User login
Electrocardiography: Flecainide Toxicity
Case
An 86-year-old woman, who recently had been seen in the same facility after a ground level fall, presented to the ED with to a 2- to 3-day history of vague abdominal pain, increasing weakness, nausea, and dry heaves.
Upon examination, the patient was unable to stand due to generalized weakness She arrived at the ED via emergency medical services. Her vital signs at presentation were significant for a systolic blood pressure (BP) of 90 mm Hg with a wide complex tachycardia concerning for ventricular tachycardia. The patient’s other vital signers were: heart rate, 136 beats/min; respiratory rate 20 breaths/min; and pulse oximetry was 94% on 4 liters/min of oxygen via nasal cannula.
The patient’s medical history was significant for atrial fibrillation and an indwelling pacemaker, for which she was chronically on flecai
The initial electrocardiogram (ECG) revealed a wide complex rhythm with pacemaker spikes (Figure 1). Based on these findings, electrodes were placed on the patient in the event she required cardioversion. The patient was started on an amiodarone intravenous (IV) drip for presumptive ventricular tachycardia.
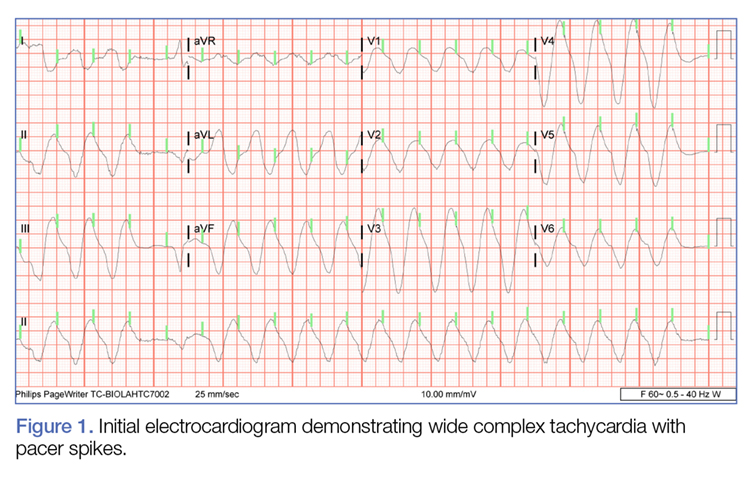
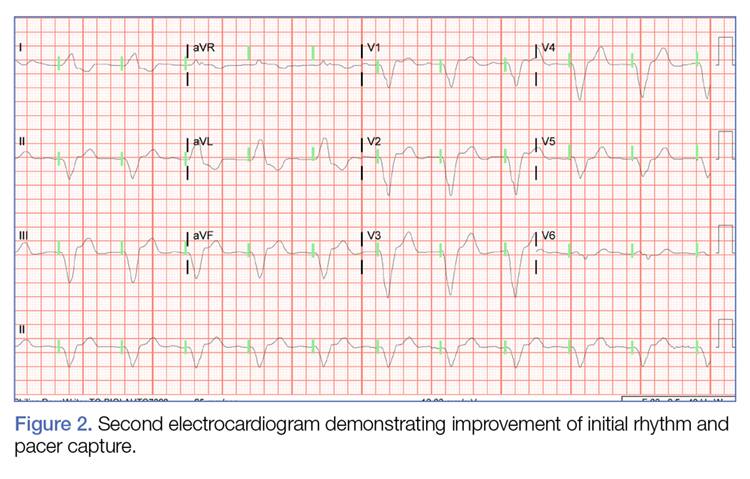
During the patient’s evaluation in the ED, she experienced transient drops in BP, which were responsive to an IV fluid bolus of normal saline, and the amiodarone drip was discontinued. The patient’s ECG findings were compared to previous ECG studies, as was her current medication list and prior health issues. After ruling-out other causes, flecainide toxicity was considered high in the differential, and she was given 1 ampule of bicarbonate IV, after which a second ECG showed heart rhythm converted from a wide-complex tachycardia to a paced rhythm, markedly improved from the initial ECG (Figure 2). Similarly, there was a marked improvement in BP.
An interrogation of the patient’s pacemaker revealed an atrial flutter with a rate below detection for mode switch, with one-to-one tracking/pacing. The pacemaker was reprogrammed to divide the DDIR mode with detection rate at 120 mm Hg with mode switch activated. This was felt to be consistent with flecainide toxicity precipitating the cardiac conduction issues.
Laboratory studies showed an elevated flecainide level at 1.39 mcg/mL (upper limits of normal of 1 mcg/mL). Other studies showed worsening congestive heart failure, with a brain natriuretic peptide of 8,057 pg/mL and mild dehydration, with serum creatinine increased from her baseline of 0.9 to 1.38 mg/dL.
The patient’s abdominal pain was further evaluated and she was found to have acute cholecystitis. She was admitted to the intensive care unit with cardiology and general surgery consulting.
Discussion
Flecainide acetate was approved by the Food and Drug Administration in 1984.1It is a Vaughan-Williams class IC antiarrhythmic with a sodium channel blocker action used to treat supra ventricular arrhythmias. The CAST trial in 1989 investigated the efficacy of this class of antiarrhythmics, which resulted in a revision of its role.2 Based on this study, flecainide is not recommended for patients with structural heart disease or coronary artery disease.2,3 However, it is recommended as a first-line therapy for pharmacologic cardioversion and maintenance of normal sinus rhythm in patients with atrial fibrillation and supraventricular tachycardia4,5 without the above caveats.
Class IC agents produce a selective block at the sodium (Na+) channels, resulting in the slowing of cardiac conduction.6,7 This high affinity for Na+ channels combined with slow unbinding kinetics during diastole explain the slowing of recovery time and prolongation of the refractory period.6,8,9 These electrophysiologic properties all can increase the PR, QRS, and QT interval duration. The QT interval is not significantly affected, as most of the QT prolongation is due to the QRS widening.6,10,11 Widening of the QRS by greater than 25% as compared to the baseline value is used as the threshold to decrease dosing or discontinue the use of flecainide.3The toxic effects of flecainide on cardiac conduction can produce prolonged QRS duration of up to 50%, and PR interval up to 30%, especially in rapid heart rates. Signs of intoxication are difficult to discern owing to its nonspecific presentation. A well-documented, but under-recognized, presentation of flecainide toxicity is the transformation of atrial fibrillation to atrial flutter.5,7,9,11-13 The reported rate of this pro arrhythmic effect can be as high as 3.5% to 5%.14,15Flecainide toxicity can occur secondary to chronic ingestion and may be precipitated in mild renal failure. The majority of flecainide is renally excreted and the half-life is 20 hours. Maximum therapeutic effect is seen between levels of 0.2 to 1 mcg/mL with levels greater than 0.7 to 1 mcg/mL associated with adverse effects.9 Systemic effects include dizziness and visual disturbances. A high degree of suspicion for flecainide toxicity is required when the patient’s initial presentation is nonspecific. In this circumstance, real-time bedside interrogation of the pacemaker is invaluable. Early diagnosis and treatment minimizes the risk for adverse sequelae, including death. Treatment includes increasing the excretion of flecainide, symptomatic support (including pacemaker placement, intravenous fat emulsion, or extracorporeal circulatory support) and administration of sodium bicarbonate, to transiently reverse the effect of the sodium channel blockade, in severe cases.15-17
1. Hudak JM, Banitt EH, Schmid JR. Discovery and development of flecainide. Am J Cardiol. 1984;53(5):17B-20B.
2. Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. The Cardiac Arrhythmia Suppression Trial (CAST). N Engl J Med. 1989;321(6):406-412. doi:10.1056/NEJM198908103210629.
3. Andrikopoulos GK, Pastromas S, Tzeis S. Flecainide: Current status and perspectives in arrhythmia management. World J Cardiol. 2015;7(2):76-85. doi:10.4330/wjc.v7.i2.76.
4. Camm AJ, Lip GY, De Caterina R, et al; ESC Committee for Practice Guidelines (CPG). 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur Heart J. 2012;33(21):2719-2747. doi:10.1093/eurheartj/ehs253.
5. Courand PY, Sibellas F, Ranc S, Mullier A, Kirkorian G, Bonnefoy E. Arrhythmogenic effect of flecainide toxicity. Cardiol J. 2013;20:203-205. doi:10.5603/CJ.2013.0035.
6. Holmes B, Heel RC. Flecainide. A preliminary review of its pharmacodynamic properties and therapeutic efficacy. Drugs. 1985;29(1):1-33.
7. Taylor R, Gandhi MM, Lloyd G. Tachycardia due to atrial flutter with rapid 1:1 conduction following treatment of atrial fibrillation with flecainide. Br Med J. 2010;340:b4684.
8. Roden DM, Woosley RL. Drug therapy. Flecainide. N Engl J Med. 1986;315(1):36-41.
9. Levis JT. ECG diagnosis: flecainide toxicity. Perm J. 2012;16(4):53.
10. Hellestrand KJ, Bexton RS, Nathan AW, Spurrell RA, Camm AJ. Acute electrophysiological effects of flecainide acetate on cardiac conduction and refractoriness in man. Br Heart J. 1982;48(2):140-148.
11. Rognoni A, Bertolazzi M, Peron M, et al. Electrocardiographic changes in a rare case of flecainide poisoning: a case report. Cases J. 2009;2:9137. doi:10.1186/1757-1626-2-9137.
12. Nabar A, Rodriguez LM, Timmermans C, Smeets JL, Wellens HJ. Radiofrequency ablation of “class IC atrial flutter” in patients with resistant atrial fibrillation. Am J Cardiol. 1999;83(5):785-787, A10.
13. Kola S, Mahata I, Kocheril AG. A case of flecainide toxicity. EP Lab Digest. 2015;15(5).
14. Falk RH. Proarrhythmia in patients treated for atrial fibrillation or flutter. Ann Intern Med. 1992;117(2):141-150.
15. Lloyd T, Zimmerman J, Griffin GD. Irreversible third-degree heart block and pacemaker implant in a case of flecainide toxicity. Am J Emerg Med. 2013;31(9):1418.e1-e2. doi:10.1016/j.ajem.2013.04.025.
16. Corkeron MA, van Heerden PV, Newman SM, Dusci L. Extracorporeal circulatory support in near-fatal flecainide overdose. Anaesth Intensive Care. 1999;27(4):405-408.
17. Ellsworth H, Stellpflug SJ, Cole JB, Dolan JA, Harris CR. A life-threatening flecainide overdose treated with intravenous fat emulsion. Pacing Clin Electrophysiol. 2013;36(3):e87-e89. doi:10.1111/j.1540-8159.2012.03485.x.
Case
An 86-year-old woman, who recently had been seen in the same facility after a ground level fall, presented to the ED with to a 2- to 3-day history of vague abdominal pain, increasing weakness, nausea, and dry heaves.
Upon examination, the patient was unable to stand due to generalized weakness She arrived at the ED via emergency medical services. Her vital signs at presentation were significant for a systolic blood pressure (BP) of 90 mm Hg with a wide complex tachycardia concerning for ventricular tachycardia. The patient’s other vital signers were: heart rate, 136 beats/min; respiratory rate 20 breaths/min; and pulse oximetry was 94% on 4 liters/min of oxygen via nasal cannula.
The patient’s medical history was significant for atrial fibrillation and an indwelling pacemaker, for which she was chronically on flecai
The initial electrocardiogram (ECG) revealed a wide complex rhythm with pacemaker spikes (Figure 1). Based on these findings, electrodes were placed on the patient in the event she required cardioversion. The patient was started on an amiodarone intravenous (IV) drip for presumptive ventricular tachycardia.
During the patient’s evaluation in the ED, she experienced transient drops in BP, which were responsive to an IV fluid bolus of normal saline, and the amiodarone drip was discontinued. The patient’s ECG findings were compared to previous ECG studies, as was her current medication list and prior health issues. After ruling-out other causes, flecainide toxicity was considered high in the differential, and she was given 1 ampule of bicarbonate IV, after which a second ECG showed heart rhythm converted from a wide-complex tachycardia to a paced rhythm, markedly improved from the initial ECG (Figure 2). Similarly, there was a marked improvement in BP.
An interrogation of the patient’s pacemaker revealed an atrial flutter with a rate below detection for mode switch, with one-to-one tracking/pacing. The pacemaker was reprogrammed to divide the DDIR mode with detection rate at 120 mm Hg with mode switch activated. This was felt to be consistent with flecainide toxicity precipitating the cardiac conduction issues.
Laboratory studies showed an elevated flecainide level at 1.39 mcg/mL (upper limits of normal of 1 mcg/mL). Other studies showed worsening congestive heart failure, with a brain natriuretic peptide of 8,057 pg/mL and mild dehydration, with serum creatinine increased from her baseline of 0.9 to 1.38 mg/dL.
The patient’s abdominal pain was further evaluated and she was found to have acute cholecystitis. She was admitted to the intensive care unit with cardiology and general surgery consulting.
Discussion
Flecainide acetate was approved by the Food and Drug Administration in 1984.1It is a Vaughan-Williams class IC antiarrhythmic with a sodium channel blocker action used to treat supra ventricular arrhythmias. The CAST trial in 1989 investigated the efficacy of this class of antiarrhythmics, which resulted in a revision of its role.2 Based on this study, flecainide is not recommended for patients with structural heart disease or coronary artery disease.2,3 However, it is recommended as a first-line therapy for pharmacologic cardioversion and maintenance of normal sinus rhythm in patients with atrial fibrillation and supraventricular tachycardia4,5 without the above caveats.
Class IC agents produce a selective block at the sodium (Na+) channels, resulting in the slowing of cardiac conduction.6,7 This high affinity for Na+ channels combined with slow unbinding kinetics during diastole explain the slowing of recovery time and prolongation of the refractory period.6,8,9 These electrophysiologic properties all can increase the PR, QRS, and QT interval duration. The QT interval is not significantly affected, as most of the QT prolongation is due to the QRS widening.6,10,11 Widening of the QRS by greater than 25% as compared to the baseline value is used as the threshold to decrease dosing or discontinue the use of flecainide.3The toxic effects of flecainide on cardiac conduction can produce prolonged QRS duration of up to 50%, and PR interval up to 30%, especially in rapid heart rates. Signs of intoxication are difficult to discern owing to its nonspecific presentation. A well-documented, but under-recognized, presentation of flecainide toxicity is the transformation of atrial fibrillation to atrial flutter.5,7,9,11-13 The reported rate of this pro arrhythmic effect can be as high as 3.5% to 5%.14,15Flecainide toxicity can occur secondary to chronic ingestion and may be precipitated in mild renal failure. The majority of flecainide is renally excreted and the half-life is 20 hours. Maximum therapeutic effect is seen between levels of 0.2 to 1 mcg/mL with levels greater than 0.7 to 1 mcg/mL associated with adverse effects.9 Systemic effects include dizziness and visual disturbances. A high degree of suspicion for flecainide toxicity is required when the patient’s initial presentation is nonspecific. In this circumstance, real-time bedside interrogation of the pacemaker is invaluable. Early diagnosis and treatment minimizes the risk for adverse sequelae, including death. Treatment includes increasing the excretion of flecainide, symptomatic support (including pacemaker placement, intravenous fat emulsion, or extracorporeal circulatory support) and administration of sodium bicarbonate, to transiently reverse the effect of the sodium channel blockade, in severe cases.15-17
Case
An 86-year-old woman, who recently had been seen in the same facility after a ground level fall, presented to the ED with to a 2- to 3-day history of vague abdominal pain, increasing weakness, nausea, and dry heaves.
Upon examination, the patient was unable to stand due to generalized weakness She arrived at the ED via emergency medical services. Her vital signs at presentation were significant for a systolic blood pressure (BP) of 90 mm Hg with a wide complex tachycardia concerning for ventricular tachycardia. The patient’s other vital signers were: heart rate, 136 beats/min; respiratory rate 20 breaths/min; and pulse oximetry was 94% on 4 liters/min of oxygen via nasal cannula.
The patient’s medical history was significant for atrial fibrillation and an indwelling pacemaker, for which she was chronically on flecai
The initial electrocardiogram (ECG) revealed a wide complex rhythm with pacemaker spikes (Figure 1). Based on these findings, electrodes were placed on the patient in the event she required cardioversion. The patient was started on an amiodarone intravenous (IV) drip for presumptive ventricular tachycardia.
During the patient’s evaluation in the ED, she experienced transient drops in BP, which were responsive to an IV fluid bolus of normal saline, and the amiodarone drip was discontinued. The patient’s ECG findings were compared to previous ECG studies, as was her current medication list and prior health issues. After ruling-out other causes, flecainide toxicity was considered high in the differential, and she was given 1 ampule of bicarbonate IV, after which a second ECG showed heart rhythm converted from a wide-complex tachycardia to a paced rhythm, markedly improved from the initial ECG (Figure 2). Similarly, there was a marked improvement in BP.
An interrogation of the patient’s pacemaker revealed an atrial flutter with a rate below detection for mode switch, with one-to-one tracking/pacing. The pacemaker was reprogrammed to divide the DDIR mode with detection rate at 120 mm Hg with mode switch activated. This was felt to be consistent with flecainide toxicity precipitating the cardiac conduction issues.
Laboratory studies showed an elevated flecainide level at 1.39 mcg/mL (upper limits of normal of 1 mcg/mL). Other studies showed worsening congestive heart failure, with a brain natriuretic peptide of 8,057 pg/mL and mild dehydration, with serum creatinine increased from her baseline of 0.9 to 1.38 mg/dL.
The patient’s abdominal pain was further evaluated and she was found to have acute cholecystitis. She was admitted to the intensive care unit with cardiology and general surgery consulting.
Discussion
Flecainide acetate was approved by the Food and Drug Administration in 1984.1It is a Vaughan-Williams class IC antiarrhythmic with a sodium channel blocker action used to treat supra ventricular arrhythmias. The CAST trial in 1989 investigated the efficacy of this class of antiarrhythmics, which resulted in a revision of its role.2 Based on this study, flecainide is not recommended for patients with structural heart disease or coronary artery disease.2,3 However, it is recommended as a first-line therapy for pharmacologic cardioversion and maintenance of normal sinus rhythm in patients with atrial fibrillation and supraventricular tachycardia4,5 without the above caveats.
Class IC agents produce a selective block at the sodium (Na+) channels, resulting in the slowing of cardiac conduction.6,7 This high affinity for Na+ channels combined with slow unbinding kinetics during diastole explain the slowing of recovery time and prolongation of the refractory period.6,8,9 These electrophysiologic properties all can increase the PR, QRS, and QT interval duration. The QT interval is not significantly affected, as most of the QT prolongation is due to the QRS widening.6,10,11 Widening of the QRS by greater than 25% as compared to the baseline value is used as the threshold to decrease dosing or discontinue the use of flecainide.3The toxic effects of flecainide on cardiac conduction can produce prolonged QRS duration of up to 50%, and PR interval up to 30%, especially in rapid heart rates. Signs of intoxication are difficult to discern owing to its nonspecific presentation. A well-documented, but under-recognized, presentation of flecainide toxicity is the transformation of atrial fibrillation to atrial flutter.5,7,9,11-13 The reported rate of this pro arrhythmic effect can be as high as 3.5% to 5%.14,15Flecainide toxicity can occur secondary to chronic ingestion and may be precipitated in mild renal failure. The majority of flecainide is renally excreted and the half-life is 20 hours. Maximum therapeutic effect is seen between levels of 0.2 to 1 mcg/mL with levels greater than 0.7 to 1 mcg/mL associated with adverse effects.9 Systemic effects include dizziness and visual disturbances. A high degree of suspicion for flecainide toxicity is required when the patient’s initial presentation is nonspecific. In this circumstance, real-time bedside interrogation of the pacemaker is invaluable. Early diagnosis and treatment minimizes the risk for adverse sequelae, including death. Treatment includes increasing the excretion of flecainide, symptomatic support (including pacemaker placement, intravenous fat emulsion, or extracorporeal circulatory support) and administration of sodium bicarbonate, to transiently reverse the effect of the sodium channel blockade, in severe cases.15-17
1. Hudak JM, Banitt EH, Schmid JR. Discovery and development of flecainide. Am J Cardiol. 1984;53(5):17B-20B.
2. Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. The Cardiac Arrhythmia Suppression Trial (CAST). N Engl J Med. 1989;321(6):406-412. doi:10.1056/NEJM198908103210629.
3. Andrikopoulos GK, Pastromas S, Tzeis S. Flecainide: Current status and perspectives in arrhythmia management. World J Cardiol. 2015;7(2):76-85. doi:10.4330/wjc.v7.i2.76.
4. Camm AJ, Lip GY, De Caterina R, et al; ESC Committee for Practice Guidelines (CPG). 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur Heart J. 2012;33(21):2719-2747. doi:10.1093/eurheartj/ehs253.
5. Courand PY, Sibellas F, Ranc S, Mullier A, Kirkorian G, Bonnefoy E. Arrhythmogenic effect of flecainide toxicity. Cardiol J. 2013;20:203-205. doi:10.5603/CJ.2013.0035.
6. Holmes B, Heel RC. Flecainide. A preliminary review of its pharmacodynamic properties and therapeutic efficacy. Drugs. 1985;29(1):1-33.
7. Taylor R, Gandhi MM, Lloyd G. Tachycardia due to atrial flutter with rapid 1:1 conduction following treatment of atrial fibrillation with flecainide. Br Med J. 2010;340:b4684.
8. Roden DM, Woosley RL. Drug therapy. Flecainide. N Engl J Med. 1986;315(1):36-41.
9. Levis JT. ECG diagnosis: flecainide toxicity. Perm J. 2012;16(4):53.
10. Hellestrand KJ, Bexton RS, Nathan AW, Spurrell RA, Camm AJ. Acute electrophysiological effects of flecainide acetate on cardiac conduction and refractoriness in man. Br Heart J. 1982;48(2):140-148.
11. Rognoni A, Bertolazzi M, Peron M, et al. Electrocardiographic changes in a rare case of flecainide poisoning: a case report. Cases J. 2009;2:9137. doi:10.1186/1757-1626-2-9137.
12. Nabar A, Rodriguez LM, Timmermans C, Smeets JL, Wellens HJ. Radiofrequency ablation of “class IC atrial flutter” in patients with resistant atrial fibrillation. Am J Cardiol. 1999;83(5):785-787, A10.
13. Kola S, Mahata I, Kocheril AG. A case of flecainide toxicity. EP Lab Digest. 2015;15(5).
14. Falk RH. Proarrhythmia in patients treated for atrial fibrillation or flutter. Ann Intern Med. 1992;117(2):141-150.
15. Lloyd T, Zimmerman J, Griffin GD. Irreversible third-degree heart block and pacemaker implant in a case of flecainide toxicity. Am J Emerg Med. 2013;31(9):1418.e1-e2. doi:10.1016/j.ajem.2013.04.025.
16. Corkeron MA, van Heerden PV, Newman SM, Dusci L. Extracorporeal circulatory support in near-fatal flecainide overdose. Anaesth Intensive Care. 1999;27(4):405-408.
17. Ellsworth H, Stellpflug SJ, Cole JB, Dolan JA, Harris CR. A life-threatening flecainide overdose treated with intravenous fat emulsion. Pacing Clin Electrophysiol. 2013;36(3):e87-e89. doi:10.1111/j.1540-8159.2012.03485.x.
1. Hudak JM, Banitt EH, Schmid JR. Discovery and development of flecainide. Am J Cardiol. 1984;53(5):17B-20B.
2. Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. The Cardiac Arrhythmia Suppression Trial (CAST). N Engl J Med. 1989;321(6):406-412. doi:10.1056/NEJM198908103210629.
3. Andrikopoulos GK, Pastromas S, Tzeis S. Flecainide: Current status and perspectives in arrhythmia management. World J Cardiol. 2015;7(2):76-85. doi:10.4330/wjc.v7.i2.76.
4. Camm AJ, Lip GY, De Caterina R, et al; ESC Committee for Practice Guidelines (CPG). 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur Heart J. 2012;33(21):2719-2747. doi:10.1093/eurheartj/ehs253.
5. Courand PY, Sibellas F, Ranc S, Mullier A, Kirkorian G, Bonnefoy E. Arrhythmogenic effect of flecainide toxicity. Cardiol J. 2013;20:203-205. doi:10.5603/CJ.2013.0035.
6. Holmes B, Heel RC. Flecainide. A preliminary review of its pharmacodynamic properties and therapeutic efficacy. Drugs. 1985;29(1):1-33.
7. Taylor R, Gandhi MM, Lloyd G. Tachycardia due to atrial flutter with rapid 1:1 conduction following treatment of atrial fibrillation with flecainide. Br Med J. 2010;340:b4684.
8. Roden DM, Woosley RL. Drug therapy. Flecainide. N Engl J Med. 1986;315(1):36-41.
9. Levis JT. ECG diagnosis: flecainide toxicity. Perm J. 2012;16(4):53.
10. Hellestrand KJ, Bexton RS, Nathan AW, Spurrell RA, Camm AJ. Acute electrophysiological effects of flecainide acetate on cardiac conduction and refractoriness in man. Br Heart J. 1982;48(2):140-148.
11. Rognoni A, Bertolazzi M, Peron M, et al. Electrocardiographic changes in a rare case of flecainide poisoning: a case report. Cases J. 2009;2:9137. doi:10.1186/1757-1626-2-9137.
12. Nabar A, Rodriguez LM, Timmermans C, Smeets JL, Wellens HJ. Radiofrequency ablation of “class IC atrial flutter” in patients with resistant atrial fibrillation. Am J Cardiol. 1999;83(5):785-787, A10.
13. Kola S, Mahata I, Kocheril AG. A case of flecainide toxicity. EP Lab Digest. 2015;15(5).
14. Falk RH. Proarrhythmia in patients treated for atrial fibrillation or flutter. Ann Intern Med. 1992;117(2):141-150.
15. Lloyd T, Zimmerman J, Griffin GD. Irreversible third-degree heart block and pacemaker implant in a case of flecainide toxicity. Am J Emerg Med. 2013;31(9):1418.e1-e2. doi:10.1016/j.ajem.2013.04.025.
16. Corkeron MA, van Heerden PV, Newman SM, Dusci L. Extracorporeal circulatory support in near-fatal flecainide overdose. Anaesth Intensive Care. 1999;27(4):405-408.
17. Ellsworth H, Stellpflug SJ, Cole JB, Dolan JA, Harris CR. A life-threatening flecainide overdose treated with intravenous fat emulsion. Pacing Clin Electrophysiol. 2013;36(3):e87-e89. doi:10.1111/j.1540-8159.2012.03485.x.
Rapid Development of Life-Threatening Emamectin Benzoate Poisoning
Emamectin benzoate (EB) is a semisynthetic derivative of avermectin that has acaricidal, nematicidal, and insecticidal action. Avermectin analogs are natural products from soil fungi (Streptomyces avermitilis).1 Emamectin benzoate was initially developed to eradicate lepidopteran larvae, particularly armyworms, and is registered in the United States and Japan for use on vegetable crops.2-4 In addition to its agricultural use, EB also has antiparasitic effects on sea lice (Lepeophtheirus salmonis) that affect Atlantic salmon, and has been registered for use in several countries since 1999.5-7 Although a few studies have evaluated the toxic effects of avermectin on humans, there is a paucity of information regarding human toxicity associated with EB.7 This case report describes rapid deterioration of a patient following ingestion of EB.
Case
A 75-year-old man presented to the ED 20 minutes after intentionally ingesting an agricultural insecticide. Upon presentation, the patient stated that he drank a whole bottle (100 mL) of insecticide after consuming alcohol, but denied coingestion of other toxic substances or any medications. The patient provided the empty bottle upon presentation, and the ingested product was identified as Affirm, an insecticide containing 2.15% EB as the active ingredient.
The patient’s medical history was significant for major depressive disorder, for which he was on alprazolam, donepezil, paroxetine, and quetiapine. The patient stated that he also suffered from chronic back pain, noting that he only took analgesics intermittently as needed.
On examination, the patient was alert and oriented to time and place. Initially, he did not experience any physical discomfort. His vital signs were: blood pressure (BP), 126/74 mm Hg; pulse rate, 67 beats/minute; respiratory rate, mildly tachypneic at 23 breaths/minute; and temperature, 97.9°F. Oxygen saturation was 96% on room air.
Ocular examination revealed both pupils to be equally round, 3 mm in diameter, and reactive to light. Examination of the oropharynx was normal and without signs of mucosal injury. The lung sounds were clear bilaterally, and the heart was a regular rate and rhythm and without murmur. The patient’s abdomen was soft and nontender. No deficits, such as ataxia, dysarthria, or tremor were found on the neurological examination.
Prompt gastric lavage via a nasogastric tube was performed, and activated charcoal was administered. Laboratory evaluation was significant for the following: white blood cell count, 22.77 x 109/L with 78% neutrophils and 16% lymphocytes; sodium, 138 mEq/L; potassium, 3.1 mEq/L; chloride, 109 mEq/L; blood urea nitrogen, 19 mg/dL; and creatinine, 0.7 mg/dL. Arterial blood gas (ABG) results revealed a pH, 7.37; partial pressure of carbon dioxide, 25 mm Hg; partial pressure of oxygen, 93 mm Hg; bicarbonate, 14.5 mEq/L; base excess, –8.9 mEq/L; and an oxygen saturation, 97%. Serum creatine kinase (CK), CK-MB and troponin levels were both within normal range. Lactic acid, serum osmolality, and serum ethanol levels were not obtained. The patient’s electrocardiogram (ECG) and chest radiograph findings were normal.
Approximately 1 hour after presentation, the patient complained of an epigastric burning sensation and continued to exhibit mild tachypnea. A subsequent ABG test revealed progressive metabolic acidosis (Table). Although the patient was given a total of 800 mL of normal saline intravenously (IV) upon arrival at the ED, his total urinary output was less than 100 mL 7 hours afterward. Attempts to increase urinary output with IV furosemide were ineffective.
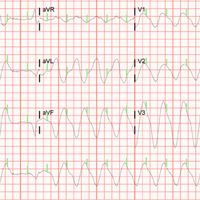
Along with the progressive metabolic acidosis, the patient became hypotensive, and did not respond to IV fluid resuscitation. A norepinephrine infusion was started to improve BP, but this was likewise ineffective. Serial ECGs did not show any specific abnormalities such as dysrhythmia or ischemia.
The patient was admitted to the intensive care unit approximately 10.5 hours after presentation where he received continuous renal replacement therapy (CRRT) to correct the severe metabolic acidosis and poor circulation. Metabolic acidosis persisted despite CRRT, and the patient remained hypotensive even after receiving high-dose IV norepinephrine (Figure).
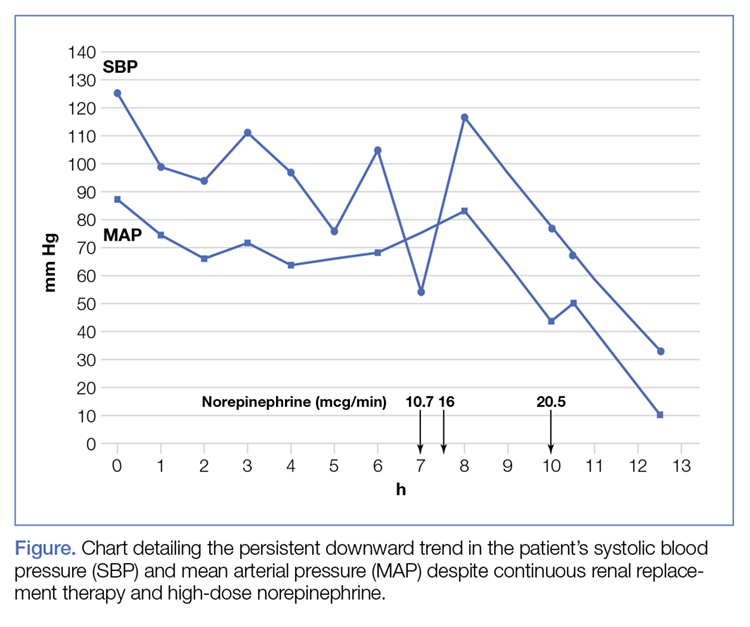
About 12.5 hours after his presentation to the ED, the patient began to vomit profusely and went into cardiac arrest. The cardiac monitor demonstrated pulseless ventricular tachycardia. Aggressive resuscitative efforts were initiated, but failed to restore spontaneous circulation.
Discussion
As an avermectin analog, EB interacts with γ-aminobutyric acid (GABA) receptors and enhances membrane chloride permeability.8 In mammals, GABA-containing neurons and receptors are found in the central nervous system (CNS), but not in the peripheral nervous system. In cases of high-dose avermectin ingestion in humans, CNS toxicity, including agitation and depressed mental status, have been reported, as well as death resulting from respiratory failure.9
With respect to human EB toxicity, there is only one other documented case in the literature by Yen and Lin.7 In their case, the authors report on a patient who ingested 100 mL of Proclaim, which contained 2.15% EB diluted with 400 mL of tap water.7 They note that the patient in their case presented with mild confusion and gastrointestinal (GI) symptoms of nausea, vomiting, and cramping discomfort. Following laboratory and radiological investigation, the patient was found to have aspiration pneumonitis and admitted to the inpatient hospital. On hospital day 2, the patient’s GI symptoms abated and he became alert and oriented. He was discharged 1 week from initial presentation and experienced no sequelae.
In our case, the patient ingested 100 mL of 2.15% EB without dilution. He also experienced GI symptoms, but did not have any CNS depression. The metabolic acidosis rapidly worsened, and could not be corrected, even with intensive therapy. This rapid life-threatening course has not previously been reported with avermectin or EB poisoning. In the avermectin poisoning cases in the literature, seven out of 19 patients (37%) exhibited severe effects, such as hypotension, coma, and aspiration with respiratory failure.9 Six of the seven patients experienced a full recovery; the remaining patient died 18 days after ingestion from multiple organ failure.
The reason for our patient’s rapid progression to metabolic acidosis and progressive deterioration (hypotension and hypoxemia) is not clear. One possible theory is that the solvents or other additives aside from EB in the ingested insecticide might make EB more toxic. In our case, the patient’s rapid deterioration alone or asphyxia by vomitus might have been the cause of the cardiac arrest. Future reports and studies about EB toxicity in humans are warranted to investigate the pathogenesis of toxicity and appropriate treatment.
Conclusion
This is the first report of a human death caused by EB poisoning; the patient experienced severe metabolic acidosis without CNS depression, ultimately leading to death. Emergency physicians should be aware of the possibility of rapid deterioration in patients who present after ingestion of EB and related substances.
1. Lasota JA, Dybas RA. Avermectins, a novel class of compounds: implications for use in arthropod pest control. Annu Rev Entomol. 1991;36:91-117. doi:10.1146/annurev.en.36.010191.000515.
2. Kuo JN, Buday C, van Aggelen G, Ikonomou MG, Pasternak J. Acute toxicity of emamectin benzoate and its desmethyl metabolite to Eohaustorius estuarius. Environ Toxicol Chem. 2010;29(8):1816-1820. doi:10.1002/etc.209.
3. Takai K, Soejima T, Suzuki T, et al. Development of a water-soluble preparation of emamectin benzoate and its preventative effect against the wilting of pot-grown pine trees inoculated with the pine wood nematode, Bursaphelenchus xylophilus. Pest Manag Sci. 2001;57(5):463-466. doi:10.1002/ps.30.
4. Chukwudebe AC, Beavers JB, Jaber M, Wislocki PG. Toxicity of emamectin benzoate to mallard duck and northern bobwhite quail. Environ Toxicol and Chem. 1998;17(6):1118-1123. doi:10.1002/etc.5620170619.
5. Armstrong R, MacPhee D, Katz T, Endris R. A field efficacy evaluation of emamectin benzoate for the control of sea lice on Atlantic salmon. Can Vet J. 2000;41(8):607-612.
6. Ramstad A, Colquhoun DJ, Nordmo R, Sutherland IH, Simmons R. Field trials in Norway with SLICE (0.2% emamectin benzoate) for the oral treatment of sea lice infestation in farmed Atlantic salmon Salmo salar. Dis Aquat Organ. 2002;21;50(1):29-33. doi:10.3354/dao050029.
7. Yen TH, Lin JL. Acute poisoning with emamectin benzoate. J Toxicol Clin Toxicol. 2004;42(5):657-661.
8. Campbell WC, Fisher MH, Stapley EO, Albers-Schönberg G, Jacob TA. Ivermectin: a potent new antiparasitic agent. Science. 1983; 221(4613):823-838.
9. Chung K, Yang CC, Wu ML, Deng JF, Tsai WJ. Agricultural avermectins: an uncommon but potentially fatal cause of pesticide poisoning. Ann Emerg Med. 1999;34(1):51-57.
Emamectin benzoate (EB) is a semisynthetic derivative of avermectin that has acaricidal, nematicidal, and insecticidal action. Avermectin analogs are natural products from soil fungi (Streptomyces avermitilis).1 Emamectin benzoate was initially developed to eradicate lepidopteran larvae, particularly armyworms, and is registered in the United States and Japan for use on vegetable crops.2-4 In addition to its agricultural use, EB also has antiparasitic effects on sea lice (Lepeophtheirus salmonis) that affect Atlantic salmon, and has been registered for use in several countries since 1999.5-7 Although a few studies have evaluated the toxic effects of avermectin on humans, there is a paucity of information regarding human toxicity associated with EB.7 This case report describes rapid deterioration of a patient following ingestion of EB.
Case
A 75-year-old man presented to the ED 20 minutes after intentionally ingesting an agricultural insecticide. Upon presentation, the patient stated that he drank a whole bottle (100 mL) of insecticide after consuming alcohol, but denied coingestion of other toxic substances or any medications. The patient provided the empty bottle upon presentation, and the ingested product was identified as Affirm, an insecticide containing 2.15% EB as the active ingredient.
The patient’s medical history was significant for major depressive disorder, for which he was on alprazolam, donepezil, paroxetine, and quetiapine. The patient stated that he also suffered from chronic back pain, noting that he only took analgesics intermittently as needed.
On examination, the patient was alert and oriented to time and place. Initially, he did not experience any physical discomfort. His vital signs were: blood pressure (BP), 126/74 mm Hg; pulse rate, 67 beats/minute; respiratory rate, mildly tachypneic at 23 breaths/minute; and temperature, 97.9°F. Oxygen saturation was 96% on room air.
Ocular examination revealed both pupils to be equally round, 3 mm in diameter, and reactive to light. Examination of the oropharynx was normal and without signs of mucosal injury. The lung sounds were clear bilaterally, and the heart was a regular rate and rhythm and without murmur. The patient’s abdomen was soft and nontender. No deficits, such as ataxia, dysarthria, or tremor were found on the neurological examination.
Prompt gastric lavage via a nasogastric tube was performed, and activated charcoal was administered. Laboratory evaluation was significant for the following: white blood cell count, 22.77 x 109/L with 78% neutrophils and 16% lymphocytes; sodium, 138 mEq/L; potassium, 3.1 mEq/L; chloride, 109 mEq/L; blood urea nitrogen, 19 mg/dL; and creatinine, 0.7 mg/dL. Arterial blood gas (ABG) results revealed a pH, 7.37; partial pressure of carbon dioxide, 25 mm Hg; partial pressure of oxygen, 93 mm Hg; bicarbonate, 14.5 mEq/L; base excess, –8.9 mEq/L; and an oxygen saturation, 97%. Serum creatine kinase (CK), CK-MB and troponin levels were both within normal range. Lactic acid, serum osmolality, and serum ethanol levels were not obtained. The patient’s electrocardiogram (ECG) and chest radiograph findings were normal.
Approximately 1 hour after presentation, the patient complained of an epigastric burning sensation and continued to exhibit mild tachypnea. A subsequent ABG test revealed progressive metabolic acidosis (Table). Although the patient was given a total of 800 mL of normal saline intravenously (IV) upon arrival at the ED, his total urinary output was less than 100 mL 7 hours afterward. Attempts to increase urinary output with IV furosemide were ineffective.
Along with the progressive metabolic acidosis, the patient became hypotensive, and did not respond to IV fluid resuscitation. A norepinephrine infusion was started to improve BP, but this was likewise ineffective. Serial ECGs did not show any specific abnormalities such as dysrhythmia or ischemia.
The patient was admitted to the intensive care unit approximately 10.5 hours after presentation where he received continuous renal replacement therapy (CRRT) to correct the severe metabolic acidosis and poor circulation. Metabolic acidosis persisted despite CRRT, and the patient remained hypotensive even after receiving high-dose IV norepinephrine (Figure).
About 12.5 hours after his presentation to the ED, the patient began to vomit profusely and went into cardiac arrest. The cardiac monitor demonstrated pulseless ventricular tachycardia. Aggressive resuscitative efforts were initiated, but failed to restore spontaneous circulation.
Discussion
As an avermectin analog, EB interacts with γ-aminobutyric acid (GABA) receptors and enhances membrane chloride permeability.8 In mammals, GABA-containing neurons and receptors are found in the central nervous system (CNS), but not in the peripheral nervous system. In cases of high-dose avermectin ingestion in humans, CNS toxicity, including agitation and depressed mental status, have been reported, as well as death resulting from respiratory failure.9
With respect to human EB toxicity, there is only one other documented case in the literature by Yen and Lin.7 In their case, the authors report on a patient who ingested 100 mL of Proclaim, which contained 2.15% EB diluted with 400 mL of tap water.7 They note that the patient in their case presented with mild confusion and gastrointestinal (GI) symptoms of nausea, vomiting, and cramping discomfort. Following laboratory and radiological investigation, the patient was found to have aspiration pneumonitis and admitted to the inpatient hospital. On hospital day 2, the patient’s GI symptoms abated and he became alert and oriented. He was discharged 1 week from initial presentation and experienced no sequelae.
In our case, the patient ingested 100 mL of 2.15% EB without dilution. He also experienced GI symptoms, but did not have any CNS depression. The metabolic acidosis rapidly worsened, and could not be corrected, even with intensive therapy. This rapid life-threatening course has not previously been reported with avermectin or EB poisoning. In the avermectin poisoning cases in the literature, seven out of 19 patients (37%) exhibited severe effects, such as hypotension, coma, and aspiration with respiratory failure.9 Six of the seven patients experienced a full recovery; the remaining patient died 18 days after ingestion from multiple organ failure.
The reason for our patient’s rapid progression to metabolic acidosis and progressive deterioration (hypotension and hypoxemia) is not clear. One possible theory is that the solvents or other additives aside from EB in the ingested insecticide might make EB more toxic. In our case, the patient’s rapid deterioration alone or asphyxia by vomitus might have been the cause of the cardiac arrest. Future reports and studies about EB toxicity in humans are warranted to investigate the pathogenesis of toxicity and appropriate treatment.
Conclusion
This is the first report of a human death caused by EB poisoning; the patient experienced severe metabolic acidosis without CNS depression, ultimately leading to death. Emergency physicians should be aware of the possibility of rapid deterioration in patients who present after ingestion of EB and related substances.
Emamectin benzoate (EB) is a semisynthetic derivative of avermectin that has acaricidal, nematicidal, and insecticidal action. Avermectin analogs are natural products from soil fungi (Streptomyces avermitilis).1 Emamectin benzoate was initially developed to eradicate lepidopteran larvae, particularly armyworms, and is registered in the United States and Japan for use on vegetable crops.2-4 In addition to its agricultural use, EB also has antiparasitic effects on sea lice (Lepeophtheirus salmonis) that affect Atlantic salmon, and has been registered for use in several countries since 1999.5-7 Although a few studies have evaluated the toxic effects of avermectin on humans, there is a paucity of information regarding human toxicity associated with EB.7 This case report describes rapid deterioration of a patient following ingestion of EB.
Case
A 75-year-old man presented to the ED 20 minutes after intentionally ingesting an agricultural insecticide. Upon presentation, the patient stated that he drank a whole bottle (100 mL) of insecticide after consuming alcohol, but denied coingestion of other toxic substances or any medications. The patient provided the empty bottle upon presentation, and the ingested product was identified as Affirm, an insecticide containing 2.15% EB as the active ingredient.
The patient’s medical history was significant for major depressive disorder, for which he was on alprazolam, donepezil, paroxetine, and quetiapine. The patient stated that he also suffered from chronic back pain, noting that he only took analgesics intermittently as needed.
On examination, the patient was alert and oriented to time and place. Initially, he did not experience any physical discomfort. His vital signs were: blood pressure (BP), 126/74 mm Hg; pulse rate, 67 beats/minute; respiratory rate, mildly tachypneic at 23 breaths/minute; and temperature, 97.9°F. Oxygen saturation was 96% on room air.
Ocular examination revealed both pupils to be equally round, 3 mm in diameter, and reactive to light. Examination of the oropharynx was normal and without signs of mucosal injury. The lung sounds were clear bilaterally, and the heart was a regular rate and rhythm and without murmur. The patient’s abdomen was soft and nontender. No deficits, such as ataxia, dysarthria, or tremor were found on the neurological examination.
Prompt gastric lavage via a nasogastric tube was performed, and activated charcoal was administered. Laboratory evaluation was significant for the following: white blood cell count, 22.77 x 109/L with 78% neutrophils and 16% lymphocytes; sodium, 138 mEq/L; potassium, 3.1 mEq/L; chloride, 109 mEq/L; blood urea nitrogen, 19 mg/dL; and creatinine, 0.7 mg/dL. Arterial blood gas (ABG) results revealed a pH, 7.37; partial pressure of carbon dioxide, 25 mm Hg; partial pressure of oxygen, 93 mm Hg; bicarbonate, 14.5 mEq/L; base excess, –8.9 mEq/L; and an oxygen saturation, 97%. Serum creatine kinase (CK), CK-MB and troponin levels were both within normal range. Lactic acid, serum osmolality, and serum ethanol levels were not obtained. The patient’s electrocardiogram (ECG) and chest radiograph findings were normal.
Approximately 1 hour after presentation, the patient complained of an epigastric burning sensation and continued to exhibit mild tachypnea. A subsequent ABG test revealed progressive metabolic acidosis (Table). Although the patient was given a total of 800 mL of normal saline intravenously (IV) upon arrival at the ED, his total urinary output was less than 100 mL 7 hours afterward. Attempts to increase urinary output with IV furosemide were ineffective.
Along with the progressive metabolic acidosis, the patient became hypotensive, and did not respond to IV fluid resuscitation. A norepinephrine infusion was started to improve BP, but this was likewise ineffective. Serial ECGs did not show any specific abnormalities such as dysrhythmia or ischemia.
The patient was admitted to the intensive care unit approximately 10.5 hours after presentation where he received continuous renal replacement therapy (CRRT) to correct the severe metabolic acidosis and poor circulation. Metabolic acidosis persisted despite CRRT, and the patient remained hypotensive even after receiving high-dose IV norepinephrine (Figure).
About 12.5 hours after his presentation to the ED, the patient began to vomit profusely and went into cardiac arrest. The cardiac monitor demonstrated pulseless ventricular tachycardia. Aggressive resuscitative efforts were initiated, but failed to restore spontaneous circulation.
Discussion
As an avermectin analog, EB interacts with γ-aminobutyric acid (GABA) receptors and enhances membrane chloride permeability.8 In mammals, GABA-containing neurons and receptors are found in the central nervous system (CNS), but not in the peripheral nervous system. In cases of high-dose avermectin ingestion in humans, CNS toxicity, including agitation and depressed mental status, have been reported, as well as death resulting from respiratory failure.9
With respect to human EB toxicity, there is only one other documented case in the literature by Yen and Lin.7 In their case, the authors report on a patient who ingested 100 mL of Proclaim, which contained 2.15% EB diluted with 400 mL of tap water.7 They note that the patient in their case presented with mild confusion and gastrointestinal (GI) symptoms of nausea, vomiting, and cramping discomfort. Following laboratory and radiological investigation, the patient was found to have aspiration pneumonitis and admitted to the inpatient hospital. On hospital day 2, the patient’s GI symptoms abated and he became alert and oriented. He was discharged 1 week from initial presentation and experienced no sequelae.
In our case, the patient ingested 100 mL of 2.15% EB without dilution. He also experienced GI symptoms, but did not have any CNS depression. The metabolic acidosis rapidly worsened, and could not be corrected, even with intensive therapy. This rapid life-threatening course has not previously been reported with avermectin or EB poisoning. In the avermectin poisoning cases in the literature, seven out of 19 patients (37%) exhibited severe effects, such as hypotension, coma, and aspiration with respiratory failure.9 Six of the seven patients experienced a full recovery; the remaining patient died 18 days after ingestion from multiple organ failure.
The reason for our patient’s rapid progression to metabolic acidosis and progressive deterioration (hypotension and hypoxemia) is not clear. One possible theory is that the solvents or other additives aside from EB in the ingested insecticide might make EB more toxic. In our case, the patient’s rapid deterioration alone or asphyxia by vomitus might have been the cause of the cardiac arrest. Future reports and studies about EB toxicity in humans are warranted to investigate the pathogenesis of toxicity and appropriate treatment.
Conclusion
This is the first report of a human death caused by EB poisoning; the patient experienced severe metabolic acidosis without CNS depression, ultimately leading to death. Emergency physicians should be aware of the possibility of rapid deterioration in patients who present after ingestion of EB and related substances.
1. Lasota JA, Dybas RA. Avermectins, a novel class of compounds: implications for use in arthropod pest control. Annu Rev Entomol. 1991;36:91-117. doi:10.1146/annurev.en.36.010191.000515.
2. Kuo JN, Buday C, van Aggelen G, Ikonomou MG, Pasternak J. Acute toxicity of emamectin benzoate and its desmethyl metabolite to Eohaustorius estuarius. Environ Toxicol Chem. 2010;29(8):1816-1820. doi:10.1002/etc.209.
3. Takai K, Soejima T, Suzuki T, et al. Development of a water-soluble preparation of emamectin benzoate and its preventative effect against the wilting of pot-grown pine trees inoculated with the pine wood nematode, Bursaphelenchus xylophilus. Pest Manag Sci. 2001;57(5):463-466. doi:10.1002/ps.30.
4. Chukwudebe AC, Beavers JB, Jaber M, Wislocki PG. Toxicity of emamectin benzoate to mallard duck and northern bobwhite quail. Environ Toxicol and Chem. 1998;17(6):1118-1123. doi:10.1002/etc.5620170619.
5. Armstrong R, MacPhee D, Katz T, Endris R. A field efficacy evaluation of emamectin benzoate for the control of sea lice on Atlantic salmon. Can Vet J. 2000;41(8):607-612.
6. Ramstad A, Colquhoun DJ, Nordmo R, Sutherland IH, Simmons R. Field trials in Norway with SLICE (0.2% emamectin benzoate) for the oral treatment of sea lice infestation in farmed Atlantic salmon Salmo salar. Dis Aquat Organ. 2002;21;50(1):29-33. doi:10.3354/dao050029.
7. Yen TH, Lin JL. Acute poisoning with emamectin benzoate. J Toxicol Clin Toxicol. 2004;42(5):657-661.
8. Campbell WC, Fisher MH, Stapley EO, Albers-Schönberg G, Jacob TA. Ivermectin: a potent new antiparasitic agent. Science. 1983; 221(4613):823-838.
9. Chung K, Yang CC, Wu ML, Deng JF, Tsai WJ. Agricultural avermectins: an uncommon but potentially fatal cause of pesticide poisoning. Ann Emerg Med. 1999;34(1):51-57.
1. Lasota JA, Dybas RA. Avermectins, a novel class of compounds: implications for use in arthropod pest control. Annu Rev Entomol. 1991;36:91-117. doi:10.1146/annurev.en.36.010191.000515.
2. Kuo JN, Buday C, van Aggelen G, Ikonomou MG, Pasternak J. Acute toxicity of emamectin benzoate and its desmethyl metabolite to Eohaustorius estuarius. Environ Toxicol Chem. 2010;29(8):1816-1820. doi:10.1002/etc.209.
3. Takai K, Soejima T, Suzuki T, et al. Development of a water-soluble preparation of emamectin benzoate and its preventative effect against the wilting of pot-grown pine trees inoculated with the pine wood nematode, Bursaphelenchus xylophilus. Pest Manag Sci. 2001;57(5):463-466. doi:10.1002/ps.30.
4. Chukwudebe AC, Beavers JB, Jaber M, Wislocki PG. Toxicity of emamectin benzoate to mallard duck and northern bobwhite quail. Environ Toxicol and Chem. 1998;17(6):1118-1123. doi:10.1002/etc.5620170619.
5. Armstrong R, MacPhee D, Katz T, Endris R. A field efficacy evaluation of emamectin benzoate for the control of sea lice on Atlantic salmon. Can Vet J. 2000;41(8):607-612.
6. Ramstad A, Colquhoun DJ, Nordmo R, Sutherland IH, Simmons R. Field trials in Norway with SLICE (0.2% emamectin benzoate) for the oral treatment of sea lice infestation in farmed Atlantic salmon Salmo salar. Dis Aquat Organ. 2002;21;50(1):29-33. doi:10.3354/dao050029.
7. Yen TH, Lin JL. Acute poisoning with emamectin benzoate. J Toxicol Clin Toxicol. 2004;42(5):657-661.
8. Campbell WC, Fisher MH, Stapley EO, Albers-Schönberg G, Jacob TA. Ivermectin: a potent new antiparasitic agent. Science. 1983; 221(4613):823-838.
9. Chung K, Yang CC, Wu ML, Deng JF, Tsai WJ. Agricultural avermectins: an uncommon but potentially fatal cause of pesticide poisoning. Ann Emerg Med. 1999;34(1):51-57.
The Use of Bolus-Dose Vasopressors in the Emergency Department
The use of bolus-dose vasopressors in anesthesiology and other areas of critical care medicine is well known. This common medical intervention, however, is not often employed in emergency medicine (EM). Bolus-dose vasopressors are defined as the administration of small bolus doses of vasopressor agents, such as epinephrine or phenylephrine, to patients with compromised perfusion who continue to have a pulse (ie, these patients are not in cardiac arrest). This intervention is considered as a temporizing measure for transient hypotension or as a bridge to more definitive therapy.
Clinical Application
Bolus-dose vasopressive therapy is also referred to as push-dose pressor (PDP) therapy—a term coined by Weingart.1-3 Theoretically, any vasopressor could be used in a mini-dose, bolus fashion, though in current clinical practice, anesthesiologists primarily employ ephedrine, epinephrine, and phenylephrine. Two of these agents are likely more appropriate for the ED, including epinephrine and phenylephrine. Both of these agents have a short half-life and therefore an abbreviated period of effect. In addition, dosing and related administration of epinephrine and phenylephrine is relatively straightforward. Moreover, most emergency physicians and nurses are quite familiar with both agents.
With respect to ephedrine, due to its longer half-life, complex dosing regimen, and associated higher-incidence of cardiovascular (CV) complications, its use is likely not appropriate in the ED as a bolus-dose vasopressor.
Epinephrine and Phenylephrine
Epinephrine is a potent sympathomimetic agent with alpha- and beta-receptor activity. In addition to its vasopressor effects, epinephrine is also an inotropic and chronotropic agent, increasing cardiac output, heart rate (HR), and systemic vascular resistance, which can markedly improve perfusion. Epinephrine also can be given to patients with hypoperfusion and/or shock due to low-cardiac output with or without vasodilation, lacking significant tachycardia.
Phenylephrine is a pure alpha agonist and therefore does not appreciably affect cardiac output and HR, but does significantly increase systemic vascular resistance and thus systemic perfusion. Phenylephrine can be used to treat patients with hypoperfusion and/or shock states due to vasodilation with coexistent, significant tachycardia.
Preparation and Administration
The preparation and dosing of push-dose epinephrine and phenylephrine are not particularly complex. Many clinicians recommend the pre-mixed, manufacturer-prepared agents for PDP therapy. These premixed formulations not only facilitate administration, but also reduce the chance of a preparation error that can result in incorrect dosing.3-5 If pre-mixed formulations are not available, clinicians can readily prepare epinephrine and phenylephrine for PDP use.
Push-Dose Epinephrine. Clinicians can prepare epinephrine for push-dose administration as follows:1-3
- Obtain 1 mL of epinephrine 1:10,000 (ie, 0.1 mg/mL or 100 mcg/mL);
- Obtain a 10 mL syringe of normal saline and remove 1 mL;
- Inject the 1 mL of epinephrine 1:10,000 (100 mcg/mL) into this syringe containing 9 mL of normal saline; and
- Result: 10 mL of epinephrine (10 mcg/mL), with each 1 mL of this solution containing 10 mcg of epinephrine.
Administration of push-dose epinephrine (10 mcg/mL) produces effect within 1 minute of use with a duration of approximately 5 to 10 minutes. Dosing at this concentration ranges from 0.5 to 2.0 mL every 2 to 5 minutes, delivering 5 to 20 mcg.1-3Push-Dose Phenylephrine. To prepare phenylephrine for push-dose administration, clinicians may use the following approach:1-3
- Obtain 1 mL of phenylephrine (10 mg/mL concentration);
- Inject this 1 mL of phenylephrine (10 mg/mL) into a 100 mL bag of normal saline; and
- Result: 100 mL of phenylephrine (100 mcg/mL), with each 1 mL of this solution containing 100 mcg of phenylephrine.
Administration of push-dose phenylephrine (100 mcg/mL) produces effect within 1 minute of use with a duration of approximately 10 to 20 minutes. Dosing at this concentration ranges from 0.5 to 2.0 mL every 2 to 5 minutes, delivering 50 to 200 mcg.1-3Alternative Push-Dose Preparations for Phenylephrine. Two other methods of preparing phenylephrine for bolus-dose administration include the following: (1) the addition of phenylephrine 20 mg to a bag of 250 cc of normal saline, resulting in an 80 mcg/mL concentration; and/or (2) phenylephrine (20 mg) is commercially available for continuous infusion in a 250 mL bag of normal saline, yielding the same concentration of 80 mcg/mL; in either case, medication can be drawn up and administered. Dosing at this concentration ranges from 0.5 to 2.5 mL every 2 to 5 minutes, delivering 40 to 200 mcg. Lastly, phenylephrine is also commercially available in pre-made mixtures, specifically manufactured for bolus-dose therapy.
Indications
Both epinephrine and phenylephrine can be considered in the management of significant transient or sustained hypoperfusion. Although the definition of significant hypotension is complex, Brunauer et al6 have suggested that a mean arterial pressure (MAP) of approximately 35 mm Hg is associated with a significant risk of CV collapse. Of course, a MAP of 40 to 50 mm Hg is also very concerning clinically, with significant risk of deterioration and CV collapse.
Procedural events, such as conscious sedation or rapid sequence intubation (RSI), can produce significant hypotension; PDP can rapidly correct hypotension. In other clinical scenarios in which sustained hypotension is likely and not transient (eg, sepsis with shock), PDP can be used as a bridge to definitive care (eg, volume replacement, continuous vasopressor infusion). It is important to note, however, that PDP administration must occur in conjunction with or after the patient has received other appropriate therapies such as a normal saline bolus and continuous vasopressor infusions. Push-dose pressors are not a replacement for these proven interventions, but rather are an important augmentation to these therapies.
Emergency Medicine Literature
As previously noted, the literature base describing and supporting the clinical use of PDP in EM is extremely limited. The few articles that comprise this literature base address significant hypotension in periendotracheal intubation intervention, post-return of spontaneous circulation (ROSC) management, and shock management with preload augmentation.7-9In addition, there are several articles in the literature that address safety concerns surrounding the use of PDP in the ED.4,5
Panchal et al10 investigated the use of phenylephrine in hypotensive patients undergoing RSI-assisted endotracheal intubation. The authors performed a 1-year retrospective review of hypotensive patients managed with endotracheal intubation for a range of clinical conditions that required clinical care intervention. In this study, 20 of the 119 patients received phenylephrine in the peri-intubation period. A range of clinical conditions requiring critical care intervention were encountered; in addition, almost three-quarters of these patients were receiving at least one other vasopressor infusion. Further differences were seen in the timing of PDP administration. In those patients receiving bolus-dose phenylephrine, blood pressure (BP) improved without change in HR. Panchal et al10 concluded that while push-dose phenylephrine improved hemodynamic status, there was significant variation among clinicians regarding dosing, timing of use, and overall clinical situation The significant variation in PDP management in this study was noted to be a potential source of medical error, thus increasing the chance of adverse clinical event.
Push-dose pressor therapy can be employed for significant hypotension while more definitive therapy is being readied and applied. For instance, patients with significant hypotension requiring continuous vasopressor infusion can be managed with PDP while appropriate venous access is established, intravenous fluids are administered, and medications are prepared. The immediate period after resuscitation from cardiac arrest can be complicated by shock of many types. In fact, hypotension following ROSC in the cardiac arrest patient is not uncommon and has been identified as a risk issue associated with poor outcome. Prompt treatment of this altered perfusion may improve outcome. Gottlieb8 described three patients with ROSC after cardiac arrest. All three patients experienced significant, sustained hypotension with systolic blood pressure reading in the 50 to 60 mm Hg range; bolus-dose epinephrine was administered with significant improvement in the hemodynamic status while central venous access was established.
In a related clinical scenario, Schwartz et al9 considered the impact of PDP on central venous line (CVL) placement with continuous vasopressor infusion. In this ED study, although patients experienced an increase in BP, this impact was transient with approximately half of these individuals ultimately requiring CVL. In addition, serious adverse effect was noted more commonly in the phenylephrine-treated patients with “reactive” hypertension and ventricular tachycardia occurring in study patients.
Patient-Safety Considerations
In addition to the limited literature base supporting PDP use in the ED, another major significant issue focuses on safety concerns and adverse effects. Extremely limited data is available describing adverse events related to ED-administered PDP. Extrapolating from other EM and critical care administrations of peripheral epinephrine, both local and systemic adverse effects have been reported.11,12 The range of adverse events noted in these studies are considerable, including local skin and soft-tissue injury (necrosis), end-organ tissue ischemia (eg, digits, tip of nose), acute hypertension, cardiac ischemic events, and left ventricular (LV) dysfunction.11,12
When comparing peripheral infusion with central infusion, the risk of extravasation with resultant local tissue injury is markedly greater with peripheral vasopressor administration. In a systematic review of this issue, Loubani and Green11 noted that such local adverse events were much more commonly associated with peripheral administration.
In another report of vasopressor use in the ED, Kanwar et al12 described apparent confusion with epinephrine dosing and route of administration, resulting in very significant, systemic CV maladies, including severe elevations in BP, acute LV dysfunction, and chest pain associated with ST segment elevation.
It must be stressed that the publications by Loubani and Green11 and Kanwar et al12 described peripheral vasopressor administration: neither study included PDP therapy. Therefore, as previously noted, the aforementioned statements are extrapolated from when applied to PDP strategy.
Acquisto et al4 describe several errors in medication administration of PDP in the ED and other critical care areas of the hospital. In this report, all treating physicians were present at the patients’ bedside, either administering the medication or directly supervising its use. Agents involved included epinephrine and phenylephrine, delivered at exceedingly high doses. In their study, the authors noted several issues which they believe contributed to medication errors, including heterogeneity of pathology treated in these patients, apparent “earlier-than-appropriate” use of vasopressors (ie, prior to giving an appropriate fluid bolus), and medication preparation at the bedside by clinicians who may not possess the experience and training to mix these agents.
From a patient-safety perspective, Holden et al5 noted the potential for dosing error with significant adverse medical consequence related to PDP, as well as several contributing issues. First, they highlight the lack of a solid literature base to support administration of PDP in the ED and the development of decision-making guidelines for use in the ED. They also observed an inconsistency in approach to patient selection, medication choice, agent preparation, dosing, and other therapies. As seen in the Acquisto et al4 report, the patient-care scenarios are high risk and quite dynamic.
Conclusion
Bolus-dose vasopressor therapy is a potentially very useful treatment in the ED and other emergency/critical care settings. However, despite its benefits in treating patients in shock or with hypoperfusion, PDP is not widely used in EM due to the lack of studies, reviews, and guidelines in the literature to support its use in the ED. Such a literature base is required to provide an appropriate, safe means of patient selection, medication choice, dosing, and administration. Continued educational and research efforts are needed to more fully explore the use of PDP therapy in the ED.
When used correctly and appropriately, PDP has promise to be an important aid in the management of shock in the ED. Although bolus-dose therapy is appropriate for select clinical scenarios involving significant shock states which have the potential for progression to complete CV collapse without timely therapy, it is an adjunct to, not a replacement for commonly employed and medically indicated therapies such as crystalloid bolus or continuous vasopressor infusions.
1. Weingart S. EMCrit podcast 6—push-dose pressors. EMCrit RACC Web site. July 2009. https://emcrit.org/racc/bolus-dose-pressors. Accessed March 12, 2018.
2. Weingart S. EMCrit podcast 205—push-dose pressors update. EMCrit RACC Web site. August 2017. https://emcrit.org/racc/push-dose-pressor-update/. March 12, 2018.
3. Weingart S. Push-dose pressors for immediate blood pressure control. Clin Exp Emerg Med. 2015;2(2):131-132. doi:10.15441/ceem.15.010.
4. Acquisto NM, Bodkin RP, Johnstone C. Medication errors with push dose pressors in the emergency department and intensive care units. Am J Emerg Med. 2017;35(12):1964-1965. doi:10.1016/j.ajem.2017.06.013.
5. Holden D, Ramich J, Timm E, Pauze D, Lesar T. Safety considerations and guideline-based safe use recommendations for “bolus-dose” vasopressors in the emergency department. Ann Emerg Med. 2018;71(1):83-92. doi:10.1016/j.annemergmed.2017.04.021.
6. Brunauer A, Koköfer A, Bataar O, Gradwohl-Matis I, Dankl D, Dünser MW. The arterial blood pressure associated with terminal cardiovascular collapse in critically ill patients: a retrospective cohort study. Crit Care. 2014;18(6):719. doi:10.1186/s13054-014-0719-2.
7. Panchal AR, Satyanarayan A, Bahadir JD, Hays D, Mosier J. Efficacy of bolus-dose phenylephrine for peri-intubation hypotension. J Emerg Med. 2015;49(4):488-494. doi:10.1016/j.jemermed.2015.04.033.
8. Gottlieb M. Bolus dose of epinephrine for refractory post-arrest hypotension. Can J Emerg Med. 2017;409:1-5. doi:10.1017/cem.2016.409.
9. Schwartz MB, Ferreira JA, Aaronson PM. The impact of push-dose phenylephrine use on subsequent preload expansion in the ED setting. Am J Emerg Med. 2016;34(12):2419-2422. doi:10.1016/j.ajem.2016.09.041.
10. Panchal AR, Satyanarayan A, Bahadir JD, Hays D, Mosier J. Efficacy of bolus-dose phenylephrine for peri-intubation hypotension. J Emerg Med. 2015;49(4):488-494. doi:10.1016/j.jemermed.2015.04.033.
11. Loubani OM, Green RS. A systematic review of extravasation and local tissue injury from administration of vasopressors through peripheral intravenous catheters and central venous catheters. J Crit Care. 2015;30:653.e9-e17.
12. Kanwar M, Irvin CB, Frank JJ, et al. Confusion about epinephrine dosing leading to iatrogenic overdose: A life-threatening problem with a potential solution. Ann Emerg Med. 2010;55:341-344.
The use of bolus-dose vasopressors in anesthesiology and other areas of critical care medicine is well known. This common medical intervention, however, is not often employed in emergency medicine (EM). Bolus-dose vasopressors are defined as the administration of small bolus doses of vasopressor agents, such as epinephrine or phenylephrine, to patients with compromised perfusion who continue to have a pulse (ie, these patients are not in cardiac arrest). This intervention is considered as a temporizing measure for transient hypotension or as a bridge to more definitive therapy.
Clinical Application
Bolus-dose vasopressive therapy is also referred to as push-dose pressor (PDP) therapy—a term coined by Weingart.1-3 Theoretically, any vasopressor could be used in a mini-dose, bolus fashion, though in current clinical practice, anesthesiologists primarily employ ephedrine, epinephrine, and phenylephrine. Two of these agents are likely more appropriate for the ED, including epinephrine and phenylephrine. Both of these agents have a short half-life and therefore an abbreviated period of effect. In addition, dosing and related administration of epinephrine and phenylephrine is relatively straightforward. Moreover, most emergency physicians and nurses are quite familiar with both agents.
With respect to ephedrine, due to its longer half-life, complex dosing regimen, and associated higher-incidence of cardiovascular (CV) complications, its use is likely not appropriate in the ED as a bolus-dose vasopressor.
Epinephrine and Phenylephrine
Epinephrine is a potent sympathomimetic agent with alpha- and beta-receptor activity. In addition to its vasopressor effects, epinephrine is also an inotropic and chronotropic agent, increasing cardiac output, heart rate (HR), and systemic vascular resistance, which can markedly improve perfusion. Epinephrine also can be given to patients with hypoperfusion and/or shock due to low-cardiac output with or without vasodilation, lacking significant tachycardia.
Phenylephrine is a pure alpha agonist and therefore does not appreciably affect cardiac output and HR, but does significantly increase systemic vascular resistance and thus systemic perfusion. Phenylephrine can be used to treat patients with hypoperfusion and/or shock states due to vasodilation with coexistent, significant tachycardia.
Preparation and Administration
The preparation and dosing of push-dose epinephrine and phenylephrine are not particularly complex. Many clinicians recommend the pre-mixed, manufacturer-prepared agents for PDP therapy. These premixed formulations not only facilitate administration, but also reduce the chance of a preparation error that can result in incorrect dosing.3-5 If pre-mixed formulations are not available, clinicians can readily prepare epinephrine and phenylephrine for PDP use.
Push-Dose Epinephrine. Clinicians can prepare epinephrine for push-dose administration as follows:1-3
- Obtain 1 mL of epinephrine 1:10,000 (ie, 0.1 mg/mL or 100 mcg/mL);
- Obtain a 10 mL syringe of normal saline and remove 1 mL;
- Inject the 1 mL of epinephrine 1:10,000 (100 mcg/mL) into this syringe containing 9 mL of normal saline; and
- Result: 10 mL of epinephrine (10 mcg/mL), with each 1 mL of this solution containing 10 mcg of epinephrine.
Administration of push-dose epinephrine (10 mcg/mL) produces effect within 1 minute of use with a duration of approximately 5 to 10 minutes. Dosing at this concentration ranges from 0.5 to 2.0 mL every 2 to 5 minutes, delivering 5 to 20 mcg.1-3Push-Dose Phenylephrine. To prepare phenylephrine for push-dose administration, clinicians may use the following approach:1-3
- Obtain 1 mL of phenylephrine (10 mg/mL concentration);
- Inject this 1 mL of phenylephrine (10 mg/mL) into a 100 mL bag of normal saline; and
- Result: 100 mL of phenylephrine (100 mcg/mL), with each 1 mL of this solution containing 100 mcg of phenylephrine.
Administration of push-dose phenylephrine (100 mcg/mL) produces effect within 1 minute of use with a duration of approximately 10 to 20 minutes. Dosing at this concentration ranges from 0.5 to 2.0 mL every 2 to 5 minutes, delivering 50 to 200 mcg.1-3Alternative Push-Dose Preparations for Phenylephrine. Two other methods of preparing phenylephrine for bolus-dose administration include the following: (1) the addition of phenylephrine 20 mg to a bag of 250 cc of normal saline, resulting in an 80 mcg/mL concentration; and/or (2) phenylephrine (20 mg) is commercially available for continuous infusion in a 250 mL bag of normal saline, yielding the same concentration of 80 mcg/mL; in either case, medication can be drawn up and administered. Dosing at this concentration ranges from 0.5 to 2.5 mL every 2 to 5 minutes, delivering 40 to 200 mcg. Lastly, phenylephrine is also commercially available in pre-made mixtures, specifically manufactured for bolus-dose therapy.
Indications
Both epinephrine and phenylephrine can be considered in the management of significant transient or sustained hypoperfusion. Although the definition of significant hypotension is complex, Brunauer et al6 have suggested that a mean arterial pressure (MAP) of approximately 35 mm Hg is associated with a significant risk of CV collapse. Of course, a MAP of 40 to 50 mm Hg is also very concerning clinically, with significant risk of deterioration and CV collapse.
Procedural events, such as conscious sedation or rapid sequence intubation (RSI), can produce significant hypotension; PDP can rapidly correct hypotension. In other clinical scenarios in which sustained hypotension is likely and not transient (eg, sepsis with shock), PDP can be used as a bridge to definitive care (eg, volume replacement, continuous vasopressor infusion). It is important to note, however, that PDP administration must occur in conjunction with or after the patient has received other appropriate therapies such as a normal saline bolus and continuous vasopressor infusions. Push-dose pressors are not a replacement for these proven interventions, but rather are an important augmentation to these therapies.
Emergency Medicine Literature
As previously noted, the literature base describing and supporting the clinical use of PDP in EM is extremely limited. The few articles that comprise this literature base address significant hypotension in periendotracheal intubation intervention, post-return of spontaneous circulation (ROSC) management, and shock management with preload augmentation.7-9In addition, there are several articles in the literature that address safety concerns surrounding the use of PDP in the ED.4,5
Panchal et al10 investigated the use of phenylephrine in hypotensive patients undergoing RSI-assisted endotracheal intubation. The authors performed a 1-year retrospective review of hypotensive patients managed with endotracheal intubation for a range of clinical conditions that required clinical care intervention. In this study, 20 of the 119 patients received phenylephrine in the peri-intubation period. A range of clinical conditions requiring critical care intervention were encountered; in addition, almost three-quarters of these patients were receiving at least one other vasopressor infusion. Further differences were seen in the timing of PDP administration. In those patients receiving bolus-dose phenylephrine, blood pressure (BP) improved without change in HR. Panchal et al10 concluded that while push-dose phenylephrine improved hemodynamic status, there was significant variation among clinicians regarding dosing, timing of use, and overall clinical situation The significant variation in PDP management in this study was noted to be a potential source of medical error, thus increasing the chance of adverse clinical event.
Push-dose pressor therapy can be employed for significant hypotension while more definitive therapy is being readied and applied. For instance, patients with significant hypotension requiring continuous vasopressor infusion can be managed with PDP while appropriate venous access is established, intravenous fluids are administered, and medications are prepared. The immediate period after resuscitation from cardiac arrest can be complicated by shock of many types. In fact, hypotension following ROSC in the cardiac arrest patient is not uncommon and has been identified as a risk issue associated with poor outcome. Prompt treatment of this altered perfusion may improve outcome. Gottlieb8 described three patients with ROSC after cardiac arrest. All three patients experienced significant, sustained hypotension with systolic blood pressure reading in the 50 to 60 mm Hg range; bolus-dose epinephrine was administered with significant improvement in the hemodynamic status while central venous access was established.
In a related clinical scenario, Schwartz et al9 considered the impact of PDP on central venous line (CVL) placement with continuous vasopressor infusion. In this ED study, although patients experienced an increase in BP, this impact was transient with approximately half of these individuals ultimately requiring CVL. In addition, serious adverse effect was noted more commonly in the phenylephrine-treated patients with “reactive” hypertension and ventricular tachycardia occurring in study patients.
Patient-Safety Considerations
In addition to the limited literature base supporting PDP use in the ED, another major significant issue focuses on safety concerns and adverse effects. Extremely limited data is available describing adverse events related to ED-administered PDP. Extrapolating from other EM and critical care administrations of peripheral epinephrine, both local and systemic adverse effects have been reported.11,12 The range of adverse events noted in these studies are considerable, including local skin and soft-tissue injury (necrosis), end-organ tissue ischemia (eg, digits, tip of nose), acute hypertension, cardiac ischemic events, and left ventricular (LV) dysfunction.11,12
When comparing peripheral infusion with central infusion, the risk of extravasation with resultant local tissue injury is markedly greater with peripheral vasopressor administration. In a systematic review of this issue, Loubani and Green11 noted that such local adverse events were much more commonly associated with peripheral administration.
In another report of vasopressor use in the ED, Kanwar et al12 described apparent confusion with epinephrine dosing and route of administration, resulting in very significant, systemic CV maladies, including severe elevations in BP, acute LV dysfunction, and chest pain associated with ST segment elevation.
It must be stressed that the publications by Loubani and Green11 and Kanwar et al12 described peripheral vasopressor administration: neither study included PDP therapy. Therefore, as previously noted, the aforementioned statements are extrapolated from when applied to PDP strategy.
Acquisto et al4 describe several errors in medication administration of PDP in the ED and other critical care areas of the hospital. In this report, all treating physicians were present at the patients’ bedside, either administering the medication or directly supervising its use. Agents involved included epinephrine and phenylephrine, delivered at exceedingly high doses. In their study, the authors noted several issues which they believe contributed to medication errors, including heterogeneity of pathology treated in these patients, apparent “earlier-than-appropriate” use of vasopressors (ie, prior to giving an appropriate fluid bolus), and medication preparation at the bedside by clinicians who may not possess the experience and training to mix these agents.
From a patient-safety perspective, Holden et al5 noted the potential for dosing error with significant adverse medical consequence related to PDP, as well as several contributing issues. First, they highlight the lack of a solid literature base to support administration of PDP in the ED and the development of decision-making guidelines for use in the ED. They also observed an inconsistency in approach to patient selection, medication choice, agent preparation, dosing, and other therapies. As seen in the Acquisto et al4 report, the patient-care scenarios are high risk and quite dynamic.
Conclusion
Bolus-dose vasopressor therapy is a potentially very useful treatment in the ED and other emergency/critical care settings. However, despite its benefits in treating patients in shock or with hypoperfusion, PDP is not widely used in EM due to the lack of studies, reviews, and guidelines in the literature to support its use in the ED. Such a literature base is required to provide an appropriate, safe means of patient selection, medication choice, dosing, and administration. Continued educational and research efforts are needed to more fully explore the use of PDP therapy in the ED.
When used correctly and appropriately, PDP has promise to be an important aid in the management of shock in the ED. Although bolus-dose therapy is appropriate for select clinical scenarios involving significant shock states which have the potential for progression to complete CV collapse without timely therapy, it is an adjunct to, not a replacement for commonly employed and medically indicated therapies such as crystalloid bolus or continuous vasopressor infusions.
The use of bolus-dose vasopressors in anesthesiology and other areas of critical care medicine is well known. This common medical intervention, however, is not often employed in emergency medicine (EM). Bolus-dose vasopressors are defined as the administration of small bolus doses of vasopressor agents, such as epinephrine or phenylephrine, to patients with compromised perfusion who continue to have a pulse (ie, these patients are not in cardiac arrest). This intervention is considered as a temporizing measure for transient hypotension or as a bridge to more definitive therapy.
Clinical Application
Bolus-dose vasopressive therapy is also referred to as push-dose pressor (PDP) therapy—a term coined by Weingart.1-3 Theoretically, any vasopressor could be used in a mini-dose, bolus fashion, though in current clinical practice, anesthesiologists primarily employ ephedrine, epinephrine, and phenylephrine. Two of these agents are likely more appropriate for the ED, including epinephrine and phenylephrine. Both of these agents have a short half-life and therefore an abbreviated period of effect. In addition, dosing and related administration of epinephrine and phenylephrine is relatively straightforward. Moreover, most emergency physicians and nurses are quite familiar with both agents.
With respect to ephedrine, due to its longer half-life, complex dosing regimen, and associated higher-incidence of cardiovascular (CV) complications, its use is likely not appropriate in the ED as a bolus-dose vasopressor.
Epinephrine and Phenylephrine
Epinephrine is a potent sympathomimetic agent with alpha- and beta-receptor activity. In addition to its vasopressor effects, epinephrine is also an inotropic and chronotropic agent, increasing cardiac output, heart rate (HR), and systemic vascular resistance, which can markedly improve perfusion. Epinephrine also can be given to patients with hypoperfusion and/or shock due to low-cardiac output with or without vasodilation, lacking significant tachycardia.
Phenylephrine is a pure alpha agonist and therefore does not appreciably affect cardiac output and HR, but does significantly increase systemic vascular resistance and thus systemic perfusion. Phenylephrine can be used to treat patients with hypoperfusion and/or shock states due to vasodilation with coexistent, significant tachycardia.
Preparation and Administration
The preparation and dosing of push-dose epinephrine and phenylephrine are not particularly complex. Many clinicians recommend the pre-mixed, manufacturer-prepared agents for PDP therapy. These premixed formulations not only facilitate administration, but also reduce the chance of a preparation error that can result in incorrect dosing.3-5 If pre-mixed formulations are not available, clinicians can readily prepare epinephrine and phenylephrine for PDP use.
Push-Dose Epinephrine. Clinicians can prepare epinephrine for push-dose administration as follows:1-3
- Obtain 1 mL of epinephrine 1:10,000 (ie, 0.1 mg/mL or 100 mcg/mL);
- Obtain a 10 mL syringe of normal saline and remove 1 mL;
- Inject the 1 mL of epinephrine 1:10,000 (100 mcg/mL) into this syringe containing 9 mL of normal saline; and
- Result: 10 mL of epinephrine (10 mcg/mL), with each 1 mL of this solution containing 10 mcg of epinephrine.
Administration of push-dose epinephrine (10 mcg/mL) produces effect within 1 minute of use with a duration of approximately 5 to 10 minutes. Dosing at this concentration ranges from 0.5 to 2.0 mL every 2 to 5 minutes, delivering 5 to 20 mcg.1-3Push-Dose Phenylephrine. To prepare phenylephrine for push-dose administration, clinicians may use the following approach:1-3
- Obtain 1 mL of phenylephrine (10 mg/mL concentration);
- Inject this 1 mL of phenylephrine (10 mg/mL) into a 100 mL bag of normal saline; and
- Result: 100 mL of phenylephrine (100 mcg/mL), with each 1 mL of this solution containing 100 mcg of phenylephrine.
Administration of push-dose phenylephrine (100 mcg/mL) produces effect within 1 minute of use with a duration of approximately 10 to 20 minutes. Dosing at this concentration ranges from 0.5 to 2.0 mL every 2 to 5 minutes, delivering 50 to 200 mcg.1-3Alternative Push-Dose Preparations for Phenylephrine. Two other methods of preparing phenylephrine for bolus-dose administration include the following: (1) the addition of phenylephrine 20 mg to a bag of 250 cc of normal saline, resulting in an 80 mcg/mL concentration; and/or (2) phenylephrine (20 mg) is commercially available for continuous infusion in a 250 mL bag of normal saline, yielding the same concentration of 80 mcg/mL; in either case, medication can be drawn up and administered. Dosing at this concentration ranges from 0.5 to 2.5 mL every 2 to 5 minutes, delivering 40 to 200 mcg. Lastly, phenylephrine is also commercially available in pre-made mixtures, specifically manufactured for bolus-dose therapy.
Indications
Both epinephrine and phenylephrine can be considered in the management of significant transient or sustained hypoperfusion. Although the definition of significant hypotension is complex, Brunauer et al6 have suggested that a mean arterial pressure (MAP) of approximately 35 mm Hg is associated with a significant risk of CV collapse. Of course, a MAP of 40 to 50 mm Hg is also very concerning clinically, with significant risk of deterioration and CV collapse.
Procedural events, such as conscious sedation or rapid sequence intubation (RSI), can produce significant hypotension; PDP can rapidly correct hypotension. In other clinical scenarios in which sustained hypotension is likely and not transient (eg, sepsis with shock), PDP can be used as a bridge to definitive care (eg, volume replacement, continuous vasopressor infusion). It is important to note, however, that PDP administration must occur in conjunction with or after the patient has received other appropriate therapies such as a normal saline bolus and continuous vasopressor infusions. Push-dose pressors are not a replacement for these proven interventions, but rather are an important augmentation to these therapies.
Emergency Medicine Literature
As previously noted, the literature base describing and supporting the clinical use of PDP in EM is extremely limited. The few articles that comprise this literature base address significant hypotension in periendotracheal intubation intervention, post-return of spontaneous circulation (ROSC) management, and shock management with preload augmentation.7-9In addition, there are several articles in the literature that address safety concerns surrounding the use of PDP in the ED.4,5
Panchal et al10 investigated the use of phenylephrine in hypotensive patients undergoing RSI-assisted endotracheal intubation. The authors performed a 1-year retrospective review of hypotensive patients managed with endotracheal intubation for a range of clinical conditions that required clinical care intervention. In this study, 20 of the 119 patients received phenylephrine in the peri-intubation period. A range of clinical conditions requiring critical care intervention were encountered; in addition, almost three-quarters of these patients were receiving at least one other vasopressor infusion. Further differences were seen in the timing of PDP administration. In those patients receiving bolus-dose phenylephrine, blood pressure (BP) improved without change in HR. Panchal et al10 concluded that while push-dose phenylephrine improved hemodynamic status, there was significant variation among clinicians regarding dosing, timing of use, and overall clinical situation The significant variation in PDP management in this study was noted to be a potential source of medical error, thus increasing the chance of adverse clinical event.
Push-dose pressor therapy can be employed for significant hypotension while more definitive therapy is being readied and applied. For instance, patients with significant hypotension requiring continuous vasopressor infusion can be managed with PDP while appropriate venous access is established, intravenous fluids are administered, and medications are prepared. The immediate period after resuscitation from cardiac arrest can be complicated by shock of many types. In fact, hypotension following ROSC in the cardiac arrest patient is not uncommon and has been identified as a risk issue associated with poor outcome. Prompt treatment of this altered perfusion may improve outcome. Gottlieb8 described three patients with ROSC after cardiac arrest. All three patients experienced significant, sustained hypotension with systolic blood pressure reading in the 50 to 60 mm Hg range; bolus-dose epinephrine was administered with significant improvement in the hemodynamic status while central venous access was established.
In a related clinical scenario, Schwartz et al9 considered the impact of PDP on central venous line (CVL) placement with continuous vasopressor infusion. In this ED study, although patients experienced an increase in BP, this impact was transient with approximately half of these individuals ultimately requiring CVL. In addition, serious adverse effect was noted more commonly in the phenylephrine-treated patients with “reactive” hypertension and ventricular tachycardia occurring in study patients.
Patient-Safety Considerations
In addition to the limited literature base supporting PDP use in the ED, another major significant issue focuses on safety concerns and adverse effects. Extremely limited data is available describing adverse events related to ED-administered PDP. Extrapolating from other EM and critical care administrations of peripheral epinephrine, both local and systemic adverse effects have been reported.11,12 The range of adverse events noted in these studies are considerable, including local skin and soft-tissue injury (necrosis), end-organ tissue ischemia (eg, digits, tip of nose), acute hypertension, cardiac ischemic events, and left ventricular (LV) dysfunction.11,12
When comparing peripheral infusion with central infusion, the risk of extravasation with resultant local tissue injury is markedly greater with peripheral vasopressor administration. In a systematic review of this issue, Loubani and Green11 noted that such local adverse events were much more commonly associated with peripheral administration.
In another report of vasopressor use in the ED, Kanwar et al12 described apparent confusion with epinephrine dosing and route of administration, resulting in very significant, systemic CV maladies, including severe elevations in BP, acute LV dysfunction, and chest pain associated with ST segment elevation.
It must be stressed that the publications by Loubani and Green11 and Kanwar et al12 described peripheral vasopressor administration: neither study included PDP therapy. Therefore, as previously noted, the aforementioned statements are extrapolated from when applied to PDP strategy.
Acquisto et al4 describe several errors in medication administration of PDP in the ED and other critical care areas of the hospital. In this report, all treating physicians were present at the patients’ bedside, either administering the medication or directly supervising its use. Agents involved included epinephrine and phenylephrine, delivered at exceedingly high doses. In their study, the authors noted several issues which they believe contributed to medication errors, including heterogeneity of pathology treated in these patients, apparent “earlier-than-appropriate” use of vasopressors (ie, prior to giving an appropriate fluid bolus), and medication preparation at the bedside by clinicians who may not possess the experience and training to mix these agents.
From a patient-safety perspective, Holden et al5 noted the potential for dosing error with significant adverse medical consequence related to PDP, as well as several contributing issues. First, they highlight the lack of a solid literature base to support administration of PDP in the ED and the development of decision-making guidelines for use in the ED. They also observed an inconsistency in approach to patient selection, medication choice, agent preparation, dosing, and other therapies. As seen in the Acquisto et al4 report, the patient-care scenarios are high risk and quite dynamic.
Conclusion
Bolus-dose vasopressor therapy is a potentially very useful treatment in the ED and other emergency/critical care settings. However, despite its benefits in treating patients in shock or with hypoperfusion, PDP is not widely used in EM due to the lack of studies, reviews, and guidelines in the literature to support its use in the ED. Such a literature base is required to provide an appropriate, safe means of patient selection, medication choice, dosing, and administration. Continued educational and research efforts are needed to more fully explore the use of PDP therapy in the ED.
When used correctly and appropriately, PDP has promise to be an important aid in the management of shock in the ED. Although bolus-dose therapy is appropriate for select clinical scenarios involving significant shock states which have the potential for progression to complete CV collapse without timely therapy, it is an adjunct to, not a replacement for commonly employed and medically indicated therapies such as crystalloid bolus or continuous vasopressor infusions.
1. Weingart S. EMCrit podcast 6—push-dose pressors. EMCrit RACC Web site. July 2009. https://emcrit.org/racc/bolus-dose-pressors. Accessed March 12, 2018.
2. Weingart S. EMCrit podcast 205—push-dose pressors update. EMCrit RACC Web site. August 2017. https://emcrit.org/racc/push-dose-pressor-update/. March 12, 2018.
3. Weingart S. Push-dose pressors for immediate blood pressure control. Clin Exp Emerg Med. 2015;2(2):131-132. doi:10.15441/ceem.15.010.
4. Acquisto NM, Bodkin RP, Johnstone C. Medication errors with push dose pressors in the emergency department and intensive care units. Am J Emerg Med. 2017;35(12):1964-1965. doi:10.1016/j.ajem.2017.06.013.
5. Holden D, Ramich J, Timm E, Pauze D, Lesar T. Safety considerations and guideline-based safe use recommendations for “bolus-dose” vasopressors in the emergency department. Ann Emerg Med. 2018;71(1):83-92. doi:10.1016/j.annemergmed.2017.04.021.
6. Brunauer A, Koköfer A, Bataar O, Gradwohl-Matis I, Dankl D, Dünser MW. The arterial blood pressure associated with terminal cardiovascular collapse in critically ill patients: a retrospective cohort study. Crit Care. 2014;18(6):719. doi:10.1186/s13054-014-0719-2.
7. Panchal AR, Satyanarayan A, Bahadir JD, Hays D, Mosier J. Efficacy of bolus-dose phenylephrine for peri-intubation hypotension. J Emerg Med. 2015;49(4):488-494. doi:10.1016/j.jemermed.2015.04.033.
8. Gottlieb M. Bolus dose of epinephrine for refractory post-arrest hypotension. Can J Emerg Med. 2017;409:1-5. doi:10.1017/cem.2016.409.
9. Schwartz MB, Ferreira JA, Aaronson PM. The impact of push-dose phenylephrine use on subsequent preload expansion in the ED setting. Am J Emerg Med. 2016;34(12):2419-2422. doi:10.1016/j.ajem.2016.09.041.
10. Panchal AR, Satyanarayan A, Bahadir JD, Hays D, Mosier J. Efficacy of bolus-dose phenylephrine for peri-intubation hypotension. J Emerg Med. 2015;49(4):488-494. doi:10.1016/j.jemermed.2015.04.033.
11. Loubani OM, Green RS. A systematic review of extravasation and local tissue injury from administration of vasopressors through peripheral intravenous catheters and central venous catheters. J Crit Care. 2015;30:653.e9-e17.
12. Kanwar M, Irvin CB, Frank JJ, et al. Confusion about epinephrine dosing leading to iatrogenic overdose: A life-threatening problem with a potential solution. Ann Emerg Med. 2010;55:341-344.
1. Weingart S. EMCrit podcast 6—push-dose pressors. EMCrit RACC Web site. July 2009. https://emcrit.org/racc/bolus-dose-pressors. Accessed March 12, 2018.
2. Weingart S. EMCrit podcast 205—push-dose pressors update. EMCrit RACC Web site. August 2017. https://emcrit.org/racc/push-dose-pressor-update/. March 12, 2018.
3. Weingart S. Push-dose pressors for immediate blood pressure control. Clin Exp Emerg Med. 2015;2(2):131-132. doi:10.15441/ceem.15.010.
4. Acquisto NM, Bodkin RP, Johnstone C. Medication errors with push dose pressors in the emergency department and intensive care units. Am J Emerg Med. 2017;35(12):1964-1965. doi:10.1016/j.ajem.2017.06.013.
5. Holden D, Ramich J, Timm E, Pauze D, Lesar T. Safety considerations and guideline-based safe use recommendations for “bolus-dose” vasopressors in the emergency department. Ann Emerg Med. 2018;71(1):83-92. doi:10.1016/j.annemergmed.2017.04.021.
6. Brunauer A, Koköfer A, Bataar O, Gradwohl-Matis I, Dankl D, Dünser MW. The arterial blood pressure associated with terminal cardiovascular collapse in critically ill patients: a retrospective cohort study. Crit Care. 2014;18(6):719. doi:10.1186/s13054-014-0719-2.
7. Panchal AR, Satyanarayan A, Bahadir JD, Hays D, Mosier J. Efficacy of bolus-dose phenylephrine for peri-intubation hypotension. J Emerg Med. 2015;49(4):488-494. doi:10.1016/j.jemermed.2015.04.033.
8. Gottlieb M. Bolus dose of epinephrine for refractory post-arrest hypotension. Can J Emerg Med. 2017;409:1-5. doi:10.1017/cem.2016.409.
9. Schwartz MB, Ferreira JA, Aaronson PM. The impact of push-dose phenylephrine use on subsequent preload expansion in the ED setting. Am J Emerg Med. 2016;34(12):2419-2422. doi:10.1016/j.ajem.2016.09.041.
10. Panchal AR, Satyanarayan A, Bahadir JD, Hays D, Mosier J. Efficacy of bolus-dose phenylephrine for peri-intubation hypotension. J Emerg Med. 2015;49(4):488-494. doi:10.1016/j.jemermed.2015.04.033.
11. Loubani OM, Green RS. A systematic review of extravasation and local tissue injury from administration of vasopressors through peripheral intravenous catheters and central venous catheters. J Crit Care. 2015;30:653.e9-e17.
12. Kanwar M, Irvin CB, Frank JJ, et al. Confusion about epinephrine dosing leading to iatrogenic overdose: A life-threatening problem with a potential solution. Ann Emerg Med. 2010;55:341-344.
Single-Dose Niacin-Induced Hepatitis
Niacin, also known as vitamin B3, is an important cofactor in many metabolic processes necessary to life. Over the past 15 to 20 years, niacin has been prescribed to patients with hyperlipidemia to increase high-density lipoprotein and lower low-density lipoprotein.1 As a naturally occurring vitamin, niacin is also available over-the-counter (OTC) as a dietary supplement, and is also a common ingredient in energy drinks and multivitamins.2
In addition to treating hyperlipidemia and as a nutritional supplement, some anecdotal reports amongst lay-persons suggests that niacin offers other health benefits, such as promoting weight loss and expediting the elimination of alcohol and illicit drugs from one’s system (eg, marijuana).3,4 The increased use of niacin supplementation in the general population for all of the aforementioned reasons has resulted in an increased incidence of niacin toxicity.
Formulations
Niacin is available in three formulations: extended-release (ER, also referred to as intermediate-release), immediate-release (IR), and sustained-release (SR).
The ER formulations of niacin are typically prescribed to treat hyperlipidemia. Patients are usually started on ER niacin at an initial dose of 250 mg once daily. The dose is gradually increased, as tolerated or necessary, to 2 g per day, taken in three doses. It is not uncommon for patients with hyperlipidemia to take more than 1 g of niacin per day after titration by their primary physicians.
Side Effects
Since niacin increases the release of arachidonic acid from cell membranes that metabolizes into prostaglandins, specifically prostaglandins E2 and D2, many patients taking niacin experience uncomfortable flushing and itching.5 Nonsteroidal anti-inflammatory drugs (NSAIDs) prevent this side effect by inhibiting the metabolism of arachidonic acid into those vasodilatory prostaglandins. The newer ER and SR formulations of niacin, which are approved for OTC use as a dietary supplement, are less likely to cause flushing.5
Extended-release niacin, however, is associated with a higher incidence of hepatotoxicity than the other prescription formulations of niacin.6 Toxicity has been well recognized in patients taking niacin chronically for hyperlipidemia, with reports of such cases dating back to the 1980s.7,8 We report a unique case of niacin toxicity following a single-dose ingestion in a young man.
Case
A 22-year-old man presented to the ED for evaluation of a 2-week history of intermittent periumbilical abdominal pain. This visit represented the patient’s second visit to the ED over the past week for the same complaint.
Upon presentation the patient’s vital signs were: blood pressure (BP), 113/64 mm Hg; heart rate, 82 beats/min; respiratory rate, 16 breaths/min; and temperature 36.6°C. Oxygen saturation was 100% on room air. The patient was otherwise healthy and had no significant recent or remote medical history. He denied taking any medications prior to his initial presentation, and reported only occasional alcohol use.
At the patient’s initial presentation 1 week earlier, he was diagnosed with acute gastroenteritis and treated with famotidine and ondansetron in the ED. The patient appeared well clinically at this visit, and laboratory values were within normal limits, including normal blood glucose and urinalysis.
The patient was discharged home from this first visit with prescriptions of famotidine and ondansetron, and was advised to follow up with his primary care physician in 1 week. Throughout the week after discharge from the ED, the patient experienced worsening abdominal pain, and he developed frequent nonbloody emesis, prompting his second presentation to the ED. At this second visit, the patient stated that he had taken one dose of ondansetron at home, without effect. He also noted subjective fevers, but had no diarrhea or melena.
Vital signs remained within normal limits with BPs ranging from 115 to 130 mm Hg systolic and 50 to 89 mm Hg diastolic. The patient was never tachycardic, tachypneic, febrile, or hypoxic. Physical examination was remarkable for periumbilical tenderness. The patient had no jaundice. A more thorough laboratory evaluation revealed elevated anion gap and blood urea nitrogen/creatinine values, and leukocytosis. The patient’s hepatic enzymes were also elevated, with aspartate aminotransferase (AST) over 2,000 U/L and alanine aminotransferase (ALT) of 1,698 U/L. Lipase, bilirubin, and alkaline phosphatase were all within normal limits. The patient’s prothrombin time (PT) was elevated at 14 seconds, and the international normalized ratio (INR) was elevated at 1.28. Laboratory analysis for acetaminophen and alcohol was negative.
A computed tomography (CT) scan of the abdomen/pelvis with intravenous (IV) contrast was unremarkable, demonstrating a liver devoid of any masses, portal or biliary dilation, or cirrhotic changes.
The patient received IV famotidine and ondansetron, and morphine for pain control, and was admitted to the general medical floor for hepatitis of uncertain etiology. A viral hepatitis panel was negative.
On the recommendation of the toxicology service, the patient was given N-acetylcysteine (NAC), and his hepatic enzymes trended down to an AST of 642 U/L and an ALT of 456 U/L by hospital day 2. (The patient essentially completed a positive dechallenge test).9
A gastroenterology consult was ordered, during which additional history-taking and chart review noted that the patient admitted to taking one or two tablets of OTC niacin as a dietary supplement the day before his initial presentation. Although the patient could not recall the exact dosage, he stated that he had been taking supplemental niacin approximately once a month over the past several years without any issues. Since OTC niacin is most commonly available in 500-mg tablets, this suggested the patient’s recent one-time ingested dose was approximately 500 to 1,000 mg.
Based on the patient’s admission to niacin use, additional studies were ordered, including an abdominal ultrasound and a urine drug screen. Ultrasound findings were unremarkable for portal venous thrombosis. The urine drug screen, however, was positive for marijuana and opiates. While the patient denied any history of opioid use, the positive opiate assay could have been attributed to the morphine given in the ED.
Throughout the patient’s hospital course, he remained normotensive and had no change in mental status. His liver enzymes, PT, and INR continued to normalize, and he was discharged home after 3 days, with instructions to follow up with the gastrointestinal clinic within 11 days. An appointment was made for him, which he did not attend.
Given the patient’s negative autoimmune and viral workup, and rapid resolution of symptoms after discontinuing niacin use, it is believed that he had an acute drug-induced hepatitis due to niacin ingestion. Regarding any coingestants that could have contributed to the hepatitis, the patient denied taking other common coingestants such as alcohol and acetaminophen; this assertion was supported by laboratory results.
Since we were unable to attain a qualitative measurement of the patient’s niacin concentration, our diagnosis was primarily based on the patient’s reported history.10 It is possible the patient had been taking more niacin than that to which he admitted, or that he was taking another hepatotoxic substance not detected on our toxicology workup. As previously noted, there are many medications and/or dietary supplements that could cause or contribute to a synergistic effect of drug-induced hepatitis for which the patient was not tested at his initial presentation. The patient could have co-ingested this large dose of niacin with acetaminophen and/or other supplements, energy drinks, or alcohol. A combination such as this could have contributed to his hepatitis, and the metabolites of these other substances would have been eliminated by the time of his second ED presentation.
Discussion
There are over 900 different drugs, toxins, and supplements known to cause hepatic injury.11,12 Clinical manifestations of toxicity range from asymptomatic incidental elevations in transaminases to fulminant liver failure causing mortality. Ingestion of commonly used medications such as statins (although not in overdose quantities) can cause transient asymptomatic transaminitis.13 These elevations are usually mild—ie, less than twice the upper limit of normal. Patients who experience such elevations can usually continue to take the medications with frequent and vigilant monitoring of hepatic function.
Signs and Symptoms
Acute Liver Injury. Acute liver injury is diagnosed when AST and ALT levels are greater than twice the upper limit of normal. Patients also typically have mild-to-moderate abdominal findings, such as pain, nausea, and vomiting—as was experienced by our patient. Along with niacin, angiotensin-converting enzyme inhibitors, NSAIDs, and antifungal medications are examples of other medications that can cause this degree of drug-induced hepatitis.
Severe Liver Injury. Severe liver injury features elevations in not only AST and ALT, but also alkaline phosphate and bilirubin. Patients with severe hepatic injury appear clinically ill and may exhibit altered mental status and jaundice. This type of subfulminant hepatic failure commonly results from acetaminophen toxicity, anesthetic gases, iron toxicity, phosphorus toxicity, and cocaine toxicity. Examples of drugs that result in massive liver necrosis and fulminant hepatitis are acetaminophen, isoniazid, phenelzine, phenytoin, propylthiouracil, and sertraline. Patients with massive hepatic necrosis and hepatitis may require liver transplantation.
Etiology
Identifying the etiology of liver injury is made largely through the patient’s history because there are simply too many possible hepatotoxic agents to test for them all. Diagnostic suspicion of hepatic toxicity should be increased with signs of more serious disease; however, drug-induced liver injury should be included in the differential diagnosis for all cases of abdominal pain.
With respect to the patient in our case, obtaining a more complete history involving supplement and vitamin use would have allowed us to make the diagnosis in the ED. Unfortunately, these subtle aspects of a patient’s history are often overlooked in the emergent care setting.
Treatment
The treatment of niacin-induced liver injury is similar to the guidelines for treating most other drug-induced pathology.14 Removal of the offending agent and providing supportive care is the primary treatment modality.15 In addition, it is important that the clinician exclude and rule-out other causes of hepatitis such as those of viral, autoimmune, or ischemic etiology.
N-acetylcysteine. A medication classically used in patients with acetaminophen overdose, NAC is a safe and effective treatment for non-acetaminophen-induced liver injury, and was given to treat our patient.16L-carnitine. L-carnitine has been shown to be effective in cases of chronic steatosis from hepatitis C and in valproic acid induced hepatitis.17Since L-carnitine is not included on our hospital’s formulary, it was not a treatment option for our patient.Glucocorticoid Therapy. Although glucocorticoids are occasionally given to patients with systemic symptoms of drug reactions, its effectiveness has not been adequately studied.18
Prognosis
The prognosis of patients with acute drug-induced hepatitis is generally good, and most patients fully recover once the offending agent is removed. Poor prognostic factors include the presence of jaundice, requirement for dialysis, underlying chronic liver conditions, or elevated serum creatinine. While most patients will experience a complete recovery, approximately 5% to 10% will develop chronic hepatitis and/or cirrhosis.
Conclusion
Niacin is now available as prescription and OTC formulations and is a potentially hepatotoxic medication and dietary supplement. Niacin can cause an acute hepatitis, especially when taken in conjunction with other hepatotoxic substances. Drug-induced liver injury from niacin ingestion will improve quickly following removal, and the prognosis in otherwise healthy individuals is good.
Patients, especially young, healthy patients who present with symptoms concerning for hepatitis, should be asked specifically about any nutritional, herbal, or other supplement usage. During the history intake, many patients do not consider vitamins or other nutritional or herbal supplements as “medication” or as being significant, and only report prescription and OTC medications.
1. Berglund L, Brunzell JD, Goldberg AC, et al. Evaluation and treatment of hypertriglyceridemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97(9):2969-2989. doi:10.1210/jc.2011-3213.
2. Vivekanandarajah A, Ni S, Waked A. Acute hepatitis in a woman following excessive ingestion of an energy drink: a case report. J Med Case Rep. 2011;5:227. doi:10.1186/1752-1947-5-227.
3. Niacin. U.S. National Library of Medicine. https://medlineplus.gov/druginfo/meds/a682518.html. Updated July 15, 2017. Accessed February 21, 2018.
4. Addiction Resource. Niacin flush for drug detox. https://addictionresource.com/drug-testing/niacin-drug-test/. Accessed February 20, 2018.
5. Kamanna VS, Ganji SH, Kashyap ML. The mechanism and mitigation of niacin-induced flushing. Int J Clin Pract. 2009;63(9):1369-1377. doi:10.1111/j.1742-1241.2009.02099.x.
6. Etchason JA, Miller TD, Squires RW, et al. Niacin-induced hepatitis: a potential side effect with low-dose time-release niacin. Mayo Clin Proc. 1991;66(1):23-28.
7. Patterson DJ, Dew EW, Gyorkey F, Graham DY. Niacin hepatitis. South Med J. 1983;76(2):239-241.
8. Ferenchick G, Rovner D. Hepatitis and hematemesis complicating nicotinic acid use. Am J Med Sci. 1989;298(3):191-193.
9. Henkin Y, Johnson KC, Segrest JP. Rechallenge with crystalline niacin after drug-induced hepatitis from sustained-release niacin. JAMA. 1990;264(2):241-243.
10. Barritt AS 4th, Lee J, Hayashi PH. Detective work in drug-induced liver injury: sometimes it is all about interviewing the right witness. Clin Gastroenterol Hepatol. 2010;8(7):635-637. doi:10.1016/j.cgh.2010.03.020.
11. Chitturi S, Teoh NC, Farrell GC. Hepatic drug metabolism and liver disease caused by drugs. In: Feldman M, Friedman LS, Brandt LJ, eds. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease. 10th ed. Philadelphia, PA: Elsevier Saunders; 2016:1442-1477.
12. National Institutes of Health. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Niacin. https://livertox.nih.gov/Niacin.htm. Updated January 18, 2018. Accessed February 21, 2018.
13. Chalasani N. Statins and hepatotoxicity: focus on patients with fatty liver. Hepatology. 2005;41(4):690-695.
14. Chalasani NP, Hayashi PH, Bonkovsky HL, et al. ACG Clinical Guideline: the diagnosis and management of idiosyncratic drug-induced liver injury. Am J Gastroenterol. 2014;109(7):950-966. doi:10.1038/ajg.2014.131.
15. Larson AM. Hepatotoxicity due to herbal medication and dietary supplements. UpToDate Web site. https://www.uptodate.com/contents/hepatotoxicity-due-to-herbal-medications-and-dietary-supplements?source=search_result&search=niacin%20induced%20hepatitis&selectedTitle=3~150. Updated December 21, 2017. Accessed February 21, 2018.
16. Mumtaz K, Azam Z, Hamid S, et al. Role of N-acetylcysteine in adults with non-acetaminophen-induced acute liver failure in a center without the facility of liver transplantation. Hepatol Int. 2009;3(4):563-570. doi:10.1007/s12072-009-9151-0.
17. Perrott J, Murphy NG, Zed PJ. L-carnitine for acute valproic acid overdose: a systematic review of published cases. Ann Pharmacother. 2010;44(7-8):1287-1293. doi:10.1345/aph.1P135.
18. O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology. 1989;97(2):439-445.
Niacin, also known as vitamin B3, is an important cofactor in many metabolic processes necessary to life. Over the past 15 to 20 years, niacin has been prescribed to patients with hyperlipidemia to increase high-density lipoprotein and lower low-density lipoprotein.1 As a naturally occurring vitamin, niacin is also available over-the-counter (OTC) as a dietary supplement, and is also a common ingredient in energy drinks and multivitamins.2
In addition to treating hyperlipidemia and as a nutritional supplement, some anecdotal reports amongst lay-persons suggests that niacin offers other health benefits, such as promoting weight loss and expediting the elimination of alcohol and illicit drugs from one’s system (eg, marijuana).3,4 The increased use of niacin supplementation in the general population for all of the aforementioned reasons has resulted in an increased incidence of niacin toxicity.
Formulations
Niacin is available in three formulations: extended-release (ER, also referred to as intermediate-release), immediate-release (IR), and sustained-release (SR).
The ER formulations of niacin are typically prescribed to treat hyperlipidemia. Patients are usually started on ER niacin at an initial dose of 250 mg once daily. The dose is gradually increased, as tolerated or necessary, to 2 g per day, taken in three doses. It is not uncommon for patients with hyperlipidemia to take more than 1 g of niacin per day after titration by their primary physicians.
Side Effects
Since niacin increases the release of arachidonic acid from cell membranes that metabolizes into prostaglandins, specifically prostaglandins E2 and D2, many patients taking niacin experience uncomfortable flushing and itching.5 Nonsteroidal anti-inflammatory drugs (NSAIDs) prevent this side effect by inhibiting the metabolism of arachidonic acid into those vasodilatory prostaglandins. The newer ER and SR formulations of niacin, which are approved for OTC use as a dietary supplement, are less likely to cause flushing.5
Extended-release niacin, however, is associated with a higher incidence of hepatotoxicity than the other prescription formulations of niacin.6 Toxicity has been well recognized in patients taking niacin chronically for hyperlipidemia, with reports of such cases dating back to the 1980s.7,8 We report a unique case of niacin toxicity following a single-dose ingestion in a young man.
Case
A 22-year-old man presented to the ED for evaluation of a 2-week history of intermittent periumbilical abdominal pain. This visit represented the patient’s second visit to the ED over the past week for the same complaint.
Upon presentation the patient’s vital signs were: blood pressure (BP), 113/64 mm Hg; heart rate, 82 beats/min; respiratory rate, 16 breaths/min; and temperature 36.6°C. Oxygen saturation was 100% on room air. The patient was otherwise healthy and had no significant recent or remote medical history. He denied taking any medications prior to his initial presentation, and reported only occasional alcohol use.
At the patient’s initial presentation 1 week earlier, he was diagnosed with acute gastroenteritis and treated with famotidine and ondansetron in the ED. The patient appeared well clinically at this visit, and laboratory values were within normal limits, including normal blood glucose and urinalysis.
The patient was discharged home from this first visit with prescriptions of famotidine and ondansetron, and was advised to follow up with his primary care physician in 1 week. Throughout the week after discharge from the ED, the patient experienced worsening abdominal pain, and he developed frequent nonbloody emesis, prompting his second presentation to the ED. At this second visit, the patient stated that he had taken one dose of ondansetron at home, without effect. He also noted subjective fevers, but had no diarrhea or melena.
Vital signs remained within normal limits with BPs ranging from 115 to 130 mm Hg systolic and 50 to 89 mm Hg diastolic. The patient was never tachycardic, tachypneic, febrile, or hypoxic. Physical examination was remarkable for periumbilical tenderness. The patient had no jaundice. A more thorough laboratory evaluation revealed elevated anion gap and blood urea nitrogen/creatinine values, and leukocytosis. The patient’s hepatic enzymes were also elevated, with aspartate aminotransferase (AST) over 2,000 U/L and alanine aminotransferase (ALT) of 1,698 U/L. Lipase, bilirubin, and alkaline phosphatase were all within normal limits. The patient’s prothrombin time (PT) was elevated at 14 seconds, and the international normalized ratio (INR) was elevated at 1.28. Laboratory analysis for acetaminophen and alcohol was negative.
A computed tomography (CT) scan of the abdomen/pelvis with intravenous (IV) contrast was unremarkable, demonstrating a liver devoid of any masses, portal or biliary dilation, or cirrhotic changes.
The patient received IV famotidine and ondansetron, and morphine for pain control, and was admitted to the general medical floor for hepatitis of uncertain etiology. A viral hepatitis panel was negative.
On the recommendation of the toxicology service, the patient was given N-acetylcysteine (NAC), and his hepatic enzymes trended down to an AST of 642 U/L and an ALT of 456 U/L by hospital day 2. (The patient essentially completed a positive dechallenge test).9
A gastroenterology consult was ordered, during which additional history-taking and chart review noted that the patient admitted to taking one or two tablets of OTC niacin as a dietary supplement the day before his initial presentation. Although the patient could not recall the exact dosage, he stated that he had been taking supplemental niacin approximately once a month over the past several years without any issues. Since OTC niacin is most commonly available in 500-mg tablets, this suggested the patient’s recent one-time ingested dose was approximately 500 to 1,000 mg.
Based on the patient’s admission to niacin use, additional studies were ordered, including an abdominal ultrasound and a urine drug screen. Ultrasound findings were unremarkable for portal venous thrombosis. The urine drug screen, however, was positive for marijuana and opiates. While the patient denied any history of opioid use, the positive opiate assay could have been attributed to the morphine given in the ED.
Throughout the patient’s hospital course, he remained normotensive and had no change in mental status. His liver enzymes, PT, and INR continued to normalize, and he was discharged home after 3 days, with instructions to follow up with the gastrointestinal clinic within 11 days. An appointment was made for him, which he did not attend.
Given the patient’s negative autoimmune and viral workup, and rapid resolution of symptoms after discontinuing niacin use, it is believed that he had an acute drug-induced hepatitis due to niacin ingestion. Regarding any coingestants that could have contributed to the hepatitis, the patient denied taking other common coingestants such as alcohol and acetaminophen; this assertion was supported by laboratory results.
Since we were unable to attain a qualitative measurement of the patient’s niacin concentration, our diagnosis was primarily based on the patient’s reported history.10 It is possible the patient had been taking more niacin than that to which he admitted, or that he was taking another hepatotoxic substance not detected on our toxicology workup. As previously noted, there are many medications and/or dietary supplements that could cause or contribute to a synergistic effect of drug-induced hepatitis for which the patient was not tested at his initial presentation. The patient could have co-ingested this large dose of niacin with acetaminophen and/or other supplements, energy drinks, or alcohol. A combination such as this could have contributed to his hepatitis, and the metabolites of these other substances would have been eliminated by the time of his second ED presentation.
Discussion
There are over 900 different drugs, toxins, and supplements known to cause hepatic injury.11,12 Clinical manifestations of toxicity range from asymptomatic incidental elevations in transaminases to fulminant liver failure causing mortality. Ingestion of commonly used medications such as statins (although not in overdose quantities) can cause transient asymptomatic transaminitis.13 These elevations are usually mild—ie, less than twice the upper limit of normal. Patients who experience such elevations can usually continue to take the medications with frequent and vigilant monitoring of hepatic function.
Signs and Symptoms
Acute Liver Injury. Acute liver injury is diagnosed when AST and ALT levels are greater than twice the upper limit of normal. Patients also typically have mild-to-moderate abdominal findings, such as pain, nausea, and vomiting—as was experienced by our patient. Along with niacin, angiotensin-converting enzyme inhibitors, NSAIDs, and antifungal medications are examples of other medications that can cause this degree of drug-induced hepatitis.
Severe Liver Injury. Severe liver injury features elevations in not only AST and ALT, but also alkaline phosphate and bilirubin. Patients with severe hepatic injury appear clinically ill and may exhibit altered mental status and jaundice. This type of subfulminant hepatic failure commonly results from acetaminophen toxicity, anesthetic gases, iron toxicity, phosphorus toxicity, and cocaine toxicity. Examples of drugs that result in massive liver necrosis and fulminant hepatitis are acetaminophen, isoniazid, phenelzine, phenytoin, propylthiouracil, and sertraline. Patients with massive hepatic necrosis and hepatitis may require liver transplantation.
Etiology
Identifying the etiology of liver injury is made largely through the patient’s history because there are simply too many possible hepatotoxic agents to test for them all. Diagnostic suspicion of hepatic toxicity should be increased with signs of more serious disease; however, drug-induced liver injury should be included in the differential diagnosis for all cases of abdominal pain.
With respect to the patient in our case, obtaining a more complete history involving supplement and vitamin use would have allowed us to make the diagnosis in the ED. Unfortunately, these subtle aspects of a patient’s history are often overlooked in the emergent care setting.
Treatment
The treatment of niacin-induced liver injury is similar to the guidelines for treating most other drug-induced pathology.14 Removal of the offending agent and providing supportive care is the primary treatment modality.15 In addition, it is important that the clinician exclude and rule-out other causes of hepatitis such as those of viral, autoimmune, or ischemic etiology.
N-acetylcysteine. A medication classically used in patients with acetaminophen overdose, NAC is a safe and effective treatment for non-acetaminophen-induced liver injury, and was given to treat our patient.16L-carnitine. L-carnitine has been shown to be effective in cases of chronic steatosis from hepatitis C and in valproic acid induced hepatitis.17Since L-carnitine is not included on our hospital’s formulary, it was not a treatment option for our patient.Glucocorticoid Therapy. Although glucocorticoids are occasionally given to patients with systemic symptoms of drug reactions, its effectiveness has not been adequately studied.18
Prognosis
The prognosis of patients with acute drug-induced hepatitis is generally good, and most patients fully recover once the offending agent is removed. Poor prognostic factors include the presence of jaundice, requirement for dialysis, underlying chronic liver conditions, or elevated serum creatinine. While most patients will experience a complete recovery, approximately 5% to 10% will develop chronic hepatitis and/or cirrhosis.
Conclusion
Niacin is now available as prescription and OTC formulations and is a potentially hepatotoxic medication and dietary supplement. Niacin can cause an acute hepatitis, especially when taken in conjunction with other hepatotoxic substances. Drug-induced liver injury from niacin ingestion will improve quickly following removal, and the prognosis in otherwise healthy individuals is good.
Patients, especially young, healthy patients who present with symptoms concerning for hepatitis, should be asked specifically about any nutritional, herbal, or other supplement usage. During the history intake, many patients do not consider vitamins or other nutritional or herbal supplements as “medication” or as being significant, and only report prescription and OTC medications.
Niacin, also known as vitamin B3, is an important cofactor in many metabolic processes necessary to life. Over the past 15 to 20 years, niacin has been prescribed to patients with hyperlipidemia to increase high-density lipoprotein and lower low-density lipoprotein.1 As a naturally occurring vitamin, niacin is also available over-the-counter (OTC) as a dietary supplement, and is also a common ingredient in energy drinks and multivitamins.2
In addition to treating hyperlipidemia and as a nutritional supplement, some anecdotal reports amongst lay-persons suggests that niacin offers other health benefits, such as promoting weight loss and expediting the elimination of alcohol and illicit drugs from one’s system (eg, marijuana).3,4 The increased use of niacin supplementation in the general population for all of the aforementioned reasons has resulted in an increased incidence of niacin toxicity.
Formulations
Niacin is available in three formulations: extended-release (ER, also referred to as intermediate-release), immediate-release (IR), and sustained-release (SR).
The ER formulations of niacin are typically prescribed to treat hyperlipidemia. Patients are usually started on ER niacin at an initial dose of 250 mg once daily. The dose is gradually increased, as tolerated or necessary, to 2 g per day, taken in three doses. It is not uncommon for patients with hyperlipidemia to take more than 1 g of niacin per day after titration by their primary physicians.
Side Effects
Since niacin increases the release of arachidonic acid from cell membranes that metabolizes into prostaglandins, specifically prostaglandins E2 and D2, many patients taking niacin experience uncomfortable flushing and itching.5 Nonsteroidal anti-inflammatory drugs (NSAIDs) prevent this side effect by inhibiting the metabolism of arachidonic acid into those vasodilatory prostaglandins. The newer ER and SR formulations of niacin, which are approved for OTC use as a dietary supplement, are less likely to cause flushing.5
Extended-release niacin, however, is associated with a higher incidence of hepatotoxicity than the other prescription formulations of niacin.6 Toxicity has been well recognized in patients taking niacin chronically for hyperlipidemia, with reports of such cases dating back to the 1980s.7,8 We report a unique case of niacin toxicity following a single-dose ingestion in a young man.
Case
A 22-year-old man presented to the ED for evaluation of a 2-week history of intermittent periumbilical abdominal pain. This visit represented the patient’s second visit to the ED over the past week for the same complaint.
Upon presentation the patient’s vital signs were: blood pressure (BP), 113/64 mm Hg; heart rate, 82 beats/min; respiratory rate, 16 breaths/min; and temperature 36.6°C. Oxygen saturation was 100% on room air. The patient was otherwise healthy and had no significant recent or remote medical history. He denied taking any medications prior to his initial presentation, and reported only occasional alcohol use.
At the patient’s initial presentation 1 week earlier, he was diagnosed with acute gastroenteritis and treated with famotidine and ondansetron in the ED. The patient appeared well clinically at this visit, and laboratory values were within normal limits, including normal blood glucose and urinalysis.
The patient was discharged home from this first visit with prescriptions of famotidine and ondansetron, and was advised to follow up with his primary care physician in 1 week. Throughout the week after discharge from the ED, the patient experienced worsening abdominal pain, and he developed frequent nonbloody emesis, prompting his second presentation to the ED. At this second visit, the patient stated that he had taken one dose of ondansetron at home, without effect. He also noted subjective fevers, but had no diarrhea or melena.
Vital signs remained within normal limits with BPs ranging from 115 to 130 mm Hg systolic and 50 to 89 mm Hg diastolic. The patient was never tachycardic, tachypneic, febrile, or hypoxic. Physical examination was remarkable for periumbilical tenderness. The patient had no jaundice. A more thorough laboratory evaluation revealed elevated anion gap and blood urea nitrogen/creatinine values, and leukocytosis. The patient’s hepatic enzymes were also elevated, with aspartate aminotransferase (AST) over 2,000 U/L and alanine aminotransferase (ALT) of 1,698 U/L. Lipase, bilirubin, and alkaline phosphatase were all within normal limits. The patient’s prothrombin time (PT) was elevated at 14 seconds, and the international normalized ratio (INR) was elevated at 1.28. Laboratory analysis for acetaminophen and alcohol was negative.
A computed tomography (CT) scan of the abdomen/pelvis with intravenous (IV) contrast was unremarkable, demonstrating a liver devoid of any masses, portal or biliary dilation, or cirrhotic changes.
The patient received IV famotidine and ondansetron, and morphine for pain control, and was admitted to the general medical floor for hepatitis of uncertain etiology. A viral hepatitis panel was negative.
On the recommendation of the toxicology service, the patient was given N-acetylcysteine (NAC), and his hepatic enzymes trended down to an AST of 642 U/L and an ALT of 456 U/L by hospital day 2. (The patient essentially completed a positive dechallenge test).9
A gastroenterology consult was ordered, during which additional history-taking and chart review noted that the patient admitted to taking one or two tablets of OTC niacin as a dietary supplement the day before his initial presentation. Although the patient could not recall the exact dosage, he stated that he had been taking supplemental niacin approximately once a month over the past several years without any issues. Since OTC niacin is most commonly available in 500-mg tablets, this suggested the patient’s recent one-time ingested dose was approximately 500 to 1,000 mg.
Based on the patient’s admission to niacin use, additional studies were ordered, including an abdominal ultrasound and a urine drug screen. Ultrasound findings were unremarkable for portal venous thrombosis. The urine drug screen, however, was positive for marijuana and opiates. While the patient denied any history of opioid use, the positive opiate assay could have been attributed to the morphine given in the ED.
Throughout the patient’s hospital course, he remained normotensive and had no change in mental status. His liver enzymes, PT, and INR continued to normalize, and he was discharged home after 3 days, with instructions to follow up with the gastrointestinal clinic within 11 days. An appointment was made for him, which he did not attend.
Given the patient’s negative autoimmune and viral workup, and rapid resolution of symptoms after discontinuing niacin use, it is believed that he had an acute drug-induced hepatitis due to niacin ingestion. Regarding any coingestants that could have contributed to the hepatitis, the patient denied taking other common coingestants such as alcohol and acetaminophen; this assertion was supported by laboratory results.
Since we were unable to attain a qualitative measurement of the patient’s niacin concentration, our diagnosis was primarily based on the patient’s reported history.10 It is possible the patient had been taking more niacin than that to which he admitted, or that he was taking another hepatotoxic substance not detected on our toxicology workup. As previously noted, there are many medications and/or dietary supplements that could cause or contribute to a synergistic effect of drug-induced hepatitis for which the patient was not tested at his initial presentation. The patient could have co-ingested this large dose of niacin with acetaminophen and/or other supplements, energy drinks, or alcohol. A combination such as this could have contributed to his hepatitis, and the metabolites of these other substances would have been eliminated by the time of his second ED presentation.
Discussion
There are over 900 different drugs, toxins, and supplements known to cause hepatic injury.11,12 Clinical manifestations of toxicity range from asymptomatic incidental elevations in transaminases to fulminant liver failure causing mortality. Ingestion of commonly used medications such as statins (although not in overdose quantities) can cause transient asymptomatic transaminitis.13 These elevations are usually mild—ie, less than twice the upper limit of normal. Patients who experience such elevations can usually continue to take the medications with frequent and vigilant monitoring of hepatic function.
Signs and Symptoms
Acute Liver Injury. Acute liver injury is diagnosed when AST and ALT levels are greater than twice the upper limit of normal. Patients also typically have mild-to-moderate abdominal findings, such as pain, nausea, and vomiting—as was experienced by our patient. Along with niacin, angiotensin-converting enzyme inhibitors, NSAIDs, and antifungal medications are examples of other medications that can cause this degree of drug-induced hepatitis.
Severe Liver Injury. Severe liver injury features elevations in not only AST and ALT, but also alkaline phosphate and bilirubin. Patients with severe hepatic injury appear clinically ill and may exhibit altered mental status and jaundice. This type of subfulminant hepatic failure commonly results from acetaminophen toxicity, anesthetic gases, iron toxicity, phosphorus toxicity, and cocaine toxicity. Examples of drugs that result in massive liver necrosis and fulminant hepatitis are acetaminophen, isoniazid, phenelzine, phenytoin, propylthiouracil, and sertraline. Patients with massive hepatic necrosis and hepatitis may require liver transplantation.
Etiology
Identifying the etiology of liver injury is made largely through the patient’s history because there are simply too many possible hepatotoxic agents to test for them all. Diagnostic suspicion of hepatic toxicity should be increased with signs of more serious disease; however, drug-induced liver injury should be included in the differential diagnosis for all cases of abdominal pain.
With respect to the patient in our case, obtaining a more complete history involving supplement and vitamin use would have allowed us to make the diagnosis in the ED. Unfortunately, these subtle aspects of a patient’s history are often overlooked in the emergent care setting.
Treatment
The treatment of niacin-induced liver injury is similar to the guidelines for treating most other drug-induced pathology.14 Removal of the offending agent and providing supportive care is the primary treatment modality.15 In addition, it is important that the clinician exclude and rule-out other causes of hepatitis such as those of viral, autoimmune, or ischemic etiology.
N-acetylcysteine. A medication classically used in patients with acetaminophen overdose, NAC is a safe and effective treatment for non-acetaminophen-induced liver injury, and was given to treat our patient.16L-carnitine. L-carnitine has been shown to be effective in cases of chronic steatosis from hepatitis C and in valproic acid induced hepatitis.17Since L-carnitine is not included on our hospital’s formulary, it was not a treatment option for our patient.Glucocorticoid Therapy. Although glucocorticoids are occasionally given to patients with systemic symptoms of drug reactions, its effectiveness has not been adequately studied.18
Prognosis
The prognosis of patients with acute drug-induced hepatitis is generally good, and most patients fully recover once the offending agent is removed. Poor prognostic factors include the presence of jaundice, requirement for dialysis, underlying chronic liver conditions, or elevated serum creatinine. While most patients will experience a complete recovery, approximately 5% to 10% will develop chronic hepatitis and/or cirrhosis.
Conclusion
Niacin is now available as prescription and OTC formulations and is a potentially hepatotoxic medication and dietary supplement. Niacin can cause an acute hepatitis, especially when taken in conjunction with other hepatotoxic substances. Drug-induced liver injury from niacin ingestion will improve quickly following removal, and the prognosis in otherwise healthy individuals is good.
Patients, especially young, healthy patients who present with symptoms concerning for hepatitis, should be asked specifically about any nutritional, herbal, or other supplement usage. During the history intake, many patients do not consider vitamins or other nutritional or herbal supplements as “medication” or as being significant, and only report prescription and OTC medications.
1. Berglund L, Brunzell JD, Goldberg AC, et al. Evaluation and treatment of hypertriglyceridemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97(9):2969-2989. doi:10.1210/jc.2011-3213.
2. Vivekanandarajah A, Ni S, Waked A. Acute hepatitis in a woman following excessive ingestion of an energy drink: a case report. J Med Case Rep. 2011;5:227. doi:10.1186/1752-1947-5-227.
3. Niacin. U.S. National Library of Medicine. https://medlineplus.gov/druginfo/meds/a682518.html. Updated July 15, 2017. Accessed February 21, 2018.
4. Addiction Resource. Niacin flush for drug detox. https://addictionresource.com/drug-testing/niacin-drug-test/. Accessed February 20, 2018.
5. Kamanna VS, Ganji SH, Kashyap ML. The mechanism and mitigation of niacin-induced flushing. Int J Clin Pract. 2009;63(9):1369-1377. doi:10.1111/j.1742-1241.2009.02099.x.
6. Etchason JA, Miller TD, Squires RW, et al. Niacin-induced hepatitis: a potential side effect with low-dose time-release niacin. Mayo Clin Proc. 1991;66(1):23-28.
7. Patterson DJ, Dew EW, Gyorkey F, Graham DY. Niacin hepatitis. South Med J. 1983;76(2):239-241.
8. Ferenchick G, Rovner D. Hepatitis and hematemesis complicating nicotinic acid use. Am J Med Sci. 1989;298(3):191-193.
9. Henkin Y, Johnson KC, Segrest JP. Rechallenge with crystalline niacin after drug-induced hepatitis from sustained-release niacin. JAMA. 1990;264(2):241-243.
10. Barritt AS 4th, Lee J, Hayashi PH. Detective work in drug-induced liver injury: sometimes it is all about interviewing the right witness. Clin Gastroenterol Hepatol. 2010;8(7):635-637. doi:10.1016/j.cgh.2010.03.020.
11. Chitturi S, Teoh NC, Farrell GC. Hepatic drug metabolism and liver disease caused by drugs. In: Feldman M, Friedman LS, Brandt LJ, eds. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease. 10th ed. Philadelphia, PA: Elsevier Saunders; 2016:1442-1477.
12. National Institutes of Health. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Niacin. https://livertox.nih.gov/Niacin.htm. Updated January 18, 2018. Accessed February 21, 2018.
13. Chalasani N. Statins and hepatotoxicity: focus on patients with fatty liver. Hepatology. 2005;41(4):690-695.
14. Chalasani NP, Hayashi PH, Bonkovsky HL, et al. ACG Clinical Guideline: the diagnosis and management of idiosyncratic drug-induced liver injury. Am J Gastroenterol. 2014;109(7):950-966. doi:10.1038/ajg.2014.131.
15. Larson AM. Hepatotoxicity due to herbal medication and dietary supplements. UpToDate Web site. https://www.uptodate.com/contents/hepatotoxicity-due-to-herbal-medications-and-dietary-supplements?source=search_result&search=niacin%20induced%20hepatitis&selectedTitle=3~150. Updated December 21, 2017. Accessed February 21, 2018.
16. Mumtaz K, Azam Z, Hamid S, et al. Role of N-acetylcysteine in adults with non-acetaminophen-induced acute liver failure in a center without the facility of liver transplantation. Hepatol Int. 2009;3(4):563-570. doi:10.1007/s12072-009-9151-0.
17. Perrott J, Murphy NG, Zed PJ. L-carnitine for acute valproic acid overdose: a systematic review of published cases. Ann Pharmacother. 2010;44(7-8):1287-1293. doi:10.1345/aph.1P135.
18. O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology. 1989;97(2):439-445.
1. Berglund L, Brunzell JD, Goldberg AC, et al. Evaluation and treatment of hypertriglyceridemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97(9):2969-2989. doi:10.1210/jc.2011-3213.
2. Vivekanandarajah A, Ni S, Waked A. Acute hepatitis in a woman following excessive ingestion of an energy drink: a case report. J Med Case Rep. 2011;5:227. doi:10.1186/1752-1947-5-227.
3. Niacin. U.S. National Library of Medicine. https://medlineplus.gov/druginfo/meds/a682518.html. Updated July 15, 2017. Accessed February 21, 2018.
4. Addiction Resource. Niacin flush for drug detox. https://addictionresource.com/drug-testing/niacin-drug-test/. Accessed February 20, 2018.
5. Kamanna VS, Ganji SH, Kashyap ML. The mechanism and mitigation of niacin-induced flushing. Int J Clin Pract. 2009;63(9):1369-1377. doi:10.1111/j.1742-1241.2009.02099.x.
6. Etchason JA, Miller TD, Squires RW, et al. Niacin-induced hepatitis: a potential side effect with low-dose time-release niacin. Mayo Clin Proc. 1991;66(1):23-28.
7. Patterson DJ, Dew EW, Gyorkey F, Graham DY. Niacin hepatitis. South Med J. 1983;76(2):239-241.
8. Ferenchick G, Rovner D. Hepatitis and hematemesis complicating nicotinic acid use. Am J Med Sci. 1989;298(3):191-193.
9. Henkin Y, Johnson KC, Segrest JP. Rechallenge with crystalline niacin after drug-induced hepatitis from sustained-release niacin. JAMA. 1990;264(2):241-243.
10. Barritt AS 4th, Lee J, Hayashi PH. Detective work in drug-induced liver injury: sometimes it is all about interviewing the right witness. Clin Gastroenterol Hepatol. 2010;8(7):635-637. doi:10.1016/j.cgh.2010.03.020.
11. Chitturi S, Teoh NC, Farrell GC. Hepatic drug metabolism and liver disease caused by drugs. In: Feldman M, Friedman LS, Brandt LJ, eds. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease. 10th ed. Philadelphia, PA: Elsevier Saunders; 2016:1442-1477.
12. National Institutes of Health. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Niacin. https://livertox.nih.gov/Niacin.htm. Updated January 18, 2018. Accessed February 21, 2018.
13. Chalasani N. Statins and hepatotoxicity: focus on patients with fatty liver. Hepatology. 2005;41(4):690-695.
14. Chalasani NP, Hayashi PH, Bonkovsky HL, et al. ACG Clinical Guideline: the diagnosis and management of idiosyncratic drug-induced liver injury. Am J Gastroenterol. 2014;109(7):950-966. doi:10.1038/ajg.2014.131.
15. Larson AM. Hepatotoxicity due to herbal medication and dietary supplements. UpToDate Web site. https://www.uptodate.com/contents/hepatotoxicity-due-to-herbal-medications-and-dietary-supplements?source=search_result&search=niacin%20induced%20hepatitis&selectedTitle=3~150. Updated December 21, 2017. Accessed February 21, 2018.
16. Mumtaz K, Azam Z, Hamid S, et al. Role of N-acetylcysteine in adults with non-acetaminophen-induced acute liver failure in a center without the facility of liver transplantation. Hepatol Int. 2009;3(4):563-570. doi:10.1007/s12072-009-9151-0.
17. Perrott J, Murphy NG, Zed PJ. L-carnitine for acute valproic acid overdose: a systematic review of published cases. Ann Pharmacother. 2010;44(7-8):1287-1293. doi:10.1345/aph.1P135.
18. O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology. 1989;97(2):439-445.
Drugs identified after suicide deaths
Antidepressants and alcohol were each identified in more than 40% of tests performed after suicide deaths in 2014, according to data from the Centers for Disease Control and Prevention’s National Violent Death Reporting System.
Of the 14,834 suicide deaths reported that year in the system’s 18 participating states, tests for alcohol – conducted for 53% (7,883) of decedents – were the most commonly performed and were the second most likely to be positive among drugs with data available: The rate was 40.2%. Among the tests for blood alcohol concentration, a level of 0.08 g/dL or higher, which is over the legal limit in all states, was seen in almost 70% of positive results, Katherine A. Fowler, PhD, and her associates at the CDC’s National Center for Injury Prevention and Control reported in Morbidity and Mortality Weekly Report Surveillance Summaries.
The 18 states that collected statewide data for 2014 – Alaska, Colorado, Georgia, Kentucky, Maryland, Massachusetts, Michigan, New Jersey, New Mexico, North Carolina, Ohio, Oklahoma, Oregon, Rhode Island, South Carolina, Utah, Virginia, and Wisconsin – represent just over 33% of the U.S. population.
SOURCE: Fowler KA et al. MMWR Surveill Summ. 2018;67(SS-2):1-36. doi: 10.15585/mmwr.ss6702a1.
Antidepressants and alcohol were each identified in more than 40% of tests performed after suicide deaths in 2014, according to data from the Centers for Disease Control and Prevention’s National Violent Death Reporting System.
Of the 14,834 suicide deaths reported that year in the system’s 18 participating states, tests for alcohol – conducted for 53% (7,883) of decedents – were the most commonly performed and were the second most likely to be positive among drugs with data available: The rate was 40.2%. Among the tests for blood alcohol concentration, a level of 0.08 g/dL or higher, which is over the legal limit in all states, was seen in almost 70% of positive results, Katherine A. Fowler, PhD, and her associates at the CDC’s National Center for Injury Prevention and Control reported in Morbidity and Mortality Weekly Report Surveillance Summaries.
The 18 states that collected statewide data for 2014 – Alaska, Colorado, Georgia, Kentucky, Maryland, Massachusetts, Michigan, New Jersey, New Mexico, North Carolina, Ohio, Oklahoma, Oregon, Rhode Island, South Carolina, Utah, Virginia, and Wisconsin – represent just over 33% of the U.S. population.
SOURCE: Fowler KA et al. MMWR Surveill Summ. 2018;67(SS-2):1-36. doi: 10.15585/mmwr.ss6702a1.
Antidepressants and alcohol were each identified in more than 40% of tests performed after suicide deaths in 2014, according to data from the Centers for Disease Control and Prevention’s National Violent Death Reporting System.
Of the 14,834 suicide deaths reported that year in the system’s 18 participating states, tests for alcohol – conducted for 53% (7,883) of decedents – were the most commonly performed and were the second most likely to be positive among drugs with data available: The rate was 40.2%. Among the tests for blood alcohol concentration, a level of 0.08 g/dL or higher, which is over the legal limit in all states, was seen in almost 70% of positive results, Katherine A. Fowler, PhD, and her associates at the CDC’s National Center for Injury Prevention and Control reported in Morbidity and Mortality Weekly Report Surveillance Summaries.
The 18 states that collected statewide data for 2014 – Alaska, Colorado, Georgia, Kentucky, Maryland, Massachusetts, Michigan, New Jersey, New Mexico, North Carolina, Ohio, Oklahoma, Oregon, Rhode Island, South Carolina, Utah, Virginia, and Wisconsin – represent just over 33% of the U.S. population.
SOURCE: Fowler KA et al. MMWR Surveill Summ. 2018;67(SS-2):1-36. doi: 10.15585/mmwr.ss6702a1.
FROM MMWR
Ethanol Intoxication From Hand Sanitizer Ingestion
Case
A 29-year-old man presented to the ED several hours after ingesting what he described as a “hefty” bottle of hand sanitizer. The patient stated that he ingested such a considerable quantity of liquid hand sanitizer because he was unable to obtain beer or liquor. He further admitted to drinking two 40-ounce beers daily for the past several years, noting that he last consumed drinking alcohol the preceding day.
The patient denied any other coingestants. He also denied nausea, vomiting, abdominal pain, or other somatic complaints. The patient’s medical history was significant for hypertension and hepatitis C, and his social history was significant for daily alcohol consumption, tobacco abuse, and former benzodiazepine, marijuana, and intravenous heroin abuse. His psychiatric history was significant for borderline personality disorder, major depression, and bulimia. The patient’s home medications included a daily multivitamin, folate, thiamine, sertraline, mirtazapine, and prazosin.
Initial vital signs at presentation were: blood pressure, 124/77 mm Hg; heart rate, 86 beats/min; respiratory rate, 15 breaths/min; and temperature, 98.0°F. On physical examination, he was noted to have slurred speech and nystagmus. His pupils were equal and reactive, without scleral icterus. The abdomen was nontender and nondistended, with regular bowel sounds, and without ascites or varicosities visualized. The rest of the examination was unremarkable. The patient did express thoughts of suicidality, but denied any homicidal ideation.
Laboratory studies revealed a serum ethanol concentration of 446 mg/dL. The patient’s basic metabolic panel was unremarkable, and liver function test results showed mildly elevated enzymes. The coagulation panel was within normal limits.
Is alcohol-based hand sanitizer consumption an emerging public health concern?
Excessive alcohol consumption is a recognized public health problem in the United States and is associated with an average of 88,000 deaths per year.1 In a select population of patients, an untoward effect has developed from another public health target—that of hand hygiene.
Alcohol-based liquid hand sanitizers have become ubiquitous as a weapon in the antimicrobial arsenal with recommendations for its use as an alternative to soap and water in certain clinical settings. Liquid hand sanitizers are ideal for hospital or community use as they are faster, more effective, and less irritating to the skin than traditional hand-washing techniques.2
The downside to the widespread availability of hand sanitizers is that they offer easy access to individuals in search of clandestine sources of alcohol. Prior case reports have discussed the practice of consuming alcohol-based hand sanitizers for the purpose of intoxication in institutionalized persons, such as prisoners or patients in psychiatric facilities who are restricted to conventional sources of alcohol.
Children and confused elderly patients are also at risk for unintentional ingestions.3,4 An article reviewed exposures reported to the American Association of Poison Control Center’s National Poison Data System over a 5-year period from 2005 to 2009.3 Of the 68,712 reported cases in this cohort, 80.5% were in children younger than 6 years of age. The investigators also noted an increased incidence of exposure over this period with an average of 1,894 additional cases per year.3There were 17,154 children aged 12 years and younger reported in 2017 to poison centers with exposures to hand sanitizers. Young children may be enticed by the bright colorful packaging and similarity to food and candy smells.5
What are the clinical manifestations of alcohol-based hand sanitizer ingestion?
Significant hazards exist from ingesting liquid hand sanitizer, including the high alcohol content, which varies from 40% to 85%.2 Because isopropanol is commonly one of the components (if not the sole component) of many hand-sanitizer preparations, isopropanol toxicity may occur when ingested. The effects of isopropanol are similar to those of ethanol, with clinical effects reported after ingestion of as little as 100 mL of 70% isopropanol solution.4
Hand sanitizer formulations vary by manufacturer and contain different concentrations of ethanol and/or isopropanol, as well as additional potential inactive ingredients such as acetone, 1-propanol, 2-propanol, benzyl alcohol, hydrogen peroxide, glycerin, water, and different perfumes.3,4
Persons who consume hand sanitizers recreationally are often unaware of the large alcohol content by volume that they are consuming. Recreational ingestion of hand sanitizer is believed to be the cause of at least one case of lethal ethanol intoxication. An articlereported a case of a male patient who suffered respiratory arrest after consuming an ethanol-based hand sanitizer.6 This patient was noted to have a serum ethanol of 536 mg/dL after consuming an unknown quantity of a 354 mL container of a 62% ethanol by volume hand sanitizer.6
Institutionalized individuals seeking alcohol through this source have discovered novel ways to yield a stronger product. Through the use of table salt and a cotton sock, it is possible to extract a liquid from a gel hand sanitizer preparation, yielding an alcohol context 30% higher by volume than the parent mixture.7
Alcohol intoxication poses a host of health effects. In nonhabituated individuals, a lethal load of alcohol can be achieved by consuming a volume of as little as 400 mL of an 80% alcohol-based solution.4 Symptoms from ingestion of an alcohol-based liquid hand sanitizer typically appear 1 to 2 hours after ingestion and mirror that of the alcohol toxidrome. Most commonly, this includes nausea, vomiting, epigastric pain, and varying degrees of central nervous system (CNS) depression.4 The life-threatening clinical manifestation of alcohol intoxication includes severe CNS and respiratory depression resulting in respiratory arrest, hypothermia, cardiac dysrhythmias with possible cardiac arrest, hypoglycemia, ketoacidosis, and hypotension.3
How is alcohol-based hand sanitizer ingestion managed?
The management of patients with alcohol-based hand sanitizer ingestion is the same as the management of alcohol ingestion from more socially acceptable sources and is mainly supportive.3,4 These measures are directed at managing the patient’s airway with intubation and mechanical ventilation when appropriate, as well as supportive measures to address any underlying metabolic derangement or hypotension.2 While hemodialysis has been used in some patients who had severe organ dysfunction and did not respond to supportive measures, it is usually not necessary.1,3
Case Conclusion
The patient in this case was subsequently admitted to an intermediate level of care. He did not require intubation or further hemodynamic support during his initial acute intoxication. Later in the patient’s hospital course, he was noted to be in alcohol withdrawal, and proper management was initiated. He also required therapeutic one-to-one supervision after members of the nursing staff observed the patient consuming the hand sanitizer gel present in patient-care areas. He was later seen by psychiatry services. The psychiatrist recommended transfer to an inpatient psychiatric facility upon medical clearance for treatment of his psychiatric illness as well as alcohol dependence.
1. Esser MB, Hedden SL, Kanny D, Brewer RD, Gfroerer JC, Naimi TS. Prevalence of alcohol dependence among US adult drinkers, 2009-2011. Prev Chronic Dis. 2014;11:E206. doi:10.5888/pcd11.140329.
2. Pittet D, Boyce JM. Revolutionizing hand hygiene in health-care settings: guidelines revisted. Lancet Infect Dis. 2003;3(5):269-270.
3. Gormley NJ, Bronstein AC, Rasimas JJ, et al. The rising incidence of intentional ingestion of ethanol-containing hand sanitizers. Crit Care Med. 2012:40(1):290-294. doi:10.1097/CCM.0b013e31822f09c0.
4. Archer JR, Wood DM, Tizzard Z, Jones AL, Dargan PI. Alcohol hand rubs: hygiene and hazard. BMJ. 2007;335(7630):1154-1155.
5. Hand sanitizer. American Association of Poison Control Centers Web site. http://www.aapcc.org/alerts/hand-sanitizer/. Accessed December 27, 2017.
6. Schneir AB, Clark RF. Death caused by ingestion of an ethanol-based hand sanitizer. J Emerg Med. 2013;45(3):358-360. doi:10.1016/j.jemermed.2013.03.018.
7. Darracq MA, Ghafouri N, Pesce A, Cantrell FL. Hand sanitizer intoxication following a crude extraction method. Am J Drug Alcohol Abuse. 2013;39(3):217-218. doi:10.3109/00952990.2013.773335.
Case
A 29-year-old man presented to the ED several hours after ingesting what he described as a “hefty” bottle of hand sanitizer. The patient stated that he ingested such a considerable quantity of liquid hand sanitizer because he was unable to obtain beer or liquor. He further admitted to drinking two 40-ounce beers daily for the past several years, noting that he last consumed drinking alcohol the preceding day.
The patient denied any other coingestants. He also denied nausea, vomiting, abdominal pain, or other somatic complaints. The patient’s medical history was significant for hypertension and hepatitis C, and his social history was significant for daily alcohol consumption, tobacco abuse, and former benzodiazepine, marijuana, and intravenous heroin abuse. His psychiatric history was significant for borderline personality disorder, major depression, and bulimia. The patient’s home medications included a daily multivitamin, folate, thiamine, sertraline, mirtazapine, and prazosin.
Initial vital signs at presentation were: blood pressure, 124/77 mm Hg; heart rate, 86 beats/min; respiratory rate, 15 breaths/min; and temperature, 98.0°F. On physical examination, he was noted to have slurred speech and nystagmus. His pupils were equal and reactive, without scleral icterus. The abdomen was nontender and nondistended, with regular bowel sounds, and without ascites or varicosities visualized. The rest of the examination was unremarkable. The patient did express thoughts of suicidality, but denied any homicidal ideation.
Laboratory studies revealed a serum ethanol concentration of 446 mg/dL. The patient’s basic metabolic panel was unremarkable, and liver function test results showed mildly elevated enzymes. The coagulation panel was within normal limits.
Is alcohol-based hand sanitizer consumption an emerging public health concern?
Excessive alcohol consumption is a recognized public health problem in the United States and is associated with an average of 88,000 deaths per year.1 In a select population of patients, an untoward effect has developed from another public health target—that of hand hygiene.
Alcohol-based liquid hand sanitizers have become ubiquitous as a weapon in the antimicrobial arsenal with recommendations for its use as an alternative to soap and water in certain clinical settings. Liquid hand sanitizers are ideal for hospital or community use as they are faster, more effective, and less irritating to the skin than traditional hand-washing techniques.2
The downside to the widespread availability of hand sanitizers is that they offer easy access to individuals in search of clandestine sources of alcohol. Prior case reports have discussed the practice of consuming alcohol-based hand sanitizers for the purpose of intoxication in institutionalized persons, such as prisoners or patients in psychiatric facilities who are restricted to conventional sources of alcohol.
Children and confused elderly patients are also at risk for unintentional ingestions.3,4 An article reviewed exposures reported to the American Association of Poison Control Center’s National Poison Data System over a 5-year period from 2005 to 2009.3 Of the 68,712 reported cases in this cohort, 80.5% were in children younger than 6 years of age. The investigators also noted an increased incidence of exposure over this period with an average of 1,894 additional cases per year.3There were 17,154 children aged 12 years and younger reported in 2017 to poison centers with exposures to hand sanitizers. Young children may be enticed by the bright colorful packaging and similarity to food and candy smells.5
What are the clinical manifestations of alcohol-based hand sanitizer ingestion?
Significant hazards exist from ingesting liquid hand sanitizer, including the high alcohol content, which varies from 40% to 85%.2 Because isopropanol is commonly one of the components (if not the sole component) of many hand-sanitizer preparations, isopropanol toxicity may occur when ingested. The effects of isopropanol are similar to those of ethanol, with clinical effects reported after ingestion of as little as 100 mL of 70% isopropanol solution.4
Hand sanitizer formulations vary by manufacturer and contain different concentrations of ethanol and/or isopropanol, as well as additional potential inactive ingredients such as acetone, 1-propanol, 2-propanol, benzyl alcohol, hydrogen peroxide, glycerin, water, and different perfumes.3,4
Persons who consume hand sanitizers recreationally are often unaware of the large alcohol content by volume that they are consuming. Recreational ingestion of hand sanitizer is believed to be the cause of at least one case of lethal ethanol intoxication. An articlereported a case of a male patient who suffered respiratory arrest after consuming an ethanol-based hand sanitizer.6 This patient was noted to have a serum ethanol of 536 mg/dL after consuming an unknown quantity of a 354 mL container of a 62% ethanol by volume hand sanitizer.6
Institutionalized individuals seeking alcohol through this source have discovered novel ways to yield a stronger product. Through the use of table salt and a cotton sock, it is possible to extract a liquid from a gel hand sanitizer preparation, yielding an alcohol context 30% higher by volume than the parent mixture.7
Alcohol intoxication poses a host of health effects. In nonhabituated individuals, a lethal load of alcohol can be achieved by consuming a volume of as little as 400 mL of an 80% alcohol-based solution.4 Symptoms from ingestion of an alcohol-based liquid hand sanitizer typically appear 1 to 2 hours after ingestion and mirror that of the alcohol toxidrome. Most commonly, this includes nausea, vomiting, epigastric pain, and varying degrees of central nervous system (CNS) depression.4 The life-threatening clinical manifestation of alcohol intoxication includes severe CNS and respiratory depression resulting in respiratory arrest, hypothermia, cardiac dysrhythmias with possible cardiac arrest, hypoglycemia, ketoacidosis, and hypotension.3
How is alcohol-based hand sanitizer ingestion managed?
The management of patients with alcohol-based hand sanitizer ingestion is the same as the management of alcohol ingestion from more socially acceptable sources and is mainly supportive.3,4 These measures are directed at managing the patient’s airway with intubation and mechanical ventilation when appropriate, as well as supportive measures to address any underlying metabolic derangement or hypotension.2 While hemodialysis has been used in some patients who had severe organ dysfunction and did not respond to supportive measures, it is usually not necessary.1,3
Case Conclusion
The patient in this case was subsequently admitted to an intermediate level of care. He did not require intubation or further hemodynamic support during his initial acute intoxication. Later in the patient’s hospital course, he was noted to be in alcohol withdrawal, and proper management was initiated. He also required therapeutic one-to-one supervision after members of the nursing staff observed the patient consuming the hand sanitizer gel present in patient-care areas. He was later seen by psychiatry services. The psychiatrist recommended transfer to an inpatient psychiatric facility upon medical clearance for treatment of his psychiatric illness as well as alcohol dependence.
Case
A 29-year-old man presented to the ED several hours after ingesting what he described as a “hefty” bottle of hand sanitizer. The patient stated that he ingested such a considerable quantity of liquid hand sanitizer because he was unable to obtain beer or liquor. He further admitted to drinking two 40-ounce beers daily for the past several years, noting that he last consumed drinking alcohol the preceding day.
The patient denied any other coingestants. He also denied nausea, vomiting, abdominal pain, or other somatic complaints. The patient’s medical history was significant for hypertension and hepatitis C, and his social history was significant for daily alcohol consumption, tobacco abuse, and former benzodiazepine, marijuana, and intravenous heroin abuse. His psychiatric history was significant for borderline personality disorder, major depression, and bulimia. The patient’s home medications included a daily multivitamin, folate, thiamine, sertraline, mirtazapine, and prazosin.
Initial vital signs at presentation were: blood pressure, 124/77 mm Hg; heart rate, 86 beats/min; respiratory rate, 15 breaths/min; and temperature, 98.0°F. On physical examination, he was noted to have slurred speech and nystagmus. His pupils were equal and reactive, without scleral icterus. The abdomen was nontender and nondistended, with regular bowel sounds, and without ascites or varicosities visualized. The rest of the examination was unremarkable. The patient did express thoughts of suicidality, but denied any homicidal ideation.
Laboratory studies revealed a serum ethanol concentration of 446 mg/dL. The patient’s basic metabolic panel was unremarkable, and liver function test results showed mildly elevated enzymes. The coagulation panel was within normal limits.
Is alcohol-based hand sanitizer consumption an emerging public health concern?
Excessive alcohol consumption is a recognized public health problem in the United States and is associated with an average of 88,000 deaths per year.1 In a select population of patients, an untoward effect has developed from another public health target—that of hand hygiene.
Alcohol-based liquid hand sanitizers have become ubiquitous as a weapon in the antimicrobial arsenal with recommendations for its use as an alternative to soap and water in certain clinical settings. Liquid hand sanitizers are ideal for hospital or community use as they are faster, more effective, and less irritating to the skin than traditional hand-washing techniques.2
The downside to the widespread availability of hand sanitizers is that they offer easy access to individuals in search of clandestine sources of alcohol. Prior case reports have discussed the practice of consuming alcohol-based hand sanitizers for the purpose of intoxication in institutionalized persons, such as prisoners or patients in psychiatric facilities who are restricted to conventional sources of alcohol.
Children and confused elderly patients are also at risk for unintentional ingestions.3,4 An article reviewed exposures reported to the American Association of Poison Control Center’s National Poison Data System over a 5-year period from 2005 to 2009.3 Of the 68,712 reported cases in this cohort, 80.5% were in children younger than 6 years of age. The investigators also noted an increased incidence of exposure over this period with an average of 1,894 additional cases per year.3There were 17,154 children aged 12 years and younger reported in 2017 to poison centers with exposures to hand sanitizers. Young children may be enticed by the bright colorful packaging and similarity to food and candy smells.5
What are the clinical manifestations of alcohol-based hand sanitizer ingestion?
Significant hazards exist from ingesting liquid hand sanitizer, including the high alcohol content, which varies from 40% to 85%.2 Because isopropanol is commonly one of the components (if not the sole component) of many hand-sanitizer preparations, isopropanol toxicity may occur when ingested. The effects of isopropanol are similar to those of ethanol, with clinical effects reported after ingestion of as little as 100 mL of 70% isopropanol solution.4
Hand sanitizer formulations vary by manufacturer and contain different concentrations of ethanol and/or isopropanol, as well as additional potential inactive ingredients such as acetone, 1-propanol, 2-propanol, benzyl alcohol, hydrogen peroxide, glycerin, water, and different perfumes.3,4
Persons who consume hand sanitizers recreationally are often unaware of the large alcohol content by volume that they are consuming. Recreational ingestion of hand sanitizer is believed to be the cause of at least one case of lethal ethanol intoxication. An articlereported a case of a male patient who suffered respiratory arrest after consuming an ethanol-based hand sanitizer.6 This patient was noted to have a serum ethanol of 536 mg/dL after consuming an unknown quantity of a 354 mL container of a 62% ethanol by volume hand sanitizer.6
Institutionalized individuals seeking alcohol through this source have discovered novel ways to yield a stronger product. Through the use of table salt and a cotton sock, it is possible to extract a liquid from a gel hand sanitizer preparation, yielding an alcohol context 30% higher by volume than the parent mixture.7
Alcohol intoxication poses a host of health effects. In nonhabituated individuals, a lethal load of alcohol can be achieved by consuming a volume of as little as 400 mL of an 80% alcohol-based solution.4 Symptoms from ingestion of an alcohol-based liquid hand sanitizer typically appear 1 to 2 hours after ingestion and mirror that of the alcohol toxidrome. Most commonly, this includes nausea, vomiting, epigastric pain, and varying degrees of central nervous system (CNS) depression.4 The life-threatening clinical manifestation of alcohol intoxication includes severe CNS and respiratory depression resulting in respiratory arrest, hypothermia, cardiac dysrhythmias with possible cardiac arrest, hypoglycemia, ketoacidosis, and hypotension.3
How is alcohol-based hand sanitizer ingestion managed?
The management of patients with alcohol-based hand sanitizer ingestion is the same as the management of alcohol ingestion from more socially acceptable sources and is mainly supportive.3,4 These measures are directed at managing the patient’s airway with intubation and mechanical ventilation when appropriate, as well as supportive measures to address any underlying metabolic derangement or hypotension.2 While hemodialysis has been used in some patients who had severe organ dysfunction and did not respond to supportive measures, it is usually not necessary.1,3
Case Conclusion
The patient in this case was subsequently admitted to an intermediate level of care. He did not require intubation or further hemodynamic support during his initial acute intoxication. Later in the patient’s hospital course, he was noted to be in alcohol withdrawal, and proper management was initiated. He also required therapeutic one-to-one supervision after members of the nursing staff observed the patient consuming the hand sanitizer gel present in patient-care areas. He was later seen by psychiatry services. The psychiatrist recommended transfer to an inpatient psychiatric facility upon medical clearance for treatment of his psychiatric illness as well as alcohol dependence.
1. Esser MB, Hedden SL, Kanny D, Brewer RD, Gfroerer JC, Naimi TS. Prevalence of alcohol dependence among US adult drinkers, 2009-2011. Prev Chronic Dis. 2014;11:E206. doi:10.5888/pcd11.140329.
2. Pittet D, Boyce JM. Revolutionizing hand hygiene in health-care settings: guidelines revisted. Lancet Infect Dis. 2003;3(5):269-270.
3. Gormley NJ, Bronstein AC, Rasimas JJ, et al. The rising incidence of intentional ingestion of ethanol-containing hand sanitizers. Crit Care Med. 2012:40(1):290-294. doi:10.1097/CCM.0b013e31822f09c0.
4. Archer JR, Wood DM, Tizzard Z, Jones AL, Dargan PI. Alcohol hand rubs: hygiene and hazard. BMJ. 2007;335(7630):1154-1155.
5. Hand sanitizer. American Association of Poison Control Centers Web site. http://www.aapcc.org/alerts/hand-sanitizer/. Accessed December 27, 2017.
6. Schneir AB, Clark RF. Death caused by ingestion of an ethanol-based hand sanitizer. J Emerg Med. 2013;45(3):358-360. doi:10.1016/j.jemermed.2013.03.018.
7. Darracq MA, Ghafouri N, Pesce A, Cantrell FL. Hand sanitizer intoxication following a crude extraction method. Am J Drug Alcohol Abuse. 2013;39(3):217-218. doi:10.3109/00952990.2013.773335.
1. Esser MB, Hedden SL, Kanny D, Brewer RD, Gfroerer JC, Naimi TS. Prevalence of alcohol dependence among US adult drinkers, 2009-2011. Prev Chronic Dis. 2014;11:E206. doi:10.5888/pcd11.140329.
2. Pittet D, Boyce JM. Revolutionizing hand hygiene in health-care settings: guidelines revisted. Lancet Infect Dis. 2003;3(5):269-270.
3. Gormley NJ, Bronstein AC, Rasimas JJ, et al. The rising incidence of intentional ingestion of ethanol-containing hand sanitizers. Crit Care Med. 2012:40(1):290-294. doi:10.1097/CCM.0b013e31822f09c0.
4. Archer JR, Wood DM, Tizzard Z, Jones AL, Dargan PI. Alcohol hand rubs: hygiene and hazard. BMJ. 2007;335(7630):1154-1155.
5. Hand sanitizer. American Association of Poison Control Centers Web site. http://www.aapcc.org/alerts/hand-sanitizer/. Accessed December 27, 2017.
6. Schneir AB, Clark RF. Death caused by ingestion of an ethanol-based hand sanitizer. J Emerg Med. 2013;45(3):358-360. doi:10.1016/j.jemermed.2013.03.018.
7. Darracq MA, Ghafouri N, Pesce A, Cantrell FL. Hand sanitizer intoxication following a crude extraction method. Am J Drug Alcohol Abuse. 2013;39(3):217-218. doi:10.3109/00952990.2013.773335.

Case Studies in Toxicology: Start Low and Go Slow
Case
A woman in her third decade with no known medical history was dropped off at the waiting area of the ED for evaluation of depressed mental status. Upon arrival, the patient was unresponsive and cyanotic, with a pulse oximetry of 65% on room air. Bag-valve mask (BVM) ventilation rapidly improved oxygen saturation to 90%. The patient’s other vital signs were: heart rate, 141 beats/min; blood pressure (BP), 117/65 mm Hg; and temperature, afebrile.
Upon examination, the patient’s pupils were pinpoint and her ventilatory effort was shallow, leading the emergency physician (EP) to suspect the patient’s depressed mental status was due to an opioid overdose.
The patient was given 2 mg of intravenous (IV) naloxone, after which she became more alert and responsive, with improved respiratory effort. After receiving naloxone, the patient vomited copiously. Pulmonary examination revealed diffuse rales, most prominently at the right lung base, and a cough productive of thick sputum.
During the patient’s course in the ED, she became increasingly hypotensive with systolic BP readings around 70 mm Hg; tachycardia, fluctuating at around 120 beats/min; and persistent hypoxia of 90% saturation on a nonrebreather mask. A chest X-ray demonstrated pulmonary edema with a continuous diaphragm sign suggesting pneumomediastinum. A computed tomography (CT) scan of the chest confirmed pulmonary edema with extensive pneumomediastinum, and the patient was admitted to the intensive care unit (ICU).
What is naloxone and why is it used?
Naloxone is a nonselective, short-acting, pure opioid antagonist that works at the mu, kappa, and sigma receptors, with the highest affinity for the mu receptor. It is a competitive opioid receptor antagonist that has an elimination half-life of approximately 30 minutes. Though naloxone was originally developed to reverse the effects of anesthesia postoperatively,1 today it is more commonly used to treat ventilatory depression in patients whose clinical findings are most likely due to an opioid overdose.
Opioid-dependent individuals who abstain from use for more than a few hours generally develop opioid withdrawal syndrome (OWS). The effects of OWS include mild-to-moderate tachycardia and hypertension, nausea, vomiting, piloerection, rhinorrhea, and agitated behavior. However, when opioid-dependent patients receive naloxone, OWS develops at a much faster rate (ie, seconds after naloxone administration) and is often more severe.
Findings of naloxone-precipitated OWS include pronounced vital sign abnormalities, seizures,pulmonary edema, and cardiac arrhythmias such as ventricular tachycardia.2 These latter findings are primarily due to the sudden release of catecholamines.3 In addition, patients suffer the psychological pangs of withdrawal, including dysphoria and drug craving, which often leads to poor decision-making as they search for additional opioids to alleviate these troubling effects.
What determines response to naloxone and development of OWS?
The severity of precipitated OWS following naloxone administration is determined by both the degree of the patient’s opioid dependency and the dosage and rate at which naloxone is given. The depth of opioid dependence is determined to a large extent by the quantity of opioid regularly used and the frequency of exposure. For example, a patient who takes 30 mg of oxycodone daily will likely demonstrate mild OWS, while one who uses 300 mg daily will demonstrate more severe OWS—whether due to abstinence or naloxone.
In addition, longer exposure time of the patient’s brain to opioids increases the dependency level. Continuous use of extended-release opioids or methadone, which are both of long duration, essentially “bathe” the brain receptors in opioid around the clock, whereas short-acting opioids, such as fentanyl or heroin, cause peaks and troughs in brain concentrations throughout the day. These trough periods reduce dependency, but increase the abuse liability of the opioid. Patients who only use opioids on the weekend, for example, will have minimal or no OWS following naloxone administration, nor will the toddler with an exploratory ingestion of an opioid medication found in the home. It is therefore important to gauge the extent of a patient’s opioid use to improve the safe use of naloxone in the ED.
What is the optimal dosing of naloxone and proper patient management?
It is essential for clinicians to remember that the ultimate goal of naloxone administration in the ED is to reverse ventilatory depression—not to restore a patient to a normal mental status.4 In fact, full awakening, in addition to precipitating OWS, may lead to difficult interpersonal situations in the ED, since such patients often insist on leaving the ED before the effects of naloxone wear off. This situation places the EP in the undesirable position of discharging a patient who may predictably relapse—though unlikely to die—after release.5
Management in the Hospital Setting. Given the advanced medical care environment in a hospital, the approach to opioid overdose patients can be metered. This means providing temporary noninvasive mechanical ventilatory support through BVM or laryngeal mask airways, which allow both oxygenation and ventilation (reducing the patient’s partial pressure of carbon dioxide), prior to giving naloxone.6 Studies on animal models have shown that lowering the partial pressure of carbon dioxide reduces the catecholamine response to naloxone.7
Although recent literature and textbook recommendations regarding naloxone dosages vary,1 the safest initial dose of naloxone in the hospital setting is 0.04 mg (40 mcg) IV, or 0.08 mg (80 mcg) intramuscularly (IM).8 Whether given by IV or IM route, frequent reassessment of the adequacy of spontaneous ventilatory effort and oxygenation are required.
While the rate of opioid reversal is slower when giving lower doses of naloxone, this approach reduces the severity of precipitated OWS. In fact, in most patients who receive low-dose naloxone administration will not awaken but will develop life-sustaining spontaneous ventilation.8
By monitoring of the patient’s ventilatory rate and depth, along with capnometry and pulse oximetry (without providing exogenous oxygen), the EP can identify the need for additional naloxone. Since the half-life of naloxone is shorter than that of many opioids, proper ventilatory monitoring is essential to assess for the waning of naloxone’s effects and return of respiratory depression.
Treatment in the Nonhospital Setting. Emergency medical service (EMS) workers typically, and often by situational necessity, approach opioid overdose patients more aggressively than do EPs in the ED. Although some EMS systems utilize the IV route, most EMS workers, like laypersons, administer an initial naloxone dose of 0.4 mg IM or 2 or 4 mg intranasally (IN). Due to the slower rate of absorption and lower bioavailability (with IN administration), both IM and IN naloxone equate to roughly 0.08 mg IV.
For patients in whom there is no risk for opioid dependence, the initial dose of naloxone is relatively inconsequential, and higher doses can be safely administered. However, for most patients, including those in the ED setting, in whom one cannot be certain of their depth of dependence, the safest approach is to “start low and go slow” with naloxone administration, while providing supportive care.
Case Conclusion
The patient was not opioid-naïve, explaining the catecholamine surge and related cardiovascular dysfunction and pulmonary edema. The pneumomediastinum and pulmonary aspiration were due to the violent retching and vomiting. After being admitted to the ICU, the patient was started on vancomycin and piperacillin/tazobactam for empiric coverage for mediastinal emphysema. She was kept NPO, assessed by cardiothoracic surgery, and treated with gentle fluid hydration.
A repeat CT showed a stable pneumomediastinum. Her hypoxia, tachycardia, and hypotension gradually improved over about 6 hours. The following day, the patient’s mental status normalized, and she discharged herself from the hospital against medical advice.
1. Connors NJ, Nelson LS. The evolution of recommended naloxone dosing for opioid overdose by medical specialty. J Med Toxicol. 2016;12(3):276-281. doi:10.1007/s13181-016-0559-3.
2. Lameijer, H, Azizi N, Ligtenberg JJ, Ter Maaten JC. Ventricular tachycardia after naloxone administration: a drug related complication? Case report and literature review. Drug Saf Case Rep. 2014;1(1):2. doi:10.1007/s40800-014-0002-0.
3. Kienbaum P, Thürauf N, Michel MC, Scherbaum N, Gastpar M, Peters J. Profound increase in epinephrine concentration in plasma and cardiovascular stimulation after mu-opioid receptor blockade in opioid-addicted patients during barbiturate-induced anesthesia for acute detoxification. Anesthesiology. 1998;88(5):1154-1161.
4. Kim HK, Nelson LS. Reducing the harm of opioid overdose with the safe use of naloxone: a pharmacologic review. Expert Opin Drug Saf. 2015;14 (7 ):1137-1146. doi:10.1517/14740338.2015.1037274.
5. Willman MW, Liss DB, Schwarz ES, Mullins ME. Do heroin overdose patients require observation after receiving naloxone? Clin Toxicol (Phila). 2017;55(2):81-87. doi:10.1080/15563650.2016.1253846.
6. Boyer EW. Management of opioid analgesic overdose. N Engl J Med. 2012;367(2):146-155. doi:10.1056/NEJMra1202561.
7. Mills CA, Flacke JW, Miller JD, Davis LJ, Bloor BC, Flacke WE. Cardiovascular effects of fentanyl reversal by naloxone at varying arterial carbon dioxide tensions in dogs. Anesth Analg. 1988;67(8):730-736.
8. Kim HK, Nelson LS. Reversal of opioid-induced ventilatory depression using low-dose naloxone (0.04 mg): a case series. J Med Toxicol. 2015;12(1):107-110. doi:10.1007/s13181-015-0499-3.
Case
A woman in her third decade with no known medical history was dropped off at the waiting area of the ED for evaluation of depressed mental status. Upon arrival, the patient was unresponsive and cyanotic, with a pulse oximetry of 65% on room air. Bag-valve mask (BVM) ventilation rapidly improved oxygen saturation to 90%. The patient’s other vital signs were: heart rate, 141 beats/min; blood pressure (BP), 117/65 mm Hg; and temperature, afebrile.
Upon examination, the patient’s pupils were pinpoint and her ventilatory effort was shallow, leading the emergency physician (EP) to suspect the patient’s depressed mental status was due to an opioid overdose.
The patient was given 2 mg of intravenous (IV) naloxone, after which she became more alert and responsive, with improved respiratory effort. After receiving naloxone, the patient vomited copiously. Pulmonary examination revealed diffuse rales, most prominently at the right lung base, and a cough productive of thick sputum.
During the patient’s course in the ED, she became increasingly hypotensive with systolic BP readings around 70 mm Hg; tachycardia, fluctuating at around 120 beats/min; and persistent hypoxia of 90% saturation on a nonrebreather mask. A chest X-ray demonstrated pulmonary edema with a continuous diaphragm sign suggesting pneumomediastinum. A computed tomography (CT) scan of the chest confirmed pulmonary edema with extensive pneumomediastinum, and the patient was admitted to the intensive care unit (ICU).
What is naloxone and why is it used?
Naloxone is a nonselective, short-acting, pure opioid antagonist that works at the mu, kappa, and sigma receptors, with the highest affinity for the mu receptor. It is a competitive opioid receptor antagonist that has an elimination half-life of approximately 30 minutes. Though naloxone was originally developed to reverse the effects of anesthesia postoperatively,1 today it is more commonly used to treat ventilatory depression in patients whose clinical findings are most likely due to an opioid overdose.
Opioid-dependent individuals who abstain from use for more than a few hours generally develop opioid withdrawal syndrome (OWS). The effects of OWS include mild-to-moderate tachycardia and hypertension, nausea, vomiting, piloerection, rhinorrhea, and agitated behavior. However, when opioid-dependent patients receive naloxone, OWS develops at a much faster rate (ie, seconds after naloxone administration) and is often more severe.
Findings of naloxone-precipitated OWS include pronounced vital sign abnormalities, seizures,pulmonary edema, and cardiac arrhythmias such as ventricular tachycardia.2 These latter findings are primarily due to the sudden release of catecholamines.3 In addition, patients suffer the psychological pangs of withdrawal, including dysphoria and drug craving, which often leads to poor decision-making as they search for additional opioids to alleviate these troubling effects.
What determines response to naloxone and development of OWS?
The severity of precipitated OWS following naloxone administration is determined by both the degree of the patient’s opioid dependency and the dosage and rate at which naloxone is given. The depth of opioid dependence is determined to a large extent by the quantity of opioid regularly used and the frequency of exposure. For example, a patient who takes 30 mg of oxycodone daily will likely demonstrate mild OWS, while one who uses 300 mg daily will demonstrate more severe OWS—whether due to abstinence or naloxone.
In addition, longer exposure time of the patient’s brain to opioids increases the dependency level. Continuous use of extended-release opioids or methadone, which are both of long duration, essentially “bathe” the brain receptors in opioid around the clock, whereas short-acting opioids, such as fentanyl or heroin, cause peaks and troughs in brain concentrations throughout the day. These trough periods reduce dependency, but increase the abuse liability of the opioid. Patients who only use opioids on the weekend, for example, will have minimal or no OWS following naloxone administration, nor will the toddler with an exploratory ingestion of an opioid medication found in the home. It is therefore important to gauge the extent of a patient’s opioid use to improve the safe use of naloxone in the ED.
What is the optimal dosing of naloxone and proper patient management?
It is essential for clinicians to remember that the ultimate goal of naloxone administration in the ED is to reverse ventilatory depression—not to restore a patient to a normal mental status.4 In fact, full awakening, in addition to precipitating OWS, may lead to difficult interpersonal situations in the ED, since such patients often insist on leaving the ED before the effects of naloxone wear off. This situation places the EP in the undesirable position of discharging a patient who may predictably relapse—though unlikely to die—after release.5
Management in the Hospital Setting. Given the advanced medical care environment in a hospital, the approach to opioid overdose patients can be metered. This means providing temporary noninvasive mechanical ventilatory support through BVM or laryngeal mask airways, which allow both oxygenation and ventilation (reducing the patient’s partial pressure of carbon dioxide), prior to giving naloxone.6 Studies on animal models have shown that lowering the partial pressure of carbon dioxide reduces the catecholamine response to naloxone.7
Although recent literature and textbook recommendations regarding naloxone dosages vary,1 the safest initial dose of naloxone in the hospital setting is 0.04 mg (40 mcg) IV, or 0.08 mg (80 mcg) intramuscularly (IM).8 Whether given by IV or IM route, frequent reassessment of the adequacy of spontaneous ventilatory effort and oxygenation are required.
While the rate of opioid reversal is slower when giving lower doses of naloxone, this approach reduces the severity of precipitated OWS. In fact, in most patients who receive low-dose naloxone administration will not awaken but will develop life-sustaining spontaneous ventilation.8
By monitoring of the patient’s ventilatory rate and depth, along with capnometry and pulse oximetry (without providing exogenous oxygen), the EP can identify the need for additional naloxone. Since the half-life of naloxone is shorter than that of many opioids, proper ventilatory monitoring is essential to assess for the waning of naloxone’s effects and return of respiratory depression.
Treatment in the Nonhospital Setting. Emergency medical service (EMS) workers typically, and often by situational necessity, approach opioid overdose patients more aggressively than do EPs in the ED. Although some EMS systems utilize the IV route, most EMS workers, like laypersons, administer an initial naloxone dose of 0.4 mg IM or 2 or 4 mg intranasally (IN). Due to the slower rate of absorption and lower bioavailability (with IN administration), both IM and IN naloxone equate to roughly 0.08 mg IV.
For patients in whom there is no risk for opioid dependence, the initial dose of naloxone is relatively inconsequential, and higher doses can be safely administered. However, for most patients, including those in the ED setting, in whom one cannot be certain of their depth of dependence, the safest approach is to “start low and go slow” with naloxone administration, while providing supportive care.
Case Conclusion
The patient was not opioid-naïve, explaining the catecholamine surge and related cardiovascular dysfunction and pulmonary edema. The pneumomediastinum and pulmonary aspiration were due to the violent retching and vomiting. After being admitted to the ICU, the patient was started on vancomycin and piperacillin/tazobactam for empiric coverage for mediastinal emphysema. She was kept NPO, assessed by cardiothoracic surgery, and treated with gentle fluid hydration.
A repeat CT showed a stable pneumomediastinum. Her hypoxia, tachycardia, and hypotension gradually improved over about 6 hours. The following day, the patient’s mental status normalized, and she discharged herself from the hospital against medical advice.
Case
A woman in her third decade with no known medical history was dropped off at the waiting area of the ED for evaluation of depressed mental status. Upon arrival, the patient was unresponsive and cyanotic, with a pulse oximetry of 65% on room air. Bag-valve mask (BVM) ventilation rapidly improved oxygen saturation to 90%. The patient’s other vital signs were: heart rate, 141 beats/min; blood pressure (BP), 117/65 mm Hg; and temperature, afebrile.
Upon examination, the patient’s pupils were pinpoint and her ventilatory effort was shallow, leading the emergency physician (EP) to suspect the patient’s depressed mental status was due to an opioid overdose.
The patient was given 2 mg of intravenous (IV) naloxone, after which she became more alert and responsive, with improved respiratory effort. After receiving naloxone, the patient vomited copiously. Pulmonary examination revealed diffuse rales, most prominently at the right lung base, and a cough productive of thick sputum.
During the patient’s course in the ED, she became increasingly hypotensive with systolic BP readings around 70 mm Hg; tachycardia, fluctuating at around 120 beats/min; and persistent hypoxia of 90% saturation on a nonrebreather mask. A chest X-ray demonstrated pulmonary edema with a continuous diaphragm sign suggesting pneumomediastinum. A computed tomography (CT) scan of the chest confirmed pulmonary edema with extensive pneumomediastinum, and the patient was admitted to the intensive care unit (ICU).
What is naloxone and why is it used?
Naloxone is a nonselective, short-acting, pure opioid antagonist that works at the mu, kappa, and sigma receptors, with the highest affinity for the mu receptor. It is a competitive opioid receptor antagonist that has an elimination half-life of approximately 30 minutes. Though naloxone was originally developed to reverse the effects of anesthesia postoperatively,1 today it is more commonly used to treat ventilatory depression in patients whose clinical findings are most likely due to an opioid overdose.
Opioid-dependent individuals who abstain from use for more than a few hours generally develop opioid withdrawal syndrome (OWS). The effects of OWS include mild-to-moderate tachycardia and hypertension, nausea, vomiting, piloerection, rhinorrhea, and agitated behavior. However, when opioid-dependent patients receive naloxone, OWS develops at a much faster rate (ie, seconds after naloxone administration) and is often more severe.
Findings of naloxone-precipitated OWS include pronounced vital sign abnormalities, seizures,pulmonary edema, and cardiac arrhythmias such as ventricular tachycardia.2 These latter findings are primarily due to the sudden release of catecholamines.3 In addition, patients suffer the psychological pangs of withdrawal, including dysphoria and drug craving, which often leads to poor decision-making as they search for additional opioids to alleviate these troubling effects.
What determines response to naloxone and development of OWS?
The severity of precipitated OWS following naloxone administration is determined by both the degree of the patient’s opioid dependency and the dosage and rate at which naloxone is given. The depth of opioid dependence is determined to a large extent by the quantity of opioid regularly used and the frequency of exposure. For example, a patient who takes 30 mg of oxycodone daily will likely demonstrate mild OWS, while one who uses 300 mg daily will demonstrate more severe OWS—whether due to abstinence or naloxone.
In addition, longer exposure time of the patient’s brain to opioids increases the dependency level. Continuous use of extended-release opioids or methadone, which are both of long duration, essentially “bathe” the brain receptors in opioid around the clock, whereas short-acting opioids, such as fentanyl or heroin, cause peaks and troughs in brain concentrations throughout the day. These trough periods reduce dependency, but increase the abuse liability of the opioid. Patients who only use opioids on the weekend, for example, will have minimal or no OWS following naloxone administration, nor will the toddler with an exploratory ingestion of an opioid medication found in the home. It is therefore important to gauge the extent of a patient’s opioid use to improve the safe use of naloxone in the ED.
What is the optimal dosing of naloxone and proper patient management?
It is essential for clinicians to remember that the ultimate goal of naloxone administration in the ED is to reverse ventilatory depression—not to restore a patient to a normal mental status.4 In fact, full awakening, in addition to precipitating OWS, may lead to difficult interpersonal situations in the ED, since such patients often insist on leaving the ED before the effects of naloxone wear off. This situation places the EP in the undesirable position of discharging a patient who may predictably relapse—though unlikely to die—after release.5
Management in the Hospital Setting. Given the advanced medical care environment in a hospital, the approach to opioid overdose patients can be metered. This means providing temporary noninvasive mechanical ventilatory support through BVM or laryngeal mask airways, which allow both oxygenation and ventilation (reducing the patient’s partial pressure of carbon dioxide), prior to giving naloxone.6 Studies on animal models have shown that lowering the partial pressure of carbon dioxide reduces the catecholamine response to naloxone.7
Although recent literature and textbook recommendations regarding naloxone dosages vary,1 the safest initial dose of naloxone in the hospital setting is 0.04 mg (40 mcg) IV, or 0.08 mg (80 mcg) intramuscularly (IM).8 Whether given by IV or IM route, frequent reassessment of the adequacy of spontaneous ventilatory effort and oxygenation are required.
While the rate of opioid reversal is slower when giving lower doses of naloxone, this approach reduces the severity of precipitated OWS. In fact, in most patients who receive low-dose naloxone administration will not awaken but will develop life-sustaining spontaneous ventilation.8
By monitoring of the patient’s ventilatory rate and depth, along with capnometry and pulse oximetry (without providing exogenous oxygen), the EP can identify the need for additional naloxone. Since the half-life of naloxone is shorter than that of many opioids, proper ventilatory monitoring is essential to assess for the waning of naloxone’s effects and return of respiratory depression.
Treatment in the Nonhospital Setting. Emergency medical service (EMS) workers typically, and often by situational necessity, approach opioid overdose patients more aggressively than do EPs in the ED. Although some EMS systems utilize the IV route, most EMS workers, like laypersons, administer an initial naloxone dose of 0.4 mg IM or 2 or 4 mg intranasally (IN). Due to the slower rate of absorption and lower bioavailability (with IN administration), both IM and IN naloxone equate to roughly 0.08 mg IV.
For patients in whom there is no risk for opioid dependence, the initial dose of naloxone is relatively inconsequential, and higher doses can be safely administered. However, for most patients, including those in the ED setting, in whom one cannot be certain of their depth of dependence, the safest approach is to “start low and go slow” with naloxone administration, while providing supportive care.
Case Conclusion
The patient was not opioid-naïve, explaining the catecholamine surge and related cardiovascular dysfunction and pulmonary edema. The pneumomediastinum and pulmonary aspiration were due to the violent retching and vomiting. After being admitted to the ICU, the patient was started on vancomycin and piperacillin/tazobactam for empiric coverage for mediastinal emphysema. She was kept NPO, assessed by cardiothoracic surgery, and treated with gentle fluid hydration.
A repeat CT showed a stable pneumomediastinum. Her hypoxia, tachycardia, and hypotension gradually improved over about 6 hours. The following day, the patient’s mental status normalized, and she discharged herself from the hospital against medical advice.
1. Connors NJ, Nelson LS. The evolution of recommended naloxone dosing for opioid overdose by medical specialty. J Med Toxicol. 2016;12(3):276-281. doi:10.1007/s13181-016-0559-3.
2. Lameijer, H, Azizi N, Ligtenberg JJ, Ter Maaten JC. Ventricular tachycardia after naloxone administration: a drug related complication? Case report and literature review. Drug Saf Case Rep. 2014;1(1):2. doi:10.1007/s40800-014-0002-0.
3. Kienbaum P, Thürauf N, Michel MC, Scherbaum N, Gastpar M, Peters J. Profound increase in epinephrine concentration in plasma and cardiovascular stimulation after mu-opioid receptor blockade in opioid-addicted patients during barbiturate-induced anesthesia for acute detoxification. Anesthesiology. 1998;88(5):1154-1161.
4. Kim HK, Nelson LS. Reducing the harm of opioid overdose with the safe use of naloxone: a pharmacologic review. Expert Opin Drug Saf. 2015;14 (7 ):1137-1146. doi:10.1517/14740338.2015.1037274.
5. Willman MW, Liss DB, Schwarz ES, Mullins ME. Do heroin overdose patients require observation after receiving naloxone? Clin Toxicol (Phila). 2017;55(2):81-87. doi:10.1080/15563650.2016.1253846.
6. Boyer EW. Management of opioid analgesic overdose. N Engl J Med. 2012;367(2):146-155. doi:10.1056/NEJMra1202561.
7. Mills CA, Flacke JW, Miller JD, Davis LJ, Bloor BC, Flacke WE. Cardiovascular effects of fentanyl reversal by naloxone at varying arterial carbon dioxide tensions in dogs. Anesth Analg. 1988;67(8):730-736.
8. Kim HK, Nelson LS. Reversal of opioid-induced ventilatory depression using low-dose naloxone (0.04 mg): a case series. J Med Toxicol. 2015;12(1):107-110. doi:10.1007/s13181-015-0499-3.
1. Connors NJ, Nelson LS. The evolution of recommended naloxone dosing for opioid overdose by medical specialty. J Med Toxicol. 2016;12(3):276-281. doi:10.1007/s13181-016-0559-3.
2. Lameijer, H, Azizi N, Ligtenberg JJ, Ter Maaten JC. Ventricular tachycardia after naloxone administration: a drug related complication? Case report and literature review. Drug Saf Case Rep. 2014;1(1):2. doi:10.1007/s40800-014-0002-0.
3. Kienbaum P, Thürauf N, Michel MC, Scherbaum N, Gastpar M, Peters J. Profound increase in epinephrine concentration in plasma and cardiovascular stimulation after mu-opioid receptor blockade in opioid-addicted patients during barbiturate-induced anesthesia for acute detoxification. Anesthesiology. 1998;88(5):1154-1161.
4. Kim HK, Nelson LS. Reducing the harm of opioid overdose with the safe use of naloxone: a pharmacologic review. Expert Opin Drug Saf. 2015;14 (7 ):1137-1146. doi:10.1517/14740338.2015.1037274.
5. Willman MW, Liss DB, Schwarz ES, Mullins ME. Do heroin overdose patients require observation after receiving naloxone? Clin Toxicol (Phila). 2017;55(2):81-87. doi:10.1080/15563650.2016.1253846.
6. Boyer EW. Management of opioid analgesic overdose. N Engl J Med. 2012;367(2):146-155. doi:10.1056/NEJMra1202561.
7. Mills CA, Flacke JW, Miller JD, Davis LJ, Bloor BC, Flacke WE. Cardiovascular effects of fentanyl reversal by naloxone at varying arterial carbon dioxide tensions in dogs. Anesth Analg. 1988;67(8):730-736.
8. Kim HK, Nelson LS. Reversal of opioid-induced ventilatory depression using low-dose naloxone (0.04 mg): a case series. J Med Toxicol. 2015;12(1):107-110. doi:10.1007/s13181-015-0499-3.

Case Studies in Toxicology: DILI Dally
Case
A 50-year-old Hispanic woman with a history of rheumatoid arthritis (RA), for which she was not currently taking medication, was referred to the ED by her primary care physician (PCP) for evaluation of generalized pruritus and jaundice, and an abnormal hepatic function panel.
The patient’s recent history was significant for a positive tuberculosis test (purified protein derivative [PPD], 13 mm), for which she had been on prophylactic medication. Laboratory evaluation taken during the patient’s recent follow-up visit with her PCP revealed the following significant hepatic abnormalities: total bilirubin, 20.0 mg/dL; direct bilirubin, 16.4 mg/dL; international normalized ratio, 2.9; aspartate aminotransferase, greater than 2,000 IU/L; and alanine aminotransferase, greater than 2,000 IU/L. The patient had no history of hepatic disease, and a hepatitis panel obtained in the ED was unremarkable.
Can this be drug-induced liver injury?
Drug-induced liver injury (DILI) accounts for nearly 50% of cases of acute liver failure in the United States.1 According to the National Institutes of Health database of drugs, supplements, and herbal medications acetaminophen is the most common drug associated with hepatotoxicity in the United States, whereas amoxicillin-clavulanate is the most common implicated drug worldwide.1,2 The histological pattern of DILI varies by drug (Table).3
Who is susceptible to drug-induced liver injury?
The factors that help predict DILI include drug pharmacokinetics and metabolism, as well as patient age, sex, and comorbidities. Although some patients are at an increased risk of DILI, it is extraordinarily difficult to accurately predict which patients will develop it. In general, there is a positive correlation between age and risk of developing DILI. For example, in a large US-based tuberculosis study, the incidence of isoniazid (INH)-induced hepatotoxicity was 4.4 per 1,000 patients aged 25 to 34 years. Patients older than age 50 years had a 20.83 per 1,000 incidence of DILI, and women also appear to be at increased risk.4
Pharmacogenetic factors affecting drug metabolism such as the specific cytochrome profile and acetylator status of an individual also influence a patient’s risk of developing DILI. Although our understanding of these issues is growing rapidly, our ability to apply this knowledge to the clinical venue is limited by the available technology, regulatory requirements, and cost.
Case Continuation
A detailed, careful history-taking in the ED revealed that, 2 months prior, the patient had been started on INH, rifampin, and pyridoxine for latent tuberculosis. She had been taking methotrexate for the RA but discontinued it 3 months ago because of the positive PPD. When routine outpatient laboratory testing results demonstrated significant hepatic dysfunction, the patient’s PCP advised her to immediately discontinue her medications and referred her to the ED for further evaluation and management.
By what mechanism does INH cause DILI?
Acute INH-associated hepatitis primarily results from the direct hepatotoxic effects of INH metabolites. Isoniazid is metabolized in the liver via N-acetylation to acetylisoniazid (Figure). Oxidation of this compound in the liver leads to an accumulation of the hepatotoxic metabolites acetylhydrazine and hydrazine.5,6
Is there a role for N-acetylcysteine in INH hepatotoxicity?
No antidote is specifically designed to treat INH-induced hepatotoxicity, and management is largely supportive. Observation for progressive liver failure is indicated and evaluation for liver transplant may become necessary.
N-acetylcysteine (NAC) has a clear role in preventing hepatotoxicity from acetaminophen overdose through its ability to act as a precursor for the synthesis of glutathione—a compound that protects hepatocytes from oxidative damage. In advanced acetaminophen-toxic patients and those with non-acetaminophen toxicity, NAC has nonspecific effects that promote healing through several mechanisms, including anti-inflammatory effect and enhanced hepatic perfusion. Though there are no studies that specifically evaluate the role of NAC in patients with INH-induced hepatotoxicity, it is commonly and appropriately administered for its aforementioned nonspecific effects.8 Common side effects from NAC administration include nausea, vomiting, and diarrhea, which are generally treatable with symptomatic and supportive care.
Case Conclusion
The patient was admitted to the hepatology service for continued clinical care. Although she received NAC, hepatic function testing showed only mild improvement. Additional etiologies of liver failure were investigated, including a computed tomography scan of the abdomen/pelvis and an abdominal ultrasound with Doppler. Both studies were negative for any pathology, and autoimmune laboratory studies were likewise unremarkable.
The patient underwent a liver biopsy, which revealed inflammation and scattered eosinophils suggestive of drug-induced hepatic injury. Her clinical condition continued to deteriorate, and she was transferred to another hospital for transplant evaluation.
1. Lee WM. Drug-induced acute liver failure. Clin Liver Dis. 2013;17(4):575-586, viii. doi:10.1016/j.cld.2013.07.001.
2. National Institutes of Health Web site. LiverTox: Clinical and research information on drug-induced liver injury. https://livertox.nlm.nih.gov/. Updated February 10, 2017. Accessed October 12, 2017.
3. Ansari JA, Sayyed M, Sayeed F. Management of non alcoholic fatty liver diseases and their complications. Int J Pharmacol. 2011;7:579-588. doi:10.3923/ijp.2011.579.588.
4. Fountain FF, Tolley E, Chrisman CR, Self TH. Isoniazid hepatotoxicity associated with treatment of latent tuberculosis infection: a 7-year evaluation from a public health tuberculosis clinic. Chest. 2005;128(1):116-123. doi:10.1378/chest.128.1.116.
5. Hernon CH. Antituberculous medications. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR, eds. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015:787-796.
6. Teixeira RL, Morato RG, Cabello PH, et al. Genetic polymorphisms of NAT2, CYP2E1 and GST enzymes and the occurrence of antituberculosis drug-induced hepatitis in Brazilian TB patients. Mem Inst Oswaldo Cruz. 2011;106(6):716-724.
7. Mitchell JR, Thorgeirsson UP, Black M, et al. Increased incidence of isoniazid hepatitis in rapid acetylators: possible relation to hydranize metabolites. Clin Pharmacol Ther. 1975;18(1):70-79.
8. Lee WM, Hynan LS, Rossaro L, et al; Acute Liver Failure Study Group. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology. 2009;137(3):856-864. doi:10.1053/j.gastro.2009.06.006.
Case
A 50-year-old Hispanic woman with a history of rheumatoid arthritis (RA), for which she was not currently taking medication, was referred to the ED by her primary care physician (PCP) for evaluation of generalized pruritus and jaundice, and an abnormal hepatic function panel.
The patient’s recent history was significant for a positive tuberculosis test (purified protein derivative [PPD], 13 mm), for which she had been on prophylactic medication. Laboratory evaluation taken during the patient’s recent follow-up visit with her PCP revealed the following significant hepatic abnormalities: total bilirubin, 20.0 mg/dL; direct bilirubin, 16.4 mg/dL; international normalized ratio, 2.9; aspartate aminotransferase, greater than 2,000 IU/L; and alanine aminotransferase, greater than 2,000 IU/L. The patient had no history of hepatic disease, and a hepatitis panel obtained in the ED was unremarkable.
Can this be drug-induced liver injury?
Drug-induced liver injury (DILI) accounts for nearly 50% of cases of acute liver failure in the United States.1 According to the National Institutes of Health database of drugs, supplements, and herbal medications acetaminophen is the most common drug associated with hepatotoxicity in the United States, whereas amoxicillin-clavulanate is the most common implicated drug worldwide.1,2 The histological pattern of DILI varies by drug (Table).3
Who is susceptible to drug-induced liver injury?
The factors that help predict DILI include drug pharmacokinetics and metabolism, as well as patient age, sex, and comorbidities. Although some patients are at an increased risk of DILI, it is extraordinarily difficult to accurately predict which patients will develop it. In general, there is a positive correlation between age and risk of developing DILI. For example, in a large US-based tuberculosis study, the incidence of isoniazid (INH)-induced hepatotoxicity was 4.4 per 1,000 patients aged 25 to 34 years. Patients older than age 50 years had a 20.83 per 1,000 incidence of DILI, and women also appear to be at increased risk.4
Pharmacogenetic factors affecting drug metabolism such as the specific cytochrome profile and acetylator status of an individual also influence a patient’s risk of developing DILI. Although our understanding of these issues is growing rapidly, our ability to apply this knowledge to the clinical venue is limited by the available technology, regulatory requirements, and cost.
Case Continuation
A detailed, careful history-taking in the ED revealed that, 2 months prior, the patient had been started on INH, rifampin, and pyridoxine for latent tuberculosis. She had been taking methotrexate for the RA but discontinued it 3 months ago because of the positive PPD. When routine outpatient laboratory testing results demonstrated significant hepatic dysfunction, the patient’s PCP advised her to immediately discontinue her medications and referred her to the ED for further evaluation and management.
By what mechanism does INH cause DILI?
Acute INH-associated hepatitis primarily results from the direct hepatotoxic effects of INH metabolites. Isoniazid is metabolized in the liver via N-acetylation to acetylisoniazid (Figure). Oxidation of this compound in the liver leads to an accumulation of the hepatotoxic metabolites acetylhydrazine and hydrazine.5,6
Is there a role for N-acetylcysteine in INH hepatotoxicity?
No antidote is specifically designed to treat INH-induced hepatotoxicity, and management is largely supportive. Observation for progressive liver failure is indicated and evaluation for liver transplant may become necessary.
N-acetylcysteine (NAC) has a clear role in preventing hepatotoxicity from acetaminophen overdose through its ability to act as a precursor for the synthesis of glutathione—a compound that protects hepatocytes from oxidative damage. In advanced acetaminophen-toxic patients and those with non-acetaminophen toxicity, NAC has nonspecific effects that promote healing through several mechanisms, including anti-inflammatory effect and enhanced hepatic perfusion. Though there are no studies that specifically evaluate the role of NAC in patients with INH-induced hepatotoxicity, it is commonly and appropriately administered for its aforementioned nonspecific effects.8 Common side effects from NAC administration include nausea, vomiting, and diarrhea, which are generally treatable with symptomatic and supportive care.
Case Conclusion
The patient was admitted to the hepatology service for continued clinical care. Although she received NAC, hepatic function testing showed only mild improvement. Additional etiologies of liver failure were investigated, including a computed tomography scan of the abdomen/pelvis and an abdominal ultrasound with Doppler. Both studies were negative for any pathology, and autoimmune laboratory studies were likewise unremarkable.
The patient underwent a liver biopsy, which revealed inflammation and scattered eosinophils suggestive of drug-induced hepatic injury. Her clinical condition continued to deteriorate, and she was transferred to another hospital for transplant evaluation.
Case
A 50-year-old Hispanic woman with a history of rheumatoid arthritis (RA), for which she was not currently taking medication, was referred to the ED by her primary care physician (PCP) for evaluation of generalized pruritus and jaundice, and an abnormal hepatic function panel.
The patient’s recent history was significant for a positive tuberculosis test (purified protein derivative [PPD], 13 mm), for which she had been on prophylactic medication. Laboratory evaluation taken during the patient’s recent follow-up visit with her PCP revealed the following significant hepatic abnormalities: total bilirubin, 20.0 mg/dL; direct bilirubin, 16.4 mg/dL; international normalized ratio, 2.9; aspartate aminotransferase, greater than 2,000 IU/L; and alanine aminotransferase, greater than 2,000 IU/L. The patient had no history of hepatic disease, and a hepatitis panel obtained in the ED was unremarkable.
Can this be drug-induced liver injury?
Drug-induced liver injury (DILI) accounts for nearly 50% of cases of acute liver failure in the United States.1 According to the National Institutes of Health database of drugs, supplements, and herbal medications acetaminophen is the most common drug associated with hepatotoxicity in the United States, whereas amoxicillin-clavulanate is the most common implicated drug worldwide.1,2 The histological pattern of DILI varies by drug (Table).3
Who is susceptible to drug-induced liver injury?
The factors that help predict DILI include drug pharmacokinetics and metabolism, as well as patient age, sex, and comorbidities. Although some patients are at an increased risk of DILI, it is extraordinarily difficult to accurately predict which patients will develop it. In general, there is a positive correlation between age and risk of developing DILI. For example, in a large US-based tuberculosis study, the incidence of isoniazid (INH)-induced hepatotoxicity was 4.4 per 1,000 patients aged 25 to 34 years. Patients older than age 50 years had a 20.83 per 1,000 incidence of DILI, and women also appear to be at increased risk.4
Pharmacogenetic factors affecting drug metabolism such as the specific cytochrome profile and acetylator status of an individual also influence a patient’s risk of developing DILI. Although our understanding of these issues is growing rapidly, our ability to apply this knowledge to the clinical venue is limited by the available technology, regulatory requirements, and cost.
Case Continuation
A detailed, careful history-taking in the ED revealed that, 2 months prior, the patient had been started on INH, rifampin, and pyridoxine for latent tuberculosis. She had been taking methotrexate for the RA but discontinued it 3 months ago because of the positive PPD. When routine outpatient laboratory testing results demonstrated significant hepatic dysfunction, the patient’s PCP advised her to immediately discontinue her medications and referred her to the ED for further evaluation and management.
By what mechanism does INH cause DILI?
Acute INH-associated hepatitis primarily results from the direct hepatotoxic effects of INH metabolites. Isoniazid is metabolized in the liver via N-acetylation to acetylisoniazid (Figure). Oxidation of this compound in the liver leads to an accumulation of the hepatotoxic metabolites acetylhydrazine and hydrazine.5,6
Is there a role for N-acetylcysteine in INH hepatotoxicity?
No antidote is specifically designed to treat INH-induced hepatotoxicity, and management is largely supportive. Observation for progressive liver failure is indicated and evaluation for liver transplant may become necessary.
N-acetylcysteine (NAC) has a clear role in preventing hepatotoxicity from acetaminophen overdose through its ability to act as a precursor for the synthesis of glutathione—a compound that protects hepatocytes from oxidative damage. In advanced acetaminophen-toxic patients and those with non-acetaminophen toxicity, NAC has nonspecific effects that promote healing through several mechanisms, including anti-inflammatory effect and enhanced hepatic perfusion. Though there are no studies that specifically evaluate the role of NAC in patients with INH-induced hepatotoxicity, it is commonly and appropriately administered for its aforementioned nonspecific effects.8 Common side effects from NAC administration include nausea, vomiting, and diarrhea, which are generally treatable with symptomatic and supportive care.
Case Conclusion
The patient was admitted to the hepatology service for continued clinical care. Although she received NAC, hepatic function testing showed only mild improvement. Additional etiologies of liver failure were investigated, including a computed tomography scan of the abdomen/pelvis and an abdominal ultrasound with Doppler. Both studies were negative for any pathology, and autoimmune laboratory studies were likewise unremarkable.
The patient underwent a liver biopsy, which revealed inflammation and scattered eosinophils suggestive of drug-induced hepatic injury. Her clinical condition continued to deteriorate, and she was transferred to another hospital for transplant evaluation.
1. Lee WM. Drug-induced acute liver failure. Clin Liver Dis. 2013;17(4):575-586, viii. doi:10.1016/j.cld.2013.07.001.
2. National Institutes of Health Web site. LiverTox: Clinical and research information on drug-induced liver injury. https://livertox.nlm.nih.gov/. Updated February 10, 2017. Accessed October 12, 2017.
3. Ansari JA, Sayyed M, Sayeed F. Management of non alcoholic fatty liver diseases and their complications. Int J Pharmacol. 2011;7:579-588. doi:10.3923/ijp.2011.579.588.
4. Fountain FF, Tolley E, Chrisman CR, Self TH. Isoniazid hepatotoxicity associated with treatment of latent tuberculosis infection: a 7-year evaluation from a public health tuberculosis clinic. Chest. 2005;128(1):116-123. doi:10.1378/chest.128.1.116.
5. Hernon CH. Antituberculous medications. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR, eds. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015:787-796.
6. Teixeira RL, Morato RG, Cabello PH, et al. Genetic polymorphisms of NAT2, CYP2E1 and GST enzymes and the occurrence of antituberculosis drug-induced hepatitis in Brazilian TB patients. Mem Inst Oswaldo Cruz. 2011;106(6):716-724.
7. Mitchell JR, Thorgeirsson UP, Black M, et al. Increased incidence of isoniazid hepatitis in rapid acetylators: possible relation to hydranize metabolites. Clin Pharmacol Ther. 1975;18(1):70-79.
8. Lee WM, Hynan LS, Rossaro L, et al; Acute Liver Failure Study Group. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology. 2009;137(3):856-864. doi:10.1053/j.gastro.2009.06.006.
1. Lee WM. Drug-induced acute liver failure. Clin Liver Dis. 2013;17(4):575-586, viii. doi:10.1016/j.cld.2013.07.001.
2. National Institutes of Health Web site. LiverTox: Clinical and research information on drug-induced liver injury. https://livertox.nlm.nih.gov/. Updated February 10, 2017. Accessed October 12, 2017.
3. Ansari JA, Sayyed M, Sayeed F. Management of non alcoholic fatty liver diseases and their complications. Int J Pharmacol. 2011;7:579-588. doi:10.3923/ijp.2011.579.588.
4. Fountain FF, Tolley E, Chrisman CR, Self TH. Isoniazid hepatotoxicity associated with treatment of latent tuberculosis infection: a 7-year evaluation from a public health tuberculosis clinic. Chest. 2005;128(1):116-123. doi:10.1378/chest.128.1.116.
5. Hernon CH. Antituberculous medications. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR, eds. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015:787-796.
6. Teixeira RL, Morato RG, Cabello PH, et al. Genetic polymorphisms of NAT2, CYP2E1 and GST enzymes and the occurrence of antituberculosis drug-induced hepatitis in Brazilian TB patients. Mem Inst Oswaldo Cruz. 2011;106(6):716-724.
7. Mitchell JR, Thorgeirsson UP, Black M, et al. Increased incidence of isoniazid hepatitis in rapid acetylators: possible relation to hydranize metabolites. Clin Pharmacol Ther. 1975;18(1):70-79.
8. Lee WM, Hynan LS, Rossaro L, et al; Acute Liver Failure Study Group. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology. 2009;137(3):856-864. doi:10.1053/j.gastro.2009.06.006.
Carbon Monoxide: The Other Silent Killer
Case Presentations
Case 1: Smoke Inhalation (Carbon Monoxide and Cyanide)
A 50-year-old woman was pulled from the window of a burning building and found to be in cardiac arrest with pulseless electrical activity. Standard advanced cardiac life-support was started, and infusion of intra-osseous hydroxocobalamin (OHCob) was administered at the time of intubation because of the concern for cyanide (CN) gas exposure during smoke inhalation. Return of spontaneous circulation occurred before arrival at the hospital.
Upon presentation to the ED, the patient’s vital signs were: initial blood pressure (BP), 92/47 mm Hg; heart rate (HR), 112 beats/min; respiratory rate (RR), 31 breaths/min; and temperature (T), 99.7°F. Following intubation, the patient’s oxygen saturation (SaO2) on pulse oximetry (POX) was 93%, and her fraction of inspired oxygen (FiO2) was 100%.
On physical examination, the patient’s face was covered with soot. The lung sounds were equal and clear to auscultation bilaterally. The neurological examination was significant for a Glasgow Coma Scale of 3, without administered sedation, and there were no signs of dermal burns. Initial arterial blood gas (ABG) results were: pH, 7.06; carbon dioxide partial pressure (PCO2), 58 mm Hg; partial pressure of oxygen (PO2), 152 mm Hg; bicarbonate (HCO3), 17 mm Hg; SaO2, 98% (after intubation); FiO2, 100%; carboxyhemoglobin (COHb), 30%; and lactate, 14 mmol/L.
Case 2: Household Misadventure (Carbon Monoxide)
Several days after disabling the carbon monoxide (CO) detector in his home to silence the alarm that had continued to sound, a 67-year-old man developed weakness and called his local fire department. Upon arrival at the man’s home, the fire department confirmed an ambient air CO gas concentration over 200 ppm. Emergency medical services (EMS) promptly brought the patient to the local ED for evaluation and treatment.
Shortly after arrival at the ED, the patient’s weakness had resolved. His vital signs at examination were: BP, 154/85 mm Hg; HR, 79 beats/min; RR, 15 breaths/min; and T, 98.8°F. The patient’s COHb level was 28% with administration of 100% oxygen (O2) via a nonrebreather mask (NRBM).
Carbon Monoxide Toxicity
Carbon monoxide is a toxin of considerable importance to emergency physicians (EPs). The diagnosis at times can be challenging, the interpretation of COHb can be confusing, and the role of hyperbaric oxygen (HBO) therapy in the treatment of CO poisoning remains controversial.
Natural Sources
Carbon monoxide is formed from the incomplete combustion of organic (carbonaceous) fuels, such as charcoal, wood, petroleum distillates (gasoline, kerosene, diesel fuel), and natural gas. Though the majority of atmospheric CO comes from natural sources (eg, volcanoes, forest fires, marsh gases), poisoning exposures are primarily due to man-made CO.
Man-Made Sources
Motor vehicle exhaust is the most abundant source of man-made CO, and exposures to exhaust fumes are common causes of both intentional and unintentional poisonings and death. Other frequent sources of CO poisoning include smoke inhalation from house fires; inadequate ventilation during use of kerosene space heaters; charcoal grills or hibachis; burning wood or charcoal; fuel-powered tools such as generators, fork lifts, and chain saws; or faulty (natural or bottled) gas appliances, such as stoves, furnaces, or water heaters (Table 1). Though propane is known to burn more cleanly than natural gas (ie, less harmful to the environment), it still can produce CO.

Though neither electrical appliances nor “gas leaks” are sources of CO, like CO, natural gas (mostly methane) and bottle gas (propane) are odorless, tasteless, and colorless. Utility companies add sulfur containing mercaptans to natural gas so that leaks can be detected, but CO is only formed when the fuel is burned in a gas-powered appliance.
Endogenous Carbon Monoxide
Endogenous CO production can occur from catabolism of heme or from hepatic metabolism of methylene chloride, but exposures to this solvent are unlikely to generate COHb concentrations above 10%.
Epidemiology
The incidence of CO poisoning is likely more frequent than documented since many cases of minor exposures are unreported due to self-limiting effects and/or the vague, nonspecific nature of symptoms associated with minor exposures. In 2015, US Poison Control Centers reported over 14,000 cases of CO poisoning, only 43% of which were treated in a health care facility.1 The vast majority of exposures (97%) were unintentional and resulted in 52 deaths (0.398%).1
Data from hospitalized patients in 2007 revealed that over 200,000 ED visits and 22,000 hospitalizations were possibly associated with unintentional, non-fire-related CO exposures.2 Approximately 10% of the exposures in each of these populations were confirmed by specific International Classification of Diseases Medical E codes.2
Regardless of dataset, ED visits due to CO exposure are most common in young adults and women, occur in winter months from exposure in and around homes, and result in discharge from the ED. Elderly patients have the highest rate of hospital admission.
Carbon monoxide poisoning has long been considered a leading cause of poisoning death, though numbers appear to be declining, and CO was responsible for fewer deaths than opioids in 2017.2 The National Center for Health Statistics reported 56,133 CO-related deaths from 1979 through 1988—an average of 5,600 per year.3,4 Of these, 46% were from suicide; 28% were related to burns or house fires; and 21% (11,547) were characterized as unintentional. Motor vehicle exhaust was associated with 57% of the unintentional deaths. A more recent analysis of unintentional exposures reported 2,244 deaths during the period of 2010 to 2015—an average of 374 deaths per year (393 in 2015).5
Preventive measures are likely responsible for the significant decline in non-fire-related CO poisoning deaths from the early 1970s through the 1990s. The introduction of catalytic converters in automobiles in 1975 and O2 sensors in 1981 eventually reduced automotive CO emissions by 95% compared to pre-1975 vehicles.6 Both unintentional death and suicide rates associated with CO from motor vehicles subsequently declined by 81% and 43%, respectively. The lower decline in suicidal deaths serves as a reminder that intentional exposure to motor vehicles remains dangerous and potentially lethal.
Pathophysiology/Mechanisms of Toxicity
Carbon monoxide is a colorless, odorless gas that readily reaches the bloodstream during alveolar gas exchange. Since absorption is rapid, exposures to high CO concentrations can produce toxicity within minutes, though exposure severity is related to both inspired CO concentration and duration of exposure.
Endogenous Elimination
Carbon monoxide is eliminated from the body in expired air, with an elimination half-life dependent on FiO2 and atmospheric pressure. Accordingly, COHb decreases with a half-life (all approximate) of 4 to 6 hours when patients are breathing room air (21% O2), 60 to 90 minutes with O2 delivery at 95% to 100%, and 20 to 40 minutes under hyperbaric conditions (2.5-3.0 atmospheres absolute [ATA]).
Effect on Hemoglobin
Once absorbed, CO has an affinity for hemoglobin (Hb) that is over 200 times greater than does O2.7 The formation of COHb results in both a decreased O2-carrying capacity of Hb at the sites where O2 would have been, and because of its new configuration, COHb does not allow currently bound O2 to be offloaded. This is graphically represented by a shift of the O2-Hb dissociation curve to the left. In addition, CO continues to be bound by other intracellular heme molecules in myoglobin of skeletal and myocardial muscle, and the cytochrome oxidase system in mitochondria.8
Immunologic and Inflammatory Effects
Carbon monoxide poisoning results in a cascade of immunologic and inflammatory effects, such as generation of nitric oxide, lipid peroxidation from neutrophils, mitochondrial oxidative stress, and apoptosis. These effects result in cellular asphyxia in all organs, but the most emergent life-threatening concerns are ischemia to the brain and heart.
Severity of Toxicity and Exposure
As previously noted, the severity of CO poisoning is dose-dependent, meaning that it is related to the concentration of CO in inspired air and the duration of the exposure. Carbon monoxide is typically absent in fresh air, but levels may approach 2 to 5 ppm due to cooking, wood burning, mild air pollution, etc. The source of levels above 5 ppm should generally be investigated.
Maximum safe exposure levels for workers over an 8-hour period range from 25 to 50 ppm. Exposures to CO levels above 50 to 100 ppm are likely to elicit symptoms in most patients, depending on duration of the exposure. Carbon monoxide levels of 200 ppm may result in a mild headache after 2 to 3 hours of exposure, and a more severe headache and nausea after 1 to 2 hours of exposures to 400 ppm of CO.
Accordingly, home CO detectors use a combination of ppm and time for alarms, and they may not sound an alarm at 40 ppm until the level persists for 8 or so hours. Home CO detectors, however, will sound an alarm immediately when a level of 80 to 100 ppm is reached.
Clinical Presentation
Acute Exposure
Acute exposure to CO causes a variety of effects that are largely nonspecific, as there is no toxic syndrome (toxidrome) considered pathognomonic for CO poisoning. Ambient CO levels, duration of exposure, minute ventilation, presence of other toxic gases, and patient comorbidities can all contribute to the severity of exposure and presenting signs and symptoms. Effects associated with mild poisoning include headache, dizziness, blurred vision, fatigue or weakness, nausea, and shortness of breath. Patients with pre-existing respiratory, cardiovascular (CV), or neurological compromise are likely to present with more pronounced symptoms. In either case, these complaints may easily be confused with a viral illness, emphasizing the importance of eliciting a history of potential exposure to CO, particularly when multiple patients are involved.
As the concentration of COHb increases, more significant clinical effects can be expected, including tachycardia, chest pain, hypotension, dysrhythmias, lethargy, coma, apnea, and seizures. Hypoxia can result in myocardial injury, cerebral edema, stroke, and acute pulmonary and kidney injury.
Following acute exposure, the severity of effects correlates with the peak pretreatment COHb concentration. However, the peak concentration is usually unknown, since most patients with significant exposures will have some time period elapsed between the exposure and the determination of COHb, and the COHb will have declined at a rate depending on FiO2 and minute ventilation. In these circumstances, COHb is a poor indicator for HBO therapy and outcome.
Delayed Neurological Sequelae
Persistent, recurrent, or delayed (following period of no symptoms) neurological effects can occur in up to 40% of cases, and patients with significant exposures (eg, loss of consciousness) appear to be at greatest risk. These effects most often occur within the first 3 weeks following exposure, and have been known to persist for months to years. Such effects include headache, dizziness, impaired memory or cognition, and emotional lability. Predicting which factors in CO exposure and/or treatments can be modified to prevent neurological sequelae remains challenging.
Diagnostic Testing
Pulse Co-oximetry
Prehospital care POX typically reads COHb as oxyhemoglobin, thereby displaying a normal SaO2.9 Noninvasive CO pulse co-oximetry using a pulse oximeter (Rad-57, Masimo Corporation) provides a reading between –6 to +4 of the true COHb with a false-positive rate of 11% and false-negative rate of 46%.10 This high false-negative rate makes noninvasive CO pulse co-oximetry a poor tool to rule out a CO exposure.11 If OHCob has been administered due to concerns for CN poisoning (smoke inhalation), concentrations of COHb detected by a co-oximeter medical device may be decreased, as noted by a mean decrease of 1% in healthy volunteers exposed to OHCob only.12
Venous and Arterial Blood Gas Testing
For a patient in the hospital, exposure to CO can rapidly be determined using co-oximetry to measure COHb in a venous or arterial sample. Obtaining a venous sample may be a more practical approach, as other venous measurements will likely also be obtained. Baseline “normal” COHb levels should be less than 5%, but may be up to 8% to 10% in tobacco smokers.
Other Laboratory Studies
Other important laboratory tests that should be obtained are a complete blood count, lactate level, venous blood gas, and basic metabolic panel (to assess acid/base status). In two retrospective studies of patients exposed to CO, elevated lactate levels were associated with altered mental status.13,14 However, elevated lactate levels were not seen in a majority of patients with CO poisoning.
In addition, CN exposure should be considered when the lactate level is greater than 8 mmol/L, particularly in patients with smoke inhalation.15 Troponin I and creatinine phosphokinase tests can be used to screen for myocardial or skeletal muscle injury.
Effect of Hydroxocobalamin on Laboratory Evaluation
It is important to be cautious when interpreting the results of laboratory studies in patients who have been given OHCob due to the potential co-exposure to CN (smoke inhalation). The red discoloration of body fluids after OHCob administration makes laboratory evaluation by spectrophotometric techniques erroneous.16 Of greatest concern is the accuracy of COHb concentrations.17 In a study using rabbit models by Lee et al,17 OHCob administration was shown to falsely increase COHb concentrations.

Livshits et al18 reported conflicting effects on COHb in two human cases. In the first case, the patient’s true COHb was 93% lower (2.5% vs 34.9%) following administration of 5 g of OHCob, as measured with a rapid blood gas analyzer. In the second patient, COHb was 76% lower (10.7% vs 44%) following OHCob administration, which was also measured by a blood gas analyzer. Both of these cases illustrate lower true COHb concentrations than would be expected following the administration of only supplemental O2.
In a controlled experiment by Pace et al19 examining the effects of OHCob on measurement of COHb at both physiological (3%) and pathological (30% and 50%) concentrations in human blood samples, the degree of interference depended on the type of co-oximeter used, the degree of COHb elevation (at pathological levels only), and the concentration of OHCob added. Other studies, including an evaluation of OHCob interference by Carlsson et al20 using nine different analyzers have confirmed the interference of OHCob on photometric assays. Of particular clinical importance, a falsely increased lactate level was seen after true lactate levels were found to be below 4.8 mmol/L (but not greater) using spectrophotometric or electrochemical detection.21 This increase in the false-positive assessment of the degree of toxicity could lead unnecessary escalation of care.
These studies emphasize the need to exercise caution when interpreting laboratory test results following OHCob administration. Ideally, it would be best if blood samples were obtained prior to OHCob administration by EMS or in the ED, if the clinical scenario allows it.
Imaging Studies
For patients presenting after a closed-space fire, a chest radiograph will help assess for pulmonary injury. The classic finding of CO poisoning on head computed tomography (CT) and magnetic resonance imaging scans is evidence of ischemia in the basal ganglia. The radiographic findings may help determine the diagnosis of the altered mental status patient who presents without a history. An electrocardiogram (ECG) is also useful for detection of myocardial ischemia or dysrhythmias, when signs or symptoms of either are possible from history and physical or cardiac monitoring.
Intentional Inhalation
Intentional inhalation of fumes containing CO is a relatively common mechanism for suicide. In patients who survive, it is important for EPs and other providers to suspect additional means of self-harm. For example, at our institution, we have encountered several patients with self-inflicted trauma after remaining conscious following a medication overdose. Accordingly, patients who have intentionally inhaled CO should also be evaluated for occult medication poisoning (and trauma).
Treatment
The first step in treating a patient with CO poisoning occurs prior to arrival at the ED, when he or she is removed from continued exposure. The second step is assessing whether this is only a CO exposure, or a mixture of gases from combustion in a closed space, that might also contain CN. When CO and CN are combined OHCob is indicated to treat CN toxicity.
Additionally, if the patient is brought to the ED via EMS, O2 therapy will most likely have been initiated en route. In either case, the concentration of COHb may not accurately reflect the magnitude of exposure or prognosis, and should not be used to dictate the level of therapy or disposition. The patient’s vital signs and clinical findings of end organ toxicity should guide the appropriate supportive care.
Supplemental Oxygen Therapy
Initial administration of 100% O2 during assessment of airway, breathing, and circulation is the first step in accelerating the removal of CO from Hb. For patients suffering from smoke inhalation, assessment and establishment of a secure airway when there are signs of soot or burns in the airway must always take precedence over other actions. Continuous cardiac monitoring, POX, observation, and establishment of intravenous access are often needed for detection and management of CV instability or change in mental status in cases of moderate-to-severe CO exposures. Mild exposures with headache, nausea, and flu-like symptoms can be managed with symptomatic treatment and normobaric O2 until resolution of symptoms and improvement in COHb occur.
Hyperbaric Oxygen Therapy
Hyperbaric oxygen therapy involves the delivery of high-flow O2 (typically at 100%) under increased atmospheric pressure (2.5 -3.0 ATA). Oxygen delivered at ambient air pressure (1.0 ATA) is often referred to as normobaric oxygen. Although HBO is best known for its ability to enhance CO elimination, research points to a much more eloquent mitigation of CO toxicity on the molecular level. These mechanisms include an increased amount of dissolved O2 in blood, regeneration of cytochrome oxidase, decreased leukocyte adhesion to microvascular endothelium in the brain, decreased lipid peroxidation in the brain after loss of consciousness, and preservation of adenosine triphosphate.22
For most patients, the majority—if not all—of COHb will be eliminated by the time they present to a suitable HBO chamber. Despite the knowledge that HBO therapy has a positive toxicokinetic effect by increasing the elimination of CO, all of the major, prospective studies on the usefulness of HBO are related to prevention of neuropsychiatric sequelae mediated by immunological and inflammatory effects. The role of HBO in the treatment of CO poisoning has been debated for decades. Multiple studies that differ in methodology, patient populations, delivery of HBO treatments, and assessment of benefits fail to provide a consensus on the role of HBO therapy (Table 2).23-30
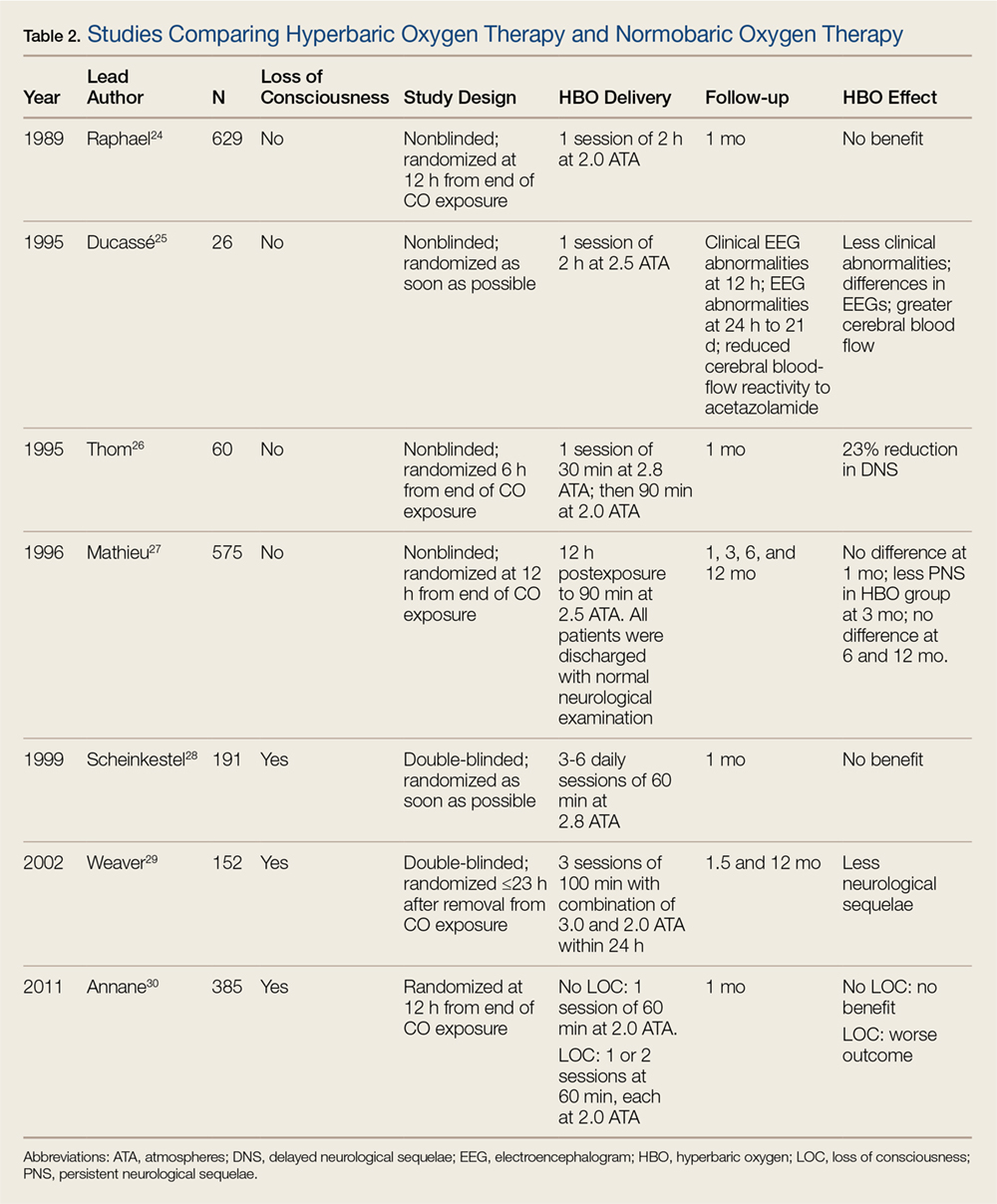
Before transferring a patient to a facility for HBO therapy, the potential risks and benefits of transport must be considered. In a 10-year retrospective study by Sloan et al31 of 297 CO-poisoned patients (mean COHb, 38%) 46% of patients had cardiopulmonary and neurological complications prior to HBO therapy at some point in the transfer pathway. During HBO therapy, 18% of patients had complications that included emesis, agitation requiring sedation, seizures, hypotension, tension pneumothorax, cardiac arrest, cardiac arrhythmias, and myocardial ischemia. It is therefore incumbent that personnel attending patients undergoing HBO therapy for CO poisoning be aware of, and able to manage, this variety of serious effects.
When an HBO chamber is at a clinical site with experts in the field and staff available 24 hours a day, the decision to utilize HBO may easily be made without obstacles. For most EPs, however, this is not the case. Locating and transferring a patient to an HBO center is typically a considerable logistical challenge. For many rural facilities, HBO is just not a timely therapeutic option. Two studies state the benefit of HBO therapy is greatest when starting within 6 hours from the end of the CO exposure.24,26
Identifying those CO-poisoned patients who meet evidence-based criteria for HBO is difficult. Patients with mild CO poisoning will do well without HBO, and critically ill patients will probably not consistently benefit from HBO. However, a pragmatic solution must be considered when efficacy studies are incongruent with conflicting results. When signs of end-organ toxicity from CO are present, but cardiac arrest has not yet occurred and the logistics are streamlined, the benefit of HBO may outweigh the risk.
Signs of end-organ toxicity include syncope, seizures, coma, ischemic changes on ECG, and pregnancy with unresolved maternal distress or fetal distress. Although a COHb level greater than 25% or 15% (pregnant) alone is commonly used as an indication for HBO, this is largely based on opinion. Conversely, HBO is unlikely to be helpful in patients who have been resuscitated after CO-related cardiac arrest.32
Treatment Guidelines
The American College of Emergency Physicians recently developed a position statement regarding the management and treatment of CO poisoning.33 The clinical policy addresses several of the controversies discussed in this review, and provides a level of evidence for each response (Table 3).

Case Conclusions
Case 1 (Smoke Inhalation Due to CO and Cyanide Poisoning)
The patient in this case suffered severe CO and CN toxicity. A head CT scan revealed diffuse edema consistent with anoxic brain injury. After conferring with the family regarding the patient’s condition and prognosis, the decision was made to withdraw life-sustaining therapy and support, and the patient died.
Case 2 (Household Misadventure)
The patient in this case was successfully treated with 100% O2 via a NRBM and was subsequently discharged home within 4 hours from presentation.
Conclusion
Exposures to CO are ubiquitous due to our heavy reliance on carbon combustion, and the manifestations of CO toxicity are protean. Therefore, CO poisoning must be considered more frequently in the differential diagnosis of indiscriminant symptoms affecting the neurological, cardiac, pulmonary, and gastrointestinal systems, especially when multiple patients have similar symptoms.
The diagnosis of CO poisoning is straightforward when a serum COHb level is obtained on a venous or arterial blood sample. Treatment starts when the patient is removed from further CO exposure and breaths normobaric oxygen at ambient levels or supplemented. Because there is no clear evidenced-based indication for HBO therapy, further treatment with HBO is naturally limited by rational constraints.
1. Mowry JB, Spyker DA, Brooks DE, Zimmerman A, Schauben JL. 2015 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 33rd Annual Report. Clin Toxicol. 2016;54(10):924-1109. doi:10.1080/15563650.2016.1245421.
2. Iqbal S, Law HZ, Clower JH, Yip FY, Elixhauser A. Hospital burden of unintentional carbon monoxide poisoning in the United States, 2007. Am J Emerg Med. 2012;30(5):657-664. doi:10.1016/j.ajem.2011.03.003.
3. Cobb N, Etzel RA. Unintentional carbon monoxide-related deaths in the United States, 1979 through 1988. JAMA. 1991;266(5):659-663.
4. Sircar K, Clower J, Shin MK, Bailey C, King M, Yip F. Carbon monoxide poisoning deaths in the United States, 1999 to 2012. Am J Emerg Med. 2015;33(9):1140-1145. doi:10.1016/j.ajem.2015.05.002.
5. Centers for Disease Control and Prevention. Environmental Public Health Tracking Network. Carbon monoxide poisoning emergency department visits. https://ephtracking.cdc.gov/showHome.action. Updated September 8, 2017. Accessed October 18, 2017.
6. Mott JA, Wolfe MI, Alverson CJ, et al. National vehicle emissions policies and practices and declining US carbon monoxide-related mortality. JAMA. 2002;288(8):988-995.
7. Buckley NA, Juurlink DN, Isbister G, Bennett MH, Lavonas EJ. Hyperbaric oxygen for carbon monoxide poisoning. Cochrane Database Syst Rev. 2011;13(4):CD002041. doi:10.1002/14651858.CD002041.pub3.
8. Hampson NB, Piantadosi CA, Thom SR, Weaver LK. Practice recommendations in the diagnosis, management, and prevention of carbon monoxide poisoning. Am J Respir Crit Care Med. 2012;186(11):1095-1101. doi:10.1164/rccm.201207-1284CI.
9. Bozeman WP, Myers RA, Barish RA. Confirmation of the pulse oximetry gap in carbon monoxide poisoning. Ann of Emerg Med. 1997;30(5):608-611.
10. Zaouter C, Zavorsky GS. The measurement of carboxyhemoglobin and methemoglobin using a non-invasive pulse CO-oximeter. Respir Physiol Neurobiol. 2012;182(2-3):88-92. doi:10.1016/j.resp.2012.05.010.
11. Shamir MY, Avramovich A, Smaka T. The current status of continuous noninvasive measurement of total, carboxy, and methemoglobin concentration. Anesth Analg. 2012;114(5);972-978. doi:10.1213/ANE.0b013e318233041a.
12. Cashin BV, Matlock AG, Kang C, Reynolds PS, Wills BK. Effect of hydroxocobalamin on surface oximetry in nonexposed humans. Prehosp Disaster Med. 2013;28(4):367-369. doi:10.1017/S1049023X13003518.
13. Moon JM, Shin MH, Chun BJ. The value of initial lactate in patients with carbon monoxide intoxication: in the emergency department. Hum Exp Toxicol. 2011;30(8):836-843. doi:10.1177/0960327110384527.
14. Doğan NÖ, Savrun A, Levent S, et al. Can initial lactate levels predict the severity of unintentional carbon monoxide poisoning? Hum Exp Toxicol. 2015;34(3):324-329. doi:10.1177/0960327114538986.
15. Baud FJ, Borron SW, Mégarbane B, et al. Value of lactic acidosis in the assessment of the severity of acute cyanide poisoning. Crit Care Med. 2002;30(9):2044-2050. doi:10.1097/01.CCM.0000026325.65944.7D.
16. Fueyo L, Robles J, Aguilar I, Yáñez AM, Socias M, Parera M. Hemolysis index to detect degree of hydroxocobalamin interference with common laboratory tests. J Clin Lab Anal. 2017;31(5):e22089. doi:10.1002/jcla.22089.
17. Lee J, Mukai D, Kreuter K, Mahon S, Tromberg B, Brenner M. Potential interference by hydroxocobalamin on cooximetry hemoglobin measurements during cyanide and smoke inhalation treatments. Ann Emerg Med. 2007;49(6):802-805. doi:10.
1016/j.annemergmed.2006.11.016.
18. Livshits Z, Lugassy DM, Shawn LK, Hoffman RS. Falsely Low Carboxyhemoglobin after Hydroxocobalamin Therapy [Letter]. N Engl J Med. 2012;367(13):1270-1271. doi:10.1056/NEJMc1114820.
19. Pace R, Bon Homme M, Hoffman RS, Lugassy D. Effects of hydroxocobalamin on carboxyhemoglobin measured under physiologic and pathologic conditions. Clin Toxicol (Phila). 2014;52(7):647-650. doi:10.3109/15563650.2014.939659.
20. Carlsson CJ, Hansen HE, Hilsted L, Malm J, Ødum L, Szecsi PB. An evaluation of the interference of hydroxycobalamin with chemistry and co-oximetry tests on nine commonly used instruments. Scand J Clin Lab Invest. 2011;71(5):378-386. doi:10.3109/00365513.2011.573573.
21. Fueyo L, Robles J, Aguilar I, Yáñez AM, Socias M, Parera M. Hemolysis index to detect degree of hydroxocobalamin interference with common laboratory tests. J Clin Lab Anal. 2017;31(5). doi:10.1002/jcla.22089.
22. Tomaszewski C. Carbon monoxide. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR, eds. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015:1581-1593.
23. Hampson NB, Mathieu D, Piantodosi CA et al. Carbon monoxide poisoning: interpretation of randomized clinical trials and unresolved treatment issues. Undersea Hyperb Med. 2001;28(3):157-164.
24. Raphael JC, Elkharrat D, Jars-Guincestre MC, et al. Trial of normobaric and hyperbaric oxygen for acute carbon monoxide intoxication. Lancet. 1989;2(8660):414-419.
25. Ducassé JL, Celsis P, Marc-Vergnes JP. Non-comatose patients with acute carbon monoxide poisoning: hyperbaric or normobaric oxygenation? Undersea Hyperb Med. 1995;22(1):9-15.
26. Thom SR, Taber RL, Mendiguren II, Clark JM, Hardy KR, Fisher AB. Delayed neuropsychologic sequelae after carbon monoxide poisoning: prevention by treatment with hyperbaric oxygen. Ann Emerg Med. 1995;25(4):474-480.
27. Mathieu D, Wattel F, Mathieu-Nolf M, et al. Randomized prospective study comparing the effects of HBO versus 12 hours of nbp in non comatose CO poisoned patients: results of the interim analysis. Undersea Hyperb Med. 1996;23(Suppl:7-8).
28. Scheinkestel CD, Bailey M, Myles PS, et al. Hyperbaric or normobaric oxygen for acute carbon monoxide poisoning: a randomized controlled clinical trial. Med J Aust. 1999;170(5):203-210.
29. Weaver LK, Hopkins RO, Chan KJ, et al. Hyperbaric oxygen for acute carbon monoxide poisoning. N Engl J Med. 2002;347(14):1057-1067. doi:10.1056/NEJMoa013121.
30. Annane D, Chadda K, Gajdos P, Jars-Guincestre MC, Chevret S, Raphael JC. Hyperbaric oxygen therapy for acute domestic carbon monoxide poisoning: two randomized controlled trials. Intensive Care Med. 2011;37(3):486-492. doi:10.1007/s00134-010-2093-0.
31. Sloan EP, Murphy DG, Hart R, et al. Complications and protocol considerations in carbon monoxide-poisoned patients who require hyperbaric oxygen therapy: report from a ten-year experience. Ann Emerg Med. 1989;18(6):629-634.
32. Hampson NB, Zmaeff JL. Outcome of patients experiencing cardiac arrest with carbon monoxide poisoning treated with hyperbaric oxygen. Ann Emerg Med. 2001;38(1):36-41. doi:10.1067/mem.2001.115532.
33. Wolf SJ, Maloney GE, Shih RD, Shy BD, Brown MD; American College of Emergency Physicians. Clinical policy: critical issues in the evaluation and management of adult patients presenting to the emergency department with acute carbon monoxide poisoning. Ann Emerg Med. pii:S0196-0644(17)30332-3. doi:10.1016/j.annemergmed.2017.03.036.
Case Presentations
Case 1: Smoke Inhalation (Carbon Monoxide and Cyanide)
A 50-year-old woman was pulled from the window of a burning building and found to be in cardiac arrest with pulseless electrical activity. Standard advanced cardiac life-support was started, and infusion of intra-osseous hydroxocobalamin (OHCob) was administered at the time of intubation because of the concern for cyanide (CN) gas exposure during smoke inhalation. Return of spontaneous circulation occurred before arrival at the hospital.
Upon presentation to the ED, the patient’s vital signs were: initial blood pressure (BP), 92/47 mm Hg; heart rate (HR), 112 beats/min; respiratory rate (RR), 31 breaths/min; and temperature (T), 99.7°F. Following intubation, the patient’s oxygen saturation (SaO2) on pulse oximetry (POX) was 93%, and her fraction of inspired oxygen (FiO2) was 100%.
On physical examination, the patient’s face was covered with soot. The lung sounds were equal and clear to auscultation bilaterally. The neurological examination was significant for a Glasgow Coma Scale of 3, without administered sedation, and there were no signs of dermal burns. Initial arterial blood gas (ABG) results were: pH, 7.06; carbon dioxide partial pressure (PCO2), 58 mm Hg; partial pressure of oxygen (PO2), 152 mm Hg; bicarbonate (HCO3), 17 mm Hg; SaO2, 98% (after intubation); FiO2, 100%; carboxyhemoglobin (COHb), 30%; and lactate, 14 mmol/L.
Case 2: Household Misadventure (Carbon Monoxide)
Several days after disabling the carbon monoxide (CO) detector in his home to silence the alarm that had continued to sound, a 67-year-old man developed weakness and called his local fire department. Upon arrival at the man’s home, the fire department confirmed an ambient air CO gas concentration over 200 ppm. Emergency medical services (EMS) promptly brought the patient to the local ED for evaluation and treatment.
Shortly after arrival at the ED, the patient’s weakness had resolved. His vital signs at examination were: BP, 154/85 mm Hg; HR, 79 beats/min; RR, 15 breaths/min; and T, 98.8°F. The patient’s COHb level was 28% with administration of 100% oxygen (O2) via a nonrebreather mask (NRBM).
Carbon Monoxide Toxicity
Carbon monoxide is a toxin of considerable importance to emergency physicians (EPs). The diagnosis at times can be challenging, the interpretation of COHb can be confusing, and the role of hyperbaric oxygen (HBO) therapy in the treatment of CO poisoning remains controversial.
Natural Sources
Carbon monoxide is formed from the incomplete combustion of organic (carbonaceous) fuels, such as charcoal, wood, petroleum distillates (gasoline, kerosene, diesel fuel), and natural gas. Though the majority of atmospheric CO comes from natural sources (eg, volcanoes, forest fires, marsh gases), poisoning exposures are primarily due to man-made CO.
Man-Made Sources
Motor vehicle exhaust is the most abundant source of man-made CO, and exposures to exhaust fumes are common causes of both intentional and unintentional poisonings and death. Other frequent sources of CO poisoning include smoke inhalation from house fires; inadequate ventilation during use of kerosene space heaters; charcoal grills or hibachis; burning wood or charcoal; fuel-powered tools such as generators, fork lifts, and chain saws; or faulty (natural or bottled) gas appliances, such as stoves, furnaces, or water heaters (Table 1). Though propane is known to burn more cleanly than natural gas (ie, less harmful to the environment), it still can produce CO.
Though neither electrical appliances nor “gas leaks” are sources of CO, like CO, natural gas (mostly methane) and bottle gas (propane) are odorless, tasteless, and colorless. Utility companies add sulfur containing mercaptans to natural gas so that leaks can be detected, but CO is only formed when the fuel is burned in a gas-powered appliance.
Endogenous Carbon Monoxide
Endogenous CO production can occur from catabolism of heme or from hepatic metabolism of methylene chloride, but exposures to this solvent are unlikely to generate COHb concentrations above 10%.
Epidemiology
The incidence of CO poisoning is likely more frequent than documented since many cases of minor exposures are unreported due to self-limiting effects and/or the vague, nonspecific nature of symptoms associated with minor exposures. In 2015, US Poison Control Centers reported over 14,000 cases of CO poisoning, only 43% of which were treated in a health care facility.1 The vast majority of exposures (97%) were unintentional and resulted in 52 deaths (0.398%).1
Data from hospitalized patients in 2007 revealed that over 200,000 ED visits and 22,000 hospitalizations were possibly associated with unintentional, non-fire-related CO exposures.2 Approximately 10% of the exposures in each of these populations were confirmed by specific International Classification of Diseases Medical E codes.2
Regardless of dataset, ED visits due to CO exposure are most common in young adults and women, occur in winter months from exposure in and around homes, and result in discharge from the ED. Elderly patients have the highest rate of hospital admission.
Carbon monoxide poisoning has long been considered a leading cause of poisoning death, though numbers appear to be declining, and CO was responsible for fewer deaths than opioids in 2017.2 The National Center for Health Statistics reported 56,133 CO-related deaths from 1979 through 1988—an average of 5,600 per year.3,4 Of these, 46% were from suicide; 28% were related to burns or house fires; and 21% (11,547) were characterized as unintentional. Motor vehicle exhaust was associated with 57% of the unintentional deaths. A more recent analysis of unintentional exposures reported 2,244 deaths during the period of 2010 to 2015—an average of 374 deaths per year (393 in 2015).5
Preventive measures are likely responsible for the significant decline in non-fire-related CO poisoning deaths from the early 1970s through the 1990s. The introduction of catalytic converters in automobiles in 1975 and O2 sensors in 1981 eventually reduced automotive CO emissions by 95% compared to pre-1975 vehicles.6 Both unintentional death and suicide rates associated with CO from motor vehicles subsequently declined by 81% and 43%, respectively. The lower decline in suicidal deaths serves as a reminder that intentional exposure to motor vehicles remains dangerous and potentially lethal.
Pathophysiology/Mechanisms of Toxicity
Carbon monoxide is a colorless, odorless gas that readily reaches the bloodstream during alveolar gas exchange. Since absorption is rapid, exposures to high CO concentrations can produce toxicity within minutes, though exposure severity is related to both inspired CO concentration and duration of exposure.
Endogenous Elimination
Carbon monoxide is eliminated from the body in expired air, with an elimination half-life dependent on FiO2 and atmospheric pressure. Accordingly, COHb decreases with a half-life (all approximate) of 4 to 6 hours when patients are breathing room air (21% O2), 60 to 90 minutes with O2 delivery at 95% to 100%, and 20 to 40 minutes under hyperbaric conditions (2.5-3.0 atmospheres absolute [ATA]).
Effect on Hemoglobin
Once absorbed, CO has an affinity for hemoglobin (Hb) that is over 200 times greater than does O2.7 The formation of COHb results in both a decreased O2-carrying capacity of Hb at the sites where O2 would have been, and because of its new configuration, COHb does not allow currently bound O2 to be offloaded. This is graphically represented by a shift of the O2-Hb dissociation curve to the left. In addition, CO continues to be bound by other intracellular heme molecules in myoglobin of skeletal and myocardial muscle, and the cytochrome oxidase system in mitochondria.8
Immunologic and Inflammatory Effects
Carbon monoxide poisoning results in a cascade of immunologic and inflammatory effects, such as generation of nitric oxide, lipid peroxidation from neutrophils, mitochondrial oxidative stress, and apoptosis. These effects result in cellular asphyxia in all organs, but the most emergent life-threatening concerns are ischemia to the brain and heart.
Severity of Toxicity and Exposure
As previously noted, the severity of CO poisoning is dose-dependent, meaning that it is related to the concentration of CO in inspired air and the duration of the exposure. Carbon monoxide is typically absent in fresh air, but levels may approach 2 to 5 ppm due to cooking, wood burning, mild air pollution, etc. The source of levels above 5 ppm should generally be investigated.
Maximum safe exposure levels for workers over an 8-hour period range from 25 to 50 ppm. Exposures to CO levels above 50 to 100 ppm are likely to elicit symptoms in most patients, depending on duration of the exposure. Carbon monoxide levels of 200 ppm may result in a mild headache after 2 to 3 hours of exposure, and a more severe headache and nausea after 1 to 2 hours of exposures to 400 ppm of CO.
Accordingly, home CO detectors use a combination of ppm and time for alarms, and they may not sound an alarm at 40 ppm until the level persists for 8 or so hours. Home CO detectors, however, will sound an alarm immediately when a level of 80 to 100 ppm is reached.
Clinical Presentation
Acute Exposure
Acute exposure to CO causes a variety of effects that are largely nonspecific, as there is no toxic syndrome (toxidrome) considered pathognomonic for CO poisoning. Ambient CO levels, duration of exposure, minute ventilation, presence of other toxic gases, and patient comorbidities can all contribute to the severity of exposure and presenting signs and symptoms. Effects associated with mild poisoning include headache, dizziness, blurred vision, fatigue or weakness, nausea, and shortness of breath. Patients with pre-existing respiratory, cardiovascular (CV), or neurological compromise are likely to present with more pronounced symptoms. In either case, these complaints may easily be confused with a viral illness, emphasizing the importance of eliciting a history of potential exposure to CO, particularly when multiple patients are involved.
As the concentration of COHb increases, more significant clinical effects can be expected, including tachycardia, chest pain, hypotension, dysrhythmias, lethargy, coma, apnea, and seizures. Hypoxia can result in myocardial injury, cerebral edema, stroke, and acute pulmonary and kidney injury.
Following acute exposure, the severity of effects correlates with the peak pretreatment COHb concentration. However, the peak concentration is usually unknown, since most patients with significant exposures will have some time period elapsed between the exposure and the determination of COHb, and the COHb will have declined at a rate depending on FiO2 and minute ventilation. In these circumstances, COHb is a poor indicator for HBO therapy and outcome.
Delayed Neurological Sequelae
Persistent, recurrent, or delayed (following period of no symptoms) neurological effects can occur in up to 40% of cases, and patients with significant exposures (eg, loss of consciousness) appear to be at greatest risk. These effects most often occur within the first 3 weeks following exposure, and have been known to persist for months to years. Such effects include headache, dizziness, impaired memory or cognition, and emotional lability. Predicting which factors in CO exposure and/or treatments can be modified to prevent neurological sequelae remains challenging.
Diagnostic Testing
Pulse Co-oximetry
Prehospital care POX typically reads COHb as oxyhemoglobin, thereby displaying a normal SaO2.9 Noninvasive CO pulse co-oximetry using a pulse oximeter (Rad-57, Masimo Corporation) provides a reading between –6 to +4 of the true COHb with a false-positive rate of 11% and false-negative rate of 46%.10 This high false-negative rate makes noninvasive CO pulse co-oximetry a poor tool to rule out a CO exposure.11 If OHCob has been administered due to concerns for CN poisoning (smoke inhalation), concentrations of COHb detected by a co-oximeter medical device may be decreased, as noted by a mean decrease of 1% in healthy volunteers exposed to OHCob only.12
Venous and Arterial Blood Gas Testing
For a patient in the hospital, exposure to CO can rapidly be determined using co-oximetry to measure COHb in a venous or arterial sample. Obtaining a venous sample may be a more practical approach, as other venous measurements will likely also be obtained. Baseline “normal” COHb levels should be less than 5%, but may be up to 8% to 10% in tobacco smokers.
Other Laboratory Studies
Other important laboratory tests that should be obtained are a complete blood count, lactate level, venous blood gas, and basic metabolic panel (to assess acid/base status). In two retrospective studies of patients exposed to CO, elevated lactate levels were associated with altered mental status.13,14 However, elevated lactate levels were not seen in a majority of patients with CO poisoning.
In addition, CN exposure should be considered when the lactate level is greater than 8 mmol/L, particularly in patients with smoke inhalation.15 Troponin I and creatinine phosphokinase tests can be used to screen for myocardial or skeletal muscle injury.
Effect of Hydroxocobalamin on Laboratory Evaluation
It is important to be cautious when interpreting the results of laboratory studies in patients who have been given OHCob due to the potential co-exposure to CN (smoke inhalation). The red discoloration of body fluids after OHCob administration makes laboratory evaluation by spectrophotometric techniques erroneous.16 Of greatest concern is the accuracy of COHb concentrations.17 In a study using rabbit models by Lee et al,17 OHCob administration was shown to falsely increase COHb concentrations.
Livshits et al18 reported conflicting effects on COHb in two human cases. In the first case, the patient’s true COHb was 93% lower (2.5% vs 34.9%) following administration of 5 g of OHCob, as measured with a rapid blood gas analyzer. In the second patient, COHb was 76% lower (10.7% vs 44%) following OHCob administration, which was also measured by a blood gas analyzer. Both of these cases illustrate lower true COHb concentrations than would be expected following the administration of only supplemental O2.
In a controlled experiment by Pace et al19 examining the effects of OHCob on measurement of COHb at both physiological (3%) and pathological (30% and 50%) concentrations in human blood samples, the degree of interference depended on the type of co-oximeter used, the degree of COHb elevation (at pathological levels only), and the concentration of OHCob added. Other studies, including an evaluation of OHCob interference by Carlsson et al20 using nine different analyzers have confirmed the interference of OHCob on photometric assays. Of particular clinical importance, a falsely increased lactate level was seen after true lactate levels were found to be below 4.8 mmol/L (but not greater) using spectrophotometric or electrochemical detection.21 This increase in the false-positive assessment of the degree of toxicity could lead unnecessary escalation of care.
These studies emphasize the need to exercise caution when interpreting laboratory test results following OHCob administration. Ideally, it would be best if blood samples were obtained prior to OHCob administration by EMS or in the ED, if the clinical scenario allows it.
Imaging Studies
For patients presenting after a closed-space fire, a chest radiograph will help assess for pulmonary injury. The classic finding of CO poisoning on head computed tomography (CT) and magnetic resonance imaging scans is evidence of ischemia in the basal ganglia. The radiographic findings may help determine the diagnosis of the altered mental status patient who presents without a history. An electrocardiogram (ECG) is also useful for detection of myocardial ischemia or dysrhythmias, when signs or symptoms of either are possible from history and physical or cardiac monitoring.
Intentional Inhalation
Intentional inhalation of fumes containing CO is a relatively common mechanism for suicide. In patients who survive, it is important for EPs and other providers to suspect additional means of self-harm. For example, at our institution, we have encountered several patients with self-inflicted trauma after remaining conscious following a medication overdose. Accordingly, patients who have intentionally inhaled CO should also be evaluated for occult medication poisoning (and trauma).
Treatment
The first step in treating a patient with CO poisoning occurs prior to arrival at the ED, when he or she is removed from continued exposure. The second step is assessing whether this is only a CO exposure, or a mixture of gases from combustion in a closed space, that might also contain CN. When CO and CN are combined OHCob is indicated to treat CN toxicity.
Additionally, if the patient is brought to the ED via EMS, O2 therapy will most likely have been initiated en route. In either case, the concentration of COHb may not accurately reflect the magnitude of exposure or prognosis, and should not be used to dictate the level of therapy or disposition. The patient’s vital signs and clinical findings of end organ toxicity should guide the appropriate supportive care.
Supplemental Oxygen Therapy
Initial administration of 100% O2 during assessment of airway, breathing, and circulation is the first step in accelerating the removal of CO from Hb. For patients suffering from smoke inhalation, assessment and establishment of a secure airway when there are signs of soot or burns in the airway must always take precedence over other actions. Continuous cardiac monitoring, POX, observation, and establishment of intravenous access are often needed for detection and management of CV instability or change in mental status in cases of moderate-to-severe CO exposures. Mild exposures with headache, nausea, and flu-like symptoms can be managed with symptomatic treatment and normobaric O2 until resolution of symptoms and improvement in COHb occur.
Hyperbaric Oxygen Therapy
Hyperbaric oxygen therapy involves the delivery of high-flow O2 (typically at 100%) under increased atmospheric pressure (2.5 -3.0 ATA). Oxygen delivered at ambient air pressure (1.0 ATA) is often referred to as normobaric oxygen. Although HBO is best known for its ability to enhance CO elimination, research points to a much more eloquent mitigation of CO toxicity on the molecular level. These mechanisms include an increased amount of dissolved O2 in blood, regeneration of cytochrome oxidase, decreased leukocyte adhesion to microvascular endothelium in the brain, decreased lipid peroxidation in the brain after loss of consciousness, and preservation of adenosine triphosphate.22
For most patients, the majority—if not all—of COHb will be eliminated by the time they present to a suitable HBO chamber. Despite the knowledge that HBO therapy has a positive toxicokinetic effect by increasing the elimination of CO, all of the major, prospective studies on the usefulness of HBO are related to prevention of neuropsychiatric sequelae mediated by immunological and inflammatory effects. The role of HBO in the treatment of CO poisoning has been debated for decades. Multiple studies that differ in methodology, patient populations, delivery of HBO treatments, and assessment of benefits fail to provide a consensus on the role of HBO therapy (Table 2).23-30
Before transferring a patient to a facility for HBO therapy, the potential risks and benefits of transport must be considered. In a 10-year retrospective study by Sloan et al31 of 297 CO-poisoned patients (mean COHb, 38%) 46% of patients had cardiopulmonary and neurological complications prior to HBO therapy at some point in the transfer pathway. During HBO therapy, 18% of patients had complications that included emesis, agitation requiring sedation, seizures, hypotension, tension pneumothorax, cardiac arrest, cardiac arrhythmias, and myocardial ischemia. It is therefore incumbent that personnel attending patients undergoing HBO therapy for CO poisoning be aware of, and able to manage, this variety of serious effects.
When an HBO chamber is at a clinical site with experts in the field and staff available 24 hours a day, the decision to utilize HBO may easily be made without obstacles. For most EPs, however, this is not the case. Locating and transferring a patient to an HBO center is typically a considerable logistical challenge. For many rural facilities, HBO is just not a timely therapeutic option. Two studies state the benefit of HBO therapy is greatest when starting within 6 hours from the end of the CO exposure.24,26
Identifying those CO-poisoned patients who meet evidence-based criteria for HBO is difficult. Patients with mild CO poisoning will do well without HBO, and critically ill patients will probably not consistently benefit from HBO. However, a pragmatic solution must be considered when efficacy studies are incongruent with conflicting results. When signs of end-organ toxicity from CO are present, but cardiac arrest has not yet occurred and the logistics are streamlined, the benefit of HBO may outweigh the risk.
Signs of end-organ toxicity include syncope, seizures, coma, ischemic changes on ECG, and pregnancy with unresolved maternal distress or fetal distress. Although a COHb level greater than 25% or 15% (pregnant) alone is commonly used as an indication for HBO, this is largely based on opinion. Conversely, HBO is unlikely to be helpful in patients who have been resuscitated after CO-related cardiac arrest.32
Treatment Guidelines
The American College of Emergency Physicians recently developed a position statement regarding the management and treatment of CO poisoning.33 The clinical policy addresses several of the controversies discussed in this review, and provides a level of evidence for each response (Table 3).
Case Conclusions
Case 1 (Smoke Inhalation Due to CO and Cyanide Poisoning)
The patient in this case suffered severe CO and CN toxicity. A head CT scan revealed diffuse edema consistent with anoxic brain injury. After conferring with the family regarding the patient’s condition and prognosis, the decision was made to withdraw life-sustaining therapy and support, and the patient died.
Case 2 (Household Misadventure)
The patient in this case was successfully treated with 100% O2 via a NRBM and was subsequently discharged home within 4 hours from presentation.
Conclusion
Exposures to CO are ubiquitous due to our heavy reliance on carbon combustion, and the manifestations of CO toxicity are protean. Therefore, CO poisoning must be considered more frequently in the differential diagnosis of indiscriminant symptoms affecting the neurological, cardiac, pulmonary, and gastrointestinal systems, especially when multiple patients have similar symptoms.
The diagnosis of CO poisoning is straightforward when a serum COHb level is obtained on a venous or arterial blood sample. Treatment starts when the patient is removed from further CO exposure and breaths normobaric oxygen at ambient levels or supplemented. Because there is no clear evidenced-based indication for HBO therapy, further treatment with HBO is naturally limited by rational constraints.
Case Presentations
Case 1: Smoke Inhalation (Carbon Monoxide and Cyanide)
A 50-year-old woman was pulled from the window of a burning building and found to be in cardiac arrest with pulseless electrical activity. Standard advanced cardiac life-support was started, and infusion of intra-osseous hydroxocobalamin (OHCob) was administered at the time of intubation because of the concern for cyanide (CN) gas exposure during smoke inhalation. Return of spontaneous circulation occurred before arrival at the hospital.
Upon presentation to the ED, the patient’s vital signs were: initial blood pressure (BP), 92/47 mm Hg; heart rate (HR), 112 beats/min; respiratory rate (RR), 31 breaths/min; and temperature (T), 99.7°F. Following intubation, the patient’s oxygen saturation (SaO2) on pulse oximetry (POX) was 93%, and her fraction of inspired oxygen (FiO2) was 100%.
On physical examination, the patient’s face was covered with soot. The lung sounds were equal and clear to auscultation bilaterally. The neurological examination was significant for a Glasgow Coma Scale of 3, without administered sedation, and there were no signs of dermal burns. Initial arterial blood gas (ABG) results were: pH, 7.06; carbon dioxide partial pressure (PCO2), 58 mm Hg; partial pressure of oxygen (PO2), 152 mm Hg; bicarbonate (HCO3), 17 mm Hg; SaO2, 98% (after intubation); FiO2, 100%; carboxyhemoglobin (COHb), 30%; and lactate, 14 mmol/L.
Case 2: Household Misadventure (Carbon Monoxide)
Several days after disabling the carbon monoxide (CO) detector in his home to silence the alarm that had continued to sound, a 67-year-old man developed weakness and called his local fire department. Upon arrival at the man’s home, the fire department confirmed an ambient air CO gas concentration over 200 ppm. Emergency medical services (EMS) promptly brought the patient to the local ED for evaluation and treatment.
Shortly after arrival at the ED, the patient’s weakness had resolved. His vital signs at examination were: BP, 154/85 mm Hg; HR, 79 beats/min; RR, 15 breaths/min; and T, 98.8°F. The patient’s COHb level was 28% with administration of 100% oxygen (O2) via a nonrebreather mask (NRBM).
Carbon Monoxide Toxicity
Carbon monoxide is a toxin of considerable importance to emergency physicians (EPs). The diagnosis at times can be challenging, the interpretation of COHb can be confusing, and the role of hyperbaric oxygen (HBO) therapy in the treatment of CO poisoning remains controversial.
Natural Sources
Carbon monoxide is formed from the incomplete combustion of organic (carbonaceous) fuels, such as charcoal, wood, petroleum distillates (gasoline, kerosene, diesel fuel), and natural gas. Though the majority of atmospheric CO comes from natural sources (eg, volcanoes, forest fires, marsh gases), poisoning exposures are primarily due to man-made CO.
Man-Made Sources
Motor vehicle exhaust is the most abundant source of man-made CO, and exposures to exhaust fumes are common causes of both intentional and unintentional poisonings and death. Other frequent sources of CO poisoning include smoke inhalation from house fires; inadequate ventilation during use of kerosene space heaters; charcoal grills or hibachis; burning wood or charcoal; fuel-powered tools such as generators, fork lifts, and chain saws; or faulty (natural or bottled) gas appliances, such as stoves, furnaces, or water heaters (Table 1). Though propane is known to burn more cleanly than natural gas (ie, less harmful to the environment), it still can produce CO.
Though neither electrical appliances nor “gas leaks” are sources of CO, like CO, natural gas (mostly methane) and bottle gas (propane) are odorless, tasteless, and colorless. Utility companies add sulfur containing mercaptans to natural gas so that leaks can be detected, but CO is only formed when the fuel is burned in a gas-powered appliance.
Endogenous Carbon Monoxide
Endogenous CO production can occur from catabolism of heme or from hepatic metabolism of methylene chloride, but exposures to this solvent are unlikely to generate COHb concentrations above 10%.
Epidemiology
The incidence of CO poisoning is likely more frequent than documented since many cases of minor exposures are unreported due to self-limiting effects and/or the vague, nonspecific nature of symptoms associated with minor exposures. In 2015, US Poison Control Centers reported over 14,000 cases of CO poisoning, only 43% of which were treated in a health care facility.1 The vast majority of exposures (97%) were unintentional and resulted in 52 deaths (0.398%).1
Data from hospitalized patients in 2007 revealed that over 200,000 ED visits and 22,000 hospitalizations were possibly associated with unintentional, non-fire-related CO exposures.2 Approximately 10% of the exposures in each of these populations were confirmed by specific International Classification of Diseases Medical E codes.2
Regardless of dataset, ED visits due to CO exposure are most common in young adults and women, occur in winter months from exposure in and around homes, and result in discharge from the ED. Elderly patients have the highest rate of hospital admission.
Carbon monoxide poisoning has long been considered a leading cause of poisoning death, though numbers appear to be declining, and CO was responsible for fewer deaths than opioids in 2017.2 The National Center for Health Statistics reported 56,133 CO-related deaths from 1979 through 1988—an average of 5,600 per year.3,4 Of these, 46% were from suicide; 28% were related to burns or house fires; and 21% (11,547) were characterized as unintentional. Motor vehicle exhaust was associated with 57% of the unintentional deaths. A more recent analysis of unintentional exposures reported 2,244 deaths during the period of 2010 to 2015—an average of 374 deaths per year (393 in 2015).5
Preventive measures are likely responsible for the significant decline in non-fire-related CO poisoning deaths from the early 1970s through the 1990s. The introduction of catalytic converters in automobiles in 1975 and O2 sensors in 1981 eventually reduced automotive CO emissions by 95% compared to pre-1975 vehicles.6 Both unintentional death and suicide rates associated with CO from motor vehicles subsequently declined by 81% and 43%, respectively. The lower decline in suicidal deaths serves as a reminder that intentional exposure to motor vehicles remains dangerous and potentially lethal.
Pathophysiology/Mechanisms of Toxicity
Carbon monoxide is a colorless, odorless gas that readily reaches the bloodstream during alveolar gas exchange. Since absorption is rapid, exposures to high CO concentrations can produce toxicity within minutes, though exposure severity is related to both inspired CO concentration and duration of exposure.
Endogenous Elimination
Carbon monoxide is eliminated from the body in expired air, with an elimination half-life dependent on FiO2 and atmospheric pressure. Accordingly, COHb decreases with a half-life (all approximate) of 4 to 6 hours when patients are breathing room air (21% O2), 60 to 90 minutes with O2 delivery at 95% to 100%, and 20 to 40 minutes under hyperbaric conditions (2.5-3.0 atmospheres absolute [ATA]).
Effect on Hemoglobin
Once absorbed, CO has an affinity for hemoglobin (Hb) that is over 200 times greater than does O2.7 The formation of COHb results in both a decreased O2-carrying capacity of Hb at the sites where O2 would have been, and because of its new configuration, COHb does not allow currently bound O2 to be offloaded. This is graphically represented by a shift of the O2-Hb dissociation curve to the left. In addition, CO continues to be bound by other intracellular heme molecules in myoglobin of skeletal and myocardial muscle, and the cytochrome oxidase system in mitochondria.8
Immunologic and Inflammatory Effects
Carbon monoxide poisoning results in a cascade of immunologic and inflammatory effects, such as generation of nitric oxide, lipid peroxidation from neutrophils, mitochondrial oxidative stress, and apoptosis. These effects result in cellular asphyxia in all organs, but the most emergent life-threatening concerns are ischemia to the brain and heart.
Severity of Toxicity and Exposure
As previously noted, the severity of CO poisoning is dose-dependent, meaning that it is related to the concentration of CO in inspired air and the duration of the exposure. Carbon monoxide is typically absent in fresh air, but levels may approach 2 to 5 ppm due to cooking, wood burning, mild air pollution, etc. The source of levels above 5 ppm should generally be investigated.
Maximum safe exposure levels for workers over an 8-hour period range from 25 to 50 ppm. Exposures to CO levels above 50 to 100 ppm are likely to elicit symptoms in most patients, depending on duration of the exposure. Carbon monoxide levels of 200 ppm may result in a mild headache after 2 to 3 hours of exposure, and a more severe headache and nausea after 1 to 2 hours of exposures to 400 ppm of CO.
Accordingly, home CO detectors use a combination of ppm and time for alarms, and they may not sound an alarm at 40 ppm until the level persists for 8 or so hours. Home CO detectors, however, will sound an alarm immediately when a level of 80 to 100 ppm is reached.
Clinical Presentation
Acute Exposure
Acute exposure to CO causes a variety of effects that are largely nonspecific, as there is no toxic syndrome (toxidrome) considered pathognomonic for CO poisoning. Ambient CO levels, duration of exposure, minute ventilation, presence of other toxic gases, and patient comorbidities can all contribute to the severity of exposure and presenting signs and symptoms. Effects associated with mild poisoning include headache, dizziness, blurred vision, fatigue or weakness, nausea, and shortness of breath. Patients with pre-existing respiratory, cardiovascular (CV), or neurological compromise are likely to present with more pronounced symptoms. In either case, these complaints may easily be confused with a viral illness, emphasizing the importance of eliciting a history of potential exposure to CO, particularly when multiple patients are involved.
As the concentration of COHb increases, more significant clinical effects can be expected, including tachycardia, chest pain, hypotension, dysrhythmias, lethargy, coma, apnea, and seizures. Hypoxia can result in myocardial injury, cerebral edema, stroke, and acute pulmonary and kidney injury.
Following acute exposure, the severity of effects correlates with the peak pretreatment COHb concentration. However, the peak concentration is usually unknown, since most patients with significant exposures will have some time period elapsed between the exposure and the determination of COHb, and the COHb will have declined at a rate depending on FiO2 and minute ventilation. In these circumstances, COHb is a poor indicator for HBO therapy and outcome.
Delayed Neurological Sequelae
Persistent, recurrent, or delayed (following period of no symptoms) neurological effects can occur in up to 40% of cases, and patients with significant exposures (eg, loss of consciousness) appear to be at greatest risk. These effects most often occur within the first 3 weeks following exposure, and have been known to persist for months to years. Such effects include headache, dizziness, impaired memory or cognition, and emotional lability. Predicting which factors in CO exposure and/or treatments can be modified to prevent neurological sequelae remains challenging.
Diagnostic Testing
Pulse Co-oximetry
Prehospital care POX typically reads COHb as oxyhemoglobin, thereby displaying a normal SaO2.9 Noninvasive CO pulse co-oximetry using a pulse oximeter (Rad-57, Masimo Corporation) provides a reading between –6 to +4 of the true COHb with a false-positive rate of 11% and false-negative rate of 46%.10 This high false-negative rate makes noninvasive CO pulse co-oximetry a poor tool to rule out a CO exposure.11 If OHCob has been administered due to concerns for CN poisoning (smoke inhalation), concentrations of COHb detected by a co-oximeter medical device may be decreased, as noted by a mean decrease of 1% in healthy volunteers exposed to OHCob only.12
Venous and Arterial Blood Gas Testing
For a patient in the hospital, exposure to CO can rapidly be determined using co-oximetry to measure COHb in a venous or arterial sample. Obtaining a venous sample may be a more practical approach, as other venous measurements will likely also be obtained. Baseline “normal” COHb levels should be less than 5%, but may be up to 8% to 10% in tobacco smokers.
Other Laboratory Studies
Other important laboratory tests that should be obtained are a complete blood count, lactate level, venous blood gas, and basic metabolic panel (to assess acid/base status). In two retrospective studies of patients exposed to CO, elevated lactate levels were associated with altered mental status.13,14 However, elevated lactate levels were not seen in a majority of patients with CO poisoning.
In addition, CN exposure should be considered when the lactate level is greater than 8 mmol/L, particularly in patients with smoke inhalation.15 Troponin I and creatinine phosphokinase tests can be used to screen for myocardial or skeletal muscle injury.
Effect of Hydroxocobalamin on Laboratory Evaluation
It is important to be cautious when interpreting the results of laboratory studies in patients who have been given OHCob due to the potential co-exposure to CN (smoke inhalation). The red discoloration of body fluids after OHCob administration makes laboratory evaluation by spectrophotometric techniques erroneous.16 Of greatest concern is the accuracy of COHb concentrations.17 In a study using rabbit models by Lee et al,17 OHCob administration was shown to falsely increase COHb concentrations.
Livshits et al18 reported conflicting effects on COHb in two human cases. In the first case, the patient’s true COHb was 93% lower (2.5% vs 34.9%) following administration of 5 g of OHCob, as measured with a rapid blood gas analyzer. In the second patient, COHb was 76% lower (10.7% vs 44%) following OHCob administration, which was also measured by a blood gas analyzer. Both of these cases illustrate lower true COHb concentrations than would be expected following the administration of only supplemental O2.
In a controlled experiment by Pace et al19 examining the effects of OHCob on measurement of COHb at both physiological (3%) and pathological (30% and 50%) concentrations in human blood samples, the degree of interference depended on the type of co-oximeter used, the degree of COHb elevation (at pathological levels only), and the concentration of OHCob added. Other studies, including an evaluation of OHCob interference by Carlsson et al20 using nine different analyzers have confirmed the interference of OHCob on photometric assays. Of particular clinical importance, a falsely increased lactate level was seen after true lactate levels were found to be below 4.8 mmol/L (but not greater) using spectrophotometric or electrochemical detection.21 This increase in the false-positive assessment of the degree of toxicity could lead unnecessary escalation of care.
These studies emphasize the need to exercise caution when interpreting laboratory test results following OHCob administration. Ideally, it would be best if blood samples were obtained prior to OHCob administration by EMS or in the ED, if the clinical scenario allows it.
Imaging Studies
For patients presenting after a closed-space fire, a chest radiograph will help assess for pulmonary injury. The classic finding of CO poisoning on head computed tomography (CT) and magnetic resonance imaging scans is evidence of ischemia in the basal ganglia. The radiographic findings may help determine the diagnosis of the altered mental status patient who presents without a history. An electrocardiogram (ECG) is also useful for detection of myocardial ischemia or dysrhythmias, when signs or symptoms of either are possible from history and physical or cardiac monitoring.
Intentional Inhalation
Intentional inhalation of fumes containing CO is a relatively common mechanism for suicide. In patients who survive, it is important for EPs and other providers to suspect additional means of self-harm. For example, at our institution, we have encountered several patients with self-inflicted trauma after remaining conscious following a medication overdose. Accordingly, patients who have intentionally inhaled CO should also be evaluated for occult medication poisoning (and trauma).
Treatment
The first step in treating a patient with CO poisoning occurs prior to arrival at the ED, when he or she is removed from continued exposure. The second step is assessing whether this is only a CO exposure, or a mixture of gases from combustion in a closed space, that might also contain CN. When CO and CN are combined OHCob is indicated to treat CN toxicity.
Additionally, if the patient is brought to the ED via EMS, O2 therapy will most likely have been initiated en route. In either case, the concentration of COHb may not accurately reflect the magnitude of exposure or prognosis, and should not be used to dictate the level of therapy or disposition. The patient’s vital signs and clinical findings of end organ toxicity should guide the appropriate supportive care.
Supplemental Oxygen Therapy
Initial administration of 100% O2 during assessment of airway, breathing, and circulation is the first step in accelerating the removal of CO from Hb. For patients suffering from smoke inhalation, assessment and establishment of a secure airway when there are signs of soot or burns in the airway must always take precedence over other actions. Continuous cardiac monitoring, POX, observation, and establishment of intravenous access are often needed for detection and management of CV instability or change in mental status in cases of moderate-to-severe CO exposures. Mild exposures with headache, nausea, and flu-like symptoms can be managed with symptomatic treatment and normobaric O2 until resolution of symptoms and improvement in COHb occur.
Hyperbaric Oxygen Therapy
Hyperbaric oxygen therapy involves the delivery of high-flow O2 (typically at 100%) under increased atmospheric pressure (2.5 -3.0 ATA). Oxygen delivered at ambient air pressure (1.0 ATA) is often referred to as normobaric oxygen. Although HBO is best known for its ability to enhance CO elimination, research points to a much more eloquent mitigation of CO toxicity on the molecular level. These mechanisms include an increased amount of dissolved O2 in blood, regeneration of cytochrome oxidase, decreased leukocyte adhesion to microvascular endothelium in the brain, decreased lipid peroxidation in the brain after loss of consciousness, and preservation of adenosine triphosphate.22
For most patients, the majority—if not all—of COHb will be eliminated by the time they present to a suitable HBO chamber. Despite the knowledge that HBO therapy has a positive toxicokinetic effect by increasing the elimination of CO, all of the major, prospective studies on the usefulness of HBO are related to prevention of neuropsychiatric sequelae mediated by immunological and inflammatory effects. The role of HBO in the treatment of CO poisoning has been debated for decades. Multiple studies that differ in methodology, patient populations, delivery of HBO treatments, and assessment of benefits fail to provide a consensus on the role of HBO therapy (Table 2).23-30
Before transferring a patient to a facility for HBO therapy, the potential risks and benefits of transport must be considered. In a 10-year retrospective study by Sloan et al31 of 297 CO-poisoned patients (mean COHb, 38%) 46% of patients had cardiopulmonary and neurological complications prior to HBO therapy at some point in the transfer pathway. During HBO therapy, 18% of patients had complications that included emesis, agitation requiring sedation, seizures, hypotension, tension pneumothorax, cardiac arrest, cardiac arrhythmias, and myocardial ischemia. It is therefore incumbent that personnel attending patients undergoing HBO therapy for CO poisoning be aware of, and able to manage, this variety of serious effects.
When an HBO chamber is at a clinical site with experts in the field and staff available 24 hours a day, the decision to utilize HBO may easily be made without obstacles. For most EPs, however, this is not the case. Locating and transferring a patient to an HBO center is typically a considerable logistical challenge. For many rural facilities, HBO is just not a timely therapeutic option. Two studies state the benefit of HBO therapy is greatest when starting within 6 hours from the end of the CO exposure.24,26
Identifying those CO-poisoned patients who meet evidence-based criteria for HBO is difficult. Patients with mild CO poisoning will do well without HBO, and critically ill patients will probably not consistently benefit from HBO. However, a pragmatic solution must be considered when efficacy studies are incongruent with conflicting results. When signs of end-organ toxicity from CO are present, but cardiac arrest has not yet occurred and the logistics are streamlined, the benefit of HBO may outweigh the risk.
Signs of end-organ toxicity include syncope, seizures, coma, ischemic changes on ECG, and pregnancy with unresolved maternal distress or fetal distress. Although a COHb level greater than 25% or 15% (pregnant) alone is commonly used as an indication for HBO, this is largely based on opinion. Conversely, HBO is unlikely to be helpful in patients who have been resuscitated after CO-related cardiac arrest.32
Treatment Guidelines
The American College of Emergency Physicians recently developed a position statement regarding the management and treatment of CO poisoning.33 The clinical policy addresses several of the controversies discussed in this review, and provides a level of evidence for each response (Table 3).
Case Conclusions
Case 1 (Smoke Inhalation Due to CO and Cyanide Poisoning)
The patient in this case suffered severe CO and CN toxicity. A head CT scan revealed diffuse edema consistent with anoxic brain injury. After conferring with the family regarding the patient’s condition and prognosis, the decision was made to withdraw life-sustaining therapy and support, and the patient died.
Case 2 (Household Misadventure)
The patient in this case was successfully treated with 100% O2 via a NRBM and was subsequently discharged home within 4 hours from presentation.
Conclusion
Exposures to CO are ubiquitous due to our heavy reliance on carbon combustion, and the manifestations of CO toxicity are protean. Therefore, CO poisoning must be considered more frequently in the differential diagnosis of indiscriminant symptoms affecting the neurological, cardiac, pulmonary, and gastrointestinal systems, especially when multiple patients have similar symptoms.
The diagnosis of CO poisoning is straightforward when a serum COHb level is obtained on a venous or arterial blood sample. Treatment starts when the patient is removed from further CO exposure and breaths normobaric oxygen at ambient levels or supplemented. Because there is no clear evidenced-based indication for HBO therapy, further treatment with HBO is naturally limited by rational constraints.
1. Mowry JB, Spyker DA, Brooks DE, Zimmerman A, Schauben JL. 2015 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 33rd Annual Report. Clin Toxicol. 2016;54(10):924-1109. doi:10.1080/15563650.2016.1245421.
2. Iqbal S, Law HZ, Clower JH, Yip FY, Elixhauser A. Hospital burden of unintentional carbon monoxide poisoning in the United States, 2007. Am J Emerg Med. 2012;30(5):657-664. doi:10.1016/j.ajem.2011.03.003.
3. Cobb N, Etzel RA. Unintentional carbon monoxide-related deaths in the United States, 1979 through 1988. JAMA. 1991;266(5):659-663.
4. Sircar K, Clower J, Shin MK, Bailey C, King M, Yip F. Carbon monoxide poisoning deaths in the United States, 1999 to 2012. Am J Emerg Med. 2015;33(9):1140-1145. doi:10.1016/j.ajem.2015.05.002.
5. Centers for Disease Control and Prevention. Environmental Public Health Tracking Network. Carbon monoxide poisoning emergency department visits. https://ephtracking.cdc.gov/showHome.action. Updated September 8, 2017. Accessed October 18, 2017.
6. Mott JA, Wolfe MI, Alverson CJ, et al. National vehicle emissions policies and practices and declining US carbon monoxide-related mortality. JAMA. 2002;288(8):988-995.
7. Buckley NA, Juurlink DN, Isbister G, Bennett MH, Lavonas EJ. Hyperbaric oxygen for carbon monoxide poisoning. Cochrane Database Syst Rev. 2011;13(4):CD002041. doi:10.1002/14651858.CD002041.pub3.
8. Hampson NB, Piantadosi CA, Thom SR, Weaver LK. Practice recommendations in the diagnosis, management, and prevention of carbon monoxide poisoning. Am J Respir Crit Care Med. 2012;186(11):1095-1101. doi:10.1164/rccm.201207-1284CI.
9. Bozeman WP, Myers RA, Barish RA. Confirmation of the pulse oximetry gap in carbon monoxide poisoning. Ann of Emerg Med. 1997;30(5):608-611.
10. Zaouter C, Zavorsky GS. The measurement of carboxyhemoglobin and methemoglobin using a non-invasive pulse CO-oximeter. Respir Physiol Neurobiol. 2012;182(2-3):88-92. doi:10.1016/j.resp.2012.05.010.
11. Shamir MY, Avramovich A, Smaka T. The current status of continuous noninvasive measurement of total, carboxy, and methemoglobin concentration. Anesth Analg. 2012;114(5);972-978. doi:10.1213/ANE.0b013e318233041a.
12. Cashin BV, Matlock AG, Kang C, Reynolds PS, Wills BK. Effect of hydroxocobalamin on surface oximetry in nonexposed humans. Prehosp Disaster Med. 2013;28(4):367-369. doi:10.1017/S1049023X13003518.
13. Moon JM, Shin MH, Chun BJ. The value of initial lactate in patients with carbon monoxide intoxication: in the emergency department. Hum Exp Toxicol. 2011;30(8):836-843. doi:10.1177/0960327110384527.
14. Doğan NÖ, Savrun A, Levent S, et al. Can initial lactate levels predict the severity of unintentional carbon monoxide poisoning? Hum Exp Toxicol. 2015;34(3):324-329. doi:10.1177/0960327114538986.
15. Baud FJ, Borron SW, Mégarbane B, et al. Value of lactic acidosis in the assessment of the severity of acute cyanide poisoning. Crit Care Med. 2002;30(9):2044-2050. doi:10.1097/01.CCM.0000026325.65944.7D.
16. Fueyo L, Robles J, Aguilar I, Yáñez AM, Socias M, Parera M. Hemolysis index to detect degree of hydroxocobalamin interference with common laboratory tests. J Clin Lab Anal. 2017;31(5):e22089. doi:10.1002/jcla.22089.
17. Lee J, Mukai D, Kreuter K, Mahon S, Tromberg B, Brenner M. Potential interference by hydroxocobalamin on cooximetry hemoglobin measurements during cyanide and smoke inhalation treatments. Ann Emerg Med. 2007;49(6):802-805. doi:10.
1016/j.annemergmed.2006.11.016.
18. Livshits Z, Lugassy DM, Shawn LK, Hoffman RS. Falsely Low Carboxyhemoglobin after Hydroxocobalamin Therapy [Letter]. N Engl J Med. 2012;367(13):1270-1271. doi:10.1056/NEJMc1114820.
19. Pace R, Bon Homme M, Hoffman RS, Lugassy D. Effects of hydroxocobalamin on carboxyhemoglobin measured under physiologic and pathologic conditions. Clin Toxicol (Phila). 2014;52(7):647-650. doi:10.3109/15563650.2014.939659.
20. Carlsson CJ, Hansen HE, Hilsted L, Malm J, Ødum L, Szecsi PB. An evaluation of the interference of hydroxycobalamin with chemistry and co-oximetry tests on nine commonly used instruments. Scand J Clin Lab Invest. 2011;71(5):378-386. doi:10.3109/00365513.2011.573573.
21. Fueyo L, Robles J, Aguilar I, Yáñez AM, Socias M, Parera M. Hemolysis index to detect degree of hydroxocobalamin interference with common laboratory tests. J Clin Lab Anal. 2017;31(5). doi:10.1002/jcla.22089.
22. Tomaszewski C. Carbon monoxide. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR, eds. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015:1581-1593.
23. Hampson NB, Mathieu D, Piantodosi CA et al. Carbon monoxide poisoning: interpretation of randomized clinical trials and unresolved treatment issues. Undersea Hyperb Med. 2001;28(3):157-164.
24. Raphael JC, Elkharrat D, Jars-Guincestre MC, et al. Trial of normobaric and hyperbaric oxygen for acute carbon monoxide intoxication. Lancet. 1989;2(8660):414-419.
25. Ducassé JL, Celsis P, Marc-Vergnes JP. Non-comatose patients with acute carbon monoxide poisoning: hyperbaric or normobaric oxygenation? Undersea Hyperb Med. 1995;22(1):9-15.
26. Thom SR, Taber RL, Mendiguren II, Clark JM, Hardy KR, Fisher AB. Delayed neuropsychologic sequelae after carbon monoxide poisoning: prevention by treatment with hyperbaric oxygen. Ann Emerg Med. 1995;25(4):474-480.
27. Mathieu D, Wattel F, Mathieu-Nolf M, et al. Randomized prospective study comparing the effects of HBO versus 12 hours of nbp in non comatose CO poisoned patients: results of the interim analysis. Undersea Hyperb Med. 1996;23(Suppl:7-8).
28. Scheinkestel CD, Bailey M, Myles PS, et al. Hyperbaric or normobaric oxygen for acute carbon monoxide poisoning: a randomized controlled clinical trial. Med J Aust. 1999;170(5):203-210.
29. Weaver LK, Hopkins RO, Chan KJ, et al. Hyperbaric oxygen for acute carbon monoxide poisoning. N Engl J Med. 2002;347(14):1057-1067. doi:10.1056/NEJMoa013121.
30. Annane D, Chadda K, Gajdos P, Jars-Guincestre MC, Chevret S, Raphael JC. Hyperbaric oxygen therapy for acute domestic carbon monoxide poisoning: two randomized controlled trials. Intensive Care Med. 2011;37(3):486-492. doi:10.1007/s00134-010-2093-0.
31. Sloan EP, Murphy DG, Hart R, et al. Complications and protocol considerations in carbon monoxide-poisoned patients who require hyperbaric oxygen therapy: report from a ten-year experience. Ann Emerg Med. 1989;18(6):629-634.
32. Hampson NB, Zmaeff JL. Outcome of patients experiencing cardiac arrest with carbon monoxide poisoning treated with hyperbaric oxygen. Ann Emerg Med. 2001;38(1):36-41. doi:10.1067/mem.2001.115532.
33. Wolf SJ, Maloney GE, Shih RD, Shy BD, Brown MD; American College of Emergency Physicians. Clinical policy: critical issues in the evaluation and management of adult patients presenting to the emergency department with acute carbon monoxide poisoning. Ann Emerg Med. pii:S0196-0644(17)30332-3. doi:10.1016/j.annemergmed.2017.03.036.
1. Mowry JB, Spyker DA, Brooks DE, Zimmerman A, Schauben JL. 2015 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 33rd Annual Report. Clin Toxicol. 2016;54(10):924-1109. doi:10.1080/15563650.2016.1245421.
2. Iqbal S, Law HZ, Clower JH, Yip FY, Elixhauser A. Hospital burden of unintentional carbon monoxide poisoning in the United States, 2007. Am J Emerg Med. 2012;30(5):657-664. doi:10.1016/j.ajem.2011.03.003.
3. Cobb N, Etzel RA. Unintentional carbon monoxide-related deaths in the United States, 1979 through 1988. JAMA. 1991;266(5):659-663.
4. Sircar K, Clower J, Shin MK, Bailey C, King M, Yip F. Carbon monoxide poisoning deaths in the United States, 1999 to 2012. Am J Emerg Med. 2015;33(9):1140-1145. doi:10.1016/j.ajem.2015.05.002.
5. Centers for Disease Control and Prevention. Environmental Public Health Tracking Network. Carbon monoxide poisoning emergency department visits. https://ephtracking.cdc.gov/showHome.action. Updated September 8, 2017. Accessed October 18, 2017.
6. Mott JA, Wolfe MI, Alverson CJ, et al. National vehicle emissions policies and practices and declining US carbon monoxide-related mortality. JAMA. 2002;288(8):988-995.
7. Buckley NA, Juurlink DN, Isbister G, Bennett MH, Lavonas EJ. Hyperbaric oxygen for carbon monoxide poisoning. Cochrane Database Syst Rev. 2011;13(4):CD002041. doi:10.1002/14651858.CD002041.pub3.
8. Hampson NB, Piantadosi CA, Thom SR, Weaver LK. Practice recommendations in the diagnosis, management, and prevention of carbon monoxide poisoning. Am J Respir Crit Care Med. 2012;186(11):1095-1101. doi:10.1164/rccm.201207-1284CI.
9. Bozeman WP, Myers RA, Barish RA. Confirmation of the pulse oximetry gap in carbon monoxide poisoning. Ann of Emerg Med. 1997;30(5):608-611.
10. Zaouter C, Zavorsky GS. The measurement of carboxyhemoglobin and methemoglobin using a non-invasive pulse CO-oximeter. Respir Physiol Neurobiol. 2012;182(2-3):88-92. doi:10.1016/j.resp.2012.05.010.
11. Shamir MY, Avramovich A, Smaka T. The current status of continuous noninvasive measurement of total, carboxy, and methemoglobin concentration. Anesth Analg. 2012;114(5);972-978. doi:10.1213/ANE.0b013e318233041a.
12. Cashin BV, Matlock AG, Kang C, Reynolds PS, Wills BK. Effect of hydroxocobalamin on surface oximetry in nonexposed humans. Prehosp Disaster Med. 2013;28(4):367-369. doi:10.1017/S1049023X13003518.
13. Moon JM, Shin MH, Chun BJ. The value of initial lactate in patients with carbon monoxide intoxication: in the emergency department. Hum Exp Toxicol. 2011;30(8):836-843. doi:10.1177/0960327110384527.
14. Doğan NÖ, Savrun A, Levent S, et al. Can initial lactate levels predict the severity of unintentional carbon monoxide poisoning? Hum Exp Toxicol. 2015;34(3):324-329. doi:10.1177/0960327114538986.
15. Baud FJ, Borron SW, Mégarbane B, et al. Value of lactic acidosis in the assessment of the severity of acute cyanide poisoning. Crit Care Med. 2002;30(9):2044-2050. doi:10.1097/01.CCM.0000026325.65944.7D.
16. Fueyo L, Robles J, Aguilar I, Yáñez AM, Socias M, Parera M. Hemolysis index to detect degree of hydroxocobalamin interference with common laboratory tests. J Clin Lab Anal. 2017;31(5):e22089. doi:10.1002/jcla.22089.
17. Lee J, Mukai D, Kreuter K, Mahon S, Tromberg B, Brenner M. Potential interference by hydroxocobalamin on cooximetry hemoglobin measurements during cyanide and smoke inhalation treatments. Ann Emerg Med. 2007;49(6):802-805. doi:10.
1016/j.annemergmed.2006.11.016.
18. Livshits Z, Lugassy DM, Shawn LK, Hoffman RS. Falsely Low Carboxyhemoglobin after Hydroxocobalamin Therapy [Letter]. N Engl J Med. 2012;367(13):1270-1271. doi:10.1056/NEJMc1114820.
19. Pace R, Bon Homme M, Hoffman RS, Lugassy D. Effects of hydroxocobalamin on carboxyhemoglobin measured under physiologic and pathologic conditions. Clin Toxicol (Phila). 2014;52(7):647-650. doi:10.3109/15563650.2014.939659.
20. Carlsson CJ, Hansen HE, Hilsted L, Malm J, Ødum L, Szecsi PB. An evaluation of the interference of hydroxycobalamin with chemistry and co-oximetry tests on nine commonly used instruments. Scand J Clin Lab Invest. 2011;71(5):378-386. doi:10.3109/00365513.2011.573573.
21. Fueyo L, Robles J, Aguilar I, Yáñez AM, Socias M, Parera M. Hemolysis index to detect degree of hydroxocobalamin interference with common laboratory tests. J Clin Lab Anal. 2017;31(5). doi:10.1002/jcla.22089.
22. Tomaszewski C. Carbon monoxide. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR, eds. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015:1581-1593.
23. Hampson NB, Mathieu D, Piantodosi CA et al. Carbon monoxide poisoning: interpretation of randomized clinical trials and unresolved treatment issues. Undersea Hyperb Med. 2001;28(3):157-164.
24. Raphael JC, Elkharrat D, Jars-Guincestre MC, et al. Trial of normobaric and hyperbaric oxygen for acute carbon monoxide intoxication. Lancet. 1989;2(8660):414-419.
25. Ducassé JL, Celsis P, Marc-Vergnes JP. Non-comatose patients with acute carbon monoxide poisoning: hyperbaric or normobaric oxygenation? Undersea Hyperb Med. 1995;22(1):9-15.
26. Thom SR, Taber RL, Mendiguren II, Clark JM, Hardy KR, Fisher AB. Delayed neuropsychologic sequelae after carbon monoxide poisoning: prevention by treatment with hyperbaric oxygen. Ann Emerg Med. 1995;25(4):474-480.
27. Mathieu D, Wattel F, Mathieu-Nolf M, et al. Randomized prospective study comparing the effects of HBO versus 12 hours of nbp in non comatose CO poisoned patients: results of the interim analysis. Undersea Hyperb Med. 1996;23(Suppl:7-8).
28. Scheinkestel CD, Bailey M, Myles PS, et al. Hyperbaric or normobaric oxygen for acute carbon monoxide poisoning: a randomized controlled clinical trial. Med J Aust. 1999;170(5):203-210.
29. Weaver LK, Hopkins RO, Chan KJ, et al. Hyperbaric oxygen for acute carbon monoxide poisoning. N Engl J Med. 2002;347(14):1057-1067. doi:10.1056/NEJMoa013121.
30. Annane D, Chadda K, Gajdos P, Jars-Guincestre MC, Chevret S, Raphael JC. Hyperbaric oxygen therapy for acute domestic carbon monoxide poisoning: two randomized controlled trials. Intensive Care Med. 2011;37(3):486-492. doi:10.1007/s00134-010-2093-0.
31. Sloan EP, Murphy DG, Hart R, et al. Complications and protocol considerations in carbon monoxide-poisoned patients who require hyperbaric oxygen therapy: report from a ten-year experience. Ann Emerg Med. 1989;18(6):629-634.
32. Hampson NB, Zmaeff JL. Outcome of patients experiencing cardiac arrest with carbon monoxide poisoning treated with hyperbaric oxygen. Ann Emerg Med. 2001;38(1):36-41. doi:10.1067/mem.2001.115532.
33. Wolf SJ, Maloney GE, Shih RD, Shy BD, Brown MD; American College of Emergency Physicians. Clinical policy: critical issues in the evaluation and management of adult patients presenting to the emergency department with acute carbon monoxide poisoning. Ann Emerg Med. pii:S0196-0644(17)30332-3. doi:10.1016/j.annemergmed.2017.03.036.

Case Studies in Toxicology: Always Cook Your Boba
Case
A 45-year-old Chinese man with no known medical history presented to the ED with right-sided facial spasm and cheek swelling, which began immediately after he bit into a piece of taro root, approximately 2 hours prior to presentation. The patient stated that the root was an ingredient in a soup that a relative had made. According to the patient, after biting into the root, he immediately experienced a burning pain on the right side of his mouth. He further noted that he swallowed less than two bites of the root and stopped eating because the act of chewing was too painful.
Initial vital signs at presentation were: blood pressure, 140/100 mm Hg; heart rate, 84 beats/min; respiratory rate, 14 beats/min; and temperature, 97.6°F. Oxygen saturation was 98% on room air. The patient’s physical examination was remarkable for pain upon opening the mouth, as well as right-sided cheek and lip swelling and tenderness. The tongue and oropharynx were not erythematous or swollen. The patient was only able to speak in short sentences, secondary to oropharyngeal pain, but he was in no respiratory distress. No urticaria, pruritus, wheezing, or stridor was present.
During the patient’s workup, his 40-year-old wife also presented to the same ED for evaluation of burning pain and spasm on the left side of her mouth, which she stated also developed immediately after she bit into a piece of taro root contained in the same soup as that ingested by the patient.
The wife’s vital signs were unremarkable, and she was in no respiratory distress. Her physical examination was remarkable only for left-sided cheek and lip swelling and tenderness, associated with an erythematous oropharynx and pain with speaking.
What is taro? What are the manifestations of taro toxicity?
Taro commonly refers to plants from the Araceae family, usually Colocasia esculenta.1 Taro is ubiquitous in Southern Asia and Southeast India. It is a widely naturalized and perennial tropical plant primarily grown as a root vegetable, and is a common flavor in boba (bubble) tea. All members of Araceae contain calcium oxalate crystals in the form of raphides, sharp needle-shaped crystals packaged in idioblasts and contained within the waxy leaf.2 Pressure on the idioblasts, such as from mastication, triggers the release of the raphides. The needles pierce the surface of any tissue with which they come into contact, creating a gateway for proteolytic enzymes to enter the consumer.3 The leaves and root of Araceae must be cooked before eating to inactivate the raphides.
Oral exposure to uncooked taro leaves or taro root can result in mouth irritation and swelling that can progress to angioedema and airway obstruction. Although the traditional method of removing taro raphides is to soak the root in cold water overnight,4,5 this does not fully remove all of the raphides. Instead, taro root should be thoroughly cooked in boiling water to draw-out oxalates from the root into the cooking water, which must then be discarded. Consuming taro with warm milk also reduces the effect of the oxalates by about 80%.6
Many other plants of the Araceae family, such as Dieffenbachia (dumbcane), share similar toxicity and are commonly kept in the home and office.
Patients with oral exposure to taro may experience a delayed (also termed biphasic) anaphylactic reaction, ie, the development of anaphylactic symptoms more than 4 hours after the inciting event. Delayed anaphylaxis is distinct from delayed hypersensitivity, though both may be immunoglobulin E-mediated. Delayed hypersensitivity presents later (2-14 days) and with less immediately life-threatening effects, most commonly dermatitis (eg, poison ivy dermatitis).
While both of the patients in this case presented with mild symptoms, life-threatening angioedema of the oropharynx, anaphylaxis, and hypocalcemia have been reported7,8 and should be considered in any symptomatic patient with exposure to taro.
What is the differential diagnosis of plant-related mouth pain?
The oral mucosa is composed of superficial layers of mucin and epithelial cells that lie over the dermis and connective tissue. Local immune cells, including mast cells and Langerhans cells, reside in the deeper layers. The differential diagnosis of plant-based mouth pain can be divided into mechanical, chemical, and thermal causes.
Mechanical Causes. Causes of mechanical plant-based oral pain include structural damage when foreign matter, such as barbs, sharp leaves, or hard seeds, pierce the layers of the oral mucosa.
Chemical Causes. Chemical-related causes of oral pain include caustic ingestion, for example from detergents or cleaning agents that contaminate the broth. Araceae, such as taro or arum, have sharp calcium oxalate crystals tipped with phospholipases and proteases that cause mechanical pain on piercing mucous membranes, and chemical pain by enzymatically degrading epithelium and mucosa. Both chemical and mechanical irritation can lead to an inflammatory response. Raw taro can cause irritant contact stomatitis as the raphides pierce the oral mucosa. It can also cause allergic stomatitis if antigens related to the phospholipases or proteases are presented to Langerhans cells.9
Thermal Causes. The hot temperature of the ingested broth could cause thermal injury, but the injury is likely to be more diffuse.
How common is taro exposure, and how is it treated?
From 1995 to 1999, 15 cases of taro poisoning were reported to the Drug and Toxicology Information service in Zimbabwe.10 From 2005 to 2009, 21 out of 31 cases reported to the Hong Kong Poison Control Center involving gastrointestinal irritation involved the consumption of Colocasia fallax, a form of taro more common in Tibet, the Himalayas, and northern Indochina.7 Of the 31 cases, six patients were treated with diphenhydramine, epinephrine, and dexamethasone for angioedema.
From 2011 to 2013, two cases of mouth irritation and swelling after eating raw taro leaves were reported to the British Columbia Poison Control Center.11 Those two patients were observed for 6 hours without specific treatment and discharged.
Case Conclusion
Due to concerns of the potential for anaphylaxis, both patients were treated intravenously with 50 mg diphenhydramine and 10 mg dexamethasone. The husband was also given 650 mg acetaminophen orally for pain relief; his wife declined pain medication. Laboratory evaluation, including a complete blood count, basic metabolic panel, liver function panel, and urinalysis were ordered for both patients; all results were within normal limits for both patients.
After an uneventful 6-hour observation period, both patients were discharged home with instructions to return to the ED if they develop any signs of allergic reaction and to call emergency medical services for any sign of anaphylaxis.
1. Rao RV, Matthews PJ, Eyzaguirre PB, Hunter D, eds. 2010. The Global Diversity of Taro: Ethnobotany and Conservation. Rome, Italy; Biouniversity International; 2010. http://www.bioversityinternational.org/fileadmin/user_upload/online_library/publications/pdfs/1402.pdf#page=11. Accessed September 15, 2017.
2. Franceschi VR, Nakata PA. Calcium oxalate in plants: formation and function. Annu Rev Plant Biol. 2005;56:41-71. doi:10.1146/annurev.arplant.56.032604.144106.
3. Herbert DA. Stinging crystals in plants. Science. 1924;60(1548):204-205. doi:10.1126/science.60.1548.204-a.
4. Njintang YN, Mbofung CMF. Effect of precooking time and drying temperature on the physico-chemical characteristics and in-vitro carbohydrate digestibility of taro flour. LWT – Food Sci and Tech. 2006;39(6):684-691. doi.org/10.1016/j.lwt.2005.03.022.
5. Savage GP, Dubois M. The effect of soaking and cooking on the oxalate content of taro leaves. Int J Food Sci Nutr. 2006;57(5-6):376-381. doi:10.1080/09637480600855239.
6. Oscarsson, KV. Savage GP. Composition and availability of soluble and insoluble oxalates in raw and cooked taro (Colocasia esculenta var. Schott) leaves. Food Chem 101. 2007;101(2):559-562. doi:10.1016/j.foodchem.2006.02.014.
7. Pang CT, Ng HW, Lau FL. Oral mucosal irritating plant ingestion in Hong Kong, epidemiology and its clinical presentation. Hong Kong J Emerg Med. 2010;17(5):477-481.
8. Yuen E. Upper airway obstruction as a presentation of Taro poisoning. Hong Kong J Emerg Med. 2001;8(3):163-165.
9. Davis CC, Squier CA, Lilly GE. Irritant contact stomatitis: a review of the condition. J Periodontol. 1998;69(6):620-631. doi:10.1902/jop.1998.69.6.620.
10 Tagwireyi D, Ball DE. The management of Elephant’s Ear poisoning. Hum Exp Toxicol. 2001;20(4):189-192. doi:10.1191/096032701678766822.
11. Omura JD, Blake C, McIntyre L, Li D, Kosatsky T. Two cases of poisoning by raw taro leaf and how a poison control centre, food safety inspectors, and a specialty supermarket chain found a solution.” Environ Health Rev. 2014;57(3):59-64. doi.org/10.5864/d2014-027.
Case
A 45-year-old Chinese man with no known medical history presented to the ED with right-sided facial spasm and cheek swelling, which began immediately after he bit into a piece of taro root, approximately 2 hours prior to presentation. The patient stated that the root was an ingredient in a soup that a relative had made. According to the patient, after biting into the root, he immediately experienced a burning pain on the right side of his mouth. He further noted that he swallowed less than two bites of the root and stopped eating because the act of chewing was too painful.
Initial vital signs at presentation were: blood pressure, 140/100 mm Hg; heart rate, 84 beats/min; respiratory rate, 14 beats/min; and temperature, 97.6°F. Oxygen saturation was 98% on room air. The patient’s physical examination was remarkable for pain upon opening the mouth, as well as right-sided cheek and lip swelling and tenderness. The tongue and oropharynx were not erythematous or swollen. The patient was only able to speak in short sentences, secondary to oropharyngeal pain, but he was in no respiratory distress. No urticaria, pruritus, wheezing, or stridor was present.
During the patient’s workup, his 40-year-old wife also presented to the same ED for evaluation of burning pain and spasm on the left side of her mouth, which she stated also developed immediately after she bit into a piece of taro root contained in the same soup as that ingested by the patient.
The wife’s vital signs were unremarkable, and she was in no respiratory distress. Her physical examination was remarkable only for left-sided cheek and lip swelling and tenderness, associated with an erythematous oropharynx and pain with speaking.
What is taro? What are the manifestations of taro toxicity?
Taro commonly refers to plants from the Araceae family, usually Colocasia esculenta.1 Taro is ubiquitous in Southern Asia and Southeast India. It is a widely naturalized and perennial tropical plant primarily grown as a root vegetable, and is a common flavor in boba (bubble) tea. All members of Araceae contain calcium oxalate crystals in the form of raphides, sharp needle-shaped crystals packaged in idioblasts and contained within the waxy leaf.2 Pressure on the idioblasts, such as from mastication, triggers the release of the raphides. The needles pierce the surface of any tissue with which they come into contact, creating a gateway for proteolytic enzymes to enter the consumer.3 The leaves and root of Araceae must be cooked before eating to inactivate the raphides.
Oral exposure to uncooked taro leaves or taro root can result in mouth irritation and swelling that can progress to angioedema and airway obstruction. Although the traditional method of removing taro raphides is to soak the root in cold water overnight,4,5 this does not fully remove all of the raphides. Instead, taro root should be thoroughly cooked in boiling water to draw-out oxalates from the root into the cooking water, which must then be discarded. Consuming taro with warm milk also reduces the effect of the oxalates by about 80%.6
Many other plants of the Araceae family, such as Dieffenbachia (dumbcane), share similar toxicity and are commonly kept in the home and office.
Patients with oral exposure to taro may experience a delayed (also termed biphasic) anaphylactic reaction, ie, the development of anaphylactic symptoms more than 4 hours after the inciting event. Delayed anaphylaxis is distinct from delayed hypersensitivity, though both may be immunoglobulin E-mediated. Delayed hypersensitivity presents later (2-14 days) and with less immediately life-threatening effects, most commonly dermatitis (eg, poison ivy dermatitis).
While both of the patients in this case presented with mild symptoms, life-threatening angioedema of the oropharynx, anaphylaxis, and hypocalcemia have been reported7,8 and should be considered in any symptomatic patient with exposure to taro.
What is the differential diagnosis of plant-related mouth pain?
The oral mucosa is composed of superficial layers of mucin and epithelial cells that lie over the dermis and connective tissue. Local immune cells, including mast cells and Langerhans cells, reside in the deeper layers. The differential diagnosis of plant-based mouth pain can be divided into mechanical, chemical, and thermal causes.
Mechanical Causes. Causes of mechanical plant-based oral pain include structural damage when foreign matter, such as barbs, sharp leaves, or hard seeds, pierce the layers of the oral mucosa.
Chemical Causes. Chemical-related causes of oral pain include caustic ingestion, for example from detergents or cleaning agents that contaminate the broth. Araceae, such as taro or arum, have sharp calcium oxalate crystals tipped with phospholipases and proteases that cause mechanical pain on piercing mucous membranes, and chemical pain by enzymatically degrading epithelium and mucosa. Both chemical and mechanical irritation can lead to an inflammatory response. Raw taro can cause irritant contact stomatitis as the raphides pierce the oral mucosa. It can also cause allergic stomatitis if antigens related to the phospholipases or proteases are presented to Langerhans cells.9
Thermal Causes. The hot temperature of the ingested broth could cause thermal injury, but the injury is likely to be more diffuse.
How common is taro exposure, and how is it treated?
From 1995 to 1999, 15 cases of taro poisoning were reported to the Drug and Toxicology Information service in Zimbabwe.10 From 2005 to 2009, 21 out of 31 cases reported to the Hong Kong Poison Control Center involving gastrointestinal irritation involved the consumption of Colocasia fallax, a form of taro more common in Tibet, the Himalayas, and northern Indochina.7 Of the 31 cases, six patients were treated with diphenhydramine, epinephrine, and dexamethasone for angioedema.
From 2011 to 2013, two cases of mouth irritation and swelling after eating raw taro leaves were reported to the British Columbia Poison Control Center.11 Those two patients were observed for 6 hours without specific treatment and discharged.
Case Conclusion
Due to concerns of the potential for anaphylaxis, both patients were treated intravenously with 50 mg diphenhydramine and 10 mg dexamethasone. The husband was also given 650 mg acetaminophen orally for pain relief; his wife declined pain medication. Laboratory evaluation, including a complete blood count, basic metabolic panel, liver function panel, and urinalysis were ordered for both patients; all results were within normal limits for both patients.
After an uneventful 6-hour observation period, both patients were discharged home with instructions to return to the ED if they develop any signs of allergic reaction and to call emergency medical services for any sign of anaphylaxis.
Case
A 45-year-old Chinese man with no known medical history presented to the ED with right-sided facial spasm and cheek swelling, which began immediately after he bit into a piece of taro root, approximately 2 hours prior to presentation. The patient stated that the root was an ingredient in a soup that a relative had made. According to the patient, after biting into the root, he immediately experienced a burning pain on the right side of his mouth. He further noted that he swallowed less than two bites of the root and stopped eating because the act of chewing was too painful.
Initial vital signs at presentation were: blood pressure, 140/100 mm Hg; heart rate, 84 beats/min; respiratory rate, 14 beats/min; and temperature, 97.6°F. Oxygen saturation was 98% on room air. The patient’s physical examination was remarkable for pain upon opening the mouth, as well as right-sided cheek and lip swelling and tenderness. The tongue and oropharynx were not erythematous or swollen. The patient was only able to speak in short sentences, secondary to oropharyngeal pain, but he was in no respiratory distress. No urticaria, pruritus, wheezing, or stridor was present.
During the patient’s workup, his 40-year-old wife also presented to the same ED for evaluation of burning pain and spasm on the left side of her mouth, which she stated also developed immediately after she bit into a piece of taro root contained in the same soup as that ingested by the patient.
The wife’s vital signs were unremarkable, and she was in no respiratory distress. Her physical examination was remarkable only for left-sided cheek and lip swelling and tenderness, associated with an erythematous oropharynx and pain with speaking.
What is taro? What are the manifestations of taro toxicity?
Taro commonly refers to plants from the Araceae family, usually Colocasia esculenta.1 Taro is ubiquitous in Southern Asia and Southeast India. It is a widely naturalized and perennial tropical plant primarily grown as a root vegetable, and is a common flavor in boba (bubble) tea. All members of Araceae contain calcium oxalate crystals in the form of raphides, sharp needle-shaped crystals packaged in idioblasts and contained within the waxy leaf.2 Pressure on the idioblasts, such as from mastication, triggers the release of the raphides. The needles pierce the surface of any tissue with which they come into contact, creating a gateway for proteolytic enzymes to enter the consumer.3 The leaves and root of Araceae must be cooked before eating to inactivate the raphides.
Oral exposure to uncooked taro leaves or taro root can result in mouth irritation and swelling that can progress to angioedema and airway obstruction. Although the traditional method of removing taro raphides is to soak the root in cold water overnight,4,5 this does not fully remove all of the raphides. Instead, taro root should be thoroughly cooked in boiling water to draw-out oxalates from the root into the cooking water, which must then be discarded. Consuming taro with warm milk also reduces the effect of the oxalates by about 80%.6
Many other plants of the Araceae family, such as Dieffenbachia (dumbcane), share similar toxicity and are commonly kept in the home and office.
Patients with oral exposure to taro may experience a delayed (also termed biphasic) anaphylactic reaction, ie, the development of anaphylactic symptoms more than 4 hours after the inciting event. Delayed anaphylaxis is distinct from delayed hypersensitivity, though both may be immunoglobulin E-mediated. Delayed hypersensitivity presents later (2-14 days) and with less immediately life-threatening effects, most commonly dermatitis (eg, poison ivy dermatitis).
While both of the patients in this case presented with mild symptoms, life-threatening angioedema of the oropharynx, anaphylaxis, and hypocalcemia have been reported7,8 and should be considered in any symptomatic patient with exposure to taro.
What is the differential diagnosis of plant-related mouth pain?
The oral mucosa is composed of superficial layers of mucin and epithelial cells that lie over the dermis and connective tissue. Local immune cells, including mast cells and Langerhans cells, reside in the deeper layers. The differential diagnosis of plant-based mouth pain can be divided into mechanical, chemical, and thermal causes.
Mechanical Causes. Causes of mechanical plant-based oral pain include structural damage when foreign matter, such as barbs, sharp leaves, or hard seeds, pierce the layers of the oral mucosa.
Chemical Causes. Chemical-related causes of oral pain include caustic ingestion, for example from detergents or cleaning agents that contaminate the broth. Araceae, such as taro or arum, have sharp calcium oxalate crystals tipped with phospholipases and proteases that cause mechanical pain on piercing mucous membranes, and chemical pain by enzymatically degrading epithelium and mucosa. Both chemical and mechanical irritation can lead to an inflammatory response. Raw taro can cause irritant contact stomatitis as the raphides pierce the oral mucosa. It can also cause allergic stomatitis if antigens related to the phospholipases or proteases are presented to Langerhans cells.9
Thermal Causes. The hot temperature of the ingested broth could cause thermal injury, but the injury is likely to be more diffuse.
How common is taro exposure, and how is it treated?
From 1995 to 1999, 15 cases of taro poisoning were reported to the Drug and Toxicology Information service in Zimbabwe.10 From 2005 to 2009, 21 out of 31 cases reported to the Hong Kong Poison Control Center involving gastrointestinal irritation involved the consumption of Colocasia fallax, a form of taro more common in Tibet, the Himalayas, and northern Indochina.7 Of the 31 cases, six patients were treated with diphenhydramine, epinephrine, and dexamethasone for angioedema.
From 2011 to 2013, two cases of mouth irritation and swelling after eating raw taro leaves were reported to the British Columbia Poison Control Center.11 Those two patients were observed for 6 hours without specific treatment and discharged.
Case Conclusion
Due to concerns of the potential for anaphylaxis, both patients were treated intravenously with 50 mg diphenhydramine and 10 mg dexamethasone. The husband was also given 650 mg acetaminophen orally for pain relief; his wife declined pain medication. Laboratory evaluation, including a complete blood count, basic metabolic panel, liver function panel, and urinalysis were ordered for both patients; all results were within normal limits for both patients.
After an uneventful 6-hour observation period, both patients were discharged home with instructions to return to the ED if they develop any signs of allergic reaction and to call emergency medical services for any sign of anaphylaxis.
1. Rao RV, Matthews PJ, Eyzaguirre PB, Hunter D, eds. 2010. The Global Diversity of Taro: Ethnobotany and Conservation. Rome, Italy; Biouniversity International; 2010. http://www.bioversityinternational.org/fileadmin/user_upload/online_library/publications/pdfs/1402.pdf#page=11. Accessed September 15, 2017.
2. Franceschi VR, Nakata PA. Calcium oxalate in plants: formation and function. Annu Rev Plant Biol. 2005;56:41-71. doi:10.1146/annurev.arplant.56.032604.144106.
3. Herbert DA. Stinging crystals in plants. Science. 1924;60(1548):204-205. doi:10.1126/science.60.1548.204-a.
4. Njintang YN, Mbofung CMF. Effect of precooking time and drying temperature on the physico-chemical characteristics and in-vitro carbohydrate digestibility of taro flour. LWT – Food Sci and Tech. 2006;39(6):684-691. doi.org/10.1016/j.lwt.2005.03.022.
5. Savage GP, Dubois M. The effect of soaking and cooking on the oxalate content of taro leaves. Int J Food Sci Nutr. 2006;57(5-6):376-381. doi:10.1080/09637480600855239.
6. Oscarsson, KV. Savage GP. Composition and availability of soluble and insoluble oxalates in raw and cooked taro (Colocasia esculenta var. Schott) leaves. Food Chem 101. 2007;101(2):559-562. doi:10.1016/j.foodchem.2006.02.014.
7. Pang CT, Ng HW, Lau FL. Oral mucosal irritating plant ingestion in Hong Kong, epidemiology and its clinical presentation. Hong Kong J Emerg Med. 2010;17(5):477-481.
8. Yuen E. Upper airway obstruction as a presentation of Taro poisoning. Hong Kong J Emerg Med. 2001;8(3):163-165.
9. Davis CC, Squier CA, Lilly GE. Irritant contact stomatitis: a review of the condition. J Periodontol. 1998;69(6):620-631. doi:10.1902/jop.1998.69.6.620.
10 Tagwireyi D, Ball DE. The management of Elephant’s Ear poisoning. Hum Exp Toxicol. 2001;20(4):189-192. doi:10.1191/096032701678766822.
11. Omura JD, Blake C, McIntyre L, Li D, Kosatsky T. Two cases of poisoning by raw taro leaf and how a poison control centre, food safety inspectors, and a specialty supermarket chain found a solution.” Environ Health Rev. 2014;57(3):59-64. doi.org/10.5864/d2014-027.
1. Rao RV, Matthews PJ, Eyzaguirre PB, Hunter D, eds. 2010. The Global Diversity of Taro: Ethnobotany and Conservation. Rome, Italy; Biouniversity International; 2010. http://www.bioversityinternational.org/fileadmin/user_upload/online_library/publications/pdfs/1402.pdf#page=11. Accessed September 15, 2017.
2. Franceschi VR, Nakata PA. Calcium oxalate in plants: formation and function. Annu Rev Plant Biol. 2005;56:41-71. doi:10.1146/annurev.arplant.56.032604.144106.
3. Herbert DA. Stinging crystals in plants. Science. 1924;60(1548):204-205. doi:10.1126/science.60.1548.204-a.
4. Njintang YN, Mbofung CMF. Effect of precooking time and drying temperature on the physico-chemical characteristics and in-vitro carbohydrate digestibility of taro flour. LWT – Food Sci and Tech. 2006;39(6):684-691. doi.org/10.1016/j.lwt.2005.03.022.
5. Savage GP, Dubois M. The effect of soaking and cooking on the oxalate content of taro leaves. Int J Food Sci Nutr. 2006;57(5-6):376-381. doi:10.1080/09637480600855239.
6. Oscarsson, KV. Savage GP. Composition and availability of soluble and insoluble oxalates in raw and cooked taro (Colocasia esculenta var. Schott) leaves. Food Chem 101. 2007;101(2):559-562. doi:10.1016/j.foodchem.2006.02.014.
7. Pang CT, Ng HW, Lau FL. Oral mucosal irritating plant ingestion in Hong Kong, epidemiology and its clinical presentation. Hong Kong J Emerg Med. 2010;17(5):477-481.
8. Yuen E. Upper airway obstruction as a presentation of Taro poisoning. Hong Kong J Emerg Med. 2001;8(3):163-165.
9. Davis CC, Squier CA, Lilly GE. Irritant contact stomatitis: a review of the condition. J Periodontol. 1998;69(6):620-631. doi:10.1902/jop.1998.69.6.620.
10 Tagwireyi D, Ball DE. The management of Elephant’s Ear poisoning. Hum Exp Toxicol. 2001;20(4):189-192. doi:10.1191/096032701678766822.
11. Omura JD, Blake C, McIntyre L, Li D, Kosatsky T. Two cases of poisoning by raw taro leaf and how a poison control centre, food safety inspectors, and a specialty supermarket chain found a solution.” Environ Health Rev. 2014;57(3):59-64. doi.org/10.5864/d2014-027.
