User login
Cytotect®CP found to be safe and effective after allo-HCT

A small retrospective study of 23 transplant patients has confirmed that CMV hyperimmune globulin (Cytotect®CP) is a safe and effective salvage therapy for patients with cytomegalovirus (CMV) infection after allogeneic hematopoietic cell transplant (allo-HCT).
Cytotect®CP used as salvage therapy resulted in a 78% overall response rate and 70% of all patients cleared CMV infection, according to investigators.
They observed no clinically significant adverse events.
CMV is a major factor contributing to high mortality rates in allo-HCT patients.
And because Cytotect®CP is less toxic than commonly used treatments for CMV infection, investigators suggested that it be used prophylactically in patients known to have a predisposition to CMV infection.
They reported their findings in the journal Bone Marrow Transplantation.
Patient characteristics and methods
All 23 patients transplanted at 8 centers in France were CMV seropositive at the time of transplant, and 70% received the transplant from a CMV serostatus negative donor.
Recipient positivity and donor negativity, the investigators indicated, is a risk factor for developing recurrent CMV infection after allo-HCT.
The patients’ median age was 53, 11 were male and 12 female.
Five patients (22%) received a haploidentical transplant and 14 (61%) from an unrelated donor, which the investigators pointed out is also a known risk factor for developing recurrent CMV infection.
Thirteen (57%) were in complete remission from their underlying disease, 4 (17%) in partial remission, 1 (4%) had stable disease, and 5 (22%) were in relapse or had progressive disease.
Most (83%) had a peripheral blood transplant, 15 (65%) had a reduced intensity conditioning regimen, and 16 (70%) had antithymocyte globulin as part of their conditioning regimen.
All patients received valacyclovir as antiviral prophylaxis prior to receiving Cytotect®CP. The investigators mentioned in the paper that valacyclovir has not been proven to be effective in treating CMV.
They noted that other CMV treatments, such as ganciclovir, foscavir, and dicofovir, cause high levels of toxicity, frequently leading to treatment discontinuation.
Seventeen patients (74%) had a history of graft-versus-host disease (GVHD), and 11 (49%) had active GVHD at the time CMV hyperimmune globulin was administered.
Investigators used quantitative polymerase chain reaction (PCR) to quantify CMV viral load in the blood.
Treatment could begin when patients’ viral load was greater than 3-3.5 log UI/mL, according to the Francophone Society of Bone Marrow Transplantation and Cellular therapy.
Three patients received Cytotect®CP at a prophylaxis dose (200 U/kg/week) to prevent CMV recurrences and 20 as preemptive therapy (400 U/kg on days 1, 4, 8 then 200 U/kg on days 12and 16).
Seven patients (30%) received Cytotect®CP as monotherapy, 5 (22%) in combination with ganciclovir, 5 (22%) in combination with foscavir, 2 (9%) in combination with ganciclovir and foscavir, and 4 (17%) with some other combination.
Investigators restricted their analysis to 100-day overall survival (OS), starting at the beginning of Cytotect®CP treatment to death within 100 days, regardless of the cause of death.
Results
Eighteen patients (78%) responded to Cytotect®CP, and 16 of the responders converted to CMV-PCR negative.
Median time to achieve CMV-PCR response was 15 days (range, 3-51).
Four patients did not respond to therapy, and 1 patient had a non-evaluable response. The latter patient died 13 days after the introduction of Cytotect®CP due to another infection.
Eight patients died within 100 days after initiation of Cytotect®CP. Two deaths were related to CMV and 6 were unrelated.
Four patients who responded to Cytotect®CP experienced CMV relapse between 9 and 49 days after their best response to therapy.
Five responders died within 100 days due to the following causes: GVHD (n = 2), other infection (n = 1), underlying disease (n = 1), and CMV-related causes (n = 1).
Two of the 4 nonresponders died of other infection (n = 1) and GVHD (n = 1).
Investigators estimated the 100-day OS from the start of Cytotect®CP to be 69.6%. They observed no statistical difference (P=0.258) between those who responded (73.7%) and those who didn’t (50.0%).
The investigators believe that Cytotect®CP is an alternative option for treatment of CMV infection because it avoids renal and bone marrow impairment and should be considered as prophylaxis in select patients.
They recommend a large prospective study be conducted to confirm safety and efficacy results of CMV hyperimmune globulin.
Cytotect®CP is authorized in more than 15 countries for the prophylaxis of CMV infection in patients receiving immunosuppressive treatment, particularly transplant recipients.
In French transplant centers, according to the study authors, use of Cytotect®CP is limited to the salvage setting for recurrent or refractory CMV infections and sometimes in combination for CMV pneumonia.
Biotest, the commercializer of Cytotect®CP, provided a grant for this study.
A small retrospective study of 23 transplant patients has confirmed that CMV hyperimmune globulin (Cytotect®CP) is a safe and effective salvage therapy for patients with cytomegalovirus (CMV) infection after allogeneic hematopoietic cell transplant (allo-HCT).
Cytotect®CP used as salvage therapy resulted in a 78% overall response rate and 70% of all patients cleared CMV infection, according to investigators.
They observed no clinically significant adverse events.
CMV is a major factor contributing to high mortality rates in allo-HCT patients.
And because Cytotect®CP is less toxic than commonly used treatments for CMV infection, investigators suggested that it be used prophylactically in patients known to have a predisposition to CMV infection.
They reported their findings in the journal Bone Marrow Transplantation.
Patient characteristics and methods
All 23 patients transplanted at 8 centers in France were CMV seropositive at the time of transplant, and 70% received the transplant from a CMV serostatus negative donor.
Recipient positivity and donor negativity, the investigators indicated, is a risk factor for developing recurrent CMV infection after allo-HCT.
The patients’ median age was 53, 11 were male and 12 female.
Five patients (22%) received a haploidentical transplant and 14 (61%) from an unrelated donor, which the investigators pointed out is also a known risk factor for developing recurrent CMV infection.
Thirteen (57%) were in complete remission from their underlying disease, 4 (17%) in partial remission, 1 (4%) had stable disease, and 5 (22%) were in relapse or had progressive disease.
Most (83%) had a peripheral blood transplant, 15 (65%) had a reduced intensity conditioning regimen, and 16 (70%) had antithymocyte globulin as part of their conditioning regimen.
All patients received valacyclovir as antiviral prophylaxis prior to receiving Cytotect®CP. The investigators mentioned in the paper that valacyclovir has not been proven to be effective in treating CMV.
They noted that other CMV treatments, such as ganciclovir, foscavir, and dicofovir, cause high levels of toxicity, frequently leading to treatment discontinuation.
Seventeen patients (74%) had a history of graft-versus-host disease (GVHD), and 11 (49%) had active GVHD at the time CMV hyperimmune globulin was administered.
Investigators used quantitative polymerase chain reaction (PCR) to quantify CMV viral load in the blood.
Treatment could begin when patients’ viral load was greater than 3-3.5 log UI/mL, according to the Francophone Society of Bone Marrow Transplantation and Cellular therapy.
Three patients received Cytotect®CP at a prophylaxis dose (200 U/kg/week) to prevent CMV recurrences and 20 as preemptive therapy (400 U/kg on days 1, 4, 8 then 200 U/kg on days 12and 16).
Seven patients (30%) received Cytotect®CP as monotherapy, 5 (22%) in combination with ganciclovir, 5 (22%) in combination with foscavir, 2 (9%) in combination with ganciclovir and foscavir, and 4 (17%) with some other combination.
Investigators restricted their analysis to 100-day overall survival (OS), starting at the beginning of Cytotect®CP treatment to death within 100 days, regardless of the cause of death.
Results
Eighteen patients (78%) responded to Cytotect®CP, and 16 of the responders converted to CMV-PCR negative.
Median time to achieve CMV-PCR response was 15 days (range, 3-51).
Four patients did not respond to therapy, and 1 patient had a non-evaluable response. The latter patient died 13 days after the introduction of Cytotect®CP due to another infection.
Eight patients died within 100 days after initiation of Cytotect®CP. Two deaths were related to CMV and 6 were unrelated.
Four patients who responded to Cytotect®CP experienced CMV relapse between 9 and 49 days after their best response to therapy.
Five responders died within 100 days due to the following causes: GVHD (n = 2), other infection (n = 1), underlying disease (n = 1), and CMV-related causes (n = 1).
Two of the 4 nonresponders died of other infection (n = 1) and GVHD (n = 1).
Investigators estimated the 100-day OS from the start of Cytotect®CP to be 69.6%. They observed no statistical difference (P=0.258) between those who responded (73.7%) and those who didn’t (50.0%).
The investigators believe that Cytotect®CP is an alternative option for treatment of CMV infection because it avoids renal and bone marrow impairment and should be considered as prophylaxis in select patients.
They recommend a large prospective study be conducted to confirm safety and efficacy results of CMV hyperimmune globulin.
Cytotect®CP is authorized in more than 15 countries for the prophylaxis of CMV infection in patients receiving immunosuppressive treatment, particularly transplant recipients.
In French transplant centers, according to the study authors, use of Cytotect®CP is limited to the salvage setting for recurrent or refractory CMV infections and sometimes in combination for CMV pneumonia.
Biotest, the commercializer of Cytotect®CP, provided a grant for this study.
A small retrospective study of 23 transplant patients has confirmed that CMV hyperimmune globulin (Cytotect®CP) is a safe and effective salvage therapy for patients with cytomegalovirus (CMV) infection after allogeneic hematopoietic cell transplant (allo-HCT).
Cytotect®CP used as salvage therapy resulted in a 78% overall response rate and 70% of all patients cleared CMV infection, according to investigators.
They observed no clinically significant adverse events.
CMV is a major factor contributing to high mortality rates in allo-HCT patients.
And because Cytotect®CP is less toxic than commonly used treatments for CMV infection, investigators suggested that it be used prophylactically in patients known to have a predisposition to CMV infection.
They reported their findings in the journal Bone Marrow Transplantation.
Patient characteristics and methods
All 23 patients transplanted at 8 centers in France were CMV seropositive at the time of transplant, and 70% received the transplant from a CMV serostatus negative donor.
Recipient positivity and donor negativity, the investigators indicated, is a risk factor for developing recurrent CMV infection after allo-HCT.
The patients’ median age was 53, 11 were male and 12 female.
Five patients (22%) received a haploidentical transplant and 14 (61%) from an unrelated donor, which the investigators pointed out is also a known risk factor for developing recurrent CMV infection.
Thirteen (57%) were in complete remission from their underlying disease, 4 (17%) in partial remission, 1 (4%) had stable disease, and 5 (22%) were in relapse or had progressive disease.
Most (83%) had a peripheral blood transplant, 15 (65%) had a reduced intensity conditioning regimen, and 16 (70%) had antithymocyte globulin as part of their conditioning regimen.
All patients received valacyclovir as antiviral prophylaxis prior to receiving Cytotect®CP. The investigators mentioned in the paper that valacyclovir has not been proven to be effective in treating CMV.
They noted that other CMV treatments, such as ganciclovir, foscavir, and dicofovir, cause high levels of toxicity, frequently leading to treatment discontinuation.
Seventeen patients (74%) had a history of graft-versus-host disease (GVHD), and 11 (49%) had active GVHD at the time CMV hyperimmune globulin was administered.
Investigators used quantitative polymerase chain reaction (PCR) to quantify CMV viral load in the blood.
Treatment could begin when patients’ viral load was greater than 3-3.5 log UI/mL, according to the Francophone Society of Bone Marrow Transplantation and Cellular therapy.
Three patients received Cytotect®CP at a prophylaxis dose (200 U/kg/week) to prevent CMV recurrences and 20 as preemptive therapy (400 U/kg on days 1, 4, 8 then 200 U/kg on days 12and 16).
Seven patients (30%) received Cytotect®CP as monotherapy, 5 (22%) in combination with ganciclovir, 5 (22%) in combination with foscavir, 2 (9%) in combination with ganciclovir and foscavir, and 4 (17%) with some other combination.
Investigators restricted their analysis to 100-day overall survival (OS), starting at the beginning of Cytotect®CP treatment to death within 100 days, regardless of the cause of death.
Results
Eighteen patients (78%) responded to Cytotect®CP, and 16 of the responders converted to CMV-PCR negative.
Median time to achieve CMV-PCR response was 15 days (range, 3-51).
Four patients did not respond to therapy, and 1 patient had a non-evaluable response. The latter patient died 13 days after the introduction of Cytotect®CP due to another infection.
Eight patients died within 100 days after initiation of Cytotect®CP. Two deaths were related to CMV and 6 were unrelated.
Four patients who responded to Cytotect®CP experienced CMV relapse between 9 and 49 days after their best response to therapy.
Five responders died within 100 days due to the following causes: GVHD (n = 2), other infection (n = 1), underlying disease (n = 1), and CMV-related causes (n = 1).
Two of the 4 nonresponders died of other infection (n = 1) and GVHD (n = 1).
Investigators estimated the 100-day OS from the start of Cytotect®CP to be 69.6%. They observed no statistical difference (P=0.258) between those who responded (73.7%) and those who didn’t (50.0%).
The investigators believe that Cytotect®CP is an alternative option for treatment of CMV infection because it avoids renal and bone marrow impairment and should be considered as prophylaxis in select patients.
They recommend a large prospective study be conducted to confirm safety and efficacy results of CMV hyperimmune globulin.
Cytotect®CP is authorized in more than 15 countries for the prophylaxis of CMV infection in patients receiving immunosuppressive treatment, particularly transplant recipients.
In French transplant centers, according to the study authors, use of Cytotect®CP is limited to the salvage setting for recurrent or refractory CMV infections and sometimes in combination for CMV pneumonia.
Biotest, the commercializer of Cytotect®CP, provided a grant for this study.
A closer look at the BK polyomavirus
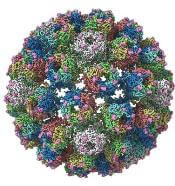
A detailed image at near-atomic levels of the BK polyomavirus (BKV), which affects kidney and bone marrow transplant patients, is now available for the first time.
This molecular-level structural visualization could allow scientists to study potential targets for antiviral therapies or drugs.
Approximately 80% to 90% of the general population are seropositive for the most prevalent BKV genotypes. Yet the virus rarely causes illness in people with healthy immune systems.
But the BKV causes nephropathy and hemorrhagic cystitis in immunosuppressed patients. Currently, no effective antiviral agents are available specifically targeting BKV.
Using high-resolution cryoelectron microscopy (cryo-EM), the research team at the University of Leeds Astbury Centre for Structural Molecular Biology determined the structure of BKV at 3.8 Å resolution.
At this resolution, they could highlight differences between human BKV and BKV pathogens that infect simian and murine hosts.
The investigators created the structures by freezing infectious BKV particles and taking thousands of images using the microscopes. The two-dimensional images were then combined computationally to produce a high-resolution, three-dimensional view of the virus.
They reported their findings in the journal Structure.
Senior investigator Neil A. Ranson, PhD, of the University of Leeds , noted that cryo-EM has been around for 30 years.
Although it’s been useful, the technology lacked the ability to routinely look at molecules at the level of detail needed, he said.
"However, the Titan Krios microscopes we have installed in Leeds are absolutely state-of-the-art and mean that these limitations have been shattered. Researchers and industry users who work with us can now image biological molecules with an incredible resolution. “
The investigators believe the quality and completeness of the cryo-EM structures they captured allows researchers to describe the various conformations of the C termini of the major capsid protein VP1, which play a fundamental role in the assembly and stability of a polyomavirus capsid.
The investigators say the images also present a more complete picture of disulfide bonding in the capsid.
Thus, the images provide insights into the structure of the human pathogen at near native conditions and give scientists a better-quality research tool to use.
“Crucially, we'll also be able to see how these molecules interact with each other," Dr Ransom said.
The BK Polyomavirus structure research was funded by Wellcome, Kidney Research UK, and Kidney Research Yorkshire.
A detailed image at near-atomic levels of the BK polyomavirus (BKV), which affects kidney and bone marrow transplant patients, is now available for the first time.
This molecular-level structural visualization could allow scientists to study potential targets for antiviral therapies or drugs.
Approximately 80% to 90% of the general population are seropositive for the most prevalent BKV genotypes. Yet the virus rarely causes illness in people with healthy immune systems.
But the BKV causes nephropathy and hemorrhagic cystitis in immunosuppressed patients. Currently, no effective antiviral agents are available specifically targeting BKV.
Using high-resolution cryoelectron microscopy (cryo-EM), the research team at the University of Leeds Astbury Centre for Structural Molecular Biology determined the structure of BKV at 3.8 Å resolution.
At this resolution, they could highlight differences between human BKV and BKV pathogens that infect simian and murine hosts.
The investigators created the structures by freezing infectious BKV particles and taking thousands of images using the microscopes. The two-dimensional images were then combined computationally to produce a high-resolution, three-dimensional view of the virus.
They reported their findings in the journal Structure.
Senior investigator Neil A. Ranson, PhD, of the University of Leeds , noted that cryo-EM has been around for 30 years.
Although it’s been useful, the technology lacked the ability to routinely look at molecules at the level of detail needed, he said.
"However, the Titan Krios microscopes we have installed in Leeds are absolutely state-of-the-art and mean that these limitations have been shattered. Researchers and industry users who work with us can now image biological molecules with an incredible resolution. “
The investigators believe the quality and completeness of the cryo-EM structures they captured allows researchers to describe the various conformations of the C termini of the major capsid protein VP1, which play a fundamental role in the assembly and stability of a polyomavirus capsid.
The investigators say the images also present a more complete picture of disulfide bonding in the capsid.
Thus, the images provide insights into the structure of the human pathogen at near native conditions and give scientists a better-quality research tool to use.
“Crucially, we'll also be able to see how these molecules interact with each other," Dr Ransom said.
The BK Polyomavirus structure research was funded by Wellcome, Kidney Research UK, and Kidney Research Yorkshire.
A detailed image at near-atomic levels of the BK polyomavirus (BKV), which affects kidney and bone marrow transplant patients, is now available for the first time.
This molecular-level structural visualization could allow scientists to study potential targets for antiviral therapies or drugs.
Approximately 80% to 90% of the general population are seropositive for the most prevalent BKV genotypes. Yet the virus rarely causes illness in people with healthy immune systems.
But the BKV causes nephropathy and hemorrhagic cystitis in immunosuppressed patients. Currently, no effective antiviral agents are available specifically targeting BKV.
Using high-resolution cryoelectron microscopy (cryo-EM), the research team at the University of Leeds Astbury Centre for Structural Molecular Biology determined the structure of BKV at 3.8 Å resolution.
At this resolution, they could highlight differences between human BKV and BKV pathogens that infect simian and murine hosts.
The investigators created the structures by freezing infectious BKV particles and taking thousands of images using the microscopes. The two-dimensional images were then combined computationally to produce a high-resolution, three-dimensional view of the virus.
They reported their findings in the journal Structure.
Senior investigator Neil A. Ranson, PhD, of the University of Leeds , noted that cryo-EM has been around for 30 years.
Although it’s been useful, the technology lacked the ability to routinely look at molecules at the level of detail needed, he said.
"However, the Titan Krios microscopes we have installed in Leeds are absolutely state-of-the-art and mean that these limitations have been shattered. Researchers and industry users who work with us can now image biological molecules with an incredible resolution. “
The investigators believe the quality and completeness of the cryo-EM structures they captured allows researchers to describe the various conformations of the C termini of the major capsid protein VP1, which play a fundamental role in the assembly and stability of a polyomavirus capsid.
The investigators say the images also present a more complete picture of disulfide bonding in the capsid.
Thus, the images provide insights into the structure of the human pathogen at near native conditions and give scientists a better-quality research tool to use.
“Crucially, we'll also be able to see how these molecules interact with each other," Dr Ransom said.
The BK Polyomavirus structure research was funded by Wellcome, Kidney Research UK, and Kidney Research Yorkshire.
Older, cheaper drug formulation to remain available

The 140 mg capsules of Imbruvica® (ibrutinib) will remain on the market, according to Pharmacyclics LLC.
Pharmacyclics (an AbbVie company) and Janssen had planned to discontinue the capsules after introducing a single-tablet formulation of Imbruvica earlier this year.
However, the companies received negative feedback about the discontinuation and decided to keep the 140 mg capsules on the market.
In February, the US Food and Drug Administration (FDA) approved a single-tablet formulation of Imbruvica that is available in 4 doses—140 mg, 280 mg, 420 mg, and 560 mg.
Pharmacyclics and Janssen introduced this formulation to enable a once-a-day dosing regimen. The companies said the goal with the new formulation was to improve adherence because some patients had to take 3 or 4 pills every day to get the recommended dose of Imbruvica.
After introducing the new formulation, Pharmacyclics and Janssen planned to discontinue the 140 mg capsules.
Critics spoke out against this change in an article published in The Cancer Letter. They noted that discontinuing the old formulation would mean price increases for some patients. That’s because the single-tablet formulation of Imbruvica has the same price regardless of dose—$400 per tablet.
Patients on lower doses of Imbruvica would experience an increase in cost if they switched from the capsules to the tablet formulation. In fact, costs could triple for patients on the 140 mg dose.
Pharmacyclics argued that most patients on Imbruvica—those taking the 420 mg and 560 mg doses—would see no increase in out-of-pocket costs when transitioning to the single-tablet formulation. And patients on the 560 mg dose would likely see a decrease in their out-of-pocket costs.
However, critics pointed to results of a recent pilot study, which indicated that the recommended dose of Imbruvica for patients with chronic lymphocytic leukemia (CLL)—420 mg—may be too high. The results suggested that CLL patients could receive lower doses of Imbruvica without a reduction in efficacy.
Therefore, keeping the 140 mg capsules on the market could mean lower costs for some CLL patients.
In addition to voicing concerns about costs, the critics pointed out that discontinuing the 140 mg capsules of Imbruvica would make it more difficult to adjust patients’ doses when needed.
Pharmacyclics said its YOU&i™ Dose Exchange Program can aid healthcare professionals in adjusting doses before patients have finished their current pack of Imbruvica. Patients would receive a “rapid shipment” of their new dose at no additional cost.
But the critics said this program “creates a barrier to optimal prescribing for some patients” and urged the FDA to review the safety of the program.
Roughly a month after the critics made this recommendation in The Cancer Letter article, Pharmacyclics announced that the 140 mg capsules of Imbruvica would remain on the market.
The 140 mg capsules of Imbruvica® (ibrutinib) will remain on the market, according to Pharmacyclics LLC.
Pharmacyclics (an AbbVie company) and Janssen had planned to discontinue the capsules after introducing a single-tablet formulation of Imbruvica earlier this year.
However, the companies received negative feedback about the discontinuation and decided to keep the 140 mg capsules on the market.
In February, the US Food and Drug Administration (FDA) approved a single-tablet formulation of Imbruvica that is available in 4 doses—140 mg, 280 mg, 420 mg, and 560 mg.
Pharmacyclics and Janssen introduced this formulation to enable a once-a-day dosing regimen. The companies said the goal with the new formulation was to improve adherence because some patients had to take 3 or 4 pills every day to get the recommended dose of Imbruvica.
After introducing the new formulation, Pharmacyclics and Janssen planned to discontinue the 140 mg capsules.
Critics spoke out against this change in an article published in The Cancer Letter. They noted that discontinuing the old formulation would mean price increases for some patients. That’s because the single-tablet formulation of Imbruvica has the same price regardless of dose—$400 per tablet.
Patients on lower doses of Imbruvica would experience an increase in cost if they switched from the capsules to the tablet formulation. In fact, costs could triple for patients on the 140 mg dose.
Pharmacyclics argued that most patients on Imbruvica—those taking the 420 mg and 560 mg doses—would see no increase in out-of-pocket costs when transitioning to the single-tablet formulation. And patients on the 560 mg dose would likely see a decrease in their out-of-pocket costs.
However, critics pointed to results of a recent pilot study, which indicated that the recommended dose of Imbruvica for patients with chronic lymphocytic leukemia (CLL)—420 mg—may be too high. The results suggested that CLL patients could receive lower doses of Imbruvica without a reduction in efficacy.
Therefore, keeping the 140 mg capsules on the market could mean lower costs for some CLL patients.
In addition to voicing concerns about costs, the critics pointed out that discontinuing the 140 mg capsules of Imbruvica would make it more difficult to adjust patients’ doses when needed.
Pharmacyclics said its YOU&i™ Dose Exchange Program can aid healthcare professionals in adjusting doses before patients have finished their current pack of Imbruvica. Patients would receive a “rapid shipment” of their new dose at no additional cost.
But the critics said this program “creates a barrier to optimal prescribing for some patients” and urged the FDA to review the safety of the program.
Roughly a month after the critics made this recommendation in The Cancer Letter article, Pharmacyclics announced that the 140 mg capsules of Imbruvica would remain on the market.
The 140 mg capsules of Imbruvica® (ibrutinib) will remain on the market, according to Pharmacyclics LLC.
Pharmacyclics (an AbbVie company) and Janssen had planned to discontinue the capsules after introducing a single-tablet formulation of Imbruvica earlier this year.
However, the companies received negative feedback about the discontinuation and decided to keep the 140 mg capsules on the market.
In February, the US Food and Drug Administration (FDA) approved a single-tablet formulation of Imbruvica that is available in 4 doses—140 mg, 280 mg, 420 mg, and 560 mg.
Pharmacyclics and Janssen introduced this formulation to enable a once-a-day dosing regimen. The companies said the goal with the new formulation was to improve adherence because some patients had to take 3 or 4 pills every day to get the recommended dose of Imbruvica.
After introducing the new formulation, Pharmacyclics and Janssen planned to discontinue the 140 mg capsules.
Critics spoke out against this change in an article published in The Cancer Letter. They noted that discontinuing the old formulation would mean price increases for some patients. That’s because the single-tablet formulation of Imbruvica has the same price regardless of dose—$400 per tablet.
Patients on lower doses of Imbruvica would experience an increase in cost if they switched from the capsules to the tablet formulation. In fact, costs could triple for patients on the 140 mg dose.
Pharmacyclics argued that most patients on Imbruvica—those taking the 420 mg and 560 mg doses—would see no increase in out-of-pocket costs when transitioning to the single-tablet formulation. And patients on the 560 mg dose would likely see a decrease in their out-of-pocket costs.
However, critics pointed to results of a recent pilot study, which indicated that the recommended dose of Imbruvica for patients with chronic lymphocytic leukemia (CLL)—420 mg—may be too high. The results suggested that CLL patients could receive lower doses of Imbruvica without a reduction in efficacy.
Therefore, keeping the 140 mg capsules on the market could mean lower costs for some CLL patients.
In addition to voicing concerns about costs, the critics pointed out that discontinuing the 140 mg capsules of Imbruvica would make it more difficult to adjust patients’ doses when needed.
Pharmacyclics said its YOU&i™ Dose Exchange Program can aid healthcare professionals in adjusting doses before patients have finished their current pack of Imbruvica. Patients would receive a “rapid shipment” of their new dose at no additional cost.
But the critics said this program “creates a barrier to optimal prescribing for some patients” and urged the FDA to review the safety of the program.
Roughly a month after the critics made this recommendation in The Cancer Letter article, Pharmacyclics announced that the 140 mg capsules of Imbruvica would remain on the market.
MSC product can treat refractory GVHD
MONTRÉAL—Results from a phase 3 trial suggest a mesenchymal stem cell (MSC) product can treat steroid-refractory, acute graft-versus-host disease (GVHD) in children.
The product, remestemcel-L (MSC-100-IV), produced an overall response rate of 69% at day 28, with complete resolution of GVHD in 29% of patients.
Adverse events (AEs) in this trial were consistent with the known safety profile of remestemcel-L.
Joanne Kurtzberg, MD, of Duke University Medical Center in Durham, North Carolina, presented these results at ISCT 2018.
The trial was sponsored by Mesoblast International Sàrl, the company developing remestemcel-L.
Remestemcel-L consists of human MSCs derived from donor bone marrow and expanded in culture.
Patients
The trial enrolled 55 patients who had acute GVHD and had failed to respond to steroid treatment. This was defined as progression within 3 days or no improvement within 7 days of consecutive treatment with at least 2 mg/kg/day of methylprednisolone or an equivalent product.
The patients had a median age of 7.6 years (range, 0.6 years to 17.9 years) at baseline, and 64% were male. Underlying diseases include acute myeloid leukemia (32.7%), acute lymphoblastic leukemia (21.8%), anemia (9.1%), chronic myeloid leukemia (7.3%), sickle cell disease (5.5%), juvenile myelomonocytic leukemia (3.6%), myelodysplastic syndromes (3.6%), and “other” disease (16.4%).
Most patients (87%) had received myeloablative conditioning, most (76%) had an unrelated donor, and roughly half (51%) received an HLA-mismatched transplant.
Fifty-five percent of patients received a bone marrow transplant, 25% received peripheral blood stem cells, and 20% received cord blood.
Forty-seven percent of patients had grade D GVHD at baseline, 42% had grade C, and 11% had grade B. Thirty-six percent of patients had multi-organ involvement (all with lower gastrointestinal), 38% had lower gastrointestinal involvement only, and 26% had skin involvement only.
Results
Fifty-four patients were treated with remestemcel-L. They received 8 injections over 4 weeks (twice weekly), consisting of 2 million cells per kg per injection.
The overall response rate at day 28 was 69%. Twenty-nine percent of patients achieved a complete response, defined as resolution of acute GVHD in all involved organs.
Forty percent of patients achieved a partial response, defined as organ-level improvement of at least one stage without worsening of any other organ.
All patients reported at least one treatment-emergent AE, and 61% had serious treatment-emergent AEs. The most common of these were infection (33%) and respiratory events (20%).
Four patients withdrew from the trial before day 100. One patient couldn’t receive treatment, 1 withdrew due to an AE (somnolence), 1 had parental consent withdrawn, and 1 was taken off study by the principal investigator.
There were 11 on-study deaths, but none were considered related to remestemcel-L. Eight deaths were due to infection, 1 due to GVHD progression, and 2 due to primary cancer relapse.
The day-100 survival analysis is pending.
MONTRÉAL—Results from a phase 3 trial suggest a mesenchymal stem cell (MSC) product can treat steroid-refractory, acute graft-versus-host disease (GVHD) in children.
The product, remestemcel-L (MSC-100-IV), produced an overall response rate of 69% at day 28, with complete resolution of GVHD in 29% of patients.
Adverse events (AEs) in this trial were consistent with the known safety profile of remestemcel-L.
Joanne Kurtzberg, MD, of Duke University Medical Center in Durham, North Carolina, presented these results at ISCT 2018.
The trial was sponsored by Mesoblast International Sàrl, the company developing remestemcel-L.
Remestemcel-L consists of human MSCs derived from donor bone marrow and expanded in culture.
Patients
The trial enrolled 55 patients who had acute GVHD and had failed to respond to steroid treatment. This was defined as progression within 3 days or no improvement within 7 days of consecutive treatment with at least 2 mg/kg/day of methylprednisolone or an equivalent product.
The patients had a median age of 7.6 years (range, 0.6 years to 17.9 years) at baseline, and 64% were male. Underlying diseases include acute myeloid leukemia (32.7%), acute lymphoblastic leukemia (21.8%), anemia (9.1%), chronic myeloid leukemia (7.3%), sickle cell disease (5.5%), juvenile myelomonocytic leukemia (3.6%), myelodysplastic syndromes (3.6%), and “other” disease (16.4%).
Most patients (87%) had received myeloablative conditioning, most (76%) had an unrelated donor, and roughly half (51%) received an HLA-mismatched transplant.
Fifty-five percent of patients received a bone marrow transplant, 25% received peripheral blood stem cells, and 20% received cord blood.
Forty-seven percent of patients had grade D GVHD at baseline, 42% had grade C, and 11% had grade B. Thirty-six percent of patients had multi-organ involvement (all with lower gastrointestinal), 38% had lower gastrointestinal involvement only, and 26% had skin involvement only.
Results
Fifty-four patients were treated with remestemcel-L. They received 8 injections over 4 weeks (twice weekly), consisting of 2 million cells per kg per injection.
The overall response rate at day 28 was 69%. Twenty-nine percent of patients achieved a complete response, defined as resolution of acute GVHD in all involved organs.
Forty percent of patients achieved a partial response, defined as organ-level improvement of at least one stage without worsening of any other organ.
All patients reported at least one treatment-emergent AE, and 61% had serious treatment-emergent AEs. The most common of these were infection (33%) and respiratory events (20%).
Four patients withdrew from the trial before day 100. One patient couldn’t receive treatment, 1 withdrew due to an AE (somnolence), 1 had parental consent withdrawn, and 1 was taken off study by the principal investigator.
There were 11 on-study deaths, but none were considered related to remestemcel-L. Eight deaths were due to infection, 1 due to GVHD progression, and 2 due to primary cancer relapse.
The day-100 survival analysis is pending.
MONTRÉAL—Results from a phase 3 trial suggest a mesenchymal stem cell (MSC) product can treat steroid-refractory, acute graft-versus-host disease (GVHD) in children.
The product, remestemcel-L (MSC-100-IV), produced an overall response rate of 69% at day 28, with complete resolution of GVHD in 29% of patients.
Adverse events (AEs) in this trial were consistent with the known safety profile of remestemcel-L.
Joanne Kurtzberg, MD, of Duke University Medical Center in Durham, North Carolina, presented these results at ISCT 2018.
The trial was sponsored by Mesoblast International Sàrl, the company developing remestemcel-L.
Remestemcel-L consists of human MSCs derived from donor bone marrow and expanded in culture.
Patients
The trial enrolled 55 patients who had acute GVHD and had failed to respond to steroid treatment. This was defined as progression within 3 days or no improvement within 7 days of consecutive treatment with at least 2 mg/kg/day of methylprednisolone or an equivalent product.
The patients had a median age of 7.6 years (range, 0.6 years to 17.9 years) at baseline, and 64% were male. Underlying diseases include acute myeloid leukemia (32.7%), acute lymphoblastic leukemia (21.8%), anemia (9.1%), chronic myeloid leukemia (7.3%), sickle cell disease (5.5%), juvenile myelomonocytic leukemia (3.6%), myelodysplastic syndromes (3.6%), and “other” disease (16.4%).
Most patients (87%) had received myeloablative conditioning, most (76%) had an unrelated donor, and roughly half (51%) received an HLA-mismatched transplant.
Fifty-five percent of patients received a bone marrow transplant, 25% received peripheral blood stem cells, and 20% received cord blood.
Forty-seven percent of patients had grade D GVHD at baseline, 42% had grade C, and 11% had grade B. Thirty-six percent of patients had multi-organ involvement (all with lower gastrointestinal), 38% had lower gastrointestinal involvement only, and 26% had skin involvement only.
Results
Fifty-four patients were treated with remestemcel-L. They received 8 injections over 4 weeks (twice weekly), consisting of 2 million cells per kg per injection.
The overall response rate at day 28 was 69%. Twenty-nine percent of patients achieved a complete response, defined as resolution of acute GVHD in all involved organs.
Forty percent of patients achieved a partial response, defined as organ-level improvement of at least one stage without worsening of any other organ.
All patients reported at least one treatment-emergent AE, and 61% had serious treatment-emergent AEs. The most common of these were infection (33%) and respiratory events (20%).
Four patients withdrew from the trial before day 100. One patient couldn’t receive treatment, 1 withdrew due to an AE (somnolence), 1 had parental consent withdrawn, and 1 was taken off study by the principal investigator.
There were 11 on-study deaths, but none were considered related to remestemcel-L. Eight deaths were due to infection, 1 due to GVHD progression, and 2 due to primary cancer relapse.
The day-100 survival analysis is pending.
CAR T-cell therapy bridges to HSCT in AML patient
A case report suggests an investigational chimeric antigen receptor (CAR) T-cell therapy can provide a bridge to transplant in relapsed/refractory acute myeloid leukemia (AML).
The therapy, known as CYAD-01, prompted a morphologic leukemia-free state in an AML patient.
This patient went on to receive an allogeneic hematopoietic stem cell transplant (allo-HSCT) and achieve a complete molecular remission, which was ongoing 6 months after transplant.
Investigators reported no adverse events related to CYAD-01.
This report was published in haematologica.
The patient is enrolled in the THINK trial (NCT03018405), which is sponsored by Celyad SA, the company developing CYAD-01.
According to Celyad, CYAD-01 consists of autologous T cells expressing a CAR based on the natural killer group 2 member D receptor (NKG2D), a transmembrane receptor expressed by natural killer cells and some T-cell subsets.
The AML patient who received CYAD-01 was 52 years old at trial enrollment. He had +8/del(7)(q22q36), FLT3/NPM1 wild-type AML.
The patient’s disease was primary refractory to 7+3 induction, so he went on to receive salvage chemotherapy with cladribine, cytarabine, G-CSF, and mitoxantrone. He achieved a complete response to this treatment and received 2 cycles of consolidation with cladribine and cytarabine.
The patient’s subsequent allo-HSCT was delayed to allow for pulmonary function test recovery. In the meantime, he relapsed.
At this point, the patient enrolled in the THINK trial. He underwent apheresis and received CYAD-01 infusions at the initial dose level of 3 x 108 cells every 2 weeks for 3 administrations.
The patient achieved a morphologic leukemia-free state at 3 months, which enabled him to undergo allo-HSCT.
The patient achieved a complete molecular remission after transplant. He remained in remission at last follow-up—9 months after his first CYAD-01 infusion and 6 months after allo-HSCT.
The investigators said CYAD-01 was well tolerated in this patient. He did not develop cytokine release syndrome or experience neurotoxic effects. The patient had only non-related grade 1 adverse events.
“The THINK study case report provides the first clinical validity of CYAD-01 as a tumor-specific antigen-receptor and AML as a disease sensitive to gene-engineered cell therapies,” said study investigator David Sallman, MD, of Moffitt Cancer Center in Tampa, Florida.
“As antigen targeting offers significant challenges in AML, this outcome brings hope for the further use of gene-engineered T cells for patients with AML [who] have run out of therapeutic options. It’s all the more striking that this outcome was observed without any prior lymphodepletion, highlighting the potential of using a physiologic antigen-receptor.”
A case report suggests an investigational chimeric antigen receptor (CAR) T-cell therapy can provide a bridge to transplant in relapsed/refractory acute myeloid leukemia (AML).
The therapy, known as CYAD-01, prompted a morphologic leukemia-free state in an AML patient.
This patient went on to receive an allogeneic hematopoietic stem cell transplant (allo-HSCT) and achieve a complete molecular remission, which was ongoing 6 months after transplant.
Investigators reported no adverse events related to CYAD-01.
This report was published in haematologica.
The patient is enrolled in the THINK trial (NCT03018405), which is sponsored by Celyad SA, the company developing CYAD-01.
According to Celyad, CYAD-01 consists of autologous T cells expressing a CAR based on the natural killer group 2 member D receptor (NKG2D), a transmembrane receptor expressed by natural killer cells and some T-cell subsets.
The AML patient who received CYAD-01 was 52 years old at trial enrollment. He had +8/del(7)(q22q36), FLT3/NPM1 wild-type AML.
The patient’s disease was primary refractory to 7+3 induction, so he went on to receive salvage chemotherapy with cladribine, cytarabine, G-CSF, and mitoxantrone. He achieved a complete response to this treatment and received 2 cycles of consolidation with cladribine and cytarabine.
The patient’s subsequent allo-HSCT was delayed to allow for pulmonary function test recovery. In the meantime, he relapsed.
At this point, the patient enrolled in the THINK trial. He underwent apheresis and received CYAD-01 infusions at the initial dose level of 3 x 108 cells every 2 weeks for 3 administrations.
The patient achieved a morphologic leukemia-free state at 3 months, which enabled him to undergo allo-HSCT.
The patient achieved a complete molecular remission after transplant. He remained in remission at last follow-up—9 months after his first CYAD-01 infusion and 6 months after allo-HSCT.
The investigators said CYAD-01 was well tolerated in this patient. He did not develop cytokine release syndrome or experience neurotoxic effects. The patient had only non-related grade 1 adverse events.
“The THINK study case report provides the first clinical validity of CYAD-01 as a tumor-specific antigen-receptor and AML as a disease sensitive to gene-engineered cell therapies,” said study investigator David Sallman, MD, of Moffitt Cancer Center in Tampa, Florida.
“As antigen targeting offers significant challenges in AML, this outcome brings hope for the further use of gene-engineered T cells for patients with AML [who] have run out of therapeutic options. It’s all the more striking that this outcome was observed without any prior lymphodepletion, highlighting the potential of using a physiologic antigen-receptor.”
A case report suggests an investigational chimeric antigen receptor (CAR) T-cell therapy can provide a bridge to transplant in relapsed/refractory acute myeloid leukemia (AML).
The therapy, known as CYAD-01, prompted a morphologic leukemia-free state in an AML patient.
This patient went on to receive an allogeneic hematopoietic stem cell transplant (allo-HSCT) and achieve a complete molecular remission, which was ongoing 6 months after transplant.
Investigators reported no adverse events related to CYAD-01.
This report was published in haematologica.
The patient is enrolled in the THINK trial (NCT03018405), which is sponsored by Celyad SA, the company developing CYAD-01.
According to Celyad, CYAD-01 consists of autologous T cells expressing a CAR based on the natural killer group 2 member D receptor (NKG2D), a transmembrane receptor expressed by natural killer cells and some T-cell subsets.
The AML patient who received CYAD-01 was 52 years old at trial enrollment. He had +8/del(7)(q22q36), FLT3/NPM1 wild-type AML.
The patient’s disease was primary refractory to 7+3 induction, so he went on to receive salvage chemotherapy with cladribine, cytarabine, G-CSF, and mitoxantrone. He achieved a complete response to this treatment and received 2 cycles of consolidation with cladribine and cytarabine.
The patient’s subsequent allo-HSCT was delayed to allow for pulmonary function test recovery. In the meantime, he relapsed.
At this point, the patient enrolled in the THINK trial. He underwent apheresis and received CYAD-01 infusions at the initial dose level of 3 x 108 cells every 2 weeks for 3 administrations.
The patient achieved a morphologic leukemia-free state at 3 months, which enabled him to undergo allo-HSCT.
The patient achieved a complete molecular remission after transplant. He remained in remission at last follow-up—9 months after his first CYAD-01 infusion and 6 months after allo-HSCT.
The investigators said CYAD-01 was well tolerated in this patient. He did not develop cytokine release syndrome or experience neurotoxic effects. The patient had only non-related grade 1 adverse events.
“The THINK study case report provides the first clinical validity of CYAD-01 as a tumor-specific antigen-receptor and AML as a disease sensitive to gene-engineered cell therapies,” said study investigator David Sallman, MD, of Moffitt Cancer Center in Tampa, Florida.
“As antigen targeting offers significant challenges in AML, this outcome brings hope for the further use of gene-engineered T cells for patients with AML [who] have run out of therapeutic options. It’s all the more striking that this outcome was observed without any prior lymphodepletion, highlighting the potential of using a physiologic antigen-receptor.”
JAK inhibitor reduces GVHD in mice

An investigational JAK1/2 inhibitor can fight graft-versus-host disease (GVHD), according to preclinical research published in Leukemia.
The inhibitor, baricitinib, reduced GVHD in mice while preserving T-cell expansion and the graft-versus-leukemia (GVL) effect.
Baricitinib proved more effective than ruxolitinib for the treatment and prevention of GVHD and enabled 100% survival in a mouse model of severe GVHD.
“We were surprised to achieve 100% survival of mice with the most severe model of graft-versus-host disease,” said study author Jaebok Choi, PhD, of the Washington University School of Medicine in St. Louis, Missouri.
“We are now studying the multi-pronged ways this drug behaves in an effort to develop an even better version for eventual use in clinical trials.”
For the current study, Dr Choi and his colleagues tested baricitinib and ruxolitinib in murine recipients of allogeneic hematopoietic stem cell transplants (allo-HSCTs). Mice received either drug for 31 days post-HSCT.
Mice treated with baricitinib had a significant reduction in intestinal GVHD compared to both ruxolitinib recipients and vehicle-treated controls (P=0.037).
In addition, 100% of baricitinib recipients were still alive at 60 days after HSCT, compared to about 60% of ruxolitinib recipients (P=0.0025) and almost none of the vehicle-treated controls.
The researchers found that baricitinib recipients had significantly better blood cell count recovery than control mice. Baricitinib recipients also had full donor chimerism and significantly higher percentages of donor bone marrow-derived B and T cells.
Furthermore, baricitinib recipients had higher levels of donor-derived regulatory T cells (Tregs) compared to vehicle- or ruxolitinib-treated mice.
However, the researchers noted that baricitinib recipients had a survival rate of about 70% even in the absence of donor Tregs. The team said this suggests baricitinib fights GVHD independently of the enhanced expansion of Tregs.
Further investigation revealed that baricitinib decreases helper T-cell 1 and 2 differentiation and reduces the expression of MHC II, CD80/86, and PD-L1 on allogeneic antigen-presenting cells.
The researchers also found that baricitinib could reverse ongoing GVHD. The team withheld baricitinib until mice developed GVHD, then tested the drug at doses of 200 μg and 400 μg per day.
Both doses reduced clinical GVHD scores and enabled 100% overall survival rates.
Finally, the researchers found that baricitinib “preserves and enhances” GVL effects. The team infused A20 cells and T-cell-depleted bone marrow cells into lethally irradiated mice, waited for the B-cell lymphoma to become established, and infused donor T cells.
The researchers found that baricitinib alone did not inhibit tumor growth, but it enhanced the GVL effects of donor T cells, significantly lowering the tumor burden.
“We don’t know yet exactly how this happens, but we’re working to understand it,” Dr Choi said. “We think at least part of the explanation is the drug strips the leukemia cells of their immune defenses, making them more vulnerable to attack by the donor T cells.”
“At the same time, the drug also stops the donor T cells from being able to make their way to important healthy tissues, such as the skin, liver, and gastrointestinal tract, where they often do the most damage.”
An investigational JAK1/2 inhibitor can fight graft-versus-host disease (GVHD), according to preclinical research published in Leukemia.
The inhibitor, baricitinib, reduced GVHD in mice while preserving T-cell expansion and the graft-versus-leukemia (GVL) effect.
Baricitinib proved more effective than ruxolitinib for the treatment and prevention of GVHD and enabled 100% survival in a mouse model of severe GVHD.
“We were surprised to achieve 100% survival of mice with the most severe model of graft-versus-host disease,” said study author Jaebok Choi, PhD, of the Washington University School of Medicine in St. Louis, Missouri.
“We are now studying the multi-pronged ways this drug behaves in an effort to develop an even better version for eventual use in clinical trials.”
For the current study, Dr Choi and his colleagues tested baricitinib and ruxolitinib in murine recipients of allogeneic hematopoietic stem cell transplants (allo-HSCTs). Mice received either drug for 31 days post-HSCT.
Mice treated with baricitinib had a significant reduction in intestinal GVHD compared to both ruxolitinib recipients and vehicle-treated controls (P=0.037).
In addition, 100% of baricitinib recipients were still alive at 60 days after HSCT, compared to about 60% of ruxolitinib recipients (P=0.0025) and almost none of the vehicle-treated controls.
The researchers found that baricitinib recipients had significantly better blood cell count recovery than control mice. Baricitinib recipients also had full donor chimerism and significantly higher percentages of donor bone marrow-derived B and T cells.
Furthermore, baricitinib recipients had higher levels of donor-derived regulatory T cells (Tregs) compared to vehicle- or ruxolitinib-treated mice.
However, the researchers noted that baricitinib recipients had a survival rate of about 70% even in the absence of donor Tregs. The team said this suggests baricitinib fights GVHD independently of the enhanced expansion of Tregs.
Further investigation revealed that baricitinib decreases helper T-cell 1 and 2 differentiation and reduces the expression of MHC II, CD80/86, and PD-L1 on allogeneic antigen-presenting cells.
The researchers also found that baricitinib could reverse ongoing GVHD. The team withheld baricitinib until mice developed GVHD, then tested the drug at doses of 200 μg and 400 μg per day.
Both doses reduced clinical GVHD scores and enabled 100% overall survival rates.
Finally, the researchers found that baricitinib “preserves and enhances” GVL effects. The team infused A20 cells and T-cell-depleted bone marrow cells into lethally irradiated mice, waited for the B-cell lymphoma to become established, and infused donor T cells.
The researchers found that baricitinib alone did not inhibit tumor growth, but it enhanced the GVL effects of donor T cells, significantly lowering the tumor burden.
“We don’t know yet exactly how this happens, but we’re working to understand it,” Dr Choi said. “We think at least part of the explanation is the drug strips the leukemia cells of their immune defenses, making them more vulnerable to attack by the donor T cells.”
“At the same time, the drug also stops the donor T cells from being able to make their way to important healthy tissues, such as the skin, liver, and gastrointestinal tract, where they often do the most damage.”
An investigational JAK1/2 inhibitor can fight graft-versus-host disease (GVHD), according to preclinical research published in Leukemia.
The inhibitor, baricitinib, reduced GVHD in mice while preserving T-cell expansion and the graft-versus-leukemia (GVL) effect.
Baricitinib proved more effective than ruxolitinib for the treatment and prevention of GVHD and enabled 100% survival in a mouse model of severe GVHD.
“We were surprised to achieve 100% survival of mice with the most severe model of graft-versus-host disease,” said study author Jaebok Choi, PhD, of the Washington University School of Medicine in St. Louis, Missouri.
“We are now studying the multi-pronged ways this drug behaves in an effort to develop an even better version for eventual use in clinical trials.”
For the current study, Dr Choi and his colleagues tested baricitinib and ruxolitinib in murine recipients of allogeneic hematopoietic stem cell transplants (allo-HSCTs). Mice received either drug for 31 days post-HSCT.
Mice treated with baricitinib had a significant reduction in intestinal GVHD compared to both ruxolitinib recipients and vehicle-treated controls (P=0.037).
In addition, 100% of baricitinib recipients were still alive at 60 days after HSCT, compared to about 60% of ruxolitinib recipients (P=0.0025) and almost none of the vehicle-treated controls.
The researchers found that baricitinib recipients had significantly better blood cell count recovery than control mice. Baricitinib recipients also had full donor chimerism and significantly higher percentages of donor bone marrow-derived B and T cells.
Furthermore, baricitinib recipients had higher levels of donor-derived regulatory T cells (Tregs) compared to vehicle- or ruxolitinib-treated mice.
However, the researchers noted that baricitinib recipients had a survival rate of about 70% even in the absence of donor Tregs. The team said this suggests baricitinib fights GVHD independently of the enhanced expansion of Tregs.
Further investigation revealed that baricitinib decreases helper T-cell 1 and 2 differentiation and reduces the expression of MHC II, CD80/86, and PD-L1 on allogeneic antigen-presenting cells.
The researchers also found that baricitinib could reverse ongoing GVHD. The team withheld baricitinib until mice developed GVHD, then tested the drug at doses of 200 μg and 400 μg per day.
Both doses reduced clinical GVHD scores and enabled 100% overall survival rates.
Finally, the researchers found that baricitinib “preserves and enhances” GVL effects. The team infused A20 cells and T-cell-depleted bone marrow cells into lethally irradiated mice, waited for the B-cell lymphoma to become established, and infused donor T cells.
The researchers found that baricitinib alone did not inhibit tumor growth, but it enhanced the GVL effects of donor T cells, significantly lowering the tumor burden.
“We don’t know yet exactly how this happens, but we’re working to understand it,” Dr Choi said. “We think at least part of the explanation is the drug strips the leukemia cells of their immune defenses, making them more vulnerable to attack by the donor T cells.”
“At the same time, the drug also stops the donor T cells from being able to make their way to important healthy tissues, such as the skin, liver, and gastrointestinal tract, where they often do the most damage.”
Drug receives breakthrough designation for HSCT-TMA
The US Food and Drug Administration (FDA) has granted a second breakthrough therapy designation to OMS721.
OMS721 is a monoclonal antibody targeting MASP-2, the effector enzyme of the lectin pathway of the complement system.
The new breakthrough designation is for OMS721 as a treatment for patients with high-risk hematopoietic stem cell transplant-associated thrombotic microangiopathy (HSCT-TMA) who have persistent TMA despite modification of immunosuppressive therapy.
OMS721 also has breakthrough designation from the FDA for the treatment of immunoglobulin A nephropathy.
Phase 2 trial
The breakthrough designation for HSCT-TMA was granted based on data from an ongoing phase 2 trial (NCT02222545). Omeros Corporation, the company developing OMS721, released some results from this study in February.
The trial is enrolling adults with HSCT-TMA persisting for at least 2 weeks following immunosuppressive regimen modification or more than 30 days post-transplant. Patients receive weekly OMS721 treatments for 4 to 8 weeks at the discretion of the investigator.
At the time of Omeros’s announcement, 18 patients had been treated on this study.
These patients had a significantly longer median overall survival than historical controls—347 days and 21 days, respectively (P<0.0001).
Omeros also reported that markers of TMA activity significantly improved following OMS721 treatment.
The mean platelet count increased from 18,100 x 106/mL at baseline to 52,300 x 106/mL (P=0.017). The mean LDH decreased from 591 U/L to 250 U/L (P<0.001). And the mean haptoglobin increased from 8 mg/dL to 141 mg/dL (P=0.003).
Mean creatinine remained stable—at approximately 120 μmol/L—but a majority of patients had co-existing conditions for which they were receiving nephrotoxic medications. These conditions included graft-versus-host disease, cytomegalovirus and human herpes virus 6 infections, prior sepsis, diffuse alveolar hemorrhage, and residual underlying malignancies.
The most commonly reported adverse events in this trial are diarrhea and neutropenia.
Four deaths occurred. One of these—due to acute renal and respiratory failure—was considered possibly related to OMS721.
The other deaths were due to progression of acute myeloid leukemia (n=1) and neutropenic sepsis (n=2).
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
The US Food and Drug Administration (FDA) has granted a second breakthrough therapy designation to OMS721.
OMS721 is a monoclonal antibody targeting MASP-2, the effector enzyme of the lectin pathway of the complement system.
The new breakthrough designation is for OMS721 as a treatment for patients with high-risk hematopoietic stem cell transplant-associated thrombotic microangiopathy (HSCT-TMA) who have persistent TMA despite modification of immunosuppressive therapy.
OMS721 also has breakthrough designation from the FDA for the treatment of immunoglobulin A nephropathy.
Phase 2 trial
The breakthrough designation for HSCT-TMA was granted based on data from an ongoing phase 2 trial (NCT02222545). Omeros Corporation, the company developing OMS721, released some results from this study in February.
The trial is enrolling adults with HSCT-TMA persisting for at least 2 weeks following immunosuppressive regimen modification or more than 30 days post-transplant. Patients receive weekly OMS721 treatments for 4 to 8 weeks at the discretion of the investigator.
At the time of Omeros’s announcement, 18 patients had been treated on this study.
These patients had a significantly longer median overall survival than historical controls—347 days and 21 days, respectively (P<0.0001).
Omeros also reported that markers of TMA activity significantly improved following OMS721 treatment.
The mean platelet count increased from 18,100 x 106/mL at baseline to 52,300 x 106/mL (P=0.017). The mean LDH decreased from 591 U/L to 250 U/L (P<0.001). And the mean haptoglobin increased from 8 mg/dL to 141 mg/dL (P=0.003).
Mean creatinine remained stable—at approximately 120 μmol/L—but a majority of patients had co-existing conditions for which they were receiving nephrotoxic medications. These conditions included graft-versus-host disease, cytomegalovirus and human herpes virus 6 infections, prior sepsis, diffuse alveolar hemorrhage, and residual underlying malignancies.
The most commonly reported adverse events in this trial are diarrhea and neutropenia.
Four deaths occurred. One of these—due to acute renal and respiratory failure—was considered possibly related to OMS721.
The other deaths were due to progression of acute myeloid leukemia (n=1) and neutropenic sepsis (n=2).
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
The US Food and Drug Administration (FDA) has granted a second breakthrough therapy designation to OMS721.
OMS721 is a monoclonal antibody targeting MASP-2, the effector enzyme of the lectin pathway of the complement system.
The new breakthrough designation is for OMS721 as a treatment for patients with high-risk hematopoietic stem cell transplant-associated thrombotic microangiopathy (HSCT-TMA) who have persistent TMA despite modification of immunosuppressive therapy.
OMS721 also has breakthrough designation from the FDA for the treatment of immunoglobulin A nephropathy.
Phase 2 trial
The breakthrough designation for HSCT-TMA was granted based on data from an ongoing phase 2 trial (NCT02222545). Omeros Corporation, the company developing OMS721, released some results from this study in February.
The trial is enrolling adults with HSCT-TMA persisting for at least 2 weeks following immunosuppressive regimen modification or more than 30 days post-transplant. Patients receive weekly OMS721 treatments for 4 to 8 weeks at the discretion of the investigator.
At the time of Omeros’s announcement, 18 patients had been treated on this study.
These patients had a significantly longer median overall survival than historical controls—347 days and 21 days, respectively (P<0.0001).
Omeros also reported that markers of TMA activity significantly improved following OMS721 treatment.
The mean platelet count increased from 18,100 x 106/mL at baseline to 52,300 x 106/mL (P=0.017). The mean LDH decreased from 591 U/L to 250 U/L (P<0.001). And the mean haptoglobin increased from 8 mg/dL to 141 mg/dL (P=0.003).
Mean creatinine remained stable—at approximately 120 μmol/L—but a majority of patients had co-existing conditions for which they were receiving nephrotoxic medications. These conditions included graft-versus-host disease, cytomegalovirus and human herpes virus 6 infections, prior sepsis, diffuse alveolar hemorrhage, and residual underlying malignancies.
The most commonly reported adverse events in this trial are diarrhea and neutropenia.
Four deaths occurred. One of these—due to acute renal and respiratory failure—was considered possibly related to OMS721.
The other deaths were due to progression of acute myeloid leukemia (n=1) and neutropenic sepsis (n=2).
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Haplo-HSCT regimen can cure SCD, team says

A haploidentical transplant regimen has led to long-term engraftment in adults with sickle cell disease (SCD), according to researchers.
Seven of 8 patients treated with this regimen were still alive at last follow-up, and 6 of them maintained engraftment.
Two patients developed graft-versus-host disease (GVHD). One of them had acute and chronic GVHD and died about 400 days after transplant. The other had acute GVHD that resolved with treatment.
Damiano Rondelli, MD, of the University of Illinois at Chicago, and his colleagues reported these results in Biology of Blood and Marrow Transplantation.
The researchers initially screened 50 adult SCD patients as candidates for haploidentical hematopoietic stem cell transplant (haplo-HSCT) between January 2014 and March 2017. Most patients were ineligible or declined the procedure.
Ultimately, 10 patients received a haplo-HSCT. Unfortunately, the first 2 patients failed to engraft. These patients had received conditioning with alemtuzumab and 3 Gy total body irradiation (TBI) as well as post-transplant cyclophosphamide.
Because of this failure, the researchers used the following regimen for the remaining 8 patients. It’s a modified version of the regimen described in Blood in 2012.
“We modified the transplant protocol by increasing the dose of radiation used before the transplant and by infusing growth factor-mobilized peripheral blood stem cells instead of bone marrow cells,” Dr Rondelli said. “These two modifications helped ensure the patient’s body could accept the healthy donor cells.”
Modified regimen
Patients received growth-factor-mobilized peripheral blood stem cells after conditioning with rabbit antithymocyte globulin (0.5 mg/kg on day -9, 2 mg/kg on day -8 and -7), cyclophosphamide (14.5 mg/kg on day -6 and -5), fludarabine (30 mg/m2 on day -6 to -2), and single-dose TBI (3 Gy on day -1).
For GVHD prophylaxis, patients received intravenous cyclophosphamide (50 mg/kg on day 3 and 4), oral mycophenolate mofetil (15 mg/kg 3 times daily from day 5 to 35), and sirolimus (from day 5 dosed for a target trough of 5 to 15 ng/mL). In patients who had T-cell chimerism greater than 50% at 1 year after HSCT and did not have signs of GVHD, sirolimus was tapered off over 3 months.
Patients stopped taking hydroxyurea on day -9. They received red blood cell exchange transfusion on day -10 (with the goal of getting hemoglobin S below 30%) and received platelet transfusions to maintain platelet counts greater than 50 x 109 cells/L.
Patients also received penicillin V (250 mg twice daily) in addition to standard antimicrobial prophylaxis.
Results
All 8 patients on the modified regimen engrafted. The median time to neutrophil engraftment was 22 days (range, 18 to 23 days). One patient experienced secondary graft failure on day 90.
Seven neutropenic patients with hemoglobin S less than 30% received G-CSF after transplant. They received a median of 7 doses (range, 3 to 14) at 5 μg/kg, starting at day 12 post-HSCT. One of these patients experienced mild bone pain in the lower extremities.
Two patients developed GVHD. At day 83, one patient developed acute or chronic GVHD involving the skin, liver, and eyes. Steroids and sirolimus improved eye and liver symptoms, but the patient died at home on day 407.
The other patient had grade 2, gastrointestinal, acute GVHD that resolved with steroid therapy.
Three patients had grade 2 or higher mucositis, and 2 had cytomegalovirus (CMV) reactivation without CMV infection.
Two patients had small subarachnoid hemorrhages. One of these patients had a history of multiple red blood cell antibodies, became refractory to platelet transfusions, and developed small subarachnoid hemorrhages day 10. The patient’s symptoms and brain imaging resolved after platelet counts were maintained above 50 x 109 cells/L with cross-matched platelets.
The second patient had a history of stroke, experienced a seizure when the platelet count was 68 x 109 cells/L, and was found to have a subarachnoid hemorrhage on day 12. Symptoms and imaging results improved once the patient began levetiracetam and platelet levels were maintained above 100 x 109 cells/L.
At a median follow-up of 17 months (range, 12 to 30), 7 of the 8 patients were still alive.
Six patients had maintained greater than 95% stable donor engraftment with improvements in their hemoglobin concentrations. Three of these patients have stopped immunosuppression, and 3 are being tapered off it.
“These patients are cured of sickle cell disease,” Dr Rondelli said. “The takeaway message is two-fold. First, this transplant protocol may cure many more adult patients with advanced sickle cell disease.”
“Second, despite the increasing safety of the transplant protocols and new compatibility of HLA half-matched donors, many sickle cell patients still face barriers to care. Of the patients we screened, only 20% underwent a transplant.”
Dr Rondelli noted that 20% of the patients screened could not undergo transplant because of insurance denial. Other patients were ineligible because they had high rates of donor-specific antigens, and still others declined transplant.
A haploidentical transplant regimen has led to long-term engraftment in adults with sickle cell disease (SCD), according to researchers.
Seven of 8 patients treated with this regimen were still alive at last follow-up, and 6 of them maintained engraftment.
Two patients developed graft-versus-host disease (GVHD). One of them had acute and chronic GVHD and died about 400 days after transplant. The other had acute GVHD that resolved with treatment.
Damiano Rondelli, MD, of the University of Illinois at Chicago, and his colleagues reported these results in Biology of Blood and Marrow Transplantation.
The researchers initially screened 50 adult SCD patients as candidates for haploidentical hematopoietic stem cell transplant (haplo-HSCT) between January 2014 and March 2017. Most patients were ineligible or declined the procedure.
Ultimately, 10 patients received a haplo-HSCT. Unfortunately, the first 2 patients failed to engraft. These patients had received conditioning with alemtuzumab and 3 Gy total body irradiation (TBI) as well as post-transplant cyclophosphamide.
Because of this failure, the researchers used the following regimen for the remaining 8 patients. It’s a modified version of the regimen described in Blood in 2012.
“We modified the transplant protocol by increasing the dose of radiation used before the transplant and by infusing growth factor-mobilized peripheral blood stem cells instead of bone marrow cells,” Dr Rondelli said. “These two modifications helped ensure the patient’s body could accept the healthy donor cells.”
Modified regimen
Patients received growth-factor-mobilized peripheral blood stem cells after conditioning with rabbit antithymocyte globulin (0.5 mg/kg on day -9, 2 mg/kg on day -8 and -7), cyclophosphamide (14.5 mg/kg on day -6 and -5), fludarabine (30 mg/m2 on day -6 to -2), and single-dose TBI (3 Gy on day -1).
For GVHD prophylaxis, patients received intravenous cyclophosphamide (50 mg/kg on day 3 and 4), oral mycophenolate mofetil (15 mg/kg 3 times daily from day 5 to 35), and sirolimus (from day 5 dosed for a target trough of 5 to 15 ng/mL). In patients who had T-cell chimerism greater than 50% at 1 year after HSCT and did not have signs of GVHD, sirolimus was tapered off over 3 months.
Patients stopped taking hydroxyurea on day -9. They received red blood cell exchange transfusion on day -10 (with the goal of getting hemoglobin S below 30%) and received platelet transfusions to maintain platelet counts greater than 50 x 109 cells/L.
Patients also received penicillin V (250 mg twice daily) in addition to standard antimicrobial prophylaxis.
Results
All 8 patients on the modified regimen engrafted. The median time to neutrophil engraftment was 22 days (range, 18 to 23 days). One patient experienced secondary graft failure on day 90.
Seven neutropenic patients with hemoglobin S less than 30% received G-CSF after transplant. They received a median of 7 doses (range, 3 to 14) at 5 μg/kg, starting at day 12 post-HSCT. One of these patients experienced mild bone pain in the lower extremities.
Two patients developed GVHD. At day 83, one patient developed acute or chronic GVHD involving the skin, liver, and eyes. Steroids and sirolimus improved eye and liver symptoms, but the patient died at home on day 407.
The other patient had grade 2, gastrointestinal, acute GVHD that resolved with steroid therapy.
Three patients had grade 2 or higher mucositis, and 2 had cytomegalovirus (CMV) reactivation without CMV infection.
Two patients had small subarachnoid hemorrhages. One of these patients had a history of multiple red blood cell antibodies, became refractory to platelet transfusions, and developed small subarachnoid hemorrhages day 10. The patient’s symptoms and brain imaging resolved after platelet counts were maintained above 50 x 109 cells/L with cross-matched platelets.
The second patient had a history of stroke, experienced a seizure when the platelet count was 68 x 109 cells/L, and was found to have a subarachnoid hemorrhage on day 12. Symptoms and imaging results improved once the patient began levetiracetam and platelet levels were maintained above 100 x 109 cells/L.
At a median follow-up of 17 months (range, 12 to 30), 7 of the 8 patients were still alive.
Six patients had maintained greater than 95% stable donor engraftment with improvements in their hemoglobin concentrations. Three of these patients have stopped immunosuppression, and 3 are being tapered off it.
“These patients are cured of sickle cell disease,” Dr Rondelli said. “The takeaway message is two-fold. First, this transplant protocol may cure many more adult patients with advanced sickle cell disease.”
“Second, despite the increasing safety of the transplant protocols and new compatibility of HLA half-matched donors, many sickle cell patients still face barriers to care. Of the patients we screened, only 20% underwent a transplant.”
Dr Rondelli noted that 20% of the patients screened could not undergo transplant because of insurance denial. Other patients were ineligible because they had high rates of donor-specific antigens, and still others declined transplant.
A haploidentical transplant regimen has led to long-term engraftment in adults with sickle cell disease (SCD), according to researchers.
Seven of 8 patients treated with this regimen were still alive at last follow-up, and 6 of them maintained engraftment.
Two patients developed graft-versus-host disease (GVHD). One of them had acute and chronic GVHD and died about 400 days after transplant. The other had acute GVHD that resolved with treatment.
Damiano Rondelli, MD, of the University of Illinois at Chicago, and his colleagues reported these results in Biology of Blood and Marrow Transplantation.
The researchers initially screened 50 adult SCD patients as candidates for haploidentical hematopoietic stem cell transplant (haplo-HSCT) between January 2014 and March 2017. Most patients were ineligible or declined the procedure.
Ultimately, 10 patients received a haplo-HSCT. Unfortunately, the first 2 patients failed to engraft. These patients had received conditioning with alemtuzumab and 3 Gy total body irradiation (TBI) as well as post-transplant cyclophosphamide.
Because of this failure, the researchers used the following regimen for the remaining 8 patients. It’s a modified version of the regimen described in Blood in 2012.
“We modified the transplant protocol by increasing the dose of radiation used before the transplant and by infusing growth factor-mobilized peripheral blood stem cells instead of bone marrow cells,” Dr Rondelli said. “These two modifications helped ensure the patient’s body could accept the healthy donor cells.”
Modified regimen
Patients received growth-factor-mobilized peripheral blood stem cells after conditioning with rabbit antithymocyte globulin (0.5 mg/kg on day -9, 2 mg/kg on day -8 and -7), cyclophosphamide (14.5 mg/kg on day -6 and -5), fludarabine (30 mg/m2 on day -6 to -2), and single-dose TBI (3 Gy on day -1).
For GVHD prophylaxis, patients received intravenous cyclophosphamide (50 mg/kg on day 3 and 4), oral mycophenolate mofetil (15 mg/kg 3 times daily from day 5 to 35), and sirolimus (from day 5 dosed for a target trough of 5 to 15 ng/mL). In patients who had T-cell chimerism greater than 50% at 1 year after HSCT and did not have signs of GVHD, sirolimus was tapered off over 3 months.
Patients stopped taking hydroxyurea on day -9. They received red blood cell exchange transfusion on day -10 (with the goal of getting hemoglobin S below 30%) and received platelet transfusions to maintain platelet counts greater than 50 x 109 cells/L.
Patients also received penicillin V (250 mg twice daily) in addition to standard antimicrobial prophylaxis.
Results
All 8 patients on the modified regimen engrafted. The median time to neutrophil engraftment was 22 days (range, 18 to 23 days). One patient experienced secondary graft failure on day 90.
Seven neutropenic patients with hemoglobin S less than 30% received G-CSF after transplant. They received a median of 7 doses (range, 3 to 14) at 5 μg/kg, starting at day 12 post-HSCT. One of these patients experienced mild bone pain in the lower extremities.
Two patients developed GVHD. At day 83, one patient developed acute or chronic GVHD involving the skin, liver, and eyes. Steroids and sirolimus improved eye and liver symptoms, but the patient died at home on day 407.
The other patient had grade 2, gastrointestinal, acute GVHD that resolved with steroid therapy.
Three patients had grade 2 or higher mucositis, and 2 had cytomegalovirus (CMV) reactivation without CMV infection.
Two patients had small subarachnoid hemorrhages. One of these patients had a history of multiple red blood cell antibodies, became refractory to platelet transfusions, and developed small subarachnoid hemorrhages day 10. The patient’s symptoms and brain imaging resolved after platelet counts were maintained above 50 x 109 cells/L with cross-matched platelets.
The second patient had a history of stroke, experienced a seizure when the platelet count was 68 x 109 cells/L, and was found to have a subarachnoid hemorrhage on day 12. Symptoms and imaging results improved once the patient began levetiracetam and platelet levels were maintained above 100 x 109 cells/L.
At a median follow-up of 17 months (range, 12 to 30), 7 of the 8 patients were still alive.
Six patients had maintained greater than 95% stable donor engraftment with improvements in their hemoglobin concentrations. Three of these patients have stopped immunosuppression, and 3 are being tapered off it.
“These patients are cured of sickle cell disease,” Dr Rondelli said. “The takeaway message is two-fold. First, this transplant protocol may cure many more adult patients with advanced sickle cell disease.”
“Second, despite the increasing safety of the transplant protocols and new compatibility of HLA half-matched donors, many sickle cell patients still face barriers to care. Of the patients we screened, only 20% underwent a transplant.”
Dr Rondelli noted that 20% of the patients screened could not undergo transplant because of insurance denial. Other patients were ineligible because they had high rates of donor-specific antigens, and still others declined transplant.
Art education benefits blood cancer patients

New research suggests a bedside visual art intervention (BVAI) can reduce pain and anxiety in inpatients with hematologic malignancies, including those undergoing transplant.
The BVAI involved an educator teaching patients art technique one-on-one for approximately 30 minutes.
After a single session, patients had significant improvements in positive mood and pain scores, as well as decreases in negative mood and anxiety.
Alexandra P. Wolanskyj, MD, of Mayo Clinic in Rochester, Minnesota, and her colleagues reported these results in the European Journal of Cancer Care.
The study included 21 patients, 19 of them female. Their median age was 53.5 (range, 19-75). Six patients were undergoing hematopoietic stem cell transplant.
The patients had multiple myeloma (n=5), acute myeloid leukemia (n=5), non-Hodgkin lymphoma (n=3), Hodgkin lymphoma (n=2), acute lymphoblastic leukemia (n=1), chronic lymphocytic leukemia (n=1), amyloidosis (n=1), Gardner-Diamond syndrome (n=1), myelodysplastic syndrome (n=1), and Waldenstrom’s macroglobulinemia (n=1).
Nearly half of patients had relapsed disease (47.6%), 23.8% had active and new disease, 19.0% had active disease with primary resistance on chemotherapy, and 9.5% of patients were in remission.
Intervention
The researchers recruited an educator from a community art center to teach art at the patients’ bedsides. Sessions were intended to be about 30 minutes. However, patients could stop at any time or continue beyond 30 minutes.
Patients and their families could make art or just observe. Materials used included watercolors, oil pastels, colored pencils, and clay (all non-toxic and odorless). The materials were left with patients so they could continue to use them after the sessions.
Results
The researchers assessed patients’ pain, anxiety, and mood at baseline and after the patients had a session with the art educator.
After the BVAI, patients had a significant decrease in pain, according to the Visual Analog Scale (VAS). The 14 patients who reported any pain at baseline had a mean reduction in VAS score of 1.5, or a 35.1% reduction in pain (P=0.017).
Patients had a 21.6% reduction in anxiety after the BVAI. Among the 20 patients who completed this assessment, there was a mean 9.2-point decrease in State-Trait Anxiety Inventory (STAI) score (P=0.001).
In addition, patients had a significant increase in positive mood and a significant decrease in negative mood after the BVAI. Mood was assessed in 20 patients using the Positive and Negative Affect Schedule (PANAS) scale.
Positive mood increased 14.6% (P=0.003), and negative mood decreased 18.0% (P=0.015) after the BVAI. Patients’ mean PANAS scores increased 4.6 points for positive mood and decreased 3.3 points for negative mood.
All 21 patients completed a questionnaire on the BVAI. All but 1 patient (95%) said the intervention was positive overall, and 85% of patients (n=18) said they would be interested in participating in future art-based interventions.
The researchers said these results suggest experiences provided by artists in the community may be an adjunct to conventional treatments in patients with cancer-related mood symptoms and pain.
New research suggests a bedside visual art intervention (BVAI) can reduce pain and anxiety in inpatients with hematologic malignancies, including those undergoing transplant.
The BVAI involved an educator teaching patients art technique one-on-one for approximately 30 minutes.
After a single session, patients had significant improvements in positive mood and pain scores, as well as decreases in negative mood and anxiety.
Alexandra P. Wolanskyj, MD, of Mayo Clinic in Rochester, Minnesota, and her colleagues reported these results in the European Journal of Cancer Care.
The study included 21 patients, 19 of them female. Their median age was 53.5 (range, 19-75). Six patients were undergoing hematopoietic stem cell transplant.
The patients had multiple myeloma (n=5), acute myeloid leukemia (n=5), non-Hodgkin lymphoma (n=3), Hodgkin lymphoma (n=2), acute lymphoblastic leukemia (n=1), chronic lymphocytic leukemia (n=1), amyloidosis (n=1), Gardner-Diamond syndrome (n=1), myelodysplastic syndrome (n=1), and Waldenstrom’s macroglobulinemia (n=1).
Nearly half of patients had relapsed disease (47.6%), 23.8% had active and new disease, 19.0% had active disease with primary resistance on chemotherapy, and 9.5% of patients were in remission.
Intervention
The researchers recruited an educator from a community art center to teach art at the patients’ bedsides. Sessions were intended to be about 30 minutes. However, patients could stop at any time or continue beyond 30 minutes.
Patients and their families could make art or just observe. Materials used included watercolors, oil pastels, colored pencils, and clay (all non-toxic and odorless). The materials were left with patients so they could continue to use them after the sessions.
Results
The researchers assessed patients’ pain, anxiety, and mood at baseline and after the patients had a session with the art educator.
After the BVAI, patients had a significant decrease in pain, according to the Visual Analog Scale (VAS). The 14 patients who reported any pain at baseline had a mean reduction in VAS score of 1.5, or a 35.1% reduction in pain (P=0.017).
Patients had a 21.6% reduction in anxiety after the BVAI. Among the 20 patients who completed this assessment, there was a mean 9.2-point decrease in State-Trait Anxiety Inventory (STAI) score (P=0.001).
In addition, patients had a significant increase in positive mood and a significant decrease in negative mood after the BVAI. Mood was assessed in 20 patients using the Positive and Negative Affect Schedule (PANAS) scale.
Positive mood increased 14.6% (P=0.003), and negative mood decreased 18.0% (P=0.015) after the BVAI. Patients’ mean PANAS scores increased 4.6 points for positive mood and decreased 3.3 points for negative mood.
All 21 patients completed a questionnaire on the BVAI. All but 1 patient (95%) said the intervention was positive overall, and 85% of patients (n=18) said they would be interested in participating in future art-based interventions.
The researchers said these results suggest experiences provided by artists in the community may be an adjunct to conventional treatments in patients with cancer-related mood symptoms and pain.
New research suggests a bedside visual art intervention (BVAI) can reduce pain and anxiety in inpatients with hematologic malignancies, including those undergoing transplant.
The BVAI involved an educator teaching patients art technique one-on-one for approximately 30 minutes.
After a single session, patients had significant improvements in positive mood and pain scores, as well as decreases in negative mood and anxiety.
Alexandra P. Wolanskyj, MD, of Mayo Clinic in Rochester, Minnesota, and her colleagues reported these results in the European Journal of Cancer Care.
The study included 21 patients, 19 of them female. Their median age was 53.5 (range, 19-75). Six patients were undergoing hematopoietic stem cell transplant.
The patients had multiple myeloma (n=5), acute myeloid leukemia (n=5), non-Hodgkin lymphoma (n=3), Hodgkin lymphoma (n=2), acute lymphoblastic leukemia (n=1), chronic lymphocytic leukemia (n=1), amyloidosis (n=1), Gardner-Diamond syndrome (n=1), myelodysplastic syndrome (n=1), and Waldenstrom’s macroglobulinemia (n=1).
Nearly half of patients had relapsed disease (47.6%), 23.8% had active and new disease, 19.0% had active disease with primary resistance on chemotherapy, and 9.5% of patients were in remission.
Intervention
The researchers recruited an educator from a community art center to teach art at the patients’ bedsides. Sessions were intended to be about 30 minutes. However, patients could stop at any time or continue beyond 30 minutes.
Patients and their families could make art or just observe. Materials used included watercolors, oil pastels, colored pencils, and clay (all non-toxic and odorless). The materials were left with patients so they could continue to use them after the sessions.
Results
The researchers assessed patients’ pain, anxiety, and mood at baseline and after the patients had a session with the art educator.
After the BVAI, patients had a significant decrease in pain, according to the Visual Analog Scale (VAS). The 14 patients who reported any pain at baseline had a mean reduction in VAS score of 1.5, or a 35.1% reduction in pain (P=0.017).
Patients had a 21.6% reduction in anxiety after the BVAI. Among the 20 patients who completed this assessment, there was a mean 9.2-point decrease in State-Trait Anxiety Inventory (STAI) score (P=0.001).
In addition, patients had a significant increase in positive mood and a significant decrease in negative mood after the BVAI. Mood was assessed in 20 patients using the Positive and Negative Affect Schedule (PANAS) scale.
Positive mood increased 14.6% (P=0.003), and negative mood decreased 18.0% (P=0.015) after the BVAI. Patients’ mean PANAS scores increased 4.6 points for positive mood and decreased 3.3 points for negative mood.
All 21 patients completed a questionnaire on the BVAI. All but 1 patient (95%) said the intervention was positive overall, and 85% of patients (n=18) said they would be interested in participating in future art-based interventions.
The researchers said these results suggest experiences provided by artists in the community may be an adjunct to conventional treatments in patients with cancer-related mood symptoms and pain.
FDA lifts hold on trials of BPX-501
The US Food and Drug Administration (FDA) has lifted the clinical hold on studies of BPX-501 in the US.
BPX-501 is an adjunct T-cell therapy intended to be given after allogeneic hematopoietic stem cell transplant (HSCT).
The product is being tested in phase 1/2 trials of children and adults with hematologic disorders.
The FDA placed all US trials of BPX-501 on hold earlier this year due to 3 cases of encephalopathy that were considered possibly related to BPX-501.
The hold did not affect a registrational trial of BPX-501—known as BP-004—that is underway in Europe.
Now, the FDA has lifted the hold on the US trials after discussions with Bellicum Pharmaceuticals, Inc., the company developing BPX-501.
Bellicum agreed to amend study protocols to include guidance on monitoring and management of neurologic adverse events. The company said it will be working with US clinical sites to resume patient recruitment in BPX-501 trials based on the amended protocols.
About BPX-501
BPX-501 is intended to speed immune reconstitution, fight infection, and enhance the graft-versus-leukemia effect of HSCT while also minimizing graft-vs-host disease (GVHD) and other T-cell-mediated transplant complications.
BPX-501 contains a safety switch, CaspaCIDe®, that can be activated with the drug rimiducid. The drug can be used to eliminate alloreactive BPX-501 T cells if uncontrollable GVHD or other T-cell-mediated complications occur.
The FDA and the European Commission granted BPX-501 and rimiducid orphan drug designation for use in HSCT recipients.
BPX-501 and rimiducid have demonstrated efficacy in the BP-004 trial. Results from this trial have been presented at the EBMT Annual Meeting in 2016 (abstract WP16 and abstract O007), the 2017 EHA Congress, and the 2017 ASH Annual Meeting.
The US Food and Drug Administration (FDA) has lifted the clinical hold on studies of BPX-501 in the US.
BPX-501 is an adjunct T-cell therapy intended to be given after allogeneic hematopoietic stem cell transplant (HSCT).
The product is being tested in phase 1/2 trials of children and adults with hematologic disorders.
The FDA placed all US trials of BPX-501 on hold earlier this year due to 3 cases of encephalopathy that were considered possibly related to BPX-501.
The hold did not affect a registrational trial of BPX-501—known as BP-004—that is underway in Europe.
Now, the FDA has lifted the hold on the US trials after discussions with Bellicum Pharmaceuticals, Inc., the company developing BPX-501.
Bellicum agreed to amend study protocols to include guidance on monitoring and management of neurologic adverse events. The company said it will be working with US clinical sites to resume patient recruitment in BPX-501 trials based on the amended protocols.
About BPX-501
BPX-501 is intended to speed immune reconstitution, fight infection, and enhance the graft-versus-leukemia effect of HSCT while also minimizing graft-vs-host disease (GVHD) and other T-cell-mediated transplant complications.
BPX-501 contains a safety switch, CaspaCIDe®, that can be activated with the drug rimiducid. The drug can be used to eliminate alloreactive BPX-501 T cells if uncontrollable GVHD or other T-cell-mediated complications occur.
The FDA and the European Commission granted BPX-501 and rimiducid orphan drug designation for use in HSCT recipients.
BPX-501 and rimiducid have demonstrated efficacy in the BP-004 trial. Results from this trial have been presented at the EBMT Annual Meeting in 2016 (abstract WP16 and abstract O007), the 2017 EHA Congress, and the 2017 ASH Annual Meeting.
The US Food and Drug Administration (FDA) has lifted the clinical hold on studies of BPX-501 in the US.
BPX-501 is an adjunct T-cell therapy intended to be given after allogeneic hematopoietic stem cell transplant (HSCT).
The product is being tested in phase 1/2 trials of children and adults with hematologic disorders.
The FDA placed all US trials of BPX-501 on hold earlier this year due to 3 cases of encephalopathy that were considered possibly related to BPX-501.
The hold did not affect a registrational trial of BPX-501—known as BP-004—that is underway in Europe.
Now, the FDA has lifted the hold on the US trials after discussions with Bellicum Pharmaceuticals, Inc., the company developing BPX-501.
Bellicum agreed to amend study protocols to include guidance on monitoring and management of neurologic adverse events. The company said it will be working with US clinical sites to resume patient recruitment in BPX-501 trials based on the amended protocols.
About BPX-501
BPX-501 is intended to speed immune reconstitution, fight infection, and enhance the graft-versus-leukemia effect of HSCT while also minimizing graft-vs-host disease (GVHD) and other T-cell-mediated transplant complications.
BPX-501 contains a safety switch, CaspaCIDe®, that can be activated with the drug rimiducid. The drug can be used to eliminate alloreactive BPX-501 T cells if uncontrollable GVHD or other T-cell-mediated complications occur.
The FDA and the European Commission granted BPX-501 and rimiducid orphan drug designation for use in HSCT recipients.
BPX-501 and rimiducid have demonstrated efficacy in the BP-004 trial. Results from this trial have been presented at the EBMT Annual Meeting in 2016 (abstract WP16 and abstract O007), the 2017 EHA Congress, and the 2017 ASH Annual Meeting.