User login
Studies point to potential for huge HCV rise in MSM
WASHINGTON – Two studies of men having sex with men suggest that hepatitis C transmission in that group might be on the cusp of a big increase, in part because of risky behavior such as unprotected anal intercourse and injection drug use.
One study was conducted among MSM in Switzerland, while another was a retrospective look at HIV-negative men in several sexual health clinics across London.

In the Swiss study, researchers wanted to assess knowledge of HCV in MSM and also the prevalence of HCV in a local population. Regular screening for HCV in the MSM population was recommended by the European AIDS Clinical Society in June, but actual screening and knowledge is thought to be low, said Dr. Matthias Cavassini of Lausanne (Switzerland) Hospital.
He and his colleagues approached 918 MSM from June 2011 to August 2012 and asked them to participate in an anonymous questionnaire and, subsequently, to undergo HCV testing. Seventy-one percent – 654 – agreed. The vast majority who agreed – 82% – had been approached at a clinic for MSM that screens for sexually transmitted illnesses. The rest came from a sauna (13%), sex clubs (3.2%), a gay pride event (1.4%), and parks (0.3%).
The median age was 33; 512 identified as homosexual (78%) and 140 as bisexual (21%). Most (431) were HIV negative, although 62 preferred not to disclose HIV status. The median number of sexual partners was 5; the range was 0 to 400. Fifty-two percent (337) had a history of STI screening within the past year. When asked about HCV screening, 154 said they’d been tested, while 380 (58%) said they did not know.
Fifty-five percent (357) had unprotected anal intercourse – a primary HCV risk factor – in the past year. Thirty-three percent (219) had a history of drug use or were current users, but only four (0.6%) reported injection drug use.
Half had no knowledge about HCV. Of the 302 who did report knowledge, a striking 84% said they were aware that HCV could be transmitted through anal sex. Only 26% knew that it could not be transmitted through oral sex. There seemed to be no particular factor that predicted knowledge.
Of the 654 who consented to HCV screening, only one (0.15%) tested positive, said Dr. Cavassini. The man’s primary risk factor was unprotected anal intercourse in the previous year.
One case does not make an epidemic, but the high prevalence of unprotected anal intercourse argues for screening in the MSM population, he said. "Is this an accident waiting to happen?" asked Dr. Cavassini.
The British study was conducted by Dr. Katie McFaul and her colleagues at Chelsea and Westminster Hospital. They retrospectively reviewed charts for all HIV-negative MSM at three clinics between Jan. 2010 and May 2014, and identified just 44 who met the criteria for acute HCV.
Of those, 15 had a previous negative HCV test within the past year, 11 had significant ALT elevation, and 18 were clinically diagnosed with HCV from risk exposure, history, and symptoms. The median age was 37, and the median number of partners was 2, with a range of 1 to 100.
Looking more closely at behavior, Dr. McFaul found that 82% reported that they engaged in unprotected anal intercourse. Many also engaged in equally risky behavior, including group sex (28%), fisting (25%), or using drugs during sex, with about 20% of those reporting injection drug use.
Of the 44 with HCV, 29 knew the status of their partner or partners; 2 had a partner with HCV, 13 had a partner with HIV, and 6 had a partner with HIV and HCV. Thirteen had an STI at the time of HCV diagnosis.
Eight had received postexposure prophylaxis for HIV before their HCV diagnosis. Overall, 15 of the 44 had a spontaneous clearance of the HCV. Eleven received treatment. Eleven percent were lost to follow-up and 30% are being observed.
While HCV prevalence seemed low, it is likely not accurate, said Dr. McFaul, noting that she and her colleagues found that HCV screening in HIV-negative MSM was rarely conducted. A one-month snapshot at the three clinics found that 15% (565) of 3,811 HIV negative men who came for sexual health screening were tested.
Extrapolating the data to all 623,350 visits during the 2010-2014 study period, Dr. McFaul estimated that about 261,036 HIV-negative MSM visited. At the current 15% screening rate, 34,657 would have been screened for HCV. But that means that almost 200,000 were not screened for HCV.
"We suggest that hepatitis C screening should be performed at routine sexual health screening, particularly in individuals with risk factors, and in environments where there is a high hepatitis C prevalence," she said. Clinicians also "need to ask penetrating questions about penetrating activities.
"If you don’t take a temperature you won’t find a fever," she said. "I think we’ll find a higher prevalence then we currently see."
On Twitter @aliciaault
WASHINGTON – Two studies of men having sex with men suggest that hepatitis C transmission in that group might be on the cusp of a big increase, in part because of risky behavior such as unprotected anal intercourse and injection drug use.
One study was conducted among MSM in Switzerland, while another was a retrospective look at HIV-negative men in several sexual health clinics across London.
In the Swiss study, researchers wanted to assess knowledge of HCV in MSM and also the prevalence of HCV in a local population. Regular screening for HCV in the MSM population was recommended by the European AIDS Clinical Society in June, but actual screening and knowledge is thought to be low, said Dr. Matthias Cavassini of Lausanne (Switzerland) Hospital.
He and his colleagues approached 918 MSM from June 2011 to August 2012 and asked them to participate in an anonymous questionnaire and, subsequently, to undergo HCV testing. Seventy-one percent – 654 – agreed. The vast majority who agreed – 82% – had been approached at a clinic for MSM that screens for sexually transmitted illnesses. The rest came from a sauna (13%), sex clubs (3.2%), a gay pride event (1.4%), and parks (0.3%).
The median age was 33; 512 identified as homosexual (78%) and 140 as bisexual (21%). Most (431) were HIV negative, although 62 preferred not to disclose HIV status. The median number of sexual partners was 5; the range was 0 to 400. Fifty-two percent (337) had a history of STI screening within the past year. When asked about HCV screening, 154 said they’d been tested, while 380 (58%) said they did not know.
Fifty-five percent (357) had unprotected anal intercourse – a primary HCV risk factor – in the past year. Thirty-three percent (219) had a history of drug use or were current users, but only four (0.6%) reported injection drug use.
Half had no knowledge about HCV. Of the 302 who did report knowledge, a striking 84% said they were aware that HCV could be transmitted through anal sex. Only 26% knew that it could not be transmitted through oral sex. There seemed to be no particular factor that predicted knowledge.
Of the 654 who consented to HCV screening, only one (0.15%) tested positive, said Dr. Cavassini. The man’s primary risk factor was unprotected anal intercourse in the previous year.
One case does not make an epidemic, but the high prevalence of unprotected anal intercourse argues for screening in the MSM population, he said. "Is this an accident waiting to happen?" asked Dr. Cavassini.
The British study was conducted by Dr. Katie McFaul and her colleagues at Chelsea and Westminster Hospital. They retrospectively reviewed charts for all HIV-negative MSM at three clinics between Jan. 2010 and May 2014, and identified just 44 who met the criteria for acute HCV.
Of those, 15 had a previous negative HCV test within the past year, 11 had significant ALT elevation, and 18 were clinically diagnosed with HCV from risk exposure, history, and symptoms. The median age was 37, and the median number of partners was 2, with a range of 1 to 100.
Looking more closely at behavior, Dr. McFaul found that 82% reported that they engaged in unprotected anal intercourse. Many also engaged in equally risky behavior, including group sex (28%), fisting (25%), or using drugs during sex, with about 20% of those reporting injection drug use.
Of the 44 with HCV, 29 knew the status of their partner or partners; 2 had a partner with HCV, 13 had a partner with HIV, and 6 had a partner with HIV and HCV. Thirteen had an STI at the time of HCV diagnosis.
Eight had received postexposure prophylaxis for HIV before their HCV diagnosis. Overall, 15 of the 44 had a spontaneous clearance of the HCV. Eleven received treatment. Eleven percent were lost to follow-up and 30% are being observed.
While HCV prevalence seemed low, it is likely not accurate, said Dr. McFaul, noting that she and her colleagues found that HCV screening in HIV-negative MSM was rarely conducted. A one-month snapshot at the three clinics found that 15% (565) of 3,811 HIV negative men who came for sexual health screening were tested.
Extrapolating the data to all 623,350 visits during the 2010-2014 study period, Dr. McFaul estimated that about 261,036 HIV-negative MSM visited. At the current 15% screening rate, 34,657 would have been screened for HCV. But that means that almost 200,000 were not screened for HCV.
"We suggest that hepatitis C screening should be performed at routine sexual health screening, particularly in individuals with risk factors, and in environments where there is a high hepatitis C prevalence," she said. Clinicians also "need to ask penetrating questions about penetrating activities.
"If you don’t take a temperature you won’t find a fever," she said. "I think we’ll find a higher prevalence then we currently see."
On Twitter @aliciaault
WASHINGTON – Two studies of men having sex with men suggest that hepatitis C transmission in that group might be on the cusp of a big increase, in part because of risky behavior such as unprotected anal intercourse and injection drug use.
One study was conducted among MSM in Switzerland, while another was a retrospective look at HIV-negative men in several sexual health clinics across London.
In the Swiss study, researchers wanted to assess knowledge of HCV in MSM and also the prevalence of HCV in a local population. Regular screening for HCV in the MSM population was recommended by the European AIDS Clinical Society in June, but actual screening and knowledge is thought to be low, said Dr. Matthias Cavassini of Lausanne (Switzerland) Hospital.
He and his colleagues approached 918 MSM from June 2011 to August 2012 and asked them to participate in an anonymous questionnaire and, subsequently, to undergo HCV testing. Seventy-one percent – 654 – agreed. The vast majority who agreed – 82% – had been approached at a clinic for MSM that screens for sexually transmitted illnesses. The rest came from a sauna (13%), sex clubs (3.2%), a gay pride event (1.4%), and parks (0.3%).
The median age was 33; 512 identified as homosexual (78%) and 140 as bisexual (21%). Most (431) were HIV negative, although 62 preferred not to disclose HIV status. The median number of sexual partners was 5; the range was 0 to 400. Fifty-two percent (337) had a history of STI screening within the past year. When asked about HCV screening, 154 said they’d been tested, while 380 (58%) said they did not know.
Fifty-five percent (357) had unprotected anal intercourse – a primary HCV risk factor – in the past year. Thirty-three percent (219) had a history of drug use or were current users, but only four (0.6%) reported injection drug use.
Half had no knowledge about HCV. Of the 302 who did report knowledge, a striking 84% said they were aware that HCV could be transmitted through anal sex. Only 26% knew that it could not be transmitted through oral sex. There seemed to be no particular factor that predicted knowledge.
Of the 654 who consented to HCV screening, only one (0.15%) tested positive, said Dr. Cavassini. The man’s primary risk factor was unprotected anal intercourse in the previous year.
One case does not make an epidemic, but the high prevalence of unprotected anal intercourse argues for screening in the MSM population, he said. "Is this an accident waiting to happen?" asked Dr. Cavassini.
The British study was conducted by Dr. Katie McFaul and her colleagues at Chelsea and Westminster Hospital. They retrospectively reviewed charts for all HIV-negative MSM at three clinics between Jan. 2010 and May 2014, and identified just 44 who met the criteria for acute HCV.
Of those, 15 had a previous negative HCV test within the past year, 11 had significant ALT elevation, and 18 were clinically diagnosed with HCV from risk exposure, history, and symptoms. The median age was 37, and the median number of partners was 2, with a range of 1 to 100.
Looking more closely at behavior, Dr. McFaul found that 82% reported that they engaged in unprotected anal intercourse. Many also engaged in equally risky behavior, including group sex (28%), fisting (25%), or using drugs during sex, with about 20% of those reporting injection drug use.
Of the 44 with HCV, 29 knew the status of their partner or partners; 2 had a partner with HCV, 13 had a partner with HIV, and 6 had a partner with HIV and HCV. Thirteen had an STI at the time of HCV diagnosis.
Eight had received postexposure prophylaxis for HIV before their HCV diagnosis. Overall, 15 of the 44 had a spontaneous clearance of the HCV. Eleven received treatment. Eleven percent were lost to follow-up and 30% are being observed.
While HCV prevalence seemed low, it is likely not accurate, said Dr. McFaul, noting that she and her colleagues found that HCV screening in HIV-negative MSM was rarely conducted. A one-month snapshot at the three clinics found that 15% (565) of 3,811 HIV negative men who came for sexual health screening were tested.
Extrapolating the data to all 623,350 visits during the 2010-2014 study period, Dr. McFaul estimated that about 261,036 HIV-negative MSM visited. At the current 15% screening rate, 34,657 would have been screened for HCV. But that means that almost 200,000 were not screened for HCV.
"We suggest that hepatitis C screening should be performed at routine sexual health screening, particularly in individuals with risk factors, and in environments where there is a high hepatitis C prevalence," she said. Clinicians also "need to ask penetrating questions about penetrating activities.
"If you don’t take a temperature you won’t find a fever," she said. "I think we’ll find a higher prevalence then we currently see."
On Twitter @aliciaault
FROM ICAAC 2014
Key clinical point: Physicians should ask men who have sex with men very specific questions about sexual activity and injection drug use.
Major finding: Half to a majority of MSM in two studies engaged in unprotected anal intercourse, a primary risk factor for HCV transmission.
Data source: A prospective survey of MSM in Switzerland and a retrospective review of HIV-negative MSM in London.
Disclosures: Dr. Cavassini received travel grants from Bristol-Myers Squibb, Boehringer-Ingelheim, and Gilead. Dr. McFaul reported no conflicts. The studies were sponsored by their respective institutions.

FDA advisers want more data on sunscreen safety
SILVER SPRING, MD. – The widespread and long-term use of sunscreens demands that manufacturers provide more long-term safety data and information on how the active ingredients affect the health of special populations including infants, children, the elderly, and those with skin conditions, according to an expert panel called by the Food and Drug Administration.
Sunscreen safety has become an increasingly more important concern, especially since they are intended to be applied over the entire body, multiple times a day, and for a lifetime, Dr. Theresa Michele, director of the FDA’s division of nonprescription clinical evaluation, said Sept. 5 at a meeting of the agency’s Nonprescription Drugs Advisory Committee. "We’re essentially talking about a cradle-to-grave product."
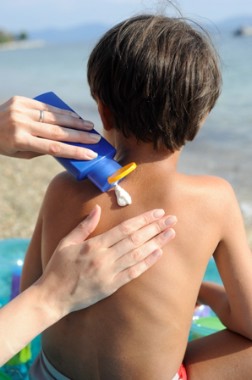
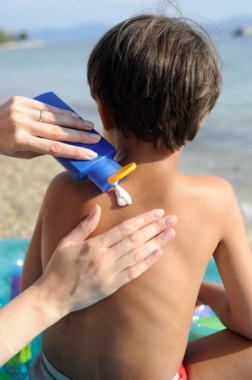
FDA has been under pressure from physicians, consumers, manufacturers, and Congress to approve a backlog of eight ingredients that are generally available outside of the United States and to improve the process for reviewing new ingredients. The agency proposes to add carcinogenicity and developmental and reproductive toxicity to the required safety testing prior to approval.
Speaking at the meeting, a coalition of sunscreen manufacturers said that most of what was being requested by the FDA is redundant or not necessary. "Today’s sunscreen ingredients have a wealth of data to support their safety," said the Personal Care Products Council (PCPC) in a statement. The group also said that many of the ingredients had been used for decades, without any indication that they caused cancer or led to any significant safety issues.
Advisory committee members, however, said that their concerns were not allayed.
"If you don’t look for a signal you’re not going to find a signal," said Dr. David J. Margolis, professor of dermatology at the University of Pennsylvania, Philadelphia. Dr. Margolis said he was concerned about safety over the long term, and about perhaps too much reliance on rodent models.
"The penetration through rodent skin is different than the penetration through human skin," he said.
Dr. Geoffrey L. Rosenthal, professor of pediatrics at the University of Maryland, Baltimore, said that he was bothered by the lack of specific studies in children. "Unless you study them in kids, you just don’t know," he said.
Dr. Marissa J. Perman, a pediatric dermatologist at the Children’s Hospital of Philadelphia, agreed and said that the agency should also consider requiring studies in pregnant and lactating women.
Dr. Perman, who spoke on behalf of the American Academy of Pediatrics during the public section of the meeting, said that AAP also would like to see studies based on actual – not directed – sunscreen use and investigations of the effects of spray sunscreens and of nanoparticles on children.
Several panel members also said that they were concerned about the disconnect between real-world use of sunscreens and the amounts studied during trials. The PCPC said in its background materials that market data indicate that many people use sunscreen only once a week, and that among users, only about 22% reapply as often as directed on the label.
Some committee members said that they viewed sunscreens as drugs that should always be subject to the same review and approval process.
"It’s really shocking to me that these are not being regulated through the process by which drugs are regulated," said Lorraine Gudas, Ph.D., chairman of pharmacology at Cornell University, New York. She noted that little was known about the potential systemic effects of sunscreen ingredients.
The FDA is not required to follow the panel’s advice. Any change to how sunscreens are regulated would require issuing a new rule and going through a public comment period. An FDA official declined to say when that process might occur.
On Twitter @aliciaault
SILVER SPRING, MD. – The widespread and long-term use of sunscreens demands that manufacturers provide more long-term safety data and information on how the active ingredients affect the health of special populations including infants, children, the elderly, and those with skin conditions, according to an expert panel called by the Food and Drug Administration.
Sunscreen safety has become an increasingly more important concern, especially since they are intended to be applied over the entire body, multiple times a day, and for a lifetime, Dr. Theresa Michele, director of the FDA’s division of nonprescription clinical evaluation, said Sept. 5 at a meeting of the agency’s Nonprescription Drugs Advisory Committee. "We’re essentially talking about a cradle-to-grave product."
FDA has been under pressure from physicians, consumers, manufacturers, and Congress to approve a backlog of eight ingredients that are generally available outside of the United States and to improve the process for reviewing new ingredients. The agency proposes to add carcinogenicity and developmental and reproductive toxicity to the required safety testing prior to approval.
Speaking at the meeting, a coalition of sunscreen manufacturers said that most of what was being requested by the FDA is redundant or not necessary. "Today’s sunscreen ingredients have a wealth of data to support their safety," said the Personal Care Products Council (PCPC) in a statement. The group also said that many of the ingredients had been used for decades, without any indication that they caused cancer or led to any significant safety issues.
Advisory committee members, however, said that their concerns were not allayed.
"If you don’t look for a signal you’re not going to find a signal," said Dr. David J. Margolis, professor of dermatology at the University of Pennsylvania, Philadelphia. Dr. Margolis said he was concerned about safety over the long term, and about perhaps too much reliance on rodent models.
"The penetration through rodent skin is different than the penetration through human skin," he said.
Dr. Geoffrey L. Rosenthal, professor of pediatrics at the University of Maryland, Baltimore, said that he was bothered by the lack of specific studies in children. "Unless you study them in kids, you just don’t know," he said.
Dr. Marissa J. Perman, a pediatric dermatologist at the Children’s Hospital of Philadelphia, agreed and said that the agency should also consider requiring studies in pregnant and lactating women.
Dr. Perman, who spoke on behalf of the American Academy of Pediatrics during the public section of the meeting, said that AAP also would like to see studies based on actual – not directed – sunscreen use and investigations of the effects of spray sunscreens and of nanoparticles on children.
Several panel members also said that they were concerned about the disconnect between real-world use of sunscreens and the amounts studied during trials. The PCPC said in its background materials that market data indicate that many people use sunscreen only once a week, and that among users, only about 22% reapply as often as directed on the label.
Some committee members said that they viewed sunscreens as drugs that should always be subject to the same review and approval process.
"It’s really shocking to me that these are not being regulated through the process by which drugs are regulated," said Lorraine Gudas, Ph.D., chairman of pharmacology at Cornell University, New York. She noted that little was known about the potential systemic effects of sunscreen ingredients.
The FDA is not required to follow the panel’s advice. Any change to how sunscreens are regulated would require issuing a new rule and going through a public comment period. An FDA official declined to say when that process might occur.
On Twitter @aliciaault
SILVER SPRING, MD. – The widespread and long-term use of sunscreens demands that manufacturers provide more long-term safety data and information on how the active ingredients affect the health of special populations including infants, children, the elderly, and those with skin conditions, according to an expert panel called by the Food and Drug Administration.
Sunscreen safety has become an increasingly more important concern, especially since they are intended to be applied over the entire body, multiple times a day, and for a lifetime, Dr. Theresa Michele, director of the FDA’s division of nonprescription clinical evaluation, said Sept. 5 at a meeting of the agency’s Nonprescription Drugs Advisory Committee. "We’re essentially talking about a cradle-to-grave product."
FDA has been under pressure from physicians, consumers, manufacturers, and Congress to approve a backlog of eight ingredients that are generally available outside of the United States and to improve the process for reviewing new ingredients. The agency proposes to add carcinogenicity and developmental and reproductive toxicity to the required safety testing prior to approval.
Speaking at the meeting, a coalition of sunscreen manufacturers said that most of what was being requested by the FDA is redundant or not necessary. "Today’s sunscreen ingredients have a wealth of data to support their safety," said the Personal Care Products Council (PCPC) in a statement. The group also said that many of the ingredients had been used for decades, without any indication that they caused cancer or led to any significant safety issues.
Advisory committee members, however, said that their concerns were not allayed.
"If you don’t look for a signal you’re not going to find a signal," said Dr. David J. Margolis, professor of dermatology at the University of Pennsylvania, Philadelphia. Dr. Margolis said he was concerned about safety over the long term, and about perhaps too much reliance on rodent models.
"The penetration through rodent skin is different than the penetration through human skin," he said.
Dr. Geoffrey L. Rosenthal, professor of pediatrics at the University of Maryland, Baltimore, said that he was bothered by the lack of specific studies in children. "Unless you study them in kids, you just don’t know," he said.
Dr. Marissa J. Perman, a pediatric dermatologist at the Children’s Hospital of Philadelphia, agreed and said that the agency should also consider requiring studies in pregnant and lactating women.
Dr. Perman, who spoke on behalf of the American Academy of Pediatrics during the public section of the meeting, said that AAP also would like to see studies based on actual – not directed – sunscreen use and investigations of the effects of spray sunscreens and of nanoparticles on children.
Several panel members also said that they were concerned about the disconnect between real-world use of sunscreens and the amounts studied during trials. The PCPC said in its background materials that market data indicate that many people use sunscreen only once a week, and that among users, only about 22% reapply as often as directed on the label.
Some committee members said that they viewed sunscreens as drugs that should always be subject to the same review and approval process.
"It’s really shocking to me that these are not being regulated through the process by which drugs are regulated," said Lorraine Gudas, Ph.D., chairman of pharmacology at Cornell University, New York. She noted that little was known about the potential systemic effects of sunscreen ingredients.
The FDA is not required to follow the panel’s advice. Any change to how sunscreens are regulated would require issuing a new rule and going through a public comment period. An FDA official declined to say when that process might occur.
On Twitter @aliciaault
AT AN FDA ADVISORY COMMITTEE
Health spending growth still low, but expected to speed up over next decade
WASHINGTON – The current trend of low growth in health care spending is expected to end in 2016 as more patients gain insurance coverage and the U.S. population continues to age, according to an analysis from the Centers for Medicare & Medicaid Services.
For the fifth year in a row, overall health care spending in 2013 grew less than 4% – significantly lower than historical growth rates, Andrea Sisko, an economist in the Office of the Actuary at the CMS, said at a press briefing. Spending growth was down, in part, because of sequestration’s 2% Medicare cut and because Medicare beneficiaries used fewer physician and hospital services. Lingering effects of the 2009 recession also meant that privately insured patients spent less on health care.

The Affordable Care Act had some impact on the growth in health spending in 2013, Ms. Sisko said. The ACA’s temporary increase in Medicaid primary care payments helped to almost double the growth in that program’s spending. Overall Medicaid spending grew almost 7% in 2013 (vs. 3% in 2012); physician payment increases by the states and enhanced benefits contributed to the overall spending growth. The analysis was published Sept. 3 in the journal Health Affairs (doi: 10.1377/hlthaff.2014.0560).
The change in Medicaid was one of few areas where the economists definitively could say that the ACA either fueled or restrained spending growth.
"We are no longer estimating or quantifying the impacts of the Affordable Care Act on national health spending," said Ms. Sisko, who noted that after 4 years, it has become difficult to tease out the law’s effects.
Overall spending on physician services grew 3.3% in 2013, but could grow by almost 6% in 2014. After a slight decline in 2015, growth is expected to be about 6% yearly from 2016 to 2023, according to the analysis.
Future growth in spending on physician services will come as more Americans gain health insurance coverage. The projected dip in 2015 is tied to the end of the temporary Medicaid pay boost, which ends in December2014. For 2016-2023, physician spending is expected to rebound as more U.S. residents become eligible for Medicare.
Spending for physician services is expected to make up about 20% of U.S. health care spending through 2023, with hospitals consuming 32% and prescription drugs 9%.
While future health care spending growth is expected to slightly outpace economic growth (6% for health care spending vs. 5% for overall spending), that growth rate is still lower than the 7% yearly increases seen in the 1990s and through the mid-2000s, Ms. Sisko said. The growth rates will be lower in part because they will be based on a historically low baseline of the last 4-5 years, Ms. Sisko added.
Drug costs, for instance, will not have the double-digit growth seen in the late 1990s, said coauthor Sean Keehan, also an economist at the CMS Office of the Actuary. Unlike in the 1990s, payers now seem to have more leverage to negotiate lower rates from physicians and hospitals, and thus keep costs and spending down.
The physician spending figures assume that Congress will override the fee cuts mandated by the Sustainable Growth Rate formula and instead keep fees flat in 2015, and then give a 0.6% increase from 2016 to 2023. They also assume that the budget sequestration will continue.
On Twitter @aliciaault
WASHINGTON – The current trend of low growth in health care spending is expected to end in 2016 as more patients gain insurance coverage and the U.S. population continues to age, according to an analysis from the Centers for Medicare & Medicaid Services.
For the fifth year in a row, overall health care spending in 2013 grew less than 4% – significantly lower than historical growth rates, Andrea Sisko, an economist in the Office of the Actuary at the CMS, said at a press briefing. Spending growth was down, in part, because of sequestration’s 2% Medicare cut and because Medicare beneficiaries used fewer physician and hospital services. Lingering effects of the 2009 recession also meant that privately insured patients spent less on health care.
The Affordable Care Act had some impact on the growth in health spending in 2013, Ms. Sisko said. The ACA’s temporary increase in Medicaid primary care payments helped to almost double the growth in that program’s spending. Overall Medicaid spending grew almost 7% in 2013 (vs. 3% in 2012); physician payment increases by the states and enhanced benefits contributed to the overall spending growth. The analysis was published Sept. 3 in the journal Health Affairs (doi: 10.1377/hlthaff.2014.0560).
The change in Medicaid was one of few areas where the economists definitively could say that the ACA either fueled or restrained spending growth.
"We are no longer estimating or quantifying the impacts of the Affordable Care Act on national health spending," said Ms. Sisko, who noted that after 4 years, it has become difficult to tease out the law’s effects.
Overall spending on physician services grew 3.3% in 2013, but could grow by almost 6% in 2014. After a slight decline in 2015, growth is expected to be about 6% yearly from 2016 to 2023, according to the analysis.
Future growth in spending on physician services will come as more Americans gain health insurance coverage. The projected dip in 2015 is tied to the end of the temporary Medicaid pay boost, which ends in December2014. For 2016-2023, physician spending is expected to rebound as more U.S. residents become eligible for Medicare.
Spending for physician services is expected to make up about 20% of U.S. health care spending through 2023, with hospitals consuming 32% and prescription drugs 9%.
While future health care spending growth is expected to slightly outpace economic growth (6% for health care spending vs. 5% for overall spending), that growth rate is still lower than the 7% yearly increases seen in the 1990s and through the mid-2000s, Ms. Sisko said. The growth rates will be lower in part because they will be based on a historically low baseline of the last 4-5 years, Ms. Sisko added.
Drug costs, for instance, will not have the double-digit growth seen in the late 1990s, said coauthor Sean Keehan, also an economist at the CMS Office of the Actuary. Unlike in the 1990s, payers now seem to have more leverage to negotiate lower rates from physicians and hospitals, and thus keep costs and spending down.
The physician spending figures assume that Congress will override the fee cuts mandated by the Sustainable Growth Rate formula and instead keep fees flat in 2015, and then give a 0.6% increase from 2016 to 2023. They also assume that the budget sequestration will continue.
On Twitter @aliciaault
WASHINGTON – The current trend of low growth in health care spending is expected to end in 2016 as more patients gain insurance coverage and the U.S. population continues to age, according to an analysis from the Centers for Medicare & Medicaid Services.
For the fifth year in a row, overall health care spending in 2013 grew less than 4% – significantly lower than historical growth rates, Andrea Sisko, an economist in the Office of the Actuary at the CMS, said at a press briefing. Spending growth was down, in part, because of sequestration’s 2% Medicare cut and because Medicare beneficiaries used fewer physician and hospital services. Lingering effects of the 2009 recession also meant that privately insured patients spent less on health care.
The Affordable Care Act had some impact on the growth in health spending in 2013, Ms. Sisko said. The ACA’s temporary increase in Medicaid primary care payments helped to almost double the growth in that program’s spending. Overall Medicaid spending grew almost 7% in 2013 (vs. 3% in 2012); physician payment increases by the states and enhanced benefits contributed to the overall spending growth. The analysis was published Sept. 3 in the journal Health Affairs (doi: 10.1377/hlthaff.2014.0560).
The change in Medicaid was one of few areas where the economists definitively could say that the ACA either fueled or restrained spending growth.
"We are no longer estimating or quantifying the impacts of the Affordable Care Act on national health spending," said Ms. Sisko, who noted that after 4 years, it has become difficult to tease out the law’s effects.
Overall spending on physician services grew 3.3% in 2013, but could grow by almost 6% in 2014. After a slight decline in 2015, growth is expected to be about 6% yearly from 2016 to 2023, according to the analysis.
Future growth in spending on physician services will come as more Americans gain health insurance coverage. The projected dip in 2015 is tied to the end of the temporary Medicaid pay boost, which ends in December2014. For 2016-2023, physician spending is expected to rebound as more U.S. residents become eligible for Medicare.
Spending for physician services is expected to make up about 20% of U.S. health care spending through 2023, with hospitals consuming 32% and prescription drugs 9%.
While future health care spending growth is expected to slightly outpace economic growth (6% for health care spending vs. 5% for overall spending), that growth rate is still lower than the 7% yearly increases seen in the 1990s and through the mid-2000s, Ms. Sisko said. The growth rates will be lower in part because they will be based on a historically low baseline of the last 4-5 years, Ms. Sisko added.
Drug costs, for instance, will not have the double-digit growth seen in the late 1990s, said coauthor Sean Keehan, also an economist at the CMS Office of the Actuary. Unlike in the 1990s, payers now seem to have more leverage to negotiate lower rates from physicians and hospitals, and thus keep costs and spending down.
The physician spending figures assume that Congress will override the fee cuts mandated by the Sustainable Growth Rate formula and instead keep fees flat in 2015, and then give a 0.6% increase from 2016 to 2023. They also assume that the budget sequestration will continue.
On Twitter @aliciaault
AT A HEALTH AFFAIRS BRIEFING

It’s tough to find a good fecal donor
Finding healthy stool donors for fecal transplant may be a tough prospect.
That’s what Australian researchers have discovered in the course of the FOCUS trial, which aims to determine whether fecal microbiota transplantation (FMT) is safe and efficacious in the treatment of chronic active ulcerative colitis and in the induction of remission.

Dr. Sudarshan Paramsothy and his colleagues at the University of New South Wales, Sydney, and the University of Melbourne, reported findings from donor recruitment for the FOCUS (Faecal Microbiota Transplantation in Ulcerative Colitis) trial at the American Gastroenterological Association’s 2014 James W. Freston Conference in Chicago.
The FOCUS study began enrolling patients in November, and is continuing to enroll, said Dr. Paramsothy. He and his colleagues also are continuing to recruit fecal donors. The data presented in Chicago were on an initial recruitment effort.
Overall, after screening, only 10% of recruits were considered eligible donors.
The researchers recruited donors through letters, newspaper ads, and online solicitations. They were told that they would be reimbursed for their time and for the transportation of their stool donations to the study site.
After responding, recruits were told that they would be expected to make stool donations five times a week for a minimum of 6 weeks.
The researchers had 116 potential donors over a 7-month recruitment period. Forty-seven declined off the bat because of the 5-day-a-week donation requirement.
Twenty-seven had other issues, including medical cormibidities (13), risk factors for variant Crueztfeldt-Jakob disease (6), and recent antibiotic use (1), that disqualified them from the study.
Thirty-eight potentially healthy donors underwent stool and blood testing. Fifteen of those donors were found to have a variety of parasites or indications of active infection that excluded them from donation: 5 had Dientamoeba fragilis, 5 had Blastocystis hominis, 1 had B. hominis and D. fragilis, 1 had Giardia intestinalis and D. fragilis, and 1 had norovirus and Clostridium difficile toxin, and 2 had leukocytes or erythrocytes on stool microscopy. One donor had indeterminate hepatitis C serology.
While it is not uncommon for people to have asymptomatic parasite carriage in the gastrointestinal tract, "we did not expect it in such a high proportion," said Dr. Paramsothy. "Our screened donor population was not an at-risk group," he said, adding that they were otherwise healthy and had no risk factors or gastrointestinal symptoms.
"Our detection rates may have been slightly higher as donor stool samples were sent to a pathology center with expert, specialized GI parasitologists for review," Dr. Paramsothy said.
There’s also some question as to whether some parasites, such as Blastocystis and Dientamoeba, "are truly pathogenic or rather commensal organisms," he said, adding that it was thought better to exclude patients with these parasites if there were any doubt.
That left 22 potential donors. Further questioning found that two had used antibiotics in between recruitment and stool testing, and one was living with a household member who was positive for D. fragilis.
Of the 19 remaining, 1 dropped out and 18 were screened again. Three were excluded because of a body mass index over 30 kg/m2, 1 because of illicit drug use, 1 because of irregular bowel movements after starting a new medication, and 1 because of uncontrolled anxiety and depression. Dr. Paramsothy said that high-BMI donors were excluded because some studies have shown that gut microbiota potentially influence insulin sensitivity and obesity. Illicit drug use is a red flag because it is potentially associated with blood-borne disease acquisition, he said.
At the end, there were only 12 healthy donors, 10% of the starting 116. Dr. Paramsothy said that it was not necessary to have a single donor for every single patient in the trial. He said he could not disclose currently the number needed for the study, however.
The donor results "suggest that while FMT is an exciting new therapy, it is difficult to identify appropriate and willing anonymous donors," Dr. Paramsothy said. But that should not have an overall impact on FMT as a therapy, he said – rather, it might just make it harder for a small practice to establish an in-house FMT program.
Dr. Paramsothy reported no relevant financial conflicts.
On Twitter @aliciaault
Finding healthy stool donors for fecal transplant may be a tough prospect.
That’s what Australian researchers have discovered in the course of the FOCUS trial, which aims to determine whether fecal microbiota transplantation (FMT) is safe and efficacious in the treatment of chronic active ulcerative colitis and in the induction of remission.
Dr. Sudarshan Paramsothy and his colleagues at the University of New South Wales, Sydney, and the University of Melbourne, reported findings from donor recruitment for the FOCUS (Faecal Microbiota Transplantation in Ulcerative Colitis) trial at the American Gastroenterological Association’s 2014 James W. Freston Conference in Chicago.
The FOCUS study began enrolling patients in November, and is continuing to enroll, said Dr. Paramsothy. He and his colleagues also are continuing to recruit fecal donors. The data presented in Chicago were on an initial recruitment effort.
Overall, after screening, only 10% of recruits were considered eligible donors.
The researchers recruited donors through letters, newspaper ads, and online solicitations. They were told that they would be reimbursed for their time and for the transportation of their stool donations to the study site.
After responding, recruits were told that they would be expected to make stool donations five times a week for a minimum of 6 weeks.
The researchers had 116 potential donors over a 7-month recruitment period. Forty-seven declined off the bat because of the 5-day-a-week donation requirement.
Twenty-seven had other issues, including medical cormibidities (13), risk factors for variant Crueztfeldt-Jakob disease (6), and recent antibiotic use (1), that disqualified them from the study.
Thirty-eight potentially healthy donors underwent stool and blood testing. Fifteen of those donors were found to have a variety of parasites or indications of active infection that excluded them from donation: 5 had Dientamoeba fragilis, 5 had Blastocystis hominis, 1 had B. hominis and D. fragilis, 1 had Giardia intestinalis and D. fragilis, and 1 had norovirus and Clostridium difficile toxin, and 2 had leukocytes or erythrocytes on stool microscopy. One donor had indeterminate hepatitis C serology.
While it is not uncommon for people to have asymptomatic parasite carriage in the gastrointestinal tract, "we did not expect it in such a high proportion," said Dr. Paramsothy. "Our screened donor population was not an at-risk group," he said, adding that they were otherwise healthy and had no risk factors or gastrointestinal symptoms.
"Our detection rates may have been slightly higher as donor stool samples were sent to a pathology center with expert, specialized GI parasitologists for review," Dr. Paramsothy said.
There’s also some question as to whether some parasites, such as Blastocystis and Dientamoeba, "are truly pathogenic or rather commensal organisms," he said, adding that it was thought better to exclude patients with these parasites if there were any doubt.
That left 22 potential donors. Further questioning found that two had used antibiotics in between recruitment and stool testing, and one was living with a household member who was positive for D. fragilis.
Of the 19 remaining, 1 dropped out and 18 were screened again. Three were excluded because of a body mass index over 30 kg/m2, 1 because of illicit drug use, 1 because of irregular bowel movements after starting a new medication, and 1 because of uncontrolled anxiety and depression. Dr. Paramsothy said that high-BMI donors were excluded because some studies have shown that gut microbiota potentially influence insulin sensitivity and obesity. Illicit drug use is a red flag because it is potentially associated with blood-borne disease acquisition, he said.
At the end, there were only 12 healthy donors, 10% of the starting 116. Dr. Paramsothy said that it was not necessary to have a single donor for every single patient in the trial. He said he could not disclose currently the number needed for the study, however.
The donor results "suggest that while FMT is an exciting new therapy, it is difficult to identify appropriate and willing anonymous donors," Dr. Paramsothy said. But that should not have an overall impact on FMT as a therapy, he said – rather, it might just make it harder for a small practice to establish an in-house FMT program.
Dr. Paramsothy reported no relevant financial conflicts.
On Twitter @aliciaault
Finding healthy stool donors for fecal transplant may be a tough prospect.
That’s what Australian researchers have discovered in the course of the FOCUS trial, which aims to determine whether fecal microbiota transplantation (FMT) is safe and efficacious in the treatment of chronic active ulcerative colitis and in the induction of remission.
Dr. Sudarshan Paramsothy and his colleagues at the University of New South Wales, Sydney, and the University of Melbourne, reported findings from donor recruitment for the FOCUS (Faecal Microbiota Transplantation in Ulcerative Colitis) trial at the American Gastroenterological Association’s 2014 James W. Freston Conference in Chicago.
The FOCUS study began enrolling patients in November, and is continuing to enroll, said Dr. Paramsothy. He and his colleagues also are continuing to recruit fecal donors. The data presented in Chicago were on an initial recruitment effort.
Overall, after screening, only 10% of recruits were considered eligible donors.
The researchers recruited donors through letters, newspaper ads, and online solicitations. They were told that they would be reimbursed for their time and for the transportation of their stool donations to the study site.
After responding, recruits were told that they would be expected to make stool donations five times a week for a minimum of 6 weeks.
The researchers had 116 potential donors over a 7-month recruitment period. Forty-seven declined off the bat because of the 5-day-a-week donation requirement.
Twenty-seven had other issues, including medical cormibidities (13), risk factors for variant Crueztfeldt-Jakob disease (6), and recent antibiotic use (1), that disqualified them from the study.
Thirty-eight potentially healthy donors underwent stool and blood testing. Fifteen of those donors were found to have a variety of parasites or indications of active infection that excluded them from donation: 5 had Dientamoeba fragilis, 5 had Blastocystis hominis, 1 had B. hominis and D. fragilis, 1 had Giardia intestinalis and D. fragilis, and 1 had norovirus and Clostridium difficile toxin, and 2 had leukocytes or erythrocytes on stool microscopy. One donor had indeterminate hepatitis C serology.
While it is not uncommon for people to have asymptomatic parasite carriage in the gastrointestinal tract, "we did not expect it in such a high proportion," said Dr. Paramsothy. "Our screened donor population was not an at-risk group," he said, adding that they were otherwise healthy and had no risk factors or gastrointestinal symptoms.
"Our detection rates may have been slightly higher as donor stool samples were sent to a pathology center with expert, specialized GI parasitologists for review," Dr. Paramsothy said.
There’s also some question as to whether some parasites, such as Blastocystis and Dientamoeba, "are truly pathogenic or rather commensal organisms," he said, adding that it was thought better to exclude patients with these parasites if there were any doubt.
That left 22 potential donors. Further questioning found that two had used antibiotics in between recruitment and stool testing, and one was living with a household member who was positive for D. fragilis.
Of the 19 remaining, 1 dropped out and 18 were screened again. Three were excluded because of a body mass index over 30 kg/m2, 1 because of illicit drug use, 1 because of irregular bowel movements after starting a new medication, and 1 because of uncontrolled anxiety and depression. Dr. Paramsothy said that high-BMI donors were excluded because some studies have shown that gut microbiota potentially influence insulin sensitivity and obesity. Illicit drug use is a red flag because it is potentially associated with blood-borne disease acquisition, he said.
At the end, there were only 12 healthy donors, 10% of the starting 116. Dr. Paramsothy said that it was not necessary to have a single donor for every single patient in the trial. He said he could not disclose currently the number needed for the study, however.
The donor results "suggest that while FMT is an exciting new therapy, it is difficult to identify appropriate and willing anonymous donors," Dr. Paramsothy said. But that should not have an overall impact on FMT as a therapy, he said – rather, it might just make it harder for a small practice to establish an in-house FMT program.
Dr. Paramsothy reported no relevant financial conflicts.
On Twitter @aliciaault
FROM THE 2014 JAMES W. FRESTON CONFERENCE
Key clinical point: Finding fecal transplant donors is not as simple as once thought.
Major finding: Only 10% of people recruited to be donors for a fecal microbiota transplant study were healthy enough to be eligible.
Data source: Donors recruited for the FOCUS study.
Disclosures: The study is sponsored by the University of New South Wales, Sydney. The investigators reported no relevant financial conflicts.

VIDEO: Federal health IT chief DeSalvo talks meaningful use
Dr. Karen DeSalvo is the fifth person to serve as National Coordinator for Health Information Technology at the Health and Human Services Department, but perhaps more than any of her predecessors, she is truly in the thick of the struggle to bring doctors, medical practices, and hospitals into the digital age.
Physicians face a major deadline this year: It’s the last year to sign up for the meaningful use incentive payment program created by the Health Information Technology for Economic and Clinical Health Act (HITECH). If they don’t participate, they lose out on the potential to recoup from the federal government at least a small portion of the money they’ve spent on electronic health record systems. And it’s becoming inevitable that not participating could mean being left behind by insurers, hospitals, and patients.
That’s causing a lot of anxiety. Dr. DeSalvo – a practicing internist – says that she feels doctors’ pain. She recently completed a national listening tour and says that what she learned from those sessions will help inform how the Office of the National Coordinator moves forward.
Dr. Karen DeSalvo is the fifth person to serve as National Coordinator for Health Information Technology at the Health and Human Services Department, but perhaps more than any of her predecessors, she is truly in the thick of the struggle to bring doctors, medical practices, and hospitals into the digital age.
Physicians face a major deadline this year: It’s the last year to sign up for the meaningful use incentive payment program created by the Health Information Technology for Economic and Clinical Health Act (HITECH). If they don’t participate, they lose out on the potential to recoup from the federal government at least a small portion of the money they’ve spent on electronic health record systems. And it’s becoming inevitable that not participating could mean being left behind by insurers, hospitals, and patients.
That’s causing a lot of anxiety. Dr. DeSalvo – a practicing internist – says that she feels doctors’ pain. She recently completed a national listening tour and says that what she learned from those sessions will help inform how the Office of the National Coordinator moves forward.
Dr. Karen DeSalvo is the fifth person to serve as National Coordinator for Health Information Technology at the Health and Human Services Department, but perhaps more than any of her predecessors, she is truly in the thick of the struggle to bring doctors, medical practices, and hospitals into the digital age.
Physicians face a major deadline this year: It’s the last year to sign up for the meaningful use incentive payment program created by the Health Information Technology for Economic and Clinical Health Act (HITECH). If they don’t participate, they lose out on the potential to recoup from the federal government at least a small portion of the money they’ve spent on electronic health record systems. And it’s becoming inevitable that not participating could mean being left behind by insurers, hospitals, and patients.
That’s causing a lot of anxiety. Dr. DeSalvo – a practicing internist – says that she feels doctors’ pain. She recently completed a national listening tour and says that what she learned from those sessions will help inform how the Office of the National Coordinator moves forward.

HHS appoints chief executive to run healthcare.gov
The U.S. Department of Health & Human Services has named a new chief executive officer to run the federal marketplace, which includes the healthcare.gov website.
Kevin Counihan, currently the CEO for Connecticut’s marketplace, has been named to run the federal marketplace. He also will be in charge of the Center for Consumer Information and Insurance Oversight (CCIIO), which regulates health insurance. HHS Secretary Sylvia Mathews Burwell announced in June that the department would be looking for a new CEO.

Mr. Counihan has served as CEO for Health Access CT since June 2012, after having served as chief marketing officer from 2006 to 2011 for the Commonwealth of Massachusetts Health Insurance Connector Authority, which administered that state’s health insurance exchange. Before that, he was a sales and marketing executive for Tufts Health Plan of Massachusetts and regional vice president for CIGNA.
"We are committed to instilling ongoing accountability for reaching milestones, measuring results, and ensuring a successful open enrollment period," Ms. Burwell said in a statement. Mr. Counihan "brings additional operational and technological expertise to the position and will be a clear, single point of contact for streamlined decision making."
Mr. Counihan will report to Centers for Medicare & Medicaid Services Administrator Marilyn Tavenner.
On Twitter @aliciaault
The U.S. Department of Health & Human Services has named a new chief executive officer to run the federal marketplace, which includes the healthcare.gov website.
Kevin Counihan, currently the CEO for Connecticut’s marketplace, has been named to run the federal marketplace. He also will be in charge of the Center for Consumer Information and Insurance Oversight (CCIIO), which regulates health insurance. HHS Secretary Sylvia Mathews Burwell announced in June that the department would be looking for a new CEO.
Mr. Counihan has served as CEO for Health Access CT since June 2012, after having served as chief marketing officer from 2006 to 2011 for the Commonwealth of Massachusetts Health Insurance Connector Authority, which administered that state’s health insurance exchange. Before that, he was a sales and marketing executive for Tufts Health Plan of Massachusetts and regional vice president for CIGNA.
"We are committed to instilling ongoing accountability for reaching milestones, measuring results, and ensuring a successful open enrollment period," Ms. Burwell said in a statement. Mr. Counihan "brings additional operational and technological expertise to the position and will be a clear, single point of contact for streamlined decision making."
Mr. Counihan will report to Centers for Medicare & Medicaid Services Administrator Marilyn Tavenner.
On Twitter @aliciaault
The U.S. Department of Health & Human Services has named a new chief executive officer to run the federal marketplace, which includes the healthcare.gov website.
Kevin Counihan, currently the CEO for Connecticut’s marketplace, has been named to run the federal marketplace. He also will be in charge of the Center for Consumer Information and Insurance Oversight (CCIIO), which regulates health insurance. HHS Secretary Sylvia Mathews Burwell announced in June that the department would be looking for a new CEO.
Mr. Counihan has served as CEO for Health Access CT since June 2012, after having served as chief marketing officer from 2006 to 2011 for the Commonwealth of Massachusetts Health Insurance Connector Authority, which administered that state’s health insurance exchange. Before that, he was a sales and marketing executive for Tufts Health Plan of Massachusetts and regional vice president for CIGNA.
"We are committed to instilling ongoing accountability for reaching milestones, measuring results, and ensuring a successful open enrollment period," Ms. Burwell said in a statement. Mr. Counihan "brings additional operational and technological expertise to the position and will be a clear, single point of contact for streamlined decision making."
Mr. Counihan will report to Centers for Medicare & Medicaid Services Administrator Marilyn Tavenner.
On Twitter @aliciaault

ABIM responds to maintenance of certification concerns of grandfathered physicians
Physicians who are grandfathered from maintenance of certification requirements may soon be listed differently on the American Board of Internal Medicine website.
The ABIM site currently reports publicly whether or not physicians are "Meeting Maintenance of Certification Requirements," and that status means grandfathered physicians do not appear to be meeting MOC requirements on the American Board of Medical Specialties’ Certification Matters website.

"Not meeting MOC requirements’ is, in essence, a scarlet letter meant to pressure grandparents into enrolling in the current flawed MOC system," Dr. Mack Harrell, president of the American Association of Clinical Endocrinologists, said in a June 30 letter to the ABIM. The Endocrine Society and AACE have asked that no information be publicly reported until the ABIM addresses the MOC concerns of internal medicine specialists and subspecialists.
At an August meeting, ABIM agreed that the language "is causing legitimate confusion." Grandfathered physicians are encouraged but are not required to participate in MOC, yet they are still being listed as either meeting or not meeting the MOC requirements, according to an Aug. 15 statement from the board.
The board is "exploring what changes to the reporting language can be made," according to the statement. The issue is making sure that "reporting of certification status is clear and consistent across the community of specialty boards."
The nature and timing of the change has not yet been decided by the board, ABIM spokesperson Lorie Slass said. "We want to work with ABMS to make sure there is consistency and clarity in web reporting."
On Twitter @aliciaault
Physicians who are grandfathered from maintenance of certification requirements may soon be listed differently on the American Board of Internal Medicine website.
The ABIM site currently reports publicly whether or not physicians are "Meeting Maintenance of Certification Requirements," and that status means grandfathered physicians do not appear to be meeting MOC requirements on the American Board of Medical Specialties’ Certification Matters website.
"Not meeting MOC requirements’ is, in essence, a scarlet letter meant to pressure grandparents into enrolling in the current flawed MOC system," Dr. Mack Harrell, president of the American Association of Clinical Endocrinologists, said in a June 30 letter to the ABIM. The Endocrine Society and AACE have asked that no information be publicly reported until the ABIM addresses the MOC concerns of internal medicine specialists and subspecialists.
At an August meeting, ABIM agreed that the language "is causing legitimate confusion." Grandfathered physicians are encouraged but are not required to participate in MOC, yet they are still being listed as either meeting or not meeting the MOC requirements, according to an Aug. 15 statement from the board.
The board is "exploring what changes to the reporting language can be made," according to the statement. The issue is making sure that "reporting of certification status is clear and consistent across the community of specialty boards."
The nature and timing of the change has not yet been decided by the board, ABIM spokesperson Lorie Slass said. "We want to work with ABMS to make sure there is consistency and clarity in web reporting."
On Twitter @aliciaault
Physicians who are grandfathered from maintenance of certification requirements may soon be listed differently on the American Board of Internal Medicine website.
The ABIM site currently reports publicly whether or not physicians are "Meeting Maintenance of Certification Requirements," and that status means grandfathered physicians do not appear to be meeting MOC requirements on the American Board of Medical Specialties’ Certification Matters website.
"Not meeting MOC requirements’ is, in essence, a scarlet letter meant to pressure grandparents into enrolling in the current flawed MOC system," Dr. Mack Harrell, president of the American Association of Clinical Endocrinologists, said in a June 30 letter to the ABIM. The Endocrine Society and AACE have asked that no information be publicly reported until the ABIM addresses the MOC concerns of internal medicine specialists and subspecialists.
At an August meeting, ABIM agreed that the language "is causing legitimate confusion." Grandfathered physicians are encouraged but are not required to participate in MOC, yet they are still being listed as either meeting or not meeting the MOC requirements, according to an Aug. 15 statement from the board.
The board is "exploring what changes to the reporting language can be made," according to the statement. The issue is making sure that "reporting of certification status is clear and consistent across the community of specialty boards."
The nature and timing of the change has not yet been decided by the board, ABIM spokesperson Lorie Slass said. "We want to work with ABMS to make sure there is consistency and clarity in web reporting."
On Twitter @aliciaault

DEA moves hydrocodone combination products to schedule II
The Drug Enforcement Administration is making it harder to prescribe hydrocodone combination products.
The move was expected, as the agency proposed in February to move hydrocodone combinations from schedule III to schedule II in response to requests from both the U.S. Department of Health & Human Services and the Food and Drug Administration.
Some physician groups have opposed the move, saying that it will lead to more administrative burdens, do nothing to curb abuse and diversion, and potentially decrease access to medications.
The DEA will publish the final rule on the rescheduling in the Federal Register on Aug. 22. Manufacturers, distributors, and prescribers will have to comply by Oct. 13.
The agency said it is time to rein in opioid prescribing and that rescheduling will help accomplish that goal.
"Almost 7 million Americans abuse controlled-substance prescription medications, including opioid painkillers, resulting in more deaths from prescription drug overdoses than auto accidents," DEA administrator Michele Leonhart said in a statement. "Today’s action recognizes that these products are some of the most addictive and potentially dangerous prescription medications available."
Hydrocodone combination products were placed on schedule III by Congress in 1970 when it created the Controlled Substances Act, in part because it was believed that adding acetaminophen or other non-narcotics might lessen the abuse potential. Hydrocodone itself was placed on schedule II.
Now, "the scientific, medical, and epidemiological data are robust and support rescheduling [of hydrocodone combination products] into schedule II," according to the final rule.
Data show that the products are widely diverted and abused at rates similar to that of oxycodone products, which are schedule II, said the agency, which added that abuse is associated with severe psychological or physical dependence, and many are being admitted to addiction treatment.
The hydrocodone combinations are also associated with large numbers of deaths, said the agency. More than 16,000 deaths in 2010 were due to abuse of opioids, including hydrocodone combinations, according to the DEA.
About 137 million prescriptions for hydrocodone combinations were dispensed in 2013, the agency said. The most frequently prescribed combination is hydrocodone/acetaminophen.
In comments to the proposed rule in April, the American Medical Association, along with a group of organizations and companies in the long-term care field, asked the agency to delay the final rule until an exception was made for nursing homes and other long-term care facilities. The AMA’s House of Delegates had also voted in 2013 to oppose rescheduling.
The American College of Emergency Physicians also urged against rescheduling, telling the DEA in April that it would not likely solve the abuse and diversion problems, but would lead to a greater administrative hassle for physicians.
The DEA said it received 573 comments after it proposed rescheduling, with 52% in favor, 41% opposed, and 7% taking no position. The agency received the most comments from the general public (44%; 250 comments) and pharmacists and pharmacy students (21%; 122 comments), physicians (13%; 73 comments), patients (6%; 35 comments), and midlevel practitioners (5%; 31 comments). Just over half of the physician comments supported, or supported with qualification, rescheduling.
Most of the commenters who were opposed to rescheduling were pharmacists, pharmacy students, and patients. Those opposed were concerned about how it would affect prescribing practices and patient access to medicine, and how it might impact long-term care facilities. Commenters also said that it would not prevent abuse or diversion.
The DEA said that although moving to schedule II does, for instance, prohibit refills, it would not necessarily block physicians from writing prescriptions for supplies of longer than 30 days, or from writing multiple prescriptions at once. State laws might have limits, however, and those will take precedence over the DEA rule.
The DEA rescheduling follows an FDA advisory committee recommendation in Jan. 2013 to do so, and the FDA’s backing of the proposal in Oct. 2013. HHS followed with its own recommendation to the DEA.
In comments on the proposed rule, manufacturers and pharmacies asked for more time to implement the rescheduling, but the DEA said no, citing high rates of abuse, overdose, and deaths relating to hydrocodone combination products.
The rescheduling goes into effect on Oct. 13, 45 days from the date of the rule’s publication in the Federal Register.
On Twitter @aliciaault
The Drug Enforcement Administration is making it harder to prescribe hydrocodone combination products.
The move was expected, as the agency proposed in February to move hydrocodone combinations from schedule III to schedule II in response to requests from both the U.S. Department of Health & Human Services and the Food and Drug Administration.
Some physician groups have opposed the move, saying that it will lead to more administrative burdens, do nothing to curb abuse and diversion, and potentially decrease access to medications.
The DEA will publish the final rule on the rescheduling in the Federal Register on Aug. 22. Manufacturers, distributors, and prescribers will have to comply by Oct. 13.
The agency said it is time to rein in opioid prescribing and that rescheduling will help accomplish that goal.
"Almost 7 million Americans abuse controlled-substance prescription medications, including opioid painkillers, resulting in more deaths from prescription drug overdoses than auto accidents," DEA administrator Michele Leonhart said in a statement. "Today’s action recognizes that these products are some of the most addictive and potentially dangerous prescription medications available."
Hydrocodone combination products were placed on schedule III by Congress in 1970 when it created the Controlled Substances Act, in part because it was believed that adding acetaminophen or other non-narcotics might lessen the abuse potential. Hydrocodone itself was placed on schedule II.
Now, "the scientific, medical, and epidemiological data are robust and support rescheduling [of hydrocodone combination products] into schedule II," according to the final rule.
Data show that the products are widely diverted and abused at rates similar to that of oxycodone products, which are schedule II, said the agency, which added that abuse is associated with severe psychological or physical dependence, and many are being admitted to addiction treatment.
The hydrocodone combinations are also associated with large numbers of deaths, said the agency. More than 16,000 deaths in 2010 were due to abuse of opioids, including hydrocodone combinations, according to the DEA.
About 137 million prescriptions for hydrocodone combinations were dispensed in 2013, the agency said. The most frequently prescribed combination is hydrocodone/acetaminophen.
In comments to the proposed rule in April, the American Medical Association, along with a group of organizations and companies in the long-term care field, asked the agency to delay the final rule until an exception was made for nursing homes and other long-term care facilities. The AMA’s House of Delegates had also voted in 2013 to oppose rescheduling.
The American College of Emergency Physicians also urged against rescheduling, telling the DEA in April that it would not likely solve the abuse and diversion problems, but would lead to a greater administrative hassle for physicians.
The DEA said it received 573 comments after it proposed rescheduling, with 52% in favor, 41% opposed, and 7% taking no position. The agency received the most comments from the general public (44%; 250 comments) and pharmacists and pharmacy students (21%; 122 comments), physicians (13%; 73 comments), patients (6%; 35 comments), and midlevel practitioners (5%; 31 comments). Just over half of the physician comments supported, or supported with qualification, rescheduling.
Most of the commenters who were opposed to rescheduling were pharmacists, pharmacy students, and patients. Those opposed were concerned about how it would affect prescribing practices and patient access to medicine, and how it might impact long-term care facilities. Commenters also said that it would not prevent abuse or diversion.
The DEA said that although moving to schedule II does, for instance, prohibit refills, it would not necessarily block physicians from writing prescriptions for supplies of longer than 30 days, or from writing multiple prescriptions at once. State laws might have limits, however, and those will take precedence over the DEA rule.
The DEA rescheduling follows an FDA advisory committee recommendation in Jan. 2013 to do so, and the FDA’s backing of the proposal in Oct. 2013. HHS followed with its own recommendation to the DEA.
In comments on the proposed rule, manufacturers and pharmacies asked for more time to implement the rescheduling, but the DEA said no, citing high rates of abuse, overdose, and deaths relating to hydrocodone combination products.
The rescheduling goes into effect on Oct. 13, 45 days from the date of the rule’s publication in the Federal Register.
On Twitter @aliciaault
The Drug Enforcement Administration is making it harder to prescribe hydrocodone combination products.
The move was expected, as the agency proposed in February to move hydrocodone combinations from schedule III to schedule II in response to requests from both the U.S. Department of Health & Human Services and the Food and Drug Administration.
Some physician groups have opposed the move, saying that it will lead to more administrative burdens, do nothing to curb abuse and diversion, and potentially decrease access to medications.
The DEA will publish the final rule on the rescheduling in the Federal Register on Aug. 22. Manufacturers, distributors, and prescribers will have to comply by Oct. 13.
The agency said it is time to rein in opioid prescribing and that rescheduling will help accomplish that goal.
"Almost 7 million Americans abuse controlled-substance prescription medications, including opioid painkillers, resulting in more deaths from prescription drug overdoses than auto accidents," DEA administrator Michele Leonhart said in a statement. "Today’s action recognizes that these products are some of the most addictive and potentially dangerous prescription medications available."
Hydrocodone combination products were placed on schedule III by Congress in 1970 when it created the Controlled Substances Act, in part because it was believed that adding acetaminophen or other non-narcotics might lessen the abuse potential. Hydrocodone itself was placed on schedule II.
Now, "the scientific, medical, and epidemiological data are robust and support rescheduling [of hydrocodone combination products] into schedule II," according to the final rule.
Data show that the products are widely diverted and abused at rates similar to that of oxycodone products, which are schedule II, said the agency, which added that abuse is associated with severe psychological or physical dependence, and many are being admitted to addiction treatment.
The hydrocodone combinations are also associated with large numbers of deaths, said the agency. More than 16,000 deaths in 2010 were due to abuse of opioids, including hydrocodone combinations, according to the DEA.
About 137 million prescriptions for hydrocodone combinations were dispensed in 2013, the agency said. The most frequently prescribed combination is hydrocodone/acetaminophen.
In comments to the proposed rule in April, the American Medical Association, along with a group of organizations and companies in the long-term care field, asked the agency to delay the final rule until an exception was made for nursing homes and other long-term care facilities. The AMA’s House of Delegates had also voted in 2013 to oppose rescheduling.
The American College of Emergency Physicians also urged against rescheduling, telling the DEA in April that it would not likely solve the abuse and diversion problems, but would lead to a greater administrative hassle for physicians.
The DEA said it received 573 comments after it proposed rescheduling, with 52% in favor, 41% opposed, and 7% taking no position. The agency received the most comments from the general public (44%; 250 comments) and pharmacists and pharmacy students (21%; 122 comments), physicians (13%; 73 comments), patients (6%; 35 comments), and midlevel practitioners (5%; 31 comments). Just over half of the physician comments supported, or supported with qualification, rescheduling.
Most of the commenters who were opposed to rescheduling were pharmacists, pharmacy students, and patients. Those opposed were concerned about how it would affect prescribing practices and patient access to medicine, and how it might impact long-term care facilities. Commenters also said that it would not prevent abuse or diversion.
The DEA said that although moving to schedule II does, for instance, prohibit refills, it would not necessarily block physicians from writing prescriptions for supplies of longer than 30 days, or from writing multiple prescriptions at once. State laws might have limits, however, and those will take precedence over the DEA rule.
The DEA rescheduling follows an FDA advisory committee recommendation in Jan. 2013 to do so, and the FDA’s backing of the proposal in Oct. 2013. HHS followed with its own recommendation to the DEA.
In comments on the proposed rule, manufacturers and pharmacies asked for more time to implement the rescheduling, but the DEA said no, citing high rates of abuse, overdose, and deaths relating to hydrocodone combination products.
The rescheduling goes into effect on Oct. 13, 45 days from the date of the rule’s publication in the Federal Register.
On Twitter @aliciaault
FDA approves novel insomnia drug suvorexant
The Food and Drug Administration has approved a new type of insomnia drug, the orexin receptor antagonist suvorexant (Belsomra).
It is the first drug in the class to be approved. Suvorexant blocks the binding of orexin A and B neuropeptides to orexin receptors, presumably inhibiting activation of neurons to the arousal system.
But it will not be available until the Drug Enforcement Administration makes a final determination of where it should fit on the schedule of controlled products. The FDA recommended scheduling, and the DEA proposed earlier this year that it receive a schedule IV classification under the Controlled Substances Act, according to suvorexant maker Merck.
The company said it expects suvorexant to be available in late 2014 or early 2015.
Merck had applied for approval in 2012. In May 2013, the FDA’s Peripheral and Central Nervous System Drugs Advisory Committee had questions about the safety of the company’s proposed starting doses. A few months later, the FDA advised Merck that it should make 10 mg the starting dose for most patients.
The company had not been prepared to manufacture that dose, so went back to work and created several new tablets.
Now, according to Dr. Ellis Unger, director of the Office of Drug Evaluation I in the FDA’s Center for Drug Evaluation and Research, "the FDA has approved Belsomra in four different strengths – 5, 10, 15, and 20 milligrams." Dr. Unger, in a statement, noted that "Using the lowest effective dose can reduce the risk of side effects, such as next-morning drowsiness."
The agency and the company say that suvorexant should only be taken once per night, within a half hour of going to bed and at least 7 hours before the person plans to wake up. Patients should not take more than 20 mg a day.
Drowsiness was the most common side effect. In FDA-required studies of next-day driving performance, patients who took 20 mg had impaired performance. Patients who take 20 mg should be warned about driving or conducting any activities that require mental alertness the next day, said the agency.
The drug also has the potential to lead to what the FDA calls "sleep-driving and other complex behaviors while not being fully awake," including preparing and eating food, making phone calls, or having sex. Patients and their families are urged to call their physician if that type of behavior is observed.
Suvorexant will be dispensed with a Medication Guide for patients that details safety information.
Although the drug was studied in three trials, none involved a comparison with other approved insomnia drugs, so its comparative effectiveness is not known, the FDA said.
On Twitter @aliciaault
The Food and Drug Administration has approved a new type of insomnia drug, the orexin receptor antagonist suvorexant (Belsomra).
It is the first drug in the class to be approved. Suvorexant blocks the binding of orexin A and B neuropeptides to orexin receptors, presumably inhibiting activation of neurons to the arousal system.
But it will not be available until the Drug Enforcement Administration makes a final determination of where it should fit on the schedule of controlled products. The FDA recommended scheduling, and the DEA proposed earlier this year that it receive a schedule IV classification under the Controlled Substances Act, according to suvorexant maker Merck.
The company said it expects suvorexant to be available in late 2014 or early 2015.
Merck had applied for approval in 2012. In May 2013, the FDA’s Peripheral and Central Nervous System Drugs Advisory Committee had questions about the safety of the company’s proposed starting doses. A few months later, the FDA advised Merck that it should make 10 mg the starting dose for most patients.
The company had not been prepared to manufacture that dose, so went back to work and created several new tablets.
Now, according to Dr. Ellis Unger, director of the Office of Drug Evaluation I in the FDA’s Center for Drug Evaluation and Research, "the FDA has approved Belsomra in four different strengths – 5, 10, 15, and 20 milligrams." Dr. Unger, in a statement, noted that "Using the lowest effective dose can reduce the risk of side effects, such as next-morning drowsiness."
The agency and the company say that suvorexant should only be taken once per night, within a half hour of going to bed and at least 7 hours before the person plans to wake up. Patients should not take more than 20 mg a day.
Drowsiness was the most common side effect. In FDA-required studies of next-day driving performance, patients who took 20 mg had impaired performance. Patients who take 20 mg should be warned about driving or conducting any activities that require mental alertness the next day, said the agency.
The drug also has the potential to lead to what the FDA calls "sleep-driving and other complex behaviors while not being fully awake," including preparing and eating food, making phone calls, or having sex. Patients and their families are urged to call their physician if that type of behavior is observed.
Suvorexant will be dispensed with a Medication Guide for patients that details safety information.
Although the drug was studied in three trials, none involved a comparison with other approved insomnia drugs, so its comparative effectiveness is not known, the FDA said.
On Twitter @aliciaault
The Food and Drug Administration has approved a new type of insomnia drug, the orexin receptor antagonist suvorexant (Belsomra).
It is the first drug in the class to be approved. Suvorexant blocks the binding of orexin A and B neuropeptides to orexin receptors, presumably inhibiting activation of neurons to the arousal system.
But it will not be available until the Drug Enforcement Administration makes a final determination of where it should fit on the schedule of controlled products. The FDA recommended scheduling, and the DEA proposed earlier this year that it receive a schedule IV classification under the Controlled Substances Act, according to suvorexant maker Merck.
The company said it expects suvorexant to be available in late 2014 or early 2015.
Merck had applied for approval in 2012. In May 2013, the FDA’s Peripheral and Central Nervous System Drugs Advisory Committee had questions about the safety of the company’s proposed starting doses. A few months later, the FDA advised Merck that it should make 10 mg the starting dose for most patients.
The company had not been prepared to manufacture that dose, so went back to work and created several new tablets.
Now, according to Dr. Ellis Unger, director of the Office of Drug Evaluation I in the FDA’s Center for Drug Evaluation and Research, "the FDA has approved Belsomra in four different strengths – 5, 10, 15, and 20 milligrams." Dr. Unger, in a statement, noted that "Using the lowest effective dose can reduce the risk of side effects, such as next-morning drowsiness."
The agency and the company say that suvorexant should only be taken once per night, within a half hour of going to bed and at least 7 hours before the person plans to wake up. Patients should not take more than 20 mg a day.
Drowsiness was the most common side effect. In FDA-required studies of next-day driving performance, patients who took 20 mg had impaired performance. Patients who take 20 mg should be warned about driving or conducting any activities that require mental alertness the next day, said the agency.
The drug also has the potential to lead to what the FDA calls "sleep-driving and other complex behaviors while not being fully awake," including preparing and eating food, making phone calls, or having sex. Patients and their families are urged to call their physician if that type of behavior is observed.
Suvorexant will be dispensed with a Medication Guide for patients that details safety information.
Although the drug was studied in three trials, none involved a comparison with other approved insomnia drugs, so its comparative effectiveness is not known, the FDA said.
On Twitter @aliciaault
Endocrinologists explore their own MOC pathway
Expressing continuing frustration with the American Board of Internal Medicine, the American Association of Clinical Endocrinologists is exploring its own avenue for maintenance of certification.
In a video message e-mailed to members Aug. 13, Dr. R. Mack Harrell, AACE president, discussed events from a July 15 meeting between ABIM board members and representatives from the 26 subspecialty organizations that receive certification from the ABIM.

AACE representatives told the ABIM that the secure exam is outmoded and not reflective of how medicine is practiced, and that its members questioned the utility and applicability of the practice modules. The organization also said it wanted the ABIM to describe physicians as either "participating" or "not participating" in MOC on its Certification Matters website.
ABIM representatives said that all this and more would be considered at an Aug. 5 board meeting.
Now, AACE is waiting to see what kind of response it will get, Dr. Harrell said in the video.
"If we get the wrong answers after the ABIM board meets, we’re going to have to entertain forming alternative pathways for certification," he said. "It’s our belief that with the right coalition of subspecialty societies, we can put together an alternative pathway for lifelong learning."
He also told members that foregoing any kind of MOC is not an option. Going back to a system in which physicians get their initial certification and then show a certain amount of continuing education credits each year "is not going to fly in the current American health care environment," he said in the video.
The old system won’t work in part, because so much CME is either "not documented, or quite frankly, bad," Dr. Harrell said in an interview, adding that insurers, health care organizations, and other stakeholders needed more. "We have to come up with some way to be very clear about how we document that people actually attended CME and learned something."
As far as AACE creating its own pathway for MOC, "it’s something that’s doable, though it will require a good deal of work," he added. "If it’s necessary, we’ll do it."
Dr. Richard J. Baron, president and CEO of ABIM, said that setting up an MOC process is not easy, nor for the faint of heart. "If AACE wants to offer a separate credentialing pathway, I’m in a great position to acknowledge how complicated a task that is, how challenging it is to do it well, and I wish them luck," he said in an interview.
He noted that the ABIM was created very deliberately by the American College of Physicians and the American Medical Association because they believed that a society might not be able to set credible standards as a membership organization.
Dr. Baron said that he could not currently disclose what was discussed at the ABIM board meeting on Aug. 5, but that the ABIM would be issuing a communication shortly.
On Twitter @aliciaault
Expressing continuing frustration with the American Board of Internal Medicine, the American Association of Clinical Endocrinologists is exploring its own avenue for maintenance of certification.
In a video message e-mailed to members Aug. 13, Dr. R. Mack Harrell, AACE president, discussed events from a July 15 meeting between ABIM board members and representatives from the 26 subspecialty organizations that receive certification from the ABIM.
AACE representatives told the ABIM that the secure exam is outmoded and not reflective of how medicine is practiced, and that its members questioned the utility and applicability of the practice modules. The organization also said it wanted the ABIM to describe physicians as either "participating" or "not participating" in MOC on its Certification Matters website.
ABIM representatives said that all this and more would be considered at an Aug. 5 board meeting.
Now, AACE is waiting to see what kind of response it will get, Dr. Harrell said in the video.
"If we get the wrong answers after the ABIM board meets, we’re going to have to entertain forming alternative pathways for certification," he said. "It’s our belief that with the right coalition of subspecialty societies, we can put together an alternative pathway for lifelong learning."
He also told members that foregoing any kind of MOC is not an option. Going back to a system in which physicians get their initial certification and then show a certain amount of continuing education credits each year "is not going to fly in the current American health care environment," he said in the video.
The old system won’t work in part, because so much CME is either "not documented, or quite frankly, bad," Dr. Harrell said in an interview, adding that insurers, health care organizations, and other stakeholders needed more. "We have to come up with some way to be very clear about how we document that people actually attended CME and learned something."
As far as AACE creating its own pathway for MOC, "it’s something that’s doable, though it will require a good deal of work," he added. "If it’s necessary, we’ll do it."
Dr. Richard J. Baron, president and CEO of ABIM, said that setting up an MOC process is not easy, nor for the faint of heart. "If AACE wants to offer a separate credentialing pathway, I’m in a great position to acknowledge how complicated a task that is, how challenging it is to do it well, and I wish them luck," he said in an interview.
He noted that the ABIM was created very deliberately by the American College of Physicians and the American Medical Association because they believed that a society might not be able to set credible standards as a membership organization.
Dr. Baron said that he could not currently disclose what was discussed at the ABIM board meeting on Aug. 5, but that the ABIM would be issuing a communication shortly.
On Twitter @aliciaault
Expressing continuing frustration with the American Board of Internal Medicine, the American Association of Clinical Endocrinologists is exploring its own avenue for maintenance of certification.
In a video message e-mailed to members Aug. 13, Dr. R. Mack Harrell, AACE president, discussed events from a July 15 meeting between ABIM board members and representatives from the 26 subspecialty organizations that receive certification from the ABIM.
AACE representatives told the ABIM that the secure exam is outmoded and not reflective of how medicine is practiced, and that its members questioned the utility and applicability of the practice modules. The organization also said it wanted the ABIM to describe physicians as either "participating" or "not participating" in MOC on its Certification Matters website.
ABIM representatives said that all this and more would be considered at an Aug. 5 board meeting.
Now, AACE is waiting to see what kind of response it will get, Dr. Harrell said in the video.
"If we get the wrong answers after the ABIM board meets, we’re going to have to entertain forming alternative pathways for certification," he said. "It’s our belief that with the right coalition of subspecialty societies, we can put together an alternative pathway for lifelong learning."
He also told members that foregoing any kind of MOC is not an option. Going back to a system in which physicians get their initial certification and then show a certain amount of continuing education credits each year "is not going to fly in the current American health care environment," he said in the video.
The old system won’t work in part, because so much CME is either "not documented, or quite frankly, bad," Dr. Harrell said in an interview, adding that insurers, health care organizations, and other stakeholders needed more. "We have to come up with some way to be very clear about how we document that people actually attended CME and learned something."
As far as AACE creating its own pathway for MOC, "it’s something that’s doable, though it will require a good deal of work," he added. "If it’s necessary, we’ll do it."
Dr. Richard J. Baron, president and CEO of ABIM, said that setting up an MOC process is not easy, nor for the faint of heart. "If AACE wants to offer a separate credentialing pathway, I’m in a great position to acknowledge how complicated a task that is, how challenging it is to do it well, and I wish them luck," he said in an interview.
He noted that the ABIM was created very deliberately by the American College of Physicians and the American Medical Association because they believed that a society might not be able to set credible standards as a membership organization.
Dr. Baron said that he could not currently disclose what was discussed at the ABIM board meeting on Aug. 5, but that the ABIM would be issuing a communication shortly.
On Twitter @aliciaault
