User login
Purpuric Eruption in a Patient With Hairy Cell Leukemia
The Diagnosis: Purpuric Drug Eruption
Histopathology revealed interface dermatitis, spongiosis, and a perivascular lymphocytic infiltrate with extravasated red blood cells consistent with a purpuric drug eruption. Our patient achieved remission of hairy cell leukemia after receiving only 2 of 5 expected doses of cladribine. The rash resolved completely in 3 weeks following a prednisone taper (Figure).
Hairy cell leukemia is a rare indolent lymphoproliferative disorder of B cells that accounts for approximately 2% of adult leukemias in the United States. Cladribine, a purine nucleoside analog that impairs DNA synthesis and repair, has become the mainstay of therapy, demonstrating a 95% complete response rate.1 Although few reports have addressed the cutaneous reactions seen with cladribine therapy, they can occur in more than 50% of patients.1,2 The most common skin manifestation associated with cladribine therapy is a morbilliform rash, but Stevens-Johnson syndrome and toxic epidermal necrolysis (TEN) have been reported.1
Few cases of purpuric eruption secondary to cladribine treatment have been described, and nearly all reports involve concomitant medications such as allopurinol, which our patient was taking, and antibiotics including trimethoprim-sulfamethoxazole and penicillins.1,3,4 In a cohort of 35 patients receiving cladribine,1 only concomitant treatment with cladribine and allopurinol caused cutaneous reactions, further supporting the hypothesis of cladribine-induced drug sensitivity. Allopurinol often is prescribed during induction therapy for prophylaxis against tumor lysis syndrome; similarly, antibiotics frequently are given prophylactically and therapeutically for neutropenic fever. It is believed that T-cell imbalance and profound lymphopenia induced by cladribine increase susceptibility to drug hypersensitivity reactions.1,3
The typical purpuric eruption develops within 2 days of starting cladribine therapy. Diascopy will reveal petechiae, and biopsy should be performed to rule out other serious drug-induced reactions, such as erythema multiforme, Stevens-Johnson syndrome, and TEN. A cladribine-induced purpuric eruption typically is self-resolving and carries a favorable prognosis, though high-dose corticosteroids often are prescribed to hasten recovery. The rare reports of serious cutaneous reactions secondary to cladribine therapy have been with maculopapular, not purpuric eruptions.2 Based on limited available data, cladribine-induced purpura should not be a limitation to continued treatment in patients who need it.1 Careful consideration of concomitant drug use is necessary, as the current literature demonstrates resolution of rash with withdrawal of other therapies, namely allopurinol.2-4 Future studies are needed to examine the safety of withholding offending medications and to further elucidate the mechanisms contributing to drug hypersensitivity due to cladribine.
Widespread purpura and petechiae can pose a wide differential; the patient’s recent history of cladribine administration pointed to a classic purpuric eruption. Other diagnoses such as toxic erythema of chemotherapy (TEC) and TEN are not purpuric, though plaques can be violaceous. Lack of bullae, blisters, and facial or mucosal surface involvement suggest TEN.5 Thrombotic thrombocytopenic purpura and disseminated intravascular coagulation do manifest with petechiae and purpura, though such a robust eruption in the context of recent cladribine therapy is less likely. The classic retiform purpura and necrosis were not present to suggest purpura fulminans from disseminated intravascular coagulation.
Several of the proposed diagnoses as well as a purpuric drug eruption would demonstrate extravasated red blood cells on histopathology, but the presence of interface dermatitis narrows the differential to a purpuric drug eruption. Necrotic keratinocytes and full-thickness necrosis were not present on biopsy to support a diagnosis of TEN in our patient. Characteristic features of TEC—including eccrine squamous syringometaplasia, dermal edema, and keratinocyte atypia—were not present on biopsy.6 Finally, although TEN should resolve with steroid treatment, TEC is self-limited and thrombotic thrombocytopenic purpura and disseminated intravascular coagulation would not resolve with use of steroids alone.
- Ganzel C, Gatt ME, Maly A, et al. High incidence of skin rash in patients with hairy cell leukemia treated with cladribine. Leuk Lymphoma. 2012;53:1169-1173. doi:10.3109/10428194.2011.635864
- Chubar Y, Bennett M. Cutaneous reactions in hairy cell leukaemia treated with 2-chlorodeoxyadenosine and allopurinol. Br J Haematol. 2003;122:768-770. doi:10.1046/j.1365-2141.2003.04506.x
- Espinosa Lara P, Quirós Redondo V, Aguado Lobo M, et al. Purpuric exanthema in a patient with hairy cell leukemia treated with cladribine and allopurinol. Ann Hematol. 2017;96:1209-1210. doi:10.1007 /s00277-017-2992-z
- Hendrick A. Purpuric rash following treatment with 2-chlorodeoxyadenosine. Clin Lab Haematol. 2001;23:67-68. doi:10.1046 /j.1365-2257.2001.0346b.x
- Kang S, Amagai M, Bruckner AL, et al, eds. Fitzpatrick’s Dermatology. 9th ed. McGraw-Hill Education; 2019.
- Bolognia JL, Cooper DL, Glusac EJ. Toxic erythema of chemotherapy: a useful clinical term. J Am Acad Dermatol. 2008;59:524-529.
The Diagnosis: Purpuric Drug Eruption
Histopathology revealed interface dermatitis, spongiosis, and a perivascular lymphocytic infiltrate with extravasated red blood cells consistent with a purpuric drug eruption. Our patient achieved remission of hairy cell leukemia after receiving only 2 of 5 expected doses of cladribine. The rash resolved completely in 3 weeks following a prednisone taper (Figure).
Hairy cell leukemia is a rare indolent lymphoproliferative disorder of B cells that accounts for approximately 2% of adult leukemias in the United States. Cladribine, a purine nucleoside analog that impairs DNA synthesis and repair, has become the mainstay of therapy, demonstrating a 95% complete response rate.1 Although few reports have addressed the cutaneous reactions seen with cladribine therapy, they can occur in more than 50% of patients.1,2 The most common skin manifestation associated with cladribine therapy is a morbilliform rash, but Stevens-Johnson syndrome and toxic epidermal necrolysis (TEN) have been reported.1
Few cases of purpuric eruption secondary to cladribine treatment have been described, and nearly all reports involve concomitant medications such as allopurinol, which our patient was taking, and antibiotics including trimethoprim-sulfamethoxazole and penicillins.1,3,4 In a cohort of 35 patients receiving cladribine,1 only concomitant treatment with cladribine and allopurinol caused cutaneous reactions, further supporting the hypothesis of cladribine-induced drug sensitivity. Allopurinol often is prescribed during induction therapy for prophylaxis against tumor lysis syndrome; similarly, antibiotics frequently are given prophylactically and therapeutically for neutropenic fever. It is believed that T-cell imbalance and profound lymphopenia induced by cladribine increase susceptibility to drug hypersensitivity reactions.1,3
The typical purpuric eruption develops within 2 days of starting cladribine therapy. Diascopy will reveal petechiae, and biopsy should be performed to rule out other serious drug-induced reactions, such as erythema multiforme, Stevens-Johnson syndrome, and TEN. A cladribine-induced purpuric eruption typically is self-resolving and carries a favorable prognosis, though high-dose corticosteroids often are prescribed to hasten recovery. The rare reports of serious cutaneous reactions secondary to cladribine therapy have been with maculopapular, not purpuric eruptions.2 Based on limited available data, cladribine-induced purpura should not be a limitation to continued treatment in patients who need it.1 Careful consideration of concomitant drug use is necessary, as the current literature demonstrates resolution of rash with withdrawal of other therapies, namely allopurinol.2-4 Future studies are needed to examine the safety of withholding offending medications and to further elucidate the mechanisms contributing to drug hypersensitivity due to cladribine.
Widespread purpura and petechiae can pose a wide differential; the patient’s recent history of cladribine administration pointed to a classic purpuric eruption. Other diagnoses such as toxic erythema of chemotherapy (TEC) and TEN are not purpuric, though plaques can be violaceous. Lack of bullae, blisters, and facial or mucosal surface involvement suggest TEN.5 Thrombotic thrombocytopenic purpura and disseminated intravascular coagulation do manifest with petechiae and purpura, though such a robust eruption in the context of recent cladribine therapy is less likely. The classic retiform purpura and necrosis were not present to suggest purpura fulminans from disseminated intravascular coagulation.
Several of the proposed diagnoses as well as a purpuric drug eruption would demonstrate extravasated red blood cells on histopathology, but the presence of interface dermatitis narrows the differential to a purpuric drug eruption. Necrotic keratinocytes and full-thickness necrosis were not present on biopsy to support a diagnosis of TEN in our patient. Characteristic features of TEC—including eccrine squamous syringometaplasia, dermal edema, and keratinocyte atypia—were not present on biopsy.6 Finally, although TEN should resolve with steroid treatment, TEC is self-limited and thrombotic thrombocytopenic purpura and disseminated intravascular coagulation would not resolve with use of steroids alone.
The Diagnosis: Purpuric Drug Eruption
Histopathology revealed interface dermatitis, spongiosis, and a perivascular lymphocytic infiltrate with extravasated red blood cells consistent with a purpuric drug eruption. Our patient achieved remission of hairy cell leukemia after receiving only 2 of 5 expected doses of cladribine. The rash resolved completely in 3 weeks following a prednisone taper (Figure).
Hairy cell leukemia is a rare indolent lymphoproliferative disorder of B cells that accounts for approximately 2% of adult leukemias in the United States. Cladribine, a purine nucleoside analog that impairs DNA synthesis and repair, has become the mainstay of therapy, demonstrating a 95% complete response rate.1 Although few reports have addressed the cutaneous reactions seen with cladribine therapy, they can occur in more than 50% of patients.1,2 The most common skin manifestation associated with cladribine therapy is a morbilliform rash, but Stevens-Johnson syndrome and toxic epidermal necrolysis (TEN) have been reported.1
Few cases of purpuric eruption secondary to cladribine treatment have been described, and nearly all reports involve concomitant medications such as allopurinol, which our patient was taking, and antibiotics including trimethoprim-sulfamethoxazole and penicillins.1,3,4 In a cohort of 35 patients receiving cladribine,1 only concomitant treatment with cladribine and allopurinol caused cutaneous reactions, further supporting the hypothesis of cladribine-induced drug sensitivity. Allopurinol often is prescribed during induction therapy for prophylaxis against tumor lysis syndrome; similarly, antibiotics frequently are given prophylactically and therapeutically for neutropenic fever. It is believed that T-cell imbalance and profound lymphopenia induced by cladribine increase susceptibility to drug hypersensitivity reactions.1,3
The typical purpuric eruption develops within 2 days of starting cladribine therapy. Diascopy will reveal petechiae, and biopsy should be performed to rule out other serious drug-induced reactions, such as erythema multiforme, Stevens-Johnson syndrome, and TEN. A cladribine-induced purpuric eruption typically is self-resolving and carries a favorable prognosis, though high-dose corticosteroids often are prescribed to hasten recovery. The rare reports of serious cutaneous reactions secondary to cladribine therapy have been with maculopapular, not purpuric eruptions.2 Based on limited available data, cladribine-induced purpura should not be a limitation to continued treatment in patients who need it.1 Careful consideration of concomitant drug use is necessary, as the current literature demonstrates resolution of rash with withdrawal of other therapies, namely allopurinol.2-4 Future studies are needed to examine the safety of withholding offending medications and to further elucidate the mechanisms contributing to drug hypersensitivity due to cladribine.
Widespread purpura and petechiae can pose a wide differential; the patient’s recent history of cladribine administration pointed to a classic purpuric eruption. Other diagnoses such as toxic erythema of chemotherapy (TEC) and TEN are not purpuric, though plaques can be violaceous. Lack of bullae, blisters, and facial or mucosal surface involvement suggest TEN.5 Thrombotic thrombocytopenic purpura and disseminated intravascular coagulation do manifest with petechiae and purpura, though such a robust eruption in the context of recent cladribine therapy is less likely. The classic retiform purpura and necrosis were not present to suggest purpura fulminans from disseminated intravascular coagulation.
Several of the proposed diagnoses as well as a purpuric drug eruption would demonstrate extravasated red blood cells on histopathology, but the presence of interface dermatitis narrows the differential to a purpuric drug eruption. Necrotic keratinocytes and full-thickness necrosis were not present on biopsy to support a diagnosis of TEN in our patient. Characteristic features of TEC—including eccrine squamous syringometaplasia, dermal edema, and keratinocyte atypia—were not present on biopsy.6 Finally, although TEN should resolve with steroid treatment, TEC is self-limited and thrombotic thrombocytopenic purpura and disseminated intravascular coagulation would not resolve with use of steroids alone.
- Ganzel C, Gatt ME, Maly A, et al. High incidence of skin rash in patients with hairy cell leukemia treated with cladribine. Leuk Lymphoma. 2012;53:1169-1173. doi:10.3109/10428194.2011.635864
- Chubar Y, Bennett M. Cutaneous reactions in hairy cell leukaemia treated with 2-chlorodeoxyadenosine and allopurinol. Br J Haematol. 2003;122:768-770. doi:10.1046/j.1365-2141.2003.04506.x
- Espinosa Lara P, Quirós Redondo V, Aguado Lobo M, et al. Purpuric exanthema in a patient with hairy cell leukemia treated with cladribine and allopurinol. Ann Hematol. 2017;96:1209-1210. doi:10.1007 /s00277-017-2992-z
- Hendrick A. Purpuric rash following treatment with 2-chlorodeoxyadenosine. Clin Lab Haematol. 2001;23:67-68. doi:10.1046 /j.1365-2257.2001.0346b.x
- Kang S, Amagai M, Bruckner AL, et al, eds. Fitzpatrick’s Dermatology. 9th ed. McGraw-Hill Education; 2019.
- Bolognia JL, Cooper DL, Glusac EJ. Toxic erythema of chemotherapy: a useful clinical term. J Am Acad Dermatol. 2008;59:524-529.
- Ganzel C, Gatt ME, Maly A, et al. High incidence of skin rash in patients with hairy cell leukemia treated with cladribine. Leuk Lymphoma. 2012;53:1169-1173. doi:10.3109/10428194.2011.635864
- Chubar Y, Bennett M. Cutaneous reactions in hairy cell leukaemia treated with 2-chlorodeoxyadenosine and allopurinol. Br J Haematol. 2003;122:768-770. doi:10.1046/j.1365-2141.2003.04506.x
- Espinosa Lara P, Quirós Redondo V, Aguado Lobo M, et al. Purpuric exanthema in a patient with hairy cell leukemia treated with cladribine and allopurinol. Ann Hematol. 2017;96:1209-1210. doi:10.1007 /s00277-017-2992-z
- Hendrick A. Purpuric rash following treatment with 2-chlorodeoxyadenosine. Clin Lab Haematol. 2001;23:67-68. doi:10.1046 /j.1365-2257.2001.0346b.x
- Kang S, Amagai M, Bruckner AL, et al, eds. Fitzpatrick’s Dermatology. 9th ed. McGraw-Hill Education; 2019.
- Bolognia JL, Cooper DL, Glusac EJ. Toxic erythema of chemotherapy: a useful clinical term. J Am Acad Dermatol. 2008;59:524-529.
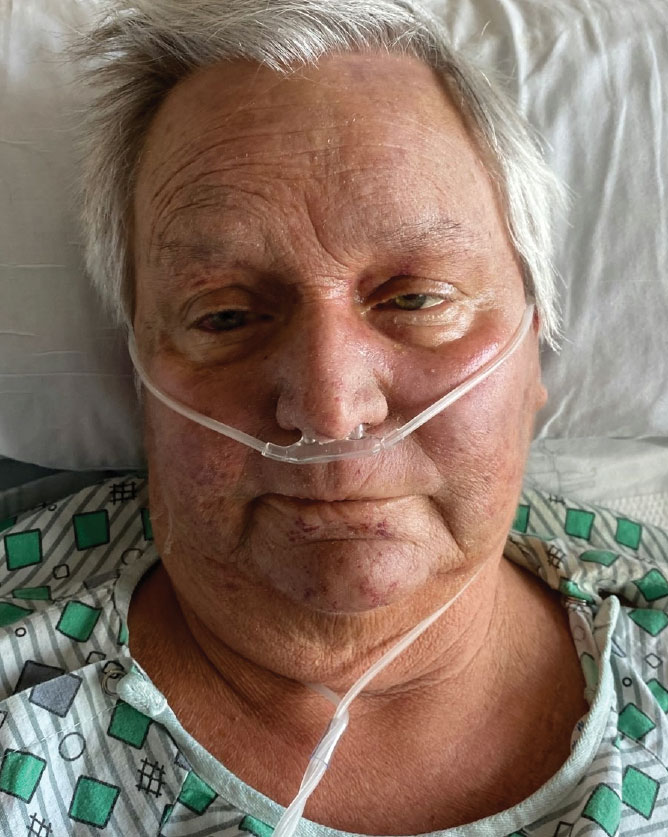
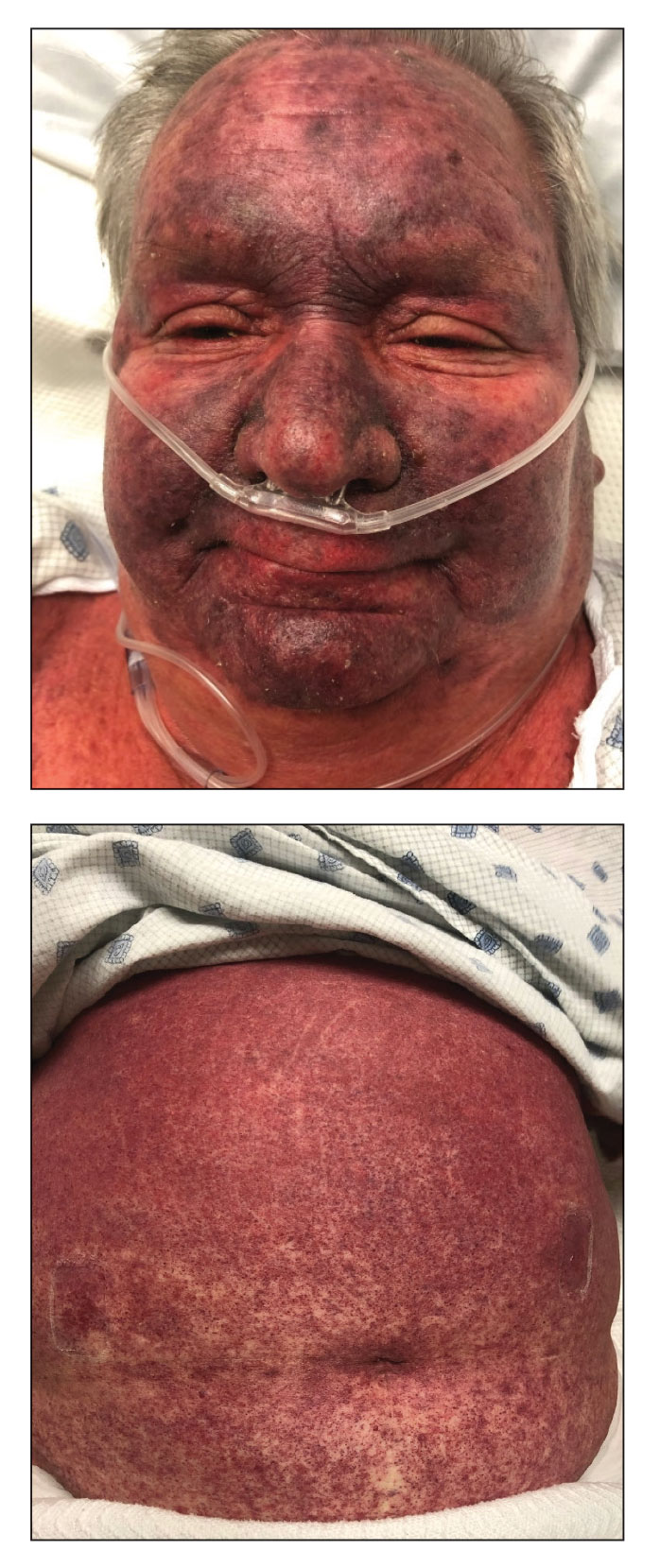
A 68-year-old woman presented to the emergency department with neutropenic fever and a rash over the body after receiving 2 doses of cladribine therapy for hairy cell leukemia. Physical examination demonstrated marked facial (top), lip, and tongue swelling, as well as a diffuse dusky nonpalpable purpuric rash on the abdomen (bottom) and back involving 90% of the body surface area. Bilateral ear edema was appreciated with accentuation of the earlobe crease. The patient exhibited subconjunctival hemorrhage, ectropion, and scleral injection. A punch biopsy of the thigh was performed.
Shiny Indurated Plaques on the Legs
The Diagnosis: Pretibial Myxedema
Histopathology showed superficial and deep mucin deposition with proliferation of fibroblasts and thin wiry collagen bundles that were consistent with a diagnosis of pretibial myxedema. The patient was treated with clobetasol ointment 0.05% twice daily for 3 months, followed by a trial of pentoxifylline 400 mg 3 times daily for 3 months. After this treatment failed, she was started on rituximab infusions of 1 g biweekly for 1 month, followed by 500 mg at 6 months, with marked improvement after the first 2 doses of 1 g.
Pretibial myxedema is an uncommon cutaneous manifestation of autoimmune thyroid disease, occurring in 1% to 5% of patients with Graves disease. It usually occurs in older adult women on the pretibial regions and less commonly on the upper extremities, face, and areas of prior trauma.1-3 Although typically asymptomatic, it can be painful and ulcerate.3 The clinical presentation consists of bilateral nonpitting edema with overlying indurated skin as well as flesh-colored, yellow-brown, violaceous, or peau d’orange papules and plaques.2,3 Lesions develop over months and often have been associated with hyperhidrosis and hypertrichosis.2 Many variants have been identified including nodular, plaquelike, diffuse swelling (ie, nonpitting edema), tumor, mixture, polypoid, and elephantiasis; severe cases with acral involvement are termed thyroid acropachy.1-3 Pathogenesis likely involves the activation of thyrotropin receptors on fibroblasts by the circulating thyrotropin autoantibodies found in Graves disease. Activated fibroblasts upregulate glycosaminoglycan production, which osmotically drives the accumulation of dermal and subdermal fluid.1,3
This diagnosis should be considered in any patient with pretibial edema or edema in areas of trauma. Graves disease most commonly is diagnosed 1 to 2 years prior to the development of pretibial myxedema; other extrathyroidal manifestations, most commonly ophthalmopathies, almost always are found in patients with pretibial myxedema. If a diagnosis of Graves disease has not been established, thyroid studies, including thyrotropin receptor antibody serum levels, should be obtained. Histopathology showing increased mucin in the dermis and increased fibroblasts can aid in diagnosis.2,3
The differential diagnosis includes inflammatory dermatoses, such as stasis dermatitis and lipodermatosclerosis. Stasis dermatitis is characterized by lichenified yellowbrown plaques that present on the lower extremities; lipodermatosclerosis then can develop and present as atrophic sclerotic plaques with a champagne bottle–like appearance. Necrobiosis lipoidica demonstrates atrophic, shiny, yellow plaques with telangiectases and ulcerations. Hypertrophic lichen planus presents with hyperkeratotic hyperpigmented plaques on the shins.1,2 Other diseases of cutaneous mucin deposition, namely scleromyxedema, demonstrate similar physical findings but more commonly are located on the trunk, face, and dorsal hands rather than the lower extremities.1-3
Treatment of pretibial myxedema is difficult; normalization of thyroid function, weight reduction, and compression stockings can help reduce edema. Medical therapies aim to decrease glycosaminoglycan production by fibroblasts. First-line treatment includes topical steroids under occlusion, and second-line therapies include intralesional steroids, systemic corticosteroids, pentoxifylline, and octreotide.2,3 Therapies for refractory disease include plasmapheresis, surgical excision, radiotherapy, and intravenous immunoglobulin; more recent studies also endorse the use of isotretinoin, intralesional hyaluronidase, and rituximab.2,4 Success also has been observed with the insulin growth factor 1 receptor inhibitor teprotumumab in active thyroid eye disease, in which insulin growth factor 1 receptor is overexpressed by fibroblasts. Given the similar pathogenesis of thyroid ophthalmopathy with other extrathyroidal manifestations, teprotumumab is a promising option for refractory cases of pretibial myxedema and has led to disease resolution in several patients.4
- Fatourechi V, Pajouhi M, Fransway AF. Dermopathy of Graves disease (pretibial myxedema). review of 150 cases. Medicine (Baltimore). 1994;73:1-7. doi:10.1097/00005792-199401000-00001
- Ai J, Leonhardt JM, Heymann WR. Autoimmune thyroid diseases: etiology, pathogenesis, and dermatologic manifestations. J Am Acad Dermatol. 2003;48:641-662. doi:10.1067/mjd.2003.257
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446. doi:10.1210/jcem.87.2.8220
- Varma A, Rheeman C, Levitt J. Resolution of pretibial myxedema with teprotumumab in a patient with Graves disease. JAAD Case Reports. 2020;6:1281-1282. doi:10.1016/j.jdcr.2020.09.003
The Diagnosis: Pretibial Myxedema
Histopathology showed superficial and deep mucin deposition with proliferation of fibroblasts and thin wiry collagen bundles that were consistent with a diagnosis of pretibial myxedema. The patient was treated with clobetasol ointment 0.05% twice daily for 3 months, followed by a trial of pentoxifylline 400 mg 3 times daily for 3 months. After this treatment failed, she was started on rituximab infusions of 1 g biweekly for 1 month, followed by 500 mg at 6 months, with marked improvement after the first 2 doses of 1 g.
Pretibial myxedema is an uncommon cutaneous manifestation of autoimmune thyroid disease, occurring in 1% to 5% of patients with Graves disease. It usually occurs in older adult women on the pretibial regions and less commonly on the upper extremities, face, and areas of prior trauma.1-3 Although typically asymptomatic, it can be painful and ulcerate.3 The clinical presentation consists of bilateral nonpitting edema with overlying indurated skin as well as flesh-colored, yellow-brown, violaceous, or peau d’orange papules and plaques.2,3 Lesions develop over months and often have been associated with hyperhidrosis and hypertrichosis.2 Many variants have been identified including nodular, plaquelike, diffuse swelling (ie, nonpitting edema), tumor, mixture, polypoid, and elephantiasis; severe cases with acral involvement are termed thyroid acropachy.1-3 Pathogenesis likely involves the activation of thyrotropin receptors on fibroblasts by the circulating thyrotropin autoantibodies found in Graves disease. Activated fibroblasts upregulate glycosaminoglycan production, which osmotically drives the accumulation of dermal and subdermal fluid.1,3
This diagnosis should be considered in any patient with pretibial edema or edema in areas of trauma. Graves disease most commonly is diagnosed 1 to 2 years prior to the development of pretibial myxedema; other extrathyroidal manifestations, most commonly ophthalmopathies, almost always are found in patients with pretibial myxedema. If a diagnosis of Graves disease has not been established, thyroid studies, including thyrotropin receptor antibody serum levels, should be obtained. Histopathology showing increased mucin in the dermis and increased fibroblasts can aid in diagnosis.2,3
The differential diagnosis includes inflammatory dermatoses, such as stasis dermatitis and lipodermatosclerosis. Stasis dermatitis is characterized by lichenified yellowbrown plaques that present on the lower extremities; lipodermatosclerosis then can develop and present as atrophic sclerotic plaques with a champagne bottle–like appearance. Necrobiosis lipoidica demonstrates atrophic, shiny, yellow plaques with telangiectases and ulcerations. Hypertrophic lichen planus presents with hyperkeratotic hyperpigmented plaques on the shins.1,2 Other diseases of cutaneous mucin deposition, namely scleromyxedema, demonstrate similar physical findings but more commonly are located on the trunk, face, and dorsal hands rather than the lower extremities.1-3
Treatment of pretibial myxedema is difficult; normalization of thyroid function, weight reduction, and compression stockings can help reduce edema. Medical therapies aim to decrease glycosaminoglycan production by fibroblasts. First-line treatment includes topical steroids under occlusion, and second-line therapies include intralesional steroids, systemic corticosteroids, pentoxifylline, and octreotide.2,3 Therapies for refractory disease include plasmapheresis, surgical excision, radiotherapy, and intravenous immunoglobulin; more recent studies also endorse the use of isotretinoin, intralesional hyaluronidase, and rituximab.2,4 Success also has been observed with the insulin growth factor 1 receptor inhibitor teprotumumab in active thyroid eye disease, in which insulin growth factor 1 receptor is overexpressed by fibroblasts. Given the similar pathogenesis of thyroid ophthalmopathy with other extrathyroidal manifestations, teprotumumab is a promising option for refractory cases of pretibial myxedema and has led to disease resolution in several patients.4
The Diagnosis: Pretibial Myxedema
Histopathology showed superficial and deep mucin deposition with proliferation of fibroblasts and thin wiry collagen bundles that were consistent with a diagnosis of pretibial myxedema. The patient was treated with clobetasol ointment 0.05% twice daily for 3 months, followed by a trial of pentoxifylline 400 mg 3 times daily for 3 months. After this treatment failed, she was started on rituximab infusions of 1 g biweekly for 1 month, followed by 500 mg at 6 months, with marked improvement after the first 2 doses of 1 g.
Pretibial myxedema is an uncommon cutaneous manifestation of autoimmune thyroid disease, occurring in 1% to 5% of patients with Graves disease. It usually occurs in older adult women on the pretibial regions and less commonly on the upper extremities, face, and areas of prior trauma.1-3 Although typically asymptomatic, it can be painful and ulcerate.3 The clinical presentation consists of bilateral nonpitting edema with overlying indurated skin as well as flesh-colored, yellow-brown, violaceous, or peau d’orange papules and plaques.2,3 Lesions develop over months and often have been associated with hyperhidrosis and hypertrichosis.2 Many variants have been identified including nodular, plaquelike, diffuse swelling (ie, nonpitting edema), tumor, mixture, polypoid, and elephantiasis; severe cases with acral involvement are termed thyroid acropachy.1-3 Pathogenesis likely involves the activation of thyrotropin receptors on fibroblasts by the circulating thyrotropin autoantibodies found in Graves disease. Activated fibroblasts upregulate glycosaminoglycan production, which osmotically drives the accumulation of dermal and subdermal fluid.1,3
This diagnosis should be considered in any patient with pretibial edema or edema in areas of trauma. Graves disease most commonly is diagnosed 1 to 2 years prior to the development of pretibial myxedema; other extrathyroidal manifestations, most commonly ophthalmopathies, almost always are found in patients with pretibial myxedema. If a diagnosis of Graves disease has not been established, thyroid studies, including thyrotropin receptor antibody serum levels, should be obtained. Histopathology showing increased mucin in the dermis and increased fibroblasts can aid in diagnosis.2,3
The differential diagnosis includes inflammatory dermatoses, such as stasis dermatitis and lipodermatosclerosis. Stasis dermatitis is characterized by lichenified yellowbrown plaques that present on the lower extremities; lipodermatosclerosis then can develop and present as atrophic sclerotic plaques with a champagne bottle–like appearance. Necrobiosis lipoidica demonstrates atrophic, shiny, yellow plaques with telangiectases and ulcerations. Hypertrophic lichen planus presents with hyperkeratotic hyperpigmented plaques on the shins.1,2 Other diseases of cutaneous mucin deposition, namely scleromyxedema, demonstrate similar physical findings but more commonly are located on the trunk, face, and dorsal hands rather than the lower extremities.1-3
Treatment of pretibial myxedema is difficult; normalization of thyroid function, weight reduction, and compression stockings can help reduce edema. Medical therapies aim to decrease glycosaminoglycan production by fibroblasts. First-line treatment includes topical steroids under occlusion, and second-line therapies include intralesional steroids, systemic corticosteroids, pentoxifylline, and octreotide.2,3 Therapies for refractory disease include plasmapheresis, surgical excision, radiotherapy, and intravenous immunoglobulin; more recent studies also endorse the use of isotretinoin, intralesional hyaluronidase, and rituximab.2,4 Success also has been observed with the insulin growth factor 1 receptor inhibitor teprotumumab in active thyroid eye disease, in which insulin growth factor 1 receptor is overexpressed by fibroblasts. Given the similar pathogenesis of thyroid ophthalmopathy with other extrathyroidal manifestations, teprotumumab is a promising option for refractory cases of pretibial myxedema and has led to disease resolution in several patients.4
- Fatourechi V, Pajouhi M, Fransway AF. Dermopathy of Graves disease (pretibial myxedema). review of 150 cases. Medicine (Baltimore). 1994;73:1-7. doi:10.1097/00005792-199401000-00001
- Ai J, Leonhardt JM, Heymann WR. Autoimmune thyroid diseases: etiology, pathogenesis, and dermatologic manifestations. J Am Acad Dermatol. 2003;48:641-662. doi:10.1067/mjd.2003.257
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446. doi:10.1210/jcem.87.2.8220
- Varma A, Rheeman C, Levitt J. Resolution of pretibial myxedema with teprotumumab in a patient with Graves disease. JAAD Case Reports. 2020;6:1281-1282. doi:10.1016/j.jdcr.2020.09.003
- Fatourechi V, Pajouhi M, Fransway AF. Dermopathy of Graves disease (pretibial myxedema). review of 150 cases. Medicine (Baltimore). 1994;73:1-7. doi:10.1097/00005792-199401000-00001
- Ai J, Leonhardt JM, Heymann WR. Autoimmune thyroid diseases: etiology, pathogenesis, and dermatologic manifestations. J Am Acad Dermatol. 2003;48:641-662. doi:10.1067/mjd.2003.257
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446. doi:10.1210/jcem.87.2.8220
- Varma A, Rheeman C, Levitt J. Resolution of pretibial myxedema with teprotumumab in a patient with Graves disease. JAAD Case Reports. 2020;6:1281-1282. doi:10.1016/j.jdcr.2020.09.003
A 70-year-old woman presented with pain and swelling in both legs of many years’ duration. She had no history of skin disease. Physical examination revealed shiny indurated plaques on the legs, ankles, and toes with limited range of motion in the ankles (top). Marked thickening of the hands and index fingers also was noted (bottom). A punch biopsy of the distal pretibial region was performed.

