User login
Type VII Collagen Disorders Simplified
Type VII Collagen Disorders Simplified
There are 3 uncommon types of mechanobullous skin diseases caused by relative reduction or complete loss of functional type VII collagen, which is the main component of anchoring fibrils in the lamina densa of the basement membrane zone (BMZ) of the skin and mucous membrane epithelium.1 The function of the anchoring fibrils is to maintain adherence of the basement membrane of the epithelium to the connective tissue of the papillary dermis and submucosa.1 The mechanism of action of the loss of type VII collagen function is via autoimmunity in epidermolysis bullosa acquisita (EBA)2 and
Epidermolysis Bullosa
Epidermolysis bullosa consists of a heterogeneous family of 4 major genetic mechanobullous diseases that affect the skin and mucous membranes with more than 30 subtypes.1 Dystrophic EB is caused by mutations in the COL7A1 gene, which encodes for the α-1 chain of collagen type VII. Classically, EB is divided into 4 main variants based on the location of the cleavage plane or split occurring in the epithelium, which in turn helps to predict the severity of the illness.
Epidermolysis bullosa may be inherited in an autosomal-dominant or autosomal-recessive fashion, or it may occur as a spontaneous mutation. All sexes and races are affected equally. Patients present at birth or in early childhood with fragile skin and mucous membranes that may develop blisters, erosions, and ulcerations after minor trauma.7 These lesions are marked by slow healing and scar formation and often are associated with itching and pain.
Dystrophic Epidermolysis Bullosa
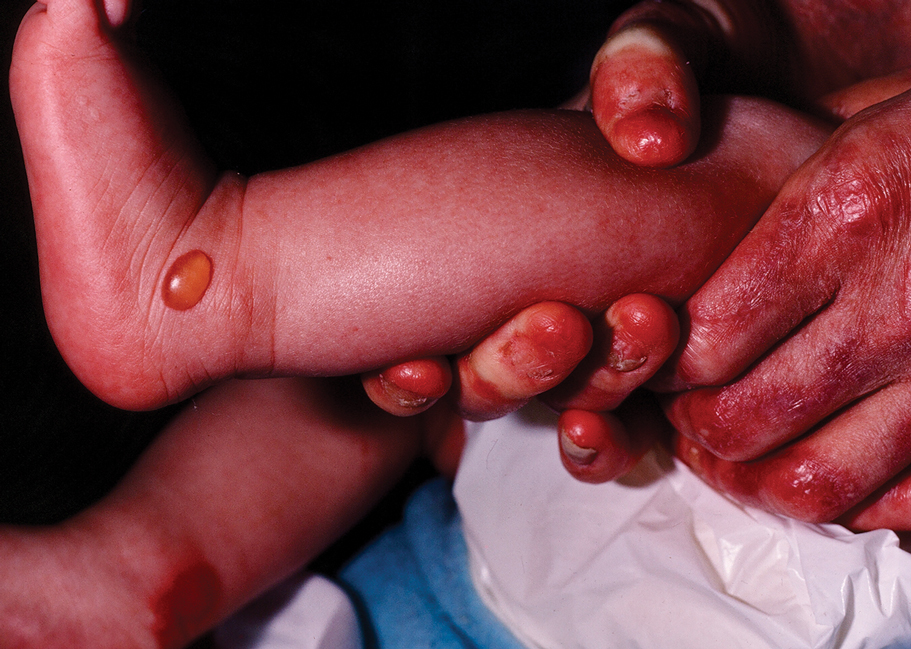
Dystrophic EB accounts for approximately 25%6 of all EB cases in the United States and may be inherited as either a dominant or recessive trait. Hundreds of different pathogenic mutations have been discovered in the COL7A1 gene in the subtypes of DEB.4,8 Dominant DEB tends to cause milder disease because the patients retain one normal COL7A1 allele and produce some type VII collagen (Figure 1), whereas patients with recessive DEB lack type VII collagen completely.9 The cleavage plane is between the lamina densa and the superficial dermis or submucosa. Severity is variable and ranges from localization to the hands and feet to severe generalized blistering and painful ulcerations depending on which of the many possible gene mutations have been inherited. Sequelae include mitten deformities, malalignment and tooth decay, and the development of early aggressive squamous cell carcinomas, which may be fatal. The most severe cases of recessive DEB also may have internal organ involvement.
Epidermolysis Bullosa Simplex
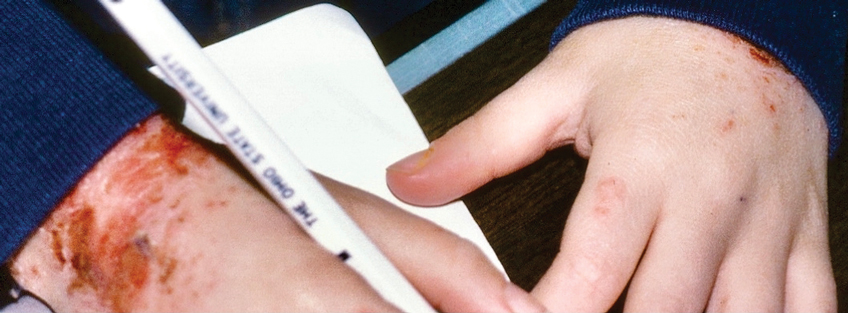
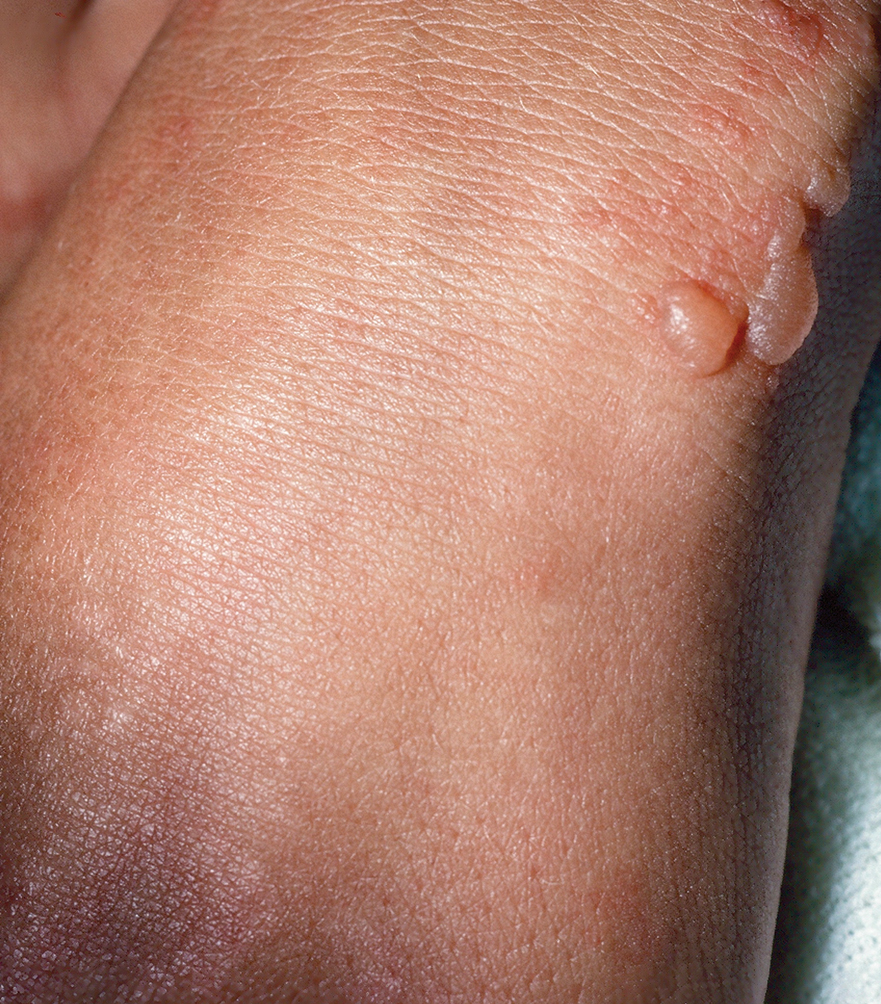
Epidermolysis bullosa simplex is the most common variant, comprising approximately 70%of EB cases in the United States.6 Epidermolysis bullosa simplex usually is inherited as autosomal-dominant mutations in the keratin 5 or keratin 14 genes,10 not COL7A1. Skin blistering results from cleavage within the basal cell layer where the keratin genes are primarily expressed. Blisters tend to occur in acral areas such as hands and feet and may heal without scarring in the localized form of epidermolysis bullosa simplex (Figure 2).
Junctional Epidermolysis Bullosa and Kindler Syndrome
Junctional epidermolysis bullosa (JEB) and Kindler syndrome11 are the rarest of the autosomal-recessive EB variants.6 The plane of cleavage in JEB is through the lamina lucida of the BMZ. Junctional epidermolysis bullosa is caused by mutations of the genes that encode for the 3 chains of laminin 332 protein and type XVII collagen,5,12 not to be confused with type VII collagen. As with DEB, there is a wide range of severity in JEB, from localized effects on the eyes, oral cavity, and tooth enamel to widespread blistering and skin cancers. In JEB cases involving newborns, nonhealing wounds on the face, buttocks, fingers, and toes may be seen, with devastating complications in the oral cavity, esophagus, and larynx. Life expectancy is limited to 2 years or less.6 There have only been approximately 40013 cases of Kindler syndrome reported worldwide6 and there is clinical overlap with DEB. Patients also may demonstrate poikiloderma and photosensitivity. Kindler syndrome is caused by mutations in the FERMT1 gene which encodes for kindlin-1. This protein mediates anchorage between the actin cytoskeleton and the extracellular matrix.5,11 Loss of function produces variable cleavage planes around the dermoepidermal junction.
Clinical management of all EB variants, especially the severe recessive types, traditionally has been limited to the prevention of trauma to the skin and mucous membranes and supportive care, including dressing changes to erosions and ulcerations, antibiotic ointments as needed, and amelioration of pain and pruritus. Bone marrow and pluripotential stem cell transplants have been attempted.12 Complications of EB, such as deformities of the hands and feet caused by excessive scarring, esophageal strictures, poor dentition, and squamous cell carcinomas, must be addressed by a multidisciplinary team of specialists, including plastic surgery, gastroenterology, dentistry/oral surgery, ophthalmology, and dermatology/Mohs surgery.
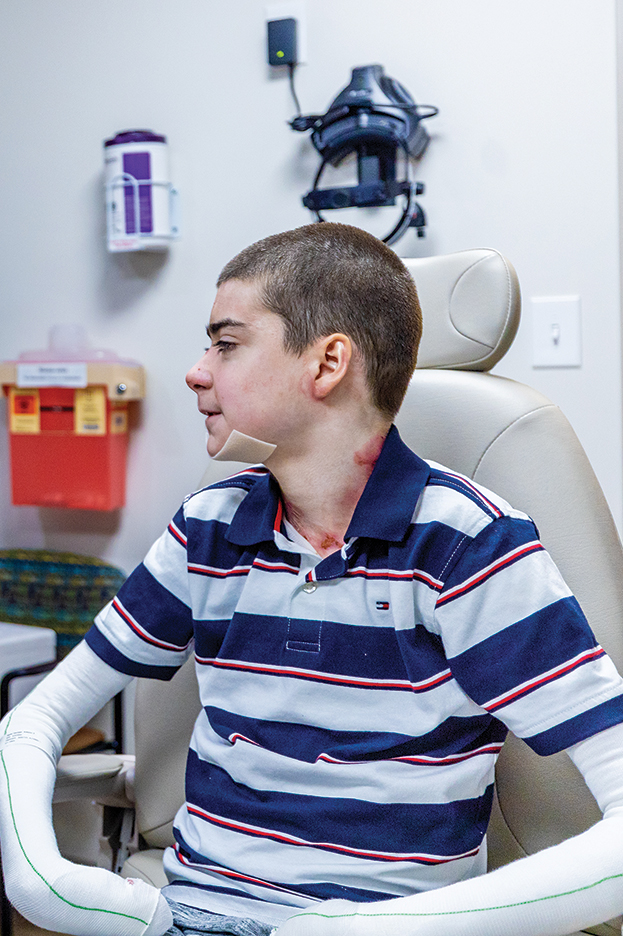
Until recently, there were no medications approved by the US Food and Drug Administration (FDA) specifically indicated for EB. In 2023, topical gene therapy was approved by the FDA for both recessive and dominant forms of DEB. Normal COL7A1 sequences are delivered by an attenuated herpes simplex virus 1 vector (beremagene geperpavec) in a gel applied directly to the wounds of patients with DEB. In a clinical trial, matching wounds on 31 patients (62 wounds total) were treated with the active agent or placebo gel. After 6 months, complete wound closure was observed in 67% (21/31) of those treated with the active agent and 22% (7/31) of those treated with placebo (P=.002).14 In a single case report, a patient with recessive DEB and cicatrizing conjunctivitis (Figure 3) was given ophthalmic beremagene geperpavec after surgery and had improved visual acuity.15 A topical gel consisting of birch triterpenes to promote healing of partial-thickness wounds also was approved for patients with DEB and JEB by the FDA and the European Commission. In a study of 223 patients, 41% of those using active gel and 29% of those using placebo gel achieved the primary end point of percentage of target wounds that had first complete closure at 45 days.16
The most recent FDA approval for DEB involves transferring the functional COL7A1 gene to the patient’s skin cells, then expanding the gene-corrected cells into sheets of keratinocytes that can be surgically applied to the chronic wound sites. In a phase 3 trial of prademagene zamikeracel (pz-cel), 11 patients with 86 matched wounds were randomized to receive pz-cel (50%) or standard wound care (50%). After 24 weeks, 35 wounds treated with pz-cel were at least 50% healed compared to 7 control wounds.17 The results for healing and reduction of pain were statistically significant (P<.0001 and P<.0002, respectively).17 Recombinant collagen VII as replacement therapy also is under study to be given by intravenous infusion to increase tissue collagen VII where it is lacking. This treatment has shown early biologic and therapeutic effects.9,18 Larger long-term follow-up studies are necessary to confirm persistence of the gene-corrected skin cells, the functionality of the replacement collagen VII, and the potential risk for the development of autoantibodies to type VII collagen.
Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita is a rare autoimmune subepithelial bullous disease that primarily affects middle-aged adults but also has been reported in children.19 Epidermolysis bullosa acquisita is caused by circulating pathogenic IgG autoantibodies that target and bind to type VII collagen in the anchoring fibrils,20-22 thereby disrupting the attachment of the epithelium to its underlying connective tissue.
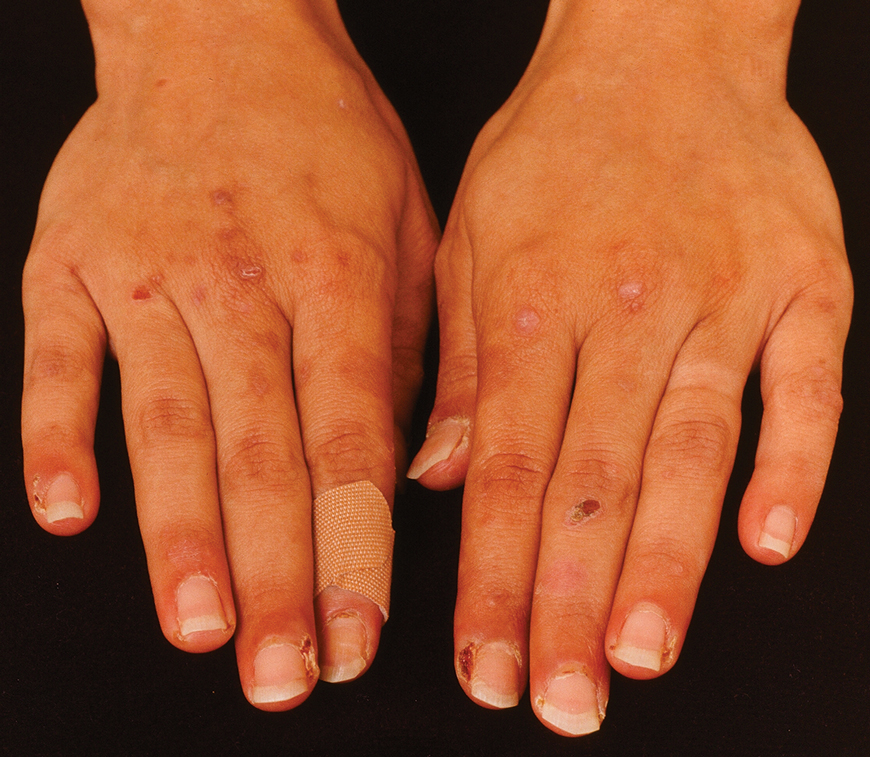
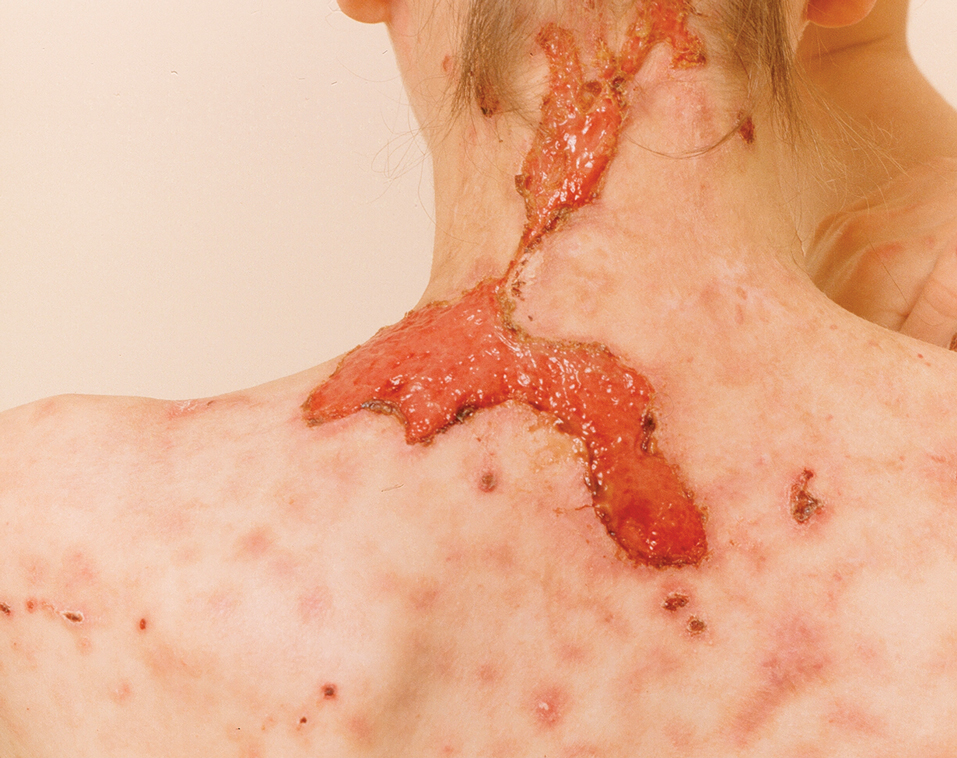
The 2 major clinical manifestations of EBA include a mechanobullous disease resembling inherited forms of DEB (Figure 4) and an inflammatory bullous pemphigoid (BP)–like disease,23 as well as a combination of both types of skin lesions (Figure 5). The skin and mucous membranes of the oral cavity, esophagus, eyes, and urogenital areas are affected in both types; scarring may cause functional disabilities. In the mechanobullous type of EBA, it is common for blisters and erosions to develop in trauma-prone areas such as the hands, feet, elbows, and knees. The blisters tend to heal with scarring and milia formation as might be seen in porphyria cutanea tarda or cicatricial pemphigoid, which are in the differential diagnosis. Dystrophy of the fingernails or complete nail loss may be observed, resembling DEB. In the BP-like presentation, tense blisters arise upon inflamed or urticarial skin and mucous membranes, which may then become generalized.
Histopathology in both forms of EBA demonstrates subepithelial separation as clefts or blisters. The mechanobullous type shows a sparse inflammatory infiltrate compared to large collections of neutrophils and eosinophils in the blister cavity and in the superficial dermis in the BP-like cases. The final diagnosis rests on the results of immunopathology testing.24 Direct immunofluorescence of perilesional skin and mucosa shows a linear-granular band of IgG and C3 and other conjugates along the BMZ. Deposits of IgA alone in EBA occur in only about 2.4% of cases and are observed more often when there is mucous membrane involvement.2 Indirect immunofluorescence of sera against salt-split skin substrates detects immunoreactants in the floor of the blister rather than in the roof, as would be seen in BP. Highly specific and sensitive enzyme-linked immunosorbent assay (ELISA) kits now are commercially available and can detect autoantibodies against the N-terminal domain of type VII collagen in more than 90% of cases of EBA.25
Inflammatory bowel disease (IBD), particularly Crohn disease (CD), precedes the onset of EBA in approximately 25% of cases.26,27 Ulcerative colitis is much less common. Type VII collagen is normally present in the basement membrane of intestinal epithelium. In a survey of patients with IBD, 68% of those with CD and 13% of those with ulcerative colitis had circulating anti–type VII collagen antibodies detected by ELISA without having symptoms of EBA.28 A case report of a patient with both well-proven EBA and CD highlighted the clinical difficulty of controlling EBA: treatment with prednisolone and sulfasalazine improved the CD but had little effect on the skin blisters.29 A variety of malignancies have been reported in association with EBA, including cancers of the uterine cervix,30 thyroid, and pancreas,31 lymphoma, and chronic lymphatic leukemia. Some of these cases have met the criteria for classification as paraneoplastic, whereas others may have been coincidental.
Treatment for chronic EBA generally has been limited.2,24 Putative antineutrophil drugs such as dapsone and colchicine combined with systemic corticosteroids may be useful in milder or juvenile cases, which tend to have a better prognosis than adult cases.19 In more severe EBA, systemic corticosteroids and/or immunosuppressive drugs such as azathioprine,23 cyclophosphamide,23 mycophenolate mofetil,31 methotrexate,23 cyclosporine,33 and infliximab23 have been used. More recently, rituximab infusion monotherapy33 and rituximab combined with intravenous immunoglobulin or
Bullous Systemic Lupus Erythematosus
Bullous systemic lupus erythematosus is a rare and specific autoimmune skin complication that mostly is seen in patients with an established diagnosis of systemic lupus erythematosus (SLE) who are experiencing a disease flare. Although more common in women, it has been reported in all sexes and races as well as in children. Vesicles and bullae may arise on sun-exposed (Figure 6) and sun-protected areas of skin.
Histopathology shows subepidermal separation with collections of neutrophils and nuclear fragments in the blister cavity. The differential diagnosis of BSLE includes EBA, BP, dermatitis herpetiformis, and linear IgA bullous dermatosis. Direct immunofluorescence testing shows linear-granular deposits of IgG and/or IgM and IgA along the BMZ.34 When utilizing the indirect immunofluorescence split-skin assay, the autoantibody to type VII collagen would be detected in the floor of the blister if the serum titer was sufficiently high.3 Proposed criteria for the diagnosis of BSLE have been published: 1) diagnosis of SLE now based on the 2019 European League Against Rheumatism/American College of Rheumatology classification35; 2) vesicles and bullae arising upon but not limited to sun-exposed skin; 3) histopathology featuring neutrophil-rich subepithelial bullae; 4) positive indirect immunofluorescence for circulating BMZ antibodies using separated human skin as substrate; 5) and direct immunofluorescence showing IgG and/or IgM and often IgA at the BMZ.36 Using ELISA to detect circulating antibodies against type VII collagen24 should now be added to the criteria. The new criteria for SLE34 do not include BSLE, perhaps because it occurs in less than 1% of patients with SLE.37
Further investigation by Gammon et al3 confirmed that the autoantibodies in BSLE are identical to those found in EBA (ie, directed against type VII collagen in the lamina densa). Bullous systemic lupus erythematosus is not considered to be the coexistence of EBA with SLE but rather a specific entity wherein type VII collagen autoantibodies are expressed in the autoimmune spectrum of SLE. It is especially important to make the diagnosis of BSLE because it is predictive of more serious systemic complications of SLE (eg, hematologic and renal disease is found in up to 90% of cases).38
The natural course of BSLE is variable. Treatments include systemic corticosteroids, dapsone, and immunosuppressive drugs such as azathioprine, methotrexate, mycophenolate mofetil, and cyclophosphamide, especially in cases with nephritis.37 There may be spontaneous resolution of the rash as the inflammatory activity of SLE subsides. Rituximab has been used effectively in several refractory cases of BSLE that failed to respond to all other conventional treatments.39
Conclusion
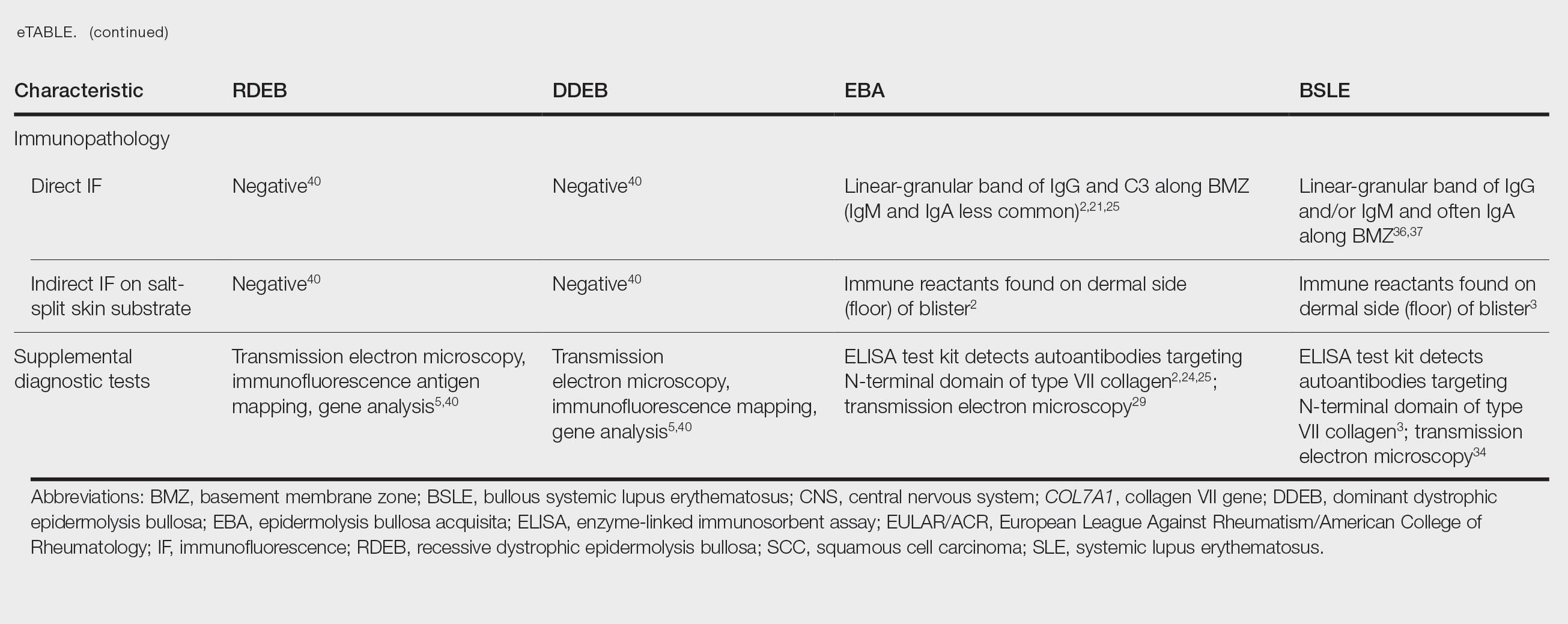
Anchoring fibrils are composed primarily of type VII collagen. Their role is to maintain the attachment of epithelium to the upper dermis and submucosa. The reduction or complete loss of type VII collagen caused by mutations of the COL7A1 gene results in dominant DEB or recessive DEB, respectively. Two distinct non-heritable immunobullous diseases, EBA and BSLE, are caused by autoantibodies that target type VII collagen. A comparison of the 4 type VII collagen disorders can be found in the eTable.
- Bardhan A, Bruckner-Tuderman L, Chapple ILC, et al. Epidermolysis bullosa. Nat Rev Dis Primers. 2020;6:78. doi:10.1038/s41572-020-0210-0
- Miyamoto D, Gordilho JO, Santi CG, et al. Epidermolysis bullosa acquisita. An Bras Dermatol. 2022;97:409-423. doi:10.1016/j.abd.2021.09.010.
- Gammon WR, Woodley DT, Dole KC, et al. Evidence that anti-basement membrane zone antibodies in bullous eruption of systemic lupus erythematosus recognize epidermolysis bullosa acquisita autoantigen. J Invest Dermatol. 1985;84:472-476. doi:10.1111/1523-1747.ep12272402.
- Yadav RS, Jaswal A, Shrestha S, et al. Dystrophic epidermolysis bullosa. J Nepal Med Assoc. 2018;56:879-882. doi:10.31729/jnma.3791
- Mariath LM, Santin JT, Schuler-Faccini L, et al. Inherited epidermolysis bullosa: update on the clinical and genetic aspects. An Bras Dermatol. 2020;95:551-569. doi:10.1016/j.abd.2020.05.001
- Understanding epidermolysis bullosa (EB). DEBRA website. Accessed August 17, 2025. https://www.debra.org/about-eb/understanding-epidermolysis-bullosa-eb
- Hon KL, Chu S, Leung AKC. Epidermolysis bullosa: pediatric perspectives. Curr Pediatr Rev. 2022;18:182-190. doi:10.2174/1573396317666210525161252
- Dang N, Klingberg S, Marr P, et al. Review of collagen VII sequence variants found in Australasian patients with dystrophic epidermolysis bullosa reveals nine COL7A1 variants. J Dermatol Sci. 2007;46:169-178. doi:10.1016/j.jdermsci.2007.02.006
- Payne AS. Topical gene therapy for epidermolysis bullosa. N Engl J Med. 2022;387:2281-2284. doi:10.1056/NEJMe2213203
- Khani P, Ghazi F, Zekri A, et al. Keratins and epidermolysis bullosa simplex. J Cell Physiol. 2018;234:289-297. doi:10.1002/jcp.26898
- Lai-Cheong JE, Tanaka A, Hawche G, et al. Kindler syndrome: a focal adhesion genodermatosis. Br J Dermatol. 2009;160:233-242. doi:10.1111/j.1365-2133.2008.08976.x
- Hou P-C, Wang H-T, Abhee S, et al. Investigational treatments for epidermolysis bullosa. Am J Clin Dermatol. 2021;22:801-817. doi:10.1007/s40257-021-00626-3
- Youseffian L, Vahidnezhad H, Uitto J. Kindler Syndrome. GeneReviews [Internet]. Updated January 6, 2022. Accessed August 21, 2025.
- Guide SV, Gonzalez ME, Bagci S, et al. Trial of beremagene geperpavec (B-VEC) for dystrophic epidermolysis bullosa. N Engl J Med. 2022;387:2211-2219. doi:10.1056/NEJMoa2206663
- Vetencourt AT, Sayed-Ahmed I, Gomez J, et al. Ocular gene therapy in a patient with dystrophic epidermolysis bullosa. N Engl J Med. 2024;390:530-535. doi:10.1056/NEJMoa2301244
- Kern JS, Sprecher E, Fernandez MF, et al. Efficacy and safety of Oleogel-S10 (birch triterpenes for epidermolysis bullosa: results from the phase III randomized double-blind phase of the EASE study. Br J Dermatol. 2023;188:12-21. doi:10.1093/bjd/ljac001
- Tang JY, Marinkovich MP, Wiss K, et al. Prademagene zamikeracel for recessive dystrophic epidermolysis bullosa wounds (VIITAL): a two-centre, randomized, open-label, intrapatient-controlled phase 3 trial. Lancet. 2025;406:163-173. doi:10.1016/S0140-6736(25)00778-0
- Gretzmeier C, Pin D, Kern JS, et al. Systemic collagen VII replacement therapy for advanced recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2022;142:1094-1102. doi:10.1016/j.jid.2021.09.008
- Hignett E, Sami N. Pediatric epidermolysis bullosa acquisita. A review. Pediatr Dermatol. 2021;38:1047-1050. doi:10.1111/pde.14722
- Chen M, Kim GH, Prakash L, et al. Autoimmunity to anchoring fibril collagen. Autoimmunity. 2012;45:91-101. doi:10.1007/s12016-007-0027-6.
- Kridin K, Kneiber D, Kowalski EH, et al. Epidermolysis bullosa acquisita: a comprehensive review. Autoimmun Rev. 2019;18:786-795. doi:10.1016/j.autrev.2019.06.007
- Hofmann SC, Weidinger A. Epidermolysis bullosa acquisita. Hautarzt. 2019;70:265-270. doi:10.1007/s00105-019-4387-7
- Ishi N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37:220-230. doi:10.1111/j.1346-8138.2009.00799.x
- Iwata H, Vorobyev A, Koga H, et al. Meta-analysis of the clinical and immunopathological characteristics and treatment outcomes in epidermolysis bullosa acquisita patients. Orphanet J Rare Dis. 2018;13:153. doi:10.1186/s13023-018-0896-1
- Komorowski L, Muller R, Vorobyev A, et al. Sensitive and specific assays for routine serological diagnosis of epidermolysis bullosa acquisita. J Am Acad Dermatol. 2013;68:e89-95. doi:10.1016/j.jaad.2011.12.032
- Antonelli E, Bassotti G, Tramontana M, et al. Dermatological manifestations in inflammatory bowel diseases. J Clin Med. 2021;10:364-390. doi:10.3390/jcm10020364
- Bezzio C, Della Corte C, Vernero M, et al. Inflammatory bowel disease and immune-mediated inflammatory diseases: looking at less frequent associations. Therap Adv Gastroenterol. 2022;15:17562848221115312. doi:10.1177/17562848221115312
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have antibodies to type VII collagen. J Invest Dermatol. 2002;118:1059-1064. doi:10.1046/j.1523-1747.2002.01772.x
- Labeille B, Gineston JL, Denoeux JP, et al. Epidermolysis bullosa acquisita and Crohn’s disease. A case report with immunological and electron microscopic studies. Arch Intern Med. 1988;148:1457-1459.
- Etienne A, Ruffieux P, Didierjean L, et al. Epidermolysis bullosa acquisita and metastatic cancer of the uterine cervix. Ann Dermatol Venereol. 1998;125:321-323.
- Busch J-O, Sticherling M. Epidermolysis bullosa acquisita and neuroendocrine pancreatic cancer-Coincidence or patho-genetic relationship? J Dtsch Dermatol Ges. 2007;5:916-918. doi:10.111/j.1610-0387.2007.06338.x
- Bevans SL, Sami N. The use of rituximab in treatment of epidermolysis bullosa acquisita: three new cases and a review of the literature. Dermatol Ther. 2018;31:e12726. doi:10.1111/j.1610-0387.2007.06338.x
- Yang A, Kim M, Craig P, et al. A case report of the use of rituximab and the epidermolysis bullosa disease activity scoring index (EBDASI) in a patient with epidermolysis bullosa acquisita with extensive esophageal involvement. Arch Dermatovenerol Croat. 2018;26:325-328.
- Burrows NP, Bhogal BS, Black MM, et al. Bullous eruption of systemic lupus erythematosus: a clinicopathological study of four cases. Br J Dermatol. 1993;128:332-338. doi:10.1111/j.1365-2133.1993.tb00180.x
- Aringer M, Leuchten N, Johnson SR. New criteria for lupus. Curr Rheum Rep. 2020;22:18. doi:10.1007/s11926-020-00896-6
- Camisa C. Vesiculobullous systemic lupus erythematosus. A report of four cases. J Am Acad Dermatol. 1988;18:93-100. doi:10.1016/s0190-9622(88)70014-6
- Duan L, Chen L, Zhong S, et al. Treatment of bullous systemic lupus erythematosus. J Immunol Res. 2015;2015:167064. doi:10.1155/2015/167064
- Sprow G, Afarideh M, Dan J, et al. Bullous systemic lupus erythematosus in females. Int J Womens Dermatol. 2022;8:e034. doi:10.1097/JW9.0000000000000034
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524. doi:10.1007/s40257-014-0098-0
- Fine JD, Mellerio JE. Epidermolysis bullosa. In: Bolognia JL, Jorizzo JL, Schaffer JV (eds), Dermatology (ed 3), Elsevier Saunders; 2012: 501-513.
There are 3 uncommon types of mechanobullous skin diseases caused by relative reduction or complete loss of functional type VII collagen, which is the main component of anchoring fibrils in the lamina densa of the basement membrane zone (BMZ) of the skin and mucous membrane epithelium.1 The function of the anchoring fibrils is to maintain adherence of the basement membrane of the epithelium to the connective tissue of the papillary dermis and submucosa.1 The mechanism of action of the loss of type VII collagen function is via autoimmunity in epidermolysis bullosa acquisita (EBA)2 and
Epidermolysis Bullosa
Epidermolysis bullosa consists of a heterogeneous family of 4 major genetic mechanobullous diseases that affect the skin and mucous membranes with more than 30 subtypes.1 Dystrophic EB is caused by mutations in the COL7A1 gene, which encodes for the α-1 chain of collagen type VII. Classically, EB is divided into 4 main variants based on the location of the cleavage plane or split occurring in the epithelium, which in turn helps to predict the severity of the illness.
Epidermolysis bullosa may be inherited in an autosomal-dominant or autosomal-recessive fashion, or it may occur as a spontaneous mutation. All sexes and races are affected equally. Patients present at birth or in early childhood with fragile skin and mucous membranes that may develop blisters, erosions, and ulcerations after minor trauma.7 These lesions are marked by slow healing and scar formation and often are associated with itching and pain.
Dystrophic Epidermolysis Bullosa
Dystrophic EB accounts for approximately 25%6 of all EB cases in the United States and may be inherited as either a dominant or recessive trait. Hundreds of different pathogenic mutations have been discovered in the COL7A1 gene in the subtypes of DEB.4,8 Dominant DEB tends to cause milder disease because the patients retain one normal COL7A1 allele and produce some type VII collagen (Figure 1), whereas patients with recessive DEB lack type VII collagen completely.9 The cleavage plane is between the lamina densa and the superficial dermis or submucosa. Severity is variable and ranges from localization to the hands and feet to severe generalized blistering and painful ulcerations depending on which of the many possible gene mutations have been inherited. Sequelae include mitten deformities, malalignment and tooth decay, and the development of early aggressive squamous cell carcinomas, which may be fatal. The most severe cases of recessive DEB also may have internal organ involvement.
Epidermolysis Bullosa Simplex
Epidermolysis bullosa simplex is the most common variant, comprising approximately 70%of EB cases in the United States.6 Epidermolysis bullosa simplex usually is inherited as autosomal-dominant mutations in the keratin 5 or keratin 14 genes,10 not COL7A1. Skin blistering results from cleavage within the basal cell layer where the keratin genes are primarily expressed. Blisters tend to occur in acral areas such as hands and feet and may heal without scarring in the localized form of epidermolysis bullosa simplex (Figure 2).
Junctional Epidermolysis Bullosa and Kindler Syndrome
Junctional epidermolysis bullosa (JEB) and Kindler syndrome11 are the rarest of the autosomal-recessive EB variants.6 The plane of cleavage in JEB is through the lamina lucida of the BMZ. Junctional epidermolysis bullosa is caused by mutations of the genes that encode for the 3 chains of laminin 332 protein and type XVII collagen,5,12 not to be confused with type VII collagen. As with DEB, there is a wide range of severity in JEB, from localized effects on the eyes, oral cavity, and tooth enamel to widespread blistering and skin cancers. In JEB cases involving newborns, nonhealing wounds on the face, buttocks, fingers, and toes may be seen, with devastating complications in the oral cavity, esophagus, and larynx. Life expectancy is limited to 2 years or less.6 There have only been approximately 40013 cases of Kindler syndrome reported worldwide6 and there is clinical overlap with DEB. Patients also may demonstrate poikiloderma and photosensitivity. Kindler syndrome is caused by mutations in the FERMT1 gene which encodes for kindlin-1. This protein mediates anchorage between the actin cytoskeleton and the extracellular matrix.5,11 Loss of function produces variable cleavage planes around the dermoepidermal junction.
Clinical management of all EB variants, especially the severe recessive types, traditionally has been limited to the prevention of trauma to the skin and mucous membranes and supportive care, including dressing changes to erosions and ulcerations, antibiotic ointments as needed, and amelioration of pain and pruritus. Bone marrow and pluripotential stem cell transplants have been attempted.12 Complications of EB, such as deformities of the hands and feet caused by excessive scarring, esophageal strictures, poor dentition, and squamous cell carcinomas, must be addressed by a multidisciplinary team of specialists, including plastic surgery, gastroenterology, dentistry/oral surgery, ophthalmology, and dermatology/Mohs surgery.
Until recently, there were no medications approved by the US Food and Drug Administration (FDA) specifically indicated for EB. In 2023, topical gene therapy was approved by the FDA for both recessive and dominant forms of DEB. Normal COL7A1 sequences are delivered by an attenuated herpes simplex virus 1 vector (beremagene geperpavec) in a gel applied directly to the wounds of patients with DEB. In a clinical trial, matching wounds on 31 patients (62 wounds total) were treated with the active agent or placebo gel. After 6 months, complete wound closure was observed in 67% (21/31) of those treated with the active agent and 22% (7/31) of those treated with placebo (P=.002).14 In a single case report, a patient with recessive DEB and cicatrizing conjunctivitis (Figure 3) was given ophthalmic beremagene geperpavec after surgery and had improved visual acuity.15 A topical gel consisting of birch triterpenes to promote healing of partial-thickness wounds also was approved for patients with DEB and JEB by the FDA and the European Commission. In a study of 223 patients, 41% of those using active gel and 29% of those using placebo gel achieved the primary end point of percentage of target wounds that had first complete closure at 45 days.16
The most recent FDA approval for DEB involves transferring the functional COL7A1 gene to the patient’s skin cells, then expanding the gene-corrected cells into sheets of keratinocytes that can be surgically applied to the chronic wound sites. In a phase 3 trial of prademagene zamikeracel (pz-cel), 11 patients with 86 matched wounds were randomized to receive pz-cel (50%) or standard wound care (50%). After 24 weeks, 35 wounds treated with pz-cel were at least 50% healed compared to 7 control wounds.17 The results for healing and reduction of pain were statistically significant (P<.0001 and P<.0002, respectively).17 Recombinant collagen VII as replacement therapy also is under study to be given by intravenous infusion to increase tissue collagen VII where it is lacking. This treatment has shown early biologic and therapeutic effects.9,18 Larger long-term follow-up studies are necessary to confirm persistence of the gene-corrected skin cells, the functionality of the replacement collagen VII, and the potential risk for the development of autoantibodies to type VII collagen.
Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita is a rare autoimmune subepithelial bullous disease that primarily affects middle-aged adults but also has been reported in children.19 Epidermolysis bullosa acquisita is caused by circulating pathogenic IgG autoantibodies that target and bind to type VII collagen in the anchoring fibrils,20-22 thereby disrupting the attachment of the epithelium to its underlying connective tissue.
The 2 major clinical manifestations of EBA include a mechanobullous disease resembling inherited forms of DEB (Figure 4) and an inflammatory bullous pemphigoid (BP)–like disease,23 as well as a combination of both types of skin lesions (Figure 5). The skin and mucous membranes of the oral cavity, esophagus, eyes, and urogenital areas are affected in both types; scarring may cause functional disabilities. In the mechanobullous type of EBA, it is common for blisters and erosions to develop in trauma-prone areas such as the hands, feet, elbows, and knees. The blisters tend to heal with scarring and milia formation as might be seen in porphyria cutanea tarda or cicatricial pemphigoid, which are in the differential diagnosis. Dystrophy of the fingernails or complete nail loss may be observed, resembling DEB. In the BP-like presentation, tense blisters arise upon inflamed or urticarial skin and mucous membranes, which may then become generalized.
Histopathology in both forms of EBA demonstrates subepithelial separation as clefts or blisters. The mechanobullous type shows a sparse inflammatory infiltrate compared to large collections of neutrophils and eosinophils in the blister cavity and in the superficial dermis in the BP-like cases. The final diagnosis rests on the results of immunopathology testing.24 Direct immunofluorescence of perilesional skin and mucosa shows a linear-granular band of IgG and C3 and other conjugates along the BMZ. Deposits of IgA alone in EBA occur in only about 2.4% of cases and are observed more often when there is mucous membrane involvement.2 Indirect immunofluorescence of sera against salt-split skin substrates detects immunoreactants in the floor of the blister rather than in the roof, as would be seen in BP. Highly specific and sensitive enzyme-linked immunosorbent assay (ELISA) kits now are commercially available and can detect autoantibodies against the N-terminal domain of type VII collagen in more than 90% of cases of EBA.25
Inflammatory bowel disease (IBD), particularly Crohn disease (CD), precedes the onset of EBA in approximately 25% of cases.26,27 Ulcerative colitis is much less common. Type VII collagen is normally present in the basement membrane of intestinal epithelium. In a survey of patients with IBD, 68% of those with CD and 13% of those with ulcerative colitis had circulating anti–type VII collagen antibodies detected by ELISA without having symptoms of EBA.28 A case report of a patient with both well-proven EBA and CD highlighted the clinical difficulty of controlling EBA: treatment with prednisolone and sulfasalazine improved the CD but had little effect on the skin blisters.29 A variety of malignancies have been reported in association with EBA, including cancers of the uterine cervix,30 thyroid, and pancreas,31 lymphoma, and chronic lymphatic leukemia. Some of these cases have met the criteria for classification as paraneoplastic, whereas others may have been coincidental.
Treatment for chronic EBA generally has been limited.2,24 Putative antineutrophil drugs such as dapsone and colchicine combined with systemic corticosteroids may be useful in milder or juvenile cases, which tend to have a better prognosis than adult cases.19 In more severe EBA, systemic corticosteroids and/or immunosuppressive drugs such as azathioprine,23 cyclophosphamide,23 mycophenolate mofetil,31 methotrexate,23 cyclosporine,33 and infliximab23 have been used. More recently, rituximab infusion monotherapy33 and rituximab combined with intravenous immunoglobulin or
Bullous Systemic Lupus Erythematosus
Bullous systemic lupus erythematosus is a rare and specific autoimmune skin complication that mostly is seen in patients with an established diagnosis of systemic lupus erythematosus (SLE) who are experiencing a disease flare. Although more common in women, it has been reported in all sexes and races as well as in children. Vesicles and bullae may arise on sun-exposed (Figure 6) and sun-protected areas of skin.
Histopathology shows subepidermal separation with collections of neutrophils and nuclear fragments in the blister cavity. The differential diagnosis of BSLE includes EBA, BP, dermatitis herpetiformis, and linear IgA bullous dermatosis. Direct immunofluorescence testing shows linear-granular deposits of IgG and/or IgM and IgA along the BMZ.34 When utilizing the indirect immunofluorescence split-skin assay, the autoantibody to type VII collagen would be detected in the floor of the blister if the serum titer was sufficiently high.3 Proposed criteria for the diagnosis of BSLE have been published: 1) diagnosis of SLE now based on the 2019 European League Against Rheumatism/American College of Rheumatology classification35; 2) vesicles and bullae arising upon but not limited to sun-exposed skin; 3) histopathology featuring neutrophil-rich subepithelial bullae; 4) positive indirect immunofluorescence for circulating BMZ antibodies using separated human skin as substrate; 5) and direct immunofluorescence showing IgG and/or IgM and often IgA at the BMZ.36 Using ELISA to detect circulating antibodies against type VII collagen24 should now be added to the criteria. The new criteria for SLE34 do not include BSLE, perhaps because it occurs in less than 1% of patients with SLE.37
Further investigation by Gammon et al3 confirmed that the autoantibodies in BSLE are identical to those found in EBA (ie, directed against type VII collagen in the lamina densa). Bullous systemic lupus erythematosus is not considered to be the coexistence of EBA with SLE but rather a specific entity wherein type VII collagen autoantibodies are expressed in the autoimmune spectrum of SLE. It is especially important to make the diagnosis of BSLE because it is predictive of more serious systemic complications of SLE (eg, hematologic and renal disease is found in up to 90% of cases).38
The natural course of BSLE is variable. Treatments include systemic corticosteroids, dapsone, and immunosuppressive drugs such as azathioprine, methotrexate, mycophenolate mofetil, and cyclophosphamide, especially in cases with nephritis.37 There may be spontaneous resolution of the rash as the inflammatory activity of SLE subsides. Rituximab has been used effectively in several refractory cases of BSLE that failed to respond to all other conventional treatments.39
Conclusion
Anchoring fibrils are composed primarily of type VII collagen. Their role is to maintain the attachment of epithelium to the upper dermis and submucosa. The reduction or complete loss of type VII collagen caused by mutations of the COL7A1 gene results in dominant DEB or recessive DEB, respectively. Two distinct non-heritable immunobullous diseases, EBA and BSLE, are caused by autoantibodies that target type VII collagen. A comparison of the 4 type VII collagen disorders can be found in the eTable.
There are 3 uncommon types of mechanobullous skin diseases caused by relative reduction or complete loss of functional type VII collagen, which is the main component of anchoring fibrils in the lamina densa of the basement membrane zone (BMZ) of the skin and mucous membrane epithelium.1 The function of the anchoring fibrils is to maintain adherence of the basement membrane of the epithelium to the connective tissue of the papillary dermis and submucosa.1 The mechanism of action of the loss of type VII collagen function is via autoimmunity in epidermolysis bullosa acquisita (EBA)2 and
Epidermolysis Bullosa
Epidermolysis bullosa consists of a heterogeneous family of 4 major genetic mechanobullous diseases that affect the skin and mucous membranes with more than 30 subtypes.1 Dystrophic EB is caused by mutations in the COL7A1 gene, which encodes for the α-1 chain of collagen type VII. Classically, EB is divided into 4 main variants based on the location of the cleavage plane or split occurring in the epithelium, which in turn helps to predict the severity of the illness.
Epidermolysis bullosa may be inherited in an autosomal-dominant or autosomal-recessive fashion, or it may occur as a spontaneous mutation. All sexes and races are affected equally. Patients present at birth or in early childhood with fragile skin and mucous membranes that may develop blisters, erosions, and ulcerations after minor trauma.7 These lesions are marked by slow healing and scar formation and often are associated with itching and pain.
Dystrophic Epidermolysis Bullosa
Dystrophic EB accounts for approximately 25%6 of all EB cases in the United States and may be inherited as either a dominant or recessive trait. Hundreds of different pathogenic mutations have been discovered in the COL7A1 gene in the subtypes of DEB.4,8 Dominant DEB tends to cause milder disease because the patients retain one normal COL7A1 allele and produce some type VII collagen (Figure 1), whereas patients with recessive DEB lack type VII collagen completely.9 The cleavage plane is between the lamina densa and the superficial dermis or submucosa. Severity is variable and ranges from localization to the hands and feet to severe generalized blistering and painful ulcerations depending on which of the many possible gene mutations have been inherited. Sequelae include mitten deformities, malalignment and tooth decay, and the development of early aggressive squamous cell carcinomas, which may be fatal. The most severe cases of recessive DEB also may have internal organ involvement.
Epidermolysis Bullosa Simplex
Epidermolysis bullosa simplex is the most common variant, comprising approximately 70%of EB cases in the United States.6 Epidermolysis bullosa simplex usually is inherited as autosomal-dominant mutations in the keratin 5 or keratin 14 genes,10 not COL7A1. Skin blistering results from cleavage within the basal cell layer where the keratin genes are primarily expressed. Blisters tend to occur in acral areas such as hands and feet and may heal without scarring in the localized form of epidermolysis bullosa simplex (Figure 2).
Junctional Epidermolysis Bullosa and Kindler Syndrome
Junctional epidermolysis bullosa (JEB) and Kindler syndrome11 are the rarest of the autosomal-recessive EB variants.6 The plane of cleavage in JEB is through the lamina lucida of the BMZ. Junctional epidermolysis bullosa is caused by mutations of the genes that encode for the 3 chains of laminin 332 protein and type XVII collagen,5,12 not to be confused with type VII collagen. As with DEB, there is a wide range of severity in JEB, from localized effects on the eyes, oral cavity, and tooth enamel to widespread blistering and skin cancers. In JEB cases involving newborns, nonhealing wounds on the face, buttocks, fingers, and toes may be seen, with devastating complications in the oral cavity, esophagus, and larynx. Life expectancy is limited to 2 years or less.6 There have only been approximately 40013 cases of Kindler syndrome reported worldwide6 and there is clinical overlap with DEB. Patients also may demonstrate poikiloderma and photosensitivity. Kindler syndrome is caused by mutations in the FERMT1 gene which encodes for kindlin-1. This protein mediates anchorage between the actin cytoskeleton and the extracellular matrix.5,11 Loss of function produces variable cleavage planes around the dermoepidermal junction.
Clinical management of all EB variants, especially the severe recessive types, traditionally has been limited to the prevention of trauma to the skin and mucous membranes and supportive care, including dressing changes to erosions and ulcerations, antibiotic ointments as needed, and amelioration of pain and pruritus. Bone marrow and pluripotential stem cell transplants have been attempted.12 Complications of EB, such as deformities of the hands and feet caused by excessive scarring, esophageal strictures, poor dentition, and squamous cell carcinomas, must be addressed by a multidisciplinary team of specialists, including plastic surgery, gastroenterology, dentistry/oral surgery, ophthalmology, and dermatology/Mohs surgery.
Until recently, there were no medications approved by the US Food and Drug Administration (FDA) specifically indicated for EB. In 2023, topical gene therapy was approved by the FDA for both recessive and dominant forms of DEB. Normal COL7A1 sequences are delivered by an attenuated herpes simplex virus 1 vector (beremagene geperpavec) in a gel applied directly to the wounds of patients with DEB. In a clinical trial, matching wounds on 31 patients (62 wounds total) were treated with the active agent or placebo gel. After 6 months, complete wound closure was observed in 67% (21/31) of those treated with the active agent and 22% (7/31) of those treated with placebo (P=.002).14 In a single case report, a patient with recessive DEB and cicatrizing conjunctivitis (Figure 3) was given ophthalmic beremagene geperpavec after surgery and had improved visual acuity.15 A topical gel consisting of birch triterpenes to promote healing of partial-thickness wounds also was approved for patients with DEB and JEB by the FDA and the European Commission. In a study of 223 patients, 41% of those using active gel and 29% of those using placebo gel achieved the primary end point of percentage of target wounds that had first complete closure at 45 days.16
The most recent FDA approval for DEB involves transferring the functional COL7A1 gene to the patient’s skin cells, then expanding the gene-corrected cells into sheets of keratinocytes that can be surgically applied to the chronic wound sites. In a phase 3 trial of prademagene zamikeracel (pz-cel), 11 patients with 86 matched wounds were randomized to receive pz-cel (50%) or standard wound care (50%). After 24 weeks, 35 wounds treated with pz-cel were at least 50% healed compared to 7 control wounds.17 The results for healing and reduction of pain were statistically significant (P<.0001 and P<.0002, respectively).17 Recombinant collagen VII as replacement therapy also is under study to be given by intravenous infusion to increase tissue collagen VII where it is lacking. This treatment has shown early biologic and therapeutic effects.9,18 Larger long-term follow-up studies are necessary to confirm persistence of the gene-corrected skin cells, the functionality of the replacement collagen VII, and the potential risk for the development of autoantibodies to type VII collagen.
Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita is a rare autoimmune subepithelial bullous disease that primarily affects middle-aged adults but also has been reported in children.19 Epidermolysis bullosa acquisita is caused by circulating pathogenic IgG autoantibodies that target and bind to type VII collagen in the anchoring fibrils,20-22 thereby disrupting the attachment of the epithelium to its underlying connective tissue.
The 2 major clinical manifestations of EBA include a mechanobullous disease resembling inherited forms of DEB (Figure 4) and an inflammatory bullous pemphigoid (BP)–like disease,23 as well as a combination of both types of skin lesions (Figure 5). The skin and mucous membranes of the oral cavity, esophagus, eyes, and urogenital areas are affected in both types; scarring may cause functional disabilities. In the mechanobullous type of EBA, it is common for blisters and erosions to develop in trauma-prone areas such as the hands, feet, elbows, and knees. The blisters tend to heal with scarring and milia formation as might be seen in porphyria cutanea tarda or cicatricial pemphigoid, which are in the differential diagnosis. Dystrophy of the fingernails or complete nail loss may be observed, resembling DEB. In the BP-like presentation, tense blisters arise upon inflamed or urticarial skin and mucous membranes, which may then become generalized.
Histopathology in both forms of EBA demonstrates subepithelial separation as clefts or blisters. The mechanobullous type shows a sparse inflammatory infiltrate compared to large collections of neutrophils and eosinophils in the blister cavity and in the superficial dermis in the BP-like cases. The final diagnosis rests on the results of immunopathology testing.24 Direct immunofluorescence of perilesional skin and mucosa shows a linear-granular band of IgG and C3 and other conjugates along the BMZ. Deposits of IgA alone in EBA occur in only about 2.4% of cases and are observed more often when there is mucous membrane involvement.2 Indirect immunofluorescence of sera against salt-split skin substrates detects immunoreactants in the floor of the blister rather than in the roof, as would be seen in BP. Highly specific and sensitive enzyme-linked immunosorbent assay (ELISA) kits now are commercially available and can detect autoantibodies against the N-terminal domain of type VII collagen in more than 90% of cases of EBA.25
Inflammatory bowel disease (IBD), particularly Crohn disease (CD), precedes the onset of EBA in approximately 25% of cases.26,27 Ulcerative colitis is much less common. Type VII collagen is normally present in the basement membrane of intestinal epithelium. In a survey of patients with IBD, 68% of those with CD and 13% of those with ulcerative colitis had circulating anti–type VII collagen antibodies detected by ELISA without having symptoms of EBA.28 A case report of a patient with both well-proven EBA and CD highlighted the clinical difficulty of controlling EBA: treatment with prednisolone and sulfasalazine improved the CD but had little effect on the skin blisters.29 A variety of malignancies have been reported in association with EBA, including cancers of the uterine cervix,30 thyroid, and pancreas,31 lymphoma, and chronic lymphatic leukemia. Some of these cases have met the criteria for classification as paraneoplastic, whereas others may have been coincidental.
Treatment for chronic EBA generally has been limited.2,24 Putative antineutrophil drugs such as dapsone and colchicine combined with systemic corticosteroids may be useful in milder or juvenile cases, which tend to have a better prognosis than adult cases.19 In more severe EBA, systemic corticosteroids and/or immunosuppressive drugs such as azathioprine,23 cyclophosphamide,23 mycophenolate mofetil,31 methotrexate,23 cyclosporine,33 and infliximab23 have been used. More recently, rituximab infusion monotherapy33 and rituximab combined with intravenous immunoglobulin or
Bullous Systemic Lupus Erythematosus
Bullous systemic lupus erythematosus is a rare and specific autoimmune skin complication that mostly is seen in patients with an established diagnosis of systemic lupus erythematosus (SLE) who are experiencing a disease flare. Although more common in women, it has been reported in all sexes and races as well as in children. Vesicles and bullae may arise on sun-exposed (Figure 6) and sun-protected areas of skin.
Histopathology shows subepidermal separation with collections of neutrophils and nuclear fragments in the blister cavity. The differential diagnosis of BSLE includes EBA, BP, dermatitis herpetiformis, and linear IgA bullous dermatosis. Direct immunofluorescence testing shows linear-granular deposits of IgG and/or IgM and IgA along the BMZ.34 When utilizing the indirect immunofluorescence split-skin assay, the autoantibody to type VII collagen would be detected in the floor of the blister if the serum titer was sufficiently high.3 Proposed criteria for the diagnosis of BSLE have been published: 1) diagnosis of SLE now based on the 2019 European League Against Rheumatism/American College of Rheumatology classification35; 2) vesicles and bullae arising upon but not limited to sun-exposed skin; 3) histopathology featuring neutrophil-rich subepithelial bullae; 4) positive indirect immunofluorescence for circulating BMZ antibodies using separated human skin as substrate; 5) and direct immunofluorescence showing IgG and/or IgM and often IgA at the BMZ.36 Using ELISA to detect circulating antibodies against type VII collagen24 should now be added to the criteria. The new criteria for SLE34 do not include BSLE, perhaps because it occurs in less than 1% of patients with SLE.37
Further investigation by Gammon et al3 confirmed that the autoantibodies in BSLE are identical to those found in EBA (ie, directed against type VII collagen in the lamina densa). Bullous systemic lupus erythematosus is not considered to be the coexistence of EBA with SLE but rather a specific entity wherein type VII collagen autoantibodies are expressed in the autoimmune spectrum of SLE. It is especially important to make the diagnosis of BSLE because it is predictive of more serious systemic complications of SLE (eg, hematologic and renal disease is found in up to 90% of cases).38
The natural course of BSLE is variable. Treatments include systemic corticosteroids, dapsone, and immunosuppressive drugs such as azathioprine, methotrexate, mycophenolate mofetil, and cyclophosphamide, especially in cases with nephritis.37 There may be spontaneous resolution of the rash as the inflammatory activity of SLE subsides. Rituximab has been used effectively in several refractory cases of BSLE that failed to respond to all other conventional treatments.39
Conclusion
Anchoring fibrils are composed primarily of type VII collagen. Their role is to maintain the attachment of epithelium to the upper dermis and submucosa. The reduction or complete loss of type VII collagen caused by mutations of the COL7A1 gene results in dominant DEB or recessive DEB, respectively. Two distinct non-heritable immunobullous diseases, EBA and BSLE, are caused by autoantibodies that target type VII collagen. A comparison of the 4 type VII collagen disorders can be found in the eTable.
- Bardhan A, Bruckner-Tuderman L, Chapple ILC, et al. Epidermolysis bullosa. Nat Rev Dis Primers. 2020;6:78. doi:10.1038/s41572-020-0210-0
- Miyamoto D, Gordilho JO, Santi CG, et al. Epidermolysis bullosa acquisita. An Bras Dermatol. 2022;97:409-423. doi:10.1016/j.abd.2021.09.010.
- Gammon WR, Woodley DT, Dole KC, et al. Evidence that anti-basement membrane zone antibodies in bullous eruption of systemic lupus erythematosus recognize epidermolysis bullosa acquisita autoantigen. J Invest Dermatol. 1985;84:472-476. doi:10.1111/1523-1747.ep12272402.
- Yadav RS, Jaswal A, Shrestha S, et al. Dystrophic epidermolysis bullosa. J Nepal Med Assoc. 2018;56:879-882. doi:10.31729/jnma.3791
- Mariath LM, Santin JT, Schuler-Faccini L, et al. Inherited epidermolysis bullosa: update on the clinical and genetic aspects. An Bras Dermatol. 2020;95:551-569. doi:10.1016/j.abd.2020.05.001
- Understanding epidermolysis bullosa (EB). DEBRA website. Accessed August 17, 2025. https://www.debra.org/about-eb/understanding-epidermolysis-bullosa-eb
- Hon KL, Chu S, Leung AKC. Epidermolysis bullosa: pediatric perspectives. Curr Pediatr Rev. 2022;18:182-190. doi:10.2174/1573396317666210525161252
- Dang N, Klingberg S, Marr P, et al. Review of collagen VII sequence variants found in Australasian patients with dystrophic epidermolysis bullosa reveals nine COL7A1 variants. J Dermatol Sci. 2007;46:169-178. doi:10.1016/j.jdermsci.2007.02.006
- Payne AS. Topical gene therapy for epidermolysis bullosa. N Engl J Med. 2022;387:2281-2284. doi:10.1056/NEJMe2213203
- Khani P, Ghazi F, Zekri A, et al. Keratins and epidermolysis bullosa simplex. J Cell Physiol. 2018;234:289-297. doi:10.1002/jcp.26898
- Lai-Cheong JE, Tanaka A, Hawche G, et al. Kindler syndrome: a focal adhesion genodermatosis. Br J Dermatol. 2009;160:233-242. doi:10.1111/j.1365-2133.2008.08976.x
- Hou P-C, Wang H-T, Abhee S, et al. Investigational treatments for epidermolysis bullosa. Am J Clin Dermatol. 2021;22:801-817. doi:10.1007/s40257-021-00626-3
- Youseffian L, Vahidnezhad H, Uitto J. Kindler Syndrome. GeneReviews [Internet]. Updated January 6, 2022. Accessed August 21, 2025.
- Guide SV, Gonzalez ME, Bagci S, et al. Trial of beremagene geperpavec (B-VEC) for dystrophic epidermolysis bullosa. N Engl J Med. 2022;387:2211-2219. doi:10.1056/NEJMoa2206663
- Vetencourt AT, Sayed-Ahmed I, Gomez J, et al. Ocular gene therapy in a patient with dystrophic epidermolysis bullosa. N Engl J Med. 2024;390:530-535. doi:10.1056/NEJMoa2301244
- Kern JS, Sprecher E, Fernandez MF, et al. Efficacy and safety of Oleogel-S10 (birch triterpenes for epidermolysis bullosa: results from the phase III randomized double-blind phase of the EASE study. Br J Dermatol. 2023;188:12-21. doi:10.1093/bjd/ljac001
- Tang JY, Marinkovich MP, Wiss K, et al. Prademagene zamikeracel for recessive dystrophic epidermolysis bullosa wounds (VIITAL): a two-centre, randomized, open-label, intrapatient-controlled phase 3 trial. Lancet. 2025;406:163-173. doi:10.1016/S0140-6736(25)00778-0
- Gretzmeier C, Pin D, Kern JS, et al. Systemic collagen VII replacement therapy for advanced recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2022;142:1094-1102. doi:10.1016/j.jid.2021.09.008
- Hignett E, Sami N. Pediatric epidermolysis bullosa acquisita. A review. Pediatr Dermatol. 2021;38:1047-1050. doi:10.1111/pde.14722
- Chen M, Kim GH, Prakash L, et al. Autoimmunity to anchoring fibril collagen. Autoimmunity. 2012;45:91-101. doi:10.1007/s12016-007-0027-6.
- Kridin K, Kneiber D, Kowalski EH, et al. Epidermolysis bullosa acquisita: a comprehensive review. Autoimmun Rev. 2019;18:786-795. doi:10.1016/j.autrev.2019.06.007
- Hofmann SC, Weidinger A. Epidermolysis bullosa acquisita. Hautarzt. 2019;70:265-270. doi:10.1007/s00105-019-4387-7
- Ishi N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37:220-230. doi:10.1111/j.1346-8138.2009.00799.x
- Iwata H, Vorobyev A, Koga H, et al. Meta-analysis of the clinical and immunopathological characteristics and treatment outcomes in epidermolysis bullosa acquisita patients. Orphanet J Rare Dis. 2018;13:153. doi:10.1186/s13023-018-0896-1
- Komorowski L, Muller R, Vorobyev A, et al. Sensitive and specific assays for routine serological diagnosis of epidermolysis bullosa acquisita. J Am Acad Dermatol. 2013;68:e89-95. doi:10.1016/j.jaad.2011.12.032
- Antonelli E, Bassotti G, Tramontana M, et al. Dermatological manifestations in inflammatory bowel diseases. J Clin Med. 2021;10:364-390. doi:10.3390/jcm10020364
- Bezzio C, Della Corte C, Vernero M, et al. Inflammatory bowel disease and immune-mediated inflammatory diseases: looking at less frequent associations. Therap Adv Gastroenterol. 2022;15:17562848221115312. doi:10.1177/17562848221115312
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have antibodies to type VII collagen. J Invest Dermatol. 2002;118:1059-1064. doi:10.1046/j.1523-1747.2002.01772.x
- Labeille B, Gineston JL, Denoeux JP, et al. Epidermolysis bullosa acquisita and Crohn’s disease. A case report with immunological and electron microscopic studies. Arch Intern Med. 1988;148:1457-1459.
- Etienne A, Ruffieux P, Didierjean L, et al. Epidermolysis bullosa acquisita and metastatic cancer of the uterine cervix. Ann Dermatol Venereol. 1998;125:321-323.
- Busch J-O, Sticherling M. Epidermolysis bullosa acquisita and neuroendocrine pancreatic cancer-Coincidence or patho-genetic relationship? J Dtsch Dermatol Ges. 2007;5:916-918. doi:10.111/j.1610-0387.2007.06338.x
- Bevans SL, Sami N. The use of rituximab in treatment of epidermolysis bullosa acquisita: three new cases and a review of the literature. Dermatol Ther. 2018;31:e12726. doi:10.1111/j.1610-0387.2007.06338.x
- Yang A, Kim M, Craig P, et al. A case report of the use of rituximab and the epidermolysis bullosa disease activity scoring index (EBDASI) in a patient with epidermolysis bullosa acquisita with extensive esophageal involvement. Arch Dermatovenerol Croat. 2018;26:325-328.
- Burrows NP, Bhogal BS, Black MM, et al. Bullous eruption of systemic lupus erythematosus: a clinicopathological study of four cases. Br J Dermatol. 1993;128:332-338. doi:10.1111/j.1365-2133.1993.tb00180.x
- Aringer M, Leuchten N, Johnson SR. New criteria for lupus. Curr Rheum Rep. 2020;22:18. doi:10.1007/s11926-020-00896-6
- Camisa C. Vesiculobullous systemic lupus erythematosus. A report of four cases. J Am Acad Dermatol. 1988;18:93-100. doi:10.1016/s0190-9622(88)70014-6
- Duan L, Chen L, Zhong S, et al. Treatment of bullous systemic lupus erythematosus. J Immunol Res. 2015;2015:167064. doi:10.1155/2015/167064
- Sprow G, Afarideh M, Dan J, et al. Bullous systemic lupus erythematosus in females. Int J Womens Dermatol. 2022;8:e034. doi:10.1097/JW9.0000000000000034
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524. doi:10.1007/s40257-014-0098-0
- Fine JD, Mellerio JE. Epidermolysis bullosa. In: Bolognia JL, Jorizzo JL, Schaffer JV (eds), Dermatology (ed 3), Elsevier Saunders; 2012: 501-513.
- Bardhan A, Bruckner-Tuderman L, Chapple ILC, et al. Epidermolysis bullosa. Nat Rev Dis Primers. 2020;6:78. doi:10.1038/s41572-020-0210-0
- Miyamoto D, Gordilho JO, Santi CG, et al. Epidermolysis bullosa acquisita. An Bras Dermatol. 2022;97:409-423. doi:10.1016/j.abd.2021.09.010.
- Gammon WR, Woodley DT, Dole KC, et al. Evidence that anti-basement membrane zone antibodies in bullous eruption of systemic lupus erythematosus recognize epidermolysis bullosa acquisita autoantigen. J Invest Dermatol. 1985;84:472-476. doi:10.1111/1523-1747.ep12272402.
- Yadav RS, Jaswal A, Shrestha S, et al. Dystrophic epidermolysis bullosa. J Nepal Med Assoc. 2018;56:879-882. doi:10.31729/jnma.3791
- Mariath LM, Santin JT, Schuler-Faccini L, et al. Inherited epidermolysis bullosa: update on the clinical and genetic aspects. An Bras Dermatol. 2020;95:551-569. doi:10.1016/j.abd.2020.05.001
- Understanding epidermolysis bullosa (EB). DEBRA website. Accessed August 17, 2025. https://www.debra.org/about-eb/understanding-epidermolysis-bullosa-eb
- Hon KL, Chu S, Leung AKC. Epidermolysis bullosa: pediatric perspectives. Curr Pediatr Rev. 2022;18:182-190. doi:10.2174/1573396317666210525161252
- Dang N, Klingberg S, Marr P, et al. Review of collagen VII sequence variants found in Australasian patients with dystrophic epidermolysis bullosa reveals nine COL7A1 variants. J Dermatol Sci. 2007;46:169-178. doi:10.1016/j.jdermsci.2007.02.006
- Payne AS. Topical gene therapy for epidermolysis bullosa. N Engl J Med. 2022;387:2281-2284. doi:10.1056/NEJMe2213203
- Khani P, Ghazi F, Zekri A, et al. Keratins and epidermolysis bullosa simplex. J Cell Physiol. 2018;234:289-297. doi:10.1002/jcp.26898
- Lai-Cheong JE, Tanaka A, Hawche G, et al. Kindler syndrome: a focal adhesion genodermatosis. Br J Dermatol. 2009;160:233-242. doi:10.1111/j.1365-2133.2008.08976.x
- Hou P-C, Wang H-T, Abhee S, et al. Investigational treatments for epidermolysis bullosa. Am J Clin Dermatol. 2021;22:801-817. doi:10.1007/s40257-021-00626-3
- Youseffian L, Vahidnezhad H, Uitto J. Kindler Syndrome. GeneReviews [Internet]. Updated January 6, 2022. Accessed August 21, 2025.
- Guide SV, Gonzalez ME, Bagci S, et al. Trial of beremagene geperpavec (B-VEC) for dystrophic epidermolysis bullosa. N Engl J Med. 2022;387:2211-2219. doi:10.1056/NEJMoa2206663
- Vetencourt AT, Sayed-Ahmed I, Gomez J, et al. Ocular gene therapy in a patient with dystrophic epidermolysis bullosa. N Engl J Med. 2024;390:530-535. doi:10.1056/NEJMoa2301244
- Kern JS, Sprecher E, Fernandez MF, et al. Efficacy and safety of Oleogel-S10 (birch triterpenes for epidermolysis bullosa: results from the phase III randomized double-blind phase of the EASE study. Br J Dermatol. 2023;188:12-21. doi:10.1093/bjd/ljac001
- Tang JY, Marinkovich MP, Wiss K, et al. Prademagene zamikeracel for recessive dystrophic epidermolysis bullosa wounds (VIITAL): a two-centre, randomized, open-label, intrapatient-controlled phase 3 trial. Lancet. 2025;406:163-173. doi:10.1016/S0140-6736(25)00778-0
- Gretzmeier C, Pin D, Kern JS, et al. Systemic collagen VII replacement therapy for advanced recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2022;142:1094-1102. doi:10.1016/j.jid.2021.09.008
- Hignett E, Sami N. Pediatric epidermolysis bullosa acquisita. A review. Pediatr Dermatol. 2021;38:1047-1050. doi:10.1111/pde.14722
- Chen M, Kim GH, Prakash L, et al. Autoimmunity to anchoring fibril collagen. Autoimmunity. 2012;45:91-101. doi:10.1007/s12016-007-0027-6.
- Kridin K, Kneiber D, Kowalski EH, et al. Epidermolysis bullosa acquisita: a comprehensive review. Autoimmun Rev. 2019;18:786-795. doi:10.1016/j.autrev.2019.06.007
- Hofmann SC, Weidinger A. Epidermolysis bullosa acquisita. Hautarzt. 2019;70:265-270. doi:10.1007/s00105-019-4387-7
- Ishi N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37:220-230. doi:10.1111/j.1346-8138.2009.00799.x
- Iwata H, Vorobyev A, Koga H, et al. Meta-analysis of the clinical and immunopathological characteristics and treatment outcomes in epidermolysis bullosa acquisita patients. Orphanet J Rare Dis. 2018;13:153. doi:10.1186/s13023-018-0896-1
- Komorowski L, Muller R, Vorobyev A, et al. Sensitive and specific assays for routine serological diagnosis of epidermolysis bullosa acquisita. J Am Acad Dermatol. 2013;68:e89-95. doi:10.1016/j.jaad.2011.12.032
- Antonelli E, Bassotti G, Tramontana M, et al. Dermatological manifestations in inflammatory bowel diseases. J Clin Med. 2021;10:364-390. doi:10.3390/jcm10020364
- Bezzio C, Della Corte C, Vernero M, et al. Inflammatory bowel disease and immune-mediated inflammatory diseases: looking at less frequent associations. Therap Adv Gastroenterol. 2022;15:17562848221115312. doi:10.1177/17562848221115312
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have antibodies to type VII collagen. J Invest Dermatol. 2002;118:1059-1064. doi:10.1046/j.1523-1747.2002.01772.x
- Labeille B, Gineston JL, Denoeux JP, et al. Epidermolysis bullosa acquisita and Crohn’s disease. A case report with immunological and electron microscopic studies. Arch Intern Med. 1988;148:1457-1459.
- Etienne A, Ruffieux P, Didierjean L, et al. Epidermolysis bullosa acquisita and metastatic cancer of the uterine cervix. Ann Dermatol Venereol. 1998;125:321-323.
- Busch J-O, Sticherling M. Epidermolysis bullosa acquisita and neuroendocrine pancreatic cancer-Coincidence or patho-genetic relationship? J Dtsch Dermatol Ges. 2007;5:916-918. doi:10.111/j.1610-0387.2007.06338.x
- Bevans SL, Sami N. The use of rituximab in treatment of epidermolysis bullosa acquisita: three new cases and a review of the literature. Dermatol Ther. 2018;31:e12726. doi:10.1111/j.1610-0387.2007.06338.x
- Yang A, Kim M, Craig P, et al. A case report of the use of rituximab and the epidermolysis bullosa disease activity scoring index (EBDASI) in a patient with epidermolysis bullosa acquisita with extensive esophageal involvement. Arch Dermatovenerol Croat. 2018;26:325-328.
- Burrows NP, Bhogal BS, Black MM, et al. Bullous eruption of systemic lupus erythematosus: a clinicopathological study of four cases. Br J Dermatol. 1993;128:332-338. doi:10.1111/j.1365-2133.1993.tb00180.x
- Aringer M, Leuchten N, Johnson SR. New criteria for lupus. Curr Rheum Rep. 2020;22:18. doi:10.1007/s11926-020-00896-6
- Camisa C. Vesiculobullous systemic lupus erythematosus. A report of four cases. J Am Acad Dermatol. 1988;18:93-100. doi:10.1016/s0190-9622(88)70014-6
- Duan L, Chen L, Zhong S, et al. Treatment of bullous systemic lupus erythematosus. J Immunol Res. 2015;2015:167064. doi:10.1155/2015/167064
- Sprow G, Afarideh M, Dan J, et al. Bullous systemic lupus erythematosus in females. Int J Womens Dermatol. 2022;8:e034. doi:10.1097/JW9.0000000000000034
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524. doi:10.1007/s40257-014-0098-0
- Fine JD, Mellerio JE. Epidermolysis bullosa. In: Bolognia JL, Jorizzo JL, Schaffer JV (eds), Dermatology (ed 3), Elsevier Saunders; 2012: 501-513.
Type VII Collagen Disorders Simplified
Type VII Collagen Disorders Simplified
PRACTICE POINTS
- The full complement of type VII collagen is required for the normal assembly of anchoring fibrils, whose function is to adhere the basement membrane to the underlying connective tissue of skin and mucous membranes.
- In the heritable epidermolysis bullosa (EB) family of diseases, only dominant and recessive dystrophic epidermolysis bullosa are caused by partial or total loss of type VII collagen function.
- New treatments that have been approved for EB include topical gene therapy with COL7A1, topical birch triterpene gel, and skin cells from patients that are genetically corrected with a functional COL7A1 gene.
- Epidermolysis bullosa acquisita and bullous systemic lupus erythematosus are rare distinct autoimmune subepithelial bullous diseases caused by IgG antibodies that target type VII collagen in the anchoring fibrils.
