User login
Conservative Approach to Treatment of Cyclosporine-Induced Gingival Hyperplasia With Azithromycin and Chlorhexidine
Conservative Approach to Treatment of Cyclosporine-Induced Gingival Hyperplasia With Azithromycin and Chlorhexidine
Cyclosporine is a calcineurin inhibitor and immunosuppressive medication with several indications, including prevention of parenchymal organ and bone marrow transplant rejection as well as treatment of numerous dermatologic conditions (eg, psoriasis, atopic dermatitis). Although it is an effective medication, there are many known adverse effects including nephrotoxicity, hypertension, and gingival hyperplasia.1 Addressing symptomatic cyclosporine-induced gingival hyperplasia can be challenging, especially if continued use of cyclosporine is necessary for adequate control of the underlying disease. We present a simplified approach for conservative management of cyclosporine-induced gingival hyperplasia that allows for continued use of cyclosporine.
Practice Gap
Cyclosporine-induced gingival hyperplasia is a fibrous overgrowth of the interdental papilla and labial gingiva that may lead to gum pain, difficulty eating, gingivitis, and/ or tooth decay or loss.2 The condition usually occurs 3 to 6 months after starting cyclosporine but may occur as soon as 1 month later.1,3 The pathophysiology of this adverse effect is incompletely understood, but several mechanisms have been implicated, including upregulation of the salivary proinflammatory cytokines IL-1α, IL-8, and IL-6.1 Additionally, patients with cyclosporine-induced gingival hyperplasia have increased bacterial colonization with species such as Porphyromonas gingivalis.4 Risk factors for cyclosporine- induced gingival hyperplasia include higher serum concentrations (>400 ng/mL) of cyclosporine, history of gingival hyperplasia, concomitant use of calcium channel blockers, and insufficient oral hygiene.2,3 A study by Seymour and Smith5 found that proper oral hygiene leads to less severe cases of cyclosporine-induced gingival hyperplasia but does not prevent gingival overgrowth. Treatment of cyclosporine-induced gingival hyperplasia traditionally involves targeting oral bacteria and reducing inflammation. Decreasing dental plaque through regular tooth-brushing and interdental cleaning may reduce symptoms such as bleeding and discomfort of the gums.
The intensity of cyclosporine-induced gingival hyperplasia can be reduced with chlorhexidine or azithromycin. Individually, each therapy has been shown to clinically improve cyclosporine-induced gingival hyperplasia; however, to our knowledge the combination of these treatments has not been reported.1 We present a simplified approach to treating cyclosporine-induced gingival hyperplasia using both azithromycin and chlorhexidine. This conservative approach results in effective and sustained improvement of gingival hyperplasia while allowing patients to continue cyclosporine therapy to control underlying disease with minimal adverse effects.
Technique
Before initiating treatment, it is important to confirm that the etiology of gingival hyperplasia is due to cyclosporine use and rule out nutritional deficiencies and autoimmune conditions as potential causes. Be sure to inquire about nutritional intake, systemic symptoms, and family history of autoimmune conditions. Our approach includes the use of azithromycin 500 mg once daily for 7 days followed by chlorhexidine 0.12% oral solution 15 mL twice daily (swish undiluted for 30 seconds, then spit) for at least 3 months for optimal management of gingival hyperplasia. Chlorhexidine should be continued for at least 6 months to maintain symptom resolution. While cyclosporine therapy may be continued throughout the duration of this regimen, consider switching to other immunosuppressive medications that are not associated with gingival hyperplasia (eg, tacrolimus) if symptoms are severe and/or resistant to therapy.1,6
We applied this technique to treat cyclosporine-induced gingival hyperplasia in a 28-year-old woman with a 3-year history of primary aplastic anemia. The patient initially presented with pain and bleeding of the gums of several months’ duration and reported experiencing gum pain when eating solid foods. Her medications included cyclosporine 225 mg daily for aplastic anemia and dapsone 100 mg daily for pneumocystis pneumonia prophylaxis, both of which were taken for the past 6 months. Oral examination revealed pink to bright red hyperplastic gingivae (Figure). She had no other symptoms associated with aplastic anemia and no signs of vitamin or nutritional deficiencies. She denied pre-existing periodontitis prior to starting cyclosporine and reported that the symptoms started several months after initiating cyclosporine therapy. Thus, the clinical diagnosis of cyclosporine-induced gingival hyperplasia was made, and treatment with azithromycin and chlorhexidine was initiated with marked reduction in symptoms.

Conservative management of gingival hyperplasia with oral hygiene including regular tooth-brushing and flossing and antimicrobial therapies was preferred in this patient to reduce gum pain and minimize the risk for tooth loss while also limiting the use of surgically invasive interventions. Due to limited therapeutic options for aplastic anemia, continued administration of cyclosporine was necessary in our patient to prevent further complications.
Practice Implications
The precise mechanism by which azithromycin treats gingival hyperplasia is unclear but may involve its antimicrobial and anti-inflammatory properties. Small concentrations of azithromycin have been shown to persist in macrophages and fibroblasts of the gingiva even with short-term administration of 3 to 5 days.7 Chlorhexidine is another antimicrobial agent often used in oral rinse solutions to decrease plaque formation and prevent gingivitis. Chlorhexidine can reduce cyclosporine-induced gingival overgrowth when used twice daily.8 After rinsing with chlorhexidine, saliva exhibits antibacterial activity for up to 5 hours; however, tooth and gum discoloration may occur.8
Recurrence of gingival hyperplasia is likely if cyclosporine is not discontinued or maintained with treatment.3 Conventional gingivectomy should be considered for cases in which conservative treatment is ineffective, aesthetic concerns arise, or gingival hyperplasia persists for more than 6 to 12 months after discontinuing cyclosporine.1
We theorize that the microbial properties of azithromycin and chlorhexidine help reduce periodontal inflammation and bacterial overgrowth in patients with cyclosporine-induced gingival hyperplasia, which allows for restoration of gingival health. Our case highlights the efficacy of our treatment approach using a 7-day course of azithromycin followed by twice-daily use of chlorhexidine oral rinse in the treatment of cyclosporine-induced gingival hyperplasia with continued use of cyclosporine.
- Chojnacka-Purpurowicz J, Wygonowska E, Placek W, et al. Cyclosporine-induced gingival overgrowth—review. Dermatol Ther. 2022;35:E15912.
- Greenburg KV, Armitage GC, Shiboski CH. Gingival enlargement among renal transplant recipients in the era of new-generation immunosuppressants. J Periodontol. 2008;79:453-460.
- Cyclosporine (ciclosporin)(systemic): drug information. UpToDate. Accessed December 19, 2023. https://www.uptodate.com/contents/table-of-contents/drug-information/general-drug-information
- Gong Y, Bi W, Cao L, et al. Association of CD14-260 polymorphisms, red-complex periodontopathogens and gingival crevicular fluid cytokine levels with cyclosporine A-induced gingival overgrowth in renal transplant patients. J Periodontal Res. 2013;48:203-212.
- Seymour RA, Smith DG. The effect of a plaque control programme on the incidence and severity of cyclosporin-induced gingival changes. J Clin Periodontol. 1991;18:107-110.
- Nash MM, Zaltzman JS. Efficacy of azithromycin in the treatment of cyclosporine-induced gingival hyperplasia in renal transplant recipients. Transplantation. 1998;65:1611-1615.
- Martín JM, Mateo E, Jordá E. Utilidad de la azitromicina en la hyperplasia gingival inducida por ciclosporina [azithromycin for the treatment of ciclosporin-induced gingival hyperplasia]. Actas Dermosifiliogr. 2016;107:780.
- Gau CH, Tu HS, Chin YT, et al. Can chlorhexidine mouthwash twice daily ameliorate cyclosporine-induced gingival overgrowth? J Formos Med Assoc. 2013;112:131-137.
Cyclosporine is a calcineurin inhibitor and immunosuppressive medication with several indications, including prevention of parenchymal organ and bone marrow transplant rejection as well as treatment of numerous dermatologic conditions (eg, psoriasis, atopic dermatitis). Although it is an effective medication, there are many known adverse effects including nephrotoxicity, hypertension, and gingival hyperplasia.1 Addressing symptomatic cyclosporine-induced gingival hyperplasia can be challenging, especially if continued use of cyclosporine is necessary for adequate control of the underlying disease. We present a simplified approach for conservative management of cyclosporine-induced gingival hyperplasia that allows for continued use of cyclosporine.
Practice Gap
Cyclosporine-induced gingival hyperplasia is a fibrous overgrowth of the interdental papilla and labial gingiva that may lead to gum pain, difficulty eating, gingivitis, and/ or tooth decay or loss.2 The condition usually occurs 3 to 6 months after starting cyclosporine but may occur as soon as 1 month later.1,3 The pathophysiology of this adverse effect is incompletely understood, but several mechanisms have been implicated, including upregulation of the salivary proinflammatory cytokines IL-1α, IL-8, and IL-6.1 Additionally, patients with cyclosporine-induced gingival hyperplasia have increased bacterial colonization with species such as Porphyromonas gingivalis.4 Risk factors for cyclosporine- induced gingival hyperplasia include higher serum concentrations (>400 ng/mL) of cyclosporine, history of gingival hyperplasia, concomitant use of calcium channel blockers, and insufficient oral hygiene.2,3 A study by Seymour and Smith5 found that proper oral hygiene leads to less severe cases of cyclosporine-induced gingival hyperplasia but does not prevent gingival overgrowth. Treatment of cyclosporine-induced gingival hyperplasia traditionally involves targeting oral bacteria and reducing inflammation. Decreasing dental plaque through regular tooth-brushing and interdental cleaning may reduce symptoms such as bleeding and discomfort of the gums.
The intensity of cyclosporine-induced gingival hyperplasia can be reduced with chlorhexidine or azithromycin. Individually, each therapy has been shown to clinically improve cyclosporine-induced gingival hyperplasia; however, to our knowledge the combination of these treatments has not been reported.1 We present a simplified approach to treating cyclosporine-induced gingival hyperplasia using both azithromycin and chlorhexidine. This conservative approach results in effective and sustained improvement of gingival hyperplasia while allowing patients to continue cyclosporine therapy to control underlying disease with minimal adverse effects.
Technique
Before initiating treatment, it is important to confirm that the etiology of gingival hyperplasia is due to cyclosporine use and rule out nutritional deficiencies and autoimmune conditions as potential causes. Be sure to inquire about nutritional intake, systemic symptoms, and family history of autoimmune conditions. Our approach includes the use of azithromycin 500 mg once daily for 7 days followed by chlorhexidine 0.12% oral solution 15 mL twice daily (swish undiluted for 30 seconds, then spit) for at least 3 months for optimal management of gingival hyperplasia. Chlorhexidine should be continued for at least 6 months to maintain symptom resolution. While cyclosporine therapy may be continued throughout the duration of this regimen, consider switching to other immunosuppressive medications that are not associated with gingival hyperplasia (eg, tacrolimus) if symptoms are severe and/or resistant to therapy.1,6
We applied this technique to treat cyclosporine-induced gingival hyperplasia in a 28-year-old woman with a 3-year history of primary aplastic anemia. The patient initially presented with pain and bleeding of the gums of several months’ duration and reported experiencing gum pain when eating solid foods. Her medications included cyclosporine 225 mg daily for aplastic anemia and dapsone 100 mg daily for pneumocystis pneumonia prophylaxis, both of which were taken for the past 6 months. Oral examination revealed pink to bright red hyperplastic gingivae (Figure). She had no other symptoms associated with aplastic anemia and no signs of vitamin or nutritional deficiencies. She denied pre-existing periodontitis prior to starting cyclosporine and reported that the symptoms started several months after initiating cyclosporine therapy. Thus, the clinical diagnosis of cyclosporine-induced gingival hyperplasia was made, and treatment with azithromycin and chlorhexidine was initiated with marked reduction in symptoms.
Conservative management of gingival hyperplasia with oral hygiene including regular tooth-brushing and flossing and antimicrobial therapies was preferred in this patient to reduce gum pain and minimize the risk for tooth loss while also limiting the use of surgically invasive interventions. Due to limited therapeutic options for aplastic anemia, continued administration of cyclosporine was necessary in our patient to prevent further complications.
Practice Implications
The precise mechanism by which azithromycin treats gingival hyperplasia is unclear but may involve its antimicrobial and anti-inflammatory properties. Small concentrations of azithromycin have been shown to persist in macrophages and fibroblasts of the gingiva even with short-term administration of 3 to 5 days.7 Chlorhexidine is another antimicrobial agent often used in oral rinse solutions to decrease plaque formation and prevent gingivitis. Chlorhexidine can reduce cyclosporine-induced gingival overgrowth when used twice daily.8 After rinsing with chlorhexidine, saliva exhibits antibacterial activity for up to 5 hours; however, tooth and gum discoloration may occur.8
Recurrence of gingival hyperplasia is likely if cyclosporine is not discontinued or maintained with treatment.3 Conventional gingivectomy should be considered for cases in which conservative treatment is ineffective, aesthetic concerns arise, or gingival hyperplasia persists for more than 6 to 12 months after discontinuing cyclosporine.1
We theorize that the microbial properties of azithromycin and chlorhexidine help reduce periodontal inflammation and bacterial overgrowth in patients with cyclosporine-induced gingival hyperplasia, which allows for restoration of gingival health. Our case highlights the efficacy of our treatment approach using a 7-day course of azithromycin followed by twice-daily use of chlorhexidine oral rinse in the treatment of cyclosporine-induced gingival hyperplasia with continued use of cyclosporine.
Cyclosporine is a calcineurin inhibitor and immunosuppressive medication with several indications, including prevention of parenchymal organ and bone marrow transplant rejection as well as treatment of numerous dermatologic conditions (eg, psoriasis, atopic dermatitis). Although it is an effective medication, there are many known adverse effects including nephrotoxicity, hypertension, and gingival hyperplasia.1 Addressing symptomatic cyclosporine-induced gingival hyperplasia can be challenging, especially if continued use of cyclosporine is necessary for adequate control of the underlying disease. We present a simplified approach for conservative management of cyclosporine-induced gingival hyperplasia that allows for continued use of cyclosporine.
Practice Gap
Cyclosporine-induced gingival hyperplasia is a fibrous overgrowth of the interdental papilla and labial gingiva that may lead to gum pain, difficulty eating, gingivitis, and/ or tooth decay or loss.2 The condition usually occurs 3 to 6 months after starting cyclosporine but may occur as soon as 1 month later.1,3 The pathophysiology of this adverse effect is incompletely understood, but several mechanisms have been implicated, including upregulation of the salivary proinflammatory cytokines IL-1α, IL-8, and IL-6.1 Additionally, patients with cyclosporine-induced gingival hyperplasia have increased bacterial colonization with species such as Porphyromonas gingivalis.4 Risk factors for cyclosporine- induced gingival hyperplasia include higher serum concentrations (>400 ng/mL) of cyclosporine, history of gingival hyperplasia, concomitant use of calcium channel blockers, and insufficient oral hygiene.2,3 A study by Seymour and Smith5 found that proper oral hygiene leads to less severe cases of cyclosporine-induced gingival hyperplasia but does not prevent gingival overgrowth. Treatment of cyclosporine-induced gingival hyperplasia traditionally involves targeting oral bacteria and reducing inflammation. Decreasing dental plaque through regular tooth-brushing and interdental cleaning may reduce symptoms such as bleeding and discomfort of the gums.
The intensity of cyclosporine-induced gingival hyperplasia can be reduced with chlorhexidine or azithromycin. Individually, each therapy has been shown to clinically improve cyclosporine-induced gingival hyperplasia; however, to our knowledge the combination of these treatments has not been reported.1 We present a simplified approach to treating cyclosporine-induced gingival hyperplasia using both azithromycin and chlorhexidine. This conservative approach results in effective and sustained improvement of gingival hyperplasia while allowing patients to continue cyclosporine therapy to control underlying disease with minimal adverse effects.
Technique
Before initiating treatment, it is important to confirm that the etiology of gingival hyperplasia is due to cyclosporine use and rule out nutritional deficiencies and autoimmune conditions as potential causes. Be sure to inquire about nutritional intake, systemic symptoms, and family history of autoimmune conditions. Our approach includes the use of azithromycin 500 mg once daily for 7 days followed by chlorhexidine 0.12% oral solution 15 mL twice daily (swish undiluted for 30 seconds, then spit) for at least 3 months for optimal management of gingival hyperplasia. Chlorhexidine should be continued for at least 6 months to maintain symptom resolution. While cyclosporine therapy may be continued throughout the duration of this regimen, consider switching to other immunosuppressive medications that are not associated with gingival hyperplasia (eg, tacrolimus) if symptoms are severe and/or resistant to therapy.1,6
We applied this technique to treat cyclosporine-induced gingival hyperplasia in a 28-year-old woman with a 3-year history of primary aplastic anemia. The patient initially presented with pain and bleeding of the gums of several months’ duration and reported experiencing gum pain when eating solid foods. Her medications included cyclosporine 225 mg daily for aplastic anemia and dapsone 100 mg daily for pneumocystis pneumonia prophylaxis, both of which were taken for the past 6 months. Oral examination revealed pink to bright red hyperplastic gingivae (Figure). She had no other symptoms associated with aplastic anemia and no signs of vitamin or nutritional deficiencies. She denied pre-existing periodontitis prior to starting cyclosporine and reported that the symptoms started several months after initiating cyclosporine therapy. Thus, the clinical diagnosis of cyclosporine-induced gingival hyperplasia was made, and treatment with azithromycin and chlorhexidine was initiated with marked reduction in symptoms.
Conservative management of gingival hyperplasia with oral hygiene including regular tooth-brushing and flossing and antimicrobial therapies was preferred in this patient to reduce gum pain and minimize the risk for tooth loss while also limiting the use of surgically invasive interventions. Due to limited therapeutic options for aplastic anemia, continued administration of cyclosporine was necessary in our patient to prevent further complications.
Practice Implications
The precise mechanism by which azithromycin treats gingival hyperplasia is unclear but may involve its antimicrobial and anti-inflammatory properties. Small concentrations of azithromycin have been shown to persist in macrophages and fibroblasts of the gingiva even with short-term administration of 3 to 5 days.7 Chlorhexidine is another antimicrobial agent often used in oral rinse solutions to decrease plaque formation and prevent gingivitis. Chlorhexidine can reduce cyclosporine-induced gingival overgrowth when used twice daily.8 After rinsing with chlorhexidine, saliva exhibits antibacterial activity for up to 5 hours; however, tooth and gum discoloration may occur.8
Recurrence of gingival hyperplasia is likely if cyclosporine is not discontinued or maintained with treatment.3 Conventional gingivectomy should be considered for cases in which conservative treatment is ineffective, aesthetic concerns arise, or gingival hyperplasia persists for more than 6 to 12 months after discontinuing cyclosporine.1
We theorize that the microbial properties of azithromycin and chlorhexidine help reduce periodontal inflammation and bacterial overgrowth in patients with cyclosporine-induced gingival hyperplasia, which allows for restoration of gingival health. Our case highlights the efficacy of our treatment approach using a 7-day course of azithromycin followed by twice-daily use of chlorhexidine oral rinse in the treatment of cyclosporine-induced gingival hyperplasia with continued use of cyclosporine.
- Chojnacka-Purpurowicz J, Wygonowska E, Placek W, et al. Cyclosporine-induced gingival overgrowth—review. Dermatol Ther. 2022;35:E15912.
- Greenburg KV, Armitage GC, Shiboski CH. Gingival enlargement among renal transplant recipients in the era of new-generation immunosuppressants. J Periodontol. 2008;79:453-460.
- Cyclosporine (ciclosporin)(systemic): drug information. UpToDate. Accessed December 19, 2023. https://www.uptodate.com/contents/table-of-contents/drug-information/general-drug-information
- Gong Y, Bi W, Cao L, et al. Association of CD14-260 polymorphisms, red-complex periodontopathogens and gingival crevicular fluid cytokine levels with cyclosporine A-induced gingival overgrowth in renal transplant patients. J Periodontal Res. 2013;48:203-212.
- Seymour RA, Smith DG. The effect of a plaque control programme on the incidence and severity of cyclosporin-induced gingival changes. J Clin Periodontol. 1991;18:107-110.
- Nash MM, Zaltzman JS. Efficacy of azithromycin in the treatment of cyclosporine-induced gingival hyperplasia in renal transplant recipients. Transplantation. 1998;65:1611-1615.
- Martín JM, Mateo E, Jordá E. Utilidad de la azitromicina en la hyperplasia gingival inducida por ciclosporina [azithromycin for the treatment of ciclosporin-induced gingival hyperplasia]. Actas Dermosifiliogr. 2016;107:780.
- Gau CH, Tu HS, Chin YT, et al. Can chlorhexidine mouthwash twice daily ameliorate cyclosporine-induced gingival overgrowth? J Formos Med Assoc. 2013;112:131-137.
- Chojnacka-Purpurowicz J, Wygonowska E, Placek W, et al. Cyclosporine-induced gingival overgrowth—review. Dermatol Ther. 2022;35:E15912.
- Greenburg KV, Armitage GC, Shiboski CH. Gingival enlargement among renal transplant recipients in the era of new-generation immunosuppressants. J Periodontol. 2008;79:453-460.
- Cyclosporine (ciclosporin)(systemic): drug information. UpToDate. Accessed December 19, 2023. https://www.uptodate.com/contents/table-of-contents/drug-information/general-drug-information
- Gong Y, Bi W, Cao L, et al. Association of CD14-260 polymorphisms, red-complex periodontopathogens and gingival crevicular fluid cytokine levels with cyclosporine A-induced gingival overgrowth in renal transplant patients. J Periodontal Res. 2013;48:203-212.
- Seymour RA, Smith DG. The effect of a plaque control programme on the incidence and severity of cyclosporin-induced gingival changes. J Clin Periodontol. 1991;18:107-110.
- Nash MM, Zaltzman JS. Efficacy of azithromycin in the treatment of cyclosporine-induced gingival hyperplasia in renal transplant recipients. Transplantation. 1998;65:1611-1615.
- Martín JM, Mateo E, Jordá E. Utilidad de la azitromicina en la hyperplasia gingival inducida por ciclosporina [azithromycin for the treatment of ciclosporin-induced gingival hyperplasia]. Actas Dermosifiliogr. 2016;107:780.
- Gau CH, Tu HS, Chin YT, et al. Can chlorhexidine mouthwash twice daily ameliorate cyclosporine-induced gingival overgrowth? J Formos Med Assoc. 2013;112:131-137.
Conservative Approach to Treatment of Cyclosporine-Induced Gingival Hyperplasia With Azithromycin and Chlorhexidine
Conservative Approach to Treatment of Cyclosporine-Induced Gingival Hyperplasia With Azithromycin and Chlorhexidine

Need a Wood Lamp Alternative? Grab Your Smartphone
Practice Gap
The Wood lamp commonly is used as a diagnostic tool for pigmentary skin conditions (eg, vitiligo) or skin conditions that exhibit fluorescence (eg, erythrasma).1 Recently, its diagnostic efficacy has extended to scabies, in which it unveils a distinctive wavy, bluish-white, linear fluorescence upon illumination.2
Functionally, the Wood lamp operates by subjecting phosphors to UV light within the wavelength range of 320 to 400 nm, inducing fluorescence in substances such as collagen and elastin. In the context of vitiligo, this process manifests as a preferential chalk white fluorescence in areas lacking melanin.1
Despite its demonstrated effectiveness, the Wood lamp is not without limitations. It comes with a notable financial investment ranging from $70 to $500, requires periodic maintenance such as light bulb replacements, and can be unwieldy.3 Furthermore, its reliance on a power source poses a challenge in settings where immediate access to convenient power outlets is limited, such as inpatient and rural dermatology clinics. These limitations underscore the need for alternative solutions and innovations to address challenges and ensure accessibility in diverse health care environments.
The Tools
Free smartphone applications (apps), such as Ultraviolet Light-UV Lamp by AppBrain or Blacklight UV Light Simulator by That Smile, can simulate UV light and functionally serve as a Wood lamp.
The Technique
UV light apps use LED or organic LED screen pixels to emit a blue light equivalent at 467 nm.4 Although these apps are not designed specifically for dermatologic uses, they are mostly free, widely available for Android and iPhone users, and portable. Importantly, they can demonstrate good performance in visualizing vitiligo, as shown in Figure 1—albeit perhaps not reaching the same level as the Wood lamp (Figure 2).
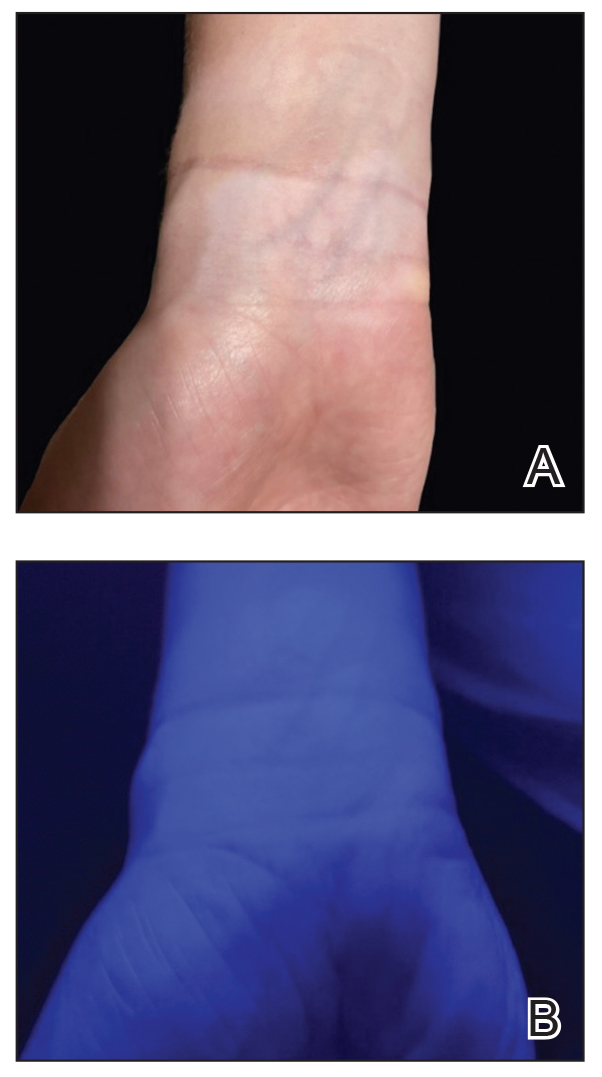
Because these UV light apps are not regulated and their efficacy for medical use has not been firmly established, the Wood lamp remains the gold standard. Therefore, we propose the use of UV light apps in situations when a Wood lamp is not available or convenient, such as in rural, inpatient, or international health care settings.
Practice Implications
Exploring and adopting these free alternatives can contribute to improved accessibility and diagnostic capabilities in diverse health care environments, particularly for communities facing financial constraints. Continued research and validation of these apps in clinical settings will be essential to establish their reliability and effectiveness in enhancing diagnostic practices.
- Dyer JM, Foy VM. Revealing the unseen: a review of Wood’s lamp in dermatology. J Clin Aesthet Dermatol. 2022;15:25-30.
- Scanni G. Facilitations in the clinical diagnosis of human scabies through the use of ultraviolet light (UV-scab scanning): a case-series study. Trop Med Infect Dis. 2022;7:422. doi:10.3390/tropicalmed7120422
- USA Medical and Surgical Supplies. Top 9 medical diagnostic applications for a Woods lamp. February 26, 2019. Accessed May 20, 2024.
- Huang Y, Hsiang E-L, Deng M-Y, et al. Mini-led, micro-led and OLED displays: present status and future perspectives. Light Sci Appl. 2020;9:105. doi:10.1038/s41377-020-0341-9
Practice Gap
The Wood lamp commonly is used as a diagnostic tool for pigmentary skin conditions (eg, vitiligo) or skin conditions that exhibit fluorescence (eg, erythrasma).1 Recently, its diagnostic efficacy has extended to scabies, in which it unveils a distinctive wavy, bluish-white, linear fluorescence upon illumination.2
Functionally, the Wood lamp operates by subjecting phosphors to UV light within the wavelength range of 320 to 400 nm, inducing fluorescence in substances such as collagen and elastin. In the context of vitiligo, this process manifests as a preferential chalk white fluorescence in areas lacking melanin.1
Despite its demonstrated effectiveness, the Wood lamp is not without limitations. It comes with a notable financial investment ranging from $70 to $500, requires periodic maintenance such as light bulb replacements, and can be unwieldy.3 Furthermore, its reliance on a power source poses a challenge in settings where immediate access to convenient power outlets is limited, such as inpatient and rural dermatology clinics. These limitations underscore the need for alternative solutions and innovations to address challenges and ensure accessibility in diverse health care environments.
The Tools
Free smartphone applications (apps), such as Ultraviolet Light-UV Lamp by AppBrain or Blacklight UV Light Simulator by That Smile, can simulate UV light and functionally serve as a Wood lamp.
The Technique
UV light apps use LED or organic LED screen pixels to emit a blue light equivalent at 467 nm.4 Although these apps are not designed specifically for dermatologic uses, they are mostly free, widely available for Android and iPhone users, and portable. Importantly, they can demonstrate good performance in visualizing vitiligo, as shown in Figure 1—albeit perhaps not reaching the same level as the Wood lamp (Figure 2).
Because these UV light apps are not regulated and their efficacy for medical use has not been firmly established, the Wood lamp remains the gold standard. Therefore, we propose the use of UV light apps in situations when a Wood lamp is not available or convenient, such as in rural, inpatient, or international health care settings.
Practice Implications
Exploring and adopting these free alternatives can contribute to improved accessibility and diagnostic capabilities in diverse health care environments, particularly for communities facing financial constraints. Continued research and validation of these apps in clinical settings will be essential to establish their reliability and effectiveness in enhancing diagnostic practices.
Practice Gap
The Wood lamp commonly is used as a diagnostic tool for pigmentary skin conditions (eg, vitiligo) or skin conditions that exhibit fluorescence (eg, erythrasma).1 Recently, its diagnostic efficacy has extended to scabies, in which it unveils a distinctive wavy, bluish-white, linear fluorescence upon illumination.2
Functionally, the Wood lamp operates by subjecting phosphors to UV light within the wavelength range of 320 to 400 nm, inducing fluorescence in substances such as collagen and elastin. In the context of vitiligo, this process manifests as a preferential chalk white fluorescence in areas lacking melanin.1
Despite its demonstrated effectiveness, the Wood lamp is not without limitations. It comes with a notable financial investment ranging from $70 to $500, requires periodic maintenance such as light bulb replacements, and can be unwieldy.3 Furthermore, its reliance on a power source poses a challenge in settings where immediate access to convenient power outlets is limited, such as inpatient and rural dermatology clinics. These limitations underscore the need for alternative solutions and innovations to address challenges and ensure accessibility in diverse health care environments.
The Tools
Free smartphone applications (apps), such as Ultraviolet Light-UV Lamp by AppBrain or Blacklight UV Light Simulator by That Smile, can simulate UV light and functionally serve as a Wood lamp.
The Technique
UV light apps use LED or organic LED screen pixels to emit a blue light equivalent at 467 nm.4 Although these apps are not designed specifically for dermatologic uses, they are mostly free, widely available for Android and iPhone users, and portable. Importantly, they can demonstrate good performance in visualizing vitiligo, as shown in Figure 1—albeit perhaps not reaching the same level as the Wood lamp (Figure 2).
Because these UV light apps are not regulated and their efficacy for medical use has not been firmly established, the Wood lamp remains the gold standard. Therefore, we propose the use of UV light apps in situations when a Wood lamp is not available or convenient, such as in rural, inpatient, or international health care settings.
Practice Implications
Exploring and adopting these free alternatives can contribute to improved accessibility and diagnostic capabilities in diverse health care environments, particularly for communities facing financial constraints. Continued research and validation of these apps in clinical settings will be essential to establish their reliability and effectiveness in enhancing diagnostic practices.
- Dyer JM, Foy VM. Revealing the unseen: a review of Wood’s lamp in dermatology. J Clin Aesthet Dermatol. 2022;15:25-30.
- Scanni G. Facilitations in the clinical diagnosis of human scabies through the use of ultraviolet light (UV-scab scanning): a case-series study. Trop Med Infect Dis. 2022;7:422. doi:10.3390/tropicalmed7120422
- USA Medical and Surgical Supplies. Top 9 medical diagnostic applications for a Woods lamp. February 26, 2019. Accessed May 20, 2024.
- Huang Y, Hsiang E-L, Deng M-Y, et al. Mini-led, micro-led and OLED displays: present status and future perspectives. Light Sci Appl. 2020;9:105. doi:10.1038/s41377-020-0341-9
- Dyer JM, Foy VM. Revealing the unseen: a review of Wood’s lamp in dermatology. J Clin Aesthet Dermatol. 2022;15:25-30.
- Scanni G. Facilitations in the clinical diagnosis of human scabies through the use of ultraviolet light (UV-scab scanning): a case-series study. Trop Med Infect Dis. 2022;7:422. doi:10.3390/tropicalmed7120422
- USA Medical and Surgical Supplies. Top 9 medical diagnostic applications for a Woods lamp. February 26, 2019. Accessed May 20, 2024.
- Huang Y, Hsiang E-L, Deng M-Y, et al. Mini-led, micro-led and OLED displays: present status and future perspectives. Light Sci Appl. 2020;9:105. doi:10.1038/s41377-020-0341-9
Purpuric Eruption in a Patient With Hairy Cell Leukemia
The Diagnosis: Purpuric Drug Eruption
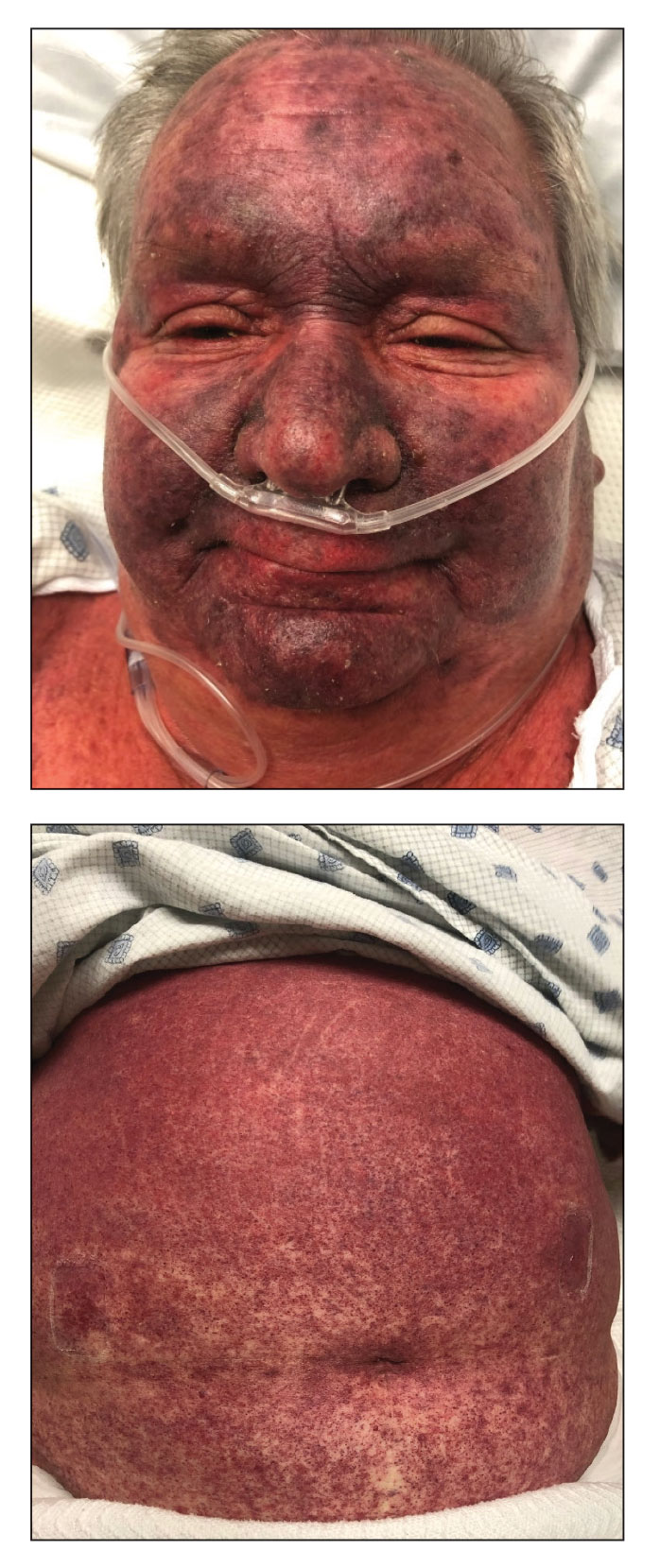
Histopathology revealed interface dermatitis, spongiosis, and a perivascular lymphocytic infiltrate with extravasated red blood cells consistent with a purpuric drug eruption. Our patient achieved remission of hairy cell leukemia after receiving only 2 of 5 expected doses of cladribine. The rash resolved completely in 3 weeks following a prednisone taper (Figure).
Hairy cell leukemia is a rare indolent lymphoproliferative disorder of B cells that accounts for approximately 2% of adult leukemias in the United States. Cladribine, a purine nucleoside analog that impairs DNA synthesis and repair, has become the mainstay of therapy, demonstrating a 95% complete response rate.1 Although few reports have addressed the cutaneous reactions seen with cladribine therapy, they can occur in more than 50% of patients.1,2 The most common skin manifestation associated with cladribine therapy is a morbilliform rash, but Stevens-Johnson syndrome and toxic epidermal necrolysis (TEN) have been reported.1
Few cases of purpuric eruption secondary to cladribine treatment have been described, and nearly all reports involve concomitant medications such as allopurinol, which our patient was taking, and antibiotics including trimethoprim-sulfamethoxazole and penicillins.1,3,4 In a cohort of 35 patients receiving cladribine,1 only concomitant treatment with cladribine and allopurinol caused cutaneous reactions, further supporting the hypothesis of cladribine-induced drug sensitivity. Allopurinol often is prescribed during induction therapy for prophylaxis against tumor lysis syndrome; similarly, antibiotics frequently are given prophylactically and therapeutically for neutropenic fever. It is believed that T-cell imbalance and profound lymphopenia induced by cladribine increase susceptibility to drug hypersensitivity reactions.1,3
The typical purpuric eruption develops within 2 days of starting cladribine therapy. Diascopy will reveal petechiae, and biopsy should be performed to rule out other serious drug-induced reactions, such as erythema multiforme, Stevens-Johnson syndrome, and TEN. A cladribine-induced purpuric eruption typically is self-resolving and carries a favorable prognosis, though high-dose corticosteroids often are prescribed to hasten recovery. The rare reports of serious cutaneous reactions secondary to cladribine therapy have been with maculopapular, not purpuric eruptions.2 Based on limited available data, cladribine-induced purpura should not be a limitation to continued treatment in patients who need it.1 Careful consideration of concomitant drug use is necessary, as the current literature demonstrates resolution of rash with withdrawal of other therapies, namely allopurinol.2-4 Future studies are needed to examine the safety of withholding offending medications and to further elucidate the mechanisms contributing to drug hypersensitivity due to cladribine.
Widespread purpura and petechiae can pose a wide differential; the patient’s recent history of cladribine administration pointed to a classic purpuric eruption. Other diagnoses such as toxic erythema of chemotherapy (TEC) and TEN are not purpuric, though plaques can be violaceous. Lack of bullae, blisters, and facial or mucosal surface involvement suggest TEN.5 Thrombotic thrombocytopenic purpura and disseminated intravascular coagulation do manifest with petechiae and purpura, though such a robust eruption in the context of recent cladribine therapy is less likely. The classic retiform purpura and necrosis were not present to suggest purpura fulminans from disseminated intravascular coagulation.
Several of the proposed diagnoses as well as a purpuric drug eruption would demonstrate extravasated red blood cells on histopathology, but the presence of interface dermatitis narrows the differential to a purpuric drug eruption. Necrotic keratinocytes and full-thickness necrosis were not present on biopsy to support a diagnosis of TEN in our patient. Characteristic features of TEC—including eccrine squamous syringometaplasia, dermal edema, and keratinocyte atypia—were not present on biopsy.6 Finally, although TEN should resolve with steroid treatment, TEC is self-limited and thrombotic thrombocytopenic purpura and disseminated intravascular coagulation would not resolve with use of steroids alone.
- Ganzel C, Gatt ME, Maly A, et al. High incidence of skin rash in patients with hairy cell leukemia treated with cladribine. Leuk Lymphoma. 2012;53:1169-1173. doi:10.3109/10428194.2011.635864
- Chubar Y, Bennett M. Cutaneous reactions in hairy cell leukaemia treated with 2-chlorodeoxyadenosine and allopurinol. Br J Haematol. 2003;122:768-770. doi:10.1046/j.1365-2141.2003.04506.x
- Espinosa Lara P, Quirós Redondo V, Aguado Lobo M, et al. Purpuric exanthema in a patient with hairy cell leukemia treated with cladribine and allopurinol. Ann Hematol. 2017;96:1209-1210. doi:10.1007 /s00277-017-2992-z
- Hendrick A. Purpuric rash following treatment with 2-chlorodeoxyadenosine. Clin Lab Haematol. 2001;23:67-68. doi:10.1046 /j.1365-2257.2001.0346b.x
- Kang S, Amagai M, Bruckner AL, et al, eds. Fitzpatrick’s Dermatology. 9th ed. McGraw-Hill Education; 2019.
- Bolognia JL, Cooper DL, Glusac EJ. Toxic erythema of chemotherapy: a useful clinical term. J Am Acad Dermatol. 2008;59:524-529.
The Diagnosis: Purpuric Drug Eruption
Histopathology revealed interface dermatitis, spongiosis, and a perivascular lymphocytic infiltrate with extravasated red blood cells consistent with a purpuric drug eruption. Our patient achieved remission of hairy cell leukemia after receiving only 2 of 5 expected doses of cladribine. The rash resolved completely in 3 weeks following a prednisone taper (Figure).
Hairy cell leukemia is a rare indolent lymphoproliferative disorder of B cells that accounts for approximately 2% of adult leukemias in the United States. Cladribine, a purine nucleoside analog that impairs DNA synthesis and repair, has become the mainstay of therapy, demonstrating a 95% complete response rate.1 Although few reports have addressed the cutaneous reactions seen with cladribine therapy, they can occur in more than 50% of patients.1,2 The most common skin manifestation associated with cladribine therapy is a morbilliform rash, but Stevens-Johnson syndrome and toxic epidermal necrolysis (TEN) have been reported.1
Few cases of purpuric eruption secondary to cladribine treatment have been described, and nearly all reports involve concomitant medications such as allopurinol, which our patient was taking, and antibiotics including trimethoprim-sulfamethoxazole and penicillins.1,3,4 In a cohort of 35 patients receiving cladribine,1 only concomitant treatment with cladribine and allopurinol caused cutaneous reactions, further supporting the hypothesis of cladribine-induced drug sensitivity. Allopurinol often is prescribed during induction therapy for prophylaxis against tumor lysis syndrome; similarly, antibiotics frequently are given prophylactically and therapeutically for neutropenic fever. It is believed that T-cell imbalance and profound lymphopenia induced by cladribine increase susceptibility to drug hypersensitivity reactions.1,3
The typical purpuric eruption develops within 2 days of starting cladribine therapy. Diascopy will reveal petechiae, and biopsy should be performed to rule out other serious drug-induced reactions, such as erythema multiforme, Stevens-Johnson syndrome, and TEN. A cladribine-induced purpuric eruption typically is self-resolving and carries a favorable prognosis, though high-dose corticosteroids often are prescribed to hasten recovery. The rare reports of serious cutaneous reactions secondary to cladribine therapy have been with maculopapular, not purpuric eruptions.2 Based on limited available data, cladribine-induced purpura should not be a limitation to continued treatment in patients who need it.1 Careful consideration of concomitant drug use is necessary, as the current literature demonstrates resolution of rash with withdrawal of other therapies, namely allopurinol.2-4 Future studies are needed to examine the safety of withholding offending medications and to further elucidate the mechanisms contributing to drug hypersensitivity due to cladribine.
Widespread purpura and petechiae can pose a wide differential; the patient’s recent history of cladribine administration pointed to a classic purpuric eruption. Other diagnoses such as toxic erythema of chemotherapy (TEC) and TEN are not purpuric, though plaques can be violaceous. Lack of bullae, blisters, and facial or mucosal surface involvement suggest TEN.5 Thrombotic thrombocytopenic purpura and disseminated intravascular coagulation do manifest with petechiae and purpura, though such a robust eruption in the context of recent cladribine therapy is less likely. The classic retiform purpura and necrosis were not present to suggest purpura fulminans from disseminated intravascular coagulation.
Several of the proposed diagnoses as well as a purpuric drug eruption would demonstrate extravasated red blood cells on histopathology, but the presence of interface dermatitis narrows the differential to a purpuric drug eruption. Necrotic keratinocytes and full-thickness necrosis were not present on biopsy to support a diagnosis of TEN in our patient. Characteristic features of TEC—including eccrine squamous syringometaplasia, dermal edema, and keratinocyte atypia—were not present on biopsy.6 Finally, although TEN should resolve with steroid treatment, TEC is self-limited and thrombotic thrombocytopenic purpura and disseminated intravascular coagulation would not resolve with use of steroids alone.
The Diagnosis: Purpuric Drug Eruption
Histopathology revealed interface dermatitis, spongiosis, and a perivascular lymphocytic infiltrate with extravasated red blood cells consistent with a purpuric drug eruption. Our patient achieved remission of hairy cell leukemia after receiving only 2 of 5 expected doses of cladribine. The rash resolved completely in 3 weeks following a prednisone taper (Figure).
Hairy cell leukemia is a rare indolent lymphoproliferative disorder of B cells that accounts for approximately 2% of adult leukemias in the United States. Cladribine, a purine nucleoside analog that impairs DNA synthesis and repair, has become the mainstay of therapy, demonstrating a 95% complete response rate.1 Although few reports have addressed the cutaneous reactions seen with cladribine therapy, they can occur in more than 50% of patients.1,2 The most common skin manifestation associated with cladribine therapy is a morbilliform rash, but Stevens-Johnson syndrome and toxic epidermal necrolysis (TEN) have been reported.1
Few cases of purpuric eruption secondary to cladribine treatment have been described, and nearly all reports involve concomitant medications such as allopurinol, which our patient was taking, and antibiotics including trimethoprim-sulfamethoxazole and penicillins.1,3,4 In a cohort of 35 patients receiving cladribine,1 only concomitant treatment with cladribine and allopurinol caused cutaneous reactions, further supporting the hypothesis of cladribine-induced drug sensitivity. Allopurinol often is prescribed during induction therapy for prophylaxis against tumor lysis syndrome; similarly, antibiotics frequently are given prophylactically and therapeutically for neutropenic fever. It is believed that T-cell imbalance and profound lymphopenia induced by cladribine increase susceptibility to drug hypersensitivity reactions.1,3
The typical purpuric eruption develops within 2 days of starting cladribine therapy. Diascopy will reveal petechiae, and biopsy should be performed to rule out other serious drug-induced reactions, such as erythema multiforme, Stevens-Johnson syndrome, and TEN. A cladribine-induced purpuric eruption typically is self-resolving and carries a favorable prognosis, though high-dose corticosteroids often are prescribed to hasten recovery. The rare reports of serious cutaneous reactions secondary to cladribine therapy have been with maculopapular, not purpuric eruptions.2 Based on limited available data, cladribine-induced purpura should not be a limitation to continued treatment in patients who need it.1 Careful consideration of concomitant drug use is necessary, as the current literature demonstrates resolution of rash with withdrawal of other therapies, namely allopurinol.2-4 Future studies are needed to examine the safety of withholding offending medications and to further elucidate the mechanisms contributing to drug hypersensitivity due to cladribine.
Widespread purpura and petechiae can pose a wide differential; the patient’s recent history of cladribine administration pointed to a classic purpuric eruption. Other diagnoses such as toxic erythema of chemotherapy (TEC) and TEN are not purpuric, though plaques can be violaceous. Lack of bullae, blisters, and facial or mucosal surface involvement suggest TEN.5 Thrombotic thrombocytopenic purpura and disseminated intravascular coagulation do manifest with petechiae and purpura, though such a robust eruption in the context of recent cladribine therapy is less likely. The classic retiform purpura and necrosis were not present to suggest purpura fulminans from disseminated intravascular coagulation.
Several of the proposed diagnoses as well as a purpuric drug eruption would demonstrate extravasated red blood cells on histopathology, but the presence of interface dermatitis narrows the differential to a purpuric drug eruption. Necrotic keratinocytes and full-thickness necrosis were not present on biopsy to support a diagnosis of TEN in our patient. Characteristic features of TEC—including eccrine squamous syringometaplasia, dermal edema, and keratinocyte atypia—were not present on biopsy.6 Finally, although TEN should resolve with steroid treatment, TEC is self-limited and thrombotic thrombocytopenic purpura and disseminated intravascular coagulation would not resolve with use of steroids alone.
- Ganzel C, Gatt ME, Maly A, et al. High incidence of skin rash in patients with hairy cell leukemia treated with cladribine. Leuk Lymphoma. 2012;53:1169-1173. doi:10.3109/10428194.2011.635864
- Chubar Y, Bennett M. Cutaneous reactions in hairy cell leukaemia treated with 2-chlorodeoxyadenosine and allopurinol. Br J Haematol. 2003;122:768-770. doi:10.1046/j.1365-2141.2003.04506.x
- Espinosa Lara P, Quirós Redondo V, Aguado Lobo M, et al. Purpuric exanthema in a patient with hairy cell leukemia treated with cladribine and allopurinol. Ann Hematol. 2017;96:1209-1210. doi:10.1007 /s00277-017-2992-z
- Hendrick A. Purpuric rash following treatment with 2-chlorodeoxyadenosine. Clin Lab Haematol. 2001;23:67-68. doi:10.1046 /j.1365-2257.2001.0346b.x
- Kang S, Amagai M, Bruckner AL, et al, eds. Fitzpatrick’s Dermatology. 9th ed. McGraw-Hill Education; 2019.
- Bolognia JL, Cooper DL, Glusac EJ. Toxic erythema of chemotherapy: a useful clinical term. J Am Acad Dermatol. 2008;59:524-529.
- Ganzel C, Gatt ME, Maly A, et al. High incidence of skin rash in patients with hairy cell leukemia treated with cladribine. Leuk Lymphoma. 2012;53:1169-1173. doi:10.3109/10428194.2011.635864
- Chubar Y, Bennett M. Cutaneous reactions in hairy cell leukaemia treated with 2-chlorodeoxyadenosine and allopurinol. Br J Haematol. 2003;122:768-770. doi:10.1046/j.1365-2141.2003.04506.x
- Espinosa Lara P, Quirós Redondo V, Aguado Lobo M, et al. Purpuric exanthema in a patient with hairy cell leukemia treated with cladribine and allopurinol. Ann Hematol. 2017;96:1209-1210. doi:10.1007 /s00277-017-2992-z
- Hendrick A. Purpuric rash following treatment with 2-chlorodeoxyadenosine. Clin Lab Haematol. 2001;23:67-68. doi:10.1046 /j.1365-2257.2001.0346b.x
- Kang S, Amagai M, Bruckner AL, et al, eds. Fitzpatrick’s Dermatology. 9th ed. McGraw-Hill Education; 2019.
- Bolognia JL, Cooper DL, Glusac EJ. Toxic erythema of chemotherapy: a useful clinical term. J Am Acad Dermatol. 2008;59:524-529.
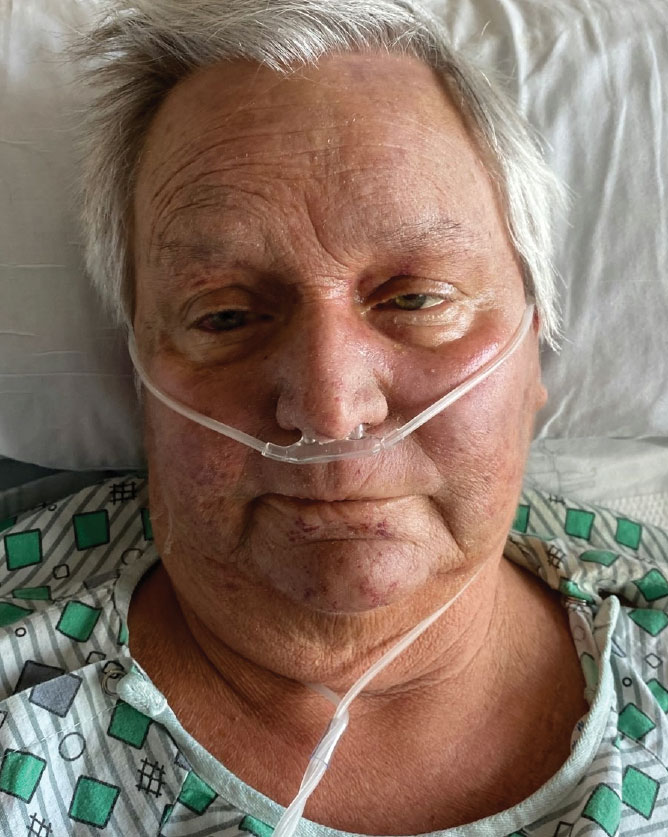
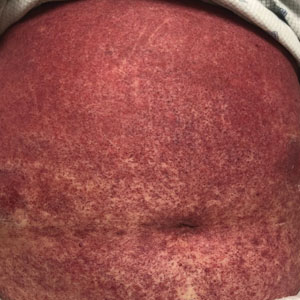
A 68-year-old woman presented to the emergency department with neutropenic fever and a rash over the body after receiving 2 doses of cladribine therapy for hairy cell leukemia. Physical examination demonstrated marked facial (top), lip, and tongue swelling, as well as a diffuse dusky nonpalpable purpuric rash on the abdomen (bottom) and back involving 90% of the body surface area. Bilateral ear edema was appreciated with accentuation of the earlobe crease. The patient exhibited subconjunctival hemorrhage, ectropion, and scleral injection. A punch biopsy of the thigh was performed.
Shiny Indurated Plaques on the Legs
The Diagnosis: Pretibial Myxedema
Histopathology showed superficial and deep mucin deposition with proliferation of fibroblasts and thin wiry collagen bundles that were consistent with a diagnosis of pretibial myxedema. The patient was treated with clobetasol ointment 0.05% twice daily for 3 months, followed by a trial of pentoxifylline 400 mg 3 times daily for 3 months. After this treatment failed, she was started on rituximab infusions of 1 g biweekly for 1 month, followed by 500 mg at 6 months, with marked improvement after the first 2 doses of 1 g.
Pretibial myxedema is an uncommon cutaneous manifestation of autoimmune thyroid disease, occurring in 1% to 5% of patients with Graves disease. It usually occurs in older adult women on the pretibial regions and less commonly on the upper extremities, face, and areas of prior trauma.1-3 Although typically asymptomatic, it can be painful and ulcerate.3 The clinical presentation consists of bilateral nonpitting edema with overlying indurated skin as well as flesh-colored, yellow-brown, violaceous, or peau d’orange papules and plaques.2,3 Lesions develop over months and often have been associated with hyperhidrosis and hypertrichosis.2 Many variants have been identified including nodular, plaquelike, diffuse swelling (ie, nonpitting edema), tumor, mixture, polypoid, and elephantiasis; severe cases with acral involvement are termed thyroid acropachy.1-3 Pathogenesis likely involves the activation of thyrotropin receptors on fibroblasts by the circulating thyrotropin autoantibodies found in Graves disease. Activated fibroblasts upregulate glycosaminoglycan production, which osmotically drives the accumulation of dermal and subdermal fluid.1,3
This diagnosis should be considered in any patient with pretibial edema or edema in areas of trauma. Graves disease most commonly is diagnosed 1 to 2 years prior to the development of pretibial myxedema; other extrathyroidal manifestations, most commonly ophthalmopathies, almost always are found in patients with pretibial myxedema. If a diagnosis of Graves disease has not been established, thyroid studies, including thyrotropin receptor antibody serum levels, should be obtained. Histopathology showing increased mucin in the dermis and increased fibroblasts can aid in diagnosis.2,3
The differential diagnosis includes inflammatory dermatoses, such as stasis dermatitis and lipodermatosclerosis. Stasis dermatitis is characterized by lichenified yellowbrown plaques that present on the lower extremities; lipodermatosclerosis then can develop and present as atrophic sclerotic plaques with a champagne bottle–like appearance. Necrobiosis lipoidica demonstrates atrophic, shiny, yellow plaques with telangiectases and ulcerations. Hypertrophic lichen planus presents with hyperkeratotic hyperpigmented plaques on the shins.1,2 Other diseases of cutaneous mucin deposition, namely scleromyxedema, demonstrate similar physical findings but more commonly are located on the trunk, face, and dorsal hands rather than the lower extremities.1-3
Treatment of pretibial myxedema is difficult; normalization of thyroid function, weight reduction, and compression stockings can help reduce edema. Medical therapies aim to decrease glycosaminoglycan production by fibroblasts. First-line treatment includes topical steroids under occlusion, and second-line therapies include intralesional steroids, systemic corticosteroids, pentoxifylline, and octreotide.2,3 Therapies for refractory disease include plasmapheresis, surgical excision, radiotherapy, and intravenous immunoglobulin; more recent studies also endorse the use of isotretinoin, intralesional hyaluronidase, and rituximab.2,4 Success also has been observed with the insulin growth factor 1 receptor inhibitor teprotumumab in active thyroid eye disease, in which insulin growth factor 1 receptor is overexpressed by fibroblasts. Given the similar pathogenesis of thyroid ophthalmopathy with other extrathyroidal manifestations, teprotumumab is a promising option for refractory cases of pretibial myxedema and has led to disease resolution in several patients.4
- Fatourechi V, Pajouhi M, Fransway AF. Dermopathy of Graves disease (pretibial myxedema). review of 150 cases. Medicine (Baltimore). 1994;73:1-7. doi:10.1097/00005792-199401000-00001
- Ai J, Leonhardt JM, Heymann WR. Autoimmune thyroid diseases: etiology, pathogenesis, and dermatologic manifestations. J Am Acad Dermatol. 2003;48:641-662. doi:10.1067/mjd.2003.257
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446. doi:10.1210/jcem.87.2.8220
- Varma A, Rheeman C, Levitt J. Resolution of pretibial myxedema with teprotumumab in a patient with Graves disease. JAAD Case Reports. 2020;6:1281-1282. doi:10.1016/j.jdcr.2020.09.003
The Diagnosis: Pretibial Myxedema
Histopathology showed superficial and deep mucin deposition with proliferation of fibroblasts and thin wiry collagen bundles that were consistent with a diagnosis of pretibial myxedema. The patient was treated with clobetasol ointment 0.05% twice daily for 3 months, followed by a trial of pentoxifylline 400 mg 3 times daily for 3 months. After this treatment failed, she was started on rituximab infusions of 1 g biweekly for 1 month, followed by 500 mg at 6 months, with marked improvement after the first 2 doses of 1 g.
Pretibial myxedema is an uncommon cutaneous manifestation of autoimmune thyroid disease, occurring in 1% to 5% of patients with Graves disease. It usually occurs in older adult women on the pretibial regions and less commonly on the upper extremities, face, and areas of prior trauma.1-3 Although typically asymptomatic, it can be painful and ulcerate.3 The clinical presentation consists of bilateral nonpitting edema with overlying indurated skin as well as flesh-colored, yellow-brown, violaceous, or peau d’orange papules and plaques.2,3 Lesions develop over months and often have been associated with hyperhidrosis and hypertrichosis.2 Many variants have been identified including nodular, plaquelike, diffuse swelling (ie, nonpitting edema), tumor, mixture, polypoid, and elephantiasis; severe cases with acral involvement are termed thyroid acropachy.1-3 Pathogenesis likely involves the activation of thyrotropin receptors on fibroblasts by the circulating thyrotropin autoantibodies found in Graves disease. Activated fibroblasts upregulate glycosaminoglycan production, which osmotically drives the accumulation of dermal and subdermal fluid.1,3
This diagnosis should be considered in any patient with pretibial edema or edema in areas of trauma. Graves disease most commonly is diagnosed 1 to 2 years prior to the development of pretibial myxedema; other extrathyroidal manifestations, most commonly ophthalmopathies, almost always are found in patients with pretibial myxedema. If a diagnosis of Graves disease has not been established, thyroid studies, including thyrotropin receptor antibody serum levels, should be obtained. Histopathology showing increased mucin in the dermis and increased fibroblasts can aid in diagnosis.2,3
The differential diagnosis includes inflammatory dermatoses, such as stasis dermatitis and lipodermatosclerosis. Stasis dermatitis is characterized by lichenified yellowbrown plaques that present on the lower extremities; lipodermatosclerosis then can develop and present as atrophic sclerotic plaques with a champagne bottle–like appearance. Necrobiosis lipoidica demonstrates atrophic, shiny, yellow plaques with telangiectases and ulcerations. Hypertrophic lichen planus presents with hyperkeratotic hyperpigmented plaques on the shins.1,2 Other diseases of cutaneous mucin deposition, namely scleromyxedema, demonstrate similar physical findings but more commonly are located on the trunk, face, and dorsal hands rather than the lower extremities.1-3
Treatment of pretibial myxedema is difficult; normalization of thyroid function, weight reduction, and compression stockings can help reduce edema. Medical therapies aim to decrease glycosaminoglycan production by fibroblasts. First-line treatment includes topical steroids under occlusion, and second-line therapies include intralesional steroids, systemic corticosteroids, pentoxifylline, and octreotide.2,3 Therapies for refractory disease include plasmapheresis, surgical excision, radiotherapy, and intravenous immunoglobulin; more recent studies also endorse the use of isotretinoin, intralesional hyaluronidase, and rituximab.2,4 Success also has been observed with the insulin growth factor 1 receptor inhibitor teprotumumab in active thyroid eye disease, in which insulin growth factor 1 receptor is overexpressed by fibroblasts. Given the similar pathogenesis of thyroid ophthalmopathy with other extrathyroidal manifestations, teprotumumab is a promising option for refractory cases of pretibial myxedema and has led to disease resolution in several patients.4
The Diagnosis: Pretibial Myxedema
Histopathology showed superficial and deep mucin deposition with proliferation of fibroblasts and thin wiry collagen bundles that were consistent with a diagnosis of pretibial myxedema. The patient was treated with clobetasol ointment 0.05% twice daily for 3 months, followed by a trial of pentoxifylline 400 mg 3 times daily for 3 months. After this treatment failed, she was started on rituximab infusions of 1 g biweekly for 1 month, followed by 500 mg at 6 months, with marked improvement after the first 2 doses of 1 g.
Pretibial myxedema is an uncommon cutaneous manifestation of autoimmune thyroid disease, occurring in 1% to 5% of patients with Graves disease. It usually occurs in older adult women on the pretibial regions and less commonly on the upper extremities, face, and areas of prior trauma.1-3 Although typically asymptomatic, it can be painful and ulcerate.3 The clinical presentation consists of bilateral nonpitting edema with overlying indurated skin as well as flesh-colored, yellow-brown, violaceous, or peau d’orange papules and plaques.2,3 Lesions develop over months and often have been associated with hyperhidrosis and hypertrichosis.2 Many variants have been identified including nodular, plaquelike, diffuse swelling (ie, nonpitting edema), tumor, mixture, polypoid, and elephantiasis; severe cases with acral involvement are termed thyroid acropachy.1-3 Pathogenesis likely involves the activation of thyrotropin receptors on fibroblasts by the circulating thyrotropin autoantibodies found in Graves disease. Activated fibroblasts upregulate glycosaminoglycan production, which osmotically drives the accumulation of dermal and subdermal fluid.1,3
This diagnosis should be considered in any patient with pretibial edema or edema in areas of trauma. Graves disease most commonly is diagnosed 1 to 2 years prior to the development of pretibial myxedema; other extrathyroidal manifestations, most commonly ophthalmopathies, almost always are found in patients with pretibial myxedema. If a diagnosis of Graves disease has not been established, thyroid studies, including thyrotropin receptor antibody serum levels, should be obtained. Histopathology showing increased mucin in the dermis and increased fibroblasts can aid in diagnosis.2,3
The differential diagnosis includes inflammatory dermatoses, such as stasis dermatitis and lipodermatosclerosis. Stasis dermatitis is characterized by lichenified yellowbrown plaques that present on the lower extremities; lipodermatosclerosis then can develop and present as atrophic sclerotic plaques with a champagne bottle–like appearance. Necrobiosis lipoidica demonstrates atrophic, shiny, yellow plaques with telangiectases and ulcerations. Hypertrophic lichen planus presents with hyperkeratotic hyperpigmented plaques on the shins.1,2 Other diseases of cutaneous mucin deposition, namely scleromyxedema, demonstrate similar physical findings but more commonly are located on the trunk, face, and dorsal hands rather than the lower extremities.1-3
Treatment of pretibial myxedema is difficult; normalization of thyroid function, weight reduction, and compression stockings can help reduce edema. Medical therapies aim to decrease glycosaminoglycan production by fibroblasts. First-line treatment includes topical steroids under occlusion, and second-line therapies include intralesional steroids, systemic corticosteroids, pentoxifylline, and octreotide.2,3 Therapies for refractory disease include plasmapheresis, surgical excision, radiotherapy, and intravenous immunoglobulin; more recent studies also endorse the use of isotretinoin, intralesional hyaluronidase, and rituximab.2,4 Success also has been observed with the insulin growth factor 1 receptor inhibitor teprotumumab in active thyroid eye disease, in which insulin growth factor 1 receptor is overexpressed by fibroblasts. Given the similar pathogenesis of thyroid ophthalmopathy with other extrathyroidal manifestations, teprotumumab is a promising option for refractory cases of pretibial myxedema and has led to disease resolution in several patients.4
- Fatourechi V, Pajouhi M, Fransway AF. Dermopathy of Graves disease (pretibial myxedema). review of 150 cases. Medicine (Baltimore). 1994;73:1-7. doi:10.1097/00005792-199401000-00001
- Ai J, Leonhardt JM, Heymann WR. Autoimmune thyroid diseases: etiology, pathogenesis, and dermatologic manifestations. J Am Acad Dermatol. 2003;48:641-662. doi:10.1067/mjd.2003.257
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446. doi:10.1210/jcem.87.2.8220
- Varma A, Rheeman C, Levitt J. Resolution of pretibial myxedema with teprotumumab in a patient with Graves disease. JAAD Case Reports. 2020;6:1281-1282. doi:10.1016/j.jdcr.2020.09.003
- Fatourechi V, Pajouhi M, Fransway AF. Dermopathy of Graves disease (pretibial myxedema). review of 150 cases. Medicine (Baltimore). 1994;73:1-7. doi:10.1097/00005792-199401000-00001
- Ai J, Leonhardt JM, Heymann WR. Autoimmune thyroid diseases: etiology, pathogenesis, and dermatologic manifestations. J Am Acad Dermatol. 2003;48:641-662. doi:10.1067/mjd.2003.257
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446. doi:10.1210/jcem.87.2.8220
- Varma A, Rheeman C, Levitt J. Resolution of pretibial myxedema with teprotumumab in a patient with Graves disease. JAAD Case Reports. 2020;6:1281-1282. doi:10.1016/j.jdcr.2020.09.003
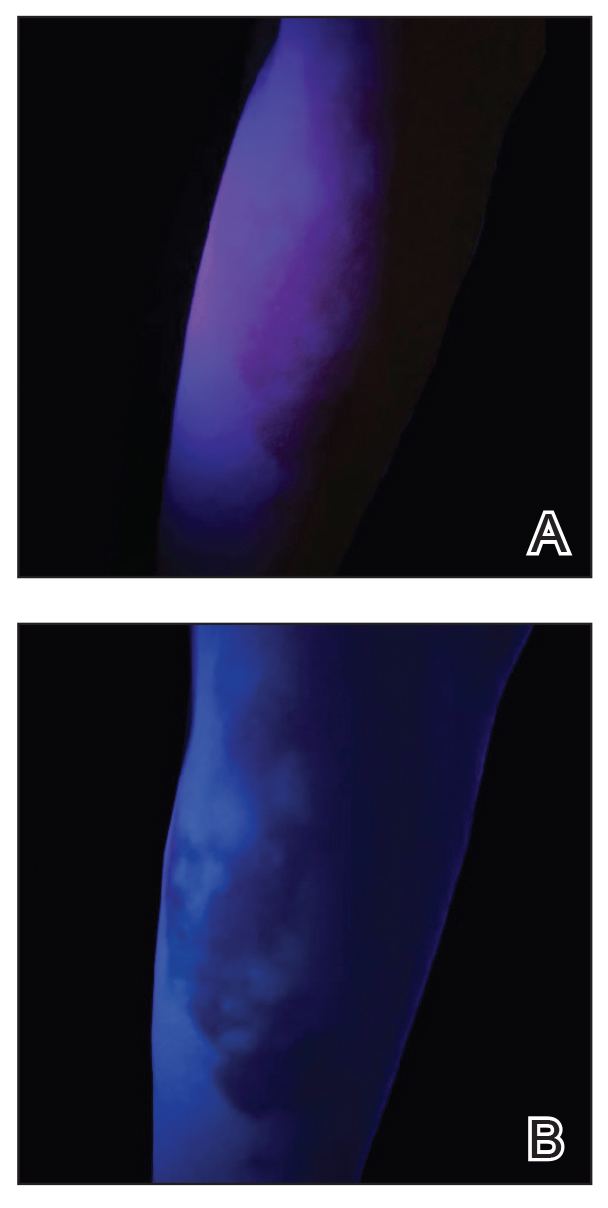
A 70-year-old woman presented with pain and swelling in both legs of many years’ duration. She had no history of skin disease. Physical examination revealed shiny indurated plaques on the legs, ankles, and toes with limited range of motion in the ankles (top). Marked thickening of the hands and index fingers also was noted (bottom). A punch biopsy of the distal pretibial region was performed.


Oval Brown Plaque on the Palm
The Diagnosis: Poroma
Histopathology showed an endophytic expansion of the epidermis by bland, uniform, basaloid epithelial cells with focal ductal differentiation and an abrupt transition with surrounding epidermal keratinocytes (Figure), consistent with a diagnosis of poroma. The patient elected to monitor the lesion rather than to have it excised.

Eccrine poroma, used interchangeably with the term poroma, is a rare benign adnexal tumor of the eccrine sweat glands resulting from proliferation of the acrosyringium.1,2 It often occurs on the palms or soles, though it also can arise anywhere sweat glands are present.1 Eccrine poromas often appear in middle-aged individuals as singular, well-circumscribed, red-brown papules or nodules.3 A characteristic feature is a shallow, cup-shaped depression within the larger papule or nodule.1
Because the condition is benign and often asymptomatic, it can be safely monitored for progression.1 However, if the lesion is symptomatic or located in a sensitive area, complete excision is curative.4 Eccrine poromas can recur, making close monitoring following excision important.5 The development of bleeding, itching, or pain in a previously asymptomatic lesion may indicate possible malignant transformation, which occurs in only 18% of cases.6
The differential diagnosis includes basal cell carcinoma, circumscribed acral hypokeratosis, Kaposi sarcoma, and pyogenic granuloma. Basal cell carcinoma is the most common type of skin cancer.7 In rare cases it has been shown to present on the palms or soles as a slowgrowing, reddish-pink papule or plaque with central ulceration. It typically is asymptomatic. Histopathology shows dermal nests of basaloid cells with peripheral palisading, stromal mucin, and peritumoral clefts. Treatment is surgical excision.7
Circumscribed acral hypokeratosis presents on the palms or soles as a solitary, shallow, well-defined lesion with a flat base and raised border.8 It often is red-pink in color and most frequently occurs in middle-aged women. Although the cause of the condition is unknown, it is thought to be the result of trauma or human papillomavirus infection.8 Biopsy results characteristically show hypokeratosis demarcated by a sharp and frayed cutoff from uninvolved acral skin with discrete hypogranulosis, dilated blood vessels in the papillary dermis, and slightly thickened collagen fibers in the reticular dermis.9 Surgical excision is a potential treatment option, as topical corticosteroids, retinoids, and calcipotriene have not been shown to be effective; spontaneous resolution has been reported.8
Kaposi sarcoma is a vascular neoplasm that is associated with human herpesvirus 8 infection.10 It typically presents on mucocutaneous sites and the lower extremities. Palmar involvement has been reported in rare cases, occurring as a solitary, well-demarcated, violaceous macule or patch that may be painful.10-12 Characteristic histopathologic features include a proliferation in the dermis of slitlike vascular spaces and spindle cell proliferation.13 Treatment options include cryosurgery; pulsed dye laser; and topical, intralesional, or systemic chemotherapy agents, depending on the stage of the patient’s disease. Antiretroviral therapy is indicated for patients with Kaposi sarcoma secondary to AIDS.14
Pyogenic granuloma presents as a solitary red-brown or bluish-black papule or nodule that bleeds easily when manipulated.15 It commonly occurs following trauma, typically on the fingers, feet, and lips.6 Although benign, potential complications include ulceration and blood loss. Pyogenic granulomas can be treated via curettage and cautery, excision, cryosurgery, or pulsed dye laser.15
- Wankhade V, Singh R, Sadhwani V, et al. Eccrine poroma. Indian Dermatol Online J. 2015;6:304-305.
- Yorulmaz A, Aksoy GG, Ozhamam EU. A growing mass under the nail: subungual eccrine poroma. Skin Appendage Disord. 2020;6:254-257.
- Wang Y, Liu M, Zheng Y, et al. Eccrine poroma presented as spindleshaped plaque: a case report. Medicine (Baltimore). 2021;100:E25971. doi:10.1097/MD.0000000000025971
- Sharma M, Singh M, Gupta K, et al. Eccrine poroma of the eyelid. Indian J Ophthalmol. 2020;68:2522.
- Rasool MN, Hawary MB. Benign eccrine poroma in the palm of the hand. Ann Saudi Med. 2004;24:46-47.
- Sawaya JL, Khachemoune A. Poroma: a review of eccrine, apocrine, and malignant forms [published online April 2, 2014]. Int J Dermatol. 2014;53:1053-1061. doi:10.1111/ijd.12448
- López-Sánchez C, Ferguson P, Collgros H. Basal cell carcinoma of the palm: an unusual presentation of a common tumour [published online August 6, 2019]. Australas J Dermatol. 2020;61:69-70. doi:10.1111/ajd.13129
- Berk DR, Böer A, Bauschard FD, et al. Circumscribed acral hypokeratosis [published online April 6, 2007]. J Am Acad Dermatol. 2007;57:292-296. doi:10.1016/j.jaad.2007.02.022
- Majluf-Cáceres P, Vera-Kellet C, González-Bombardiere S. New dermoscopic keys for circumscribed acral hypokeratosis: report of four cases. Dermatol Pract Concept. 2021;11:E2021010. doi:10.5826/dpc.1102a10
- Simonart T, De Dobbeleer G, Stallenberg B. Classic Kaposi’s sarcoma of the palm in a metallurgist: role of iron filings in its development? Br J Dermatol. 2003;148:1061-1063. doi:10.1046/j.1365-2133.2003.05331.x
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294. doi:10.5858/arpa.2012-0101-RS
- Al Zolibani AA, Al Robaee AA. Primary palmoplantar Kaposi’s sarcoma: an unusual presentation. Skinmed. 2006;5:248-249. doi:10.1111/j.1540-9740.2006.04662.x
- Cesarman E, Damania B, Krown SE, et al. Kaposi sarcoma. Nat Rev Dis Primers. 2019;5:9. doi:10.1038/s41572-019-0060-9
- Etemad SA, Dewan AK. Kaposi sarcoma updates [published online July 10, 2019]. Dermatol Clin. 2019;37:505-517. doi:10.1016/j. det.2019.05.008
- Murthy SC, Nagaraj A. Pyogenic granuloma. Indian Pediatr. 2012;49:855. doi:10.1007/s13312-012-0184-4
The Diagnosis: Poroma
Histopathology showed an endophytic expansion of the epidermis by bland, uniform, basaloid epithelial cells with focal ductal differentiation and an abrupt transition with surrounding epidermal keratinocytes (Figure), consistent with a diagnosis of poroma. The patient elected to monitor the lesion rather than to have it excised.
Eccrine poroma, used interchangeably with the term poroma, is a rare benign adnexal tumor of the eccrine sweat glands resulting from proliferation of the acrosyringium.1,2 It often occurs on the palms or soles, though it also can arise anywhere sweat glands are present.1 Eccrine poromas often appear in middle-aged individuals as singular, well-circumscribed, red-brown papules or nodules.3 A characteristic feature is a shallow, cup-shaped depression within the larger papule or nodule.1
Because the condition is benign and often asymptomatic, it can be safely monitored for progression.1 However, if the lesion is symptomatic or located in a sensitive area, complete excision is curative.4 Eccrine poromas can recur, making close monitoring following excision important.5 The development of bleeding, itching, or pain in a previously asymptomatic lesion may indicate possible malignant transformation, which occurs in only 18% of cases.6
The differential diagnosis includes basal cell carcinoma, circumscribed acral hypokeratosis, Kaposi sarcoma, and pyogenic granuloma. Basal cell carcinoma is the most common type of skin cancer.7 In rare cases it has been shown to present on the palms or soles as a slowgrowing, reddish-pink papule or plaque with central ulceration. It typically is asymptomatic. Histopathology shows dermal nests of basaloid cells with peripheral palisading, stromal mucin, and peritumoral clefts. Treatment is surgical excision.7
Circumscribed acral hypokeratosis presents on the palms or soles as a solitary, shallow, well-defined lesion with a flat base and raised border.8 It often is red-pink in color and most frequently occurs in middle-aged women. Although the cause of the condition is unknown, it is thought to be the result of trauma or human papillomavirus infection.8 Biopsy results characteristically show hypokeratosis demarcated by a sharp and frayed cutoff from uninvolved acral skin with discrete hypogranulosis, dilated blood vessels in the papillary dermis, and slightly thickened collagen fibers in the reticular dermis.9 Surgical excision is a potential treatment option, as topical corticosteroids, retinoids, and calcipotriene have not been shown to be effective; spontaneous resolution has been reported.8
Kaposi sarcoma is a vascular neoplasm that is associated with human herpesvirus 8 infection.10 It typically presents on mucocutaneous sites and the lower extremities. Palmar involvement has been reported in rare cases, occurring as a solitary, well-demarcated, violaceous macule or patch that may be painful.10-12 Characteristic histopathologic features include a proliferation in the dermis of slitlike vascular spaces and spindle cell proliferation.13 Treatment options include cryosurgery; pulsed dye laser; and topical, intralesional, or systemic chemotherapy agents, depending on the stage of the patient’s disease. Antiretroviral therapy is indicated for patients with Kaposi sarcoma secondary to AIDS.14
Pyogenic granuloma presents as a solitary red-brown or bluish-black papule or nodule that bleeds easily when manipulated.15 It commonly occurs following trauma, typically on the fingers, feet, and lips.6 Although benign, potential complications include ulceration and blood loss. Pyogenic granulomas can be treated via curettage and cautery, excision, cryosurgery, or pulsed dye laser.15
The Diagnosis: Poroma
Histopathology showed an endophytic expansion of the epidermis by bland, uniform, basaloid epithelial cells with focal ductal differentiation and an abrupt transition with surrounding epidermal keratinocytes (Figure), consistent with a diagnosis of poroma. The patient elected to monitor the lesion rather than to have it excised.
Eccrine poroma, used interchangeably with the term poroma, is a rare benign adnexal tumor of the eccrine sweat glands resulting from proliferation of the acrosyringium.1,2 It often occurs on the palms or soles, though it also can arise anywhere sweat glands are present.1 Eccrine poromas often appear in middle-aged individuals as singular, well-circumscribed, red-brown papules or nodules.3 A characteristic feature is a shallow, cup-shaped depression within the larger papule or nodule.1
Because the condition is benign and often asymptomatic, it can be safely monitored for progression.1 However, if the lesion is symptomatic or located in a sensitive area, complete excision is curative.4 Eccrine poromas can recur, making close monitoring following excision important.5 The development of bleeding, itching, or pain in a previously asymptomatic lesion may indicate possible malignant transformation, which occurs in only 18% of cases.6
The differential diagnosis includes basal cell carcinoma, circumscribed acral hypokeratosis, Kaposi sarcoma, and pyogenic granuloma. Basal cell carcinoma is the most common type of skin cancer.7 In rare cases it has been shown to present on the palms or soles as a slowgrowing, reddish-pink papule or plaque with central ulceration. It typically is asymptomatic. Histopathology shows dermal nests of basaloid cells with peripheral palisading, stromal mucin, and peritumoral clefts. Treatment is surgical excision.7
Circumscribed acral hypokeratosis presents on the palms or soles as a solitary, shallow, well-defined lesion with a flat base and raised border.8 It often is red-pink in color and most frequently occurs in middle-aged women. Although the cause of the condition is unknown, it is thought to be the result of trauma or human papillomavirus infection.8 Biopsy results characteristically show hypokeratosis demarcated by a sharp and frayed cutoff from uninvolved acral skin with discrete hypogranulosis, dilated blood vessels in the papillary dermis, and slightly thickened collagen fibers in the reticular dermis.9 Surgical excision is a potential treatment option, as topical corticosteroids, retinoids, and calcipotriene have not been shown to be effective; spontaneous resolution has been reported.8
Kaposi sarcoma is a vascular neoplasm that is associated with human herpesvirus 8 infection.10 It typically presents on mucocutaneous sites and the lower extremities. Palmar involvement has been reported in rare cases, occurring as a solitary, well-demarcated, violaceous macule or patch that may be painful.10-12 Characteristic histopathologic features include a proliferation in the dermis of slitlike vascular spaces and spindle cell proliferation.13 Treatment options include cryosurgery; pulsed dye laser; and topical, intralesional, or systemic chemotherapy agents, depending on the stage of the patient’s disease. Antiretroviral therapy is indicated for patients with Kaposi sarcoma secondary to AIDS.14
Pyogenic granuloma presents as a solitary red-brown or bluish-black papule or nodule that bleeds easily when manipulated.15 It commonly occurs following trauma, typically on the fingers, feet, and lips.6 Although benign, potential complications include ulceration and blood loss. Pyogenic granulomas can be treated via curettage and cautery, excision, cryosurgery, or pulsed dye laser.15
- Wankhade V, Singh R, Sadhwani V, et al. Eccrine poroma. Indian Dermatol Online J. 2015;6:304-305.
- Yorulmaz A, Aksoy GG, Ozhamam EU. A growing mass under the nail: subungual eccrine poroma. Skin Appendage Disord. 2020;6:254-257.
- Wang Y, Liu M, Zheng Y, et al. Eccrine poroma presented as spindleshaped plaque: a case report. Medicine (Baltimore). 2021;100:E25971. doi:10.1097/MD.0000000000025971
- Sharma M, Singh M, Gupta K, et al. Eccrine poroma of the eyelid. Indian J Ophthalmol. 2020;68:2522.
- Rasool MN, Hawary MB. Benign eccrine poroma in the palm of the hand. Ann Saudi Med. 2004;24:46-47.
- Sawaya JL, Khachemoune A. Poroma: a review of eccrine, apocrine, and malignant forms [published online April 2, 2014]. Int J Dermatol. 2014;53:1053-1061. doi:10.1111/ijd.12448
- López-Sánchez C, Ferguson P, Collgros H. Basal cell carcinoma of the palm: an unusual presentation of a common tumour [published online August 6, 2019]. Australas J Dermatol. 2020;61:69-70. doi:10.1111/ajd.13129
- Berk DR, Böer A, Bauschard FD, et al. Circumscribed acral hypokeratosis [published online April 6, 2007]. J Am Acad Dermatol. 2007;57:292-296. doi:10.1016/j.jaad.2007.02.022
- Majluf-Cáceres P, Vera-Kellet C, González-Bombardiere S. New dermoscopic keys for circumscribed acral hypokeratosis: report of four cases. Dermatol Pract Concept. 2021;11:E2021010. doi:10.5826/dpc.1102a10
- Simonart T, De Dobbeleer G, Stallenberg B. Classic Kaposi’s sarcoma of the palm in a metallurgist: role of iron filings in its development? Br J Dermatol. 2003;148:1061-1063. doi:10.1046/j.1365-2133.2003.05331.x
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294. doi:10.5858/arpa.2012-0101-RS
- Al Zolibani AA, Al Robaee AA. Primary palmoplantar Kaposi’s sarcoma: an unusual presentation. Skinmed. 2006;5:248-249. doi:10.1111/j.1540-9740.2006.04662.x
- Cesarman E, Damania B, Krown SE, et al. Kaposi sarcoma. Nat Rev Dis Primers. 2019;5:9. doi:10.1038/s41572-019-0060-9
- Etemad SA, Dewan AK. Kaposi sarcoma updates [published online July 10, 2019]. Dermatol Clin. 2019;37:505-517. doi:10.1016/j. det.2019.05.008
- Murthy SC, Nagaraj A. Pyogenic granuloma. Indian Pediatr. 2012;49:855. doi:10.1007/s13312-012-0184-4
- Wankhade V, Singh R, Sadhwani V, et al. Eccrine poroma. Indian Dermatol Online J. 2015;6:304-305.
- Yorulmaz A, Aksoy GG, Ozhamam EU. A growing mass under the nail: subungual eccrine poroma. Skin Appendage Disord. 2020;6:254-257.
- Wang Y, Liu M, Zheng Y, et al. Eccrine poroma presented as spindleshaped plaque: a case report. Medicine (Baltimore). 2021;100:E25971. doi:10.1097/MD.0000000000025971
- Sharma M, Singh M, Gupta K, et al. Eccrine poroma of the eyelid. Indian J Ophthalmol. 2020;68:2522.
- Rasool MN, Hawary MB. Benign eccrine poroma in the palm of the hand. Ann Saudi Med. 2004;24:46-47.
- Sawaya JL, Khachemoune A. Poroma: a review of eccrine, apocrine, and malignant forms [published online April 2, 2014]. Int J Dermatol. 2014;53:1053-1061. doi:10.1111/ijd.12448
- López-Sánchez C, Ferguson P, Collgros H. Basal cell carcinoma of the palm: an unusual presentation of a common tumour [published online August 6, 2019]. Australas J Dermatol. 2020;61:69-70. doi:10.1111/ajd.13129
- Berk DR, Böer A, Bauschard FD, et al. Circumscribed acral hypokeratosis [published online April 6, 2007]. J Am Acad Dermatol. 2007;57:292-296. doi:10.1016/j.jaad.2007.02.022
- Majluf-Cáceres P, Vera-Kellet C, González-Bombardiere S. New dermoscopic keys for circumscribed acral hypokeratosis: report of four cases. Dermatol Pract Concept. 2021;11:E2021010. doi:10.5826/dpc.1102a10
- Simonart T, De Dobbeleer G, Stallenberg B. Classic Kaposi’s sarcoma of the palm in a metallurgist: role of iron filings in its development? Br J Dermatol. 2003;148:1061-1063. doi:10.1046/j.1365-2133.2003.05331.x
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294. doi:10.5858/arpa.2012-0101-RS
- Al Zolibani AA, Al Robaee AA. Primary palmoplantar Kaposi’s sarcoma: an unusual presentation. Skinmed. 2006;5:248-249. doi:10.1111/j.1540-9740.2006.04662.x
- Cesarman E, Damania B, Krown SE, et al. Kaposi sarcoma. Nat Rev Dis Primers. 2019;5:9. doi:10.1038/s41572-019-0060-9
- Etemad SA, Dewan AK. Kaposi sarcoma updates [published online July 10, 2019]. Dermatol Clin. 2019;37:505-517. doi:10.1016/j. det.2019.05.008
- Murthy SC, Nagaraj A. Pyogenic granuloma. Indian Pediatr. 2012;49:855. doi:10.1007/s13312-012-0184-4
A 43-year-old woman presented with a painful lesion on the palm of 30 years’ duration that had grown in size. Physical examination revealed an oval, brown, lobulated plaque with a hyperkeratotic rim on the left palm. She reported bleeding and pain. A shallow cup-shaped depression was noted within the plaque. A 4-mm punch biopsy was performed.


Retiform Purpura on the Lower Legs
The Diagnosis: Type I Cryoglobulinemia
Retiform purpura with overlying necrosis subsequently developed over the course of a week following presentation (Figure 1). A skin biopsy showed fibrin thrombi and congestion of small- and medium-sized blood vessels, consistent with vasculopathy (Figure 2). Urinalysis revealed hematuria and proteinuria. A renal biopsy performed due to a continually elevated serum creatinine level revealed glomerulonephritis with numerous IgG1 lambda–restricted glomerular capillary hyaline thrombi, compatible with a lymphoproliferative disorder–associated type I cryoglobulinemia. A serum cryoglobulin immunofixation test confirmed type I cryoglobulinemia involving monoclonal IgG lambda. The combination of cutaneous, renal, and hematologic findings was consistent with type I cryoglobulinemia. A subsequent bone marrow biopsy demonstrated a CD20+ lambda–restricted plasma cell neoplasm. Initial treatment with high-dose corticosteroids followed by targeted treatment of the underlying hematologic condition with bortezomib, rituximab, and dexamethasone improved the skin disease.

Cryoglobulins are abnormal immunoglobulins that precipitate at temperatures below 37 °C. The persistent presence of cryoglobulins in the serum is termed cryoglobulinemia.1 Type I cryoglobulinemia is distinguished from mixed cryoglobulinemia—types II and III—by the presence of a single monoclonal immunoglobulin, typically IgM or IgG. It is associated with lymphoproliferative disorders, most commonly monoclonal gammopathy of undetermined significance and B-cell malignancies such as Waldenström macroglobulinemia, multiple myeloma, or chronic lymphocytic leukemia. Histopathology shows occlusion of small vessel lumina with homogenous eosinophilic material containing the monoclonal cryoprecipitate.2 Disease manifestations are caused by small vessel occlusion, which leads to ischemia and tissue damage.
.")
Retiform purpura, livedo reticularis/racemosa, and necrosis leading to ulcers are the most common cutaneous clinical findings. Extracutaneous signs include peripheral neuropathy, arthralgia, Raynaud phenomenon, and acrocyanosis. Renal involvement, most commonly glomerulonephritis with associated proteinuria, is noted in 14% to 20% of cases.3,4 An elevated cryocrit can lead to symptoms of hyperviscosity syndrome.2
Treatment is difficult and primarily is focused on addressing the underlying hematologic condition, which is responsible for synthesis of the cryoglobulin. Decreasing cryoglobulin production leads to decreased occlusion of blood vessels, thus alleviating the ischemia and skin damage. Monoclonal gammopathy of undetermined significance–related type I cryoglobulinemia initially is treated with corticosteroids followed by rituximab if a CD20+ B-cell clone is identified.2 Bortezomib is recommended for cases associated with Waldenström macroglobulinemia and cases associated with multiple myeloma with concurrent renal failure. In patients with neuropathy, a lenalidomide-based treatment can be employed. Patients should be instructed to keep extremities warm.2 Diabetic foot care guidelines should be followed to prevent wound complications. The differential diagnosis for type I cryoglobulinemia includes other causes of retiform purpura–like angioinvasive fungal infection, antiphospholipid antibody syndrome, calciphylaxis, and livedoid vasculopathy.5 Angioinvasive fungal infections are caused by Candida, Aspergillus, and Mucorales species, as well as other hyaline molds. They typically occur in immunocompromised patients and invade the blood vessels via direct inoculation or dissemination.6 Patients present with retiform purpura but typically will be acutely ill with fevers and vital sign abnormalities. Histopathology with special stains often will identify the fungal organisms in the dermis or inside blood vessel walls with vessel wall destruction and hemorrhage.7 Accurate diagnosis is essential to selecting appropriate antifungal agents. If angioinvasive fungal infection is clinically suspected, treatment should begin before culture and histopathologic data are available.7
Antiphospholipid antibody syndrome is an autoimmune thrombophilia that can occur as primary disease or in association with other autoimmune conditions, most commonly systemic lupus erythematosus. Diagnosis requires the presence of antiphospholipid antibodies, such as lupus anticoagulant, anticardiolipin antibody, anti–β2-glycoprotein-1 antibody, with arterial or venous thrombosis and/or recurrent pregnancy loss. Paraproteinemia is not seen. The most common cutaneous finding is livedo reticularis, with livedo racemosa being a more distinctive finding.8 Small vessel thrombosis is seen histopathologically. Treatment includes antiplatelet and anticoagulant medications. Patients with refractory disease may benefit from additional therapy with hydroxychloroquine or intravenous immunoglobulins.8
Calciphylaxis is a rare depositional vasculopathy that often occurs in patients with end-stage renal disease on dialysis. Patients present with painful and poor-healing skin lesions including indurated nodules, violaceous plaques, and retiform purpura that typically affect areas of high adiposity such as the thighs, abdomen, and buttocks.9 Ulceration and superimposed infections are common complications. Histopathologically, small dermal and subcutaneous vessels demonstrate calcification, microthrombosis, and fibrointimal hyperplasia.9 Wound management is critically important in patients with calciphylaxis. Treatment with intravenous sodium thiosulfate is typical, but prognosis remains poor. Although livedoid vasculopathy may present with retiform purpura in the ankles, paraproteinemia is not seen and patients frequently present with punched-out ulcerations that tend to heal into atrophie blanche.10 Livedoid vasculopathy has been associated with underlying hypercoagulable states, connective tissue diseases, and chronic venous hypertension. Hypercoagulability and endothelial cell damage contribute to the formation of fibrin thrombi in the superficial dermal blood vessels. Histopathology demonstrates thickening of vessel walls and intraluminal hyaline thrombi. Successful treatment in most cases is achieved with anticoagulation therapy, typically rivaroxaban, especially in patients with underlying hypercoagulability. Antiplatelet therapy also may be considered, while anabolic agents have been shown to be helpful in patients with connective tissue disease.10
- Desbois AC, Cacoub P, Saadoun D. Cryoglobulinemia: an update in 2019. Joint Bone Spine. 2019;86:707-713. doi:10.1016/j .jbspin.2019.01.016
- Muchtar E, Magen H, Gertz MA. How I treat cryoglobulinemia. Blood. 2017;129:289-298. doi:10.1182/blood-2016-09-719773
- Sidana S, Rajkumar SV, Dispenzieri A, et al. Clinical presentation and outcomes of patients with type 1 monoclonal cryoglobulinemia. Am J Hematol. 2017;92:668-673. doi:10.1002/ajh.24745
- Harel S, Mohr M, Jahn I, et al. Clinico-biological characteristics and treatment of type I monoclonal cryoglobulinaemia: a study of 64 cases. Br J Haematol. 2015;168:671-678. doi:10.1111/bjh.13196
- Georgesen C, Fox LP, Harp J. Retiform purpura: a diagnostic approach. J Am Acad Dermatol. 2020;82:783-796. doi:10.1016/j.jaad.2019.07.112
- Shields BE, Rosenbach M, Brown-Joel Z, et al. Angioinvasive fungal infections impacting the skin: background, epidemiology, and clinical presentation. J Am Acad Dermatol. 2019;80:869-880.e5. doi:10.1016/j.jaad.2018.04.059
- Berger AP, Ford BA, Brown-Joel Z, et al. Angioinvasive fungal infections impacting the skin: diagnosis, management, and complications. J Am Acad Dermatol. 2019;80:883-898.e2. doi:10.1016/j.jaad.2018.04.058
- Negrini S, Pappalardo F, Murdaca G, et al. The antiphospholipid syndrome: from pathophysiology to treatment. Clin Exp Med. 2017;17:257-267. doi:10.1007/s10238-016-0430-5
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146. doi:10.1053/j.ajkd.2015.01.034
- Georgesen C, Fox LP, Harp J. Retiform purpura: workup and therapeutic considerations in select conditions. J Am Acad Dermatol. 2020;82:799-816. doi:10.1016/j.jaad.2019.07.113
The Diagnosis: Type I Cryoglobulinemia
Retiform purpura with overlying necrosis subsequently developed over the course of a week following presentation (Figure 1). A skin biopsy showed fibrin thrombi and congestion of small- and medium-sized blood vessels, consistent with vasculopathy (Figure 2). Urinalysis revealed hematuria and proteinuria. A renal biopsy performed due to a continually elevated serum creatinine level revealed glomerulonephritis with numerous IgG1 lambda–restricted glomerular capillary hyaline thrombi, compatible with a lymphoproliferative disorder–associated type I cryoglobulinemia. A serum cryoglobulin immunofixation test confirmed type I cryoglobulinemia involving monoclonal IgG lambda. The combination of cutaneous, renal, and hematologic findings was consistent with type I cryoglobulinemia. A subsequent bone marrow biopsy demonstrated a CD20+ lambda–restricted plasma cell neoplasm. Initial treatment with high-dose corticosteroids followed by targeted treatment of the underlying hematologic condition with bortezomib, rituximab, and dexamethasone improved the skin disease.
Cryoglobulins are abnormal immunoglobulins that precipitate at temperatures below 37 °C. The persistent presence of cryoglobulins in the serum is termed cryoglobulinemia.1 Type I cryoglobulinemia is distinguished from mixed cryoglobulinemia—types II and III—by the presence of a single monoclonal immunoglobulin, typically IgM or IgG. It is associated with lymphoproliferative disorders, most commonly monoclonal gammopathy of undetermined significance and B-cell malignancies such as Waldenström macroglobulinemia, multiple myeloma, or chronic lymphocytic leukemia. Histopathology shows occlusion of small vessel lumina with homogenous eosinophilic material containing the monoclonal cryoprecipitate.2 Disease manifestations are caused by small vessel occlusion, which leads to ischemia and tissue damage.
Retiform purpura, livedo reticularis/racemosa, and necrosis leading to ulcers are the most common cutaneous clinical findings. Extracutaneous signs include peripheral neuropathy, arthralgia, Raynaud phenomenon, and acrocyanosis. Renal involvement, most commonly glomerulonephritis with associated proteinuria, is noted in 14% to 20% of cases.3,4 An elevated cryocrit can lead to symptoms of hyperviscosity syndrome.2
Treatment is difficult and primarily is focused on addressing the underlying hematologic condition, which is responsible for synthesis of the cryoglobulin. Decreasing cryoglobulin production leads to decreased occlusion of blood vessels, thus alleviating the ischemia and skin damage. Monoclonal gammopathy of undetermined significance–related type I cryoglobulinemia initially is treated with corticosteroids followed by rituximab if a CD20+ B-cell clone is identified.2 Bortezomib is recommended for cases associated with Waldenström macroglobulinemia and cases associated with multiple myeloma with concurrent renal failure. In patients with neuropathy, a lenalidomide-based treatment can be employed. Patients should be instructed to keep extremities warm.2 Diabetic foot care guidelines should be followed to prevent wound complications. The differential diagnosis for type I cryoglobulinemia includes other causes of retiform purpura–like angioinvasive fungal infection, antiphospholipid antibody syndrome, calciphylaxis, and livedoid vasculopathy.5 Angioinvasive fungal infections are caused by Candida, Aspergillus, and Mucorales species, as well as other hyaline molds. They typically occur in immunocompromised patients and invade the blood vessels via direct inoculation or dissemination.6 Patients present with retiform purpura but typically will be acutely ill with fevers and vital sign abnormalities. Histopathology with special stains often will identify the fungal organisms in the dermis or inside blood vessel walls with vessel wall destruction and hemorrhage.7 Accurate diagnosis is essential to selecting appropriate antifungal agents. If angioinvasive fungal infection is clinically suspected, treatment should begin before culture and histopathologic data are available.7
Antiphospholipid antibody syndrome is an autoimmune thrombophilia that can occur as primary disease or in association with other autoimmune conditions, most commonly systemic lupus erythematosus. Diagnosis requires the presence of antiphospholipid antibodies, such as lupus anticoagulant, anticardiolipin antibody, anti–β2-glycoprotein-1 antibody, with arterial or venous thrombosis and/or recurrent pregnancy loss. Paraproteinemia is not seen. The most common cutaneous finding is livedo reticularis, with livedo racemosa being a more distinctive finding.8 Small vessel thrombosis is seen histopathologically. Treatment includes antiplatelet and anticoagulant medications. Patients with refractory disease may benefit from additional therapy with hydroxychloroquine or intravenous immunoglobulins.8
Calciphylaxis is a rare depositional vasculopathy that often occurs in patients with end-stage renal disease on dialysis. Patients present with painful and poor-healing skin lesions including indurated nodules, violaceous plaques, and retiform purpura that typically affect areas of high adiposity such as the thighs, abdomen, and buttocks.9 Ulceration and superimposed infections are common complications. Histopathologically, small dermal and subcutaneous vessels demonstrate calcification, microthrombosis, and fibrointimal hyperplasia.9 Wound management is critically important in patients with calciphylaxis. Treatment with intravenous sodium thiosulfate is typical, but prognosis remains poor. Although livedoid vasculopathy may present with retiform purpura in the ankles, paraproteinemia is not seen and patients frequently present with punched-out ulcerations that tend to heal into atrophie blanche.10 Livedoid vasculopathy has been associated with underlying hypercoagulable states, connective tissue diseases, and chronic venous hypertension. Hypercoagulability and endothelial cell damage contribute to the formation of fibrin thrombi in the superficial dermal blood vessels. Histopathology demonstrates thickening of vessel walls and intraluminal hyaline thrombi. Successful treatment in most cases is achieved with anticoagulation therapy, typically rivaroxaban, especially in patients with underlying hypercoagulability. Antiplatelet therapy also may be considered, while anabolic agents have been shown to be helpful in patients with connective tissue disease.10
The Diagnosis: Type I Cryoglobulinemia
Retiform purpura with overlying necrosis subsequently developed over the course of a week following presentation (Figure 1). A skin biopsy showed fibrin thrombi and congestion of small- and medium-sized blood vessels, consistent with vasculopathy (Figure 2). Urinalysis revealed hematuria and proteinuria. A renal biopsy performed due to a continually elevated serum creatinine level revealed glomerulonephritis with numerous IgG1 lambda–restricted glomerular capillary hyaline thrombi, compatible with a lymphoproliferative disorder–associated type I cryoglobulinemia. A serum cryoglobulin immunofixation test confirmed type I cryoglobulinemia involving monoclonal IgG lambda. The combination of cutaneous, renal, and hematologic findings was consistent with type I cryoglobulinemia. A subsequent bone marrow biopsy demonstrated a CD20+ lambda–restricted plasma cell neoplasm. Initial treatment with high-dose corticosteroids followed by targeted treatment of the underlying hematologic condition with bortezomib, rituximab, and dexamethasone improved the skin disease.
Cryoglobulins are abnormal immunoglobulins that precipitate at temperatures below 37 °C. The persistent presence of cryoglobulins in the serum is termed cryoglobulinemia.1 Type I cryoglobulinemia is distinguished from mixed cryoglobulinemia—types II and III—by the presence of a single monoclonal immunoglobulin, typically IgM or IgG. It is associated with lymphoproliferative disorders, most commonly monoclonal gammopathy of undetermined significance and B-cell malignancies such as Waldenström macroglobulinemia, multiple myeloma, or chronic lymphocytic leukemia. Histopathology shows occlusion of small vessel lumina with homogenous eosinophilic material containing the monoclonal cryoprecipitate.2 Disease manifestations are caused by small vessel occlusion, which leads to ischemia and tissue damage.
Retiform purpura, livedo reticularis/racemosa, and necrosis leading to ulcers are the most common cutaneous clinical findings. Extracutaneous signs include peripheral neuropathy, arthralgia, Raynaud phenomenon, and acrocyanosis. Renal involvement, most commonly glomerulonephritis with associated proteinuria, is noted in 14% to 20% of cases.3,4 An elevated cryocrit can lead to symptoms of hyperviscosity syndrome.2
Treatment is difficult and primarily is focused on addressing the underlying hematologic condition, which is responsible for synthesis of the cryoglobulin. Decreasing cryoglobulin production leads to decreased occlusion of blood vessels, thus alleviating the ischemia and skin damage. Monoclonal gammopathy of undetermined significance–related type I cryoglobulinemia initially is treated with corticosteroids followed by rituximab if a CD20+ B-cell clone is identified.2 Bortezomib is recommended for cases associated with Waldenström macroglobulinemia and cases associated with multiple myeloma with concurrent renal failure. In patients with neuropathy, a lenalidomide-based treatment can be employed. Patients should be instructed to keep extremities warm.2 Diabetic foot care guidelines should be followed to prevent wound complications. The differential diagnosis for type I cryoglobulinemia includes other causes of retiform purpura–like angioinvasive fungal infection, antiphospholipid antibody syndrome, calciphylaxis, and livedoid vasculopathy.5 Angioinvasive fungal infections are caused by Candida, Aspergillus, and Mucorales species, as well as other hyaline molds. They typically occur in immunocompromised patients and invade the blood vessels via direct inoculation or dissemination.6 Patients present with retiform purpura but typically will be acutely ill with fevers and vital sign abnormalities. Histopathology with special stains often will identify the fungal organisms in the dermis or inside blood vessel walls with vessel wall destruction and hemorrhage.7 Accurate diagnosis is essential to selecting appropriate antifungal agents. If angioinvasive fungal infection is clinically suspected, treatment should begin before culture and histopathologic data are available.7
Antiphospholipid antibody syndrome is an autoimmune thrombophilia that can occur as primary disease or in association with other autoimmune conditions, most commonly systemic lupus erythematosus. Diagnosis requires the presence of antiphospholipid antibodies, such as lupus anticoagulant, anticardiolipin antibody, anti–β2-glycoprotein-1 antibody, with arterial or venous thrombosis and/or recurrent pregnancy loss. Paraproteinemia is not seen. The most common cutaneous finding is livedo reticularis, with livedo racemosa being a more distinctive finding.8 Small vessel thrombosis is seen histopathologically. Treatment includes antiplatelet and anticoagulant medications. Patients with refractory disease may benefit from additional therapy with hydroxychloroquine or intravenous immunoglobulins.8
Calciphylaxis is a rare depositional vasculopathy that often occurs in patients with end-stage renal disease on dialysis. Patients present with painful and poor-healing skin lesions including indurated nodules, violaceous plaques, and retiform purpura that typically affect areas of high adiposity such as the thighs, abdomen, and buttocks.9 Ulceration and superimposed infections are common complications. Histopathologically, small dermal and subcutaneous vessels demonstrate calcification, microthrombosis, and fibrointimal hyperplasia.9 Wound management is critically important in patients with calciphylaxis. Treatment with intravenous sodium thiosulfate is typical, but prognosis remains poor. Although livedoid vasculopathy may present with retiform purpura in the ankles, paraproteinemia is not seen and patients frequently present with punched-out ulcerations that tend to heal into atrophie blanche.10 Livedoid vasculopathy has been associated with underlying hypercoagulable states, connective tissue diseases, and chronic venous hypertension. Hypercoagulability and endothelial cell damage contribute to the formation of fibrin thrombi in the superficial dermal blood vessels. Histopathology demonstrates thickening of vessel walls and intraluminal hyaline thrombi. Successful treatment in most cases is achieved with anticoagulation therapy, typically rivaroxaban, especially in patients with underlying hypercoagulability. Antiplatelet therapy also may be considered, while anabolic agents have been shown to be helpful in patients with connective tissue disease.10
- Desbois AC, Cacoub P, Saadoun D. Cryoglobulinemia: an update in 2019. Joint Bone Spine. 2019;86:707-713. doi:10.1016/j .jbspin.2019.01.016
- Muchtar E, Magen H, Gertz MA. How I treat cryoglobulinemia. Blood. 2017;129:289-298. doi:10.1182/blood-2016-09-719773
- Sidana S, Rajkumar SV, Dispenzieri A, et al. Clinical presentation and outcomes of patients with type 1 monoclonal cryoglobulinemia. Am J Hematol. 2017;92:668-673. doi:10.1002/ajh.24745
- Harel S, Mohr M, Jahn I, et al. Clinico-biological characteristics and treatment of type I monoclonal cryoglobulinaemia: a study of 64 cases. Br J Haematol. 2015;168:671-678. doi:10.1111/bjh.13196
- Georgesen C, Fox LP, Harp J. Retiform purpura: a diagnostic approach. J Am Acad Dermatol. 2020;82:783-796. doi:10.1016/j.jaad.2019.07.112
- Shields BE, Rosenbach M, Brown-Joel Z, et al. Angioinvasive fungal infections impacting the skin: background, epidemiology, and clinical presentation. J Am Acad Dermatol. 2019;80:869-880.e5. doi:10.1016/j.jaad.2018.04.059
- Berger AP, Ford BA, Brown-Joel Z, et al. Angioinvasive fungal infections impacting the skin: diagnosis, management, and complications. J Am Acad Dermatol. 2019;80:883-898.e2. doi:10.1016/j.jaad.2018.04.058
- Negrini S, Pappalardo F, Murdaca G, et al. The antiphospholipid syndrome: from pathophysiology to treatment. Clin Exp Med. 2017;17:257-267. doi:10.1007/s10238-016-0430-5
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146. doi:10.1053/j.ajkd.2015.01.034
- Georgesen C, Fox LP, Harp J. Retiform purpura: workup and therapeutic considerations in select conditions. J Am Acad Dermatol. 2020;82:799-816. doi:10.1016/j.jaad.2019.07.113
- Desbois AC, Cacoub P, Saadoun D. Cryoglobulinemia: an update in 2019. Joint Bone Spine. 2019;86:707-713. doi:10.1016/j .jbspin.2019.01.016
- Muchtar E, Magen H, Gertz MA. How I treat cryoglobulinemia. Blood. 2017;129:289-298. doi:10.1182/blood-2016-09-719773
- Sidana S, Rajkumar SV, Dispenzieri A, et al. Clinical presentation and outcomes of patients with type 1 monoclonal cryoglobulinemia. Am J Hematol. 2017;92:668-673. doi:10.1002/ajh.24745
- Harel S, Mohr M, Jahn I, et al. Clinico-biological characteristics and treatment of type I monoclonal cryoglobulinaemia: a study of 64 cases. Br J Haematol. 2015;168:671-678. doi:10.1111/bjh.13196
- Georgesen C, Fox LP, Harp J. Retiform purpura: a diagnostic approach. J Am Acad Dermatol. 2020;82:783-796. doi:10.1016/j.jaad.2019.07.112
- Shields BE, Rosenbach M, Brown-Joel Z, et al. Angioinvasive fungal infections impacting the skin: background, epidemiology, and clinical presentation. J Am Acad Dermatol. 2019;80:869-880.e5. doi:10.1016/j.jaad.2018.04.059
- Berger AP, Ford BA, Brown-Joel Z, et al. Angioinvasive fungal infections impacting the skin: diagnosis, management, and complications. J Am Acad Dermatol. 2019;80:883-898.e2. doi:10.1016/j.jaad.2018.04.058
- Negrini S, Pappalardo F, Murdaca G, et al. The antiphospholipid syndrome: from pathophysiology to treatment. Clin Exp Med. 2017;17:257-267. doi:10.1007/s10238-016-0430-5
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146. doi:10.1053/j.ajkd.2015.01.034
- Georgesen C, Fox LP, Harp J. Retiform purpura: workup and therapeutic considerations in select conditions. J Am Acad Dermatol. 2020;82:799-816. doi:10.1016/j.jaad.2019.07.113
A 58-year-old man presented with a petechial and purpuric rash limited to the lower extremities. He reported that the rash had been present for months but worsened acutely over the last 3 days with new-onset dark urine, joint pain, and edema limiting his ability to walk. Physical examination showed areas of violaceous macules and papules on the legs and dorsal feet in a reticular distribution. Laboratory findings were remarkable for an elevated serum creatinine level of 2.75 mg/dL (reference range, 0.70–1.30 mg/dL), and serum immunofixation revealed the presence of markedly elevated IgG lambda monoclonal proteins. He was afebrile and his vital signs were stable. Dermatology, nephrology, and rheumatology services were consulted.


Spreading Painful Lesions on the Legs
The Diagnosis: Cutaneous Leishmaniasis
A punch biopsy of the skin showed pseudoepitheliomatous hyperplasia of the epidermis with dermal granulomatous and suppurative inflammation; tissue cultures remained sterile. Polymerase chain reaction testing of the skin revealed the presence of Leishmania guyanensis complex. Leishmaniasis is a widespread parasitic disease transmitted via sandflies that often is seen in children and young adults.1 Although leishmaniasis is endemic to several countries within Southeast Asia, East Africa, and Latin America, an increase in international travel has brought the disease to nonendemic regions. Therefore, it is crucial to obtain a detailed history of travel and exposure to sandflies in patients who have recently returned from endemic regions.
Leishmaniasis may present in 3 forms: cutaneous, mucocutaneous, or visceral. Cutaneous clinical findings vary depending on disease stage, causative species, and host immune activation. Presentation following a sandfly bite typically includes a papule that progresses to an erythematous nodule. Cutaneous leishmaniasis commonly occurs in areas of the body that are easily accessible to sandflies, such as the face, neck, and limbs. Mucocutaneous leishmaniasis presents with nasal or oral involvement several years after the onset of cutaneous leishmaniasis; however, it can coexist with cutaneous involvement. Without treatment, mucocutaneous leishmaniasis may lead to perforation of the nasal septum, destruction of the mouth, and life-threatening airway obstruction.1 Determining the specific species is important due to the variation in treatment options and prognosis. Because Leishmania organisms are fastidious, obtaining a positive culture often is challenging. Polymerase chain reaction can be utilized for identification, with detection rates of 97%.1 Systemic treatment is indicated for patients with multiple or large lesions; lesions on the hands, feet, face, or joints; or immunocompromised patients. Antimonial drugs are the first-line treatment for most forms of leishmaniasis, though increasing resistance has led to a decrease in efficacy.1 Our patient ultimately was treated with 4 weeks of miltefosine 50 mg 3 times daily. She obtained full resolution of the lesions with no further treatment indicated.
Pemphigus vegetans may present with various clinical manifestations that often can lead to a delay in diagnosis. The Hallopeau subtype typically presents as pustular lesions, while the Neumann subtype may present as large vesiculobullous erosive lesions that rupture and form verrucous, crusted, vegetative plaques. The groin, inguinal folds, axillae, thighs, and flexural areas commonly are affected, but reports of nasal, vaginal, and conjunctival involvement also exist.2
Granuloma inguinale is a sexually transmitted ulcerative disease that is caused by infection with Klebsiella granulomatis. It typically is found in tropical and subtropical climates, including Australia, Brazil, India, and South Africa. The initial presentation includes a single papule or multiple papules or nodules in the genital area that progress to a painless ulcer. It can be diagnosed via biopsies or tissue smears, which will demonstrate the presence of inclusion bodies known as Donovan bodies.3
Cutaneous tuberculosis (TB) can have variable clinical presentations and may be acquired exogenously or endogenously. Cutaneous TB can be divided into 2 categories: exogenous TB caused by inoculation and endogenous TB due to direct spread or autoinoculation. Exogenous TB subtypes include tuberculous chancre and TB verrucosa cutis, while endogenous TB includes scrofuloderma, orificial TB, and lupus vulgaris.4 Patches and plaques are found in patients with lupus vulgaris and TB verrucosa cutis. Scrofuloderma, tuberculous chancre, and orificial TB can present as ulcerative or erosive lesions. Cutaneous TB infection can be diagnosed through a smear, culture, or polymerase chain reaction.4
Deep cutaneous fungal infections most commonly present in immunocompromised individuals, particularly those who are severely neutropenic and are receiving broad-spectrum systemic antimicrobial agents. Deep cutaneous fungal infections initially present as a papule and evolve into a pustule followed by a necrotic ulcer. The lesions typically are accompanied by a fever and/or vital sign abnormalities.5
- Pace D. Leishmaniasis [published online September 17, 2014]. J Infect. 2014;69(suppl 1):S10-S18. doi:10.1016/j.jinf.2014.07.016
- Messersmith L, Krauland K. Pemphigus vegetans. StatPearls. StatPearls Publishing; 2022.
- Ornelas J, Kiuru M, Konia T, et al. Granuloma inguinale in a 51-year-old man. Dermatol Online J. 2016;22:13030/qt52k0c4hj.
- Chen Q, Chen W, Hao F. Cutaneous tuberculosis: a great imitator. Clin Dermatol. 2019;37:192-199.
- Marcoux D, Jafarian F, Joncas V, et al. Deep cutaneous fungal infections in immunocompromised children. J Am Acad Dermatol. 2009;61:857-864.
The Diagnosis: Cutaneous Leishmaniasis
A punch biopsy of the skin showed pseudoepitheliomatous hyperplasia of the epidermis with dermal granulomatous and suppurative inflammation; tissue cultures remained sterile. Polymerase chain reaction testing of the skin revealed the presence of Leishmania guyanensis complex. Leishmaniasis is a widespread parasitic disease transmitted via sandflies that often is seen in children and young adults.1 Although leishmaniasis is endemic to several countries within Southeast Asia, East Africa, and Latin America, an increase in international travel has brought the disease to nonendemic regions. Therefore, it is crucial to obtain a detailed history of travel and exposure to sandflies in patients who have recently returned from endemic regions.
Leishmaniasis may present in 3 forms: cutaneous, mucocutaneous, or visceral. Cutaneous clinical findings vary depending on disease stage, causative species, and host immune activation. Presentation following a sandfly bite typically includes a papule that progresses to an erythematous nodule. Cutaneous leishmaniasis commonly occurs in areas of the body that are easily accessible to sandflies, such as the face, neck, and limbs. Mucocutaneous leishmaniasis presents with nasal or oral involvement several years after the onset of cutaneous leishmaniasis; however, it can coexist with cutaneous involvement. Without treatment, mucocutaneous leishmaniasis may lead to perforation of the nasal septum, destruction of the mouth, and life-threatening airway obstruction.1 Determining the specific species is important due to the variation in treatment options and prognosis. Because Leishmania organisms are fastidious, obtaining a positive culture often is challenging. Polymerase chain reaction can be utilized for identification, with detection rates of 97%.1 Systemic treatment is indicated for patients with multiple or large lesions; lesions on the hands, feet, face, or joints; or immunocompromised patients. Antimonial drugs are the first-line treatment for most forms of leishmaniasis, though increasing resistance has led to a decrease in efficacy.1 Our patient ultimately was treated with 4 weeks of miltefosine 50 mg 3 times daily. She obtained full resolution of the lesions with no further treatment indicated.
Pemphigus vegetans may present with various clinical manifestations that often can lead to a delay in diagnosis. The Hallopeau subtype typically presents as pustular lesions, while the Neumann subtype may present as large vesiculobullous erosive lesions that rupture and form verrucous, crusted, vegetative plaques. The groin, inguinal folds, axillae, thighs, and flexural areas commonly are affected, but reports of nasal, vaginal, and conjunctival involvement also exist.2
Granuloma inguinale is a sexually transmitted ulcerative disease that is caused by infection with Klebsiella granulomatis. It typically is found in tropical and subtropical climates, including Australia, Brazil, India, and South Africa. The initial presentation includes a single papule or multiple papules or nodules in the genital area that progress to a painless ulcer. It can be diagnosed via biopsies or tissue smears, which will demonstrate the presence of inclusion bodies known as Donovan bodies.3
Cutaneous tuberculosis (TB) can have variable clinical presentations and may be acquired exogenously or endogenously. Cutaneous TB can be divided into 2 categories: exogenous TB caused by inoculation and endogenous TB due to direct spread or autoinoculation. Exogenous TB subtypes include tuberculous chancre and TB verrucosa cutis, while endogenous TB includes scrofuloderma, orificial TB, and lupus vulgaris.4 Patches and plaques are found in patients with lupus vulgaris and TB verrucosa cutis. Scrofuloderma, tuberculous chancre, and orificial TB can present as ulcerative or erosive lesions. Cutaneous TB infection can be diagnosed through a smear, culture, or polymerase chain reaction.4
Deep cutaneous fungal infections most commonly present in immunocompromised individuals, particularly those who are severely neutropenic and are receiving broad-spectrum systemic antimicrobial agents. Deep cutaneous fungal infections initially present as a papule and evolve into a pustule followed by a necrotic ulcer. The lesions typically are accompanied by a fever and/or vital sign abnormalities.5
The Diagnosis: Cutaneous Leishmaniasis
A punch biopsy of the skin showed pseudoepitheliomatous hyperplasia of the epidermis with dermal granulomatous and suppurative inflammation; tissue cultures remained sterile. Polymerase chain reaction testing of the skin revealed the presence of Leishmania guyanensis complex. Leishmaniasis is a widespread parasitic disease transmitted via sandflies that often is seen in children and young adults.1 Although leishmaniasis is endemic to several countries within Southeast Asia, East Africa, and Latin America, an increase in international travel has brought the disease to nonendemic regions. Therefore, it is crucial to obtain a detailed history of travel and exposure to sandflies in patients who have recently returned from endemic regions.
Leishmaniasis may present in 3 forms: cutaneous, mucocutaneous, or visceral. Cutaneous clinical findings vary depending on disease stage, causative species, and host immune activation. Presentation following a sandfly bite typically includes a papule that progresses to an erythematous nodule. Cutaneous leishmaniasis commonly occurs in areas of the body that are easily accessible to sandflies, such as the face, neck, and limbs. Mucocutaneous leishmaniasis presents with nasal or oral involvement several years after the onset of cutaneous leishmaniasis; however, it can coexist with cutaneous involvement. Without treatment, mucocutaneous leishmaniasis may lead to perforation of the nasal septum, destruction of the mouth, and life-threatening airway obstruction.1 Determining the specific species is important due to the variation in treatment options and prognosis. Because Leishmania organisms are fastidious, obtaining a positive culture often is challenging. Polymerase chain reaction can be utilized for identification, with detection rates of 97%.1 Systemic treatment is indicated for patients with multiple or large lesions; lesions on the hands, feet, face, or joints; or immunocompromised patients. Antimonial drugs are the first-line treatment for most forms of leishmaniasis, though increasing resistance has led to a decrease in efficacy.1 Our patient ultimately was treated with 4 weeks of miltefosine 50 mg 3 times daily. She obtained full resolution of the lesions with no further treatment indicated.
Pemphigus vegetans may present with various clinical manifestations that often can lead to a delay in diagnosis. The Hallopeau subtype typically presents as pustular lesions, while the Neumann subtype may present as large vesiculobullous erosive lesions that rupture and form verrucous, crusted, vegetative plaques. The groin, inguinal folds, axillae, thighs, and flexural areas commonly are affected, but reports of nasal, vaginal, and conjunctival involvement also exist.2
Granuloma inguinale is a sexually transmitted ulcerative disease that is caused by infection with Klebsiella granulomatis. It typically is found in tropical and subtropical climates, including Australia, Brazil, India, and South Africa. The initial presentation includes a single papule or multiple papules or nodules in the genital area that progress to a painless ulcer. It can be diagnosed via biopsies or tissue smears, which will demonstrate the presence of inclusion bodies known as Donovan bodies.3
Cutaneous tuberculosis (TB) can have variable clinical presentations and may be acquired exogenously or endogenously. Cutaneous TB can be divided into 2 categories: exogenous TB caused by inoculation and endogenous TB due to direct spread or autoinoculation. Exogenous TB subtypes include tuberculous chancre and TB verrucosa cutis, while endogenous TB includes scrofuloderma, orificial TB, and lupus vulgaris.4 Patches and plaques are found in patients with lupus vulgaris and TB verrucosa cutis. Scrofuloderma, tuberculous chancre, and orificial TB can present as ulcerative or erosive lesions. Cutaneous TB infection can be diagnosed through a smear, culture, or polymerase chain reaction.4
Deep cutaneous fungal infections most commonly present in immunocompromised individuals, particularly those who are severely neutropenic and are receiving broad-spectrum systemic antimicrobial agents. Deep cutaneous fungal infections initially present as a papule and evolve into a pustule followed by a necrotic ulcer. The lesions typically are accompanied by a fever and/or vital sign abnormalities.5
- Pace D. Leishmaniasis [published online September 17, 2014]. J Infect. 2014;69(suppl 1):S10-S18. doi:10.1016/j.jinf.2014.07.016
- Messersmith L, Krauland K. Pemphigus vegetans. StatPearls. StatPearls Publishing; 2022.
- Ornelas J, Kiuru M, Konia T, et al. Granuloma inguinale in a 51-year-old man. Dermatol Online J. 2016;22:13030/qt52k0c4hj.
- Chen Q, Chen W, Hao F. Cutaneous tuberculosis: a great imitator. Clin Dermatol. 2019;37:192-199.
- Marcoux D, Jafarian F, Joncas V, et al. Deep cutaneous fungal infections in immunocompromised children. J Am Acad Dermatol. 2009;61:857-864.
- Pace D. Leishmaniasis [published online September 17, 2014]. J Infect. 2014;69(suppl 1):S10-S18. doi:10.1016/j.jinf.2014.07.016
- Messersmith L, Krauland K. Pemphigus vegetans. StatPearls. StatPearls Publishing; 2022.
- Ornelas J, Kiuru M, Konia T, et al. Granuloma inguinale in a 51-year-old man. Dermatol Online J. 2016;22:13030/qt52k0c4hj.
- Chen Q, Chen W, Hao F. Cutaneous tuberculosis: a great imitator. Clin Dermatol. 2019;37:192-199.
- Marcoux D, Jafarian F, Joncas V, et al. Deep cutaneous fungal infections in immunocompromised children. J Am Acad Dermatol. 2009;61:857-864.
A 14-year-old adolescent girl presented with spreading painful lesions on the legs and left forearm of 2 years’ duration. Her travel history included several countries in South and Central America, traversing the Colombian jungle on foot. Near the end of the jungle trip, she noted a skin lesion on the left forearm around the site of an insect bite. Within 1 month, the lesions spread to the legs. She was treated with topical corticosteroids without improvement. Physical examination revealed verrucous, reddish-brown plaques on the legs and left forearm. Intranasal examination revealed a red rounded lesion inside the left nostril.


Diffuse Urticarial Rash in a Pregnant Patient
The Diagnosis: Pemphigoid Gestationis
A lesional biopsy showed a subepidermal split with eosinophils and neutrophils. Perilesional biopsy for direct immunofluorescence (DIF) showed linear deposition of 3+ C3 along the basement membrane zone. The clinical, histopathologic, and immunofluorescent findings were consistent with pemphigoid gestationis (PG). Prednisone 1 mg/kg daily was initiated. Her condition continued to worsen, and cyclosporine 250 mg daily was added while prednisone was tapered, with remission of disease.
Pemphigoid gestationis is an autoimmune bullous dermatosis that occurs in the second or third trimester of pregnancy, with an incidence of 1 in 50,000 to 60,000 pregnancies.1 In terms of pathogenesis, aberrant expression of major histocompatibility complex class II molecules on placental tissues causes the loss of immune tolerance of the placenta, which leads to the production of antibodies against the placental protein bullous pemphigoid 180.2 Bullous pemphigoid 180 also is a hemidesmosomal protein found in the skin of the mother; therefore, the presence of the circulating antibodies leads to separation at the dermoepidermal junction and vesiculation.
Pemphigoid gestationis is characterized by the sudden eruption of intensely pruritic urticarial papules and plaques, classically with periumbilical involvement. Tense vesicles and bullae can develop. Women with PG have an increased risk for development of Graves disease. Histopathology shows subepidermal vesiculation with a predominance of eosinophils. Direct immunofluorescence classically shows linear deposition of C3 along the basement membrane zone. Fetal complications include prematurity and small size for gestational age. Additionally, blisters can be seen in 5% to 10% of neonates due to placental transmission of autoantibodies.3
Frequently PG flares shortly postpartum. Pemphigoid gestationis resolves within 6 months postdelivery but frequently reoccurs in subsequent pregnancies. Mild disease can be treated with mid- to high-potency topical corticosteroids. Severe disease is managed with oral corticosteroids, most commonly prednisone. Refractory disease is managed with azathioprine, cyclosporine, intravenous immunoglobulin, or plasmapheresis.
The differential diagnosis of PG includes other pregnancy-associated dermatoses such as atopic eruption of pregnancy, impetigo herpetiformis, intrahepatic cholestasis of pregnancy, and polymorphous eruption of pregnancy. Atopic eruption of pregnancy is the most common dermatosis of pregnancy and is characterized by an eczematous eruption in patients with an atopic history, typically in the first trimester. Blisters are not seen, and DIF is negative. Impetigo herpetiformis, or pustular psoriasis of pregnancy, is a variant of generalized pustular psoriasis that occurs during pregnancy. Diffuse erythematous patches studded with pustules, rather than vesicles, are seen; DIF is negative. Intrahepatic cholestasis of pregnancy presents without primary skin findings and severe pruritus predominantly on the palms and soles, often with secondary excoriations. Polymorphous eruption of pregnancy presents as a polymorphous eruption of urticarial to erythematous papules and plaques commonly originating in striae. In contrast to PG, there is periumbilical sparing, vesiculation is rare, and DIF is negative.
- Shornick JK, Bangert JL, Freeman RG, et al. Herpes gestationis: clinical and histologic features of twenty-eight cases. J Am Acad Dermatol. 1983;8:214-224.
- Sadik CD, Lima AL, Zillikens D. Pemphigoid gestationis: toward a better understanding of the etiopathogenesis. Clin Dermatol. 2016;34:378-382.
- Shornick JK, Black MM. Fetal risks in herpes gestationis. J Am Acad Dermatol. 1992;26:63-68.
The Diagnosis: Pemphigoid Gestationis
A lesional biopsy showed a subepidermal split with eosinophils and neutrophils. Perilesional biopsy for direct immunofluorescence (DIF) showed linear deposition of 3+ C3 along the basement membrane zone. The clinical, histopathologic, and immunofluorescent findings were consistent with pemphigoid gestationis (PG). Prednisone 1 mg/kg daily was initiated. Her condition continued to worsen, and cyclosporine 250 mg daily was added while prednisone was tapered, with remission of disease.
Pemphigoid gestationis is an autoimmune bullous dermatosis that occurs in the second or third trimester of pregnancy, with an incidence of 1 in 50,000 to 60,000 pregnancies.1 In terms of pathogenesis, aberrant expression of major histocompatibility complex class II molecules on placental tissues causes the loss of immune tolerance of the placenta, which leads to the production of antibodies against the placental protein bullous pemphigoid 180.2 Bullous pemphigoid 180 also is a hemidesmosomal protein found in the skin of the mother; therefore, the presence of the circulating antibodies leads to separation at the dermoepidermal junction and vesiculation.
Pemphigoid gestationis is characterized by the sudden eruption of intensely pruritic urticarial papules and plaques, classically with periumbilical involvement. Tense vesicles and bullae can develop. Women with PG have an increased risk for development of Graves disease. Histopathology shows subepidermal vesiculation with a predominance of eosinophils. Direct immunofluorescence classically shows linear deposition of C3 along the basement membrane zone. Fetal complications include prematurity and small size for gestational age. Additionally, blisters can be seen in 5% to 10% of neonates due to placental transmission of autoantibodies.3
Frequently PG flares shortly postpartum. Pemphigoid gestationis resolves within 6 months postdelivery but frequently reoccurs in subsequent pregnancies. Mild disease can be treated with mid- to high-potency topical corticosteroids. Severe disease is managed with oral corticosteroids, most commonly prednisone. Refractory disease is managed with azathioprine, cyclosporine, intravenous immunoglobulin, or plasmapheresis.
The differential diagnosis of PG includes other pregnancy-associated dermatoses such as atopic eruption of pregnancy, impetigo herpetiformis, intrahepatic cholestasis of pregnancy, and polymorphous eruption of pregnancy. Atopic eruption of pregnancy is the most common dermatosis of pregnancy and is characterized by an eczematous eruption in patients with an atopic history, typically in the first trimester. Blisters are not seen, and DIF is negative. Impetigo herpetiformis, or pustular psoriasis of pregnancy, is a variant of generalized pustular psoriasis that occurs during pregnancy. Diffuse erythematous patches studded with pustules, rather than vesicles, are seen; DIF is negative. Intrahepatic cholestasis of pregnancy presents without primary skin findings and severe pruritus predominantly on the palms and soles, often with secondary excoriations. Polymorphous eruption of pregnancy presents as a polymorphous eruption of urticarial to erythematous papules and plaques commonly originating in striae. In contrast to PG, there is periumbilical sparing, vesiculation is rare, and DIF is negative.
The Diagnosis: Pemphigoid Gestationis
A lesional biopsy showed a subepidermal split with eosinophils and neutrophils. Perilesional biopsy for direct immunofluorescence (DIF) showed linear deposition of 3+ C3 along the basement membrane zone. The clinical, histopathologic, and immunofluorescent findings were consistent with pemphigoid gestationis (PG). Prednisone 1 mg/kg daily was initiated. Her condition continued to worsen, and cyclosporine 250 mg daily was added while prednisone was tapered, with remission of disease.
Pemphigoid gestationis is an autoimmune bullous dermatosis that occurs in the second or third trimester of pregnancy, with an incidence of 1 in 50,000 to 60,000 pregnancies.1 In terms of pathogenesis, aberrant expression of major histocompatibility complex class II molecules on placental tissues causes the loss of immune tolerance of the placenta, which leads to the production of antibodies against the placental protein bullous pemphigoid 180.2 Bullous pemphigoid 180 also is a hemidesmosomal protein found in the skin of the mother; therefore, the presence of the circulating antibodies leads to separation at the dermoepidermal junction and vesiculation.
Pemphigoid gestationis is characterized by the sudden eruption of intensely pruritic urticarial papules and plaques, classically with periumbilical involvement. Tense vesicles and bullae can develop. Women with PG have an increased risk for development of Graves disease. Histopathology shows subepidermal vesiculation with a predominance of eosinophils. Direct immunofluorescence classically shows linear deposition of C3 along the basement membrane zone. Fetal complications include prematurity and small size for gestational age. Additionally, blisters can be seen in 5% to 10% of neonates due to placental transmission of autoantibodies.3
Frequently PG flares shortly postpartum. Pemphigoid gestationis resolves within 6 months postdelivery but frequently reoccurs in subsequent pregnancies. Mild disease can be treated with mid- to high-potency topical corticosteroids. Severe disease is managed with oral corticosteroids, most commonly prednisone. Refractory disease is managed with azathioprine, cyclosporine, intravenous immunoglobulin, or plasmapheresis.
The differential diagnosis of PG includes other pregnancy-associated dermatoses such as atopic eruption of pregnancy, impetigo herpetiformis, intrahepatic cholestasis of pregnancy, and polymorphous eruption of pregnancy. Atopic eruption of pregnancy is the most common dermatosis of pregnancy and is characterized by an eczematous eruption in patients with an atopic history, typically in the first trimester. Blisters are not seen, and DIF is negative. Impetigo herpetiformis, or pustular psoriasis of pregnancy, is a variant of generalized pustular psoriasis that occurs during pregnancy. Diffuse erythematous patches studded with pustules, rather than vesicles, are seen; DIF is negative. Intrahepatic cholestasis of pregnancy presents without primary skin findings and severe pruritus predominantly on the palms and soles, often with secondary excoriations. Polymorphous eruption of pregnancy presents as a polymorphous eruption of urticarial to erythematous papules and plaques commonly originating in striae. In contrast to PG, there is periumbilical sparing, vesiculation is rare, and DIF is negative.
- Shornick JK, Bangert JL, Freeman RG, et al. Herpes gestationis: clinical and histologic features of twenty-eight cases. J Am Acad Dermatol. 1983;8:214-224.
- Sadik CD, Lima AL, Zillikens D. Pemphigoid gestationis: toward a better understanding of the etiopathogenesis. Clin Dermatol. 2016;34:378-382.
- Shornick JK, Black MM. Fetal risks in herpes gestationis. J Am Acad Dermatol. 1992;26:63-68.
- Shornick JK, Bangert JL, Freeman RG, et al. Herpes gestationis: clinical and histologic features of twenty-eight cases. J Am Acad Dermatol. 1983;8:214-224.
- Sadik CD, Lima AL, Zillikens D. Pemphigoid gestationis: toward a better understanding of the etiopathogenesis. Clin Dermatol. 2016;34:378-382.
- Shornick JK, Black MM. Fetal risks in herpes gestationis. J Am Acad Dermatol. 1992;26:63-68.
A 29-year-old pregnant woman at 18 weeks and 5 days of gestation presented with a diffuse, pruritic, blistering rash of 5 weeks’ duration that started on the forearms and generalized to affect the trunk, legs, palms, and soles. Physical examination showed diffuse urticarial papules and plaques with small tense vesicles with an annular configuration on the abdomen and marked periumbilical involvement.

