User login
Hemophilia A drug approved in Europe

The European Commission has granted marketing authorization for lonoctocog alfa (Afstyla), a recombinant factor VIII (FVIII) single-chain therapy.
Lonoctocog alfa is indicated for the treatment and prophylaxis of bleeding in hemophilia A patients of all ages.
Lonoctocog alfa is the first and only single-chain recombinant FVIII therapy for hemophilia A specifically designed to provide long-lasting protection from bleeds with 2- to 3-times weekly dosing, according to CSL Behring, the company developing the product.
The company says lonoctocog alfa uses a covalent bond that forms one structural entity—a single polypeptide chain—to improve the stability of FVIII and provide FVIII activity with the option of twice-weekly dosing.
According to CSL Behring, lonoctocog alfa will be launched in European markets in the coming months, as market access is obtained.
Lonoctocog alfa is also approved for use in the US and Canada.
Regulatory submissions for lonoctocog alfa are based on results from the AFFINITY clinical development program, which includes a trial of children (n=84) and a trial of adolescents and adults (n=175).
Among patients who received lonoctocog alfa prophylactically in these trials, the median annualized bleeding rate was 1.14 in the adults/adolescents and 3.69 in children younger than 12.
In all, there were 1195 bleeding events—848 in the adults/adolescents and 347 in the children.
Ninety-four percent of bleeds in adults/adolescents and 96% of bleeds in pediatric patients were effectively controlled with no more than 2 infusions of lonoctocog alfa weekly.
Eighty-one percent of bleeds in adults/adolescents and 86% of bleeds in pediatric patients were controlled by a single infusion.
Researchers assessed safety in 258 patients from both studies. Adverse reactions occurred in 14 patients and included hypersensitivity (n=4), dizziness (n=2), paresthesia (n=1), rash (n=1), erythema (n=1), pruritus (n=1), pyrexia (n=1), injection-site pain (n=1), chills (n=1), and feeling hot (n=1).
One patient withdrew from treatment due to hypersensitivity.
None of the patients developed neutralizing antibodies to FVIII or antibodies to host cell proteins. There were no reports of anaphylaxis or thrombosis.
Results from the trial of adolescents/adults were published in Blood in August 2016. Results from the trial of children were presented at the World Federation of Hemophilia 2016 World Congress in July 2016.* ![]()
*Stasyshyn
O et al; rVIII-SingleChain, results of the pivotal efficacy data from a
phase III PK, efficacy and safety clinical study in children less than
12 years of age with severe hemophilia A; WFH 2016 World Congress, July
2016.
The European Commission has granted marketing authorization for lonoctocog alfa (Afstyla), a recombinant factor VIII (FVIII) single-chain therapy.
Lonoctocog alfa is indicated for the treatment and prophylaxis of bleeding in hemophilia A patients of all ages.
Lonoctocog alfa is the first and only single-chain recombinant FVIII therapy for hemophilia A specifically designed to provide long-lasting protection from bleeds with 2- to 3-times weekly dosing, according to CSL Behring, the company developing the product.
The company says lonoctocog alfa uses a covalent bond that forms one structural entity—a single polypeptide chain—to improve the stability of FVIII and provide FVIII activity with the option of twice-weekly dosing.
According to CSL Behring, lonoctocog alfa will be launched in European markets in the coming months, as market access is obtained.
Lonoctocog alfa is also approved for use in the US and Canada.
Regulatory submissions for lonoctocog alfa are based on results from the AFFINITY clinical development program, which includes a trial of children (n=84) and a trial of adolescents and adults (n=175).
Among patients who received lonoctocog alfa prophylactically in these trials, the median annualized bleeding rate was 1.14 in the adults/adolescents and 3.69 in children younger than 12.
In all, there were 1195 bleeding events—848 in the adults/adolescents and 347 in the children.
Ninety-four percent of bleeds in adults/adolescents and 96% of bleeds in pediatric patients were effectively controlled with no more than 2 infusions of lonoctocog alfa weekly.
Eighty-one percent of bleeds in adults/adolescents and 86% of bleeds in pediatric patients were controlled by a single infusion.
Researchers assessed safety in 258 patients from both studies. Adverse reactions occurred in 14 patients and included hypersensitivity (n=4), dizziness (n=2), paresthesia (n=1), rash (n=1), erythema (n=1), pruritus (n=1), pyrexia (n=1), injection-site pain (n=1), chills (n=1), and feeling hot (n=1).
One patient withdrew from treatment due to hypersensitivity.
None of the patients developed neutralizing antibodies to FVIII or antibodies to host cell proteins. There were no reports of anaphylaxis or thrombosis.
Results from the trial of adolescents/adults were published in Blood in August 2016. Results from the trial of children were presented at the World Federation of Hemophilia 2016 World Congress in July 2016.* ![]()
*Stasyshyn
O et al; rVIII-SingleChain, results of the pivotal efficacy data from a
phase III PK, efficacy and safety clinical study in children less than
12 years of age with severe hemophilia A; WFH 2016 World Congress, July
2016.
The European Commission has granted marketing authorization for lonoctocog alfa (Afstyla), a recombinant factor VIII (FVIII) single-chain therapy.
Lonoctocog alfa is indicated for the treatment and prophylaxis of bleeding in hemophilia A patients of all ages.
Lonoctocog alfa is the first and only single-chain recombinant FVIII therapy for hemophilia A specifically designed to provide long-lasting protection from bleeds with 2- to 3-times weekly dosing, according to CSL Behring, the company developing the product.
The company says lonoctocog alfa uses a covalent bond that forms one structural entity—a single polypeptide chain—to improve the stability of FVIII and provide FVIII activity with the option of twice-weekly dosing.
According to CSL Behring, lonoctocog alfa will be launched in European markets in the coming months, as market access is obtained.
Lonoctocog alfa is also approved for use in the US and Canada.
Regulatory submissions for lonoctocog alfa are based on results from the AFFINITY clinical development program, which includes a trial of children (n=84) and a trial of adolescents and adults (n=175).
Among patients who received lonoctocog alfa prophylactically in these trials, the median annualized bleeding rate was 1.14 in the adults/adolescents and 3.69 in children younger than 12.
In all, there were 1195 bleeding events—848 in the adults/adolescents and 347 in the children.
Ninety-four percent of bleeds in adults/adolescents and 96% of bleeds in pediatric patients were effectively controlled with no more than 2 infusions of lonoctocog alfa weekly.
Eighty-one percent of bleeds in adults/adolescents and 86% of bleeds in pediatric patients were controlled by a single infusion.
Researchers assessed safety in 258 patients from both studies. Adverse reactions occurred in 14 patients and included hypersensitivity (n=4), dizziness (n=2), paresthesia (n=1), rash (n=1), erythema (n=1), pruritus (n=1), pyrexia (n=1), injection-site pain (n=1), chills (n=1), and feeling hot (n=1).
One patient withdrew from treatment due to hypersensitivity.
None of the patients developed neutralizing antibodies to FVIII or antibodies to host cell proteins. There were no reports of anaphylaxis or thrombosis.
Results from the trial of adolescents/adults were published in Blood in August 2016. Results from the trial of children were presented at the World Federation of Hemophilia 2016 World Congress in July 2016.* ![]()
*Stasyshyn
O et al; rVIII-SingleChain, results of the pivotal efficacy data from a
phase III PK, efficacy and safety clinical study in children less than
12 years of age with severe hemophilia A; WFH 2016 World Congress, July
2016.
Yoga may improve QOL in kids with cancer

Photo by Bill Branson
A yoga program for children with cancer can be carried out during cancer treatment and has quality of life (QOL) benefits for the children as well as their caregivers, according to research published in Rehabilitation Oncology.
However, the program was not feasible for all patients. More than half of those initially enrolled could not complete the study due to treatment toxicity or scheduling conflicts.
Andrea Orsey, MD, of Connecticut Children’s Medical Center in Hartford, and her colleagues conducted this research to evaluate the feasibility and effectiveness of a yoga intervention for children with cancer and their families.
The team began by conducting a survey of 20 children and adolescents with cancer and their parents/guardians.
Survey respondents expressed interest in a yoga program. But they also perceived several barriers to such a program, including concerns about side effects, pain/discomfort, and physical limitations.
With these barriers in mind, Dr Orsey and her colleagues developed a yoga intervention for pediatric cancer patients, delivered by certified yoga instructors.
The program was designed to be performed in a variety of settings and tailored to the children’s physical condition or mobility issues.
A pilot evaluation included 10 children with cancer and their caregivers. Twenty-two patient/caregiver pairs were actually enrolled, but 6 pairs withdrew because of treatment toxicity, and 6 had the study window lapse due to scheduling conflicts.
Although limited by its small size, the study suggested that yoga improved health-related QOL for both caregivers and children.
The children had significant improvements in both social and emotional QOL. They had an overall improvement in fatigue, but this was not statistically significant.
Caregivers had a significant improvement in mental health but not physical health or caregiver burden.
Both caregivers and children said they were satisfied with the yoga program and would recommend it to others.
Dr Orsey and her colleagues hope this pilot study will help guide future efforts to provide yoga to children with cancer and their families.
The researchers noted that a key issue will be coordinating yoga sessions with the medical demands of chemotherapy. ![]()
Photo by Bill Branson
A yoga program for children with cancer can be carried out during cancer treatment and has quality of life (QOL) benefits for the children as well as their caregivers, according to research published in Rehabilitation Oncology.
However, the program was not feasible for all patients. More than half of those initially enrolled could not complete the study due to treatment toxicity or scheduling conflicts.
Andrea Orsey, MD, of Connecticut Children’s Medical Center in Hartford, and her colleagues conducted this research to evaluate the feasibility and effectiveness of a yoga intervention for children with cancer and their families.
The team began by conducting a survey of 20 children and adolescents with cancer and their parents/guardians.
Survey respondents expressed interest in a yoga program. But they also perceived several barriers to such a program, including concerns about side effects, pain/discomfort, and physical limitations.
With these barriers in mind, Dr Orsey and her colleagues developed a yoga intervention for pediatric cancer patients, delivered by certified yoga instructors.
The program was designed to be performed in a variety of settings and tailored to the children’s physical condition or mobility issues.
A pilot evaluation included 10 children with cancer and their caregivers. Twenty-two patient/caregiver pairs were actually enrolled, but 6 pairs withdrew because of treatment toxicity, and 6 had the study window lapse due to scheduling conflicts.
Although limited by its small size, the study suggested that yoga improved health-related QOL for both caregivers and children.
The children had significant improvements in both social and emotional QOL. They had an overall improvement in fatigue, but this was not statistically significant.
Caregivers had a significant improvement in mental health but not physical health or caregiver burden.
Both caregivers and children said they were satisfied with the yoga program and would recommend it to others.
Dr Orsey and her colleagues hope this pilot study will help guide future efforts to provide yoga to children with cancer and their families.
The researchers noted that a key issue will be coordinating yoga sessions with the medical demands of chemotherapy. ![]()
Photo by Bill Branson
A yoga program for children with cancer can be carried out during cancer treatment and has quality of life (QOL) benefits for the children as well as their caregivers, according to research published in Rehabilitation Oncology.
However, the program was not feasible for all patients. More than half of those initially enrolled could not complete the study due to treatment toxicity or scheduling conflicts.
Andrea Orsey, MD, of Connecticut Children’s Medical Center in Hartford, and her colleagues conducted this research to evaluate the feasibility and effectiveness of a yoga intervention for children with cancer and their families.
The team began by conducting a survey of 20 children and adolescents with cancer and their parents/guardians.
Survey respondents expressed interest in a yoga program. But they also perceived several barriers to such a program, including concerns about side effects, pain/discomfort, and physical limitations.
With these barriers in mind, Dr Orsey and her colleagues developed a yoga intervention for pediatric cancer patients, delivered by certified yoga instructors.
The program was designed to be performed in a variety of settings and tailored to the children’s physical condition or mobility issues.
A pilot evaluation included 10 children with cancer and their caregivers. Twenty-two patient/caregiver pairs were actually enrolled, but 6 pairs withdrew because of treatment toxicity, and 6 had the study window lapse due to scheduling conflicts.
Although limited by its small size, the study suggested that yoga improved health-related QOL for both caregivers and children.
The children had significant improvements in both social and emotional QOL. They had an overall improvement in fatigue, but this was not statistically significant.
Caregivers had a significant improvement in mental health but not physical health or caregiver burden.
Both caregivers and children said they were satisfied with the yoga program and would recommend it to others.
Dr Orsey and her colleagues hope this pilot study will help guide future efforts to provide yoga to children with cancer and their families.
The researchers noted that a key issue will be coordinating yoga sessions with the medical demands of chemotherapy. ![]()
US cancer cases may near 1.7 million in 2017

patient and her father
Photo by Rhoda Baer
The US may see nearly 1.7 million new cancer cases in 2017 and more than 600,000 cancer-related deaths, according to a report from the American Cancer Society (ACS).
In addition to estimates for 2017, the report, “Cancer Statistics 2017,” includes the most recent data on cancer incidence, mortality, and survival in the US.
The report was published in CA: A Cancer Journal for Clinicians.
The report projects there will be 1,688,780 new cancer cases and 600,920 cancer deaths in the US this year.
This includes:
- 80,500 new cases of lymphoma and 21,210 lymphoma deaths
- 62,130 new cases of leukemia and 24,500 leukemia deaths
- 30,280 new cases of myeloma and 12,590 myeloma deaths.
The report also shows that, from 2004 to 2013, the overall cancer incidence rate was stable in women and declined by about 2% per year in men. From 2005 to 2014, the cancer death rate declined by about 1.5% annually in both men and women.
Overall, the cancer death rate dropped 25% from its peak of 215.1 (per 100,000 population) in 1991 to 161.2 (per 100,000 population) in 2014, the latest year for which data was available. This translates to about 2,143,200 fewer cancer deaths.
“The continuing drops in the cancer death rate are a powerful sign of the potential we have to reduce cancer’s deadly toll,” said Otis W. Brawley, MD, chief medical officer of the ACS.
He said the decrease in cancer death rates is the result of steady reductions in smoking and advances in early detection and treatment. The decrease is driven by decreasing death rates for the 4 major cancer sites—lung, breast, colorectal, and prostate.
The report also shows that racial disparities in cancer death rates continue to decline. The excess risk of cancer death in black men has dropped from 47% in 1990 to 21% in 2014. The black/white disparity declined similarly in women, from a peak of 20% in 1998 to 13% in 2014.
On the other hand, significant gender disparities persist for both cancer incidence and death in the US. For all cancer sites combined, the incidence rate is 20% higher in men than in women, and the cancer death rate is 40% higher in men.
Dr Brawley said the gender gap in cancer mortality largely reflects variation in the distribution of cancers that occur in men and women, much of which is due to differences in the prevalence of cancer risk factors.
The yearly “Cancer Statistics” reports have been published by ACS researchers since 1967 to inform and guide clinicians, investigators, and others in public health in prioritizing efforts to reduce the burden of cancer.
Cancer incidence data for the current report were collected by the Surveillance, Epidemiology, and End Results Program; the National Program of Cancer Registries; and the North American Association of Central Cancer Registries. Mortality data were collected by the National Center for Health Statistics. ![]()
patient and her father
Photo by Rhoda Baer
The US may see nearly 1.7 million new cancer cases in 2017 and more than 600,000 cancer-related deaths, according to a report from the American Cancer Society (ACS).
In addition to estimates for 2017, the report, “Cancer Statistics 2017,” includes the most recent data on cancer incidence, mortality, and survival in the US.
The report was published in CA: A Cancer Journal for Clinicians.
The report projects there will be 1,688,780 new cancer cases and 600,920 cancer deaths in the US this year.
This includes:
- 80,500 new cases of lymphoma and 21,210 lymphoma deaths
- 62,130 new cases of leukemia and 24,500 leukemia deaths
- 30,280 new cases of myeloma and 12,590 myeloma deaths.
The report also shows that, from 2004 to 2013, the overall cancer incidence rate was stable in women and declined by about 2% per year in men. From 2005 to 2014, the cancer death rate declined by about 1.5% annually in both men and women.
Overall, the cancer death rate dropped 25% from its peak of 215.1 (per 100,000 population) in 1991 to 161.2 (per 100,000 population) in 2014, the latest year for which data was available. This translates to about 2,143,200 fewer cancer deaths.
“The continuing drops in the cancer death rate are a powerful sign of the potential we have to reduce cancer’s deadly toll,” said Otis W. Brawley, MD, chief medical officer of the ACS.
He said the decrease in cancer death rates is the result of steady reductions in smoking and advances in early detection and treatment. The decrease is driven by decreasing death rates for the 4 major cancer sites—lung, breast, colorectal, and prostate.
The report also shows that racial disparities in cancer death rates continue to decline. The excess risk of cancer death in black men has dropped from 47% in 1990 to 21% in 2014. The black/white disparity declined similarly in women, from a peak of 20% in 1998 to 13% in 2014.
On the other hand, significant gender disparities persist for both cancer incidence and death in the US. For all cancer sites combined, the incidence rate is 20% higher in men than in women, and the cancer death rate is 40% higher in men.
Dr Brawley said the gender gap in cancer mortality largely reflects variation in the distribution of cancers that occur in men and women, much of which is due to differences in the prevalence of cancer risk factors.
The yearly “Cancer Statistics” reports have been published by ACS researchers since 1967 to inform and guide clinicians, investigators, and others in public health in prioritizing efforts to reduce the burden of cancer.
Cancer incidence data for the current report were collected by the Surveillance, Epidemiology, and End Results Program; the National Program of Cancer Registries; and the North American Association of Central Cancer Registries. Mortality data were collected by the National Center for Health Statistics. ![]()
patient and her father
Photo by Rhoda Baer
The US may see nearly 1.7 million new cancer cases in 2017 and more than 600,000 cancer-related deaths, according to a report from the American Cancer Society (ACS).
In addition to estimates for 2017, the report, “Cancer Statistics 2017,” includes the most recent data on cancer incidence, mortality, and survival in the US.
The report was published in CA: A Cancer Journal for Clinicians.
The report projects there will be 1,688,780 new cancer cases and 600,920 cancer deaths in the US this year.
This includes:
- 80,500 new cases of lymphoma and 21,210 lymphoma deaths
- 62,130 new cases of leukemia and 24,500 leukemia deaths
- 30,280 new cases of myeloma and 12,590 myeloma deaths.
The report also shows that, from 2004 to 2013, the overall cancer incidence rate was stable in women and declined by about 2% per year in men. From 2005 to 2014, the cancer death rate declined by about 1.5% annually in both men and women.
Overall, the cancer death rate dropped 25% from its peak of 215.1 (per 100,000 population) in 1991 to 161.2 (per 100,000 population) in 2014, the latest year for which data was available. This translates to about 2,143,200 fewer cancer deaths.
“The continuing drops in the cancer death rate are a powerful sign of the potential we have to reduce cancer’s deadly toll,” said Otis W. Brawley, MD, chief medical officer of the ACS.
He said the decrease in cancer death rates is the result of steady reductions in smoking and advances in early detection and treatment. The decrease is driven by decreasing death rates for the 4 major cancer sites—lung, breast, colorectal, and prostate.
The report also shows that racial disparities in cancer death rates continue to decline. The excess risk of cancer death in black men has dropped from 47% in 1990 to 21% in 2014. The black/white disparity declined similarly in women, from a peak of 20% in 1998 to 13% in 2014.
On the other hand, significant gender disparities persist for both cancer incidence and death in the US. For all cancer sites combined, the incidence rate is 20% higher in men than in women, and the cancer death rate is 40% higher in men.
Dr Brawley said the gender gap in cancer mortality largely reflects variation in the distribution of cancers that occur in men and women, much of which is due to differences in the prevalence of cancer risk factors.
The yearly “Cancer Statistics” reports have been published by ACS researchers since 1967 to inform and guide clinicians, investigators, and others in public health in prioritizing efforts to reduce the burden of cancer.
Cancer incidence data for the current report were collected by the Surveillance, Epidemiology, and End Results Program; the National Program of Cancer Registries; and the North American Association of Central Cancer Registries. Mortality data were collected by the National Center for Health Statistics. ![]()
Cancer genomic data released to public

Photo courtesy of the
National Institute of
General Medical Sciences
The American Association for Cancer Research (AACR) has announced the first public release of cancer genomic data aggregated through the AACR Project Genomics Evidence Neoplasia Information Exchange (GENIE).
The data set includes nearly 19,000 de-identified genomic records collected from patients who were treated at 8 international institutions, making it one of the largest public cancer genomic data sets released to date.
The release includes data for 59 major cancer types, including leukemias, lymphomas, and multiple myeloma.
The genomic data and a limited amount of linked clinical data for each patient can be accessed via the AACR Project GENIE cBioPortal or from Sage Bionetworks. (Users must create an account for either site to access the data.)
“We are excited to make publicly available this very large set of clinical-grade, next-generation sequencing data obtained during routine patient care,” said Charles L. Sawyers, MD, AACR Project GENIE Steering Committee chairperson.
“These data were generated as part of routine patient care and, without AACR Project GENIE, they would likely never have been shared with the global cancer research community.”
AACR Project GENIE is a multi-phase, international data-sharing project aimed at catalyzing precision oncology through the development of a registry that aggregates and links clinical-grade cancer genomic data with clinical outcomes from tens of thousands of cancer patients treated at multiple institutions.
The newly released data are fully de-identified in compliance with the Health Insurance Portability and Accountability Act (HIPAA).
The data are derived from patients whose tumors were genetically sequenced as part of their care at any of the 8 institutions that participated in the first phase of AACR Project GENIE.
The goal of releasing these data to the cancer research community is to aid new research that will accelerate the pace of progress against cancer.
According to AACR, the data can be used to validate gene signatures of drug response or prognosis, identify new patient populations for drugs that are currently available, and uncover new drug targets and biomarkers.
“I am extremely proud that the American Association for Cancer Research, as the coordinating center for AACR Project GENIE, is delivering on its promise to make these important data publicly available just over a year after unveiling the initiative,” said Margaret Foti, PhD, MD, chief executive officer of the AACR.
To expand the AACR Project GENIE registry, the consortium is accepting applications for new participating centers. Any nonprofit institution that meets certain criteria can submit an application to become a project participant.
For more information on AACR Project GENIE, visit the project website or send an email to [email protected]. ![]()
Photo courtesy of the
National Institute of
General Medical Sciences
The American Association for Cancer Research (AACR) has announced the first public release of cancer genomic data aggregated through the AACR Project Genomics Evidence Neoplasia Information Exchange (GENIE).
The data set includes nearly 19,000 de-identified genomic records collected from patients who were treated at 8 international institutions, making it one of the largest public cancer genomic data sets released to date.
The release includes data for 59 major cancer types, including leukemias, lymphomas, and multiple myeloma.
The genomic data and a limited amount of linked clinical data for each patient can be accessed via the AACR Project GENIE cBioPortal or from Sage Bionetworks. (Users must create an account for either site to access the data.)
“We are excited to make publicly available this very large set of clinical-grade, next-generation sequencing data obtained during routine patient care,” said Charles L. Sawyers, MD, AACR Project GENIE Steering Committee chairperson.
“These data were generated as part of routine patient care and, without AACR Project GENIE, they would likely never have been shared with the global cancer research community.”
AACR Project GENIE is a multi-phase, international data-sharing project aimed at catalyzing precision oncology through the development of a registry that aggregates and links clinical-grade cancer genomic data with clinical outcomes from tens of thousands of cancer patients treated at multiple institutions.
The newly released data are fully de-identified in compliance with the Health Insurance Portability and Accountability Act (HIPAA).
The data are derived from patients whose tumors were genetically sequenced as part of their care at any of the 8 institutions that participated in the first phase of AACR Project GENIE.
The goal of releasing these data to the cancer research community is to aid new research that will accelerate the pace of progress against cancer.
According to AACR, the data can be used to validate gene signatures of drug response or prognosis, identify new patient populations for drugs that are currently available, and uncover new drug targets and biomarkers.
“I am extremely proud that the American Association for Cancer Research, as the coordinating center for AACR Project GENIE, is delivering on its promise to make these important data publicly available just over a year after unveiling the initiative,” said Margaret Foti, PhD, MD, chief executive officer of the AACR.
To expand the AACR Project GENIE registry, the consortium is accepting applications for new participating centers. Any nonprofit institution that meets certain criteria can submit an application to become a project participant.
For more information on AACR Project GENIE, visit the project website or send an email to [email protected]. ![]()
Photo courtesy of the
National Institute of
General Medical Sciences
The American Association for Cancer Research (AACR) has announced the first public release of cancer genomic data aggregated through the AACR Project Genomics Evidence Neoplasia Information Exchange (GENIE).
The data set includes nearly 19,000 de-identified genomic records collected from patients who were treated at 8 international institutions, making it one of the largest public cancer genomic data sets released to date.
The release includes data for 59 major cancer types, including leukemias, lymphomas, and multiple myeloma.
The genomic data and a limited amount of linked clinical data for each patient can be accessed via the AACR Project GENIE cBioPortal or from Sage Bionetworks. (Users must create an account for either site to access the data.)
“We are excited to make publicly available this very large set of clinical-grade, next-generation sequencing data obtained during routine patient care,” said Charles L. Sawyers, MD, AACR Project GENIE Steering Committee chairperson.
“These data were generated as part of routine patient care and, without AACR Project GENIE, they would likely never have been shared with the global cancer research community.”
AACR Project GENIE is a multi-phase, international data-sharing project aimed at catalyzing precision oncology through the development of a registry that aggregates and links clinical-grade cancer genomic data with clinical outcomes from tens of thousands of cancer patients treated at multiple institutions.
The newly released data are fully de-identified in compliance with the Health Insurance Portability and Accountability Act (HIPAA).
The data are derived from patients whose tumors were genetically sequenced as part of their care at any of the 8 institutions that participated in the first phase of AACR Project GENIE.
The goal of releasing these data to the cancer research community is to aid new research that will accelerate the pace of progress against cancer.
According to AACR, the data can be used to validate gene signatures of drug response or prognosis, identify new patient populations for drugs that are currently available, and uncover new drug targets and biomarkers.
“I am extremely proud that the American Association for Cancer Research, as the coordinating center for AACR Project GENIE, is delivering on its promise to make these important data publicly available just over a year after unveiling the initiative,” said Margaret Foti, PhD, MD, chief executive officer of the AACR.
To expand the AACR Project GENIE registry, the consortium is accepting applications for new participating centers. Any nonprofit institution that meets certain criteria can submit an application to become a project participant.
For more information on AACR Project GENIE, visit the project website or send an email to [email protected]. ![]()
FDA lifts full clinical hold on pacritinib

The US Food and Drug Administration (FDA) has lifted the full clinical hold placed on all clinical trials conducted under the investigational new drug application for pacritinib, an oral kinase inhibitor being developed as a treatment for myelofibrosis (MF).
The FDA placed the hold on pacritinib trials in February 2016 after results from 2 phase 3 trials, PERSIST-1 and PERSIST-2, showed excess mortality in patients who received pacritinib.
Both trials suggested pacritinib can be more effective than the best available therapy for MF.
However, interim overall survival results from PERSIST-2 indicated that pacritinib had a detrimental effect on survival, which was consistent with the results from PERSIST-1.
Due to the full clinical hold, CTI BioPharma withdrew the new drug application for pacritinib while reviewing data from PERSIST-2.
When the FDA enacted the hold, the agency recommended that CTI BioPharma conduct dose-exploration studies for pacritinib, submit final study reports and data sets for PERSIST-1 and PERSIST-2, make certain modifications to protocols and study-related documents, and request a meeting with the FDA prior to submitting a response to the full clinical hold.
CTI BioPharma followed the FDA’s recommendations and submitted a response to the hold that included final clinical study reports for PERSIST-1 and PERSIST-2 and a protocol for a dose-exploration trial.
The trial, PAC203, should enroll up to approximately 105 patients with primary MF who have failed prior ruxolitinib therapy. The goal is to evaluate the safety and the dose-response relationship for efficacy (spleen volume reduction at 24 weeks) of pacritinib at 3 doses: 100 mg once daily, 100 mg twice daily (BID), and 200 mg BID. The 200 mg BID dose regimen was used in PERSIST-2.
CTI BioPharma said it expects to start the trial in the second quarter of 2017.
“We are pleased to resolve the full clinical hold through working diligently with the FDA to provide a comprehensive response to their requests,” said Richard Love, interim president and chief executive officer of CTI BioPharma.
“We look forward to discussing with the FDA the future development of pacritinib. We believe pacritinib can ultimately address the unmet need of patients with myelofibrosis who are ineligible to receive or are not benefitting from the approved JAK1/JAK2 inhibitor, ruxolitinib, as these patients have limited treatment options.” ![]()
The US Food and Drug Administration (FDA) has lifted the full clinical hold placed on all clinical trials conducted under the investigational new drug application for pacritinib, an oral kinase inhibitor being developed as a treatment for myelofibrosis (MF).
The FDA placed the hold on pacritinib trials in February 2016 after results from 2 phase 3 trials, PERSIST-1 and PERSIST-2, showed excess mortality in patients who received pacritinib.
Both trials suggested pacritinib can be more effective than the best available therapy for MF.
However, interim overall survival results from PERSIST-2 indicated that pacritinib had a detrimental effect on survival, which was consistent with the results from PERSIST-1.
Due to the full clinical hold, CTI BioPharma withdrew the new drug application for pacritinib while reviewing data from PERSIST-2.
When the FDA enacted the hold, the agency recommended that CTI BioPharma conduct dose-exploration studies for pacritinib, submit final study reports and data sets for PERSIST-1 and PERSIST-2, make certain modifications to protocols and study-related documents, and request a meeting with the FDA prior to submitting a response to the full clinical hold.
CTI BioPharma followed the FDA’s recommendations and submitted a response to the hold that included final clinical study reports for PERSIST-1 and PERSIST-2 and a protocol for a dose-exploration trial.
The trial, PAC203, should enroll up to approximately 105 patients with primary MF who have failed prior ruxolitinib therapy. The goal is to evaluate the safety and the dose-response relationship for efficacy (spleen volume reduction at 24 weeks) of pacritinib at 3 doses: 100 mg once daily, 100 mg twice daily (BID), and 200 mg BID. The 200 mg BID dose regimen was used in PERSIST-2.
CTI BioPharma said it expects to start the trial in the second quarter of 2017.
“We are pleased to resolve the full clinical hold through working diligently with the FDA to provide a comprehensive response to their requests,” said Richard Love, interim president and chief executive officer of CTI BioPharma.
“We look forward to discussing with the FDA the future development of pacritinib. We believe pacritinib can ultimately address the unmet need of patients with myelofibrosis who are ineligible to receive or are not benefitting from the approved JAK1/JAK2 inhibitor, ruxolitinib, as these patients have limited treatment options.” ![]()
The US Food and Drug Administration (FDA) has lifted the full clinical hold placed on all clinical trials conducted under the investigational new drug application for pacritinib, an oral kinase inhibitor being developed as a treatment for myelofibrosis (MF).
The FDA placed the hold on pacritinib trials in February 2016 after results from 2 phase 3 trials, PERSIST-1 and PERSIST-2, showed excess mortality in patients who received pacritinib.
Both trials suggested pacritinib can be more effective than the best available therapy for MF.
However, interim overall survival results from PERSIST-2 indicated that pacritinib had a detrimental effect on survival, which was consistent with the results from PERSIST-1.
Due to the full clinical hold, CTI BioPharma withdrew the new drug application for pacritinib while reviewing data from PERSIST-2.
When the FDA enacted the hold, the agency recommended that CTI BioPharma conduct dose-exploration studies for pacritinib, submit final study reports and data sets for PERSIST-1 and PERSIST-2, make certain modifications to protocols and study-related documents, and request a meeting with the FDA prior to submitting a response to the full clinical hold.
CTI BioPharma followed the FDA’s recommendations and submitted a response to the hold that included final clinical study reports for PERSIST-1 and PERSIST-2 and a protocol for a dose-exploration trial.
The trial, PAC203, should enroll up to approximately 105 patients with primary MF who have failed prior ruxolitinib therapy. The goal is to evaluate the safety and the dose-response relationship for efficacy (spleen volume reduction at 24 weeks) of pacritinib at 3 doses: 100 mg once daily, 100 mg twice daily (BID), and 200 mg BID. The 200 mg BID dose regimen was used in PERSIST-2.
CTI BioPharma said it expects to start the trial in the second quarter of 2017.
“We are pleased to resolve the full clinical hold through working diligently with the FDA to provide a comprehensive response to their requests,” said Richard Love, interim president and chief executive officer of CTI BioPharma.
“We look forward to discussing with the FDA the future development of pacritinib. We believe pacritinib can ultimately address the unmet need of patients with myelofibrosis who are ineligible to receive or are not benefitting from the approved JAK1/JAK2 inhibitor, ruxolitinib, as these patients have limited treatment options.” ![]()
Obesity-associated protein linked to AML

Photo courtesy of
University of Cincinnati
Preclinical research indicates that a protein associated with obesity is also involved in the development of acute myeloid leukemia (AML) and may affect AML patients’ response to treatment.
Researchers found evidence to suggest that the fat mass- and obesity-associated protein (FTO) regulates the expression of a set of genes through a mechanism involving RNA modification, thereby increasing the reproduction of leukemia cells and prohibiting drug response.
Jianjun Chen, PhD, of the University of Cincinnati in Ohio, and his colleagues conducted this research and reported the findings in Cancer Cell.
The team noted that N6-methyladenosine (m6A) RNA methylation is the most prevalent internal modification in messenger RNAs (mRNAs) in genes. And they found that FTO, an m6A demethylase, plays a critical oncogenic role in AML.
The researchers made this discovery by analyzing 2 microarray datasets of samples from AML as well as samples from control subjects.
The team found that FTO was highly expressed in AMLs with t(11q23)/MLL rearrangements, t(15;17)/PML-RARA, FLT3-ITD, and/or NPM1 mutations.
The high level of FTO expression contributed to leukemia cells multiplying and surviving and also promoted the development of AML in animal models and the non-response of AML cells to therapeutic agents.
Additionally, the researchers found that genes like ASB2 and RARA, which were reported to inhibit leukemia cell growth and/or mediate the response of leukemia cells to therapeutic agents, were suppressed in the AML samples with higher FTO expression.
The suppression of these genes was attributed to FTO-controlled decreased stability of their mRNA and was connected to FTO’s m6A demethylase activity.
“Our study shows, for the first time, the functional importance of the m6A modification machinery in leukemia,” Dr Chen said. “In addition, given the functional importance of FTO in the formation of leukemia and drug response, targeting FTO signaling may present a new therapeutic strategy to treat leukemia.”
“As FTO may also play a cancer-promoting role in various types of solid tumors, besides leukemia, our discoveries may have a broad impact in cancer biology and cancer therapy. Further studies are needed to advance our understanding of the critical role of FTO in various types of cancers and to develop more effective novel therapeutic strategies based on such understanding to treat cancers.” ![]()
Photo courtesy of
University of Cincinnati
Preclinical research indicates that a protein associated with obesity is also involved in the development of acute myeloid leukemia (AML) and may affect AML patients’ response to treatment.
Researchers found evidence to suggest that the fat mass- and obesity-associated protein (FTO) regulates the expression of a set of genes through a mechanism involving RNA modification, thereby increasing the reproduction of leukemia cells and prohibiting drug response.
Jianjun Chen, PhD, of the University of Cincinnati in Ohio, and his colleagues conducted this research and reported the findings in Cancer Cell.
The team noted that N6-methyladenosine (m6A) RNA methylation is the most prevalent internal modification in messenger RNAs (mRNAs) in genes. And they found that FTO, an m6A demethylase, plays a critical oncogenic role in AML.
The researchers made this discovery by analyzing 2 microarray datasets of samples from AML as well as samples from control subjects.
The team found that FTO was highly expressed in AMLs with t(11q23)/MLL rearrangements, t(15;17)/PML-RARA, FLT3-ITD, and/or NPM1 mutations.
The high level of FTO expression contributed to leukemia cells multiplying and surviving and also promoted the development of AML in animal models and the non-response of AML cells to therapeutic agents.
Additionally, the researchers found that genes like ASB2 and RARA, which were reported to inhibit leukemia cell growth and/or mediate the response of leukemia cells to therapeutic agents, were suppressed in the AML samples with higher FTO expression.
The suppression of these genes was attributed to FTO-controlled decreased stability of their mRNA and was connected to FTO’s m6A demethylase activity.
“Our study shows, for the first time, the functional importance of the m6A modification machinery in leukemia,” Dr Chen said. “In addition, given the functional importance of FTO in the formation of leukemia and drug response, targeting FTO signaling may present a new therapeutic strategy to treat leukemia.”
“As FTO may also play a cancer-promoting role in various types of solid tumors, besides leukemia, our discoveries may have a broad impact in cancer biology and cancer therapy. Further studies are needed to advance our understanding of the critical role of FTO in various types of cancers and to develop more effective novel therapeutic strategies based on such understanding to treat cancers.” ![]()
Photo courtesy of
University of Cincinnati
Preclinical research indicates that a protein associated with obesity is also involved in the development of acute myeloid leukemia (AML) and may affect AML patients’ response to treatment.
Researchers found evidence to suggest that the fat mass- and obesity-associated protein (FTO) regulates the expression of a set of genes through a mechanism involving RNA modification, thereby increasing the reproduction of leukemia cells and prohibiting drug response.
Jianjun Chen, PhD, of the University of Cincinnati in Ohio, and his colleagues conducted this research and reported the findings in Cancer Cell.
The team noted that N6-methyladenosine (m6A) RNA methylation is the most prevalent internal modification in messenger RNAs (mRNAs) in genes. And they found that FTO, an m6A demethylase, plays a critical oncogenic role in AML.
The researchers made this discovery by analyzing 2 microarray datasets of samples from AML as well as samples from control subjects.
The team found that FTO was highly expressed in AMLs with t(11q23)/MLL rearrangements, t(15;17)/PML-RARA, FLT3-ITD, and/or NPM1 mutations.
The high level of FTO expression contributed to leukemia cells multiplying and surviving and also promoted the development of AML in animal models and the non-response of AML cells to therapeutic agents.
Additionally, the researchers found that genes like ASB2 and RARA, which were reported to inhibit leukemia cell growth and/or mediate the response of leukemia cells to therapeutic agents, were suppressed in the AML samples with higher FTO expression.
The suppression of these genes was attributed to FTO-controlled decreased stability of their mRNA and was connected to FTO’s m6A demethylase activity.
“Our study shows, for the first time, the functional importance of the m6A modification machinery in leukemia,” Dr Chen said. “In addition, given the functional importance of FTO in the formation of leukemia and drug response, targeting FTO signaling may present a new therapeutic strategy to treat leukemia.”
“As FTO may also play a cancer-promoting role in various types of solid tumors, besides leukemia, our discoveries may have a broad impact in cancer biology and cancer therapy. Further studies are needed to advance our understanding of the critical role of FTO in various types of cancers and to develop more effective novel therapeutic strategies based on such understanding to treat cancers.” ![]()
Syringes recalled due to infection risk
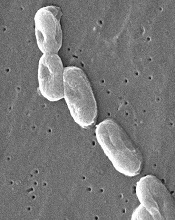
Image courtesy of
CDC/Janice Carr
The US Food and Drug Administration (FDA) has announced a Class I, nationwide recall of Nurse Assist Inc.’s normal saline flush syringes due to incidents of Burkholderia cepacia contamination.
Nurse Assist reported that patients developed B cepacia bloodstream infections while receiving intravenous care using the company’s prepackaged saline flush syringes, so the company voluntarily recalled all unexpired lots of the product.
This recall was first announced last October, but, yesterday, the FDA classified it as a Class I recall and said use of these syringes may cause serious injuries or death.
Nurse Assist said it is investigating the link between the syringes and the infections with the FDA, the US Centers for Disease Control, and various state health departments.
Until the investigation can be completed, all healthcare facilities with affected product should discontinue use and return the product to the supplier.
The normal saline flush is a plastic syringe filled with 0.9% sodium chloride (a 12 mL IV flush syringe with a 3 mL, 5 mL, or 10 mL fill volume). It is used to clear out medical devices that deliver medicine directly into the veins of a patient through a needle or catheter.
All unexpired lots of this product are covered by the recall. Product Numbers 1203, 1205, and 1210 are packaged 30 syringes to an inner carton and 6 inner cartons in a case (180 syringes).
For product number 1210-BP, 100 syringes are packaged in an inner carton with 4 inner cartons in a case (400 syringes). Lot code information can be found on the outer case panel, the back panel of the inner carton, and on each syringe label.
The lots being recalled were distributed to customers and distributors between February 16, 2016, and September 30, 2016. Product can be identified by the labeling on the packaging and device.
Customers with questions can contact Nurse Assist Inc. at 1-800-649-6800 ext. 10, Monday through Friday, between the hours of 8 am and 5 pm, Central Time, or via email at [email protected].
Healthcare professionals and consumers can report adverse reactions or quality problems they experienced using these devices to MedWatch: The FDA Safety Information and Adverse Event Reporting Program. ![]()
Image courtesy of
CDC/Janice Carr
The US Food and Drug Administration (FDA) has announced a Class I, nationwide recall of Nurse Assist Inc.’s normal saline flush syringes due to incidents of Burkholderia cepacia contamination.
Nurse Assist reported that patients developed B cepacia bloodstream infections while receiving intravenous care using the company’s prepackaged saline flush syringes, so the company voluntarily recalled all unexpired lots of the product.
This recall was first announced last October, but, yesterday, the FDA classified it as a Class I recall and said use of these syringes may cause serious injuries or death.
Nurse Assist said it is investigating the link between the syringes and the infections with the FDA, the US Centers for Disease Control, and various state health departments.
Until the investigation can be completed, all healthcare facilities with affected product should discontinue use and return the product to the supplier.
The normal saline flush is a plastic syringe filled with 0.9% sodium chloride (a 12 mL IV flush syringe with a 3 mL, 5 mL, or 10 mL fill volume). It is used to clear out medical devices that deliver medicine directly into the veins of a patient through a needle or catheter.
All unexpired lots of this product are covered by the recall. Product Numbers 1203, 1205, and 1210 are packaged 30 syringes to an inner carton and 6 inner cartons in a case (180 syringes).
For product number 1210-BP, 100 syringes are packaged in an inner carton with 4 inner cartons in a case (400 syringes). Lot code information can be found on the outer case panel, the back panel of the inner carton, and on each syringe label.
The lots being recalled were distributed to customers and distributors between February 16, 2016, and September 30, 2016. Product can be identified by the labeling on the packaging and device.
Customers with questions can contact Nurse Assist Inc. at 1-800-649-6800 ext. 10, Monday through Friday, between the hours of 8 am and 5 pm, Central Time, or via email at [email protected].
Healthcare professionals and consumers can report adverse reactions or quality problems they experienced using these devices to MedWatch: The FDA Safety Information and Adverse Event Reporting Program. ![]()
Image courtesy of
CDC/Janice Carr
The US Food and Drug Administration (FDA) has announced a Class I, nationwide recall of Nurse Assist Inc.’s normal saline flush syringes due to incidents of Burkholderia cepacia contamination.
Nurse Assist reported that patients developed B cepacia bloodstream infections while receiving intravenous care using the company’s prepackaged saline flush syringes, so the company voluntarily recalled all unexpired lots of the product.
This recall was first announced last October, but, yesterday, the FDA classified it as a Class I recall and said use of these syringes may cause serious injuries or death.
Nurse Assist said it is investigating the link between the syringes and the infections with the FDA, the US Centers for Disease Control, and various state health departments.
Until the investigation can be completed, all healthcare facilities with affected product should discontinue use and return the product to the supplier.
The normal saline flush is a plastic syringe filled with 0.9% sodium chloride (a 12 mL IV flush syringe with a 3 mL, 5 mL, or 10 mL fill volume). It is used to clear out medical devices that deliver medicine directly into the veins of a patient through a needle or catheter.
All unexpired lots of this product are covered by the recall. Product Numbers 1203, 1205, and 1210 are packaged 30 syringes to an inner carton and 6 inner cartons in a case (180 syringes).
For product number 1210-BP, 100 syringes are packaged in an inner carton with 4 inner cartons in a case (400 syringes). Lot code information can be found on the outer case panel, the back panel of the inner carton, and on each syringe label.
The lots being recalled were distributed to customers and distributors between February 16, 2016, and September 30, 2016. Product can be identified by the labeling on the packaging and device.
Customers with questions can contact Nurse Assist Inc. at 1-800-649-6800 ext. 10, Monday through Friday, between the hours of 8 am and 5 pm, Central Time, or via email at [email protected].
Healthcare professionals and consumers can report adverse reactions or quality problems they experienced using these devices to MedWatch: The FDA Safety Information and Adverse Event Reporting Program.
Antiplatelet agent may pose lower risk of bleeding

Photo by Sakurai Midori
Researchers say they have developed an antiplatelet agent that has demonstrated considerable
antithrombotic activity and low bleeding liability in monkeys.
The agent, known as BMS-986120, is a PAR4 antagonist.
Experiments in cynomolgus monkeys indicated that BMS-986120 can effectively fight thrombosis but poses a lower risk of bleeding than clopidogrel.
The researchers described these experiments in Science Translational Medicine.
The research was supported by Bristol-Myers Squibb but also benefited from funding from the “Fonds pour un Québec Innovant et en Santé” from the Ministry of Economy, Science and Innovation of Québec.
To investigate the effects of PAR4 inhibition, the researchers developed PAR4-targeted antibodies and tested them in guinea pigs. The antibodies exhibited antithrombotic activity in the animals, while posing a low risk of bleeding.
Because of these positive results, the researchers performed a high-throughput screen of more than 1 million molecules to find a PAR4 inhibitor. They identified BMS-986120 as a “highly potent, selective, and reversible” inhibitor of PAR4 in human platelets.
The researchers wanted to test BMS-986120 in cynomolgus monkey models of occlusive arterial thrombosis and provoked bleeding time (BT). But the team first had to validate the translatability of the models (for clinical prediction of efficacy and bleeding liability) by studying clopidogrel in the animals.
When compared to vehicle control, clopidogrel slowed vascular occlusion and reduced thrombus weight in the monkeys in a dose-dependent manner. But clopidogrel also had “strong effects” on hemostasis.
When the researchers tested BMS-986120 (vs vehicle control) in these models, they observed a wider therapeutic window with BMS-986120 than with clopidogrel.
The researchers said BMS-986120 was “highly effective” in preserving vascular patency during the induction of occlusive thrombosis. When given at 1 mg/kg, BMS-986120 prevented vascular occlusion in all monkeys.
BMS-986120 also decreased thrombus weight in a dose-dependent manner. When given at 1 mg/kg, BMS-986120 reduced thrombus formation by 82%.
BMS-986120 had a limited, though still statistically significant, impact on hemostasis. BMS-986120 at 1 mg/kg significantly increased kidney and mesenteric BT when compared to vehicle control (P<0.0001 for both).
The researchers plotted the relative antithrombotic (thrombus weight reduction) and BT effects of clopidogrel and BMS-986120 as a function of the dose of each drug.
They found that, at doses resulting in about 50% to 80% antithrombotic activity, clopidogrel produced about 8- to 9-fold increases in kidney and mesenteric BT. BMS-986120, on the other hand, produced increases of about 2-fold or less in kidney and mesenteric BT.
The researchers said these preclinical findings suggest that targeting PAR4 is an attractive antiplatelet strategy, and BMS-986120 has potential as an antiplatelet agent. The drug is currently being evaluated in clinical trials.
Photo by Sakurai Midori
Researchers say they have developed an antiplatelet agent that has demonstrated considerable
antithrombotic activity and low bleeding liability in monkeys.
The agent, known as BMS-986120, is a PAR4 antagonist.
Experiments in cynomolgus monkeys indicated that BMS-986120 can effectively fight thrombosis but poses a lower risk of bleeding than clopidogrel.
The researchers described these experiments in Science Translational Medicine.
The research was supported by Bristol-Myers Squibb but also benefited from funding from the “Fonds pour un Québec Innovant et en Santé” from the Ministry of Economy, Science and Innovation of Québec.
To investigate the effects of PAR4 inhibition, the researchers developed PAR4-targeted antibodies and tested them in guinea pigs. The antibodies exhibited antithrombotic activity in the animals, while posing a low risk of bleeding.
Because of these positive results, the researchers performed a high-throughput screen of more than 1 million molecules to find a PAR4 inhibitor. They identified BMS-986120 as a “highly potent, selective, and reversible” inhibitor of PAR4 in human platelets.
The researchers wanted to test BMS-986120 in cynomolgus monkey models of occlusive arterial thrombosis and provoked bleeding time (BT). But the team first had to validate the translatability of the models (for clinical prediction of efficacy and bleeding liability) by studying clopidogrel in the animals.
When compared to vehicle control, clopidogrel slowed vascular occlusion and reduced thrombus weight in the monkeys in a dose-dependent manner. But clopidogrel also had “strong effects” on hemostasis.
When the researchers tested BMS-986120 (vs vehicle control) in these models, they observed a wider therapeutic window with BMS-986120 than with clopidogrel.
The researchers said BMS-986120 was “highly effective” in preserving vascular patency during the induction of occlusive thrombosis. When given at 1 mg/kg, BMS-986120 prevented vascular occlusion in all monkeys.
BMS-986120 also decreased thrombus weight in a dose-dependent manner. When given at 1 mg/kg, BMS-986120 reduced thrombus formation by 82%.
BMS-986120 had a limited, though still statistically significant, impact on hemostasis. BMS-986120 at 1 mg/kg significantly increased kidney and mesenteric BT when compared to vehicle control (P<0.0001 for both).
The researchers plotted the relative antithrombotic (thrombus weight reduction) and BT effects of clopidogrel and BMS-986120 as a function of the dose of each drug.
They found that, at doses resulting in about 50% to 80% antithrombotic activity, clopidogrel produced about 8- to 9-fold increases in kidney and mesenteric BT. BMS-986120, on the other hand, produced increases of about 2-fold or less in kidney and mesenteric BT.
The researchers said these preclinical findings suggest that targeting PAR4 is an attractive antiplatelet strategy, and BMS-986120 has potential as an antiplatelet agent. The drug is currently being evaluated in clinical trials.
Photo by Sakurai Midori
Researchers say they have developed an antiplatelet agent that has demonstrated considerable
antithrombotic activity and low bleeding liability in monkeys.
The agent, known as BMS-986120, is a PAR4 antagonist.
Experiments in cynomolgus monkeys indicated that BMS-986120 can effectively fight thrombosis but poses a lower risk of bleeding than clopidogrel.
The researchers described these experiments in Science Translational Medicine.
The research was supported by Bristol-Myers Squibb but also benefited from funding from the “Fonds pour un Québec Innovant et en Santé” from the Ministry of Economy, Science and Innovation of Québec.
To investigate the effects of PAR4 inhibition, the researchers developed PAR4-targeted antibodies and tested them in guinea pigs. The antibodies exhibited antithrombotic activity in the animals, while posing a low risk of bleeding.
Because of these positive results, the researchers performed a high-throughput screen of more than 1 million molecules to find a PAR4 inhibitor. They identified BMS-986120 as a “highly potent, selective, and reversible” inhibitor of PAR4 in human platelets.
The researchers wanted to test BMS-986120 in cynomolgus monkey models of occlusive arterial thrombosis and provoked bleeding time (BT). But the team first had to validate the translatability of the models (for clinical prediction of efficacy and bleeding liability) by studying clopidogrel in the animals.
When compared to vehicle control, clopidogrel slowed vascular occlusion and reduced thrombus weight in the monkeys in a dose-dependent manner. But clopidogrel also had “strong effects” on hemostasis.
When the researchers tested BMS-986120 (vs vehicle control) in these models, they observed a wider therapeutic window with BMS-986120 than with clopidogrel.
The researchers said BMS-986120 was “highly effective” in preserving vascular patency during the induction of occlusive thrombosis. When given at 1 mg/kg, BMS-986120 prevented vascular occlusion in all monkeys.
BMS-986120 also decreased thrombus weight in a dose-dependent manner. When given at 1 mg/kg, BMS-986120 reduced thrombus formation by 82%.
BMS-986120 had a limited, though still statistically significant, impact on hemostasis. BMS-986120 at 1 mg/kg significantly increased kidney and mesenteric BT when compared to vehicle control (P<0.0001 for both).
The researchers plotted the relative antithrombotic (thrombus weight reduction) and BT effects of clopidogrel and BMS-986120 as a function of the dose of each drug.
They found that, at doses resulting in about 50% to 80% antithrombotic activity, clopidogrel produced about 8- to 9-fold increases in kidney and mesenteric BT. BMS-986120, on the other hand, produced increases of about 2-fold or less in kidney and mesenteric BT.
The researchers said these preclinical findings suggest that targeting PAR4 is an attractive antiplatelet strategy, and BMS-986120 has potential as an antiplatelet agent. The drug is currently being evaluated in clinical trials.
GAP malaria vaccine shows early promise
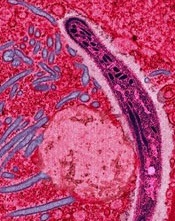
Image from Margaret Shear
and Ute Frevert
A next-generation malaria vaccine that uses genetically attenuated parasites (GAPs) has demonstrated promise in a phase 1 trial and in experiments with mice, according to researchers.
The team generated a genetically attenuated Plasmodium falciparum parasite by knocking out 3 genes that are required for the parasite to cause malaria in humans.
Healthy volunteers infected with this parasite, Pf GAP3KO, exhibited strong antibody responses, according to researchers, and experienced only mild or moderate adverse events (AEs).
When antibodies from these patients were transferred to humanized mice, they inhibited malaria infection in the liver.
The researchers described this work in Science Translational Medicine.
“This most recent publication builds on our previous work,” said study author Sebastian Mikolajczak, PhD, of the Center for Infectious Disease Research in Seattle, Washington.
“We had already good indicators in preclinical studies that this new ‘triple knock-out’ GAP (GAP3KO), which has 3 genes removed, is completely attenuated. The clinical study now shows that the GAP3KO vaccine is completely attenuated in humans and also shows that, even after only a single administration, it elicits a robust immune response against the malaria parasite. Together, these findings are critical milestones for malaria vaccine development.”
Dr Mikolajczak and his colleagues created GAP3KO by knocking out 3 genes in P falciparum that are required for the parasite to successfully infect and cause disease in humans—Pf p52−/p36−/sap1−.
The researchers then tested Pf GAP3KO sporozoites in 10 healthy human volunteers. The sporozoites were delivered via bites from infected mosquitoes.
All subjects experienced grade 1 AEs, and 70% had grade 2 AEs. There were no grade 3 or higher AEs.
Grade 2 AEs included 1 case of fatigue and several cases of administration-site reactions, including erythema (n=6), pruritus (n=2), and swelling (n=3).
All 10 volunteers completed the 28-day study period without exhibiting any symptoms of malaria. They also remained negative for blood-stage parasitemia throughout the study period.
All subjects experienced antibody responses as well. The researchers collected sera from the volunteers on day 0, 7, 13, and 28 after immunization and analyzed it for anti-circumsporozoite protein (CSP) antibody responses.
The team measured anti-CSP immunoglobulin G (IgG) by enzyme-linked immunosorbent assay (ELISA) using full-length recombinant PfCSP protein.
At day 0, volunteers’ sera showed an average of 1436 ± 257.3 arbitrary units (AU) of anti-CSP titers by ELISA. However, 3 volunteers were slightly above the 2000 AU cutoff for positivity.
At day 7, 8 of the 10 volunteers were positive for anti-CSP IgG, with an average titer of 3821 ± 808.1 AU. At day 13, all volunteers were positive, with an average of 11,547 ± 2084 AU. Positivity was maintained through day 28, with an average of 5774± 840 AU.
To determine whether these antibodies could inhibit Pf sporozoite invasion in vivo, the researchers transferred them to FRG huHep liver chimeric humanized mice, which allow for complete development of liver-stage parasites.
The researchers chose 5 volunteers whose immune serum showed high levels of invasion inhibition in vitro and varying CSP IgG titers. The mice received purified IgG from these volunteers from day 0 and day 13 immune samples (5 mice per volunteer per time point).
The mice were then challenged with bites from mosquitoes infected with sporozoites of a green fluorescent protein–luciferase–expressing P falciparum strain.
The researchers calculated inhibition of infection by comparing the liver-stage burden of mice receiving day 13 IgG samples to the mean value of liver-stage burden in mice receiving day 0 IgG samples.
The day 13 IgG samples inhibited liver infection by an average of 23.18 ± 57%, 32.23 ± 29.16%, 69.38 ± 18.64%, 76.63 ± 19.98%, and 87.99 ± 15.56% (per volunteer).
The 3 volunteers whose serum exhibited the highest inhibition had the lowest Pf CSP titers of the 5 samples.
The researchers said these promising results in mice and humans pave the way to a phase 1b trial of the GAP3KO vaccine candidate using controlled human malaria infection.
Image from Margaret Shear
and Ute Frevert
A next-generation malaria vaccine that uses genetically attenuated parasites (GAPs) has demonstrated promise in a phase 1 trial and in experiments with mice, according to researchers.
The team generated a genetically attenuated Plasmodium falciparum parasite by knocking out 3 genes that are required for the parasite to cause malaria in humans.
Healthy volunteers infected with this parasite, Pf GAP3KO, exhibited strong antibody responses, according to researchers, and experienced only mild or moderate adverse events (AEs).
When antibodies from these patients were transferred to humanized mice, they inhibited malaria infection in the liver.
The researchers described this work in Science Translational Medicine.
“This most recent publication builds on our previous work,” said study author Sebastian Mikolajczak, PhD, of the Center for Infectious Disease Research in Seattle, Washington.
“We had already good indicators in preclinical studies that this new ‘triple knock-out’ GAP (GAP3KO), which has 3 genes removed, is completely attenuated. The clinical study now shows that the GAP3KO vaccine is completely attenuated in humans and also shows that, even after only a single administration, it elicits a robust immune response against the malaria parasite. Together, these findings are critical milestones for malaria vaccine development.”
Dr Mikolajczak and his colleagues created GAP3KO by knocking out 3 genes in P falciparum that are required for the parasite to successfully infect and cause disease in humans—Pf p52−/p36−/sap1−.
The researchers then tested Pf GAP3KO sporozoites in 10 healthy human volunteers. The sporozoites were delivered via bites from infected mosquitoes.
All subjects experienced grade 1 AEs, and 70% had grade 2 AEs. There were no grade 3 or higher AEs.
Grade 2 AEs included 1 case of fatigue and several cases of administration-site reactions, including erythema (n=6), pruritus (n=2), and swelling (n=3).
All 10 volunteers completed the 28-day study period without exhibiting any symptoms of malaria. They also remained negative for blood-stage parasitemia throughout the study period.
All subjects experienced antibody responses as well. The researchers collected sera from the volunteers on day 0, 7, 13, and 28 after immunization and analyzed it for anti-circumsporozoite protein (CSP) antibody responses.
The team measured anti-CSP immunoglobulin G (IgG) by enzyme-linked immunosorbent assay (ELISA) using full-length recombinant PfCSP protein.
At day 0, volunteers’ sera showed an average of 1436 ± 257.3 arbitrary units (AU) of anti-CSP titers by ELISA. However, 3 volunteers were slightly above the 2000 AU cutoff for positivity.
At day 7, 8 of the 10 volunteers were positive for anti-CSP IgG, with an average titer of 3821 ± 808.1 AU. At day 13, all volunteers were positive, with an average of 11,547 ± 2084 AU. Positivity was maintained through day 28, with an average of 5774± 840 AU.
To determine whether these antibodies could inhibit Pf sporozoite invasion in vivo, the researchers transferred them to FRG huHep liver chimeric humanized mice, which allow for complete development of liver-stage parasites.
The researchers chose 5 volunteers whose immune serum showed high levels of invasion inhibition in vitro and varying CSP IgG titers. The mice received purified IgG from these volunteers from day 0 and day 13 immune samples (5 mice per volunteer per time point).
The mice were then challenged with bites from mosquitoes infected with sporozoites of a green fluorescent protein–luciferase–expressing P falciparum strain.
The researchers calculated inhibition of infection by comparing the liver-stage burden of mice receiving day 13 IgG samples to the mean value of liver-stage burden in mice receiving day 0 IgG samples.
The day 13 IgG samples inhibited liver infection by an average of 23.18 ± 57%, 32.23 ± 29.16%, 69.38 ± 18.64%, 76.63 ± 19.98%, and 87.99 ± 15.56% (per volunteer).
The 3 volunteers whose serum exhibited the highest inhibition had the lowest Pf CSP titers of the 5 samples.
The researchers said these promising results in mice and humans pave the way to a phase 1b trial of the GAP3KO vaccine candidate using controlled human malaria infection.
Image from Margaret Shear
and Ute Frevert
A next-generation malaria vaccine that uses genetically attenuated parasites (GAPs) has demonstrated promise in a phase 1 trial and in experiments with mice, according to researchers.
The team generated a genetically attenuated Plasmodium falciparum parasite by knocking out 3 genes that are required for the parasite to cause malaria in humans.
Healthy volunteers infected with this parasite, Pf GAP3KO, exhibited strong antibody responses, according to researchers, and experienced only mild or moderate adverse events (AEs).
When antibodies from these patients were transferred to humanized mice, they inhibited malaria infection in the liver.
The researchers described this work in Science Translational Medicine.
“This most recent publication builds on our previous work,” said study author Sebastian Mikolajczak, PhD, of the Center for Infectious Disease Research in Seattle, Washington.
“We had already good indicators in preclinical studies that this new ‘triple knock-out’ GAP (GAP3KO), which has 3 genes removed, is completely attenuated. The clinical study now shows that the GAP3KO vaccine is completely attenuated in humans and also shows that, even after only a single administration, it elicits a robust immune response against the malaria parasite. Together, these findings are critical milestones for malaria vaccine development.”
Dr Mikolajczak and his colleagues created GAP3KO by knocking out 3 genes in P falciparum that are required for the parasite to successfully infect and cause disease in humans—Pf p52−/p36−/sap1−.
The researchers then tested Pf GAP3KO sporozoites in 10 healthy human volunteers. The sporozoites were delivered via bites from infected mosquitoes.
All subjects experienced grade 1 AEs, and 70% had grade 2 AEs. There were no grade 3 or higher AEs.
Grade 2 AEs included 1 case of fatigue and several cases of administration-site reactions, including erythema (n=6), pruritus (n=2), and swelling (n=3).
All 10 volunteers completed the 28-day study period without exhibiting any symptoms of malaria. They also remained negative for blood-stage parasitemia throughout the study period.
All subjects experienced antibody responses as well. The researchers collected sera from the volunteers on day 0, 7, 13, and 28 after immunization and analyzed it for anti-circumsporozoite protein (CSP) antibody responses.
The team measured anti-CSP immunoglobulin G (IgG) by enzyme-linked immunosorbent assay (ELISA) using full-length recombinant PfCSP protein.
At day 0, volunteers’ sera showed an average of 1436 ± 257.3 arbitrary units (AU) of anti-CSP titers by ELISA. However, 3 volunteers were slightly above the 2000 AU cutoff for positivity.
At day 7, 8 of the 10 volunteers were positive for anti-CSP IgG, with an average titer of 3821 ± 808.1 AU. At day 13, all volunteers were positive, with an average of 11,547 ± 2084 AU. Positivity was maintained through day 28, with an average of 5774± 840 AU.
To determine whether these antibodies could inhibit Pf sporozoite invasion in vivo, the researchers transferred them to FRG huHep liver chimeric humanized mice, which allow for complete development of liver-stage parasites.
The researchers chose 5 volunteers whose immune serum showed high levels of invasion inhibition in vitro and varying CSP IgG titers. The mice received purified IgG from these volunteers from day 0 and day 13 immune samples (5 mice per volunteer per time point).
The mice were then challenged with bites from mosquitoes infected with sporozoites of a green fluorescent protein–luciferase–expressing P falciparum strain.
The researchers calculated inhibition of infection by comparing the liver-stage burden of mice receiving day 13 IgG samples to the mean value of liver-stage burden in mice receiving day 0 IgG samples.
The day 13 IgG samples inhibited liver infection by an average of 23.18 ± 57%, 32.23 ± 29.16%, 69.38 ± 18.64%, 76.63 ± 19.98%, and 87.99 ± 15.56% (per volunteer).
The 3 volunteers whose serum exhibited the highest inhibition had the lowest Pf CSP titers of the 5 samples.
The researchers said these promising results in mice and humans pave the way to a phase 1b trial of the GAP3KO vaccine candidate using controlled human malaria infection.
Intervention relieves distress in cancer patients

chemotherapy
Photo by Rhoda Baer
Results of a small study suggest a single dose of the hallucinogenic drug psilocybin, when combined with counseling, can significantly lessen psychological distress in cancer patients for months at a time.
The study showed that psychological counseling and a single dose of psilocybin brought relief from distress that lasted for more than 6 months in a majority of the subjects monitored.
This was based on clinical evaluation scores for anxiety and depression.
“Our results represent the strongest evidence to date of a clinical benefit from psilocybin therapy, with the potential to transform care for patients with cancer-related psychological distress,” said study author Stephen Ross, MD, of New York University School of Medicine in New York, New York.
“If larger clinical trials prove successful, then we could ultimately have available a safe, effective, and inexpensive medication—dispensed under strict control—to alleviate the distress that increases suicide rates among cancer patients.”
Dr Ross and his colleagues reported the results of their study in the Journal of Psychopharmacology alongside a related study and 11 accompanying editorials.
Dr Ross’s study included 29 patients with cancer-related anxiety and depression. Their mean age was 56, and 62% were female. Ninety percent were Caucasian, and 10% were classified as “other” race.
Patients had breast cancer (31%), reproductive cancers (28%), digestive cancers (17%), leukemia/lymphoma (14%), and other cancers (10%).
All patients had been diagnosed as suffering from serious psychological distress related to their disease.
Treatment
Half of the patients were randomly assigned to receive a 0.3 mg/kg dose of psilocybin, and half received a vitamin placebo (250 mg of niacin) known to produce a “rush” that mimics a hallucinogenic drug experience.
Approximately half way through the study’s monitoring period (after 7 weeks), all patients switched treatments. Those who initially received psilocybin took a single dose of niacin, and vice-versa. Neither patients nor researchers knew who had first received psilocybin or placebo.
All patients were provided with tailored counseling from a psychiatrist, psychologist, nurse, or social worker. And the patients were monitored for side effects and improvements in their mental state.
Safety
The researchers said there were no serious adverse events (AEs), either medical or psychiatric, that were attributed to psilocybin.
The most common medical AEs that were attributable to psilocybin were non-clinically significant elevations in blood pressure and heart rate (76%), headaches/migraines (28%), and nausea (14%).
The most common psychiatric AEs attributable to psilocybin were transient anxiety (17%) and transient psychotic-like symptoms (7%; 1 case of transient paranoid ideation and 1 case of transient thought disorder).
Efficacy
The researchers said that, prior to the crossover, psilocybin produced immediate, substantial, and sustained improvements in anxiety and depression.
Specifically, patients who received psilocybin first had significant improvements in responses on the Hospital Anxiety and Depression Scale and the Beck Depression Inventory, when compared to patients who received niacin first.
The differences were significant 1 day after the patients’ first session and 7 weeks after the first session (P≤0.01 for all).
At the 6.5-month follow-up, 60% to 80% of participants continued with clinically significant reductions in depression or anxiety.
The researchers said a key finding of this study was that improvements in clinical evaluation scores for anxiety and depression lasted for the study’s extended monitoring period, which was 8 months for those who took psilocybin first.
Patients also reported post-psilocybin improvements in their quality of life, such as going out more, greater energy, getting along better with family members, and doing well at work. Some reported variations of spirituality, unusual peacefulness, and increased feelings of altruism.
“Our study showed that psilocybin facilitated experiences that drove reductions in psychological distress,” said study author Anthony Bossis, PhD, of New York University School of Medicine. “And if it’s true for cancer care, then it could apply to other stressful medical conditions.”
He cautioned that patients should not consume psilocybin on their own or without supervision from a physician and a trained counselor.
“Psilocybin therapy may not work for everyone,” he noted. “And some groups, such as people with schizophrenia, as well as adolescents, should not be treated with it.”
chemotherapy
Photo by Rhoda Baer
Results of a small study suggest a single dose of the hallucinogenic drug psilocybin, when combined with counseling, can significantly lessen psychological distress in cancer patients for months at a time.
The study showed that psychological counseling and a single dose of psilocybin brought relief from distress that lasted for more than 6 months in a majority of the subjects monitored.
This was based on clinical evaluation scores for anxiety and depression.
“Our results represent the strongest evidence to date of a clinical benefit from psilocybin therapy, with the potential to transform care for patients with cancer-related psychological distress,” said study author Stephen Ross, MD, of New York University School of Medicine in New York, New York.
“If larger clinical trials prove successful, then we could ultimately have available a safe, effective, and inexpensive medication—dispensed under strict control—to alleviate the distress that increases suicide rates among cancer patients.”
Dr Ross and his colleagues reported the results of their study in the Journal of Psychopharmacology alongside a related study and 11 accompanying editorials.
Dr Ross’s study included 29 patients with cancer-related anxiety and depression. Their mean age was 56, and 62% were female. Ninety percent were Caucasian, and 10% were classified as “other” race.
Patients had breast cancer (31%), reproductive cancers (28%), digestive cancers (17%), leukemia/lymphoma (14%), and other cancers (10%).
All patients had been diagnosed as suffering from serious psychological distress related to their disease.
Treatment
Half of the patients were randomly assigned to receive a 0.3 mg/kg dose of psilocybin, and half received a vitamin placebo (250 mg of niacin) known to produce a “rush” that mimics a hallucinogenic drug experience.
Approximately half way through the study’s monitoring period (after 7 weeks), all patients switched treatments. Those who initially received psilocybin took a single dose of niacin, and vice-versa. Neither patients nor researchers knew who had first received psilocybin or placebo.
All patients were provided with tailored counseling from a psychiatrist, psychologist, nurse, or social worker. And the patients were monitored for side effects and improvements in their mental state.
Safety
The researchers said there were no serious adverse events (AEs), either medical or psychiatric, that were attributed to psilocybin.
The most common medical AEs that were attributable to psilocybin were non-clinically significant elevations in blood pressure and heart rate (76%), headaches/migraines (28%), and nausea (14%).
The most common psychiatric AEs attributable to psilocybin were transient anxiety (17%) and transient psychotic-like symptoms (7%; 1 case of transient paranoid ideation and 1 case of transient thought disorder).
Efficacy
The researchers said that, prior to the crossover, psilocybin produced immediate, substantial, and sustained improvements in anxiety and depression.
Specifically, patients who received psilocybin first had significant improvements in responses on the Hospital Anxiety and Depression Scale and the Beck Depression Inventory, when compared to patients who received niacin first.
The differences were significant 1 day after the patients’ first session and 7 weeks after the first session (P≤0.01 for all).
At the 6.5-month follow-up, 60% to 80% of participants continued with clinically significant reductions in depression or anxiety.
The researchers said a key finding of this study was that improvements in clinical evaluation scores for anxiety and depression lasted for the study’s extended monitoring period, which was 8 months for those who took psilocybin first.
Patients also reported post-psilocybin improvements in their quality of life, such as going out more, greater energy, getting along better with family members, and doing well at work. Some reported variations of spirituality, unusual peacefulness, and increased feelings of altruism.
“Our study showed that psilocybin facilitated experiences that drove reductions in psychological distress,” said study author Anthony Bossis, PhD, of New York University School of Medicine. “And if it’s true for cancer care, then it could apply to other stressful medical conditions.”
He cautioned that patients should not consume psilocybin on their own or without supervision from a physician and a trained counselor.
“Psilocybin therapy may not work for everyone,” he noted. “And some groups, such as people with schizophrenia, as well as adolescents, should not be treated with it.”
chemotherapy
Photo by Rhoda Baer
Results of a small study suggest a single dose of the hallucinogenic drug psilocybin, when combined with counseling, can significantly lessen psychological distress in cancer patients for months at a time.
The study showed that psychological counseling and a single dose of psilocybin brought relief from distress that lasted for more than 6 months in a majority of the subjects monitored.
This was based on clinical evaluation scores for anxiety and depression.
“Our results represent the strongest evidence to date of a clinical benefit from psilocybin therapy, with the potential to transform care for patients with cancer-related psychological distress,” said study author Stephen Ross, MD, of New York University School of Medicine in New York, New York.
“If larger clinical trials prove successful, then we could ultimately have available a safe, effective, and inexpensive medication—dispensed under strict control—to alleviate the distress that increases suicide rates among cancer patients.”
Dr Ross and his colleagues reported the results of their study in the Journal of Psychopharmacology alongside a related study and 11 accompanying editorials.
Dr Ross’s study included 29 patients with cancer-related anxiety and depression. Their mean age was 56, and 62% were female. Ninety percent were Caucasian, and 10% were classified as “other” race.
Patients had breast cancer (31%), reproductive cancers (28%), digestive cancers (17%), leukemia/lymphoma (14%), and other cancers (10%).
All patients had been diagnosed as suffering from serious psychological distress related to their disease.
Treatment
Half of the patients were randomly assigned to receive a 0.3 mg/kg dose of psilocybin, and half received a vitamin placebo (250 mg of niacin) known to produce a “rush” that mimics a hallucinogenic drug experience.
Approximately half way through the study’s monitoring period (after 7 weeks), all patients switched treatments. Those who initially received psilocybin took a single dose of niacin, and vice-versa. Neither patients nor researchers knew who had first received psilocybin or placebo.
All patients were provided with tailored counseling from a psychiatrist, psychologist, nurse, or social worker. And the patients were monitored for side effects and improvements in their mental state.
Safety
The researchers said there were no serious adverse events (AEs), either medical or psychiatric, that were attributed to psilocybin.
The most common medical AEs that were attributable to psilocybin were non-clinically significant elevations in blood pressure and heart rate (76%), headaches/migraines (28%), and nausea (14%).
The most common psychiatric AEs attributable to psilocybin were transient anxiety (17%) and transient psychotic-like symptoms (7%; 1 case of transient paranoid ideation and 1 case of transient thought disorder).
Efficacy
The researchers said that, prior to the crossover, psilocybin produced immediate, substantial, and sustained improvements in anxiety and depression.
Specifically, patients who received psilocybin first had significant improvements in responses on the Hospital Anxiety and Depression Scale and the Beck Depression Inventory, when compared to patients who received niacin first.
The differences were significant 1 day after the patients’ first session and 7 weeks after the first session (P≤0.01 for all).
At the 6.5-month follow-up, 60% to 80% of participants continued with clinically significant reductions in depression or anxiety.
The researchers said a key finding of this study was that improvements in clinical evaluation scores for anxiety and depression lasted for the study’s extended monitoring period, which was 8 months for those who took psilocybin first.
Patients also reported post-psilocybin improvements in their quality of life, such as going out more, greater energy, getting along better with family members, and doing well at work. Some reported variations of spirituality, unusual peacefulness, and increased feelings of altruism.
“Our study showed that psilocybin facilitated experiences that drove reductions in psychological distress,” said study author Anthony Bossis, PhD, of New York University School of Medicine. “And if it’s true for cancer care, then it could apply to other stressful medical conditions.”
He cautioned that patients should not consume psilocybin on their own or without supervision from a physician and a trained counselor.
“Psilocybin therapy may not work for everyone,” he noted. “And some groups, such as people with schizophrenia, as well as adolescents, should not be treated with it.”