017 suggested mAbs are “not quite ready” for this use.
MAbs, particularly daratumumab, have shown single-agent activity in refractory MM and have been feasibly added to proteasome inhibitors and immunomodulatory drugs.
MAbs may even have the potential to enhance induction and shorten the time to minimal residual disease negativity.
“So the tendency is to simply add them to frontline therapy,” said the speaker, Joseph Mikhael, MD, from Mayo Clinic Arizona in Scottsdale.
However, he noted that there is little long-term experience with these agents.
“I’m going to suggest to you that they’re not quite ready [for upfront use] but will likely be ready in the future,” Dr Mikhael said. “We’ve had such a revolution in myeloma the last decade that it’s just easy for us to say, ‘Oh, throw it in there, just like we did, frankly, with rituximab in the lymphoma days. We added it to CVP, we added it to CHOP, we added it to bendamustine. It didn’t matter what we added it to, it just upgraded the response.”
“And, sometimes, I think we have the same approach with daratumumab or elotuzumab or some of the other mAbs that we have. I think we just have to do so cautiously.”
At present, the combination of a proteasome inhibitor and an immunomodulatory drug are the standard of care upfront in transplant-eligible and -ineligible MM patients.
Daratumumab plus KRd
Dr Mikhael described the experience of daratumumab added to carfilzomib, lenalidomide, and dexamethasone (KRd) in patients with newly diagnosed MM in the MMY1001 study.
Twenty-two patients were enrolled on the study, 91% achieved a very good partial response (VGPR) or better, and 43% achieved a complete response (CR). The depth of response improved with the duration of treatment.
Eight patients (36%) discontinued treatment.
Dr Mikhael emphasized that the preliminary data included very small numbers.
“There is a little bit of a yellow flag that pops up here,” he added, “when I see that 36% discontinued treatment, even in small numbers.”
The safety profile was consistent with previous reports for daratumumab or KRd.
The most common hematologic grade 3-4 treatment-emergent adverse events (AEs) occurring in 30% or more of patients were lymphopenia (64%), thrombocytopenia (9%), anemia (9%), leukopenia (9%), and neutropenia (14%).
Diarrhea (14%), cough (5%), fatigue (5%), insomnia (5%), and increased ALT (9%) were the most common grade 3-4 nonhematologic treatment-emergent AEs occurring in 30% or more of patients.
The treatment had no adverse impact on stem cell collection.
Elotuzumab plus VRd
Turning to elotuzumab in combination with bortezomib, lenalidomide, and dexamethasone (VRd), Dr Mikhael reviewed the phase 2a study (NCT02375555) presented at ASCO 2017 (abstract 8002).
Forty-one patients were enrolled on the study.
The overall response rate after 4 cycles was 100%, with 24% achieving a CR, 47% achieving a VGPR, and 29% a partial response.
Fatigue (60%), neuropathy (55%), musculoskeletal/joint pain (55%), infection (50%), back/neck pain (48%), diarrhea (45%), edema (38%), constipation (38%), cough (35%), mood alteration (35%), rash (35%), and insomnia (30%) occurred in 30% or more of patients.
“So again, not shocking,” Dr Mikhael said, “there was fatigue, there was neuropathy, and there were infections in 50% of patients.”
Grade 4 or greater AEs included thrombocytopenia, hyperglycemia, sepsis, cardiac arrest, and respiratory failure.
“However, here, [we have] maybe not even a yellow flag but a red flag of caution that there were 2 patients who died,” Dr Mikhael noted.
One patient died on study due to respiratory failure and sepsis that arose during cycle 2.
The other patient died more than 30 days after discontinuing study therapy due to febrile neutropenia and hypotension related to sepsis, followed by renal failure.
“Again, in a study that has such small numbers, I don’t want to overstate the case . . ., we don’t want to overreact, but whenever there is death involved, obviously, we have to be particularly cautious,” Dr Mikhael said.
Put into the context of 3 other VRd studies, he noted, the response rate with elotuzumab and VRd is relatively similar but not as good as the phase 3 study of VRd, which was a much larger study of 350 patients.
The situation with daratumumab and KRd is similar to elotuzumab, Dr Mikhael pointed out.
The initial response rates are impressive, but, when compared to other studies, “71% VGPR is good, only after 4 cycles, but we know that, in other studies, after a few more cycles, it was significantly higher.”
Cost
Dr Mikhael also considered cost in his assessment of daratumumab and elotuzumab integrated into frontline regimens.
Adding elotuzumab to VRd would almost double the cost of 12 weeks of therapy. And adding daratumumab to KRd would increase the cost even more.
“These costs are real,” Dr Mikhael said, “and, ultimately, if it’s the best thing for our patients, that’s what we are going to do. But until we have that convincing evidence, I think it’s critical to keep that in perspective. I would suggest that VRd, in many respects, is the standard of care for most patients.”
In terms of adding a mAb upfront, he said, “I don’t think we’re there yet. Do I think, in time, we will be? Quite likely, but I don’t think we are there yet.”
NEW YORK, NY—Despite the attraction of incorporating monoclonal antibodies (mAbs) into upfront therapy for multiple myeloma (MM), a speaker at Lymphoma & Myeloma 2017 suggested mAbs are “not quite ready” for this use.
MAbs, particularly daratumumab, have shown single-agent activity in refractory MM and have been feasibly added to proteasome inhibitors and immunomodulatory drugs.
MAbs may even have the potential to enhance induction and shorten the time to minimal residual disease negativity.
“So the tendency is to simply add them to frontline therapy,” said the speaker, Joseph Mikhael, MD, from Mayo Clinic Arizona in Scottsdale.
However, he noted that there is little long-term experience with these agents.
“I’m going to suggest to you that they’re not quite ready [for upfront use] but will likely be ready in the future,” Dr Mikhael said. “We’ve had such a revolution in myeloma the last decade that it’s just easy for us to say, ‘Oh, throw it in there, just like we did, frankly, with rituximab in the lymphoma days. We added it to CVP, we added it to CHOP, we added it to bendamustine. It didn’t matter what we added it to, it just upgraded the response.”
“And, sometimes, I think we have the same approach with daratumumab or elotuzumab or some of the other mAbs that we have. I think we just have to do so cautiously.”
At present, the combination of a proteasome inhibitor and an immunomodulatory drug are the standard of care upfront in transplant-eligible and -ineligible MM patients.
Daratumumab plus KRd
Dr Mikhael described the experience of daratumumab added to carfilzomib, lenalidomide, and dexamethasone (KRd) in patients with newly diagnosed MM in the MMY1001 study.
Twenty-two patients were enrolled on the study, 91% achieved a very good partial response (VGPR) or better, and 43% achieved a complete response (CR). The depth of response improved with the duration of treatment.
Eight patients (36%) discontinued treatment.
Dr Mikhael emphasized that the preliminary data included very small numbers.
“There is a little bit of a yellow flag that pops up here,” he added, “when I see that 36% discontinued treatment, even in small numbers.”
The safety profile was consistent with previous reports for daratumumab or KRd.
The most common hematologic grade 3-4 treatment-emergent adverse events (AEs) occurring in 30% or more of patients were lymphopenia (64%), thrombocytopenia (9%), anemia (9%), leukopenia (9%), and neutropenia (14%).
Diarrhea (14%), cough (5%), fatigue (5%), insomnia (5%), and increased ALT (9%) were the most common grade 3-4 nonhematologic treatment-emergent AEs occurring in 30% or more of patients.
The treatment had no adverse impact on stem cell collection.
Elotuzumab plus VRd
Turning to elotuzumab in combination with bortezomib, lenalidomide, and dexamethasone (VRd), Dr Mikhael reviewed the phase 2a study (NCT02375555) presented at ASCO 2017 (abstract 8002).
Forty-one patients were enrolled on the study.
The overall response rate after 4 cycles was 100%, with 24% achieving a CR, 47% achieving a VGPR, and 29% a partial response.
Fatigue (60%), neuropathy (55%), musculoskeletal/joint pain (55%), infection (50%), back/neck pain (48%), diarrhea (45%), edema (38%), constipation (38%), cough (35%), mood alteration (35%), rash (35%), and insomnia (30%) occurred in 30% or more of patients.
“So again, not shocking,” Dr Mikhael said, “there was fatigue, there was neuropathy, and there were infections in 50% of patients.”
Grade 4 or greater AEs included thrombocytopenia, hyperglycemia, sepsis, cardiac arrest, and respiratory failure.
“However, here, [we have] maybe not even a yellow flag but a red flag of caution that there were 2 patients who died,” Dr Mikhael noted.
One patient died on study due to respiratory failure and sepsis that arose during cycle 2.
The other patient died more than 30 days after discontinuing study therapy due to febrile neutropenia and hypotension related to sepsis, followed by renal failure.
“Again, in a study that has such small numbers, I don’t want to overstate the case . . ., we don’t want to overreact, but whenever there is death involved, obviously, we have to be particularly cautious,” Dr Mikhael said.
Put into the context of 3 other VRd studies, he noted, the response rate with elotuzumab and VRd is relatively similar but not as good as the phase 3 study of VRd, which was a much larger study of 350 patients.
The situation with daratumumab and KRd is similar to elotuzumab, Dr Mikhael pointed out.
The initial response rates are impressive, but, when compared to other studies, “71% VGPR is good, only after 4 cycles, but we know that, in other studies, after a few more cycles, it was significantly higher.”
Cost
Dr Mikhael also considered cost in his assessment of daratumumab and elotuzumab integrated into frontline regimens.
Adding elotuzumab to VRd would almost double the cost of 12 weeks of therapy. And adding daratumumab to KRd would increase the cost even more.
“These costs are real,” Dr Mikhael said, “and, ultimately, if it’s the best thing for our patients, that’s what we are going to do. But until we have that convincing evidence, I think it’s critical to keep that in perspective. I would suggest that VRd, in many respects, is the standard of care for most patients.”
In terms of adding a mAb upfront, he said, “I don’t think we’re there yet. Do I think, in time, we will be? Quite likely, but I don’t think we are there yet.”
X-GEM finds drug to have economic value in MM
NEW YORK, NY—Investigators have developed a model that suggests the clinical benefits of denosumab translate into economic value.
The investigators designed their model, X-GEM (Exgeva-Global Economic Model), using results from a large multiple myeloma (MM) trial that showed denosumab to be non-inferior to zoledronic acid (ZA) for skeletal-related events (SRE).
Noopur S. Raje, MD, of Massachusetts General Hospital Cancer Center in Boston, discussed this work at Lymphoma & Myeloma 2017.
The abstract was selected as the best clinical myeloma abstract of the meeting.
“I do think this is a very timely topic because people are not just concerned about the clinical outcomes in patients,” Dr Raje said. “[T]here’s a lot of buzz around the economic evaluation of what we do.”
Trial results
The phase 3 study (NCT01345019) on which X-GEM is based enrolled 1718 MM patients and randomized them to either denosumab or ZA.
The results, reported this year at ASCO, showed denosumab to be non-inferior to ZA in time to first on-study SRE, the primary endpoint.
Denosumab was also found to be non-inferior to ZA for the secondary endpoint of overall survival.
For the exploratory endpoint of progression-free survival (PFS), denosumab-treated patients experienced a significant benefit in terms of PFS. The median PFS was 46.09 months with denosumab and 35.38 months with ZA (difference, 10.71 months).
“Now, we’ve never really seen a survival difference or progression-free survival difference in patients, even in treatment studies, amounting to about 10.7 months,” Dr Raje said. “So this, we found, was quite remarkable in the study.”
Dr Raje also highlighted some safety features from the study. There was significantly less renal toxicity with denosumab than with ZA.
“And in patients who had a creatinine clearance of less than 60 [mL/min], there was almost a doubling of renal toxicity in patients getting zoledronic acid,” she said.
“[D]enosumab may, in fact, be the safer alternative, specifically, in our patients with multiple myeloma who we all know have this problem of renal toxicity throughout the course of their lifetime with myeloma.”
X-GEM
Investigators based X-GEM on the original model published by Stopeck et al. in 2012, which evaluated the cost-effectiveness of denosumab to prevent SREs compared with ZA in patients with solid tumors—prostate, breast, and lung cancer.
“[Stopeck’s] data did show that denosumab was, in fact, cost-effective when compared to zoledronic acid in respect to SREs,” Dr Raje noted.
The timeline for the economic analysis in MM patients spanned the time from diagnosis to death, and patients were evaluated for SREs every 4 weeks on study.
Investigators used 2 cost scenarios. The first was based on an average sales price for 28 days, which was $1928 for denosumab and $45 for ZA. The second was based on the wholesale acquisition cost, which was $2155 for denosumab and $922 for ZA.
“No surprise to anybody,” Dr Raje noted, “denosumab is a lot more expensive. But obviously, this does not tell the whole story. Built into the X-GEM model are a whole host of other factors.”
These include the costs of administration, adverse events, the number of SREs, treatment of an MM patient, and the quality-adjusted life year (QALY) gain with either denosumab or zoledronic acid.
“When you count up all these costs and calculate them based on the data set from the 1800 patients, we found that there was really a difference of zoledronic acid costing a little bit more than denosumab,” Dr Raje said.
From the payer perspective, when clinical outcomes were monetized, the net monetary benefit of denosumab compared with ZA was $5959.
And from a societal perspective, the model calculated the net monetary benefit for denosumab to be $10,259.
The societal perspective included SRE direct costs (hospital, outpatient, and emergency department visits, long-term care, hospice, physical therapy and devices, skilled nursing facility, and strong opioids), direct non-medical costs (driving for treatment, parking, and caregiver costs), and indirect costs (short-term disability and productivity loss).
The investigators concluded that denosumab is cost-effective below a willingness-to-pay threshold of $150,000/QALY regardless of ZA price, whether wholesale or average sales price.
“The bone-specific benefits and observed prolongation of progression-free survival in combination with the economic analysis provides denosumab as a valuable option for patients with multiple myeloma,” Dr Raje said. “In total, we still think it’s more cost-effective to use denosumab when compared to zoledronic acid in this patient population.”
At present, treatment options to prevent bone complications in MM patients are limited to bisphosphonates, including ZA. Denosumab is under review by the US Food and Drug Administration for an expanded indication to include MM.
Both the phase 3 and cost-effectiveness studies were funded by Amgen, Inc. Dr Raje and co-investigators of the study have either consulted for or are employed by Amgen, Inc.
NEW YORK, NY—Investigators have developed a model that suggests the clinical benefits of denosumab translate into economic value.
The investigators designed their model, X-GEM (Exgeva-Global Economic Model), using results from a large multiple myeloma (MM) trial that showed denosumab to be non-inferior to zoledronic acid (ZA) for skeletal-related events (SRE).
Noopur S. Raje, MD, of Massachusetts General Hospital Cancer Center in Boston, discussed this work at Lymphoma & Myeloma 2017.
The abstract was selected as the best clinical myeloma abstract of the meeting.
“I do think this is a very timely topic because people are not just concerned about the clinical outcomes in patients,” Dr Raje said. “[T]here’s a lot of buzz around the economic evaluation of what we do.”
Trial results
The phase 3 study (NCT01345019) on which X-GEM is based enrolled 1718 MM patients and randomized them to either denosumab or ZA.
The results, reported this year at ASCO, showed denosumab to be non-inferior to ZA in time to first on-study SRE, the primary endpoint.
Denosumab was also found to be non-inferior to ZA for the secondary endpoint of overall survival.
For the exploratory endpoint of progression-free survival (PFS), denosumab-treated patients experienced a significant benefit in terms of PFS. The median PFS was 46.09 months with denosumab and 35.38 months with ZA (difference, 10.71 months).
“Now, we’ve never really seen a survival difference or progression-free survival difference in patients, even in treatment studies, amounting to about 10.7 months,” Dr Raje said. “So this, we found, was quite remarkable in the study.”
Dr Raje also highlighted some safety features from the study. There was significantly less renal toxicity with denosumab than with ZA.
“And in patients who had a creatinine clearance of less than 60 [mL/min], there was almost a doubling of renal toxicity in patients getting zoledronic acid,” she said.
“[D]enosumab may, in fact, be the safer alternative, specifically, in our patients with multiple myeloma who we all know have this problem of renal toxicity throughout the course of their lifetime with myeloma.”
X-GEM
Investigators based X-GEM on the original model published by Stopeck et al. in 2012, which evaluated the cost-effectiveness of denosumab to prevent SREs compared with ZA in patients with solid tumors—prostate, breast, and lung cancer.
“[Stopeck’s] data did show that denosumab was, in fact, cost-effective when compared to zoledronic acid in respect to SREs,” Dr Raje noted.
The timeline for the economic analysis in MM patients spanned the time from diagnosis to death, and patients were evaluated for SREs every 4 weeks on study.
Investigators used 2 cost scenarios. The first was based on an average sales price for 28 days, which was $1928 for denosumab and $45 for ZA. The second was based on the wholesale acquisition cost, which was $2155 for denosumab and $922 for ZA.
“No surprise to anybody,” Dr Raje noted, “denosumab is a lot more expensive. But obviously, this does not tell the whole story. Built into the X-GEM model are a whole host of other factors.”
These include the costs of administration, adverse events, the number of SREs, treatment of an MM patient, and the quality-adjusted life year (QALY) gain with either denosumab or zoledronic acid.
“When you count up all these costs and calculate them based on the data set from the 1800 patients, we found that there was really a difference of zoledronic acid costing a little bit more than denosumab,” Dr Raje said.
From the payer perspective, when clinical outcomes were monetized, the net monetary benefit of denosumab compared with ZA was $5959.
And from a societal perspective, the model calculated the net monetary benefit for denosumab to be $10,259.
The societal perspective included SRE direct costs (hospital, outpatient, and emergency department visits, long-term care, hospice, physical therapy and devices, skilled nursing facility, and strong opioids), direct non-medical costs (driving for treatment, parking, and caregiver costs), and indirect costs (short-term disability and productivity loss).
The investigators concluded that denosumab is cost-effective below a willingness-to-pay threshold of $150,000/QALY regardless of ZA price, whether wholesale or average sales price.
“The bone-specific benefits and observed prolongation of progression-free survival in combination with the economic analysis provides denosumab as a valuable option for patients with multiple myeloma,” Dr Raje said. “In total, we still think it’s more cost-effective to use denosumab when compared to zoledronic acid in this patient population.”
At present, treatment options to prevent bone complications in MM patients are limited to bisphosphonates, including ZA. Denosumab is under review by the US Food and Drug Administration for an expanded indication to include MM.
Both the phase 3 and cost-effectiveness studies were funded by Amgen, Inc. Dr Raje and co-investigators of the study have either consulted for or are employed by Amgen, Inc.
NEW YORK, NY—Investigators have developed a model that suggests the clinical benefits of denosumab translate into economic value.
The investigators designed their model, X-GEM (Exgeva-Global Economic Model), using results from a large multiple myeloma (MM) trial that showed denosumab to be non-inferior to zoledronic acid (ZA) for skeletal-related events (SRE).
Noopur S. Raje, MD, of Massachusetts General Hospital Cancer Center in Boston, discussed this work at Lymphoma & Myeloma 2017.
The abstract was selected as the best clinical myeloma abstract of the meeting.
“I do think this is a very timely topic because people are not just concerned about the clinical outcomes in patients,” Dr Raje said. “[T]here’s a lot of buzz around the economic evaluation of what we do.”
Trial results
The phase 3 study (NCT01345019) on which X-GEM is based enrolled 1718 MM patients and randomized them to either denosumab or ZA.
The results, reported this year at ASCO, showed denosumab to be non-inferior to ZA in time to first on-study SRE, the primary endpoint.
Denosumab was also found to be non-inferior to ZA for the secondary endpoint of overall survival.
For the exploratory endpoint of progression-free survival (PFS), denosumab-treated patients experienced a significant benefit in terms of PFS. The median PFS was 46.09 months with denosumab and 35.38 months with ZA (difference, 10.71 months).
“Now, we’ve never really seen a survival difference or progression-free survival difference in patients, even in treatment studies, amounting to about 10.7 months,” Dr Raje said. “So this, we found, was quite remarkable in the study.”
Dr Raje also highlighted some safety features from the study. There was significantly less renal toxicity with denosumab than with ZA.
“And in patients who had a creatinine clearance of less than 60 [mL/min], there was almost a doubling of renal toxicity in patients getting zoledronic acid,” she said.
“[D]enosumab may, in fact, be the safer alternative, specifically, in our patients with multiple myeloma who we all know have this problem of renal toxicity throughout the course of their lifetime with myeloma.”
X-GEM
Investigators based X-GEM on the original model published by Stopeck et al. in 2012, which evaluated the cost-effectiveness of denosumab to prevent SREs compared with ZA in patients with solid tumors—prostate, breast, and lung cancer.
“[Stopeck’s] data did show that denosumab was, in fact, cost-effective when compared to zoledronic acid in respect to SREs,” Dr Raje noted.
The timeline for the economic analysis in MM patients spanned the time from diagnosis to death, and patients were evaluated for SREs every 4 weeks on study.
Investigators used 2 cost scenarios. The first was based on an average sales price for 28 days, which was $1928 for denosumab and $45 for ZA. The second was based on the wholesale acquisition cost, which was $2155 for denosumab and $922 for ZA.
“No surprise to anybody,” Dr Raje noted, “denosumab is a lot more expensive. But obviously, this does not tell the whole story. Built into the X-GEM model are a whole host of other factors.”
These include the costs of administration, adverse events, the number of SREs, treatment of an MM patient, and the quality-adjusted life year (QALY) gain with either denosumab or zoledronic acid.
“When you count up all these costs and calculate them based on the data set from the 1800 patients, we found that there was really a difference of zoledronic acid costing a little bit more than denosumab,” Dr Raje said.
From the payer perspective, when clinical outcomes were monetized, the net monetary benefit of denosumab compared with ZA was $5959.
And from a societal perspective, the model calculated the net monetary benefit for denosumab to be $10,259.
The societal perspective included SRE direct costs (hospital, outpatient, and emergency department visits, long-term care, hospice, physical therapy and devices, skilled nursing facility, and strong opioids), direct non-medical costs (driving for treatment, parking, and caregiver costs), and indirect costs (short-term disability and productivity loss).
The investigators concluded that denosumab is cost-effective below a willingness-to-pay threshold of $150,000/QALY regardless of ZA price, whether wholesale or average sales price.
“The bone-specific benefits and observed prolongation of progression-free survival in combination with the economic analysis provides denosumab as a valuable option for patients with multiple myeloma,” Dr Raje said. “In total, we still think it’s more cost-effective to use denosumab when compared to zoledronic acid in this patient population.”
At present, treatment options to prevent bone complications in MM patients are limited to bisphosphonates, including ZA. Denosumab is under review by the US Food and Drug Administration for an expanded indication to include MM.
Both the phase 3 and cost-effectiveness studies were funded by Amgen, Inc. Dr Raje and co-investigators of the study have either consulted for or are employed by Amgen, Inc.
Protein may be therapeutic target for MM

Preclinical research suggests the protein BMI-1 may be a therapeutic target for multiple myeloma (MM).
Researchers found that BMI-1 inhibition, with a drug called PTC-209, induced apoptosis in MM cell lines and primary cells.
In addition, PTC-209’s anti-myeloma activity was enhanced by inhibitors targeting EZH2 and BET bromodomains.
The researchers reported these results in Oncotarget.
The team noted that previous studies have suggested epigenetic alterations are involved in the development of MM. They chose to focus the current study on BMI-1 because it’s part of a protein complex involved in epigenetic regulation and could therefore be a potential target for influencing MM development.
“We used the substance PTC-209, which we know inhibits BMI-1, and treated cultivated multiple myeloma cells,” said study author Mohammad Alzrigat, PhD, of Uppsala University in Uppsala, Sweden.
“We used both cell lines that are continuously kept as cultivated cells and cells that were purified from multiple myeloma patients, either newly diagnosed or at relapse. In all cases, we found that [PTC-209] decreased cell survival, which indicates that PTC-209 has an anti-myeloma effect.”
The researchers said PTC-209 demonstrated “potent anti-myeloma activity, reducing the viability of MM cell lines at concentrations ranging up to 1.6 μM over 48 hours of treatment.” INA-6 was the cell line that proved most responsive to PTC-209, and U266-1970 was the least responsive.
The team also found that a 10 μM concentration of PTC-209 reduced the viability of all primary MM cells. This anti-myeloma activity was independent of disease state (whether patients were newly diagnosed or had relapsed disease) and cytogenetic karyotype.
“Our study showed that PTC-209 most likely functions as an anti-myeloma agent by inhibiting the production of BMI-1,” said study author Helena Jernberg-Wiklund, PhD, also of Uppsala University.
“We also saw that when PTC-209 was combined with other substances that inhibit epigenetic alterations, the myeloma cells’ survival was reduced even further compared to when only PTC-209 was used.”
Specifically, the researchers observed synergistic and additive activity when PTC-209 was combined with the EZH2 inhibitor UNC1999 and the BET inhibitor JQ1.
“Our results have both increased our understanding of how epigenetic alterations affect cancer development and shown how inhibiting these mechanisms in combination could potentially be utilized for future treatment of multiple myeloma patients, especially at relapse,” Dr Jernberg-Wiklund said.
Preclinical research suggests the protein BMI-1 may be a therapeutic target for multiple myeloma (MM).
Researchers found that BMI-1 inhibition, with a drug called PTC-209, induced apoptosis in MM cell lines and primary cells.
In addition, PTC-209’s anti-myeloma activity was enhanced by inhibitors targeting EZH2 and BET bromodomains.
The researchers reported these results in Oncotarget.
The team noted that previous studies have suggested epigenetic alterations are involved in the development of MM. They chose to focus the current study on BMI-1 because it’s part of a protein complex involved in epigenetic regulation and could therefore be a potential target for influencing MM development.
“We used the substance PTC-209, which we know inhibits BMI-1, and treated cultivated multiple myeloma cells,” said study author Mohammad Alzrigat, PhD, of Uppsala University in Uppsala, Sweden.
“We used both cell lines that are continuously kept as cultivated cells and cells that were purified from multiple myeloma patients, either newly diagnosed or at relapse. In all cases, we found that [PTC-209] decreased cell survival, which indicates that PTC-209 has an anti-myeloma effect.”
The researchers said PTC-209 demonstrated “potent anti-myeloma activity, reducing the viability of MM cell lines at concentrations ranging up to 1.6 μM over 48 hours of treatment.” INA-6 was the cell line that proved most responsive to PTC-209, and U266-1970 was the least responsive.
The team also found that a 10 μM concentration of PTC-209 reduced the viability of all primary MM cells. This anti-myeloma activity was independent of disease state (whether patients were newly diagnosed or had relapsed disease) and cytogenetic karyotype.
“Our study showed that PTC-209 most likely functions as an anti-myeloma agent by inhibiting the production of BMI-1,” said study author Helena Jernberg-Wiklund, PhD, also of Uppsala University.
“We also saw that when PTC-209 was combined with other substances that inhibit epigenetic alterations, the myeloma cells’ survival was reduced even further compared to when only PTC-209 was used.”
Specifically, the researchers observed synergistic and additive activity when PTC-209 was combined with the EZH2 inhibitor UNC1999 and the BET inhibitor JQ1.
“Our results have both increased our understanding of how epigenetic alterations affect cancer development and shown how inhibiting these mechanisms in combination could potentially be utilized for future treatment of multiple myeloma patients, especially at relapse,” Dr Jernberg-Wiklund said.
Preclinical research suggests the protein BMI-1 may be a therapeutic target for multiple myeloma (MM).
Researchers found that BMI-1 inhibition, with a drug called PTC-209, induced apoptosis in MM cell lines and primary cells.
In addition, PTC-209’s anti-myeloma activity was enhanced by inhibitors targeting EZH2 and BET bromodomains.
The researchers reported these results in Oncotarget.
The team noted that previous studies have suggested epigenetic alterations are involved in the development of MM. They chose to focus the current study on BMI-1 because it’s part of a protein complex involved in epigenetic regulation and could therefore be a potential target for influencing MM development.
“We used the substance PTC-209, which we know inhibits BMI-1, and treated cultivated multiple myeloma cells,” said study author Mohammad Alzrigat, PhD, of Uppsala University in Uppsala, Sweden.
“We used both cell lines that are continuously kept as cultivated cells and cells that were purified from multiple myeloma patients, either newly diagnosed or at relapse. In all cases, we found that [PTC-209] decreased cell survival, which indicates that PTC-209 has an anti-myeloma effect.”
The researchers said PTC-209 demonstrated “potent anti-myeloma activity, reducing the viability of MM cell lines at concentrations ranging up to 1.6 μM over 48 hours of treatment.” INA-6 was the cell line that proved most responsive to PTC-209, and U266-1970 was the least responsive.
The team also found that a 10 μM concentration of PTC-209 reduced the viability of all primary MM cells. This anti-myeloma activity was independent of disease state (whether patients were newly diagnosed or had relapsed disease) and cytogenetic karyotype.
“Our study showed that PTC-209 most likely functions as an anti-myeloma agent by inhibiting the production of BMI-1,” said study author Helena Jernberg-Wiklund, PhD, also of Uppsala University.
“We also saw that when PTC-209 was combined with other substances that inhibit epigenetic alterations, the myeloma cells’ survival was reduced even further compared to when only PTC-209 was used.”
Specifically, the researchers observed synergistic and additive activity when PTC-209 was combined with the EZH2 inhibitor UNC1999 and the BET inhibitor JQ1.
“Our results have both increased our understanding of how epigenetic alterations affect cancer development and shown how inhibiting these mechanisms in combination could potentially be utilized for future treatment of multiple myeloma patients, especially at relapse,” Dr Jernberg-Wiklund said.