User login
Phacomatosis Pigmentokeratotica Associated With Raynaud Phenomenon, Segmental Nevi, Hyperhidrosis, and Scoliosis
To the Editor:
Phacomatosis pigmentokeratotica (PPK) is a rare epidermal nevus syndrome complicated by multiple extracutaneous anomalies, including skeletal defects and neurologic anomalies. Less common associations include lateral curvature of the spine and hyperhidrosis. We present a patient with PPK and unilateral Raynaud phenomenon in addition to a segmental distribution of melanocytic nevi, hyperhidrosis, and scoliosis.
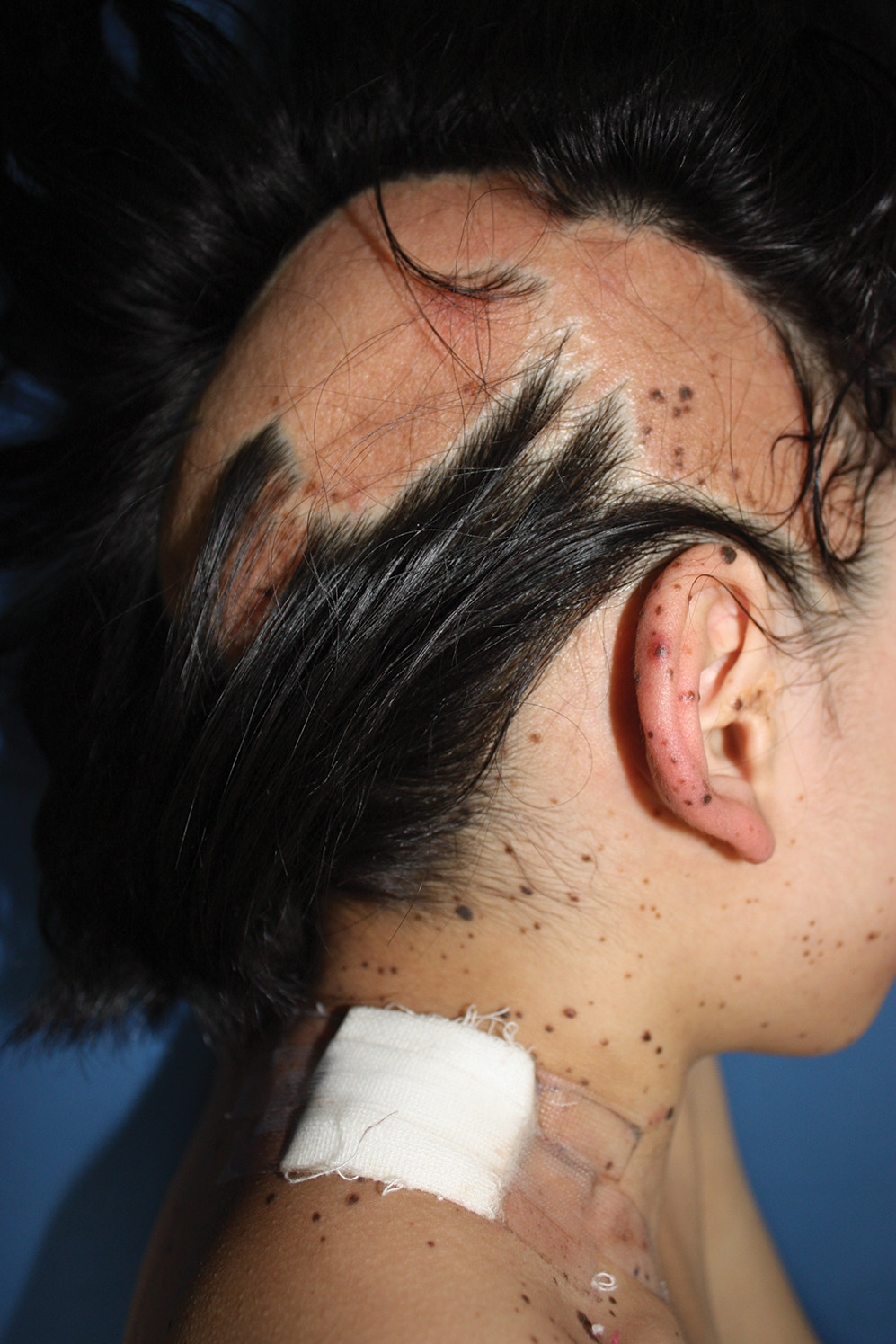
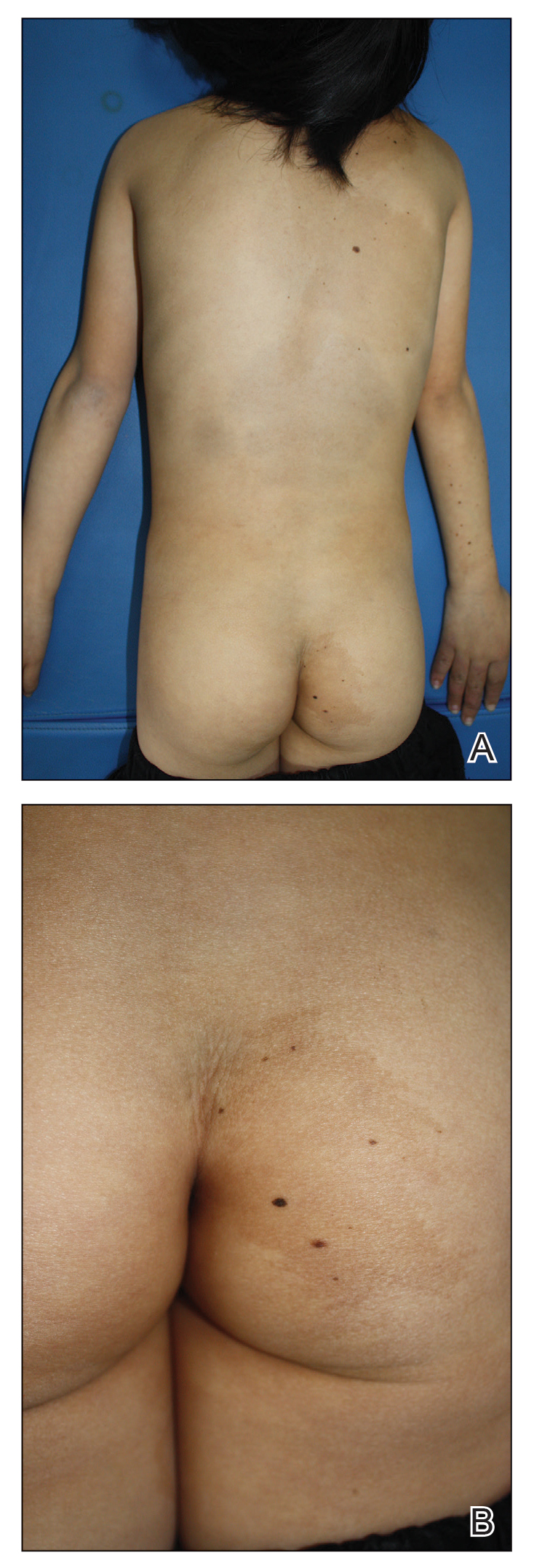
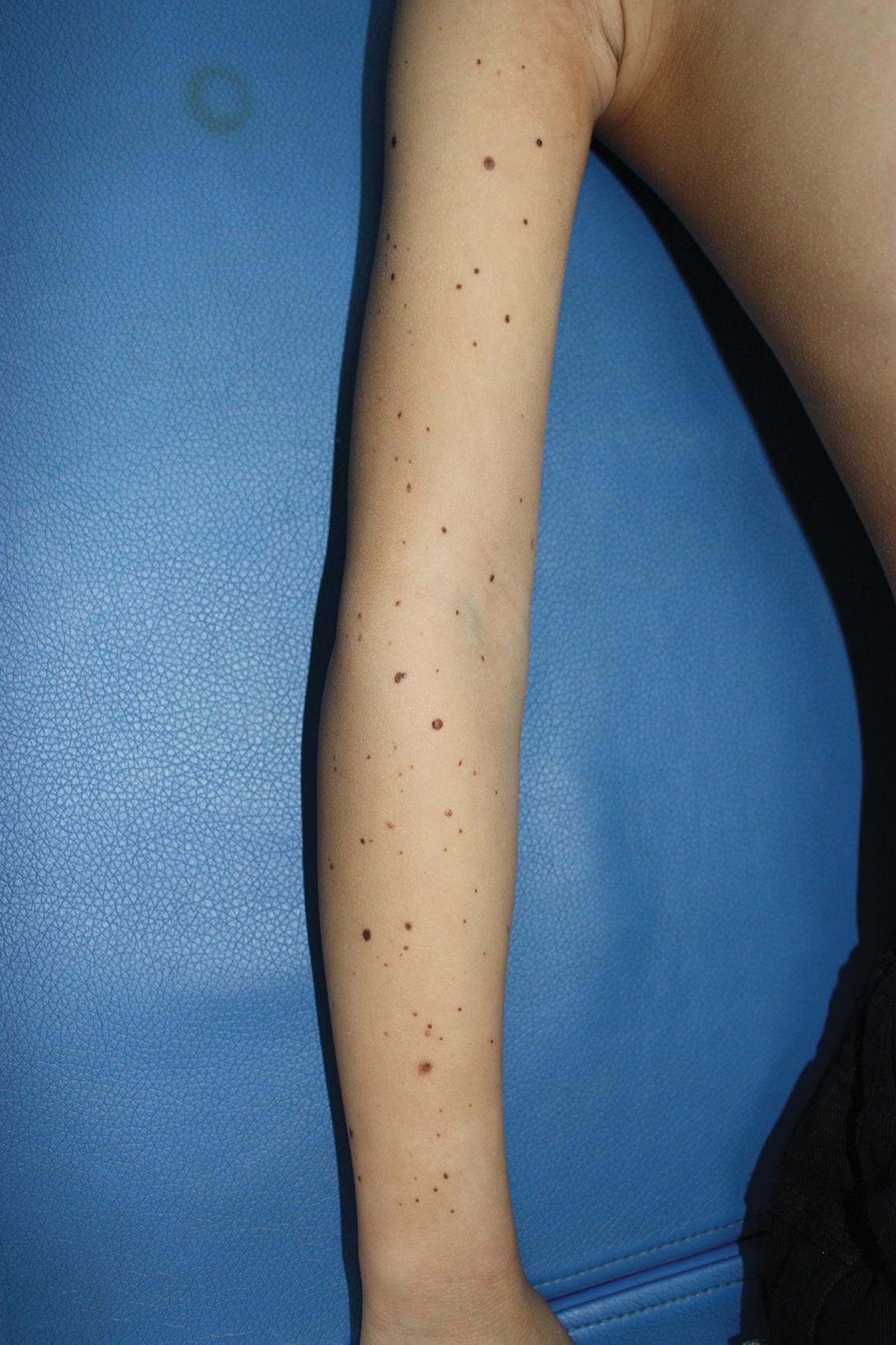
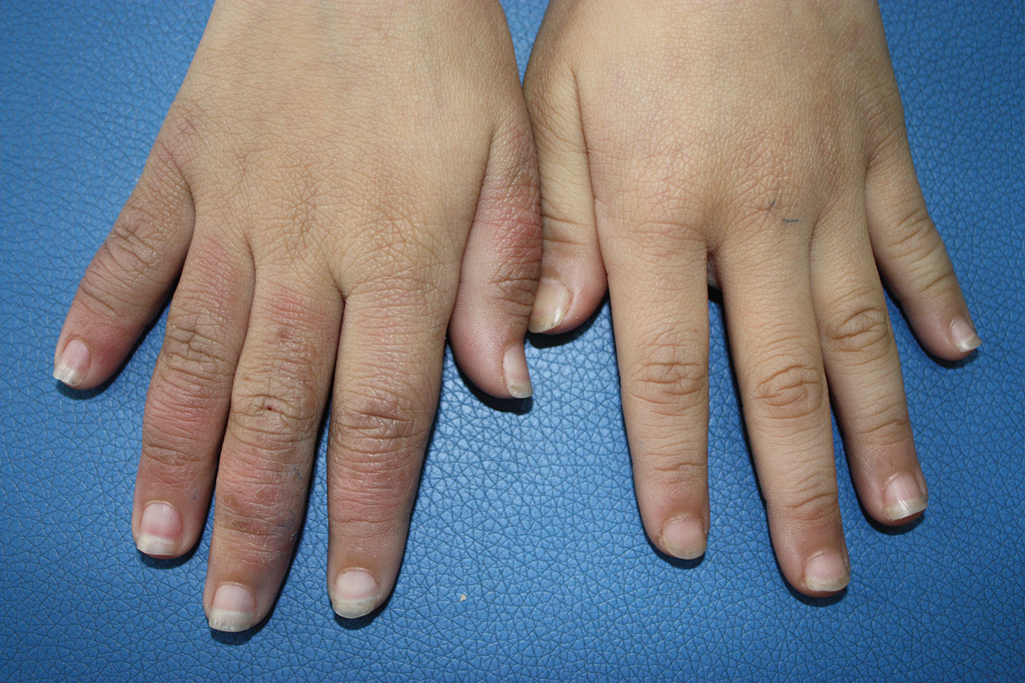
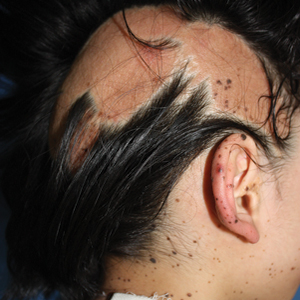
A 9-year-old girl was born with a yellow-orange alopecic plaque on the right side of the scalp (Figure 1). There also were 2 large, irregularly pigmented patches localized on the right side of the upper back and buttock. Over 3 years, numerous papular nevi developed within these pigmented patches and were diagnosed as speckled lentiginous nevi (Figure 2). In addition, numerous nevi of various sizes affected the right face, right shoulder, right arm (Figure 3), and right neck and were clearly demarcated along the midline. Several nevi also were noted within the nevus sebaceous on the right scalp. These skin lesions expanded progressively with age. At 6 years of age, she was diagnosed with hyperhidrosis of the right half of the body, which was most pronounced on the face. Raynaud phenomenon restricted to the right hand also was noted (Figure 4). Upon cold exposure, the digits become pale white, cold, and numb; then blue; and finally red. She lacked other features of connective tissue disease, and autoantibody testing was negative. She also was noted to have an abnormal lateral curvature of the spine (scoliosis). Auditory, ocular, and neurologic examinations were normal. Cranial and cerebral magnetic resonance imaging showed no central nervous system abnormalities. Her family history was negative for nevus spilus, nevus sebaceous, and neurofibromatosis. The clinical findings in our patient led to the diagnosis of PPK.
Phacomatosis pigmentokeratotica is a distinctive epidermal nevus syndrome characterized by the coexistence of a speckled lentiginous nevus, also known as a nevus spilus, and a nevus sebaceous1; PPK frequently is complicated by skeletal, ophthalmic, or neurologic abnormalities.2 Most cases reported are sporadic, and a postzygotic mosaic HRas proto-oncogene, GTPase, HRAS, mutation has been demonstrated in some patients and may contribute to the phenotype of PPK.3,4
Other anomalies have included ichthyosislike diffuse hyperkeratosis, laxity of the hands, pelvic hypoplasia, glaucoma, psychomotor retardation, and hypophosphatemic rickets. These patients also should be monitored for the development of malignant neoplasms within the nevus sebaceous.5 Segmental hyperhidrosis may be seen in association with the nevus spilus component.2
Raynaud phenomenon involving only the right hand was a unique finding in our patient. In 3 years of follow-up, our patient developed no evidence of connective tissue disease or other systemic illness. We speculate that Raynaud phenomenon of the right hand along with hyperhidrosis of the right side of the body could be a result of dysfunction of the autonomic nervous system. We propose that Raynaud phenomenon represents an unusual manifestation of PPK and may broaden the spectrum of extracutaneous anomalies associated with the disease. The finding of segmental nevi outside of the confines of the nevus spilus was another unusual manifestation of mosaicism.
- Happle R, Hoffmann R, Restano L, et al. Phacomatosis pigmentokeratotica: a melanocytic-epidermal twin nevus syndrome. Am J Med Genet. 1996;65:363-365.
- Happle R. The group of epidermal nevus syndromes part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22, 23-24.
- Groesser L, Herschberger E, Sagrera A, et al. Phacomatosis pigmentokeratotica is caused by a postzygotic HRAS mutation in a multipotent progenitor cell. J Invest Dermatol. 2013;133:1998-2003.
- Martin RJ, Arefi M, Splitt M, et al. Phacomatosis pigmentokeratotica and precocious puberty associated with HRAS mutation. Br J Dermatol. 2018;178:289-291.
- Chu GY, Wu CY. Phacomatosis pigmentokeratotica: a follow-up report with fatal outcome. Acta Derm Venereol. 2014;94:467-468.
To the Editor:
Phacomatosis pigmentokeratotica (PPK) is a rare epidermal nevus syndrome complicated by multiple extracutaneous anomalies, including skeletal defects and neurologic anomalies. Less common associations include lateral curvature of the spine and hyperhidrosis. We present a patient with PPK and unilateral Raynaud phenomenon in addition to a segmental distribution of melanocytic nevi, hyperhidrosis, and scoliosis.
A 9-year-old girl was born with a yellow-orange alopecic plaque on the right side of the scalp (Figure 1). There also were 2 large, irregularly pigmented patches localized on the right side of the upper back and buttock. Over 3 years, numerous papular nevi developed within these pigmented patches and were diagnosed as speckled lentiginous nevi (Figure 2). In addition, numerous nevi of various sizes affected the right face, right shoulder, right arm (Figure 3), and right neck and were clearly demarcated along the midline. Several nevi also were noted within the nevus sebaceous on the right scalp. These skin lesions expanded progressively with age. At 6 years of age, she was diagnosed with hyperhidrosis of the right half of the body, which was most pronounced on the face. Raynaud phenomenon restricted to the right hand also was noted (Figure 4). Upon cold exposure, the digits become pale white, cold, and numb; then blue; and finally red. She lacked other features of connective tissue disease, and autoantibody testing was negative. She also was noted to have an abnormal lateral curvature of the spine (scoliosis). Auditory, ocular, and neurologic examinations were normal. Cranial and cerebral magnetic resonance imaging showed no central nervous system abnormalities. Her family history was negative for nevus spilus, nevus sebaceous, and neurofibromatosis. The clinical findings in our patient led to the diagnosis of PPK.
Phacomatosis pigmentokeratotica is a distinctive epidermal nevus syndrome characterized by the coexistence of a speckled lentiginous nevus, also known as a nevus spilus, and a nevus sebaceous1; PPK frequently is complicated by skeletal, ophthalmic, or neurologic abnormalities.2 Most cases reported are sporadic, and a postzygotic mosaic HRas proto-oncogene, GTPase, HRAS, mutation has been demonstrated in some patients and may contribute to the phenotype of PPK.3,4
Other anomalies have included ichthyosislike diffuse hyperkeratosis, laxity of the hands, pelvic hypoplasia, glaucoma, psychomotor retardation, and hypophosphatemic rickets. These patients also should be monitored for the development of malignant neoplasms within the nevus sebaceous.5 Segmental hyperhidrosis may be seen in association with the nevus spilus component.2
Raynaud phenomenon involving only the right hand was a unique finding in our patient. In 3 years of follow-up, our patient developed no evidence of connective tissue disease or other systemic illness. We speculate that Raynaud phenomenon of the right hand along with hyperhidrosis of the right side of the body could be a result of dysfunction of the autonomic nervous system. We propose that Raynaud phenomenon represents an unusual manifestation of PPK and may broaden the spectrum of extracutaneous anomalies associated with the disease. The finding of segmental nevi outside of the confines of the nevus spilus was another unusual manifestation of mosaicism.
To the Editor:
Phacomatosis pigmentokeratotica (PPK) is a rare epidermal nevus syndrome complicated by multiple extracutaneous anomalies, including skeletal defects and neurologic anomalies. Less common associations include lateral curvature of the spine and hyperhidrosis. We present a patient with PPK and unilateral Raynaud phenomenon in addition to a segmental distribution of melanocytic nevi, hyperhidrosis, and scoliosis.
A 9-year-old girl was born with a yellow-orange alopecic plaque on the right side of the scalp (Figure 1). There also were 2 large, irregularly pigmented patches localized on the right side of the upper back and buttock. Over 3 years, numerous papular nevi developed within these pigmented patches and were diagnosed as speckled lentiginous nevi (Figure 2). In addition, numerous nevi of various sizes affected the right face, right shoulder, right arm (Figure 3), and right neck and were clearly demarcated along the midline. Several nevi also were noted within the nevus sebaceous on the right scalp. These skin lesions expanded progressively with age. At 6 years of age, she was diagnosed with hyperhidrosis of the right half of the body, which was most pronounced on the face. Raynaud phenomenon restricted to the right hand also was noted (Figure 4). Upon cold exposure, the digits become pale white, cold, and numb; then blue; and finally red. She lacked other features of connective tissue disease, and autoantibody testing was negative. She also was noted to have an abnormal lateral curvature of the spine (scoliosis). Auditory, ocular, and neurologic examinations were normal. Cranial and cerebral magnetic resonance imaging showed no central nervous system abnormalities. Her family history was negative for nevus spilus, nevus sebaceous, and neurofibromatosis. The clinical findings in our patient led to the diagnosis of PPK.
Phacomatosis pigmentokeratotica is a distinctive epidermal nevus syndrome characterized by the coexistence of a speckled lentiginous nevus, also known as a nevus spilus, and a nevus sebaceous1; PPK frequently is complicated by skeletal, ophthalmic, or neurologic abnormalities.2 Most cases reported are sporadic, and a postzygotic mosaic HRas proto-oncogene, GTPase, HRAS, mutation has been demonstrated in some patients and may contribute to the phenotype of PPK.3,4
Other anomalies have included ichthyosislike diffuse hyperkeratosis, laxity of the hands, pelvic hypoplasia, glaucoma, psychomotor retardation, and hypophosphatemic rickets. These patients also should be monitored for the development of malignant neoplasms within the nevus sebaceous.5 Segmental hyperhidrosis may be seen in association with the nevus spilus component.2
Raynaud phenomenon involving only the right hand was a unique finding in our patient. In 3 years of follow-up, our patient developed no evidence of connective tissue disease or other systemic illness. We speculate that Raynaud phenomenon of the right hand along with hyperhidrosis of the right side of the body could be a result of dysfunction of the autonomic nervous system. We propose that Raynaud phenomenon represents an unusual manifestation of PPK and may broaden the spectrum of extracutaneous anomalies associated with the disease. The finding of segmental nevi outside of the confines of the nevus spilus was another unusual manifestation of mosaicism.
- Happle R, Hoffmann R, Restano L, et al. Phacomatosis pigmentokeratotica: a melanocytic-epidermal twin nevus syndrome. Am J Med Genet. 1996;65:363-365.
- Happle R. The group of epidermal nevus syndromes part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22, 23-24.
- Groesser L, Herschberger E, Sagrera A, et al. Phacomatosis pigmentokeratotica is caused by a postzygotic HRAS mutation in a multipotent progenitor cell. J Invest Dermatol. 2013;133:1998-2003.
- Martin RJ, Arefi M, Splitt M, et al. Phacomatosis pigmentokeratotica and precocious puberty associated with HRAS mutation. Br J Dermatol. 2018;178:289-291.
- Chu GY, Wu CY. Phacomatosis pigmentokeratotica: a follow-up report with fatal outcome. Acta Derm Venereol. 2014;94:467-468.
- Happle R, Hoffmann R, Restano L, et al. Phacomatosis pigmentokeratotica: a melanocytic-epidermal twin nevus syndrome. Am J Med Genet. 1996;65:363-365.
- Happle R. The group of epidermal nevus syndromes part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22, 23-24.
- Groesser L, Herschberger E, Sagrera A, et al. Phacomatosis pigmentokeratotica is caused by a postzygotic HRAS mutation in a multipotent progenitor cell. J Invest Dermatol. 2013;133:1998-2003.
- Martin RJ, Arefi M, Splitt M, et al. Phacomatosis pigmentokeratotica and precocious puberty associated with HRAS mutation. Br J Dermatol. 2018;178:289-291.
- Chu GY, Wu CY. Phacomatosis pigmentokeratotica: a follow-up report with fatal outcome. Acta Derm Venereol. 2014;94:467-468.
Practice Points
- Phacomatosis pigmentokeratotica (PPK) is characterized by the coexistence of speckled lentiginous nevus and nevus sebaceous.
- Raynaud phenomenon may be an unreported association with PPK.
Systemic Epstein-Barr Virus–Positive T-cell Lymphoma of Childhood
Case Report
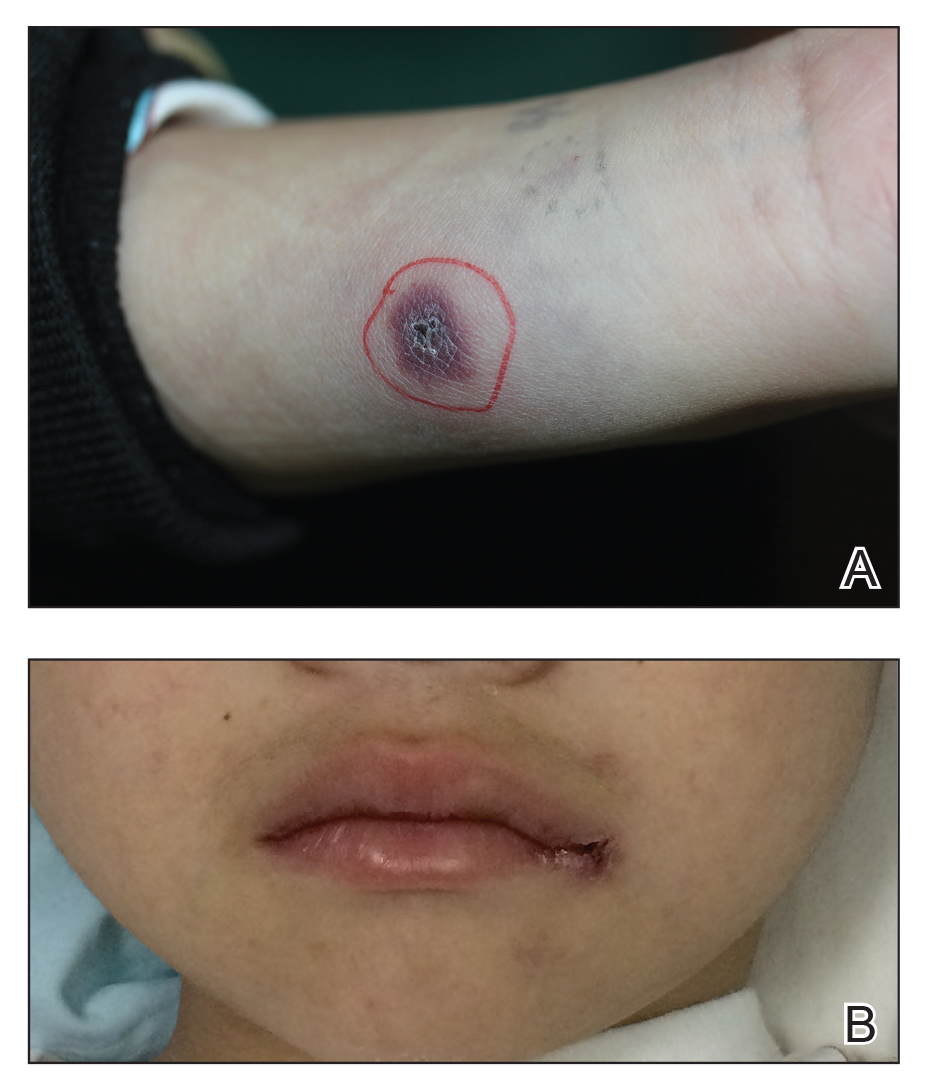
A 7-year-old Chinese boy presented with multiple painful oral and tongue ulcers of 2 weeks’ duration as well as acute onset of moderate to high fever (highest temperature, 39.3°C) for 5 days. The fever was reported to have run a relapsing course, accompanied by rigors but without convulsions or cognitive changes. At times, the patient had nasal congestion, nasal discharge, and cough. He also had a transient eruption on the back and hands as well as an indurated red nodule on the left forearm.
Before the patient was hospitalized, antibiotic therapy was administered by other physicians, but the condition of fever and oral ulcers did not improve. After the patient was hospitalized, new tender nodules emerged on the scalp, buttocks, and lower extremities. New ulcers also appeared on the palate.
History
Two months earlier, the patient had presented with a painful perioral skin ulcer that resolved after being treated as contagious eczema. Another dermatologist previously had considered a diagnosis of hand-foot-and-mouth disease.
The patient was born by normal spontaneous vaginal delivery, without abnormality. He was breastfed; feeding, growth, and the developmental history showed no abnormality. He was the family’s eldest child, with a healthy brother and sister. There was no history of familial illness. He received bacillus Calmette-Guérin and poliomyelitis vaccines after birth; the rest of the vaccine history was unclear. There was no history of immunologic abnormality.
Physical Examination
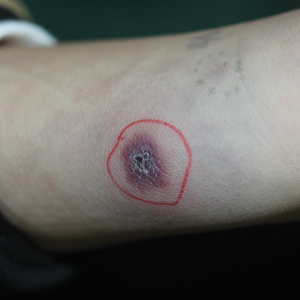
A 1.5×1.5-cm, warm, red nodule with a central black crust was noted on the left forearm (Figure 1A). Several similar lesions were noted on the buttocks, scalp, and lower extremities. Multiple ulcers, as large as 1 cm, were present on the tongue, palate, and left angle of the mouth (Figure 1B). The pharynx was congested, and the tonsils were mildly enlarged. Multiple enlarged, movable, nontender lymph nodes could be palpated in the cervical basins, axillae, and groin. No purpura or ecchymosis was detected.
Laboratory Results
Laboratory testing revealed a normal total white blood cell count (4.26×109/L [reference range, 4.0–12.0×109/L]), with normal neutrophils (1.36×109/L [reference range, 1.32–7.90×109/L]), lymphocytes (2.77×109/L [reference range, 1.20–6.00×109/L]), and monocytes (0.13×109/L [reference range, 0.08–0.80×109/L]); a mildly decreased hemoglobin level (115 g/L [reference range, 120–160 g/L]); a normal platelet count (102×109/L [reference range, 100–380×109/L]); an elevated lactate dehydrogenase level (614 U/L [reference range, 110–330 U/L]); an elevated α-hydroxybutyrate dehydrogenase level (483 U/L [reference range, 120–270 U/L]); elevated prothrombin time (15.3 s [reference range, 9–14 s]); elevated activated partial thromboplastin time (59.8 s [reference range, 20.6–39.6 s]); and an elevated D-dimer level (1.51 mg/L [reference range, <0.73 mg/L]). In addition, autoantibody testing revealed a positive antinuclear antibody titer of 1:320 and a strong positive anti–Ro-52 level.
The peripheral blood lymphocyte classification demonstrated a prominent elevated percentage of T lymphocytes, with predominantly CD8+ cells (CD3, 94.87%; CD8, 71.57%; CD4, 24.98%; CD4:CD8 ratio, 0.35) and a diminished percentage of B lymphocytes and natural killer (NK) cells. Epstein-Barr virus (EBV) antibody testing was positive for anti–viral capsid antigen (VCA) IgG and negative for anti-VCA IgM.
Smears of the ulcer on the tongue demonstrated gram-positive cocci, gram-negative bacilli, and diplococci. Culture of sputum showed methicillin-resistant Staphylococcus aureus. Inspection for acid-fast bacilli in sputum yielded negative results 3 times. A purified protein derivative skin test for Mycobacterium tuberculosis infection was negative.
Imaging and Other Studies
Computed tomography of the chest and abdomen demonstrated 2 nodular opacities on the lower right lung; spotted opacities on the upper right lung; floccular opacities on the rest area of the lung; mild pleural effusion; enlargement of lymph nodes on the mediastinum, the bilateral hilum of the lung, and mesentery; and hepatosplenomegaly. Electrocardiography showed sinus tachycardia. Nasal cavity endoscopy showed sinusitis. Fundus examination showed vasculopathy of the left retina. A colonoscopy showed normal mucosa.
Histopathology
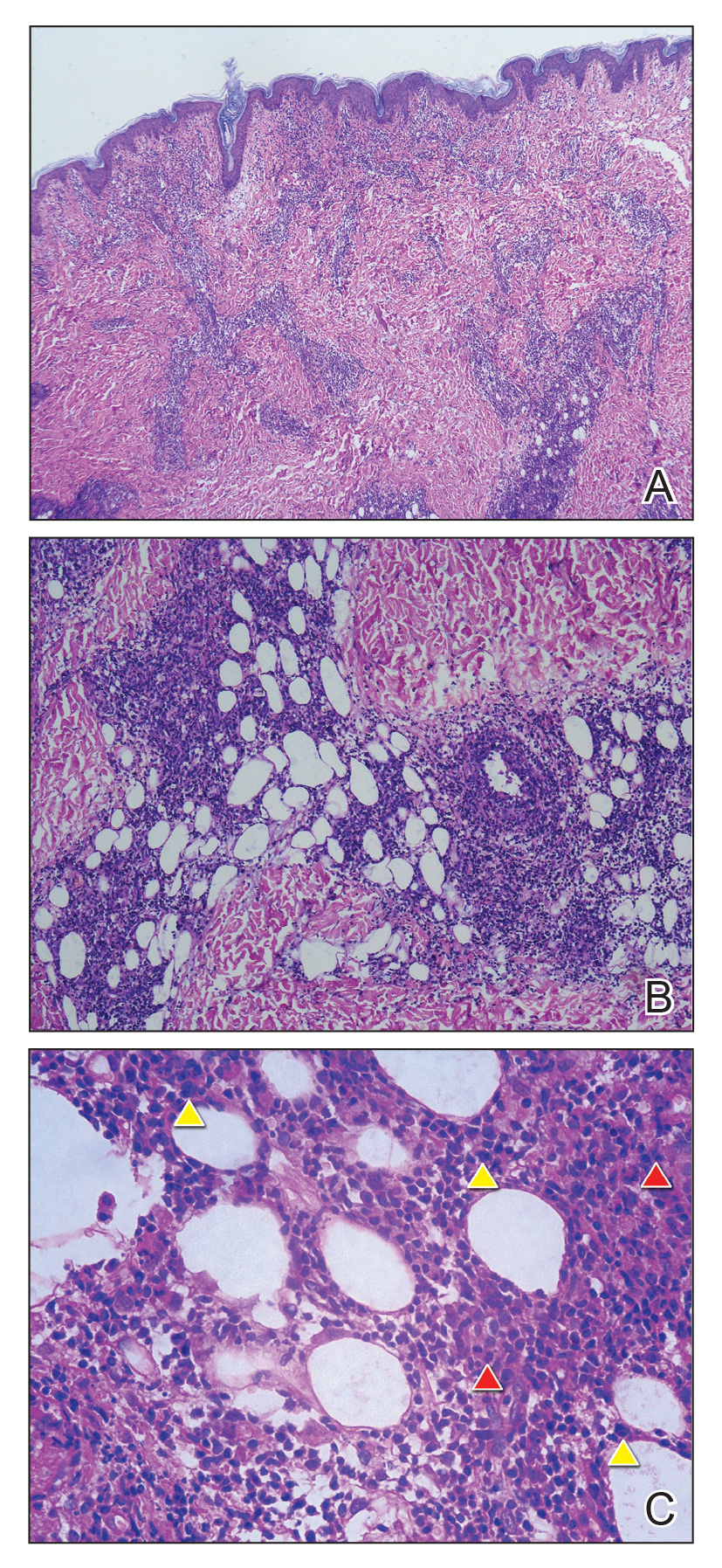
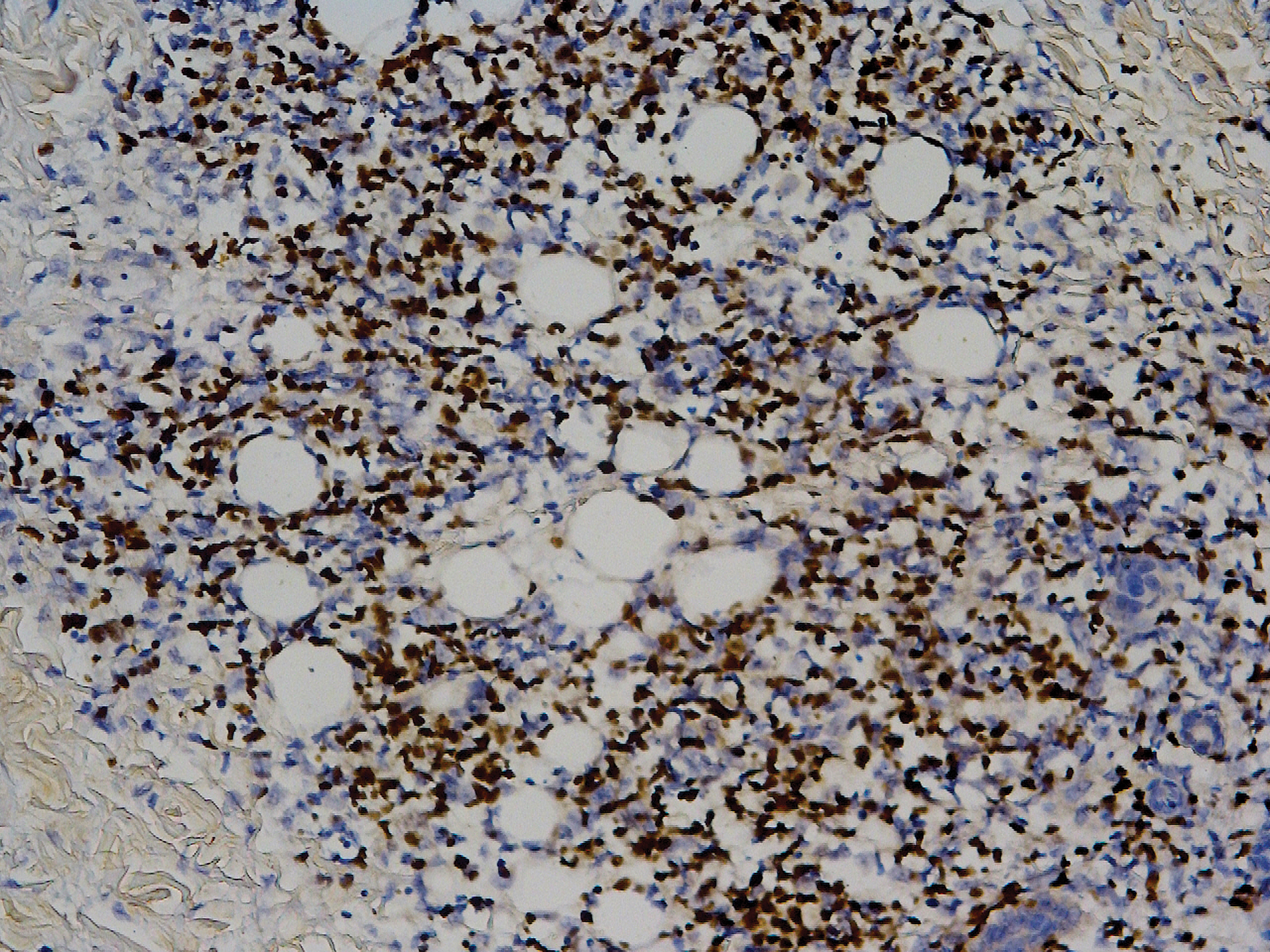
Biopsy of the nodule on the left arm showed dense, superficial to deep perivascular, periadnexal, perineural, and panniculitislike lymphoid infiltrates, as well as a sparse interstitial infiltrate with irregular and pleomorphic medium to large nuclei. Lymphoid cells showed mild epidermotropism, with tagging to the basal layer. Some vessel walls were infiltrated by similar cells (Figure 2). Infiltrative atypical lymphoid cells expressed CD3 and CD7 and were mostly CD8+, with a few CD4+ cells and most cells negative for CD5, CD20, CD30, CD56, and anaplastic lymphoma kinase. Cytotoxic markers granzyme B and T-cell intracellular antigen protein 1 were scattered positive. Immunostaining for Ki-67 protein highlighted an increased proliferative rate of 80% in malignant cells. In situ hybridization for EBV-encoded RNA (EBER) demonstrated EBV-positive atypical lymphoid cells (Figure 3). Analysis for T-cell receptor (TCR) γ gene rearrangement revealed a monoclonal pattern. Bone marrow aspirate showed proliferation of the 3 cell lines. The percentage of T lymphocytes was increased (20% of all nucleated cells). No hemophagocytic activity was found.
Diagnosis
A diagnosis of systemic EBV-positive T-cell lymphoma was made. Before the final diagnosis was made, the patient was treated by rheumatologists with antibiotics, antiviral drugs, nonsteroidal anti-inflammatory drugs, and other symptomatic treatments. Following antibiotic therapy, a sputum culture reverted to normal flora, the coagulation index (ie, prothrombin time, activated partial thromboplastin time) returned to normal, and the D-dimer level decreased to 1.19 mg/L.
The patient’s parents refused to accept chemotherapy for him. Instead, they chose herbal therapy only; 5 months later, they reported that all of his symptoms had resolved; however, the disease suddenly relapsed after another 7 months, with multiple skin nodules and fever. The patient died, even with chemotherapy in another hospital.
Comment
Prevalence and Presentation
Epstein-Barr virus is a ubiquitous γ-herpesvirus with tropism for B cells, affecting more than 90% of the adult population worldwide. In addition to infecting B cells, EBV is capable of infecting T and NK cells, leading to various EBV-related lymphoproliferative disorders (LPDs). The frequency and clinical presentation of infection varies based on the type of EBV-infected cells and the state of host immunity.1-3
Primary infection usually is asymptomatic and occurs early in life; when symptomatic, the disease usually presents as infectious mononucleosis (IM), characterized by polyclonal expansion of infected B cells and subsequent cytotoxic T-cell response. A diagnosis of EBV infection can be made by testing for specific IgM and IgG antibodies against VCA, early antigens, and EBV nuclear antigen proteins.3,4
Associated LPDs
Although most symptoms associated with IM resolve within weeks or months, persistent or recurrent IM-like symptoms or even lasting disease occasionally occur, particularly in children and young adults. This complication is known as chronic active EBV infection (CAEBV), frequently associated with EBV-infected T-cell or NK-cell proliferation, especially in East Asian populations.3,5
Epstein-Barr virus–positive T-cell and NK-cell LPDs of childhood include CAEBV infection of T-cell and NK-cell types and systemic EBV-positive T-cell lymphoma of childhood. The former includes hydroa vacciniforme–like LPD and severe mosquito bite allergy.3
Systemic EBV-Positive T-cell Lymphoma of Childhood
This entity occurs not only in children but also in adolescents and young adults. A fulminant illness characterized by clonal proliferation of EBV-infected cytotoxic T cells, it can develop shortly after primary EBV infection or is linked to CAEBV infection. The disorder is rare and has a racial predilection for Asian (ie, Japanese, Chinese, Korean) populations and indigenous populations of Mexico and Central and South America.6-8
Complications
Systemic EBV-positive T-cell lymphoma of childhood is often complicated by hemophagocytic syndrome, coagulopathy, sepsis, and multiorgan failure. Other signs and symptoms include high fever, rash, jaundice, diarrhea, pancytopenia, and hepatosplenomegaly. The liver, spleen, lymph nodes, and bone marrow are commonly involved, and the disease can involve skin, the heart, and the lungs.9,10
Diagnosis
When systemic EBV-positive T-cell lymphoma occurs shortly after IM, serology shows low or absent anti-VCA IgM and positive anti-VCA IgG. Infiltrating T cells usually are small and lack cytologic atypia; however, cases with pleomorphic, medium to large lymphoid cells, irregular nuclei, and frequent mitoses have been described. Hemophagocytosis can be seen in the liver, spleen, and bone marrow.3,11
The most typical phenotype of systemic EBV-positive T-cell lymphoma is CD2+CD3+CD8+CD20−CD56−, with expression of the cytotoxic granules known as T-cell intracellular antigen 1 and granzyme B. Rare cases of CD4+ and mixed CD4+/CD8+ phenotypes have been described, usually in the setting of CAEBV infection.3,12 Neoplastic cells have monoclonally rearranged TCR-γ genes and consistent EBER positivity with in situ hybridization.13 A final diagnosis is based on a comprehensive analysis of clinical, morphological, immunohistochemical, and molecular biological aspects.
Clinical Course and Prognosis
Most patients with systemic EBV-positive T-cell lymphoma have an aggressive clinical course with high mortality. In a few cases, patients were reported to respond to a regimen of etoposide and dexamethasone, followed by allogeneic hematopoietic stem cell transplantation.3
In recognition of the aggressive clinical behavior and desire to clearly distinguish systemic EBV-positive T-cell lymphoma from CAEBV infection, the older term systemic EBV-positive T-cell LPD of childhood, which had been introduced in 2008 to the World Health Organization classification, was changed to systemic EBV-positive T-cell lymphoma of childhood in the revised 2016 World Health Organization classification.6,12 However, Kim et al14 reported a case with excellent response to corticosteroid administration, suggesting that systemic EBV-positive T-cell lymphoma of childhood may be more heterogeneous in terms of prognosis.
Our patient presented with acute IM-like symptoms, including high fever, tonsillar enlargement, lymphadenopathy, and hepatosplenomegaly, as well as uncommon oral ulcers and skin lesions, including indurated nodules. Histopathologic changes in the skin nodule, proliferation in bone marrow, immunohistochemical phenotype, and positivity of EBER and TCR-γ monoclonal rearrangement were all consistent with systemic EBV-positive T-cell lymphoma of childhood. The patient was positive for VCA IgG and negative for VCA IgM, compatible with systemic EBV-positive T-cell lymphoma of childhood occurring shortly after IM. Neither pancytopenia, hemophagocytic syndrome, nor multiorgan failure occurred during the course.
Differential Diagnosis
It is important to distinguish IM from systemic EBV-positive T-cell lymphoma of childhood and CAEBV infection. Detection of anti–VCA IgM in the early stage, its disappearance during the clinical course, and appearance of anti-EBV–determined nuclear antigen is useful to distinguish IM from the neoplasms, as systemic EBV-positive T-cell lymphoma of childhood is negative for anti-EBV–determined nuclear antigen. Carefully following the clinical course also is important.3,15
Epstein-Barr virus–associated hemophagocytic lymphohistiocytosis can occur in association with systemic EBV-positive T-cell lymphoma of childhood and might represent a continuum of disease rather than distinct entities.14 The most useful marker for differentiating EBV-associated hemophagocytic lymphohistiocytosis and systemic EBV-positive T-cell lymphoma of childhood is an abnormal karyotype rather than molecular clonality.16
Outcome
Mortality risk in EBV-associated T-cell and NK-cell LPD is not primarily dependent on whether the lesion has progressed to lymphoma but instead is related to associated complications.17
Conclusion
Although systemic EBV-positive T-cell lymphoma of childhood is a rare disorder and has race predilection, dermatologists should be aware due to the aggressive clinical source and poor prognosis. Histopathology and in situ hybridization for EBER and TCR gene rearrangements are critical for final diagnosis. Although rare cases can show temporary resolution, the final outcome of this disease is not optimistic.
- Ameli F, Ghafourian F, Masir N. Systematic Epstein-Barr virus-positive T-cell lymphoproliferative disease presenting as a persistent fever and cough: a case report. J Med Case Rep. 2014;8:288.
- Kim HJ, Ko YH, Kim JE, et al. Epstein-Barr virus-associated lympho-proliferative disorders: review and update on 2016 WHO classification. J Pathol Transl Med. 2017;51:352-358.
- Dojcinov SD, Fend F, Quintanilla-Martinez L. EBV-positive lymphoproliferations of B- T- and NK-cell derivation in non-immunocompromised hosts [published online March 7, 2018]. Pathogens. doi:10.3390/pathogens7010028.
- Luzuriaga K, Sullivan JL. Infectious mononucleosis. N Engl J Med. 2010;362:1993-2000.
- Cohen JI, Kimura H, Nakamura S, et al. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8-9 September 2008. Ann Oncol. 2009;20:1472-1482.
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375-2390.
- Kim WY, Montes-Mojarro IA, Fend F, et al. Epstein-Barr virus-associated T and NK-cell lymphoproliferative diseases. Front Pediatr. 2019;7:71.
- Hong M, Ko YH, Yoo KH, et al. EBV-positive T/NK-cell lymphoproliferative disease of childhood. Korean J Pathol. 2013;47:137-147.
- Quintanilla-Martinez L, Kumar S, Fend F, et al. Fulminant EBV(+) T-cell lymphoproliferative disorder following acute/chronic EBV infection: a distinct clinicopathologic syndrome. Blood. 2000;96:443-451.
- Chen G, Chen L, Qin X, et al. Systemic Epstein-Barr virus positive T-cell lymphoproliferative disease of childhood with hemophagocytic syndrome. Int J Clin Exp Pathol. 2014;7:7110-7113.
- Grywalska E, Rolinski J. Epstein-Barr virus-associated lymphomas. Semin Oncol. 2015;42:291-303.
- Huang W, Lv N, Ying J, et al. Clinicopathological characteristics of four cases of EBV positive T-cell lymphoproliferative disorders of childhood in China. Int J Clin Exp Pathol. 2014;7:4991-4999.
- Tabanelli V, Agostinelli C, Sabattini E, et al. Systemic Epstein-Barr-virus-positive T cell lymphoproliferative childhood disease in a 22-year-old Caucasian man: a case report and review of the literature. J Med Case Rep. 2011;5:218.
- Kim DH, Kim M, Kim Y, et al. Systemic Epstein-Barr virus-positive T-cell lymphoproliferative disease of childhood with good response to steroid therapy. J Pediatr Hematol Oncol. 2017;39:e497-e500.
- Arai A, Yamaguchi T, Komatsu H, et al. Infectious mononucleosis accompanied by clonal proliferation of EBV-infected cells and infection of CD8-positive cells. Int J Hematol. 2014;99:671-675.
- Smith MC, Cohen DN, Greig B, et al. The ambiguous boundary between EBV-related hemophagocytic lymphohistiocytosis and systemic EBV-driven T cell lymphoproliferative disorder. Int J Clin Exp Pathol. 2014;7:5738-5749.
- Paik JH, Choe JY, Kim H, et al. Clinicopathological categorization of Epstein-Barr virus-positive T/NK-cell lymphoproliferative disease: an analysis of 42 cases with an emphasis on prognostic implications. Leuk Lymphoma. 2017;58:53-63.
Case Report
A 7-year-old Chinese boy presented with multiple painful oral and tongue ulcers of 2 weeks’ duration as well as acute onset of moderate to high fever (highest temperature, 39.3°C) for 5 days. The fever was reported to have run a relapsing course, accompanied by rigors but without convulsions or cognitive changes. At times, the patient had nasal congestion, nasal discharge, and cough. He also had a transient eruption on the back and hands as well as an indurated red nodule on the left forearm.
Before the patient was hospitalized, antibiotic therapy was administered by other physicians, but the condition of fever and oral ulcers did not improve. After the patient was hospitalized, new tender nodules emerged on the scalp, buttocks, and lower extremities. New ulcers also appeared on the palate.
History
Two months earlier, the patient had presented with a painful perioral skin ulcer that resolved after being treated as contagious eczema. Another dermatologist previously had considered a diagnosis of hand-foot-and-mouth disease.
The patient was born by normal spontaneous vaginal delivery, without abnormality. He was breastfed; feeding, growth, and the developmental history showed no abnormality. He was the family’s eldest child, with a healthy brother and sister. There was no history of familial illness. He received bacillus Calmette-Guérin and poliomyelitis vaccines after birth; the rest of the vaccine history was unclear. There was no history of immunologic abnormality.
Physical Examination
A 1.5×1.5-cm, warm, red nodule with a central black crust was noted on the left forearm (Figure 1A). Several similar lesions were noted on the buttocks, scalp, and lower extremities. Multiple ulcers, as large as 1 cm, were present on the tongue, palate, and left angle of the mouth (Figure 1B). The pharynx was congested, and the tonsils were mildly enlarged. Multiple enlarged, movable, nontender lymph nodes could be palpated in the cervical basins, axillae, and groin. No purpura or ecchymosis was detected.
Laboratory Results
Laboratory testing revealed a normal total white blood cell count (4.26×109/L [reference range, 4.0–12.0×109/L]), with normal neutrophils (1.36×109/L [reference range, 1.32–7.90×109/L]), lymphocytes (2.77×109/L [reference range, 1.20–6.00×109/L]), and monocytes (0.13×109/L [reference range, 0.08–0.80×109/L]); a mildly decreased hemoglobin level (115 g/L [reference range, 120–160 g/L]); a normal platelet count (102×109/L [reference range, 100–380×109/L]); an elevated lactate dehydrogenase level (614 U/L [reference range, 110–330 U/L]); an elevated α-hydroxybutyrate dehydrogenase level (483 U/L [reference range, 120–270 U/L]); elevated prothrombin time (15.3 s [reference range, 9–14 s]); elevated activated partial thromboplastin time (59.8 s [reference range, 20.6–39.6 s]); and an elevated D-dimer level (1.51 mg/L [reference range, <0.73 mg/L]). In addition, autoantibody testing revealed a positive antinuclear antibody titer of 1:320 and a strong positive anti–Ro-52 level.
The peripheral blood lymphocyte classification demonstrated a prominent elevated percentage of T lymphocytes, with predominantly CD8+ cells (CD3, 94.87%; CD8, 71.57%; CD4, 24.98%; CD4:CD8 ratio, 0.35) and a diminished percentage of B lymphocytes and natural killer (NK) cells. Epstein-Barr virus (EBV) antibody testing was positive for anti–viral capsid antigen (VCA) IgG and negative for anti-VCA IgM.
Smears of the ulcer on the tongue demonstrated gram-positive cocci, gram-negative bacilli, and diplococci. Culture of sputum showed methicillin-resistant Staphylococcus aureus. Inspection for acid-fast bacilli in sputum yielded negative results 3 times. A purified protein derivative skin test for Mycobacterium tuberculosis infection was negative.
Imaging and Other Studies
Computed tomography of the chest and abdomen demonstrated 2 nodular opacities on the lower right lung; spotted opacities on the upper right lung; floccular opacities on the rest area of the lung; mild pleural effusion; enlargement of lymph nodes on the mediastinum, the bilateral hilum of the lung, and mesentery; and hepatosplenomegaly. Electrocardiography showed sinus tachycardia. Nasal cavity endoscopy showed sinusitis. Fundus examination showed vasculopathy of the left retina. A colonoscopy showed normal mucosa.
Histopathology
Biopsy of the nodule on the left arm showed dense, superficial to deep perivascular, periadnexal, perineural, and panniculitislike lymphoid infiltrates, as well as a sparse interstitial infiltrate with irregular and pleomorphic medium to large nuclei. Lymphoid cells showed mild epidermotropism, with tagging to the basal layer. Some vessel walls were infiltrated by similar cells (Figure 2). Infiltrative atypical lymphoid cells expressed CD3 and CD7 and were mostly CD8+, with a few CD4+ cells and most cells negative for CD5, CD20, CD30, CD56, and anaplastic lymphoma kinase. Cytotoxic markers granzyme B and T-cell intracellular antigen protein 1 were scattered positive. Immunostaining for Ki-67 protein highlighted an increased proliferative rate of 80% in malignant cells. In situ hybridization for EBV-encoded RNA (EBER) demonstrated EBV-positive atypical lymphoid cells (Figure 3). Analysis for T-cell receptor (TCR) γ gene rearrangement revealed a monoclonal pattern. Bone marrow aspirate showed proliferation of the 3 cell lines. The percentage of T lymphocytes was increased (20% of all nucleated cells). No hemophagocytic activity was found.
Diagnosis
A diagnosis of systemic EBV-positive T-cell lymphoma was made. Before the final diagnosis was made, the patient was treated by rheumatologists with antibiotics, antiviral drugs, nonsteroidal anti-inflammatory drugs, and other symptomatic treatments. Following antibiotic therapy, a sputum culture reverted to normal flora, the coagulation index (ie, prothrombin time, activated partial thromboplastin time) returned to normal, and the D-dimer level decreased to 1.19 mg/L.
The patient’s parents refused to accept chemotherapy for him. Instead, they chose herbal therapy only; 5 months later, they reported that all of his symptoms had resolved; however, the disease suddenly relapsed after another 7 months, with multiple skin nodules and fever. The patient died, even with chemotherapy in another hospital.
Comment
Prevalence and Presentation
Epstein-Barr virus is a ubiquitous γ-herpesvirus with tropism for B cells, affecting more than 90% of the adult population worldwide. In addition to infecting B cells, EBV is capable of infecting T and NK cells, leading to various EBV-related lymphoproliferative disorders (LPDs). The frequency and clinical presentation of infection varies based on the type of EBV-infected cells and the state of host immunity.1-3
Primary infection usually is asymptomatic and occurs early in life; when symptomatic, the disease usually presents as infectious mononucleosis (IM), characterized by polyclonal expansion of infected B cells and subsequent cytotoxic T-cell response. A diagnosis of EBV infection can be made by testing for specific IgM and IgG antibodies against VCA, early antigens, and EBV nuclear antigen proteins.3,4
Associated LPDs
Although most symptoms associated with IM resolve within weeks or months, persistent or recurrent IM-like symptoms or even lasting disease occasionally occur, particularly in children and young adults. This complication is known as chronic active EBV infection (CAEBV), frequently associated with EBV-infected T-cell or NK-cell proliferation, especially in East Asian populations.3,5
Epstein-Barr virus–positive T-cell and NK-cell LPDs of childhood include CAEBV infection of T-cell and NK-cell types and systemic EBV-positive T-cell lymphoma of childhood. The former includes hydroa vacciniforme–like LPD and severe mosquito bite allergy.3
Systemic EBV-Positive T-cell Lymphoma of Childhood
This entity occurs not only in children but also in adolescents and young adults. A fulminant illness characterized by clonal proliferation of EBV-infected cytotoxic T cells, it can develop shortly after primary EBV infection or is linked to CAEBV infection. The disorder is rare and has a racial predilection for Asian (ie, Japanese, Chinese, Korean) populations and indigenous populations of Mexico and Central and South America.6-8
Complications
Systemic EBV-positive T-cell lymphoma of childhood is often complicated by hemophagocytic syndrome, coagulopathy, sepsis, and multiorgan failure. Other signs and symptoms include high fever, rash, jaundice, diarrhea, pancytopenia, and hepatosplenomegaly. The liver, spleen, lymph nodes, and bone marrow are commonly involved, and the disease can involve skin, the heart, and the lungs.9,10
Diagnosis
When systemic EBV-positive T-cell lymphoma occurs shortly after IM, serology shows low or absent anti-VCA IgM and positive anti-VCA IgG. Infiltrating T cells usually are small and lack cytologic atypia; however, cases with pleomorphic, medium to large lymphoid cells, irregular nuclei, and frequent mitoses have been described. Hemophagocytosis can be seen in the liver, spleen, and bone marrow.3,11
The most typical phenotype of systemic EBV-positive T-cell lymphoma is CD2+CD3+CD8+CD20−CD56−, with expression of the cytotoxic granules known as T-cell intracellular antigen 1 and granzyme B. Rare cases of CD4+ and mixed CD4+/CD8+ phenotypes have been described, usually in the setting of CAEBV infection.3,12 Neoplastic cells have monoclonally rearranged TCR-γ genes and consistent EBER positivity with in situ hybridization.13 A final diagnosis is based on a comprehensive analysis of clinical, morphological, immunohistochemical, and molecular biological aspects.
Clinical Course and Prognosis
Most patients with systemic EBV-positive T-cell lymphoma have an aggressive clinical course with high mortality. In a few cases, patients were reported to respond to a regimen of etoposide and dexamethasone, followed by allogeneic hematopoietic stem cell transplantation.3
In recognition of the aggressive clinical behavior and desire to clearly distinguish systemic EBV-positive T-cell lymphoma from CAEBV infection, the older term systemic EBV-positive T-cell LPD of childhood, which had been introduced in 2008 to the World Health Organization classification, was changed to systemic EBV-positive T-cell lymphoma of childhood in the revised 2016 World Health Organization classification.6,12 However, Kim et al14 reported a case with excellent response to corticosteroid administration, suggesting that systemic EBV-positive T-cell lymphoma of childhood may be more heterogeneous in terms of prognosis.
Our patient presented with acute IM-like symptoms, including high fever, tonsillar enlargement, lymphadenopathy, and hepatosplenomegaly, as well as uncommon oral ulcers and skin lesions, including indurated nodules. Histopathologic changes in the skin nodule, proliferation in bone marrow, immunohistochemical phenotype, and positivity of EBER and TCR-γ monoclonal rearrangement were all consistent with systemic EBV-positive T-cell lymphoma of childhood. The patient was positive for VCA IgG and negative for VCA IgM, compatible with systemic EBV-positive T-cell lymphoma of childhood occurring shortly after IM. Neither pancytopenia, hemophagocytic syndrome, nor multiorgan failure occurred during the course.
Differential Diagnosis
It is important to distinguish IM from systemic EBV-positive T-cell lymphoma of childhood and CAEBV infection. Detection of anti–VCA IgM in the early stage, its disappearance during the clinical course, and appearance of anti-EBV–determined nuclear antigen is useful to distinguish IM from the neoplasms, as systemic EBV-positive T-cell lymphoma of childhood is negative for anti-EBV–determined nuclear antigen. Carefully following the clinical course also is important.3,15
Epstein-Barr virus–associated hemophagocytic lymphohistiocytosis can occur in association with systemic EBV-positive T-cell lymphoma of childhood and might represent a continuum of disease rather than distinct entities.14 The most useful marker for differentiating EBV-associated hemophagocytic lymphohistiocytosis and systemic EBV-positive T-cell lymphoma of childhood is an abnormal karyotype rather than molecular clonality.16
Outcome
Mortality risk in EBV-associated T-cell and NK-cell LPD is not primarily dependent on whether the lesion has progressed to lymphoma but instead is related to associated complications.17
Conclusion
Although systemic EBV-positive T-cell lymphoma of childhood is a rare disorder and has race predilection, dermatologists should be aware due to the aggressive clinical source and poor prognosis. Histopathology and in situ hybridization for EBER and TCR gene rearrangements are critical for final diagnosis. Although rare cases can show temporary resolution, the final outcome of this disease is not optimistic.
Case Report
A 7-year-old Chinese boy presented with multiple painful oral and tongue ulcers of 2 weeks’ duration as well as acute onset of moderate to high fever (highest temperature, 39.3°C) for 5 days. The fever was reported to have run a relapsing course, accompanied by rigors but without convulsions or cognitive changes. At times, the patient had nasal congestion, nasal discharge, and cough. He also had a transient eruption on the back and hands as well as an indurated red nodule on the left forearm.
Before the patient was hospitalized, antibiotic therapy was administered by other physicians, but the condition of fever and oral ulcers did not improve. After the patient was hospitalized, new tender nodules emerged on the scalp, buttocks, and lower extremities. New ulcers also appeared on the palate.
History
Two months earlier, the patient had presented with a painful perioral skin ulcer that resolved after being treated as contagious eczema. Another dermatologist previously had considered a diagnosis of hand-foot-and-mouth disease.
The patient was born by normal spontaneous vaginal delivery, without abnormality. He was breastfed; feeding, growth, and the developmental history showed no abnormality. He was the family’s eldest child, with a healthy brother and sister. There was no history of familial illness. He received bacillus Calmette-Guérin and poliomyelitis vaccines after birth; the rest of the vaccine history was unclear. There was no history of immunologic abnormality.
Physical Examination
A 1.5×1.5-cm, warm, red nodule with a central black crust was noted on the left forearm (Figure 1A). Several similar lesions were noted on the buttocks, scalp, and lower extremities. Multiple ulcers, as large as 1 cm, were present on the tongue, palate, and left angle of the mouth (Figure 1B). The pharynx was congested, and the tonsils were mildly enlarged. Multiple enlarged, movable, nontender lymph nodes could be palpated in the cervical basins, axillae, and groin. No purpura or ecchymosis was detected.
Laboratory Results
Laboratory testing revealed a normal total white blood cell count (4.26×109/L [reference range, 4.0–12.0×109/L]), with normal neutrophils (1.36×109/L [reference range, 1.32–7.90×109/L]), lymphocytes (2.77×109/L [reference range, 1.20–6.00×109/L]), and monocytes (0.13×109/L [reference range, 0.08–0.80×109/L]); a mildly decreased hemoglobin level (115 g/L [reference range, 120–160 g/L]); a normal platelet count (102×109/L [reference range, 100–380×109/L]); an elevated lactate dehydrogenase level (614 U/L [reference range, 110–330 U/L]); an elevated α-hydroxybutyrate dehydrogenase level (483 U/L [reference range, 120–270 U/L]); elevated prothrombin time (15.3 s [reference range, 9–14 s]); elevated activated partial thromboplastin time (59.8 s [reference range, 20.6–39.6 s]); and an elevated D-dimer level (1.51 mg/L [reference range, <0.73 mg/L]). In addition, autoantibody testing revealed a positive antinuclear antibody titer of 1:320 and a strong positive anti–Ro-52 level.
The peripheral blood lymphocyte classification demonstrated a prominent elevated percentage of T lymphocytes, with predominantly CD8+ cells (CD3, 94.87%; CD8, 71.57%; CD4, 24.98%; CD4:CD8 ratio, 0.35) and a diminished percentage of B lymphocytes and natural killer (NK) cells. Epstein-Barr virus (EBV) antibody testing was positive for anti–viral capsid antigen (VCA) IgG and negative for anti-VCA IgM.
Smears of the ulcer on the tongue demonstrated gram-positive cocci, gram-negative bacilli, and diplococci. Culture of sputum showed methicillin-resistant Staphylococcus aureus. Inspection for acid-fast bacilli in sputum yielded negative results 3 times. A purified protein derivative skin test for Mycobacterium tuberculosis infection was negative.
Imaging and Other Studies
Computed tomography of the chest and abdomen demonstrated 2 nodular opacities on the lower right lung; spotted opacities on the upper right lung; floccular opacities on the rest area of the lung; mild pleural effusion; enlargement of lymph nodes on the mediastinum, the bilateral hilum of the lung, and mesentery; and hepatosplenomegaly. Electrocardiography showed sinus tachycardia. Nasal cavity endoscopy showed sinusitis. Fundus examination showed vasculopathy of the left retina. A colonoscopy showed normal mucosa.
Histopathology
Biopsy of the nodule on the left arm showed dense, superficial to deep perivascular, periadnexal, perineural, and panniculitislike lymphoid infiltrates, as well as a sparse interstitial infiltrate with irregular and pleomorphic medium to large nuclei. Lymphoid cells showed mild epidermotropism, with tagging to the basal layer. Some vessel walls were infiltrated by similar cells (Figure 2). Infiltrative atypical lymphoid cells expressed CD3 and CD7 and were mostly CD8+, with a few CD4+ cells and most cells negative for CD5, CD20, CD30, CD56, and anaplastic lymphoma kinase. Cytotoxic markers granzyme B and T-cell intracellular antigen protein 1 were scattered positive. Immunostaining for Ki-67 protein highlighted an increased proliferative rate of 80% in malignant cells. In situ hybridization for EBV-encoded RNA (EBER) demonstrated EBV-positive atypical lymphoid cells (Figure 3). Analysis for T-cell receptor (TCR) γ gene rearrangement revealed a monoclonal pattern. Bone marrow aspirate showed proliferation of the 3 cell lines. The percentage of T lymphocytes was increased (20% of all nucleated cells). No hemophagocytic activity was found.
Diagnosis
A diagnosis of systemic EBV-positive T-cell lymphoma was made. Before the final diagnosis was made, the patient was treated by rheumatologists with antibiotics, antiviral drugs, nonsteroidal anti-inflammatory drugs, and other symptomatic treatments. Following antibiotic therapy, a sputum culture reverted to normal flora, the coagulation index (ie, prothrombin time, activated partial thromboplastin time) returned to normal, and the D-dimer level decreased to 1.19 mg/L.
The patient’s parents refused to accept chemotherapy for him. Instead, they chose herbal therapy only; 5 months later, they reported that all of his symptoms had resolved; however, the disease suddenly relapsed after another 7 months, with multiple skin nodules and fever. The patient died, even with chemotherapy in another hospital.
Comment
Prevalence and Presentation
Epstein-Barr virus is a ubiquitous γ-herpesvirus with tropism for B cells, affecting more than 90% of the adult population worldwide. In addition to infecting B cells, EBV is capable of infecting T and NK cells, leading to various EBV-related lymphoproliferative disorders (LPDs). The frequency and clinical presentation of infection varies based on the type of EBV-infected cells and the state of host immunity.1-3
Primary infection usually is asymptomatic and occurs early in life; when symptomatic, the disease usually presents as infectious mononucleosis (IM), characterized by polyclonal expansion of infected B cells and subsequent cytotoxic T-cell response. A diagnosis of EBV infection can be made by testing for specific IgM and IgG antibodies against VCA, early antigens, and EBV nuclear antigen proteins.3,4
Associated LPDs
Although most symptoms associated with IM resolve within weeks or months, persistent or recurrent IM-like symptoms or even lasting disease occasionally occur, particularly in children and young adults. This complication is known as chronic active EBV infection (CAEBV), frequently associated with EBV-infected T-cell or NK-cell proliferation, especially in East Asian populations.3,5
Epstein-Barr virus–positive T-cell and NK-cell LPDs of childhood include CAEBV infection of T-cell and NK-cell types and systemic EBV-positive T-cell lymphoma of childhood. The former includes hydroa vacciniforme–like LPD and severe mosquito bite allergy.3
Systemic EBV-Positive T-cell Lymphoma of Childhood
This entity occurs not only in children but also in adolescents and young adults. A fulminant illness characterized by clonal proliferation of EBV-infected cytotoxic T cells, it can develop shortly after primary EBV infection or is linked to CAEBV infection. The disorder is rare and has a racial predilection for Asian (ie, Japanese, Chinese, Korean) populations and indigenous populations of Mexico and Central and South America.6-8
Complications
Systemic EBV-positive T-cell lymphoma of childhood is often complicated by hemophagocytic syndrome, coagulopathy, sepsis, and multiorgan failure. Other signs and symptoms include high fever, rash, jaundice, diarrhea, pancytopenia, and hepatosplenomegaly. The liver, spleen, lymph nodes, and bone marrow are commonly involved, and the disease can involve skin, the heart, and the lungs.9,10
Diagnosis
When systemic EBV-positive T-cell lymphoma occurs shortly after IM, serology shows low or absent anti-VCA IgM and positive anti-VCA IgG. Infiltrating T cells usually are small and lack cytologic atypia; however, cases with pleomorphic, medium to large lymphoid cells, irregular nuclei, and frequent mitoses have been described. Hemophagocytosis can be seen in the liver, spleen, and bone marrow.3,11
The most typical phenotype of systemic EBV-positive T-cell lymphoma is CD2+CD3+CD8+CD20−CD56−, with expression of the cytotoxic granules known as T-cell intracellular antigen 1 and granzyme B. Rare cases of CD4+ and mixed CD4+/CD8+ phenotypes have been described, usually in the setting of CAEBV infection.3,12 Neoplastic cells have monoclonally rearranged TCR-γ genes and consistent EBER positivity with in situ hybridization.13 A final diagnosis is based on a comprehensive analysis of clinical, morphological, immunohistochemical, and molecular biological aspects.
Clinical Course and Prognosis
Most patients with systemic EBV-positive T-cell lymphoma have an aggressive clinical course with high mortality. In a few cases, patients were reported to respond to a regimen of etoposide and dexamethasone, followed by allogeneic hematopoietic stem cell transplantation.3
In recognition of the aggressive clinical behavior and desire to clearly distinguish systemic EBV-positive T-cell lymphoma from CAEBV infection, the older term systemic EBV-positive T-cell LPD of childhood, which had been introduced in 2008 to the World Health Organization classification, was changed to systemic EBV-positive T-cell lymphoma of childhood in the revised 2016 World Health Organization classification.6,12 However, Kim et al14 reported a case with excellent response to corticosteroid administration, suggesting that systemic EBV-positive T-cell lymphoma of childhood may be more heterogeneous in terms of prognosis.
Our patient presented with acute IM-like symptoms, including high fever, tonsillar enlargement, lymphadenopathy, and hepatosplenomegaly, as well as uncommon oral ulcers and skin lesions, including indurated nodules. Histopathologic changes in the skin nodule, proliferation in bone marrow, immunohistochemical phenotype, and positivity of EBER and TCR-γ monoclonal rearrangement were all consistent with systemic EBV-positive T-cell lymphoma of childhood. The patient was positive for VCA IgG and negative for VCA IgM, compatible with systemic EBV-positive T-cell lymphoma of childhood occurring shortly after IM. Neither pancytopenia, hemophagocytic syndrome, nor multiorgan failure occurred during the course.
Differential Diagnosis
It is important to distinguish IM from systemic EBV-positive T-cell lymphoma of childhood and CAEBV infection. Detection of anti–VCA IgM in the early stage, its disappearance during the clinical course, and appearance of anti-EBV–determined nuclear antigen is useful to distinguish IM from the neoplasms, as systemic EBV-positive T-cell lymphoma of childhood is negative for anti-EBV–determined nuclear antigen. Carefully following the clinical course also is important.3,15
Epstein-Barr virus–associated hemophagocytic lymphohistiocytosis can occur in association with systemic EBV-positive T-cell lymphoma of childhood and might represent a continuum of disease rather than distinct entities.14 The most useful marker for differentiating EBV-associated hemophagocytic lymphohistiocytosis and systemic EBV-positive T-cell lymphoma of childhood is an abnormal karyotype rather than molecular clonality.16
Outcome
Mortality risk in EBV-associated T-cell and NK-cell LPD is not primarily dependent on whether the lesion has progressed to lymphoma but instead is related to associated complications.17
Conclusion
Although systemic EBV-positive T-cell lymphoma of childhood is a rare disorder and has race predilection, dermatologists should be aware due to the aggressive clinical source and poor prognosis. Histopathology and in situ hybridization for EBER and TCR gene rearrangements are critical for final diagnosis. Although rare cases can show temporary resolution, the final outcome of this disease is not optimistic.
- Ameli F, Ghafourian F, Masir N. Systematic Epstein-Barr virus-positive T-cell lymphoproliferative disease presenting as a persistent fever and cough: a case report. J Med Case Rep. 2014;8:288.
- Kim HJ, Ko YH, Kim JE, et al. Epstein-Barr virus-associated lympho-proliferative disorders: review and update on 2016 WHO classification. J Pathol Transl Med. 2017;51:352-358.
- Dojcinov SD, Fend F, Quintanilla-Martinez L. EBV-positive lymphoproliferations of B- T- and NK-cell derivation in non-immunocompromised hosts [published online March 7, 2018]. Pathogens. doi:10.3390/pathogens7010028.
- Luzuriaga K, Sullivan JL. Infectious mononucleosis. N Engl J Med. 2010;362:1993-2000.
- Cohen JI, Kimura H, Nakamura S, et al. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8-9 September 2008. Ann Oncol. 2009;20:1472-1482.
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375-2390.
- Kim WY, Montes-Mojarro IA, Fend F, et al. Epstein-Barr virus-associated T and NK-cell lymphoproliferative diseases. Front Pediatr. 2019;7:71.
- Hong M, Ko YH, Yoo KH, et al. EBV-positive T/NK-cell lymphoproliferative disease of childhood. Korean J Pathol. 2013;47:137-147.
- Quintanilla-Martinez L, Kumar S, Fend F, et al. Fulminant EBV(+) T-cell lymphoproliferative disorder following acute/chronic EBV infection: a distinct clinicopathologic syndrome. Blood. 2000;96:443-451.
- Chen G, Chen L, Qin X, et al. Systemic Epstein-Barr virus positive T-cell lymphoproliferative disease of childhood with hemophagocytic syndrome. Int J Clin Exp Pathol. 2014;7:7110-7113.
- Grywalska E, Rolinski J. Epstein-Barr virus-associated lymphomas. Semin Oncol. 2015;42:291-303.
- Huang W, Lv N, Ying J, et al. Clinicopathological characteristics of four cases of EBV positive T-cell lymphoproliferative disorders of childhood in China. Int J Clin Exp Pathol. 2014;7:4991-4999.
- Tabanelli V, Agostinelli C, Sabattini E, et al. Systemic Epstein-Barr-virus-positive T cell lymphoproliferative childhood disease in a 22-year-old Caucasian man: a case report and review of the literature. J Med Case Rep. 2011;5:218.
- Kim DH, Kim M, Kim Y, et al. Systemic Epstein-Barr virus-positive T-cell lymphoproliferative disease of childhood with good response to steroid therapy. J Pediatr Hematol Oncol. 2017;39:e497-e500.
- Arai A, Yamaguchi T, Komatsu H, et al. Infectious mononucleosis accompanied by clonal proliferation of EBV-infected cells and infection of CD8-positive cells. Int J Hematol. 2014;99:671-675.
- Smith MC, Cohen DN, Greig B, et al. The ambiguous boundary between EBV-related hemophagocytic lymphohistiocytosis and systemic EBV-driven T cell lymphoproliferative disorder. Int J Clin Exp Pathol. 2014;7:5738-5749.
- Paik JH, Choe JY, Kim H, et al. Clinicopathological categorization of Epstein-Barr virus-positive T/NK-cell lymphoproliferative disease: an analysis of 42 cases with an emphasis on prognostic implications. Leuk Lymphoma. 2017;58:53-63.
- Ameli F, Ghafourian F, Masir N. Systematic Epstein-Barr virus-positive T-cell lymphoproliferative disease presenting as a persistent fever and cough: a case report. J Med Case Rep. 2014;8:288.
- Kim HJ, Ko YH, Kim JE, et al. Epstein-Barr virus-associated lympho-proliferative disorders: review and update on 2016 WHO classification. J Pathol Transl Med. 2017;51:352-358.
- Dojcinov SD, Fend F, Quintanilla-Martinez L. EBV-positive lymphoproliferations of B- T- and NK-cell derivation in non-immunocompromised hosts [published online March 7, 2018]. Pathogens. doi:10.3390/pathogens7010028.
- Luzuriaga K, Sullivan JL. Infectious mononucleosis. N Engl J Med. 2010;362:1993-2000.
- Cohen JI, Kimura H, Nakamura S, et al. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8-9 September 2008. Ann Oncol. 2009;20:1472-1482.
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375-2390.
- Kim WY, Montes-Mojarro IA, Fend F, et al. Epstein-Barr virus-associated T and NK-cell lymphoproliferative diseases. Front Pediatr. 2019;7:71.
- Hong M, Ko YH, Yoo KH, et al. EBV-positive T/NK-cell lymphoproliferative disease of childhood. Korean J Pathol. 2013;47:137-147.
- Quintanilla-Martinez L, Kumar S, Fend F, et al. Fulminant EBV(+) T-cell lymphoproliferative disorder following acute/chronic EBV infection: a distinct clinicopathologic syndrome. Blood. 2000;96:443-451.
- Chen G, Chen L, Qin X, et al. Systemic Epstein-Barr virus positive T-cell lymphoproliferative disease of childhood with hemophagocytic syndrome. Int J Clin Exp Pathol. 2014;7:7110-7113.
- Grywalska E, Rolinski J. Epstein-Barr virus-associated lymphomas. Semin Oncol. 2015;42:291-303.
- Huang W, Lv N, Ying J, et al. Clinicopathological characteristics of four cases of EBV positive T-cell lymphoproliferative disorders of childhood in China. Int J Clin Exp Pathol. 2014;7:4991-4999.
- Tabanelli V, Agostinelli C, Sabattini E, et al. Systemic Epstein-Barr-virus-positive T cell lymphoproliferative childhood disease in a 22-year-old Caucasian man: a case report and review of the literature. J Med Case Rep. 2011;5:218.
- Kim DH, Kim M, Kim Y, et al. Systemic Epstein-Barr virus-positive T-cell lymphoproliferative disease of childhood with good response to steroid therapy. J Pediatr Hematol Oncol. 2017;39:e497-e500.
- Arai A, Yamaguchi T, Komatsu H, et al. Infectious mononucleosis accompanied by clonal proliferation of EBV-infected cells and infection of CD8-positive cells. Int J Hematol. 2014;99:671-675.
- Smith MC, Cohen DN, Greig B, et al. The ambiguous boundary between EBV-related hemophagocytic lymphohistiocytosis and systemic EBV-driven T cell lymphoproliferative disorder. Int J Clin Exp Pathol. 2014;7:5738-5749.
- Paik JH, Choe JY, Kim H, et al. Clinicopathological categorization of Epstein-Barr virus-positive T/NK-cell lymphoproliferative disease: an analysis of 42 cases with an emphasis on prognostic implications. Leuk Lymphoma. 2017;58:53-63.
Practice Points
- Systemic Epstein-Barr virus (EBV)–positive T-cell lymphoma of childhood is a fulminant illness with a predilection for Asians and indigenous populations from Mexico and Central and South America. In most patients, the disease has an aggressive clinical course with high mortality.
- The disease often is complicated by hemophagocytic syndrome, coagulopathy, sepsis, and multiorgan failure. When these severe complications are absent, the prognosis might be better.
- In situ hybridization for EBV-encoded RNA and for T-cell receptor gene rearrangements is an important tool to establish the diagnosis as well as for treatment options and predicting the prognosis.