User login
Ascites in a 42-year-old woman
A 42-year-old woman is admitted to the hospital with worsening shortness of breath on exertion, poor exercise tolerance, leg edema, and swelling of the abdomen. Her symptoms have been getting worse over the last 4 months. She reports no history of fever, chills, night sweats, bleeding disorder, joint pain, weight loss, or loss of appetite.
She has type 2 diabetes mellitus and hypothyroidism. She had rheumatoid arthritis but said it was “inactive,” not requiring treatment for the last 18 years. Three months ago, she underwent a total hysterectomy and salpingo-oophorectomy for a complex adnexal mass, biopsy of which revealed a benign mucinous ovarian cyst.
Her current medications include furosemide, levothyroxine, and metformin. She is an ex-smoker with a 7 pack-year history. She drinks a glass of wine on social occasions only. Her family history is unremarkable.
On examination, she is not in distress and she has no fever. She has jugular venous distention of 5 cm, tense ascites, and marked edema of the legs, as well as hyperpigmented patches and erythematous plaques over both shins. Neck palpation reveals no lymphadenopathy or thyromegaly.
Her liver and the tip of the spleen are palpable following paracentesis, once ascitic fluid is removed.
The cardiovascular examination is normal. Chest auscultation reveals decreased breath sounds at the right lung base with bibasilar crackles. No focal neurologic deficit is noted on clinical examination.
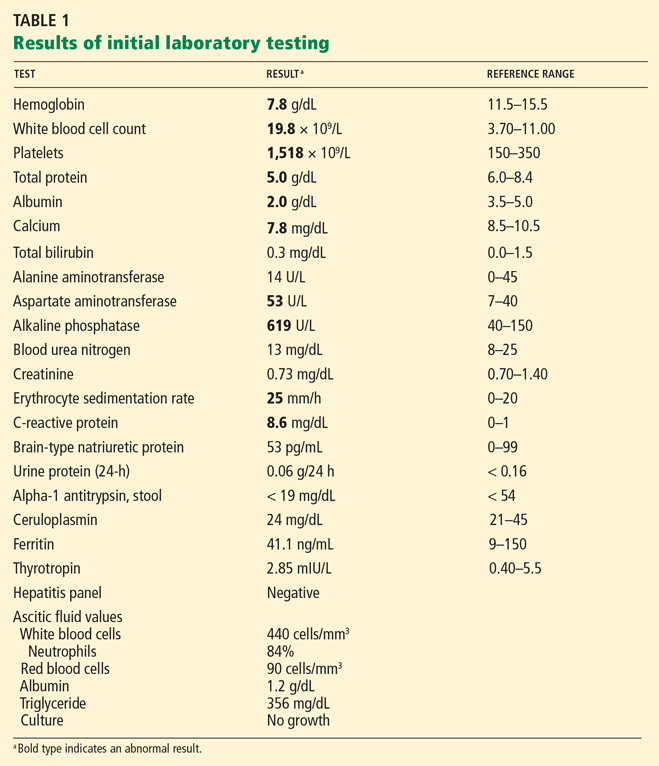
Laboratory testing at the time of hospital admission (Table 1) includes a hepatitis panel (negative for exposure to hepatitis A, B, and C) and ascitic fluid studies. Chest radiography shows a right pleural effusion. Echocardiography demonstrates moderate pericardial effusion without tamponade; left and right ventricular function is normal. Cardiac magnetic resonance imaging finds no evidence of pericardial constriction or restrictive cardiomyopathy. Pressures are normal on pulmonary artery catheterization.
FINDING THE CAUSE OF ASCITES
1. What is the most likely cause of ascites in this patient?
- Cirrhosis
- Recent abdominal surgery
- Congestive heart failure
- Abdominal malignancy
- Nephrotic syndrome
The serum-ascites albumin gradient—ie, the serum albumin concentration minus the ascitic fluid albumin concentration—helps determine whether ascites is related to portal hypertension.1 A high gradient (ie, above 1.1 g/dL) is seen in cirrhosis, alcoholic hepatitis, congestive heart failure, vascular occlusion syndromes (eg, Budd-Chiari syndrome), and metastatic liver disease.
From the values in Table 1, our patient’s gradient is 0.8 g/dL, which is considered low. However, we cannot completely rule out cirrhosis as the cause of her ascites because she was taking a diuretic, and diuretics can falsely decrease the gradient. Heart failure is unlikely, based on the results of echocardiography and catheterization. In addition, the 24-hour urinary protein concentration is normal, as is alpha-1 antitrypsin secretion in the stool, ruling out protein-losing nephropathy or enteropathy as the cause of her low albumin and ascites.
A high triglyceride content in her ascitic fluid (> 150 mg/dL) is consistent with chylous ascites, which is seen in patients with previous abdominal surgery or with lymphatic obstruction due to malignancy. A high neutrophil count in the ascitic fluid and a negative culture are also consistent with chylous ascites. However, in this patient, recent surgery as the cause of chylous ascites does not explain the systemic features of hepatosplenomegaly, anemia, thrombocytosis, and low albumin. Moreover, her high C-reactive protein value suggests an ongoing inflammatory process, although her erythrocyte sedimentation rate is not significantly elevated.
Therefore, the most likely cause of ascites in this patient is abdominal malignancy.
WHAT SHOULD BE DONE NEXT?
2. Which of the following studies is reasonable in this patient at this point?
- Serum protein electrophoresis
- Computed tomography (CT) of the chest, abdomen, and pelvis
- Liver biopsy
- Cytologic study of the ascitic fluid
All of these studies would be reasonable and in fact were done in this patient.
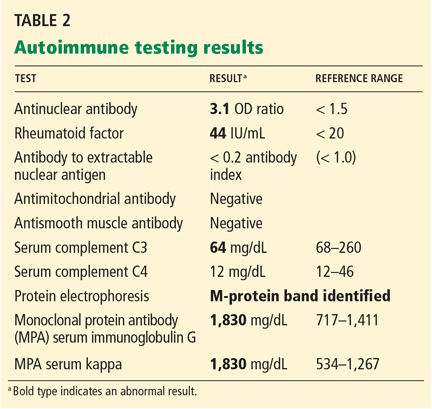
Serum protein electrophoresis (Table 2) identified a monoclonal protein band in the immunoglobulin G (IgG) kappa region.
Cytologic study of the ascitic fluid was negative for malignant cells.
Chest CT revealed bilateral pleural effusions, pericardial effusion, and bilateral axillary lymphadenopathy. CT of the abdomen and pelvis was normal, except for ascites, and no pelvic tumor was noted.
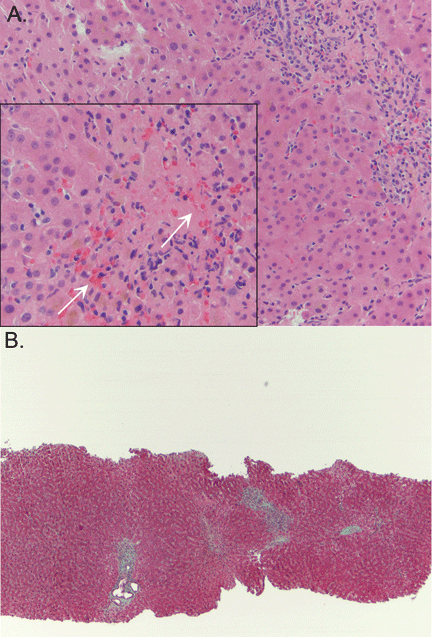
Liver biopsy was done to look for the source of her unexplained ascites with elevated alkaline phosphatase, as all other investigations so far were normal. It revealed mild centrilobular scarring, but the rest of the parenchymal architecture was normal, with no evidence of bridging fibrosis or nodular regenerative hyperplasia (Figure 1).
Transjugular measurement of the hepatic vein pressure revealed a hepatic vein pressure gradient of 9 mm Hg, indicating mild portal hypertension. Venography showed widely patent hepatic and portal veins. Her high inflammatory marker levels could have been caused by smoldering rheumatoid arthritis; however, since the patient has had no joint symptoms for 18 years, this is very unlikely. It is more likely to be caused by a plasma cell disorder, as suggested by a monoclonal protein on electrophoresis.
WHAT IS THE DIAGNOSIS?
3. What is the most likely diagnosis in our patient?
- Rheumatoid arthritis
- Cryoglobulinemia
- Capillary leak syndrome
- Hematologic malignancy
- Syndrome of polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes (POEMS syndrome)
Rheumatoid arthritis can present with hepatosplenomegaly, lymphadenopathy, ascites, and skin rash, particularly if antinuclear antibody and rheumatoid factor are elevated. Ascites is known to occur in association with rheumatoid arthritis in the setting of Felty syndrome or nodular regenerative hyperplasia of the liver.2 However, our patient did not have leukopenia or evidence of regenerative hyperplasia on liver biopsy. Moreover, her rheumatoid arthritis had remained clinically inactive for a long time.
Cryoglobulinemia was possible, given her ascites, neuropathy, and splenomegaly, but her serum hepatic antibody and C4 complement values were normal.3 Also, the appearance of her rash was not typical of cryoglobulinemia.
Capillary leak syndrome was ruled out by the absence of hypotensive episodes, edema of the face or upper extremities, or renal failure.4
Lymphoma was excluded by flow cytometry.
A monoclonal protein on serum electrophoresis may suggest multiple myeloma, but this patient had multisystem involvement including organomegaly, endocrinopathy, and skin abnormalities. Thus, POEMS syndrome is the most likely diagnosis.
4. Which test should be done at this time to confirm the diagnosis of POEMS syndrome?
- Bone marrow biopsy
- Vascular endothelial growth factor testing
- Nerve conduction study
- Complete x-ray bone survey
A test for vascular endothelial growth factor should be done. This growth factor is almost always elevated in POEMS, and a positive test helps confirm the diagnosis of POEMS. Our patient’s level was elevated at 1,664 pg/mL (reference range 31–86).
POEMS is thought to be a variant of plasma cell dyscrasia, and all patients with POEMS have a monoclonal protein on electrophoresis. On this background, multiple myeloma is an important consideration.
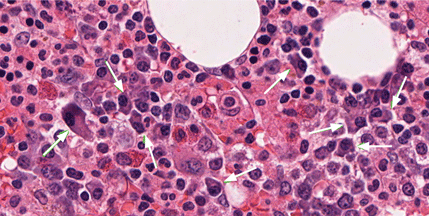
Our patient underwent bone marrow biopsy, which revealed mild plasmacytosis (< 10%) (Figure 2). A complete bone survey showed generalized osteopenia without blastic or lytic lesions. To complete the workup for POEMS syndrome, a nerve conduction study was done to look for neuropathy; it showed bilateral sensory motor neuropathy with features of both a demyelinating process and axonal loss.
POEMS SYNDROME
POEMS syndrome is a constellation of features such as organomegaly and endocrine and skin abnormalities in association with neuropathy and a monoclonal protein on electrophoresis.5 In 2003, Dispenzieri et al6 described the major and minor diagnostic criteria based on a retrospective analysis of 99 patients with POEMS syndrome.6 Later, elevated vascular endothelial growth factor was added as a confirmatory diagnostic criterion.7 This growth factor is also an indicator of prognosis in POEMS syndrome, and its level can be used to monitor the response to treatment.7
Our patient met both major criteria for POEMS syndrome, ie, polyneuropathy (based on nerve conduction studies) and a monoclonal protein. Polyneuropathy in POEMS syndrome usually occurs as sensorimotor peripheral neuropathy of insidious onset and is seldom painful. Nerve biopsy study reveals demyelination with features of axonal loss. Interestingly, although our patient had neuropathy as diagnosed by electromyography, she remained clinically asymptomatic.
The monoclonal protein in POEMS syndrome is commonly IgA or IgG. Light chains are always present and are mainly the lambda type; kappa light chains are also reported in rare cases. Our patient had IgG kappa light chains.
Our patient met a number of the minor criteria for POEMS syndrome: ie, organomegaly (hepatosplenomegaly, lymphadenopathy), endocrinopathy (hypothyroidism, diabetes), skin changes (hyperpigmentation and plaques of the lower extremities), edema, pleural effusion, and ascites.
Endocrine disorders in POEMS syndrome
The endocrine abnormalities most often described in POEMS syndrome are hypogonadism, hypothyroidism, and diabetes mellitus. But because hypothyroidism and diabetes are common in the general population, it is debatable whether either of these could constitute the endocrine component of POEMS syndrome. Nevertheless, in three large series,6,7 occurrences of these two disorders were common, although less specific than adrenal or pituitary involvement.
In the analysis by Dispenzieri et al,6 67% of patients had at least one endocrine abnormality. Our patient had no evidence of an adrenal disorder.
Skin, skeletal, and other changes
The skin changes in POEMS syndrome are often nonspecific and include hyperpigmentation, sclerodema-like thickening, and plaques.
Skeletal changes are noted in up to 97% of patients. A skeletal survey in our patient revealed generalized osteopenia as opposed to osteosclerotic lesions, which are common in POEMS syndrome.
Anemia and thrombocytosis (as in our patient) are usually seen in POEMS syndrome and are induced by cytokines.6 POEMS syndrome also leads to increased thrombotic complications from the release of inflammatory cytokines.
Hypoalbuminemia and anasarca including ascites are often seen in POEMS syndrome (prevalence 29% to 89%) and are attributed to cytokine-induced increased vascular permeability. In POEMS syndrome, the serum-ascites albumin gradient is usually less than 1.1 g/dL, as in our patient.
Stepani et al8 reported one case of culture-negative neutrocytic ascites with portal hypertension in POEMS syndrome.8 (Culture-negative neutrocytic ascites is defined as an ascitic fluid polymorphonuclear count greater than 250/mm3 and a negative ascitic fluid culture in the absence of previous antibiotic therapy.) Chylous ascites has not yet been described in POEMS syndrome. However, chylous ascites is predominantly lymphocytic, whereas our patient had neutrocytic ascites.
We concluded that the cause of our patient’s ascites was multifactorial and included previous surgery and POEMS syndrome.
Nonclassic presentation
In addition to its classic presentation, POEMS syndrome is often reported in association with other “unusual features” such as cardiomyopathy, pulmonary hypertension, and cryoglobulinemia.6
So far, very few cases of portal hypertension in POEMS syndrome have been reported. Stepani et al8 described a patient who had POEMS syndrome and portal hypertension with extensive portal fibrosis without cirrhosis on liver biopsy. Inoue et al9 reported a liver biopsy feature consistent with idiopathic portal hypertension, also noting a case with mild fibrosis and few lymphocytic infiltrates in the portal tract.9
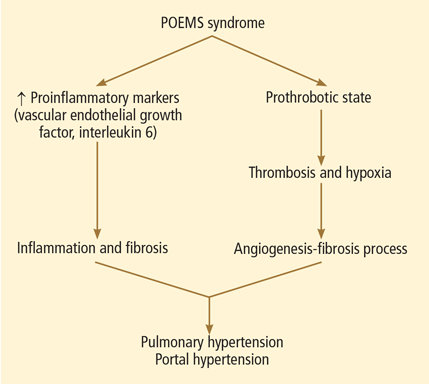
The etiopathogenesis of POEMS syndrome is attributed to proangiogenic vascular endothelial growth factor, and other inflammatory cytokines (interleukin 6, interleukin 1 beta, tumor necrosis factor alpha) also play a key role in pulmonary hypertension.10,11 A similar pathogenesis could also contribute to the development of portal hypertension (Figure 3).
CASE CONCLUDED
We started our patient on oral prednisone 60 mg daily for a month, tapered to a maintenance dose of 15 mg to suppress clonal proliferation of plasma cells. Her symptoms improved. Her vascular endothelial growth factor level decreased from 1,664 to 624 pg/mL. She was enrolled in a National Institutes of Health study to evaluate the effect of a potential new immunomodulator treatment for POEMS syndrome.
In conclusion, POEMS syndrome is rare and can present with many atypical features. A high index of suspicion is needed to detect it in a patient who has noncirrhotic portal hypertension with ascites and multisystem involvement.
- Runyon BA, Montano AA, Akriviadis EA, Antillon MR, Irving MA, McHutchison JG. The serum-ascites albumin gradient is superior to the exudate-transudate concept in the differential diagnosis of ascites. Ann Intern Med 1992; 117:215–220.
- Harris M, Rash RM, Dymock IW. Nodular, non-cirrhotic liver associated with portal hypertension in a patient with rheumatoid arthritis. J Clin Pathol 1974; 27:963–966.
- Ramos-Casals M, Stone JH, Cid MC, Bosch X. The cryoglobulinaemias. Lancet 2012; 379:348–360.
- Druey KM, Greipp PR. Narrative review: the systemic capillary leak syndrome. Ann Intern Med 2010; 153:90–98.
- Bardwick PA, Zvaifler NJ, Gill GN, Newman D, Greenway GD, Resnick DL. Plasma cell dyscrasia with polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes: the POEMS syndrome. Report on two cases and a review of the literature. Medicine (Baltimore) 1980; 59:311–322.
- Dispenzieri A, Kyle RA, Lacy MQ, et al. POEMS syndrome: definitions and long-term outcome. Blood 2003; 101:2496–2506.
- Dispenzieri A. POEMS syndrome. Blood Rev 2007; 21:285–299.
- Stepani P, Courouble Y, Postel P, et al. Portal hypertension and neutrocytic ascites in POEMS syndrome. Gastroenterol Clin Biol 1998; 22:1095–1097. Article in French.
- Inoue R, Nakazawa A, Tsukada N, et al. POEMS syndrome with idiopathic portal hypertension: autopsy case and review of the literature. Pathol Int 2010; 60:316–320.
- Gherardi RK, Bélec L, Soubrier M, et al. Overproduction of proinflammatory cytokines imbalanced by their antagonists in POEMS syndrome. Blood 1996; 87:1458–1465.
- Mukerjee D, Kingdon E, Vanderpump M, Coghlan JG. Pathophysiological insights from a case of reversible pulmonary arterial hypertension. J R Soc Med 2003; 96:403–404.
A 42-year-old woman is admitted to the hospital with worsening shortness of breath on exertion, poor exercise tolerance, leg edema, and swelling of the abdomen. Her symptoms have been getting worse over the last 4 months. She reports no history of fever, chills, night sweats, bleeding disorder, joint pain, weight loss, or loss of appetite.
She has type 2 diabetes mellitus and hypothyroidism. She had rheumatoid arthritis but said it was “inactive,” not requiring treatment for the last 18 years. Three months ago, she underwent a total hysterectomy and salpingo-oophorectomy for a complex adnexal mass, biopsy of which revealed a benign mucinous ovarian cyst.
Her current medications include furosemide, levothyroxine, and metformin. She is an ex-smoker with a 7 pack-year history. She drinks a glass of wine on social occasions only. Her family history is unremarkable.
On examination, she is not in distress and she has no fever. She has jugular venous distention of 5 cm, tense ascites, and marked edema of the legs, as well as hyperpigmented patches and erythematous plaques over both shins. Neck palpation reveals no lymphadenopathy or thyromegaly.
Her liver and the tip of the spleen are palpable following paracentesis, once ascitic fluid is removed.
The cardiovascular examination is normal. Chest auscultation reveals decreased breath sounds at the right lung base with bibasilar crackles. No focal neurologic deficit is noted on clinical examination.
Laboratory testing at the time of hospital admission (Table 1) includes a hepatitis panel (negative for exposure to hepatitis A, B, and C) and ascitic fluid studies. Chest radiography shows a right pleural effusion. Echocardiography demonstrates moderate pericardial effusion without tamponade; left and right ventricular function is normal. Cardiac magnetic resonance imaging finds no evidence of pericardial constriction or restrictive cardiomyopathy. Pressures are normal on pulmonary artery catheterization.
FINDING THE CAUSE OF ASCITES
1. What is the most likely cause of ascites in this patient?
- Cirrhosis
- Recent abdominal surgery
- Congestive heart failure
- Abdominal malignancy
- Nephrotic syndrome
The serum-ascites albumin gradient—ie, the serum albumin concentration minus the ascitic fluid albumin concentration—helps determine whether ascites is related to portal hypertension.1 A high gradient (ie, above 1.1 g/dL) is seen in cirrhosis, alcoholic hepatitis, congestive heart failure, vascular occlusion syndromes (eg, Budd-Chiari syndrome), and metastatic liver disease.
From the values in Table 1, our patient’s gradient is 0.8 g/dL, which is considered low. However, we cannot completely rule out cirrhosis as the cause of her ascites because she was taking a diuretic, and diuretics can falsely decrease the gradient. Heart failure is unlikely, based on the results of echocardiography and catheterization. In addition, the 24-hour urinary protein concentration is normal, as is alpha-1 antitrypsin secretion in the stool, ruling out protein-losing nephropathy or enteropathy as the cause of her low albumin and ascites.
A high triglyceride content in her ascitic fluid (> 150 mg/dL) is consistent with chylous ascites, which is seen in patients with previous abdominal surgery or with lymphatic obstruction due to malignancy. A high neutrophil count in the ascitic fluid and a negative culture are also consistent with chylous ascites. However, in this patient, recent surgery as the cause of chylous ascites does not explain the systemic features of hepatosplenomegaly, anemia, thrombocytosis, and low albumin. Moreover, her high C-reactive protein value suggests an ongoing inflammatory process, although her erythrocyte sedimentation rate is not significantly elevated.
Therefore, the most likely cause of ascites in this patient is abdominal malignancy.
WHAT SHOULD BE DONE NEXT?
2. Which of the following studies is reasonable in this patient at this point?
- Serum protein electrophoresis
- Computed tomography (CT) of the chest, abdomen, and pelvis
- Liver biopsy
- Cytologic study of the ascitic fluid
All of these studies would be reasonable and in fact were done in this patient.
Serum protein electrophoresis (Table 2) identified a monoclonal protein band in the immunoglobulin G (IgG) kappa region.
Cytologic study of the ascitic fluid was negative for malignant cells.
Chest CT revealed bilateral pleural effusions, pericardial effusion, and bilateral axillary lymphadenopathy. CT of the abdomen and pelvis was normal, except for ascites, and no pelvic tumor was noted.
Liver biopsy was done to look for the source of her unexplained ascites with elevated alkaline phosphatase, as all other investigations so far were normal. It revealed mild centrilobular scarring, but the rest of the parenchymal architecture was normal, with no evidence of bridging fibrosis or nodular regenerative hyperplasia (Figure 1).
Transjugular measurement of the hepatic vein pressure revealed a hepatic vein pressure gradient of 9 mm Hg, indicating mild portal hypertension. Venography showed widely patent hepatic and portal veins. Her high inflammatory marker levels could have been caused by smoldering rheumatoid arthritis; however, since the patient has had no joint symptoms for 18 years, this is very unlikely. It is more likely to be caused by a plasma cell disorder, as suggested by a monoclonal protein on electrophoresis.
WHAT IS THE DIAGNOSIS?
3. What is the most likely diagnosis in our patient?
- Rheumatoid arthritis
- Cryoglobulinemia
- Capillary leak syndrome
- Hematologic malignancy
- Syndrome of polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes (POEMS syndrome)
Rheumatoid arthritis can present with hepatosplenomegaly, lymphadenopathy, ascites, and skin rash, particularly if antinuclear antibody and rheumatoid factor are elevated. Ascites is known to occur in association with rheumatoid arthritis in the setting of Felty syndrome or nodular regenerative hyperplasia of the liver.2 However, our patient did not have leukopenia or evidence of regenerative hyperplasia on liver biopsy. Moreover, her rheumatoid arthritis had remained clinically inactive for a long time.
Cryoglobulinemia was possible, given her ascites, neuropathy, and splenomegaly, but her serum hepatic antibody and C4 complement values were normal.3 Also, the appearance of her rash was not typical of cryoglobulinemia.
Capillary leak syndrome was ruled out by the absence of hypotensive episodes, edema of the face or upper extremities, or renal failure.4
Lymphoma was excluded by flow cytometry.
A monoclonal protein on serum electrophoresis may suggest multiple myeloma, but this patient had multisystem involvement including organomegaly, endocrinopathy, and skin abnormalities. Thus, POEMS syndrome is the most likely diagnosis.
4. Which test should be done at this time to confirm the diagnosis of POEMS syndrome?
- Bone marrow biopsy
- Vascular endothelial growth factor testing
- Nerve conduction study
- Complete x-ray bone survey
A test for vascular endothelial growth factor should be done. This growth factor is almost always elevated in POEMS, and a positive test helps confirm the diagnosis of POEMS. Our patient’s level was elevated at 1,664 pg/mL (reference range 31–86).
POEMS is thought to be a variant of plasma cell dyscrasia, and all patients with POEMS have a monoclonal protein on electrophoresis. On this background, multiple myeloma is an important consideration.
Our patient underwent bone marrow biopsy, which revealed mild plasmacytosis (< 10%) (Figure 2). A complete bone survey showed generalized osteopenia without blastic or lytic lesions. To complete the workup for POEMS syndrome, a nerve conduction study was done to look for neuropathy; it showed bilateral sensory motor neuropathy with features of both a demyelinating process and axonal loss.
POEMS SYNDROME
POEMS syndrome is a constellation of features such as organomegaly and endocrine and skin abnormalities in association with neuropathy and a monoclonal protein on electrophoresis.5 In 2003, Dispenzieri et al6 described the major and minor diagnostic criteria based on a retrospective analysis of 99 patients with POEMS syndrome.6 Later, elevated vascular endothelial growth factor was added as a confirmatory diagnostic criterion.7 This growth factor is also an indicator of prognosis in POEMS syndrome, and its level can be used to monitor the response to treatment.7
Our patient met both major criteria for POEMS syndrome, ie, polyneuropathy (based on nerve conduction studies) and a monoclonal protein. Polyneuropathy in POEMS syndrome usually occurs as sensorimotor peripheral neuropathy of insidious onset and is seldom painful. Nerve biopsy study reveals demyelination with features of axonal loss. Interestingly, although our patient had neuropathy as diagnosed by electromyography, she remained clinically asymptomatic.
The monoclonal protein in POEMS syndrome is commonly IgA or IgG. Light chains are always present and are mainly the lambda type; kappa light chains are also reported in rare cases. Our patient had IgG kappa light chains.
Our patient met a number of the minor criteria for POEMS syndrome: ie, organomegaly (hepatosplenomegaly, lymphadenopathy), endocrinopathy (hypothyroidism, diabetes), skin changes (hyperpigmentation and plaques of the lower extremities), edema, pleural effusion, and ascites.
Endocrine disorders in POEMS syndrome
The endocrine abnormalities most often described in POEMS syndrome are hypogonadism, hypothyroidism, and diabetes mellitus. But because hypothyroidism and diabetes are common in the general population, it is debatable whether either of these could constitute the endocrine component of POEMS syndrome. Nevertheless, in three large series,6,7 occurrences of these two disorders were common, although less specific than adrenal or pituitary involvement.
In the analysis by Dispenzieri et al,6 67% of patients had at least one endocrine abnormality. Our patient had no evidence of an adrenal disorder.
Skin, skeletal, and other changes
The skin changes in POEMS syndrome are often nonspecific and include hyperpigmentation, sclerodema-like thickening, and plaques.
Skeletal changes are noted in up to 97% of patients. A skeletal survey in our patient revealed generalized osteopenia as opposed to osteosclerotic lesions, which are common in POEMS syndrome.
Anemia and thrombocytosis (as in our patient) are usually seen in POEMS syndrome and are induced by cytokines.6 POEMS syndrome also leads to increased thrombotic complications from the release of inflammatory cytokines.
Hypoalbuminemia and anasarca including ascites are often seen in POEMS syndrome (prevalence 29% to 89%) and are attributed to cytokine-induced increased vascular permeability. In POEMS syndrome, the serum-ascites albumin gradient is usually less than 1.1 g/dL, as in our patient.
Stepani et al8 reported one case of culture-negative neutrocytic ascites with portal hypertension in POEMS syndrome.8 (Culture-negative neutrocytic ascites is defined as an ascitic fluid polymorphonuclear count greater than 250/mm3 and a negative ascitic fluid culture in the absence of previous antibiotic therapy.) Chylous ascites has not yet been described in POEMS syndrome. However, chylous ascites is predominantly lymphocytic, whereas our patient had neutrocytic ascites.
We concluded that the cause of our patient’s ascites was multifactorial and included previous surgery and POEMS syndrome.
Nonclassic presentation
In addition to its classic presentation, POEMS syndrome is often reported in association with other “unusual features” such as cardiomyopathy, pulmonary hypertension, and cryoglobulinemia.6
So far, very few cases of portal hypertension in POEMS syndrome have been reported. Stepani et al8 described a patient who had POEMS syndrome and portal hypertension with extensive portal fibrosis without cirrhosis on liver biopsy. Inoue et al9 reported a liver biopsy feature consistent with idiopathic portal hypertension, also noting a case with mild fibrosis and few lymphocytic infiltrates in the portal tract.9
The etiopathogenesis of POEMS syndrome is attributed to proangiogenic vascular endothelial growth factor, and other inflammatory cytokines (interleukin 6, interleukin 1 beta, tumor necrosis factor alpha) also play a key role in pulmonary hypertension.10,11 A similar pathogenesis could also contribute to the development of portal hypertension (Figure 3).
CASE CONCLUDED
We started our patient on oral prednisone 60 mg daily for a month, tapered to a maintenance dose of 15 mg to suppress clonal proliferation of plasma cells. Her symptoms improved. Her vascular endothelial growth factor level decreased from 1,664 to 624 pg/mL. She was enrolled in a National Institutes of Health study to evaluate the effect of a potential new immunomodulator treatment for POEMS syndrome.
In conclusion, POEMS syndrome is rare and can present with many atypical features. A high index of suspicion is needed to detect it in a patient who has noncirrhotic portal hypertension with ascites and multisystem involvement.
A 42-year-old woman is admitted to the hospital with worsening shortness of breath on exertion, poor exercise tolerance, leg edema, and swelling of the abdomen. Her symptoms have been getting worse over the last 4 months. She reports no history of fever, chills, night sweats, bleeding disorder, joint pain, weight loss, or loss of appetite.
She has type 2 diabetes mellitus and hypothyroidism. She had rheumatoid arthritis but said it was “inactive,” not requiring treatment for the last 18 years. Three months ago, she underwent a total hysterectomy and salpingo-oophorectomy for a complex adnexal mass, biopsy of which revealed a benign mucinous ovarian cyst.
Her current medications include furosemide, levothyroxine, and metformin. She is an ex-smoker with a 7 pack-year history. She drinks a glass of wine on social occasions only. Her family history is unremarkable.
On examination, she is not in distress and she has no fever. She has jugular venous distention of 5 cm, tense ascites, and marked edema of the legs, as well as hyperpigmented patches and erythematous plaques over both shins. Neck palpation reveals no lymphadenopathy or thyromegaly.
Her liver and the tip of the spleen are palpable following paracentesis, once ascitic fluid is removed.
The cardiovascular examination is normal. Chest auscultation reveals decreased breath sounds at the right lung base with bibasilar crackles. No focal neurologic deficit is noted on clinical examination.
Laboratory testing at the time of hospital admission (Table 1) includes a hepatitis panel (negative for exposure to hepatitis A, B, and C) and ascitic fluid studies. Chest radiography shows a right pleural effusion. Echocardiography demonstrates moderate pericardial effusion without tamponade; left and right ventricular function is normal. Cardiac magnetic resonance imaging finds no evidence of pericardial constriction or restrictive cardiomyopathy. Pressures are normal on pulmonary artery catheterization.
FINDING THE CAUSE OF ASCITES
1. What is the most likely cause of ascites in this patient?
- Cirrhosis
- Recent abdominal surgery
- Congestive heart failure
- Abdominal malignancy
- Nephrotic syndrome
The serum-ascites albumin gradient—ie, the serum albumin concentration minus the ascitic fluid albumin concentration—helps determine whether ascites is related to portal hypertension.1 A high gradient (ie, above 1.1 g/dL) is seen in cirrhosis, alcoholic hepatitis, congestive heart failure, vascular occlusion syndromes (eg, Budd-Chiari syndrome), and metastatic liver disease.
From the values in Table 1, our patient’s gradient is 0.8 g/dL, which is considered low. However, we cannot completely rule out cirrhosis as the cause of her ascites because she was taking a diuretic, and diuretics can falsely decrease the gradient. Heart failure is unlikely, based on the results of echocardiography and catheterization. In addition, the 24-hour urinary protein concentration is normal, as is alpha-1 antitrypsin secretion in the stool, ruling out protein-losing nephropathy or enteropathy as the cause of her low albumin and ascites.
A high triglyceride content in her ascitic fluid (> 150 mg/dL) is consistent with chylous ascites, which is seen in patients with previous abdominal surgery or with lymphatic obstruction due to malignancy. A high neutrophil count in the ascitic fluid and a negative culture are also consistent with chylous ascites. However, in this patient, recent surgery as the cause of chylous ascites does not explain the systemic features of hepatosplenomegaly, anemia, thrombocytosis, and low albumin. Moreover, her high C-reactive protein value suggests an ongoing inflammatory process, although her erythrocyte sedimentation rate is not significantly elevated.
Therefore, the most likely cause of ascites in this patient is abdominal malignancy.
WHAT SHOULD BE DONE NEXT?
2. Which of the following studies is reasonable in this patient at this point?
- Serum protein electrophoresis
- Computed tomography (CT) of the chest, abdomen, and pelvis
- Liver biopsy
- Cytologic study of the ascitic fluid
All of these studies would be reasonable and in fact were done in this patient.
Serum protein electrophoresis (Table 2) identified a monoclonal protein band in the immunoglobulin G (IgG) kappa region.
Cytologic study of the ascitic fluid was negative for malignant cells.
Chest CT revealed bilateral pleural effusions, pericardial effusion, and bilateral axillary lymphadenopathy. CT of the abdomen and pelvis was normal, except for ascites, and no pelvic tumor was noted.
Liver biopsy was done to look for the source of her unexplained ascites with elevated alkaline phosphatase, as all other investigations so far were normal. It revealed mild centrilobular scarring, but the rest of the parenchymal architecture was normal, with no evidence of bridging fibrosis or nodular regenerative hyperplasia (Figure 1).
Transjugular measurement of the hepatic vein pressure revealed a hepatic vein pressure gradient of 9 mm Hg, indicating mild portal hypertension. Venography showed widely patent hepatic and portal veins. Her high inflammatory marker levels could have been caused by smoldering rheumatoid arthritis; however, since the patient has had no joint symptoms for 18 years, this is very unlikely. It is more likely to be caused by a plasma cell disorder, as suggested by a monoclonal protein on electrophoresis.
WHAT IS THE DIAGNOSIS?
3. What is the most likely diagnosis in our patient?
- Rheumatoid arthritis
- Cryoglobulinemia
- Capillary leak syndrome
- Hematologic malignancy
- Syndrome of polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes (POEMS syndrome)
Rheumatoid arthritis can present with hepatosplenomegaly, lymphadenopathy, ascites, and skin rash, particularly if antinuclear antibody and rheumatoid factor are elevated. Ascites is known to occur in association with rheumatoid arthritis in the setting of Felty syndrome or nodular regenerative hyperplasia of the liver.2 However, our patient did not have leukopenia or evidence of regenerative hyperplasia on liver biopsy. Moreover, her rheumatoid arthritis had remained clinically inactive for a long time.
Cryoglobulinemia was possible, given her ascites, neuropathy, and splenomegaly, but her serum hepatic antibody and C4 complement values were normal.3 Also, the appearance of her rash was not typical of cryoglobulinemia.
Capillary leak syndrome was ruled out by the absence of hypotensive episodes, edema of the face or upper extremities, or renal failure.4
Lymphoma was excluded by flow cytometry.
A monoclonal protein on serum electrophoresis may suggest multiple myeloma, but this patient had multisystem involvement including organomegaly, endocrinopathy, and skin abnormalities. Thus, POEMS syndrome is the most likely diagnosis.
4. Which test should be done at this time to confirm the diagnosis of POEMS syndrome?
- Bone marrow biopsy
- Vascular endothelial growth factor testing
- Nerve conduction study
- Complete x-ray bone survey
A test for vascular endothelial growth factor should be done. This growth factor is almost always elevated in POEMS, and a positive test helps confirm the diagnosis of POEMS. Our patient’s level was elevated at 1,664 pg/mL (reference range 31–86).
POEMS is thought to be a variant of plasma cell dyscrasia, and all patients with POEMS have a monoclonal protein on electrophoresis. On this background, multiple myeloma is an important consideration.
Our patient underwent bone marrow biopsy, which revealed mild plasmacytosis (< 10%) (Figure 2). A complete bone survey showed generalized osteopenia without blastic or lytic lesions. To complete the workup for POEMS syndrome, a nerve conduction study was done to look for neuropathy; it showed bilateral sensory motor neuropathy with features of both a demyelinating process and axonal loss.
POEMS SYNDROME
POEMS syndrome is a constellation of features such as organomegaly and endocrine and skin abnormalities in association with neuropathy and a monoclonal protein on electrophoresis.5 In 2003, Dispenzieri et al6 described the major and minor diagnostic criteria based on a retrospective analysis of 99 patients with POEMS syndrome.6 Later, elevated vascular endothelial growth factor was added as a confirmatory diagnostic criterion.7 This growth factor is also an indicator of prognosis in POEMS syndrome, and its level can be used to monitor the response to treatment.7
Our patient met both major criteria for POEMS syndrome, ie, polyneuropathy (based on nerve conduction studies) and a monoclonal protein. Polyneuropathy in POEMS syndrome usually occurs as sensorimotor peripheral neuropathy of insidious onset and is seldom painful. Nerve biopsy study reveals demyelination with features of axonal loss. Interestingly, although our patient had neuropathy as diagnosed by electromyography, she remained clinically asymptomatic.
The monoclonal protein in POEMS syndrome is commonly IgA or IgG. Light chains are always present and are mainly the lambda type; kappa light chains are also reported in rare cases. Our patient had IgG kappa light chains.
Our patient met a number of the minor criteria for POEMS syndrome: ie, organomegaly (hepatosplenomegaly, lymphadenopathy), endocrinopathy (hypothyroidism, diabetes), skin changes (hyperpigmentation and plaques of the lower extremities), edema, pleural effusion, and ascites.
Endocrine disorders in POEMS syndrome
The endocrine abnormalities most often described in POEMS syndrome are hypogonadism, hypothyroidism, and diabetes mellitus. But because hypothyroidism and diabetes are common in the general population, it is debatable whether either of these could constitute the endocrine component of POEMS syndrome. Nevertheless, in three large series,6,7 occurrences of these two disorders were common, although less specific than adrenal or pituitary involvement.
In the analysis by Dispenzieri et al,6 67% of patients had at least one endocrine abnormality. Our patient had no evidence of an adrenal disorder.
Skin, skeletal, and other changes
The skin changes in POEMS syndrome are often nonspecific and include hyperpigmentation, sclerodema-like thickening, and plaques.
Skeletal changes are noted in up to 97% of patients. A skeletal survey in our patient revealed generalized osteopenia as opposed to osteosclerotic lesions, which are common in POEMS syndrome.
Anemia and thrombocytosis (as in our patient) are usually seen in POEMS syndrome and are induced by cytokines.6 POEMS syndrome also leads to increased thrombotic complications from the release of inflammatory cytokines.
Hypoalbuminemia and anasarca including ascites are often seen in POEMS syndrome (prevalence 29% to 89%) and are attributed to cytokine-induced increased vascular permeability. In POEMS syndrome, the serum-ascites albumin gradient is usually less than 1.1 g/dL, as in our patient.
Stepani et al8 reported one case of culture-negative neutrocytic ascites with portal hypertension in POEMS syndrome.8 (Culture-negative neutrocytic ascites is defined as an ascitic fluid polymorphonuclear count greater than 250/mm3 and a negative ascitic fluid culture in the absence of previous antibiotic therapy.) Chylous ascites has not yet been described in POEMS syndrome. However, chylous ascites is predominantly lymphocytic, whereas our patient had neutrocytic ascites.
We concluded that the cause of our patient’s ascites was multifactorial and included previous surgery and POEMS syndrome.
Nonclassic presentation
In addition to its classic presentation, POEMS syndrome is often reported in association with other “unusual features” such as cardiomyopathy, pulmonary hypertension, and cryoglobulinemia.6
So far, very few cases of portal hypertension in POEMS syndrome have been reported. Stepani et al8 described a patient who had POEMS syndrome and portal hypertension with extensive portal fibrosis without cirrhosis on liver biopsy. Inoue et al9 reported a liver biopsy feature consistent with idiopathic portal hypertension, also noting a case with mild fibrosis and few lymphocytic infiltrates in the portal tract.9
The etiopathogenesis of POEMS syndrome is attributed to proangiogenic vascular endothelial growth factor, and other inflammatory cytokines (interleukin 6, interleukin 1 beta, tumor necrosis factor alpha) also play a key role in pulmonary hypertension.10,11 A similar pathogenesis could also contribute to the development of portal hypertension (Figure 3).
CASE CONCLUDED
We started our patient on oral prednisone 60 mg daily for a month, tapered to a maintenance dose of 15 mg to suppress clonal proliferation of plasma cells. Her symptoms improved. Her vascular endothelial growth factor level decreased from 1,664 to 624 pg/mL. She was enrolled in a National Institutes of Health study to evaluate the effect of a potential new immunomodulator treatment for POEMS syndrome.
In conclusion, POEMS syndrome is rare and can present with many atypical features. A high index of suspicion is needed to detect it in a patient who has noncirrhotic portal hypertension with ascites and multisystem involvement.
- Runyon BA, Montano AA, Akriviadis EA, Antillon MR, Irving MA, McHutchison JG. The serum-ascites albumin gradient is superior to the exudate-transudate concept in the differential diagnosis of ascites. Ann Intern Med 1992; 117:215–220.
- Harris M, Rash RM, Dymock IW. Nodular, non-cirrhotic liver associated with portal hypertension in a patient with rheumatoid arthritis. J Clin Pathol 1974; 27:963–966.
- Ramos-Casals M, Stone JH, Cid MC, Bosch X. The cryoglobulinaemias. Lancet 2012; 379:348–360.
- Druey KM, Greipp PR. Narrative review: the systemic capillary leak syndrome. Ann Intern Med 2010; 153:90–98.
- Bardwick PA, Zvaifler NJ, Gill GN, Newman D, Greenway GD, Resnick DL. Plasma cell dyscrasia with polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes: the POEMS syndrome. Report on two cases and a review of the literature. Medicine (Baltimore) 1980; 59:311–322.
- Dispenzieri A, Kyle RA, Lacy MQ, et al. POEMS syndrome: definitions and long-term outcome. Blood 2003; 101:2496–2506.
- Dispenzieri A. POEMS syndrome. Blood Rev 2007; 21:285–299.
- Stepani P, Courouble Y, Postel P, et al. Portal hypertension and neutrocytic ascites in POEMS syndrome. Gastroenterol Clin Biol 1998; 22:1095–1097. Article in French.
- Inoue R, Nakazawa A, Tsukada N, et al. POEMS syndrome with idiopathic portal hypertension: autopsy case and review of the literature. Pathol Int 2010; 60:316–320.
- Gherardi RK, Bélec L, Soubrier M, et al. Overproduction of proinflammatory cytokines imbalanced by their antagonists in POEMS syndrome. Blood 1996; 87:1458–1465.
- Mukerjee D, Kingdon E, Vanderpump M, Coghlan JG. Pathophysiological insights from a case of reversible pulmonary arterial hypertension. J R Soc Med 2003; 96:403–404.
- Runyon BA, Montano AA, Akriviadis EA, Antillon MR, Irving MA, McHutchison JG. The serum-ascites albumin gradient is superior to the exudate-transudate concept in the differential diagnosis of ascites. Ann Intern Med 1992; 117:215–220.
- Harris M, Rash RM, Dymock IW. Nodular, non-cirrhotic liver associated with portal hypertension in a patient with rheumatoid arthritis. J Clin Pathol 1974; 27:963–966.
- Ramos-Casals M, Stone JH, Cid MC, Bosch X. The cryoglobulinaemias. Lancet 2012; 379:348–360.
- Druey KM, Greipp PR. Narrative review: the systemic capillary leak syndrome. Ann Intern Med 2010; 153:90–98.
- Bardwick PA, Zvaifler NJ, Gill GN, Newman D, Greenway GD, Resnick DL. Plasma cell dyscrasia with polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes: the POEMS syndrome. Report on two cases and a review of the literature. Medicine (Baltimore) 1980; 59:311–322.
- Dispenzieri A, Kyle RA, Lacy MQ, et al. POEMS syndrome: definitions and long-term outcome. Blood 2003; 101:2496–2506.
- Dispenzieri A. POEMS syndrome. Blood Rev 2007; 21:285–299.
- Stepani P, Courouble Y, Postel P, et al. Portal hypertension and neutrocytic ascites in POEMS syndrome. Gastroenterol Clin Biol 1998; 22:1095–1097. Article in French.
- Inoue R, Nakazawa A, Tsukada N, et al. POEMS syndrome with idiopathic portal hypertension: autopsy case and review of the literature. Pathol Int 2010; 60:316–320.
- Gherardi RK, Bélec L, Soubrier M, et al. Overproduction of proinflammatory cytokines imbalanced by their antagonists in POEMS syndrome. Blood 1996; 87:1458–1465.
- Mukerjee D, Kingdon E, Vanderpump M, Coghlan JG. Pathophysiological insights from a case of reversible pulmonary arterial hypertension. J R Soc Med 2003; 96:403–404.
Not all joint pain is arthritis
A 47-year-old man who had been diagnosed with rheumatoid arthritis 5 years previously was referred to us for management of bilateral pleural effusions.
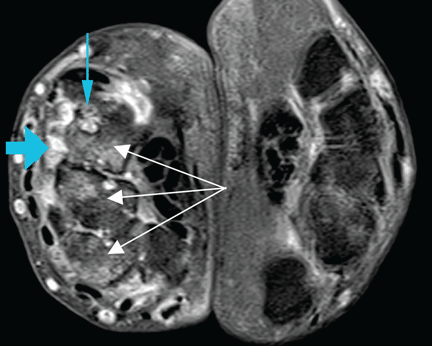
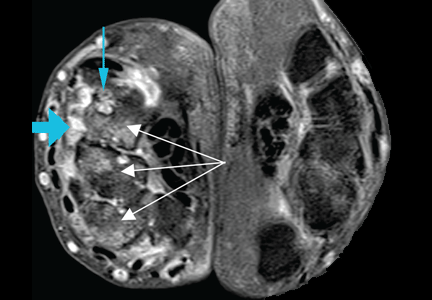
At the time of his diagnosis, his symptoms included pain and swelling of both wrists and the metacarpal joints of both hands. His serum C-reactive protein level had been elevated at that time, but he had no detectable rheumatoid factor. Findings on magnetic resonance imaging of the hand were very suggestive of rheumatoid arthritis (Figure 1).
He had been started on the anti-tumor necrosis factor agent etanercept but his symptoms improved only slightly, and therefore a glucocorticoid had been added.
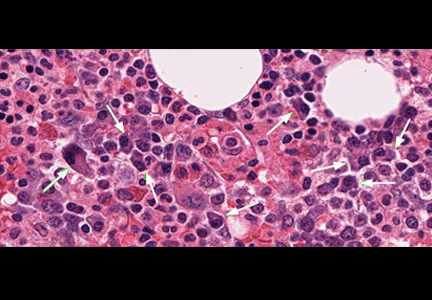
Two years later, he developed abdominal pain, for which he underwent cholecystectomy. However, he continued to have chronic, generalized abdominal pain, and over the next 4 years he lost 25 lb. Upper endoscopy showed no mucosal changes, and multiple random biopsy samples were obtained for histologic evaluation (FIGURE 2) as part of his workup for chronic abdominal pain.
Q: What is the diagnosis?
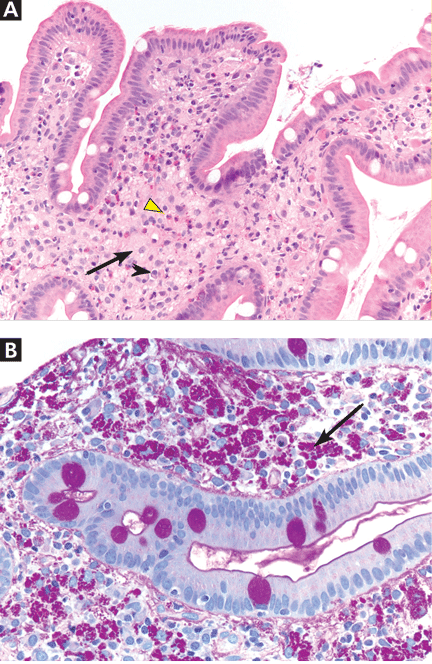
A: As shown in Figure 2, staining of duodenal specimens showed intact villous architecture, with focal expansion of the lamina propria by “foamy” macrophages, rare plasma cells, and eosinophils, a key feature of Whipple disease. Periodic acid-Schiff staining showed numerous bacilli within the macrophages, thus confirming the diagnosis of Whipple disease. The diagnosis was also confirmed by polymerase chain reaction testing. Staining for acid-fast bacilli was negative.
WHEN TO CONSIDER WHIPPLE DISEASE
Whipple disease is a rare systemic disease with a very low incidence rate worldwide. Thus, its prevalence is difficult to estimate accurately. It is caused by a gram-positive bacterium, Tropheryma whippelii.1,2 The typical clinical manifestations are diarrhea, abdominal pain, weight loss, and fever. In most patients, these are often preceded by articular symptoms,3 as in our patient, who had articular symptoms for 5 years before he was diagnosed with Whipple disease.
Interestingly, our patient also had pleural effusion, which is uncommon in Whipple disease.4
The pathogenesis of Whipple disease is thought to be related to bacterial replication within macrophages, which leads to a systemic immune response and tissue infiltration by the organism.5 Histologic evaluation is the most common way to confirm the diagnosis.
As our patient’s disease course illustrates, Whipple disease should be part of the differential diagnosis of arthritis, as antibiotic therapy alone leads to a dramatic clinical response.
Our patient was started on a 2-week course of intravenous ceftriaxone followed by oral sulfamethoxazole and trimethoprim, and his abdominal and articular symptoms completely resolved within 4 weeks.
- Dutly F, Altwegg M. Whipple’s disease and ‘Tropheryma whippelii.’ Clin Microbiol Rev 2001; 14:561–583.
- Raoult D, Birg ML, La Scola B, et al. Cultivation of the bacillus of Whipple’s disease. N Engl J Med 2000; 342:620–625.
- Relman DA, Schmidt TM, MacDermott RP, Falkow S. Identification of the uncultured bacillus of Whipple’s disease. N Engl J Med 1992; 327:293–301.
- Durand DV, Lecomte C, Cathébras P, Rousset H, Godeau P. Whipple disease. Clinical review of 52 cases. The SNFMI Research Group on Whipple disease. Société Nationale Française de Médecine Interne. Medicine (Baltimore) 1997; 76:170–184.
- Dobbins WO, Ruffin JM. A light- and electron-microscopic study of bacterial invasion in Whipple’s disease. Am J Pathol 1967; 51:225–242.
A 47-year-old man who had been diagnosed with rheumatoid arthritis 5 years previously was referred to us for management of bilateral pleural effusions.
At the time of his diagnosis, his symptoms included pain and swelling of both wrists and the metacarpal joints of both hands. His serum C-reactive protein level had been elevated at that time, but he had no detectable rheumatoid factor. Findings on magnetic resonance imaging of the hand were very suggestive of rheumatoid arthritis (Figure 1).
He had been started on the anti-tumor necrosis factor agent etanercept but his symptoms improved only slightly, and therefore a glucocorticoid had been added.
Two years later, he developed abdominal pain, for which he underwent cholecystectomy. However, he continued to have chronic, generalized abdominal pain, and over the next 4 years he lost 25 lb. Upper endoscopy showed no mucosal changes, and multiple random biopsy samples were obtained for histologic evaluation (FIGURE 2) as part of his workup for chronic abdominal pain.
Q: What is the diagnosis?
A: As shown in Figure 2, staining of duodenal specimens showed intact villous architecture, with focal expansion of the lamina propria by “foamy” macrophages, rare plasma cells, and eosinophils, a key feature of Whipple disease. Periodic acid-Schiff staining showed numerous bacilli within the macrophages, thus confirming the diagnosis of Whipple disease. The diagnosis was also confirmed by polymerase chain reaction testing. Staining for acid-fast bacilli was negative.
WHEN TO CONSIDER WHIPPLE DISEASE
Whipple disease is a rare systemic disease with a very low incidence rate worldwide. Thus, its prevalence is difficult to estimate accurately. It is caused by a gram-positive bacterium, Tropheryma whippelii.1,2 The typical clinical manifestations are diarrhea, abdominal pain, weight loss, and fever. In most patients, these are often preceded by articular symptoms,3 as in our patient, who had articular symptoms for 5 years before he was diagnosed with Whipple disease.
Interestingly, our patient also had pleural effusion, which is uncommon in Whipple disease.4
The pathogenesis of Whipple disease is thought to be related to bacterial replication within macrophages, which leads to a systemic immune response and tissue infiltration by the organism.5 Histologic evaluation is the most common way to confirm the diagnosis.
As our patient’s disease course illustrates, Whipple disease should be part of the differential diagnosis of arthritis, as antibiotic therapy alone leads to a dramatic clinical response.
Our patient was started on a 2-week course of intravenous ceftriaxone followed by oral sulfamethoxazole and trimethoprim, and his abdominal and articular symptoms completely resolved within 4 weeks.
A 47-year-old man who had been diagnosed with rheumatoid arthritis 5 years previously was referred to us for management of bilateral pleural effusions.
At the time of his diagnosis, his symptoms included pain and swelling of both wrists and the metacarpal joints of both hands. His serum C-reactive protein level had been elevated at that time, but he had no detectable rheumatoid factor. Findings on magnetic resonance imaging of the hand were very suggestive of rheumatoid arthritis (Figure 1).
He had been started on the anti-tumor necrosis factor agent etanercept but his symptoms improved only slightly, and therefore a glucocorticoid had been added.
Two years later, he developed abdominal pain, for which he underwent cholecystectomy. However, he continued to have chronic, generalized abdominal pain, and over the next 4 years he lost 25 lb. Upper endoscopy showed no mucosal changes, and multiple random biopsy samples were obtained for histologic evaluation (FIGURE 2) as part of his workup for chronic abdominal pain.
Q: What is the diagnosis?
A: As shown in Figure 2, staining of duodenal specimens showed intact villous architecture, with focal expansion of the lamina propria by “foamy” macrophages, rare plasma cells, and eosinophils, a key feature of Whipple disease. Periodic acid-Schiff staining showed numerous bacilli within the macrophages, thus confirming the diagnosis of Whipple disease. The diagnosis was also confirmed by polymerase chain reaction testing. Staining for acid-fast bacilli was negative.
WHEN TO CONSIDER WHIPPLE DISEASE
Whipple disease is a rare systemic disease with a very low incidence rate worldwide. Thus, its prevalence is difficult to estimate accurately. It is caused by a gram-positive bacterium, Tropheryma whippelii.1,2 The typical clinical manifestations are diarrhea, abdominal pain, weight loss, and fever. In most patients, these are often preceded by articular symptoms,3 as in our patient, who had articular symptoms for 5 years before he was diagnosed with Whipple disease.
Interestingly, our patient also had pleural effusion, which is uncommon in Whipple disease.4
The pathogenesis of Whipple disease is thought to be related to bacterial replication within macrophages, which leads to a systemic immune response and tissue infiltration by the organism.5 Histologic evaluation is the most common way to confirm the diagnosis.
As our patient’s disease course illustrates, Whipple disease should be part of the differential diagnosis of arthritis, as antibiotic therapy alone leads to a dramatic clinical response.
Our patient was started on a 2-week course of intravenous ceftriaxone followed by oral sulfamethoxazole and trimethoprim, and his abdominal and articular symptoms completely resolved within 4 weeks.
- Dutly F, Altwegg M. Whipple’s disease and ‘Tropheryma whippelii.’ Clin Microbiol Rev 2001; 14:561–583.
- Raoult D, Birg ML, La Scola B, et al. Cultivation of the bacillus of Whipple’s disease. N Engl J Med 2000; 342:620–625.
- Relman DA, Schmidt TM, MacDermott RP, Falkow S. Identification of the uncultured bacillus of Whipple’s disease. N Engl J Med 1992; 327:293–301.
- Durand DV, Lecomte C, Cathébras P, Rousset H, Godeau P. Whipple disease. Clinical review of 52 cases. The SNFMI Research Group on Whipple disease. Société Nationale Française de Médecine Interne. Medicine (Baltimore) 1997; 76:170–184.
- Dobbins WO, Ruffin JM. A light- and electron-microscopic study of bacterial invasion in Whipple’s disease. Am J Pathol 1967; 51:225–242.
- Dutly F, Altwegg M. Whipple’s disease and ‘Tropheryma whippelii.’ Clin Microbiol Rev 2001; 14:561–583.
- Raoult D, Birg ML, La Scola B, et al. Cultivation of the bacillus of Whipple’s disease. N Engl J Med 2000; 342:620–625.
- Relman DA, Schmidt TM, MacDermott RP, Falkow S. Identification of the uncultured bacillus of Whipple’s disease. N Engl J Med 1992; 327:293–301.
- Durand DV, Lecomte C, Cathébras P, Rousset H, Godeau P. Whipple disease. Clinical review of 52 cases. The SNFMI Research Group on Whipple disease. Société Nationale Française de Médecine Interne. Medicine (Baltimore) 1997; 76:170–184.
- Dobbins WO, Ruffin JM. A light- and electron-microscopic study of bacterial invasion in Whipple’s disease. Am J Pathol 1967; 51:225–242.