User login
FDA clears use of glucose testing system for critically ill
For the first time, a blood glucose monitoring system has been cleared for use in critically ill hospitalized patients, the Food and Drug Administration announced on Sept. 24.
The device is the Nova StatStrip Glucose Hospital Meter System, which was cleared for use in 2006 for use in hospitals, but not in critically ill patients. It is “the first FDA clearance of a device specifically indicated for use in all types of hospital patients, including critically ill patients,” according to the FDA statement.

The expanded use applies to indications using arterial or venous whole blood, from patients “in all areas of a hospital with various conditions, including: trauma, cancer, sepsis and infection; cardiac, kidney, neurological, obstetric, gynecological, gastroenterological, endocrine, and lung issues; and people recovering from general or cardiothoracic surgery,” the statement said.
The system is manufactured by Nova Biomedical.
For the first time, a blood glucose monitoring system has been cleared for use in critically ill hospitalized patients, the Food and Drug Administration announced on Sept. 24.
The device is the Nova StatStrip Glucose Hospital Meter System, which was cleared for use in 2006 for use in hospitals, but not in critically ill patients. It is “the first FDA clearance of a device specifically indicated for use in all types of hospital patients, including critically ill patients,” according to the FDA statement.
The expanded use applies to indications using arterial or venous whole blood, from patients “in all areas of a hospital with various conditions, including: trauma, cancer, sepsis and infection; cardiac, kidney, neurological, obstetric, gynecological, gastroenterological, endocrine, and lung issues; and people recovering from general or cardiothoracic surgery,” the statement said.
The system is manufactured by Nova Biomedical.
For the first time, a blood glucose monitoring system has been cleared for use in critically ill hospitalized patients, the Food and Drug Administration announced on Sept. 24.
The device is the Nova StatStrip Glucose Hospital Meter System, which was cleared for use in 2006 for use in hospitals, but not in critically ill patients. It is “the first FDA clearance of a device specifically indicated for use in all types of hospital patients, including critically ill patients,” according to the FDA statement.
The expanded use applies to indications using arterial or venous whole blood, from patients “in all areas of a hospital with various conditions, including: trauma, cancer, sepsis and infection; cardiac, kidney, neurological, obstetric, gynecological, gastroenterological, endocrine, and lung issues; and people recovering from general or cardiothoracic surgery,” the statement said.
The system is manufactured by Nova Biomedical.

Apremilast approval expanded to include plaque psoriasis
The oral phosphodiesterase-4 inhibitor apremilast is now indicated for the treatment of moderate to severe plaque psoriasis.
On Sept. 23, the manufacturer, Celgene, announced that the Food and Drug Administration had approved the expanded indication for apremilast, which was initially approved in March 2014 for treating psoriatic arthritis. The new indication is for the treatment of patients with moderate to severe plaque psoriasis who are candidates for phototherapy or systemic therapy.
Approval was primarily based on the results of two multicenter, randomized, double-blind, placebo-controlled studies of adults with moderate to severe plaque psoriasis, according to Celgene. At 16 weeks, 33% and 29% of those randomized to the 30-mg, twice daily dose of apremilast had achieved at least a 75% reduction in the Psoriasis Area and Severity Index (PASI 75), compared with 5%-6% of those on placebo, according to the prescribing information.
The most common adverse events reported in studies, which affected at least 1% of treated patients and were more common than in patients on placebo, included diarrhea in 17% and nausea in 17%; other adverse events included upper respiratory infection, tension headache, and headache. The warnings and precautions section of the label includes the recommendation to be alert for the emergence or worsening of depression, suicidal thoughts, or other mood changes in patients treated with the drug, and to monitor weight regularly for significant weight loss. In studies, treatment with apremilast has been associated with an increase in reports of depression and significant weight loss.
Celgene markets apremilast as Otezla.
Serious adverse events associated with treatment should be reported to the FDA at 800-332-1088 or www.fda.gov/medwatch.
Information about the pregnancy registry of women exposed to the drug during pregnancy can be obtained at 877-311-8972.
The updated prescribing information is available at http://www.celgene.com/content/uploads/2014/09/psoriasis-pi.pdf
The oral phosphodiesterase-4 inhibitor apremilast is now indicated for the treatment of moderate to severe plaque psoriasis.
On Sept. 23, the manufacturer, Celgene, announced that the Food and Drug Administration had approved the expanded indication for apremilast, which was initially approved in March 2014 for treating psoriatic arthritis. The new indication is for the treatment of patients with moderate to severe plaque psoriasis who are candidates for phototherapy or systemic therapy.
Approval was primarily based on the results of two multicenter, randomized, double-blind, placebo-controlled studies of adults with moderate to severe plaque psoriasis, according to Celgene. At 16 weeks, 33% and 29% of those randomized to the 30-mg, twice daily dose of apremilast had achieved at least a 75% reduction in the Psoriasis Area and Severity Index (PASI 75), compared with 5%-6% of those on placebo, according to the prescribing information.
The most common adverse events reported in studies, which affected at least 1% of treated patients and were more common than in patients on placebo, included diarrhea in 17% and nausea in 17%; other adverse events included upper respiratory infection, tension headache, and headache. The warnings and precautions section of the label includes the recommendation to be alert for the emergence or worsening of depression, suicidal thoughts, or other mood changes in patients treated with the drug, and to monitor weight regularly for significant weight loss. In studies, treatment with apremilast has been associated with an increase in reports of depression and significant weight loss.
Celgene markets apremilast as Otezla.
Serious adverse events associated with treatment should be reported to the FDA at 800-332-1088 or www.fda.gov/medwatch.
Information about the pregnancy registry of women exposed to the drug during pregnancy can be obtained at 877-311-8972.
The updated prescribing information is available at http://www.celgene.com/content/uploads/2014/09/psoriasis-pi.pdf
The oral phosphodiesterase-4 inhibitor apremilast is now indicated for the treatment of moderate to severe plaque psoriasis.
On Sept. 23, the manufacturer, Celgene, announced that the Food and Drug Administration had approved the expanded indication for apremilast, which was initially approved in March 2014 for treating psoriatic arthritis. The new indication is for the treatment of patients with moderate to severe plaque psoriasis who are candidates for phototherapy or systemic therapy.
Approval was primarily based on the results of two multicenter, randomized, double-blind, placebo-controlled studies of adults with moderate to severe plaque psoriasis, according to Celgene. At 16 weeks, 33% and 29% of those randomized to the 30-mg, twice daily dose of apremilast had achieved at least a 75% reduction in the Psoriasis Area and Severity Index (PASI 75), compared with 5%-6% of those on placebo, according to the prescribing information.
The most common adverse events reported in studies, which affected at least 1% of treated patients and were more common than in patients on placebo, included diarrhea in 17% and nausea in 17%; other adverse events included upper respiratory infection, tension headache, and headache. The warnings and precautions section of the label includes the recommendation to be alert for the emergence or worsening of depression, suicidal thoughts, or other mood changes in patients treated with the drug, and to monitor weight regularly for significant weight loss. In studies, treatment with apremilast has been associated with an increase in reports of depression and significant weight loss.
Celgene markets apremilast as Otezla.
Serious adverse events associated with treatment should be reported to the FDA at 800-332-1088 or www.fda.gov/medwatch.
Information about the pregnancy registry of women exposed to the drug during pregnancy can be obtained at 877-311-8972.
The updated prescribing information is available at http://www.celgene.com/content/uploads/2014/09/psoriasis-pi.pdf
Specific pattern of male baldness linked to aggressive prostate cancer
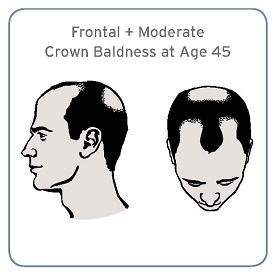
Men with a combination of frontal and moderate vertex baldness at age 45 were at a significantly increased risk of having aggressive prostate cancer in a large prospective cohort study, according to investigators.
“Although the effect is moderate, it supports the possibility of overlapping pathogenesis between male pattern baldness and prostate cancer,” Cindy Ke Zhou of the National Cancer Institute, Bethesda, Md., and her associates wrote in their analysis (Sept. 15, J. Clin. Onc. 2014 Sept. 15 [doi: 10.1200/JCO.2014.55.4279])
The results of previous studies evaluating the association between male pattern baldness and prostate cancer – mostly small cross-sectional or case-control studies – have been inconsistent. Unlike those studies, this study looked at subtypes of prostate cancer among 39,070 mostly white men enrolled at aged 55-74 in a large prospective cohort cancer screening study, who had not been diagnosed with cancer. About half reported being bald at age 45.
Over a median of almost 3 years, 1,138 prostate cancers were diagnosed, including 571 that were aggressive. There was no significant association between having frontal plus moderate vertex baldness (affecting both the front and the crown of the head) at age 45 years and prostate cancer overall (hazard ratio, 1.19) or with prostate cancer that was not aggressive (HR, 0.97), when compared with men who were not bald at that age.
However, compared with men who were not bald, this particular pattern of male baldness was associated with a nearly 40% increased risk of aggressive prostate cancer (HR, 1.39), which was statistically significant. There was no association with an increased risk for overall or any subtype of prostate cancer and other hair loss patterns.
“While our data show a strong possibility for a link between the development of baldness and aggressive prostate cancer, it’s too soon to apply these findings to patient care,” the study’s senior author, Michael Cook, Ph.D., of the NCI’s Division of Cancer Epidemiology and Genetics, said in a statement issued by the American Society of Clinical Oncology, the publisher of the journal. But if the results are confirmed, he added, baldness may be a possible way to help identify men who could be at an increased risk of having aggressive prostate cancer.
The study was funded by the NCI’s intramural program of the Division of Cancer Epidemiology and Genetics. None of the authors had disclosures.
Men with a combination of frontal and moderate vertex baldness at age 45 were at a significantly increased risk of having aggressive prostate cancer in a large prospective cohort study, according to investigators.
“Although the effect is moderate, it supports the possibility of overlapping pathogenesis between male pattern baldness and prostate cancer,” Cindy Ke Zhou of the National Cancer Institute, Bethesda, Md., and her associates wrote in their analysis (Sept. 15, J. Clin. Onc. 2014 Sept. 15 [doi: 10.1200/JCO.2014.55.4279])
The results of previous studies evaluating the association between male pattern baldness and prostate cancer – mostly small cross-sectional or case-control studies – have been inconsistent. Unlike those studies, this study looked at subtypes of prostate cancer among 39,070 mostly white men enrolled at aged 55-74 in a large prospective cohort cancer screening study, who had not been diagnosed with cancer. About half reported being bald at age 45.
Over a median of almost 3 years, 1,138 prostate cancers were diagnosed, including 571 that were aggressive. There was no significant association between having frontal plus moderate vertex baldness (affecting both the front and the crown of the head) at age 45 years and prostate cancer overall (hazard ratio, 1.19) or with prostate cancer that was not aggressive (HR, 0.97), when compared with men who were not bald at that age.
However, compared with men who were not bald, this particular pattern of male baldness was associated with a nearly 40% increased risk of aggressive prostate cancer (HR, 1.39), which was statistically significant. There was no association with an increased risk for overall or any subtype of prostate cancer and other hair loss patterns.
“While our data show a strong possibility for a link between the development of baldness and aggressive prostate cancer, it’s too soon to apply these findings to patient care,” the study’s senior author, Michael Cook, Ph.D., of the NCI’s Division of Cancer Epidemiology and Genetics, said in a statement issued by the American Society of Clinical Oncology, the publisher of the journal. But if the results are confirmed, he added, baldness may be a possible way to help identify men who could be at an increased risk of having aggressive prostate cancer.
The study was funded by the NCI’s intramural program of the Division of Cancer Epidemiology and Genetics. None of the authors had disclosures.
Men with a combination of frontal and moderate vertex baldness at age 45 were at a significantly increased risk of having aggressive prostate cancer in a large prospective cohort study, according to investigators.
“Although the effect is moderate, it supports the possibility of overlapping pathogenesis between male pattern baldness and prostate cancer,” Cindy Ke Zhou of the National Cancer Institute, Bethesda, Md., and her associates wrote in their analysis (Sept. 15, J. Clin. Onc. 2014 Sept. 15 [doi: 10.1200/JCO.2014.55.4279])
The results of previous studies evaluating the association between male pattern baldness and prostate cancer – mostly small cross-sectional or case-control studies – have been inconsistent. Unlike those studies, this study looked at subtypes of prostate cancer among 39,070 mostly white men enrolled at aged 55-74 in a large prospective cohort cancer screening study, who had not been diagnosed with cancer. About half reported being bald at age 45.
Over a median of almost 3 years, 1,138 prostate cancers were diagnosed, including 571 that were aggressive. There was no significant association between having frontal plus moderate vertex baldness (affecting both the front and the crown of the head) at age 45 years and prostate cancer overall (hazard ratio, 1.19) or with prostate cancer that was not aggressive (HR, 0.97), when compared with men who were not bald at that age.
However, compared with men who were not bald, this particular pattern of male baldness was associated with a nearly 40% increased risk of aggressive prostate cancer (HR, 1.39), which was statistically significant. There was no association with an increased risk for overall or any subtype of prostate cancer and other hair loss patterns.
“While our data show a strong possibility for a link between the development of baldness and aggressive prostate cancer, it’s too soon to apply these findings to patient care,” the study’s senior author, Michael Cook, Ph.D., of the NCI’s Division of Cancer Epidemiology and Genetics, said in a statement issued by the American Society of Clinical Oncology, the publisher of the journal. But if the results are confirmed, he added, baldness may be a possible way to help identify men who could be at an increased risk of having aggressive prostate cancer.
The study was funded by the NCI’s intramural program of the Division of Cancer Epidemiology and Genetics. None of the authors had disclosures.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: If the association between this specific pattern of male baldness and aggressive prostate cancer is confirmed, this feature could eventually be used to help identify men at increased risk for aggressive disease.
Major finding: Men with a combination of frontal and moderate vertex baldness were at a 40% increased risk of being diagnosed with aggressive prostate cancer.
Data source: Data on 39,070 men who had no cancer at baseline and remembered their hair loss pattern at age 45 years, who were enrolled in one of the arms of the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial.
Disclosures: The study was funded by National Cancer Institute’s intramural program of the Division of Cancer Epidemiology and Genetics. None of the authors had disclosures.
Dulaglutide injection approved for type 2 diabetes
Dulaglutide, a glucagon-like peptide-1 receptor agonist administered subcutaneously once a week, has been approved for treatment of adults with type 2 diabetes, the Food and Drug Administration announced Sept. 18.
It was approved with a Risk Evaluation and Mitigation Strategy (REMS) aimed at informing health care professionals about the serious risks of treatment and requirements for postmarketing studies that address some of those risks, the FDA said in a statement.
Approval was based on six studies of about 3,300 patients with type 2 diabetes who were treated with dulaglutide as monotherapy, and in combination with other approved type 2 diabetes treatments, including metformin. It will be marketed as Trulicity by Eli Lilly.
The drug’s label has a boxed warning that thyroid C-cell tumors have been observed in rodent studies of dulaglutide, “but that it is unknown whether Trulicity causes thyroid C-cell tumors,” including medullary thyroid carcinoma (MTC) in humans, the statement said. It should not be used to treat patients who have had, or have, a family history of MTC, or in patients who have multiple endocrine neoplasia syndrome type 2, who are predisposed to MTC, the statement noted.
Eli Lilly is required to conduct several postmarketing studies, including a cardiovascular outcomes study in patients at a high risk of cardiovascular disease at baseline, a pediatric study, and a registry study of MTC cases to help determine whether there is an increase in the incidence of MTC associated with the use of the drug, for at least 15 years. Another postmarketing study will compare dulaglutide to insulin glargine for glycemic control in patients with type 2 diabetes and moderate or severe renal impairment.
Dulaglutide, a glucagon-like peptide-1 receptor agonist administered subcutaneously once a week, has been approved for treatment of adults with type 2 diabetes, the Food and Drug Administration announced Sept. 18.
It was approved with a Risk Evaluation and Mitigation Strategy (REMS) aimed at informing health care professionals about the serious risks of treatment and requirements for postmarketing studies that address some of those risks, the FDA said in a statement.
Approval was based on six studies of about 3,300 patients with type 2 diabetes who were treated with dulaglutide as monotherapy, and in combination with other approved type 2 diabetes treatments, including metformin. It will be marketed as Trulicity by Eli Lilly.
The drug’s label has a boxed warning that thyroid C-cell tumors have been observed in rodent studies of dulaglutide, “but that it is unknown whether Trulicity causes thyroid C-cell tumors,” including medullary thyroid carcinoma (MTC) in humans, the statement said. It should not be used to treat patients who have had, or have, a family history of MTC, or in patients who have multiple endocrine neoplasia syndrome type 2, who are predisposed to MTC, the statement noted.
Eli Lilly is required to conduct several postmarketing studies, including a cardiovascular outcomes study in patients at a high risk of cardiovascular disease at baseline, a pediatric study, and a registry study of MTC cases to help determine whether there is an increase in the incidence of MTC associated with the use of the drug, for at least 15 years. Another postmarketing study will compare dulaglutide to insulin glargine for glycemic control in patients with type 2 diabetes and moderate or severe renal impairment.
Dulaglutide, a glucagon-like peptide-1 receptor agonist administered subcutaneously once a week, has been approved for treatment of adults with type 2 diabetes, the Food and Drug Administration announced Sept. 18.
It was approved with a Risk Evaluation and Mitigation Strategy (REMS) aimed at informing health care professionals about the serious risks of treatment and requirements for postmarketing studies that address some of those risks, the FDA said in a statement.
Approval was based on six studies of about 3,300 patients with type 2 diabetes who were treated with dulaglutide as monotherapy, and in combination with other approved type 2 diabetes treatments, including metformin. It will be marketed as Trulicity by Eli Lilly.
The drug’s label has a boxed warning that thyroid C-cell tumors have been observed in rodent studies of dulaglutide, “but that it is unknown whether Trulicity causes thyroid C-cell tumors,” including medullary thyroid carcinoma (MTC) in humans, the statement said. It should not be used to treat patients who have had, or have, a family history of MTC, or in patients who have multiple endocrine neoplasia syndrome type 2, who are predisposed to MTC, the statement noted.
Eli Lilly is required to conduct several postmarketing studies, including a cardiovascular outcomes study in patients at a high risk of cardiovascular disease at baseline, a pediatric study, and a registry study of MTC cases to help determine whether there is an increase in the incidence of MTC associated with the use of the drug, for at least 15 years. Another postmarketing study will compare dulaglutide to insulin glargine for glycemic control in patients with type 2 diabetes and moderate or severe renal impairment.
FDA panel recommends against approval of oral testosterone replacement therapy
HYATTSVILLE, MD. – In an 18-3 vote, the majority of a Food and Drug Administration advisory panel agreed that the overall risk-benefit profile of an oral formulation of testosterone undecanoate did not support approval at this time, citing issues with the pivotal trial, safety, and wide dietary-dependent variations in absorption, at a meeting on Sept. 18.

The results of two phase III pharmacokinetic studies in hypogonadal adult men, whose mean age was about 55 years, with a total serum testosterone of 300 ng/dL or less, were reviewed at a joint meeting of the FDA’s Bone, Reproductive, and Urologic Drugs Advisory Committee, and Drug Safety and Risk Management Advisory Committee. The manufacturer, Clarus Therapeutics, has proposed that testosterone undecanoate capsules be approved for replacement therapy in men for conditions associated with a deficiency or absence of endogenous testosterone, primary hypogonadism (congenital or acquired), and hypogonadotropic hypogonadism (congenital or acquired), the standard indication for testosterone replacement therapy (TRT). For approval of TRT products, the FDA requires a pharmacokinetic study.
Because of excessive testosterone spikes in the first study, a less aggressive dosing algorithm was evaluated in the pivotal open-label study of 116 patients, evaluating a starting dose of 200 mg twice a day, titrated in increments of 50 mg twice a day based on the serum testosterone levels at least 7 days after the initial or adjusted dose, with a minimum dose of 100 mg twice a day and a maximum dose of 300 mg twice a day.
The study just met the prespecified primary endpoint, which was that at least 75% of patients achieve a “Cavg” of testosterone (a measure that divides total exposure to testosterone, based on pharmacokinetic sampling over 24 hours, by 24) in the eugonadal range (300-1,000 ng/dL) at the end of treatment, at 114 days. However, almost 20% of patients were lost to follow-up, a major issue cited by the FDA and panelists. In this study, four patients had a testosterone measurement that exceeded 2,500 ng/dL, and one patient (less than 1%) had a serious cardiovascular event. But in the initial study with the higher dose, the rate of adverse CV events was almost 4%, and there were increases in CV biomarkers, such as C-reactive protein and blood pressure.
The panels voted 12-8, with 1 abstention, that these data did not provide sufficient evidence that oral testosterone undecanoate was effective, for reasons that included concerns about the effect the high dropout rate had on the results, and the failure to meet any of the secondary endpoints in the study. The panelists also cited concerns about the effect dietary changes had on systemic absorption, particularly increased fat content.
Like other panelists, Dr. Richard Alexander, a professor in the urology division at the University of Maryland, Baltimore, agreed there was a great need for an oral formulation of testosterone, but he voted against approval because the way the drug works is ”highly dependent on patient behavior ... which is very problematic.” Referring to a marked increase in bioavailability with a high-fat diet, he said, “I’m afraid that in the real world,“ men would be at a heightened risk of overdoses with this product.
Another concern mentioned by several panelists was the increased likelihood that the availability of an oral TRT that is easier to administer than currently available topical and injectable formulations could further increase the widespread off-label use among men with age-related low testosterone, the most common use of TRT products, despite no conclusive data TRT is effective in this population and some concerns about safety – the topic of the panel meeting the previous day.
If the product is approved, the company would market it as Rextoro, and it would the first oral testosterone product available, other than the rarely used oral methyltestosterone formulation.
The FDA usually follows the recommendations of its advisory panels. In a statement issued by Clarus after the meeting, Dr. Robert Dudley, the chief executive officer, said that the company “will work closely with the FDA to respond to the panel’s concerns, and remain committed to bringing Rextoro to the market as soon as possible.” The FDA is expected to make a decision on approval by Nov. 3, 2014.
Usually, members of advisory panels have no financial conflicts, but in some cases, a panelist is granted a waiver, and at this meeting, one panelist who owned stock in an affected company was granted a waiver.
HYATTSVILLE, MD. – In an 18-3 vote, the majority of a Food and Drug Administration advisory panel agreed that the overall risk-benefit profile of an oral formulation of testosterone undecanoate did not support approval at this time, citing issues with the pivotal trial, safety, and wide dietary-dependent variations in absorption, at a meeting on Sept. 18.
The results of two phase III pharmacokinetic studies in hypogonadal adult men, whose mean age was about 55 years, with a total serum testosterone of 300 ng/dL or less, were reviewed at a joint meeting of the FDA’s Bone, Reproductive, and Urologic Drugs Advisory Committee, and Drug Safety and Risk Management Advisory Committee. The manufacturer, Clarus Therapeutics, has proposed that testosterone undecanoate capsules be approved for replacement therapy in men for conditions associated with a deficiency or absence of endogenous testosterone, primary hypogonadism (congenital or acquired), and hypogonadotropic hypogonadism (congenital or acquired), the standard indication for testosterone replacement therapy (TRT). For approval of TRT products, the FDA requires a pharmacokinetic study.
Because of excessive testosterone spikes in the first study, a less aggressive dosing algorithm was evaluated in the pivotal open-label study of 116 patients, evaluating a starting dose of 200 mg twice a day, titrated in increments of 50 mg twice a day based on the serum testosterone levels at least 7 days after the initial or adjusted dose, with a minimum dose of 100 mg twice a day and a maximum dose of 300 mg twice a day.
The study just met the prespecified primary endpoint, which was that at least 75% of patients achieve a “Cavg” of testosterone (a measure that divides total exposure to testosterone, based on pharmacokinetic sampling over 24 hours, by 24) in the eugonadal range (300-1,000 ng/dL) at the end of treatment, at 114 days. However, almost 20% of patients were lost to follow-up, a major issue cited by the FDA and panelists. In this study, four patients had a testosterone measurement that exceeded 2,500 ng/dL, and one patient (less than 1%) had a serious cardiovascular event. But in the initial study with the higher dose, the rate of adverse CV events was almost 4%, and there were increases in CV biomarkers, such as C-reactive protein and blood pressure.
The panels voted 12-8, with 1 abstention, that these data did not provide sufficient evidence that oral testosterone undecanoate was effective, for reasons that included concerns about the effect the high dropout rate had on the results, and the failure to meet any of the secondary endpoints in the study. The panelists also cited concerns about the effect dietary changes had on systemic absorption, particularly increased fat content.
Like other panelists, Dr. Richard Alexander, a professor in the urology division at the University of Maryland, Baltimore, agreed there was a great need for an oral formulation of testosterone, but he voted against approval because the way the drug works is ”highly dependent on patient behavior ... which is very problematic.” Referring to a marked increase in bioavailability with a high-fat diet, he said, “I’m afraid that in the real world,“ men would be at a heightened risk of overdoses with this product.
Another concern mentioned by several panelists was the increased likelihood that the availability of an oral TRT that is easier to administer than currently available topical and injectable formulations could further increase the widespread off-label use among men with age-related low testosterone, the most common use of TRT products, despite no conclusive data TRT is effective in this population and some concerns about safety – the topic of the panel meeting the previous day.
If the product is approved, the company would market it as Rextoro, and it would the first oral testosterone product available, other than the rarely used oral methyltestosterone formulation.
The FDA usually follows the recommendations of its advisory panels. In a statement issued by Clarus after the meeting, Dr. Robert Dudley, the chief executive officer, said that the company “will work closely with the FDA to respond to the panel’s concerns, and remain committed to bringing Rextoro to the market as soon as possible.” The FDA is expected to make a decision on approval by Nov. 3, 2014.
Usually, members of advisory panels have no financial conflicts, but in some cases, a panelist is granted a waiver, and at this meeting, one panelist who owned stock in an affected company was granted a waiver.
HYATTSVILLE, MD. – In an 18-3 vote, the majority of a Food and Drug Administration advisory panel agreed that the overall risk-benefit profile of an oral formulation of testosterone undecanoate did not support approval at this time, citing issues with the pivotal trial, safety, and wide dietary-dependent variations in absorption, at a meeting on Sept. 18.
The results of two phase III pharmacokinetic studies in hypogonadal adult men, whose mean age was about 55 years, with a total serum testosterone of 300 ng/dL or less, were reviewed at a joint meeting of the FDA’s Bone, Reproductive, and Urologic Drugs Advisory Committee, and Drug Safety and Risk Management Advisory Committee. The manufacturer, Clarus Therapeutics, has proposed that testosterone undecanoate capsules be approved for replacement therapy in men for conditions associated with a deficiency or absence of endogenous testosterone, primary hypogonadism (congenital or acquired), and hypogonadotropic hypogonadism (congenital or acquired), the standard indication for testosterone replacement therapy (TRT). For approval of TRT products, the FDA requires a pharmacokinetic study.
Because of excessive testosterone spikes in the first study, a less aggressive dosing algorithm was evaluated in the pivotal open-label study of 116 patients, evaluating a starting dose of 200 mg twice a day, titrated in increments of 50 mg twice a day based on the serum testosterone levels at least 7 days after the initial or adjusted dose, with a minimum dose of 100 mg twice a day and a maximum dose of 300 mg twice a day.
The study just met the prespecified primary endpoint, which was that at least 75% of patients achieve a “Cavg” of testosterone (a measure that divides total exposure to testosterone, based on pharmacokinetic sampling over 24 hours, by 24) in the eugonadal range (300-1,000 ng/dL) at the end of treatment, at 114 days. However, almost 20% of patients were lost to follow-up, a major issue cited by the FDA and panelists. In this study, four patients had a testosterone measurement that exceeded 2,500 ng/dL, and one patient (less than 1%) had a serious cardiovascular event. But in the initial study with the higher dose, the rate of adverse CV events was almost 4%, and there were increases in CV biomarkers, such as C-reactive protein and blood pressure.
The panels voted 12-8, with 1 abstention, that these data did not provide sufficient evidence that oral testosterone undecanoate was effective, for reasons that included concerns about the effect the high dropout rate had on the results, and the failure to meet any of the secondary endpoints in the study. The panelists also cited concerns about the effect dietary changes had on systemic absorption, particularly increased fat content.
Like other panelists, Dr. Richard Alexander, a professor in the urology division at the University of Maryland, Baltimore, agreed there was a great need for an oral formulation of testosterone, but he voted against approval because the way the drug works is ”highly dependent on patient behavior ... which is very problematic.” Referring to a marked increase in bioavailability with a high-fat diet, he said, “I’m afraid that in the real world,“ men would be at a heightened risk of overdoses with this product.
Another concern mentioned by several panelists was the increased likelihood that the availability of an oral TRT that is easier to administer than currently available topical and injectable formulations could further increase the widespread off-label use among men with age-related low testosterone, the most common use of TRT products, despite no conclusive data TRT is effective in this population and some concerns about safety – the topic of the panel meeting the previous day.
If the product is approved, the company would market it as Rextoro, and it would the first oral testosterone product available, other than the rarely used oral methyltestosterone formulation.
The FDA usually follows the recommendations of its advisory panels. In a statement issued by Clarus after the meeting, Dr. Robert Dudley, the chief executive officer, said that the company “will work closely with the FDA to respond to the panel’s concerns, and remain committed to bringing Rextoro to the market as soon as possible.” The FDA is expected to make a decision on approval by Nov. 3, 2014.
Usually, members of advisory panels have no financial conflicts, but in some cases, a panelist is granted a waiver, and at this meeting, one panelist who owned stock in an affected company was granted a waiver.
AT AN FDA ADVISORY COMMITTEE MEETING

FDA panel recommends changes in testosterone indications
HYATTSVILLE, MD.* – The majority of a Food and Drug Administration advisory panel recommended that the indication for testosterone replacement therapy should be revised to reflect the lack of efficacy data for age-related low testosterone and the possible increased cardiovascular risks associated with treatment.
At a meeting Sept. 17 the FDA’s Bone, Reproductive, and Urologic Drugs Advisory Committee and the Drug Safety and Risk Management Advisory Committee voted 20-1 that the current indication, as worded in the labeling for all testosterone products, should be tightened to make it clear that testosterone therapy is not indicated for men with age-related reductions in testosterone. Other recommendations included adding diagnostic criteria to the indications section and specifying testosterone levels needed for a diagnosis of low testosterone. The meeting was held by the FDA to address whether the labeling for testosterone products reflects the appropriate indicated population for testosterone replacement therapy and to discuss the potential for adverse cardiovascular outcomes associated with this use.
The panel agreed there was a signal of a possible increased cardiovascular risk, which they described as “inconclusive” and “weak.” Several panelists noted that the risk was biologically plausible, adding that the label should include a statement with this information.
The majority of panelists (16) voted that the manufacturers of testosterone products should conduct randomized, controlled studies to evaluate the possible cardiovascular risks of testosterone therapy associated with certain indications. Four panelists voted that manufacturers should be required to conduct these studies regardless of the indication, largely because all testosterone products are being used off label for treating age-related low testosterone. Panelists pointed out that for the classic indications, such as primary hypogonadism, the benefit-risk profile of testosterone replacement therapy (TRT) was clear. Evidence that it is beneficial for treating older men with age-related low testosterone, however, was inconclusive, they noted.
“We don’t really know whether aging-associated low testosterone is in fact a disease at all,” said panelist Dr. A. Michael Lincoff, vice-chairman of cardiovascular medicine and professor of medicine at the Cleveland Clinic. Randomized controlled trials to “determine once and for all whether there really are benefits in this population are needed ... and whatever magnitude of efficacy is found, assuming efficacy is shown, will have to be balanced against safety,” he added.
The results of observational studies and meta-analyses have found mixed results regarding risk for cardiovascular adverse events associated with testosterone therapy, and the overall cardiovascular risks and benefits of testosterone therapy risks are unclear, according to the FDA.
According to national data presented by the FDA at the meeting, 2.3 million patients in 2013 received a prescription for testosterone, up from 1.3 million in 2010, and about 70% of the prescriptions are for men aged 40-64 years. The largest increase in prescriptions for TRT during 2010-2013 was in this age group, increasing from 850,000 to 1.5 million prescriptions. In a U.S. office-based survey of physicians, ”testicular hypofunction, not elsewhere classified” was the most common diagnosis for TRT use in men of all age groups.
The results of coordinated randomized, placebo-controlled studies on testosterone therapy in 800 elderly men with low testosterone are expected to be available by the summer of 2015. Outcomes will include physical, sexual, and cognitive function; vitality; and cardiovascular effects.
The FDA usually follows the recommendations of its advisory panels. In most cases, members of advisory panels have no financial conflicts, but in some cases, a panelist is granted waiver. At, this meeting, one panelist who owned stock in an affected company was granted a waiver.
*Correction, 9/19/14: An earlier version of this article contained an incorrect dateline.
HYATTSVILLE, MD.* – The majority of a Food and Drug Administration advisory panel recommended that the indication for testosterone replacement therapy should be revised to reflect the lack of efficacy data for age-related low testosterone and the possible increased cardiovascular risks associated with treatment.
At a meeting Sept. 17 the FDA’s Bone, Reproductive, and Urologic Drugs Advisory Committee and the Drug Safety and Risk Management Advisory Committee voted 20-1 that the current indication, as worded in the labeling for all testosterone products, should be tightened to make it clear that testosterone therapy is not indicated for men with age-related reductions in testosterone. Other recommendations included adding diagnostic criteria to the indications section and specifying testosterone levels needed for a diagnosis of low testosterone. The meeting was held by the FDA to address whether the labeling for testosterone products reflects the appropriate indicated population for testosterone replacement therapy and to discuss the potential for adverse cardiovascular outcomes associated with this use.
The panel agreed there was a signal of a possible increased cardiovascular risk, which they described as “inconclusive” and “weak.” Several panelists noted that the risk was biologically plausible, adding that the label should include a statement with this information.
The majority of panelists (16) voted that the manufacturers of testosterone products should conduct randomized, controlled studies to evaluate the possible cardiovascular risks of testosterone therapy associated with certain indications. Four panelists voted that manufacturers should be required to conduct these studies regardless of the indication, largely because all testosterone products are being used off label for treating age-related low testosterone. Panelists pointed out that for the classic indications, such as primary hypogonadism, the benefit-risk profile of testosterone replacement therapy (TRT) was clear. Evidence that it is beneficial for treating older men with age-related low testosterone, however, was inconclusive, they noted.
“We don’t really know whether aging-associated low testosterone is in fact a disease at all,” said panelist Dr. A. Michael Lincoff, vice-chairman of cardiovascular medicine and professor of medicine at the Cleveland Clinic. Randomized controlled trials to “determine once and for all whether there really are benefits in this population are needed ... and whatever magnitude of efficacy is found, assuming efficacy is shown, will have to be balanced against safety,” he added.
The results of observational studies and meta-analyses have found mixed results regarding risk for cardiovascular adverse events associated with testosterone therapy, and the overall cardiovascular risks and benefits of testosterone therapy risks are unclear, according to the FDA.
According to national data presented by the FDA at the meeting, 2.3 million patients in 2013 received a prescription for testosterone, up from 1.3 million in 2010, and about 70% of the prescriptions are for men aged 40-64 years. The largest increase in prescriptions for TRT during 2010-2013 was in this age group, increasing from 850,000 to 1.5 million prescriptions. In a U.S. office-based survey of physicians, ”testicular hypofunction, not elsewhere classified” was the most common diagnosis for TRT use in men of all age groups.
The results of coordinated randomized, placebo-controlled studies on testosterone therapy in 800 elderly men with low testosterone are expected to be available by the summer of 2015. Outcomes will include physical, sexual, and cognitive function; vitality; and cardiovascular effects.
The FDA usually follows the recommendations of its advisory panels. In most cases, members of advisory panels have no financial conflicts, but in some cases, a panelist is granted waiver. At, this meeting, one panelist who owned stock in an affected company was granted a waiver.
*Correction, 9/19/14: An earlier version of this article contained an incorrect dateline.
HYATTSVILLE, MD.* – The majority of a Food and Drug Administration advisory panel recommended that the indication for testosterone replacement therapy should be revised to reflect the lack of efficacy data for age-related low testosterone and the possible increased cardiovascular risks associated with treatment.
At a meeting Sept. 17 the FDA’s Bone, Reproductive, and Urologic Drugs Advisory Committee and the Drug Safety and Risk Management Advisory Committee voted 20-1 that the current indication, as worded in the labeling for all testosterone products, should be tightened to make it clear that testosterone therapy is not indicated for men with age-related reductions in testosterone. Other recommendations included adding diagnostic criteria to the indications section and specifying testosterone levels needed for a diagnosis of low testosterone. The meeting was held by the FDA to address whether the labeling for testosterone products reflects the appropriate indicated population for testosterone replacement therapy and to discuss the potential for adverse cardiovascular outcomes associated with this use.
The panel agreed there was a signal of a possible increased cardiovascular risk, which they described as “inconclusive” and “weak.” Several panelists noted that the risk was biologically plausible, adding that the label should include a statement with this information.
The majority of panelists (16) voted that the manufacturers of testosterone products should conduct randomized, controlled studies to evaluate the possible cardiovascular risks of testosterone therapy associated with certain indications. Four panelists voted that manufacturers should be required to conduct these studies regardless of the indication, largely because all testosterone products are being used off label for treating age-related low testosterone. Panelists pointed out that for the classic indications, such as primary hypogonadism, the benefit-risk profile of testosterone replacement therapy (TRT) was clear. Evidence that it is beneficial for treating older men with age-related low testosterone, however, was inconclusive, they noted.
“We don’t really know whether aging-associated low testosterone is in fact a disease at all,” said panelist Dr. A. Michael Lincoff, vice-chairman of cardiovascular medicine and professor of medicine at the Cleveland Clinic. Randomized controlled trials to “determine once and for all whether there really are benefits in this population are needed ... and whatever magnitude of efficacy is found, assuming efficacy is shown, will have to be balanced against safety,” he added.
The results of observational studies and meta-analyses have found mixed results regarding risk for cardiovascular adverse events associated with testosterone therapy, and the overall cardiovascular risks and benefits of testosterone therapy risks are unclear, according to the FDA.
According to national data presented by the FDA at the meeting, 2.3 million patients in 2013 received a prescription for testosterone, up from 1.3 million in 2010, and about 70% of the prescriptions are for men aged 40-64 years. The largest increase in prescriptions for TRT during 2010-2013 was in this age group, increasing from 850,000 to 1.5 million prescriptions. In a U.S. office-based survey of physicians, ”testicular hypofunction, not elsewhere classified” was the most common diagnosis for TRT use in men of all age groups.
The results of coordinated randomized, placebo-controlled studies on testosterone therapy in 800 elderly men with low testosterone are expected to be available by the summer of 2015. Outcomes will include physical, sexual, and cognitive function; vitality; and cardiovascular effects.
The FDA usually follows the recommendations of its advisory panels. In most cases, members of advisory panels have no financial conflicts, but in some cases, a panelist is granted waiver. At, this meeting, one panelist who owned stock in an affected company was granted a waiver.
*Correction, 9/19/14: An earlier version of this article contained an incorrect dateline.
AT AN FDA ADVISORY COMMITTEE MEETING
FDA approves drug for opioid-induced constipation in chronic noncancer pain population
Naloxegol, an orally administered peripherally acting mu-opioid receptor antagonist, has been approved by the Food and Drug Administration as a treatment for opioid-induced constipation in adults with chronic noncancer pain, the agency announced on Sept. 17.
The drug will be marketed by AstraZeneca Pharmaceuticals as Movantik and is expected to be available during the first half of 2015, according to a company statement. A company petition for the drug to be descheduled is under review at the US Drug Enforcement Administration. Because the drug is structurally related to noroxymorphone, naloxegol is considered a schedule II drug, but the FDA-approved labeling states that it has “no risk of abuse or dependency,” the company statement said.
Approval of the drug, which is taken once a day in a tablet formulation, was based on two 12-week studies of 1,352 patients who had taken opioids for noncancer related pain for at least 4 weeks and had opioid-induced constipation, according to a FDA statement. The two studies, KODIAC-04 and KODIAC-05, were published in June (N. Engl. J. Med. 2014;370:2387-96).
Patients with noncancer-related pain and opioid-induced constipation were randomized to receive one of two doses of naloxegol or placebo, once a day for 12 weeks. The 12-week endpoint was at least three spontaneous bowel movements per week and an increase of at least one spontaneous bowel movement per week from baseline for at least 9 of the 12 weeks and for at least 3 of the final 4 weeks of the study. In one study, 44% of those on the 25-mg dose and 41% of those on the lower dose met this endpoint, vs. 29% of those on placebo. In the second study, 40% of those on the higher dose and 35% of those on the lower dose met this endpoint, vs. 29% of those on placebo.
Abdominal pain, diarrhea, headache, and flatulence were among the common side effects of the drug, according to the FDA.
The manufacturer is required to conduct a postmarketing study to evaluate the possible cardiovascular risks of the drug in patients, according to the agency.
Naloxegol is under review by the European Medicines Agency in Europe, according to the company.
Naloxegol, an orally administered peripherally acting mu-opioid receptor antagonist, has been approved by the Food and Drug Administration as a treatment for opioid-induced constipation in adults with chronic noncancer pain, the agency announced on Sept. 17.
The drug will be marketed by AstraZeneca Pharmaceuticals as Movantik and is expected to be available during the first half of 2015, according to a company statement. A company petition for the drug to be descheduled is under review at the US Drug Enforcement Administration. Because the drug is structurally related to noroxymorphone, naloxegol is considered a schedule II drug, but the FDA-approved labeling states that it has “no risk of abuse or dependency,” the company statement said.
Approval of the drug, which is taken once a day in a tablet formulation, was based on two 12-week studies of 1,352 patients who had taken opioids for noncancer related pain for at least 4 weeks and had opioid-induced constipation, according to a FDA statement. The two studies, KODIAC-04 and KODIAC-05, were published in June (N. Engl. J. Med. 2014;370:2387-96).
Patients with noncancer-related pain and opioid-induced constipation were randomized to receive one of two doses of naloxegol or placebo, once a day for 12 weeks. The 12-week endpoint was at least three spontaneous bowel movements per week and an increase of at least one spontaneous bowel movement per week from baseline for at least 9 of the 12 weeks and for at least 3 of the final 4 weeks of the study. In one study, 44% of those on the 25-mg dose and 41% of those on the lower dose met this endpoint, vs. 29% of those on placebo. In the second study, 40% of those on the higher dose and 35% of those on the lower dose met this endpoint, vs. 29% of those on placebo.
Abdominal pain, diarrhea, headache, and flatulence were among the common side effects of the drug, according to the FDA.
The manufacturer is required to conduct a postmarketing study to evaluate the possible cardiovascular risks of the drug in patients, according to the agency.
Naloxegol is under review by the European Medicines Agency in Europe, according to the company.
Naloxegol, an orally administered peripherally acting mu-opioid receptor antagonist, has been approved by the Food and Drug Administration as a treatment for opioid-induced constipation in adults with chronic noncancer pain, the agency announced on Sept. 17.
The drug will be marketed by AstraZeneca Pharmaceuticals as Movantik and is expected to be available during the first half of 2015, according to a company statement. A company petition for the drug to be descheduled is under review at the US Drug Enforcement Administration. Because the drug is structurally related to noroxymorphone, naloxegol is considered a schedule II drug, but the FDA-approved labeling states that it has “no risk of abuse or dependency,” the company statement said.
Approval of the drug, which is taken once a day in a tablet formulation, was based on two 12-week studies of 1,352 patients who had taken opioids for noncancer related pain for at least 4 weeks and had opioid-induced constipation, according to a FDA statement. The two studies, KODIAC-04 and KODIAC-05, were published in June (N. Engl. J. Med. 2014;370:2387-96).
Patients with noncancer-related pain and opioid-induced constipation were randomized to receive one of two doses of naloxegol or placebo, once a day for 12 weeks. The 12-week endpoint was at least three spontaneous bowel movements per week and an increase of at least one spontaneous bowel movement per week from baseline for at least 9 of the 12 weeks and for at least 3 of the final 4 weeks of the study. In one study, 44% of those on the 25-mg dose and 41% of those on the lower dose met this endpoint, vs. 29% of those on placebo. In the second study, 40% of those on the higher dose and 35% of those on the lower dose met this endpoint, vs. 29% of those on placebo.
Abdominal pain, diarrhea, headache, and flatulence were among the common side effects of the drug, according to the FDA.
The manufacturer is required to conduct a postmarketing study to evaluate the possible cardiovascular risks of the drug in patients, according to the agency.
Naloxegol is under review by the European Medicines Agency in Europe, according to the company.
Successful Bariatric Surgery Also May Improve Urinary Incontinence
WASHINGTON – The majority of obese women who had urinary incontinence before bariatric surgery had complete or near-complete resolution of symptoms for up to 3 years after surgery in a study of more than 1,500 women, Dr. Leslee Subak reported at the scientific meetings of the American Urogynecologic Society and the International Urogynecological Association.
These results indicate that "improvement in incontinence may be another long-term benefit of weight loss, in this case surgical weight loss," said Dr. Subak, professor of obstetrics and gynecology and reproductive sciences, epidemiology and biostatistics, and urology at the University of California, San Francisco.
The study evaluated the effect of surgery on urinary incontinence in 1,565 severely obese women who were part of the multicenter Longitudinal Cohort Study of Bariatric Surgery-2 and had completed self-administered questionnaires about urinary incontinence episodes before surgery and at one or more annual follow-up assessments within 3 years of surgery.
The results were based on outcomes of the 772 women who reported experiencing episodes of incontinence at least weekly, with an average of about 11 incontinence episodes per week. Their median age was 46 years and most were white; about 7% had undergone previous incontinence surgery and about 8% had received or were receiving behavioral treatment or medication for incontinence.
Most of the patients underwent a Roux-en-Y gastric bypass (71%) or laparoscopic adjustable gastric banding (25%). After the first year, they had lost a median of about 30% of their baseline weight, which was maintained through the third year.
At all follow-up times after surgery, there were significantly fewer incontinence episodes, compared with baseline, with a remission rate of 60%-65%, Dr. Subak said.
Urinary incontinence episodes dropped from an average of about 11 episodes per week before surgery to an average of almost 3 episodes per week at 1 year and 4 episodes per week at 2 and 3 years, she noted. Episodes of stress incontinence also decreased from an average of about 5 episodes per week at baseline to about 1 episode per week at 1 year and almost 2 episodes per week at 2 and 3 years.
The remission rate – defined as less than 1 weekly urinary incontinence episode over the past 3 months – was 70% at 1 year, dropping to and stabilizing at about 61%-62% at 2 and 3 years. Moreover, 25% of the women had a complete remission (no episodes of incontinence during the past 3 months) at 3 years, a slight increase from almost 27% at 1 year, Dr. Subak said.
"The magnitude of weight loss was the strongest predictor of improvement in incontinence over time," she noted. "Incontinence and BMI [body mass index] seemed to track together, as [whenever] there’s a reduction in BMI ... there’s a reduction in urinary incontinence episode frequency."
A younger age also was significantly associated with a reduction in the frequency or a remission of urinary incontinence, while being pregnant in the previous year and having had a hysterectomy were associated with a lower likelihood of having a remission.
Dr. Subak noted that limitations of the study included the observational design and the lack of a control group, as well as the fact that data were based on self-reports.
She referred to urinary incontinence and obesity as "twin epidemics," with about a fourfold increased risk of urinary incontinence associated with obesity. About one-third of women in the United States are obese and about 70% of women with incontinence are obese, she pointed out.
The Longitudinal Cohort Study of Bariatric Surgery-2 is funded by the National Institute of Diabetes and Digestive and Kidney Diseases. Dr. Subak received additional funding from the NIDDK; she had no other disclosures.
WASHINGTON – The majority of obese women who had urinary incontinence before bariatric surgery had complete or near-complete resolution of symptoms for up to 3 years after surgery in a study of more than 1,500 women, Dr. Leslee Subak reported at the scientific meetings of the American Urogynecologic Society and the International Urogynecological Association.
These results indicate that "improvement in incontinence may be another long-term benefit of weight loss, in this case surgical weight loss," said Dr. Subak, professor of obstetrics and gynecology and reproductive sciences, epidemiology and biostatistics, and urology at the University of California, San Francisco.
The study evaluated the effect of surgery on urinary incontinence in 1,565 severely obese women who were part of the multicenter Longitudinal Cohort Study of Bariatric Surgery-2 and had completed self-administered questionnaires about urinary incontinence episodes before surgery and at one or more annual follow-up assessments within 3 years of surgery.
The results were based on outcomes of the 772 women who reported experiencing episodes of incontinence at least weekly, with an average of about 11 incontinence episodes per week. Their median age was 46 years and most were white; about 7% had undergone previous incontinence surgery and about 8% had received or were receiving behavioral treatment or medication for incontinence.
Most of the patients underwent a Roux-en-Y gastric bypass (71%) or laparoscopic adjustable gastric banding (25%). After the first year, they had lost a median of about 30% of their baseline weight, which was maintained through the third year.
At all follow-up times after surgery, there were significantly fewer incontinence episodes, compared with baseline, with a remission rate of 60%-65%, Dr. Subak said.
Urinary incontinence episodes dropped from an average of about 11 episodes per week before surgery to an average of almost 3 episodes per week at 1 year and 4 episodes per week at 2 and 3 years, she noted. Episodes of stress incontinence also decreased from an average of about 5 episodes per week at baseline to about 1 episode per week at 1 year and almost 2 episodes per week at 2 and 3 years.
The remission rate – defined as less than 1 weekly urinary incontinence episode over the past 3 months – was 70% at 1 year, dropping to and stabilizing at about 61%-62% at 2 and 3 years. Moreover, 25% of the women had a complete remission (no episodes of incontinence during the past 3 months) at 3 years, a slight increase from almost 27% at 1 year, Dr. Subak said.
"The magnitude of weight loss was the strongest predictor of improvement in incontinence over time," she noted. "Incontinence and BMI [body mass index] seemed to track together, as [whenever] there’s a reduction in BMI ... there’s a reduction in urinary incontinence episode frequency."
A younger age also was significantly associated with a reduction in the frequency or a remission of urinary incontinence, while being pregnant in the previous year and having had a hysterectomy were associated with a lower likelihood of having a remission.
Dr. Subak noted that limitations of the study included the observational design and the lack of a control group, as well as the fact that data were based on self-reports.
She referred to urinary incontinence and obesity as "twin epidemics," with about a fourfold increased risk of urinary incontinence associated with obesity. About one-third of women in the United States are obese and about 70% of women with incontinence are obese, she pointed out.
The Longitudinal Cohort Study of Bariatric Surgery-2 is funded by the National Institute of Diabetes and Digestive and Kidney Diseases. Dr. Subak received additional funding from the NIDDK; she had no other disclosures.
WASHINGTON – The majority of obese women who had urinary incontinence before bariatric surgery had complete or near-complete resolution of symptoms for up to 3 years after surgery in a study of more than 1,500 women, Dr. Leslee Subak reported at the scientific meetings of the American Urogynecologic Society and the International Urogynecological Association.
These results indicate that "improvement in incontinence may be another long-term benefit of weight loss, in this case surgical weight loss," said Dr. Subak, professor of obstetrics and gynecology and reproductive sciences, epidemiology and biostatistics, and urology at the University of California, San Francisco.
The study evaluated the effect of surgery on urinary incontinence in 1,565 severely obese women who were part of the multicenter Longitudinal Cohort Study of Bariatric Surgery-2 and had completed self-administered questionnaires about urinary incontinence episodes before surgery and at one or more annual follow-up assessments within 3 years of surgery.
The results were based on outcomes of the 772 women who reported experiencing episodes of incontinence at least weekly, with an average of about 11 incontinence episodes per week. Their median age was 46 years and most were white; about 7% had undergone previous incontinence surgery and about 8% had received or were receiving behavioral treatment or medication for incontinence.
Most of the patients underwent a Roux-en-Y gastric bypass (71%) or laparoscopic adjustable gastric banding (25%). After the first year, they had lost a median of about 30% of their baseline weight, which was maintained through the third year.
At all follow-up times after surgery, there were significantly fewer incontinence episodes, compared with baseline, with a remission rate of 60%-65%, Dr. Subak said.
Urinary incontinence episodes dropped from an average of about 11 episodes per week before surgery to an average of almost 3 episodes per week at 1 year and 4 episodes per week at 2 and 3 years, she noted. Episodes of stress incontinence also decreased from an average of about 5 episodes per week at baseline to about 1 episode per week at 1 year and almost 2 episodes per week at 2 and 3 years.
The remission rate – defined as less than 1 weekly urinary incontinence episode over the past 3 months – was 70% at 1 year, dropping to and stabilizing at about 61%-62% at 2 and 3 years. Moreover, 25% of the women had a complete remission (no episodes of incontinence during the past 3 months) at 3 years, a slight increase from almost 27% at 1 year, Dr. Subak said.
"The magnitude of weight loss was the strongest predictor of improvement in incontinence over time," she noted. "Incontinence and BMI [body mass index] seemed to track together, as [whenever] there’s a reduction in BMI ... there’s a reduction in urinary incontinence episode frequency."
A younger age also was significantly associated with a reduction in the frequency or a remission of urinary incontinence, while being pregnant in the previous year and having had a hysterectomy were associated with a lower likelihood of having a remission.
Dr. Subak noted that limitations of the study included the observational design and the lack of a control group, as well as the fact that data were based on self-reports.
She referred to urinary incontinence and obesity as "twin epidemics," with about a fourfold increased risk of urinary incontinence associated with obesity. About one-third of women in the United States are obese and about 70% of women with incontinence are obese, she pointed out.
The Longitudinal Cohort Study of Bariatric Surgery-2 is funded by the National Institute of Diabetes and Digestive and Kidney Diseases. Dr. Subak received additional funding from the NIDDK; she had no other disclosures.
AT AUGS/IUGA 2014

Ex vivo lung perfusion device preserves donor organs
A device that preserves less-than-ideal donor lungs until they are cleared for transplantation has been approved, the Food and Drug Administration announced on Aug. 12.
The ex vivo perfusion device preserves donated lungs that initially do not meet all the criteria for a transplantable lung. The device does this by warming the donor lungs to "near normal body temperature," continuously flushing the lung with a sterile solution, and ventilating them, "which oxygenates the cells and makes it possible for the transplant team to examine the lung’s’ airways with a bronchoscope," according to the FDA statement.
The lungs can remain in the machine for up to 4 hours, providing time for the transplant team to evaluate the lungs to determine if they meet the criteria; donor lungs that meet the criteria are then transplanted into a patient.
The device, the XVIVO Perfusion System (XPS) with STEEN Solution, is manufactured by XVIVO Perfusion.
"With this approval, there may be more lungs available for transplant, which could allow more people with end-stage lung disease who have exhausted all other treatment options to be able to receive a lung transplant," Christy Foreman, director of the Office of Device Evaluation in the FDA’s Center for Devices and Radiological Health, Silver Spring, Md., said in the statement.
About 1 in 5 donor lungs meets the standard transplantation criteria. In the United States, 1,754 lung transplants were performed in 2012 and 1,616 potential recipients were on the lung transplant waiting list at the end of 2012, according to the FDA.
In two studies, outcomes for lung-transplant recipients were similar among those who received a donor lung preserved with the device and those who received donor lungs that were considered ideal and were preserved in cold storage.
"Both trials showed that recipients of the ideal and non-ideal lungs had similar survival rates up to 12 months after transplant and similar rates of organ rejection," the FDA statement said.
The manufacturer is required to conduct a long-term study of the effects of the device as a condition of approval.

|
|
Dr. Jennifer Cox, FCCP, comments: This is exciting news given the shortage of available lungs that meet the current transplant criteria. Early studies showing similar 12-month survival rates and rates of organ rejection are encouraging. I would like to know if there were similar hospital lengths of stay and if there was a difference in post operative complications. Also, how significant will the financial impact be using the device. I look forward to the results of long- term studies. Hopefully this will be a viable option for our patients.
Dr. Jennifer D. Cox, FCCP, is an Assistant Professor of Pulmonary and Critical Care Medicine and clerkship director for the fourth-year medical student Critical Care Selective, Morsani College of Medicine, University of South Florida, in Tampa, Florida.
|
|
|
Dr. Jennifer Cox, FCCP, comments: This is exciting news given the shortage of available lungs that meet the current transplant criteria. Early studies showing similar 12-month survival rates and rates of organ rejection are encouraging. I would like to know if there were similar hospital lengths of stay and if there was a difference in post operative complications. Also, how significant will the financial impact be using the device. I look forward to the results of long- term studies. Hopefully this will be a viable option for our patients.
Dr. Jennifer D. Cox, FCCP, is an Assistant Professor of Pulmonary and Critical Care Medicine and clerkship director for the fourth-year medical student Critical Care Selective, Morsani College of Medicine, University of South Florida, in Tampa, Florida.
|
|
|
Dr. Jennifer Cox, FCCP, comments: This is exciting news given the shortage of available lungs that meet the current transplant criteria. Early studies showing similar 12-month survival rates and rates of organ rejection are encouraging. I would like to know if there were similar hospital lengths of stay and if there was a difference in post operative complications. Also, how significant will the financial impact be using the device. I look forward to the results of long- term studies. Hopefully this will be a viable option for our patients.
Dr. Jennifer D. Cox, FCCP, is an Assistant Professor of Pulmonary and Critical Care Medicine and clerkship director for the fourth-year medical student Critical Care Selective, Morsani College of Medicine, University of South Florida, in Tampa, Florida.
A device that preserves less-than-ideal donor lungs until they are cleared for transplantation has been approved, the Food and Drug Administration announced on Aug. 12.
The ex vivo perfusion device preserves donated lungs that initially do not meet all the criteria for a transplantable lung. The device does this by warming the donor lungs to "near normal body temperature," continuously flushing the lung with a sterile solution, and ventilating them, "which oxygenates the cells and makes it possible for the transplant team to examine the lung’s’ airways with a bronchoscope," according to the FDA statement.
The lungs can remain in the machine for up to 4 hours, providing time for the transplant team to evaluate the lungs to determine if they meet the criteria; donor lungs that meet the criteria are then transplanted into a patient.
The device, the XVIVO Perfusion System (XPS) with STEEN Solution, is manufactured by XVIVO Perfusion.
"With this approval, there may be more lungs available for transplant, which could allow more people with end-stage lung disease who have exhausted all other treatment options to be able to receive a lung transplant," Christy Foreman, director of the Office of Device Evaluation in the FDA’s Center for Devices and Radiological Health, Silver Spring, Md., said in the statement.
About 1 in 5 donor lungs meets the standard transplantation criteria. In the United States, 1,754 lung transplants were performed in 2012 and 1,616 potential recipients were on the lung transplant waiting list at the end of 2012, according to the FDA.
In two studies, outcomes for lung-transplant recipients were similar among those who received a donor lung preserved with the device and those who received donor lungs that were considered ideal and were preserved in cold storage.
"Both trials showed that recipients of the ideal and non-ideal lungs had similar survival rates up to 12 months after transplant and similar rates of organ rejection," the FDA statement said.
The manufacturer is required to conduct a long-term study of the effects of the device as a condition of approval.
A device that preserves less-than-ideal donor lungs until they are cleared for transplantation has been approved, the Food and Drug Administration announced on Aug. 12.
The ex vivo perfusion device preserves donated lungs that initially do not meet all the criteria for a transplantable lung. The device does this by warming the donor lungs to "near normal body temperature," continuously flushing the lung with a sterile solution, and ventilating them, "which oxygenates the cells and makes it possible for the transplant team to examine the lung’s’ airways with a bronchoscope," according to the FDA statement.
The lungs can remain in the machine for up to 4 hours, providing time for the transplant team to evaluate the lungs to determine if they meet the criteria; donor lungs that meet the criteria are then transplanted into a patient.
The device, the XVIVO Perfusion System (XPS) with STEEN Solution, is manufactured by XVIVO Perfusion.
"With this approval, there may be more lungs available for transplant, which could allow more people with end-stage lung disease who have exhausted all other treatment options to be able to receive a lung transplant," Christy Foreman, director of the Office of Device Evaluation in the FDA’s Center for Devices and Radiological Health, Silver Spring, Md., said in the statement.
About 1 in 5 donor lungs meets the standard transplantation criteria. In the United States, 1,754 lung transplants were performed in 2012 and 1,616 potential recipients were on the lung transplant waiting list at the end of 2012, according to the FDA.
In two studies, outcomes for lung-transplant recipients were similar among those who received a donor lung preserved with the device and those who received donor lungs that were considered ideal and were preserved in cold storage.
"Both trials showed that recipients of the ideal and non-ideal lungs had similar survival rates up to 12 months after transplant and similar rates of organ rejection," the FDA statement said.
The manufacturer is required to conduct a long-term study of the effects of the device as a condition of approval.
Autism Sibling Studies Beginning to Yield Data
WASHINGTON – Studies of the younger siblings of children with autism spectrum disorders are beginning to yield information that has important implications for screening, reducing the age at diagnosis, and identifying the window of time during which still unidentified interventions might be able to affect the course, according to experts who spoke at the annual convention of the American Psychological Association.
During a symposium on early development and predictors of outcomes in infant siblings of children with autism, several experts summarized the results of studies evaluating the emergence of autism spectrum disorders (ASD) and ASD-like symptoms in the younger siblings of children with ASD, as well as tools that are being studied in high-risk sibling studies as potential methods of diagnosing ASD earlier.

The risk of ASD is 1 in 68 in the general population, but is almost 1 in 5 among the siblings of children diagnosed with ASD. An additional one in five siblings will have "shadow symptoms" of autism, which refers to children with some of the symptomatology but not enough to merit a diagnosis. In addition, another 1 in 10 will have nonautism developmental delays.
Therefore, about 50% of infant siblings of children with ASD "are vulnerable in their development to some degree, so that’s why we’re studying them so closely," said one of the speakers, Celine Saulnier, Ph.D., clinical director for research at the Marcus Autism Center, Emory University, Atlanta.
During the symposium, Alice Carter, Ph.D., professor of clinical psychology at the University of Massachusetts, Boston, provided the results of a prospective study that identified high-risk siblings who exhibited some ASD-like behaviors early on but were not diagnosed with autism at age 3.
A growing body of literature focusing on milder ASD symptoms in relatives of people diagnosed with ASD indicates that about 20%-30% of children who are at high genetic risk develop some symptoms of autism but not enough symptoms or symptoms that are severe enough to make the diagnosis, she said. This is often referred to as the broader autism phenotype (BAP), which affects an estimated 19% of unaffected siblings at age 12 months.
The prospective study followed 26 infants who had older siblings with ASD, the high-risk group, to 33 infants whose older siblings had typical development. None of these infants met the diagnostic criteria for ASD at 18, 24, and 36 months. Parents completed a questionnaire, the Brief Infant-Toddler Social Emotional Assessment (BITSEA), when the child was 12, 18, and 24 months.
Based on the results of these parent reports, unaffected siblings of the children with autism "displayed more ASD-relevant problem behaviors than the unaffected infants with typically developing siblings," Dr. Carter said. The former also appear to show "a somewhat different trajectory of these behaviors over time," with scores that were significantly different at 18 months from the low-risk group but "converging" by the time they were aged 24 months, when they appeared to be comparable.
These results support the importance of monitoring younger siblings of children diagnosed with ASD for the emergence of ASD symptoms with routine developmental screening and surveillance, "as they may benefit from early intervention services," Dr. Carter said. But even without such services, children may follow the "self-righting trajectory that takes them back on a normative developmental course," which was evident in the study, she added. (She noted that the limitations of the study included the small sample size in the Boston area. In addition, the participants, were mostly white and had higher than average annual family incomes above $65,000.)
Dr. Carter said future challenges will be to distinguish between three different groups of infants who do not meet the full criteria for an ASD diagnosis: Those manifesting symptoms that mark the beginning of the pathway toward ASD or the emergence of ASD (prodromal ASD); those whose symptoms are likely to resolve; and those who show signs of ASD that "will endure and reflect the broader autism phenotype." The primary target for early screening and detection is the identification of the prodromal group, she added.
Other studies involving high-risk siblings include the UC Davis Infant Sibling Study, which is prospectively following infants with an older sibling who has autism and infants with an older sibling who is developing typically, following the infants up to age 3 years, through the window of autism risk. The second phase of the study followed a small group of infants, enrolled between 6 and 9 months, to age 3 years, with testing that included retrospective and prospective parent ratings, and prospective examiner ratings. Most (89%) of the children who developed ASD showed a regressive onset in terms of their social communication behaviors.
Parents were able to identify those trajectories, indicating impending ASD, with prospective reports, "but not as well retrospectively" with the Autism Diagnostic Interview-Revised (ADI-R), which was accurate in only 30% of cases," said Meghan Miller, Ph.D., a postdoctoral fellow at the University of California, Davis, MIND Institute. These results suggest that while parental reports of regression on the ADI-R can be accurate, they might only pick up "the tip of the iceberg," she noted.
The findings, based on a small sample, raise concerns about the use of the ADI-R and other retrospective measures in research, but do provide "some hope for early screening using parent report data," Dr. Miller pointed out. Such reports would be more feasible than neuroimaging, eye tracking, and other sophisticated testing that could be used over several well child visits, to detect declines and help determine which children should be further evaluated further or referred for interventions, she added. Phase III of the study is currently underway.
In other sibling studies, Dr. Saulnier discussed the use of eye-tracking testing in studies that prospectively followed high-risk infants whose siblings have been diagnosed with ASD and low-risk infants with typically developing siblings.
Under eye-tracking methodology, the child watches prerecorded movies on a monitor of women acting as caregivers, singing, and talking to an infant. It is well established that when viewing a social scene, typically developing adults focus on the eye region of the face, but those with autism look less at the eyes and significantly more at the mouth, body, and objects in the social scene. Those findings have been replicated in school-aged children, adolescents, and toddlers down to aged 2 years, she noted.
In infant studies, eye-tracking testing is performed at least 10 times from birth to age 3, with vocal recordings and clinical assessments at different periods.
Summarizing the findings, Dr. Saulnier said infants at low risk who have a typical outcome show a steady fixation of eye gaze from birth. But infants who are at high risk for ASD and "develop ASD by age 3 show a rapid decline in eye fixation between 2 and 6 months of age that is more predictive of ASD than any clinical measure."
Autism cannot be diagnosed with this test alone, she said. (Most of this work was conducted at the Yale Child Study Center, before she moved to Emory, she said.)
She provided examples of a 5-month-old low-risk infant, who developed typically and focused at the eye region of the face, contrasting with another 5-month old infant, who went on to develop autism by agd 3, who was not looking at the eye region of the face, focusing on the mouth, body, and objects.
Another example she showed was an infant who had normal results at 3 and 6 months, but by 9 months, the researchers started to see a shift, which she described as the "unfolding of autism." This child was diagnosed with autism at 12 months. She added, however, that it is rare to see that symptomatology so early and pronounced.
While it would be assumed that the children who go on to develop autism manifest the lack of eye fixation from birth, another finding from eye fixation studies is that the opposite is true, and "babies who went on to develop autism actually had significantly more eye fixation in the first 2 months of life," Dr. Saulnier said.
Typically developing infants maintain this focus on the eyes, "but whatever is happening in autism to make social monitoring not as salient, they’re trailing off, and you’re seeing this derailment in this eye fixation."
Other results of eye-tracking sibling studies might provide information about a window of opportunity – at about age 9-12 months – for an intervention "that could capitalize on the potential for resilience, she noted.
In infants whose older siblings have ASD and are either unaffected or have "shadow symptoms" of autism at age 3 years, eye-tracking results indicate that after reduced eye fixation, they exhibit a "course correction" in increased eye fixation that appears to start around 9 months of age.
Why this occurs is not understood, but this observation sheds some light on a possible window of time during which something could be done, before autism fully unfolds, such as coaching strategies for parents on keeping their child engaged "to produce a course correction if it wasn’t naturally going to occur," Dr. Saulnier said.
She and her associates are now conducting a randomized, controlled study of infants at risk at 12 months, evaluating a potential way to correct the course.
Among the goals in the future is to reduce the mean age of an autism diagnosis so that, once recognized, effective interventions can be started that could affect outcomes, she said. Although autism can be reliably diagnosed by 18-24 months, the mean age of ASD diagnosis is currently about 4.5 years, she noted.
Dr. Carter disclosed that she is one of the developers of the BITSEA screening tool, used in the study she presented. Dr. Saulnier had no disclosures; the studies she presented received support from the National Institute of Mental Health, the National Institute of Child Health and Human Development, the Marcus Foundation, and the Whitehead and Woodruff Foundations. Dr. Miller said the UCSD study has been funded by NIMH, Autism Speaks, and the MINDlist.
A tutorial on recognizing the early signs of ASD, developed by the Kennedy Krieger Institute, Baltimore,is available at https://www.youtube.com/watch?v=YtvP5A5OHpU.
WASHINGTON – Studies of the younger siblings of children with autism spectrum disorders are beginning to yield information that has important implications for screening, reducing the age at diagnosis, and identifying the window of time during which still unidentified interventions might be able to affect the course, according to experts who spoke at the annual convention of the American Psychological Association.
During a symposium on early development and predictors of outcomes in infant siblings of children with autism, several experts summarized the results of studies evaluating the emergence of autism spectrum disorders (ASD) and ASD-like symptoms in the younger siblings of children with ASD, as well as tools that are being studied in high-risk sibling studies as potential methods of diagnosing ASD earlier.
The risk of ASD is 1 in 68 in the general population, but is almost 1 in 5 among the siblings of children diagnosed with ASD. An additional one in five siblings will have "shadow symptoms" of autism, which refers to children with some of the symptomatology but not enough to merit a diagnosis. In addition, another 1 in 10 will have nonautism developmental delays.
Therefore, about 50% of infant siblings of children with ASD "are vulnerable in their development to some degree, so that’s why we’re studying them so closely," said one of the speakers, Celine Saulnier, Ph.D., clinical director for research at the Marcus Autism Center, Emory University, Atlanta.
During the symposium, Alice Carter, Ph.D., professor of clinical psychology at the University of Massachusetts, Boston, provided the results of a prospective study that identified high-risk siblings who exhibited some ASD-like behaviors early on but were not diagnosed with autism at age 3.
A growing body of literature focusing on milder ASD symptoms in relatives of people diagnosed with ASD indicates that about 20%-30% of children who are at high genetic risk develop some symptoms of autism but not enough symptoms or symptoms that are severe enough to make the diagnosis, she said. This is often referred to as the broader autism phenotype (BAP), which affects an estimated 19% of unaffected siblings at age 12 months.
The prospective study followed 26 infants who had older siblings with ASD, the high-risk group, to 33 infants whose older siblings had typical development. None of these infants met the diagnostic criteria for ASD at 18, 24, and 36 months. Parents completed a questionnaire, the Brief Infant-Toddler Social Emotional Assessment (BITSEA), when the child was 12, 18, and 24 months.
Based on the results of these parent reports, unaffected siblings of the children with autism "displayed more ASD-relevant problem behaviors than the unaffected infants with typically developing siblings," Dr. Carter said. The former also appear to show "a somewhat different trajectory of these behaviors over time," with scores that were significantly different at 18 months from the low-risk group but "converging" by the time they were aged 24 months, when they appeared to be comparable.
These results support the importance of monitoring younger siblings of children diagnosed with ASD for the emergence of ASD symptoms with routine developmental screening and surveillance, "as they may benefit from early intervention services," Dr. Carter said. But even without such services, children may follow the "self-righting trajectory that takes them back on a normative developmental course," which was evident in the study, she added. (She noted that the limitations of the study included the small sample size in the Boston area. In addition, the participants, were mostly white and had higher than average annual family incomes above $65,000.)
Dr. Carter said future challenges will be to distinguish between three different groups of infants who do not meet the full criteria for an ASD diagnosis: Those manifesting symptoms that mark the beginning of the pathway toward ASD or the emergence of ASD (prodromal ASD); those whose symptoms are likely to resolve; and those who show signs of ASD that "will endure and reflect the broader autism phenotype." The primary target for early screening and detection is the identification of the prodromal group, she added.
Other studies involving high-risk siblings include the UC Davis Infant Sibling Study, which is prospectively following infants with an older sibling who has autism and infants with an older sibling who is developing typically, following the infants up to age 3 years, through the window of autism risk. The second phase of the study followed a small group of infants, enrolled between 6 and 9 months, to age 3 years, with testing that included retrospective and prospective parent ratings, and prospective examiner ratings. Most (89%) of the children who developed ASD showed a regressive onset in terms of their social communication behaviors.
Parents were able to identify those trajectories, indicating impending ASD, with prospective reports, "but not as well retrospectively" with the Autism Diagnostic Interview-Revised (ADI-R), which was accurate in only 30% of cases," said Meghan Miller, Ph.D., a postdoctoral fellow at the University of California, Davis, MIND Institute. These results suggest that while parental reports of regression on the ADI-R can be accurate, they might only pick up "the tip of the iceberg," she noted.
The findings, based on a small sample, raise concerns about the use of the ADI-R and other retrospective measures in research, but do provide "some hope for early screening using parent report data," Dr. Miller pointed out. Such reports would be more feasible than neuroimaging, eye tracking, and other sophisticated testing that could be used over several well child visits, to detect declines and help determine which children should be further evaluated further or referred for interventions, she added. Phase III of the study is currently underway.
In other sibling studies, Dr. Saulnier discussed the use of eye-tracking testing in studies that prospectively followed high-risk infants whose siblings have been diagnosed with ASD and low-risk infants with typically developing siblings.
Under eye-tracking methodology, the child watches prerecorded movies on a monitor of women acting as caregivers, singing, and talking to an infant. It is well established that when viewing a social scene, typically developing adults focus on the eye region of the face, but those with autism look less at the eyes and significantly more at the mouth, body, and objects in the social scene. Those findings have been replicated in school-aged children, adolescents, and toddlers down to aged 2 years, she noted.
In infant studies, eye-tracking testing is performed at least 10 times from birth to age 3, with vocal recordings and clinical assessments at different periods.
Summarizing the findings, Dr. Saulnier said infants at low risk who have a typical outcome show a steady fixation of eye gaze from birth. But infants who are at high risk for ASD and "develop ASD by age 3 show a rapid decline in eye fixation between 2 and 6 months of age that is more predictive of ASD than any clinical measure."
Autism cannot be diagnosed with this test alone, she said. (Most of this work was conducted at the Yale Child Study Center, before she moved to Emory, she said.)
She provided examples of a 5-month-old low-risk infant, who developed typically and focused at the eye region of the face, contrasting with another 5-month old infant, who went on to develop autism by agd 3, who was not looking at the eye region of the face, focusing on the mouth, body, and objects.
Another example she showed was an infant who had normal results at 3 and 6 months, but by 9 months, the researchers started to see a shift, which she described as the "unfolding of autism." This child was diagnosed with autism at 12 months. She added, however, that it is rare to see that symptomatology so early and pronounced.
While it would be assumed that the children who go on to develop autism manifest the lack of eye fixation from birth, another finding from eye fixation studies is that the opposite is true, and "babies who went on to develop autism actually had significantly more eye fixation in the first 2 months of life," Dr. Saulnier said.
Typically developing infants maintain this focus on the eyes, "but whatever is happening in autism to make social monitoring not as salient, they’re trailing off, and you’re seeing this derailment in this eye fixation."
Other results of eye-tracking sibling studies might provide information about a window of opportunity – at about age 9-12 months – for an intervention "that could capitalize on the potential for resilience, she noted.
In infants whose older siblings have ASD and are either unaffected or have "shadow symptoms" of autism at age 3 years, eye-tracking results indicate that after reduced eye fixation, they exhibit a "course correction" in increased eye fixation that appears to start around 9 months of age.
Why this occurs is not understood, but this observation sheds some light on a possible window of time during which something could be done, before autism fully unfolds, such as coaching strategies for parents on keeping their child engaged "to produce a course correction if it wasn’t naturally going to occur," Dr. Saulnier said.
She and her associates are now conducting a randomized, controlled study of infants at risk at 12 months, evaluating a potential way to correct the course.
Among the goals in the future is to reduce the mean age of an autism diagnosis so that, once recognized, effective interventions can be started that could affect outcomes, she said. Although autism can be reliably diagnosed by 18-24 months, the mean age of ASD diagnosis is currently about 4.5 years, she noted.
Dr. Carter disclosed that she is one of the developers of the BITSEA screening tool, used in the study she presented. Dr. Saulnier had no disclosures; the studies she presented received support from the National Institute of Mental Health, the National Institute of Child Health and Human Development, the Marcus Foundation, and the Whitehead and Woodruff Foundations. Dr. Miller said the UCSD study has been funded by NIMH, Autism Speaks, and the MINDlist.
A tutorial on recognizing the early signs of ASD, developed by the Kennedy Krieger Institute, Baltimore,is available at https://www.youtube.com/watch?v=YtvP5A5OHpU.
WASHINGTON – Studies of the younger siblings of children with autism spectrum disorders are beginning to yield information that has important implications for screening, reducing the age at diagnosis, and identifying the window of time during which still unidentified interventions might be able to affect the course, according to experts who spoke at the annual convention of the American Psychological Association.
During a symposium on early development and predictors of outcomes in infant siblings of children with autism, several experts summarized the results of studies evaluating the emergence of autism spectrum disorders (ASD) and ASD-like symptoms in the younger siblings of children with ASD, as well as tools that are being studied in high-risk sibling studies as potential methods of diagnosing ASD earlier.
The risk of ASD is 1 in 68 in the general population, but is almost 1 in 5 among the siblings of children diagnosed with ASD. An additional one in five siblings will have "shadow symptoms" of autism, which refers to children with some of the symptomatology but not enough to merit a diagnosis. In addition, another 1 in 10 will have nonautism developmental delays.
Therefore, about 50% of infant siblings of children with ASD "are vulnerable in their development to some degree, so that’s why we’re studying them so closely," said one of the speakers, Celine Saulnier, Ph.D., clinical director for research at the Marcus Autism Center, Emory University, Atlanta.
During the symposium, Alice Carter, Ph.D., professor of clinical psychology at the University of Massachusetts, Boston, provided the results of a prospective study that identified high-risk siblings who exhibited some ASD-like behaviors early on but were not diagnosed with autism at age 3.
A growing body of literature focusing on milder ASD symptoms in relatives of people diagnosed with ASD indicates that about 20%-30% of children who are at high genetic risk develop some symptoms of autism but not enough symptoms or symptoms that are severe enough to make the diagnosis, she said. This is often referred to as the broader autism phenotype (BAP), which affects an estimated 19% of unaffected siblings at age 12 months.
The prospective study followed 26 infants who had older siblings with ASD, the high-risk group, to 33 infants whose older siblings had typical development. None of these infants met the diagnostic criteria for ASD at 18, 24, and 36 months. Parents completed a questionnaire, the Brief Infant-Toddler Social Emotional Assessment (BITSEA), when the child was 12, 18, and 24 months.
Based on the results of these parent reports, unaffected siblings of the children with autism "displayed more ASD-relevant problem behaviors than the unaffected infants with typically developing siblings," Dr. Carter said. The former also appear to show "a somewhat different trajectory of these behaviors over time," with scores that were significantly different at 18 months from the low-risk group but "converging" by the time they were aged 24 months, when they appeared to be comparable.
These results support the importance of monitoring younger siblings of children diagnosed with ASD for the emergence of ASD symptoms with routine developmental screening and surveillance, "as they may benefit from early intervention services," Dr. Carter said. But even without such services, children may follow the "self-righting trajectory that takes them back on a normative developmental course," which was evident in the study, she added. (She noted that the limitations of the study included the small sample size in the Boston area. In addition, the participants, were mostly white and had higher than average annual family incomes above $65,000.)
Dr. Carter said future challenges will be to distinguish between three different groups of infants who do not meet the full criteria for an ASD diagnosis: Those manifesting symptoms that mark the beginning of the pathway toward ASD or the emergence of ASD (prodromal ASD); those whose symptoms are likely to resolve; and those who show signs of ASD that "will endure and reflect the broader autism phenotype." The primary target for early screening and detection is the identification of the prodromal group, she added.
Other studies involving high-risk siblings include the UC Davis Infant Sibling Study, which is prospectively following infants with an older sibling who has autism and infants with an older sibling who is developing typically, following the infants up to age 3 years, through the window of autism risk. The second phase of the study followed a small group of infants, enrolled between 6 and 9 months, to age 3 years, with testing that included retrospective and prospective parent ratings, and prospective examiner ratings. Most (89%) of the children who developed ASD showed a regressive onset in terms of their social communication behaviors.
Parents were able to identify those trajectories, indicating impending ASD, with prospective reports, "but not as well retrospectively" with the Autism Diagnostic Interview-Revised (ADI-R), which was accurate in only 30% of cases," said Meghan Miller, Ph.D., a postdoctoral fellow at the University of California, Davis, MIND Institute. These results suggest that while parental reports of regression on the ADI-R can be accurate, they might only pick up "the tip of the iceberg," she noted.
The findings, based on a small sample, raise concerns about the use of the ADI-R and other retrospective measures in research, but do provide "some hope for early screening using parent report data," Dr. Miller pointed out. Such reports would be more feasible than neuroimaging, eye tracking, and other sophisticated testing that could be used over several well child visits, to detect declines and help determine which children should be further evaluated further or referred for interventions, she added. Phase III of the study is currently underway.
In other sibling studies, Dr. Saulnier discussed the use of eye-tracking testing in studies that prospectively followed high-risk infants whose siblings have been diagnosed with ASD and low-risk infants with typically developing siblings.
Under eye-tracking methodology, the child watches prerecorded movies on a monitor of women acting as caregivers, singing, and talking to an infant. It is well established that when viewing a social scene, typically developing adults focus on the eye region of the face, but those with autism look less at the eyes and significantly more at the mouth, body, and objects in the social scene. Those findings have been replicated in school-aged children, adolescents, and toddlers down to aged 2 years, she noted.
In infant studies, eye-tracking testing is performed at least 10 times from birth to age 3, with vocal recordings and clinical assessments at different periods.
Summarizing the findings, Dr. Saulnier said infants at low risk who have a typical outcome show a steady fixation of eye gaze from birth. But infants who are at high risk for ASD and "develop ASD by age 3 show a rapid decline in eye fixation between 2 and 6 months of age that is more predictive of ASD than any clinical measure."
Autism cannot be diagnosed with this test alone, she said. (Most of this work was conducted at the Yale Child Study Center, before she moved to Emory, she said.)
She provided examples of a 5-month-old low-risk infant, who developed typically and focused at the eye region of the face, contrasting with another 5-month old infant, who went on to develop autism by agd 3, who was not looking at the eye region of the face, focusing on the mouth, body, and objects.
Another example she showed was an infant who had normal results at 3 and 6 months, but by 9 months, the researchers started to see a shift, which she described as the "unfolding of autism." This child was diagnosed with autism at 12 months. She added, however, that it is rare to see that symptomatology so early and pronounced.
While it would be assumed that the children who go on to develop autism manifest the lack of eye fixation from birth, another finding from eye fixation studies is that the opposite is true, and "babies who went on to develop autism actually had significantly more eye fixation in the first 2 months of life," Dr. Saulnier said.
Typically developing infants maintain this focus on the eyes, "but whatever is happening in autism to make social monitoring not as salient, they’re trailing off, and you’re seeing this derailment in this eye fixation."
Other results of eye-tracking sibling studies might provide information about a window of opportunity – at about age 9-12 months – for an intervention "that could capitalize on the potential for resilience, she noted.
In infants whose older siblings have ASD and are either unaffected or have "shadow symptoms" of autism at age 3 years, eye-tracking results indicate that after reduced eye fixation, they exhibit a "course correction" in increased eye fixation that appears to start around 9 months of age.
Why this occurs is not understood, but this observation sheds some light on a possible window of time during which something could be done, before autism fully unfolds, such as coaching strategies for parents on keeping their child engaged "to produce a course correction if it wasn’t naturally going to occur," Dr. Saulnier said.
She and her associates are now conducting a randomized, controlled study of infants at risk at 12 months, evaluating a potential way to correct the course.
Among the goals in the future is to reduce the mean age of an autism diagnosis so that, once recognized, effective interventions can be started that could affect outcomes, she said. Although autism can be reliably diagnosed by 18-24 months, the mean age of ASD diagnosis is currently about 4.5 years, she noted.
Dr. Carter disclosed that she is one of the developers of the BITSEA screening tool, used in the study she presented. Dr. Saulnier had no disclosures; the studies she presented received support from the National Institute of Mental Health, the National Institute of Child Health and Human Development, the Marcus Foundation, and the Whitehead and Woodruff Foundations. Dr. Miller said the UCSD study has been funded by NIMH, Autism Speaks, and the MINDlist.
A tutorial on recognizing the early signs of ASD, developed by the Kennedy Krieger Institute, Baltimore,is available at https://www.youtube.com/watch?v=YtvP5A5OHpU.
EXPERT ANAYSIS FROM THE 2014 APA CONVENTION
