User login
Perfect storm of SARS-CoV-2 during flu season
COVID-19 now. The urban phase of the U.S. pandemic is leveling somewhat, while the rural phase is accelerating – in part because of food processing and handling industries. The pediatric burden has been surprisingly small, with the multisystem inflammatory disease (MIS-c) in children noted in several hundred cases now being seen across the country.
Next wave? Given ongoing COVID-19 disease, controversy rages about when and how to re-open the country. Regardless how more reopening occurs over the next months, we should expect a next or ongoing COVID-19 wave, particularly given loss of social distancing during social justice protests. A sawtooth disease prevalence pattern is predicted by many experts: a drop in prevalence leading to reopening, leading to scattered prevalence increases and regional if not local restriction tightening, followed by another drop in prevalence. Then “rinse and repeat” until 70% of the population is immune either by disease experience or vaccine-induced immunity, likely sometime in 2021.
Influenza too. A COVID-19 up-cycle is likely during influenza season, although influenza season’s onset could be altered because of whatever social distancing rules are in place in November and December. That said, we need to consider the worst. We have seen what happens if we fail to prepare and then react only after a prevalent respiratory infection has surged into the overall population. Best estimates are that at most 20% of the U.S. population is currently immune to SARS-CoV-2. Given that at least some of that 20% of individuals currently immune to SARS-CoV-2 will lose their neutralizing antibody over the next 4-6 months, we can still expect 70%-80% of the U.S. population to be susceptible to SARS-CoV-2 infection in the fall of 2020.
Pediatric preparedness. As pediatric providers, we have struggled with lower patient loads and dramatic income losses/declines. Many clinics/offices’ attendance remain less than 50% of pre–COVID-19 levels, with necessary furloughs of personnel and spotty office hours. But influenza is coming, and SARS-CoV-2 will not be gone yet. How do we prepare for concurrent influenza and COVID-19?

The annual purchase/administration of influenza vaccine in summer/fall is expensive, time consuming, and logistically difficult even in the best times. Given the loss of income, likely reluctance of patients to come to clinics/offices if COVID-19 is still circulating, and likely need for some form of social distancing during late summer and early fall, how will providers, health departments, and hospitals implement influenza vaccine administration this year?
Minimize double whammy infections. It is easy to understand why we should maximize influenza protection in SARS-CoV-2 vulnerables (elderly or persons with existing comorbidities). But is it as critical for otherwise healthy children? My answer is yes.
Children are not currently known as SARS-CoV-2 vectors, but children are excellent influenza vectors, shedding higher titers for longer than other age groups. As with SARS-CoV-2, influenza exposure is cumulative, i.e., the more intense and more frequently a person is exposed, the more likely that infection/disease will result. So, the fewer who get and can transmit influenza during the COVID-19 pandemic, the fewer people are likely to get a double whammy of SARS-CoV-2 concurrent or in tandem with influenza. Double whammy infections likely would further increase the medical care burden and return us to March-April crisis mode.
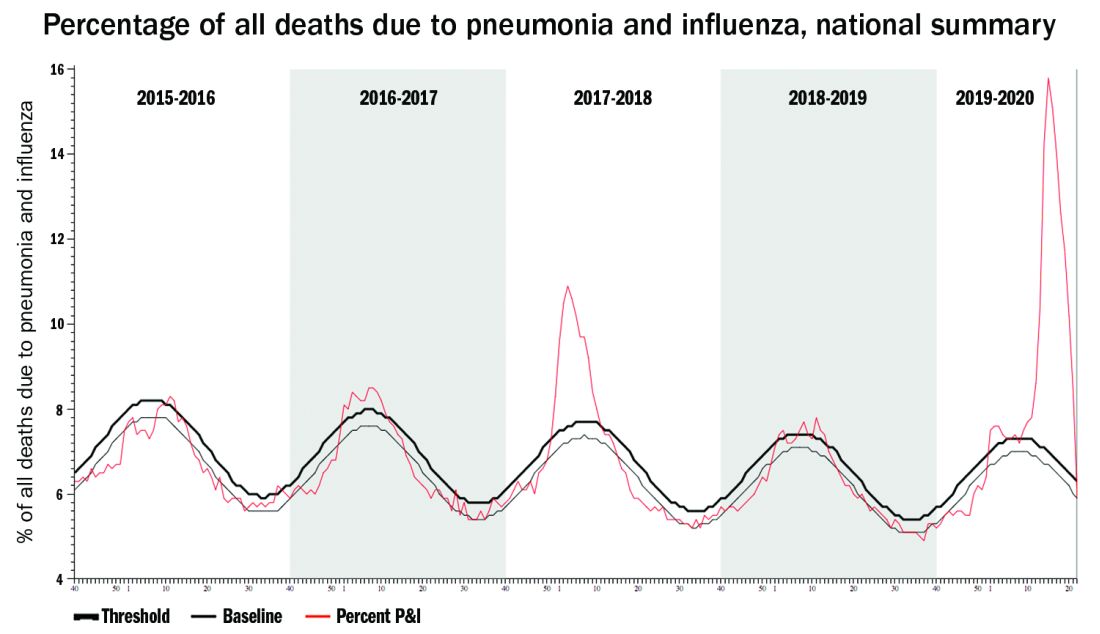
One alarming new question is whether recent influenza could make children vulnerable to SARS-CoV-2 and trigger hospitalizations. A surge in pediatric plus adult COVID-19 disease plus a surge in all-ages influenza disease would likely break the medical care system, at least in some areas.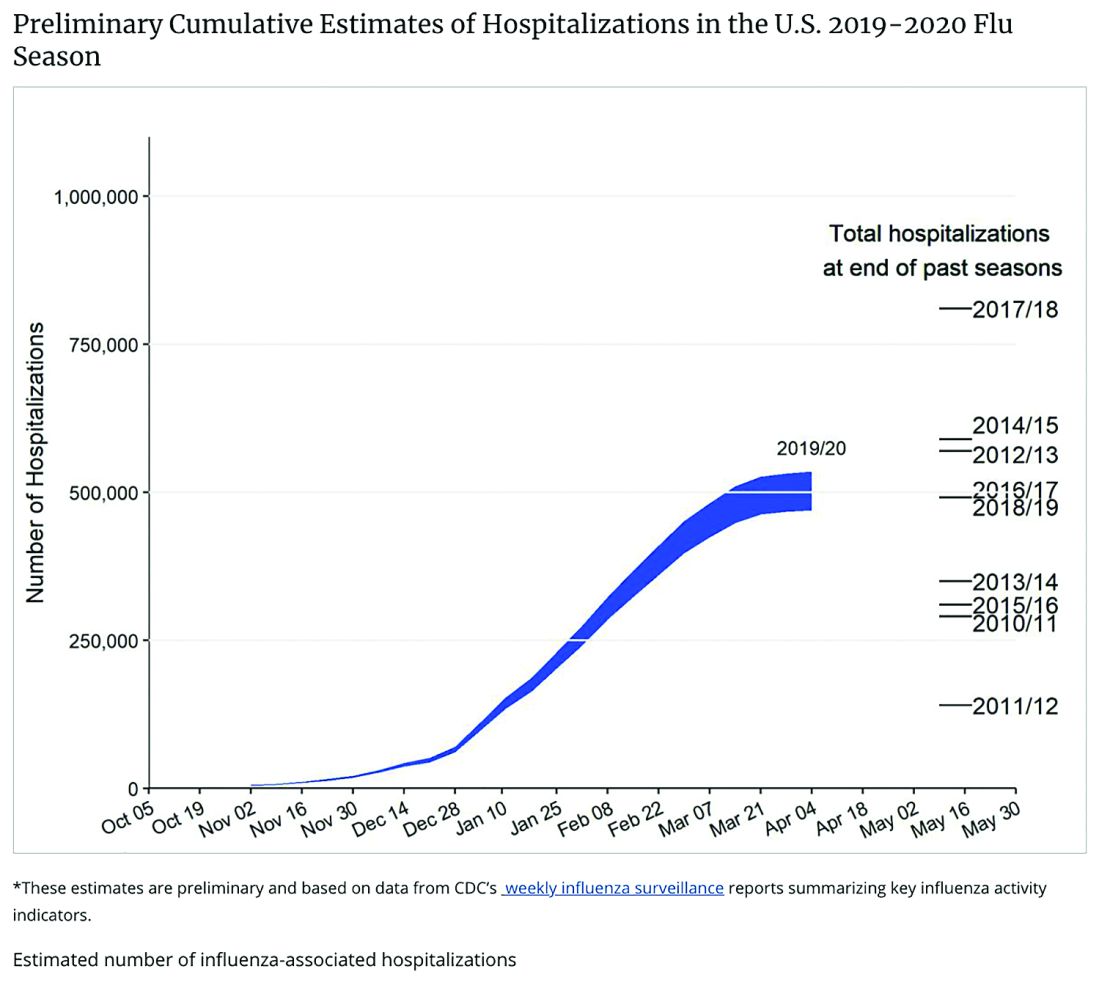
Staggering COVID-19 burden. As of June 8, we have had approximately 2 million SARS-CoV-2 cases with 500,000 hospitalizations and 120,000 deaths. Over the past 10 years, total annual U.S. influenza hospitalizations ranged from 180,000 (2011-2012) to 825,000 (2017-2018). The interquartile range for hospitalization length of stay for influenza is 4-6 days1 vs. 15-23 days2 for SARS-CoV-2. One COVID-19 hospitalization uses hospital resources roughly equal to four influenza hospitalizations. To date COVID-19 hospitalizations have used resources equal to an estimated 1.9 million influenza hospitalizations – over twice the worst influenza season in this century – and we are still on the rise. We are likely not even halfway to truly controlling the U.S. pandemic, so expect another 500,000 hospitalizations – equal to another 1.9 million influenza hospitalizations. Further, pneumonia deaths have skyrocketed this year when COVID-19 was superimposed on the last third of influenza season. One hope is that widespread use of antivirals (for example, new antivirals, convalescent plasma, or other interventions) can reduce length of stay by 30% for COVID-19 hospitalizations, yet even with that the numbers remain grim.
Less influenza disease can free up medical resources. Planning ahead could prevent a bad influenza season (for example, up to 850,000 hospitalizations just for influenza). Can we preemptively use vaccine to reduce influenza hospitalizations below 2011-2012 levels – less than 150,000 hospitalizations? Perhaps, if we start by reducing pediatric influenza.
1. Aim to exceed 75% influenza vaccine uptake in your patients.
a. It is ambitious, but if there was ever a year that needed influenza herd immunity, it is 2020-2021.
2. Review practice/group/institution plans for vaccine purchase and ensure adequate personnel to administer vaccine.
3. Plan safe and efficient processes to vaccinate large numbers in August through November.
a. Consider that routine and influenza vaccines can be given concurrently with the annual uptick in school and sports physical examinations.
b. What social distancing and masking rules will be needed?
i. Will patients need to bring their own masks, or will you supply them?
c. What extra supplies and efforts are needed, e.g. hand sanitizer, new signage, 6-foot interval markings on floors or sidewalks, families calling from parking lot to announce their arrivals, etc.?
d. Remember younger patients need two doses before Dec 1, 2020.
e. Be creative, for example, are parking-lot tents for influenza vaccination feasible?
f. Can we partner with other providers to implement influenza vaccine–specific mass clinics?
Ramping up to give seasonal influenza vaccine in 2020 is daunting. But if we do not prepare, it will be even more difficult. Let’s make this the mildest influenza season in memory by vaccinating more than any time in memory – and by doing so, we can hope to blunt medical care burdens despite ongoing COVID-19 disease.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Kansas City (Mo.). Children’s Mercy receives funding from GlaxoSmithKline, Merck, and Pfizer for vaccine research studies on which Dr. Harrison is an investigator. Email him at [email protected].
References
1.. HCUP Statistical Brief #253. 2019 Oct.
2. medrxiv. 2020 Apr 10. doi: 10.1101/2020.04.07.20057299.
COVID-19 now. The urban phase of the U.S. pandemic is leveling somewhat, while the rural phase is accelerating – in part because of food processing and handling industries. The pediatric burden has been surprisingly small, with the multisystem inflammatory disease (MIS-c) in children noted in several hundred cases now being seen across the country.
Next wave? Given ongoing COVID-19 disease, controversy rages about when and how to re-open the country. Regardless how more reopening occurs over the next months, we should expect a next or ongoing COVID-19 wave, particularly given loss of social distancing during social justice protests. A sawtooth disease prevalence pattern is predicted by many experts: a drop in prevalence leading to reopening, leading to scattered prevalence increases and regional if not local restriction tightening, followed by another drop in prevalence. Then “rinse and repeat” until 70% of the population is immune either by disease experience or vaccine-induced immunity, likely sometime in 2021.
Influenza too. A COVID-19 up-cycle is likely during influenza season, although influenza season’s onset could be altered because of whatever social distancing rules are in place in November and December. That said, we need to consider the worst. We have seen what happens if we fail to prepare and then react only after a prevalent respiratory infection has surged into the overall population. Best estimates are that at most 20% of the U.S. population is currently immune to SARS-CoV-2. Given that at least some of that 20% of individuals currently immune to SARS-CoV-2 will lose their neutralizing antibody over the next 4-6 months, we can still expect 70%-80% of the U.S. population to be susceptible to SARS-CoV-2 infection in the fall of 2020.
Pediatric preparedness. As pediatric providers, we have struggled with lower patient loads and dramatic income losses/declines. Many clinics/offices’ attendance remain less than 50% of pre–COVID-19 levels, with necessary furloughs of personnel and spotty office hours. But influenza is coming, and SARS-CoV-2 will not be gone yet. How do we prepare for concurrent influenza and COVID-19?
The annual purchase/administration of influenza vaccine in summer/fall is expensive, time consuming, and logistically difficult even in the best times. Given the loss of income, likely reluctance of patients to come to clinics/offices if COVID-19 is still circulating, and likely need for some form of social distancing during late summer and early fall, how will providers, health departments, and hospitals implement influenza vaccine administration this year?
Minimize double whammy infections. It is easy to understand why we should maximize influenza protection in SARS-CoV-2 vulnerables (elderly or persons with existing comorbidities). But is it as critical for otherwise healthy children? My answer is yes.
Children are not currently known as SARS-CoV-2 vectors, but children are excellent influenza vectors, shedding higher titers for longer than other age groups. As with SARS-CoV-2, influenza exposure is cumulative, i.e., the more intense and more frequently a person is exposed, the more likely that infection/disease will result. So, the fewer who get and can transmit influenza during the COVID-19 pandemic, the fewer people are likely to get a double whammy of SARS-CoV-2 concurrent or in tandem with influenza. Double whammy infections likely would further increase the medical care burden and return us to March-April crisis mode.
One alarming new question is whether recent influenza could make children vulnerable to SARS-CoV-2 and trigger hospitalizations. A surge in pediatric plus adult COVID-19 disease plus a surge in all-ages influenza disease would likely break the medical care system, at least in some areas.
Staggering COVID-19 burden. As of June 8, we have had approximately 2 million SARS-CoV-2 cases with 500,000 hospitalizations and 120,000 deaths. Over the past 10 years, total annual U.S. influenza hospitalizations ranged from 180,000 (2011-2012) to 825,000 (2017-2018). The interquartile range for hospitalization length of stay for influenza is 4-6 days1 vs. 15-23 days2 for SARS-CoV-2. One COVID-19 hospitalization uses hospital resources roughly equal to four influenza hospitalizations. To date COVID-19 hospitalizations have used resources equal to an estimated 1.9 million influenza hospitalizations – over twice the worst influenza season in this century – and we are still on the rise. We are likely not even halfway to truly controlling the U.S. pandemic, so expect another 500,000 hospitalizations – equal to another 1.9 million influenza hospitalizations. Further, pneumonia deaths have skyrocketed this year when COVID-19 was superimposed on the last third of influenza season. One hope is that widespread use of antivirals (for example, new antivirals, convalescent plasma, or other interventions) can reduce length of stay by 30% for COVID-19 hospitalizations, yet even with that the numbers remain grim.
Less influenza disease can free up medical resources. Planning ahead could prevent a bad influenza season (for example, up to 850,000 hospitalizations just for influenza). Can we preemptively use vaccine to reduce influenza hospitalizations below 2011-2012 levels – less than 150,000 hospitalizations? Perhaps, if we start by reducing pediatric influenza.
1. Aim to exceed 75% influenza vaccine uptake in your patients.
a. It is ambitious, but if there was ever a year that needed influenza herd immunity, it is 2020-2021.
2. Review practice/group/institution plans for vaccine purchase and ensure adequate personnel to administer vaccine.
3. Plan safe and efficient processes to vaccinate large numbers in August through November.
a. Consider that routine and influenza vaccines can be given concurrently with the annual uptick in school and sports physical examinations.
b. What social distancing and masking rules will be needed?
i. Will patients need to bring their own masks, or will you supply them?
c. What extra supplies and efforts are needed, e.g. hand sanitizer, new signage, 6-foot interval markings on floors or sidewalks, families calling from parking lot to announce their arrivals, etc.?
d. Remember younger patients need two doses before Dec 1, 2020.
e. Be creative, for example, are parking-lot tents for influenza vaccination feasible?
f. Can we partner with other providers to implement influenza vaccine–specific mass clinics?
Ramping up to give seasonal influenza vaccine in 2020 is daunting. But if we do not prepare, it will be even more difficult. Let’s make this the mildest influenza season in memory by vaccinating more than any time in memory – and by doing so, we can hope to blunt medical care burdens despite ongoing COVID-19 disease.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Kansas City (Mo.). Children’s Mercy receives funding from GlaxoSmithKline, Merck, and Pfizer for vaccine research studies on which Dr. Harrison is an investigator. Email him at [email protected].
References
1.. HCUP Statistical Brief #253. 2019 Oct.
2. medrxiv. 2020 Apr 10. doi: 10.1101/2020.04.07.20057299.
COVID-19 now. The urban phase of the U.S. pandemic is leveling somewhat, while the rural phase is accelerating – in part because of food processing and handling industries. The pediatric burden has been surprisingly small, with the multisystem inflammatory disease (MIS-c) in children noted in several hundred cases now being seen across the country.
Next wave? Given ongoing COVID-19 disease, controversy rages about when and how to re-open the country. Regardless how more reopening occurs over the next months, we should expect a next or ongoing COVID-19 wave, particularly given loss of social distancing during social justice protests. A sawtooth disease prevalence pattern is predicted by many experts: a drop in prevalence leading to reopening, leading to scattered prevalence increases and regional if not local restriction tightening, followed by another drop in prevalence. Then “rinse and repeat” until 70% of the population is immune either by disease experience or vaccine-induced immunity, likely sometime in 2021.
Influenza too. A COVID-19 up-cycle is likely during influenza season, although influenza season’s onset could be altered because of whatever social distancing rules are in place in November and December. That said, we need to consider the worst. We have seen what happens if we fail to prepare and then react only after a prevalent respiratory infection has surged into the overall population. Best estimates are that at most 20% of the U.S. population is currently immune to SARS-CoV-2. Given that at least some of that 20% of individuals currently immune to SARS-CoV-2 will lose their neutralizing antibody over the next 4-6 months, we can still expect 70%-80% of the U.S. population to be susceptible to SARS-CoV-2 infection in the fall of 2020.
Pediatric preparedness. As pediatric providers, we have struggled with lower patient loads and dramatic income losses/declines. Many clinics/offices’ attendance remain less than 50% of pre–COVID-19 levels, with necessary furloughs of personnel and spotty office hours. But influenza is coming, and SARS-CoV-2 will not be gone yet. How do we prepare for concurrent influenza and COVID-19?
The annual purchase/administration of influenza vaccine in summer/fall is expensive, time consuming, and logistically difficult even in the best times. Given the loss of income, likely reluctance of patients to come to clinics/offices if COVID-19 is still circulating, and likely need for some form of social distancing during late summer and early fall, how will providers, health departments, and hospitals implement influenza vaccine administration this year?
Minimize double whammy infections. It is easy to understand why we should maximize influenza protection in SARS-CoV-2 vulnerables (elderly or persons with existing comorbidities). But is it as critical for otherwise healthy children? My answer is yes.
Children are not currently known as SARS-CoV-2 vectors, but children are excellent influenza vectors, shedding higher titers for longer than other age groups. As with SARS-CoV-2, influenza exposure is cumulative, i.e., the more intense and more frequently a person is exposed, the more likely that infection/disease will result. So, the fewer who get and can transmit influenza during the COVID-19 pandemic, the fewer people are likely to get a double whammy of SARS-CoV-2 concurrent or in tandem with influenza. Double whammy infections likely would further increase the medical care burden and return us to March-April crisis mode.
One alarming new question is whether recent influenza could make children vulnerable to SARS-CoV-2 and trigger hospitalizations. A surge in pediatric plus adult COVID-19 disease plus a surge in all-ages influenza disease would likely break the medical care system, at least in some areas.
Staggering COVID-19 burden. As of June 8, we have had approximately 2 million SARS-CoV-2 cases with 500,000 hospitalizations and 120,000 deaths. Over the past 10 years, total annual U.S. influenza hospitalizations ranged from 180,000 (2011-2012) to 825,000 (2017-2018). The interquartile range for hospitalization length of stay for influenza is 4-6 days1 vs. 15-23 days2 for SARS-CoV-2. One COVID-19 hospitalization uses hospital resources roughly equal to four influenza hospitalizations. To date COVID-19 hospitalizations have used resources equal to an estimated 1.9 million influenza hospitalizations – over twice the worst influenza season in this century – and we are still on the rise. We are likely not even halfway to truly controlling the U.S. pandemic, so expect another 500,000 hospitalizations – equal to another 1.9 million influenza hospitalizations. Further, pneumonia deaths have skyrocketed this year when COVID-19 was superimposed on the last third of influenza season. One hope is that widespread use of antivirals (for example, new antivirals, convalescent plasma, or other interventions) can reduce length of stay by 30% for COVID-19 hospitalizations, yet even with that the numbers remain grim.
Less influenza disease can free up medical resources. Planning ahead could prevent a bad influenza season (for example, up to 850,000 hospitalizations just for influenza). Can we preemptively use vaccine to reduce influenza hospitalizations below 2011-2012 levels – less than 150,000 hospitalizations? Perhaps, if we start by reducing pediatric influenza.
1. Aim to exceed 75% influenza vaccine uptake in your patients.
a. It is ambitious, but if there was ever a year that needed influenza herd immunity, it is 2020-2021.
2. Review practice/group/institution plans for vaccine purchase and ensure adequate personnel to administer vaccine.
3. Plan safe and efficient processes to vaccinate large numbers in August through November.
a. Consider that routine and influenza vaccines can be given concurrently with the annual uptick in school and sports physical examinations.
b. What social distancing and masking rules will be needed?
i. Will patients need to bring their own masks, or will you supply them?
c. What extra supplies and efforts are needed, e.g. hand sanitizer, new signage, 6-foot interval markings on floors or sidewalks, families calling from parking lot to announce their arrivals, etc.?
d. Remember younger patients need two doses before Dec 1, 2020.
e. Be creative, for example, are parking-lot tents for influenza vaccination feasible?
f. Can we partner with other providers to implement influenza vaccine–specific mass clinics?
Ramping up to give seasonal influenza vaccine in 2020 is daunting. But if we do not prepare, it will be even more difficult. Let’s make this the mildest influenza season in memory by vaccinating more than any time in memory – and by doing so, we can hope to blunt medical care burdens despite ongoing COVID-19 disease.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Kansas City (Mo.). Children’s Mercy receives funding from GlaxoSmithKline, Merck, and Pfizer for vaccine research studies on which Dr. Harrison is an investigator. Email him at [email protected].
References
1.. HCUP Statistical Brief #253. 2019 Oct.
2. medrxiv. 2020 Apr 10. doi: 10.1101/2020.04.07.20057299.
Young children with neuromuscular disease are vulnerable to respiratory viruses
This highlights the need for new vaccines
Influenza gets a lot of attention each winter, but respiratory syncytial virus (RSV) and other respiratory viruses have as much or more impact on pediatric populations, particularly certain high-risk groups. But currently there are no vaccines for noninfluenza respiratory viruses. That said, several are under development, for RSV and parainfluenza.
Which groups are likely to get the most benefit from these newer vaccines?
We all are aware of the extra vulnerability to respiratory viruses (RSV being the most frequent) in premature infants, those with chronic lung disease, or those with congenital heart syndromes; such vulnerable patients are not infrequently seen in routine practice. A recent report shined a brighter light on such a group.
Real-world data from a nationwide Canadian surveillance system (CARESS) was used to analyze relative risks of categories of young children who are thought to be vulnerable to respiratory viruses, with a particular focus on those with neuromuscular disease. The CARESS investigators analyzed 12 years’ data on respiratory hospitalizations from among palivizumab-prophylaxed patients (including specific data on RSV when patients were tested for RSV per standard of care).1 Unfortunately, RSV testing was not universal despite hospitalization, so the true incidence of RSV-specific hospitalizations was likely underestimated.
Nevertheless, more than 25,000 children from 2005 through 2017 were grouped into three categories of palivizumab-prophylaxed high-risk children: standard indications (SI), n = 20,335; chronic medical conditions (CMD), n = 4,063; and neuromuscular disease (NMD), n = 605. This study is notable for having a relatively large number of neuromuscular disease subjects. Two-thirds of each group were fully palivizumab adherent.
The SI group included the standard American Academy of Pediatrics–recommended groups, such as premature infants, congenital heart disease, etc.
The CMD group included conditions that lead clinicians to use palivizumab off label, such as cystic fibrosis, congenital airway anomalies, immunodeficiency, and pulmonary disorders.
The NMD participants were subdivided into two groups. Group 1 comprised general hypotonic neuromuscular diseases such as hypoxic-ischemic encephalopathy, Prader-Willi syndrome, chromosomal disorders, and migration/demyelinating diseases. Group 2 included more severe infantile neuromuscular disorders, such as spinal muscular atrophy, myotonic dystrophy, centronuclear and nemaline myopathy, mitochondrial and glycogen storage myopathies, or arthrogryposis.
Overall, 6.9% of CARESS RSV-prophylaxed subjects were hospitalized. About one in five hospitalized patients from each group was hospitalized more than once. Specific respiratory hospitalization rates for each group were 6% (n = 1,228) for SI subjects and 9.4% (n = 380) for CMD, compared with 19.2% (n = 116) for NMD subjects.
It is unclear what proportion underwent RSV testing, but a total of 334 were confirmed RSV positive: 261 were SI, 54 were CMD and 19 were NMD. The RSV-test-positive rate was 1.5% for SI, 1.6% for CMD and 3.3% for NMD; so while a higher number of SI children were RSV positive, the rate of RSV positivity was actually highest with NMD.
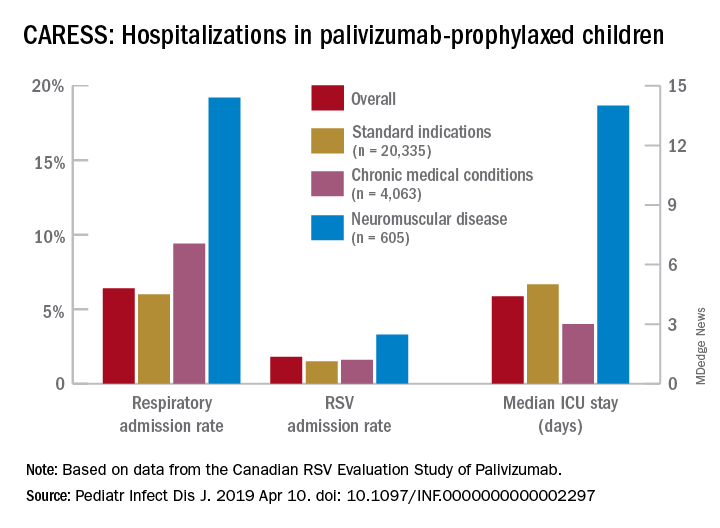
RSV-positive subjects needing ICU care among NMD patients also had longer ICU stays (median 14 days), compared with RSV-positive CMD or SI subjects (median 3 and 5 days, respectively). Further, hospitalized RSV-positive NMD subjects presented more frequently with pneumonia (42% vs. 30% for CMD and 20% for SI) while hospitalized RSV-positive SI subjects more often had apnea (17% vs. 10% for NMD and 5% for CMD, P less than .05).
These differences in the courses of NMD patients raise the question as to whether the NMD group was somehow different from the SI and CMD groups, other than muscular weakness that likely leads to less ability to clear secretions and a less efficient cough. It turns out that NMD children were older and had worse neonatal medical courses (longer hospital stays, more often ventilated, and used oxygen longer). It could be argued that these differences may have been in part due to the muscular weakness inherent in their underlying disease, but they appear to be predictors of worse respiratory infectious disease than other vulnerable populations as the NMD children get older.
Indeed, the overall risk of any respiratory admission among NMD subjects was nearly twice as high, compared with SI (hazard ratio, 1.90, P less than .0005); but the somewhat higher risk for NMD vs. CMD was not significant (HR, 1.33, P = .090). However, when looking specifically at RSV confirmed admissions, NMD had more than twice the hospitalization risk than either other group (HR, 2.26, P = .001 vs. SI; and HR, 2.74, P = .001 vs. CMD).
Further, an NMD subgroup analysis showed 1.69 times the overall respiratory hospitalization risk among the more severe vs. less severe NMD group, but a similar risk of RSV admission. The authors point out that one reason for this discrepancy may be a higher probability of aspiration causing hospitalization because of more dramatic acute events during respiratory infections in patients with more severe NMD. It also may be that palivizumab evened the playing field for RSV but not for other viruses such as parainfluenza, adenovirus, or even rhinovirus.
Nevertheless, these data tell us that risk of respiratory disease severe enough to need hospitalization continues to an older age in NMD than SI or CMD patients, well past 2 years of age. And the risk is not only from RSV. That said, RSV remains a player in some patients (particularly NMD patients) despite palivizumab prophylaxis, highlighting the need for RSV as well as parainfluenza vaccines. While these vaccines should help all young children, they seem likely to be even more beneficial for high-risk children including those with NMD, and particularly those with more severe NMD.
Eleven among 60 total candidate RSV vaccines (live attenuated, particle based, or vector based) are currently in clinical trials.2 Fewer parainfluenza vaccines are in the pipeline, but clinical trials also are underway.3-5 Approval of such vaccines is not expected until the mid-2020s, so at present we are left with providing palivizumab to our vulnerable patients while emphasizing nonmedical strategies that may help prevent respiratory viruses. These only partially successful preventive interventions include breastfeeding, avoiding secondhand smoke, and avoiding known high-risk exposures, such as large day care centers.
My hope is for quicker than projected progress on the vaccine front so that winter admissions for respiratory viruses might decrease in numbers similar to the decrease we have noted with another vaccine successful against a seasonally active pathogen – rotavirus.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospital–Kansas City, Mo. Children’s Mercy Hospital receives grant funding to study two candidate RSV vaccines. The hospital also receives CDC funding under the New Vaccine Surveillance Network for multicenter surveillance of acute respiratory infections, including influenza, RSV, and parainfluenza virus. Email Dr. Harrison at [email protected].
References
1. Pediatr Infect Dis J. 2019 Apr 10. doi: 10.1097/INF.0000000000002297.
2. “Advances in RSV Vaccine Research and Development – A Global Agenda.”
3. J Pediatric Infect Dis Soc. 2015 Dec;4(4): e143-6.
4. J Virol. 2015 Oct;89(20):10319-32.
5. Vaccine. 2017 Dec 18;35(51):7139-46.
This highlights the need for new vaccines
This highlights the need for new vaccines
Influenza gets a lot of attention each winter, but respiratory syncytial virus (RSV) and other respiratory viruses have as much or more impact on pediatric populations, particularly certain high-risk groups. But currently there are no vaccines for noninfluenza respiratory viruses. That said, several are under development, for RSV and parainfluenza.
Which groups are likely to get the most benefit from these newer vaccines?
We all are aware of the extra vulnerability to respiratory viruses (RSV being the most frequent) in premature infants, those with chronic lung disease, or those with congenital heart syndromes; such vulnerable patients are not infrequently seen in routine practice. A recent report shined a brighter light on such a group.
Real-world data from a nationwide Canadian surveillance system (CARESS) was used to analyze relative risks of categories of young children who are thought to be vulnerable to respiratory viruses, with a particular focus on those with neuromuscular disease. The CARESS investigators analyzed 12 years’ data on respiratory hospitalizations from among palivizumab-prophylaxed patients (including specific data on RSV when patients were tested for RSV per standard of care).1 Unfortunately, RSV testing was not universal despite hospitalization, so the true incidence of RSV-specific hospitalizations was likely underestimated.
Nevertheless, more than 25,000 children from 2005 through 2017 were grouped into three categories of palivizumab-prophylaxed high-risk children: standard indications (SI), n = 20,335; chronic medical conditions (CMD), n = 4,063; and neuromuscular disease (NMD), n = 605. This study is notable for having a relatively large number of neuromuscular disease subjects. Two-thirds of each group were fully palivizumab adherent.
The SI group included the standard American Academy of Pediatrics–recommended groups, such as premature infants, congenital heart disease, etc.
The CMD group included conditions that lead clinicians to use palivizumab off label, such as cystic fibrosis, congenital airway anomalies, immunodeficiency, and pulmonary disorders.
The NMD participants were subdivided into two groups. Group 1 comprised general hypotonic neuromuscular diseases such as hypoxic-ischemic encephalopathy, Prader-Willi syndrome, chromosomal disorders, and migration/demyelinating diseases. Group 2 included more severe infantile neuromuscular disorders, such as spinal muscular atrophy, myotonic dystrophy, centronuclear and nemaline myopathy, mitochondrial and glycogen storage myopathies, or arthrogryposis.
Overall, 6.9% of CARESS RSV-prophylaxed subjects were hospitalized. About one in five hospitalized patients from each group was hospitalized more than once. Specific respiratory hospitalization rates for each group were 6% (n = 1,228) for SI subjects and 9.4% (n = 380) for CMD, compared with 19.2% (n = 116) for NMD subjects.
It is unclear what proportion underwent RSV testing, but a total of 334 were confirmed RSV positive: 261 were SI, 54 were CMD and 19 were NMD. The RSV-test-positive rate was 1.5% for SI, 1.6% for CMD and 3.3% for NMD; so while a higher number of SI children were RSV positive, the rate of RSV positivity was actually highest with NMD.
RSV-positive subjects needing ICU care among NMD patients also had longer ICU stays (median 14 days), compared with RSV-positive CMD or SI subjects (median 3 and 5 days, respectively). Further, hospitalized RSV-positive NMD subjects presented more frequently with pneumonia (42% vs. 30% for CMD and 20% for SI) while hospitalized RSV-positive SI subjects more often had apnea (17% vs. 10% for NMD and 5% for CMD, P less than .05).
These differences in the courses of NMD patients raise the question as to whether the NMD group was somehow different from the SI and CMD groups, other than muscular weakness that likely leads to less ability to clear secretions and a less efficient cough. It turns out that NMD children were older and had worse neonatal medical courses (longer hospital stays, more often ventilated, and used oxygen longer). It could be argued that these differences may have been in part due to the muscular weakness inherent in their underlying disease, but they appear to be predictors of worse respiratory infectious disease than other vulnerable populations as the NMD children get older.
Indeed, the overall risk of any respiratory admission among NMD subjects was nearly twice as high, compared with SI (hazard ratio, 1.90, P less than .0005); but the somewhat higher risk for NMD vs. CMD was not significant (HR, 1.33, P = .090). However, when looking specifically at RSV confirmed admissions, NMD had more than twice the hospitalization risk than either other group (HR, 2.26, P = .001 vs. SI; and HR, 2.74, P = .001 vs. CMD).
Further, an NMD subgroup analysis showed 1.69 times the overall respiratory hospitalization risk among the more severe vs. less severe NMD group, but a similar risk of RSV admission. The authors point out that one reason for this discrepancy may be a higher probability of aspiration causing hospitalization because of more dramatic acute events during respiratory infections in patients with more severe NMD. It also may be that palivizumab evened the playing field for RSV but not for other viruses such as parainfluenza, adenovirus, or even rhinovirus.
Nevertheless, these data tell us that risk of respiratory disease severe enough to need hospitalization continues to an older age in NMD than SI or CMD patients, well past 2 years of age. And the risk is not only from RSV. That said, RSV remains a player in some patients (particularly NMD patients) despite palivizumab prophylaxis, highlighting the need for RSV as well as parainfluenza vaccines. While these vaccines should help all young children, they seem likely to be even more beneficial for high-risk children including those with NMD, and particularly those with more severe NMD.
Eleven among 60 total candidate RSV vaccines (live attenuated, particle based, or vector based) are currently in clinical trials.2 Fewer parainfluenza vaccines are in the pipeline, but clinical trials also are underway.3-5 Approval of such vaccines is not expected until the mid-2020s, so at present we are left with providing palivizumab to our vulnerable patients while emphasizing nonmedical strategies that may help prevent respiratory viruses. These only partially successful preventive interventions include breastfeeding, avoiding secondhand smoke, and avoiding known high-risk exposures, such as large day care centers.
My hope is for quicker than projected progress on the vaccine front so that winter admissions for respiratory viruses might decrease in numbers similar to the decrease we have noted with another vaccine successful against a seasonally active pathogen – rotavirus.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospital–Kansas City, Mo. Children’s Mercy Hospital receives grant funding to study two candidate RSV vaccines. The hospital also receives CDC funding under the New Vaccine Surveillance Network for multicenter surveillance of acute respiratory infections, including influenza, RSV, and parainfluenza virus. Email Dr. Harrison at [email protected].
References
1. Pediatr Infect Dis J. 2019 Apr 10. doi: 10.1097/INF.0000000000002297.
2. “Advances in RSV Vaccine Research and Development – A Global Agenda.”
3. J Pediatric Infect Dis Soc. 2015 Dec;4(4): e143-6.
4. J Virol. 2015 Oct;89(20):10319-32.
5. Vaccine. 2017 Dec 18;35(51):7139-46.
Influenza gets a lot of attention each winter, but respiratory syncytial virus (RSV) and other respiratory viruses have as much or more impact on pediatric populations, particularly certain high-risk groups. But currently there are no vaccines for noninfluenza respiratory viruses. That said, several are under development, for RSV and parainfluenza.
Which groups are likely to get the most benefit from these newer vaccines?
We all are aware of the extra vulnerability to respiratory viruses (RSV being the most frequent) in premature infants, those with chronic lung disease, or those with congenital heart syndromes; such vulnerable patients are not infrequently seen in routine practice. A recent report shined a brighter light on such a group.
Real-world data from a nationwide Canadian surveillance system (CARESS) was used to analyze relative risks of categories of young children who are thought to be vulnerable to respiratory viruses, with a particular focus on those with neuromuscular disease. The CARESS investigators analyzed 12 years’ data on respiratory hospitalizations from among palivizumab-prophylaxed patients (including specific data on RSV when patients were tested for RSV per standard of care).1 Unfortunately, RSV testing was not universal despite hospitalization, so the true incidence of RSV-specific hospitalizations was likely underestimated.
Nevertheless, more than 25,000 children from 2005 through 2017 were grouped into three categories of palivizumab-prophylaxed high-risk children: standard indications (SI), n = 20,335; chronic medical conditions (CMD), n = 4,063; and neuromuscular disease (NMD), n = 605. This study is notable for having a relatively large number of neuromuscular disease subjects. Two-thirds of each group were fully palivizumab adherent.
The SI group included the standard American Academy of Pediatrics–recommended groups, such as premature infants, congenital heart disease, etc.
The CMD group included conditions that lead clinicians to use palivizumab off label, such as cystic fibrosis, congenital airway anomalies, immunodeficiency, and pulmonary disorders.
The NMD participants were subdivided into two groups. Group 1 comprised general hypotonic neuromuscular diseases such as hypoxic-ischemic encephalopathy, Prader-Willi syndrome, chromosomal disorders, and migration/demyelinating diseases. Group 2 included more severe infantile neuromuscular disorders, such as spinal muscular atrophy, myotonic dystrophy, centronuclear and nemaline myopathy, mitochondrial and glycogen storage myopathies, or arthrogryposis.
Overall, 6.9% of CARESS RSV-prophylaxed subjects were hospitalized. About one in five hospitalized patients from each group was hospitalized more than once. Specific respiratory hospitalization rates for each group were 6% (n = 1,228) for SI subjects and 9.4% (n = 380) for CMD, compared with 19.2% (n = 116) for NMD subjects.
It is unclear what proportion underwent RSV testing, but a total of 334 were confirmed RSV positive: 261 were SI, 54 were CMD and 19 were NMD. The RSV-test-positive rate was 1.5% for SI, 1.6% for CMD and 3.3% for NMD; so while a higher number of SI children were RSV positive, the rate of RSV positivity was actually highest with NMD.
RSV-positive subjects needing ICU care among NMD patients also had longer ICU stays (median 14 days), compared with RSV-positive CMD or SI subjects (median 3 and 5 days, respectively). Further, hospitalized RSV-positive NMD subjects presented more frequently with pneumonia (42% vs. 30% for CMD and 20% for SI) while hospitalized RSV-positive SI subjects more often had apnea (17% vs. 10% for NMD and 5% for CMD, P less than .05).
These differences in the courses of NMD patients raise the question as to whether the NMD group was somehow different from the SI and CMD groups, other than muscular weakness that likely leads to less ability to clear secretions and a less efficient cough. It turns out that NMD children were older and had worse neonatal medical courses (longer hospital stays, more often ventilated, and used oxygen longer). It could be argued that these differences may have been in part due to the muscular weakness inherent in their underlying disease, but they appear to be predictors of worse respiratory infectious disease than other vulnerable populations as the NMD children get older.
Indeed, the overall risk of any respiratory admission among NMD subjects was nearly twice as high, compared with SI (hazard ratio, 1.90, P less than .0005); but the somewhat higher risk for NMD vs. CMD was not significant (HR, 1.33, P = .090). However, when looking specifically at RSV confirmed admissions, NMD had more than twice the hospitalization risk than either other group (HR, 2.26, P = .001 vs. SI; and HR, 2.74, P = .001 vs. CMD).
Further, an NMD subgroup analysis showed 1.69 times the overall respiratory hospitalization risk among the more severe vs. less severe NMD group, but a similar risk of RSV admission. The authors point out that one reason for this discrepancy may be a higher probability of aspiration causing hospitalization because of more dramatic acute events during respiratory infections in patients with more severe NMD. It also may be that palivizumab evened the playing field for RSV but not for other viruses such as parainfluenza, adenovirus, or even rhinovirus.
Nevertheless, these data tell us that risk of respiratory disease severe enough to need hospitalization continues to an older age in NMD than SI or CMD patients, well past 2 years of age. And the risk is not only from RSV. That said, RSV remains a player in some patients (particularly NMD patients) despite palivizumab prophylaxis, highlighting the need for RSV as well as parainfluenza vaccines. While these vaccines should help all young children, they seem likely to be even more beneficial for high-risk children including those with NMD, and particularly those with more severe NMD.
Eleven among 60 total candidate RSV vaccines (live attenuated, particle based, or vector based) are currently in clinical trials.2 Fewer parainfluenza vaccines are in the pipeline, but clinical trials also are underway.3-5 Approval of such vaccines is not expected until the mid-2020s, so at present we are left with providing palivizumab to our vulnerable patients while emphasizing nonmedical strategies that may help prevent respiratory viruses. These only partially successful preventive interventions include breastfeeding, avoiding secondhand smoke, and avoiding known high-risk exposures, such as large day care centers.
My hope is for quicker than projected progress on the vaccine front so that winter admissions for respiratory viruses might decrease in numbers similar to the decrease we have noted with another vaccine successful against a seasonally active pathogen – rotavirus.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospital–Kansas City, Mo. Children’s Mercy Hospital receives grant funding to study two candidate RSV vaccines. The hospital also receives CDC funding under the New Vaccine Surveillance Network for multicenter surveillance of acute respiratory infections, including influenza, RSV, and parainfluenza virus. Email Dr. Harrison at [email protected].
References
1. Pediatr Infect Dis J. 2019 Apr 10. doi: 10.1097/INF.0000000000002297.
2. “Advances in RSV Vaccine Research and Development – A Global Agenda.”
3. J Pediatric Infect Dis Soc. 2015 Dec;4(4): e143-6.
4. J Virol. 2015 Oct;89(20):10319-32.
5. Vaccine. 2017 Dec 18;35(51):7139-46.
Uptick in adult syphilis means congenital syphilis may be lurking
While many pediatric clinicians have not frequently managed newborns of mothers with reactive syphilis serology, increased adult syphilis may change that.1
Diagnosing/managing congenital syphilis is not always clear cut. A positive rapid plasma reagin (RPR) titer in a newborn may not indicate congenital infection but merely may reflect transplacental, passively acquired maternal IgG from the mother’s current or previous infection rather than antibodies produced by the newborn. Because currently no IgM assay for syphilis is recommended by the Centers for Disease Control and Prevention for newborn testing, we must deal with IgG test results.
Often initial management decisions are needed while the infant’s status is evolving. The questions to answer to make final decisions include the following2:
- Was the mother actively infected with Treponema pallidum during pregnancy?
- If so, was the mother appropriately treated and when?
- Does the infant have any clinical, laboratory, or radiographic evidence of syphilis?
- How do the mother’s and infant’s nontreponemal serologic titers (NTT) compare at delivery using the same test?
Note: All infants assessed for congenital syphilis need a full evaluation for HIV.
Managing the infant of a mother with positive tests3,4
All such neonates need an examination for evidence of congenital syphilis. The clinical signs of congenital syphilis in neonates include nonimmune hydrops, jaundice, hepatosplenomegaly, rhinitis, skin rash, and pseudoparalysis of extremity. Also, consider dark-field examination or polymerase chain reaction (PCR) of lesions (such as bullae) or secretions (nasal). If available, have the placenta examined histologically (silver stain) or by PCR (Clinical Laboratory Improvement Amendments–validated test). Skeletal radiographic surveys are more useful for stillborn than live born infants. (The complete algorithm can be found in Figure 3.10 of reference 4.)
Order a quantitative NTT, using the Venereal Disease Research Laboratory (VDRL) test or RPR test on neonatal serum. Umbilical cord blood is not appropriate because of potential maternal blood contamination, which could give a false-positive result, or Wharton’s jelly, which could give a false-negative result. Use of treponemal-specific tests that are used for maternal diagnosis – such as T. pallidum particle agglutination (TP-PA), T. pallidum enzyme-linked immunosorbent assay (TP-EIA), fluorescent treponemal antibody absorption (FTA-ABS) test, or T. pallidum chemiluminescence immunoassay (TP-CIA) – on neonatal serum is not recommended because of difficulties in interpretation.
Diagnostic results allow designation of an infant into one of four CDC categories: proven/highly probable syphilis; possible syphilis; syphilis less likely; and syphilis unlikely. Treatment recommendations are based on these categories.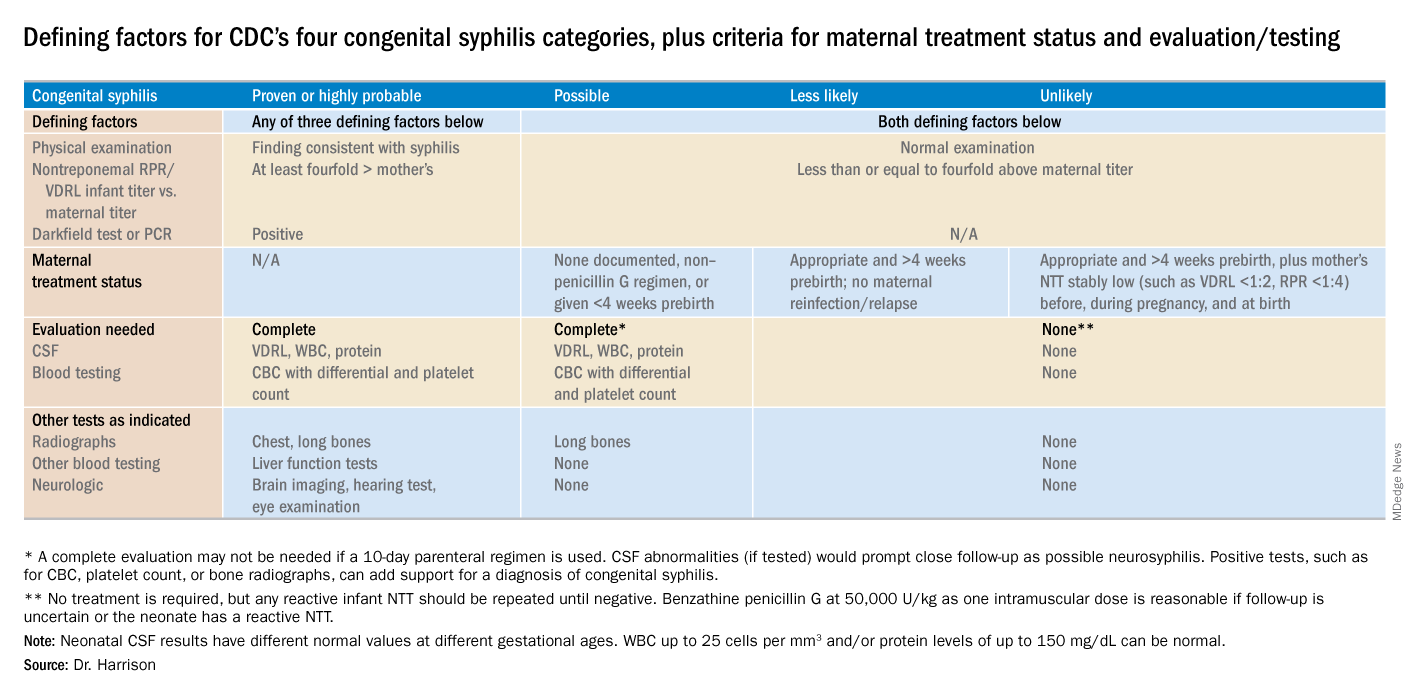
Proven or highly probable syphilis
There are two alternative recommended 10-day treatment regimens.
A. Aqueous crystalline penicillin G 100,000-150,000 U/kg per day by IV at 50,000 U/kg per dose, given every 12 hours through 7 days of age or every 8 hours if greater than 7 days old.
B. Procaine penicillin G at 50,000 U/kg per dose intramuscularly in one dose each day.
More than 1 day of missed therapy requires restarting a new 10-day course. Use of other antimicrobial agents (such as ampicillin) is not validated, so any empiric ampicillin initially given for possible sepsis does not count toward the 10-day penicillin regimen. If nonpenicillin drugs must be used, close serologic follow-up must occur to ensure adequacy of response to therapy.
Possible syphilis
There are three alternative regimens, the same two as in proven/highly probable syphilis (above) plus a single-dose option
A. Aqueous crystalline penicillin G, as described above.
B. Procaine penicillin G, as described above.
C. Benzathine penicillin G at 50,000 U/kg per dose intramuscularly in a single dose.
Note: To be eligible for regimen C, an infant must have a complete evaluation that is normal (cerebrospinal fluid [CSF] examination, long-bone radiographs, and complete blood count with platelet count) and follow-up must be assured. Exception: Neonates born to mothers with untreated early syphilis at the time of delivery are at increased risk for congenital syphilis, and the 10-day course of penicillin G may be considered even if the complete evaluation is normal and follow-up is certain.
Less likely syphilis
One antibiotic regimen is available, but no treatment also may be an option.
A. Benzathine penicillin G as described above.
B. If mother’s NTT has decreased at least fourfold after appropriate early syphilis therapy or remained stably low, which indicates latent syphilis (VDRL less than 1:2; RPR less than 1:4), no treatment is an option but requires repeat serology every 2-3 months until infant is 6 months old.
Unlikely syphilis
No treatment is recommended unless follow-up is uncertain, in which case it is appropriate to give the infant benzathine penicillin G as described above.
Infant with positive NTT at birth
All neonates with reactive NTT need careful follow-up examinations and repeat NTT every 2-3 months until nonreactive. NTT in infants who are not treated because of less likely or unlikely syphilis status should drop by 3 months and be nonreactive by 6 months; this indicates NTT was passively transferred maternal IgG. If NTT remains reactive at 6 months, the infant is likely infected and needs treatment. Persistent NTT at 6-12 months in treated neonates should trigger repeat CSF examination and infectious diseases consultation about a possible repeat of the 10-day penicillin G regimen. If the mother was seroreactive, but the newborn’s NTT was negative at birth, testing of the infant’s NTT needs repeating at 3 months to exclude the possibility that the congenital syphilis was incubating when prior testing occurred at birth. Note: Treponemal-specific tests are not useful in assessing treatment because detectable maternal IgG treponemal antibody can persist at least 15 months.
Neonates with abnormal CSF at birth
Repeat cerebrospinal fluid evaluation every 6 months until results normalize. Persistently reactive CSF VDRL or abnormal CSF indexes not caused by another known cause requires retreatment for possible neurosyphilis, as well as consultation with an expert.
Summary
NTT are the essential test for newborns and some degree of laboratory or imaging work up often are needed. Consider consulting an expert in infectious diseases and/or perinatology if the gray areas do not readily become clear. Treatment of the correct patients with the right drug for the right duration remains the goal, as usual.
Dr. Harrison is a professor of pediatrics at University of Missouri-Kansas City and Director of Research Affairs in the pediatric infectious diseases division at Children’s Mercy Hospital – Kansas City. He said he had no relevant financial disclosures. Email him at [email protected].
References
1. MMWR. 2015 Nov 13;64(44);1241-5.
2. “Congenital Syphilis,” 2015 Sexually Transmitted Diseases Treatment Guidelines.
3. “Syphilis During Pregnancy,” 2015 Sexually Transmitted Diseases Treatment Guidelines.
4. Syphilis – Section 3: Summaries of Infectious Diseases. Red Book Online. 2018.
While many pediatric clinicians have not frequently managed newborns of mothers with reactive syphilis serology, increased adult syphilis may change that.1
Diagnosing/managing congenital syphilis is not always clear cut. A positive rapid plasma reagin (RPR) titer in a newborn may not indicate congenital infection but merely may reflect transplacental, passively acquired maternal IgG from the mother’s current or previous infection rather than antibodies produced by the newborn. Because currently no IgM assay for syphilis is recommended by the Centers for Disease Control and Prevention for newborn testing, we must deal with IgG test results.
Often initial management decisions are needed while the infant’s status is evolving. The questions to answer to make final decisions include the following2:
- Was the mother actively infected with Treponema pallidum during pregnancy?
- If so, was the mother appropriately treated and when?
- Does the infant have any clinical, laboratory, or radiographic evidence of syphilis?
- How do the mother’s and infant’s nontreponemal serologic titers (NTT) compare at delivery using the same test?
Note: All infants assessed for congenital syphilis need a full evaluation for HIV.
Managing the infant of a mother with positive tests3,4
All such neonates need an examination for evidence of congenital syphilis. The clinical signs of congenital syphilis in neonates include nonimmune hydrops, jaundice, hepatosplenomegaly, rhinitis, skin rash, and pseudoparalysis of extremity. Also, consider dark-field examination or polymerase chain reaction (PCR) of lesions (such as bullae) or secretions (nasal). If available, have the placenta examined histologically (silver stain) or by PCR (Clinical Laboratory Improvement Amendments–validated test). Skeletal radiographic surveys are more useful for stillborn than live born infants. (The complete algorithm can be found in Figure 3.10 of reference 4.)
Order a quantitative NTT, using the Venereal Disease Research Laboratory (VDRL) test or RPR test on neonatal serum. Umbilical cord blood is not appropriate because of potential maternal blood contamination, which could give a false-positive result, or Wharton’s jelly, which could give a false-negative result. Use of treponemal-specific tests that are used for maternal diagnosis – such as T. pallidum particle agglutination (TP-PA), T. pallidum enzyme-linked immunosorbent assay (TP-EIA), fluorescent treponemal antibody absorption (FTA-ABS) test, or T. pallidum chemiluminescence immunoassay (TP-CIA) – on neonatal serum is not recommended because of difficulties in interpretation.
Diagnostic results allow designation of an infant into one of four CDC categories: proven/highly probable syphilis; possible syphilis; syphilis less likely; and syphilis unlikely. Treatment recommendations are based on these categories.
Proven or highly probable syphilis
There are two alternative recommended 10-day treatment regimens.
A. Aqueous crystalline penicillin G 100,000-150,000 U/kg per day by IV at 50,000 U/kg per dose, given every 12 hours through 7 days of age or every 8 hours if greater than 7 days old.
B. Procaine penicillin G at 50,000 U/kg per dose intramuscularly in one dose each day.
More than 1 day of missed therapy requires restarting a new 10-day course. Use of other antimicrobial agents (such as ampicillin) is not validated, so any empiric ampicillin initially given for possible sepsis does not count toward the 10-day penicillin regimen. If nonpenicillin drugs must be used, close serologic follow-up must occur to ensure adequacy of response to therapy.
Possible syphilis
There are three alternative regimens, the same two as in proven/highly probable syphilis (above) plus a single-dose option
A. Aqueous crystalline penicillin G, as described above.
B. Procaine penicillin G, as described above.
C. Benzathine penicillin G at 50,000 U/kg per dose intramuscularly in a single dose.
Note: To be eligible for regimen C, an infant must have a complete evaluation that is normal (cerebrospinal fluid [CSF] examination, long-bone radiographs, and complete blood count with platelet count) and follow-up must be assured. Exception: Neonates born to mothers with untreated early syphilis at the time of delivery are at increased risk for congenital syphilis, and the 10-day course of penicillin G may be considered even if the complete evaluation is normal and follow-up is certain.
Less likely syphilis
One antibiotic regimen is available, but no treatment also may be an option.
A. Benzathine penicillin G as described above.
B. If mother’s NTT has decreased at least fourfold after appropriate early syphilis therapy or remained stably low, which indicates latent syphilis (VDRL less than 1:2; RPR less than 1:4), no treatment is an option but requires repeat serology every 2-3 months until infant is 6 months old.
Unlikely syphilis
No treatment is recommended unless follow-up is uncertain, in which case it is appropriate to give the infant benzathine penicillin G as described above.
Infant with positive NTT at birth
All neonates with reactive NTT need careful follow-up examinations and repeat NTT every 2-3 months until nonreactive. NTT in infants who are not treated because of less likely or unlikely syphilis status should drop by 3 months and be nonreactive by 6 months; this indicates NTT was passively transferred maternal IgG. If NTT remains reactive at 6 months, the infant is likely infected and needs treatment. Persistent NTT at 6-12 months in treated neonates should trigger repeat CSF examination and infectious diseases consultation about a possible repeat of the 10-day penicillin G regimen. If the mother was seroreactive, but the newborn’s NTT was negative at birth, testing of the infant’s NTT needs repeating at 3 months to exclude the possibility that the congenital syphilis was incubating when prior testing occurred at birth. Note: Treponemal-specific tests are not useful in assessing treatment because detectable maternal IgG treponemal antibody can persist at least 15 months.
Neonates with abnormal CSF at birth
Repeat cerebrospinal fluid evaluation every 6 months until results normalize. Persistently reactive CSF VDRL or abnormal CSF indexes not caused by another known cause requires retreatment for possible neurosyphilis, as well as consultation with an expert.
Summary
NTT are the essential test for newborns and some degree of laboratory or imaging work up often are needed. Consider consulting an expert in infectious diseases and/or perinatology if the gray areas do not readily become clear. Treatment of the correct patients with the right drug for the right duration remains the goal, as usual.
Dr. Harrison is a professor of pediatrics at University of Missouri-Kansas City and Director of Research Affairs in the pediatric infectious diseases division at Children’s Mercy Hospital – Kansas City. He said he had no relevant financial disclosures. Email him at [email protected].
References
1. MMWR. 2015 Nov 13;64(44);1241-5.
2. “Congenital Syphilis,” 2015 Sexually Transmitted Diseases Treatment Guidelines.
3. “Syphilis During Pregnancy,” 2015 Sexually Transmitted Diseases Treatment Guidelines.
4. Syphilis – Section 3: Summaries of Infectious Diseases. Red Book Online. 2018.
While many pediatric clinicians have not frequently managed newborns of mothers with reactive syphilis serology, increased adult syphilis may change that.1
Diagnosing/managing congenital syphilis is not always clear cut. A positive rapid plasma reagin (RPR) titer in a newborn may not indicate congenital infection but merely may reflect transplacental, passively acquired maternal IgG from the mother’s current or previous infection rather than antibodies produced by the newborn. Because currently no IgM assay for syphilis is recommended by the Centers for Disease Control and Prevention for newborn testing, we must deal with IgG test results.
Often initial management decisions are needed while the infant’s status is evolving. The questions to answer to make final decisions include the following2:
- Was the mother actively infected with Treponema pallidum during pregnancy?
- If so, was the mother appropriately treated and when?
- Does the infant have any clinical, laboratory, or radiographic evidence of syphilis?
- How do the mother’s and infant’s nontreponemal serologic titers (NTT) compare at delivery using the same test?
Note: All infants assessed for congenital syphilis need a full evaluation for HIV.
Managing the infant of a mother with positive tests3,4
All such neonates need an examination for evidence of congenital syphilis. The clinical signs of congenital syphilis in neonates include nonimmune hydrops, jaundice, hepatosplenomegaly, rhinitis, skin rash, and pseudoparalysis of extremity. Also, consider dark-field examination or polymerase chain reaction (PCR) of lesions (such as bullae) or secretions (nasal). If available, have the placenta examined histologically (silver stain) or by PCR (Clinical Laboratory Improvement Amendments–validated test). Skeletal radiographic surveys are more useful for stillborn than live born infants. (The complete algorithm can be found in Figure 3.10 of reference 4.)
Order a quantitative NTT, using the Venereal Disease Research Laboratory (VDRL) test or RPR test on neonatal serum. Umbilical cord blood is not appropriate because of potential maternal blood contamination, which could give a false-positive result, or Wharton’s jelly, which could give a false-negative result. Use of treponemal-specific tests that are used for maternal diagnosis – such as T. pallidum particle agglutination (TP-PA), T. pallidum enzyme-linked immunosorbent assay (TP-EIA), fluorescent treponemal antibody absorption (FTA-ABS) test, or T. pallidum chemiluminescence immunoassay (TP-CIA) – on neonatal serum is not recommended because of difficulties in interpretation.
Diagnostic results allow designation of an infant into one of four CDC categories: proven/highly probable syphilis; possible syphilis; syphilis less likely; and syphilis unlikely. Treatment recommendations are based on these categories.
Proven or highly probable syphilis
There are two alternative recommended 10-day treatment regimens.
A. Aqueous crystalline penicillin G 100,000-150,000 U/kg per day by IV at 50,000 U/kg per dose, given every 12 hours through 7 days of age or every 8 hours if greater than 7 days old.
B. Procaine penicillin G at 50,000 U/kg per dose intramuscularly in one dose each day.
More than 1 day of missed therapy requires restarting a new 10-day course. Use of other antimicrobial agents (such as ampicillin) is not validated, so any empiric ampicillin initially given for possible sepsis does not count toward the 10-day penicillin regimen. If nonpenicillin drugs must be used, close serologic follow-up must occur to ensure adequacy of response to therapy.
Possible syphilis
There are three alternative regimens, the same two as in proven/highly probable syphilis (above) plus a single-dose option
A. Aqueous crystalline penicillin G, as described above.
B. Procaine penicillin G, as described above.
C. Benzathine penicillin G at 50,000 U/kg per dose intramuscularly in a single dose.
Note: To be eligible for regimen C, an infant must have a complete evaluation that is normal (cerebrospinal fluid [CSF] examination, long-bone radiographs, and complete blood count with platelet count) and follow-up must be assured. Exception: Neonates born to mothers with untreated early syphilis at the time of delivery are at increased risk for congenital syphilis, and the 10-day course of penicillin G may be considered even if the complete evaluation is normal and follow-up is certain.
Less likely syphilis
One antibiotic regimen is available, but no treatment also may be an option.
A. Benzathine penicillin G as described above.
B. If mother’s NTT has decreased at least fourfold after appropriate early syphilis therapy or remained stably low, which indicates latent syphilis (VDRL less than 1:2; RPR less than 1:4), no treatment is an option but requires repeat serology every 2-3 months until infant is 6 months old.
Unlikely syphilis
No treatment is recommended unless follow-up is uncertain, in which case it is appropriate to give the infant benzathine penicillin G as described above.
Infant with positive NTT at birth
All neonates with reactive NTT need careful follow-up examinations and repeat NTT every 2-3 months until nonreactive. NTT in infants who are not treated because of less likely or unlikely syphilis status should drop by 3 months and be nonreactive by 6 months; this indicates NTT was passively transferred maternal IgG. If NTT remains reactive at 6 months, the infant is likely infected and needs treatment. Persistent NTT at 6-12 months in treated neonates should trigger repeat CSF examination and infectious diseases consultation about a possible repeat of the 10-day penicillin G regimen. If the mother was seroreactive, but the newborn’s NTT was negative at birth, testing of the infant’s NTT needs repeating at 3 months to exclude the possibility that the congenital syphilis was incubating when prior testing occurred at birth. Note: Treponemal-specific tests are not useful in assessing treatment because detectable maternal IgG treponemal antibody can persist at least 15 months.
Neonates with abnormal CSF at birth
Repeat cerebrospinal fluid evaluation every 6 months until results normalize. Persistently reactive CSF VDRL or abnormal CSF indexes not caused by another known cause requires retreatment for possible neurosyphilis, as well as consultation with an expert.
Summary
NTT are the essential test for newborns and some degree of laboratory or imaging work up often are needed. Consider consulting an expert in infectious diseases and/or perinatology if the gray areas do not readily become clear. Treatment of the correct patients with the right drug for the right duration remains the goal, as usual.
Dr. Harrison is a professor of pediatrics at University of Missouri-Kansas City and Director of Research Affairs in the pediatric infectious diseases division at Children’s Mercy Hospital – Kansas City. He said he had no relevant financial disclosures. Email him at [email protected].
References
1. MMWR. 2015 Nov 13;64(44);1241-5.
2. “Congenital Syphilis,” 2015 Sexually Transmitted Diseases Treatment Guidelines.
3. “Syphilis During Pregnancy,” 2015 Sexually Transmitted Diseases Treatment Guidelines.
4. Syphilis – Section 3: Summaries of Infectious Diseases. Red Book Online. 2018.
Kawasaki disease: New info to enhance our index of suspicion
Most U.S. mainland pediatric practitioners will see only one or two cases of Kawasaki disease (KD) in their careers, but no one wants to miss even one case.
Making the diagnosis as early as possible is important to reduce the chance of sequelae, particularly the coronary artery aneurysms that will eventually lead to 5% of overall acute coronary syndromes in adults. And because there is no “KD test,” or sometimes incomplete KD. And there are some new data that complicate this. Despite the recently updated 2017 guideline,1 most cases end up being confirmed and managed by regional “experts.” But nearly all of the approximately 6,000 KD cases/year in U.S. children younger than 5 years old start out with one or more primary care, urgent care, or ED visits.
This means that every clinician in the trenches not only needs a high index of suspicion but also needs to be at least a partial expert, too. What raises our index of suspicion? Classic data tell us we need 5 consecutive days of fever plus four or five other principal clinical findings for a KD diagnosis. The principal findings are:
1. Eyes: Bilateral bulbar nonexudative conjunctival injection.
2. Mouth: Erythema of oral/pharyngeal mucosa or cracked lips or strawberry tongue or oral mucositis.
3. Rash.
4. Hands or feet findings: Swelling/erythema or later periungual desquamation.
5. Cervical adenopathy greater than 1.4 cm, usually unilateral.
Other factors that have classically increased suspicion are winter/early spring presentation in North America, male gender (1.5:1 ratio to females), and Asian (particularly Japanese) ancestry. The importance of genetics was initially based on epidemiology (Japan/Asian risk) but lately has been further associated with six gene polymorphisms. However, molecular genetic testing is not currently a practical tool.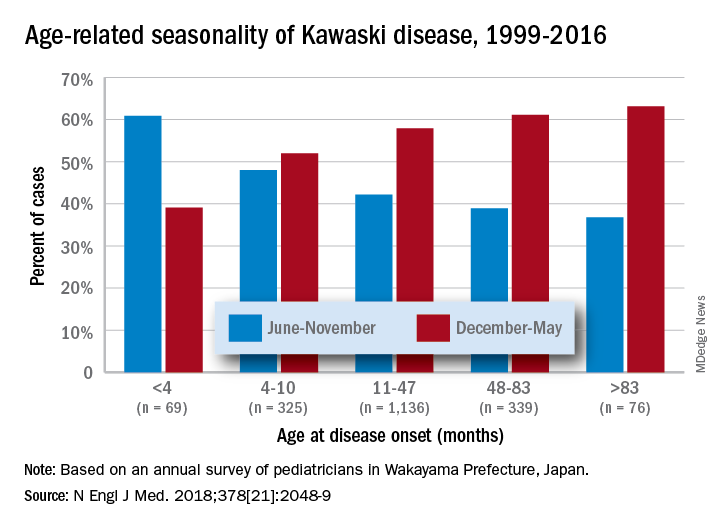
Clinical scenarios that also should raise suspicion include less-than-6-month-old infants with prolonged fever/irritability (may be the only clinical manifestations of KD) and children over 4 years old who more often may have incomplete KD. Both groups have higher prevalence of coronary artery abnormalities. Other high suspicion scenarios include prolonged fever with unexplained/culture-negative shock, or antibiotic treatment failure for cervical adenitis or retro/parapharyngeal phlegmon. Consultation with or referral to a regional KD expert may be needed.
Fuzzy KD math
Current guidelines list an exception to the 5-day fever requirement in that only 4 days of fever are needed with four or more principal clinical features, particularly when hand and feet findings exist. Some call this the “4X4 exception.” Then there is a sub-caveat: “Experienced clinicians who have treated many patients with KD may establish the diagnosis with 3 days of fever in rare cases.”1
Incomplete KD
This is another exception, which seems to be a more frequent diagnosis in the past decade. Incomplete KD requires the 5 days of fever duration plus an elevated C-reactive protein or erythrocyte sedimentation rate. But one needs only two or three of the five principal clinical KD criteria plus three or more of six other laboratory findings (anemia, low albumin, leukocytosis, thrombocytosis, pyuria, or elevated alanine aminotransferase). Incomplete KD can be confirmed by an abnormal echocardiogram – usually not until after 7 days of KD symptoms.1
New KD nuances
In a recent report on 20 years of data from Japan (n = 1,945 KD cases), more granularity on age, seasonal epidemiology, and outcome were seen.2 There was an inverse correlation of male predominance to age, i.e. as age groups got older, there was a gradual shift to female predominance by 7 years of age. The winter/spring predominance (60% of overall cases) did not hold true in younger age groups where summer/fall was the peak season (65% of cases).
With the goal of not missing any KD and diagnosing as early as possible to limit sequelae, we all need to be relative experts and keep alert for clinical scenarios that warrant our raising our index of suspicion. But now the seasonality trends appear blurred in the youngest cases and the male predominance is blurred in the oldest cases. And remember that fever and irritability for longer than 7 days in young infants may be the only clue to KD.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. He said he had no relevant financial disclosures. Email him at [email protected].
References
1. Circulation. 2017 Mar 29. doi: 10.1161/CIR.0000000000000484
2. N Engl J Med. 2018 May 24. doi: 10.1056/NEJMc1804312.
Most U.S. mainland pediatric practitioners will see only one or two cases of Kawasaki disease (KD) in their careers, but no one wants to miss even one case.
Making the diagnosis as early as possible is important to reduce the chance of sequelae, particularly the coronary artery aneurysms that will eventually lead to 5% of overall acute coronary syndromes in adults. And because there is no “KD test,” or sometimes incomplete KD. And there are some new data that complicate this. Despite the recently updated 2017 guideline,1 most cases end up being confirmed and managed by regional “experts.” But nearly all of the approximately 6,000 KD cases/year in U.S. children younger than 5 years old start out with one or more primary care, urgent care, or ED visits.
This means that every clinician in the trenches not only needs a high index of suspicion but also needs to be at least a partial expert, too. What raises our index of suspicion? Classic data tell us we need 5 consecutive days of fever plus four or five other principal clinical findings for a KD diagnosis. The principal findings are:
1. Eyes: Bilateral bulbar nonexudative conjunctival injection.
2. Mouth: Erythema of oral/pharyngeal mucosa or cracked lips or strawberry tongue or oral mucositis.
3. Rash.
4. Hands or feet findings: Swelling/erythema or later periungual desquamation.
5. Cervical adenopathy greater than 1.4 cm, usually unilateral.
Other factors that have classically increased suspicion are winter/early spring presentation in North America, male gender (1.5:1 ratio to females), and Asian (particularly Japanese) ancestry. The importance of genetics was initially based on epidemiology (Japan/Asian risk) but lately has been further associated with six gene polymorphisms. However, molecular genetic testing is not currently a practical tool.
Clinical scenarios that also should raise suspicion include less-than-6-month-old infants with prolonged fever/irritability (may be the only clinical manifestations of KD) and children over 4 years old who more often may have incomplete KD. Both groups have higher prevalence of coronary artery abnormalities. Other high suspicion scenarios include prolonged fever with unexplained/culture-negative shock, or antibiotic treatment failure for cervical adenitis or retro/parapharyngeal phlegmon. Consultation with or referral to a regional KD expert may be needed.
Fuzzy KD math
Current guidelines list an exception to the 5-day fever requirement in that only 4 days of fever are needed with four or more principal clinical features, particularly when hand and feet findings exist. Some call this the “4X4 exception.” Then there is a sub-caveat: “Experienced clinicians who have treated many patients with KD may establish the diagnosis with 3 days of fever in rare cases.”1
Incomplete KD
This is another exception, which seems to be a more frequent diagnosis in the past decade. Incomplete KD requires the 5 days of fever duration plus an elevated C-reactive protein or erythrocyte sedimentation rate. But one needs only two or three of the five principal clinical KD criteria plus three or more of six other laboratory findings (anemia, low albumin, leukocytosis, thrombocytosis, pyuria, or elevated alanine aminotransferase). Incomplete KD can be confirmed by an abnormal echocardiogram – usually not until after 7 days of KD symptoms.1
New KD nuances
In a recent report on 20 years of data from Japan (n = 1,945 KD cases), more granularity on age, seasonal epidemiology, and outcome were seen.2 There was an inverse correlation of male predominance to age, i.e. as age groups got older, there was a gradual shift to female predominance by 7 years of age. The winter/spring predominance (60% of overall cases) did not hold true in younger age groups where summer/fall was the peak season (65% of cases).
With the goal of not missing any KD and diagnosing as early as possible to limit sequelae, we all need to be relative experts and keep alert for clinical scenarios that warrant our raising our index of suspicion. But now the seasonality trends appear blurred in the youngest cases and the male predominance is blurred in the oldest cases. And remember that fever and irritability for longer than 7 days in young infants may be the only clue to KD.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. He said he had no relevant financial disclosures. Email him at [email protected].
References
1. Circulation. 2017 Mar 29. doi: 10.1161/CIR.0000000000000484
2. N Engl J Med. 2018 May 24. doi: 10.1056/NEJMc1804312.
Most U.S. mainland pediatric practitioners will see only one or two cases of Kawasaki disease (KD) in their careers, but no one wants to miss even one case.
Making the diagnosis as early as possible is important to reduce the chance of sequelae, particularly the coronary artery aneurysms that will eventually lead to 5% of overall acute coronary syndromes in adults. And because there is no “KD test,” or sometimes incomplete KD. And there are some new data that complicate this. Despite the recently updated 2017 guideline,1 most cases end up being confirmed and managed by regional “experts.” But nearly all of the approximately 6,000 KD cases/year in U.S. children younger than 5 years old start out with one or more primary care, urgent care, or ED visits.
This means that every clinician in the trenches not only needs a high index of suspicion but also needs to be at least a partial expert, too. What raises our index of suspicion? Classic data tell us we need 5 consecutive days of fever plus four or five other principal clinical findings for a KD diagnosis. The principal findings are:
1. Eyes: Bilateral bulbar nonexudative conjunctival injection.
2. Mouth: Erythema of oral/pharyngeal mucosa or cracked lips or strawberry tongue or oral mucositis.
3. Rash.
4. Hands or feet findings: Swelling/erythema or later periungual desquamation.
5. Cervical adenopathy greater than 1.4 cm, usually unilateral.
Other factors that have classically increased suspicion are winter/early spring presentation in North America, male gender (1.5:1 ratio to females), and Asian (particularly Japanese) ancestry. The importance of genetics was initially based on epidemiology (Japan/Asian risk) but lately has been further associated with six gene polymorphisms. However, molecular genetic testing is not currently a practical tool.
Clinical scenarios that also should raise suspicion include less-than-6-month-old infants with prolonged fever/irritability (may be the only clinical manifestations of KD) and children over 4 years old who more often may have incomplete KD. Both groups have higher prevalence of coronary artery abnormalities. Other high suspicion scenarios include prolonged fever with unexplained/culture-negative shock, or antibiotic treatment failure for cervical adenitis or retro/parapharyngeal phlegmon. Consultation with or referral to a regional KD expert may be needed.
Fuzzy KD math
Current guidelines list an exception to the 5-day fever requirement in that only 4 days of fever are needed with four or more principal clinical features, particularly when hand and feet findings exist. Some call this the “4X4 exception.” Then there is a sub-caveat: “Experienced clinicians who have treated many patients with KD may establish the diagnosis with 3 days of fever in rare cases.”1
Incomplete KD
This is another exception, which seems to be a more frequent diagnosis in the past decade. Incomplete KD requires the 5 days of fever duration plus an elevated C-reactive protein or erythrocyte sedimentation rate. But one needs only two or three of the five principal clinical KD criteria plus three or more of six other laboratory findings (anemia, low albumin, leukocytosis, thrombocytosis, pyuria, or elevated alanine aminotransferase). Incomplete KD can be confirmed by an abnormal echocardiogram – usually not until after 7 days of KD symptoms.1
New KD nuances
In a recent report on 20 years of data from Japan (n = 1,945 KD cases), more granularity on age, seasonal epidemiology, and outcome were seen.2 There was an inverse correlation of male predominance to age, i.e. as age groups got older, there was a gradual shift to female predominance by 7 years of age. The winter/spring predominance (60% of overall cases) did not hold true in younger age groups where summer/fall was the peak season (65% of cases).
With the goal of not missing any KD and diagnosing as early as possible to limit sequelae, we all need to be relative experts and keep alert for clinical scenarios that warrant our raising our index of suspicion. But now the seasonality trends appear blurred in the youngest cases and the male predominance is blurred in the oldest cases. And remember that fever and irritability for longer than 7 days in young infants may be the only clue to KD.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. He said he had no relevant financial disclosures. Email him at [email protected].
References
1. Circulation. 2017 Mar 29. doi: 10.1161/CIR.0000000000000484
2. N Engl J Med. 2018 May 24. doi: 10.1056/NEJMc1804312.
Vaccines: Effectiveness vs. efficacy
During the influenza portion of the Feb. 21, 2018, Centers for Diseases Control and Prevention’s Advisory Committee on Immunization Practices meeting, two pleas from the audience asked the CDC/ACIP to make messages very clear about how protective influenza vaccine really is.
We hear apparently conflicting percentages from Australia, Canada, Europe, and the United States from the many stories/press releases in the news media and from official public health outlets. And the gloomiest ones get the most exposure.1 It can be confusing even for medical care providers who are supposed to advise families on such matters.
A key misunderstanding in many medical and lay news stories is about what vaccine effectiveness and vaccine efficacy really mean. What? Aren’t those the same thing? Nope. They are quite different. And are we sure of what those 95% confidence intervals (CI) mean? Let’s review the “math” so we can explain this to families.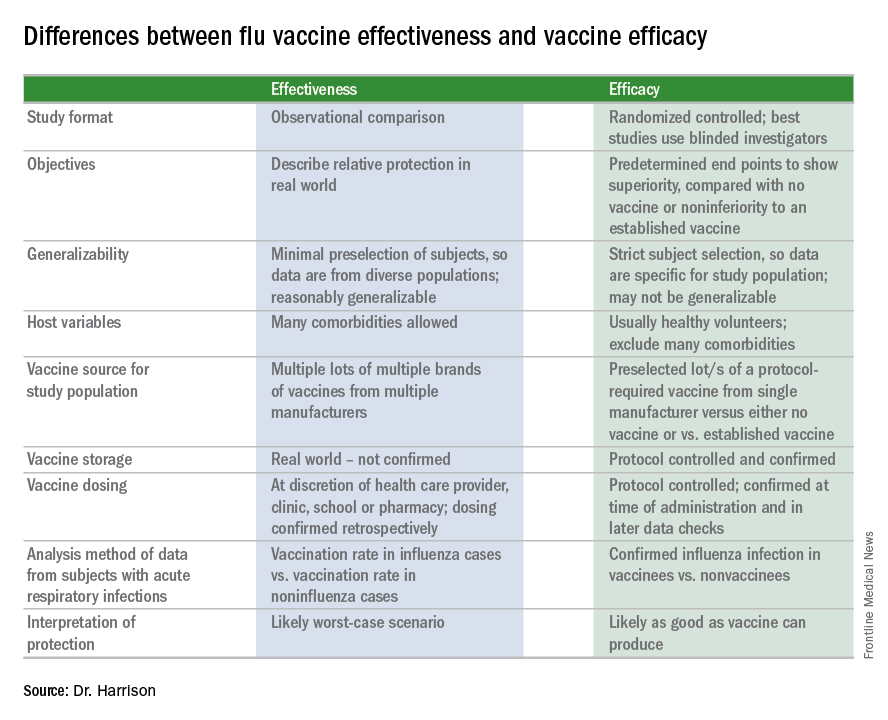
Vaccine effectiveness (VE)2,3
The first thing to know is that the CDC and similar public health agencies in other countries do not report vaccine efficacy. Instead, the percentage reported is VE during (interim estimated VE) and just after (final adjusted VE) each influenza season. This means that VE is generally a retrospective analysis of data, most of which were collected prospectively. Further, VE is likely the worst case scenario. VE is a measure of real-world benefit to patients for whom vaccine is recommended, by analyzing specific geographically diverse populations (population-based) without excluding most underlying illness or comorbidities (note that immunosuppressed persons are excluded). Subjects in VE studies receive their vaccine in the real world and, therefore, vaccinees may receive their vaccines from any number of the usual outlets (e.g., primary care provider, urgent care or emergency department, public health department, pharmacy, school, or nursing home). There are multiple lots of multiple brands from multiple vaccine manufacturers. Children who need two doses of influenza vaccine do not necessarily receive those doses according to the package insert’s schedule. VE studies do not have the capability to confirm that vaccine was stored, handled, and administered in a precisely correct manner according to manufacturer’s and CDC’s recommendations.
VE is calculated using a “test-negative” (case-control) analysis of patients presenting with acute respiratory infections (ARIs). People who are not in vaccine research can find this methodology confusing. Briefly, the VE compares the odds of vaccination in ARIs due to confirmed influenza to the odds of vaccination in ARIs not due to influenza. Additional statistical tools can adjust VE for specific factors. VE is also calculated by factors of interest, such as age, gender, pregnancy, influenza type, region of the country, presence of asthma or other comorbidity, etc. Whether the VE value is the “truth in the universe” is related to having enough subjects in each analyzed group and the degree to which the studied populations actually represent the whole country. So, VE is more accurate when there are large subject numbers.
Remember also that VE is usually calculated from outpatients, so it does not really measure all the benefits of vaccination. Prevention rates for severe influenza (such as influenza hospitalizations) are higher but usually unavailable until after the entire season.
VE studies generally measure real-world and likely worst-case-scenario benefit for the overall population being protected against outpatient influenza medical visits.
Vaccine efficacy2,3
Vaccine efficacy measures how the vaccine performs under ideal circumstances in a regimented protocol in relatively normal hosts – likely the best-case-scenario benefit. Vaccine efficacy is the percent difference in confirmed influenza episodes in vaccinees getting the “experimental” vaccine vs. episodes in nonvaccinees (or vaccinees getting an established vaccine). Vaccine efficacy, therefore, is usually calculated based on prospective well-controlled studies under ideal circumstances in subjects who received their vaccines on time per the recommended schedule. Most such studies are performed on otherwise healthy children or adults, with most comorbidities excluded. The “experimental” vaccine is generally from a single manufacturer from a single lot, and chain-of-custody is well controlled. The vaccine is administered at selected research sites according to a strict protocol; vaccine storage is ensured to be as recommended.
Confidence intervals
To assess whether the “protection” is “significant,” the calculations derive 95% confidence intervals (CI). If the 95% CI range is wide, such as many tens of percents, then there is less confidence that the calculation is correct. And if the lower CI is less than 0, then the result is not significant. For example, a VE of 20% is not highly protective, but can be significant if the 95% CI ranges from 10 to 28 (the lower value of 10 is above zero). It would not be significant if the 95% CI lower limit was –10. Values for seasons 2004-2005 and 2005-2006 were similar to this. Consider however that a VE of 55% seems great, but may not be significant if the 95% CI range is –20 to 89 (the lower value is less than zero). In the ideal world, the VE would be greater than 50% and the 95% CI range would be tight with the lower CI value far above zero; for example, VE of 70% with 95% CI ranging from 60 to 80. The 2010-2011 season was close to this.
Type and age-specific VE
Aside from overall VE, there are subset analyses that can be revealing. This year there are the concerning mid-season VE estimates of approximately 25% for the United States and 17% in Canada, for one specific type, H3N2, which unfortunately has been the dominant circulating U.S. type. That number is what everybody seems to have focused on. But remember influenza B becomes dominant late in most seasons (increasing at the time of writing this article). Interim 2017-2018 VE for influenza B was in the mid 60% range, making the box plot near 40% overall.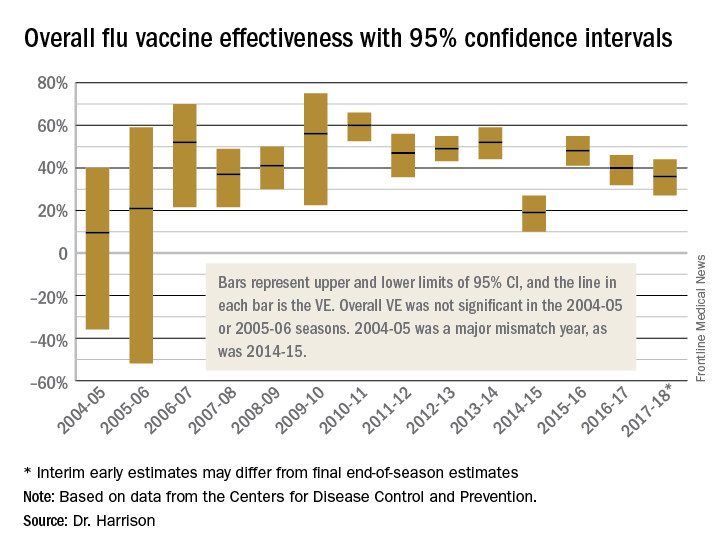
Age-related VE analysis can show difference; for example, the best benefit for H3N2 this season has been in young children and the worst in elderly and 9- to 17-year-olds.
Take-home message
The simplest way to think of overall VE is that it is the real-world, worst-case-scenario value for influenza protection by vaccine against the several circulating types of influenza.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. Children’s Mercy Hospital receives grant funding for Dr. Harrison’s work as an investigator from GSK for MMR and rotavirus vaccine studies, from Merck for in vitro and clinical antibiotic studies, from Allergan for clinical antibiotic studies, from Pfizer for pneumococcal seroepidemiology studies, and from Regeneron for RSV studies. Dr. Harrison received support for travel and to present seroepidemiology data at one meeting. Email him at [email protected].
References
1. MMWR Weekly. 2017 Feb 17;66(6):167-71.
2. Dev Biol Stand. 1998;95:195-201.
3. Lancet Infect Dis. 2012 Jan;12(1):36-44.
During the influenza portion of the Feb. 21, 2018, Centers for Diseases Control and Prevention’s Advisory Committee on Immunization Practices meeting, two pleas from the audience asked the CDC/ACIP to make messages very clear about how protective influenza vaccine really is.
We hear apparently conflicting percentages from Australia, Canada, Europe, and the United States from the many stories/press releases in the news media and from official public health outlets. And the gloomiest ones get the most exposure.1 It can be confusing even for medical care providers who are supposed to advise families on such matters.
A key misunderstanding in many medical and lay news stories is about what vaccine effectiveness and vaccine efficacy really mean. What? Aren’t those the same thing? Nope. They are quite different. And are we sure of what those 95% confidence intervals (CI) mean? Let’s review the “math” so we can explain this to families.
Vaccine effectiveness (VE)2,3
The first thing to know is that the CDC and similar public health agencies in other countries do not report vaccine efficacy. Instead, the percentage reported is VE during (interim estimated VE) and just after (final adjusted VE) each influenza season. This means that VE is generally a retrospective analysis of data, most of which were collected prospectively. Further, VE is likely the worst case scenario. VE is a measure of real-world benefit to patients for whom vaccine is recommended, by analyzing specific geographically diverse populations (population-based) without excluding most underlying illness or comorbidities (note that immunosuppressed persons are excluded). Subjects in VE studies receive their vaccine in the real world and, therefore, vaccinees may receive their vaccines from any number of the usual outlets (e.g., primary care provider, urgent care or emergency department, public health department, pharmacy, school, or nursing home). There are multiple lots of multiple brands from multiple vaccine manufacturers. Children who need two doses of influenza vaccine do not necessarily receive those doses according to the package insert’s schedule. VE studies do not have the capability to confirm that vaccine was stored, handled, and administered in a precisely correct manner according to manufacturer’s and CDC’s recommendations.
VE is calculated using a “test-negative” (case-control) analysis of patients presenting with acute respiratory infections (ARIs). People who are not in vaccine research can find this methodology confusing. Briefly, the VE compares the odds of vaccination in ARIs due to confirmed influenza to the odds of vaccination in ARIs not due to influenza. Additional statistical tools can adjust VE for specific factors. VE is also calculated by factors of interest, such as age, gender, pregnancy, influenza type, region of the country, presence of asthma or other comorbidity, etc. Whether the VE value is the “truth in the universe” is related to having enough subjects in each analyzed group and the degree to which the studied populations actually represent the whole country. So, VE is more accurate when there are large subject numbers.
Remember also that VE is usually calculated from outpatients, so it does not really measure all the benefits of vaccination. Prevention rates for severe influenza (such as influenza hospitalizations) are higher but usually unavailable until after the entire season.
VE studies generally measure real-world and likely worst-case-scenario benefit for the overall population being protected against outpatient influenza medical visits.
Vaccine efficacy2,3
Vaccine efficacy measures how the vaccine performs under ideal circumstances in a regimented protocol in relatively normal hosts – likely the best-case-scenario benefit. Vaccine efficacy is the percent difference in confirmed influenza episodes in vaccinees getting the “experimental” vaccine vs. episodes in nonvaccinees (or vaccinees getting an established vaccine). Vaccine efficacy, therefore, is usually calculated based on prospective well-controlled studies under ideal circumstances in subjects who received their vaccines on time per the recommended schedule. Most such studies are performed on otherwise healthy children or adults, with most comorbidities excluded. The “experimental” vaccine is generally from a single manufacturer from a single lot, and chain-of-custody is well controlled. The vaccine is administered at selected research sites according to a strict protocol; vaccine storage is ensured to be as recommended.
Confidence intervals
To assess whether the “protection” is “significant,” the calculations derive 95% confidence intervals (CI). If the 95% CI range is wide, such as many tens of percents, then there is less confidence that the calculation is correct. And if the lower CI is less than 0, then the result is not significant. For example, a VE of 20% is not highly protective, but can be significant if the 95% CI ranges from 10 to 28 (the lower value of 10 is above zero). It would not be significant if the 95% CI lower limit was –10. Values for seasons 2004-2005 and 2005-2006 were similar to this. Consider however that a VE of 55% seems great, but may not be significant if the 95% CI range is –20 to 89 (the lower value is less than zero). In the ideal world, the VE would be greater than 50% and the 95% CI range would be tight with the lower CI value far above zero; for example, VE of 70% with 95% CI ranging from 60 to 80. The 2010-2011 season was close to this.
Type and age-specific VE
Aside from overall VE, there are subset analyses that can be revealing. This year there are the concerning mid-season VE estimates of approximately 25% for the United States and 17% in Canada, for one specific type, H3N2, which unfortunately has been the dominant circulating U.S. type. That number is what everybody seems to have focused on. But remember influenza B becomes dominant late in most seasons (increasing at the time of writing this article). Interim 2017-2018 VE for influenza B was in the mid 60% range, making the box plot near 40% overall.
Age-related VE analysis can show difference; for example, the best benefit for H3N2 this season has been in young children and the worst in elderly and 9- to 17-year-olds.
Take-home message
The simplest way to think of overall VE is that it is the real-world, worst-case-scenario value for influenza protection by vaccine against the several circulating types of influenza.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. Children’s Mercy Hospital receives grant funding for Dr. Harrison’s work as an investigator from GSK for MMR and rotavirus vaccine studies, from Merck for in vitro and clinical antibiotic studies, from Allergan for clinical antibiotic studies, from Pfizer for pneumococcal seroepidemiology studies, and from Regeneron for RSV studies. Dr. Harrison received support for travel and to present seroepidemiology data at one meeting. Email him at [email protected].
References
1. MMWR Weekly. 2017 Feb 17;66(6):167-71.
2. Dev Biol Stand. 1998;95:195-201.
3. Lancet Infect Dis. 2012 Jan;12(1):36-44.
During the influenza portion of the Feb. 21, 2018, Centers for Diseases Control and Prevention’s Advisory Committee on Immunization Practices meeting, two pleas from the audience asked the CDC/ACIP to make messages very clear about how protective influenza vaccine really is.
We hear apparently conflicting percentages from Australia, Canada, Europe, and the United States from the many stories/press releases in the news media and from official public health outlets. And the gloomiest ones get the most exposure.1 It can be confusing even for medical care providers who are supposed to advise families on such matters.
A key misunderstanding in many medical and lay news stories is about what vaccine effectiveness and vaccine efficacy really mean. What? Aren’t those the same thing? Nope. They are quite different. And are we sure of what those 95% confidence intervals (CI) mean? Let’s review the “math” so we can explain this to families.
Vaccine effectiveness (VE)2,3
The first thing to know is that the CDC and similar public health agencies in other countries do not report vaccine efficacy. Instead, the percentage reported is VE during (interim estimated VE) and just after (final adjusted VE) each influenza season. This means that VE is generally a retrospective analysis of data, most of which were collected prospectively. Further, VE is likely the worst case scenario. VE is a measure of real-world benefit to patients for whom vaccine is recommended, by analyzing specific geographically diverse populations (population-based) without excluding most underlying illness or comorbidities (note that immunosuppressed persons are excluded). Subjects in VE studies receive their vaccine in the real world and, therefore, vaccinees may receive their vaccines from any number of the usual outlets (e.g., primary care provider, urgent care or emergency department, public health department, pharmacy, school, or nursing home). There are multiple lots of multiple brands from multiple vaccine manufacturers. Children who need two doses of influenza vaccine do not necessarily receive those doses according to the package insert’s schedule. VE studies do not have the capability to confirm that vaccine was stored, handled, and administered in a precisely correct manner according to manufacturer’s and CDC’s recommendations.
VE is calculated using a “test-negative” (case-control) analysis of patients presenting with acute respiratory infections (ARIs). People who are not in vaccine research can find this methodology confusing. Briefly, the VE compares the odds of vaccination in ARIs due to confirmed influenza to the odds of vaccination in ARIs not due to influenza. Additional statistical tools can adjust VE for specific factors. VE is also calculated by factors of interest, such as age, gender, pregnancy, influenza type, region of the country, presence of asthma or other comorbidity, etc. Whether the VE value is the “truth in the universe” is related to having enough subjects in each analyzed group and the degree to which the studied populations actually represent the whole country. So, VE is more accurate when there are large subject numbers.
Remember also that VE is usually calculated from outpatients, so it does not really measure all the benefits of vaccination. Prevention rates for severe influenza (such as influenza hospitalizations) are higher but usually unavailable until after the entire season.
VE studies generally measure real-world and likely worst-case-scenario benefit for the overall population being protected against outpatient influenza medical visits.
Vaccine efficacy2,3
Vaccine efficacy measures how the vaccine performs under ideal circumstances in a regimented protocol in relatively normal hosts – likely the best-case-scenario benefit. Vaccine efficacy is the percent difference in confirmed influenza episodes in vaccinees getting the “experimental” vaccine vs. episodes in nonvaccinees (or vaccinees getting an established vaccine). Vaccine efficacy, therefore, is usually calculated based on prospective well-controlled studies under ideal circumstances in subjects who received their vaccines on time per the recommended schedule. Most such studies are performed on otherwise healthy children or adults, with most comorbidities excluded. The “experimental” vaccine is generally from a single manufacturer from a single lot, and chain-of-custody is well controlled. The vaccine is administered at selected research sites according to a strict protocol; vaccine storage is ensured to be as recommended.
Confidence intervals
To assess whether the “protection” is “significant,” the calculations derive 95% confidence intervals (CI). If the 95% CI range is wide, such as many tens of percents, then there is less confidence that the calculation is correct. And if the lower CI is less than 0, then the result is not significant. For example, a VE of 20% is not highly protective, but can be significant if the 95% CI ranges from 10 to 28 (the lower value of 10 is above zero). It would not be significant if the 95% CI lower limit was –10. Values for seasons 2004-2005 and 2005-2006 were similar to this. Consider however that a VE of 55% seems great, but may not be significant if the 95% CI range is –20 to 89 (the lower value is less than zero). In the ideal world, the VE would be greater than 50% and the 95% CI range would be tight with the lower CI value far above zero; for example, VE of 70% with 95% CI ranging from 60 to 80. The 2010-2011 season was close to this.
Type and age-specific VE
Aside from overall VE, there are subset analyses that can be revealing. This year there are the concerning mid-season VE estimates of approximately 25% for the United States and 17% in Canada, for one specific type, H3N2, which unfortunately has been the dominant circulating U.S. type. That number is what everybody seems to have focused on. But remember influenza B becomes dominant late in most seasons (increasing at the time of writing this article). Interim 2017-2018 VE for influenza B was in the mid 60% range, making the box plot near 40% overall.
Age-related VE analysis can show difference; for example, the best benefit for H3N2 this season has been in young children and the worst in elderly and 9- to 17-year-olds.
Take-home message
The simplest way to think of overall VE is that it is the real-world, worst-case-scenario value for influenza protection by vaccine against the several circulating types of influenza.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. Children’s Mercy Hospital receives grant funding for Dr. Harrison’s work as an investigator from GSK for MMR and rotavirus vaccine studies, from Merck for in vitro and clinical antibiotic studies, from Allergan for clinical antibiotic studies, from Pfizer for pneumococcal seroepidemiology studies, and from Regeneron for RSV studies. Dr. Harrison received support for travel and to present seroepidemiology data at one meeting. Email him at [email protected].
References
1. MMWR Weekly. 2017 Feb 17;66(6):167-71.
2. Dev Biol Stand. 1998;95:195-201.
3. Lancet Infect Dis. 2012 Jan;12(1):36-44.
Foodborne illnesses of foreign, domestic origin: On the rise?
Are foodborne illness outbreaks more common now, or are we simply better at detection? Have the foods and sources associated with foodborne illness changed? Two recent Centers for Disease Control & Prevention reports provide insight.1,2 In 2016, the Foodborne Diseases Active Surveillance Network (FoodNet) detected 24,029 infections, 5,212 hospitalizations, and 98 fatalities.1 FoodNet has 10 sites serving 49 million people (15% of the U.S. population). These 2016 numbers changed only modestly from the 3 prior years.
The big two
, detected by traditional cultures or culture-independent diagnostic tests (CIDTs). (See table.) CIDTs are relatively new molecular-based, mostly multiplex assays that test for more than a dozen pathogens in one assay.
Overall, Salmonella originated from diverse sources (eggs, poultry, meat, unpasteurized milk/juice/cheese, or raw fruits/vegetables/spices/nuts). But, in 2016, U.S. Salmonella outbreaks were from eggs, alfalfa sprouts, poultry, pistachios, and organic shake/meal products.
The runners-up
Most of the remainder of the 2016 foodborne illnesses were caused by Shigella, with nearly 3,000 cases; shigatoxin-producing Escherichia coli (STEC), with nearly 2,000 cases; and Cryptosporidium, also with nearly 2,000 cases. (See table.)
Hemolytic uremic syndrome (HUS)
HUS rates, mostly resulting from E. coli 0157 H7 in meat, did not vary from 2013 to 2016, with a total 62 pediatric HUS cases in FoodNet (0.56 /100,000 population). Slightly over half (56%) occurred in children under 5 years old at 1.18 per 100,000 population.
Does CIDT increase detection rates?
Detection of the “big two” did not change from 2013 to 2016 or over the past 2 decades. That said, Campylobacter detection was actually down 11% if considering only culture-confirmed cases. That is, if we do not count detections made exclusively by CIDT.
This is important because CIDT – now supplanting culture in many laboratories – identifies pathogens not likely detected by standard culture because culture is generally selective and CIDT is more sensitive. CIDT can increase detection rates (solo and multiple pathogens), even if illnesses do not really increase. The CDC suggested that this contributed to increased STEC and Yersinia detection in 2016. Some would not have been detected if only culture had been utilized.
Viable bacterial/viral isolates are not available from CIDT. A replicating pathogen is needed to characterize shifting/emerging pathogen strains (for example, analysis for mutations or new pathogens via sequencing or antimicrobial susceptibility testing).
To compensate, some CIDT-using laboratories perform “reflex cultures.” CIDT positive specimens also are cultured to provide viable isolates. However, this adds cost to an already costly CIDT test.
The role of imported food
Surveillance systems, such as the Foodborne Disease Outbreak Surveillance System, also track imported foodborne illness. Despite an approximately 50% decrease in overall U.S. foodborne outbreaks since 2000, imported food-related outbreaks increased to 195 during 2006-2014 from 54 during 1996-2004, with 10,685 illnesses, 1,017 hospitalizations, and 19 deaths since 2009. Also, imported food-related outbreaks rose from a mean 3 per year pre-2000 to a mean 18 per year during 2009-2014. Most imported food outbreaks (86% of total) had three causes: scombroid toxin (42% of total), Salmonella (33%), and hepatitis A virus (11%).

Most imported food illnesses were from Salmonella (4,421 from 52 outbreaks), Cyclospora (2,533 from 33 outbreaks), hepatitis A virus (1,150 from 11 outbreaks), and Shigella (625 from 6 outbreaks). While eggs, ice cream, and poultry are notorious origins for Salmonella in domestic food, most imported Salmonella were from produce: fruits (26%), seeded vegetables (20%), sprouts (11%), nuts/seeds (10%), spices (7%), and herbs (2%).
Seafood/fish caused 55% of outbreaks but few illnesses per outbreak (median 3 illnesses/outbreak), so only 11% of total illnesses were caused by seafood/fish. In contrast, fresh produce caused only 33% of outbreaks but 84% of illnesses (median 40 illnesses/outbreak).
Geographic source, outbreak locations
The origin was known in 91% of outbreaks. Latin America and the Caribbean were most common, followed by Asia.3 Main contributing countries were Mexico (42 outbreaks), Indonesia (17) and Canada (11).
Contaminated fish/shellfish originated from all regions except Europe, most commonly from Asia (the majority of fish/shellfish outbreaks were from Indonesia, Vietnam, China, Philippines, Taiwan, and Thailand) with smaller contributions from the Bahamas and Ecuador.
Contaminated produce originated from all regions, mostly (64%) from Mexico and the Americas (Chile, Guatemala, and Honduras). All but one dairy outbreak originated in Latin America/the Caribbean.3 Outbreaks occurred in 31 states, most commonly California (30), Florida (25), and New York (16). Additionally, 43 (22%) were multistate outbreaks.
Conclusions
Outbreaks from domestic foods decreased, but those from imported foods increased. This makes sense given recent increases in outbreak-prone food imports, such as seafood/fish and produce.
To reduce overall foodborne illness outbreaks, governmental agencies need to:
- Develop/enforce regulations that promote proper growing, handling, and processing of foods.
- Strengthen surveillance networks and share standard culture and molecular detection/characterization protocols to identify outbreaks as close to real time as possible.
- Ensure rapid traceability not only to country of origin but to an exact farm or seafood/fish harvesting entity.
- Provide rapid public knowledge of outbreaks and origins, plus outbreak-specific recommendations to control/minimize resultant illnesses.
Individuals can help protect themselves by avoiding inadequately washed or incompletely cooked foods or foods of uncertain origin.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. He said he had no relevant financial disclosures. Email him at [email protected].
References
1. MMWR. 2017 Apr 21;66(15):397-403.
2. Emerg Infect Dis. 2017 Mar;23(3):525-8.
3. Technical appendix in Emerg Infect Dis. 2017 Mar;23(3):525-8.
Are foodborne illness outbreaks more common now, or are we simply better at detection? Have the foods and sources associated with foodborne illness changed? Two recent Centers for Disease Control & Prevention reports provide insight.1,2 In 2016, the Foodborne Diseases Active Surveillance Network (FoodNet) detected 24,029 infections, 5,212 hospitalizations, and 98 fatalities.1 FoodNet has 10 sites serving 49 million people (15% of the U.S. population). These 2016 numbers changed only modestly from the 3 prior years.
The big two
, detected by traditional cultures or culture-independent diagnostic tests (CIDTs). (See table.) CIDTs are relatively new molecular-based, mostly multiplex assays that test for more than a dozen pathogens in one assay.
Overall, Salmonella originated from diverse sources (eggs, poultry, meat, unpasteurized milk/juice/cheese, or raw fruits/vegetables/spices/nuts). But, in 2016, U.S. Salmonella outbreaks were from eggs, alfalfa sprouts, poultry, pistachios, and organic shake/meal products.
The runners-up
Most of the remainder of the 2016 foodborne illnesses were caused by Shigella, with nearly 3,000 cases; shigatoxin-producing Escherichia coli (STEC), with nearly 2,000 cases; and Cryptosporidium, also with nearly 2,000 cases. (See table.)
Hemolytic uremic syndrome (HUS)
HUS rates, mostly resulting from E. coli 0157 H7 in meat, did not vary from 2013 to 2016, with a total 62 pediatric HUS cases in FoodNet (0.56 /100,000 population). Slightly over half (56%) occurred in children under 5 years old at 1.18 per 100,000 population.
Does CIDT increase detection rates?
Detection of the “big two” did not change from 2013 to 2016 or over the past 2 decades. That said, Campylobacter detection was actually down 11% if considering only culture-confirmed cases. That is, if we do not count detections made exclusively by CIDT.
This is important because CIDT – now supplanting culture in many laboratories – identifies pathogens not likely detected by standard culture because culture is generally selective and CIDT is more sensitive. CIDT can increase detection rates (solo and multiple pathogens), even if illnesses do not really increase. The CDC suggested that this contributed to increased STEC and Yersinia detection in 2016. Some would not have been detected if only culture had been utilized.
Viable bacterial/viral isolates are not available from CIDT. A replicating pathogen is needed to characterize shifting/emerging pathogen strains (for example, analysis for mutations or new pathogens via sequencing or antimicrobial susceptibility testing).
To compensate, some CIDT-using laboratories perform “reflex cultures.” CIDT positive specimens also are cultured to provide viable isolates. However, this adds cost to an already costly CIDT test.
The role of imported food
Surveillance systems, such as the Foodborne Disease Outbreak Surveillance System, also track imported foodborne illness. Despite an approximately 50% decrease in overall U.S. foodborne outbreaks since 2000, imported food-related outbreaks increased to 195 during 2006-2014 from 54 during 1996-2004, with 10,685 illnesses, 1,017 hospitalizations, and 19 deaths since 2009. Also, imported food-related outbreaks rose from a mean 3 per year pre-2000 to a mean 18 per year during 2009-2014. Most imported food outbreaks (86% of total) had three causes: scombroid toxin (42% of total), Salmonella (33%), and hepatitis A virus (11%).
Most imported food illnesses were from Salmonella (4,421 from 52 outbreaks), Cyclospora (2,533 from 33 outbreaks), hepatitis A virus (1,150 from 11 outbreaks), and Shigella (625 from 6 outbreaks). While eggs, ice cream, and poultry are notorious origins for Salmonella in domestic food, most imported Salmonella were from produce: fruits (26%), seeded vegetables (20%), sprouts (11%), nuts/seeds (10%), spices (7%), and herbs (2%).
Seafood/fish caused 55% of outbreaks but few illnesses per outbreak (median 3 illnesses/outbreak), so only 11% of total illnesses were caused by seafood/fish. In contrast, fresh produce caused only 33% of outbreaks but 84% of illnesses (median 40 illnesses/outbreak).
Geographic source, outbreak locations
The origin was known in 91% of outbreaks. Latin America and the Caribbean were most common, followed by Asia.3 Main contributing countries were Mexico (42 outbreaks), Indonesia (17) and Canada (11).
Contaminated fish/shellfish originated from all regions except Europe, most commonly from Asia (the majority of fish/shellfish outbreaks were from Indonesia, Vietnam, China, Philippines, Taiwan, and Thailand) with smaller contributions from the Bahamas and Ecuador.
Contaminated produce originated from all regions, mostly (64%) from Mexico and the Americas (Chile, Guatemala, and Honduras). All but one dairy outbreak originated in Latin America/the Caribbean.3 Outbreaks occurred in 31 states, most commonly California (30), Florida (25), and New York (16). Additionally, 43 (22%) were multistate outbreaks.
Conclusions
Outbreaks from domestic foods decreased, but those from imported foods increased. This makes sense given recent increases in outbreak-prone food imports, such as seafood/fish and produce.
To reduce overall foodborne illness outbreaks, governmental agencies need to:
- Develop/enforce regulations that promote proper growing, handling, and processing of foods.
- Strengthen surveillance networks and share standard culture and molecular detection/characterization protocols to identify outbreaks as close to real time as possible.
- Ensure rapid traceability not only to country of origin but to an exact farm or seafood/fish harvesting entity.
- Provide rapid public knowledge of outbreaks and origins, plus outbreak-specific recommendations to control/minimize resultant illnesses.
Individuals can help protect themselves by avoiding inadequately washed or incompletely cooked foods or foods of uncertain origin.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. He said he had no relevant financial disclosures. Email him at [email protected].
References
1. MMWR. 2017 Apr 21;66(15):397-403.
2. Emerg Infect Dis. 2017 Mar;23(3):525-8.
3. Technical appendix in Emerg Infect Dis. 2017 Mar;23(3):525-8.
Are foodborne illness outbreaks more common now, or are we simply better at detection? Have the foods and sources associated with foodborne illness changed? Two recent Centers for Disease Control & Prevention reports provide insight.1,2 In 2016, the Foodborne Diseases Active Surveillance Network (FoodNet) detected 24,029 infections, 5,212 hospitalizations, and 98 fatalities.1 FoodNet has 10 sites serving 49 million people (15% of the U.S. population). These 2016 numbers changed only modestly from the 3 prior years.
The big two
, detected by traditional cultures or culture-independent diagnostic tests (CIDTs). (See table.) CIDTs are relatively new molecular-based, mostly multiplex assays that test for more than a dozen pathogens in one assay.
Overall, Salmonella originated from diverse sources (eggs, poultry, meat, unpasteurized milk/juice/cheese, or raw fruits/vegetables/spices/nuts). But, in 2016, U.S. Salmonella outbreaks were from eggs, alfalfa sprouts, poultry, pistachios, and organic shake/meal products.
The runners-up
Most of the remainder of the 2016 foodborne illnesses were caused by Shigella, with nearly 3,000 cases; shigatoxin-producing Escherichia coli (STEC), with nearly 2,000 cases; and Cryptosporidium, also with nearly 2,000 cases. (See table.)
Hemolytic uremic syndrome (HUS)
HUS rates, mostly resulting from E. coli 0157 H7 in meat, did not vary from 2013 to 2016, with a total 62 pediatric HUS cases in FoodNet (0.56 /100,000 population). Slightly over half (56%) occurred in children under 5 years old at 1.18 per 100,000 population.
Does CIDT increase detection rates?
Detection of the “big two” did not change from 2013 to 2016 or over the past 2 decades. That said, Campylobacter detection was actually down 11% if considering only culture-confirmed cases. That is, if we do not count detections made exclusively by CIDT.
This is important because CIDT – now supplanting culture in many laboratories – identifies pathogens not likely detected by standard culture because culture is generally selective and CIDT is more sensitive. CIDT can increase detection rates (solo and multiple pathogens), even if illnesses do not really increase. The CDC suggested that this contributed to increased STEC and Yersinia detection in 2016. Some would not have been detected if only culture had been utilized.
Viable bacterial/viral isolates are not available from CIDT. A replicating pathogen is needed to characterize shifting/emerging pathogen strains (for example, analysis for mutations or new pathogens via sequencing or antimicrobial susceptibility testing).
To compensate, some CIDT-using laboratories perform “reflex cultures.” CIDT positive specimens also are cultured to provide viable isolates. However, this adds cost to an already costly CIDT test.
The role of imported food
Surveillance systems, such as the Foodborne Disease Outbreak Surveillance System, also track imported foodborne illness. Despite an approximately 50% decrease in overall U.S. foodborne outbreaks since 2000, imported food-related outbreaks increased to 195 during 2006-2014 from 54 during 1996-2004, with 10,685 illnesses, 1,017 hospitalizations, and 19 deaths since 2009. Also, imported food-related outbreaks rose from a mean 3 per year pre-2000 to a mean 18 per year during 2009-2014. Most imported food outbreaks (86% of total) had three causes: scombroid toxin (42% of total), Salmonella (33%), and hepatitis A virus (11%).
Most imported food illnesses were from Salmonella (4,421 from 52 outbreaks), Cyclospora (2,533 from 33 outbreaks), hepatitis A virus (1,150 from 11 outbreaks), and Shigella (625 from 6 outbreaks). While eggs, ice cream, and poultry are notorious origins for Salmonella in domestic food, most imported Salmonella were from produce: fruits (26%), seeded vegetables (20%), sprouts (11%), nuts/seeds (10%), spices (7%), and herbs (2%).
Seafood/fish caused 55% of outbreaks but few illnesses per outbreak (median 3 illnesses/outbreak), so only 11% of total illnesses were caused by seafood/fish. In contrast, fresh produce caused only 33% of outbreaks but 84% of illnesses (median 40 illnesses/outbreak).
Geographic source, outbreak locations
The origin was known in 91% of outbreaks. Latin America and the Caribbean were most common, followed by Asia.3 Main contributing countries were Mexico (42 outbreaks), Indonesia (17) and Canada (11).
Contaminated fish/shellfish originated from all regions except Europe, most commonly from Asia (the majority of fish/shellfish outbreaks were from Indonesia, Vietnam, China, Philippines, Taiwan, and Thailand) with smaller contributions from the Bahamas and Ecuador.
Contaminated produce originated from all regions, mostly (64%) from Mexico and the Americas (Chile, Guatemala, and Honduras). All but one dairy outbreak originated in Latin America/the Caribbean.3 Outbreaks occurred in 31 states, most commonly California (30), Florida (25), and New York (16). Additionally, 43 (22%) were multistate outbreaks.
Conclusions
Outbreaks from domestic foods decreased, but those from imported foods increased. This makes sense given recent increases in outbreak-prone food imports, such as seafood/fish and produce.
To reduce overall foodborne illness outbreaks, governmental agencies need to:
- Develop/enforce regulations that promote proper growing, handling, and processing of foods.
- Strengthen surveillance networks and share standard culture and molecular detection/characterization protocols to identify outbreaks as close to real time as possible.
- Ensure rapid traceability not only to country of origin but to an exact farm or seafood/fish harvesting entity.
- Provide rapid public knowledge of outbreaks and origins, plus outbreak-specific recommendations to control/minimize resultant illnesses.
Individuals can help protect themselves by avoiding inadequately washed or incompletely cooked foods or foods of uncertain origin.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. He said he had no relevant financial disclosures. Email him at [email protected].
References
1. MMWR. 2017 Apr 21;66(15):397-403.
2. Emerg Infect Dis. 2017 Mar;23(3):525-8.
3. Technical appendix in Emerg Infect Dis. 2017 Mar;23(3):525-8.
Bacterial colonizer vs. pathogen
Although acute otitis media (AOM) has decreased in number, and especially the more severe difficult to treat versions, I was reminded that this still is a problem for young children based on personal experience with grandchildren. What can be baffling to some families is the fact that some strains of the same organism species cause AOM and some simply colonize the nasopharynx (NP) of children without causing any disease at all. These organisms include Streptococcus pneumoniae (SPN) and nontypeable Haemophilus influenzae (ntHi).
Recent studies have uncovered several molecular reasons for the pathogen vs. colonizer dichotomy:
• Strains within a species can have variants of a gene that make them more disease producing.
• Some usually colonizing strains produce disease after acquiring new genes.
• Some strains have native genes with on-off switches that convert them from a colonizing to disease producing under selected circumstances.
• Molecular targets in the respiratory tract increase, allowing more dense colonization that increases chances of AOM.
Variant gene
J.R. Gilsdorf, MD, and his group at the University of Michigan,1 Ann Arbor, recently showed that among the various high-molecular-weight molecules (HMW) produced by 170 ntHi from three different geographically diverse countries, one variant in particular (HMW-A) was more likely to be found in strains producing AOM than strains simply colonizing the nasopharynx. The protein product of this gene allows better adherence to respiratory epithelia. So more bacteria sticking in the NP near the eustachian tube opening make development of AOM more likely. Some call this the “more barbarians at the gate” phenomenon.

Gene acquisition
SPN inherently has a somewhat incomplete arginine synthesis pathway. Because arginine is essential for growth of SPN, S. pneumoniae utilizes some host factors to compensate; but this compensation is inefficient. However, SPN strains can acquire new genes – usually from other gram-positive organisms in their environment – by a process called conjugation.
One recently reported acquired gene set is that which completes functionality of SPN’s arginine synthesis pathway.2 Investigators showed that SPN that acquire these arginine synthesis genes replicate more readily in bodily fluids, such as serum or cerebrospinal fluid, making these strains more aggressive, more virulent, and more likely to produce disease. More efficient replication makes it very difficult for host immune responses to handle these SPN. This is not limited to AOM alone, but seems important in invasive disease (such as meningitis) from SPN type 7, which had recently become more frequent after introduction of pneumococcal conjugate vaccines.
On-off gene switches
Another group of investigators reported that a thing called “phasevarion,” which is fancy lingo for an on-off switch is at the root of more virulence in ntHi.3 It seems that some strains of ntHi have a version of the ModA2 gene, which is always turned off, while other strains have a gene that is always on. Then, there is a third version in which the gene is usually off, but turns on when in places like the middle ear. The ModA2 gene appears to affect several other downstream protein groups that include HMW-A, antibiotic susceptibility, and biofilm formation. When inoculated into the middle ear in a chinchilla AOM model, the ntHi strains that can turn on their ModA2 gene were much more likely to produce AOM than either version that could not change. Interestingly, the authors postulate that preventing the switch capability could be a novel way to prevent ntHi disease, such as pediatric AOM, acute bacterial sinusitis, or some bronchitis in adults.
Molecular environment becomes more favorable
Another group4 reported that adherence receptor for ntHi is intercellular adhesion molecule 1 (ICAM1), a molecule found in modest quantities on respiratory epithelium. You may know it as the attachment molecule for rhinovirus and enteroviruses. What makes this interesting is that adenovirus, respiratory syncytial virus, and exposure to cigarette smoke5 markedly increase expression of ICAM1 on respiratory epithelium, predisposing to more ntHi adhering and more likely to produce an inflammatory process, such as AOM. This is another version of the barbarians at the gate phenomenon.
So when families ask why SPN or ntHi sometimes exist quietly (colonize) the nasopharynx and sometimes they cause AOM or acute bacterial sinusitis, you can hopefully use these four examples as partial explanations of why the same bacterial species has strains that can be either colonizers or pathogens.
References
1. Infect Genet Evol. 2014 Dec;28:223-32
2. J Infect Dis. 2014 Jun 1;209:1781-91.
3. J Infect Dis. 2016 Jun 10. pii: jiw243. [Epub ahead of print]
4. Cell Microbiol. 2016 Feb 9. doi: 10.1111/cmi.12575. [Epub ahead of print]
5. Am J Respir Cell Mol Biol. 2003 Oct;29:472-82.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. He said he had no relevant financial disclosures. Email him at [email protected].
Although acute otitis media (AOM) has decreased in number, and especially the more severe difficult to treat versions, I was reminded that this still is a problem for young children based on personal experience with grandchildren. What can be baffling to some families is the fact that some strains of the same organism species cause AOM and some simply colonize the nasopharynx (NP) of children without causing any disease at all. These organisms include Streptococcus pneumoniae (SPN) and nontypeable Haemophilus influenzae (ntHi).
Recent studies have uncovered several molecular reasons for the pathogen vs. colonizer dichotomy:
• Strains within a species can have variants of a gene that make them more disease producing.
• Some usually colonizing strains produce disease after acquiring new genes.
• Some strains have native genes with on-off switches that convert them from a colonizing to disease producing under selected circumstances.
• Molecular targets in the respiratory tract increase, allowing more dense colonization that increases chances of AOM.
Variant gene
J.R. Gilsdorf, MD, and his group at the University of Michigan,1 Ann Arbor, recently showed that among the various high-molecular-weight molecules (HMW) produced by 170 ntHi from three different geographically diverse countries, one variant in particular (HMW-A) was more likely to be found in strains producing AOM than strains simply colonizing the nasopharynx. The protein product of this gene allows better adherence to respiratory epithelia. So more bacteria sticking in the NP near the eustachian tube opening make development of AOM more likely. Some call this the “more barbarians at the gate” phenomenon.
Gene acquisition
SPN inherently has a somewhat incomplete arginine synthesis pathway. Because arginine is essential for growth of SPN, S. pneumoniae utilizes some host factors to compensate; but this compensation is inefficient. However, SPN strains can acquire new genes – usually from other gram-positive organisms in their environment – by a process called conjugation.
One recently reported acquired gene set is that which completes functionality of SPN’s arginine synthesis pathway.2 Investigators showed that SPN that acquire these arginine synthesis genes replicate more readily in bodily fluids, such as serum or cerebrospinal fluid, making these strains more aggressive, more virulent, and more likely to produce disease. More efficient replication makes it very difficult for host immune responses to handle these SPN. This is not limited to AOM alone, but seems important in invasive disease (such as meningitis) from SPN type 7, which had recently become more frequent after introduction of pneumococcal conjugate vaccines.
On-off gene switches
Another group of investigators reported that a thing called “phasevarion,” which is fancy lingo for an on-off switch is at the root of more virulence in ntHi.3 It seems that some strains of ntHi have a version of the ModA2 gene, which is always turned off, while other strains have a gene that is always on. Then, there is a third version in which the gene is usually off, but turns on when in places like the middle ear. The ModA2 gene appears to affect several other downstream protein groups that include HMW-A, antibiotic susceptibility, and biofilm formation. When inoculated into the middle ear in a chinchilla AOM model, the ntHi strains that can turn on their ModA2 gene were much more likely to produce AOM than either version that could not change. Interestingly, the authors postulate that preventing the switch capability could be a novel way to prevent ntHi disease, such as pediatric AOM, acute bacterial sinusitis, or some bronchitis in adults.
Molecular environment becomes more favorable
Another group4 reported that adherence receptor for ntHi is intercellular adhesion molecule 1 (ICAM1), a molecule found in modest quantities on respiratory epithelium. You may know it as the attachment molecule for rhinovirus and enteroviruses. What makes this interesting is that adenovirus, respiratory syncytial virus, and exposure to cigarette smoke5 markedly increase expression of ICAM1 on respiratory epithelium, predisposing to more ntHi adhering and more likely to produce an inflammatory process, such as AOM. This is another version of the barbarians at the gate phenomenon.
So when families ask why SPN or ntHi sometimes exist quietly (colonize) the nasopharynx and sometimes they cause AOM or acute bacterial sinusitis, you can hopefully use these four examples as partial explanations of why the same bacterial species has strains that can be either colonizers or pathogens.
References
1. Infect Genet Evol. 2014 Dec;28:223-32
2. J Infect Dis. 2014 Jun 1;209:1781-91.
3. J Infect Dis. 2016 Jun 10. pii: jiw243. [Epub ahead of print]
4. Cell Microbiol. 2016 Feb 9. doi: 10.1111/cmi.12575. [Epub ahead of print]
5. Am J Respir Cell Mol Biol. 2003 Oct;29:472-82.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. He said he had no relevant financial disclosures. Email him at [email protected].
Although acute otitis media (AOM) has decreased in number, and especially the more severe difficult to treat versions, I was reminded that this still is a problem for young children based on personal experience with grandchildren. What can be baffling to some families is the fact that some strains of the same organism species cause AOM and some simply colonize the nasopharynx (NP) of children without causing any disease at all. These organisms include Streptococcus pneumoniae (SPN) and nontypeable Haemophilus influenzae (ntHi).
Recent studies have uncovered several molecular reasons for the pathogen vs. colonizer dichotomy:
• Strains within a species can have variants of a gene that make them more disease producing.
• Some usually colonizing strains produce disease after acquiring new genes.
• Some strains have native genes with on-off switches that convert them from a colonizing to disease producing under selected circumstances.
• Molecular targets in the respiratory tract increase, allowing more dense colonization that increases chances of AOM.
Variant gene
J.R. Gilsdorf, MD, and his group at the University of Michigan,1 Ann Arbor, recently showed that among the various high-molecular-weight molecules (HMW) produced by 170 ntHi from three different geographically diverse countries, one variant in particular (HMW-A) was more likely to be found in strains producing AOM than strains simply colonizing the nasopharynx. The protein product of this gene allows better adherence to respiratory epithelia. So more bacteria sticking in the NP near the eustachian tube opening make development of AOM more likely. Some call this the “more barbarians at the gate” phenomenon.
Gene acquisition
SPN inherently has a somewhat incomplete arginine synthesis pathway. Because arginine is essential for growth of SPN, S. pneumoniae utilizes some host factors to compensate; but this compensation is inefficient. However, SPN strains can acquire new genes – usually from other gram-positive organisms in their environment – by a process called conjugation.
One recently reported acquired gene set is that which completes functionality of SPN’s arginine synthesis pathway.2 Investigators showed that SPN that acquire these arginine synthesis genes replicate more readily in bodily fluids, such as serum or cerebrospinal fluid, making these strains more aggressive, more virulent, and more likely to produce disease. More efficient replication makes it very difficult for host immune responses to handle these SPN. This is not limited to AOM alone, but seems important in invasive disease (such as meningitis) from SPN type 7, which had recently become more frequent after introduction of pneumococcal conjugate vaccines.
On-off gene switches
Another group of investigators reported that a thing called “phasevarion,” which is fancy lingo for an on-off switch is at the root of more virulence in ntHi.3 It seems that some strains of ntHi have a version of the ModA2 gene, which is always turned off, while other strains have a gene that is always on. Then, there is a third version in which the gene is usually off, but turns on when in places like the middle ear. The ModA2 gene appears to affect several other downstream protein groups that include HMW-A, antibiotic susceptibility, and biofilm formation. When inoculated into the middle ear in a chinchilla AOM model, the ntHi strains that can turn on their ModA2 gene were much more likely to produce AOM than either version that could not change. Interestingly, the authors postulate that preventing the switch capability could be a novel way to prevent ntHi disease, such as pediatric AOM, acute bacterial sinusitis, or some bronchitis in adults.
Molecular environment becomes more favorable
Another group4 reported that adherence receptor for ntHi is intercellular adhesion molecule 1 (ICAM1), a molecule found in modest quantities on respiratory epithelium. You may know it as the attachment molecule for rhinovirus and enteroviruses. What makes this interesting is that adenovirus, respiratory syncytial virus, and exposure to cigarette smoke5 markedly increase expression of ICAM1 on respiratory epithelium, predisposing to more ntHi adhering and more likely to produce an inflammatory process, such as AOM. This is another version of the barbarians at the gate phenomenon.
So when families ask why SPN or ntHi sometimes exist quietly (colonize) the nasopharynx and sometimes they cause AOM or acute bacterial sinusitis, you can hopefully use these four examples as partial explanations of why the same bacterial species has strains that can be either colonizers or pathogens.
References
1. Infect Genet Evol. 2014 Dec;28:223-32
2. J Infect Dis. 2014 Jun 1;209:1781-91.
3. J Infect Dis. 2016 Jun 10. pii: jiw243. [Epub ahead of print]
4. Cell Microbiol. 2016 Feb 9. doi: 10.1111/cmi.12575. [Epub ahead of print]
5. Am J Respir Cell Mol Biol. 2003 Oct;29:472-82.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. He said he had no relevant financial disclosures. Email him at [email protected].

Why so many pertussis outbreaks despite acellular pertussis vaccine? A call to action
There has been a justified re-examination of acellular pertussis vaccine (aP)1,2 in light of the multiple large outbreaks of pertussis since 2000, particularly the two large California outbreaks in 2010 and 2014.
Lessons learned: aP protection is less durable than originally thought, and much pertussis is not in infants, but in the school-age and adolescent populations.

aP appears to produce reasonable protection (approximately 84% overall) for infants and preschool children, plus a much improved adverse effect profile, compared with whole cell pertussis vaccine (WCP), which provided approximately 94% protection.1 This 10% difference in aP versus WCP, however, means that herd immunity is more difficult to attain. The accepted pertussis immunization rate needed to provide herd immunity is 92%-94%. Because our current tools (DTaP and Tdap) provide only 84% protection at least in infants and preschoolers, even 100% uptake may leave us 6% to 8% short of the threshold for complete herd immunity.
The California outbreak data from school-age and teenage populations show protection rates drop each year post aP booster. That means that by the fourth year after the last dose, protection is less than 10%. So despite a Tdap dose at 11- to 12-years-of-age, protection gaps occur in 8-to 10-year-olds and 14- to 18-year-olds. These vulnerable periods in older children add to aP’s 84% vs. WCP’s 94% protection for those greater than 3 years of age, explaining more frequent pertussis outbreaks as the pool of WCP-immunized children among older populations decreased.
But before we place all blame on switching to aP, consider that we can now confirm more pertussis infections with molecular assays than was possible with culture and fluorescent assay testing in the WCP era. So improved testing sensitivity means more reports of minimally symptomatic cases that may have been missed before. So WCP, if still used today, might not show 94% protection either.
Additionally, aPs rely heavily on pertactin as a target antigen,3 likely less than WCP, given that WCP contained all pertussis antigens rather than just the 3-5 purified antigens in aPs. So the emergence of pertactin-altered pertussis strains could disproportionately affect protection from aP, compared with WCP.
There seem to be no quick fixes to preventing outbreaks using aPs as our vaccine. One suggestion by the authors of the California outbreak report is to use aP mostly to terminate outbreaks rather than routinely in late childhood. My concern is that if we do not continue routine use in 4-to 6-year-olds, 10-to 11-year-olds, and in early adulthood, the vulnerable proportion of the population during outbreaks would be larger, making outbreaks more difficult to terminate. So continuing to produce some protection, albeit short-lived, with current schedules of aP vaccines seems important.
Also remember that T cells, particularly TH 17 pertussis-specific cells, may be as important as pertussis antibody. Therefore, crafting pertussis vaccines that yield improved antibody plus T cell responses is the current goal. Disease and WCP seem to elicit more T-17 response than aP. One method to craft a better vaccine is to use antigen blends that differ from those in the current vaccines, such as antigens derived from circulating pertussis strains instead of the standard laboratory strain. Another option is to use current antigens but with more potent adjuvants. Such vaccines are likely 5 years away.
But we need to have reasonable expectations for pertussis vaccines. Pertussis infection begins in respiratory epithelium. Many of the most obvious signs and symptoms are due to destruction of ciliated respiratory epithelium plus increased tenacity/volume of secretions. Can a parenterally administered vaccine that induces mostly serum antibody protect against infection of epithelium where antibody concentrations are likely 10% or less than in serum? The short answer is – likely not. We should expect neither aP nor WCP to consistently protect against pertussis infection, but it does seem reasonable to expect aP to reduce disease severity. Preventing infections awaits a vaccine that induces surface IgA. Mucosally administered vaccines produce surface IgA – for example, rotavirus vaccine – but no mucosal pertussis vaccine appears imminent.
A key question is whether our most vulnerable populations, young children, have increased morbidity and mortality. Data from the California suggest an increase but mostly in infants under 6 months of age, the group not old enough to benefit from even the most effective of infant vaccines. Protecting young infants depends on vaccine administered prenatally to mothers. The over-representation of the Hispanic infants among fatalities shows a population on which to focus with maternal immunization. Hopefully, the recent universal TdaP recommendation in pregnancy will help when maternal immunization is higher than current approximately 50% rates.4
Despite the problems, it seems clear that we must continue to use current aP vaccines according to the current schedules, attempting to get as close to 100% uptake as possible. While the current, nearly 10% unimmunized rates add to the likelihood that we are losing complete herd immunity, partial herd immunity is better than no herd immunity.
Expectations: There will be ongoing outbreaks. Continue to be alert for signs of pertussis. They are often less obvious in older patients, and may be as subtle as more than 2 weeks of persistent cough. During outbreaks, we may be called upon to give aP doses at intervals shorter than the usual schedule.
Our responsibility: Do not become discouraged or lose enthusiasm for aP, but explain to parents that because aP is less reactogenic, it produces less protection and is less durable, particularly in school-age children. But please emphasize that modest protection is best in the youngest and modest protection of older children is better than none. Emphasize that the adverse effect profile of current aPs puts the harm/benefit balance heavily in favor of aP.
Bottom line: We can hopefully do better than the current 88% to 92% rate of aP vaccine uptake. We need to get as close to 100% uptake as possible until new vaccines or new strategies become available.
1. Clin Infect Dis. 2016 Feb 7; doi: 10.1093/cid/ciw051.
2. Pediatrics. 2016 Feb 5; doi: 10.1542/peds.2015-3326.
3. Expert Rev Vaccines. 2007 Feb;6(1):47-56.
4. Vaccine. 2016 Feb 10;34(7):968-73.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. He disclosed that his institution received grant support for a study on hexavalent infant vaccine containing pertussis from GlaxoSmithKline, and he was the local primary investigator.*
*Correction, 2/17/2016: An earlier version of this article incompletely stated Dr. Harrison's disclosure information.
There has been a justified re-examination of acellular pertussis vaccine (aP)1,2 in light of the multiple large outbreaks of pertussis since 2000, particularly the two large California outbreaks in 2010 and 2014.
Lessons learned: aP protection is less durable than originally thought, and much pertussis is not in infants, but in the school-age and adolescent populations.
aP appears to produce reasonable protection (approximately 84% overall) for infants and preschool children, plus a much improved adverse effect profile, compared with whole cell pertussis vaccine (WCP), which provided approximately 94% protection.1 This 10% difference in aP versus WCP, however, means that herd immunity is more difficult to attain. The accepted pertussis immunization rate needed to provide herd immunity is 92%-94%. Because our current tools (DTaP and Tdap) provide only 84% protection at least in infants and preschoolers, even 100% uptake may leave us 6% to 8% short of the threshold for complete herd immunity.
The California outbreak data from school-age and teenage populations show protection rates drop each year post aP booster. That means that by the fourth year after the last dose, protection is less than 10%. So despite a Tdap dose at 11- to 12-years-of-age, protection gaps occur in 8-to 10-year-olds and 14- to 18-year-olds. These vulnerable periods in older children add to aP’s 84% vs. WCP’s 94% protection for those greater than 3 years of age, explaining more frequent pertussis outbreaks as the pool of WCP-immunized children among older populations decreased.
But before we place all blame on switching to aP, consider that we can now confirm more pertussis infections with molecular assays than was possible with culture and fluorescent assay testing in the WCP era. So improved testing sensitivity means more reports of minimally symptomatic cases that may have been missed before. So WCP, if still used today, might not show 94% protection either.
Additionally, aPs rely heavily on pertactin as a target antigen,3 likely less than WCP, given that WCP contained all pertussis antigens rather than just the 3-5 purified antigens in aPs. So the emergence of pertactin-altered pertussis strains could disproportionately affect protection from aP, compared with WCP.
There seem to be no quick fixes to preventing outbreaks using aPs as our vaccine. One suggestion by the authors of the California outbreak report is to use aP mostly to terminate outbreaks rather than routinely in late childhood. My concern is that if we do not continue routine use in 4-to 6-year-olds, 10-to 11-year-olds, and in early adulthood, the vulnerable proportion of the population during outbreaks would be larger, making outbreaks more difficult to terminate. So continuing to produce some protection, albeit short-lived, with current schedules of aP vaccines seems important.
Also remember that T cells, particularly TH 17 pertussis-specific cells, may be as important as pertussis antibody. Therefore, crafting pertussis vaccines that yield improved antibody plus T cell responses is the current goal. Disease and WCP seem to elicit more T-17 response than aP. One method to craft a better vaccine is to use antigen blends that differ from those in the current vaccines, such as antigens derived from circulating pertussis strains instead of the standard laboratory strain. Another option is to use current antigens but with more potent adjuvants. Such vaccines are likely 5 years away.
But we need to have reasonable expectations for pertussis vaccines. Pertussis infection begins in respiratory epithelium. Many of the most obvious signs and symptoms are due to destruction of ciliated respiratory epithelium plus increased tenacity/volume of secretions. Can a parenterally administered vaccine that induces mostly serum antibody protect against infection of epithelium where antibody concentrations are likely 10% or less than in serum? The short answer is – likely not. We should expect neither aP nor WCP to consistently protect against pertussis infection, but it does seem reasonable to expect aP to reduce disease severity. Preventing infections awaits a vaccine that induces surface IgA. Mucosally administered vaccines produce surface IgA – for example, rotavirus vaccine – but no mucosal pertussis vaccine appears imminent.
A key question is whether our most vulnerable populations, young children, have increased morbidity and mortality. Data from the California suggest an increase but mostly in infants under 6 months of age, the group not old enough to benefit from even the most effective of infant vaccines. Protecting young infants depends on vaccine administered prenatally to mothers. The over-representation of the Hispanic infants among fatalities shows a population on which to focus with maternal immunization. Hopefully, the recent universal TdaP recommendation in pregnancy will help when maternal immunization is higher than current approximately 50% rates.4
Despite the problems, it seems clear that we must continue to use current aP vaccines according to the current schedules, attempting to get as close to 100% uptake as possible. While the current, nearly 10% unimmunized rates add to the likelihood that we are losing complete herd immunity, partial herd immunity is better than no herd immunity.
Expectations: There will be ongoing outbreaks. Continue to be alert for signs of pertussis. They are often less obvious in older patients, and may be as subtle as more than 2 weeks of persistent cough. During outbreaks, we may be called upon to give aP doses at intervals shorter than the usual schedule.
Our responsibility: Do not become discouraged or lose enthusiasm for aP, but explain to parents that because aP is less reactogenic, it produces less protection and is less durable, particularly in school-age children. But please emphasize that modest protection is best in the youngest and modest protection of older children is better than none. Emphasize that the adverse effect profile of current aPs puts the harm/benefit balance heavily in favor of aP.
Bottom line: We can hopefully do better than the current 88% to 92% rate of aP vaccine uptake. We need to get as close to 100% uptake as possible until new vaccines or new strategies become available.
1. Clin Infect Dis. 2016 Feb 7; doi: 10.1093/cid/ciw051.
2. Pediatrics. 2016 Feb 5; doi: 10.1542/peds.2015-3326.
3. Expert Rev Vaccines. 2007 Feb;6(1):47-56.
4. Vaccine. 2016 Feb 10;34(7):968-73.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. He disclosed that his institution received grant support for a study on hexavalent infant vaccine containing pertussis from GlaxoSmithKline, and he was the local primary investigator.*
*Correction, 2/17/2016: An earlier version of this article incompletely stated Dr. Harrison's disclosure information.
There has been a justified re-examination of acellular pertussis vaccine (aP)1,2 in light of the multiple large outbreaks of pertussis since 2000, particularly the two large California outbreaks in 2010 and 2014.
Lessons learned: aP protection is less durable than originally thought, and much pertussis is not in infants, but in the school-age and adolescent populations.
aP appears to produce reasonable protection (approximately 84% overall) for infants and preschool children, plus a much improved adverse effect profile, compared with whole cell pertussis vaccine (WCP), which provided approximately 94% protection.1 This 10% difference in aP versus WCP, however, means that herd immunity is more difficult to attain. The accepted pertussis immunization rate needed to provide herd immunity is 92%-94%. Because our current tools (DTaP and Tdap) provide only 84% protection at least in infants and preschoolers, even 100% uptake may leave us 6% to 8% short of the threshold for complete herd immunity.
The California outbreak data from school-age and teenage populations show protection rates drop each year post aP booster. That means that by the fourth year after the last dose, protection is less than 10%. So despite a Tdap dose at 11- to 12-years-of-age, protection gaps occur in 8-to 10-year-olds and 14- to 18-year-olds. These vulnerable periods in older children add to aP’s 84% vs. WCP’s 94% protection for those greater than 3 years of age, explaining more frequent pertussis outbreaks as the pool of WCP-immunized children among older populations decreased.
But before we place all blame on switching to aP, consider that we can now confirm more pertussis infections with molecular assays than was possible with culture and fluorescent assay testing in the WCP era. So improved testing sensitivity means more reports of minimally symptomatic cases that may have been missed before. So WCP, if still used today, might not show 94% protection either.
Additionally, aPs rely heavily on pertactin as a target antigen,3 likely less than WCP, given that WCP contained all pertussis antigens rather than just the 3-5 purified antigens in aPs. So the emergence of pertactin-altered pertussis strains could disproportionately affect protection from aP, compared with WCP.
There seem to be no quick fixes to preventing outbreaks using aPs as our vaccine. One suggestion by the authors of the California outbreak report is to use aP mostly to terminate outbreaks rather than routinely in late childhood. My concern is that if we do not continue routine use in 4-to 6-year-olds, 10-to 11-year-olds, and in early adulthood, the vulnerable proportion of the population during outbreaks would be larger, making outbreaks more difficult to terminate. So continuing to produce some protection, albeit short-lived, with current schedules of aP vaccines seems important.
Also remember that T cells, particularly TH 17 pertussis-specific cells, may be as important as pertussis antibody. Therefore, crafting pertussis vaccines that yield improved antibody plus T cell responses is the current goal. Disease and WCP seem to elicit more T-17 response than aP. One method to craft a better vaccine is to use antigen blends that differ from those in the current vaccines, such as antigens derived from circulating pertussis strains instead of the standard laboratory strain. Another option is to use current antigens but with more potent adjuvants. Such vaccines are likely 5 years away.
But we need to have reasonable expectations for pertussis vaccines. Pertussis infection begins in respiratory epithelium. Many of the most obvious signs and symptoms are due to destruction of ciliated respiratory epithelium plus increased tenacity/volume of secretions. Can a parenterally administered vaccine that induces mostly serum antibody protect against infection of epithelium where antibody concentrations are likely 10% or less than in serum? The short answer is – likely not. We should expect neither aP nor WCP to consistently protect against pertussis infection, but it does seem reasonable to expect aP to reduce disease severity. Preventing infections awaits a vaccine that induces surface IgA. Mucosally administered vaccines produce surface IgA – for example, rotavirus vaccine – but no mucosal pertussis vaccine appears imminent.
A key question is whether our most vulnerable populations, young children, have increased morbidity and mortality. Data from the California suggest an increase but mostly in infants under 6 months of age, the group not old enough to benefit from even the most effective of infant vaccines. Protecting young infants depends on vaccine administered prenatally to mothers. The over-representation of the Hispanic infants among fatalities shows a population on which to focus with maternal immunization. Hopefully, the recent universal TdaP recommendation in pregnancy will help when maternal immunization is higher than current approximately 50% rates.4
Despite the problems, it seems clear that we must continue to use current aP vaccines according to the current schedules, attempting to get as close to 100% uptake as possible. While the current, nearly 10% unimmunized rates add to the likelihood that we are losing complete herd immunity, partial herd immunity is better than no herd immunity.
Expectations: There will be ongoing outbreaks. Continue to be alert for signs of pertussis. They are often less obvious in older patients, and may be as subtle as more than 2 weeks of persistent cough. During outbreaks, we may be called upon to give aP doses at intervals shorter than the usual schedule.
Our responsibility: Do not become discouraged or lose enthusiasm for aP, but explain to parents that because aP is less reactogenic, it produces less protection and is less durable, particularly in school-age children. But please emphasize that modest protection is best in the youngest and modest protection of older children is better than none. Emphasize that the adverse effect profile of current aPs puts the harm/benefit balance heavily in favor of aP.
Bottom line: We can hopefully do better than the current 88% to 92% rate of aP vaccine uptake. We need to get as close to 100% uptake as possible until new vaccines or new strategies become available.
1. Clin Infect Dis. 2016 Feb 7; doi: 10.1093/cid/ciw051.
2. Pediatrics. 2016 Feb 5; doi: 10.1542/peds.2015-3326.
3. Expert Rev Vaccines. 2007 Feb;6(1):47-56.
4. Vaccine. 2016 Feb 10;34(7):968-73.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. He disclosed that his institution received grant support for a study on hexavalent infant vaccine containing pertussis from GlaxoSmithKline, and he was the local primary investigator.*
*Correction, 2/17/2016: An earlier version of this article incompletely stated Dr. Harrison's disclosure information.

The importance of UA in diagnosing UTIs in infants under 2 months
A 28-day-old uncircumcised male infant presents to the emergency department with fever of 38.9° C, decreased feeding, and irritability. The physical examination is normal with the exception of the irritability and your assessment of mild dehydration. The infant undergoes a sepsis work-up. The CBC is remarkable for a WBC of 16,500/mm3 with 44% neutrophils, 52% lymphocytes, and 4% monocytes. Platelet count is normal. Cerebrospinal fluid (CSF) shows no white or red blood cells with normal glucose and protein. The urinalysis (UA) has a positive 1+ leukocyte esterase (LE) with 10 WBC per high-power field (HPF), but negative nitrite and 1+ bacteria microscopically. The child is admitted to the hospital for empiric antibiotics pending blood, urine, and CSF cultures. What are the chances that a urinary tract infection (UTI) is the origin of the febrile presentation?

UTIs are currently the most common serious bacterial infection (SBI) in < 2-year-old febrile children without an apparent source of fever (Pediatrics 2011;128:595-610). Since 2000, the prevalence of UTIs in all febrile infants and young children without an apparent source is unchanged, being approximately 5%. The rate of UTIs in fever-without-apparent-source presentations at < 90 days of age is higher, ranging from 6%-15% in different studies.
Meanwhile bacteremia, sepsis, meningitis, and other previously common SBIs, mostly caused by Haemophilus influenzae type b (Hib) or pneumococcus, have decreased. We recognize these reductions as effects of universal implementation of Hib (mid-1990s) and pneumococcal (2000 and 2010) conjugate vaccines.
Given the case above, other pertinent facts are that uncircumcised males have more UTIs in the first months of life (J. Pediatr. 1996;128:23-7) and approximately 5% of young infants with UTIs also are concurrently bacteremic (Pediatrics 1999;104:79-86;J. Pediatr. 1994;124:513-9)
The elephant in the room is the fact that we also need to be cognizant of asymptomatic bacteriuria (AB). AB is colonization of the lower urinary tract without infection. Patients with AB may meet culture criteria for UTI (whether we consider > 50,000 or > 100,000 colony-forming units/mL), but there is no evidence of true infection, that is no inflammation or mucosal injury. So children with AB are not at risk for renal injury or later renal damage and do not require antibiotic treatment.
But when AB patients develop fever, for example with an enterovirus infection, their urine cultures (together with the fever) can do a good imitation of a UTI, unless we focus on the UA results. It not only remains critical to detect true UTIs in infants < 90 days old, such as the one in our case above, but also to distinguish UTI from AB.
The 1999 American Academy of Pediatrics’ UTI guidelines (Pediatrics 1999;103:843-52) included UA results as suggestive of UTI. They stated that a positive LE or nitrite test or > 5 WBC/HPF in a spun urine, or bacteria visualized in unspun gram-stained specimen suggest, but cannot be diagnostic of a UTI. Recommendation five in the guidelines states that UTI diagnosis required 100,000 CFU/mL in culture of sterilely obtained catheterized urine as the threshold criterion (strength of evidence: strong). However, AB was not fully considered because, in part, data defining AB was incomplete in 1999.
The 1999 guidelines also stated, “The urinalysis … can be valuable in selecting individuals for prompt initiation of treatment while waiting for the results of the urine culture.” So, UA was considered adjunctive. UA’s main function was to allow empiric therapy of sufficiently ill children, given positive results for LE, nitrites, or microscopic visualization of > 5 WBC/HPF or bacteria in the spun urine.
In the 2011 AAP guidelines for UTI, things have changed (Pediatrics 2011;128:595-610). The third action statement tells us that both the UA and culture taken together are necessary for UTI diagnosis. To paraphrase: The diagnosis of UTI requires urinalysis results suggesting infection (pyuria or microscopic bacteriuria) plus > 50,000 CFU/mL of a uropathogen in urine from catheterization or suprapubic aspiration. But remember that these guidelines do not apply specifically to the youngest of infants, that is < 2 months old.
Both of these criteria were changes from the 1999 UTI guidelines. Previously pyuria or microscopic bacteriuria were not considered necessary to diagnose UTI, and >100,000 CFU/mL rather than > 50,000 CFU/mL of a single pathogen species was the critical diagnostic result for catheterized urine. For suprapubic aspiration urine samples, > 10,000 CFU/HPF were considered adequate for UTI diagnoses in 1999.
Now, a recent study of children < 90 days of age (including those < 2 months of age) reports that pyuria (> 3 WBC/HPF) plus > 50,000 CFU/mL are the keys to diagnosing UTI (Pediatrics 2015;135:965-71). One caveat is that the study population was febrile infants < 90 days old with concurrent bacteremia (bacteremic UTI). Bacteremic UTI was studied to reduce as much as possible the chance that AB patients might be inadvertently included in the study. One other conclusion of this new study is that microscopic bacteriuria did not add significantly to the either sensitivity or specificity.
These data in an overall younger population than that covered by the 2011 guidelines adds evidence that pyuria (but not microscopic bacteriuria) is critical to diagnosing UTI. Pyuria plus positive culture has been a combination for the pediatric infectious diseases practitioner’s toolkit for decades. Likewise, it seems to me that primary care pediatric clinicians also often decide whether to undertake the expense of culture based on UA results. For example, a completely normal UA may obviate need for culture except in selected unusual cases.
Requiring UA evidence of inflammation to diagnose UTI (per the 2011 guidelines and the recommendations of the authors of the recent 2015 study) makes sense because most UTIs in otherwise healthy children are caused by gram-negative organisms (> 90% from Escherichia coli) (J. Pediatr. 1994;124:513-9). Why are UA results so important?
A positive nitrite test strongly suggests UTI because nitrites in the urine indicate viable gram-negative organisms also are present in the urine. Nitrates in the urine are converted to nitrites by metabolic activity of gram-negative pathogens. For WBCs or LE in the urine, their presence indicates inflammation in the urinary tract, Consider that lipopolysaccharide (LPS), also known as gram-negative endotoxin, is a major component of the cell membrane of > 90% of uropathogens like E. coli. Moreover, LPS elicits about the strongest innate immune response via toll-like receptor 4 (TLR4) from monocytes/macrophages, inducing a large pro-inflammatory and chemotactic response – interleukin-6, interleukin-8, tetrahydrofuran-alpha. Remember that LPS is also a major cause of fever and of shock during gram-negative sepsis.
So a UTI diagnosis based on a “positive” culture without evidence of metabolic products of gram negatives (nitrites) or without inflammation (no pyuria or negative LE) should be questioned. The combination of > 50,000 CFU/mL with no detectable LE or < 3-5 WBC/HPF in a febrile child is most likely evidence for AB in a child with the fever caused by some non-UTI process.
In contrast, selected SBIs may occur when the culture is “positive” without inflammation or nitrites. The first of three examples is a renal parenchymal abscess, where bacteria enter the urine sporadically in only small numbers, and do not actually infect the urinary tract mucosa. The scenario of no inflammation but “positive” culture also may occur when a large bacteremic load causes results in organisms filtering through the kidney into the urine, again without urinary mucosal infection, such as Staphylococcus aureus, group A streptococcus, or group B streptococcus bacteremia/sepsis. The third scenario with a “positive” culture and no pyuria can be with organisms that have blunted abilities to induce inflammation, such as enterococcus. Enterococcal cell components have weak inflammatory and chemotactic capability. So a urinary mucosal infection in the collecting system or bladder may occur without much if any pyuria. In fact, the patients from the recent study with insufficient evidence of pyuria/inflammation were those who had either gram-positive organisms or considerably less than 50,000 CFU/mL of gram-negative organisms.
The sensitivity and specificity of the LE or pyuria was higher in the recent study (Pediatrics 2015;135:965-71) than any prior study. The authors comment that they had not expected such a high sensitivity of 97.6% (94.5-99.2) for LE in confirmed bacteremic UTI, nor did they expect the high specificity of 93.9% (87.9-97.5). The presence of microscopic pyuria defined as > 3 WBC/HPF was nearly as sensitive, 96%, and specific, 91.3%. Disappointingly, positive nitrite testing was only 39.5% sensitive, but it was 100% specific. This likely reflects the short time that urine resides in the bladder of infants < 90 days of age, so there is insufficient time for the pathogens to metabolically convert the nitrates to nitrites.
So how would the UA help with our example case? There is microscopic bacteriuria, pyuria, and positive LE, but negative nitrites. Using the suggestions of the authors of the recent report (Pediatrics 2015;135:965-71) and those of another report on the utility of UA results (Acta Paediatr. 2010;99:581-4), the UA in our case indicates that we should be highly suspicious of a UTI in this child < 2 months old for whom the 2011 guidelines do not directly apply. But remember that these impressive sensitivity and specificity values relate to bacteremic UTI. Whether they apply to nonbacteremic UTI is not known. Likewise, the authors caution that their study design did not allow calculation of positive or negative predictive values – aspects that would clarify things even further.
So we still cannot be more than highly suspicious. Without a positive predictive value, we do not know the odds of this case having a UTI with mathematical precision. The authors do point out that only one of their subjects had a completely normal UA and actually had a bacteremic UTI. If you guessed that it was a gram-positive pathogen, you win the prize. So it seems reasonable to predict that a normal UA has a high specificity for not being a UTI (87.8%), but a positive UA remains only highly suggestive. It is still not clear if a negative UA statistically justifies not submitting the culture of the sterilely obtained urine because we still don’t have a negative predictive value.
Bottom line: The 2011 UTI guidelines provide good advice on diagnosing UTIs.
1. We have more data that evidence of inflammation is essential for diagnosing gram-negative UTIs.
2. We also have more evidence that 50,000 CFU/mL is a good threshold for diagnosing UTIs.
3. It appears that microscopic bacteriuria did not add significantly to the either sensitivity or specificity.
4. And we now have more evidence that these criteria also apply to infants < 2 months of age.
To close the loop on our case, the child’s CSF and blood cultures were negative, but the urine culture revealed > 100,000 CFU/mL of E. coli susceptible to second- and third-generation cephalosporins, ciprofloxacin, and nitrofurantoin, but resistant to trimethoprim-sulfamethoxazole.
Have a great summer and watch for UTIs in your young patients < 90 days old and fever without apparent focus.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. He said he had no relevant financial disclosures. E-mail Dr. Harrison at [email protected].
A 28-day-old uncircumcised male infant presents to the emergency department with fever of 38.9° C, decreased feeding, and irritability. The physical examination is normal with the exception of the irritability and your assessment of mild dehydration. The infant undergoes a sepsis work-up. The CBC is remarkable for a WBC of 16,500/mm3 with 44% neutrophils, 52% lymphocytes, and 4% monocytes. Platelet count is normal. Cerebrospinal fluid (CSF) shows no white or red blood cells with normal glucose and protein. The urinalysis (UA) has a positive 1+ leukocyte esterase (LE) with 10 WBC per high-power field (HPF), but negative nitrite and 1+ bacteria microscopically. The child is admitted to the hospital for empiric antibiotics pending blood, urine, and CSF cultures. What are the chances that a urinary tract infection (UTI) is the origin of the febrile presentation?
UTIs are currently the most common serious bacterial infection (SBI) in < 2-year-old febrile children without an apparent source of fever (Pediatrics 2011;128:595-610). Since 2000, the prevalence of UTIs in all febrile infants and young children without an apparent source is unchanged, being approximately 5%. The rate of UTIs in fever-without-apparent-source presentations at < 90 days of age is higher, ranging from 6%-15% in different studies.
Meanwhile bacteremia, sepsis, meningitis, and other previously common SBIs, mostly caused by Haemophilus influenzae type b (Hib) or pneumococcus, have decreased. We recognize these reductions as effects of universal implementation of Hib (mid-1990s) and pneumococcal (2000 and 2010) conjugate vaccines.
Given the case above, other pertinent facts are that uncircumcised males have more UTIs in the first months of life (J. Pediatr. 1996;128:23-7) and approximately 5% of young infants with UTIs also are concurrently bacteremic (Pediatrics 1999;104:79-86;J. Pediatr. 1994;124:513-9)
The elephant in the room is the fact that we also need to be cognizant of asymptomatic bacteriuria (AB). AB is colonization of the lower urinary tract without infection. Patients with AB may meet culture criteria for UTI (whether we consider > 50,000 or > 100,000 colony-forming units/mL), but there is no evidence of true infection, that is no inflammation or mucosal injury. So children with AB are not at risk for renal injury or later renal damage and do not require antibiotic treatment.
But when AB patients develop fever, for example with an enterovirus infection, their urine cultures (together with the fever) can do a good imitation of a UTI, unless we focus on the UA results. It not only remains critical to detect true UTIs in infants < 90 days old, such as the one in our case above, but also to distinguish UTI from AB.
The 1999 American Academy of Pediatrics’ UTI guidelines (Pediatrics 1999;103:843-52) included UA results as suggestive of UTI. They stated that a positive LE or nitrite test or > 5 WBC/HPF in a spun urine, or bacteria visualized in unspun gram-stained specimen suggest, but cannot be diagnostic of a UTI. Recommendation five in the guidelines states that UTI diagnosis required 100,000 CFU/mL in culture of sterilely obtained catheterized urine as the threshold criterion (strength of evidence: strong). However, AB was not fully considered because, in part, data defining AB was incomplete in 1999.
The 1999 guidelines also stated, “The urinalysis … can be valuable in selecting individuals for prompt initiation of treatment while waiting for the results of the urine culture.” So, UA was considered adjunctive. UA’s main function was to allow empiric therapy of sufficiently ill children, given positive results for LE, nitrites, or microscopic visualization of > 5 WBC/HPF or bacteria in the spun urine.
In the 2011 AAP guidelines for UTI, things have changed (Pediatrics 2011;128:595-610). The third action statement tells us that both the UA and culture taken together are necessary for UTI diagnosis. To paraphrase: The diagnosis of UTI requires urinalysis results suggesting infection (pyuria or microscopic bacteriuria) plus > 50,000 CFU/mL of a uropathogen in urine from catheterization or suprapubic aspiration. But remember that these guidelines do not apply specifically to the youngest of infants, that is < 2 months old.
Both of these criteria were changes from the 1999 UTI guidelines. Previously pyuria or microscopic bacteriuria were not considered necessary to diagnose UTI, and >100,000 CFU/mL rather than > 50,000 CFU/mL of a single pathogen species was the critical diagnostic result for catheterized urine. For suprapubic aspiration urine samples, > 10,000 CFU/HPF were considered adequate for UTI diagnoses in 1999.
Now, a recent study of children < 90 days of age (including those < 2 months of age) reports that pyuria (> 3 WBC/HPF) plus > 50,000 CFU/mL are the keys to diagnosing UTI (Pediatrics 2015;135:965-71). One caveat is that the study population was febrile infants < 90 days old with concurrent bacteremia (bacteremic UTI). Bacteremic UTI was studied to reduce as much as possible the chance that AB patients might be inadvertently included in the study. One other conclusion of this new study is that microscopic bacteriuria did not add significantly to the either sensitivity or specificity.
These data in an overall younger population than that covered by the 2011 guidelines adds evidence that pyuria (but not microscopic bacteriuria) is critical to diagnosing UTI. Pyuria plus positive culture has been a combination for the pediatric infectious diseases practitioner’s toolkit for decades. Likewise, it seems to me that primary care pediatric clinicians also often decide whether to undertake the expense of culture based on UA results. For example, a completely normal UA may obviate need for culture except in selected unusual cases.
Requiring UA evidence of inflammation to diagnose UTI (per the 2011 guidelines and the recommendations of the authors of the recent 2015 study) makes sense because most UTIs in otherwise healthy children are caused by gram-negative organisms (> 90% from Escherichia coli) (J. Pediatr. 1994;124:513-9). Why are UA results so important?
A positive nitrite test strongly suggests UTI because nitrites in the urine indicate viable gram-negative organisms also are present in the urine. Nitrates in the urine are converted to nitrites by metabolic activity of gram-negative pathogens. For WBCs or LE in the urine, their presence indicates inflammation in the urinary tract, Consider that lipopolysaccharide (LPS), also known as gram-negative endotoxin, is a major component of the cell membrane of > 90% of uropathogens like E. coli. Moreover, LPS elicits about the strongest innate immune response via toll-like receptor 4 (TLR4) from monocytes/macrophages, inducing a large pro-inflammatory and chemotactic response – interleukin-6, interleukin-8, tetrahydrofuran-alpha. Remember that LPS is also a major cause of fever and of shock during gram-negative sepsis.
So a UTI diagnosis based on a “positive” culture without evidence of metabolic products of gram negatives (nitrites) or without inflammation (no pyuria or negative LE) should be questioned. The combination of > 50,000 CFU/mL with no detectable LE or < 3-5 WBC/HPF in a febrile child is most likely evidence for AB in a child with the fever caused by some non-UTI process.
In contrast, selected SBIs may occur when the culture is “positive” without inflammation or nitrites. The first of three examples is a renal parenchymal abscess, where bacteria enter the urine sporadically in only small numbers, and do not actually infect the urinary tract mucosa. The scenario of no inflammation but “positive” culture also may occur when a large bacteremic load causes results in organisms filtering through the kidney into the urine, again without urinary mucosal infection, such as Staphylococcus aureus, group A streptococcus, or group B streptococcus bacteremia/sepsis. The third scenario with a “positive” culture and no pyuria can be with organisms that have blunted abilities to induce inflammation, such as enterococcus. Enterococcal cell components have weak inflammatory and chemotactic capability. So a urinary mucosal infection in the collecting system or bladder may occur without much if any pyuria. In fact, the patients from the recent study with insufficient evidence of pyuria/inflammation were those who had either gram-positive organisms or considerably less than 50,000 CFU/mL of gram-negative organisms.
The sensitivity and specificity of the LE or pyuria was higher in the recent study (Pediatrics 2015;135:965-71) than any prior study. The authors comment that they had not expected such a high sensitivity of 97.6% (94.5-99.2) for LE in confirmed bacteremic UTI, nor did they expect the high specificity of 93.9% (87.9-97.5). The presence of microscopic pyuria defined as > 3 WBC/HPF was nearly as sensitive, 96%, and specific, 91.3%. Disappointingly, positive nitrite testing was only 39.5% sensitive, but it was 100% specific. This likely reflects the short time that urine resides in the bladder of infants < 90 days of age, so there is insufficient time for the pathogens to metabolically convert the nitrates to nitrites.
So how would the UA help with our example case? There is microscopic bacteriuria, pyuria, and positive LE, but negative nitrites. Using the suggestions of the authors of the recent report (Pediatrics 2015;135:965-71) and those of another report on the utility of UA results (Acta Paediatr. 2010;99:581-4), the UA in our case indicates that we should be highly suspicious of a UTI in this child < 2 months old for whom the 2011 guidelines do not directly apply. But remember that these impressive sensitivity and specificity values relate to bacteremic UTI. Whether they apply to nonbacteremic UTI is not known. Likewise, the authors caution that their study design did not allow calculation of positive or negative predictive values – aspects that would clarify things even further.
So we still cannot be more than highly suspicious. Without a positive predictive value, we do not know the odds of this case having a UTI with mathematical precision. The authors do point out that only one of their subjects had a completely normal UA and actually had a bacteremic UTI. If you guessed that it was a gram-positive pathogen, you win the prize. So it seems reasonable to predict that a normal UA has a high specificity for not being a UTI (87.8%), but a positive UA remains only highly suggestive. It is still not clear if a negative UA statistically justifies not submitting the culture of the sterilely obtained urine because we still don’t have a negative predictive value.
Bottom line: The 2011 UTI guidelines provide good advice on diagnosing UTIs.
1. We have more data that evidence of inflammation is essential for diagnosing gram-negative UTIs.
2. We also have more evidence that 50,000 CFU/mL is a good threshold for diagnosing UTIs.
3. It appears that microscopic bacteriuria did not add significantly to the either sensitivity or specificity.
4. And we now have more evidence that these criteria also apply to infants < 2 months of age.
To close the loop on our case, the child’s CSF and blood cultures were negative, but the urine culture revealed > 100,000 CFU/mL of E. coli susceptible to second- and third-generation cephalosporins, ciprofloxacin, and nitrofurantoin, but resistant to trimethoprim-sulfamethoxazole.
Have a great summer and watch for UTIs in your young patients < 90 days old and fever without apparent focus.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. He said he had no relevant financial disclosures. E-mail Dr. Harrison at [email protected].
A 28-day-old uncircumcised male infant presents to the emergency department with fever of 38.9° C, decreased feeding, and irritability. The physical examination is normal with the exception of the irritability and your assessment of mild dehydration. The infant undergoes a sepsis work-up. The CBC is remarkable for a WBC of 16,500/mm3 with 44% neutrophils, 52% lymphocytes, and 4% monocytes. Platelet count is normal. Cerebrospinal fluid (CSF) shows no white or red blood cells with normal glucose and protein. The urinalysis (UA) has a positive 1+ leukocyte esterase (LE) with 10 WBC per high-power field (HPF), but negative nitrite and 1+ bacteria microscopically. The child is admitted to the hospital for empiric antibiotics pending blood, urine, and CSF cultures. What are the chances that a urinary tract infection (UTI) is the origin of the febrile presentation?
UTIs are currently the most common serious bacterial infection (SBI) in < 2-year-old febrile children without an apparent source of fever (Pediatrics 2011;128:595-610). Since 2000, the prevalence of UTIs in all febrile infants and young children without an apparent source is unchanged, being approximately 5%. The rate of UTIs in fever-without-apparent-source presentations at < 90 days of age is higher, ranging from 6%-15% in different studies.
Meanwhile bacteremia, sepsis, meningitis, and other previously common SBIs, mostly caused by Haemophilus influenzae type b (Hib) or pneumococcus, have decreased. We recognize these reductions as effects of universal implementation of Hib (mid-1990s) and pneumococcal (2000 and 2010) conjugate vaccines.
Given the case above, other pertinent facts are that uncircumcised males have more UTIs in the first months of life (J. Pediatr. 1996;128:23-7) and approximately 5% of young infants with UTIs also are concurrently bacteremic (Pediatrics 1999;104:79-86;J. Pediatr. 1994;124:513-9)
The elephant in the room is the fact that we also need to be cognizant of asymptomatic bacteriuria (AB). AB is colonization of the lower urinary tract without infection. Patients with AB may meet culture criteria for UTI (whether we consider > 50,000 or > 100,000 colony-forming units/mL), but there is no evidence of true infection, that is no inflammation or mucosal injury. So children with AB are not at risk for renal injury or later renal damage and do not require antibiotic treatment.
But when AB patients develop fever, for example with an enterovirus infection, their urine cultures (together with the fever) can do a good imitation of a UTI, unless we focus on the UA results. It not only remains critical to detect true UTIs in infants < 90 days old, such as the one in our case above, but also to distinguish UTI from AB.
The 1999 American Academy of Pediatrics’ UTI guidelines (Pediatrics 1999;103:843-52) included UA results as suggestive of UTI. They stated that a positive LE or nitrite test or > 5 WBC/HPF in a spun urine, or bacteria visualized in unspun gram-stained specimen suggest, but cannot be diagnostic of a UTI. Recommendation five in the guidelines states that UTI diagnosis required 100,000 CFU/mL in culture of sterilely obtained catheterized urine as the threshold criterion (strength of evidence: strong). However, AB was not fully considered because, in part, data defining AB was incomplete in 1999.
The 1999 guidelines also stated, “The urinalysis … can be valuable in selecting individuals for prompt initiation of treatment while waiting for the results of the urine culture.” So, UA was considered adjunctive. UA’s main function was to allow empiric therapy of sufficiently ill children, given positive results for LE, nitrites, or microscopic visualization of > 5 WBC/HPF or bacteria in the spun urine.
In the 2011 AAP guidelines for UTI, things have changed (Pediatrics 2011;128:595-610). The third action statement tells us that both the UA and culture taken together are necessary for UTI diagnosis. To paraphrase: The diagnosis of UTI requires urinalysis results suggesting infection (pyuria or microscopic bacteriuria) plus > 50,000 CFU/mL of a uropathogen in urine from catheterization or suprapubic aspiration. But remember that these guidelines do not apply specifically to the youngest of infants, that is < 2 months old.
Both of these criteria were changes from the 1999 UTI guidelines. Previously pyuria or microscopic bacteriuria were not considered necessary to diagnose UTI, and >100,000 CFU/mL rather than > 50,000 CFU/mL of a single pathogen species was the critical diagnostic result for catheterized urine. For suprapubic aspiration urine samples, > 10,000 CFU/HPF were considered adequate for UTI diagnoses in 1999.
Now, a recent study of children < 90 days of age (including those < 2 months of age) reports that pyuria (> 3 WBC/HPF) plus > 50,000 CFU/mL are the keys to diagnosing UTI (Pediatrics 2015;135:965-71). One caveat is that the study population was febrile infants < 90 days old with concurrent bacteremia (bacteremic UTI). Bacteremic UTI was studied to reduce as much as possible the chance that AB patients might be inadvertently included in the study. One other conclusion of this new study is that microscopic bacteriuria did not add significantly to the either sensitivity or specificity.
These data in an overall younger population than that covered by the 2011 guidelines adds evidence that pyuria (but not microscopic bacteriuria) is critical to diagnosing UTI. Pyuria plus positive culture has been a combination for the pediatric infectious diseases practitioner’s toolkit for decades. Likewise, it seems to me that primary care pediatric clinicians also often decide whether to undertake the expense of culture based on UA results. For example, a completely normal UA may obviate need for culture except in selected unusual cases.
Requiring UA evidence of inflammation to diagnose UTI (per the 2011 guidelines and the recommendations of the authors of the recent 2015 study) makes sense because most UTIs in otherwise healthy children are caused by gram-negative organisms (> 90% from Escherichia coli) (J. Pediatr. 1994;124:513-9). Why are UA results so important?
A positive nitrite test strongly suggests UTI because nitrites in the urine indicate viable gram-negative organisms also are present in the urine. Nitrates in the urine are converted to nitrites by metabolic activity of gram-negative pathogens. For WBCs or LE in the urine, their presence indicates inflammation in the urinary tract, Consider that lipopolysaccharide (LPS), also known as gram-negative endotoxin, is a major component of the cell membrane of > 90% of uropathogens like E. coli. Moreover, LPS elicits about the strongest innate immune response via toll-like receptor 4 (TLR4) from monocytes/macrophages, inducing a large pro-inflammatory and chemotactic response – interleukin-6, interleukin-8, tetrahydrofuran-alpha. Remember that LPS is also a major cause of fever and of shock during gram-negative sepsis.
So a UTI diagnosis based on a “positive” culture without evidence of metabolic products of gram negatives (nitrites) or without inflammation (no pyuria or negative LE) should be questioned. The combination of > 50,000 CFU/mL with no detectable LE or < 3-5 WBC/HPF in a febrile child is most likely evidence for AB in a child with the fever caused by some non-UTI process.
In contrast, selected SBIs may occur when the culture is “positive” without inflammation or nitrites. The first of three examples is a renal parenchymal abscess, where bacteria enter the urine sporadically in only small numbers, and do not actually infect the urinary tract mucosa. The scenario of no inflammation but “positive” culture also may occur when a large bacteremic load causes results in organisms filtering through the kidney into the urine, again without urinary mucosal infection, such as Staphylococcus aureus, group A streptococcus, or group B streptococcus bacteremia/sepsis. The third scenario with a “positive” culture and no pyuria can be with organisms that have blunted abilities to induce inflammation, such as enterococcus. Enterococcal cell components have weak inflammatory and chemotactic capability. So a urinary mucosal infection in the collecting system or bladder may occur without much if any pyuria. In fact, the patients from the recent study with insufficient evidence of pyuria/inflammation were those who had either gram-positive organisms or considerably less than 50,000 CFU/mL of gram-negative organisms.
The sensitivity and specificity of the LE or pyuria was higher in the recent study (Pediatrics 2015;135:965-71) than any prior study. The authors comment that they had not expected such a high sensitivity of 97.6% (94.5-99.2) for LE in confirmed bacteremic UTI, nor did they expect the high specificity of 93.9% (87.9-97.5). The presence of microscopic pyuria defined as > 3 WBC/HPF was nearly as sensitive, 96%, and specific, 91.3%. Disappointingly, positive nitrite testing was only 39.5% sensitive, but it was 100% specific. This likely reflects the short time that urine resides in the bladder of infants < 90 days of age, so there is insufficient time for the pathogens to metabolically convert the nitrates to nitrites.
So how would the UA help with our example case? There is microscopic bacteriuria, pyuria, and positive LE, but negative nitrites. Using the suggestions of the authors of the recent report (Pediatrics 2015;135:965-71) and those of another report on the utility of UA results (Acta Paediatr. 2010;99:581-4), the UA in our case indicates that we should be highly suspicious of a UTI in this child < 2 months old for whom the 2011 guidelines do not directly apply. But remember that these impressive sensitivity and specificity values relate to bacteremic UTI. Whether they apply to nonbacteremic UTI is not known. Likewise, the authors caution that their study design did not allow calculation of positive or negative predictive values – aspects that would clarify things even further.
So we still cannot be more than highly suspicious. Without a positive predictive value, we do not know the odds of this case having a UTI with mathematical precision. The authors do point out that only one of their subjects had a completely normal UA and actually had a bacteremic UTI. If you guessed that it was a gram-positive pathogen, you win the prize. So it seems reasonable to predict that a normal UA has a high specificity for not being a UTI (87.8%), but a positive UA remains only highly suggestive. It is still not clear if a negative UA statistically justifies not submitting the culture of the sterilely obtained urine because we still don’t have a negative predictive value.
Bottom line: The 2011 UTI guidelines provide good advice on diagnosing UTIs.
1. We have more data that evidence of inflammation is essential for diagnosing gram-negative UTIs.
2. We also have more evidence that 50,000 CFU/mL is a good threshold for diagnosing UTIs.
3. It appears that microscopic bacteriuria did not add significantly to the either sensitivity or specificity.
4. And we now have more evidence that these criteria also apply to infants < 2 months of age.
To close the loop on our case, the child’s CSF and blood cultures were negative, but the urine culture revealed > 100,000 CFU/mL of E. coli susceptible to second- and third-generation cephalosporins, ciprofloxacin, and nitrofurantoin, but resistant to trimethoprim-sulfamethoxazole.
Have a great summer and watch for UTIs in your young patients < 90 days old and fever without apparent focus.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. He said he had no relevant financial disclosures. E-mail Dr. Harrison at [email protected].

Pertussis persists
The Centers for Disease Control and Prevention suggests that recurring pertussis outbreaks may be the “new normal.” Such outbreaks show that some of what we “know” about pertussis is still correct, but some things are evolving. So in this new year, what do we need to know about patient vulnerability post vaccine as well as the clinical course, diagnosis, and treatment of this stubborn persisting disease?
Vulnerability after acellular pertussis vaccine
The recent large 2014 California outbreak surpassed the record numbers for the previously highest incidence year, 2010 (MMWR 2014;63:1129-32). This is scary because more cases had been reported in California in 2010 than in any prior year since the 1940s. The overall 2014 California pertussis rate (26/100,000 population) was approximately 10 times the national average for the first 9 years of this century, Are there clues as to who is most vulnerable and why?

No age group was spared, but certain age groups did appear more vulnerable. Infants had a startling 174.6/100,000 incidence (six times the rate for the overall population). It is not surprising to any clinician that infants less than 1 year of age were hardest hit. Infants have the most evident symptoms with pertussis. Infants also have 5-7 months of their first year in which they are incompletely immunized. Therefore, many are not expected to be protected until about 7-9 months of age. This vulnerability could be partly remedied if all pregnant women got Tdap boosters as recommended during pregnancy.
Of note, 15-year-olds had an incidence similar to that of infants (137.8/100,000). Ethnically, non-Hispanic whites had the highest incidence among adolescents (166.2/100,000), compared with Hispanics (64.2/100,000), Asian/Pacific Islanders (43.9/100,000), and non-Hispanic blacks (23.7/100,000). Disturbingly, 87% of cases among 15-year-olds had received a prior Tdap booster dose (median time since booster Tdap = 3 years, range = 0-7 years). Previous data from the 2010 outbreak suggested that immunity to pertussis wanes 3-4 years after receipt of the last acellular pertussis (aP)–containing vaccine. This is likely part of the explanation in 2014 as well. However, waning immunity after the booster does not explain why non-Hispanic whites had two to six times the incidence of other ethnicities. Non-Hispanic whites are thought to be the demographic with the most vaccine refusal and vaccine delay in California, so this may partially explain excess cases. Racial differences in access to care or genetic differences in disease susceptibility also may play a role.
Why is this biphasic increase in incidence in California a microcosm of the new epidemiology of pertussis in the United States? A kinder, gentler DTaP vaccine replaced the whole-cell DTP in the late 1990s. This occurred in response to the public’s concern about potential central nervous system adverse effects associated with the whole-cell DTP vaccine. Immunogenicity studies seemed to show equivalent immune responses in infants and toddlers receiving DTaP, compared with those who received DTP. It has only been in the last 5 years that we now know that the new DTaP and Tdap are not working as well as we had hoped.
The two aspects to the lesser protection provided by aP vaccines are pertactin-deficient pertussis strains and quicker waning of aP vaccine–induced immunity. Antibody to pertactin appears to be important in protection against clinical pertussis. New circulating clinical strains of pertussis may not have pertactin (N. Engl. J. Med. 2013;368:583-4). The strains used in our current DTaP and Tdap were designed to protect against pertactin-containing strains and were tested for this. This means that a proportion of the antibodies induced by vaccine strains are not useful against pertactin-deficient strains. The aP vaccine still induces antibody to the pertussis toxin and other pertussis components in the vaccines, so they will likely still reduce the severity of disease. But the vaccines are not likely to prevent infections from pertactin-deficient strains. This is similar to the partial vaccine mismatch that we are seeing with the current seasonal H3N2 influenza vaccine strain.
The second aspect is that protection appears to wane approximately 3-5 years after the last dose of aP-containing vaccine. This contrasts sharply with expectations in the past of 7-10 years of protection from whole cell pertussis–containing vaccines. The less reactive aP vaccine produces fewer adverse effects by not producing as much inflammation as DPT. The problem is that part of the reason the DPT has such good protective responses is the amount of inflammation it produces. So with less aP vaccine–induced inflammation comes less robust antibody and T-cell responses.
Nevertheless, the current acellular pertussis vaccines remain the most effective available tools to reduce pertussis disease (Cochrane Database Syst. Rev. 2014;9:CD001478]). But until we have new versions of pertussis vaccines that address these two issues, we clinicians need to remain vigilant for signs and symptoms of pertussis.
Clinical course
Remember that a whoop is rarely seen in young children and often also not seen when older patients present. The many outbreaks over the last 10 years have confirmed that paroxysmal cough with/without apnea in an infant/toddler should raise our index of suspicion. Likewise, older children, adolescents, and adults with persistent cough beyond 2 weeks are potential pertussis cases. Once the diagnosis is made, treatment is not expected to have a major impact on the clinical course, in part because the diagnosis is usually delayed (more than 10 days into symptoms). This delay allows more injury to the respiratory mucosa and cilia so that healing can require 6-12 weeks after bacterial replication ceases. This prolonged healing process is what is mostly responsible for the syndrome known as the “100-day cough.” So the clinical course of pertussis has not changed in the last 10 years. However, there have been changes in the commonly used diagnostic approach.
Pertussis diagnosis and contagion
During the last 5 years, polymerase chain reaction (PCR) testing has become the preferred technology to detect pertussis. This is due to the sensitivity and quick turnaround time of the assay. The gold standard for pertussis diagnosis remains culture, but it is expensive, cumbersome, and slow (up to a week to provide results). An ongoing debate arose about how long PCR testing remains positive after the onset of symptoms or treatment. This was not the problem when culture was the diagnostic tool of choice. Data from the 1970s and 1980s indicated that cultures were rarely positive after the third week of symptoms even without treatment. Furthermore, macrolides eliminated both contagion and positive culture results of infected patients after 5 days of treatment.
So now that we use PCR most often for diagnosis, what is the outer limit of positivity? A recent prospective cohort study from Salt Lake City suggests that PCR may detect pertussis DNA way beyond 3 weeks after symptom onset (J. Ped. Infect. Dis. 2014;3:347-9). Among patients hospitalized with laboratory-confirmed Bordetella pertussis infection, half had persistently positive pertussis PCR testing more than 50 days after symptom onset, despite antibiotic treatment and clinical improvement. The median (range) for the last day for a positive test after symptom onset was 58 days (4-172 days).
This raises the question as to whether there are viable pertussis organisms in the respiratory tract beyond the traditional 3 weeks defined by culture data. It is likely that DNA persists in the thick mucus of the respiratory tract way beyond viability of the last pertussis organisms. Put another way, PCR likely detects bacterial corpses or components way beyond the time that the patient is contagious. Unfortunately, current PCR data do not tell us how long patients remain contagious with the current strains of pertussis as infecting agents. Some institutions appear to be extending the isolation time for patients treated for pertussis beyond the traditional 5 days post initiation of effective treatment. Until more data are available, we are somewhat in the dark. But I would take comfort in the fact that it is unlikely the “new” data will be much different from those derived from the traditional studies that use culture to define infectivity. The American Academy of Pediatrics Committee on Infectious Diseases Red Book appears to agree.
For hospitalized pertussis patients, the AAP Committee on Infectious Diseases Red Book recommends standard and droplet precautions for 5 days after starting effective therapy, or 3 weeks after cough onset if appropriate antimicrobial therapy has not been given.
In addition, the CDC states: “PCR has optimal sensitivity during the first 3 weeks of cough when bacterial DNA is still present in the nasopharynx. After the fourth week of cough, the amount of bacterial DNA rapidly diminishes, which increases the risk of obtaining falsely negative results.” Later in the same document, the CDC says: “PCR testing following antibiotic therapy also can result in falsely negative findings. The exact duration of positivity following antibiotic use is not well understood, but PCR testing after 5 days of antibiotic use is unlikely to be of benefit and is generally not recommended.”
So what do we know? Not all PCR assays use the same primers, so some variance from the usual experience of up to 4 weeks of positive PCR results may be due to differences in the assays. But this raises concern that the PCR that you order may be positive at times when the patient is no longer contagious.
Pertussis treatment
If strains of pertussis have changed their pertactin antigen, are they changing their antibiotic susceptibility patterns? While there have been reports of macrolide resistance in a few pertussis strains, these still remain rare. The most recent comprehensive review of treatment efficacy was a Cochrane review performed in 2005 and published in 2007 (Cochrane Database Syst. Rev. 2007;3:CD004404). They evaluated 10 trials from 1969 to 2004 in which microbiologic eradication was defined by negative results from repeat pertussis culture. While meta-analysis of microbiologic eradication was not possible because of differences in antibiotic use, the investigators did conclude that antibiotic treatment “is effective in eliminating B. pertussis from patients with the disease to render them noninfectious, but does not alter the subsequent clinical course of the illness.”
Further, they state that “the best regimens for microbiologic clearance, with fewer side effects,” are 3 days of azithromycin (a single 10-mg/kg dose on 3 consecutive days) or 7 days of clarithromycin (7.5-mg/kg dose twice daily).
Another effective regimen is 14 days of erythromycin ethylsuccinate (60 mg/kg per day in 3 divided doses) .
CDC treatment recommendations include azithromycin or erythromycin, with trimethoprim-sulfamethoxazole as a possibility for macrolide-intolerant patients, although there are less data and success rates may not be as high.
Conclusion
So what do we know now about pertussis?
• Outbreaks are ongoing and likely will continue until newer more effective vaccines are produced, including those that circumvent the problem of pertactin-deficient strains.
• Pertussis is likely contagious up to 5 days on effective therapy, and for as long as 3 weeks if effective therapy has not been administered.
• PCR is a sensitive test that may remain positive for many weeks beyond contagion.
• Treatment with macrolides appears to be the most effective way to eradicate replicating pertussis pathogens.
• Treatment is not likely to have a major impact on the clinical course of disease because most of the damage to the respiratory tract is done prior to diagnosis and treatment. Treatment does reduce infectivity and subsequent cases.
• Current aP vaccines currently are our best preventative tools – including use in pregnant women to protect young infants.
As clinicians, our best course is to continue to immunize with the current vaccines, and remain vigilant for symptoms and signs of pertussis infection of patients so that early diagnosis and treatment can prevent further spread.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. Children’s Mercy Hospitals receives funds from GlaxoSmithKline for Dr. Harrison being principal investigator on a multicenter research study of a hexavalent pertussis-containing infant vaccine. E-mail Dr. Harrison at [email protected].
The Centers for Disease Control and Prevention suggests that recurring pertussis outbreaks may be the “new normal.” Such outbreaks show that some of what we “know” about pertussis is still correct, but some things are evolving. So in this new year, what do we need to know about patient vulnerability post vaccine as well as the clinical course, diagnosis, and treatment of this stubborn persisting disease?
Vulnerability after acellular pertussis vaccine
The recent large 2014 California outbreak surpassed the record numbers for the previously highest incidence year, 2010 (MMWR 2014;63:1129-32). This is scary because more cases had been reported in California in 2010 than in any prior year since the 1940s. The overall 2014 California pertussis rate (26/100,000 population) was approximately 10 times the national average for the first 9 years of this century, Are there clues as to who is most vulnerable and why?
No age group was spared, but certain age groups did appear more vulnerable. Infants had a startling 174.6/100,000 incidence (six times the rate for the overall population). It is not surprising to any clinician that infants less than 1 year of age were hardest hit. Infants have the most evident symptoms with pertussis. Infants also have 5-7 months of their first year in which they are incompletely immunized. Therefore, many are not expected to be protected until about 7-9 months of age. This vulnerability could be partly remedied if all pregnant women got Tdap boosters as recommended during pregnancy.
Of note, 15-year-olds had an incidence similar to that of infants (137.8/100,000). Ethnically, non-Hispanic whites had the highest incidence among adolescents (166.2/100,000), compared with Hispanics (64.2/100,000), Asian/Pacific Islanders (43.9/100,000), and non-Hispanic blacks (23.7/100,000). Disturbingly, 87% of cases among 15-year-olds had received a prior Tdap booster dose (median time since booster Tdap = 3 years, range = 0-7 years). Previous data from the 2010 outbreak suggested that immunity to pertussis wanes 3-4 years after receipt of the last acellular pertussis (aP)–containing vaccine. This is likely part of the explanation in 2014 as well. However, waning immunity after the booster does not explain why non-Hispanic whites had two to six times the incidence of other ethnicities. Non-Hispanic whites are thought to be the demographic with the most vaccine refusal and vaccine delay in California, so this may partially explain excess cases. Racial differences in access to care or genetic differences in disease susceptibility also may play a role.
Why is this biphasic increase in incidence in California a microcosm of the new epidemiology of pertussis in the United States? A kinder, gentler DTaP vaccine replaced the whole-cell DTP in the late 1990s. This occurred in response to the public’s concern about potential central nervous system adverse effects associated with the whole-cell DTP vaccine. Immunogenicity studies seemed to show equivalent immune responses in infants and toddlers receiving DTaP, compared with those who received DTP. It has only been in the last 5 years that we now know that the new DTaP and Tdap are not working as well as we had hoped.
The two aspects to the lesser protection provided by aP vaccines are pertactin-deficient pertussis strains and quicker waning of aP vaccine–induced immunity. Antibody to pertactin appears to be important in protection against clinical pertussis. New circulating clinical strains of pertussis may not have pertactin (N. Engl. J. Med. 2013;368:583-4). The strains used in our current DTaP and Tdap were designed to protect against pertactin-containing strains and were tested for this. This means that a proportion of the antibodies induced by vaccine strains are not useful against pertactin-deficient strains. The aP vaccine still induces antibody to the pertussis toxin and other pertussis components in the vaccines, so they will likely still reduce the severity of disease. But the vaccines are not likely to prevent infections from pertactin-deficient strains. This is similar to the partial vaccine mismatch that we are seeing with the current seasonal H3N2 influenza vaccine strain.
The second aspect is that protection appears to wane approximately 3-5 years after the last dose of aP-containing vaccine. This contrasts sharply with expectations in the past of 7-10 years of protection from whole cell pertussis–containing vaccines. The less reactive aP vaccine produces fewer adverse effects by not producing as much inflammation as DPT. The problem is that part of the reason the DPT has such good protective responses is the amount of inflammation it produces. So with less aP vaccine–induced inflammation comes less robust antibody and T-cell responses.
Nevertheless, the current acellular pertussis vaccines remain the most effective available tools to reduce pertussis disease (Cochrane Database Syst. Rev. 2014;9:CD001478]). But until we have new versions of pertussis vaccines that address these two issues, we clinicians need to remain vigilant for signs and symptoms of pertussis.
Clinical course
Remember that a whoop is rarely seen in young children and often also not seen when older patients present. The many outbreaks over the last 10 years have confirmed that paroxysmal cough with/without apnea in an infant/toddler should raise our index of suspicion. Likewise, older children, adolescents, and adults with persistent cough beyond 2 weeks are potential pertussis cases. Once the diagnosis is made, treatment is not expected to have a major impact on the clinical course, in part because the diagnosis is usually delayed (more than 10 days into symptoms). This delay allows more injury to the respiratory mucosa and cilia so that healing can require 6-12 weeks after bacterial replication ceases. This prolonged healing process is what is mostly responsible for the syndrome known as the “100-day cough.” So the clinical course of pertussis has not changed in the last 10 years. However, there have been changes in the commonly used diagnostic approach.
Pertussis diagnosis and contagion
During the last 5 years, polymerase chain reaction (PCR) testing has become the preferred technology to detect pertussis. This is due to the sensitivity and quick turnaround time of the assay. The gold standard for pertussis diagnosis remains culture, but it is expensive, cumbersome, and slow (up to a week to provide results). An ongoing debate arose about how long PCR testing remains positive after the onset of symptoms or treatment. This was not the problem when culture was the diagnostic tool of choice. Data from the 1970s and 1980s indicated that cultures were rarely positive after the third week of symptoms even without treatment. Furthermore, macrolides eliminated both contagion and positive culture results of infected patients after 5 days of treatment.
So now that we use PCR most often for diagnosis, what is the outer limit of positivity? A recent prospective cohort study from Salt Lake City suggests that PCR may detect pertussis DNA way beyond 3 weeks after symptom onset (J. Ped. Infect. Dis. 2014;3:347-9). Among patients hospitalized with laboratory-confirmed Bordetella pertussis infection, half had persistently positive pertussis PCR testing more than 50 days after symptom onset, despite antibiotic treatment and clinical improvement. The median (range) for the last day for a positive test after symptom onset was 58 days (4-172 days).
This raises the question as to whether there are viable pertussis organisms in the respiratory tract beyond the traditional 3 weeks defined by culture data. It is likely that DNA persists in the thick mucus of the respiratory tract way beyond viability of the last pertussis organisms. Put another way, PCR likely detects bacterial corpses or components way beyond the time that the patient is contagious. Unfortunately, current PCR data do not tell us how long patients remain contagious with the current strains of pertussis as infecting agents. Some institutions appear to be extending the isolation time for patients treated for pertussis beyond the traditional 5 days post initiation of effective treatment. Until more data are available, we are somewhat in the dark. But I would take comfort in the fact that it is unlikely the “new” data will be much different from those derived from the traditional studies that use culture to define infectivity. The American Academy of Pediatrics Committee on Infectious Diseases Red Book appears to agree.
For hospitalized pertussis patients, the AAP Committee on Infectious Diseases Red Book recommends standard and droplet precautions for 5 days after starting effective therapy, or 3 weeks after cough onset if appropriate antimicrobial therapy has not been given.
In addition, the CDC states: “PCR has optimal sensitivity during the first 3 weeks of cough when bacterial DNA is still present in the nasopharynx. After the fourth week of cough, the amount of bacterial DNA rapidly diminishes, which increases the risk of obtaining falsely negative results.” Later in the same document, the CDC says: “PCR testing following antibiotic therapy also can result in falsely negative findings. The exact duration of positivity following antibiotic use is not well understood, but PCR testing after 5 days of antibiotic use is unlikely to be of benefit and is generally not recommended.”
So what do we know? Not all PCR assays use the same primers, so some variance from the usual experience of up to 4 weeks of positive PCR results may be due to differences in the assays. But this raises concern that the PCR that you order may be positive at times when the patient is no longer contagious.
Pertussis treatment
If strains of pertussis have changed their pertactin antigen, are they changing their antibiotic susceptibility patterns? While there have been reports of macrolide resistance in a few pertussis strains, these still remain rare. The most recent comprehensive review of treatment efficacy was a Cochrane review performed in 2005 and published in 2007 (Cochrane Database Syst. Rev. 2007;3:CD004404). They evaluated 10 trials from 1969 to 2004 in which microbiologic eradication was defined by negative results from repeat pertussis culture. While meta-analysis of microbiologic eradication was not possible because of differences in antibiotic use, the investigators did conclude that antibiotic treatment “is effective in eliminating B. pertussis from patients with the disease to render them noninfectious, but does not alter the subsequent clinical course of the illness.”
Further, they state that “the best regimens for microbiologic clearance, with fewer side effects,” are 3 days of azithromycin (a single 10-mg/kg dose on 3 consecutive days) or 7 days of clarithromycin (7.5-mg/kg dose twice daily).
Another effective regimen is 14 days of erythromycin ethylsuccinate (60 mg/kg per day in 3 divided doses) .
CDC treatment recommendations include azithromycin or erythromycin, with trimethoprim-sulfamethoxazole as a possibility for macrolide-intolerant patients, although there are less data and success rates may not be as high.
Conclusion
So what do we know now about pertussis?
• Outbreaks are ongoing and likely will continue until newer more effective vaccines are produced, including those that circumvent the problem of pertactin-deficient strains.
• Pertussis is likely contagious up to 5 days on effective therapy, and for as long as 3 weeks if effective therapy has not been administered.
• PCR is a sensitive test that may remain positive for many weeks beyond contagion.
• Treatment with macrolides appears to be the most effective way to eradicate replicating pertussis pathogens.
• Treatment is not likely to have a major impact on the clinical course of disease because most of the damage to the respiratory tract is done prior to diagnosis and treatment. Treatment does reduce infectivity and subsequent cases.
• Current aP vaccines currently are our best preventative tools – including use in pregnant women to protect young infants.
As clinicians, our best course is to continue to immunize with the current vaccines, and remain vigilant for symptoms and signs of pertussis infection of patients so that early diagnosis and treatment can prevent further spread.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. Children’s Mercy Hospitals receives funds from GlaxoSmithKline for Dr. Harrison being principal investigator on a multicenter research study of a hexavalent pertussis-containing infant vaccine. E-mail Dr. Harrison at [email protected].
The Centers for Disease Control and Prevention suggests that recurring pertussis outbreaks may be the “new normal.” Such outbreaks show that some of what we “know” about pertussis is still correct, but some things are evolving. So in this new year, what do we need to know about patient vulnerability post vaccine as well as the clinical course, diagnosis, and treatment of this stubborn persisting disease?
Vulnerability after acellular pertussis vaccine
The recent large 2014 California outbreak surpassed the record numbers for the previously highest incidence year, 2010 (MMWR 2014;63:1129-32). This is scary because more cases had been reported in California in 2010 than in any prior year since the 1940s. The overall 2014 California pertussis rate (26/100,000 population) was approximately 10 times the national average for the first 9 years of this century, Are there clues as to who is most vulnerable and why?
No age group was spared, but certain age groups did appear more vulnerable. Infants had a startling 174.6/100,000 incidence (six times the rate for the overall population). It is not surprising to any clinician that infants less than 1 year of age were hardest hit. Infants have the most evident symptoms with pertussis. Infants also have 5-7 months of their first year in which they are incompletely immunized. Therefore, many are not expected to be protected until about 7-9 months of age. This vulnerability could be partly remedied if all pregnant women got Tdap boosters as recommended during pregnancy.
Of note, 15-year-olds had an incidence similar to that of infants (137.8/100,000). Ethnically, non-Hispanic whites had the highest incidence among adolescents (166.2/100,000), compared with Hispanics (64.2/100,000), Asian/Pacific Islanders (43.9/100,000), and non-Hispanic blacks (23.7/100,000). Disturbingly, 87% of cases among 15-year-olds had received a prior Tdap booster dose (median time since booster Tdap = 3 years, range = 0-7 years). Previous data from the 2010 outbreak suggested that immunity to pertussis wanes 3-4 years after receipt of the last acellular pertussis (aP)–containing vaccine. This is likely part of the explanation in 2014 as well. However, waning immunity after the booster does not explain why non-Hispanic whites had two to six times the incidence of other ethnicities. Non-Hispanic whites are thought to be the demographic with the most vaccine refusal and vaccine delay in California, so this may partially explain excess cases. Racial differences in access to care or genetic differences in disease susceptibility also may play a role.
Why is this biphasic increase in incidence in California a microcosm of the new epidemiology of pertussis in the United States? A kinder, gentler DTaP vaccine replaced the whole-cell DTP in the late 1990s. This occurred in response to the public’s concern about potential central nervous system adverse effects associated with the whole-cell DTP vaccine. Immunogenicity studies seemed to show equivalent immune responses in infants and toddlers receiving DTaP, compared with those who received DTP. It has only been in the last 5 years that we now know that the new DTaP and Tdap are not working as well as we had hoped.
The two aspects to the lesser protection provided by aP vaccines are pertactin-deficient pertussis strains and quicker waning of aP vaccine–induced immunity. Antibody to pertactin appears to be important in protection against clinical pertussis. New circulating clinical strains of pertussis may not have pertactin (N. Engl. J. Med. 2013;368:583-4). The strains used in our current DTaP and Tdap were designed to protect against pertactin-containing strains and were tested for this. This means that a proportion of the antibodies induced by vaccine strains are not useful against pertactin-deficient strains. The aP vaccine still induces antibody to the pertussis toxin and other pertussis components in the vaccines, so they will likely still reduce the severity of disease. But the vaccines are not likely to prevent infections from pertactin-deficient strains. This is similar to the partial vaccine mismatch that we are seeing with the current seasonal H3N2 influenza vaccine strain.
The second aspect is that protection appears to wane approximately 3-5 years after the last dose of aP-containing vaccine. This contrasts sharply with expectations in the past of 7-10 years of protection from whole cell pertussis–containing vaccines. The less reactive aP vaccine produces fewer adverse effects by not producing as much inflammation as DPT. The problem is that part of the reason the DPT has such good protective responses is the amount of inflammation it produces. So with less aP vaccine–induced inflammation comes less robust antibody and T-cell responses.
Nevertheless, the current acellular pertussis vaccines remain the most effective available tools to reduce pertussis disease (Cochrane Database Syst. Rev. 2014;9:CD001478]). But until we have new versions of pertussis vaccines that address these two issues, we clinicians need to remain vigilant for signs and symptoms of pertussis.
Clinical course
Remember that a whoop is rarely seen in young children and often also not seen when older patients present. The many outbreaks over the last 10 years have confirmed that paroxysmal cough with/without apnea in an infant/toddler should raise our index of suspicion. Likewise, older children, adolescents, and adults with persistent cough beyond 2 weeks are potential pertussis cases. Once the diagnosis is made, treatment is not expected to have a major impact on the clinical course, in part because the diagnosis is usually delayed (more than 10 days into symptoms). This delay allows more injury to the respiratory mucosa and cilia so that healing can require 6-12 weeks after bacterial replication ceases. This prolonged healing process is what is mostly responsible for the syndrome known as the “100-day cough.” So the clinical course of pertussis has not changed in the last 10 years. However, there have been changes in the commonly used diagnostic approach.
Pertussis diagnosis and contagion
During the last 5 years, polymerase chain reaction (PCR) testing has become the preferred technology to detect pertussis. This is due to the sensitivity and quick turnaround time of the assay. The gold standard for pertussis diagnosis remains culture, but it is expensive, cumbersome, and slow (up to a week to provide results). An ongoing debate arose about how long PCR testing remains positive after the onset of symptoms or treatment. This was not the problem when culture was the diagnostic tool of choice. Data from the 1970s and 1980s indicated that cultures were rarely positive after the third week of symptoms even without treatment. Furthermore, macrolides eliminated both contagion and positive culture results of infected patients after 5 days of treatment.
So now that we use PCR most often for diagnosis, what is the outer limit of positivity? A recent prospective cohort study from Salt Lake City suggests that PCR may detect pertussis DNA way beyond 3 weeks after symptom onset (J. Ped. Infect. Dis. 2014;3:347-9). Among patients hospitalized with laboratory-confirmed Bordetella pertussis infection, half had persistently positive pertussis PCR testing more than 50 days after symptom onset, despite antibiotic treatment and clinical improvement. The median (range) for the last day for a positive test after symptom onset was 58 days (4-172 days).
This raises the question as to whether there are viable pertussis organisms in the respiratory tract beyond the traditional 3 weeks defined by culture data. It is likely that DNA persists in the thick mucus of the respiratory tract way beyond viability of the last pertussis organisms. Put another way, PCR likely detects bacterial corpses or components way beyond the time that the patient is contagious. Unfortunately, current PCR data do not tell us how long patients remain contagious with the current strains of pertussis as infecting agents. Some institutions appear to be extending the isolation time for patients treated for pertussis beyond the traditional 5 days post initiation of effective treatment. Until more data are available, we are somewhat in the dark. But I would take comfort in the fact that it is unlikely the “new” data will be much different from those derived from the traditional studies that use culture to define infectivity. The American Academy of Pediatrics Committee on Infectious Diseases Red Book appears to agree.
For hospitalized pertussis patients, the AAP Committee on Infectious Diseases Red Book recommends standard and droplet precautions for 5 days after starting effective therapy, or 3 weeks after cough onset if appropriate antimicrobial therapy has not been given.
In addition, the CDC states: “PCR has optimal sensitivity during the first 3 weeks of cough when bacterial DNA is still present in the nasopharynx. After the fourth week of cough, the amount of bacterial DNA rapidly diminishes, which increases the risk of obtaining falsely negative results.” Later in the same document, the CDC says: “PCR testing following antibiotic therapy also can result in falsely negative findings. The exact duration of positivity following antibiotic use is not well understood, but PCR testing after 5 days of antibiotic use is unlikely to be of benefit and is generally not recommended.”
So what do we know? Not all PCR assays use the same primers, so some variance from the usual experience of up to 4 weeks of positive PCR results may be due to differences in the assays. But this raises concern that the PCR that you order may be positive at times when the patient is no longer contagious.
Pertussis treatment
If strains of pertussis have changed their pertactin antigen, are they changing their antibiotic susceptibility patterns? While there have been reports of macrolide resistance in a few pertussis strains, these still remain rare. The most recent comprehensive review of treatment efficacy was a Cochrane review performed in 2005 and published in 2007 (Cochrane Database Syst. Rev. 2007;3:CD004404). They evaluated 10 trials from 1969 to 2004 in which microbiologic eradication was defined by negative results from repeat pertussis culture. While meta-analysis of microbiologic eradication was not possible because of differences in antibiotic use, the investigators did conclude that antibiotic treatment “is effective in eliminating B. pertussis from patients with the disease to render them noninfectious, but does not alter the subsequent clinical course of the illness.”
Further, they state that “the best regimens for microbiologic clearance, with fewer side effects,” are 3 days of azithromycin (a single 10-mg/kg dose on 3 consecutive days) or 7 days of clarithromycin (7.5-mg/kg dose twice daily).
Another effective regimen is 14 days of erythromycin ethylsuccinate (60 mg/kg per day in 3 divided doses) .
CDC treatment recommendations include azithromycin or erythromycin, with trimethoprim-sulfamethoxazole as a possibility for macrolide-intolerant patients, although there are less data and success rates may not be as high.
Conclusion
So what do we know now about pertussis?
• Outbreaks are ongoing and likely will continue until newer more effective vaccines are produced, including those that circumvent the problem of pertactin-deficient strains.
• Pertussis is likely contagious up to 5 days on effective therapy, and for as long as 3 weeks if effective therapy has not been administered.
• PCR is a sensitive test that may remain positive for many weeks beyond contagion.
• Treatment with macrolides appears to be the most effective way to eradicate replicating pertussis pathogens.
• Treatment is not likely to have a major impact on the clinical course of disease because most of the damage to the respiratory tract is done prior to diagnosis and treatment. Treatment does reduce infectivity and subsequent cases.
• Current aP vaccines currently are our best preventative tools – including use in pregnant women to protect young infants.
As clinicians, our best course is to continue to immunize with the current vaccines, and remain vigilant for symptoms and signs of pertussis infection of patients so that early diagnosis and treatment can prevent further spread.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. Children’s Mercy Hospitals receives funds from GlaxoSmithKline for Dr. Harrison being principal investigator on a multicenter research study of a hexavalent pertussis-containing infant vaccine. E-mail Dr. Harrison at [email protected].
