User login
FDA Panel Backs Pazopanib for Sarcoma
SILVER SPRING, MD. – A Food and Drug Administration advisory panel supports approval of the angiogenesis inhibitor pazopanib as a treatment for advanced soft-tissue sarcoma, despite some concerns about the degree of response in the main study considered for approval.
At its March 20 meeting, the Oncologic Drugs Advisory Committee (ODAC) voted 11-2 that pazopanib (Votrient) has a favorable risk-benefit profile as a treatment for advanced soft-tissue sarcoma (STS) in patients who have received prior chemotherapy, the new indication proposed by the manufacturer, GlaxoSmithKline.
In the pivotal placebo-controlled PALETTE study, the gain in median progression-free survival was 3 months, which was significant. The difference in median overall survival, a secondary end point, was 1.9 months, however, and this was not statistically significant.
Although panelists agreed that the effects of the drug were marginal, those voting in favor of approval said that they believed the effect was real. They reasoned that a 3-month benefit was meaningful to patients, as there are few options available for this rare tumor.
Several were encouraged by the fact that 14% of patients in the study remained still on treatment after 1 year (compared with 1% of those on placebo). Panelists also said that having a noncytotoxic treatment option for soft-tissue sarcomas would be beneficial.
Pazopanib, a tyrosine kinase inhibitor, targets the vascular endothelial growth factor receptor, the platelet-derived growth factor receptor, and stem cell growth factor. It was approved in 2009 for the treatment of advanced renal cell carcinoma.
In the double-blind, phase III study, patients with soft-tissue sarcoma who had progressed during or following prior chemotherapy (including anthracyclines) were randomized to pazopanib, 800 mg daily, in tablet formulation (246 patients) or placebo (123 patients). They were examined every 4 weeks during a 12-week period and then every 8 weeks until evidence of progression, death, excessive toxicity, or patient withdrawal.
A little more than half the patients were female, their median age was 51-56 years; most were from the European Union, 12% were from the United States. The most common histologies were leiomyosarcoma (40% on placebo and 44% on pazopanib) and synovial sarcoma (11% and 10%, respectively).
Median progression-free survival, the primary end point, reached 4.6 months among those on pazopanib, compared with 1.6 months among those on placebo, a statistically significant difference that represented a 65% reduced risk.
Treatment was not associated with a statistically significant improvement in overall survival, however. It reached 12.6 months among those on pazopanib and 10.7 months among those on placebo, for a reduced risk of 13%.
With a few exceptions, the safety profile in sarcoma trials was similar to the safety profile of patients in renal cell carcinoma studies, with adverse effects that included hepatoxicity, hypothyroidism, and hemorrhagic events.
"From my own experience of treating many sarcoma patients, this is a drug I would like to have in my armamentarium," panelist Dr. Lee J. Helman, scientific director for clinical research, at the Center for Cancer Research at the National Cancer Institute, Bethesda, Md.
While he would have liked to see a greater impact on progression-free survival, he said, "It’s a little step ... but it’s a little step in a field that has had nothing."
The company has proposed that the indication include the statement "Important limitations of use: The phase [III] STS trial population excluded patients with adipocytic STS or gastrointestinal stromal tumors."
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting.
Occasionally, a panelist may be given a waiver and at this meeting, Dr. Deborah K. Armstrong was granted a waiver to participate in the meeting, because of her expertise in STS. The conflict pertained to study grants received by her employer, the Sidney Kimmel Comprehensive Cancer Center in Philadelphia, according to the FDA.
SILVER SPRING, MD. – A Food and Drug Administration advisory panel supports approval of the angiogenesis inhibitor pazopanib as a treatment for advanced soft-tissue sarcoma, despite some concerns about the degree of response in the main study considered for approval.
At its March 20 meeting, the Oncologic Drugs Advisory Committee (ODAC) voted 11-2 that pazopanib (Votrient) has a favorable risk-benefit profile as a treatment for advanced soft-tissue sarcoma (STS) in patients who have received prior chemotherapy, the new indication proposed by the manufacturer, GlaxoSmithKline.
In the pivotal placebo-controlled PALETTE study, the gain in median progression-free survival was 3 months, which was significant. The difference in median overall survival, a secondary end point, was 1.9 months, however, and this was not statistically significant.
Although panelists agreed that the effects of the drug were marginal, those voting in favor of approval said that they believed the effect was real. They reasoned that a 3-month benefit was meaningful to patients, as there are few options available for this rare tumor.
Several were encouraged by the fact that 14% of patients in the study remained still on treatment after 1 year (compared with 1% of those on placebo). Panelists also said that having a noncytotoxic treatment option for soft-tissue sarcomas would be beneficial.
Pazopanib, a tyrosine kinase inhibitor, targets the vascular endothelial growth factor receptor, the platelet-derived growth factor receptor, and stem cell growth factor. It was approved in 2009 for the treatment of advanced renal cell carcinoma.
In the double-blind, phase III study, patients with soft-tissue sarcoma who had progressed during or following prior chemotherapy (including anthracyclines) were randomized to pazopanib, 800 mg daily, in tablet formulation (246 patients) or placebo (123 patients). They were examined every 4 weeks during a 12-week period and then every 8 weeks until evidence of progression, death, excessive toxicity, or patient withdrawal.
A little more than half the patients were female, their median age was 51-56 years; most were from the European Union, 12% were from the United States. The most common histologies were leiomyosarcoma (40% on placebo and 44% on pazopanib) and synovial sarcoma (11% and 10%, respectively).
Median progression-free survival, the primary end point, reached 4.6 months among those on pazopanib, compared with 1.6 months among those on placebo, a statistically significant difference that represented a 65% reduced risk.
Treatment was not associated with a statistically significant improvement in overall survival, however. It reached 12.6 months among those on pazopanib and 10.7 months among those on placebo, for a reduced risk of 13%.
With a few exceptions, the safety profile in sarcoma trials was similar to the safety profile of patients in renal cell carcinoma studies, with adverse effects that included hepatoxicity, hypothyroidism, and hemorrhagic events.
"From my own experience of treating many sarcoma patients, this is a drug I would like to have in my armamentarium," panelist Dr. Lee J. Helman, scientific director for clinical research, at the Center for Cancer Research at the National Cancer Institute, Bethesda, Md.
While he would have liked to see a greater impact on progression-free survival, he said, "It’s a little step ... but it’s a little step in a field that has had nothing."
The company has proposed that the indication include the statement "Important limitations of use: The phase [III] STS trial population excluded patients with adipocytic STS or gastrointestinal stromal tumors."
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting.
Occasionally, a panelist may be given a waiver and at this meeting, Dr. Deborah K. Armstrong was granted a waiver to participate in the meeting, because of her expertise in STS. The conflict pertained to study grants received by her employer, the Sidney Kimmel Comprehensive Cancer Center in Philadelphia, according to the FDA.
SILVER SPRING, MD. – A Food and Drug Administration advisory panel supports approval of the angiogenesis inhibitor pazopanib as a treatment for advanced soft-tissue sarcoma, despite some concerns about the degree of response in the main study considered for approval.
At its March 20 meeting, the Oncologic Drugs Advisory Committee (ODAC) voted 11-2 that pazopanib (Votrient) has a favorable risk-benefit profile as a treatment for advanced soft-tissue sarcoma (STS) in patients who have received prior chemotherapy, the new indication proposed by the manufacturer, GlaxoSmithKline.
In the pivotal placebo-controlled PALETTE study, the gain in median progression-free survival was 3 months, which was significant. The difference in median overall survival, a secondary end point, was 1.9 months, however, and this was not statistically significant.
Although panelists agreed that the effects of the drug were marginal, those voting in favor of approval said that they believed the effect was real. They reasoned that a 3-month benefit was meaningful to patients, as there are few options available for this rare tumor.
Several were encouraged by the fact that 14% of patients in the study remained still on treatment after 1 year (compared with 1% of those on placebo). Panelists also said that having a noncytotoxic treatment option for soft-tissue sarcomas would be beneficial.
Pazopanib, a tyrosine kinase inhibitor, targets the vascular endothelial growth factor receptor, the platelet-derived growth factor receptor, and stem cell growth factor. It was approved in 2009 for the treatment of advanced renal cell carcinoma.
In the double-blind, phase III study, patients with soft-tissue sarcoma who had progressed during or following prior chemotherapy (including anthracyclines) were randomized to pazopanib, 800 mg daily, in tablet formulation (246 patients) or placebo (123 patients). They were examined every 4 weeks during a 12-week period and then every 8 weeks until evidence of progression, death, excessive toxicity, or patient withdrawal.
A little more than half the patients were female, their median age was 51-56 years; most were from the European Union, 12% were from the United States. The most common histologies were leiomyosarcoma (40% on placebo and 44% on pazopanib) and synovial sarcoma (11% and 10%, respectively).
Median progression-free survival, the primary end point, reached 4.6 months among those on pazopanib, compared with 1.6 months among those on placebo, a statistically significant difference that represented a 65% reduced risk.
Treatment was not associated with a statistically significant improvement in overall survival, however. It reached 12.6 months among those on pazopanib and 10.7 months among those on placebo, for a reduced risk of 13%.
With a few exceptions, the safety profile in sarcoma trials was similar to the safety profile of patients in renal cell carcinoma studies, with adverse effects that included hepatoxicity, hypothyroidism, and hemorrhagic events.
"From my own experience of treating many sarcoma patients, this is a drug I would like to have in my armamentarium," panelist Dr. Lee J. Helman, scientific director for clinical research, at the Center for Cancer Research at the National Cancer Institute, Bethesda, Md.
While he would have liked to see a greater impact on progression-free survival, he said, "It’s a little step ... but it’s a little step in a field that has had nothing."
The company has proposed that the indication include the statement "Important limitations of use: The phase [III] STS trial population excluded patients with adipocytic STS or gastrointestinal stromal tumors."
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting.
Occasionally, a panelist may be given a waiver and at this meeting, Dr. Deborah K. Armstrong was granted a waiver to participate in the meeting, because of her expertise in STS. The conflict pertained to study grants received by her employer, the Sidney Kimmel Comprehensive Cancer Center in Philadelphia, according to the FDA.
FROM A MEETING OF THE FDA'S ONCOLOGIC DRUGS ADVISORY COMMITTEE
FDA Approves First Generic Form of Escitalopram
The first generic formulation of the selective serotonin reuptake inhibitor escitalopram has been approved by the Food and Drug Administration, the agency announced on March 14.
In a statement, the FDA said that generic escitalopram – in 5-mg, 10-mg, and 20-mg doses – manufactured by Teva Pharmaceutical Industries/IVAX Pharmaceuticals, has been approved for treating depression and generalized anxiety disorder in adults. Escitalopram initially was approved in 2002 and is marketed as Lexapro by Forest Laboratories.
Teva has 180 days of generic drug "exclusivity," during which time the FDA cannot approve any other generic formulation of escitalopram, the statement said.
For more information on generic drugs, click here to access the FDA site.
The first generic formulation of the selective serotonin reuptake inhibitor escitalopram has been approved by the Food and Drug Administration, the agency announced on March 14.
In a statement, the FDA said that generic escitalopram – in 5-mg, 10-mg, and 20-mg doses – manufactured by Teva Pharmaceutical Industries/IVAX Pharmaceuticals, has been approved for treating depression and generalized anxiety disorder in adults. Escitalopram initially was approved in 2002 and is marketed as Lexapro by Forest Laboratories.
Teva has 180 days of generic drug "exclusivity," during which time the FDA cannot approve any other generic formulation of escitalopram, the statement said.
For more information on generic drugs, click here to access the FDA site.
The first generic formulation of the selective serotonin reuptake inhibitor escitalopram has been approved by the Food and Drug Administration, the agency announced on March 14.
In a statement, the FDA said that generic escitalopram – in 5-mg, 10-mg, and 20-mg doses – manufactured by Teva Pharmaceutical Industries/IVAX Pharmaceuticals, has been approved for treating depression and generalized anxiety disorder in adults. Escitalopram initially was approved in 2002 and is marketed as Lexapro by Forest Laboratories.
Teva has 180 days of generic drug "exclusivity," during which time the FDA cannot approve any other generic formulation of escitalopram, the statement said.
For more information on generic drugs, click here to access the FDA site.
FDA: Continue Study of Antinerve Growth Factors for Pain
SILVER SPRING, MD. – The clinical development of antinerve growth factors as a treatment for pain should continue, despite reports of serious joint-related adverse effects associated with these agents in clinical trials, a Food and Drug Administration panel unanimously agreed on March 12.
At the meeting, the FDA’s Arthritis Advisory Committee voted 21-0 that there was a role for the ongoing development of antinerve growth factors (anti-NGF), which panelists agreed appeared to show promise as analgesics in studies and may eventually have a niche in the treatment of chronic pain conditions. But they strongly recommended that future studies should closely monitor patients for rapidly progressing osteoarthritis (OA) and other joint-related adverse events that have been observed in trials, and include a rigorous informed-consent process so that patients are well informed about the potential risks. The panel also recommended that the manufacturers of these products conduct studies to investigate the mechanism behind the joint-related adverse events, and that in clinical trials, the use of concomitant NSAIDs should be contraindicated or limited.
The FDA convened the panel to discuss reports of joint destruction in patients treated with two of the three agents in clinical development, which resulted in a hold on clinical trials of all three agents, tanezumab (Pfizer), fulranumab (Janssen), and REGN475 (Regeneron). Anti-NGF agents, monoclonal antibodies directed against nerve growth factor, are being studied for chronic pain caused by various conditions, primarily osteoarthritis. The greatest amounts of data are available on tanezumab, which was being studied in phase III trials of patients with OA and chronic low back pain.
The FDA put a hold on clinical trials of the three anti-NGF factors in 2010, after cases of osteonecrosis and avascular necrosis were reported in osteoarthritis patients treated with tanezumab and in those treated with fulranumab, because of concerns that these cases might be a class effect. There were no such cases among patients on placebo or comparator drugs. The risk increased further among those on an NSAID and an anti-NGF agent, compared with the anti-NGF agent alone.
Panelists agreed with the FDA reviewers that that these cases were unusual and severe.
In addition to OA, the companies are developing these agents for other chronic pain conditions, including low back pain, postherpetic neuralgia, chronic pancreatitis, endometriosis, and cancer pain.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
SILVER SPRING, MD. – The clinical development of antinerve growth factors as a treatment for pain should continue, despite reports of serious joint-related adverse effects associated with these agents in clinical trials, a Food and Drug Administration panel unanimously agreed on March 12.
At the meeting, the FDA’s Arthritis Advisory Committee voted 21-0 that there was a role for the ongoing development of antinerve growth factors (anti-NGF), which panelists agreed appeared to show promise as analgesics in studies and may eventually have a niche in the treatment of chronic pain conditions. But they strongly recommended that future studies should closely monitor patients for rapidly progressing osteoarthritis (OA) and other joint-related adverse events that have been observed in trials, and include a rigorous informed-consent process so that patients are well informed about the potential risks. The panel also recommended that the manufacturers of these products conduct studies to investigate the mechanism behind the joint-related adverse events, and that in clinical trials, the use of concomitant NSAIDs should be contraindicated or limited.
The FDA convened the panel to discuss reports of joint destruction in patients treated with two of the three agents in clinical development, which resulted in a hold on clinical trials of all three agents, tanezumab (Pfizer), fulranumab (Janssen), and REGN475 (Regeneron). Anti-NGF agents, monoclonal antibodies directed against nerve growth factor, are being studied for chronic pain caused by various conditions, primarily osteoarthritis. The greatest amounts of data are available on tanezumab, which was being studied in phase III trials of patients with OA and chronic low back pain.
The FDA put a hold on clinical trials of the three anti-NGF factors in 2010, after cases of osteonecrosis and avascular necrosis were reported in osteoarthritis patients treated with tanezumab and in those treated with fulranumab, because of concerns that these cases might be a class effect. There were no such cases among patients on placebo or comparator drugs. The risk increased further among those on an NSAID and an anti-NGF agent, compared with the anti-NGF agent alone.
Panelists agreed with the FDA reviewers that that these cases were unusual and severe.
In addition to OA, the companies are developing these agents for other chronic pain conditions, including low back pain, postherpetic neuralgia, chronic pancreatitis, endometriosis, and cancer pain.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
SILVER SPRING, MD. – The clinical development of antinerve growth factors as a treatment for pain should continue, despite reports of serious joint-related adverse effects associated with these agents in clinical trials, a Food and Drug Administration panel unanimously agreed on March 12.
At the meeting, the FDA’s Arthritis Advisory Committee voted 21-0 that there was a role for the ongoing development of antinerve growth factors (anti-NGF), which panelists agreed appeared to show promise as analgesics in studies and may eventually have a niche in the treatment of chronic pain conditions. But they strongly recommended that future studies should closely monitor patients for rapidly progressing osteoarthritis (OA) and other joint-related adverse events that have been observed in trials, and include a rigorous informed-consent process so that patients are well informed about the potential risks. The panel also recommended that the manufacturers of these products conduct studies to investigate the mechanism behind the joint-related adverse events, and that in clinical trials, the use of concomitant NSAIDs should be contraindicated or limited.
The FDA convened the panel to discuss reports of joint destruction in patients treated with two of the three agents in clinical development, which resulted in a hold on clinical trials of all three agents, tanezumab (Pfizer), fulranumab (Janssen), and REGN475 (Regeneron). Anti-NGF agents, monoclonal antibodies directed against nerve growth factor, are being studied for chronic pain caused by various conditions, primarily osteoarthritis. The greatest amounts of data are available on tanezumab, which was being studied in phase III trials of patients with OA and chronic low back pain.
The FDA put a hold on clinical trials of the three anti-NGF factors in 2010, after cases of osteonecrosis and avascular necrosis were reported in osteoarthritis patients treated with tanezumab and in those treated with fulranumab, because of concerns that these cases might be a class effect. There were no such cases among patients on placebo or comparator drugs. The risk increased further among those on an NSAID and an anti-NGF agent, compared with the anti-NGF agent alone.
Panelists agreed with the FDA reviewers that that these cases were unusual and severe.
In addition to OA, the companies are developing these agents for other chronic pain conditions, including low back pain, postherpetic neuralgia, chronic pancreatitis, endometriosis, and cancer pain.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
FROM A MEETING OF THE FDA'S ARTHRITIS ADVISORY COMMITTEE
FDA Announces Nationwide Recall of Three Cytarabine Lots
Three lots of the antineoplastic drug cytarabine have been recalled because they may not be sterile, according to a posting on the Food and Drug Administration’s MedWatch site.
The nationwide recall applies to lots of cytarabine for injection (1 gm/vial), manufactured by Bedford Laboratories. The risk for lack of sterility was determined from an investigation of the manufacturing area, after the product was released, according to the FDA.
The affected lot numbers are: 2066986, 2111675, and 2131148. Any hospital, physician, clinic, or other health care facility that has any of the affected products should not use it for patients and "should immediately quarantine any product for return," the statement said. Wholesalers, distributors, or retailers that have the recalled product should stop using it and contact the company at 800-562-4797. Click here to report the product to the FDA Medwatch program.
Bedford is one of several generic manufacturers of cytarabine.
Cytarabine is approved for use in combination with other approved cancer treatments for inducing remission of acute nonlymphocytic leukemia in children and adults, and has also been found to be useful in treating acute lymphocytic leukemia and the blast phase of chronic myelocytic leukemia. It is also indicated in the prophylaxis and treatment of meningeal leukemia, administered intrathecally for this indication.
To view the notice, visit the MedWatch site.
Three lots of the antineoplastic drug cytarabine have been recalled because they may not be sterile, according to a posting on the Food and Drug Administration’s MedWatch site.
The nationwide recall applies to lots of cytarabine for injection (1 gm/vial), manufactured by Bedford Laboratories. The risk for lack of sterility was determined from an investigation of the manufacturing area, after the product was released, according to the FDA.
The affected lot numbers are: 2066986, 2111675, and 2131148. Any hospital, physician, clinic, or other health care facility that has any of the affected products should not use it for patients and "should immediately quarantine any product for return," the statement said. Wholesalers, distributors, or retailers that have the recalled product should stop using it and contact the company at 800-562-4797. Click here to report the product to the FDA Medwatch program.
Bedford is one of several generic manufacturers of cytarabine.
Cytarabine is approved for use in combination with other approved cancer treatments for inducing remission of acute nonlymphocytic leukemia in children and adults, and has also been found to be useful in treating acute lymphocytic leukemia and the blast phase of chronic myelocytic leukemia. It is also indicated in the prophylaxis and treatment of meningeal leukemia, administered intrathecally for this indication.
To view the notice, visit the MedWatch site.
Three lots of the antineoplastic drug cytarabine have been recalled because they may not be sterile, according to a posting on the Food and Drug Administration’s MedWatch site.
The nationwide recall applies to lots of cytarabine for injection (1 gm/vial), manufactured by Bedford Laboratories. The risk for lack of sterility was determined from an investigation of the manufacturing area, after the product was released, according to the FDA.
The affected lot numbers are: 2066986, 2111675, and 2131148. Any hospital, physician, clinic, or other health care facility that has any of the affected products should not use it for patients and "should immediately quarantine any product for return," the statement said. Wholesalers, distributors, or retailers that have the recalled product should stop using it and contact the company at 800-562-4797. Click here to report the product to the FDA Medwatch program.
Bedford is one of several generic manufacturers of cytarabine.
Cytarabine is approved for use in combination with other approved cancer treatments for inducing remission of acute nonlymphocytic leukemia in children and adults, and has also been found to be useful in treating acute lymphocytic leukemia and the blast phase of chronic myelocytic leukemia. It is also indicated in the prophylaxis and treatment of meningeal leukemia, administered intrathecally for this indication.
To view the notice, visit the MedWatch site.
FDA Approves Another Surfactant for Premature Infants
A new surfactant, lucinactant, has been approved for prevention of respiratory distress syndrome in premature infants, the fifth surfactant approved in the United States, the Food and Drug Administration announced on March 6.
The safety and efficacy of lucinactant was demonstrated in a randomized active controlled multidose study of 1,294 premature infants, comparing it with two of the other approved surfactants, according to the FDA statement announcing the approval.
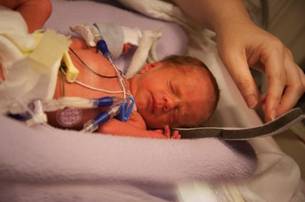
In the study, the infants were treated with lucinactant (Surfaxin), colfosceril palmitate (Exosurf), or beractant (Survanta) within 30 minutes of birth. Those treated with lucinactant had significant improvement in respiratory distress syndrome at 24 hours after birth and RDS-related mortality through 2 weeks, when compared with those treated with colfosceril, the statement said. Colfosceril, which is no longer marketed in the United States, was used as the primary comparator in the study, according to the statement.
The most common side effects associated with the lucinactant were associated with intratracheal administration, and included endotracheal tube reflux, endotracheal tube obstruction, and need for dose interruption, the statement said.
In addition to colfosceril and beractant, the other two FDA-approved surfactants are poractant alpha (Curosurf) and calfactant (Infasurf).
Lucinactant, which is a suspension formulation intended for intratracheal use only, will be marketed as Surfaxin by Discovery Laboratories Inc. A statement issued by the company said that it anticipates that the product will be available in the United States in late 2012. The company describes it as a synthetic, peptide-containing surfactant.
A new surfactant, lucinactant, has been approved for prevention of respiratory distress syndrome in premature infants, the fifth surfactant approved in the United States, the Food and Drug Administration announced on March 6.
The safety and efficacy of lucinactant was demonstrated in a randomized active controlled multidose study of 1,294 premature infants, comparing it with two of the other approved surfactants, according to the FDA statement announcing the approval.
In the study, the infants were treated with lucinactant (Surfaxin), colfosceril palmitate (Exosurf), or beractant (Survanta) within 30 minutes of birth. Those treated with lucinactant had significant improvement in respiratory distress syndrome at 24 hours after birth and RDS-related mortality through 2 weeks, when compared with those treated with colfosceril, the statement said. Colfosceril, which is no longer marketed in the United States, was used as the primary comparator in the study, according to the statement.
The most common side effects associated with the lucinactant were associated with intratracheal administration, and included endotracheal tube reflux, endotracheal tube obstruction, and need for dose interruption, the statement said.
In addition to colfosceril and beractant, the other two FDA-approved surfactants are poractant alpha (Curosurf) and calfactant (Infasurf).
Lucinactant, which is a suspension formulation intended for intratracheal use only, will be marketed as Surfaxin by Discovery Laboratories Inc. A statement issued by the company said that it anticipates that the product will be available in the United States in late 2012. The company describes it as a synthetic, peptide-containing surfactant.
A new surfactant, lucinactant, has been approved for prevention of respiratory distress syndrome in premature infants, the fifth surfactant approved in the United States, the Food and Drug Administration announced on March 6.
The safety and efficacy of lucinactant was demonstrated in a randomized active controlled multidose study of 1,294 premature infants, comparing it with two of the other approved surfactants, according to the FDA statement announcing the approval.
In the study, the infants were treated with lucinactant (Surfaxin), colfosceril palmitate (Exosurf), or beractant (Survanta) within 30 minutes of birth. Those treated with lucinactant had significant improvement in respiratory distress syndrome at 24 hours after birth and RDS-related mortality through 2 weeks, when compared with those treated with colfosceril, the statement said. Colfosceril, which is no longer marketed in the United States, was used as the primary comparator in the study, according to the statement.
The most common side effects associated with the lucinactant were associated with intratracheal administration, and included endotracheal tube reflux, endotracheal tube obstruction, and need for dose interruption, the statement said.
In addition to colfosceril and beractant, the other two FDA-approved surfactants are poractant alpha (Curosurf) and calfactant (Infasurf).
Lucinactant, which is a suspension formulation intended for intratracheal use only, will be marketed as Surfaxin by Discovery Laboratories Inc. A statement issued by the company said that it anticipates that the product will be available in the United States in late 2012. The company describes it as a synthetic, peptide-containing surfactant.
Drug-Eluting Stent Cleared for Diabetes Patients
The zotarolimus-eluting coronary stent, approved last month, can be used for the treatment of patients with symptomatic heart disease, including those with diabetes, the manufacturer announced.
The approval of the stent, which will be marketed by Medtronic as the Resolute Integrity zotarolimus-eluting coronary stent, is indicated for "improving coronary luminal diameters in patients, including those with diabetes mellitus, with symptomatic ischemic heart disease due to de novo lesions of length less than or equal to 27 mm in native coronary arteries with reference vessel diameters of 2.25 mm to 4.20 mm," according to the company website.
About one-third (1,535) of the patients who have received the Resolute Integrity drug-eluting stent (DES) in clinical trials had diabetes, according to aMedtronic press release announcing the approval. Among these trials was the RESOLUTE US study, a multicenter study of 1,402 patients (34% had diabetes) who received the Resolute Integrity DES. At 1 year, the target lesion failure rate was 4.7%, the clinically driven target lesion revascularization rate was 2.8%, and the definite/probable stent thrombosis rate was 0.1%, the release said.
Another study, the Resolute All-Comers trial, a noninferiority study of 2,292 patients with chronic, stable coronary artery disease or acute coronary syndromes, found that the Resolute Integrity DES was noninferior to the everolimus-eluting stent (Abbott Xience V). The primary end point, target lesion failure (death from cardiac causes, any MI, or clinically indicated target lesion vascularization) within 12 months was about 8% in both groups. The study had minimal exclusion criteria, about 23% of those enrolled had diabetes, and 66% had at least one off-label criterion for stent placement (N. Engl. J. Med 2010;363:136-46).
The Medtronic press release refers to a prespecified analysis of patients with diabetes who received a Resolute DES in RESOLUTE studies, which found that at 1-year follow-up, the rates of target lesion failure (6.6%), target lesion revascularization (3.4%), and definite/probable stent thrombosis (0.3%) were low.
In an interview, Dr. Bruce Bode, an endocrinologist in group practice in Atlanta, said that the approval of the Resolute Integrity stent marks the first time patients with diabetes have been included in the approval of a coronary stent – and is unique because there is now an approved coronary stent that had a similar failure rate in patients with and those without diabetes in studies.
Because of the results of the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) trial of patients with type 2 diabetes and heart disease, published in 2009, coronary artery bypass grafting has been considered preferable in patients with diabetes, because of better outcomes among those who underwent surgery in that study, he noted (N. Engl. J. Med. 2009;360:2503-15).
Dr. Bode is a consultant to Medtronic. He is affiliated with Piedmont Hospital, a RESOLUTE study site, but he was not an investigator in those studies.
The Resolute All-Comers and RESOLUTE US studies are sponsored by Medtronic.
The zotarolimus-eluting coronary stent, approved last month, can be used for the treatment of patients with symptomatic heart disease, including those with diabetes, the manufacturer announced.
The approval of the stent, which will be marketed by Medtronic as the Resolute Integrity zotarolimus-eluting coronary stent, is indicated for "improving coronary luminal diameters in patients, including those with diabetes mellitus, with symptomatic ischemic heart disease due to de novo lesions of length less than or equal to 27 mm in native coronary arteries with reference vessel diameters of 2.25 mm to 4.20 mm," according to the company website.
About one-third (1,535) of the patients who have received the Resolute Integrity drug-eluting stent (DES) in clinical trials had diabetes, according to aMedtronic press release announcing the approval. Among these trials was the RESOLUTE US study, a multicenter study of 1,402 patients (34% had diabetes) who received the Resolute Integrity DES. At 1 year, the target lesion failure rate was 4.7%, the clinically driven target lesion revascularization rate was 2.8%, and the definite/probable stent thrombosis rate was 0.1%, the release said.
Another study, the Resolute All-Comers trial, a noninferiority study of 2,292 patients with chronic, stable coronary artery disease or acute coronary syndromes, found that the Resolute Integrity DES was noninferior to the everolimus-eluting stent (Abbott Xience V). The primary end point, target lesion failure (death from cardiac causes, any MI, or clinically indicated target lesion vascularization) within 12 months was about 8% in both groups. The study had minimal exclusion criteria, about 23% of those enrolled had diabetes, and 66% had at least one off-label criterion for stent placement (N. Engl. J. Med 2010;363:136-46).
The Medtronic press release refers to a prespecified analysis of patients with diabetes who received a Resolute DES in RESOLUTE studies, which found that at 1-year follow-up, the rates of target lesion failure (6.6%), target lesion revascularization (3.4%), and definite/probable stent thrombosis (0.3%) were low.
In an interview, Dr. Bruce Bode, an endocrinologist in group practice in Atlanta, said that the approval of the Resolute Integrity stent marks the first time patients with diabetes have been included in the approval of a coronary stent – and is unique because there is now an approved coronary stent that had a similar failure rate in patients with and those without diabetes in studies.
Because of the results of the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) trial of patients with type 2 diabetes and heart disease, published in 2009, coronary artery bypass grafting has been considered preferable in patients with diabetes, because of better outcomes among those who underwent surgery in that study, he noted (N. Engl. J. Med. 2009;360:2503-15).
Dr. Bode is a consultant to Medtronic. He is affiliated with Piedmont Hospital, a RESOLUTE study site, but he was not an investigator in those studies.
The Resolute All-Comers and RESOLUTE US studies are sponsored by Medtronic.
The zotarolimus-eluting coronary stent, approved last month, can be used for the treatment of patients with symptomatic heart disease, including those with diabetes, the manufacturer announced.
The approval of the stent, which will be marketed by Medtronic as the Resolute Integrity zotarolimus-eluting coronary stent, is indicated for "improving coronary luminal diameters in patients, including those with diabetes mellitus, with symptomatic ischemic heart disease due to de novo lesions of length less than or equal to 27 mm in native coronary arteries with reference vessel diameters of 2.25 mm to 4.20 mm," according to the company website.
About one-third (1,535) of the patients who have received the Resolute Integrity drug-eluting stent (DES) in clinical trials had diabetes, according to aMedtronic press release announcing the approval. Among these trials was the RESOLUTE US study, a multicenter study of 1,402 patients (34% had diabetes) who received the Resolute Integrity DES. At 1 year, the target lesion failure rate was 4.7%, the clinically driven target lesion revascularization rate was 2.8%, and the definite/probable stent thrombosis rate was 0.1%, the release said.
Another study, the Resolute All-Comers trial, a noninferiority study of 2,292 patients with chronic, stable coronary artery disease or acute coronary syndromes, found that the Resolute Integrity DES was noninferior to the everolimus-eluting stent (Abbott Xience V). The primary end point, target lesion failure (death from cardiac causes, any MI, or clinically indicated target lesion vascularization) within 12 months was about 8% in both groups. The study had minimal exclusion criteria, about 23% of those enrolled had diabetes, and 66% had at least one off-label criterion for stent placement (N. Engl. J. Med 2010;363:136-46).
The Medtronic press release refers to a prespecified analysis of patients with diabetes who received a Resolute DES in RESOLUTE studies, which found that at 1-year follow-up, the rates of target lesion failure (6.6%), target lesion revascularization (3.4%), and definite/probable stent thrombosis (0.3%) were low.
In an interview, Dr. Bruce Bode, an endocrinologist in group practice in Atlanta, said that the approval of the Resolute Integrity stent marks the first time patients with diabetes have been included in the approval of a coronary stent – and is unique because there is now an approved coronary stent that had a similar failure rate in patients with and those without diabetes in studies.
Because of the results of the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) trial of patients with type 2 diabetes and heart disease, published in 2009, coronary artery bypass grafting has been considered preferable in patients with diabetes, because of better outcomes among those who underwent surgery in that study, he noted (N. Engl. J. Med. 2009;360:2503-15).
Dr. Bode is a consultant to Medtronic. He is affiliated with Piedmont Hospital, a RESOLUTE study site, but he was not an investigator in those studies.
The Resolute All-Comers and RESOLUTE US studies are sponsored by Medtronic.
Two More Pancreatic Enzymes Approved by FDA
The Food and Drug Administration has approved two formulations of pancrelipase for different indications and patient populations, the agency announced on March 1.
A delayed-release capsule formulation of pancrelipase, which will be marketed as Ultresa, has been approved for treating for the treatment of exocrine pancreatic insufficiency caused by cystic fibrosis or other conditions in children and adults. The second pancrelipase formulation, which will be marketed as Viokace, has been approved in combination with a proton pump inhibitor for the treatment of exocrine pancreatic insufficiency from chronic pancreatitis or pancreatectomy in adults. The safety and efficacy of Viokace in children has not been established, according to the March 1 FDA statement announcing the approval.
Both products are a combination of porcine-derived lipases, proteases, and amylases.
The approval of these two products, along with three other pancreatic enzyme products that have been approved since 2009, "allow health care providers to prescribe the product that is most appropriate for the estimated 200,000 patients in the United States who have pancreatic insufficiency," Dr. Julie Beitz, director of the Office of Drug Evaluation III in FDA’s Center for Drug Evaluation and Research, said in the statement.
Since April 2010, manufacturers have not been allowed to manufacture and distribute unapproved pancreatic enzyme products. For years, they had been marketed in the United States as unapproved products, but because of problems with consistency in the amount of the enzymes contained in these products, the FDA required that these products had to go through the regular approval process before they were marketed.
The other pancreatic enzyme products (PEPs) – which contain the active ingredient pancrelipase, a mixture of the digestive enzymes amylase, lipase, and protease – that have been approved and meet FDA standards for safety, efficacy, and product quality" are Creon and Zenpep (2009), and Pancreaze (2010), the statement said.
Ultresa and Viokace are marketed by Aptalis Pharma U.S. Inc.
The Food and Drug Administration has approved two formulations of pancrelipase for different indications and patient populations, the agency announced on March 1.
A delayed-release capsule formulation of pancrelipase, which will be marketed as Ultresa, has been approved for treating for the treatment of exocrine pancreatic insufficiency caused by cystic fibrosis or other conditions in children and adults. The second pancrelipase formulation, which will be marketed as Viokace, has been approved in combination with a proton pump inhibitor for the treatment of exocrine pancreatic insufficiency from chronic pancreatitis or pancreatectomy in adults. The safety and efficacy of Viokace in children has not been established, according to the March 1 FDA statement announcing the approval.
Both products are a combination of porcine-derived lipases, proteases, and amylases.
The approval of these two products, along with three other pancreatic enzyme products that have been approved since 2009, "allow health care providers to prescribe the product that is most appropriate for the estimated 200,000 patients in the United States who have pancreatic insufficiency," Dr. Julie Beitz, director of the Office of Drug Evaluation III in FDA’s Center for Drug Evaluation and Research, said in the statement.
Since April 2010, manufacturers have not been allowed to manufacture and distribute unapproved pancreatic enzyme products. For years, they had been marketed in the United States as unapproved products, but because of problems with consistency in the amount of the enzymes contained in these products, the FDA required that these products had to go through the regular approval process before they were marketed.
The other pancreatic enzyme products (PEPs) – which contain the active ingredient pancrelipase, a mixture of the digestive enzymes amylase, lipase, and protease – that have been approved and meet FDA standards for safety, efficacy, and product quality" are Creon and Zenpep (2009), and Pancreaze (2010), the statement said.
Ultresa and Viokace are marketed by Aptalis Pharma U.S. Inc.
The Food and Drug Administration has approved two formulations of pancrelipase for different indications and patient populations, the agency announced on March 1.
A delayed-release capsule formulation of pancrelipase, which will be marketed as Ultresa, has been approved for treating for the treatment of exocrine pancreatic insufficiency caused by cystic fibrosis or other conditions in children and adults. The second pancrelipase formulation, which will be marketed as Viokace, has been approved in combination with a proton pump inhibitor for the treatment of exocrine pancreatic insufficiency from chronic pancreatitis or pancreatectomy in adults. The safety and efficacy of Viokace in children has not been established, according to the March 1 FDA statement announcing the approval.
Both products are a combination of porcine-derived lipases, proteases, and amylases.
The approval of these two products, along with three other pancreatic enzyme products that have been approved since 2009, "allow health care providers to prescribe the product that is most appropriate for the estimated 200,000 patients in the United States who have pancreatic insufficiency," Dr. Julie Beitz, director of the Office of Drug Evaluation III in FDA’s Center for Drug Evaluation and Research, said in the statement.
Since April 2010, manufacturers have not been allowed to manufacture and distribute unapproved pancreatic enzyme products. For years, they had been marketed in the United States as unapproved products, but because of problems with consistency in the amount of the enzymes contained in these products, the FDA required that these products had to go through the regular approval process before they were marketed.
The other pancreatic enzyme products (PEPs) – which contain the active ingredient pancrelipase, a mixture of the digestive enzymes amylase, lipase, and protease – that have been approved and meet FDA standards for safety, efficacy, and product quality" are Creon and Zenpep (2009), and Pancreaze (2010), the statement said.
Ultresa and Viokace are marketed by Aptalis Pharma U.S. Inc.
FDA Approves First Quadrivalent Seasonal Flu Vaccine
The first influenza vaccine that contains a second influenza B strain, in addition to two influenza A strains and one influenza B strain, has been approved for people aged 2 through 49 years, the Food and Drug Administration announced on Feb. 29.
The quadrivalent vaccine is an intranasal vaccine, similar to FluMist, the trivalent influenza vaccine licensed as a seasonal influenza vaccine for individuals aged 2 through 49 years. The quadrivalent vaccine contains two strains of type A influenza (A/H1N1 and A/H3N2) and two type B lineage strains, "which increases the likelihood of adequate protection against circulating influenza B strains," the FDA statement announcing the approval said.
"A vaccine containing the four virus strains most likely to spread and cause illness during the influenza season offers an additional option to aid in influenza prevention efforts," Dr. Karen Midthun, director of the FDA’s Center for Biologics Evaluation and Research, said in the statement. She pointed out that influenza B-related illness "affects children, particularly young and school-aged, more than any other population."
The new vaccine will be marketed as "FluMist Quadrivalent" by MedImmune LLC, which also markets FluMist, the trivalent intranasal influenza vaccine.
The safety and effectiveness of the Flu Mist quadrivalent vaccine is supported by studies of the trivalent formulation conducted in the past, as well as three new studies of the quadrivalent vaccine in about 4,000 children and adults in the United States, which "demonstrated that the immune responses were similar between FluMist Quadrivalent and FluMist," the FDA statement said. Adverse reactions associated with both vaccines were similar. A runny or stuffy nose in children and adults and headache and sore throat in adults were the most common adverse reactions reported, according to the FDA.
The other seasonal influenza vaccines that are licensed by the FDA contain the three strains only. At a meeting of the FDA’s vaccines advisory panel, held the day before the FDA announced the FluMist Quadrivalent approval, representatives of several manufacturers said that they had completed trials of quadrivalent formulations of their trivalent vaccines and had either filed or planned to file for approval with the FDA and anticipated approval in time for the 2012-2013 influenza season.
At that meeting, held annually to recommend the two influenza A and one influenza B strains to be included in the vaccine in the United States for the upcoming season, panelists pointed out how challenging it is every year to choose which B strain to recommend, based on the different B strains that are circulating worldwide, and described choosing the B component as a "gamble" and a "crapshoot."
"Flu B has been a dilemma, and I do look forward to having a quadrivalent so that our crapshoot is not so poor," one of the panel members, Dr. Pedro Piedra, professor in the department of molecular virology and microbiology, Baylor College of Medicine, Houston, said at the meeting.
"I’m really gratified to hear of all the progress that’s been made by the manufacturers in developing the quadrivalent product and I look forward to the end of this annual conversation about which B [strain] should go into the vaccine," said another panelist, Dr. Melinda Wharton, deputy director of the Centers for Disease Control and Prevention’s National Center for Immunization and Respiratory Diseases. "I always feel like we’re having to make a decision that has a pretty high probability of being wrong on these B strains," she added. (FDA panelists are cleared of potential conflicts related to the topic of the meeting.)
At that meeting, the panel recommended that the influenza B strain be replaced with a B/Yamagata lineage strain, a switch from a Victoria lineage B strain that has been in the vaccine for several years. But they were less confident about this recommendation than they were with their recommendations for the two influenza A strains.
During this flu season in the United States, which has started later than usual and with less activity than previous years, there is evidence that influenza B viruses from both lineages have been circulating. At the panel meeting, Dr. Lisa Grohskopf of the CDC’s influenza division, said that of the 397 influenza viruses tested at the CDC this season, 48 (12%) have been influenza B strains. About half of those have been a B/Victoria lineage strains and most of those match the strain in the current influenza vaccine, but the rest have been a Yamagata lineage strain, not included in the current vaccine. But Dr. Grohskopf, who was not a panelist, added that there have not been many B viruses available for testing in the United States.
Click here for prescribing information for FluMist Quadrivalent.
The first influenza vaccine that contains a second influenza B strain, in addition to two influenza A strains and one influenza B strain, has been approved for people aged 2 through 49 years, the Food and Drug Administration announced on Feb. 29.
The quadrivalent vaccine is an intranasal vaccine, similar to FluMist, the trivalent influenza vaccine licensed as a seasonal influenza vaccine for individuals aged 2 through 49 years. The quadrivalent vaccine contains two strains of type A influenza (A/H1N1 and A/H3N2) and two type B lineage strains, "which increases the likelihood of adequate protection against circulating influenza B strains," the FDA statement announcing the approval said.
"A vaccine containing the four virus strains most likely to spread and cause illness during the influenza season offers an additional option to aid in influenza prevention efforts," Dr. Karen Midthun, director of the FDA’s Center for Biologics Evaluation and Research, said in the statement. She pointed out that influenza B-related illness "affects children, particularly young and school-aged, more than any other population."
The new vaccine will be marketed as "FluMist Quadrivalent" by MedImmune LLC, which also markets FluMist, the trivalent intranasal influenza vaccine.
The safety and effectiveness of the Flu Mist quadrivalent vaccine is supported by studies of the trivalent formulation conducted in the past, as well as three new studies of the quadrivalent vaccine in about 4,000 children and adults in the United States, which "demonstrated that the immune responses were similar between FluMist Quadrivalent and FluMist," the FDA statement said. Adverse reactions associated with both vaccines were similar. A runny or stuffy nose in children and adults and headache and sore throat in adults were the most common adverse reactions reported, according to the FDA.
The other seasonal influenza vaccines that are licensed by the FDA contain the three strains only. At a meeting of the FDA’s vaccines advisory panel, held the day before the FDA announced the FluMist Quadrivalent approval, representatives of several manufacturers said that they had completed trials of quadrivalent formulations of their trivalent vaccines and had either filed or planned to file for approval with the FDA and anticipated approval in time for the 2012-2013 influenza season.
At that meeting, held annually to recommend the two influenza A and one influenza B strains to be included in the vaccine in the United States for the upcoming season, panelists pointed out how challenging it is every year to choose which B strain to recommend, based on the different B strains that are circulating worldwide, and described choosing the B component as a "gamble" and a "crapshoot."
"Flu B has been a dilemma, and I do look forward to having a quadrivalent so that our crapshoot is not so poor," one of the panel members, Dr. Pedro Piedra, professor in the department of molecular virology and microbiology, Baylor College of Medicine, Houston, said at the meeting.
"I’m really gratified to hear of all the progress that’s been made by the manufacturers in developing the quadrivalent product and I look forward to the end of this annual conversation about which B [strain] should go into the vaccine," said another panelist, Dr. Melinda Wharton, deputy director of the Centers for Disease Control and Prevention’s National Center for Immunization and Respiratory Diseases. "I always feel like we’re having to make a decision that has a pretty high probability of being wrong on these B strains," she added. (FDA panelists are cleared of potential conflicts related to the topic of the meeting.)
At that meeting, the panel recommended that the influenza B strain be replaced with a B/Yamagata lineage strain, a switch from a Victoria lineage B strain that has been in the vaccine for several years. But they were less confident about this recommendation than they were with their recommendations for the two influenza A strains.
During this flu season in the United States, which has started later than usual and with less activity than previous years, there is evidence that influenza B viruses from both lineages have been circulating. At the panel meeting, Dr. Lisa Grohskopf of the CDC’s influenza division, said that of the 397 influenza viruses tested at the CDC this season, 48 (12%) have been influenza B strains. About half of those have been a B/Victoria lineage strains and most of those match the strain in the current influenza vaccine, but the rest have been a Yamagata lineage strain, not included in the current vaccine. But Dr. Grohskopf, who was not a panelist, added that there have not been many B viruses available for testing in the United States.
Click here for prescribing information for FluMist Quadrivalent.
The first influenza vaccine that contains a second influenza B strain, in addition to two influenza A strains and one influenza B strain, has been approved for people aged 2 through 49 years, the Food and Drug Administration announced on Feb. 29.
The quadrivalent vaccine is an intranasal vaccine, similar to FluMist, the trivalent influenza vaccine licensed as a seasonal influenza vaccine for individuals aged 2 through 49 years. The quadrivalent vaccine contains two strains of type A influenza (A/H1N1 and A/H3N2) and two type B lineage strains, "which increases the likelihood of adequate protection against circulating influenza B strains," the FDA statement announcing the approval said.
"A vaccine containing the four virus strains most likely to spread and cause illness during the influenza season offers an additional option to aid in influenza prevention efforts," Dr. Karen Midthun, director of the FDA’s Center for Biologics Evaluation and Research, said in the statement. She pointed out that influenza B-related illness "affects children, particularly young and school-aged, more than any other population."
The new vaccine will be marketed as "FluMist Quadrivalent" by MedImmune LLC, which also markets FluMist, the trivalent intranasal influenza vaccine.
The safety and effectiveness of the Flu Mist quadrivalent vaccine is supported by studies of the trivalent formulation conducted in the past, as well as three new studies of the quadrivalent vaccine in about 4,000 children and adults in the United States, which "demonstrated that the immune responses were similar between FluMist Quadrivalent and FluMist," the FDA statement said. Adverse reactions associated with both vaccines were similar. A runny or stuffy nose in children and adults and headache and sore throat in adults were the most common adverse reactions reported, according to the FDA.
The other seasonal influenza vaccines that are licensed by the FDA contain the three strains only. At a meeting of the FDA’s vaccines advisory panel, held the day before the FDA announced the FluMist Quadrivalent approval, representatives of several manufacturers said that they had completed trials of quadrivalent formulations of their trivalent vaccines and had either filed or planned to file for approval with the FDA and anticipated approval in time for the 2012-2013 influenza season.
At that meeting, held annually to recommend the two influenza A and one influenza B strains to be included in the vaccine in the United States for the upcoming season, panelists pointed out how challenging it is every year to choose which B strain to recommend, based on the different B strains that are circulating worldwide, and described choosing the B component as a "gamble" and a "crapshoot."
"Flu B has been a dilemma, and I do look forward to having a quadrivalent so that our crapshoot is not so poor," one of the panel members, Dr. Pedro Piedra, professor in the department of molecular virology and microbiology, Baylor College of Medicine, Houston, said at the meeting.
"I’m really gratified to hear of all the progress that’s been made by the manufacturers in developing the quadrivalent product and I look forward to the end of this annual conversation about which B [strain] should go into the vaccine," said another panelist, Dr. Melinda Wharton, deputy director of the Centers for Disease Control and Prevention’s National Center for Immunization and Respiratory Diseases. "I always feel like we’re having to make a decision that has a pretty high probability of being wrong on these B strains," she added. (FDA panelists are cleared of potential conflicts related to the topic of the meeting.)
At that meeting, the panel recommended that the influenza B strain be replaced with a B/Yamagata lineage strain, a switch from a Victoria lineage B strain that has been in the vaccine for several years. But they were less confident about this recommendation than they were with their recommendations for the two influenza A strains.
During this flu season in the United States, which has started later than usual and with less activity than previous years, there is evidence that influenza B viruses from both lineages have been circulating. At the panel meeting, Dr. Lisa Grohskopf of the CDC’s influenza division, said that of the 397 influenza viruses tested at the CDC this season, 48 (12%) have been influenza B strains. About half of those have been a B/Victoria lineage strains and most of those match the strain in the current influenza vaccine, but the rest have been a Yamagata lineage strain, not included in the current vaccine. But Dr. Grohskopf, who was not a panelist, added that there have not been many B viruses available for testing in the United States.
Click here for prescribing information for FluMist Quadrivalent.
National Psoriasis Foundation Launches Seal of Approval Program
The National Psoriasis Foundation has launched a "seal of approval" program for over-the-counter products that an expert panel has determined to be "helpful in the management of psoriasis and psoriatic arthritis," according to the organization’s website.
The program’s objective "is to improve the quality of life for people who live with psoriasis and psoriatic arthritis," according to the NPF.

Under the seal program, companies submit over-the-counter products for approval, with an annual, nonrefundable application fee. A panel of dermatologists and other experts review the product and determine whether it meets required criteria, including the ability to hydrate skin, reduce redness, reduce joint pain, and/or provide aid for patients with psoriatic arthritis, according to a release announcing the launch of the program.
The four-person panel also reviews the ingredients and data on its safety, toxicity, and formulation.
Once approved, the seal can remain on the product for 2 years. The application fee is $5,000, which covers honoraria and time spent reviewing the clinical information that the company provides, plus an additional fee of $10,000 per year for each product that earns the seal, according to Jackie Groah, director of strategic alliances at the NPF. The reviewers will excuse themselves from the review if they have had any dealings with the company applying for the seal, she said in an interview.
Five scalp psoriasis products manufactured by Neutrogena and a gel product for patients with plaque psoriasis, manufactured by Alva-Amco, were the first products to receive the seal. The seal can be awarded to personal care and household products, fabrics, and devices, according to the NPF.
Click here for more details about the program.
The National Psoriasis Foundation has launched a "seal of approval" program for over-the-counter products that an expert panel has determined to be "helpful in the management of psoriasis and psoriatic arthritis," according to the organization’s website.
The program’s objective "is to improve the quality of life for people who live with psoriasis and psoriatic arthritis," according to the NPF.
Under the seal program, companies submit over-the-counter products for approval, with an annual, nonrefundable application fee. A panel of dermatologists and other experts review the product and determine whether it meets required criteria, including the ability to hydrate skin, reduce redness, reduce joint pain, and/or provide aid for patients with psoriatic arthritis, according to a release announcing the launch of the program.
The four-person panel also reviews the ingredients and data on its safety, toxicity, and formulation.
Once approved, the seal can remain on the product for 2 years. The application fee is $5,000, which covers honoraria and time spent reviewing the clinical information that the company provides, plus an additional fee of $10,000 per year for each product that earns the seal, according to Jackie Groah, director of strategic alliances at the NPF. The reviewers will excuse themselves from the review if they have had any dealings with the company applying for the seal, she said in an interview.
Five scalp psoriasis products manufactured by Neutrogena and a gel product for patients with plaque psoriasis, manufactured by Alva-Amco, were the first products to receive the seal. The seal can be awarded to personal care and household products, fabrics, and devices, according to the NPF.
Click here for more details about the program.
The National Psoriasis Foundation has launched a "seal of approval" program for over-the-counter products that an expert panel has determined to be "helpful in the management of psoriasis and psoriatic arthritis," according to the organization’s website.
The program’s objective "is to improve the quality of life for people who live with psoriasis and psoriatic arthritis," according to the NPF.
Under the seal program, companies submit over-the-counter products for approval, with an annual, nonrefundable application fee. A panel of dermatologists and other experts review the product and determine whether it meets required criteria, including the ability to hydrate skin, reduce redness, reduce joint pain, and/or provide aid for patients with psoriatic arthritis, according to a release announcing the launch of the program.
The four-person panel also reviews the ingredients and data on its safety, toxicity, and formulation.
Once approved, the seal can remain on the product for 2 years. The application fee is $5,000, which covers honoraria and time spent reviewing the clinical information that the company provides, plus an additional fee of $10,000 per year for each product that earns the seal, according to Jackie Groah, director of strategic alliances at the NPF. The reviewers will excuse themselves from the review if they have had any dealings with the company applying for the seal, she said in an interview.
Five scalp psoriasis products manufactured by Neutrogena and a gel product for patients with plaque psoriasis, manufactured by Alva-Amco, were the first products to receive the seal. The seal can be awarded to personal care and household products, fabrics, and devices, according to the NPF.
Click here for more details about the program.
Packaging Error Spurs Nationwide Recall of Generic OC
Seven lots of oral contraceptives containing norgestimate and ethinyl estradiol have been recalled nationwide because of a packaging error that could result in suboptimal protection against pregnancy, according to an announcement Feb. 27 by the Food and Drug Administration.
A statement on the agency's MedWatch site said that as a result of the packaging error in these lots, “the daily regimen for these oral contraceptives may be incorrect and could leave women without adequate contraception, and at risk for unintended pregnancy.” The FDA advised that women who have been “exposed to” the affected packaging should start using a nonhormonal form of contraception immediately and those who have the product should return it to the pharmacy and contact their physician.
The affected OCs, manufactured by Glenmark Generics, were distributed to wholesalers and retail pharmacies nationwide between Sept. 21, 2011, and Dec. 30, 2011, and contain the following doses of norgestimate and ethinyl estradiol: 0.18 mg/0.035 mg, 0.215 mg/0.035 mg, and 0.25 mg/0.035 mg.
The packaging error that prompted the recall is described as select blisters that are rotated 180 degrees within the card, which reverses the weekly tablet orientation and makes the lot number and expiration date visible on the outer pouch only, according to the FDA.
A statement issued by Glenmark said the following lot numbers are affected: 04110101, 04110106, and 04110107, with the expiration date of 7/31/2013; 04110114, 04110124, and 04110129, with the expiration date of 8/31/2013; and 04110134, with the expiration date of 9/30/2013.
Adverse events that may be related to the use of these products should be reported to Glenmark Generics, 888-721-7115 (8 a.m. to 5 p.m. ET Monday through Friday), or to the FDA's MedWatch Program anytime by faxing 800-332-1078 or visiting www.fda.gov/medwatch/report.htm
{kind=link}
In the recalled lots, blisters rotated 180 degrees in the card reverse tablet orientation.
Source Courtesy U.S. Food and Drug Administration
Seven lots of oral contraceptives containing norgestimate and ethinyl estradiol have been recalled nationwide because of a packaging error that could result in suboptimal protection against pregnancy, according to an announcement Feb. 27 by the Food and Drug Administration.
A statement on the agency's MedWatch site said that as a result of the packaging error in these lots, “the daily regimen for these oral contraceptives may be incorrect and could leave women without adequate contraception, and at risk for unintended pregnancy.” The FDA advised that women who have been “exposed to” the affected packaging should start using a nonhormonal form of contraception immediately and those who have the product should return it to the pharmacy and contact their physician.
The affected OCs, manufactured by Glenmark Generics, were distributed to wholesalers and retail pharmacies nationwide between Sept. 21, 2011, and Dec. 30, 2011, and contain the following doses of norgestimate and ethinyl estradiol: 0.18 mg/0.035 mg, 0.215 mg/0.035 mg, and 0.25 mg/0.035 mg.
The packaging error that prompted the recall is described as select blisters that are rotated 180 degrees within the card, which reverses the weekly tablet orientation and makes the lot number and expiration date visible on the outer pouch only, according to the FDA.
A statement issued by Glenmark said the following lot numbers are affected: 04110101, 04110106, and 04110107, with the expiration date of 7/31/2013; 04110114, 04110124, and 04110129, with the expiration date of 8/31/2013; and 04110134, with the expiration date of 9/30/2013.
Adverse events that may be related to the use of these products should be reported to Glenmark Generics, 888-721-7115 (8 a.m. to 5 p.m. ET Monday through Friday), or to the FDA's MedWatch Program anytime by faxing 800-332-1078 or visiting www.fda.gov/medwatch/report.htm
In the recalled lots, blisters rotated 180 degrees in the card reverse tablet orientation.
Source Courtesy U.S. Food and Drug Administration
Seven lots of oral contraceptives containing norgestimate and ethinyl estradiol have been recalled nationwide because of a packaging error that could result in suboptimal protection against pregnancy, according to an announcement Feb. 27 by the Food and Drug Administration.
A statement on the agency's MedWatch site said that as a result of the packaging error in these lots, “the daily regimen for these oral contraceptives may be incorrect and could leave women without adequate contraception, and at risk for unintended pregnancy.” The FDA advised that women who have been “exposed to” the affected packaging should start using a nonhormonal form of contraception immediately and those who have the product should return it to the pharmacy and contact their physician.
The affected OCs, manufactured by Glenmark Generics, were distributed to wholesalers and retail pharmacies nationwide between Sept. 21, 2011, and Dec. 30, 2011, and contain the following doses of norgestimate and ethinyl estradiol: 0.18 mg/0.035 mg, 0.215 mg/0.035 mg, and 0.25 mg/0.035 mg.
The packaging error that prompted the recall is described as select blisters that are rotated 180 degrees within the card, which reverses the weekly tablet orientation and makes the lot number and expiration date visible on the outer pouch only, according to the FDA.
A statement issued by Glenmark said the following lot numbers are affected: 04110101, 04110106, and 04110107, with the expiration date of 7/31/2013; 04110114, 04110124, and 04110129, with the expiration date of 8/31/2013; and 04110134, with the expiration date of 9/30/2013.
Adverse events that may be related to the use of these products should be reported to Glenmark Generics, 888-721-7115 (8 a.m. to 5 p.m. ET Monday through Friday), or to the FDA's MedWatch Program anytime by faxing 800-332-1078 or visiting www.fda.gov/medwatch/report.htm
In the recalled lots, blisters rotated 180 degrees in the card reverse tablet orientation.
Source Courtesy U.S. Food and Drug Administration