User login
FDA approves idelalisib for three leukemia and lymphoma indications
The oral kinase inhibitor idelalisib has been approved for treating patients with relapsed chronic lymphocytic leukemia, follicular lymphoma, and small lymphocytic lymphoma, with a boxed warning about fatal and serious toxicities associated with treatment, the Food and Drug Administration announced on July 23.
The three approved indications for idelalisib – administered at a recommended starting dose of 150 mg, twice a day – are for:
![]()
• Relapsed chronic lymphocytic leukemia (CLL), in combination with rituximab, in patients for whom rituximab alone would be considered appropriate therapy due to other comorbidities. This is a traditional approval based on progression-free survival (PFS) data.
• Relapsed follicular B-cell non-Hodgkin’s lymphoma (FL) in patients who have received at least two prior systemic therapies.
• Relapsed small lymphocytic lymphoma (SLL) in patients who have received at least two prior systemic therapies.
The FL and SLL indications, based on objective response rates in one study, are accelerated approvals, which provides patients with serious or life-threatening diseases earlier access to a promising drug based on an effect on a surrogate endpoint considered " reasonably likely to predict clinical benefit," according to the FDA. Full approval is contingent on the manufacturer conducting trials confirming clinical benefits; if they do not, the FDA can withdraw the approval.
Idelalisib is being marketed as Zydelig, by Gilead Sciences. There is also a Risk Evaluation and Mitigation Strategy (REMS) in place for the drug, which includes a communication plan to ensure that prescribers are fully informed about treatment-associated risks. The boxed warning lists the risks of fatal and/or serious hepatoxicity (affecting 14% of treated patients); fatal and/or serious and severe diarrhea or colitis (also affecting 14% of treated patients); as well as fatal and serious pneumonitis; and fatal and serious intestinal perforation.
Idelalisib is an oral inhibitor of phosphoinositide 3-kinase (PI3K) delta, "a protein that plays a role in the activation, proliferation, and viability of B cells," according to a Gilead statement announcing the approval, which added that PI3K delta signaling "is active in many B-cell leukemias and lymphomas, and by inhibiting the protein, Zydelig blocks several cellular signaling pathways that drive B-cell viability."
The FDA granted a full approval for the CLL indication, based on a phase III study of 220 patients, which was stopped early in October 2013 at the first-specified interim analysis. Median PFS was not reached among those randomized to on idelalisib plus rituximab, but was at least 10.7 months, and was 5.5 months among those randomized to placebo plus rituximab (N. Engl. J. Med. 2014;370:997-1007). "Results from a second interim analysis continued to show a statistically significant improvement for Zydelig and Rituxan over placebo and Rituxan," the FDA statement said.
The accelerated approvals for relapsed FL and relapsed SLL were based on a single-arm phase II study of 123 patients refractory to rituximab and chemotherapy including alkylating agents, treated with idelalisib. The objective response rates were 54% among those with relapsed FL, and 58% of those with SLL in the study (N. Engl. J. Med. 2014;370:1008-18).
Common adverse events associated with treatment included diarrhea, fever, fatigue, nausea, cough, pneumonia, abdominal pain, chills, and rash; and common lab abnormalities associated with treatment included neutropenia, hypertriglyceridemia, hyperglycemia, and liver enzyme elevations, according to the FDA.
In the FDA statement, Dr. Richard Pazdur, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, pointed out that in less than a year, "We have seen considerable progress in the availability of treatments for chronic lymphocytic leukemia." The other treatments for CLL are obinutuzumab (Gazyva), approved in November 2013; ibrutinib (Imbruvica), approved in February 2014; and ofatumumab (Arzerra) approved in April 2014.
Prescribing information is available at the FDA website.
The oral kinase inhibitor idelalisib has been approved for treating patients with relapsed chronic lymphocytic leukemia, follicular lymphoma, and small lymphocytic lymphoma, with a boxed warning about fatal and serious toxicities associated with treatment, the Food and Drug Administration announced on July 23.
The three approved indications for idelalisib – administered at a recommended starting dose of 150 mg, twice a day – are for:
![]()
• Relapsed chronic lymphocytic leukemia (CLL), in combination with rituximab, in patients for whom rituximab alone would be considered appropriate therapy due to other comorbidities. This is a traditional approval based on progression-free survival (PFS) data.
• Relapsed follicular B-cell non-Hodgkin’s lymphoma (FL) in patients who have received at least two prior systemic therapies.
• Relapsed small lymphocytic lymphoma (SLL) in patients who have received at least two prior systemic therapies.
The FL and SLL indications, based on objective response rates in one study, are accelerated approvals, which provides patients with serious or life-threatening diseases earlier access to a promising drug based on an effect on a surrogate endpoint considered " reasonably likely to predict clinical benefit," according to the FDA. Full approval is contingent on the manufacturer conducting trials confirming clinical benefits; if they do not, the FDA can withdraw the approval.
Idelalisib is being marketed as Zydelig, by Gilead Sciences. There is also a Risk Evaluation and Mitigation Strategy (REMS) in place for the drug, which includes a communication plan to ensure that prescribers are fully informed about treatment-associated risks. The boxed warning lists the risks of fatal and/or serious hepatoxicity (affecting 14% of treated patients); fatal and/or serious and severe diarrhea or colitis (also affecting 14% of treated patients); as well as fatal and serious pneumonitis; and fatal and serious intestinal perforation.
Idelalisib is an oral inhibitor of phosphoinositide 3-kinase (PI3K) delta, "a protein that plays a role in the activation, proliferation, and viability of B cells," according to a Gilead statement announcing the approval, which added that PI3K delta signaling "is active in many B-cell leukemias and lymphomas, and by inhibiting the protein, Zydelig blocks several cellular signaling pathways that drive B-cell viability."
The FDA granted a full approval for the CLL indication, based on a phase III study of 220 patients, which was stopped early in October 2013 at the first-specified interim analysis. Median PFS was not reached among those randomized to on idelalisib plus rituximab, but was at least 10.7 months, and was 5.5 months among those randomized to placebo plus rituximab (N. Engl. J. Med. 2014;370:997-1007). "Results from a second interim analysis continued to show a statistically significant improvement for Zydelig and Rituxan over placebo and Rituxan," the FDA statement said.
The accelerated approvals for relapsed FL and relapsed SLL were based on a single-arm phase II study of 123 patients refractory to rituximab and chemotherapy including alkylating agents, treated with idelalisib. The objective response rates were 54% among those with relapsed FL, and 58% of those with SLL in the study (N. Engl. J. Med. 2014;370:1008-18).
Common adverse events associated with treatment included diarrhea, fever, fatigue, nausea, cough, pneumonia, abdominal pain, chills, and rash; and common lab abnormalities associated with treatment included neutropenia, hypertriglyceridemia, hyperglycemia, and liver enzyme elevations, according to the FDA.
In the FDA statement, Dr. Richard Pazdur, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, pointed out that in less than a year, "We have seen considerable progress in the availability of treatments for chronic lymphocytic leukemia." The other treatments for CLL are obinutuzumab (Gazyva), approved in November 2013; ibrutinib (Imbruvica), approved in February 2014; and ofatumumab (Arzerra) approved in April 2014.
Prescribing information is available at the FDA website.
The oral kinase inhibitor idelalisib has been approved for treating patients with relapsed chronic lymphocytic leukemia, follicular lymphoma, and small lymphocytic lymphoma, with a boxed warning about fatal and serious toxicities associated with treatment, the Food and Drug Administration announced on July 23.
The three approved indications for idelalisib – administered at a recommended starting dose of 150 mg, twice a day – are for:
![]()
• Relapsed chronic lymphocytic leukemia (CLL), in combination with rituximab, in patients for whom rituximab alone would be considered appropriate therapy due to other comorbidities. This is a traditional approval based on progression-free survival (PFS) data.
• Relapsed follicular B-cell non-Hodgkin’s lymphoma (FL) in patients who have received at least two prior systemic therapies.
• Relapsed small lymphocytic lymphoma (SLL) in patients who have received at least two prior systemic therapies.
The FL and SLL indications, based on objective response rates in one study, are accelerated approvals, which provides patients with serious or life-threatening diseases earlier access to a promising drug based on an effect on a surrogate endpoint considered " reasonably likely to predict clinical benefit," according to the FDA. Full approval is contingent on the manufacturer conducting trials confirming clinical benefits; if they do not, the FDA can withdraw the approval.
Idelalisib is being marketed as Zydelig, by Gilead Sciences. There is also a Risk Evaluation and Mitigation Strategy (REMS) in place for the drug, which includes a communication plan to ensure that prescribers are fully informed about treatment-associated risks. The boxed warning lists the risks of fatal and/or serious hepatoxicity (affecting 14% of treated patients); fatal and/or serious and severe diarrhea or colitis (also affecting 14% of treated patients); as well as fatal and serious pneumonitis; and fatal and serious intestinal perforation.
Idelalisib is an oral inhibitor of phosphoinositide 3-kinase (PI3K) delta, "a protein that plays a role in the activation, proliferation, and viability of B cells," according to a Gilead statement announcing the approval, which added that PI3K delta signaling "is active in many B-cell leukemias and lymphomas, and by inhibiting the protein, Zydelig blocks several cellular signaling pathways that drive B-cell viability."
The FDA granted a full approval for the CLL indication, based on a phase III study of 220 patients, which was stopped early in October 2013 at the first-specified interim analysis. Median PFS was not reached among those randomized to on idelalisib plus rituximab, but was at least 10.7 months, and was 5.5 months among those randomized to placebo plus rituximab (N. Engl. J. Med. 2014;370:997-1007). "Results from a second interim analysis continued to show a statistically significant improvement for Zydelig and Rituxan over placebo and Rituxan," the FDA statement said.
The accelerated approvals for relapsed FL and relapsed SLL were based on a single-arm phase II study of 123 patients refractory to rituximab and chemotherapy including alkylating agents, treated with idelalisib. The objective response rates were 54% among those with relapsed FL, and 58% of those with SLL in the study (N. Engl. J. Med. 2014;370:1008-18).
Common adverse events associated with treatment included diarrhea, fever, fatigue, nausea, cough, pneumonia, abdominal pain, chills, and rash; and common lab abnormalities associated with treatment included neutropenia, hypertriglyceridemia, hyperglycemia, and liver enzyme elevations, according to the FDA.
In the FDA statement, Dr. Richard Pazdur, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, pointed out that in less than a year, "We have seen considerable progress in the availability of treatments for chronic lymphocytic leukemia." The other treatments for CLL are obinutuzumab (Gazyva), approved in November 2013; ibrutinib (Imbruvica), approved in February 2014; and ofatumumab (Arzerra) approved in April 2014.
Prescribing information is available at the FDA website.
Study provides cancer risk estimates among women undergoing morcellation
An estimated 1 in 370 women who undergo electric power morcellation during a minimally invasive hysterectomy have uterine cancer, with the risk of cancer and endometrial hyperplasia markedly increasing with age, according to an analysis using an insurance database of more than 500 U.S. hospitals.

The estimate, published online in JAMA on July 22, is close to the Food and Drug Administration’s estimate that about 1 in 350 women undergoing hysterectomy or myomectomy for presumed fibroids have an unsuspected uterine sarcoma and is higher than historical estimates provided in the literature. The FDA’s estimate, first reported when the agency issued a safety communication in April discouraging the use of laparoscopic power morcellators (LPMs) during a hysterectomy or myomectomy because of the risk of disseminating cancerous tissue and upstaging disease, was based on 9 U.S. and international studies of women treated from 1983 to 2011. The risk of an unsuspected leiomyosarcoma was about 1 in 500.
But unlike the FDA analysis, the most recent analysis, conducted by Dr. Jason Wright and his associates at Columbia University, New York, specifically addressed the risk associated with morcellation. The estimate was based on the records of 232,882 women who underwent a minimally invasive hysterectomy from 2006 to 2012 obtained from a database that represents about 15% of U.S. hospitalizations. Morcellators were used in almost 16% (36,470) of the women, and there were 99 cases of uterine cancers, for a prevalence of 27/10,000 – about one in 370.
Among the women who underwent morcellation, the strongest risk factor for abnormal pathology, either for cancer or any of the precancerous changes, was advanced age. Compared with women under age 40 years, the prevalence ratio for uterine malignancy was 1.42 among those aged 40-44 and 2.55 for those aged 45-49, increasing to 4.97 among those aged 50-54 years, 19.37 among those aged 55-59 years, 21.36 among those aged 60-64 years – and 35.97 among those aged 65 and older.
The researchers also identified cases of endometrial hyperplasia, other gynecologic cancers, and smooth muscle tumors of uncertain malignant potential. The risk of endometrial hyperplasia also increased significantly with age, compared with women under age 40, with prevalence ratios of 1.17 among those aged 40-44 (not statistically significant) and 1.71 among those aged 45-49, to 4.07 among those aged 50-54 years, to 10.21 among those aged 65 years and older. The results are reported in a research letter (doi:10.1001/jama.2014.9005)
Despite the availability of power morcellators for 20 years, "few studies have described the prevalence of unexpected pathology at the time of hysterectomy," Dr. Wright and his associates wrote. While the analysis had limitations, including the lack of long-term follow-up and not being able to verify the pathology results, they concluded that patients who may be undergoing morcellation "should be adequately counseled about the prevalence of cancerous and precancerous conditions prior to undergoing the procedure."

One of the strengths and unique aspects of this study was being able to identify a large population of women who specifically had morcellation and a hysterectomy, providing a population-based estimate of the prevalence of cancer in this group, Dr. Wright said in an interview. This is slightly different than other estimates and studies, which were not specific to morcellation, including those that looked at the incidence of sarcoma among women who had a hysterectomy. "We could not separate out epithelial endometrial cancers from uterine sarcomas, so this is an estimate of any malignancy within the uterus," so "probably a high proportion of women who underwent morcellation underwent the procedure for fibroid uterus, so the chance of sarcomas is probably higher in these patients than in the general population."
The study did not allow evaluation of whether the use of morcellation increased the risk of dissemination of cancer, which "certainly warrants further study," added Dr. Wright, chief of the division of gynecologic oncology at Columbia.
The lack of data has been one of the major problems surrounding morcellation, with very few studies specifically looking at data that can be used to help guide patients and clinicians. Dr. Wright and his associates hope that their results can help guide patients and clinicians.
"There’s undoubtedly a risk of cancer and precancerous changes in women who undergo morcellation ... and [patients and clinicians] need to weigh that risk," he said. "But certainly morcellation may allow some women to undergo a minimally invasive surgery who otherwise require laparotomy, and the complications and recovery are much easier with minimally invasive surgery, when it’s feasible."
Dr. Charles E. Miller, director of minimally invasive gynecologic surgery at Advocate Lutheran General Hospital, Park Ridge, Ill., described these data as "compelling," with well-presented information that stratifies risk by age that can be used to help properly counsel patients.
The marked difference in risk among women under age 40 is particularly important, he said. Dr. Miller performs about 250 morcellations per year and has had only one patient with a sarcoma, a woman in her mid-40s, which he said reflects the younger age of his patients.
"If we can better identify age groups where that risk is higher and do the treatment that is appropriate in that age group, then I think we’ve come a long, long way," he said in an interview. There are always outliers, and unfortunately, women in younger age groups develop sarcomas, but there are also the risks of more invasive surgery that should be considered, he added.
"There’s always going to be risk, and there’s always going to be decision making with surgery," and while there is a need for better ways to identify those patients at risk, currently, "all we can hang our hat on now is stratifying [risk] with age," Dr. Miller said.
On July 10-11, the FDA held a meeting of its Obstetrics and Gynecology Devices panel, to review the safety of LPMs during uterine surgery for fibroids, Among the questions the panel was asked was whether there were characteristics of patients – such as age, physical exam findings, and imaging test results – that could help identify patients who might be at a higher risk of a sarcoma.
The FDA is currently reviewing the safety of LPMs in women undergoing surgery for presumably benign fibroids, an issue that has received widespread attention this year and resulted in the FDA’s safety communication in April – largely due to the case of Dr. Amy Reed, an anesthesiologist who was diagnosed with stage IV leiomyosarcoma after undergoing a hysterectomy with morcellation at age 40 last year. She and her husband, Dr. Hooman Noorchashm, a cardiothoracic surgeon, are leading a campaign to highlight these risks, including a petition on change.org calling for a halt to morcellation during minimally invasive and robotic-assisted hysterectomy and myomectomy.
The authors of the JAMA report had no disclosures. Dr. Wright and one of the other authors, are recipients of National Cancer Institute (NCI) grants; another author is a recipient of an NCI fellowship. Dr. Miller disclosed that he is a consultant to Ethicon Endo-Surgery. Ethicon is a morcellator manufacturer.
An estimated 1 in 370 women who undergo electric power morcellation during a minimally invasive hysterectomy have uterine cancer, with the risk of cancer and endometrial hyperplasia markedly increasing with age, according to an analysis using an insurance database of more than 500 U.S. hospitals.
The estimate, published online in JAMA on July 22, is close to the Food and Drug Administration’s estimate that about 1 in 350 women undergoing hysterectomy or myomectomy for presumed fibroids have an unsuspected uterine sarcoma and is higher than historical estimates provided in the literature. The FDA’s estimate, first reported when the agency issued a safety communication in April discouraging the use of laparoscopic power morcellators (LPMs) during a hysterectomy or myomectomy because of the risk of disseminating cancerous tissue and upstaging disease, was based on 9 U.S. and international studies of women treated from 1983 to 2011. The risk of an unsuspected leiomyosarcoma was about 1 in 500.
But unlike the FDA analysis, the most recent analysis, conducted by Dr. Jason Wright and his associates at Columbia University, New York, specifically addressed the risk associated with morcellation. The estimate was based on the records of 232,882 women who underwent a minimally invasive hysterectomy from 2006 to 2012 obtained from a database that represents about 15% of U.S. hospitalizations. Morcellators were used in almost 16% (36,470) of the women, and there were 99 cases of uterine cancers, for a prevalence of 27/10,000 – about one in 370.
Among the women who underwent morcellation, the strongest risk factor for abnormal pathology, either for cancer or any of the precancerous changes, was advanced age. Compared with women under age 40 years, the prevalence ratio for uterine malignancy was 1.42 among those aged 40-44 and 2.55 for those aged 45-49, increasing to 4.97 among those aged 50-54 years, 19.37 among those aged 55-59 years, 21.36 among those aged 60-64 years – and 35.97 among those aged 65 and older.
The researchers also identified cases of endometrial hyperplasia, other gynecologic cancers, and smooth muscle tumors of uncertain malignant potential. The risk of endometrial hyperplasia also increased significantly with age, compared with women under age 40, with prevalence ratios of 1.17 among those aged 40-44 (not statistically significant) and 1.71 among those aged 45-49, to 4.07 among those aged 50-54 years, to 10.21 among those aged 65 years and older. The results are reported in a research letter (doi:10.1001/jama.2014.9005)
Despite the availability of power morcellators for 20 years, "few studies have described the prevalence of unexpected pathology at the time of hysterectomy," Dr. Wright and his associates wrote. While the analysis had limitations, including the lack of long-term follow-up and not being able to verify the pathology results, they concluded that patients who may be undergoing morcellation "should be adequately counseled about the prevalence of cancerous and precancerous conditions prior to undergoing the procedure."
One of the strengths and unique aspects of this study was being able to identify a large population of women who specifically had morcellation and a hysterectomy, providing a population-based estimate of the prevalence of cancer in this group, Dr. Wright said in an interview. This is slightly different than other estimates and studies, which were not specific to morcellation, including those that looked at the incidence of sarcoma among women who had a hysterectomy. "We could not separate out epithelial endometrial cancers from uterine sarcomas, so this is an estimate of any malignancy within the uterus," so "probably a high proportion of women who underwent morcellation underwent the procedure for fibroid uterus, so the chance of sarcomas is probably higher in these patients than in the general population."
The study did not allow evaluation of whether the use of morcellation increased the risk of dissemination of cancer, which "certainly warrants further study," added Dr. Wright, chief of the division of gynecologic oncology at Columbia.
The lack of data has been one of the major problems surrounding morcellation, with very few studies specifically looking at data that can be used to help guide patients and clinicians. Dr. Wright and his associates hope that their results can help guide patients and clinicians.
"There’s undoubtedly a risk of cancer and precancerous changes in women who undergo morcellation ... and [patients and clinicians] need to weigh that risk," he said. "But certainly morcellation may allow some women to undergo a minimally invasive surgery who otherwise require laparotomy, and the complications and recovery are much easier with minimally invasive surgery, when it’s feasible."
Dr. Charles E. Miller, director of minimally invasive gynecologic surgery at Advocate Lutheran General Hospital, Park Ridge, Ill., described these data as "compelling," with well-presented information that stratifies risk by age that can be used to help properly counsel patients.
The marked difference in risk among women under age 40 is particularly important, he said. Dr. Miller performs about 250 morcellations per year and has had only one patient with a sarcoma, a woman in her mid-40s, which he said reflects the younger age of his patients.
"If we can better identify age groups where that risk is higher and do the treatment that is appropriate in that age group, then I think we’ve come a long, long way," he said in an interview. There are always outliers, and unfortunately, women in younger age groups develop sarcomas, but there are also the risks of more invasive surgery that should be considered, he added.
"There’s always going to be risk, and there’s always going to be decision making with surgery," and while there is a need for better ways to identify those patients at risk, currently, "all we can hang our hat on now is stratifying [risk] with age," Dr. Miller said.
On July 10-11, the FDA held a meeting of its Obstetrics and Gynecology Devices panel, to review the safety of LPMs during uterine surgery for fibroids, Among the questions the panel was asked was whether there were characteristics of patients – such as age, physical exam findings, and imaging test results – that could help identify patients who might be at a higher risk of a sarcoma.
The FDA is currently reviewing the safety of LPMs in women undergoing surgery for presumably benign fibroids, an issue that has received widespread attention this year and resulted in the FDA’s safety communication in April – largely due to the case of Dr. Amy Reed, an anesthesiologist who was diagnosed with stage IV leiomyosarcoma after undergoing a hysterectomy with morcellation at age 40 last year. She and her husband, Dr. Hooman Noorchashm, a cardiothoracic surgeon, are leading a campaign to highlight these risks, including a petition on change.org calling for a halt to morcellation during minimally invasive and robotic-assisted hysterectomy and myomectomy.
The authors of the JAMA report had no disclosures. Dr. Wright and one of the other authors, are recipients of National Cancer Institute (NCI) grants; another author is a recipient of an NCI fellowship. Dr. Miller disclosed that he is a consultant to Ethicon Endo-Surgery. Ethicon is a morcellator manufacturer.
An estimated 1 in 370 women who undergo electric power morcellation during a minimally invasive hysterectomy have uterine cancer, with the risk of cancer and endometrial hyperplasia markedly increasing with age, according to an analysis using an insurance database of more than 500 U.S. hospitals.
The estimate, published online in JAMA on July 22, is close to the Food and Drug Administration’s estimate that about 1 in 350 women undergoing hysterectomy or myomectomy for presumed fibroids have an unsuspected uterine sarcoma and is higher than historical estimates provided in the literature. The FDA’s estimate, first reported when the agency issued a safety communication in April discouraging the use of laparoscopic power morcellators (LPMs) during a hysterectomy or myomectomy because of the risk of disseminating cancerous tissue and upstaging disease, was based on 9 U.S. and international studies of women treated from 1983 to 2011. The risk of an unsuspected leiomyosarcoma was about 1 in 500.
But unlike the FDA analysis, the most recent analysis, conducted by Dr. Jason Wright and his associates at Columbia University, New York, specifically addressed the risk associated with morcellation. The estimate was based on the records of 232,882 women who underwent a minimally invasive hysterectomy from 2006 to 2012 obtained from a database that represents about 15% of U.S. hospitalizations. Morcellators were used in almost 16% (36,470) of the women, and there were 99 cases of uterine cancers, for a prevalence of 27/10,000 – about one in 370.
Among the women who underwent morcellation, the strongest risk factor for abnormal pathology, either for cancer or any of the precancerous changes, was advanced age. Compared with women under age 40 years, the prevalence ratio for uterine malignancy was 1.42 among those aged 40-44 and 2.55 for those aged 45-49, increasing to 4.97 among those aged 50-54 years, 19.37 among those aged 55-59 years, 21.36 among those aged 60-64 years – and 35.97 among those aged 65 and older.
The researchers also identified cases of endometrial hyperplasia, other gynecologic cancers, and smooth muscle tumors of uncertain malignant potential. The risk of endometrial hyperplasia also increased significantly with age, compared with women under age 40, with prevalence ratios of 1.17 among those aged 40-44 (not statistically significant) and 1.71 among those aged 45-49, to 4.07 among those aged 50-54 years, to 10.21 among those aged 65 years and older. The results are reported in a research letter (doi:10.1001/jama.2014.9005)
Despite the availability of power morcellators for 20 years, "few studies have described the prevalence of unexpected pathology at the time of hysterectomy," Dr. Wright and his associates wrote. While the analysis had limitations, including the lack of long-term follow-up and not being able to verify the pathology results, they concluded that patients who may be undergoing morcellation "should be adequately counseled about the prevalence of cancerous and precancerous conditions prior to undergoing the procedure."
One of the strengths and unique aspects of this study was being able to identify a large population of women who specifically had morcellation and a hysterectomy, providing a population-based estimate of the prevalence of cancer in this group, Dr. Wright said in an interview. This is slightly different than other estimates and studies, which were not specific to morcellation, including those that looked at the incidence of sarcoma among women who had a hysterectomy. "We could not separate out epithelial endometrial cancers from uterine sarcomas, so this is an estimate of any malignancy within the uterus," so "probably a high proportion of women who underwent morcellation underwent the procedure for fibroid uterus, so the chance of sarcomas is probably higher in these patients than in the general population."
The study did not allow evaluation of whether the use of morcellation increased the risk of dissemination of cancer, which "certainly warrants further study," added Dr. Wright, chief of the division of gynecologic oncology at Columbia.
The lack of data has been one of the major problems surrounding morcellation, with very few studies specifically looking at data that can be used to help guide patients and clinicians. Dr. Wright and his associates hope that their results can help guide patients and clinicians.
"There’s undoubtedly a risk of cancer and precancerous changes in women who undergo morcellation ... and [patients and clinicians] need to weigh that risk," he said. "But certainly morcellation may allow some women to undergo a minimally invasive surgery who otherwise require laparotomy, and the complications and recovery are much easier with minimally invasive surgery, when it’s feasible."
Dr. Charles E. Miller, director of minimally invasive gynecologic surgery at Advocate Lutheran General Hospital, Park Ridge, Ill., described these data as "compelling," with well-presented information that stratifies risk by age that can be used to help properly counsel patients.
The marked difference in risk among women under age 40 is particularly important, he said. Dr. Miller performs about 250 morcellations per year and has had only one patient with a sarcoma, a woman in her mid-40s, which he said reflects the younger age of his patients.
"If we can better identify age groups where that risk is higher and do the treatment that is appropriate in that age group, then I think we’ve come a long, long way," he said in an interview. There are always outliers, and unfortunately, women in younger age groups develop sarcomas, but there are also the risks of more invasive surgery that should be considered, he added.
"There’s always going to be risk, and there’s always going to be decision making with surgery," and while there is a need for better ways to identify those patients at risk, currently, "all we can hang our hat on now is stratifying [risk] with age," Dr. Miller said.
On July 10-11, the FDA held a meeting of its Obstetrics and Gynecology Devices panel, to review the safety of LPMs during uterine surgery for fibroids, Among the questions the panel was asked was whether there were characteristics of patients – such as age, physical exam findings, and imaging test results – that could help identify patients who might be at a higher risk of a sarcoma.
The FDA is currently reviewing the safety of LPMs in women undergoing surgery for presumably benign fibroids, an issue that has received widespread attention this year and resulted in the FDA’s safety communication in April – largely due to the case of Dr. Amy Reed, an anesthesiologist who was diagnosed with stage IV leiomyosarcoma after undergoing a hysterectomy with morcellation at age 40 last year. She and her husband, Dr. Hooman Noorchashm, a cardiothoracic surgeon, are leading a campaign to highlight these risks, including a petition on change.org calling for a halt to morcellation during minimally invasive and robotic-assisted hysterectomy and myomectomy.
The authors of the JAMA report had no disclosures. Dr. Wright and one of the other authors, are recipients of National Cancer Institute (NCI) grants; another author is a recipient of an NCI fellowship. Dr. Miller disclosed that he is a consultant to Ethicon Endo-Surgery. Ethicon is a morcellator manufacturer.
FROM JAMA
Key clinical point: A study that provides an estimate of malignancies specifically among women undergoing morcellation and minimally invasive hysterectomy provides valuable information on the risk overall, and risk stratified by age, that can be used in patient counseling.
Major finding: The prevalence of uterine cancer among women who underwent morcellation and a minimally invasive hysterectomy was 27/10,000 – about 1 in 370 women – a risk that significantly increased with age.
Data source: The study identified women who had a minimally invasive hysterectomy with morcellation from 2006 to 2012 in a national insurance database of over 500 hospitals, representing about 15% of hospitalizations.
Disclosures: The authors had no disclosures. Dr. Wright and one of the other authors are recipients of National Cancer Institute (NCI) grants; another author is a recipient of an NCI fellowship. Dr. Miller disclosed that he is a consultant to Ethicon Endo-Surgery. Ethicon is a morcellator manufacturer.

Study provides cancer risk estimates among women undergoing morcellation
An estimated 1 in 370 women who undergo electric power morcellation during a minimally invasive hysterectomy have uterine cancer, with the risk of cancer and endometrial hyperplasia markedly increasing with age, according to an analysis using an insurance database of more than 500 U.S. hospitals.
The estimate, published online in JAMA on July 22, is close to the Food and Drug Administration’s estimate that about 1 in 350 women undergoing hysterectomy or myomectomy for presumed fibroids have an unsuspected uterine sarcoma and is higher than historical estimates provided in the literature. The FDA’s estimate, first reported when the agency issued a safety communication in April discouraging the use of laparoscopic power morcellators (LPMs) during a hysterectomy or myomectomy because of the risk of disseminating cancerous tissue and upstaging disease, was based on 9 U.S. and international studies of women treated from 1983 to 2011. The risk of an unsuspected leiomyosarcoma was about 1 in 500.
But unlike the FDA analysis, the most recent analysis, conducted by Dr. Jason Wright and his associates at Columbia University, New York, specifically addressed the risk associated with morcellation. The estimate was based on the records of 232,882 women who underwent a minimally invasive hysterectomy from 2006 to 2012 obtained from a database that represents about 15% of U.S. hospitalizations. Morcellators were used in almost 16% (36,470) of the women, and there were 99 cases of uterine cancers, for a prevalence of 27/10,000 – about one in 370.
Among the women who underwent morcellation, the strongest risk factor for abnormal pathology, either for cancer or any of the precancerous changes, was advanced age. Compared with women under age 40 years, the prevalence ratio for uterine malignancy was 1.42 among those aged 40-44 and 2.55 for those aged 45-49, increasing to 4.97 among those aged 50-54 years, 19.37 among those aged 55-59 years, 21.36 among those aged 60-64 years – and 35.97 among those aged 65 and older.
The researchers also identified cases of endometrial hyperplasia, other gynecologic cancers, and smooth muscle tumors of uncertain malignant potential. The risk of endometrial hyperplasia also increased significantly with age, compared with women under age 40, with prevalence ratios of 1.17 among those aged 40-44 (not statistically significant) and 1.71 among those aged 45-49, to 4.07 among those aged 50-54 years, to 10.21 among those aged 65 years and older. The results are reported in a research letter (doi:10.1001/jama.2014.9005)
Despite the availability of power morcellators for 20 years, "few studies have described the prevalence of unexpected pathology at the time of hysterectomy," Dr. Wright and his associates wrote. While the analysis had limitations, including the lack of long-term follow-up and not being able to verify the pathology results, they concluded that patients who may be undergoing morcellation "should be adequately counseled about the prevalence of cancerous and precancerous conditions prior to undergoing the procedure."
One of the strengths and unique aspects of this study was being able to identify a large population of women who specifically had morcellation and a hysterectomy, providing a population-based estimate of the prevalence of cancer in this group, Dr. Wright said in an interview. This is slightly different than other estimates and studies, which were not specific to morcellation, including those that looked at the incidence of sarcoma among women who had a hysterectomy. "We could not separate out epithelial endometrial cancers from uterine sarcomas, so this is an estimate of any malignancy within the uterus," so "probably a high proportion of women who underwent morcellation underwent the procedure for fibroid uterus, so the chance of sarcomas is probably higher in these patients than in the general population."
The study did not allow evaluation of whether the use of morcellation increased the risk of dissemination of cancer, which "certainly warrants further study," added Dr. Wright, chief of the division of gynecologic oncology at Columbia.
The lack of data has been one of the major problems surrounding morcellation, with very few studies specifically looking at data that can be used to help guide patients and clinicians. Dr. Wright and his associates hope that their results can help guide patients and clinicians.
"There’s undoubtedly a risk of cancer and precancerous changes in women who undergo morcellation ... and [patients and clinicians] need to weigh that risk," he said. "But certainly morcellation may allow some women to undergo a minimally invasive surgery who otherwise require laparotomy, and the complications and recovery are much easier with minimally invasive surgery, when it’s feasible."
Dr. Charles E. Miller, director of minimally invasive gynecologic surgery at Advocate Lutheran General Hospital, Park Ridge, Ill., described these data as "compelling," with well-presented information that stratifies risk by age that can be used to help properly counsel patients.
The marked difference in risk among women under age 40 is particularly important, he said. Dr. Miller performs about 250 morcellations per year and has had only one patient with a sarcoma, a woman in her mid-40s, which he said reflects the younger age of his patients.
"If we can better identify age groups where that risk is higher and do the treatment that is appropriate in that age group, then I think we’ve come a long, long way," he said in an interview. There are always outliers, and unfortunately, women in younger age groups develop sarcomas, but there are also the risks of more invasive surgery that should be considered, he added.
"There’s always going to be risk, and there’s always going to be decision making with surgery," and while there is a need for better ways to identify those patients at risk, currently, "all we can hang our hat on now is stratifying [risk] with age," Dr. Miller said.
On July 10-11, the FDA held a meeting of its Obstetrics and Gynecology Devices panel, to review the safety of LPMs during uterine surgery for fibroids, Among the questions the panel was asked was whether there were characteristics of patients – such as age, physical exam findings, and imaging test results – that could help identify patients who might be at a higher risk of a sarcoma.
The FDA is currently reviewing the safety of LPMs in women undergoing surgery for presumably benign fibroids, an issue that has received widespread attention this year and resulted in the FDA’s safety communication in April – largely due to the case of Dr. Amy Reed, an anesthesiologist who was diagnosed with stage IV leiomyosarcoma after undergoing a hysterectomy with morcellation at age 40 last year. She and her husband, Dr. Hooman Noorchashm, a cardiothoracic surgeon, are leading a campaign to highlight these risks, including a petition on change.org calling for a halt to morcellation during minimally invasive and robotic-assisted hysterectomy and myomectomy.
The authors of the JAMA report had no disclosures. Dr. Wright and one of the other authors, are recipients of National Cancer Institute (NCI) grants; another author is a recipient of an NCI fellowship. Dr. Miller disclosed that he is a consultant to Ethicon Endo-Surgery. Ethicon is a morcellator manufacturer.
An estimated 1 in 370 women who undergo electric power morcellation during a minimally invasive hysterectomy have uterine cancer, with the risk of cancer and endometrial hyperplasia markedly increasing with age, according to an analysis using an insurance database of more than 500 U.S. hospitals.
The estimate, published online in JAMA on July 22, is close to the Food and Drug Administration’s estimate that about 1 in 350 women undergoing hysterectomy or myomectomy for presumed fibroids have an unsuspected uterine sarcoma and is higher than historical estimates provided in the literature. The FDA’s estimate, first reported when the agency issued a safety communication in April discouraging the use of laparoscopic power morcellators (LPMs) during a hysterectomy or myomectomy because of the risk of disseminating cancerous tissue and upstaging disease, was based on 9 U.S. and international studies of women treated from 1983 to 2011. The risk of an unsuspected leiomyosarcoma was about 1 in 500.
But unlike the FDA analysis, the most recent analysis, conducted by Dr. Jason Wright and his associates at Columbia University, New York, specifically addressed the risk associated with morcellation. The estimate was based on the records of 232,882 women who underwent a minimally invasive hysterectomy from 2006 to 2012 obtained from a database that represents about 15% of U.S. hospitalizations. Morcellators were used in almost 16% (36,470) of the women, and there were 99 cases of uterine cancers, for a prevalence of 27/10,000 – about one in 370.
Among the women who underwent morcellation, the strongest risk factor for abnormal pathology, either for cancer or any of the precancerous changes, was advanced age. Compared with women under age 40 years, the prevalence ratio for uterine malignancy was 1.42 among those aged 40-44 and 2.55 for those aged 45-49, increasing to 4.97 among those aged 50-54 years, 19.37 among those aged 55-59 years, 21.36 among those aged 60-64 years – and 35.97 among those aged 65 and older.
The researchers also identified cases of endometrial hyperplasia, other gynecologic cancers, and smooth muscle tumors of uncertain malignant potential. The risk of endometrial hyperplasia also increased significantly with age, compared with women under age 40, with prevalence ratios of 1.17 among those aged 40-44 (not statistically significant) and 1.71 among those aged 45-49, to 4.07 among those aged 50-54 years, to 10.21 among those aged 65 years and older. The results are reported in a research letter (doi:10.1001/jama.2014.9005)
Despite the availability of power morcellators for 20 years, "few studies have described the prevalence of unexpected pathology at the time of hysterectomy," Dr. Wright and his associates wrote. While the analysis had limitations, including the lack of long-term follow-up and not being able to verify the pathology results, they concluded that patients who may be undergoing morcellation "should be adequately counseled about the prevalence of cancerous and precancerous conditions prior to undergoing the procedure."
One of the strengths and unique aspects of this study was being able to identify a large population of women who specifically had morcellation and a hysterectomy, providing a population-based estimate of the prevalence of cancer in this group, Dr. Wright said in an interview. This is slightly different than other estimates and studies, which were not specific to morcellation, including those that looked at the incidence of sarcoma among women who had a hysterectomy. "We could not separate out epithelial endometrial cancers from uterine sarcomas, so this is an estimate of any malignancy within the uterus," so "probably a high proportion of women who underwent morcellation underwent the procedure for fibroid uterus, so the chance of sarcomas is probably higher in these patients than in the general population."
The study did not allow evaluation of whether the use of morcellation increased the risk of dissemination of cancer, which "certainly warrants further study," added Dr. Wright, chief of the division of gynecologic oncology at Columbia.
The lack of data has been one of the major problems surrounding morcellation, with very few studies specifically looking at data that can be used to help guide patients and clinicians. Dr. Wright and his associates hope that their results can help guide patients and clinicians.
"There’s undoubtedly a risk of cancer and precancerous changes in women who undergo morcellation ... and [patients and clinicians] need to weigh that risk," he said. "But certainly morcellation may allow some women to undergo a minimally invasive surgery who otherwise require laparotomy, and the complications and recovery are much easier with minimally invasive surgery, when it’s feasible."
Dr. Charles E. Miller, director of minimally invasive gynecologic surgery at Advocate Lutheran General Hospital, Park Ridge, Ill., described these data as "compelling," with well-presented information that stratifies risk by age that can be used to help properly counsel patients.
The marked difference in risk among women under age 40 is particularly important, he said. Dr. Miller performs about 250 morcellations per year and has had only one patient with a sarcoma, a woman in her mid-40s, which he said reflects the younger age of his patients.
"If we can better identify age groups where that risk is higher and do the treatment that is appropriate in that age group, then I think we’ve come a long, long way," he said in an interview. There are always outliers, and unfortunately, women in younger age groups develop sarcomas, but there are also the risks of more invasive surgery that should be considered, he added.
"There’s always going to be risk, and there’s always going to be decision making with surgery," and while there is a need for better ways to identify those patients at risk, currently, "all we can hang our hat on now is stratifying [risk] with age," Dr. Miller said.
On July 10-11, the FDA held a meeting of its Obstetrics and Gynecology Devices panel, to review the safety of LPMs during uterine surgery for fibroids, Among the questions the panel was asked was whether there were characteristics of patients – such as age, physical exam findings, and imaging test results – that could help identify patients who might be at a higher risk of a sarcoma.
The FDA is currently reviewing the safety of LPMs in women undergoing surgery for presumably benign fibroids, an issue that has received widespread attention this year and resulted in the FDA’s safety communication in April – largely due to the case of Dr. Amy Reed, an anesthesiologist who was diagnosed with stage IV leiomyosarcoma after undergoing a hysterectomy with morcellation at age 40 last year. She and her husband, Dr. Hooman Noorchashm, a cardiothoracic surgeon, are leading a campaign to highlight these risks, including a petition on change.org calling for a halt to morcellation during minimally invasive and robotic-assisted hysterectomy and myomectomy.
The authors of the JAMA report had no disclosures. Dr. Wright and one of the other authors, are recipients of National Cancer Institute (NCI) grants; another author is a recipient of an NCI fellowship. Dr. Miller disclosed that he is a consultant to Ethicon Endo-Surgery. Ethicon is a morcellator manufacturer.
An estimated 1 in 370 women who undergo electric power morcellation during a minimally invasive hysterectomy have uterine cancer, with the risk of cancer and endometrial hyperplasia markedly increasing with age, according to an analysis using an insurance database of more than 500 U.S. hospitals.
The estimate, published online in JAMA on July 22, is close to the Food and Drug Administration’s estimate that about 1 in 350 women undergoing hysterectomy or myomectomy for presumed fibroids have an unsuspected uterine sarcoma and is higher than historical estimates provided in the literature. The FDA’s estimate, first reported when the agency issued a safety communication in April discouraging the use of laparoscopic power morcellators (LPMs) during a hysterectomy or myomectomy because of the risk of disseminating cancerous tissue and upstaging disease, was based on 9 U.S. and international studies of women treated from 1983 to 2011. The risk of an unsuspected leiomyosarcoma was about 1 in 500.
But unlike the FDA analysis, the most recent analysis, conducted by Dr. Jason Wright and his associates at Columbia University, New York, specifically addressed the risk associated with morcellation. The estimate was based on the records of 232,882 women who underwent a minimally invasive hysterectomy from 2006 to 2012 obtained from a database that represents about 15% of U.S. hospitalizations. Morcellators were used in almost 16% (36,470) of the women, and there were 99 cases of uterine cancers, for a prevalence of 27/10,000 – about one in 370.
Among the women who underwent morcellation, the strongest risk factor for abnormal pathology, either for cancer or any of the precancerous changes, was advanced age. Compared with women under age 40 years, the prevalence ratio for uterine malignancy was 1.42 among those aged 40-44 and 2.55 for those aged 45-49, increasing to 4.97 among those aged 50-54 years, 19.37 among those aged 55-59 years, 21.36 among those aged 60-64 years – and 35.97 among those aged 65 and older.
The researchers also identified cases of endometrial hyperplasia, other gynecologic cancers, and smooth muscle tumors of uncertain malignant potential. The risk of endometrial hyperplasia also increased significantly with age, compared with women under age 40, with prevalence ratios of 1.17 among those aged 40-44 (not statistically significant) and 1.71 among those aged 45-49, to 4.07 among those aged 50-54 years, to 10.21 among those aged 65 years and older. The results are reported in a research letter (doi:10.1001/jama.2014.9005)
Despite the availability of power morcellators for 20 years, "few studies have described the prevalence of unexpected pathology at the time of hysterectomy," Dr. Wright and his associates wrote. While the analysis had limitations, including the lack of long-term follow-up and not being able to verify the pathology results, they concluded that patients who may be undergoing morcellation "should be adequately counseled about the prevalence of cancerous and precancerous conditions prior to undergoing the procedure."
One of the strengths and unique aspects of this study was being able to identify a large population of women who specifically had morcellation and a hysterectomy, providing a population-based estimate of the prevalence of cancer in this group, Dr. Wright said in an interview. This is slightly different than other estimates and studies, which were not specific to morcellation, including those that looked at the incidence of sarcoma among women who had a hysterectomy. "We could not separate out epithelial endometrial cancers from uterine sarcomas, so this is an estimate of any malignancy within the uterus," so "probably a high proportion of women who underwent morcellation underwent the procedure for fibroid uterus, so the chance of sarcomas is probably higher in these patients than in the general population."
The study did not allow evaluation of whether the use of morcellation increased the risk of dissemination of cancer, which "certainly warrants further study," added Dr. Wright, chief of the division of gynecologic oncology at Columbia.
The lack of data has been one of the major problems surrounding morcellation, with very few studies specifically looking at data that can be used to help guide patients and clinicians. Dr. Wright and his associates hope that their results can help guide patients and clinicians.
"There’s undoubtedly a risk of cancer and precancerous changes in women who undergo morcellation ... and [patients and clinicians] need to weigh that risk," he said. "But certainly morcellation may allow some women to undergo a minimally invasive surgery who otherwise require laparotomy, and the complications and recovery are much easier with minimally invasive surgery, when it’s feasible."
Dr. Charles E. Miller, director of minimally invasive gynecologic surgery at Advocate Lutheran General Hospital, Park Ridge, Ill., described these data as "compelling," with well-presented information that stratifies risk by age that can be used to help properly counsel patients.
The marked difference in risk among women under age 40 is particularly important, he said. Dr. Miller performs about 250 morcellations per year and has had only one patient with a sarcoma, a woman in her mid-40s, which he said reflects the younger age of his patients.
"If we can better identify age groups where that risk is higher and do the treatment that is appropriate in that age group, then I think we’ve come a long, long way," he said in an interview. There are always outliers, and unfortunately, women in younger age groups develop sarcomas, but there are also the risks of more invasive surgery that should be considered, he added.
"There’s always going to be risk, and there’s always going to be decision making with surgery," and while there is a need for better ways to identify those patients at risk, currently, "all we can hang our hat on now is stratifying [risk] with age," Dr. Miller said.
On July 10-11, the FDA held a meeting of its Obstetrics and Gynecology Devices panel, to review the safety of LPMs during uterine surgery for fibroids, Among the questions the panel was asked was whether there were characteristics of patients – such as age, physical exam findings, and imaging test results – that could help identify patients who might be at a higher risk of a sarcoma.
The FDA is currently reviewing the safety of LPMs in women undergoing surgery for presumably benign fibroids, an issue that has received widespread attention this year and resulted in the FDA’s safety communication in April – largely due to the case of Dr. Amy Reed, an anesthesiologist who was diagnosed with stage IV leiomyosarcoma after undergoing a hysterectomy with morcellation at age 40 last year. She and her husband, Dr. Hooman Noorchashm, a cardiothoracic surgeon, are leading a campaign to highlight these risks, including a petition on change.org calling for a halt to morcellation during minimally invasive and robotic-assisted hysterectomy and myomectomy.
The authors of the JAMA report had no disclosures. Dr. Wright and one of the other authors, are recipients of National Cancer Institute (NCI) grants; another author is a recipient of an NCI fellowship. Dr. Miller disclosed that he is a consultant to Ethicon Endo-Surgery. Ethicon is a morcellator manufacturer.
FROM JAMA
Key clinical point: A study that provides an estimate of malignancies specifically among women undergoing morcellation and minimally invasive hysterectomy provides valuable information on the risk overall, and risk stratified by age, that can be used in patient counseling.
Major finding: The prevalence of uterine cancer among women who underwent morcellation and a minimally invasive hysterectomy was 27/10,000 – about 1 in 370 women – a risk that significantly increased with age.
Data source: The study identified women who had a minimally invasive hysterectomy with morcellation from 2006 to 2012 in a national insurance database of over 500 hospitals, representing about 15% of hospitalizations.
Disclosures: The authors had no disclosures. Dr. Wright and one of the other authors are recipients of National Cancer Institute (NCI) grants; another author is a recipient of an NCI fellowship. Dr. Miller disclosed that he is a consultant to Ethicon Endo-Surgery. Ethicon is a morcellator manufacturer.

FDA approves C1-esterase inhibitor for hereditary angioedema
Ruconest, an injectable human recombinant C1-esterase inhibitor, has been approved by the Food and Drug Administration for treating acute angioedema attacks in adults and adolescents with hereditary angioedema.
Ruconest is the first recombinant C1-esterase inhibitor to be approved by the agency, according to the July 17 FDA statement announcing the approval. Ruconest "is intended to restore the level of functional C1-esterase inhibitor in a patient’s plasma, thereby treating the acute attack of swelling" in patients with hereditary angioedema (HAE). HAE is caused by an insufficient amount of C1-esterase inhibitor, a plasma protein, the statement said. HAE symptoms include acute attacks of swelling of the hands, feet, limbs, face, intestinal tract, or airway, which can occur spontaneously or are triggered by stress, surgery, or an infection, according to the FDA.
Ruconest is expected to be available "later in 2014," according to the press release on the approval issued by Salix Pharmaceuticals.
Santarus, a subsidiary of Salix, will distribute the drug in the United States; the drug is manufactured by the Pharming Group NV, in the Netherlands.
A Salix press release issued on July 17 provided details of the study that was the basis of the approval, a randomized, double-blind, placebo-controlled phase III trial, which included an open-label extension phase, of adults and adolescents who had 170 HAE attacks. The time to the beginning of symptom relief, based on patient reported responses, was significantly faster among those treated with Ruconest, administered at the time of the attack, compared with those on placebo – a median of 90 minutes among the 44 patients treated with Ruconest vs. a median of 152 minutes among the 31 patients who received a placebo, a statistically significant difference, according to Salix.
However, "Because of the limited number of patients with laryngeal attacks, effectiveness was not established in HAE patients with laryngeal attacks," according to the Salix statement.
Headache, nausea, and diarrhea were the most common adverse events associated with treatment, the FDA statement said.
Ruconest was designated an orphan drug because it is intended to treat a rare disease or condition; about 6,000-10,000 people in the United States have hereditary angioedema, according to the FDA.
Ruconest, an injectable human recombinant C1-esterase inhibitor, has been approved by the Food and Drug Administration for treating acute angioedema attacks in adults and adolescents with hereditary angioedema.
Ruconest is the first recombinant C1-esterase inhibitor to be approved by the agency, according to the July 17 FDA statement announcing the approval. Ruconest "is intended to restore the level of functional C1-esterase inhibitor in a patient’s plasma, thereby treating the acute attack of swelling" in patients with hereditary angioedema (HAE). HAE is caused by an insufficient amount of C1-esterase inhibitor, a plasma protein, the statement said. HAE symptoms include acute attacks of swelling of the hands, feet, limbs, face, intestinal tract, or airway, which can occur spontaneously or are triggered by stress, surgery, or an infection, according to the FDA.
Ruconest is expected to be available "later in 2014," according to the press release on the approval issued by Salix Pharmaceuticals.
Santarus, a subsidiary of Salix, will distribute the drug in the United States; the drug is manufactured by the Pharming Group NV, in the Netherlands.
A Salix press release issued on July 17 provided details of the study that was the basis of the approval, a randomized, double-blind, placebo-controlled phase III trial, which included an open-label extension phase, of adults and adolescents who had 170 HAE attacks. The time to the beginning of symptom relief, based on patient reported responses, was significantly faster among those treated with Ruconest, administered at the time of the attack, compared with those on placebo – a median of 90 minutes among the 44 patients treated with Ruconest vs. a median of 152 minutes among the 31 patients who received a placebo, a statistically significant difference, according to Salix.
However, "Because of the limited number of patients with laryngeal attacks, effectiveness was not established in HAE patients with laryngeal attacks," according to the Salix statement.
Headache, nausea, and diarrhea were the most common adverse events associated with treatment, the FDA statement said.
Ruconest was designated an orphan drug because it is intended to treat a rare disease or condition; about 6,000-10,000 people in the United States have hereditary angioedema, according to the FDA.
Ruconest, an injectable human recombinant C1-esterase inhibitor, has been approved by the Food and Drug Administration for treating acute angioedema attacks in adults and adolescents with hereditary angioedema.
Ruconest is the first recombinant C1-esterase inhibitor to be approved by the agency, according to the July 17 FDA statement announcing the approval. Ruconest "is intended to restore the level of functional C1-esterase inhibitor in a patient’s plasma, thereby treating the acute attack of swelling" in patients with hereditary angioedema (HAE). HAE is caused by an insufficient amount of C1-esterase inhibitor, a plasma protein, the statement said. HAE symptoms include acute attacks of swelling of the hands, feet, limbs, face, intestinal tract, or airway, which can occur spontaneously or are triggered by stress, surgery, or an infection, according to the FDA.
Ruconest is expected to be available "later in 2014," according to the press release on the approval issued by Salix Pharmaceuticals.
Santarus, a subsidiary of Salix, will distribute the drug in the United States; the drug is manufactured by the Pharming Group NV, in the Netherlands.
A Salix press release issued on July 17 provided details of the study that was the basis of the approval, a randomized, double-blind, placebo-controlled phase III trial, which included an open-label extension phase, of adults and adolescents who had 170 HAE attacks. The time to the beginning of symptom relief, based on patient reported responses, was significantly faster among those treated with Ruconest, administered at the time of the attack, compared with those on placebo – a median of 90 minutes among the 44 patients treated with Ruconest vs. a median of 152 minutes among the 31 patients who received a placebo, a statistically significant difference, according to Salix.
However, "Because of the limited number of patients with laryngeal attacks, effectiveness was not established in HAE patients with laryngeal attacks," according to the Salix statement.
Headache, nausea, and diarrhea were the most common adverse events associated with treatment, the FDA statement said.
Ruconest was designated an orphan drug because it is intended to treat a rare disease or condition; about 6,000-10,000 people in the United States have hereditary angioedema, according to the FDA.
FROM THE FDA
Viral reactivation common in septic patients
Critically ill patients with sepsis have a much higher prevalence of different viruses than do nonseptic critically ill patients and healthy controls, judging from the findings of a study of more than 800 patients.
These findings provide evidence that the reactivation of latent viruses "is extremely common in patients with prolonged sepsis and is consistent with development of immunosuppression," researchers concluded.

For some of the viruses, the levels detected in septic patients were comparable to the levels in organ transplant recipients, "who are pharmacologically immunosuppressed, providing further support that our findings are indicative of clinically relevant immunosuppression," Dr. Anthony Walton, of the department of anesthesiology, Washington University, St. Louis, and his coauthors wrote. The study was published online June 6 in PLoS One (2014;9:e98819 [doi: 10.1371/journal.pone.0098819]).
In what they said is the first study to evaluate the effect of sepsis on "multiple families of viruses," the investigators addressed whether sepsis progresses from a hyperinflammatory phase early in the course of sepsis to an immunosuppressive state, a "controversial hypothesis" for explaining the course of sepsis, they wrote.
The researchers compared levels of viruses that included cytomegalovirus (CMV), Epstein-Barr virus (EBV), herpes simplex virus (HSV), human herpesvirus 6 (HHV-6), and the anellovirus TTV in whole blood and plasma, and of the polyomaviruses BK and JC in the urine of 560 critically ill patients with sepsis and 161 critically ill patients who did not have sepsis, who were not immunocompromised at baseline; and 164 healthy, age-matched controls, who were ambulatory and whose blood sample was obtained before elective surgery. The median age of the patients was 63-64 years; the median APACHE II score was 18 in the septic group and 5 in the critically ill, nonseptic group; and the median length of stay in the intensive care unit was 11 days and 2 days, respectively; mortality was 26% and 6%, respectively. (Patients who were HIV-1 infected, organ transplant recipients, or on immunosuppressive medications were among those who were excluded.)
Among the investigators’ key findings were these:
• CMV seropositivity was detected in about 70% of the patients in the three groups, indicating they had been infected previously. Among these patients, CMV levels were markedly elevated in 24.2% of the septic patients, compared with 1.1% of the critically ill, nonseptic patients and none of the healthy controls.
• EBV was detected in 53.2% of septic patients, vs. 12.1% of the critically ill, nonseptic patients and 3.6% of the healthy controls. (Almost 9% of the septic patients had EBV detected at the level that would be the basis for reducing immunosuppressive medications in solid organ transplant recipients at Washington University, the authors pointed out.)
• HSV was detected in 14.1% of septic patients, vs. 1.5% of the critically ill, nonseptic patients and none of the healthy controls.
• HHV-6 was detected in 10.4% of septic patients, vs. less than 1% of the critically ill, nonseptic patients and 3.3% of the healthy controls.
• TTV was detected in almost 78% of the septic patients, close to 64% of the critically ill, nonseptic patients, and 60.1% of the healthy controls, but levels were lower among the latter two groups.
The authors said that it is "likely that viral detection in the setting of sepsis is not due to primary infection but rather to viral reactivation." Almost 43% of those with sepsis had evidence of at least two viruses, which, combined with the "magnitude of viral loads ... provides strong evidence that host immunity is impaired in sepsis," they added.

Among their other findings was that in the septic patients, the detection rate of the viruses increased for all the viruses with increasing number of days spent in the ICU, and septic patients who had CMV detected in the plasma had significantly higher 90-day mortality than did septic patients with no CMV detected.
Limitations of the study include the fact that it does not address whether the increased prevalence of viral reactivation among the septic patients "is merely a marker of impaired immunity or contributes to sepsis morbidity/mortality," they noted. But the implications of their results include the possibility that tracking the viral load of different viruses in septic patients "may be useful as a biomarker of host immunity in sepsis."
The study was funded by the National Institutes of Health. One author is an employee of Biomérieux, a company that is working on a method to monitor levels of different viruses in the blood as an indicator of immune status which is performing related research. No other author disclosures were listed.
Dr. Steven Q. Simpson, FCCP, comments: The investigators have demonstrated that reactivation of latent viral infections may well contribute to the death of critically ill septic patients. Some of the viral reactivations were associated with secondary fungal infection as well.
Although viral DNA was detected as early as 1 day into sepsis, the bulk of the manifested reactivations occurred over the subsequent 2 weeks. Viral reactivation is a clear marker that the "late" immune suppression of sepsis is a real phenomenon and leads to real sequelae.
Nevertheless, it is not yet clear exactly how this information will become useful in practice, as the cost of daily DNA screening for multiple viruses would be prohibitive, unless high-volume demand drives pricing down. One can see, under that scenario, how viral reactivation could be the signal that immune augmentation therapy is required, and that it might be beneficial. This work is not quite ready for prime time, but it is getting ever closer.
Dr. Steven Q. Simpson is professor of medicine, University of Kansas, Kansas City. He is also founder of the Kansas Sepsis Project. He had no disclosures.
Dr. Steven Q. Simpson, FCCP, comments: The investigators have demonstrated that reactivation of latent viral infections may well contribute to the death of critically ill septic patients. Some of the viral reactivations were associated with secondary fungal infection as well.
Although viral DNA was detected as early as 1 day into sepsis, the bulk of the manifested reactivations occurred over the subsequent 2 weeks. Viral reactivation is a clear marker that the "late" immune suppression of sepsis is a real phenomenon and leads to real sequelae.
Nevertheless, it is not yet clear exactly how this information will become useful in practice, as the cost of daily DNA screening for multiple viruses would be prohibitive, unless high-volume demand drives pricing down. One can see, under that scenario, how viral reactivation could be the signal that immune augmentation therapy is required, and that it might be beneficial. This work is not quite ready for prime time, but it is getting ever closer.
Dr. Steven Q. Simpson is professor of medicine, University of Kansas, Kansas City. He is also founder of the Kansas Sepsis Project. He had no disclosures.
Dr. Steven Q. Simpson, FCCP, comments: The investigators have demonstrated that reactivation of latent viral infections may well contribute to the death of critically ill septic patients. Some of the viral reactivations were associated with secondary fungal infection as well.
Although viral DNA was detected as early as 1 day into sepsis, the bulk of the manifested reactivations occurred over the subsequent 2 weeks. Viral reactivation is a clear marker that the "late" immune suppression of sepsis is a real phenomenon and leads to real sequelae.
Nevertheless, it is not yet clear exactly how this information will become useful in practice, as the cost of daily DNA screening for multiple viruses would be prohibitive, unless high-volume demand drives pricing down. One can see, under that scenario, how viral reactivation could be the signal that immune augmentation therapy is required, and that it might be beneficial. This work is not quite ready for prime time, but it is getting ever closer.
Dr. Steven Q. Simpson is professor of medicine, University of Kansas, Kansas City. He is also founder of the Kansas Sepsis Project. He had no disclosures.
Critically ill patients with sepsis have a much higher prevalence of different viruses than do nonseptic critically ill patients and healthy controls, judging from the findings of a study of more than 800 patients.
These findings provide evidence that the reactivation of latent viruses "is extremely common in patients with prolonged sepsis and is consistent with development of immunosuppression," researchers concluded.
For some of the viruses, the levels detected in septic patients were comparable to the levels in organ transplant recipients, "who are pharmacologically immunosuppressed, providing further support that our findings are indicative of clinically relevant immunosuppression," Dr. Anthony Walton, of the department of anesthesiology, Washington University, St. Louis, and his coauthors wrote. The study was published online June 6 in PLoS One (2014;9:e98819 [doi: 10.1371/journal.pone.0098819]).
In what they said is the first study to evaluate the effect of sepsis on "multiple families of viruses," the investigators addressed whether sepsis progresses from a hyperinflammatory phase early in the course of sepsis to an immunosuppressive state, a "controversial hypothesis" for explaining the course of sepsis, they wrote.
The researchers compared levels of viruses that included cytomegalovirus (CMV), Epstein-Barr virus (EBV), herpes simplex virus (HSV), human herpesvirus 6 (HHV-6), and the anellovirus TTV in whole blood and plasma, and of the polyomaviruses BK and JC in the urine of 560 critically ill patients with sepsis and 161 critically ill patients who did not have sepsis, who were not immunocompromised at baseline; and 164 healthy, age-matched controls, who were ambulatory and whose blood sample was obtained before elective surgery. The median age of the patients was 63-64 years; the median APACHE II score was 18 in the septic group and 5 in the critically ill, nonseptic group; and the median length of stay in the intensive care unit was 11 days and 2 days, respectively; mortality was 26% and 6%, respectively. (Patients who were HIV-1 infected, organ transplant recipients, or on immunosuppressive medications were among those who were excluded.)
Among the investigators’ key findings were these:
• CMV seropositivity was detected in about 70% of the patients in the three groups, indicating they had been infected previously. Among these patients, CMV levels were markedly elevated in 24.2% of the septic patients, compared with 1.1% of the critically ill, nonseptic patients and none of the healthy controls.
• EBV was detected in 53.2% of septic patients, vs. 12.1% of the critically ill, nonseptic patients and 3.6% of the healthy controls. (Almost 9% of the septic patients had EBV detected at the level that would be the basis for reducing immunosuppressive medications in solid organ transplant recipients at Washington University, the authors pointed out.)
• HSV was detected in 14.1% of septic patients, vs. 1.5% of the critically ill, nonseptic patients and none of the healthy controls.
• HHV-6 was detected in 10.4% of septic patients, vs. less than 1% of the critically ill, nonseptic patients and 3.3% of the healthy controls.
• TTV was detected in almost 78% of the septic patients, close to 64% of the critically ill, nonseptic patients, and 60.1% of the healthy controls, but levels were lower among the latter two groups.
The authors said that it is "likely that viral detection in the setting of sepsis is not due to primary infection but rather to viral reactivation." Almost 43% of those with sepsis had evidence of at least two viruses, which, combined with the "magnitude of viral loads ... provides strong evidence that host immunity is impaired in sepsis," they added.
Among their other findings was that in the septic patients, the detection rate of the viruses increased for all the viruses with increasing number of days spent in the ICU, and septic patients who had CMV detected in the plasma had significantly higher 90-day mortality than did septic patients with no CMV detected.
Limitations of the study include the fact that it does not address whether the increased prevalence of viral reactivation among the septic patients "is merely a marker of impaired immunity or contributes to sepsis morbidity/mortality," they noted. But the implications of their results include the possibility that tracking the viral load of different viruses in septic patients "may be useful as a biomarker of host immunity in sepsis."
The study was funded by the National Institutes of Health. One author is an employee of Biomérieux, a company that is working on a method to monitor levels of different viruses in the blood as an indicator of immune status which is performing related research. No other author disclosures were listed.
Critically ill patients with sepsis have a much higher prevalence of different viruses than do nonseptic critically ill patients and healthy controls, judging from the findings of a study of more than 800 patients.
These findings provide evidence that the reactivation of latent viruses "is extremely common in patients with prolonged sepsis and is consistent with development of immunosuppression," researchers concluded.
For some of the viruses, the levels detected in septic patients were comparable to the levels in organ transplant recipients, "who are pharmacologically immunosuppressed, providing further support that our findings are indicative of clinically relevant immunosuppression," Dr. Anthony Walton, of the department of anesthesiology, Washington University, St. Louis, and his coauthors wrote. The study was published online June 6 in PLoS One (2014;9:e98819 [doi: 10.1371/journal.pone.0098819]).
In what they said is the first study to evaluate the effect of sepsis on "multiple families of viruses," the investigators addressed whether sepsis progresses from a hyperinflammatory phase early in the course of sepsis to an immunosuppressive state, a "controversial hypothesis" for explaining the course of sepsis, they wrote.
The researchers compared levels of viruses that included cytomegalovirus (CMV), Epstein-Barr virus (EBV), herpes simplex virus (HSV), human herpesvirus 6 (HHV-6), and the anellovirus TTV in whole blood and plasma, and of the polyomaviruses BK and JC in the urine of 560 critically ill patients with sepsis and 161 critically ill patients who did not have sepsis, who were not immunocompromised at baseline; and 164 healthy, age-matched controls, who were ambulatory and whose blood sample was obtained before elective surgery. The median age of the patients was 63-64 years; the median APACHE II score was 18 in the septic group and 5 in the critically ill, nonseptic group; and the median length of stay in the intensive care unit was 11 days and 2 days, respectively; mortality was 26% and 6%, respectively. (Patients who were HIV-1 infected, organ transplant recipients, or on immunosuppressive medications were among those who were excluded.)
Among the investigators’ key findings were these:
• CMV seropositivity was detected in about 70% of the patients in the three groups, indicating they had been infected previously. Among these patients, CMV levels were markedly elevated in 24.2% of the septic patients, compared with 1.1% of the critically ill, nonseptic patients and none of the healthy controls.
• EBV was detected in 53.2% of septic patients, vs. 12.1% of the critically ill, nonseptic patients and 3.6% of the healthy controls. (Almost 9% of the septic patients had EBV detected at the level that would be the basis for reducing immunosuppressive medications in solid organ transplant recipients at Washington University, the authors pointed out.)
• HSV was detected in 14.1% of septic patients, vs. 1.5% of the critically ill, nonseptic patients and none of the healthy controls.
• HHV-6 was detected in 10.4% of septic patients, vs. less than 1% of the critically ill, nonseptic patients and 3.3% of the healthy controls.
• TTV was detected in almost 78% of the septic patients, close to 64% of the critically ill, nonseptic patients, and 60.1% of the healthy controls, but levels were lower among the latter two groups.
The authors said that it is "likely that viral detection in the setting of sepsis is not due to primary infection but rather to viral reactivation." Almost 43% of those with sepsis had evidence of at least two viruses, which, combined with the "magnitude of viral loads ... provides strong evidence that host immunity is impaired in sepsis," they added.
Among their other findings was that in the septic patients, the detection rate of the viruses increased for all the viruses with increasing number of days spent in the ICU, and septic patients who had CMV detected in the plasma had significantly higher 90-day mortality than did septic patients with no CMV detected.
Limitations of the study include the fact that it does not address whether the increased prevalence of viral reactivation among the septic patients "is merely a marker of impaired immunity or contributes to sepsis morbidity/mortality," they noted. But the implications of their results include the possibility that tracking the viral load of different viruses in septic patients "may be useful as a biomarker of host immunity in sepsis."
The study was funded by the National Institutes of Health. One author is an employee of Biomérieux, a company that is working on a method to monitor levels of different viruses in the blood as an indicator of immune status which is performing related research. No other author disclosures were listed.
Key clinical point: Reactivation of latent viruses may underlie the development of sepsis in critically ill patients and contribute to their death.
Major finding: Evidence of reactivation with multiple viruses in septic patients – which included almost 43% who were positive for at least two viruses – and the magnitude of viral loads in septic patients indicate that patients with sepsis are immunosuppressed.
Data source: The study compared levels of cytomegalovirus, herpes simplex, and other viruses in 560 critically ill septic patients and 161 critically ill nonseptic patients in intensive care units, and 164 healthy, age-matched controls.
Disclosures: The study was funded by the National Institutes of Health. One of the 13 authors is an employee of Biomérieux, a company that is working on a method to monitor levels of different viruses in the blood as an indicator of immune status. No other author disclosures were listed.
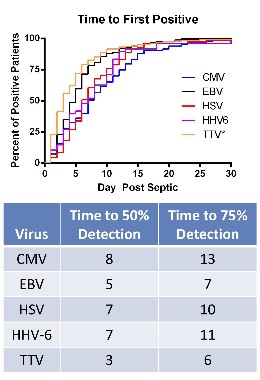
FDA panel recommends informed consent, labeling changes to address morcellator risk
SILVER SPRING, MD. – A requirement for informed consent outlining the risks of morcellation of unsuspected malignancies in women treated with laparoscopic power morcellators for presumably benign fibroids was among the recommendations made by a Food and Drug Administration advisory panel.
On July 11, the second day of a two-day meeting of the FDA’s Obstetrics and Gynecology Devices Advisory Committee, panelists also supported adding a boxed warning to the labels of laparoscopic power morcellators (LPMs). Other suggestions and recommendations included identifying characteristics of patients or fibroids, physical exam findings, and other features that could help determine patients who may have a sarcoma before treatment, as well as strategies to mitigate risks during treatment.
"There should be some labeling or special controls" to ensure that women who are being considered for treatment with this device and their physicians "get the message that we do believe there is an increased risk" and that the device is contraindicated in patients with a known or suspected malignancy, said Dr. Carol Brown, a gynecologic cancer surgeon at Memorial Sloan Kettering Cancer Center, New York.
The advisory panel did not vote on any issues and was not asked whether LPMs should be taken off the market or reclassified as class III medical devices, which require clinical data for approval.
During testimony from women and relatives of those who had been diagnosed with stage 4 LMS after treatment that included morcellation for what was thought to be uterine fibroids, "a recurring theme" was that they had not been told that morcellation could be part of their treatment, and if they had known, they may not have agreed to that treatment and chosen an alternative, she pointed out.
The FDA convened the meeting to discuss the benefits, risks, and clinical role of LPMs in the treatment of women with uterine fibroids. Panelists also were told to discuss strategies that could be used to reduce the risks of morcellation disseminating cancerous tissue into the pelvis and abdomen of women with an unsuspected uterine sarcoma or leiomyosarcoma (LMS).
Concerns about this risk have received widespread attention this year. An FDA safety advisory was issued in April recommending that the use of LPMs during a hysterectomy or myomectomy in women with uterine fibroids be discouraged. The case of Dr. Amy Reed, an anesthesiologist who was diagnosed with stage 4 LMS after undergoing a hysterectomy with morcellation at the age of 40 for what was thought to be benign fibroids, also garnered media attention. She and her husband, Dr. Hooman Noorchashm, a cardiothoracic surgeon, are leading a campaign highlighting these risks, calling for a ban on the use of LPMs. They -- along with women who have had similar experiences, and husbands and sisters of women who died of disseminated LMS after undergoing morcellation for what was thought to be benign fibroids -- also spoke at the meeting. These speakers emphasized that they are aware of at least 130 such cases, despite the small number of cases reported to the FDA (21 as of June 2014).
In April, the FDA recommended that physicians discuss alternative treatment options with women who have symptomatic uterine fibroids. If power morcellation is considered the best option, the agency advised, women should be informed that their fibroids may contain cancerous cells and, if so, morcellation could significantly worsen their prognosis. Among women who undergo a hysterectomy or myomectomy for a presumed fibroid, about one in 350 have a uterine sarcoma, and about one in 500 have a leiomyosarcoma (LMS), the FDA estimates.
The currently available LPMs that are "cleared" for gynecologic indications are regulated as moderate-risk class II devices, which require little or no clinical data. The agency is considering requiring that clinical data be provided for LPMs used for gynecologic indications.
Panelists said that features and tools that could help determine whether a patient could have a sarcoma include an older age, certain symptoms, and genetic susceptibility (a history of retinoblastoma), some imaging techniques, as well as a history of pelvic radiation.
Two panelists – a surgical oncologist and a bioethicist – said that LPMs should not be used for gynecologic indications until better data are available.
"I have not seen anything in isolation or together" that could help predict whether a woman with presumed uterine fibroids has a leiomyosarcoma, said Dr. Craig Shriver, professor of surgery and director of the John P. Murtha Cancer Center at Walter Reed National Navy Military Medical Center, Bethesda, Md.
Referring to the tenets of considering all masses as cancer until proven otherwise, he said, "I have been perplexed over the last two decades watching the introduction of laparoscopic power morcellation techniques that is totally anathema to these and my core principles as a cancer surgeon," he said. Two days of testimony have "only more strongly reaffirmed my commitment and belief that at present, there is no safe way to offer laparoscopic power morcellation as part of any minimally invasive surgery," he added. They should be withdrawn from the market, reclassified as class III devices and studied in clinical trials, he recommended.
But Dr. Brown said that while she agreed with those principles, since fibroids are so common, banning the use of morcellation could result in “hundreds of thousands” of hysterectomies.
Specimen collection bags were used when LPMs were first introduced for gynecologic indications, but dropped over time. Use has increased recently as a result of the increased attention to the risks, but there is no evidence that bags are effective in reducing the risk of disseminating malignant cells in the peritoneal cavity, in the case of an unsuspected malignancy.
“If you are going to morcellate, and it can be done safely in a bag, then that should be encouraged,” said panelist Dr. Keith Isaacson, medical director of the Center for Minimally Invasive Gynecologic Surgery at Newton (Mass.) Wellesley Hospital. More work is needed to evaluate bags and the techniques for their use, and to determine how easy it is train clinicians in how to use them safely, he added. “I still believe that intuitively – and that’s all we have is intuition here – that it more likely mitigates the risk of upstaging a tumor if you morcellate within a containment system, such as a bag.”
The FDA usually follows the recommendations of its advisory panels. Panel members have been cleared of potential conflicts, although occasionally a panelist is given a waiver, but not at this meeting.
Adverse events related to LPMs or other medical devices should be reported to the FDA.
This article has been updated 7/14/2014.
SILVER SPRING, MD. – A requirement for informed consent outlining the risks of morcellation of unsuspected malignancies in women treated with laparoscopic power morcellators for presumably benign fibroids was among the recommendations made by a Food and Drug Administration advisory panel.
On July 11, the second day of a two-day meeting of the FDA’s Obstetrics and Gynecology Devices Advisory Committee, panelists also supported adding a boxed warning to the labels of laparoscopic power morcellators (LPMs). Other suggestions and recommendations included identifying characteristics of patients or fibroids, physical exam findings, and other features that could help determine patients who may have a sarcoma before treatment, as well as strategies to mitigate risks during treatment.
"There should be some labeling or special controls" to ensure that women who are being considered for treatment with this device and their physicians "get the message that we do believe there is an increased risk" and that the device is contraindicated in patients with a known or suspected malignancy, said Dr. Carol Brown, a gynecologic cancer surgeon at Memorial Sloan Kettering Cancer Center, New York.
The advisory panel did not vote on any issues and was not asked whether LPMs should be taken off the market or reclassified as class III medical devices, which require clinical data for approval.
During testimony from women and relatives of those who had been diagnosed with stage 4 LMS after treatment that included morcellation for what was thought to be uterine fibroids, "a recurring theme" was that they had not been told that morcellation could be part of their treatment, and if they had known, they may not have agreed to that treatment and chosen an alternative, she pointed out.
The FDA convened the meeting to discuss the benefits, risks, and clinical role of LPMs in the treatment of women with uterine fibroids. Panelists also were told to discuss strategies that could be used to reduce the risks of morcellation disseminating cancerous tissue into the pelvis and abdomen of women with an unsuspected uterine sarcoma or leiomyosarcoma (LMS).
Concerns about this risk have received widespread attention this year. An FDA safety advisory was issued in April recommending that the use of LPMs during a hysterectomy or myomectomy in women with uterine fibroids be discouraged. The case of Dr. Amy Reed, an anesthesiologist who was diagnosed with stage 4 LMS after undergoing a hysterectomy with morcellation at the age of 40 for what was thought to be benign fibroids, also garnered media attention. She and her husband, Dr. Hooman Noorchashm, a cardiothoracic surgeon, are leading a campaign highlighting these risks, calling for a ban on the use of LPMs. They -- along with women who have had similar experiences, and husbands and sisters of women who died of disseminated LMS after undergoing morcellation for what was thought to be benign fibroids -- also spoke at the meeting. These speakers emphasized that they are aware of at least 130 such cases, despite the small number of cases reported to the FDA (21 as of June 2014).
In April, the FDA recommended that physicians discuss alternative treatment options with women who have symptomatic uterine fibroids. If power morcellation is considered the best option, the agency advised, women should be informed that their fibroids may contain cancerous cells and, if so, morcellation could significantly worsen their prognosis. Among women who undergo a hysterectomy or myomectomy for a presumed fibroid, about one in 350 have a uterine sarcoma, and about one in 500 have a leiomyosarcoma (LMS), the FDA estimates.
The currently available LPMs that are "cleared" for gynecologic indications are regulated as moderate-risk class II devices, which require little or no clinical data. The agency is considering requiring that clinical data be provided for LPMs used for gynecologic indications.
Panelists said that features and tools that could help determine whether a patient could have a sarcoma include an older age, certain symptoms, and genetic susceptibility (a history of retinoblastoma), some imaging techniques, as well as a history of pelvic radiation.
Two panelists – a surgical oncologist and a bioethicist – said that LPMs should not be used for gynecologic indications until better data are available.
"I have not seen anything in isolation or together" that could help predict whether a woman with presumed uterine fibroids has a leiomyosarcoma, said Dr. Craig Shriver, professor of surgery and director of the John P. Murtha Cancer Center at Walter Reed National Navy Military Medical Center, Bethesda, Md.
Referring to the tenets of considering all masses as cancer until proven otherwise, he said, "I have been perplexed over the last two decades watching the introduction of laparoscopic power morcellation techniques that is totally anathema to these and my core principles as a cancer surgeon," he said. Two days of testimony have "only more strongly reaffirmed my commitment and belief that at present, there is no safe way to offer laparoscopic power morcellation as part of any minimally invasive surgery," he added. They should be withdrawn from the market, reclassified as class III devices and studied in clinical trials, he recommended.
But Dr. Brown said that while she agreed with those principles, since fibroids are so common, banning the use of morcellation could result in “hundreds of thousands” of hysterectomies.
Specimen collection bags were used when LPMs were first introduced for gynecologic indications, but dropped over time. Use has increased recently as a result of the increased attention to the risks, but there is no evidence that bags are effective in reducing the risk of disseminating malignant cells in the peritoneal cavity, in the case of an unsuspected malignancy.
“If you are going to morcellate, and it can be done safely in a bag, then that should be encouraged,” said panelist Dr. Keith Isaacson, medical director of the Center for Minimally Invasive Gynecologic Surgery at Newton (Mass.) Wellesley Hospital. More work is needed to evaluate bags and the techniques for their use, and to determine how easy it is train clinicians in how to use them safely, he added. “I still believe that intuitively – and that’s all we have is intuition here – that it more likely mitigates the risk of upstaging a tumor if you morcellate within a containment system, such as a bag.”
The FDA usually follows the recommendations of its advisory panels. Panel members have been cleared of potential conflicts, although occasionally a panelist is given a waiver, but not at this meeting.
Adverse events related to LPMs or other medical devices should be reported to the FDA.
This article has been updated 7/14/2014.
SILVER SPRING, MD. – A requirement for informed consent outlining the risks of morcellation of unsuspected malignancies in women treated with laparoscopic power morcellators for presumably benign fibroids was among the recommendations made by a Food and Drug Administration advisory panel.
On July 11, the second day of a two-day meeting of the FDA’s Obstetrics and Gynecology Devices Advisory Committee, panelists also supported adding a boxed warning to the labels of laparoscopic power morcellators (LPMs). Other suggestions and recommendations included identifying characteristics of patients or fibroids, physical exam findings, and other features that could help determine patients who may have a sarcoma before treatment, as well as strategies to mitigate risks during treatment.
"There should be some labeling or special controls" to ensure that women who are being considered for treatment with this device and their physicians "get the message that we do believe there is an increased risk" and that the device is contraindicated in patients with a known or suspected malignancy, said Dr. Carol Brown, a gynecologic cancer surgeon at Memorial Sloan Kettering Cancer Center, New York.
The advisory panel did not vote on any issues and was not asked whether LPMs should be taken off the market or reclassified as class III medical devices, which require clinical data for approval.
During testimony from women and relatives of those who had been diagnosed with stage 4 LMS after treatment that included morcellation for what was thought to be uterine fibroids, "a recurring theme" was that they had not been told that morcellation could be part of their treatment, and if they had known, they may not have agreed to that treatment and chosen an alternative, she pointed out.
The FDA convened the meeting to discuss the benefits, risks, and clinical role of LPMs in the treatment of women with uterine fibroids. Panelists also were told to discuss strategies that could be used to reduce the risks of morcellation disseminating cancerous tissue into the pelvis and abdomen of women with an unsuspected uterine sarcoma or leiomyosarcoma (LMS).
Concerns about this risk have received widespread attention this year. An FDA safety advisory was issued in April recommending that the use of LPMs during a hysterectomy or myomectomy in women with uterine fibroids be discouraged. The case of Dr. Amy Reed, an anesthesiologist who was diagnosed with stage 4 LMS after undergoing a hysterectomy with morcellation at the age of 40 for what was thought to be benign fibroids, also garnered media attention. She and her husband, Dr. Hooman Noorchashm, a cardiothoracic surgeon, are leading a campaign highlighting these risks, calling for a ban on the use of LPMs. They -- along with women who have had similar experiences, and husbands and sisters of women who died of disseminated LMS after undergoing morcellation for what was thought to be benign fibroids -- also spoke at the meeting. These speakers emphasized that they are aware of at least 130 such cases, despite the small number of cases reported to the FDA (21 as of June 2014).
In April, the FDA recommended that physicians discuss alternative treatment options with women who have symptomatic uterine fibroids. If power morcellation is considered the best option, the agency advised, women should be informed that their fibroids may contain cancerous cells and, if so, morcellation could significantly worsen their prognosis. Among women who undergo a hysterectomy or myomectomy for a presumed fibroid, about one in 350 have a uterine sarcoma, and about one in 500 have a leiomyosarcoma (LMS), the FDA estimates.
The currently available LPMs that are "cleared" for gynecologic indications are regulated as moderate-risk class II devices, which require little or no clinical data. The agency is considering requiring that clinical data be provided for LPMs used for gynecologic indications.
Panelists said that features and tools that could help determine whether a patient could have a sarcoma include an older age, certain symptoms, and genetic susceptibility (a history of retinoblastoma), some imaging techniques, as well as a history of pelvic radiation.
Two panelists – a surgical oncologist and a bioethicist – said that LPMs should not be used for gynecologic indications until better data are available.
"I have not seen anything in isolation or together" that could help predict whether a woman with presumed uterine fibroids has a leiomyosarcoma, said Dr. Craig Shriver, professor of surgery and director of the John P. Murtha Cancer Center at Walter Reed National Navy Military Medical Center, Bethesda, Md.
Referring to the tenets of considering all masses as cancer until proven otherwise, he said, "I have been perplexed over the last two decades watching the introduction of laparoscopic power morcellation techniques that is totally anathema to these and my core principles as a cancer surgeon," he said. Two days of testimony have "only more strongly reaffirmed my commitment and belief that at present, there is no safe way to offer laparoscopic power morcellation as part of any minimally invasive surgery," he added. They should be withdrawn from the market, reclassified as class III devices and studied in clinical trials, he recommended.
But Dr. Brown said that while she agreed with those principles, since fibroids are so common, banning the use of morcellation could result in “hundreds of thousands” of hysterectomies.
Specimen collection bags were used when LPMs were first introduced for gynecologic indications, but dropped over time. Use has increased recently as a result of the increased attention to the risks, but there is no evidence that bags are effective in reducing the risk of disseminating malignant cells in the peritoneal cavity, in the case of an unsuspected malignancy.
“If you are going to morcellate, and it can be done safely in a bag, then that should be encouraged,” said panelist Dr. Keith Isaacson, medical director of the Center for Minimally Invasive Gynecologic Surgery at Newton (Mass.) Wellesley Hospital. More work is needed to evaluate bags and the techniques for their use, and to determine how easy it is train clinicians in how to use them safely, he added. “I still believe that intuitively – and that’s all we have is intuition here – that it more likely mitigates the risk of upstaging a tumor if you morcellate within a containment system, such as a bag.”
The FDA usually follows the recommendations of its advisory panels. Panel members have been cleared of potential conflicts, although occasionally a panelist is given a waiver, but not at this meeting.
Adverse events related to LPMs or other medical devices should be reported to the FDA.
This article has been updated 7/14/2014.
AT AN FDA ADVISORY COMMITTEE MEETING
VIDEO: ‘Improve – but do not abandon – power morcellation’
SILVER SPRING, MD. – Without power morcellation, the number of hysterectomies performed using an open approach would dramatically increase – and the combined mortality from laparoscopic hysterectomy and potential dissemination of leiomyosarcoma would be less than that of open hysterectomy, according to testimony given July 11 at a Food and Drug Administration expert panel meeting.
Dr. Jubilee Brown, director of gynecologic oncology at the Woman's Hospital of Texas, University of Texas M.D. Anderson Cancer Center, Houston, testified at the meeting on behalf of the AAGL, an association that promotes minimally invasive gynecologic surgery. She presented results of a decision analysis suggesting that if all U.S. cases were converted to open hysterectomy from laparoscopic hysterectomy (LH) with morcellation of fibroids, 17 more women each year would die from the open procedure than from the combination LH and morcellation.
"Improve – but do not abandon – power morcellation," Dr. Brown told the FDA Obstetrics and Gynecology Devices Advisory Committee. She discussed her testimony during this video interview.
Dr. Brown said she had no relevant financial conflicts of interest.
SILVER SPRING, MD. – Without power morcellation, the number of hysterectomies performed using an open approach would dramatically increase – and the combined mortality from laparoscopic hysterectomy and potential dissemination of leiomyosarcoma would be less than that of open hysterectomy, according to testimony given July 11 at a Food and Drug Administration expert panel meeting.
Dr. Jubilee Brown, director of gynecologic oncology at the Woman's Hospital of Texas, University of Texas M.D. Anderson Cancer Center, Houston, testified at the meeting on behalf of the AAGL, an association that promotes minimally invasive gynecologic surgery. She presented results of a decision analysis suggesting that if all U.S. cases were converted to open hysterectomy from laparoscopic hysterectomy (LH) with morcellation of fibroids, 17 more women each year would die from the open procedure than from the combination LH and morcellation.
"Improve – but do not abandon – power morcellation," Dr. Brown told the FDA Obstetrics and Gynecology Devices Advisory Committee. She discussed her testimony during this video interview.
Dr. Brown said she had no relevant financial conflicts of interest.
SILVER SPRING, MD. – Without power morcellation, the number of hysterectomies performed using an open approach would dramatically increase – and the combined mortality from laparoscopic hysterectomy and potential dissemination of leiomyosarcoma would be less than that of open hysterectomy, according to testimony given July 11 at a Food and Drug Administration expert panel meeting.
Dr. Jubilee Brown, director of gynecologic oncology at the Woman's Hospital of Texas, University of Texas M.D. Anderson Cancer Center, Houston, testified at the meeting on behalf of the AAGL, an association that promotes minimally invasive gynecologic surgery. She presented results of a decision analysis suggesting that if all U.S. cases were converted to open hysterectomy from laparoscopic hysterectomy (LH) with morcellation of fibroids, 17 more women each year would die from the open procedure than from the combination LH and morcellation.
"Improve – but do not abandon – power morcellation," Dr. Brown told the FDA Obstetrics and Gynecology Devices Advisory Committee. She discussed her testimony during this video interview.
Dr. Brown said she had no relevant financial conflicts of interest.
AT AN FDA ADVISORY COMMITTEE MEETING

FDA panel addresses risks of power morcellators in fibroid surgery
SILVER SPRING, MD. – Expert advisers expressed little confidence in the Food and Drug Administration estimate that women who undergo a hysterectomy or myomectomy for a presumed fibroid have about a 1 in 350 chance of having an unsuspected sarcoma – the first of several issues the advisers will address at a 2-day meeting on the risks of power morcellation in this setting.
Members of the FDA Obstetrics and Gynecology Devices Advisory Committee said July 10 that they considered the data weak and confusing. Several pointed out that the studies used in the FDA analysis were limited by publication bias, that many patients with fibroids were not included in these estimates, and that better estimates could be obtained with other data sources.
Based on the FDA estimates, clinicians would expect to see about one case of an undiagnosed sarcoma per year, but most do not, one panelist noted.
The estimate is based on an analysis of nine studies of unsuspected uterine sarcomas and leiomyosarcomas (LMSs) treated with a morcellator during a hysterectomy or myomectomy, published between 1990 and 2012. The agency estimated that peritoneal dissemination and/or cancer up-staging to FIGO stage III/IV occurs in about 25%-64% of cases where a uterine sarcoma is morcellated, and that these women have poorer disease-free survival and overall survival than women not treated with morcellation.
The FDA convened the advisory panel meeting to address the benefits, risks, and clinical role of laparoscopic power morcellators (LPMs) in the treatment of women with uterine fibroids, and the use of what the agency described as “potential” mitigation strategies that might reduce or eliminate the risks of disseminating cancerous tissue into the pelvis and abdomen of women with an unsuspected uterine sarcoma or LMS.
In April, the FDA issued a safety communication about the risks of spreading cancerous tissue beyond the uterus when morcellation is used in a patient with an unsuspected uterine sarcoma – and first cited the 1-in-350 risk, notably higher than previously reported rates.The FDA recommended that the use of LPMs during a hysterectomy or myomectomy in women with fibroids should be discouraged and that physicians should discuss alternative treatment options with these patients. But if power morcellation was considered the best option, patients should be informed that their fibroids “may contain unexpected cancerous tissue and that laparoscopic power morcellation may spread the cancer, significantly worsening their prognosis,” the FDA advisory said.
At the meeting, Dr. Ron Yustein, of the FDA Office of Surveillance and Biometrics, said that as of June 22, the voluntary FDA medical device adverse event reporting program had received 21 reports of dissemination of malignancy (mostly LMS) after morcellation of suspected fibroids, including seven reported to be fatal.
Family members of women who had developed disseminated disease after morcellation of an unsuspected sarcoma interrupted the panel discussion to point out they are aware of at least 130 such cases.
LPMs, also cleared for general surgery and urologic indications, are considered class II “moderate risk” devices and were cleared for use based on older devices that were already on the market, with little if any clinical testing in humans. The first LPM for a gynecologic indication was cleared in 1995. LPMs include electromechanical morcellators, which typically have spinning blades to cut up the tissue, and one electrosurgical device that uses radiofrequency energy.
Clinical data are required for class III medical devices to be approved, and the FDA is considering whether to require clinical data for LPMs used in gynecologic indications.
Dr. Craig Sobolewski, who spoke about surgical options for fibroids at the FDA’s invitation, said that the use of specimen collection bags during morcellation dropped because the varied sizes of specimens make it difficult to use bags, and it can be difficult to view a spinning blade through the bag.
The use of collection bags was “minuscule,” but has increased subsequent to the April FDA advisory, said Dr. Sobolewski, chief of the division of minimally invasive gynecologic surgery, Duke University, Durham, N.C.
One “very positive outcome” of this attention will be innovations to address this risk, which will “hopefully enable us to continue to offer this approach ... while simultaneously mitigating the risk,” he said.
The meeting continues July 11, when the panel will address labeling of these devices to reflect the risk of morcellating an unsuspected sarcoma. The panel will not be asked whether these devices should be taken off the market.
Dr. Sobolewski disclosed being a consultant and speaker for Covidien, and is a consultant for and holds stock options in TransEnterix. (Neither company has morcellation products; Covidien manufactures specimen retrieval bags.) None of the panel members has relevant disclosures.
SILVER SPRING, MD. – Expert advisers expressed little confidence in the Food and Drug Administration estimate that women who undergo a hysterectomy or myomectomy for a presumed fibroid have about a 1 in 350 chance of having an unsuspected sarcoma – the first of several issues the advisers will address at a 2-day meeting on the risks of power morcellation in this setting.
Members of the FDA Obstetrics and Gynecology Devices Advisory Committee said July 10 that they considered the data weak and confusing. Several pointed out that the studies used in the FDA analysis were limited by publication bias, that many patients with fibroids were not included in these estimates, and that better estimates could be obtained with other data sources.
Based on the FDA estimates, clinicians would expect to see about one case of an undiagnosed sarcoma per year, but most do not, one panelist noted.
The estimate is based on an analysis of nine studies of unsuspected uterine sarcomas and leiomyosarcomas (LMSs) treated with a morcellator during a hysterectomy or myomectomy, published between 1990 and 2012. The agency estimated that peritoneal dissemination and/or cancer up-staging to FIGO stage III/IV occurs in about 25%-64% of cases where a uterine sarcoma is morcellated, and that these women have poorer disease-free survival and overall survival than women not treated with morcellation.
The FDA convened the advisory panel meeting to address the benefits, risks, and clinical role of laparoscopic power morcellators (LPMs) in the treatment of women with uterine fibroids, and the use of what the agency described as “potential” mitigation strategies that might reduce or eliminate the risks of disseminating cancerous tissue into the pelvis and abdomen of women with an unsuspected uterine sarcoma or LMS.
In April, the FDA issued a safety communication about the risks of spreading cancerous tissue beyond the uterus when morcellation is used in a patient with an unsuspected uterine sarcoma – and first cited the 1-in-350 risk, notably higher than previously reported rates.The FDA recommended that the use of LPMs during a hysterectomy or myomectomy in women with fibroids should be discouraged and that physicians should discuss alternative treatment options with these patients. But if power morcellation was considered the best option, patients should be informed that their fibroids “may contain unexpected cancerous tissue and that laparoscopic power morcellation may spread the cancer, significantly worsening their prognosis,” the FDA advisory said.
At the meeting, Dr. Ron Yustein, of the FDA Office of Surveillance and Biometrics, said that as of June 22, the voluntary FDA medical device adverse event reporting program had received 21 reports of dissemination of malignancy (mostly LMS) after morcellation of suspected fibroids, including seven reported to be fatal.
Family members of women who had developed disseminated disease after morcellation of an unsuspected sarcoma interrupted the panel discussion to point out they are aware of at least 130 such cases.
LPMs, also cleared for general surgery and urologic indications, are considered class II “moderate risk” devices and were cleared for use based on older devices that were already on the market, with little if any clinical testing in humans. The first LPM for a gynecologic indication was cleared in 1995. LPMs include electromechanical morcellators, which typically have spinning blades to cut up the tissue, and one electrosurgical device that uses radiofrequency energy.
Clinical data are required for class III medical devices to be approved, and the FDA is considering whether to require clinical data for LPMs used in gynecologic indications.
Dr. Craig Sobolewski, who spoke about surgical options for fibroids at the FDA’s invitation, said that the use of specimen collection bags during morcellation dropped because the varied sizes of specimens make it difficult to use bags, and it can be difficult to view a spinning blade through the bag.
The use of collection bags was “minuscule,” but has increased subsequent to the April FDA advisory, said Dr. Sobolewski, chief of the division of minimally invasive gynecologic surgery, Duke University, Durham, N.C.
One “very positive outcome” of this attention will be innovations to address this risk, which will “hopefully enable us to continue to offer this approach ... while simultaneously mitigating the risk,” he said.
The meeting continues July 11, when the panel will address labeling of these devices to reflect the risk of morcellating an unsuspected sarcoma. The panel will not be asked whether these devices should be taken off the market.
Dr. Sobolewski disclosed being a consultant and speaker for Covidien, and is a consultant for and holds stock options in TransEnterix. (Neither company has morcellation products; Covidien manufactures specimen retrieval bags.) None of the panel members has relevant disclosures.
SILVER SPRING, MD. – Expert advisers expressed little confidence in the Food and Drug Administration estimate that women who undergo a hysterectomy or myomectomy for a presumed fibroid have about a 1 in 350 chance of having an unsuspected sarcoma – the first of several issues the advisers will address at a 2-day meeting on the risks of power morcellation in this setting.
Members of the FDA Obstetrics and Gynecology Devices Advisory Committee said July 10 that they considered the data weak and confusing. Several pointed out that the studies used in the FDA analysis were limited by publication bias, that many patients with fibroids were not included in these estimates, and that better estimates could be obtained with other data sources.
Based on the FDA estimates, clinicians would expect to see about one case of an undiagnosed sarcoma per year, but most do not, one panelist noted.
The estimate is based on an analysis of nine studies of unsuspected uterine sarcomas and leiomyosarcomas (LMSs) treated with a morcellator during a hysterectomy or myomectomy, published between 1990 and 2012. The agency estimated that peritoneal dissemination and/or cancer up-staging to FIGO stage III/IV occurs in about 25%-64% of cases where a uterine sarcoma is morcellated, and that these women have poorer disease-free survival and overall survival than women not treated with morcellation.
The FDA convened the advisory panel meeting to address the benefits, risks, and clinical role of laparoscopic power morcellators (LPMs) in the treatment of women with uterine fibroids, and the use of what the agency described as “potential” mitigation strategies that might reduce or eliminate the risks of disseminating cancerous tissue into the pelvis and abdomen of women with an unsuspected uterine sarcoma or LMS.
In April, the FDA issued a safety communication about the risks of spreading cancerous tissue beyond the uterus when morcellation is used in a patient with an unsuspected uterine sarcoma – and first cited the 1-in-350 risk, notably higher than previously reported rates.The FDA recommended that the use of LPMs during a hysterectomy or myomectomy in women with fibroids should be discouraged and that physicians should discuss alternative treatment options with these patients. But if power morcellation was considered the best option, patients should be informed that their fibroids “may contain unexpected cancerous tissue and that laparoscopic power morcellation may spread the cancer, significantly worsening their prognosis,” the FDA advisory said.
At the meeting, Dr. Ron Yustein, of the FDA Office of Surveillance and Biometrics, said that as of June 22, the voluntary FDA medical device adverse event reporting program had received 21 reports of dissemination of malignancy (mostly LMS) after morcellation of suspected fibroids, including seven reported to be fatal.
Family members of women who had developed disseminated disease after morcellation of an unsuspected sarcoma interrupted the panel discussion to point out they are aware of at least 130 such cases.
LPMs, also cleared for general surgery and urologic indications, are considered class II “moderate risk” devices and were cleared for use based on older devices that were already on the market, with little if any clinical testing in humans. The first LPM for a gynecologic indication was cleared in 1995. LPMs include electromechanical morcellators, which typically have spinning blades to cut up the tissue, and one electrosurgical device that uses radiofrequency energy.
Clinical data are required for class III medical devices to be approved, and the FDA is considering whether to require clinical data for LPMs used in gynecologic indications.
Dr. Craig Sobolewski, who spoke about surgical options for fibroids at the FDA’s invitation, said that the use of specimen collection bags during morcellation dropped because the varied sizes of specimens make it difficult to use bags, and it can be difficult to view a spinning blade through the bag.
The use of collection bags was “minuscule,” but has increased subsequent to the April FDA advisory, said Dr. Sobolewski, chief of the division of minimally invasive gynecologic surgery, Duke University, Durham, N.C.
One “very positive outcome” of this attention will be innovations to address this risk, which will “hopefully enable us to continue to offer this approach ... while simultaneously mitigating the risk,” he said.
The meeting continues July 11, when the panel will address labeling of these devices to reflect the risk of morcellating an unsuspected sarcoma. The panel will not be asked whether these devices should be taken off the market.
Dr. Sobolewski disclosed being a consultant and speaker for Covidien, and is a consultant for and holds stock options in TransEnterix. (Neither company has morcellation products; Covidien manufactures specimen retrieval bags.) None of the panel members has relevant disclosures.
FDA approves belinostat for peripheral T-cell lymphoma
Belinostat, a histone deacetylase inhibitor, has been approved for treating peripheral T-cell lymphoma, based on the results of the BELIEF study that found an overall response rate of nearly 26% among treated patients.
This is the third drug approved for this rare, aggressive form of non-Hodgkin’s lymphoma (NHL) since 2009, according to the Food and Drug Administration statement announcing the approval on July 3.
The other two drugs are pralatrexate injection (Folotyn), a folate analogue metabolic inhibitor approved in 2009 for treating relapsed or refractory peripheral T-cell lymphoma (PTCL); and romidepsin (Istodax), a histone deacetylase (HDAC) inhibitor approved in 2011 for treating PTCL in patients who have received at least one previous treatment.
This is an accelerated approval, which is based on surrogate or intermediate endpoints considered by the FDA as "reasonably likely to predict clinical benefit for patients with serious conditions with unmet medical needs." Confirmatory trials that verify the clinical benefit are required for full approval; otherwise, the approval can be withdrawn by the FDA. Belinostat will be marketed as Beleodaq by Spectrum Pharmaceuticals, which also markets Folotyn.
HDAC inhibitors "catalyze the removal of acetyl groups from the lysine residues of histones and some nonhistone proteins," and in vitro, belinostat "caused the accumulation of acetylated histones and other proteins, inducing cell cycle arrest and/or apoptosis of some transformed cells," according to a statement on the approval, issued by Spectrum on July 7. "Belinostat shows preferential cytotoxicity towards tumor cells compared to normal cells," and it "inhibited the enzymatic activity of histone deacetylases at nanomolar concentrations," the statement said.
In the BELIEF study, an open-label, single-arm, nonrandomized study, 129 patients with relapsed or refractory PTCL were treated with belinostat, administered via an IV infusion, once a day on days 1-5 of a 21-day cycle, repeated every 3 weeks until the disease progressed or adverse effects became unacceptable. The overall response rate (complete and partial responses), the primary efficacy endpoint, was 25.8%. Nausea, vomiting, fatigue, pyrexia, and anemia were the most common adverse events associated with treatment, according to the FDA.
The company said that the drug is expected to be available in less than 3 weeks of approval (before July 24). The confirmatory trial is a phase III study that will evaluate belinostat plus CHOP (cyclophosphamide, vincristine, doxorubicin, prednisone), compared with CHOP alone.
PTCL accounts for about 10%-15% of NHL cases in North America, according to the FDA, which cites National Cancer Institute estimates that 70,800 Americans will be diagnosed with NHL and 18,990 will die of NHL in 2014.
The prescribing information for belinostat is available here.
Belinostat, a histone deacetylase inhibitor, has been approved for treating peripheral T-cell lymphoma, based on the results of the BELIEF study that found an overall response rate of nearly 26% among treated patients.
This is the third drug approved for this rare, aggressive form of non-Hodgkin’s lymphoma (NHL) since 2009, according to the Food and Drug Administration statement announcing the approval on July 3.
The other two drugs are pralatrexate injection (Folotyn), a folate analogue metabolic inhibitor approved in 2009 for treating relapsed or refractory peripheral T-cell lymphoma (PTCL); and romidepsin (Istodax), a histone deacetylase (HDAC) inhibitor approved in 2011 for treating PTCL in patients who have received at least one previous treatment.
This is an accelerated approval, which is based on surrogate or intermediate endpoints considered by the FDA as "reasonably likely to predict clinical benefit for patients with serious conditions with unmet medical needs." Confirmatory trials that verify the clinical benefit are required for full approval; otherwise, the approval can be withdrawn by the FDA. Belinostat will be marketed as Beleodaq by Spectrum Pharmaceuticals, which also markets Folotyn.
HDAC inhibitors "catalyze the removal of acetyl groups from the lysine residues of histones and some nonhistone proteins," and in vitro, belinostat "caused the accumulation of acetylated histones and other proteins, inducing cell cycle arrest and/or apoptosis of some transformed cells," according to a statement on the approval, issued by Spectrum on July 7. "Belinostat shows preferential cytotoxicity towards tumor cells compared to normal cells," and it "inhibited the enzymatic activity of histone deacetylases at nanomolar concentrations," the statement said.
In the BELIEF study, an open-label, single-arm, nonrandomized study, 129 patients with relapsed or refractory PTCL were treated with belinostat, administered via an IV infusion, once a day on days 1-5 of a 21-day cycle, repeated every 3 weeks until the disease progressed or adverse effects became unacceptable. The overall response rate (complete and partial responses), the primary efficacy endpoint, was 25.8%. Nausea, vomiting, fatigue, pyrexia, and anemia were the most common adverse events associated with treatment, according to the FDA.
The company said that the drug is expected to be available in less than 3 weeks of approval (before July 24). The confirmatory trial is a phase III study that will evaluate belinostat plus CHOP (cyclophosphamide, vincristine, doxorubicin, prednisone), compared with CHOP alone.
PTCL accounts for about 10%-15% of NHL cases in North America, according to the FDA, which cites National Cancer Institute estimates that 70,800 Americans will be diagnosed with NHL and 18,990 will die of NHL in 2014.
The prescribing information for belinostat is available here.
Belinostat, a histone deacetylase inhibitor, has been approved for treating peripheral T-cell lymphoma, based on the results of the BELIEF study that found an overall response rate of nearly 26% among treated patients.
This is the third drug approved for this rare, aggressive form of non-Hodgkin’s lymphoma (NHL) since 2009, according to the Food and Drug Administration statement announcing the approval on July 3.
The other two drugs are pralatrexate injection (Folotyn), a folate analogue metabolic inhibitor approved in 2009 for treating relapsed or refractory peripheral T-cell lymphoma (PTCL); and romidepsin (Istodax), a histone deacetylase (HDAC) inhibitor approved in 2011 for treating PTCL in patients who have received at least one previous treatment.
This is an accelerated approval, which is based on surrogate or intermediate endpoints considered by the FDA as "reasonably likely to predict clinical benefit for patients with serious conditions with unmet medical needs." Confirmatory trials that verify the clinical benefit are required for full approval; otherwise, the approval can be withdrawn by the FDA. Belinostat will be marketed as Beleodaq by Spectrum Pharmaceuticals, which also markets Folotyn.
HDAC inhibitors "catalyze the removal of acetyl groups from the lysine residues of histones and some nonhistone proteins," and in vitro, belinostat "caused the accumulation of acetylated histones and other proteins, inducing cell cycle arrest and/or apoptosis of some transformed cells," according to a statement on the approval, issued by Spectrum on July 7. "Belinostat shows preferential cytotoxicity towards tumor cells compared to normal cells," and it "inhibited the enzymatic activity of histone deacetylases at nanomolar concentrations," the statement said.
In the BELIEF study, an open-label, single-arm, nonrandomized study, 129 patients with relapsed or refractory PTCL were treated with belinostat, administered via an IV infusion, once a day on days 1-5 of a 21-day cycle, repeated every 3 weeks until the disease progressed or adverse effects became unacceptable. The overall response rate (complete and partial responses), the primary efficacy endpoint, was 25.8%. Nausea, vomiting, fatigue, pyrexia, and anemia were the most common adverse events associated with treatment, according to the FDA.
The company said that the drug is expected to be available in less than 3 weeks of approval (before July 24). The confirmatory trial is a phase III study that will evaluate belinostat plus CHOP (cyclophosphamide, vincristine, doxorubicin, prednisone), compared with CHOP alone.
PTCL accounts for about 10%-15% of NHL cases in North America, according to the FDA, which cites National Cancer Institute estimates that 70,800 Americans will be diagnosed with NHL and 18,990 will die of NHL in 2014.
The prescribing information for belinostat is available here.
FDA grants "breakthrough" status to investigational T-cell therapy for relapsed/refractory ALL
A chimeric antigen receptor therapy in phase I studies for relapsed/refractory acute lymphoblastic leukemia has been granted "breakthrough therapy" status by the Food and Drug Administration, which will expedite the development and review of the product as a result, Novartis has announced.
The investigational T-cell therapy – CTL019 – is now being researched in phase I studies of pediatric and adult patients with relapsed/refractory ALL. CTL019 "uses CAR [chimeric antigen receptor] technology to reprogram a patient’s own T cells to ‘hunt’ cancer cells" that express specific proteins [CD19]. "After they have been reprogrammed, the T cells (now called CTL019) are reintroduced into the patient’s blood; they proliferate and bind to the targeted CD19+ cancer cells and destroy them," according to the statement issued by Novartis on July 7.
Researchers at the University of Pennsylvania, Philadelphia, are conducting phase I/II studies of CTL019 and are working with Novartis to study and develop CAR T-cell treatments as cancer treatments; they have submitted the filing for the breakthrough therapy status, the statement said.
If a drug is designated as breakthrough therapy, the FDA will expedite the development and review of such drug.
The breakthrough therapy designation was created by the 2012 FDA Safety and Innovation Act. The FDA defines a breakthrough therapy as a drug that is intended to treat a serious or life-threatening disease or condition, alone or in combination with other drugs, and where "preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development," according to the FDA website.
"Because of uncertainty of clinical trials, there is no guarantee that CTL019 will ever be commercially available anywhere in the world," the Novartis statement added.
A chimeric antigen receptor therapy in phase I studies for relapsed/refractory acute lymphoblastic leukemia has been granted "breakthrough therapy" status by the Food and Drug Administration, which will expedite the development and review of the product as a result, Novartis has announced.
The investigational T-cell therapy – CTL019 – is now being researched in phase I studies of pediatric and adult patients with relapsed/refractory ALL. CTL019 "uses CAR [chimeric antigen receptor] technology to reprogram a patient’s own T cells to ‘hunt’ cancer cells" that express specific proteins [CD19]. "After they have been reprogrammed, the T cells (now called CTL019) are reintroduced into the patient’s blood; they proliferate and bind to the targeted CD19+ cancer cells and destroy them," according to the statement issued by Novartis on July 7.
Researchers at the University of Pennsylvania, Philadelphia, are conducting phase I/II studies of CTL019 and are working with Novartis to study and develop CAR T-cell treatments as cancer treatments; they have submitted the filing for the breakthrough therapy status, the statement said.
If a drug is designated as breakthrough therapy, the FDA will expedite the development and review of such drug.
The breakthrough therapy designation was created by the 2012 FDA Safety and Innovation Act. The FDA defines a breakthrough therapy as a drug that is intended to treat a serious or life-threatening disease or condition, alone or in combination with other drugs, and where "preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development," according to the FDA website.
"Because of uncertainty of clinical trials, there is no guarantee that CTL019 will ever be commercially available anywhere in the world," the Novartis statement added.
A chimeric antigen receptor therapy in phase I studies for relapsed/refractory acute lymphoblastic leukemia has been granted "breakthrough therapy" status by the Food and Drug Administration, which will expedite the development and review of the product as a result, Novartis has announced.
The investigational T-cell therapy – CTL019 – is now being researched in phase I studies of pediatric and adult patients with relapsed/refractory ALL. CTL019 "uses CAR [chimeric antigen receptor] technology to reprogram a patient’s own T cells to ‘hunt’ cancer cells" that express specific proteins [CD19]. "After they have been reprogrammed, the T cells (now called CTL019) are reintroduced into the patient’s blood; they proliferate and bind to the targeted CD19+ cancer cells and destroy them," according to the statement issued by Novartis on July 7.
Researchers at the University of Pennsylvania, Philadelphia, are conducting phase I/II studies of CTL019 and are working with Novartis to study and develop CAR T-cell treatments as cancer treatments; they have submitted the filing for the breakthrough therapy status, the statement said.
If a drug is designated as breakthrough therapy, the FDA will expedite the development and review of such drug.
The breakthrough therapy designation was created by the 2012 FDA Safety and Innovation Act. The FDA defines a breakthrough therapy as a drug that is intended to treat a serious or life-threatening disease or condition, alone or in combination with other drugs, and where "preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development," according to the FDA website.
"Because of uncertainty of clinical trials, there is no guarantee that CTL019 will ever be commercially available anywhere in the world," the Novartis statement added.