User login
FDA Approves Second Drug for Rare Thyroid Cancer
Cabozantinib, a kinase inhibitor, has been approved for the treatment of progressive, metastatic medullary thyroid cancer, on the basis of the results of an international study of 330 patients, the Food and Drug Administration has announced.
This is the second drug approved by the FDA for this indication in the past 2 years; the first was vandetanib (Caprelsa), approved in April 2011. Before these approvals, "patients with this rare and difficult to treat disease had limited therapeutic treatment options," Dr. Richard Pazdur, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in the FDA’s Nov. 29 statement announcing the approval.
Cabozantinib will be marketed as Cometriq by Exelixis Inc. The recommended dose is 140 mg orally, without food (people taking the drug should not eat for at least 2 hours before and at least 1 hour after taking it). Exelixis is planning to make the drug available in late January, at an average wholesale price of $9,900 for a 28-day supply, according to the company.
In the EXAM study of 330 patients with metastatic medullary thyroid cancer, with evidence of actively progressive disease within 14 months of enrollment, the median progression-free survival was 11.2 months among those treated with cabozantinib, compared with 4 months among those on placebo, a statistically significant difference, according to the prescribing information. Partial responses were observed among 27% of those on cabozantinib but none of those on placebo, and the median duration of the objective response was 14.7 months among those on cabozantinib. At the planned interim analysis, there was no statistically significant difference in overall survival between the two groups.
The most common adverse events, reported in at least 25% of patients treated with cabozantinib, were diarrhea; stomatitis, palmar-plantar erythrodysesthesia syndrome; decreases in weight and appetite; nausea; fatigue; oral pain; hair color changes; dysgeusia; hypertension; abdominal pain; and constipation. The most common laboratory abnormalities, reported in at least 25% of patients, were increased AST, increased ALT, lymphopenia, increased alkaline phosphatase, hypocalcemia, neutropenia, thrombocytopenia, hypophosphatemia, and hyperbilirubinemia.
The prescribing information includes a boxed warning about severe and sometimes fatal hemorrhage (3%), and gastrointestinal perforations (3%) and fistulas (1%) were reported in patients treated with the drug.
The review of this drug was a priority review, completed in 6 months, which is used for drugs "that may offer major advances in treatment or that provide a treatment when no adequate therapy exists," the FDA statement said. The drug has also been designated an orphan drug, because medullary thyroid cancer is considered a rare disease.
As part of its postmarketing commitments, the FDA has requested that the company submit the final overall survival results of the study, expected to be submitted in December 2014, according to the agency’s approval letter. The letter also lists other postmarketing commitments, including the completion of a study comparing the approved dose of "a biologically active and potentially safer lower daily cabozantinib dose" in patients with progressive medullary thyroid cancer.
Cabozantinib is a kinase inhibitor that inhibits the activity of RET, MET, and other tyrosine kinases, which "are involved in both normal cellular function, and in pathologic processes such as oncogenesis, metastasis, tumor angiogenesis, and maintenance of the tumor microenvironment," according to the Exelixis statement announcing approval
The company also announced on Nov. 29 that the application for approval of cabozantinib, also based on the EXAM study, has been accepted by the European Medicines Agency for approval in the European Union.
The FDA statement cites estimates from the National Cancer Institute that 56,460 people will be diagnosed with thyroid cancer this year in the United States, and 1,780 will die from the disease, and about 4% percent of thyroid cancers are medullary thyroid cancer.
The drug’s label is available on the FDA website. Serious adverse events associated with the drug should be reported to the FDA’s MedWatch site or at 800-332-1088.
Cabozantinib, a kinase inhibitor, has been approved for the treatment of progressive, metastatic medullary thyroid cancer, on the basis of the results of an international study of 330 patients, the Food and Drug Administration has announced.
This is the second drug approved by the FDA for this indication in the past 2 years; the first was vandetanib (Caprelsa), approved in April 2011. Before these approvals, "patients with this rare and difficult to treat disease had limited therapeutic treatment options," Dr. Richard Pazdur, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in the FDA’s Nov. 29 statement announcing the approval.
Cabozantinib will be marketed as Cometriq by Exelixis Inc. The recommended dose is 140 mg orally, without food (people taking the drug should not eat for at least 2 hours before and at least 1 hour after taking it). Exelixis is planning to make the drug available in late January, at an average wholesale price of $9,900 for a 28-day supply, according to the company.
In the EXAM study of 330 patients with metastatic medullary thyroid cancer, with evidence of actively progressive disease within 14 months of enrollment, the median progression-free survival was 11.2 months among those treated with cabozantinib, compared with 4 months among those on placebo, a statistically significant difference, according to the prescribing information. Partial responses were observed among 27% of those on cabozantinib but none of those on placebo, and the median duration of the objective response was 14.7 months among those on cabozantinib. At the planned interim analysis, there was no statistically significant difference in overall survival between the two groups.
The most common adverse events, reported in at least 25% of patients treated with cabozantinib, were diarrhea; stomatitis, palmar-plantar erythrodysesthesia syndrome; decreases in weight and appetite; nausea; fatigue; oral pain; hair color changes; dysgeusia; hypertension; abdominal pain; and constipation. The most common laboratory abnormalities, reported in at least 25% of patients, were increased AST, increased ALT, lymphopenia, increased alkaline phosphatase, hypocalcemia, neutropenia, thrombocytopenia, hypophosphatemia, and hyperbilirubinemia.
The prescribing information includes a boxed warning about severe and sometimes fatal hemorrhage (3%), and gastrointestinal perforations (3%) and fistulas (1%) were reported in patients treated with the drug.
The review of this drug was a priority review, completed in 6 months, which is used for drugs "that may offer major advances in treatment or that provide a treatment when no adequate therapy exists," the FDA statement said. The drug has also been designated an orphan drug, because medullary thyroid cancer is considered a rare disease.
As part of its postmarketing commitments, the FDA has requested that the company submit the final overall survival results of the study, expected to be submitted in December 2014, according to the agency’s approval letter. The letter also lists other postmarketing commitments, including the completion of a study comparing the approved dose of "a biologically active and potentially safer lower daily cabozantinib dose" in patients with progressive medullary thyroid cancer.
Cabozantinib is a kinase inhibitor that inhibits the activity of RET, MET, and other tyrosine kinases, which "are involved in both normal cellular function, and in pathologic processes such as oncogenesis, metastasis, tumor angiogenesis, and maintenance of the tumor microenvironment," according to the Exelixis statement announcing approval
The company also announced on Nov. 29 that the application for approval of cabozantinib, also based on the EXAM study, has been accepted by the European Medicines Agency for approval in the European Union.
The FDA statement cites estimates from the National Cancer Institute that 56,460 people will be diagnosed with thyroid cancer this year in the United States, and 1,780 will die from the disease, and about 4% percent of thyroid cancers are medullary thyroid cancer.
The drug’s label is available on the FDA website. Serious adverse events associated with the drug should be reported to the FDA’s MedWatch site or at 800-332-1088.
Cabozantinib, a kinase inhibitor, has been approved for the treatment of progressive, metastatic medullary thyroid cancer, on the basis of the results of an international study of 330 patients, the Food and Drug Administration has announced.
This is the second drug approved by the FDA for this indication in the past 2 years; the first was vandetanib (Caprelsa), approved in April 2011. Before these approvals, "patients with this rare and difficult to treat disease had limited therapeutic treatment options," Dr. Richard Pazdur, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in the FDA’s Nov. 29 statement announcing the approval.
Cabozantinib will be marketed as Cometriq by Exelixis Inc. The recommended dose is 140 mg orally, without food (people taking the drug should not eat for at least 2 hours before and at least 1 hour after taking it). Exelixis is planning to make the drug available in late January, at an average wholesale price of $9,900 for a 28-day supply, according to the company.
In the EXAM study of 330 patients with metastatic medullary thyroid cancer, with evidence of actively progressive disease within 14 months of enrollment, the median progression-free survival was 11.2 months among those treated with cabozantinib, compared with 4 months among those on placebo, a statistically significant difference, according to the prescribing information. Partial responses were observed among 27% of those on cabozantinib but none of those on placebo, and the median duration of the objective response was 14.7 months among those on cabozantinib. At the planned interim analysis, there was no statistically significant difference in overall survival between the two groups.
The most common adverse events, reported in at least 25% of patients treated with cabozantinib, were diarrhea; stomatitis, palmar-plantar erythrodysesthesia syndrome; decreases in weight and appetite; nausea; fatigue; oral pain; hair color changes; dysgeusia; hypertension; abdominal pain; and constipation. The most common laboratory abnormalities, reported in at least 25% of patients, were increased AST, increased ALT, lymphopenia, increased alkaline phosphatase, hypocalcemia, neutropenia, thrombocytopenia, hypophosphatemia, and hyperbilirubinemia.
The prescribing information includes a boxed warning about severe and sometimes fatal hemorrhage (3%), and gastrointestinal perforations (3%) and fistulas (1%) were reported in patients treated with the drug.
The review of this drug was a priority review, completed in 6 months, which is used for drugs "that may offer major advances in treatment or that provide a treatment when no adequate therapy exists," the FDA statement said. The drug has also been designated an orphan drug, because medullary thyroid cancer is considered a rare disease.
As part of its postmarketing commitments, the FDA has requested that the company submit the final overall survival results of the study, expected to be submitted in December 2014, according to the agency’s approval letter. The letter also lists other postmarketing commitments, including the completion of a study comparing the approved dose of "a biologically active and potentially safer lower daily cabozantinib dose" in patients with progressive medullary thyroid cancer.
Cabozantinib is a kinase inhibitor that inhibits the activity of RET, MET, and other tyrosine kinases, which "are involved in both normal cellular function, and in pathologic processes such as oncogenesis, metastasis, tumor angiogenesis, and maintenance of the tumor microenvironment," according to the Exelixis statement announcing approval
The company also announced on Nov. 29 that the application for approval of cabozantinib, also based on the EXAM study, has been accepted by the European Medicines Agency for approval in the European Union.
The FDA statement cites estimates from the National Cancer Institute that 56,460 people will be diagnosed with thyroid cancer this year in the United States, and 1,780 will die from the disease, and about 4% percent of thyroid cancers are medullary thyroid cancer.
The drug’s label is available on the FDA website. Serious adverse events associated with the drug should be reported to the FDA’s MedWatch site or at 800-332-1088.
FDA Panel Wants Limited Nosocomial Pneumonia Indication for Telavancin
SILVER SPRING, MD. – The approval of the antibacterial drug telavancin should be expanded to include patients with nosocomial pneumonia, but only in limited situations, according to the majority of a Food and Drug Administration advisory panel.
At a meeting Nov. 29, the FDA’s Anti-Infective Drugs Advisory Committee voted 13-2 that the available data on telavancin provided substantial evidence that it was safe and effective for treating nosocomial pneumonia, "when other alternatives are not suitable." Panelists agreed that telavancin should be reserved to treat patients with nosocomial pneumonia caused by methicillin-resistant Staphylococcus aureus (MRSA) not methicillin-sensitive S. aureus (MSSA) or Streptococcus pneumoniae, because there are many other treatments available for those infections.
Most panelists did not support approval of the broader indication requested by the manufacturer: treatment of patients with nosocomial pneumonia, including ventilator-associated pneumonia (VAP), caused by susceptible isolates of the Gram-positive microorganisms, S. aureus (including methicillin-susceptible and methicillin-resistant isolates) or S. pneumoniae. The panel voted 9-6 that the data from the two studies did not provide substantial evidence that the drug was safe and effective for this indication.
Telavancin is a lipoglycopeptide antibacterial administered intravenously, with bactericidal activity that results from inhibition of cell wall synthesis and disruption of bacterial plasma membrane function, according to Theravance, the drug’s manufacturer. It was approved in 2009 for the treatment of adults with complicated skin and skin structure infections caused by susceptible Gram-positive bacteria and is marketed as Vibativ.
The current labeling for telavancin includes warnings and precautions in the prescribing information about the increased risk of nephrotoxicity associated with treatment, based on experience in patients treated for the skin infection indication. A boxed warning cites the potential fetal risks.
Among the other points made by panelists were that labeling should include statements about use in patients with reduced creatinine clearance and renal failure and that the company should aggressively collect postmarketing safety and efficacy data.
The panel considered data that included analyses of clinical cure and 28-day all-cause mortality from two non-inferiority studies. These studies comprised 1,503 patients with nosocomial pneumonia and compared telavancin for 7-21 days (10 mg/kg every 24 hours administered intravenously in patients with normal renal function and mild renal impairment or an adjusted dose for patients with moderate or severe renal insufficiency) with vancomycin (1 g IV every 12 hours).
Theravance first submitted an application for approval of the broad nosocomial pneumonia indication in 2009. That application included results of the two studies, which used clinical cure rate as the primary endpoint. Since then, the FDA has requested more data from the company, including a post hoc analysis of mortality, but subsequently declined to approve telavancin after deciding that the trials did not provide adequate evidence of non-inferiority to vancomycin. In response, the company filed a formal dispute, which was rejected by the FDA, but the agency encouraged the company to resubmit the application and held the panel meeting Nov. 29 to review the data.
Telavancin has been approved in the European Union for the treatment of nosocomial pneumonia caused by MRSA, similar to the indication backed by the FDA panel.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting, although a panelist may occasionally be given a waiver, but not at this meeting.
SILVER SPRING, MD. – The approval of the antibacterial drug telavancin should be expanded to include patients with nosocomial pneumonia, but only in limited situations, according to the majority of a Food and Drug Administration advisory panel.
At a meeting Nov. 29, the FDA’s Anti-Infective Drugs Advisory Committee voted 13-2 that the available data on telavancin provided substantial evidence that it was safe and effective for treating nosocomial pneumonia, "when other alternatives are not suitable." Panelists agreed that telavancin should be reserved to treat patients with nosocomial pneumonia caused by methicillin-resistant Staphylococcus aureus (MRSA) not methicillin-sensitive S. aureus (MSSA) or Streptococcus pneumoniae, because there are many other treatments available for those infections.
Most panelists did not support approval of the broader indication requested by the manufacturer: treatment of patients with nosocomial pneumonia, including ventilator-associated pneumonia (VAP), caused by susceptible isolates of the Gram-positive microorganisms, S. aureus (including methicillin-susceptible and methicillin-resistant isolates) or S. pneumoniae. The panel voted 9-6 that the data from the two studies did not provide substantial evidence that the drug was safe and effective for this indication.
Telavancin is a lipoglycopeptide antibacterial administered intravenously, with bactericidal activity that results from inhibition of cell wall synthesis and disruption of bacterial plasma membrane function, according to Theravance, the drug’s manufacturer. It was approved in 2009 for the treatment of adults with complicated skin and skin structure infections caused by susceptible Gram-positive bacteria and is marketed as Vibativ.
The current labeling for telavancin includes warnings and precautions in the prescribing information about the increased risk of nephrotoxicity associated with treatment, based on experience in patients treated for the skin infection indication. A boxed warning cites the potential fetal risks.
Among the other points made by panelists were that labeling should include statements about use in patients with reduced creatinine clearance and renal failure and that the company should aggressively collect postmarketing safety and efficacy data.
The panel considered data that included analyses of clinical cure and 28-day all-cause mortality from two non-inferiority studies. These studies comprised 1,503 patients with nosocomial pneumonia and compared telavancin for 7-21 days (10 mg/kg every 24 hours administered intravenously in patients with normal renal function and mild renal impairment or an adjusted dose for patients with moderate or severe renal insufficiency) with vancomycin (1 g IV every 12 hours).
Theravance first submitted an application for approval of the broad nosocomial pneumonia indication in 2009. That application included results of the two studies, which used clinical cure rate as the primary endpoint. Since then, the FDA has requested more data from the company, including a post hoc analysis of mortality, but subsequently declined to approve telavancin after deciding that the trials did not provide adequate evidence of non-inferiority to vancomycin. In response, the company filed a formal dispute, which was rejected by the FDA, but the agency encouraged the company to resubmit the application and held the panel meeting Nov. 29 to review the data.
Telavancin has been approved in the European Union for the treatment of nosocomial pneumonia caused by MRSA, similar to the indication backed by the FDA panel.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting, although a panelist may occasionally be given a waiver, but not at this meeting.
SILVER SPRING, MD. – The approval of the antibacterial drug telavancin should be expanded to include patients with nosocomial pneumonia, but only in limited situations, according to the majority of a Food and Drug Administration advisory panel.
At a meeting Nov. 29, the FDA’s Anti-Infective Drugs Advisory Committee voted 13-2 that the available data on telavancin provided substantial evidence that it was safe and effective for treating nosocomial pneumonia, "when other alternatives are not suitable." Panelists agreed that telavancin should be reserved to treat patients with nosocomial pneumonia caused by methicillin-resistant Staphylococcus aureus (MRSA) not methicillin-sensitive S. aureus (MSSA) or Streptococcus pneumoniae, because there are many other treatments available for those infections.
Most panelists did not support approval of the broader indication requested by the manufacturer: treatment of patients with nosocomial pneumonia, including ventilator-associated pneumonia (VAP), caused by susceptible isolates of the Gram-positive microorganisms, S. aureus (including methicillin-susceptible and methicillin-resistant isolates) or S. pneumoniae. The panel voted 9-6 that the data from the two studies did not provide substantial evidence that the drug was safe and effective for this indication.
Telavancin is a lipoglycopeptide antibacterial administered intravenously, with bactericidal activity that results from inhibition of cell wall synthesis and disruption of bacterial plasma membrane function, according to Theravance, the drug’s manufacturer. It was approved in 2009 for the treatment of adults with complicated skin and skin structure infections caused by susceptible Gram-positive bacteria and is marketed as Vibativ.
The current labeling for telavancin includes warnings and precautions in the prescribing information about the increased risk of nephrotoxicity associated with treatment, based on experience in patients treated for the skin infection indication. A boxed warning cites the potential fetal risks.
Among the other points made by panelists were that labeling should include statements about use in patients with reduced creatinine clearance and renal failure and that the company should aggressively collect postmarketing safety and efficacy data.
The panel considered data that included analyses of clinical cure and 28-day all-cause mortality from two non-inferiority studies. These studies comprised 1,503 patients with nosocomial pneumonia and compared telavancin for 7-21 days (10 mg/kg every 24 hours administered intravenously in patients with normal renal function and mild renal impairment or an adjusted dose for patients with moderate or severe renal insufficiency) with vancomycin (1 g IV every 12 hours).
Theravance first submitted an application for approval of the broad nosocomial pneumonia indication in 2009. That application included results of the two studies, which used clinical cure rate as the primary endpoint. Since then, the FDA has requested more data from the company, including a post hoc analysis of mortality, but subsequently declined to approve telavancin after deciding that the trials did not provide adequate evidence of non-inferiority to vancomycin. In response, the company filed a formal dispute, which was rejected by the FDA, but the agency encouraged the company to resubmit the application and held the panel meeting Nov. 29 to review the data.
Telavancin has been approved in the European Union for the treatment of nosocomial pneumonia caused by MRSA, similar to the indication backed by the FDA panel.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting, although a panelist may occasionally be given a waiver, but not at this meeting.
AT A MEETING OF THE FDA'S ANTI-INFECTIVE DRUGS ADVISORY COMMITTEE
FDA Panel Gives Nod to Drug for Multidrug-Resistant TB
SILVER SPRING, MD. – Bedaquiline, an oral antimycobacterial drug with a novel mechanism of action, received an 18 to 0 vote for approval for the treatment of multidrug-resistant pulmonary tuberculosis by a Food and Drug Administration advisory panel.
At a meeting on Nov. 28, the FDA’s Anti-Infective Drugs Advisory Committee concluded that phase II clinical data indicated the drug increased the time to sputum culture conversion, a surrogate marker for clinical effectiveness, when added to standard background treatment in adults with multidrug-resistant (MDR) TB. Sputum culture conversion was defined as two consecutive negative cultures collected at least 25 days apart that were not followed by a confirmed positive culture.
The panel voted 11 to 7 that the data provided substantial evidence that the drug was safe for this indication. There were 10 deaths among 79 patients on bedaquiline, compared with 2 deaths in 81 patients on placebo. The FDA reviewers and the company could not identify any pattern or cause that could explain the imbalance in the death rate. All but one of the deaths in the bedaquiline-treated patients occurred after treatment with the drug was completed.
The FDA is reviewing the drug as an accelerated approval, because bedaquiline addresses the unmet need for an effective therapy for MDR TB. In such cases, recommendation for approval can be based on surrogate clinical end points, with the requirement that clinical effectiveness be confirmed in a postmarketing study with hard clinical end points before full approval is granted. Panelists recommended that full approval require concrete evidence of increased cure rates with treatment in a confirmatory study. Further, more data on the drug are needed in HIV-positive and black populations, and studies also are needed to address the implications of the drug’s long half-life.
Bedaquiline, a diarylquinolone discovered at Janssen Therapeutics, a division of Janssen Products, inhibits mycobacterial adenosine triphosphate (ATP) synthetase. The drug kills both replicating and nonreplicating TB bacilli, and is active against drug-sensitive and MDR TB, according to the company.
If approved by the FDA, bedaquiline will be the first antituberculosis drug with a novel mechanism of action approved in the United States since rifampin’s approval in 1970, and the first drug approved for TB since rifapentine was approved in 1998.
"The fact that this drug is able to shorten the period the organism is in the sputum has public health implications for [reducing]transmission," said Dr. Peter Katona, of the University of California, Los Angeles.
In 2011, 98 cases of MDR TB were reported in the United States, but the condition is a far greater problem globally, with an estimated incidence in 2011 of 310,000 cases, according to the Centers for Disease Control and Prevention.
The panel reviewed two phase II studies of almost 400 patients with pulmonary MDR TB.
One study enrolled 160 nonwhite patients, most of them men from South Africa with a mean age of 35 years. All were newly diagnosed with pulmonary MDR TB; a small proportion were HIV positive. They were treated with a standard five-drug regimen that included ethionamide, pyrazinamide, ofloxacin, kanamycin, and 400 mg once daily of bedaquiline for 2 weeks, followed by 200 mg three times a week for 22 weeks, or placebo. After 24 weeks, patients continued background treatment for 12-18 months.
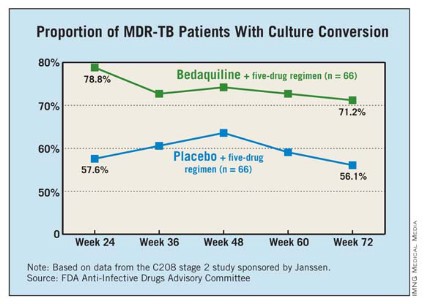
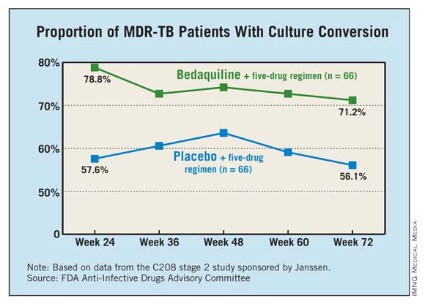
At 24 weeks, 79% of those treated with bedaquiline had a culture conversion, compared with 58% of those on placebo, a significant difference.
The second study was an open-label trial of 233 previously treated patients with sputum smear–positive pulmonary MDR TB. They received the same dosing regimen of bedaquiline combined with an individualized background regimen for MDR TB. At 24 weeks, the culture conversion rate was 80%, and sputum cultures became negative in a mean of 57 days. The faster conversion rate likely reflected the fact that most patients were already on treatment when enrolled in the trial.
The incidence of serious adverse events was higher among those on bedaquiline (6.9% vs. 1.9%). Treatment was associated with elevated transaminases in 4 cases, compared with 0 in placebo-treated patients. There was a modest increase in QT prolongation, although there were no cases of torsades de pointes or evidence that this caused any deaths.
The FDA’s deadline for making a decision on bedaquiline is by the end of December. The agency usually follows the recommendations of its advisory panels, which are not binding. Panelists were cleared of potential conflicts of interest related to the topic of the meeting.
SILVER SPRING, MD. – Bedaquiline, an oral antimycobacterial drug with a novel mechanism of action, received an 18 to 0 vote for approval for the treatment of multidrug-resistant pulmonary tuberculosis by a Food and Drug Administration advisory panel.
At a meeting on Nov. 28, the FDA’s Anti-Infective Drugs Advisory Committee concluded that phase II clinical data indicated the drug increased the time to sputum culture conversion, a surrogate marker for clinical effectiveness, when added to standard background treatment in adults with multidrug-resistant (MDR) TB. Sputum culture conversion was defined as two consecutive negative cultures collected at least 25 days apart that were not followed by a confirmed positive culture.
The panel voted 11 to 7 that the data provided substantial evidence that the drug was safe for this indication. There were 10 deaths among 79 patients on bedaquiline, compared with 2 deaths in 81 patients on placebo. The FDA reviewers and the company could not identify any pattern or cause that could explain the imbalance in the death rate. All but one of the deaths in the bedaquiline-treated patients occurred after treatment with the drug was completed.
The FDA is reviewing the drug as an accelerated approval, because bedaquiline addresses the unmet need for an effective therapy for MDR TB. In such cases, recommendation for approval can be based on surrogate clinical end points, with the requirement that clinical effectiveness be confirmed in a postmarketing study with hard clinical end points before full approval is granted. Panelists recommended that full approval require concrete evidence of increased cure rates with treatment in a confirmatory study. Further, more data on the drug are needed in HIV-positive and black populations, and studies also are needed to address the implications of the drug’s long half-life.
Bedaquiline, a diarylquinolone discovered at Janssen Therapeutics, a division of Janssen Products, inhibits mycobacterial adenosine triphosphate (ATP) synthetase. The drug kills both replicating and nonreplicating TB bacilli, and is active against drug-sensitive and MDR TB, according to the company.
If approved by the FDA, bedaquiline will be the first antituberculosis drug with a novel mechanism of action approved in the United States since rifampin’s approval in 1970, and the first drug approved for TB since rifapentine was approved in 1998.
"The fact that this drug is able to shorten the period the organism is in the sputum has public health implications for [reducing]transmission," said Dr. Peter Katona, of the University of California, Los Angeles.
In 2011, 98 cases of MDR TB were reported in the United States, but the condition is a far greater problem globally, with an estimated incidence in 2011 of 310,000 cases, according to the Centers for Disease Control and Prevention.
The panel reviewed two phase II studies of almost 400 patients with pulmonary MDR TB.
One study enrolled 160 nonwhite patients, most of them men from South Africa with a mean age of 35 years. All were newly diagnosed with pulmonary MDR TB; a small proportion were HIV positive. They were treated with a standard five-drug regimen that included ethionamide, pyrazinamide, ofloxacin, kanamycin, and 400 mg once daily of bedaquiline for 2 weeks, followed by 200 mg three times a week for 22 weeks, or placebo. After 24 weeks, patients continued background treatment for 12-18 months.
At 24 weeks, 79% of those treated with bedaquiline had a culture conversion, compared with 58% of those on placebo, a significant difference.
The second study was an open-label trial of 233 previously treated patients with sputum smear–positive pulmonary MDR TB. They received the same dosing regimen of bedaquiline combined with an individualized background regimen for MDR TB. At 24 weeks, the culture conversion rate was 80%, and sputum cultures became negative in a mean of 57 days. The faster conversion rate likely reflected the fact that most patients were already on treatment when enrolled in the trial.
The incidence of serious adverse events was higher among those on bedaquiline (6.9% vs. 1.9%). Treatment was associated with elevated transaminases in 4 cases, compared with 0 in placebo-treated patients. There was a modest increase in QT prolongation, although there were no cases of torsades de pointes or evidence that this caused any deaths.
The FDA’s deadline for making a decision on bedaquiline is by the end of December. The agency usually follows the recommendations of its advisory panels, which are not binding. Panelists were cleared of potential conflicts of interest related to the topic of the meeting.
SILVER SPRING, MD. – Bedaquiline, an oral antimycobacterial drug with a novel mechanism of action, received an 18 to 0 vote for approval for the treatment of multidrug-resistant pulmonary tuberculosis by a Food and Drug Administration advisory panel.
At a meeting on Nov. 28, the FDA’s Anti-Infective Drugs Advisory Committee concluded that phase II clinical data indicated the drug increased the time to sputum culture conversion, a surrogate marker for clinical effectiveness, when added to standard background treatment in adults with multidrug-resistant (MDR) TB. Sputum culture conversion was defined as two consecutive negative cultures collected at least 25 days apart that were not followed by a confirmed positive culture.
The panel voted 11 to 7 that the data provided substantial evidence that the drug was safe for this indication. There were 10 deaths among 79 patients on bedaquiline, compared with 2 deaths in 81 patients on placebo. The FDA reviewers and the company could not identify any pattern or cause that could explain the imbalance in the death rate. All but one of the deaths in the bedaquiline-treated patients occurred after treatment with the drug was completed.
The FDA is reviewing the drug as an accelerated approval, because bedaquiline addresses the unmet need for an effective therapy for MDR TB. In such cases, recommendation for approval can be based on surrogate clinical end points, with the requirement that clinical effectiveness be confirmed in a postmarketing study with hard clinical end points before full approval is granted. Panelists recommended that full approval require concrete evidence of increased cure rates with treatment in a confirmatory study. Further, more data on the drug are needed in HIV-positive and black populations, and studies also are needed to address the implications of the drug’s long half-life.
Bedaquiline, a diarylquinolone discovered at Janssen Therapeutics, a division of Janssen Products, inhibits mycobacterial adenosine triphosphate (ATP) synthetase. The drug kills both replicating and nonreplicating TB bacilli, and is active against drug-sensitive and MDR TB, according to the company.
If approved by the FDA, bedaquiline will be the first antituberculosis drug with a novel mechanism of action approved in the United States since rifampin’s approval in 1970, and the first drug approved for TB since rifapentine was approved in 1998.
"The fact that this drug is able to shorten the period the organism is in the sputum has public health implications for [reducing]transmission," said Dr. Peter Katona, of the University of California, Los Angeles.
In 2011, 98 cases of MDR TB were reported in the United States, but the condition is a far greater problem globally, with an estimated incidence in 2011 of 310,000 cases, according to the Centers for Disease Control and Prevention.
The panel reviewed two phase II studies of almost 400 patients with pulmonary MDR TB.
One study enrolled 160 nonwhite patients, most of them men from South Africa with a mean age of 35 years. All were newly diagnosed with pulmonary MDR TB; a small proportion were HIV positive. They were treated with a standard five-drug regimen that included ethionamide, pyrazinamide, ofloxacin, kanamycin, and 400 mg once daily of bedaquiline for 2 weeks, followed by 200 mg three times a week for 22 weeks, or placebo. After 24 weeks, patients continued background treatment for 12-18 months.
At 24 weeks, 79% of those treated with bedaquiline had a culture conversion, compared with 58% of those on placebo, a significant difference.
The second study was an open-label trial of 233 previously treated patients with sputum smear–positive pulmonary MDR TB. They received the same dosing regimen of bedaquiline combined with an individualized background regimen for MDR TB. At 24 weeks, the culture conversion rate was 80%, and sputum cultures became negative in a mean of 57 days. The faster conversion rate likely reflected the fact that most patients were already on treatment when enrolled in the trial.
The incidence of serious adverse events was higher among those on bedaquiline (6.9% vs. 1.9%). Treatment was associated with elevated transaminases in 4 cases, compared with 0 in placebo-treated patients. There was a modest increase in QT prolongation, although there were no cases of torsades de pointes or evidence that this caused any deaths.
The FDA’s deadline for making a decision on bedaquiline is by the end of December. The agency usually follows the recommendations of its advisory panels, which are not binding. Panelists were cleared of potential conflicts of interest related to the topic of the meeting.
FROM A MEETING OF THE FOOD AND DRUG ADMINISTRATION’S ANTI-INFECTIVE DRUGS ADVISORY COMMITTEE
Respiratory Symptoms Vary Throughout Menstrual Cycle
Significant variations in respiratory symptoms during different stages of the menstrual cycle were identified in a study of almost 4,000 women in Northern Europe, with patterns that varied by body mass index, asthma, and smoking status.
"The findings suggest substantial hormonal influences in interplay with metabolic factors on airway physiology and on pathophysiological processes in respiratory diseases like asthma," concluded Dr. Ferenc Macsali, of the department of gynecology and obstetrics at Haukeland University Hospital, Bergen, Norway, and his associates. The study was published online in November (Am. J. Respir. Crit. Care Med. 2012 [doi: 10.1164/rccm.201206-1112OC]).
The study obtained information from questionnaires mailed to the Nordic-Baltic population of women, with questions about respiratory symptoms, menstrual symptoms, BMI, and smoking status, from 3,926 women (mean age 39 years), with regular cycles no greater than 28 days, who were not on any hormonal medications. The study is part of Respiratory Health in Northern Europe (RHINE), a population-based multicenter questionnaire study. Almost 29% of the women were regular smokers, almost 8% said they had been diagnosed with asthma, and their mean BMI was 23 kg/m2.
Based on their analysis of the responses, the investigators identified significant variations during the menstrual cycle for each of the three symptoms analyzed – wheezing, shortness of breath, and coughing – including reports of wheezing that were higher during cycle days 10-22, with a "dramatic" drop in the middle of the cycle at about days 14-16, the "putative time of ovulation," in most subgroups. Wheezing was lower before and after menses. The daily incidence of shortness of breath was highest on days 7-21, dropping just before the middle of the cycle "in a number of subgroups." And there were peaks in the incidence of cough before and after midcycle, around the time of "putative" ovulation, and before the onset of menses; the incidence of coughs was lower after menses..
The effects of the menstrual cycle on respiratory symptoms in the general female population have not been well studied, and "our finding that respiratory symptoms vary according to the stage of the menstrual cycle is novel, as is our finding that these patterns vary according to BMI and smoking status," Dr. Macsali said in a statement issued by the American Thoracic Society, which publishes the American Journal of Respiratory and Critical Care Medicine.
"These relationships indicate a link between respiratory symptoms and hormonal changes through the menstrual cycle," he added.
"Our results point to the potential for individualizing therapy for respiratory diseases according to individual symptom patterns," Dr. Macsali said in the statement. As an example, he noted, adjusting asthma medication "according to a woman’s menstrual cycle might improve its efficacy and help reduce disability and the costs of care."
The authors acknowledged that the use of the questionnaire to obtain the data, as well as variations in the length of the menstrual cycle among those who responded, were limitations of the study.
No author disclosures were listed in the study. RHINE receives financial support from organizations that include the Swedish Heart and Lung Foundation, the Norwegian Asthma and Allergy Association, and the Danish Lung Association, according to the RHINE website.
Significant variations in respiratory symptoms during different stages of the menstrual cycle were identified in a study of almost 4,000 women in Northern Europe, with patterns that varied by body mass index, asthma, and smoking status.
"The findings suggest substantial hormonal influences in interplay with metabolic factors on airway physiology and on pathophysiological processes in respiratory diseases like asthma," concluded Dr. Ferenc Macsali, of the department of gynecology and obstetrics at Haukeland University Hospital, Bergen, Norway, and his associates. The study was published online in November (Am. J. Respir. Crit. Care Med. 2012 [doi: 10.1164/rccm.201206-1112OC]).
The study obtained information from questionnaires mailed to the Nordic-Baltic population of women, with questions about respiratory symptoms, menstrual symptoms, BMI, and smoking status, from 3,926 women (mean age 39 years), with regular cycles no greater than 28 days, who were not on any hormonal medications. The study is part of Respiratory Health in Northern Europe (RHINE), a population-based multicenter questionnaire study. Almost 29% of the women were regular smokers, almost 8% said they had been diagnosed with asthma, and their mean BMI was 23 kg/m2.
Based on their analysis of the responses, the investigators identified significant variations during the menstrual cycle for each of the three symptoms analyzed – wheezing, shortness of breath, and coughing – including reports of wheezing that were higher during cycle days 10-22, with a "dramatic" drop in the middle of the cycle at about days 14-16, the "putative time of ovulation," in most subgroups. Wheezing was lower before and after menses. The daily incidence of shortness of breath was highest on days 7-21, dropping just before the middle of the cycle "in a number of subgroups." And there were peaks in the incidence of cough before and after midcycle, around the time of "putative" ovulation, and before the onset of menses; the incidence of coughs was lower after menses..
The effects of the menstrual cycle on respiratory symptoms in the general female population have not been well studied, and "our finding that respiratory symptoms vary according to the stage of the menstrual cycle is novel, as is our finding that these patterns vary according to BMI and smoking status," Dr. Macsali said in a statement issued by the American Thoracic Society, which publishes the American Journal of Respiratory and Critical Care Medicine.
"These relationships indicate a link between respiratory symptoms and hormonal changes through the menstrual cycle," he added.
"Our results point to the potential for individualizing therapy for respiratory diseases according to individual symptom patterns," Dr. Macsali said in the statement. As an example, he noted, adjusting asthma medication "according to a woman’s menstrual cycle might improve its efficacy and help reduce disability and the costs of care."
The authors acknowledged that the use of the questionnaire to obtain the data, as well as variations in the length of the menstrual cycle among those who responded, were limitations of the study.
No author disclosures were listed in the study. RHINE receives financial support from organizations that include the Swedish Heart and Lung Foundation, the Norwegian Asthma and Allergy Association, and the Danish Lung Association, according to the RHINE website.
Significant variations in respiratory symptoms during different stages of the menstrual cycle were identified in a study of almost 4,000 women in Northern Europe, with patterns that varied by body mass index, asthma, and smoking status.
"The findings suggest substantial hormonal influences in interplay with metabolic factors on airway physiology and on pathophysiological processes in respiratory diseases like asthma," concluded Dr. Ferenc Macsali, of the department of gynecology and obstetrics at Haukeland University Hospital, Bergen, Norway, and his associates. The study was published online in November (Am. J. Respir. Crit. Care Med. 2012 [doi: 10.1164/rccm.201206-1112OC]).
The study obtained information from questionnaires mailed to the Nordic-Baltic population of women, with questions about respiratory symptoms, menstrual symptoms, BMI, and smoking status, from 3,926 women (mean age 39 years), with regular cycles no greater than 28 days, who were not on any hormonal medications. The study is part of Respiratory Health in Northern Europe (RHINE), a population-based multicenter questionnaire study. Almost 29% of the women were regular smokers, almost 8% said they had been diagnosed with asthma, and their mean BMI was 23 kg/m2.
Based on their analysis of the responses, the investigators identified significant variations during the menstrual cycle for each of the three symptoms analyzed – wheezing, shortness of breath, and coughing – including reports of wheezing that were higher during cycle days 10-22, with a "dramatic" drop in the middle of the cycle at about days 14-16, the "putative time of ovulation," in most subgroups. Wheezing was lower before and after menses. The daily incidence of shortness of breath was highest on days 7-21, dropping just before the middle of the cycle "in a number of subgroups." And there were peaks in the incidence of cough before and after midcycle, around the time of "putative" ovulation, and before the onset of menses; the incidence of coughs was lower after menses..
The effects of the menstrual cycle on respiratory symptoms in the general female population have not been well studied, and "our finding that respiratory symptoms vary according to the stage of the menstrual cycle is novel, as is our finding that these patterns vary according to BMI and smoking status," Dr. Macsali said in a statement issued by the American Thoracic Society, which publishes the American Journal of Respiratory and Critical Care Medicine.
"These relationships indicate a link between respiratory symptoms and hormonal changes through the menstrual cycle," he added.
"Our results point to the potential for individualizing therapy for respiratory diseases according to individual symptom patterns," Dr. Macsali said in the statement. As an example, he noted, adjusting asthma medication "according to a woman’s menstrual cycle might improve its efficacy and help reduce disability and the costs of care."
The authors acknowledged that the use of the questionnaire to obtain the data, as well as variations in the length of the menstrual cycle among those who responded, were limitations of the study.
No author disclosures were listed in the study. RHINE receives financial support from organizations that include the Swedish Heart and Lung Foundation, the Norwegian Asthma and Allergy Association, and the Danish Lung Association, according to the RHINE website.
FROM THE AMERICAN JOURNAL OF RESPIRATORY AND CRITICAL CARE MEDICINE
Major Finding: Significant variations in respiratory symptoms (wheezing, cough, and shortness of breath) during the menstrual cycle were identified in a general population of women with regular menstrual cycles, which has possible implications for individualizing the treatment of respiratory diseases, according to the authors.
Data Source: The study analyzed responses to a questionnaire about respiratory symptoms and menstrual cycles from 3,926 women with regular menstrual cycles, who were not taking any exogenous hormones. The women were part of a population-based postal questionnaire study (the RHINE study).
Disclosures: No author disclosures were listed in the study. RHINE receives financial support from organizations that include the Swedish Heart and Lung Foundation, the Norwegian Asthma and Allergy Association, and the Danish Lung Association, according to the RHINE website.
Rivaroxaban: Now For DVT and PE
The oral anticoagulant rivaroxaban has been approved for the treatment of deep vein thrombosis or pulmonary embolism and for reducing the recurrence of DVT and PE after initial treatment, the Food and Drug Administration announced Nov. 5.

The expanded approval was based on the results of three studies of almost 9,500 patients with a DVT or PE, which found that treatment with rivaroxaban was as effective as the combination of the low-molecular-weight heparin enoxaparin and a vitamin K antagonist in treating DVT and PE. In one of the studies, continued treatment with rivaroxaban reduced the risk of recurrent DVT and PE, according to an FDA statement.
Rivaroxaban, a factor Xa inhibitor, was initially approved in July 2011 for prophylaxis of DVT or PE in patients undergoing hip replacement or knee-replacement surgery, at a dose of 10 mg orally once a day. Marketed as Xarelto by Janssen Pharmaceuticals, it received in November 2011 a second indication for reducing the risk of stroke in patients with nonvalvular atrial fibrillation.
The FDA is currently reviewing the company’s applications to approve rivaroxaban for reducing the risk of stent thrombosis in patients with acute coronary syndrome and for reducing the risk of secondary cardiovascular events in patients with ACS, according to Janssen.
The oral anticoagulant rivaroxaban has been approved for the treatment of deep vein thrombosis or pulmonary embolism and for reducing the recurrence of DVT and PE after initial treatment, the Food and Drug Administration announced Nov. 5.
The expanded approval was based on the results of three studies of almost 9,500 patients with a DVT or PE, which found that treatment with rivaroxaban was as effective as the combination of the low-molecular-weight heparin enoxaparin and a vitamin K antagonist in treating DVT and PE. In one of the studies, continued treatment with rivaroxaban reduced the risk of recurrent DVT and PE, according to an FDA statement.
Rivaroxaban, a factor Xa inhibitor, was initially approved in July 2011 for prophylaxis of DVT or PE in patients undergoing hip replacement or knee-replacement surgery, at a dose of 10 mg orally once a day. Marketed as Xarelto by Janssen Pharmaceuticals, it received in November 2011 a second indication for reducing the risk of stroke in patients with nonvalvular atrial fibrillation.
The FDA is currently reviewing the company’s applications to approve rivaroxaban for reducing the risk of stent thrombosis in patients with acute coronary syndrome and for reducing the risk of secondary cardiovascular events in patients with ACS, according to Janssen.
The oral anticoagulant rivaroxaban has been approved for the treatment of deep vein thrombosis or pulmonary embolism and for reducing the recurrence of DVT and PE after initial treatment, the Food and Drug Administration announced Nov. 5.
The expanded approval was based on the results of three studies of almost 9,500 patients with a DVT or PE, which found that treatment with rivaroxaban was as effective as the combination of the low-molecular-weight heparin enoxaparin and a vitamin K antagonist in treating DVT and PE. In one of the studies, continued treatment with rivaroxaban reduced the risk of recurrent DVT and PE, according to an FDA statement.
Rivaroxaban, a factor Xa inhibitor, was initially approved in July 2011 for prophylaxis of DVT or PE in patients undergoing hip replacement or knee-replacement surgery, at a dose of 10 mg orally once a day. Marketed as Xarelto by Janssen Pharmaceuticals, it received in November 2011 a second indication for reducing the risk of stroke in patients with nonvalvular atrial fibrillation.
The FDA is currently reviewing the company’s applications to approve rivaroxaban for reducing the risk of stent thrombosis in patients with acute coronary syndrome and for reducing the risk of secondary cardiovascular events in patients with ACS, according to Janssen.
Panel Calls for More Safety Data for Hepatitis B Vaccine
SILVER SPRING, MD. – Despite evidence of effectiveness and enthusiasm about its potential, more safety data in thousands of people are needed before a two-dose hepatitis B vaccine can be approved for use in adults, according to the majority of a Food and Drug Administration advisory panel.
At a meeting on Nov. 15, the FDA’s Vaccines and Related Biological Products Advisory Committee voted 13 to 1 that the immunogenicity data on the Heplisav vaccine were adequate to support its effectiveness in preventing hepatitis B infection in adults aged 18-70 years. The proposed indication for the vaccine, which combines hepatitis B surface antigen (HBsAg) with a novel adjuvant to enhance the immune response, is for the active immunization against all known subtypes of the hepatitis B virus in adults aged 18-70 years.
But the panel voted 8 to 5, with one abstention, that the available data were not adequate to support the safety of the vaccine, citing the need for more data because the adjuvant, a Toll-like receptor 9 agonist, is not included in any available vaccine. Almost 4,000 people received the vaccine in two phase III studies. The manufacturer, Dynavax Technologies, also has proposed a postmarketing safety study that will enroll up to 30,000 recipients of the vaccine in a managed care organization. Panelists voting no on the safety question said that more data from a more ethnically diverse population than those enrolled in the studies would be needed in as many as 10,000 patients before approval.
While a hepatitis B vaccine that is more immunogenic in populations that do not respond as well to the hepatitis B vaccines would be beneficial, "I don’t think the safety data is sufficiently large to support a recommendation for use in the general adult population given that this vaccine contains a new adjuvant," said one of the panelists, Dr. Melinda Wharton, deputy director of the National Center for Immunization and Respiratory Diseases at the Centers for Disease Control and Prevention.
The two Heplisav doses are administered intramuscularly 1 month apart, compared with the 0, 1, and 6 month schedule for the two currently approved hepatitis B vaccines, Engerix-B and Recombivax HB.
The two phase III noninferiority studies of healthy adults aged 18-70 years compared immune response to vaccination with Heplisav (administered at 0 and 1 months, with a saline placebo administered at 6 months) in 3,778 adults, and with Engerix-B (administered at 0, 1, and 6 months) in 1,089 people. The primary immunogenicity end point was the seroprotection rate (SPR) – an anti-HBsAg level of 10 mIU/mL or greater, recognized as conferring protection against hepatitis B virus infection. In both studies, the SPR results for Heplisav met the noninferiority criteria for the studies.
In the two studies, the SPRs were higher among those who received Heplisav: 95% and 90% at 3 months (8 weeks after the last active dose), compared with 81.1% (4 weeks after the last dose) and 70.5% (8 weeks after the last dose), respectively, of those who received Engerix-B.
The most common adverse event associated with the vaccine was injection-site reaction in both groups. Rates of severe adverse events were lower among those who received Heplisav, and rates of autoimmune events and autoantibody conversions were similar in the two groups, according to Dynavax.
However, thyroid-related adverse events, which could be representative of autoimmune events, were reported in a higher proportion of people who received Heplisav. Cases of serious events, although rare, included one of Wegener’s granulomatosis and one of Guillain-Barré syndrome. Autoimmune diseases are relatively rare in the general population, and a large sample size of patients was necessary to accurately evaluate the associated risk, according to the FDA reviewer.
An increased risk of autoimmune reactions is a theoretical risk with adjuvants.
The FDA’s deadline for making a decision on the approval is Feb. 24, 2013, according to Dynavax. If approved, the company plans to market the vaccine as Heplisav. The vaccine also is under review in Europe.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting, although a panelist may be given a waiver for conflict of interest, but none were granted at this meeting.
SILVER SPRING, MD. – Despite evidence of effectiveness and enthusiasm about its potential, more safety data in thousands of people are needed before a two-dose hepatitis B vaccine can be approved for use in adults, according to the majority of a Food and Drug Administration advisory panel.
At a meeting on Nov. 15, the FDA’s Vaccines and Related Biological Products Advisory Committee voted 13 to 1 that the immunogenicity data on the Heplisav vaccine were adequate to support its effectiveness in preventing hepatitis B infection in adults aged 18-70 years. The proposed indication for the vaccine, which combines hepatitis B surface antigen (HBsAg) with a novel adjuvant to enhance the immune response, is for the active immunization against all known subtypes of the hepatitis B virus in adults aged 18-70 years.
But the panel voted 8 to 5, with one abstention, that the available data were not adequate to support the safety of the vaccine, citing the need for more data because the adjuvant, a Toll-like receptor 9 agonist, is not included in any available vaccine. Almost 4,000 people received the vaccine in two phase III studies. The manufacturer, Dynavax Technologies, also has proposed a postmarketing safety study that will enroll up to 30,000 recipients of the vaccine in a managed care organization. Panelists voting no on the safety question said that more data from a more ethnically diverse population than those enrolled in the studies would be needed in as many as 10,000 patients before approval.
While a hepatitis B vaccine that is more immunogenic in populations that do not respond as well to the hepatitis B vaccines would be beneficial, "I don’t think the safety data is sufficiently large to support a recommendation for use in the general adult population given that this vaccine contains a new adjuvant," said one of the panelists, Dr. Melinda Wharton, deputy director of the National Center for Immunization and Respiratory Diseases at the Centers for Disease Control and Prevention.
The two Heplisav doses are administered intramuscularly 1 month apart, compared with the 0, 1, and 6 month schedule for the two currently approved hepatitis B vaccines, Engerix-B and Recombivax HB.
The two phase III noninferiority studies of healthy adults aged 18-70 years compared immune response to vaccination with Heplisav (administered at 0 and 1 months, with a saline placebo administered at 6 months) in 3,778 adults, and with Engerix-B (administered at 0, 1, and 6 months) in 1,089 people. The primary immunogenicity end point was the seroprotection rate (SPR) – an anti-HBsAg level of 10 mIU/mL or greater, recognized as conferring protection against hepatitis B virus infection. In both studies, the SPR results for Heplisav met the noninferiority criteria for the studies.
In the two studies, the SPRs were higher among those who received Heplisav: 95% and 90% at 3 months (8 weeks after the last active dose), compared with 81.1% (4 weeks after the last dose) and 70.5% (8 weeks after the last dose), respectively, of those who received Engerix-B.
The most common adverse event associated with the vaccine was injection-site reaction in both groups. Rates of severe adverse events were lower among those who received Heplisav, and rates of autoimmune events and autoantibody conversions were similar in the two groups, according to Dynavax.
However, thyroid-related adverse events, which could be representative of autoimmune events, were reported in a higher proportion of people who received Heplisav. Cases of serious events, although rare, included one of Wegener’s granulomatosis and one of Guillain-Barré syndrome. Autoimmune diseases are relatively rare in the general population, and a large sample size of patients was necessary to accurately evaluate the associated risk, according to the FDA reviewer.
An increased risk of autoimmune reactions is a theoretical risk with adjuvants.
The FDA’s deadline for making a decision on the approval is Feb. 24, 2013, according to Dynavax. If approved, the company plans to market the vaccine as Heplisav. The vaccine also is under review in Europe.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting, although a panelist may be given a waiver for conflict of interest, but none were granted at this meeting.
SILVER SPRING, MD. – Despite evidence of effectiveness and enthusiasm about its potential, more safety data in thousands of people are needed before a two-dose hepatitis B vaccine can be approved for use in adults, according to the majority of a Food and Drug Administration advisory panel.
At a meeting on Nov. 15, the FDA’s Vaccines and Related Biological Products Advisory Committee voted 13 to 1 that the immunogenicity data on the Heplisav vaccine were adequate to support its effectiveness in preventing hepatitis B infection in adults aged 18-70 years. The proposed indication for the vaccine, which combines hepatitis B surface antigen (HBsAg) with a novel adjuvant to enhance the immune response, is for the active immunization against all known subtypes of the hepatitis B virus in adults aged 18-70 years.
But the panel voted 8 to 5, with one abstention, that the available data were not adequate to support the safety of the vaccine, citing the need for more data because the adjuvant, a Toll-like receptor 9 agonist, is not included in any available vaccine. Almost 4,000 people received the vaccine in two phase III studies. The manufacturer, Dynavax Technologies, also has proposed a postmarketing safety study that will enroll up to 30,000 recipients of the vaccine in a managed care organization. Panelists voting no on the safety question said that more data from a more ethnically diverse population than those enrolled in the studies would be needed in as many as 10,000 patients before approval.
While a hepatitis B vaccine that is more immunogenic in populations that do not respond as well to the hepatitis B vaccines would be beneficial, "I don’t think the safety data is sufficiently large to support a recommendation for use in the general adult population given that this vaccine contains a new adjuvant," said one of the panelists, Dr. Melinda Wharton, deputy director of the National Center for Immunization and Respiratory Diseases at the Centers for Disease Control and Prevention.
The two Heplisav doses are administered intramuscularly 1 month apart, compared with the 0, 1, and 6 month schedule for the two currently approved hepatitis B vaccines, Engerix-B and Recombivax HB.
The two phase III noninferiority studies of healthy adults aged 18-70 years compared immune response to vaccination with Heplisav (administered at 0 and 1 months, with a saline placebo administered at 6 months) in 3,778 adults, and with Engerix-B (administered at 0, 1, and 6 months) in 1,089 people. The primary immunogenicity end point was the seroprotection rate (SPR) – an anti-HBsAg level of 10 mIU/mL or greater, recognized as conferring protection against hepatitis B virus infection. In both studies, the SPR results for Heplisav met the noninferiority criteria for the studies.
In the two studies, the SPRs were higher among those who received Heplisav: 95% and 90% at 3 months (8 weeks after the last active dose), compared with 81.1% (4 weeks after the last dose) and 70.5% (8 weeks after the last dose), respectively, of those who received Engerix-B.
The most common adverse event associated with the vaccine was injection-site reaction in both groups. Rates of severe adverse events were lower among those who received Heplisav, and rates of autoimmune events and autoantibody conversions were similar in the two groups, according to Dynavax.
However, thyroid-related adverse events, which could be representative of autoimmune events, were reported in a higher proportion of people who received Heplisav. Cases of serious events, although rare, included one of Wegener’s granulomatosis and one of Guillain-Barré syndrome. Autoimmune diseases are relatively rare in the general population, and a large sample size of patients was necessary to accurately evaluate the associated risk, according to the FDA reviewer.
An increased risk of autoimmune reactions is a theoretical risk with adjuvants.
The FDA’s deadline for making a decision on the approval is Feb. 24, 2013, according to Dynavax. If approved, the company plans to market the vaccine as Heplisav. The vaccine also is under review in Europe.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting, although a panelist may be given a waiver for conflict of interest, but none were granted at this meeting.
AT A MEETING OF THE FDA'S VACCINES AND RELATED BIOLOGICAL PRODUCTS ADVISORY COMMITTEE
FDA Panel Backs Avian Flu Vaccine
SILVER SPRING, MD. – A Food and Drug Administration advisory panel gave its unanimous support Nov. 14 to an H5N1 influenza vaccine designated for a national stockpile, where it would be reserved for use during an avian influenza pandemic or outbreak.
The FDA’s Vaccines and Related Biological Products Advisory Committee voted 14-0 that the influenza A (H5N1) Virus Monovalent Vaccine should be approved based on the safety and immune responses to the vaccine in clinical studies,
GlaxoSmithKline contracted with the U.S. government to develop the vaccine, which contains an antigen-sparing adjuvant that boosts the immune response. If licensed, it will be deposited in the U.S. Strategic National Stockpile and owned by the U.S. government, which would control the distribution and use of the vaccine in the case of a pandemic. GSK has no plans to market the vaccine,
The advisory committee agreed that immunogenicity and safety data on the "Q-Pan H5N1" vaccine support licensure for use in adults at increased risk of exposure or during a pandemic. The proposed indication is for the "active immunization for the prevention of disease in persons 18 years of age and older at increased risk of exposure to the influenza A virus H5N1 subtype contained in the vaccine." The vaccine is administered in two doses about 21 days apart.
Mortality from the infection is highest among children and young adults. GSK is conducting studies in children aged 17 months and older, with plans to expand the approval.
The influenza A (H5N1) virus is highly pathogenic, contagious, and deadly among birds, particular domestic poultry, but it is relatively rare in humans. However, there are sporadic outbreaks in humans; since November 2003, there have been 608 confirmed cases in 15 countries – mostly in Asia – with a high (59%) mortality rate, according to the Centers for Disease Control and Prevention.
The vaccine was studied in two pivotal studies of 5,241 patients, including 3,574 who received the Q-Pan H5N1. In a phase III study comparing the vaccine with a saline placebo, seroconversion rates 42 days after the second dose were 90% among those aged 18-64 years and 74% of those over age 64 years. This exceeded FDA criteria for immunogenicity for a vaccine. Injection site reactions were the most common adverse reactions.
The vaccine is being considered for an accelerated approval, with the immune responses to the vaccine being considered a surrogate for clinical effectiveness. Moreover, the vaccine is manufactured using the same process as GSK’s seasonal influenza vaccine, FluLaval; full approval is dependent on postapproval studies confirming clinical benefit.
The Q-Pan H5N1 vaccine has been licensed in 30 countries, including in Europe and Australia, and is under review in Canada.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting, although a panelist may be given a waiver. No waivers for conflict of interest were granted at this meeting.
SILVER SPRING, MD. – A Food and Drug Administration advisory panel gave its unanimous support Nov. 14 to an H5N1 influenza vaccine designated for a national stockpile, where it would be reserved for use during an avian influenza pandemic or outbreak.
The FDA’s Vaccines and Related Biological Products Advisory Committee voted 14-0 that the influenza A (H5N1) Virus Monovalent Vaccine should be approved based on the safety and immune responses to the vaccine in clinical studies,
GlaxoSmithKline contracted with the U.S. government to develop the vaccine, which contains an antigen-sparing adjuvant that boosts the immune response. If licensed, it will be deposited in the U.S. Strategic National Stockpile and owned by the U.S. government, which would control the distribution and use of the vaccine in the case of a pandemic. GSK has no plans to market the vaccine,
The advisory committee agreed that immunogenicity and safety data on the "Q-Pan H5N1" vaccine support licensure for use in adults at increased risk of exposure or during a pandemic. The proposed indication is for the "active immunization for the prevention of disease in persons 18 years of age and older at increased risk of exposure to the influenza A virus H5N1 subtype contained in the vaccine." The vaccine is administered in two doses about 21 days apart.
Mortality from the infection is highest among children and young adults. GSK is conducting studies in children aged 17 months and older, with plans to expand the approval.
The influenza A (H5N1) virus is highly pathogenic, contagious, and deadly among birds, particular domestic poultry, but it is relatively rare in humans. However, there are sporadic outbreaks in humans; since November 2003, there have been 608 confirmed cases in 15 countries – mostly in Asia – with a high (59%) mortality rate, according to the Centers for Disease Control and Prevention.
The vaccine was studied in two pivotal studies of 5,241 patients, including 3,574 who received the Q-Pan H5N1. In a phase III study comparing the vaccine with a saline placebo, seroconversion rates 42 days after the second dose were 90% among those aged 18-64 years and 74% of those over age 64 years. This exceeded FDA criteria for immunogenicity for a vaccine. Injection site reactions were the most common adverse reactions.
The vaccine is being considered for an accelerated approval, with the immune responses to the vaccine being considered a surrogate for clinical effectiveness. Moreover, the vaccine is manufactured using the same process as GSK’s seasonal influenza vaccine, FluLaval; full approval is dependent on postapproval studies confirming clinical benefit.
The Q-Pan H5N1 vaccine has been licensed in 30 countries, including in Europe and Australia, and is under review in Canada.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting, although a panelist may be given a waiver. No waivers for conflict of interest were granted at this meeting.
SILVER SPRING, MD. – A Food and Drug Administration advisory panel gave its unanimous support Nov. 14 to an H5N1 influenza vaccine designated for a national stockpile, where it would be reserved for use during an avian influenza pandemic or outbreak.
The FDA’s Vaccines and Related Biological Products Advisory Committee voted 14-0 that the influenza A (H5N1) Virus Monovalent Vaccine should be approved based on the safety and immune responses to the vaccine in clinical studies,
GlaxoSmithKline contracted with the U.S. government to develop the vaccine, which contains an antigen-sparing adjuvant that boosts the immune response. If licensed, it will be deposited in the U.S. Strategic National Stockpile and owned by the U.S. government, which would control the distribution and use of the vaccine in the case of a pandemic. GSK has no plans to market the vaccine,
The advisory committee agreed that immunogenicity and safety data on the "Q-Pan H5N1" vaccine support licensure for use in adults at increased risk of exposure or during a pandemic. The proposed indication is for the "active immunization for the prevention of disease in persons 18 years of age and older at increased risk of exposure to the influenza A virus H5N1 subtype contained in the vaccine." The vaccine is administered in two doses about 21 days apart.
Mortality from the infection is highest among children and young adults. GSK is conducting studies in children aged 17 months and older, with plans to expand the approval.
The influenza A (H5N1) virus is highly pathogenic, contagious, and deadly among birds, particular domestic poultry, but it is relatively rare in humans. However, there are sporadic outbreaks in humans; since November 2003, there have been 608 confirmed cases in 15 countries – mostly in Asia – with a high (59%) mortality rate, according to the Centers for Disease Control and Prevention.
The vaccine was studied in two pivotal studies of 5,241 patients, including 3,574 who received the Q-Pan H5N1. In a phase III study comparing the vaccine with a saline placebo, seroconversion rates 42 days after the second dose were 90% among those aged 18-64 years and 74% of those over age 64 years. This exceeded FDA criteria for immunogenicity for a vaccine. Injection site reactions were the most common adverse reactions.
The vaccine is being considered for an accelerated approval, with the immune responses to the vaccine being considered a surrogate for clinical effectiveness. Moreover, the vaccine is manufactured using the same process as GSK’s seasonal influenza vaccine, FluLaval; full approval is dependent on postapproval studies confirming clinical benefit.
The Q-Pan H5N1 vaccine has been licensed in 30 countries, including in Europe and Australia, and is under review in Canada.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting, although a panelist may be given a waiver. No waivers for conflict of interest were granted at this meeting.
AT A MEETING OF THE FDA'S VACCINES AND RELATED BIOLOGICAL PRODUCTS ADVISORY COMMITTEE
Remediation, Attrition Rates High in Surgery Residents
Almost one-third of general surgery residents required remediation over an 11-year period, most often because of a deficiency in medical knowledge, judging from findings in a retrospective study of remediation and attrition rates among general surgery residents at six academic surgical residency programs in California.
The high remediation rate identified in this study "begs the question of whether we are falling short in the education of surgical residents," said Dr. Arezou Yaghoubian of the department of surgery, Harbor-UCLA Medical Center, Los Angeles, and associates (Arch. Surg. 2012;147:829-33).
They conducted the study to determine which of the six Accreditation Council for Graduate Medical Education (ACGME) competencies (patient care, medical knowledge, practice-based learning, interpersonal and communication skills, professionalism, and system-based practice), most often require remediation and to identify predictors of remediation. There is a scarcity of data on how well surgical residency programs have been achieving these competencies, and this information may provide insight into how to modify the surgical curriculum more effectively in this new era of limited hours, they said.
In the study of 348 general surgery residents at the six training programs between 1999 and 2010, the most common reason for remediation was medical knowledge in 74%, followed by interpersonal and communication skills in 24%, patient care in 22%, professionalism in 18%, system-based practice in 14%, and practice-based learning in 8%.
Of the 107 residents who required remediation, 27 required remediation more than once. Almost 16% of the residents left their programs, but most (53 of 55 residents) left voluntarily. The other two failed remediation and had to leave the program.
Monthly meetings with faculty was the most common form of remediation, in 79%, followed by specific reading assignments (72%), required attendance at review courses and/or conferences (27%), evaluation by a therapist, psychologist, or psychiatrist (12%), and having to repeat a clinical year (6.5%).
More than half of the remediations were initiated during the first 2 postgraduate years (25% in the first and 35% in the second year), followed by 21% during the third year, 16% during the fourth year, and 4% during the fifth year.
A predictor of remediation was having received honors during the third-year surgery clerkship (58% of those who were subject to remediation vs. 45% of those who were not remediated, a statistically significant difference), which, the authors noted, was counterintuitive.
United States Medical Licensing Examination (USMLE) step 1 and/or step 2 scores and American Board of Surgery In-Training Examination (ABSITE) scores at postgraduate years 1 through 4 were also predictive of remediation. The median USMLE step 1 and step 2 scores were 225 and 223 among the residents subject to remediation, vs. 232 for step 1 and step 2 scores among those who were not remediated, statistically significant differences.
The ABSITE scores during postgraduate years 1 through 4 were significantly lower among those who were subject to remediation, but the differences in median scores in years 5 through 7 were not significantly different.
But remediation was not a predictor of attrition. The only predictor of attrition was the ABSITE score at the third postgraduate year, which was a median of 34 among those who left the program and 62 among those who stayed.
Possible explanations for the high remediation rate is that residents are not well prepared for the demands of a surgical residency, they need to be more efficient with their time because of the 80-hour work week, and they may not necessarily be spending their increased time outside of the hospital studying at home, the authors said. Possible reasons for the attrition rate among the residents, an "ongoing concern" in general surgery, may be marital, family, and personal issues and a need for a less stressful environment, they added.
Acknowledging the study's limitations, including the retrospective design and lack of information on how many residents passed the American Board of Surgery boards, the authors concluded that the high remediation rate "should give surgical educators pause as we should closely examine the potential sources of these deficiencies."
They called on surgical societies to "take the initiative to encourage the restructuring of medical school education, such that future surgeons are better prepared to enter surgical residencies," and for residency programs to "determine whether current educational methods are adequate to prepare future surgeons."
The authors reported they had no financial disclosures.
The surgical residents in this study were intelligent high achievers and the mean USMLE step 1 score of the residents who were remediated was a "quite respectable 225," Dr. Karen Deveney said in an editorial. "A substantial portion of our very bright residents who have a history of great success in everything they do may have difficulty keeping up with the fast pace and high workload demands," while some may opt for a less stressful career path, and others may need more help from faculty to meet expectations and will "persevere," she said.
| Dr. Deveney |
"It is incumbent on those of us in more senior positions to create educational systems that eliminate nonessential tasks so that residents can devote more attention during the compressed work hours to learning what they need to become competent surgeons," she wrote. "Only then can we have a better chance of training and retaining the best and the brightest" (Arch. Surg. 2012;147; 833).
Dr. Karen Deveney is professor of surgery and vice chair of education and program director, department of surgery, Oregon Health and Science University, Portland. She had no financial disclosures to report.
The surgical residents in this study were intelligent high achievers and the mean USMLE step 1 score of the residents who were remediated was a "quite respectable 225," Dr. Karen Deveney said in an editorial. "A substantial portion of our very bright residents who have a history of great success in everything they do may have difficulty keeping up with the fast pace and high workload demands," while some may opt for a less stressful career path, and others may need more help from faculty to meet expectations and will "persevere," she said.
| Dr. Deveney |
"It is incumbent on those of us in more senior positions to create educational systems that eliminate nonessential tasks so that residents can devote more attention during the compressed work hours to learning what they need to become competent surgeons," she wrote. "Only then can we have a better chance of training and retaining the best and the brightest" (Arch. Surg. 2012;147; 833).
Dr. Karen Deveney is professor of surgery and vice chair of education and program director, department of surgery, Oregon Health and Science University, Portland. She had no financial disclosures to report.
The surgical residents in this study were intelligent high achievers and the mean USMLE step 1 score of the residents who were remediated was a "quite respectable 225," Dr. Karen Deveney said in an editorial. "A substantial portion of our very bright residents who have a history of great success in everything they do may have difficulty keeping up with the fast pace and high workload demands," while some may opt for a less stressful career path, and others may need more help from faculty to meet expectations and will "persevere," she said.
| Dr. Deveney |
"It is incumbent on those of us in more senior positions to create educational systems that eliminate nonessential tasks so that residents can devote more attention during the compressed work hours to learning what they need to become competent surgeons," she wrote. "Only then can we have a better chance of training and retaining the best and the brightest" (Arch. Surg. 2012;147; 833).
Dr. Karen Deveney is professor of surgery and vice chair of education and program director, department of surgery, Oregon Health and Science University, Portland. She had no financial disclosures to report.
Almost one-third of general surgery residents required remediation over an 11-year period, most often because of a deficiency in medical knowledge, judging from findings in a retrospective study of remediation and attrition rates among general surgery residents at six academic surgical residency programs in California.
The high remediation rate identified in this study "begs the question of whether we are falling short in the education of surgical residents," said Dr. Arezou Yaghoubian of the department of surgery, Harbor-UCLA Medical Center, Los Angeles, and associates (Arch. Surg. 2012;147:829-33).
They conducted the study to determine which of the six Accreditation Council for Graduate Medical Education (ACGME) competencies (patient care, medical knowledge, practice-based learning, interpersonal and communication skills, professionalism, and system-based practice), most often require remediation and to identify predictors of remediation. There is a scarcity of data on how well surgical residency programs have been achieving these competencies, and this information may provide insight into how to modify the surgical curriculum more effectively in this new era of limited hours, they said.
In the study of 348 general surgery residents at the six training programs between 1999 and 2010, the most common reason for remediation was medical knowledge in 74%, followed by interpersonal and communication skills in 24%, patient care in 22%, professionalism in 18%, system-based practice in 14%, and practice-based learning in 8%.
Of the 107 residents who required remediation, 27 required remediation more than once. Almost 16% of the residents left their programs, but most (53 of 55 residents) left voluntarily. The other two failed remediation and had to leave the program.
Monthly meetings with faculty was the most common form of remediation, in 79%, followed by specific reading assignments (72%), required attendance at review courses and/or conferences (27%), evaluation by a therapist, psychologist, or psychiatrist (12%), and having to repeat a clinical year (6.5%).
More than half of the remediations were initiated during the first 2 postgraduate years (25% in the first and 35% in the second year), followed by 21% during the third year, 16% during the fourth year, and 4% during the fifth year.
A predictor of remediation was having received honors during the third-year surgery clerkship (58% of those who were subject to remediation vs. 45% of those who were not remediated, a statistically significant difference), which, the authors noted, was counterintuitive.
United States Medical Licensing Examination (USMLE) step 1 and/or step 2 scores and American Board of Surgery In-Training Examination (ABSITE) scores at postgraduate years 1 through 4 were also predictive of remediation. The median USMLE step 1 and step 2 scores were 225 and 223 among the residents subject to remediation, vs. 232 for step 1 and step 2 scores among those who were not remediated, statistically significant differences.
The ABSITE scores during postgraduate years 1 through 4 were significantly lower among those who were subject to remediation, but the differences in median scores in years 5 through 7 were not significantly different.
But remediation was not a predictor of attrition. The only predictor of attrition was the ABSITE score at the third postgraduate year, which was a median of 34 among those who left the program and 62 among those who stayed.
Possible explanations for the high remediation rate is that residents are not well prepared for the demands of a surgical residency, they need to be more efficient with their time because of the 80-hour work week, and they may not necessarily be spending their increased time outside of the hospital studying at home, the authors said. Possible reasons for the attrition rate among the residents, an "ongoing concern" in general surgery, may be marital, family, and personal issues and a need for a less stressful environment, they added.
Acknowledging the study's limitations, including the retrospective design and lack of information on how many residents passed the American Board of Surgery boards, the authors concluded that the high remediation rate "should give surgical educators pause as we should closely examine the potential sources of these deficiencies."
They called on surgical societies to "take the initiative to encourage the restructuring of medical school education, such that future surgeons are better prepared to enter surgical residencies," and for residency programs to "determine whether current educational methods are adequate to prepare future surgeons."
The authors reported they had no financial disclosures.
Almost one-third of general surgery residents required remediation over an 11-year period, most often because of a deficiency in medical knowledge, judging from findings in a retrospective study of remediation and attrition rates among general surgery residents at six academic surgical residency programs in California.
The high remediation rate identified in this study "begs the question of whether we are falling short in the education of surgical residents," said Dr. Arezou Yaghoubian of the department of surgery, Harbor-UCLA Medical Center, Los Angeles, and associates (Arch. Surg. 2012;147:829-33).
They conducted the study to determine which of the six Accreditation Council for Graduate Medical Education (ACGME) competencies (patient care, medical knowledge, practice-based learning, interpersonal and communication skills, professionalism, and system-based practice), most often require remediation and to identify predictors of remediation. There is a scarcity of data on how well surgical residency programs have been achieving these competencies, and this information may provide insight into how to modify the surgical curriculum more effectively in this new era of limited hours, they said.
In the study of 348 general surgery residents at the six training programs between 1999 and 2010, the most common reason for remediation was medical knowledge in 74%, followed by interpersonal and communication skills in 24%, patient care in 22%, professionalism in 18%, system-based practice in 14%, and practice-based learning in 8%.
Of the 107 residents who required remediation, 27 required remediation more than once. Almost 16% of the residents left their programs, but most (53 of 55 residents) left voluntarily. The other two failed remediation and had to leave the program.
Monthly meetings with faculty was the most common form of remediation, in 79%, followed by specific reading assignments (72%), required attendance at review courses and/or conferences (27%), evaluation by a therapist, psychologist, or psychiatrist (12%), and having to repeat a clinical year (6.5%).
More than half of the remediations were initiated during the first 2 postgraduate years (25% in the first and 35% in the second year), followed by 21% during the third year, 16% during the fourth year, and 4% during the fifth year.
A predictor of remediation was having received honors during the third-year surgery clerkship (58% of those who were subject to remediation vs. 45% of those who were not remediated, a statistically significant difference), which, the authors noted, was counterintuitive.
United States Medical Licensing Examination (USMLE) step 1 and/or step 2 scores and American Board of Surgery In-Training Examination (ABSITE) scores at postgraduate years 1 through 4 were also predictive of remediation. The median USMLE step 1 and step 2 scores were 225 and 223 among the residents subject to remediation, vs. 232 for step 1 and step 2 scores among those who were not remediated, statistically significant differences.
The ABSITE scores during postgraduate years 1 through 4 were significantly lower among those who were subject to remediation, but the differences in median scores in years 5 through 7 were not significantly different.
But remediation was not a predictor of attrition. The only predictor of attrition was the ABSITE score at the third postgraduate year, which was a median of 34 among those who left the program and 62 among those who stayed.
Possible explanations for the high remediation rate is that residents are not well prepared for the demands of a surgical residency, they need to be more efficient with their time because of the 80-hour work week, and they may not necessarily be spending their increased time outside of the hospital studying at home, the authors said. Possible reasons for the attrition rate among the residents, an "ongoing concern" in general surgery, may be marital, family, and personal issues and a need for a less stressful environment, they added.
Acknowledging the study's limitations, including the retrospective design and lack of information on how many residents passed the American Board of Surgery boards, the authors concluded that the high remediation rate "should give surgical educators pause as we should closely examine the potential sources of these deficiencies."
They called on surgical societies to "take the initiative to encourage the restructuring of medical school education, such that future surgeons are better prepared to enter surgical residencies," and for residency programs to "determine whether current educational methods are adequate to prepare future surgeons."
The authors reported they had no financial disclosures.
Major Finding: Remediation was required for 31% of the general surgery residents in the study, most often initiated because of a deficiency in medical knowledge (74%). All but 2 of the 55 residents who left the program left voluntarily, not because of failed remediation.
Data Source: A retrospective study of 348 general surgery residents at six academic surgical training programs in California between 1999 and 2010, which evaluated the rates and predictors of remediation and attrition.
Disclosures: The authors of the study had no disclosures.

Insulin Degludec's Long Duration Sways FDA Panel
SILVER SPRING, MD. – A long duration of action and dosing flexibility were among the attributes of insulin degludec that a Food and Drug Administration advisory panel unanimously agreed would make it a useful addition to currently available diabetes treatments. But concerns over a cardiovascular safety signal resulted in a less than unanimous vote to recommend approval.
The FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted 8-4 on Nov. 8 to support approval of insulin degludec (IDeg), a long-acting basal insulin analogue, used alone, and in a combined formulation with the fast-acting insulin analogue aspart (IDegAsp) for the treatment of type 1 and type 2 diabetes, based on clinical trial data from 17 studies.
Panel members voting either way said it was a difficult decision because of a cardiovascular safety signal detected in a meta-analysis of the clinical trials, the major issue discussed at the meeting.
The panel unanimously recommended that Novo Nordisk, the manufacturer of IDeg and IDegAsp, conduct a cardiovascular outcomes trial to further investigate the safety signal, which was an increase in a composite major cardiovascular event (MACE) end point of unstable angina pectoris, cardiovascular death, nonfatal myocardial infarction, and nonfatal stroke. Although panelists agreed that the signal could be caused by chance, they also said that it should be investigated further.
In approving a drug, the FDA has the authority to require that a company conduct a postmarketing safety trial. Novo Nordisk has proposed a postapproval study that would compare cardiovascular outcomes in 7,500 patients with type 2 diabetes, treated with IDeg or another insulin plus standard of care, with up to 5 years’ follow-up. Designed for slower absorption, IDeg forms multihexamers after injection, resulting in a soluble depot from which there is a slow, continuous, and extended release of insulin degludec, with a flat stable profile, according to the company, which markets insulin aspart as Novolog.
In phase-III 26- and 52-week studies of about 6,500 adult patients with type 1 or type 2 diabetes, treatment with IDeg or IDegAsp was noninferior to insulin comparators in reducing hemoglobin A1c from baseline and resulted in lower fasting blood glucose levels, with a reduced risk of hypoglycemia, particularly nocturnal hypoglycemia, according to Novo Nordisk. The company has highlighted the reduced rate of nocturnal hypoglycemia, but FDA reviewers said they were not convinced that it was clinically relevant.
Panelists agreed that IDeg and IDegAsp had features that would be useful in clinical practice, including the combination with a short-acting insulin, which would reduce the number of injections; evidence that it lasts for about 24 hours; and some flexibility if a patient misses a dose – a common problem in clinical practice, several of the endocrinologists on the panel pointed out.
The company’s proposed label recommends that IDeg be taken at about the same time every day, but if a dose is missed the patient can take a catch-up dose at any time as long as 8 hours elapse between doses.
Voting in favor of approval, Dr. David Cooke of the division of pediatric endocrinology at Johns Hopkins in Baltimore, said that, although it was a difficult call, “the pharmacokinetics of this insulin are an advance for our treatment of both type 1 and type 2 diabetes and would provide an additional option that could improve our treatment of these patients.”
Like other panelists, he said that currently available basal insulins do not last for 24 hours and that a basal insulin that provides coverage over 24 hours would be an advantage. The question of whether it increases cardiovascular risk is “far from proven,” but clinicians need to be aware of these issues, he added.
Another endocrinologist on the panel, Dr. Robert Smith, professor of medicine at Brown University in Providence, R.I., agreed that IDeg addressed the need for a long-acting insulin. If the data on efficacy and lower rate of hypoglycemia hold up in clinical practice, he said, “it is likely to be useful and broadly used as a form of insulin in diabetes care.”
Dr. Smith voted against approval, however, noting that there are alternatives and that the benefit of IDeg was “not of such magnitude that it merits taking on significant level of risk.” Despite the uncertainty of the cardiovascular risk signal, a study addressing this issue further should be done before approval, “given the importance of cardiovascular disease in patients with type 1 and type 2 diabetes,” he added.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting, although one panelist at this meeting was given a waiver. If approved, the company plans to make both IDeg and the combination product available in prefilled insulin pens, which they will market as Tresiba and Ryzodeg.
The company also has submitted an application for approval of IDeg and IDegAsp in Europe, in Canada, and other countries. They were approved in Japan in late September.
UPDATED 11/12/12
SILVER SPRING, MD. – A long duration of action and dosing flexibility were among the attributes of insulin degludec that a Food and Drug Administration advisory panel unanimously agreed would make it a useful addition to currently available diabetes treatments. But concerns over a cardiovascular safety signal resulted in a less than unanimous vote to recommend approval.
The FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted 8-4 on Nov. 8 to support approval of insulin degludec (IDeg), a long-acting basal insulin analogue, used alone, and in a combined formulation with the fast-acting insulin analogue aspart (IDegAsp) for the treatment of type 1 and type 2 diabetes, based on clinical trial data from 17 studies.
Panel members voting either way said it was a difficult decision because of a cardiovascular safety signal detected in a meta-analysis of the clinical trials, the major issue discussed at the meeting.
The panel unanimously recommended that Novo Nordisk, the manufacturer of IDeg and IDegAsp, conduct a cardiovascular outcomes trial to further investigate the safety signal, which was an increase in a composite major cardiovascular event (MACE) end point of unstable angina pectoris, cardiovascular death, nonfatal myocardial infarction, and nonfatal stroke. Although panelists agreed that the signal could be caused by chance, they also said that it should be investigated further.
In approving a drug, the FDA has the authority to require that a company conduct a postmarketing safety trial. Novo Nordisk has proposed a postapproval study that would compare cardiovascular outcomes in 7,500 patients with type 2 diabetes, treated with IDeg or another insulin plus standard of care, with up to 5 years’ follow-up. Designed for slower absorption, IDeg forms multihexamers after injection, resulting in a soluble depot from which there is a slow, continuous, and extended release of insulin degludec, with a flat stable profile, according to the company, which markets insulin aspart as Novolog.
In phase-III 26- and 52-week studies of about 6,500 adult patients with type 1 or type 2 diabetes, treatment with IDeg or IDegAsp was noninferior to insulin comparators in reducing hemoglobin A1c from baseline and resulted in lower fasting blood glucose levels, with a reduced risk of hypoglycemia, particularly nocturnal hypoglycemia, according to Novo Nordisk. The company has highlighted the reduced rate of nocturnal hypoglycemia, but FDA reviewers said they were not convinced that it was clinically relevant.
Panelists agreed that IDeg and IDegAsp had features that would be useful in clinical practice, including the combination with a short-acting insulin, which would reduce the number of injections; evidence that it lasts for about 24 hours; and some flexibility if a patient misses a dose – a common problem in clinical practice, several of the endocrinologists on the panel pointed out.
The company’s proposed label recommends that IDeg be taken at about the same time every day, but if a dose is missed the patient can take a catch-up dose at any time as long as 8 hours elapse between doses.
Voting in favor of approval, Dr. David Cooke of the division of pediatric endocrinology at Johns Hopkins in Baltimore, said that, although it was a difficult call, “the pharmacokinetics of this insulin are an advance for our treatment of both type 1 and type 2 diabetes and would provide an additional option that could improve our treatment of these patients.”
Like other panelists, he said that currently available basal insulins do not last for 24 hours and that a basal insulin that provides coverage over 24 hours would be an advantage. The question of whether it increases cardiovascular risk is “far from proven,” but clinicians need to be aware of these issues, he added.
Another endocrinologist on the panel, Dr. Robert Smith, professor of medicine at Brown University in Providence, R.I., agreed that IDeg addressed the need for a long-acting insulin. If the data on efficacy and lower rate of hypoglycemia hold up in clinical practice, he said, “it is likely to be useful and broadly used as a form of insulin in diabetes care.”
Dr. Smith voted against approval, however, noting that there are alternatives and that the benefit of IDeg was “not of such magnitude that it merits taking on significant level of risk.” Despite the uncertainty of the cardiovascular risk signal, a study addressing this issue further should be done before approval, “given the importance of cardiovascular disease in patients with type 1 and type 2 diabetes,” he added.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting, although one panelist at this meeting was given a waiver. If approved, the company plans to make both IDeg and the combination product available in prefilled insulin pens, which they will market as Tresiba and Ryzodeg.
The company also has submitted an application for approval of IDeg and IDegAsp in Europe, in Canada, and other countries. They were approved in Japan in late September.
UPDATED 11/12/12
SILVER SPRING, MD. – A long duration of action and dosing flexibility were among the attributes of insulin degludec that a Food and Drug Administration advisory panel unanimously agreed would make it a useful addition to currently available diabetes treatments. But concerns over a cardiovascular safety signal resulted in a less than unanimous vote to recommend approval.
The FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted 8-4 on Nov. 8 to support approval of insulin degludec (IDeg), a long-acting basal insulin analogue, used alone, and in a combined formulation with the fast-acting insulin analogue aspart (IDegAsp) for the treatment of type 1 and type 2 diabetes, based on clinical trial data from 17 studies.
Panel members voting either way said it was a difficult decision because of a cardiovascular safety signal detected in a meta-analysis of the clinical trials, the major issue discussed at the meeting.
The panel unanimously recommended that Novo Nordisk, the manufacturer of IDeg and IDegAsp, conduct a cardiovascular outcomes trial to further investigate the safety signal, which was an increase in a composite major cardiovascular event (MACE) end point of unstable angina pectoris, cardiovascular death, nonfatal myocardial infarction, and nonfatal stroke. Although panelists agreed that the signal could be caused by chance, they also said that it should be investigated further.
In approving a drug, the FDA has the authority to require that a company conduct a postmarketing safety trial. Novo Nordisk has proposed a postapproval study that would compare cardiovascular outcomes in 7,500 patients with type 2 diabetes, treated with IDeg or another insulin plus standard of care, with up to 5 years’ follow-up. Designed for slower absorption, IDeg forms multihexamers after injection, resulting in a soluble depot from which there is a slow, continuous, and extended release of insulin degludec, with a flat stable profile, according to the company, which markets insulin aspart as Novolog.
In phase-III 26- and 52-week studies of about 6,500 adult patients with type 1 or type 2 diabetes, treatment with IDeg or IDegAsp was noninferior to insulin comparators in reducing hemoglobin A1c from baseline and resulted in lower fasting blood glucose levels, with a reduced risk of hypoglycemia, particularly nocturnal hypoglycemia, according to Novo Nordisk. The company has highlighted the reduced rate of nocturnal hypoglycemia, but FDA reviewers said they were not convinced that it was clinically relevant.
Panelists agreed that IDeg and IDegAsp had features that would be useful in clinical practice, including the combination with a short-acting insulin, which would reduce the number of injections; evidence that it lasts for about 24 hours; and some flexibility if a patient misses a dose – a common problem in clinical practice, several of the endocrinologists on the panel pointed out.
The company’s proposed label recommends that IDeg be taken at about the same time every day, but if a dose is missed the patient can take a catch-up dose at any time as long as 8 hours elapse between doses.
Voting in favor of approval, Dr. David Cooke of the division of pediatric endocrinology at Johns Hopkins in Baltimore, said that, although it was a difficult call, “the pharmacokinetics of this insulin are an advance for our treatment of both type 1 and type 2 diabetes and would provide an additional option that could improve our treatment of these patients.”
Like other panelists, he said that currently available basal insulins do not last for 24 hours and that a basal insulin that provides coverage over 24 hours would be an advantage. The question of whether it increases cardiovascular risk is “far from proven,” but clinicians need to be aware of these issues, he added.
Another endocrinologist on the panel, Dr. Robert Smith, professor of medicine at Brown University in Providence, R.I., agreed that IDeg addressed the need for a long-acting insulin. If the data on efficacy and lower rate of hypoglycemia hold up in clinical practice, he said, “it is likely to be useful and broadly used as a form of insulin in diabetes care.”
Dr. Smith voted against approval, however, noting that there are alternatives and that the benefit of IDeg was “not of such magnitude that it merits taking on significant level of risk.” Despite the uncertainty of the cardiovascular risk signal, a study addressing this issue further should be done before approval, “given the importance of cardiovascular disease in patients with type 1 and type 2 diabetes,” he added.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting, although one panelist at this meeting was given a waiver. If approved, the company plans to make both IDeg and the combination product available in prefilled insulin pens, which they will market as Tresiba and Ryzodeg.
The company also has submitted an application for approval of IDeg and IDegAsp in Europe, in Canada, and other countries. They were approved in Japan in late September.
UPDATED 11/12/12
AT A MEETING OF THE FOOD AND DRUG ADMINISTRATION ENDOCRINOLOGIC AND METABOLIC DRUGS ADVISORY COMMITTEE

Oral JAK Inhibitor's Clinical Place To Be Determined
The new oral janus kinase inhibitor approved on Nov. 6 by the Food and Drug Administration is likely to be embraced by those rheumatoid arthritis patients who have found infection and infusions of biologics to be needling.
The Janus kinase inhibitor (JAK) tofacitinib, a drug that has the promise to change the treatment experience for some patients with rheumatoid arthritis (RA), has been approved to treat adults with moderately to severely active disease who have not responded adequately to or cannot tolerate methotrexate.
Tofacitinib is a small-molecule inhibitor of the JAK pathway of inflammatory cytokines that play a role in the pathogenesis of RA, and is the first drug in this class of oral drugs to be approved for RA.
Dr. Eric L. Matteson said in an interview that he plans to offer this drug to patients with active disease. While some patients may embrace the idea of taking a pill, those who do not mind the needles because of their convenience and efficiency may opt to stay on their injected therapy. Of the nine biologic agents on the market currently for RA, four are infused and the others either are taken as subcutaneous injections or self-administered subcutaneously via a prefilled syringe. One of the four infused drugs is available as a prefilled self-administered syringe as well, with another biologic maker about to launch such a product.

Dr. Larry Greenbaum, a rheumatologist in Greenwood, Ind., recalled that before the introduction of biologics, "I thought patients would never accept parenteral medications, but almost all of them do accept these medications when they see how well they work. A pill will certainly be more welcome than an injection for most patients. But the Enbrel SureClick and the Humira Pen are very easy to use, so I don’t think patients are going to be breaking down the doors demanding this medication just so they don’t have to give themselves an injection!"
When asked where the new JAK inhibitor would fit into his own therapeutic lineup, Dr. Greenbaum noted that "the number of biologic medications is increasing all the time, and my conservative approach is usually to park the new medication at the bottom of the treatment algorithm until I have some compelling reason to use it sooner. No matter how good this medication is, it will have some very stiff competition from the available biologic drugs that work well and have long clinical track records."
In contrast, Dr. Karmela K. Chan, a rheumatologist in Pawtucket, R.I., said, "I definitely have patients who are completely opposed to any kind of injection, and, given a choice, they would rather take an oral drug. Several patients have asked me about switching from their injectable drug to an oral drug that they\'d already heard of.
"I don’t think I will switch most patients over. If something works, I tend not to want to mess with it. For new patients, I suspect my pitch will still be for the anti-TNF agent. I feel it is prudent to use agents that have been around longer. Another question will be how much more comfortable we are with how much information we have about potential adverse effects. However, I will most likely also present the option of the oral drug."
And then there is cost.
The JAK inhibitor will be expensive, just as the currently available biologic agents are. Pfizer has said that the recommended regimen of one 5-mg tablet twice a day will be priced at $2,000 a month, according to Dr. Matteson, chair of rheumatology and professor of medicine at the Mayo Clinic Medical School in Rochester, Minn. He noted that Pfizer is already offering a program to help patients cover their share of the copayment for the new drug.

Dr. Chan said that "without a doubt the cost will be an issue for patients. Cost of drug, side effect potential, efficaciousness, and convenience all factor into patients’ decisions.
"Also, yes, in our practice, I have been told multiple times that I could make more money if I put more people on infusions. We can buy and bill so we make more money that way, plus we make money off just the service of the infusion. But I think this issue of making money from infusions will perhaps not pass muster too much longer for the following reasons: Ethical physicians won’t infuse just to infuse (one would hope), and I think in the coming years fewer insurers will allow buy and bill. On top of that, I am not sure if Medicare reimbursement rates for the infusion service will change."
Dr. Matteson noted that the drug, to be marketed as Xeljanz, has shown efficacy compared with placebo in a number of studies considered by the Food and Drug Administration (FDA). The drug inhibits the protein kinase, which is important in cell-to-cell interaction and may be how the drug acts to decrease inflammation.
Only time will tell whether that decrease in inflammation will translate into reduced joint damage in RA patients or even into decreased risk for extra-articular manifestations of the disease, including cardiovascular disease, lung disease, and eye disease.

Treatment with anti–tumor necrosis factor (anti-TNF) drugs has been shown to lower the risk for cardiovascular disease associated with RA. But it took 5 years of post-marketing surveillance before rheumatologists began to recognize that benefit. Any similar effect with the JAK inhibitor may take just as long to become apparent, said Dr. Matteson.
In order to detect any effects, the FDA approved the drug with a Risk Evaluation and Mitigation Strategy (REMS) that addresses the serious risks associated with treatment, and a requirement that the manufacturer, Pfizer, conduct a post-marketing study, according to the FDA’s statement announcing the approval.
Dr. Matteson noted that safety and efficacy trials of the drugs showed that two common side effects were headaches and diarrhea, severe enough to cause the patient to discontinue the drug.
Approval was based on the results of seven studies, which found that patients with moderately to severely active RA had improvements in clinical response and physical functioning, when compared with those on placebo. Tofacitinib was associated with an increased risk of serious infections, including opportunistic infections; tuberculosis; cancers; and lymphoma, which are described in the boxed warning in the drug’s label, the FDA said.
Treatment was also associated with increases in cholesterol and liver enzymes, and decreased blood counts. The REMS consists of a Medication Guide that includes information for patients about the drug’s safety and a communication plan that will educate health care providers about the serious risks associated with the treatment.
The post-marketing study will compare two doses of tofacitinib with another approved treatment for RA.
At a meeting in May, the FDA’s Arthritis Advisory Committee voted 8 to 2 to recommend approval of tofacitinib for patients with RA, although panel members had lingering safety concerns.
Another JAK inhibitor, ruxolitinib (Jakafi), was approved to treat myelofibrosis in 2011.
The new oral janus kinase inhibitor approved on Nov. 6 by the Food and Drug Administration is likely to be embraced by those rheumatoid arthritis patients who have found infection and infusions of biologics to be needling.
The Janus kinase inhibitor (JAK) tofacitinib, a drug that has the promise to change the treatment experience for some patients with rheumatoid arthritis (RA), has been approved to treat adults with moderately to severely active disease who have not responded adequately to or cannot tolerate methotrexate.
Tofacitinib is a small-molecule inhibitor of the JAK pathway of inflammatory cytokines that play a role in the pathogenesis of RA, and is the first drug in this class of oral drugs to be approved for RA.
Dr. Eric L. Matteson said in an interview that he plans to offer this drug to patients with active disease. While some patients may embrace the idea of taking a pill, those who do not mind the needles because of their convenience and efficiency may opt to stay on their injected therapy. Of the nine biologic agents on the market currently for RA, four are infused and the others either are taken as subcutaneous injections or self-administered subcutaneously via a prefilled syringe. One of the four infused drugs is available as a prefilled self-administered syringe as well, with another biologic maker about to launch such a product.
Dr. Larry Greenbaum, a rheumatologist in Greenwood, Ind., recalled that before the introduction of biologics, "I thought patients would never accept parenteral medications, but almost all of them do accept these medications when they see how well they work. A pill will certainly be more welcome than an injection for most patients. But the Enbrel SureClick and the Humira Pen are very easy to use, so I don’t think patients are going to be breaking down the doors demanding this medication just so they don’t have to give themselves an injection!"
When asked where the new JAK inhibitor would fit into his own therapeutic lineup, Dr. Greenbaum noted that "the number of biologic medications is increasing all the time, and my conservative approach is usually to park the new medication at the bottom of the treatment algorithm until I have some compelling reason to use it sooner. No matter how good this medication is, it will have some very stiff competition from the available biologic drugs that work well and have long clinical track records."
In contrast, Dr. Karmela K. Chan, a rheumatologist in Pawtucket, R.I., said, "I definitely have patients who are completely opposed to any kind of injection, and, given a choice, they would rather take an oral drug. Several patients have asked me about switching from their injectable drug to an oral drug that they\'d already heard of.
"I don’t think I will switch most patients over. If something works, I tend not to want to mess with it. For new patients, I suspect my pitch will still be for the anti-TNF agent. I feel it is prudent to use agents that have been around longer. Another question will be how much more comfortable we are with how much information we have about potential adverse effects. However, I will most likely also present the option of the oral drug."
And then there is cost.
The JAK inhibitor will be expensive, just as the currently available biologic agents are. Pfizer has said that the recommended regimen of one 5-mg tablet twice a day will be priced at $2,000 a month, according to Dr. Matteson, chair of rheumatology and professor of medicine at the Mayo Clinic Medical School in Rochester, Minn. He noted that Pfizer is already offering a program to help patients cover their share of the copayment for the new drug.
Dr. Chan said that "without a doubt the cost will be an issue for patients. Cost of drug, side effect potential, efficaciousness, and convenience all factor into patients’ decisions.
"Also, yes, in our practice, I have been told multiple times that I could make more money if I put more people on infusions. We can buy and bill so we make more money that way, plus we make money off just the service of the infusion. But I think this issue of making money from infusions will perhaps not pass muster too much longer for the following reasons: Ethical physicians won’t infuse just to infuse (one would hope), and I think in the coming years fewer insurers will allow buy and bill. On top of that, I am not sure if Medicare reimbursement rates for the infusion service will change."
Dr. Matteson noted that the drug, to be marketed as Xeljanz, has shown efficacy compared with placebo in a number of studies considered by the Food and Drug Administration (FDA). The drug inhibits the protein kinase, which is important in cell-to-cell interaction and may be how the drug acts to decrease inflammation.
Only time will tell whether that decrease in inflammation will translate into reduced joint damage in RA patients or even into decreased risk for extra-articular manifestations of the disease, including cardiovascular disease, lung disease, and eye disease.
Treatment with anti–tumor necrosis factor (anti-TNF) drugs has been shown to lower the risk for cardiovascular disease associated with RA. But it took 5 years of post-marketing surveillance before rheumatologists began to recognize that benefit. Any similar effect with the JAK inhibitor may take just as long to become apparent, said Dr. Matteson.
In order to detect any effects, the FDA approved the drug with a Risk Evaluation and Mitigation Strategy (REMS) that addresses the serious risks associated with treatment, and a requirement that the manufacturer, Pfizer, conduct a post-marketing study, according to the FDA’s statement announcing the approval.
Dr. Matteson noted that safety and efficacy trials of the drugs showed that two common side effects were headaches and diarrhea, severe enough to cause the patient to discontinue the drug.
Approval was based on the results of seven studies, which found that patients with moderately to severely active RA had improvements in clinical response and physical functioning, when compared with those on placebo. Tofacitinib was associated with an increased risk of serious infections, including opportunistic infections; tuberculosis; cancers; and lymphoma, which are described in the boxed warning in the drug’s label, the FDA said.
Treatment was also associated with increases in cholesterol and liver enzymes, and decreased blood counts. The REMS consists of a Medication Guide that includes information for patients about the drug’s safety and a communication plan that will educate health care providers about the serious risks associated with the treatment.
The post-marketing study will compare two doses of tofacitinib with another approved treatment for RA.
At a meeting in May, the FDA’s Arthritis Advisory Committee voted 8 to 2 to recommend approval of tofacitinib for patients with RA, although panel members had lingering safety concerns.
Another JAK inhibitor, ruxolitinib (Jakafi), was approved to treat myelofibrosis in 2011.
The new oral janus kinase inhibitor approved on Nov. 6 by the Food and Drug Administration is likely to be embraced by those rheumatoid arthritis patients who have found infection and infusions of biologics to be needling.
The Janus kinase inhibitor (JAK) tofacitinib, a drug that has the promise to change the treatment experience for some patients with rheumatoid arthritis (RA), has been approved to treat adults with moderately to severely active disease who have not responded adequately to or cannot tolerate methotrexate.
Tofacitinib is a small-molecule inhibitor of the JAK pathway of inflammatory cytokines that play a role in the pathogenesis of RA, and is the first drug in this class of oral drugs to be approved for RA.
Dr. Eric L. Matteson said in an interview that he plans to offer this drug to patients with active disease. While some patients may embrace the idea of taking a pill, those who do not mind the needles because of their convenience and efficiency may opt to stay on their injected therapy. Of the nine biologic agents on the market currently for RA, four are infused and the others either are taken as subcutaneous injections or self-administered subcutaneously via a prefilled syringe. One of the four infused drugs is available as a prefilled self-administered syringe as well, with another biologic maker about to launch such a product.
Dr. Larry Greenbaum, a rheumatologist in Greenwood, Ind., recalled that before the introduction of biologics, "I thought patients would never accept parenteral medications, but almost all of them do accept these medications when they see how well they work. A pill will certainly be more welcome than an injection for most patients. But the Enbrel SureClick and the Humira Pen are very easy to use, so I don’t think patients are going to be breaking down the doors demanding this medication just so they don’t have to give themselves an injection!"
When asked where the new JAK inhibitor would fit into his own therapeutic lineup, Dr. Greenbaum noted that "the number of biologic medications is increasing all the time, and my conservative approach is usually to park the new medication at the bottom of the treatment algorithm until I have some compelling reason to use it sooner. No matter how good this medication is, it will have some very stiff competition from the available biologic drugs that work well and have long clinical track records."
In contrast, Dr. Karmela K. Chan, a rheumatologist in Pawtucket, R.I., said, "I definitely have patients who are completely opposed to any kind of injection, and, given a choice, they would rather take an oral drug. Several patients have asked me about switching from their injectable drug to an oral drug that they\'d already heard of.
"I don’t think I will switch most patients over. If something works, I tend not to want to mess with it. For new patients, I suspect my pitch will still be for the anti-TNF agent. I feel it is prudent to use agents that have been around longer. Another question will be how much more comfortable we are with how much information we have about potential adverse effects. However, I will most likely also present the option of the oral drug."
And then there is cost.
The JAK inhibitor will be expensive, just as the currently available biologic agents are. Pfizer has said that the recommended regimen of one 5-mg tablet twice a day will be priced at $2,000 a month, according to Dr. Matteson, chair of rheumatology and professor of medicine at the Mayo Clinic Medical School in Rochester, Minn. He noted that Pfizer is already offering a program to help patients cover their share of the copayment for the new drug.
Dr. Chan said that "without a doubt the cost will be an issue for patients. Cost of drug, side effect potential, efficaciousness, and convenience all factor into patients’ decisions.
"Also, yes, in our practice, I have been told multiple times that I could make more money if I put more people on infusions. We can buy and bill so we make more money that way, plus we make money off just the service of the infusion. But I think this issue of making money from infusions will perhaps not pass muster too much longer for the following reasons: Ethical physicians won’t infuse just to infuse (one would hope), and I think in the coming years fewer insurers will allow buy and bill. On top of that, I am not sure if Medicare reimbursement rates for the infusion service will change."
Dr. Matteson noted that the drug, to be marketed as Xeljanz, has shown efficacy compared with placebo in a number of studies considered by the Food and Drug Administration (FDA). The drug inhibits the protein kinase, which is important in cell-to-cell interaction and may be how the drug acts to decrease inflammation.
Only time will tell whether that decrease in inflammation will translate into reduced joint damage in RA patients or even into decreased risk for extra-articular manifestations of the disease, including cardiovascular disease, lung disease, and eye disease.
Treatment with anti–tumor necrosis factor (anti-TNF) drugs has been shown to lower the risk for cardiovascular disease associated with RA. But it took 5 years of post-marketing surveillance before rheumatologists began to recognize that benefit. Any similar effect with the JAK inhibitor may take just as long to become apparent, said Dr. Matteson.
In order to detect any effects, the FDA approved the drug with a Risk Evaluation and Mitigation Strategy (REMS) that addresses the serious risks associated with treatment, and a requirement that the manufacturer, Pfizer, conduct a post-marketing study, according to the FDA’s statement announcing the approval.
Dr. Matteson noted that safety and efficacy trials of the drugs showed that two common side effects were headaches and diarrhea, severe enough to cause the patient to discontinue the drug.
Approval was based on the results of seven studies, which found that patients with moderately to severely active RA had improvements in clinical response and physical functioning, when compared with those on placebo. Tofacitinib was associated with an increased risk of serious infections, including opportunistic infections; tuberculosis; cancers; and lymphoma, which are described in the boxed warning in the drug’s label, the FDA said.
Treatment was also associated with increases in cholesterol and liver enzymes, and decreased blood counts. The REMS consists of a Medication Guide that includes information for patients about the drug’s safety and a communication plan that will educate health care providers about the serious risks associated with the treatment.
The post-marketing study will compare two doses of tofacitinib with another approved treatment for RA.
At a meeting in May, the FDA’s Arthritis Advisory Committee voted 8 to 2 to recommend approval of tofacitinib for patients with RA, although panel members had lingering safety concerns.
Another JAK inhibitor, ruxolitinib (Jakafi), was approved to treat myelofibrosis in 2011.
