User login
FDA Panel: Add Two New Strains to 2012 Influenza Vaccine
SILVER SPRING, MD. – A Food and Drug Administration advisory panel on Feb. 28 recommended that the vaccine for the next influenza season should include two new strains and retain only one of the three strains in the current vaccine.
The FDA’s Vaccines and Related Biological Products Advisory Committee voted 18-0 that the 2012-2013 seasonal flu vaccine used in the United States should include the same influenza A (H1N1) component included in the 2011-2012 vaccine, an A/California/7/2009 (H1N1)-like virus. For the second influenza A strain in the vaccine, the panel’s vote was also unanimous, recommending that the influenza A (H3N2) component be replaced with an A/Victoria/361/2011 (H3N2)-like virus.
The panel voted 17-1 that that the influenza B strain be replaced with a B/Wisconsin/1/2010-like virus (B/Yamagata lineage). The current vaccine strain is a B/Brisbane/60/2008-like virus, a B/Victoria lineage strain. Panelists pointed out, however, that determining which B strain to select, a Victoria or Yamagata lineage B strain virus, is always challenging and said that this illustrated the utility of a quadrivalent influenza vaccine that contains B/Victoria lineage and B/Yamagata lineage viruses.
It appears that a quadrivalent influenza vaccine may soon be available, possibly as early as 2013. At the meeting, representatives of several vaccine manufacturers provided updates on the status of their quadrivalent influenza vaccines in development, including GlaxoSmithKline, which has filed for FDA approval of a quadrivalent influenza vaccine for people aged 3 and older.
The FDA panel’s recommendations are the same as the World Health Organization’s recommendations for the 2012-2013 Northern Hemisphere seasonal influenza vaccine, made at a meeting earlier in February.
The FDA panel meets at this time every year to recommend the strains to be included in the trivalent influenza vaccine in the United States in the upcoming season, considering information on the strains circulating worldwide and the WHO recommendation for the vaccine to be used in the Northern Hemisphere.
This influenza season has started late, in February, and flu activity has been low, although it is expected to increase, the Centers for Disease Control and Prevention announced last week.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
SILVER SPRING, MD. – A Food and Drug Administration advisory panel on Feb. 28 recommended that the vaccine for the next influenza season should include two new strains and retain only one of the three strains in the current vaccine.
The FDA’s Vaccines and Related Biological Products Advisory Committee voted 18-0 that the 2012-2013 seasonal flu vaccine used in the United States should include the same influenza A (H1N1) component included in the 2011-2012 vaccine, an A/California/7/2009 (H1N1)-like virus. For the second influenza A strain in the vaccine, the panel’s vote was also unanimous, recommending that the influenza A (H3N2) component be replaced with an A/Victoria/361/2011 (H3N2)-like virus.
The panel voted 17-1 that that the influenza B strain be replaced with a B/Wisconsin/1/2010-like virus (B/Yamagata lineage). The current vaccine strain is a B/Brisbane/60/2008-like virus, a B/Victoria lineage strain. Panelists pointed out, however, that determining which B strain to select, a Victoria or Yamagata lineage B strain virus, is always challenging and said that this illustrated the utility of a quadrivalent influenza vaccine that contains B/Victoria lineage and B/Yamagata lineage viruses.
It appears that a quadrivalent influenza vaccine may soon be available, possibly as early as 2013. At the meeting, representatives of several vaccine manufacturers provided updates on the status of their quadrivalent influenza vaccines in development, including GlaxoSmithKline, which has filed for FDA approval of a quadrivalent influenza vaccine for people aged 3 and older.
The FDA panel’s recommendations are the same as the World Health Organization’s recommendations for the 2012-2013 Northern Hemisphere seasonal influenza vaccine, made at a meeting earlier in February.
The FDA panel meets at this time every year to recommend the strains to be included in the trivalent influenza vaccine in the United States in the upcoming season, considering information on the strains circulating worldwide and the WHO recommendation for the vaccine to be used in the Northern Hemisphere.
This influenza season has started late, in February, and flu activity has been low, although it is expected to increase, the Centers for Disease Control and Prevention announced last week.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
SILVER SPRING, MD. – A Food and Drug Administration advisory panel on Feb. 28 recommended that the vaccine for the next influenza season should include two new strains and retain only one of the three strains in the current vaccine.
The FDA’s Vaccines and Related Biological Products Advisory Committee voted 18-0 that the 2012-2013 seasonal flu vaccine used in the United States should include the same influenza A (H1N1) component included in the 2011-2012 vaccine, an A/California/7/2009 (H1N1)-like virus. For the second influenza A strain in the vaccine, the panel’s vote was also unanimous, recommending that the influenza A (H3N2) component be replaced with an A/Victoria/361/2011 (H3N2)-like virus.
The panel voted 17-1 that that the influenza B strain be replaced with a B/Wisconsin/1/2010-like virus (B/Yamagata lineage). The current vaccine strain is a B/Brisbane/60/2008-like virus, a B/Victoria lineage strain. Panelists pointed out, however, that determining which B strain to select, a Victoria or Yamagata lineage B strain virus, is always challenging and said that this illustrated the utility of a quadrivalent influenza vaccine that contains B/Victoria lineage and B/Yamagata lineage viruses.
It appears that a quadrivalent influenza vaccine may soon be available, possibly as early as 2013. At the meeting, representatives of several vaccine manufacturers provided updates on the status of their quadrivalent influenza vaccines in development, including GlaxoSmithKline, which has filed for FDA approval of a quadrivalent influenza vaccine for people aged 3 and older.
The FDA panel’s recommendations are the same as the World Health Organization’s recommendations for the 2012-2013 Northern Hemisphere seasonal influenza vaccine, made at a meeting earlier in February.
The FDA panel meets at this time every year to recommend the strains to be included in the trivalent influenza vaccine in the United States in the upcoming season, considering information on the strains circulating worldwide and the WHO recommendation for the vaccine to be used in the Northern Hemisphere.
This influenza season has started late, in February, and flu activity has been low, although it is expected to increase, the Centers for Disease Control and Prevention announced last week.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
FROM A MEETING OF THE FDA'S VACCINES AND RELATED BIOLOGICAL PRODUCTS ADVISORY COMMITTEE
Packaging Problem Prompts Generic OC Nationwide Recall
A nationwide recall of seven lots of oral contraceptives containing norgestimate and ethinyl estradiol because of a packaging error that could result in suboptimal protection against pregnancy has been announced by the manufacturer, the Food and Drug Administration announced on Feb. 27.
A statement on the agency’s MedWatch site said that as a result of a packaging error in these lots, "the daily regimen for these oral contraceptives may be incorrect and could leave women without adequate contraception, and at risk for unintended pregnancy." The FDA advised that women who have been "exposed to" the affected packaging should start using a nonhormonal form of contraception immediately and those who have the product should return it to the pharmacy and contact their physician.
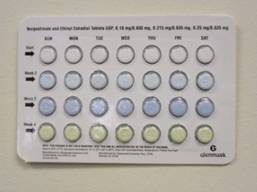
The affected OCs, manufactured by Glenmark Generics, were distributed to wholesalers and retail pharmacies nationwide between Sept. 21, 2011, and Dec. 30, 2011, and contain the following doses of norgestimate and ethinyl estradiol: 0.18 mg/0.035 mg, 0.215 mg/0.035 mg, and 0.25 mg/0.035 mg.
The packaging error that prompted the recall is described as select blisters that are rotated 180 degrees* within the card, which reverses the weekly tablet orientation and makes the lot number and expiration date visible on the outer pouch only, according to the FDA.
A statement issued by Glenmark said the following lot numbers are affected: 04110101, 04110106, and 04110107, with the expiration date of 7/31/2013; 04110114, 04110124, and 04110129, with the expiration date of 8/31/2013; and 04110134, with the expiration date of 9/30/2013.
Adverse events that may be related to the use of these products should be reported to Glenmark Generics, at 888-721-7115 (8 a.m. to 5 p.m. Monday through Friday EST), or to the FDA’s MedWatch Program anytime by faxing 800-332-1078 or visiting its website.
*CORRECTION 3/7/12: The original sentence misidentified the packaging error.
A nationwide recall of seven lots of oral contraceptives containing norgestimate and ethinyl estradiol because of a packaging error that could result in suboptimal protection against pregnancy has been announced by the manufacturer, the Food and Drug Administration announced on Feb. 27.
A statement on the agency’s MedWatch site said that as a result of a packaging error in these lots, "the daily regimen for these oral contraceptives may be incorrect and could leave women without adequate contraception, and at risk for unintended pregnancy." The FDA advised that women who have been "exposed to" the affected packaging should start using a nonhormonal form of contraception immediately and those who have the product should return it to the pharmacy and contact their physician.
The affected OCs, manufactured by Glenmark Generics, were distributed to wholesalers and retail pharmacies nationwide between Sept. 21, 2011, and Dec. 30, 2011, and contain the following doses of norgestimate and ethinyl estradiol: 0.18 mg/0.035 mg, 0.215 mg/0.035 mg, and 0.25 mg/0.035 mg.
The packaging error that prompted the recall is described as select blisters that are rotated 180 degrees* within the card, which reverses the weekly tablet orientation and makes the lot number and expiration date visible on the outer pouch only, according to the FDA.
A statement issued by Glenmark said the following lot numbers are affected: 04110101, 04110106, and 04110107, with the expiration date of 7/31/2013; 04110114, 04110124, and 04110129, with the expiration date of 8/31/2013; and 04110134, with the expiration date of 9/30/2013.
Adverse events that may be related to the use of these products should be reported to Glenmark Generics, at 888-721-7115 (8 a.m. to 5 p.m. Monday through Friday EST), or to the FDA’s MedWatch Program anytime by faxing 800-332-1078 or visiting its website.
*CORRECTION 3/7/12: The original sentence misidentified the packaging error.
A nationwide recall of seven lots of oral contraceptives containing norgestimate and ethinyl estradiol because of a packaging error that could result in suboptimal protection against pregnancy has been announced by the manufacturer, the Food and Drug Administration announced on Feb. 27.
A statement on the agency’s MedWatch site said that as a result of a packaging error in these lots, "the daily regimen for these oral contraceptives may be incorrect and could leave women without adequate contraception, and at risk for unintended pregnancy." The FDA advised that women who have been "exposed to" the affected packaging should start using a nonhormonal form of contraception immediately and those who have the product should return it to the pharmacy and contact their physician.
The affected OCs, manufactured by Glenmark Generics, were distributed to wholesalers and retail pharmacies nationwide between Sept. 21, 2011, and Dec. 30, 2011, and contain the following doses of norgestimate and ethinyl estradiol: 0.18 mg/0.035 mg, 0.215 mg/0.035 mg, and 0.25 mg/0.035 mg.
The packaging error that prompted the recall is described as select blisters that are rotated 180 degrees* within the card, which reverses the weekly tablet orientation and makes the lot number and expiration date visible on the outer pouch only, according to the FDA.
A statement issued by Glenmark said the following lot numbers are affected: 04110101, 04110106, and 04110107, with the expiration date of 7/31/2013; 04110114, 04110124, and 04110129, with the expiration date of 8/31/2013; and 04110134, with the expiration date of 9/30/2013.
Adverse events that may be related to the use of these products should be reported to Glenmark Generics, at 888-721-7115 (8 a.m. to 5 p.m. Monday through Friday EST), or to the FDA’s MedWatch Program anytime by faxing 800-332-1078 or visiting its website.
*CORRECTION 3/7/12: The original sentence misidentified the packaging error.
FDA Panel Backs Approval of Inhaled Anticholinergic for COPD
SILVER SPRING, MD. – The majority of a Food and Drug Administration advisory panel on Feb. 23 recommended that aclidinium bromide, an inhaled long-acting anticholinergic bronchodilator, should be approved as a treatment for chronic obstructive pulmonary disease, with a postmarketing study that evaluates the cardiovascular safety of the drug.
The FDA’s Pulmonary-Allergy Drugs Advisory Committee voted 12 to 2 that the efficacy and safety data from clinical trials provided "substantial evidence" to support approval of aclidinium, at a dose of 400 mcg twice a day administered in a breath-actuated dry powder inhaler, for the long-term maintenance treatment of bronchospasm associated with COPD, including chronic bronchitis and emphysema.
The panel unanimously agreed that the data in the clinical trials presented by the manufacturer provided evidence that this dose provided a clinically meaningful benefit in patients, citing the significant increases in trough forced expiratory volume in 1 second (FEV1) from baseline (the primary efficacy end point) among those treated with 400 mcg twice a day, compared with those on placebo after 12 weeks of treatment in clinical studies. Some panelists pointed out that there was also some evidence of improvements in COPD exacerbations and in patient-reported symptoms on a patient questionnaire, which were secondary end points.
If approved, it will be the second long-acting inhaled anticholinergic agent for COPD on the U.S. market, following tiotropium bromide (Spiriva HandiHaler), approved for COPD in 2004, but taken once a day. Ipratropium bromide, a short-acting inhaled anticholinergic, was approved in 1986 for COPD.
Clinical data presented by the manufacturer Forest Laboratories Inc., included two pivotal 12-week studies of almost 1,400 outpatients, most of whom were white and had stable moderate to severe COPD (mean age about 64 years). Patients received 400 mcg or 200 mcg of aclidinium twice a day or placebo. About half were still smoking; people with clinically significant cardiovascular disease were excluded. They were allowed to continue treatment with short-acting bronchodilators, inhaled corticosteroids, long-acting theophylline, oxygen for 15 hours or less, and stable-dose prednisone.
Overall, cardiovascular events were not common and were evenly distributed among the treatment arms, according to the company. This included the overall rates of major adverse cardiac events (MACE), although there were some small differences in the individual components of this score, a combination of CV deaths, nonfatal myocardial infarction, and nonfatal strokes. (There were two CV deaths among those aclidinium, and no deaths among those on placebo.) The rate of anticholinergic effects was low, which the company said might be due to low systemic exposure to the drug.
Cardiovascular safety of the drug is an issue because of some concerns raised by data on other inhaled anticholinergics over the past several years.
The panel voted 10 to 3 with 1 abstention that that the safety had been adequately evaluated. But panelists, including those who supported approval, still had concerns about cardiovascular safety and recommended that these concerns should be evaluated further in a postmarketing study.
The company plans to conduct a double-blind, randomized parallel postmarketing 3-year study of 4,000 patients with COPD, with a history of COPD exacerbations during the previous year, randomized to 400 mcg aclidinium or placebo plus standard of care, which will compare rates of moderate to severe COPD exacerbations within the first year.
The primary safety end point will be time to first MACE event; the study will also evaluate the rates of other serious cardiac events, conduction disorders and cerebrovascular disorders.
The FDA’s deadline for making an approval decision is April 23. If approved, Forest plans to market it as "Tudorza Pressair."
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
SILVER SPRING, MD. – The majority of a Food and Drug Administration advisory panel on Feb. 23 recommended that aclidinium bromide, an inhaled long-acting anticholinergic bronchodilator, should be approved as a treatment for chronic obstructive pulmonary disease, with a postmarketing study that evaluates the cardiovascular safety of the drug.
The FDA’s Pulmonary-Allergy Drugs Advisory Committee voted 12 to 2 that the efficacy and safety data from clinical trials provided "substantial evidence" to support approval of aclidinium, at a dose of 400 mcg twice a day administered in a breath-actuated dry powder inhaler, for the long-term maintenance treatment of bronchospasm associated with COPD, including chronic bronchitis and emphysema.
The panel unanimously agreed that the data in the clinical trials presented by the manufacturer provided evidence that this dose provided a clinically meaningful benefit in patients, citing the significant increases in trough forced expiratory volume in 1 second (FEV1) from baseline (the primary efficacy end point) among those treated with 400 mcg twice a day, compared with those on placebo after 12 weeks of treatment in clinical studies. Some panelists pointed out that there was also some evidence of improvements in COPD exacerbations and in patient-reported symptoms on a patient questionnaire, which were secondary end points.
If approved, it will be the second long-acting inhaled anticholinergic agent for COPD on the U.S. market, following tiotropium bromide (Spiriva HandiHaler), approved for COPD in 2004, but taken once a day. Ipratropium bromide, a short-acting inhaled anticholinergic, was approved in 1986 for COPD.
Clinical data presented by the manufacturer Forest Laboratories Inc., included two pivotal 12-week studies of almost 1,400 outpatients, most of whom were white and had stable moderate to severe COPD (mean age about 64 years). Patients received 400 mcg or 200 mcg of aclidinium twice a day or placebo. About half were still smoking; people with clinically significant cardiovascular disease were excluded. They were allowed to continue treatment with short-acting bronchodilators, inhaled corticosteroids, long-acting theophylline, oxygen for 15 hours or less, and stable-dose prednisone.
Overall, cardiovascular events were not common and were evenly distributed among the treatment arms, according to the company. This included the overall rates of major adverse cardiac events (MACE), although there were some small differences in the individual components of this score, a combination of CV deaths, nonfatal myocardial infarction, and nonfatal strokes. (There were two CV deaths among those aclidinium, and no deaths among those on placebo.) The rate of anticholinergic effects was low, which the company said might be due to low systemic exposure to the drug.
Cardiovascular safety of the drug is an issue because of some concerns raised by data on other inhaled anticholinergics over the past several years.
The panel voted 10 to 3 with 1 abstention that that the safety had been adequately evaluated. But panelists, including those who supported approval, still had concerns about cardiovascular safety and recommended that these concerns should be evaluated further in a postmarketing study.
The company plans to conduct a double-blind, randomized parallel postmarketing 3-year study of 4,000 patients with COPD, with a history of COPD exacerbations during the previous year, randomized to 400 mcg aclidinium or placebo plus standard of care, which will compare rates of moderate to severe COPD exacerbations within the first year.
The primary safety end point will be time to first MACE event; the study will also evaluate the rates of other serious cardiac events, conduction disorders and cerebrovascular disorders.
The FDA’s deadline for making an approval decision is April 23. If approved, Forest plans to market it as "Tudorza Pressair."
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
SILVER SPRING, MD. – The majority of a Food and Drug Administration advisory panel on Feb. 23 recommended that aclidinium bromide, an inhaled long-acting anticholinergic bronchodilator, should be approved as a treatment for chronic obstructive pulmonary disease, with a postmarketing study that evaluates the cardiovascular safety of the drug.
The FDA’s Pulmonary-Allergy Drugs Advisory Committee voted 12 to 2 that the efficacy and safety data from clinical trials provided "substantial evidence" to support approval of aclidinium, at a dose of 400 mcg twice a day administered in a breath-actuated dry powder inhaler, for the long-term maintenance treatment of bronchospasm associated with COPD, including chronic bronchitis and emphysema.
The panel unanimously agreed that the data in the clinical trials presented by the manufacturer provided evidence that this dose provided a clinically meaningful benefit in patients, citing the significant increases in trough forced expiratory volume in 1 second (FEV1) from baseline (the primary efficacy end point) among those treated with 400 mcg twice a day, compared with those on placebo after 12 weeks of treatment in clinical studies. Some panelists pointed out that there was also some evidence of improvements in COPD exacerbations and in patient-reported symptoms on a patient questionnaire, which were secondary end points.
If approved, it will be the second long-acting inhaled anticholinergic agent for COPD on the U.S. market, following tiotropium bromide (Spiriva HandiHaler), approved for COPD in 2004, but taken once a day. Ipratropium bromide, a short-acting inhaled anticholinergic, was approved in 1986 for COPD.
Clinical data presented by the manufacturer Forest Laboratories Inc., included two pivotal 12-week studies of almost 1,400 outpatients, most of whom were white and had stable moderate to severe COPD (mean age about 64 years). Patients received 400 mcg or 200 mcg of aclidinium twice a day or placebo. About half were still smoking; people with clinically significant cardiovascular disease were excluded. They were allowed to continue treatment with short-acting bronchodilators, inhaled corticosteroids, long-acting theophylline, oxygen for 15 hours or less, and stable-dose prednisone.
Overall, cardiovascular events were not common and were evenly distributed among the treatment arms, according to the company. This included the overall rates of major adverse cardiac events (MACE), although there were some small differences in the individual components of this score, a combination of CV deaths, nonfatal myocardial infarction, and nonfatal strokes. (There were two CV deaths among those aclidinium, and no deaths among those on placebo.) The rate of anticholinergic effects was low, which the company said might be due to low systemic exposure to the drug.
Cardiovascular safety of the drug is an issue because of some concerns raised by data on other inhaled anticholinergics over the past several years.
The panel voted 10 to 3 with 1 abstention that that the safety had been adequately evaluated. But panelists, including those who supported approval, still had concerns about cardiovascular safety and recommended that these concerns should be evaluated further in a postmarketing study.
The company plans to conduct a double-blind, randomized parallel postmarketing 3-year study of 4,000 patients with COPD, with a history of COPD exacerbations during the previous year, randomized to 400 mcg aclidinium or placebo plus standard of care, which will compare rates of moderate to severe COPD exacerbations within the first year.
The primary safety end point will be time to first MACE event; the study will also evaluate the rates of other serious cardiac events, conduction disorders and cerebrovascular disorders.
The FDA’s deadline for making an approval decision is April 23. If approved, Forest plans to market it as "Tudorza Pressair."
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
FROM A MEETING OF THE FDA'S PULMONARY-ALLERGY DRUGS ADVISORY COMMITTEE
FDA Panel Endorses Obesity Drug Qnexa After All
SILVER SPRING, MD. – The combination formulation of phentermine and topiramate should be approved as a weight-loss treatment, with a risk-management plan that addresses the teratogenic effects of topiramate and a postmarketing study that evaluates cardiovascular outcomes associated with treatment, the majority of a Food and Drug Administration panel agreed at a meeting on Feb. 22.
The FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted 20 to 2 that the benefit-risk profile of the phentermine-topiramate combination supported its approval for the treatment of obesity in people with a body mass index (BMI) of at least 30 kg/m2, or those with a BMI of at least 27 kg/m2 who also have weight-related comorbidities. The manufacturer, Vivus, has proposed that the combination product – in three fixed-dose combinations – be approved for this population, in combination with diet and exercise.
If approved, this will be the first new obesity drug treatment approved in 13 years and will be marketed as Qnexa by Vivus. The combination product contains an immediate-release formulation of phentermine, which is a sympathomimetic amine approved for short-term weight loss, on the market in the United States since 1959; and a controlled-release formulation of topiramate, an antiepileptic drug approved for treating epilepsy in 1996, for migraine prophylaxis in 2004, and for pediatric epilepsy in 2011. Qnexa is manufactured in three fixed-dose combinations: the starting low dose of 3.75 mg of phentermine and 23 mg of topiramate, the recommended dose of 7.5 mg/46 mg, and the highest dose (15 mg/92 mg) for patients not reaching their weight-loss goal.
The two separate components are available at higher doses than those contained in the combination product.
At a meeting in July 2010, the same panel had agreed that the same product had been shown to be effective as a weight-loss agent, compared with placebo in two 1-year, pivotal studies in this patient population, but the majority voted against recommending approval of the agent because of concerns over the risk-benefit profile, particularly the potential for teratogenicity and increases in heart rate associated with treatment.
The FDA advised the company in October 2010 that the cardiovascular risks and teratogenic potential associated with treatment had not been adequately assessed, and requested that the company provide evidence that an increase in heart rate (a mean of 1.6 beats/minute at the highest dose) did not increase the risk for major adverse cardiovascular events, further evaluate the potential risk for oral clefts associated with prenatal exposure to the topiramate component, provide 2-year data, and develop a Risk Evaluation and Mitigation Strategy (REMS).
Components of the REMS as now planned include a patient medication guide explaining the risk of oral clefts (cleft lip with or without cleft palate) associated with first trimester exposure in studies and pregnancy registries, a certified pharmacy network that dispenses a month’s supply at a time via mail order, and a plan to educate prescribers about the teratogenic risk and train them to prescribe the drug appropriately. The product would be a category X drug, contraindicated during pregnancy, with the recommendation to immediately stop taking the drug if a woman becomes pregnant during treatment.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
SILVER SPRING, MD. – The combination formulation of phentermine and topiramate should be approved as a weight-loss treatment, with a risk-management plan that addresses the teratogenic effects of topiramate and a postmarketing study that evaluates cardiovascular outcomes associated with treatment, the majority of a Food and Drug Administration panel agreed at a meeting on Feb. 22.
The FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted 20 to 2 that the benefit-risk profile of the phentermine-topiramate combination supported its approval for the treatment of obesity in people with a body mass index (BMI) of at least 30 kg/m2, or those with a BMI of at least 27 kg/m2 who also have weight-related comorbidities. The manufacturer, Vivus, has proposed that the combination product – in three fixed-dose combinations – be approved for this population, in combination with diet and exercise.
If approved, this will be the first new obesity drug treatment approved in 13 years and will be marketed as Qnexa by Vivus. The combination product contains an immediate-release formulation of phentermine, which is a sympathomimetic amine approved for short-term weight loss, on the market in the United States since 1959; and a controlled-release formulation of topiramate, an antiepileptic drug approved for treating epilepsy in 1996, for migraine prophylaxis in 2004, and for pediatric epilepsy in 2011. Qnexa is manufactured in three fixed-dose combinations: the starting low dose of 3.75 mg of phentermine and 23 mg of topiramate, the recommended dose of 7.5 mg/46 mg, and the highest dose (15 mg/92 mg) for patients not reaching their weight-loss goal.
The two separate components are available at higher doses than those contained in the combination product.
At a meeting in July 2010, the same panel had agreed that the same product had been shown to be effective as a weight-loss agent, compared with placebo in two 1-year, pivotal studies in this patient population, but the majority voted against recommending approval of the agent because of concerns over the risk-benefit profile, particularly the potential for teratogenicity and increases in heart rate associated with treatment.
The FDA advised the company in October 2010 that the cardiovascular risks and teratogenic potential associated with treatment had not been adequately assessed, and requested that the company provide evidence that an increase in heart rate (a mean of 1.6 beats/minute at the highest dose) did not increase the risk for major adverse cardiovascular events, further evaluate the potential risk for oral clefts associated with prenatal exposure to the topiramate component, provide 2-year data, and develop a Risk Evaluation and Mitigation Strategy (REMS).
Components of the REMS as now planned include a patient medication guide explaining the risk of oral clefts (cleft lip with or without cleft palate) associated with first trimester exposure in studies and pregnancy registries, a certified pharmacy network that dispenses a month’s supply at a time via mail order, and a plan to educate prescribers about the teratogenic risk and train them to prescribe the drug appropriately. The product would be a category X drug, contraindicated during pregnancy, with the recommendation to immediately stop taking the drug if a woman becomes pregnant during treatment.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
SILVER SPRING, MD. – The combination formulation of phentermine and topiramate should be approved as a weight-loss treatment, with a risk-management plan that addresses the teratogenic effects of topiramate and a postmarketing study that evaluates cardiovascular outcomes associated with treatment, the majority of a Food and Drug Administration panel agreed at a meeting on Feb. 22.
The FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted 20 to 2 that the benefit-risk profile of the phentermine-topiramate combination supported its approval for the treatment of obesity in people with a body mass index (BMI) of at least 30 kg/m2, or those with a BMI of at least 27 kg/m2 who also have weight-related comorbidities. The manufacturer, Vivus, has proposed that the combination product – in three fixed-dose combinations – be approved for this population, in combination with diet and exercise.
If approved, this will be the first new obesity drug treatment approved in 13 years and will be marketed as Qnexa by Vivus. The combination product contains an immediate-release formulation of phentermine, which is a sympathomimetic amine approved for short-term weight loss, on the market in the United States since 1959; and a controlled-release formulation of topiramate, an antiepileptic drug approved for treating epilepsy in 1996, for migraine prophylaxis in 2004, and for pediatric epilepsy in 2011. Qnexa is manufactured in three fixed-dose combinations: the starting low dose of 3.75 mg of phentermine and 23 mg of topiramate, the recommended dose of 7.5 mg/46 mg, and the highest dose (15 mg/92 mg) for patients not reaching their weight-loss goal.
The two separate components are available at higher doses than those contained in the combination product.
At a meeting in July 2010, the same panel had agreed that the same product had been shown to be effective as a weight-loss agent, compared with placebo in two 1-year, pivotal studies in this patient population, but the majority voted against recommending approval of the agent because of concerns over the risk-benefit profile, particularly the potential for teratogenicity and increases in heart rate associated with treatment.
The FDA advised the company in October 2010 that the cardiovascular risks and teratogenic potential associated with treatment had not been adequately assessed, and requested that the company provide evidence that an increase in heart rate (a mean of 1.6 beats/minute at the highest dose) did not increase the risk for major adverse cardiovascular events, further evaluate the potential risk for oral clefts associated with prenatal exposure to the topiramate component, provide 2-year data, and develop a Risk Evaluation and Mitigation Strategy (REMS).
Components of the REMS as now planned include a patient medication guide explaining the risk of oral clefts (cleft lip with or without cleft palate) associated with first trimester exposure in studies and pregnancy registries, a certified pharmacy network that dispenses a month’s supply at a time via mail order, and a plan to educate prescribers about the teratogenic risk and train them to prescribe the drug appropriately. The product would be a category X drug, contraindicated during pregnancy, with the recommendation to immediately stop taking the drug if a woman becomes pregnant during treatment.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
FROM A MEETING OF THE FDA'S ENDOCRINOLOGIC AND METABOLIC DRUGS ADVISORY COMMITTEE
The Oncology Report Guide to Cancer Drugs and Devices in 2011
From an immunotherapy that improves melanoma survival to a device that zaps brain tumors, 2011 was a year of firsts from the FDA. This is our second annual guide to approvals of new cancer drugs, indications, and devices – along with a review of safety warnings and other actions affecting oncology practices.
(Click here for a PDF version of The Oncology Report Guide to Cancer Drugs and Devices in 2011.)
NEW DRUG APPROVALS
• Abiraterone acetate (Zytiga Tablets, Centocor Ortho Biotech, Inc.). An androgen suppressant that decreases testosterone production for use in combination with prednisone for the treatment of patients with metastatic castration-resistant prostate cancer (mCRPC) who have received prior chemotherapy containing docetaxel. The fourth approval in late-stage prostate cancer since April 2010.
Basis: An international study of 1,195 men who had previously received chemotherapy containing docetaxel. Median overall survival was 14.8 months with abiraterone plus prednisone, vs. 10.9 months with prednisone alone.

• Asparaginase Erwinia chrysanthemi [Erwinaze for injection, EUSA Pharma (USA), Inc.] An asparagine-specific enzyme approved as a component of a multiagent chemotherapeutic regimen for the treatment of patients with acute lymphoblastic leukemia (ALL) who developed hypersensitivity to E. coli-derived asparaginase. An orphan drug, it works by the same mechanism as the two previously approved treatments that block asparagine, a protein necessary for the proliferation of neoplastic cells.
Basis: In a single-arm, multicenter, open-label study of 58 patients, all 48 patients with available samples achieved threshold trough asparaginase levels shown to correlate with asparagine depletion and with serum levels that predict clinical efficacy.
• Brentuximab vedotin (Adcetris for injection, Seattle Genetics, Inc.). A CD30-directed antibody-drug conjugate for the treatment of Hodgkin’s lymphoma after failure of autologous stem cell transplant (ASCT) or at least two prior multiagent chemotherapy regimens in patients who are not ASCT candidates, and for the treatment of systemic anaplastic large cell lymphoma (ALCL) after failure of at least one prior multiagent chemotherapy regimen. The first new treatment approved for Hodgkin’s since 1977 and the first treatment approved for ALCL.
Basis: In a single-arm study of 102 patients treated with brentuximab, 73% had either a complete or partial response. In a similarly designed study of 58 patients with ALCL, 86% achieved a partial or complete response.
Addendum: An accelerated approval, based on clinical data suggesting that treatment resulted in significant responses; conversion to full approval is contingent on confirmation of benefits in follow-up studies. (In January 2012, the FDA added a warning about the risk of a rare brain infection to the label.)
• Crizotinib (Xalkori Capsules, Pfizer, Inc.). A kinase inhibitor for the treatment of locally advanced or metastatic non–small cell lung cancer (NSCLC) that express an abnormal anaplastic lymphoma kinase (ALK) gene, as detected by the Vysis ALK Break-Apart FISH Probe Kit (Abbott Molecular, Inc.), a companion diagnostic test approved at the same time.
Basis: In two single-arm studies of 255 patients with locally advanced or metastatic, ALK-positive NSCLC, most of whom had received prior chemotherapy, the overall response rate was 50% and 61%, with a median response duration of 42 and 48 weeks, respectively.
Addendum: An accelerated approval, so conversion to full approval is contingent on follow-up studies confirming clinical benefit.
• Deferiprone (Ferriprox Tablets, ApoPharma, Inc.). An oral iron chelator for the treatment of patients with transfusional iron overload due to thalassemia syndromes when current chelation therapy is inadequate.
Basis: A prospective pooled analysis of previously conducted studies of 236 patients (most had thalassemia syndromes) with transfusion-dependent iron overload, for which previous iron chelation therapy had failed or was considered inadequate because of poor tolerance. Within 1 year of starting treatment, 50% of the patients had at least a 20% decrease in serum ferritin levels.
Addendum: An accelerated approval. Safety and effectiveness not established for the treatment of transfusional iron overload due to other chronic anemias, including sickle cell disease. As a condition of approval, ApoPharma will conduct a study of safety and efficacy in patients with sickle cell disease and transfusional iron overload not adequately treated with available chelating agents.
• Hemacord (New York Blood Center). A hematopoietic progenitor cells–cord (HPC-C) preparation for allogenic hematopoietic stem cell transplantation. The first approval of a license for an umbilical cord blood therapy.
Basis: Safety and effectiveness data submitted to a public docket and data submitted in the license application demonstrating compliance with other regulatory requirements.
• Ipilimumab injection (Yervoy, Bristol-Myers Squibb). A human cytotoxic T-lymphocyte antigen (CTLA-4)-blocking antibody for the treatment of unresectable or metastatic melanoma. The first treatment proven to prolong survival in patients with metastatic melanoma.
Basis: An international study of 676 patients with previously treated unresectable stage III or IV melanoma whose disease had progressed, comparing ipilimumab plus an experimental tumor vaccine (gp100), gp100 alone, and ipilimumab alone. Median overall survival was 10 months among those who received ipilimumab with or without the vaccine, vs. 6 months among those who received the vaccine alone.
Addendum: Approved with a Risk Evaluation and Mitigation Strategy (REMS) program addressing life-threatening toxicity and deaths associated with ipilimumab in the study. Acts slowly, and some patients may have tumor progression before therapy becomes effective.
• Ruxolitinib (Jakafi oral tablets, Incyte Corp.). A Janus-associated kinase (JAK) inhibitor for the treatment of intermediate- or high-risk myelofibrosis, including primary myelofibrosis, post–polycythemia vera myelofibrosis, and post–essential thrombocythemia myelofibrosis. An orphan drug that is the first treatment approved for the rare blood disease – and the first approved JAK inhibitor.
Basis: Two randomized controlled trials of 528 patients with intermediate- or high-risk myelofibrosis. In one study, 42% of those on ruxolitinib had more than at least a 35% reduction in spleen volume vs. 1% of those on placebo at 24 weeks, and nearly half of those on treatment had achieved at least a 50% reduction in myelofibrosis symptoms, vs. 5% of those on placebo. In the second study, 29% of those on ruxolitinib had at least a 35% reduction in spleen size, vs. none of those on best available therapy after 48 weeks.
• Vandetanib (Caprelsa, AstraZeneca Pharmaceuticals LP). A kinase inhibitor for the treatment of symptomatic or progressive medullary thyroid cancer in adults with unresectable, locally advanced, or metastatic disease. The first drug approved for this rare cancer.
Basis: An international study of 331 patients with unresectable locally advanced or metastatic medullary thyroid cancer. Median progression-free survival was at least 22.6 months with vandetanib vs. 16.4 months among with placebo, a highly significant difference.
Addendum: Approved with a REMS addressing the risk of QT interval prolongation and a note that use in patients with indolent, asymptomatic, or slowly progressing disease "should be carefully considered" because of treatment-associated risks, as well as a boxed warning and a restricted distribution program.
• Vemurafenib (Zelboraf tablets, Hoffmann-LaRoche, Inc.). A BRAF inhibitor for the treatment of patients with unresectable or metastatic melanoma with the BRAFV600E mutation, as detected by the cobas 4800 BRAF V600 Mutation Test (Roche Molecular Systems), a companion diagnostic test approved at the same time. The second therapy found to prolong survival in patients with melanoma.
Basis: A study of 675 treatment-naive patients with unresectable or metastatic melanoma, positive for the BRAFV600E mutation. At the time of review, median survival was 7.9 months in a control group treated with dacarbazine, but had not been reached among those on vemurafenib. Median progression-free survival was 1.6 and 5.3 months, respectively.
Addendum: About half of melanoma patients have the BRAF mutation, and about half of them respond to vemurafenib. Approved with a medication guide to inform patients about the need for the diagnostic test and potential risks associated with treatment, including cutaneous squamous cell carcinoma. Roche and ipilimumab manufacturer Bristol-Meyers Squibb announced collaboration on a phase I/II study to determine whether combining the two agents is safe and effective in patients with BRAF mutations.
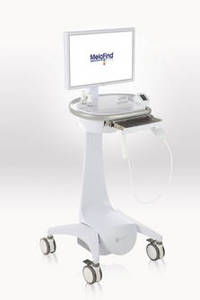
NEW DEVICES
• MelaFind (Mela Sciences). A noninvasive melanoma detection device for use "on clinically atypical cutaneous pigmented lesions with one or more clinical or historical characteristics of melanoma, excluding those with a clinical diagnosis of melanoma or likely melanoma."
Basis: The road to approval was controversial, with a split vote from an advisory panel, a citizens petition submitted by the manufacturer, and an agreement between the agency and manufacturer on how the device would be used and by whom.
• NovoTTF-100A System (NovoTTF, Novocure). A portable, battery-powered device that delivers electrical fields to the brain as a treatment for adults with glioblastoma multiforme that has recurred after chemotherapy.
Basis: In a randomized study of 237 patients whose GBM had recurred after surgery, radiation, and chemotherapy, median overall survival was 6.3 months with the approximately 6-pound device and 6.4 months with best available chemotherapy. The agency cited evidence suggesting better quality of life without chemotherapy side effects.
NEW INDICATIONS
• Cetuximab (Erbitux, ImClone LLC, a wholly owned subsidiary of Eli Lilly & Co. and Bristol-Myers Squibb). The epidermal growth factor receptor (EGFR) antagonist in combination with platinum-based therapy plus 5-fluorouracil for the first-line treatment of recurrent locoregional and/or metastatic squamous cell carcinoma of the head and neck.
Basis: A multicenter non-U.S. study of 442 patients not suitable for potentially curative treatment with surgery or radiation. Mean overall survival was 10.1 months with cetuximab and chemotherapy (cisplatin or carboplatin and 5-fluorouracil), vs. 7.4 months with chemotherapy alone.
Addendum: Cetuximab available in the United States provides about 22% greater exposure than the European Union–approved cetuximab used in this study, but the study results, pharmacokinetic data, and other clinical trial data "establish the efficacy" of cetuximab at the dose recommended, according to the prescribing information.
• Denosumab (Prolia, Amgen, Inc.). The RANK ligand (RANKL) inhibitor to increase bone mass in patients at high risk for fracture when receiving androgen deprivation therapy (ADT) for nonmetastatic prostate cancer or adjuvant aromatase inhibitor (AI) therapy for breast cancer.
Basis: Two international, randomized, double-blind placebo-controlled studies. In one study of 1,468 men with nonmetastatic prostate cancer receiving ADT, lumbar spine bone mineral density (BMD) at 2 years was significantly higher with denosumab vs. placebo (6.7% difference). At 3 years of treatment, the incidence of new vertebral fractures was 1.5% with denosumab vs. 3.9% with placebo, a risk reduction of 62%. In a study of 252 postmenopausal women with breast cancer on AI treatment, BMD at the lumbar spine was significantly higher with denosumab vs. placebo after 12 months of treatment (5.5% difference).
Addendum: Denosumab was initially approved in 2010. It is marketed as Prolia for treating postmenopausal women with osteoporosis at high risk of fracture, and as Xgeva for preventing skeletal-related events in patients with bone metastases from solid tumors; Xgeva is administered more frequently and at a higher dose for the latter indication.
• Everolimus (Afinitor Tablets, Novartis Pharmaceuticals Corp.). The mammalian target of rapamycin (mTOR) inhibitor for progressive pancreatic neuroendocrine tumors (pNETs) that are unresectable, locally advanced, or metastatic. (Safety and effectiveness of everolimus for treating carcinoid tumors have not been established). The first new drug in about 30 years for the treatment of advanced pNET.
Basis: A randomized controlled study of 410 patients, all of whom also received best supportive care. Median progression-free survival was 11 months with everolimus, vs. 4.6 months with placebo. In an interim analysis, overall survival was not different between the two groups.
• Peg-interferon alfa-2b (Sylatron, Schering Corp.). An alpha interferon for the adjuvant treatment of melanoma with microscopic or gross nodal involvement within 84 days of definitive surgical resection, including complete lymphadenectomy.
Basis: An open-label multicenter study of 1,256 patients. Median relapse-free survival was 34.8 months among those treated with peg-interferon alfa-2b, vs. 25.5 months with observation.
• Rituximab (Rituxan, Genentech, Inc.). The CD20-directed cytolytic antibody for maintenance therapy in people with previously untreated follicular, CD-20 positive, B-cell non-Hodgkin lymphoma who achieve a response to rituximab in combination with chemotherapy.
Basis: An international study of 1,217 previously untreated patients with advanced follicular lymphoma; it randomized 1,018 patients who had a complete or partial response with rituximab and chemotherapy. Maintenance treatment with rituximab reduced the risk of progression by 46% compared with observation only.
• Sunitinib (Sutent Capsules, Pfizer, Inc.). A kinase inhibitor for the treatment of progressive, well-differentiated pancreatic neuroendocrine tumors (pNETs) in patients with unresectable, locally advanced, or metastatic disease; it was approved soon after everolimus for this indication.
Basis: A randomized controlled study of 171 patients with unresectable, locally advanced or well-differentiated pNET; treatment with somatostatin analogues was allowed. Median progression-free survival was 10.2 months with sunitinib, vs. 5.4 months with placebo.
WARNINGS AND LABEL CHANGES
• Bevacizumab (Avastin, Genentech, Inc.). Revised prescribing information includes the risk of ovarian failure in premenopausal women treated with bevacizumab and chemotherapy; postmarketing reports of osteonecrosis of the jaw in patients treated with bevacizumab but not bisphosphonates; and information from a clinical trial about the risk of venous thromboembolic events and bleeding in patients receiving anticoagulation therapy after a first VTE event while receiving bevacizumab.
• Dasatinib (Sprycel, Bristol-Myers Squibb). Information on increased risk for pulmonary arterial hypertension added to prescribing information.
• Fentanyl. A classwide risk management program approved for all transmucosal immediate-release fentanyl formulations.
• IV methotrexate and proton pump inhibitors (PPIs). A warning added to the methotrexate label advises caution when high-dose methotrexate is administered to patients taking a PPI, because concomitant administration may result in elevated and prolonged serum levels of methotrexate and/or its metabolite hydroxymethotrexate, with possible toxic effects.
• Lenalidomide (Revlimid, Celgene). Advisory informed the public that the FDA is monitoring the risk of secondary cancers in patients treated for myelodysplastic syndromes or multiple myeloma.
• Ondansetron (Zofran, GlaxoSmithKline, and generic formulations). Following a review of information on the risk of QT prolongation, label now recommends ECG monitoring in patients who are treated with ondansetron and have an electrolyte abnormality, congestive heart failure, or bradyarrhythmias; and in patients taking other medications that prolong the QT interval – who are at an increased risk of developing torsades de pointes. Label recommends against use in patients with congenital long QT syndrome. Safety review ongoing.
• Opioids. Draft issued for opioid prescriber education program, as part of the Risk Evaluation and Mitigation Strategy (REMS) now in place for brand-name and generic long-acting and extended-release opioids.
• Pioglitazone. Bladder cancer warning risk added to the label for type 2 diabetes drug marketed as Actos (or in combination with metformin as Actoplus Met, and with glimepiride as Duetact).
• Romiplostim (Nplate for subcutaneous injection, Amgen, Inc.) and eltrombopag (Promacta tablets, GlaxoSmithKline LLC). Elements of the REMS for the two thrombopoietin receptor agonists were dropped, including the restricted distribution program that required health care professionals, hospitals, and patients to be enrolled in order to prescribe, dispense, or receive these drugs. Clinicians no longer need to file periodic safety forms for their patients on these treatments.
• Tumor necrosis factor (TNF) blockers. Manufacturers asked to report cases of malignancies in children, adolescents, and adults aged 30 years and younger treated with TNF blockers and to conduct in-depth follow-up of them.
OTHER ACTIONS
• Bevacizumab (Avastin, Genentech, Inc.). FDA Commissioner Margaret Hamburg withdrew bevacizumab’s indication in metastatic breast cancer. She cited a lack of data establishing benefit and serious risks associated with treatment. The decision followed a July hearing, where the Oncologic Drugs Advisory Committee unanimously voted that the indication be withdrawn despite emotional testimony from patients who said they benefited from the monoclonal antibody.
• Drug shortages. A new rule requires some manufacturers to give early warning of shortages. The agency also held a workshop on the crisis and is working to avert and/or resolve shortages.
• Office of Oncology Products. Many New Drug Applications, Biologic License Applications, and Investigational New Drug Applications for cancer drugs are being reassigned to new review divisions as part of a reorganization.
From an immunotherapy that improves melanoma survival to a device that zaps brain tumors, 2011 was a year of firsts from the FDA. This is our second annual guide to approvals of new cancer drugs, indications, and devices – along with a review of safety warnings and other actions affecting oncology practices.
(Click here for a PDF version of The Oncology Report Guide to Cancer Drugs and Devices in 2011.)
NEW DRUG APPROVALS
• Abiraterone acetate (Zytiga Tablets, Centocor Ortho Biotech, Inc.). An androgen suppressant that decreases testosterone production for use in combination with prednisone for the treatment of patients with metastatic castration-resistant prostate cancer (mCRPC) who have received prior chemotherapy containing docetaxel. The fourth approval in late-stage prostate cancer since April 2010.
Basis: An international study of 1,195 men who had previously received chemotherapy containing docetaxel. Median overall survival was 14.8 months with abiraterone plus prednisone, vs. 10.9 months with prednisone alone.
• Asparaginase Erwinia chrysanthemi [Erwinaze for injection, EUSA Pharma (USA), Inc.] An asparagine-specific enzyme approved as a component of a multiagent chemotherapeutic regimen for the treatment of patients with acute lymphoblastic leukemia (ALL) who developed hypersensitivity to E. coli-derived asparaginase. An orphan drug, it works by the same mechanism as the two previously approved treatments that block asparagine, a protein necessary for the proliferation of neoplastic cells.
Basis: In a single-arm, multicenter, open-label study of 58 patients, all 48 patients with available samples achieved threshold trough asparaginase levels shown to correlate with asparagine depletion and with serum levels that predict clinical efficacy.
• Brentuximab vedotin (Adcetris for injection, Seattle Genetics, Inc.). A CD30-directed antibody-drug conjugate for the treatment of Hodgkin’s lymphoma after failure of autologous stem cell transplant (ASCT) or at least two prior multiagent chemotherapy regimens in patients who are not ASCT candidates, and for the treatment of systemic anaplastic large cell lymphoma (ALCL) after failure of at least one prior multiagent chemotherapy regimen. The first new treatment approved for Hodgkin’s since 1977 and the first treatment approved for ALCL.
Basis: In a single-arm study of 102 patients treated with brentuximab, 73% had either a complete or partial response. In a similarly designed study of 58 patients with ALCL, 86% achieved a partial or complete response.
Addendum: An accelerated approval, based on clinical data suggesting that treatment resulted in significant responses; conversion to full approval is contingent on confirmation of benefits in follow-up studies. (In January 2012, the FDA added a warning about the risk of a rare brain infection to the label.)
• Crizotinib (Xalkori Capsules, Pfizer, Inc.). A kinase inhibitor for the treatment of locally advanced or metastatic non–small cell lung cancer (NSCLC) that express an abnormal anaplastic lymphoma kinase (ALK) gene, as detected by the Vysis ALK Break-Apart FISH Probe Kit (Abbott Molecular, Inc.), a companion diagnostic test approved at the same time.
Basis: In two single-arm studies of 255 patients with locally advanced or metastatic, ALK-positive NSCLC, most of whom had received prior chemotherapy, the overall response rate was 50% and 61%, with a median response duration of 42 and 48 weeks, respectively.
Addendum: An accelerated approval, so conversion to full approval is contingent on follow-up studies confirming clinical benefit.
• Deferiprone (Ferriprox Tablets, ApoPharma, Inc.). An oral iron chelator for the treatment of patients with transfusional iron overload due to thalassemia syndromes when current chelation therapy is inadequate.
Basis: A prospective pooled analysis of previously conducted studies of 236 patients (most had thalassemia syndromes) with transfusion-dependent iron overload, for which previous iron chelation therapy had failed or was considered inadequate because of poor tolerance. Within 1 year of starting treatment, 50% of the patients had at least a 20% decrease in serum ferritin levels.
Addendum: An accelerated approval. Safety and effectiveness not established for the treatment of transfusional iron overload due to other chronic anemias, including sickle cell disease. As a condition of approval, ApoPharma will conduct a study of safety and efficacy in patients with sickle cell disease and transfusional iron overload not adequately treated with available chelating agents.
• Hemacord (New York Blood Center). A hematopoietic progenitor cells–cord (HPC-C) preparation for allogenic hematopoietic stem cell transplantation. The first approval of a license for an umbilical cord blood therapy.
Basis: Safety and effectiveness data submitted to a public docket and data submitted in the license application demonstrating compliance with other regulatory requirements.
• Ipilimumab injection (Yervoy, Bristol-Myers Squibb). A human cytotoxic T-lymphocyte antigen (CTLA-4)-blocking antibody for the treatment of unresectable or metastatic melanoma. The first treatment proven to prolong survival in patients with metastatic melanoma.
Basis: An international study of 676 patients with previously treated unresectable stage III or IV melanoma whose disease had progressed, comparing ipilimumab plus an experimental tumor vaccine (gp100), gp100 alone, and ipilimumab alone. Median overall survival was 10 months among those who received ipilimumab with or without the vaccine, vs. 6 months among those who received the vaccine alone.
Addendum: Approved with a Risk Evaluation and Mitigation Strategy (REMS) program addressing life-threatening toxicity and deaths associated with ipilimumab in the study. Acts slowly, and some patients may have tumor progression before therapy becomes effective.
• Ruxolitinib (Jakafi oral tablets, Incyte Corp.). A Janus-associated kinase (JAK) inhibitor for the treatment of intermediate- or high-risk myelofibrosis, including primary myelofibrosis, post–polycythemia vera myelofibrosis, and post–essential thrombocythemia myelofibrosis. An orphan drug that is the first treatment approved for the rare blood disease – and the first approved JAK inhibitor.
Basis: Two randomized controlled trials of 528 patients with intermediate- or high-risk myelofibrosis. In one study, 42% of those on ruxolitinib had more than at least a 35% reduction in spleen volume vs. 1% of those on placebo at 24 weeks, and nearly half of those on treatment had achieved at least a 50% reduction in myelofibrosis symptoms, vs. 5% of those on placebo. In the second study, 29% of those on ruxolitinib had at least a 35% reduction in spleen size, vs. none of those on best available therapy after 48 weeks.
• Vandetanib (Caprelsa, AstraZeneca Pharmaceuticals LP). A kinase inhibitor for the treatment of symptomatic or progressive medullary thyroid cancer in adults with unresectable, locally advanced, or metastatic disease. The first drug approved for this rare cancer.
Basis: An international study of 331 patients with unresectable locally advanced or metastatic medullary thyroid cancer. Median progression-free survival was at least 22.6 months with vandetanib vs. 16.4 months among with placebo, a highly significant difference.
Addendum: Approved with a REMS addressing the risk of QT interval prolongation and a note that use in patients with indolent, asymptomatic, or slowly progressing disease "should be carefully considered" because of treatment-associated risks, as well as a boxed warning and a restricted distribution program.
• Vemurafenib (Zelboraf tablets, Hoffmann-LaRoche, Inc.). A BRAF inhibitor for the treatment of patients with unresectable or metastatic melanoma with the BRAFV600E mutation, as detected by the cobas 4800 BRAF V600 Mutation Test (Roche Molecular Systems), a companion diagnostic test approved at the same time. The second therapy found to prolong survival in patients with melanoma.
Basis: A study of 675 treatment-naive patients with unresectable or metastatic melanoma, positive for the BRAFV600E mutation. At the time of review, median survival was 7.9 months in a control group treated with dacarbazine, but had not been reached among those on vemurafenib. Median progression-free survival was 1.6 and 5.3 months, respectively.
Addendum: About half of melanoma patients have the BRAF mutation, and about half of them respond to vemurafenib. Approved with a medication guide to inform patients about the need for the diagnostic test and potential risks associated with treatment, including cutaneous squamous cell carcinoma. Roche and ipilimumab manufacturer Bristol-Meyers Squibb announced collaboration on a phase I/II study to determine whether combining the two agents is safe and effective in patients with BRAF mutations.
NEW DEVICES
• MelaFind (Mela Sciences). A noninvasive melanoma detection device for use "on clinically atypical cutaneous pigmented lesions with one or more clinical or historical characteristics of melanoma, excluding those with a clinical diagnosis of melanoma or likely melanoma."
Basis: The road to approval was controversial, with a split vote from an advisory panel, a citizens petition submitted by the manufacturer, and an agreement between the agency and manufacturer on how the device would be used and by whom.
• NovoTTF-100A System (NovoTTF, Novocure). A portable, battery-powered device that delivers electrical fields to the brain as a treatment for adults with glioblastoma multiforme that has recurred after chemotherapy.
Basis: In a randomized study of 237 patients whose GBM had recurred after surgery, radiation, and chemotherapy, median overall survival was 6.3 months with the approximately 6-pound device and 6.4 months with best available chemotherapy. The agency cited evidence suggesting better quality of life without chemotherapy side effects.
NEW INDICATIONS
• Cetuximab (Erbitux, ImClone LLC, a wholly owned subsidiary of Eli Lilly & Co. and Bristol-Myers Squibb). The epidermal growth factor receptor (EGFR) antagonist in combination with platinum-based therapy plus 5-fluorouracil for the first-line treatment of recurrent locoregional and/or metastatic squamous cell carcinoma of the head and neck.
Basis: A multicenter non-U.S. study of 442 patients not suitable for potentially curative treatment with surgery or radiation. Mean overall survival was 10.1 months with cetuximab and chemotherapy (cisplatin or carboplatin and 5-fluorouracil), vs. 7.4 months with chemotherapy alone.
Addendum: Cetuximab available in the United States provides about 22% greater exposure than the European Union–approved cetuximab used in this study, but the study results, pharmacokinetic data, and other clinical trial data "establish the efficacy" of cetuximab at the dose recommended, according to the prescribing information.
• Denosumab (Prolia, Amgen, Inc.). The RANK ligand (RANKL) inhibitor to increase bone mass in patients at high risk for fracture when receiving androgen deprivation therapy (ADT) for nonmetastatic prostate cancer or adjuvant aromatase inhibitor (AI) therapy for breast cancer.
Basis: Two international, randomized, double-blind placebo-controlled studies. In one study of 1,468 men with nonmetastatic prostate cancer receiving ADT, lumbar spine bone mineral density (BMD) at 2 years was significantly higher with denosumab vs. placebo (6.7% difference). At 3 years of treatment, the incidence of new vertebral fractures was 1.5% with denosumab vs. 3.9% with placebo, a risk reduction of 62%. In a study of 252 postmenopausal women with breast cancer on AI treatment, BMD at the lumbar spine was significantly higher with denosumab vs. placebo after 12 months of treatment (5.5% difference).
Addendum: Denosumab was initially approved in 2010. It is marketed as Prolia for treating postmenopausal women with osteoporosis at high risk of fracture, and as Xgeva for preventing skeletal-related events in patients with bone metastases from solid tumors; Xgeva is administered more frequently and at a higher dose for the latter indication.
• Everolimus (Afinitor Tablets, Novartis Pharmaceuticals Corp.). The mammalian target of rapamycin (mTOR) inhibitor for progressive pancreatic neuroendocrine tumors (pNETs) that are unresectable, locally advanced, or metastatic. (Safety and effectiveness of everolimus for treating carcinoid tumors have not been established). The first new drug in about 30 years for the treatment of advanced pNET.
Basis: A randomized controlled study of 410 patients, all of whom also received best supportive care. Median progression-free survival was 11 months with everolimus, vs. 4.6 months with placebo. In an interim analysis, overall survival was not different between the two groups.
• Peg-interferon alfa-2b (Sylatron, Schering Corp.). An alpha interferon for the adjuvant treatment of melanoma with microscopic or gross nodal involvement within 84 days of definitive surgical resection, including complete lymphadenectomy.
Basis: An open-label multicenter study of 1,256 patients. Median relapse-free survival was 34.8 months among those treated with peg-interferon alfa-2b, vs. 25.5 months with observation.
• Rituximab (Rituxan, Genentech, Inc.). The CD20-directed cytolytic antibody for maintenance therapy in people with previously untreated follicular, CD-20 positive, B-cell non-Hodgkin lymphoma who achieve a response to rituximab in combination with chemotherapy.
Basis: An international study of 1,217 previously untreated patients with advanced follicular lymphoma; it randomized 1,018 patients who had a complete or partial response with rituximab and chemotherapy. Maintenance treatment with rituximab reduced the risk of progression by 46% compared with observation only.
• Sunitinib (Sutent Capsules, Pfizer, Inc.). A kinase inhibitor for the treatment of progressive, well-differentiated pancreatic neuroendocrine tumors (pNETs) in patients with unresectable, locally advanced, or metastatic disease; it was approved soon after everolimus for this indication.
Basis: A randomized controlled study of 171 patients with unresectable, locally advanced or well-differentiated pNET; treatment with somatostatin analogues was allowed. Median progression-free survival was 10.2 months with sunitinib, vs. 5.4 months with placebo.
WARNINGS AND LABEL CHANGES
• Bevacizumab (Avastin, Genentech, Inc.). Revised prescribing information includes the risk of ovarian failure in premenopausal women treated with bevacizumab and chemotherapy; postmarketing reports of osteonecrosis of the jaw in patients treated with bevacizumab but not bisphosphonates; and information from a clinical trial about the risk of venous thromboembolic events and bleeding in patients receiving anticoagulation therapy after a first VTE event while receiving bevacizumab.
• Dasatinib (Sprycel, Bristol-Myers Squibb). Information on increased risk for pulmonary arterial hypertension added to prescribing information.
• Fentanyl. A classwide risk management program approved for all transmucosal immediate-release fentanyl formulations.
• IV methotrexate and proton pump inhibitors (PPIs). A warning added to the methotrexate label advises caution when high-dose methotrexate is administered to patients taking a PPI, because concomitant administration may result in elevated and prolonged serum levels of methotrexate and/or its metabolite hydroxymethotrexate, with possible toxic effects.
• Lenalidomide (Revlimid, Celgene). Advisory informed the public that the FDA is monitoring the risk of secondary cancers in patients treated for myelodysplastic syndromes or multiple myeloma.
• Ondansetron (Zofran, GlaxoSmithKline, and generic formulations). Following a review of information on the risk of QT prolongation, label now recommends ECG monitoring in patients who are treated with ondansetron and have an electrolyte abnormality, congestive heart failure, or bradyarrhythmias; and in patients taking other medications that prolong the QT interval – who are at an increased risk of developing torsades de pointes. Label recommends against use in patients with congenital long QT syndrome. Safety review ongoing.
• Opioids. Draft issued for opioid prescriber education program, as part of the Risk Evaluation and Mitigation Strategy (REMS) now in place for brand-name and generic long-acting and extended-release opioids.
• Pioglitazone. Bladder cancer warning risk added to the label for type 2 diabetes drug marketed as Actos (or in combination with metformin as Actoplus Met, and with glimepiride as Duetact).
• Romiplostim (Nplate for subcutaneous injection, Amgen, Inc.) and eltrombopag (Promacta tablets, GlaxoSmithKline LLC). Elements of the REMS for the two thrombopoietin receptor agonists were dropped, including the restricted distribution program that required health care professionals, hospitals, and patients to be enrolled in order to prescribe, dispense, or receive these drugs. Clinicians no longer need to file periodic safety forms for their patients on these treatments.
• Tumor necrosis factor (TNF) blockers. Manufacturers asked to report cases of malignancies in children, adolescents, and adults aged 30 years and younger treated with TNF blockers and to conduct in-depth follow-up of them.
OTHER ACTIONS
• Bevacizumab (Avastin, Genentech, Inc.). FDA Commissioner Margaret Hamburg withdrew bevacizumab’s indication in metastatic breast cancer. She cited a lack of data establishing benefit and serious risks associated with treatment. The decision followed a July hearing, where the Oncologic Drugs Advisory Committee unanimously voted that the indication be withdrawn despite emotional testimony from patients who said they benefited from the monoclonal antibody.
• Drug shortages. A new rule requires some manufacturers to give early warning of shortages. The agency also held a workshop on the crisis and is working to avert and/or resolve shortages.
• Office of Oncology Products. Many New Drug Applications, Biologic License Applications, and Investigational New Drug Applications for cancer drugs are being reassigned to new review divisions as part of a reorganization.
From an immunotherapy that improves melanoma survival to a device that zaps brain tumors, 2011 was a year of firsts from the FDA. This is our second annual guide to approvals of new cancer drugs, indications, and devices – along with a review of safety warnings and other actions affecting oncology practices.
(Click here for a PDF version of The Oncology Report Guide to Cancer Drugs and Devices in 2011.)
NEW DRUG APPROVALS
• Abiraterone acetate (Zytiga Tablets, Centocor Ortho Biotech, Inc.). An androgen suppressant that decreases testosterone production for use in combination with prednisone for the treatment of patients with metastatic castration-resistant prostate cancer (mCRPC) who have received prior chemotherapy containing docetaxel. The fourth approval in late-stage prostate cancer since April 2010.
Basis: An international study of 1,195 men who had previously received chemotherapy containing docetaxel. Median overall survival was 14.8 months with abiraterone plus prednisone, vs. 10.9 months with prednisone alone.
• Asparaginase Erwinia chrysanthemi [Erwinaze for injection, EUSA Pharma (USA), Inc.] An asparagine-specific enzyme approved as a component of a multiagent chemotherapeutic regimen for the treatment of patients with acute lymphoblastic leukemia (ALL) who developed hypersensitivity to E. coli-derived asparaginase. An orphan drug, it works by the same mechanism as the two previously approved treatments that block asparagine, a protein necessary for the proliferation of neoplastic cells.
Basis: In a single-arm, multicenter, open-label study of 58 patients, all 48 patients with available samples achieved threshold trough asparaginase levels shown to correlate with asparagine depletion and with serum levels that predict clinical efficacy.
• Brentuximab vedotin (Adcetris for injection, Seattle Genetics, Inc.). A CD30-directed antibody-drug conjugate for the treatment of Hodgkin’s lymphoma after failure of autologous stem cell transplant (ASCT) or at least two prior multiagent chemotherapy regimens in patients who are not ASCT candidates, and for the treatment of systemic anaplastic large cell lymphoma (ALCL) after failure of at least one prior multiagent chemotherapy regimen. The first new treatment approved for Hodgkin’s since 1977 and the first treatment approved for ALCL.
Basis: In a single-arm study of 102 patients treated with brentuximab, 73% had either a complete or partial response. In a similarly designed study of 58 patients with ALCL, 86% achieved a partial or complete response.
Addendum: An accelerated approval, based on clinical data suggesting that treatment resulted in significant responses; conversion to full approval is contingent on confirmation of benefits in follow-up studies. (In January 2012, the FDA added a warning about the risk of a rare brain infection to the label.)
• Crizotinib (Xalkori Capsules, Pfizer, Inc.). A kinase inhibitor for the treatment of locally advanced or metastatic non–small cell lung cancer (NSCLC) that express an abnormal anaplastic lymphoma kinase (ALK) gene, as detected by the Vysis ALK Break-Apart FISH Probe Kit (Abbott Molecular, Inc.), a companion diagnostic test approved at the same time.
Basis: In two single-arm studies of 255 patients with locally advanced or metastatic, ALK-positive NSCLC, most of whom had received prior chemotherapy, the overall response rate was 50% and 61%, with a median response duration of 42 and 48 weeks, respectively.
Addendum: An accelerated approval, so conversion to full approval is contingent on follow-up studies confirming clinical benefit.
• Deferiprone (Ferriprox Tablets, ApoPharma, Inc.). An oral iron chelator for the treatment of patients with transfusional iron overload due to thalassemia syndromes when current chelation therapy is inadequate.
Basis: A prospective pooled analysis of previously conducted studies of 236 patients (most had thalassemia syndromes) with transfusion-dependent iron overload, for which previous iron chelation therapy had failed or was considered inadequate because of poor tolerance. Within 1 year of starting treatment, 50% of the patients had at least a 20% decrease in serum ferritin levels.
Addendum: An accelerated approval. Safety and effectiveness not established for the treatment of transfusional iron overload due to other chronic anemias, including sickle cell disease. As a condition of approval, ApoPharma will conduct a study of safety and efficacy in patients with sickle cell disease and transfusional iron overload not adequately treated with available chelating agents.
• Hemacord (New York Blood Center). A hematopoietic progenitor cells–cord (HPC-C) preparation for allogenic hematopoietic stem cell transplantation. The first approval of a license for an umbilical cord blood therapy.
Basis: Safety and effectiveness data submitted to a public docket and data submitted in the license application demonstrating compliance with other regulatory requirements.
• Ipilimumab injection (Yervoy, Bristol-Myers Squibb). A human cytotoxic T-lymphocyte antigen (CTLA-4)-blocking antibody for the treatment of unresectable or metastatic melanoma. The first treatment proven to prolong survival in patients with metastatic melanoma.
Basis: An international study of 676 patients with previously treated unresectable stage III or IV melanoma whose disease had progressed, comparing ipilimumab plus an experimental tumor vaccine (gp100), gp100 alone, and ipilimumab alone. Median overall survival was 10 months among those who received ipilimumab with or without the vaccine, vs. 6 months among those who received the vaccine alone.
Addendum: Approved with a Risk Evaluation and Mitigation Strategy (REMS) program addressing life-threatening toxicity and deaths associated with ipilimumab in the study. Acts slowly, and some patients may have tumor progression before therapy becomes effective.
• Ruxolitinib (Jakafi oral tablets, Incyte Corp.). A Janus-associated kinase (JAK) inhibitor for the treatment of intermediate- or high-risk myelofibrosis, including primary myelofibrosis, post–polycythemia vera myelofibrosis, and post–essential thrombocythemia myelofibrosis. An orphan drug that is the first treatment approved for the rare blood disease – and the first approved JAK inhibitor.
Basis: Two randomized controlled trials of 528 patients with intermediate- or high-risk myelofibrosis. In one study, 42% of those on ruxolitinib had more than at least a 35% reduction in spleen volume vs. 1% of those on placebo at 24 weeks, and nearly half of those on treatment had achieved at least a 50% reduction in myelofibrosis symptoms, vs. 5% of those on placebo. In the second study, 29% of those on ruxolitinib had at least a 35% reduction in spleen size, vs. none of those on best available therapy after 48 weeks.
• Vandetanib (Caprelsa, AstraZeneca Pharmaceuticals LP). A kinase inhibitor for the treatment of symptomatic or progressive medullary thyroid cancer in adults with unresectable, locally advanced, or metastatic disease. The first drug approved for this rare cancer.
Basis: An international study of 331 patients with unresectable locally advanced or metastatic medullary thyroid cancer. Median progression-free survival was at least 22.6 months with vandetanib vs. 16.4 months among with placebo, a highly significant difference.
Addendum: Approved with a REMS addressing the risk of QT interval prolongation and a note that use in patients with indolent, asymptomatic, or slowly progressing disease "should be carefully considered" because of treatment-associated risks, as well as a boxed warning and a restricted distribution program.
• Vemurafenib (Zelboraf tablets, Hoffmann-LaRoche, Inc.). A BRAF inhibitor for the treatment of patients with unresectable or metastatic melanoma with the BRAFV600E mutation, as detected by the cobas 4800 BRAF V600 Mutation Test (Roche Molecular Systems), a companion diagnostic test approved at the same time. The second therapy found to prolong survival in patients with melanoma.
Basis: A study of 675 treatment-naive patients with unresectable or metastatic melanoma, positive for the BRAFV600E mutation. At the time of review, median survival was 7.9 months in a control group treated with dacarbazine, but had not been reached among those on vemurafenib. Median progression-free survival was 1.6 and 5.3 months, respectively.
Addendum: About half of melanoma patients have the BRAF mutation, and about half of them respond to vemurafenib. Approved with a medication guide to inform patients about the need for the diagnostic test and potential risks associated with treatment, including cutaneous squamous cell carcinoma. Roche and ipilimumab manufacturer Bristol-Meyers Squibb announced collaboration on a phase I/II study to determine whether combining the two agents is safe and effective in patients with BRAF mutations.
NEW DEVICES
• MelaFind (Mela Sciences). A noninvasive melanoma detection device for use "on clinically atypical cutaneous pigmented lesions with one or more clinical or historical characteristics of melanoma, excluding those with a clinical diagnosis of melanoma or likely melanoma."
Basis: The road to approval was controversial, with a split vote from an advisory panel, a citizens petition submitted by the manufacturer, and an agreement between the agency and manufacturer on how the device would be used and by whom.
• NovoTTF-100A System (NovoTTF, Novocure). A portable, battery-powered device that delivers electrical fields to the brain as a treatment for adults with glioblastoma multiforme that has recurred after chemotherapy.
Basis: In a randomized study of 237 patients whose GBM had recurred after surgery, radiation, and chemotherapy, median overall survival was 6.3 months with the approximately 6-pound device and 6.4 months with best available chemotherapy. The agency cited evidence suggesting better quality of life without chemotherapy side effects.
NEW INDICATIONS
• Cetuximab (Erbitux, ImClone LLC, a wholly owned subsidiary of Eli Lilly & Co. and Bristol-Myers Squibb). The epidermal growth factor receptor (EGFR) antagonist in combination with platinum-based therapy plus 5-fluorouracil for the first-line treatment of recurrent locoregional and/or metastatic squamous cell carcinoma of the head and neck.
Basis: A multicenter non-U.S. study of 442 patients not suitable for potentially curative treatment with surgery or radiation. Mean overall survival was 10.1 months with cetuximab and chemotherapy (cisplatin or carboplatin and 5-fluorouracil), vs. 7.4 months with chemotherapy alone.
Addendum: Cetuximab available in the United States provides about 22% greater exposure than the European Union–approved cetuximab used in this study, but the study results, pharmacokinetic data, and other clinical trial data "establish the efficacy" of cetuximab at the dose recommended, according to the prescribing information.
• Denosumab (Prolia, Amgen, Inc.). The RANK ligand (RANKL) inhibitor to increase bone mass in patients at high risk for fracture when receiving androgen deprivation therapy (ADT) for nonmetastatic prostate cancer or adjuvant aromatase inhibitor (AI) therapy for breast cancer.
Basis: Two international, randomized, double-blind placebo-controlled studies. In one study of 1,468 men with nonmetastatic prostate cancer receiving ADT, lumbar spine bone mineral density (BMD) at 2 years was significantly higher with denosumab vs. placebo (6.7% difference). At 3 years of treatment, the incidence of new vertebral fractures was 1.5% with denosumab vs. 3.9% with placebo, a risk reduction of 62%. In a study of 252 postmenopausal women with breast cancer on AI treatment, BMD at the lumbar spine was significantly higher with denosumab vs. placebo after 12 months of treatment (5.5% difference).
Addendum: Denosumab was initially approved in 2010. It is marketed as Prolia for treating postmenopausal women with osteoporosis at high risk of fracture, and as Xgeva for preventing skeletal-related events in patients with bone metastases from solid tumors; Xgeva is administered more frequently and at a higher dose for the latter indication.
• Everolimus (Afinitor Tablets, Novartis Pharmaceuticals Corp.). The mammalian target of rapamycin (mTOR) inhibitor for progressive pancreatic neuroendocrine tumors (pNETs) that are unresectable, locally advanced, or metastatic. (Safety and effectiveness of everolimus for treating carcinoid tumors have not been established). The first new drug in about 30 years for the treatment of advanced pNET.
Basis: A randomized controlled study of 410 patients, all of whom also received best supportive care. Median progression-free survival was 11 months with everolimus, vs. 4.6 months with placebo. In an interim analysis, overall survival was not different between the two groups.
• Peg-interferon alfa-2b (Sylatron, Schering Corp.). An alpha interferon for the adjuvant treatment of melanoma with microscopic or gross nodal involvement within 84 days of definitive surgical resection, including complete lymphadenectomy.
Basis: An open-label multicenter study of 1,256 patients. Median relapse-free survival was 34.8 months among those treated with peg-interferon alfa-2b, vs. 25.5 months with observation.
• Rituximab (Rituxan, Genentech, Inc.). The CD20-directed cytolytic antibody for maintenance therapy in people with previously untreated follicular, CD-20 positive, B-cell non-Hodgkin lymphoma who achieve a response to rituximab in combination with chemotherapy.
Basis: An international study of 1,217 previously untreated patients with advanced follicular lymphoma; it randomized 1,018 patients who had a complete or partial response with rituximab and chemotherapy. Maintenance treatment with rituximab reduced the risk of progression by 46% compared with observation only.
• Sunitinib (Sutent Capsules, Pfizer, Inc.). A kinase inhibitor for the treatment of progressive, well-differentiated pancreatic neuroendocrine tumors (pNETs) in patients with unresectable, locally advanced, or metastatic disease; it was approved soon after everolimus for this indication.
Basis: A randomized controlled study of 171 patients with unresectable, locally advanced or well-differentiated pNET; treatment with somatostatin analogues was allowed. Median progression-free survival was 10.2 months with sunitinib, vs. 5.4 months with placebo.
WARNINGS AND LABEL CHANGES
• Bevacizumab (Avastin, Genentech, Inc.). Revised prescribing information includes the risk of ovarian failure in premenopausal women treated with bevacizumab and chemotherapy; postmarketing reports of osteonecrosis of the jaw in patients treated with bevacizumab but not bisphosphonates; and information from a clinical trial about the risk of venous thromboembolic events and bleeding in patients receiving anticoagulation therapy after a first VTE event while receiving bevacizumab.
• Dasatinib (Sprycel, Bristol-Myers Squibb). Information on increased risk for pulmonary arterial hypertension added to prescribing information.
• Fentanyl. A classwide risk management program approved for all transmucosal immediate-release fentanyl formulations.
• IV methotrexate and proton pump inhibitors (PPIs). A warning added to the methotrexate label advises caution when high-dose methotrexate is administered to patients taking a PPI, because concomitant administration may result in elevated and prolonged serum levels of methotrexate and/or its metabolite hydroxymethotrexate, with possible toxic effects.
• Lenalidomide (Revlimid, Celgene). Advisory informed the public that the FDA is monitoring the risk of secondary cancers in patients treated for myelodysplastic syndromes or multiple myeloma.
• Ondansetron (Zofran, GlaxoSmithKline, and generic formulations). Following a review of information on the risk of QT prolongation, label now recommends ECG monitoring in patients who are treated with ondansetron and have an electrolyte abnormality, congestive heart failure, or bradyarrhythmias; and in patients taking other medications that prolong the QT interval – who are at an increased risk of developing torsades de pointes. Label recommends against use in patients with congenital long QT syndrome. Safety review ongoing.
• Opioids. Draft issued for opioid prescriber education program, as part of the Risk Evaluation and Mitigation Strategy (REMS) now in place for brand-name and generic long-acting and extended-release opioids.
• Pioglitazone. Bladder cancer warning risk added to the label for type 2 diabetes drug marketed as Actos (or in combination with metformin as Actoplus Met, and with glimepiride as Duetact).
• Romiplostim (Nplate for subcutaneous injection, Amgen, Inc.) and eltrombopag (Promacta tablets, GlaxoSmithKline LLC). Elements of the REMS for the two thrombopoietin receptor agonists were dropped, including the restricted distribution program that required health care professionals, hospitals, and patients to be enrolled in order to prescribe, dispense, or receive these drugs. Clinicians no longer need to file periodic safety forms for their patients on these treatments.
• Tumor necrosis factor (TNF) blockers. Manufacturers asked to report cases of malignancies in children, adolescents, and adults aged 30 years and younger treated with TNF blockers and to conduct in-depth follow-up of them.
OTHER ACTIONS
• Bevacizumab (Avastin, Genentech, Inc.). FDA Commissioner Margaret Hamburg withdrew bevacizumab’s indication in metastatic breast cancer. She cited a lack of data establishing benefit and serious risks associated with treatment. The decision followed a July hearing, where the Oncologic Drugs Advisory Committee unanimously voted that the indication be withdrawn despite emotional testimony from patients who said they benefited from the monoclonal antibody.
• Drug shortages. A new rule requires some manufacturers to give early warning of shortages. The agency also held a workshop on the crisis and is working to avert and/or resolve shortages.
• Office of Oncology Products. Many New Drug Applications, Biologic License Applications, and Investigational New Drug Applications for cancer drugs are being reassigned to new review divisions as part of a reorganization.
MRI Inflammation 'Marginally' Predicts AS Bone Growth
Evidence of inflammation on spinal MRIs was "marginally predictive" of new syndesmophyte formation 2 years later at the vertebral sites where inflammation was observed, in a study of over 100 patients with ankylosing spondylitis.
However, growth of syndesmophytes that were present at vertebral sites at baseline was not associated with evidence of inflammation on MRIs, reported Dr. Désirée van der Heijde, of Leiden University Medical Center, Leiden, the Netherlands, and her associates, in the March issue of the Annals of the Rheumatic Diseases (Ann. Rheum. Dis. 2012;71:369-73).
"MRI inflammation in a vertebral unit slightly increases the likelihood of finding a new syndesmophyte in the same vertebral unit 2 years later, but does not predict the growth of already existing syndesmophytes," they wrote. The finding that most of the syndesmophytes grew in the vertebral units with no MRI evidence of inflammation suggests that "the relationship between MRI inflammation and syndesmophyte formation is not straightforward," they added.
The goal of the study was to address the association between inflammation of a vertebral unit (defined as "the region between two virtual lines through the middle of each vertebra") on MRI and the subsequent formation and growth of syndesmophytes, the excessive bone formation that represents the structural damage seen in ankylosing spondylitis (AS). The processes behind the formation of syndesmophytes in AS are not well understood and are not inhibited by treatment with tumor necrosis factor (TNF) blockers, despite the dramatic effects these agents have on inflammation, they noted.
They used data that included spinal MRI activity scores, from a random sample of people enrolled in ASSERT (AS Study for the Evaluation of Recombinant Infliximab Therapy), a 24-week study published in 2005 that compared infliximab to placebo in patients with active AS and followed patients in a open extension period up to 102 weeks, during which time everyone was treated with infliximab (Arthritis Rheum. 2005;52:582-91).
They analyzed information from 1,827 to 2,070 vertebral units from 177 to 183 patients, (the numbers varied, depending on the case definition and the reader of the MRI). MRI images of all 23 vertebral units of the spine – C2 to S1 – were scored on a scale of 0 to 3 (the presence of inflammation in a vertebral unit was defined as a score greater than 0), and a score from 4 to 6 if erosions were also evident.
After adjusting for possible confounders, the risk of developing a syndesmophyte in a vertebral unit with MRI evidence of inflammation was increased 1.5- to 2-fold, a statistically significant increase, they said.
Depending on the syndesmophyte definition used, the reader of the MRI, and the MRI case definition, 6%-32% of new syndesmophytes developed at the vertebral sites with evidence of active inflammation and 68%-94% of new syndesmophytes developed at the vertebral sites that had no evidence of active inflammation. Similar findings were observed for the syndesmophytes that were present at baseline: 0%-30% of the syndesmophytes that were present at baseline and grew were at sites of active inflammation, and 70%-100% of the syndesmophytes that grew "did so in vertebral units without inflammation," they reported.
"The subtle association between MRI activity and new syndesmophytes is in conflict with the absence of an effect of TNF blockers on structural damage," the authors wrote. One explanation they proposed was that the formation of syndesmophytes is "a post-inflammatory repair reaction that may only be inhibited if a TNF blocker is started early before inflammation gives way to repair."
Based on the finding that evidence of inflammation at the vertebral unit "only marginally predicts" the formation of new syndesmophytes at that site, "if inflammation is indeed the principle trigger of repair responses, a strong case can be made for early and aggressive anti-inflammatory treatment," they speculated. However, they added, "if inflammation and repair are independent pathways triggered by common factors, new therapies targeting the pathologically enhanced repair responses need to be developed."
One of the eight authors was supported by a grant from the Fundação para a Ciência e a Tecnologia (FCT); otherwise, the authors had no disclosures to report.
In an accompanying editorial titled, "Inflammation and Ankylosis: still an enigmatic relationship in spondyloarthritis," Dr. Rik Lories and Dr. Maxime Dougados referred to TNF-blocker therapy’s apparent lack of an effect on the structural progression of disease in patients with spondyloarthritis as a "clinical conundrum," noting that anti-TNF therapy slows progression in rheumatoid arthritis.
One hypothesis that has been proposed to address this issue is that inflammation, including TNF upregulation, triggers local damage and repair, which results in the formation of new bone and ankylosis, "establishing a causal coupling between inflammation" and the ankylosing process. Another hypothesis is that inflammation and new bone formation are caused by a "common trigger," but "further develop in a largely molecular independent way."
"Taken together, recent MRI studies on the structural progression of disease in spondyloarthritis and its relation to inflammation are another important step forward in our understanding of a disease that was neglected by most of the rheumatology research community for too long," they wrote, adding: "The complex nature of the disease and the many factors that have recently been implicated in playing a role in its onset and progression indicate that further research is needed" (Ann. Rheum. Dis. 2012;71:317-8).
Dr. Lories is K. U. Leuven is research professor at Universiteit Leuven (Belgium) and Dr. Dougados is professor of rheumatology at René Descartes University in Paris. They said they had no disclosures.
In an accompanying editorial titled, "Inflammation and Ankylosis: still an enigmatic relationship in spondyloarthritis," Dr. Rik Lories and Dr. Maxime Dougados referred to TNF-blocker therapy’s apparent lack of an effect on the structural progression of disease in patients with spondyloarthritis as a "clinical conundrum," noting that anti-TNF therapy slows progression in rheumatoid arthritis.
One hypothesis that has been proposed to address this issue is that inflammation, including TNF upregulation, triggers local damage and repair, which results in the formation of new bone and ankylosis, "establishing a causal coupling between inflammation" and the ankylosing process. Another hypothesis is that inflammation and new bone formation are caused by a "common trigger," but "further develop in a largely molecular independent way."
"Taken together, recent MRI studies on the structural progression of disease in spondyloarthritis and its relation to inflammation are another important step forward in our understanding of a disease that was neglected by most of the rheumatology research community for too long," they wrote, adding: "The complex nature of the disease and the many factors that have recently been implicated in playing a role in its onset and progression indicate that further research is needed" (Ann. Rheum. Dis. 2012;71:317-8).
Dr. Lories is K. U. Leuven is research professor at Universiteit Leuven (Belgium) and Dr. Dougados is professor of rheumatology at René Descartes University in Paris. They said they had no disclosures.
In an accompanying editorial titled, "Inflammation and Ankylosis: still an enigmatic relationship in spondyloarthritis," Dr. Rik Lories and Dr. Maxime Dougados referred to TNF-blocker therapy’s apparent lack of an effect on the structural progression of disease in patients with spondyloarthritis as a "clinical conundrum," noting that anti-TNF therapy slows progression in rheumatoid arthritis.
One hypothesis that has been proposed to address this issue is that inflammation, including TNF upregulation, triggers local damage and repair, which results in the formation of new bone and ankylosis, "establishing a causal coupling between inflammation" and the ankylosing process. Another hypothesis is that inflammation and new bone formation are caused by a "common trigger," but "further develop in a largely molecular independent way."
"Taken together, recent MRI studies on the structural progression of disease in spondyloarthritis and its relation to inflammation are another important step forward in our understanding of a disease that was neglected by most of the rheumatology research community for too long," they wrote, adding: "The complex nature of the disease and the many factors that have recently been implicated in playing a role in its onset and progression indicate that further research is needed" (Ann. Rheum. Dis. 2012;71:317-8).
Dr. Lories is K. U. Leuven is research professor at Universiteit Leuven (Belgium) and Dr. Dougados is professor of rheumatology at René Descartes University in Paris. They said they had no disclosures.
Evidence of inflammation on spinal MRIs was "marginally predictive" of new syndesmophyte formation 2 years later at the vertebral sites where inflammation was observed, in a study of over 100 patients with ankylosing spondylitis.
However, growth of syndesmophytes that were present at vertebral sites at baseline was not associated with evidence of inflammation on MRIs, reported Dr. Désirée van der Heijde, of Leiden University Medical Center, Leiden, the Netherlands, and her associates, in the March issue of the Annals of the Rheumatic Diseases (Ann. Rheum. Dis. 2012;71:369-73).
"MRI inflammation in a vertebral unit slightly increases the likelihood of finding a new syndesmophyte in the same vertebral unit 2 years later, but does not predict the growth of already existing syndesmophytes," they wrote. The finding that most of the syndesmophytes grew in the vertebral units with no MRI evidence of inflammation suggests that "the relationship between MRI inflammation and syndesmophyte formation is not straightforward," they added.
The goal of the study was to address the association between inflammation of a vertebral unit (defined as "the region between two virtual lines through the middle of each vertebra") on MRI and the subsequent formation and growth of syndesmophytes, the excessive bone formation that represents the structural damage seen in ankylosing spondylitis (AS). The processes behind the formation of syndesmophytes in AS are not well understood and are not inhibited by treatment with tumor necrosis factor (TNF) blockers, despite the dramatic effects these agents have on inflammation, they noted.
They used data that included spinal MRI activity scores, from a random sample of people enrolled in ASSERT (AS Study for the Evaluation of Recombinant Infliximab Therapy), a 24-week study published in 2005 that compared infliximab to placebo in patients with active AS and followed patients in a open extension period up to 102 weeks, during which time everyone was treated with infliximab (Arthritis Rheum. 2005;52:582-91).
They analyzed information from 1,827 to 2,070 vertebral units from 177 to 183 patients, (the numbers varied, depending on the case definition and the reader of the MRI). MRI images of all 23 vertebral units of the spine – C2 to S1 – were scored on a scale of 0 to 3 (the presence of inflammation in a vertebral unit was defined as a score greater than 0), and a score from 4 to 6 if erosions were also evident.
After adjusting for possible confounders, the risk of developing a syndesmophyte in a vertebral unit with MRI evidence of inflammation was increased 1.5- to 2-fold, a statistically significant increase, they said.
Depending on the syndesmophyte definition used, the reader of the MRI, and the MRI case definition, 6%-32% of new syndesmophytes developed at the vertebral sites with evidence of active inflammation and 68%-94% of new syndesmophytes developed at the vertebral sites that had no evidence of active inflammation. Similar findings were observed for the syndesmophytes that were present at baseline: 0%-30% of the syndesmophytes that were present at baseline and grew were at sites of active inflammation, and 70%-100% of the syndesmophytes that grew "did so in vertebral units without inflammation," they reported.
"The subtle association between MRI activity and new syndesmophytes is in conflict with the absence of an effect of TNF blockers on structural damage," the authors wrote. One explanation they proposed was that the formation of syndesmophytes is "a post-inflammatory repair reaction that may only be inhibited if a TNF blocker is started early before inflammation gives way to repair."
Based on the finding that evidence of inflammation at the vertebral unit "only marginally predicts" the formation of new syndesmophytes at that site, "if inflammation is indeed the principle trigger of repair responses, a strong case can be made for early and aggressive anti-inflammatory treatment," they speculated. However, they added, "if inflammation and repair are independent pathways triggered by common factors, new therapies targeting the pathologically enhanced repair responses need to be developed."
One of the eight authors was supported by a grant from the Fundação para a Ciência e a Tecnologia (FCT); otherwise, the authors had no disclosures to report.
Evidence of inflammation on spinal MRIs was "marginally predictive" of new syndesmophyte formation 2 years later at the vertebral sites where inflammation was observed, in a study of over 100 patients with ankylosing spondylitis.
However, growth of syndesmophytes that were present at vertebral sites at baseline was not associated with evidence of inflammation on MRIs, reported Dr. Désirée van der Heijde, of Leiden University Medical Center, Leiden, the Netherlands, and her associates, in the March issue of the Annals of the Rheumatic Diseases (Ann. Rheum. Dis. 2012;71:369-73).
"MRI inflammation in a vertebral unit slightly increases the likelihood of finding a new syndesmophyte in the same vertebral unit 2 years later, but does not predict the growth of already existing syndesmophytes," they wrote. The finding that most of the syndesmophytes grew in the vertebral units with no MRI evidence of inflammation suggests that "the relationship between MRI inflammation and syndesmophyte formation is not straightforward," they added.
The goal of the study was to address the association between inflammation of a vertebral unit (defined as "the region between two virtual lines through the middle of each vertebra") on MRI and the subsequent formation and growth of syndesmophytes, the excessive bone formation that represents the structural damage seen in ankylosing spondylitis (AS). The processes behind the formation of syndesmophytes in AS are not well understood and are not inhibited by treatment with tumor necrosis factor (TNF) blockers, despite the dramatic effects these agents have on inflammation, they noted.
They used data that included spinal MRI activity scores, from a random sample of people enrolled in ASSERT (AS Study for the Evaluation of Recombinant Infliximab Therapy), a 24-week study published in 2005 that compared infliximab to placebo in patients with active AS and followed patients in a open extension period up to 102 weeks, during which time everyone was treated with infliximab (Arthritis Rheum. 2005;52:582-91).
They analyzed information from 1,827 to 2,070 vertebral units from 177 to 183 patients, (the numbers varied, depending on the case definition and the reader of the MRI). MRI images of all 23 vertebral units of the spine – C2 to S1 – were scored on a scale of 0 to 3 (the presence of inflammation in a vertebral unit was defined as a score greater than 0), and a score from 4 to 6 if erosions were also evident.
After adjusting for possible confounders, the risk of developing a syndesmophyte in a vertebral unit with MRI evidence of inflammation was increased 1.5- to 2-fold, a statistically significant increase, they said.
Depending on the syndesmophyte definition used, the reader of the MRI, and the MRI case definition, 6%-32% of new syndesmophytes developed at the vertebral sites with evidence of active inflammation and 68%-94% of new syndesmophytes developed at the vertebral sites that had no evidence of active inflammation. Similar findings were observed for the syndesmophytes that were present at baseline: 0%-30% of the syndesmophytes that were present at baseline and grew were at sites of active inflammation, and 70%-100% of the syndesmophytes that grew "did so in vertebral units without inflammation," they reported.
"The subtle association between MRI activity and new syndesmophytes is in conflict with the absence of an effect of TNF blockers on structural damage," the authors wrote. One explanation they proposed was that the formation of syndesmophytes is "a post-inflammatory repair reaction that may only be inhibited if a TNF blocker is started early before inflammation gives way to repair."
Based on the finding that evidence of inflammation at the vertebral unit "only marginally predicts" the formation of new syndesmophytes at that site, "if inflammation is indeed the principle trigger of repair responses, a strong case can be made for early and aggressive anti-inflammatory treatment," they speculated. However, they added, "if inflammation and repair are independent pathways triggered by common factors, new therapies targeting the pathologically enhanced repair responses need to be developed."
One of the eight authors was supported by a grant from the Fundação para a Ciência e a Tecnologia (FCT); otherwise, the authors had no disclosures to report.
FROM ANNALS OF THE RHEUMATIC DISEASES
Major Finding: Evidence of inflammation on spinal MRIs of patients with ankylosing spondylitis (AS) was associated with a 1.5- to 2-fold increased risk of new syndesmophyte formation at the vertebral sites of inflammation 2 years later, which was statistically significant. But the growth of syndesmophytes that were present at vertebral sites at baseline was not associated with evidence of inflammation on MRIs.
Data Source: A study using a random sample of patients enrolled in ASSERT (AS Study for the Evaluation of Recombinant Infliximab Therapy), a randomized controlled study published in 2005 that compared infliximab to placebo in patients with active AS for 24 weeks, then followed patients until 102 weeks, during which time everyone was treated with infliximab.
Disclosures: One of the eight authors was supported by a grant from the Fundação para a Ciência e a Tecnologia (FCT); otherwise, the authors had no disclosures.
FDA Panel: Pain Patch Not Yet Approvable for HIV Neuropathy
SILVER SPRING, MD. – A Food and Drug Administration advisory panel that met on Feb. 9 did not support approval of a capsaicin-containing transdermal patch as a treatment for pain associated with HIV neuropathy, because product studies had not provided adequate and compelling evidence of its efficacy for treating the condition.
At the meeting, the FDA’s Anesthetic and Analgesic Drug Products Advisory Committee voted 12 to 0 that data from two studies of about 800 people did not provide "substantial evidence of effectiveness" of the 8% capsaicin patch for the management of neuropathic pain related to HIV-associated peripheral neuropathy. While panelists agreed that the risks associated with treatment were low, the panel also voted 11 to 0, with 1 abstention, that, based on the available data, the risk-benefit profile was not acceptable for approval.
Marketed as Qutenza by San Francisco–based NeurogesX, the 8% capsaicin patch was approved in the United States in November 2009 for the treatment of neuropathic pain associated with postherpetic neuralgia, and has been approved in the European Union for the treatment of peripheral neuropathic pain in nondiabetic adults.
The proposed new indication is for the use of the patch, applied for 30 minutes, for the management of neuropathic pain related to HIV-associated peripheral neuropathy. The patch is applied to the foot after a lidocaine cream is applied to the skin; it cannot be reapplied until at least 3 months later. The patch delivers capsaicin – the most abundant component of chili peppers – to the site of pain, where it desensitizes hyperactive nociceptors, according to the company.
Panelists expressed regret that they could not support expanded approval because of the unmet need for this type of treatment for these patients – and noted some evidence that it was effective in one of the two studies presented by the company. But they agreed that the evidence did not meet the FDA’s standards for approval.
Treatment with the 8% capsaicin patch was compared to a low-dose 0.04% capsaicin patch as the control in two 12-week randomized studies of mostly white, middle-aged men with documented HIV, who had experienced HIV peripheral neuropathy for at least 2 months, with moderate to severe pain. Almost 70% of the participants were on other analgesics at baseline, including opioids.
The primary outcome end point evaluating effectiveness was the percent change in the mean daily scores in a pain rating scale from 2 to 12 weeks after the patch was applied. In the first study, which compared the effects of the 8% capsaicin patch and the control patch applied for 30, 60, or 90 minutes, there was some evidence that the 8% patch was more effective than the control patch, but most of that evidence was from post hoc analyses. In the second study, which compared the 8% patch to the low-dose patch applied for 30 or 60 minutes, there were no statistically significant reductions in pain relief at 12 weeks, compared with the control patch.
Overall, the safety profile of the 8% patch in these trials was similar to that seen in patients with postherpetic neuralgia. The most common adverse events associated with treatment were application site reactions, affecting 76% of those who had the higher-dose patch and about 50% of those who had the low-dose control patch.
One of the panelists, Dr. Thomas Giordano, an HIV specialist at Baylor College of Medicine and medical director of HIV services, Harris County Hospital District, both in Houston, said it was difficult to vote against approval, because he believed that some of his patients could benefit from this treatment. But he added that he could not support approval because of the inconsistent results in the studies and the lack of an effect in the second study
Among the issues that complicated the study results was that the low-dose active control patch also appeared to provide some degree of pain relief. In addition, there was evidence that the 8% patch may have been more effective in patients who were not on an analgesic, according to the company.*
If approved, this would be the first product approved by the FDA for this indication, which affects about 40% of people with HIV, according to the FDA and the company. Approval is unlikely though, because the FDA usually follows the recommendations of its advisory panels.
The FDA’s deadline for a decision is March 7. The review is accelerated, and is being conducted within 6 months from the time of application. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
The patch is being studied in phase II trials in patients with painful diabetic neuropathy, according to the company.
*Correction, 2/10/2012: An earlier version of this story contained an incorrect statement about use of the low-dose patch in conjunction with an opioid.
SILVER SPRING, MD. – A Food and Drug Administration advisory panel that met on Feb. 9 did not support approval of a capsaicin-containing transdermal patch as a treatment for pain associated with HIV neuropathy, because product studies had not provided adequate and compelling evidence of its efficacy for treating the condition.
At the meeting, the FDA’s Anesthetic and Analgesic Drug Products Advisory Committee voted 12 to 0 that data from two studies of about 800 people did not provide "substantial evidence of effectiveness" of the 8% capsaicin patch for the management of neuropathic pain related to HIV-associated peripheral neuropathy. While panelists agreed that the risks associated with treatment were low, the panel also voted 11 to 0, with 1 abstention, that, based on the available data, the risk-benefit profile was not acceptable for approval.
Marketed as Qutenza by San Francisco–based NeurogesX, the 8% capsaicin patch was approved in the United States in November 2009 for the treatment of neuropathic pain associated with postherpetic neuralgia, and has been approved in the European Union for the treatment of peripheral neuropathic pain in nondiabetic adults.
The proposed new indication is for the use of the patch, applied for 30 minutes, for the management of neuropathic pain related to HIV-associated peripheral neuropathy. The patch is applied to the foot after a lidocaine cream is applied to the skin; it cannot be reapplied until at least 3 months later. The patch delivers capsaicin – the most abundant component of chili peppers – to the site of pain, where it desensitizes hyperactive nociceptors, according to the company.
Panelists expressed regret that they could not support expanded approval because of the unmet need for this type of treatment for these patients – and noted some evidence that it was effective in one of the two studies presented by the company. But they agreed that the evidence did not meet the FDA’s standards for approval.
Treatment with the 8% capsaicin patch was compared to a low-dose 0.04% capsaicin patch as the control in two 12-week randomized studies of mostly white, middle-aged men with documented HIV, who had experienced HIV peripheral neuropathy for at least 2 months, with moderate to severe pain. Almost 70% of the participants were on other analgesics at baseline, including opioids.
The primary outcome end point evaluating effectiveness was the percent change in the mean daily scores in a pain rating scale from 2 to 12 weeks after the patch was applied. In the first study, which compared the effects of the 8% capsaicin patch and the control patch applied for 30, 60, or 90 minutes, there was some evidence that the 8% patch was more effective than the control patch, but most of that evidence was from post hoc analyses. In the second study, which compared the 8% patch to the low-dose patch applied for 30 or 60 minutes, there were no statistically significant reductions in pain relief at 12 weeks, compared with the control patch.
Overall, the safety profile of the 8% patch in these trials was similar to that seen in patients with postherpetic neuralgia. The most common adverse events associated with treatment were application site reactions, affecting 76% of those who had the higher-dose patch and about 50% of those who had the low-dose control patch.
One of the panelists, Dr. Thomas Giordano, an HIV specialist at Baylor College of Medicine and medical director of HIV services, Harris County Hospital District, both in Houston, said it was difficult to vote against approval, because he believed that some of his patients could benefit from this treatment. But he added that he could not support approval because of the inconsistent results in the studies and the lack of an effect in the second study
Among the issues that complicated the study results was that the low-dose active control patch also appeared to provide some degree of pain relief. In addition, there was evidence that the 8% patch may have been more effective in patients who were not on an analgesic, according to the company.*
If approved, this would be the first product approved by the FDA for this indication, which affects about 40% of people with HIV, according to the FDA and the company. Approval is unlikely though, because the FDA usually follows the recommendations of its advisory panels.
The FDA’s deadline for a decision is March 7. The review is accelerated, and is being conducted within 6 months from the time of application. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
The patch is being studied in phase II trials in patients with painful diabetic neuropathy, according to the company.
*Correction, 2/10/2012: An earlier version of this story contained an incorrect statement about use of the low-dose patch in conjunction with an opioid.
SILVER SPRING, MD. – A Food and Drug Administration advisory panel that met on Feb. 9 did not support approval of a capsaicin-containing transdermal patch as a treatment for pain associated with HIV neuropathy, because product studies had not provided adequate and compelling evidence of its efficacy for treating the condition.
At the meeting, the FDA’s Anesthetic and Analgesic Drug Products Advisory Committee voted 12 to 0 that data from two studies of about 800 people did not provide "substantial evidence of effectiveness" of the 8% capsaicin patch for the management of neuropathic pain related to HIV-associated peripheral neuropathy. While panelists agreed that the risks associated with treatment were low, the panel also voted 11 to 0, with 1 abstention, that, based on the available data, the risk-benefit profile was not acceptable for approval.
Marketed as Qutenza by San Francisco–based NeurogesX, the 8% capsaicin patch was approved in the United States in November 2009 for the treatment of neuropathic pain associated with postherpetic neuralgia, and has been approved in the European Union for the treatment of peripheral neuropathic pain in nondiabetic adults.
The proposed new indication is for the use of the patch, applied for 30 minutes, for the management of neuropathic pain related to HIV-associated peripheral neuropathy. The patch is applied to the foot after a lidocaine cream is applied to the skin; it cannot be reapplied until at least 3 months later. The patch delivers capsaicin – the most abundant component of chili peppers – to the site of pain, where it desensitizes hyperactive nociceptors, according to the company.
Panelists expressed regret that they could not support expanded approval because of the unmet need for this type of treatment for these patients – and noted some evidence that it was effective in one of the two studies presented by the company. But they agreed that the evidence did not meet the FDA’s standards for approval.
Treatment with the 8% capsaicin patch was compared to a low-dose 0.04% capsaicin patch as the control in two 12-week randomized studies of mostly white, middle-aged men with documented HIV, who had experienced HIV peripheral neuropathy for at least 2 months, with moderate to severe pain. Almost 70% of the participants were on other analgesics at baseline, including opioids.
The primary outcome end point evaluating effectiveness was the percent change in the mean daily scores in a pain rating scale from 2 to 12 weeks after the patch was applied. In the first study, which compared the effects of the 8% capsaicin patch and the control patch applied for 30, 60, or 90 minutes, there was some evidence that the 8% patch was more effective than the control patch, but most of that evidence was from post hoc analyses. In the second study, which compared the 8% patch to the low-dose patch applied for 30 or 60 minutes, there were no statistically significant reductions in pain relief at 12 weeks, compared with the control patch.
Overall, the safety profile of the 8% patch in these trials was similar to that seen in patients with postherpetic neuralgia. The most common adverse events associated with treatment were application site reactions, affecting 76% of those who had the higher-dose patch and about 50% of those who had the low-dose control patch.
One of the panelists, Dr. Thomas Giordano, an HIV specialist at Baylor College of Medicine and medical director of HIV services, Harris County Hospital District, both in Houston, said it was difficult to vote against approval, because he believed that some of his patients could benefit from this treatment. But he added that he could not support approval because of the inconsistent results in the studies and the lack of an effect in the second study
Among the issues that complicated the study results was that the low-dose active control patch also appeared to provide some degree of pain relief. In addition, there was evidence that the 8% patch may have been more effective in patients who were not on an analgesic, according to the company.*
If approved, this would be the first product approved by the FDA for this indication, which affects about 40% of people with HIV, according to the FDA and the company. Approval is unlikely though, because the FDA usually follows the recommendations of its advisory panels.
The FDA’s deadline for a decision is March 7. The review is accelerated, and is being conducted within 6 months from the time of application. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
The patch is being studied in phase II trials in patients with painful diabetic neuropathy, according to the company.
*Correction, 2/10/2012: An earlier version of this story contained an incorrect statement about use of the low-dose patch in conjunction with an opioid.
FROM A MEETING OF THE FDA'S ANESTHETIC AND ANALGESIC DRUG PRODUCTS ADVISORY COMMITTEE
FDA Panel Rejects Denosumab Against Bone Metastasis in Prostate Cancer
SILVER SPRING, MD. – A Food and Drug Administration advisory panel voted 12 to 1 on Feb. 8 that denosumab did not have a favorable risk-benefit profile as a treatment to reduce the risk of bone metastases in men who are at high risk for developing them with castrate-resistant prostate cancer.
Concern over the possibility of increased toxicity, especially osteonecrosis of the jaw, with longer drug exposure weighed heavily in the Oncologic Drugs Advisory Committee decision (ODAC). The patient representative was the only panelist to vote in favor.
Denosumab (Xgeva) was approved in 2010 for the prevention of skeletal-related events in patients with bone metastases from solid tumors. The new indication, proposed by manufacturer Amgen, would start treatment earlier to avert bone metastases before they develop in men with castrate-resistant prostate cancer (CRPC).
Amgen said that denosumab "prolongs bone metastasis-free survival by reducing the risk of developing bone metastases."
The application is based on the results of a phase III international study of 1,432 men with CRPC at high risk of developing bone metastases (defined as a prostate-specific antigen level of 8.0 ng/mL or higher or a PSA level that had doubled within 10 months or less).
The study found that survival free of bone metastases, the primary end point, was prolonged by a median of about 4 months in patients randomized to denosumab (120 mg subcutaneously every 4 weeks) compared with those on placebo – a statistically significant difference that represented a 15% reduction in risk.
But ODAC was nearly unanimous in finding that this difference in bone metastasis–free survival was modest and did not outweigh the risks of treatment, namely osteonecrosis of the jaw. In the study, almost 5% of those on denosumab developed osteonecrosis of the jaw, a known effect of treatment, compared with none of those on placebo. Panelists were concerned that this risk could increase with longer exposure to the drug.
The panel was not asked specifically whether it recommended approval for this expanded indication. The FDA must make a decision by the Prescription Drug User Fee Act (PDUFA) action date of April 26, 2012.
Amgen issued the following statement after the ODAC vote: "We look forward to further discussions with the FDA as they continue to review our application," it said. "The development of bone metastases in men with castration-resistant prostate cancer is a clinically significant event, and delaying bone metastases in these men is a clear unmet need with no approved therapies."
In the study under consideration, bone metastasis–free survival was determined by time to first occurrence of bone metastasis (symptomatic or asymptomatic at detection) or death, which was a median of 29.5 months among those on denosumab vs. 25.2 months among those on placebo.
There were no significant differences in overall survival, progression-free survival, or patient-reported outcomes between the two groups. In about two-thirds of cases, bone metastases were asymptomatic.
Denosumab, a monoclonal antibody that inhibits the RANK ligand (RANKL), is marketed as Xgeva at the same dose and schedule used in this study for prevention of skeletal events such as fractures from bone metastases. It is not indicated for the prevention of skeletal-related events in patients with multiple myeloma. By binding to RANKL, denosumab "prevents activation of RANK, thereby inhibiting osteoclast formation, activation, and survival," according to Amgen.
Denosumab is also approved as Prolia, at a lower dose administered once a year, to treat postmenopausal women with osteoporosis who are at high risk of fracture; to increase bone mass in men who are at high risk of fracture while receiving androgen deprivation therapy for nonmetastatic prostate cancer; and to increase bone mass in women at high risk of fracture while receiving adjuvant aromatase inhibitor therapy for breast cancer.
If the new indication is granted, Xgeva would be the first treatment approved to prevent or delay bone metastases in men with nonmetastatic CRPC, according to Amgen. But that appears unlikely, since the FDA usually follows the recommendations of its advisory panels.
Panelists were cleared of potential conflicts before voting on denosumab.
SILVER SPRING, MD. – A Food and Drug Administration advisory panel voted 12 to 1 on Feb. 8 that denosumab did not have a favorable risk-benefit profile as a treatment to reduce the risk of bone metastases in men who are at high risk for developing them with castrate-resistant prostate cancer.
Concern over the possibility of increased toxicity, especially osteonecrosis of the jaw, with longer drug exposure weighed heavily in the Oncologic Drugs Advisory Committee decision (ODAC). The patient representative was the only panelist to vote in favor.
Denosumab (Xgeva) was approved in 2010 for the prevention of skeletal-related events in patients with bone metastases from solid tumors. The new indication, proposed by manufacturer Amgen, would start treatment earlier to avert bone metastases before they develop in men with castrate-resistant prostate cancer (CRPC).
Amgen said that denosumab "prolongs bone metastasis-free survival by reducing the risk of developing bone metastases."
The application is based on the results of a phase III international study of 1,432 men with CRPC at high risk of developing bone metastases (defined as a prostate-specific antigen level of 8.0 ng/mL or higher or a PSA level that had doubled within 10 months or less).
The study found that survival free of bone metastases, the primary end point, was prolonged by a median of about 4 months in patients randomized to denosumab (120 mg subcutaneously every 4 weeks) compared with those on placebo – a statistically significant difference that represented a 15% reduction in risk.
But ODAC was nearly unanimous in finding that this difference in bone metastasis–free survival was modest and did not outweigh the risks of treatment, namely osteonecrosis of the jaw. In the study, almost 5% of those on denosumab developed osteonecrosis of the jaw, a known effect of treatment, compared with none of those on placebo. Panelists were concerned that this risk could increase with longer exposure to the drug.
The panel was not asked specifically whether it recommended approval for this expanded indication. The FDA must make a decision by the Prescription Drug User Fee Act (PDUFA) action date of April 26, 2012.
Amgen issued the following statement after the ODAC vote: "We look forward to further discussions with the FDA as they continue to review our application," it said. "The development of bone metastases in men with castration-resistant prostate cancer is a clinically significant event, and delaying bone metastases in these men is a clear unmet need with no approved therapies."
In the study under consideration, bone metastasis–free survival was determined by time to first occurrence of bone metastasis (symptomatic or asymptomatic at detection) or death, which was a median of 29.5 months among those on denosumab vs. 25.2 months among those on placebo.
There were no significant differences in overall survival, progression-free survival, or patient-reported outcomes between the two groups. In about two-thirds of cases, bone metastases were asymptomatic.
Denosumab, a monoclonal antibody that inhibits the RANK ligand (RANKL), is marketed as Xgeva at the same dose and schedule used in this study for prevention of skeletal events such as fractures from bone metastases. It is not indicated for the prevention of skeletal-related events in patients with multiple myeloma. By binding to RANKL, denosumab "prevents activation of RANK, thereby inhibiting osteoclast formation, activation, and survival," according to Amgen.
Denosumab is also approved as Prolia, at a lower dose administered once a year, to treat postmenopausal women with osteoporosis who are at high risk of fracture; to increase bone mass in men who are at high risk of fracture while receiving androgen deprivation therapy for nonmetastatic prostate cancer; and to increase bone mass in women at high risk of fracture while receiving adjuvant aromatase inhibitor therapy for breast cancer.
If the new indication is granted, Xgeva would be the first treatment approved to prevent or delay bone metastases in men with nonmetastatic CRPC, according to Amgen. But that appears unlikely, since the FDA usually follows the recommendations of its advisory panels.
Panelists were cleared of potential conflicts before voting on denosumab.
SILVER SPRING, MD. – A Food and Drug Administration advisory panel voted 12 to 1 on Feb. 8 that denosumab did not have a favorable risk-benefit profile as a treatment to reduce the risk of bone metastases in men who are at high risk for developing them with castrate-resistant prostate cancer.
Concern over the possibility of increased toxicity, especially osteonecrosis of the jaw, with longer drug exposure weighed heavily in the Oncologic Drugs Advisory Committee decision (ODAC). The patient representative was the only panelist to vote in favor.
Denosumab (Xgeva) was approved in 2010 for the prevention of skeletal-related events in patients with bone metastases from solid tumors. The new indication, proposed by manufacturer Amgen, would start treatment earlier to avert bone metastases before they develop in men with castrate-resistant prostate cancer (CRPC).
Amgen said that denosumab "prolongs bone metastasis-free survival by reducing the risk of developing bone metastases."
The application is based on the results of a phase III international study of 1,432 men with CRPC at high risk of developing bone metastases (defined as a prostate-specific antigen level of 8.0 ng/mL or higher or a PSA level that had doubled within 10 months or less).
The study found that survival free of bone metastases, the primary end point, was prolonged by a median of about 4 months in patients randomized to denosumab (120 mg subcutaneously every 4 weeks) compared with those on placebo – a statistically significant difference that represented a 15% reduction in risk.
But ODAC was nearly unanimous in finding that this difference in bone metastasis–free survival was modest and did not outweigh the risks of treatment, namely osteonecrosis of the jaw. In the study, almost 5% of those on denosumab developed osteonecrosis of the jaw, a known effect of treatment, compared with none of those on placebo. Panelists were concerned that this risk could increase with longer exposure to the drug.
The panel was not asked specifically whether it recommended approval for this expanded indication. The FDA must make a decision by the Prescription Drug User Fee Act (PDUFA) action date of April 26, 2012.
Amgen issued the following statement after the ODAC vote: "We look forward to further discussions with the FDA as they continue to review our application," it said. "The development of bone metastases in men with castration-resistant prostate cancer is a clinically significant event, and delaying bone metastases in these men is a clear unmet need with no approved therapies."
In the study under consideration, bone metastasis–free survival was determined by time to first occurrence of bone metastasis (symptomatic or asymptomatic at detection) or death, which was a median of 29.5 months among those on denosumab vs. 25.2 months among those on placebo.
There were no significant differences in overall survival, progression-free survival, or patient-reported outcomes between the two groups. In about two-thirds of cases, bone metastases were asymptomatic.
Denosumab, a monoclonal antibody that inhibits the RANK ligand (RANKL), is marketed as Xgeva at the same dose and schedule used in this study for prevention of skeletal events such as fractures from bone metastases. It is not indicated for the prevention of skeletal-related events in patients with multiple myeloma. By binding to RANKL, denosumab "prevents activation of RANK, thereby inhibiting osteoclast formation, activation, and survival," according to Amgen.
Denosumab is also approved as Prolia, at a lower dose administered once a year, to treat postmenopausal women with osteoporosis who are at high risk of fracture; to increase bone mass in men who are at high risk of fracture while receiving androgen deprivation therapy for nonmetastatic prostate cancer; and to increase bone mass in women at high risk of fracture while receiving adjuvant aromatase inhibitor therapy for breast cancer.
If the new indication is granted, Xgeva would be the first treatment approved to prevent or delay bone metastases in men with nonmetastatic CRPC, according to Amgen. But that appears unlikely, since the FDA usually follows the recommendations of its advisory panels.
Panelists were cleared of potential conflicts before voting on denosumab.
FDA Adds Longer GIST Therapy to Imatinib Label
Clinical trial data showing a significant increase in survival when imatinib is used for 3 years as adjuvant therapy after resection of Kit-positive gastrointestinal stromal tumors has been added to the drug’s label.
The change "highlights an increase in overall patient survival when the drug is taken for 36 months rather than the standard 12 months of treatment," the Food and Drug Administration said in a Jan. 31 statement.
The agency also announced the granting of "regular" approval to imatinib (Gleevec) as adjuvant treatment for adults "following gross resection of Kit (CD117) positive" gastrointestinal stromal tumors (GIST). This indication had received an accelerated approval in 2008; accelerated approvals are granted to provide earlier access to a drug while confirmatory trials required for full approval are under way.
Imatinib, an oral tyrosine kinase inhibitor, was initially approved by the FDA in 2001 to treat patients with advanced Philadelphia chromosome–positive chronic myeloid leukemia. It received an accelerated approval for the treatment of advanced or metastatic GIST in 2002; this was converted to a full approval in 2008 based on confirmatory trials.
"The development of Gleevec over the past decade highlights the need to further study drugs after approval to truly characterize their benefits," Dr. Richard Pazdur, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in the agency’s statement.
"Although originally approved in the metastatic disease setting, this subsequent trial has demonstrated that longer use of Gleevec can prolong patient’s lives in earlier disease settings," he added.
The data added to the label come from a large, international, randomized open-label phase III study of patients with more than a 50% estimated risk of recurrence of GIST. The overall survival rate at 5 years was 92% among patients treated for 36 months with imatinib, compared with 82% in those treated for 12 months, according to the FDA.
The longer overall survival among those who received 36 months of imatinib treatment after surgery represents a 55% reduced risk of death, according to a statement from Novartis Pharmaceuticals Corp., which markets Gleevec.
Recurrence-free survival also was longer among those treated for 36 months, with a 54% reduced risk of risk of recurrence compared with the 12-month schedule, Novartis said.
Dr. Heikki Joensuu of Helsinki University Central Hospital in Finland presented the trial results in a plenary session at the annual meeting of the American Society of Clinical Oncology in June 2011. Click here to see a full report of his presentation and a video interview with Dr. Joensuu on the findings.
In August 2011, the National Comprehensive Cancer Network recommended consideration of at least 3 years of adjuvant treatment with imatinib in patients with high-risk GIST, according to Novartis.
The most common side effects associated with treatment with imatinib include edema, nausea, vomiting, muscle cramps, bone or muscle pain, diarrhea, rash, fatigue, and abdominal pain, according to the FDA.
It identified the recommended dose of imatinib for adjuvant treatment as 400 mg/day administered with meals daily for 3 years but noted that the optimal duration of treatment is not known.
Clinical trial data showing a significant increase in survival when imatinib is used for 3 years as adjuvant therapy after resection of Kit-positive gastrointestinal stromal tumors has been added to the drug’s label.
The change "highlights an increase in overall patient survival when the drug is taken for 36 months rather than the standard 12 months of treatment," the Food and Drug Administration said in a Jan. 31 statement.
The agency also announced the granting of "regular" approval to imatinib (Gleevec) as adjuvant treatment for adults "following gross resection of Kit (CD117) positive" gastrointestinal stromal tumors (GIST). This indication had received an accelerated approval in 2008; accelerated approvals are granted to provide earlier access to a drug while confirmatory trials required for full approval are under way.
Imatinib, an oral tyrosine kinase inhibitor, was initially approved by the FDA in 2001 to treat patients with advanced Philadelphia chromosome–positive chronic myeloid leukemia. It received an accelerated approval for the treatment of advanced or metastatic GIST in 2002; this was converted to a full approval in 2008 based on confirmatory trials.
"The development of Gleevec over the past decade highlights the need to further study drugs after approval to truly characterize their benefits," Dr. Richard Pazdur, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in the agency’s statement.
"Although originally approved in the metastatic disease setting, this subsequent trial has demonstrated that longer use of Gleevec can prolong patient’s lives in earlier disease settings," he added.
The data added to the label come from a large, international, randomized open-label phase III study of patients with more than a 50% estimated risk of recurrence of GIST. The overall survival rate at 5 years was 92% among patients treated for 36 months with imatinib, compared with 82% in those treated for 12 months, according to the FDA.
The longer overall survival among those who received 36 months of imatinib treatment after surgery represents a 55% reduced risk of death, according to a statement from Novartis Pharmaceuticals Corp., which markets Gleevec.
Recurrence-free survival also was longer among those treated for 36 months, with a 54% reduced risk of risk of recurrence compared with the 12-month schedule, Novartis said.
Dr. Heikki Joensuu of Helsinki University Central Hospital in Finland presented the trial results in a plenary session at the annual meeting of the American Society of Clinical Oncology in June 2011. Click here to see a full report of his presentation and a video interview with Dr. Joensuu on the findings.
In August 2011, the National Comprehensive Cancer Network recommended consideration of at least 3 years of adjuvant treatment with imatinib in patients with high-risk GIST, according to Novartis.
The most common side effects associated with treatment with imatinib include edema, nausea, vomiting, muscle cramps, bone or muscle pain, diarrhea, rash, fatigue, and abdominal pain, according to the FDA.
It identified the recommended dose of imatinib for adjuvant treatment as 400 mg/day administered with meals daily for 3 years but noted that the optimal duration of treatment is not known.
Clinical trial data showing a significant increase in survival when imatinib is used for 3 years as adjuvant therapy after resection of Kit-positive gastrointestinal stromal tumors has been added to the drug’s label.
The change "highlights an increase in overall patient survival when the drug is taken for 36 months rather than the standard 12 months of treatment," the Food and Drug Administration said in a Jan. 31 statement.
The agency also announced the granting of "regular" approval to imatinib (Gleevec) as adjuvant treatment for adults "following gross resection of Kit (CD117) positive" gastrointestinal stromal tumors (GIST). This indication had received an accelerated approval in 2008; accelerated approvals are granted to provide earlier access to a drug while confirmatory trials required for full approval are under way.
Imatinib, an oral tyrosine kinase inhibitor, was initially approved by the FDA in 2001 to treat patients with advanced Philadelphia chromosome–positive chronic myeloid leukemia. It received an accelerated approval for the treatment of advanced or metastatic GIST in 2002; this was converted to a full approval in 2008 based on confirmatory trials.
"The development of Gleevec over the past decade highlights the need to further study drugs after approval to truly characterize their benefits," Dr. Richard Pazdur, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in the agency’s statement.
"Although originally approved in the metastatic disease setting, this subsequent trial has demonstrated that longer use of Gleevec can prolong patient’s lives in earlier disease settings," he added.
The data added to the label come from a large, international, randomized open-label phase III study of patients with more than a 50% estimated risk of recurrence of GIST. The overall survival rate at 5 years was 92% among patients treated for 36 months with imatinib, compared with 82% in those treated for 12 months, according to the FDA.
The longer overall survival among those who received 36 months of imatinib treatment after surgery represents a 55% reduced risk of death, according to a statement from Novartis Pharmaceuticals Corp., which markets Gleevec.
Recurrence-free survival also was longer among those treated for 36 months, with a 54% reduced risk of risk of recurrence compared with the 12-month schedule, Novartis said.
Dr. Heikki Joensuu of Helsinki University Central Hospital in Finland presented the trial results in a plenary session at the annual meeting of the American Society of Clinical Oncology in June 2011. Click here to see a full report of his presentation and a video interview with Dr. Joensuu on the findings.
In August 2011, the National Comprehensive Cancer Network recommended consideration of at least 3 years of adjuvant treatment with imatinib in patients with high-risk GIST, according to Novartis.
The most common side effects associated with treatment with imatinib include edema, nausea, vomiting, muscle cramps, bone or muscle pain, diarrhea, rash, fatigue, and abdominal pain, according to the FDA.
It identified the recommended dose of imatinib for adjuvant treatment as 400 mg/day administered with meals daily for 3 years but noted that the optimal duration of treatment is not known.
Once-Weekly Exenatide Approved
The first once-weekly diabetes drug has been approved by the Food and Drug Administration. An extended-release formulation of the type 2 diabetes medication exenatide, a glucagonlike peptide–1 (GLP-1) receptor, was approved last month, the manufacturer announced.
The formulation, called Bydureon, is administered once a week in a subcutaneous injection, and is indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. The regular formulation of exenatide (Byetta), approved in 2005, is administered twice a day.
Approval was based on the results of the DURATION-5 study, which compared treatment with the two formulations in 252 patients with type 2 diabetes and inadequate glycemic control (mean baseline hemoglobin A1c was 8.4%) with diet and exercise alone or with oral therapy, including metformin, a sulfonylurea, a thiazolidinedione, or a combination of two of these treatments.
The mean reduction in HbA1c was 1.6 percentage points in those treated with 2 mg of extended-release exenatide once weekly, compared with a reduction of 0.9 percentage points in those treated with the regular form of exenatide, a statistically significant difference, according to the prescribing information.
Bydureon has been approved with a Risk Evaluation and Mitigation Strategy to ensure that the benefits of the drug outweigh the risks of acute pancreatitis and the “potential” risk of medullary thyroid cancer with treatment. There have been postmarketing reports of pancreatitis associated with exenatide, including nonfatal hemorrhagic or necrotizing pancreatitis.
Bydureon is contraindicated in people with a personal or family history of medullary thyroid cancer or those with multiple endocrine neoplasia syndrome type 2.
The first once-weekly diabetes drug has been approved by the Food and Drug Administration. An extended-release formulation of the type 2 diabetes medication exenatide, a glucagonlike peptide–1 (GLP-1) receptor, was approved last month, the manufacturer announced.
The formulation, called Bydureon, is administered once a week in a subcutaneous injection, and is indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. The regular formulation of exenatide (Byetta), approved in 2005, is administered twice a day.
Approval was based on the results of the DURATION-5 study, which compared treatment with the two formulations in 252 patients with type 2 diabetes and inadequate glycemic control (mean baseline hemoglobin A1c was 8.4%) with diet and exercise alone or with oral therapy, including metformin, a sulfonylurea, a thiazolidinedione, or a combination of two of these treatments.
The mean reduction in HbA1c was 1.6 percentage points in those treated with 2 mg of extended-release exenatide once weekly, compared with a reduction of 0.9 percentage points in those treated with the regular form of exenatide, a statistically significant difference, according to the prescribing information.
Bydureon has been approved with a Risk Evaluation and Mitigation Strategy to ensure that the benefits of the drug outweigh the risks of acute pancreatitis and the “potential” risk of medullary thyroid cancer with treatment. There have been postmarketing reports of pancreatitis associated with exenatide, including nonfatal hemorrhagic or necrotizing pancreatitis.
Bydureon is contraindicated in people with a personal or family history of medullary thyroid cancer or those with multiple endocrine neoplasia syndrome type 2.
The first once-weekly diabetes drug has been approved by the Food and Drug Administration. An extended-release formulation of the type 2 diabetes medication exenatide, a glucagonlike peptide–1 (GLP-1) receptor, was approved last month, the manufacturer announced.
The formulation, called Bydureon, is administered once a week in a subcutaneous injection, and is indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. The regular formulation of exenatide (Byetta), approved in 2005, is administered twice a day.
Approval was based on the results of the DURATION-5 study, which compared treatment with the two formulations in 252 patients with type 2 diabetes and inadequate glycemic control (mean baseline hemoglobin A1c was 8.4%) with diet and exercise alone or with oral therapy, including metformin, a sulfonylurea, a thiazolidinedione, or a combination of two of these treatments.
The mean reduction in HbA1c was 1.6 percentage points in those treated with 2 mg of extended-release exenatide once weekly, compared with a reduction of 0.9 percentage points in those treated with the regular form of exenatide, a statistically significant difference, according to the prescribing information.
Bydureon has been approved with a Risk Evaluation and Mitigation Strategy to ensure that the benefits of the drug outweigh the risks of acute pancreatitis and the “potential” risk of medullary thyroid cancer with treatment. There have been postmarketing reports of pancreatitis associated with exenatide, including nonfatal hemorrhagic or necrotizing pancreatitis.
Bydureon is contraindicated in people with a personal or family history of medullary thyroid cancer or those with multiple endocrine neoplasia syndrome type 2.