User login
Multiple Myeloma and Stroke: What’s the Risk?
Related: Treating Patients With Multiple Myeloma in the VA
Patients were enrolled in Total Therapy protocols (TT2, TT3a, and TT3b), which tested varying combinations of thalidomide, bortezomib, lenalidomide, and dexamethasone. Of 1,148 patients, 46 (4%) had strokes, usually ischemic stroke (33 patients, or 72%). Hypercoagulability, atrial fibrillation and small-vessel occlusion were common mechanisms. Whereas other research has found a higher risk of arterial thrombosis from activated prothrombotic factors, especially during the early period of chemotherapy, in this study vascular events occurred months later.
Seven patients died in the hospital (15% compared with a national average of 5%). Although 6 of those deaths were stroke related, 36 patients were discharged home or to a rehabilitation facility; 2 were discharged to a long-term nursing facility. During a median follow-up of 10 years, 6 patients had another stroke. The cumulative risk of recurrent stroke was 15% compared with 5% for the general population.
Related: Link Found Between Agent Orange Exposure and Multiple Myeloma
Stage I and II cancers and renal insufficiency independently predicted stroke. Also noteworthy, according to the researchers: Patients with MM who developed renal insufficiency had worse clinical outcomes despite improvement in their renal function or lack of significant difference in their baseline renal functions between various treatment protocols. Thus, the increased risk of stroke, recurrent stroke, and mortality could partly be due to renal disease, which may or may not have resulted from myeloma.
Use of combination chemotherapy has “markedly improved” clinical outcomes for MM patients, the researchers say, but those drugs have also been associated with an increased risk of VTE, especially during the first months of chemotherapy. Thalidomide alone did not increase the risk of VTE, nor did lenalinomide on its own. However, thalidomide combined with multiagent chemotherapy increased VTE risk as much as 34% in newly diagnosed patients, and lenalinomide with dexamethasone boosted risk as high as 75%.
The researchers found no significant relationship between mortality and use of thalidomide. Median survival was 103 months for a thalidomide-based regimen and 78 months for a regimen without thalidomide.
Related: Multiple Myeloma: Updates on Diagnosis and Management
The researchers noted that the patients developed strokes despite a trend toward coagulopathy, to the extent that half were ineligible for immediate use of antiplatelet agents. The study findings “heightened our awareness,” the researchers say, that aggressive preventive measures can help reduce the incidence of stroke in patients with renal insufficiency.
Source:
Hinduja A, Limaye K, Ravilla R, et al. PLoS One. 2016;11(11): e0166627.
doi: 10.1371/journal.pone.0166627.
Related: Treating Patients With Multiple Myeloma in the VA
Patients were enrolled in Total Therapy protocols (TT2, TT3a, and TT3b), which tested varying combinations of thalidomide, bortezomib, lenalidomide, and dexamethasone. Of 1,148 patients, 46 (4%) had strokes, usually ischemic stroke (33 patients, or 72%). Hypercoagulability, atrial fibrillation and small-vessel occlusion were common mechanisms. Whereas other research has found a higher risk of arterial thrombosis from activated prothrombotic factors, especially during the early period of chemotherapy, in this study vascular events occurred months later.
Seven patients died in the hospital (15% compared with a national average of 5%). Although 6 of those deaths were stroke related, 36 patients were discharged home or to a rehabilitation facility; 2 were discharged to a long-term nursing facility. During a median follow-up of 10 years, 6 patients had another stroke. The cumulative risk of recurrent stroke was 15% compared with 5% for the general population.
Related: Link Found Between Agent Orange Exposure and Multiple Myeloma
Stage I and II cancers and renal insufficiency independently predicted stroke. Also noteworthy, according to the researchers: Patients with MM who developed renal insufficiency had worse clinical outcomes despite improvement in their renal function or lack of significant difference in their baseline renal functions between various treatment protocols. Thus, the increased risk of stroke, recurrent stroke, and mortality could partly be due to renal disease, which may or may not have resulted from myeloma.
Use of combination chemotherapy has “markedly improved” clinical outcomes for MM patients, the researchers say, but those drugs have also been associated with an increased risk of VTE, especially during the first months of chemotherapy. Thalidomide alone did not increase the risk of VTE, nor did lenalinomide on its own. However, thalidomide combined with multiagent chemotherapy increased VTE risk as much as 34% in newly diagnosed patients, and lenalinomide with dexamethasone boosted risk as high as 75%.
The researchers found no significant relationship between mortality and use of thalidomide. Median survival was 103 months for a thalidomide-based regimen and 78 months for a regimen without thalidomide.
Related: Multiple Myeloma: Updates on Diagnosis and Management
The researchers noted that the patients developed strokes despite a trend toward coagulopathy, to the extent that half were ineligible for immediate use of antiplatelet agents. The study findings “heightened our awareness,” the researchers say, that aggressive preventive measures can help reduce the incidence of stroke in patients with renal insufficiency.
Source:
Hinduja A, Limaye K, Ravilla R, et al. PLoS One. 2016;11(11): e0166627.
doi: 10.1371/journal.pone.0166627.
Related: Treating Patients With Multiple Myeloma in the VA
Patients were enrolled in Total Therapy protocols (TT2, TT3a, and TT3b), which tested varying combinations of thalidomide, bortezomib, lenalidomide, and dexamethasone. Of 1,148 patients, 46 (4%) had strokes, usually ischemic stroke (33 patients, or 72%). Hypercoagulability, atrial fibrillation and small-vessel occlusion were common mechanisms. Whereas other research has found a higher risk of arterial thrombosis from activated prothrombotic factors, especially during the early period of chemotherapy, in this study vascular events occurred months later.
Seven patients died in the hospital (15% compared with a national average of 5%). Although 6 of those deaths were stroke related, 36 patients were discharged home or to a rehabilitation facility; 2 were discharged to a long-term nursing facility. During a median follow-up of 10 years, 6 patients had another stroke. The cumulative risk of recurrent stroke was 15% compared with 5% for the general population.
Related: Link Found Between Agent Orange Exposure and Multiple Myeloma
Stage I and II cancers and renal insufficiency independently predicted stroke. Also noteworthy, according to the researchers: Patients with MM who developed renal insufficiency had worse clinical outcomes despite improvement in their renal function or lack of significant difference in their baseline renal functions between various treatment protocols. Thus, the increased risk of stroke, recurrent stroke, and mortality could partly be due to renal disease, which may or may not have resulted from myeloma.
Use of combination chemotherapy has “markedly improved” clinical outcomes for MM patients, the researchers say, but those drugs have also been associated with an increased risk of VTE, especially during the first months of chemotherapy. Thalidomide alone did not increase the risk of VTE, nor did lenalinomide on its own. However, thalidomide combined with multiagent chemotherapy increased VTE risk as much as 34% in newly diagnosed patients, and lenalinomide with dexamethasone boosted risk as high as 75%.
The researchers found no significant relationship between mortality and use of thalidomide. Median survival was 103 months for a thalidomide-based regimen and 78 months for a regimen without thalidomide.
Related: Multiple Myeloma: Updates on Diagnosis and Management
The researchers noted that the patients developed strokes despite a trend toward coagulopathy, to the extent that half were ineligible for immediate use of antiplatelet agents. The study findings “heightened our awareness,” the researchers say, that aggressive preventive measures can help reduce the incidence of stroke in patients with renal insufficiency.
Source:
Hinduja A, Limaye K, Ravilla R, et al. PLoS One. 2016;11(11): e0166627.
doi: 10.1371/journal.pone.0166627.
US cancer cases may near 1.7 million in 2017

patient and her father
Photo by Rhoda Baer
The US may see nearly 1.7 million new cancer cases in 2017 and more than 600,000 cancer-related deaths, according to a report from the American Cancer Society (ACS).
In addition to estimates for 2017, the report, “Cancer Statistics 2017,” includes the most recent data on cancer incidence, mortality, and survival in the US.
The report was published in CA: A Cancer Journal for Clinicians.
The report projects there will be 1,688,780 new cancer cases and 600,920 cancer deaths in the US this year.
This includes:
- 80,500 new cases of lymphoma and 21,210 lymphoma deaths
- 62,130 new cases of leukemia and 24,500 leukemia deaths
- 30,280 new cases of myeloma and 12,590 myeloma deaths.
The report also shows that, from 2004 to 2013, the overall cancer incidence rate was stable in women and declined by about 2% per year in men. From 2005 to 2014, the cancer death rate declined by about 1.5% annually in both men and women.
Overall, the cancer death rate dropped 25% from its peak of 215.1 (per 100,000 population) in 1991 to 161.2 (per 100,000 population) in 2014, the latest year for which data was available. This translates to about 2,143,200 fewer cancer deaths.
“The continuing drops in the cancer death rate are a powerful sign of the potential we have to reduce cancer’s deadly toll,” said Otis W. Brawley, MD, chief medical officer of the ACS.
He said the decrease in cancer death rates is the result of steady reductions in smoking and advances in early detection and treatment. The decrease is driven by decreasing death rates for the 4 major cancer sites—lung, breast, colorectal, and prostate.
The report also shows that racial disparities in cancer death rates continue to decline. The excess risk of cancer death in black men has dropped from 47% in 1990 to 21% in 2014. The black/white disparity declined similarly in women, from a peak of 20% in 1998 to 13% in 2014.
On the other hand, significant gender disparities persist for both cancer incidence and death in the US. For all cancer sites combined, the incidence rate is 20% higher in men than in women, and the cancer death rate is 40% higher in men.
Dr Brawley said the gender gap in cancer mortality largely reflects variation in the distribution of cancers that occur in men and women, much of which is due to differences in the prevalence of cancer risk factors.
The yearly “Cancer Statistics” reports have been published by ACS researchers since 1967 to inform and guide clinicians, investigators, and others in public health in prioritizing efforts to reduce the burden of cancer.
Cancer incidence data for the current report were collected by the Surveillance, Epidemiology, and End Results Program; the National Program of Cancer Registries; and the North American Association of Central Cancer Registries. Mortality data were collected by the National Center for Health Statistics. ![]()
patient and her father
Photo by Rhoda Baer
The US may see nearly 1.7 million new cancer cases in 2017 and more than 600,000 cancer-related deaths, according to a report from the American Cancer Society (ACS).
In addition to estimates for 2017, the report, “Cancer Statistics 2017,” includes the most recent data on cancer incidence, mortality, and survival in the US.
The report was published in CA: A Cancer Journal for Clinicians.
The report projects there will be 1,688,780 new cancer cases and 600,920 cancer deaths in the US this year.
This includes:
- 80,500 new cases of lymphoma and 21,210 lymphoma deaths
- 62,130 new cases of leukemia and 24,500 leukemia deaths
- 30,280 new cases of myeloma and 12,590 myeloma deaths.
The report also shows that, from 2004 to 2013, the overall cancer incidence rate was stable in women and declined by about 2% per year in men. From 2005 to 2014, the cancer death rate declined by about 1.5% annually in both men and women.
Overall, the cancer death rate dropped 25% from its peak of 215.1 (per 100,000 population) in 1991 to 161.2 (per 100,000 population) in 2014, the latest year for which data was available. This translates to about 2,143,200 fewer cancer deaths.
“The continuing drops in the cancer death rate are a powerful sign of the potential we have to reduce cancer’s deadly toll,” said Otis W. Brawley, MD, chief medical officer of the ACS.
He said the decrease in cancer death rates is the result of steady reductions in smoking and advances in early detection and treatment. The decrease is driven by decreasing death rates for the 4 major cancer sites—lung, breast, colorectal, and prostate.
The report also shows that racial disparities in cancer death rates continue to decline. The excess risk of cancer death in black men has dropped from 47% in 1990 to 21% in 2014. The black/white disparity declined similarly in women, from a peak of 20% in 1998 to 13% in 2014.
On the other hand, significant gender disparities persist for both cancer incidence and death in the US. For all cancer sites combined, the incidence rate is 20% higher in men than in women, and the cancer death rate is 40% higher in men.
Dr Brawley said the gender gap in cancer mortality largely reflects variation in the distribution of cancers that occur in men and women, much of which is due to differences in the prevalence of cancer risk factors.
The yearly “Cancer Statistics” reports have been published by ACS researchers since 1967 to inform and guide clinicians, investigators, and others in public health in prioritizing efforts to reduce the burden of cancer.
Cancer incidence data for the current report were collected by the Surveillance, Epidemiology, and End Results Program; the National Program of Cancer Registries; and the North American Association of Central Cancer Registries. Mortality data were collected by the National Center for Health Statistics. ![]()
patient and her father
Photo by Rhoda Baer
The US may see nearly 1.7 million new cancer cases in 2017 and more than 600,000 cancer-related deaths, according to a report from the American Cancer Society (ACS).
In addition to estimates for 2017, the report, “Cancer Statistics 2017,” includes the most recent data on cancer incidence, mortality, and survival in the US.
The report was published in CA: A Cancer Journal for Clinicians.
The report projects there will be 1,688,780 new cancer cases and 600,920 cancer deaths in the US this year.
This includes:
- 80,500 new cases of lymphoma and 21,210 lymphoma deaths
- 62,130 new cases of leukemia and 24,500 leukemia deaths
- 30,280 new cases of myeloma and 12,590 myeloma deaths.
The report also shows that, from 2004 to 2013, the overall cancer incidence rate was stable in women and declined by about 2% per year in men. From 2005 to 2014, the cancer death rate declined by about 1.5% annually in both men and women.
Overall, the cancer death rate dropped 25% from its peak of 215.1 (per 100,000 population) in 1991 to 161.2 (per 100,000 population) in 2014, the latest year for which data was available. This translates to about 2,143,200 fewer cancer deaths.
“The continuing drops in the cancer death rate are a powerful sign of the potential we have to reduce cancer’s deadly toll,” said Otis W. Brawley, MD, chief medical officer of the ACS.
He said the decrease in cancer death rates is the result of steady reductions in smoking and advances in early detection and treatment. The decrease is driven by decreasing death rates for the 4 major cancer sites—lung, breast, colorectal, and prostate.
The report also shows that racial disparities in cancer death rates continue to decline. The excess risk of cancer death in black men has dropped from 47% in 1990 to 21% in 2014. The black/white disparity declined similarly in women, from a peak of 20% in 1998 to 13% in 2014.
On the other hand, significant gender disparities persist for both cancer incidence and death in the US. For all cancer sites combined, the incidence rate is 20% higher in men than in women, and the cancer death rate is 40% higher in men.
Dr Brawley said the gender gap in cancer mortality largely reflects variation in the distribution of cancers that occur in men and women, much of which is due to differences in the prevalence of cancer risk factors.
The yearly “Cancer Statistics” reports have been published by ACS researchers since 1967 to inform and guide clinicians, investigators, and others in public health in prioritizing efforts to reduce the burden of cancer.
Cancer incidence data for the current report were collected by the Surveillance, Epidemiology, and End Results Program; the National Program of Cancer Registries; and the North American Association of Central Cancer Registries. Mortality data were collected by the National Center for Health Statistics. ![]()
Cancer genomic data released to public

Photo courtesy of the
National Institute of
General Medical Sciences
The American Association for Cancer Research (AACR) has announced the first public release of cancer genomic data aggregated through the AACR Project Genomics Evidence Neoplasia Information Exchange (GENIE).
The data set includes nearly 19,000 de-identified genomic records collected from patients who were treated at 8 international institutions, making it one of the largest public cancer genomic data sets released to date.
The release includes data for 59 major cancer types, including leukemias, lymphomas, and multiple myeloma.
The genomic data and a limited amount of linked clinical data for each patient can be accessed via the AACR Project GENIE cBioPortal or from Sage Bionetworks. (Users must create an account for either site to access the data.)
“We are excited to make publicly available this very large set of clinical-grade, next-generation sequencing data obtained during routine patient care,” said Charles L. Sawyers, MD, AACR Project GENIE Steering Committee chairperson.
“These data were generated as part of routine patient care and, without AACR Project GENIE, they would likely never have been shared with the global cancer research community.”
AACR Project GENIE is a multi-phase, international data-sharing project aimed at catalyzing precision oncology through the development of a registry that aggregates and links clinical-grade cancer genomic data with clinical outcomes from tens of thousands of cancer patients treated at multiple institutions.
The newly released data are fully de-identified in compliance with the Health Insurance Portability and Accountability Act (HIPAA).
The data are derived from patients whose tumors were genetically sequenced as part of their care at any of the 8 institutions that participated in the first phase of AACR Project GENIE.
The goal of releasing these data to the cancer research community is to aid new research that will accelerate the pace of progress against cancer.
According to AACR, the data can be used to validate gene signatures of drug response or prognosis, identify new patient populations for drugs that are currently available, and uncover new drug targets and biomarkers.
“I am extremely proud that the American Association for Cancer Research, as the coordinating center for AACR Project GENIE, is delivering on its promise to make these important data publicly available just over a year after unveiling the initiative,” said Margaret Foti, PhD, MD, chief executive officer of the AACR.
To expand the AACR Project GENIE registry, the consortium is accepting applications for new participating centers. Any nonprofit institution that meets certain criteria can submit an application to become a project participant.
For more information on AACR Project GENIE, visit the project website or send an email to [email protected]. ![]()
Photo courtesy of the
National Institute of
General Medical Sciences
The American Association for Cancer Research (AACR) has announced the first public release of cancer genomic data aggregated through the AACR Project Genomics Evidence Neoplasia Information Exchange (GENIE).
The data set includes nearly 19,000 de-identified genomic records collected from patients who were treated at 8 international institutions, making it one of the largest public cancer genomic data sets released to date.
The release includes data for 59 major cancer types, including leukemias, lymphomas, and multiple myeloma.
The genomic data and a limited amount of linked clinical data for each patient can be accessed via the AACR Project GENIE cBioPortal or from Sage Bionetworks. (Users must create an account for either site to access the data.)
“We are excited to make publicly available this very large set of clinical-grade, next-generation sequencing data obtained during routine patient care,” said Charles L. Sawyers, MD, AACR Project GENIE Steering Committee chairperson.
“These data were generated as part of routine patient care and, without AACR Project GENIE, they would likely never have been shared with the global cancer research community.”
AACR Project GENIE is a multi-phase, international data-sharing project aimed at catalyzing precision oncology through the development of a registry that aggregates and links clinical-grade cancer genomic data with clinical outcomes from tens of thousands of cancer patients treated at multiple institutions.
The newly released data are fully de-identified in compliance with the Health Insurance Portability and Accountability Act (HIPAA).
The data are derived from patients whose tumors were genetically sequenced as part of their care at any of the 8 institutions that participated in the first phase of AACR Project GENIE.
The goal of releasing these data to the cancer research community is to aid new research that will accelerate the pace of progress against cancer.
According to AACR, the data can be used to validate gene signatures of drug response or prognosis, identify new patient populations for drugs that are currently available, and uncover new drug targets and biomarkers.
“I am extremely proud that the American Association for Cancer Research, as the coordinating center for AACR Project GENIE, is delivering on its promise to make these important data publicly available just over a year after unveiling the initiative,” said Margaret Foti, PhD, MD, chief executive officer of the AACR.
To expand the AACR Project GENIE registry, the consortium is accepting applications for new participating centers. Any nonprofit institution that meets certain criteria can submit an application to become a project participant.
For more information on AACR Project GENIE, visit the project website or send an email to [email protected]. ![]()
Photo courtesy of the
National Institute of
General Medical Sciences
The American Association for Cancer Research (AACR) has announced the first public release of cancer genomic data aggregated through the AACR Project Genomics Evidence Neoplasia Information Exchange (GENIE).
The data set includes nearly 19,000 de-identified genomic records collected from patients who were treated at 8 international institutions, making it one of the largest public cancer genomic data sets released to date.
The release includes data for 59 major cancer types, including leukemias, lymphomas, and multiple myeloma.
The genomic data and a limited amount of linked clinical data for each patient can be accessed via the AACR Project GENIE cBioPortal or from Sage Bionetworks. (Users must create an account for either site to access the data.)
“We are excited to make publicly available this very large set of clinical-grade, next-generation sequencing data obtained during routine patient care,” said Charles L. Sawyers, MD, AACR Project GENIE Steering Committee chairperson.
“These data were generated as part of routine patient care and, without AACR Project GENIE, they would likely never have been shared with the global cancer research community.”
AACR Project GENIE is a multi-phase, international data-sharing project aimed at catalyzing precision oncology through the development of a registry that aggregates and links clinical-grade cancer genomic data with clinical outcomes from tens of thousands of cancer patients treated at multiple institutions.
The newly released data are fully de-identified in compliance with the Health Insurance Portability and Accountability Act (HIPAA).
The data are derived from patients whose tumors were genetically sequenced as part of their care at any of the 8 institutions that participated in the first phase of AACR Project GENIE.
The goal of releasing these data to the cancer research community is to aid new research that will accelerate the pace of progress against cancer.
According to AACR, the data can be used to validate gene signatures of drug response or prognosis, identify new patient populations for drugs that are currently available, and uncover new drug targets and biomarkers.
“I am extremely proud that the American Association for Cancer Research, as the coordinating center for AACR Project GENIE, is delivering on its promise to make these important data publicly available just over a year after unveiling the initiative,” said Margaret Foti, PhD, MD, chief executive officer of the AACR.
To expand the AACR Project GENIE registry, the consortium is accepting applications for new participating centers. Any nonprofit institution that meets certain criteria can submit an application to become a project participant.
For more information on AACR Project GENIE, visit the project website or send an email to [email protected]. ![]()
Daratumumab combo holds up across POLLUX myeloma subgroups
SAN DIEGO – Adding daratumumab (D) to lenalidomide and dexamethasone (Rd) significantly improved outcomes in relapsed and refractory multiple myeloma, even when patients had previously received lenalidomide, were refractory to bortezomib, or had high-risk tumor cytogenetics, based on updated analyses from the multicenter, randomized, phase III, open-label POLLUX trial.
The findings underscore the “significant benefit of combining daratumumab with lenalidomide and dexamethasone for relapsed or refractory multiple myeloma,” said lead investigator Philippe Moreau, MD, of University Hospital Hotel-Dieu in Nantes, France.
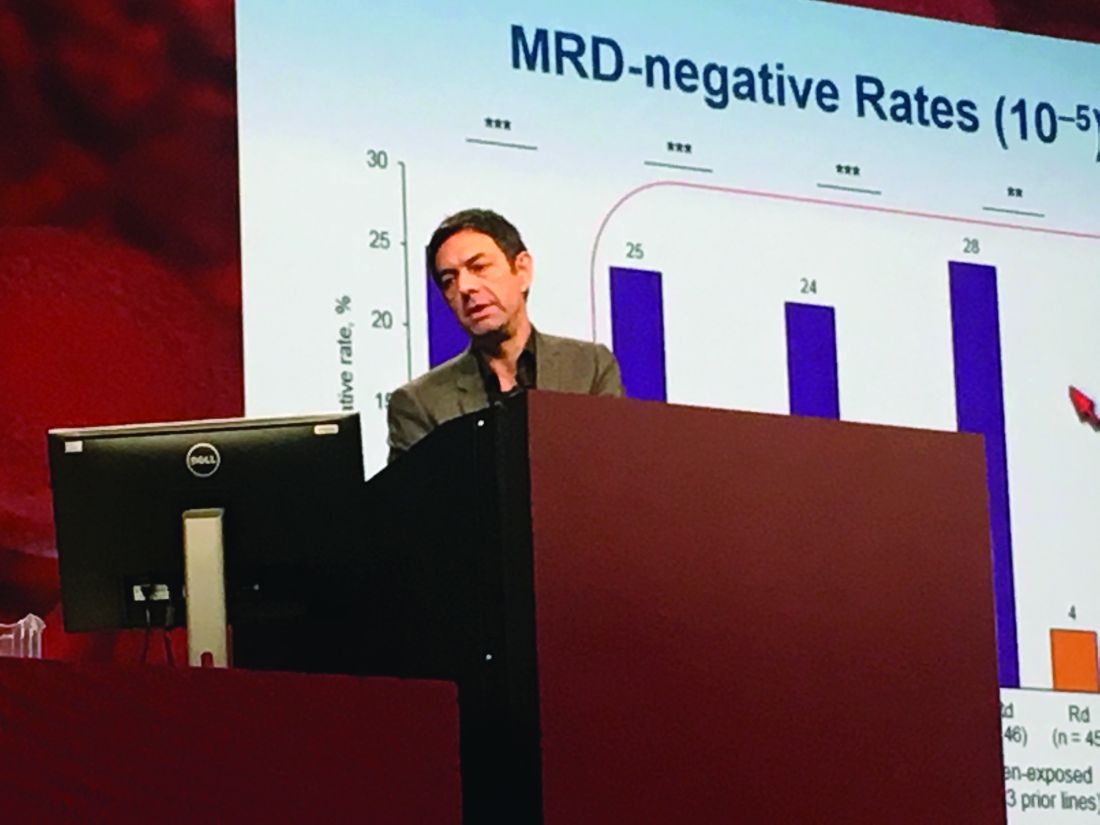
Among a large subgroup of 524 POLLUX patients who had received one to three prior lines of therapy, estimated median progression-free survival (PFS) has not been reached in the daratumumab, lenalidomide, and dexamethasone (DRd) arm, versus 18.4 months in the lenalidomide and dexamethasone (Rd) arm (hazard ratio, 0.36; 95% CI: 0.26 to 0.49; P less than .0001), Dr. Moreau said at the annual meeting of the American Society of Hematology.
That means adding daratumumab to Rd led to a 64% reduction in the risk of disease progression or death among patients with relapsed or refractory multiple myeloma, he noted. Fully 77% of DRd patients were alive without having progressed at 18 months, and responses “continued to deepen in the DRd group with longer follow-up,” he added.
Additional analyses supported the use of DRd in relapsed or refractory multiple myeloma, “irrespective of prior lenalidomide treatment or bortezomib refractoriness,” Dr. Moreau continued. He reported that DRd significantly improved PFS over Rd alone not only among 445 lenalidomide-naive patients (HR, 0.37; P less than .0001), but also among 91 lenalidomide-exposed patients (HR, 0.45; P = .04), 140 patients who were refractory to their most recent line of therapy (HR, 0.45; P = .001), and 99 bortezomib- refractory patients (HR 0.51; P = .02).
Daratumumab (Darzalex), a human CD38 IgG1k monoclonal antibody, was first approved as monotherapy for multiple myeloma in patients who had received at least three prior lines of therapy or had double-refractory disease. In 2016, results from the twin POLLUX and CASTOR studies won daratumumab a Food and Drug Administration breakthrough designation status for use with Rd in patients who had received at least one prior line.
The POLLUX trial included 569 patients with multiple myeloma who had received a median of 1 and up to 11 prior lines of therapy. Patients were randomized to either Rd alone or to Rd plus intravenous daratumumab (16 mg/kg) once a week during the first two 28-day treatment cycles, every 2 weeks during cycles 3-6, and once only on day 1 of subsequent cycles.
POLLUX patients were fairly heavily pretreated, Dr. Moreau noted. Thirteen percent had received three prior lines of therapy, 86% had received a proteasome inhibitor, 18% had received lenalidomide, 21% were refractory to bortezomib, and 28% were refractory to their most recent line of therapy.
Researchers performed “stringent, unbiased” assessments of minimal residual disease (MRD) negativity not only when a complete response was suspected, but also 3 and 6 months later, Dr. Moreau said. He emphasized that rates of MRD negativity in lenalidomide-exposed, bortezomib-refractory subgroups in POLLUX almost exactly matched those in the intent-to-treat population (25% on DRd vs. 6% on Rd; P less than .0001).
A total of 17% of DRd patients and 25% of Rd patients had high-risk cytogenetic profiles, and DRd performed well in these individuals, Dr. Moreau reported. Fully 85% of all evaluable high-risk patients had at least a partial response to DRd, and 33% had a complete response, versus only 67% and 6% of high-risk Rd patients, respectively. Among patients with standard-risk cytogenetics, rates of best overall response were 95% on DRd and 82% on Rd, and rates of complete response were 52% on DRd and 24% on Rd.
POLLUX yielded no new safety signals for DRd, Dr. Moreau said. Rates of primary and secondary malignancies were less than 2%. Neutropenia, the most common adverse effect, was managed by interrupting treatment, reducing the dose of lenalidomide, and administering growth factor.
Janssen Research & Development funded the study. Dr. Moreau had no relevant financial disclosures.
SAN DIEGO – Adding daratumumab (D) to lenalidomide and dexamethasone (Rd) significantly improved outcomes in relapsed and refractory multiple myeloma, even when patients had previously received lenalidomide, were refractory to bortezomib, or had high-risk tumor cytogenetics, based on updated analyses from the multicenter, randomized, phase III, open-label POLLUX trial.
The findings underscore the “significant benefit of combining daratumumab with lenalidomide and dexamethasone for relapsed or refractory multiple myeloma,” said lead investigator Philippe Moreau, MD, of University Hospital Hotel-Dieu in Nantes, France.
Among a large subgroup of 524 POLLUX patients who had received one to three prior lines of therapy, estimated median progression-free survival (PFS) has not been reached in the daratumumab, lenalidomide, and dexamethasone (DRd) arm, versus 18.4 months in the lenalidomide and dexamethasone (Rd) arm (hazard ratio, 0.36; 95% CI: 0.26 to 0.49; P less than .0001), Dr. Moreau said at the annual meeting of the American Society of Hematology.
That means adding daratumumab to Rd led to a 64% reduction in the risk of disease progression or death among patients with relapsed or refractory multiple myeloma, he noted. Fully 77% of DRd patients were alive without having progressed at 18 months, and responses “continued to deepen in the DRd group with longer follow-up,” he added.
Additional analyses supported the use of DRd in relapsed or refractory multiple myeloma, “irrespective of prior lenalidomide treatment or bortezomib refractoriness,” Dr. Moreau continued. He reported that DRd significantly improved PFS over Rd alone not only among 445 lenalidomide-naive patients (HR, 0.37; P less than .0001), but also among 91 lenalidomide-exposed patients (HR, 0.45; P = .04), 140 patients who were refractory to their most recent line of therapy (HR, 0.45; P = .001), and 99 bortezomib- refractory patients (HR 0.51; P = .02).
Daratumumab (Darzalex), a human CD38 IgG1k monoclonal antibody, was first approved as monotherapy for multiple myeloma in patients who had received at least three prior lines of therapy or had double-refractory disease. In 2016, results from the twin POLLUX and CASTOR studies won daratumumab a Food and Drug Administration breakthrough designation status for use with Rd in patients who had received at least one prior line.
The POLLUX trial included 569 patients with multiple myeloma who had received a median of 1 and up to 11 prior lines of therapy. Patients were randomized to either Rd alone or to Rd plus intravenous daratumumab (16 mg/kg) once a week during the first two 28-day treatment cycles, every 2 weeks during cycles 3-6, and once only on day 1 of subsequent cycles.
POLLUX patients were fairly heavily pretreated, Dr. Moreau noted. Thirteen percent had received three prior lines of therapy, 86% had received a proteasome inhibitor, 18% had received lenalidomide, 21% were refractory to bortezomib, and 28% were refractory to their most recent line of therapy.
Researchers performed “stringent, unbiased” assessments of minimal residual disease (MRD) negativity not only when a complete response was suspected, but also 3 and 6 months later, Dr. Moreau said. He emphasized that rates of MRD negativity in lenalidomide-exposed, bortezomib-refractory subgroups in POLLUX almost exactly matched those in the intent-to-treat population (25% on DRd vs. 6% on Rd; P less than .0001).
A total of 17% of DRd patients and 25% of Rd patients had high-risk cytogenetic profiles, and DRd performed well in these individuals, Dr. Moreau reported. Fully 85% of all evaluable high-risk patients had at least a partial response to DRd, and 33% had a complete response, versus only 67% and 6% of high-risk Rd patients, respectively. Among patients with standard-risk cytogenetics, rates of best overall response were 95% on DRd and 82% on Rd, and rates of complete response were 52% on DRd and 24% on Rd.
POLLUX yielded no new safety signals for DRd, Dr. Moreau said. Rates of primary and secondary malignancies were less than 2%. Neutropenia, the most common adverse effect, was managed by interrupting treatment, reducing the dose of lenalidomide, and administering growth factor.
Janssen Research & Development funded the study. Dr. Moreau had no relevant financial disclosures.
SAN DIEGO – Adding daratumumab (D) to lenalidomide and dexamethasone (Rd) significantly improved outcomes in relapsed and refractory multiple myeloma, even when patients had previously received lenalidomide, were refractory to bortezomib, or had high-risk tumor cytogenetics, based on updated analyses from the multicenter, randomized, phase III, open-label POLLUX trial.
The findings underscore the “significant benefit of combining daratumumab with lenalidomide and dexamethasone for relapsed or refractory multiple myeloma,” said lead investigator Philippe Moreau, MD, of University Hospital Hotel-Dieu in Nantes, France.
Among a large subgroup of 524 POLLUX patients who had received one to three prior lines of therapy, estimated median progression-free survival (PFS) has not been reached in the daratumumab, lenalidomide, and dexamethasone (DRd) arm, versus 18.4 months in the lenalidomide and dexamethasone (Rd) arm (hazard ratio, 0.36; 95% CI: 0.26 to 0.49; P less than .0001), Dr. Moreau said at the annual meeting of the American Society of Hematology.
That means adding daratumumab to Rd led to a 64% reduction in the risk of disease progression or death among patients with relapsed or refractory multiple myeloma, he noted. Fully 77% of DRd patients were alive without having progressed at 18 months, and responses “continued to deepen in the DRd group with longer follow-up,” he added.
Additional analyses supported the use of DRd in relapsed or refractory multiple myeloma, “irrespective of prior lenalidomide treatment or bortezomib refractoriness,” Dr. Moreau continued. He reported that DRd significantly improved PFS over Rd alone not only among 445 lenalidomide-naive patients (HR, 0.37; P less than .0001), but also among 91 lenalidomide-exposed patients (HR, 0.45; P = .04), 140 patients who were refractory to their most recent line of therapy (HR, 0.45; P = .001), and 99 bortezomib- refractory patients (HR 0.51; P = .02).
Daratumumab (Darzalex), a human CD38 IgG1k monoclonal antibody, was first approved as monotherapy for multiple myeloma in patients who had received at least three prior lines of therapy or had double-refractory disease. In 2016, results from the twin POLLUX and CASTOR studies won daratumumab a Food and Drug Administration breakthrough designation status for use with Rd in patients who had received at least one prior line.
The POLLUX trial included 569 patients with multiple myeloma who had received a median of 1 and up to 11 prior lines of therapy. Patients were randomized to either Rd alone or to Rd plus intravenous daratumumab (16 mg/kg) once a week during the first two 28-day treatment cycles, every 2 weeks during cycles 3-6, and once only on day 1 of subsequent cycles.
POLLUX patients were fairly heavily pretreated, Dr. Moreau noted. Thirteen percent had received three prior lines of therapy, 86% had received a proteasome inhibitor, 18% had received lenalidomide, 21% were refractory to bortezomib, and 28% were refractory to their most recent line of therapy.
Researchers performed “stringent, unbiased” assessments of minimal residual disease (MRD) negativity not only when a complete response was suspected, but also 3 and 6 months later, Dr. Moreau said. He emphasized that rates of MRD negativity in lenalidomide-exposed, bortezomib-refractory subgroups in POLLUX almost exactly matched those in the intent-to-treat population (25% on DRd vs. 6% on Rd; P less than .0001).
A total of 17% of DRd patients and 25% of Rd patients had high-risk cytogenetic profiles, and DRd performed well in these individuals, Dr. Moreau reported. Fully 85% of all evaluable high-risk patients had at least a partial response to DRd, and 33% had a complete response, versus only 67% and 6% of high-risk Rd patients, respectively. Among patients with standard-risk cytogenetics, rates of best overall response were 95% on DRd and 82% on Rd, and rates of complete response were 52% on DRd and 24% on Rd.
POLLUX yielded no new safety signals for DRd, Dr. Moreau said. Rates of primary and secondary malignancies were less than 2%. Neutropenia, the most common adverse effect, was managed by interrupting treatment, reducing the dose of lenalidomide, and administering growth factor.
Janssen Research & Development funded the study. Dr. Moreau had no relevant financial disclosures.
AT ASH 2016
Key clinical point: Adding daratumumab (D) to lenalidomide and dexamethasone (Rd) significantly improved outcomes in patients with relapsed and refractory multiple myeloma, regardless of factors such as bortezomib refractoriness, lenalidomide exposure, or high-risk tumor cytogenetics.
Major finding: DRd significantly improved PFS over Rd not only among 445 lenalidomide-naive patients (HR, 0.37; P less than .0001), but also among 91 lenalidomide-exposed patients (HR, 0.45; P = .04), 140 patients who were refractory to their most recent line of therapy (HR, 0.45; P = .001), and 99 bortezomib-refractory patients (HR 0.51; P = .02).
Data source: POLLUX, a multicenter, randomized, phase III, open-label trial of 569 patients with multiple myeloma who had received one or more previous lines of therapy.
Disclosures: Janssen Research & Development funded the study. Dr. Moreau had no relevant financial disclosures.
Intervention relieves distress in cancer patients

chemotherapy
Photo by Rhoda Baer
Results of a small study suggest a single dose of the hallucinogenic drug psilocybin, when combined with counseling, can significantly lessen psychological distress in cancer patients for months at a time.
The study showed that psychological counseling and a single dose of psilocybin brought relief from distress that lasted for more than 6 months in a majority of the subjects monitored.
This was based on clinical evaluation scores for anxiety and depression.
“Our results represent the strongest evidence to date of a clinical benefit from psilocybin therapy, with the potential to transform care for patients with cancer-related psychological distress,” said study author Stephen Ross, MD, of New York University School of Medicine in New York, New York.
“If larger clinical trials prove successful, then we could ultimately have available a safe, effective, and inexpensive medication—dispensed under strict control—to alleviate the distress that increases suicide rates among cancer patients.”
Dr Ross and his colleagues reported the results of their study in the Journal of Psychopharmacology alongside a related study and 11 accompanying editorials.
Dr Ross’s study included 29 patients with cancer-related anxiety and depression. Their mean age was 56, and 62% were female. Ninety percent were Caucasian, and 10% were classified as “other” race.
Patients had breast cancer (31%), reproductive cancers (28%), digestive cancers (17%), leukemia/lymphoma (14%), and other cancers (10%).
All patients had been diagnosed as suffering from serious psychological distress related to their disease.
Treatment
Half of the patients were randomly assigned to receive a 0.3 mg/kg dose of psilocybin, and half received a vitamin placebo (250 mg of niacin) known to produce a “rush” that mimics a hallucinogenic drug experience.
Approximately half way through the study’s monitoring period (after 7 weeks), all patients switched treatments. Those who initially received psilocybin took a single dose of niacin, and vice-versa. Neither patients nor researchers knew who had first received psilocybin or placebo.
All patients were provided with tailored counseling from a psychiatrist, psychologist, nurse, or social worker. And the patients were monitored for side effects and improvements in their mental state.
Safety
The researchers said there were no serious adverse events (AEs), either medical or psychiatric, that were attributed to psilocybin.
The most common medical AEs that were attributable to psilocybin were non-clinically significant elevations in blood pressure and heart rate (76%), headaches/migraines (28%), and nausea (14%).
The most common psychiatric AEs attributable to psilocybin were transient anxiety (17%) and transient psychotic-like symptoms (7%; 1 case of transient paranoid ideation and 1 case of transient thought disorder).
Efficacy
The researchers said that, prior to the crossover, psilocybin produced immediate, substantial, and sustained improvements in anxiety and depression.
Specifically, patients who received psilocybin first had significant improvements in responses on the Hospital Anxiety and Depression Scale and the Beck Depression Inventory, when compared to patients who received niacin first.
The differences were significant 1 day after the patients’ first session and 7 weeks after the first session (P≤0.01 for all).
At the 6.5-month follow-up, 60% to 80% of participants continued with clinically significant reductions in depression or anxiety.
The researchers said a key finding of this study was that improvements in clinical evaluation scores for anxiety and depression lasted for the study’s extended monitoring period, which was 8 months for those who took psilocybin first.
Patients also reported post-psilocybin improvements in their quality of life, such as going out more, greater energy, getting along better with family members, and doing well at work. Some reported variations of spirituality, unusual peacefulness, and increased feelings of altruism.
“Our study showed that psilocybin facilitated experiences that drove reductions in psychological distress,” said study author Anthony Bossis, PhD, of New York University School of Medicine. “And if it’s true for cancer care, then it could apply to other stressful medical conditions.”
He cautioned that patients should not consume psilocybin on their own or without supervision from a physician and a trained counselor.
“Psilocybin therapy may not work for everyone,” he noted. “And some groups, such as people with schizophrenia, as well as adolescents, should not be treated with it.” ![]()
chemotherapy
Photo by Rhoda Baer
Results of a small study suggest a single dose of the hallucinogenic drug psilocybin, when combined with counseling, can significantly lessen psychological distress in cancer patients for months at a time.
The study showed that psychological counseling and a single dose of psilocybin brought relief from distress that lasted for more than 6 months in a majority of the subjects monitored.
This was based on clinical evaluation scores for anxiety and depression.
“Our results represent the strongest evidence to date of a clinical benefit from psilocybin therapy, with the potential to transform care for patients with cancer-related psychological distress,” said study author Stephen Ross, MD, of New York University School of Medicine in New York, New York.
“If larger clinical trials prove successful, then we could ultimately have available a safe, effective, and inexpensive medication—dispensed under strict control—to alleviate the distress that increases suicide rates among cancer patients.”
Dr Ross and his colleagues reported the results of their study in the Journal of Psychopharmacology alongside a related study and 11 accompanying editorials.
Dr Ross’s study included 29 patients with cancer-related anxiety and depression. Their mean age was 56, and 62% were female. Ninety percent were Caucasian, and 10% were classified as “other” race.
Patients had breast cancer (31%), reproductive cancers (28%), digestive cancers (17%), leukemia/lymphoma (14%), and other cancers (10%).
All patients had been diagnosed as suffering from serious psychological distress related to their disease.
Treatment
Half of the patients were randomly assigned to receive a 0.3 mg/kg dose of psilocybin, and half received a vitamin placebo (250 mg of niacin) known to produce a “rush” that mimics a hallucinogenic drug experience.
Approximately half way through the study’s monitoring period (after 7 weeks), all patients switched treatments. Those who initially received psilocybin took a single dose of niacin, and vice-versa. Neither patients nor researchers knew who had first received psilocybin or placebo.
All patients were provided with tailored counseling from a psychiatrist, psychologist, nurse, or social worker. And the patients were monitored for side effects and improvements in their mental state.
Safety
The researchers said there were no serious adverse events (AEs), either medical or psychiatric, that were attributed to psilocybin.
The most common medical AEs that were attributable to psilocybin were non-clinically significant elevations in blood pressure and heart rate (76%), headaches/migraines (28%), and nausea (14%).
The most common psychiatric AEs attributable to psilocybin were transient anxiety (17%) and transient psychotic-like symptoms (7%; 1 case of transient paranoid ideation and 1 case of transient thought disorder).
Efficacy
The researchers said that, prior to the crossover, psilocybin produced immediate, substantial, and sustained improvements in anxiety and depression.
Specifically, patients who received psilocybin first had significant improvements in responses on the Hospital Anxiety and Depression Scale and the Beck Depression Inventory, when compared to patients who received niacin first.
The differences were significant 1 day after the patients’ first session and 7 weeks after the first session (P≤0.01 for all).
At the 6.5-month follow-up, 60% to 80% of participants continued with clinically significant reductions in depression or anxiety.
The researchers said a key finding of this study was that improvements in clinical evaluation scores for anxiety and depression lasted for the study’s extended monitoring period, which was 8 months for those who took psilocybin first.
Patients also reported post-psilocybin improvements in their quality of life, such as going out more, greater energy, getting along better with family members, and doing well at work. Some reported variations of spirituality, unusual peacefulness, and increased feelings of altruism.
“Our study showed that psilocybin facilitated experiences that drove reductions in psychological distress,” said study author Anthony Bossis, PhD, of New York University School of Medicine. “And if it’s true for cancer care, then it could apply to other stressful medical conditions.”
He cautioned that patients should not consume psilocybin on their own or without supervision from a physician and a trained counselor.
“Psilocybin therapy may not work for everyone,” he noted. “And some groups, such as people with schizophrenia, as well as adolescents, should not be treated with it.” ![]()
chemotherapy
Photo by Rhoda Baer
Results of a small study suggest a single dose of the hallucinogenic drug psilocybin, when combined with counseling, can significantly lessen psychological distress in cancer patients for months at a time.
The study showed that psychological counseling and a single dose of psilocybin brought relief from distress that lasted for more than 6 months in a majority of the subjects monitored.
This was based on clinical evaluation scores for anxiety and depression.
“Our results represent the strongest evidence to date of a clinical benefit from psilocybin therapy, with the potential to transform care for patients with cancer-related psychological distress,” said study author Stephen Ross, MD, of New York University School of Medicine in New York, New York.
“If larger clinical trials prove successful, then we could ultimately have available a safe, effective, and inexpensive medication—dispensed under strict control—to alleviate the distress that increases suicide rates among cancer patients.”
Dr Ross and his colleagues reported the results of their study in the Journal of Psychopharmacology alongside a related study and 11 accompanying editorials.
Dr Ross’s study included 29 patients with cancer-related anxiety and depression. Their mean age was 56, and 62% were female. Ninety percent were Caucasian, and 10% were classified as “other” race.
Patients had breast cancer (31%), reproductive cancers (28%), digestive cancers (17%), leukemia/lymphoma (14%), and other cancers (10%).
All patients had been diagnosed as suffering from serious psychological distress related to their disease.
Treatment
Half of the patients were randomly assigned to receive a 0.3 mg/kg dose of psilocybin, and half received a vitamin placebo (250 mg of niacin) known to produce a “rush” that mimics a hallucinogenic drug experience.
Approximately half way through the study’s monitoring period (after 7 weeks), all patients switched treatments. Those who initially received psilocybin took a single dose of niacin, and vice-versa. Neither patients nor researchers knew who had first received psilocybin or placebo.
All patients were provided with tailored counseling from a psychiatrist, psychologist, nurse, or social worker. And the patients were monitored for side effects and improvements in their mental state.
Safety
The researchers said there were no serious adverse events (AEs), either medical or psychiatric, that were attributed to psilocybin.
The most common medical AEs that were attributable to psilocybin were non-clinically significant elevations in blood pressure and heart rate (76%), headaches/migraines (28%), and nausea (14%).
The most common psychiatric AEs attributable to psilocybin were transient anxiety (17%) and transient psychotic-like symptoms (7%; 1 case of transient paranoid ideation and 1 case of transient thought disorder).
Efficacy
The researchers said that, prior to the crossover, psilocybin produced immediate, substantial, and sustained improvements in anxiety and depression.
Specifically, patients who received psilocybin first had significant improvements in responses on the Hospital Anxiety and Depression Scale and the Beck Depression Inventory, when compared to patients who received niacin first.
The differences were significant 1 day after the patients’ first session and 7 weeks after the first session (P≤0.01 for all).
At the 6.5-month follow-up, 60% to 80% of participants continued with clinically significant reductions in depression or anxiety.
The researchers said a key finding of this study was that improvements in clinical evaluation scores for anxiety and depression lasted for the study’s extended monitoring period, which was 8 months for those who took psilocybin first.
Patients also reported post-psilocybin improvements in their quality of life, such as going out more, greater energy, getting along better with family members, and doing well at work. Some reported variations of spirituality, unusual peacefulness, and increased feelings of altruism.
“Our study showed that psilocybin facilitated experiences that drove reductions in psychological distress,” said study author Anthony Bossis, PhD, of New York University School of Medicine. “And if it’s true for cancer care, then it could apply to other stressful medical conditions.”
He cautioned that patients should not consume psilocybin on their own or without supervision from a physician and a trained counselor.
“Psilocybin therapy may not work for everyone,” he noted. “And some groups, such as people with schizophrenia, as well as adolescents, should not be treated with it.” ![]()
Drug eases existential anxiety in cancer patients

chemotherapy
Photo by Rhoda Baer
One-time treatment with the hallucinogenic drug psilocybin may provide long-term relief of existential anxiety in patients with life-threatening cancers, according to a small study.
After receiving a single high dose of the drug, most of the patients studied reported decreases in depression and anxiety as well as increases in quality of life and optimism.
These improvements were sustained at 6 months of follow-up.
“The most interesting and remarkable finding is that a single dose of psilocybin, which lasts 4 to 6 hours, produced enduring decreases in depression and anxiety symptoms, and this may represent a fascinating new model for treating some psychiatric conditions,” said study author Roland Griffiths, PhD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
Dr Griffiths said this study grew out of a decade of research at Johns Hopkins on the effects of psilocybin in healthy volunteers, which showed that psilocybin can consistently produce positive changes in mood, behavior, and spirituality when administered to carefully screened and prepared participants.
The current study was designed to see if psilocybin could produce similar results in psychologically distressed cancer patients.
The results were published in the Journal of Psychopharmacology alongside a similar study and 11 accompanying editorials.
For their study, Dr Griffiths and his colleagues recruited 51 participants diagnosed with life-threatening cancers, most of which were recurrent or metastatic.
Types of cancer included breast (n=13), upper aerodigestive (n=7), gastrointestinal (n=4), genitourinary (n=18), and “other” cancers (n=1), as well as hematologic malignancies (n=8).
All participants had been given a formal psychiatric diagnosis, including an anxiety or depressive disorder.
Half of the participants were female, and they had an average age of 56. Ninety-two percent were white, 4% were black, and 2% were Asian.
Treatment
Each participant had 2 treatment sessions scheduled 5 weeks apart. In 1 session, they received a capsule containing a very low dose (1 or 3 mg per 70 kg) of psilocybin that was meant to act as a “control” because the dose was too low to produce effects.
In the other session, participants received a capsule with what is considered a moderate or high dose (22 or 30 mg per 70 kg).
To minimize expectancy effects, the participants and the staff members supervising the sessions were told that participants would receive psilocybin on both sessions, but they did not know that all participants would receive a high dose and a low dose.
Blood pressure and mood were monitored throughout the sessions.
Two monitors aided participants during each session, encouraging them to lie down, wear an eye mask, listen to music through headphones, and direct their attention on their inner experience. If anxiety or confusion arose, the monitors provided reassurance to the participants.
Participants, staff, and community observers rated participants’ moods, attitudes, and behaviors throughout the study.
The researchers assessed each participant via questionnaires and structured interviews before the first session, 7 hours after taking the psilocybin, 5 weeks after each session, and 6 months after the second session.
Adverse events
Thirty-four percent of participants had an episode of elevated systolic blood pressure (>160 mm Hg at 1 or more time-point) in the high-dose psilocybin session, and 17% of participants had such an episode in the low-dose session.
Thirteen percent and 2%, respectively, had an episode of elevated diastolic blood pressure (>100 mm Hg at 1 or more time-point). None of these episodes met criteria for medical intervention.
During the high-dose psilocybin session, 15% of patients experienced nausea or vomiting. There were no such events during the low-dose session.
Three participants reported mild to moderate headaches after the high-dose session.
Twenty-one percent of patients reported physical discomfort during the high-dose session, as did 8% of patients during the low-dose session.
Psychological discomfort occurred in 32% and 12% of participants, respectively. The researchers said there were no cases of hallucinogen persisting perception disorder or prolonged psychosis.
Efficacy outcomes
Most participants reported experiencing changes in visual perception, emotions, and thinking after taking high-dose psilocybin. They also reported experiences of psychological insight and profound, deeply meaningful experiences.
Six months after the final session of treatment, about 80% of participants continued to show clinically significant decreases in depressed mood and anxiety, according to clinician assessment.
According to the participants themselves, 83% had increases in well-being or life satisfaction at 6 months after treatment.
Sixty-seven percent of participants rated the experience as one of the top 5 meaningful experiences in their lives, and 70% rated the experience as one of their top 5 spiritually significant lifetime events.
“Before beginning the study, it wasn’t clear to me that this treatment would be helpful, since cancer patients may experience profound hopelessness in response to their diagnosis, which is often followed by multiple surgeries and prolonged chemotherapy,” Dr Griffiths said.
“I could imagine that cancer patients would receive psilocybin, look into the existential void, and come out even more fearful. However, the positive changes in attitudes, moods, and behavior that we documented in healthy volunteers were replicated in cancer patients.” ![]()
chemotherapy
Photo by Rhoda Baer
One-time treatment with the hallucinogenic drug psilocybin may provide long-term relief of existential anxiety in patients with life-threatening cancers, according to a small study.
After receiving a single high dose of the drug, most of the patients studied reported decreases in depression and anxiety as well as increases in quality of life and optimism.
These improvements were sustained at 6 months of follow-up.
“The most interesting and remarkable finding is that a single dose of psilocybin, which lasts 4 to 6 hours, produced enduring decreases in depression and anxiety symptoms, and this may represent a fascinating new model for treating some psychiatric conditions,” said study author Roland Griffiths, PhD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
Dr Griffiths said this study grew out of a decade of research at Johns Hopkins on the effects of psilocybin in healthy volunteers, which showed that psilocybin can consistently produce positive changes in mood, behavior, and spirituality when administered to carefully screened and prepared participants.
The current study was designed to see if psilocybin could produce similar results in psychologically distressed cancer patients.
The results were published in the Journal of Psychopharmacology alongside a similar study and 11 accompanying editorials.
For their study, Dr Griffiths and his colleagues recruited 51 participants diagnosed with life-threatening cancers, most of which were recurrent or metastatic.
Types of cancer included breast (n=13), upper aerodigestive (n=7), gastrointestinal (n=4), genitourinary (n=18), and “other” cancers (n=1), as well as hematologic malignancies (n=8).
All participants had been given a formal psychiatric diagnosis, including an anxiety or depressive disorder.
Half of the participants were female, and they had an average age of 56. Ninety-two percent were white, 4% were black, and 2% were Asian.
Treatment
Each participant had 2 treatment sessions scheduled 5 weeks apart. In 1 session, they received a capsule containing a very low dose (1 or 3 mg per 70 kg) of psilocybin that was meant to act as a “control” because the dose was too low to produce effects.
In the other session, participants received a capsule with what is considered a moderate or high dose (22 or 30 mg per 70 kg).
To minimize expectancy effects, the participants and the staff members supervising the sessions were told that participants would receive psilocybin on both sessions, but they did not know that all participants would receive a high dose and a low dose.
Blood pressure and mood were monitored throughout the sessions.
Two monitors aided participants during each session, encouraging them to lie down, wear an eye mask, listen to music through headphones, and direct their attention on their inner experience. If anxiety or confusion arose, the monitors provided reassurance to the participants.
Participants, staff, and community observers rated participants’ moods, attitudes, and behaviors throughout the study.
The researchers assessed each participant via questionnaires and structured interviews before the first session, 7 hours after taking the psilocybin, 5 weeks after each session, and 6 months after the second session.
Adverse events
Thirty-four percent of participants had an episode of elevated systolic blood pressure (>160 mm Hg at 1 or more time-point) in the high-dose psilocybin session, and 17% of participants had such an episode in the low-dose session.
Thirteen percent and 2%, respectively, had an episode of elevated diastolic blood pressure (>100 mm Hg at 1 or more time-point). None of these episodes met criteria for medical intervention.
During the high-dose psilocybin session, 15% of patients experienced nausea or vomiting. There were no such events during the low-dose session.
Three participants reported mild to moderate headaches after the high-dose session.
Twenty-one percent of patients reported physical discomfort during the high-dose session, as did 8% of patients during the low-dose session.
Psychological discomfort occurred in 32% and 12% of participants, respectively. The researchers said there were no cases of hallucinogen persisting perception disorder or prolonged psychosis.
Efficacy outcomes
Most participants reported experiencing changes in visual perception, emotions, and thinking after taking high-dose psilocybin. They also reported experiences of psychological insight and profound, deeply meaningful experiences.
Six months after the final session of treatment, about 80% of participants continued to show clinically significant decreases in depressed mood and anxiety, according to clinician assessment.
According to the participants themselves, 83% had increases in well-being or life satisfaction at 6 months after treatment.
Sixty-seven percent of participants rated the experience as one of the top 5 meaningful experiences in their lives, and 70% rated the experience as one of their top 5 spiritually significant lifetime events.
“Before beginning the study, it wasn’t clear to me that this treatment would be helpful, since cancer patients may experience profound hopelessness in response to their diagnosis, which is often followed by multiple surgeries and prolonged chemotherapy,” Dr Griffiths said.
“I could imagine that cancer patients would receive psilocybin, look into the existential void, and come out even more fearful. However, the positive changes in attitudes, moods, and behavior that we documented in healthy volunteers were replicated in cancer patients.” ![]()
chemotherapy
Photo by Rhoda Baer
One-time treatment with the hallucinogenic drug psilocybin may provide long-term relief of existential anxiety in patients with life-threatening cancers, according to a small study.
After receiving a single high dose of the drug, most of the patients studied reported decreases in depression and anxiety as well as increases in quality of life and optimism.
These improvements were sustained at 6 months of follow-up.
“The most interesting and remarkable finding is that a single dose of psilocybin, which lasts 4 to 6 hours, produced enduring decreases in depression and anxiety symptoms, and this may represent a fascinating new model for treating some psychiatric conditions,” said study author Roland Griffiths, PhD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
Dr Griffiths said this study grew out of a decade of research at Johns Hopkins on the effects of psilocybin in healthy volunteers, which showed that psilocybin can consistently produce positive changes in mood, behavior, and spirituality when administered to carefully screened and prepared participants.
The current study was designed to see if psilocybin could produce similar results in psychologically distressed cancer patients.
The results were published in the Journal of Psychopharmacology alongside a similar study and 11 accompanying editorials.
For their study, Dr Griffiths and his colleagues recruited 51 participants diagnosed with life-threatening cancers, most of which were recurrent or metastatic.
Types of cancer included breast (n=13), upper aerodigestive (n=7), gastrointestinal (n=4), genitourinary (n=18), and “other” cancers (n=1), as well as hematologic malignancies (n=8).
All participants had been given a formal psychiatric diagnosis, including an anxiety or depressive disorder.
Half of the participants were female, and they had an average age of 56. Ninety-two percent were white, 4% were black, and 2% were Asian.
Treatment
Each participant had 2 treatment sessions scheduled 5 weeks apart. In 1 session, they received a capsule containing a very low dose (1 or 3 mg per 70 kg) of psilocybin that was meant to act as a “control” because the dose was too low to produce effects.
In the other session, participants received a capsule with what is considered a moderate or high dose (22 or 30 mg per 70 kg).
To minimize expectancy effects, the participants and the staff members supervising the sessions were told that participants would receive psilocybin on both sessions, but they did not know that all participants would receive a high dose and a low dose.
Blood pressure and mood were monitored throughout the sessions.
Two monitors aided participants during each session, encouraging them to lie down, wear an eye mask, listen to music through headphones, and direct their attention on their inner experience. If anxiety or confusion arose, the monitors provided reassurance to the participants.
Participants, staff, and community observers rated participants’ moods, attitudes, and behaviors throughout the study.
The researchers assessed each participant via questionnaires and structured interviews before the first session, 7 hours after taking the psilocybin, 5 weeks after each session, and 6 months after the second session.
Adverse events
Thirty-four percent of participants had an episode of elevated systolic blood pressure (>160 mm Hg at 1 or more time-point) in the high-dose psilocybin session, and 17% of participants had such an episode in the low-dose session.
Thirteen percent and 2%, respectively, had an episode of elevated diastolic blood pressure (>100 mm Hg at 1 or more time-point). None of these episodes met criteria for medical intervention.
During the high-dose psilocybin session, 15% of patients experienced nausea or vomiting. There were no such events during the low-dose session.
Three participants reported mild to moderate headaches after the high-dose session.
Twenty-one percent of patients reported physical discomfort during the high-dose session, as did 8% of patients during the low-dose session.
Psychological discomfort occurred in 32% and 12% of participants, respectively. The researchers said there were no cases of hallucinogen persisting perception disorder or prolonged psychosis.
Efficacy outcomes
Most participants reported experiencing changes in visual perception, emotions, and thinking after taking high-dose psilocybin. They also reported experiences of psychological insight and profound, deeply meaningful experiences.
Six months after the final session of treatment, about 80% of participants continued to show clinically significant decreases in depressed mood and anxiety, according to clinician assessment.
According to the participants themselves, 83% had increases in well-being or life satisfaction at 6 months after treatment.
Sixty-seven percent of participants rated the experience as one of the top 5 meaningful experiences in their lives, and 70% rated the experience as one of their top 5 spiritually significant lifetime events.
“Before beginning the study, it wasn’t clear to me that this treatment would be helpful, since cancer patients may experience profound hopelessness in response to their diagnosis, which is often followed by multiple surgeries and prolonged chemotherapy,” Dr Griffiths said.
“I could imagine that cancer patients would receive psilocybin, look into the existential void, and come out even more fearful. However, the positive changes in attitudes, moods, and behavior that we documented in healthy volunteers were replicated in cancer patients.” ![]()
Drugs may be effective against hematologic, other cancers

Image courtesy of PNAS
A diabetes medication and an antihypertensive drug may prove effective in the treatment of hematologic malignancies and other cancers, according to preclinical research published in Science Advances.
Past research has shown that metformin, a drug used to treat type 2 diabetes, has anticancer properties.
However, the usual therapeutic dose is too low to effectively fight cancer, and higher doses of metformin could be too toxic.
With the current study, researchers found that the antihypertensive drug syrosingopine enhances the anticancer efficacy of metformin without harming normal blood cells.
The team screened over a thousand drugs to find one that could boost metformin’s efficacy against cancers.
They identified syrosingopine and tested it in combination with metformin—at concentrations substantially below the drugs’ therapeutic thresholds—on a range of cancer cell lines and in mouse models of liver cancer.
Thirty-five of the 43 cell lines tested were susceptible to both syrosingopine and metformin. This included leukemia, lymphoma, and multiple myeloma cell lines.
In addition, the mice given a short course of syrosingopine and metformin experienced a reduction in the number of visible liver tumors.
The researchers also tested syrosingopine and metformin in peripheral blasts from 12 patients with acute myeloid leukemia and a patient with blast crisis chronic myeloid leukemia. All 13 samples responded to the treatment.
On the other hand, syrosingopine and metformin did not affect peripheral blood cells from healthy subjects.
“[A]lmost all tumor cells were killed by this cocktail and at doses that are actually not toxic to normal cells,” said study author Don Benjamin, of the University of Basel in Switzerland.
“And the effect was exclusively confined to cancer cells, as the blood cells from healthy donors were insensitive to the treatment.”
The researchers believe metformin functions by lowering blood glucose levels for cancer cells, starving them of essential nutrients needed for their survival. However, it is not clear how syrosingopine works in conjunction with metformin.
The team emphasized the need for more research evaluating the drugs in combination.
“We have been able to show that the 2 known drugs lead to more profound effects on cancer cell proliferation than each drug alone,” Dr Benjamin said. “The data from this study support the development of combination approaches for the treatment of cancer patients.” ![]()
Image courtesy of PNAS
A diabetes medication and an antihypertensive drug may prove effective in the treatment of hematologic malignancies and other cancers, according to preclinical research published in Science Advances.
Past research has shown that metformin, a drug used to treat type 2 diabetes, has anticancer properties.
However, the usual therapeutic dose is too low to effectively fight cancer, and higher doses of metformin could be too toxic.
With the current study, researchers found that the antihypertensive drug syrosingopine enhances the anticancer efficacy of metformin without harming normal blood cells.
The team screened over a thousand drugs to find one that could boost metformin’s efficacy against cancers.
They identified syrosingopine and tested it in combination with metformin—at concentrations substantially below the drugs’ therapeutic thresholds—on a range of cancer cell lines and in mouse models of liver cancer.
Thirty-five of the 43 cell lines tested were susceptible to both syrosingopine and metformin. This included leukemia, lymphoma, and multiple myeloma cell lines.
In addition, the mice given a short course of syrosingopine and metformin experienced a reduction in the number of visible liver tumors.
The researchers also tested syrosingopine and metformin in peripheral blasts from 12 patients with acute myeloid leukemia and a patient with blast crisis chronic myeloid leukemia. All 13 samples responded to the treatment.
On the other hand, syrosingopine and metformin did not affect peripheral blood cells from healthy subjects.
“[A]lmost all tumor cells were killed by this cocktail and at doses that are actually not toxic to normal cells,” said study author Don Benjamin, of the University of Basel in Switzerland.
“And the effect was exclusively confined to cancer cells, as the blood cells from healthy donors were insensitive to the treatment.”
The researchers believe metformin functions by lowering blood glucose levels for cancer cells, starving them of essential nutrients needed for their survival. However, it is not clear how syrosingopine works in conjunction with metformin.
The team emphasized the need for more research evaluating the drugs in combination.
“We have been able to show that the 2 known drugs lead to more profound effects on cancer cell proliferation than each drug alone,” Dr Benjamin said. “The data from this study support the development of combination approaches for the treatment of cancer patients.” ![]()
Image courtesy of PNAS
A diabetes medication and an antihypertensive drug may prove effective in the treatment of hematologic malignancies and other cancers, according to preclinical research published in Science Advances.
Past research has shown that metformin, a drug used to treat type 2 diabetes, has anticancer properties.
However, the usual therapeutic dose is too low to effectively fight cancer, and higher doses of metformin could be too toxic.
With the current study, researchers found that the antihypertensive drug syrosingopine enhances the anticancer efficacy of metformin without harming normal blood cells.
The team screened over a thousand drugs to find one that could boost metformin’s efficacy against cancers.
They identified syrosingopine and tested it in combination with metformin—at concentrations substantially below the drugs’ therapeutic thresholds—on a range of cancer cell lines and in mouse models of liver cancer.
Thirty-five of the 43 cell lines tested were susceptible to both syrosingopine and metformin. This included leukemia, lymphoma, and multiple myeloma cell lines.
In addition, the mice given a short course of syrosingopine and metformin experienced a reduction in the number of visible liver tumors.
The researchers also tested syrosingopine and metformin in peripheral blasts from 12 patients with acute myeloid leukemia and a patient with blast crisis chronic myeloid leukemia. All 13 samples responded to the treatment.
On the other hand, syrosingopine and metformin did not affect peripheral blood cells from healthy subjects.
“[A]lmost all tumor cells were killed by this cocktail and at doses that are actually not toxic to normal cells,” said study author Don Benjamin, of the University of Basel in Switzerland.
“And the effect was exclusively confined to cancer cells, as the blood cells from healthy donors were insensitive to the treatment.”
The researchers believe metformin functions by lowering blood glucose levels for cancer cells, starving them of essential nutrients needed for their survival. However, it is not clear how syrosingopine works in conjunction with metformin.
The team emphasized the need for more research evaluating the drugs in combination.
“We have been able to show that the 2 known drugs lead to more profound effects on cancer cell proliferation than each drug alone,” Dr Benjamin said. “The data from this study support the development of combination approaches for the treatment of cancer patients.” ![]()
Combo improves survival in newly diagnosed MM
Photo courtesy of
Millenium Pharmaceuticals
Results of a phase 3 trial suggest a 3-drug combination improves survival in newly diagnosed multiple myeloma (MM).
The trial showed that bortezomib, lenalidomide, and dexamethasone (VRd) could significantly
improve progression-free and overall survival when compared to lenalidomide and dexamethasone (Rd).
In addition, investigators said the risk-benefit profile of VRd was acceptable.
The team noted, however, that grade 3 or higher neurologic adverse events (AEs) were more common with VRd than with Rd.
“Our results are clear,” said principal investigator Brian G.M. Durie, MD, of Cedars-Sinai Outpatient Cancer Center in Los Angeles, California.
“Using bortezomib in combination with lenalidomide and dexamethasone in frontline treatment—hitting the disease early and hard—makes a meaningful difference for myeloma patients. Our results represent a potential new standard of care.”
Dr Durie and his colleagues reported the results of this trial in The Lancet.
The research was funded by Millennium Pharmaceuticals, Takeda Oncology Company, Celgene Corporation, the National Institutes of Health, the National Cancer Institute, and the National Clinical Trial Network.
The trial enrolled 525 adults with MM. The patients ranged in age from 28 to 87, had active MM, and had not had any prior treatment for their disease.
The patients were randomized to 2 treatment groups. One group received Rd for 6 cycles over 6 months. The other group received VRd for 8 cycles over 6 months.
Results
The median follow-up was 55 months. In the VRd group, 241 patients were evaluable for safety and 216 for response. In the Rd group, 226 patients were evaluable for safety and 214 for response.
The overall response rate was 82% (176/216) in the VRd group and 72% (153/214) in the Rd group. The complete response rates were 16% (34/216) and 8% (18/214), respectively.
The median progression-free survival was 43 months in the VRd group and 30 months in the Rd group. The hazard ratio was 0.712 (P=0.0018).
The median overall survival was 75 months in the VRd group and 64 months in the Rd group. The hazard ratio was 0.709 (P=0.025).
The rate of grade 3 or higher AEs was 82% in the VRd group and 75% in the Rd group. The rate of discontinuation due to AEs was 23% and 10%, respectively.
Grade 3 or higher neurologic AEs were more frequent in the VRd group than in the Rd group—33% and 11%, respectively (P<0.0001).
There were 2 treatment-related deaths in the VRd group but none in the Rd group. And 10 patients in each group had a second primary malignancy.
“This is a landmark study that lends clarity to frontline therapy of myeloma,” said study author S. Vincent Rajkumar, MD, of Mayo Clinic in Rochester, Minnesota.
“Newer alternatives to VRd may be more expensive, cumbersome, or toxic. These regimens will therefore need to show superiority over VRd in randomized trials.”
Also worth noting, Dr Rajkumar said, is that the VRd regimen will become even more cost-effective as the drugs in this combination become generic over time. ![]()
Photo courtesy of
Millenium Pharmaceuticals
Results of a phase 3 trial suggest a 3-drug combination improves survival in newly diagnosed multiple myeloma (MM).
The trial showed that bortezomib, lenalidomide, and dexamethasone (VRd) could significantly
improve progression-free and overall survival when compared to lenalidomide and dexamethasone (Rd).
In addition, investigators said the risk-benefit profile of VRd was acceptable.
The team noted, however, that grade 3 or higher neurologic adverse events (AEs) were more common with VRd than with Rd.
“Our results are clear,” said principal investigator Brian G.M. Durie, MD, of Cedars-Sinai Outpatient Cancer Center in Los Angeles, California.
“Using bortezomib in combination with lenalidomide and dexamethasone in frontline treatment—hitting the disease early and hard—makes a meaningful difference for myeloma patients. Our results represent a potential new standard of care.”
Dr Durie and his colleagues reported the results of this trial in The Lancet.
The research was funded by Millennium Pharmaceuticals, Takeda Oncology Company, Celgene Corporation, the National Institutes of Health, the National Cancer Institute, and the National Clinical Trial Network.
The trial enrolled 525 adults with MM. The patients ranged in age from 28 to 87, had active MM, and had not had any prior treatment for their disease.
The patients were randomized to 2 treatment groups. One group received Rd for 6 cycles over 6 months. The other group received VRd for 8 cycles over 6 months.
Results
The median follow-up was 55 months. In the VRd group, 241 patients were evaluable for safety and 216 for response. In the Rd group, 226 patients were evaluable for safety and 214 for response.
The overall response rate was 82% (176/216) in the VRd group and 72% (153/214) in the Rd group. The complete response rates were 16% (34/216) and 8% (18/214), respectively.
The median progression-free survival was 43 months in the VRd group and 30 months in the Rd group. The hazard ratio was 0.712 (P=0.0018).
The median overall survival was 75 months in the VRd group and 64 months in the Rd group. The hazard ratio was 0.709 (P=0.025).
The rate of grade 3 or higher AEs was 82% in the VRd group and 75% in the Rd group. The rate of discontinuation due to AEs was 23% and 10%, respectively.
Grade 3 or higher neurologic AEs were more frequent in the VRd group than in the Rd group—33% and 11%, respectively (P<0.0001).
There were 2 treatment-related deaths in the VRd group but none in the Rd group. And 10 patients in each group had a second primary malignancy.
“This is a landmark study that lends clarity to frontline therapy of myeloma,” said study author S. Vincent Rajkumar, MD, of Mayo Clinic in Rochester, Minnesota.
“Newer alternatives to VRd may be more expensive, cumbersome, or toxic. These regimens will therefore need to show superiority over VRd in randomized trials.”
Also worth noting, Dr Rajkumar said, is that the VRd regimen will become even more cost-effective as the drugs in this combination become generic over time. ![]()
Photo courtesy of
Millenium Pharmaceuticals
Results of a phase 3 trial suggest a 3-drug combination improves survival in newly diagnosed multiple myeloma (MM).
The trial showed that bortezomib, lenalidomide, and dexamethasone (VRd) could significantly
improve progression-free and overall survival when compared to lenalidomide and dexamethasone (Rd).
In addition, investigators said the risk-benefit profile of VRd was acceptable.
The team noted, however, that grade 3 or higher neurologic adverse events (AEs) were more common with VRd than with Rd.
“Our results are clear,” said principal investigator Brian G.M. Durie, MD, of Cedars-Sinai Outpatient Cancer Center in Los Angeles, California.
“Using bortezomib in combination with lenalidomide and dexamethasone in frontline treatment—hitting the disease early and hard—makes a meaningful difference for myeloma patients. Our results represent a potential new standard of care.”
Dr Durie and his colleagues reported the results of this trial in The Lancet.
The research was funded by Millennium Pharmaceuticals, Takeda Oncology Company, Celgene Corporation, the National Institutes of Health, the National Cancer Institute, and the National Clinical Trial Network.
The trial enrolled 525 adults with MM. The patients ranged in age from 28 to 87, had active MM, and had not had any prior treatment for their disease.
The patients were randomized to 2 treatment groups. One group received Rd for 6 cycles over 6 months. The other group received VRd for 8 cycles over 6 months.
Results
The median follow-up was 55 months. In the VRd group, 241 patients were evaluable for safety and 216 for response. In the Rd group, 226 patients were evaluable for safety and 214 for response.
The overall response rate was 82% (176/216) in the VRd group and 72% (153/214) in the Rd group. The complete response rates were 16% (34/216) and 8% (18/214), respectively.
The median progression-free survival was 43 months in the VRd group and 30 months in the Rd group. The hazard ratio was 0.712 (P=0.0018).
The median overall survival was 75 months in the VRd group and 64 months in the Rd group. The hazard ratio was 0.709 (P=0.025).
The rate of grade 3 or higher AEs was 82% in the VRd group and 75% in the Rd group. The rate of discontinuation due to AEs was 23% and 10%, respectively.
Grade 3 or higher neurologic AEs were more frequent in the VRd group than in the Rd group—33% and 11%, respectively (P<0.0001).
There were 2 treatment-related deaths in the VRd group but none in the Rd group. And 10 patients in each group had a second primary malignancy.
“This is a landmark study that lends clarity to frontline therapy of myeloma,” said study author S. Vincent Rajkumar, MD, of Mayo Clinic in Rochester, Minnesota.
“Newer alternatives to VRd may be more expensive, cumbersome, or toxic. These regimens will therefore need to show superiority over VRd in randomized trials.”
Also worth noting, Dr Rajkumar said, is that the VRd regimen will become even more cost-effective as the drugs in this combination become generic over time. ![]()
Company withdraws MAA for biosimilar pegfilgrastim

Image by Volker Brinkmann
Gedeon Richter Plc. has withdrawn its marketing authorization application (MAA) for the biosimilar pegfilgrastim product Cavoley from the European Medicines Agency (EMA).
Richter was seeking approval of Cavoley for the same indications as the reference product, Neulasta, a pegylated recombinant granulocyte-colony stimulating factor used to reduce the duration of neutropenia and the occurrence of febrile neutropenia in adults receiving cytotoxic chemotherapy to treat malignancies (except chronic myeloid leukemia and myelodysplastic syndromes).
The MAA filing for Cavoley was based on data from Richter’s completed biosimilar development program.
The company presented to the EMA results of studies in healthy volunteers designed to show that Cavoley is highly similar to Neulasta in terms of chemical structure, purity, the way it works, and how the body handles the drug.
Richter also presented results of a study comparing the safety and effectiveness of Cavoley and Neulasta in breast cancer patients receiving cytotoxic chemotherapy (EudraCT 2013-003166-14).
Richter withdrew the MAA for Cavoley after the EMA’s Committee for Medicinal Products for Human Use (CHMP) evaluated the documentation provided by the company and formulated lists of questions.
After the CHMP had assessed the company’s responses to the last round of questions, there were still some unresolved issues.
Based on the review of the data and Richter’s response to the CHMP’s list of questions, at the time of the withdrawal, the CHMP had some concerns and was of the provisional opinion that Cavoley could not have been approved.
The CHMP said the company had not demonstrated that Cavoley is highly similar to Neulasta.
In its letter notifying the EMA of the MAA withdrawal, Richter said it would continue developing Cavoley and follow the CHMP’s advice to eliminate the remaining uncertainty that Cavoley is highly similar to Neulasta. ![]()
Image by Volker Brinkmann
Gedeon Richter Plc. has withdrawn its marketing authorization application (MAA) for the biosimilar pegfilgrastim product Cavoley from the European Medicines Agency (EMA).
Richter was seeking approval of Cavoley for the same indications as the reference product, Neulasta, a pegylated recombinant granulocyte-colony stimulating factor used to reduce the duration of neutropenia and the occurrence of febrile neutropenia in adults receiving cytotoxic chemotherapy to treat malignancies (except chronic myeloid leukemia and myelodysplastic syndromes).
The MAA filing for Cavoley was based on data from Richter’s completed biosimilar development program.
The company presented to the EMA results of studies in healthy volunteers designed to show that Cavoley is highly similar to Neulasta in terms of chemical structure, purity, the way it works, and how the body handles the drug.
Richter also presented results of a study comparing the safety and effectiveness of Cavoley and Neulasta in breast cancer patients receiving cytotoxic chemotherapy (EudraCT 2013-003166-14).
Richter withdrew the MAA for Cavoley after the EMA’s Committee for Medicinal Products for Human Use (CHMP) evaluated the documentation provided by the company and formulated lists of questions.
After the CHMP had assessed the company’s responses to the last round of questions, there were still some unresolved issues.
Based on the review of the data and Richter’s response to the CHMP’s list of questions, at the time of the withdrawal, the CHMP had some concerns and was of the provisional opinion that Cavoley could not have been approved.
The CHMP said the company had not demonstrated that Cavoley is highly similar to Neulasta.
In its letter notifying the EMA of the MAA withdrawal, Richter said it would continue developing Cavoley and follow the CHMP’s advice to eliminate the remaining uncertainty that Cavoley is highly similar to Neulasta. ![]()
Image by Volker Brinkmann
Gedeon Richter Plc. has withdrawn its marketing authorization application (MAA) for the biosimilar pegfilgrastim product Cavoley from the European Medicines Agency (EMA).
Richter was seeking approval of Cavoley for the same indications as the reference product, Neulasta, a pegylated recombinant granulocyte-colony stimulating factor used to reduce the duration of neutropenia and the occurrence of febrile neutropenia in adults receiving cytotoxic chemotherapy to treat malignancies (except chronic myeloid leukemia and myelodysplastic syndromes).
The MAA filing for Cavoley was based on data from Richter’s completed biosimilar development program.
The company presented to the EMA results of studies in healthy volunteers designed to show that Cavoley is highly similar to Neulasta in terms of chemical structure, purity, the way it works, and how the body handles the drug.
Richter also presented results of a study comparing the safety and effectiveness of Cavoley and Neulasta in breast cancer patients receiving cytotoxic chemotherapy (EudraCT 2013-003166-14).
Richter withdrew the MAA for Cavoley after the EMA’s Committee for Medicinal Products for Human Use (CHMP) evaluated the documentation provided by the company and formulated lists of questions.
After the CHMP had assessed the company’s responses to the last round of questions, there were still some unresolved issues.
Based on the review of the data and Richter’s response to the CHMP’s list of questions, at the time of the withdrawal, the CHMP had some concerns and was of the provisional opinion that Cavoley could not have been approved.
The CHMP said the company had not demonstrated that Cavoley is highly similar to Neulasta.
In its letter notifying the EMA of the MAA withdrawal, Richter said it would continue developing Cavoley and follow the CHMP’s advice to eliminate the remaining uncertainty that Cavoley is highly similar to Neulasta.
Old drug, new tricks possible in MM

SAN DIEGO—An antiretroviral drug used to treat the human immunodeficiency virus (HIV) may find a role in the treatment of multiple myeloma (MM) patients who are proteasome inhibitor (PI)-refractory.
According to investigators, nelfinavir may sensitize refractory patients so that PI-based treatments become an option for them.
In a phase 2 study of 34 patients, nelfinavir in combination with bortezomib and dexamethasone produced an objective response rate of 65%, which investigators called an “exceptional” response in this heavily pretreated, mostly dual-refractory patient population.
Christoph Driessen, MD, PhD, of Kantonsspital St Gallen in Switzerland, discussed the findings of this study, known as SAKK 39/13, at the 2016 ASH Annual Meeting as abstract 487.
Dr Driessen explained that downregulation of IRE1/XBP1 produces PI resistance, and this downregulation occurs in PI-refractory MM patients.
High expression of IRE1/XBP1 correlates with bortezomib sensitivity, and pharmacologic upregulation of IRE1/XBP1 re-sensitizes myeloma cells to PI treatment.
Nelfinavir, which overcomes PI resistance in vitro, is approved for oral HIV therapy.
“It’s an old drug, it’s a generic drug,” Dr Driessen said, and it’s approved at a dose of 2 x 1250 mg daily.
So the SAKK investigators undertook a phase 1 trial of nelfinavir in MM patients.
In an exploratory extension cohort, they found that 5 of 6 MM patients double-refractory to bortezomib and lenalidomide experienced clinical benefit from nelfinavir at the recommended phase 2 dose (2 x 2500 mg daily) in addition to standard treatment with bortezomib and dexamethasone.
Three patients achieved a partial response (PR) and 3 a minor response (MR).
The investigators’ objective in the phase 2 study was to determine whether the addition of nelfinavir to approved bortezomib-dexamethasone therapy is sufficiently active to merit further investigation in a randomized trial.
Study design
Patients in this prospective, single-arm, multicenter, open-label trial received the following treatment:
- Nelfinavir at 2 x 2500 mg orally on days 1–14
- Bortezomib at 1.3 mg/m2 intravenously or subcutaneously on days 1, 4, 8, and 11
- Dexamethasone at 20 mg orally on days 1-2, 4-5, 8-9, and 11-12 of each 21-day cycle.
Trial therapy lasted for a maximum of 6 cycles (18 weeks).
Dr Driessen explained that the trial “was a truly academic trial, without any finances from industry or drug support from industry. So we actually had to get a grant to buy commercial drugs for the study on the commercial drug market, and that limited the duration of treatment in this trial.”
The primary endpoint of the trial was response rate—best response of PR or better by IMWG criteria.
Investigators considered a 30% or higher response rate promising.
Secondary endpoints included adverse events, time to next new anti-myeloma therapy or death, progressive disease under trial treatment, duration of response, progression-free survival, and time to progression.
Patients were eligible to enroll if they had been exposed to or could not tolerate an immunomodulatory drug, were refractory to their most recent PI-containing regimen, had a performance status of 3 or less, had creatinine clearance of 15 mL/minute or greater, had a platelet count of 50,000/μL or more, and had a hemoglobin level of 8.0 g/dL or higher.
Patients were excluded if they had uncontrolled, clinically significant, active concurrent disease, concomitant additional systemic cancer treatment, concomitant radiotherapy, or significant neuropathy of grades 3-4 or grade 2 with pain.
Patient population
Thirty-four patients enrolled on the trial. They were a median age of 67 (range, 42–82), 62% were male, 91% had a performance status of 0 or 1, and 76% had a prior autologous stem cell transplant.
They had a median of 5 prior systemic therapies (range, 2–10), and 38% had poor-risk cytogenetics.
The time from last dose of prior therapy to enrollment on the study was a median of 27 days.
“So [it was] a truly progressive, highly refractory myeloma population,” Dr Driessen emphasized.
All 34 patients were refractory to bortezomib. All patients were also exposed to lenalidomide, and 79% were refractory to it.
Forty-four percent were refractory to pomalidomide, and 6% were refractory to carfilzomib. One patient was refractory to all 4 agents.
“Very few patients were exposed to carfilzomib because it wasn’t available in Switzerland at that time,” Dr Driessen explained.
Efficacy
Patients received a median of 4.5 cycles of therapy (range, 1–6), and the best response of PR or greater was achieved by 22 patients (65%).
Five patients (15%) achieved a very good partial response (VGPR), 17 (50%) PR, 3 (9%) MR, and 4 (12%) stable disease.
Twenty-five patients (74%) achieved a clinical benefit (VGPR+PR+MR).
Ten of the 13 patients (77%) with poor-risk cytogenetics achieved a best response of PR or greater.
Patients had a median of 16 weeks (range, 13–24) time to a new anti-myeloma therapy or death, and 13 patients (38%) had confirmed progressive disease while on trial therapy.
In 32 patients, all but 4 had a decrease from baseline in serum M protein or serum free light chain concentration.
Efficacy by prior therapy
Twenty-two of 34 patients (65%) refractory to bortezomib had a best response of PR or greater.
For patients refractory to bortezomib and lenalidomide, 70% achieved a best response of PR or greater.
For patients refractory to bortezomib, lenalidomide, and pomalidomide, 60% achieved a best response of PR or greater.
And for patients who were refractory to bortezomib, lenalidomide, and carfilzomib, 50% achieved a best response of PR or greater.
Adverse events
“The hematologic toxicity was essentially what you would expect from this heavily pretreated population,” Dr Driessen said.
“We did, however, experience 4 deaths on the trial therapy from infectious complications of sepsis and neutropenia, and we don’t know whether this is a true signal or whether this is due to the low numbers. We did not mandate antibiotic prophylaxis on the trial.”
Grade 3 or higher adverse events (AEs) occurring in 2 or more patients were anemia (n=10), febrile neutropenia (n=4, including 1 grade 5), thrombocytopenia (n=15), lung infection (n=8), sepsis (n=3, all grade 5), fatigue (n=5), peripheral sensory neuropathy (n=3), hypertension (n=6), increased creatinine (n=4), hyperglycemia (n=6) hypokalemia (n=3), and hyponatremia (n=5).
Dr Driessen indicated that with a future generic version of bortezomib, nelfinavir plus bortezomib and dexamethasone “has the potential to become a fully generic, affordable, active therapy option for PI-refractory patients.”
The investigators believe the results of their study call for further development of nelfinavir as a sensitizing drug for PI-based treatments and as a promising new agent for MM therapy.
SAN DIEGO—An antiretroviral drug used to treat the human immunodeficiency virus (HIV) may find a role in the treatment of multiple myeloma (MM) patients who are proteasome inhibitor (PI)-refractory.
According to investigators, nelfinavir may sensitize refractory patients so that PI-based treatments become an option for them.
In a phase 2 study of 34 patients, nelfinavir in combination with bortezomib and dexamethasone produced an objective response rate of 65%, which investigators called an “exceptional” response in this heavily pretreated, mostly dual-refractory patient population.
Christoph Driessen, MD, PhD, of Kantonsspital St Gallen in Switzerland, discussed the findings of this study, known as SAKK 39/13, at the 2016 ASH Annual Meeting as abstract 487.
Dr Driessen explained that downregulation of IRE1/XBP1 produces PI resistance, and this downregulation occurs in PI-refractory MM patients.
High expression of IRE1/XBP1 correlates with bortezomib sensitivity, and pharmacologic upregulation of IRE1/XBP1 re-sensitizes myeloma cells to PI treatment.
Nelfinavir, which overcomes PI resistance in vitro, is approved for oral HIV therapy.
“It’s an old drug, it’s a generic drug,” Dr Driessen said, and it’s approved at a dose of 2 x 1250 mg daily.
So the SAKK investigators undertook a phase 1 trial of nelfinavir in MM patients.
In an exploratory extension cohort, they found that 5 of 6 MM patients double-refractory to bortezomib and lenalidomide experienced clinical benefit from nelfinavir at the recommended phase 2 dose (2 x 2500 mg daily) in addition to standard treatment with bortezomib and dexamethasone.
Three patients achieved a partial response (PR) and 3 a minor response (MR).
The investigators’ objective in the phase 2 study was to determine whether the addition of nelfinavir to approved bortezomib-dexamethasone therapy is sufficiently active to merit further investigation in a randomized trial.
Study design
Patients in this prospective, single-arm, multicenter, open-label trial received the following treatment:
- Nelfinavir at 2 x 2500 mg orally on days 1–14
- Bortezomib at 1.3 mg/m2 intravenously or subcutaneously on days 1, 4, 8, and 11
- Dexamethasone at 20 mg orally on days 1-2, 4-5, 8-9, and 11-12 of each 21-day cycle.
Trial therapy lasted for a maximum of 6 cycles (18 weeks).
Dr Driessen explained that the trial “was a truly academic trial, without any finances from industry or drug support from industry. So we actually had to get a grant to buy commercial drugs for the study on the commercial drug market, and that limited the duration of treatment in this trial.”
The primary endpoint of the trial was response rate—best response of PR or better by IMWG criteria.
Investigators considered a 30% or higher response rate promising.
Secondary endpoints included adverse events, time to next new anti-myeloma therapy or death, progressive disease under trial treatment, duration of response, progression-free survival, and time to progression.
Patients were eligible to enroll if they had been exposed to or could not tolerate an immunomodulatory drug, were refractory to their most recent PI-containing regimen, had a performance status of 3 or less, had creatinine clearance of 15 mL/minute or greater, had a platelet count of 50,000/μL or more, and had a hemoglobin level of 8.0 g/dL or higher.
Patients were excluded if they had uncontrolled, clinically significant, active concurrent disease, concomitant additional systemic cancer treatment, concomitant radiotherapy, or significant neuropathy of grades 3-4 or grade 2 with pain.
Patient population
Thirty-four patients enrolled on the trial. They were a median age of 67 (range, 42–82), 62% were male, 91% had a performance status of 0 or 1, and 76% had a prior autologous stem cell transplant.
They had a median of 5 prior systemic therapies (range, 2–10), and 38% had poor-risk cytogenetics.
The time from last dose of prior therapy to enrollment on the study was a median of 27 days.
“So [it was] a truly progressive, highly refractory myeloma population,” Dr Driessen emphasized.
All 34 patients were refractory to bortezomib. All patients were also exposed to lenalidomide, and 79% were refractory to it.
Forty-four percent were refractory to pomalidomide, and 6% were refractory to carfilzomib. One patient was refractory to all 4 agents.
“Very few patients were exposed to carfilzomib because it wasn’t available in Switzerland at that time,” Dr Driessen explained.
Efficacy
Patients received a median of 4.5 cycles of therapy (range, 1–6), and the best response of PR or greater was achieved by 22 patients (65%).
Five patients (15%) achieved a very good partial response (VGPR), 17 (50%) PR, 3 (9%) MR, and 4 (12%) stable disease.
Twenty-five patients (74%) achieved a clinical benefit (VGPR+PR+MR).
Ten of the 13 patients (77%) with poor-risk cytogenetics achieved a best response of PR or greater.
Patients had a median of 16 weeks (range, 13–24) time to a new anti-myeloma therapy or death, and 13 patients (38%) had confirmed progressive disease while on trial therapy.
In 32 patients, all but 4 had a decrease from baseline in serum M protein or serum free light chain concentration.
Efficacy by prior therapy
Twenty-two of 34 patients (65%) refractory to bortezomib had a best response of PR or greater.
For patients refractory to bortezomib and lenalidomide, 70% achieved a best response of PR or greater.
For patients refractory to bortezomib, lenalidomide, and pomalidomide, 60% achieved a best response of PR or greater.
And for patients who were refractory to bortezomib, lenalidomide, and carfilzomib, 50% achieved a best response of PR or greater.
Adverse events
“The hematologic toxicity was essentially what you would expect from this heavily pretreated population,” Dr Driessen said.
“We did, however, experience 4 deaths on the trial therapy from infectious complications of sepsis and neutropenia, and we don’t know whether this is a true signal or whether this is due to the low numbers. We did not mandate antibiotic prophylaxis on the trial.”
Grade 3 or higher adverse events (AEs) occurring in 2 or more patients were anemia (n=10), febrile neutropenia (n=4, including 1 grade 5), thrombocytopenia (n=15), lung infection (n=8), sepsis (n=3, all grade 5), fatigue (n=5), peripheral sensory neuropathy (n=3), hypertension (n=6), increased creatinine (n=4), hyperglycemia (n=6) hypokalemia (n=3), and hyponatremia (n=5).
Dr Driessen indicated that with a future generic version of bortezomib, nelfinavir plus bortezomib and dexamethasone “has the potential to become a fully generic, affordable, active therapy option for PI-refractory patients.”
The investigators believe the results of their study call for further development of nelfinavir as a sensitizing drug for PI-based treatments and as a promising new agent for MM therapy.
SAN DIEGO—An antiretroviral drug used to treat the human immunodeficiency virus (HIV) may find a role in the treatment of multiple myeloma (MM) patients who are proteasome inhibitor (PI)-refractory.
According to investigators, nelfinavir may sensitize refractory patients so that PI-based treatments become an option for them.
In a phase 2 study of 34 patients, nelfinavir in combination with bortezomib and dexamethasone produced an objective response rate of 65%, which investigators called an “exceptional” response in this heavily pretreated, mostly dual-refractory patient population.
Christoph Driessen, MD, PhD, of Kantonsspital St Gallen in Switzerland, discussed the findings of this study, known as SAKK 39/13, at the 2016 ASH Annual Meeting as abstract 487.
Dr Driessen explained that downregulation of IRE1/XBP1 produces PI resistance, and this downregulation occurs in PI-refractory MM patients.
High expression of IRE1/XBP1 correlates with bortezomib sensitivity, and pharmacologic upregulation of IRE1/XBP1 re-sensitizes myeloma cells to PI treatment.
Nelfinavir, which overcomes PI resistance in vitro, is approved for oral HIV therapy.
“It’s an old drug, it’s a generic drug,” Dr Driessen said, and it’s approved at a dose of 2 x 1250 mg daily.
So the SAKK investigators undertook a phase 1 trial of nelfinavir in MM patients.
In an exploratory extension cohort, they found that 5 of 6 MM patients double-refractory to bortezomib and lenalidomide experienced clinical benefit from nelfinavir at the recommended phase 2 dose (2 x 2500 mg daily) in addition to standard treatment with bortezomib and dexamethasone.
Three patients achieved a partial response (PR) and 3 a minor response (MR).
The investigators’ objective in the phase 2 study was to determine whether the addition of nelfinavir to approved bortezomib-dexamethasone therapy is sufficiently active to merit further investigation in a randomized trial.
Study design
Patients in this prospective, single-arm, multicenter, open-label trial received the following treatment:
- Nelfinavir at 2 x 2500 mg orally on days 1–14
- Bortezomib at 1.3 mg/m2 intravenously or subcutaneously on days 1, 4, 8, and 11
- Dexamethasone at 20 mg orally on days 1-2, 4-5, 8-9, and 11-12 of each 21-day cycle.
Trial therapy lasted for a maximum of 6 cycles (18 weeks).
Dr Driessen explained that the trial “was a truly academic trial, without any finances from industry or drug support from industry. So we actually had to get a grant to buy commercial drugs for the study on the commercial drug market, and that limited the duration of treatment in this trial.”
The primary endpoint of the trial was response rate—best response of PR or better by IMWG criteria.
Investigators considered a 30% or higher response rate promising.
Secondary endpoints included adverse events, time to next new anti-myeloma therapy or death, progressive disease under trial treatment, duration of response, progression-free survival, and time to progression.
Patients were eligible to enroll if they had been exposed to or could not tolerate an immunomodulatory drug, were refractory to their most recent PI-containing regimen, had a performance status of 3 or less, had creatinine clearance of 15 mL/minute or greater, had a platelet count of 50,000/μL or more, and had a hemoglobin level of 8.0 g/dL or higher.
Patients were excluded if they had uncontrolled, clinically significant, active concurrent disease, concomitant additional systemic cancer treatment, concomitant radiotherapy, or significant neuropathy of grades 3-4 or grade 2 with pain.
Patient population
Thirty-four patients enrolled on the trial. They were a median age of 67 (range, 42–82), 62% were male, 91% had a performance status of 0 or 1, and 76% had a prior autologous stem cell transplant.
They had a median of 5 prior systemic therapies (range, 2–10), and 38% had poor-risk cytogenetics.
The time from last dose of prior therapy to enrollment on the study was a median of 27 days.
“So [it was] a truly progressive, highly refractory myeloma population,” Dr Driessen emphasized.
All 34 patients were refractory to bortezomib. All patients were also exposed to lenalidomide, and 79% were refractory to it.
Forty-four percent were refractory to pomalidomide, and 6% were refractory to carfilzomib. One patient was refractory to all 4 agents.
“Very few patients were exposed to carfilzomib because it wasn’t available in Switzerland at that time,” Dr Driessen explained.
Efficacy
Patients received a median of 4.5 cycles of therapy (range, 1–6), and the best response of PR or greater was achieved by 22 patients (65%).
Five patients (15%) achieved a very good partial response (VGPR), 17 (50%) PR, 3 (9%) MR, and 4 (12%) stable disease.
Twenty-five patients (74%) achieved a clinical benefit (VGPR+PR+MR).
Ten of the 13 patients (77%) with poor-risk cytogenetics achieved a best response of PR or greater.
Patients had a median of 16 weeks (range, 13–24) time to a new anti-myeloma therapy or death, and 13 patients (38%) had confirmed progressive disease while on trial therapy.
In 32 patients, all but 4 had a decrease from baseline in serum M protein or serum free light chain concentration.
Efficacy by prior therapy
Twenty-two of 34 patients (65%) refractory to bortezomib had a best response of PR or greater.
For patients refractory to bortezomib and lenalidomide, 70% achieved a best response of PR or greater.
For patients refractory to bortezomib, lenalidomide, and pomalidomide, 60% achieved a best response of PR or greater.
And for patients who were refractory to bortezomib, lenalidomide, and carfilzomib, 50% achieved a best response of PR or greater.
Adverse events
“The hematologic toxicity was essentially what you would expect from this heavily pretreated population,” Dr Driessen said.
“We did, however, experience 4 deaths on the trial therapy from infectious complications of sepsis and neutropenia, and we don’t know whether this is a true signal or whether this is due to the low numbers. We did not mandate antibiotic prophylaxis on the trial.”
Grade 3 or higher adverse events (AEs) occurring in 2 or more patients were anemia (n=10), febrile neutropenia (n=4, including 1 grade 5), thrombocytopenia (n=15), lung infection (n=8), sepsis (n=3, all grade 5), fatigue (n=5), peripheral sensory neuropathy (n=3), hypertension (n=6), increased creatinine (n=4), hyperglycemia (n=6) hypokalemia (n=3), and hyponatremia (n=5).
Dr Driessen indicated that with a future generic version of bortezomib, nelfinavir plus bortezomib and dexamethasone “has the potential to become a fully generic, affordable, active therapy option for PI-refractory patients.”
The investigators believe the results of their study call for further development of nelfinavir as a sensitizing drug for PI-based treatments and as a promising new agent for MM therapy.