User login
Lobular-Appearing Nodule on the Scalp
The Diagnosis: Dermal Cylindroma
Microsopic evaluation of a tangential biopsy revealed findings of a dermal process consisting of well-circumscribed islands of pale and darker blue cells with little cytoplasm outlined by a hyaline basement membrane (Figure). These cellular islands were arranged in a jigsawlike configuration. These findings were thought to be consistent with a diagnosis of cylindroma.
|
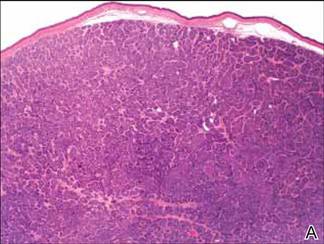
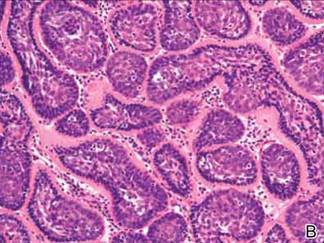
Cylindromas are benign appendageal neoplasms with a somewhat controversial histogenesis. Munger and colleagues1 investigated the pattern of acid mucopolysaccharide secretion by these tumors in association with prosecretory vacuoles in proximity to the Golgi apparatus, which led to their impression that cylindromas most resemble eccrine rather than apocrine sweat glands. Other researchers, however, have concluded that cylindromas are of apocrine derivation.2
Clinically, cylindromas appear most often in 2 settings: isolated or as a manifestation of one of several inherited familial syndromes. One such syndrome is familial cylindromatosis, a rare autosomal-dominant disorder in which affected individuals develop multiple cylindromas, usually on the head and neck. The merging of multiple lesions gives rise to the often-employed term turban tumor.3 This syndrome has been linked to mutations in the cylindromatosis gene, CYLD.4 Brooke-Spiegler syndrome also has been associated with the development of multiple cylindromas. Similar to familial cylindromatosis, it is inherited in an autosomal-dominant fashion. Brooke-Spiegler syndrome is typified by the appearance of multiple cylindromas, trichoepitheliomas, and less commonly spiradenomas. Mutations in the CYLD gene also have been linked to Brooke-Spiegler syndrome in some cases.5
Although considered a benign entity, in rare cases cylindromas have shown evidence of malignant transformation to cylindrocarcinoma. This more aggressive tumor may occur in the setting of isolated cylindromas or more commonly in individuals with numerous lesions, as with both familial cylindromatosis and Brooke-Spiegler syndrome. These lesions may appear to grow rapidly, ulcerate, or bleed, traits that are not associated with their benign counterparts.
Diagnosis of cylindromas rests on histopathologic confirmation, which demonstrates well-defined dermal islands of epithelial cells comprised of dark- and pale-staining nuclei. These tumor islands are surrounded by a hyaline basement membrane and often take on the appearance of a jigsaw puzzle. Cylindrocarcinomas exhibit greater cellular pleomorphism and higher mitotic rates.
Dermal cylindromas require no further treatment but can be electively excised, while treatment of cylindrocarcinoma with excision is curative.6 Definitive excision was offered to our patient, but she declined treatment.
1. Munger BL, Graham JH, Helwig EB. Ultrastructure and histochemical characteristics of dermal eccrine cylindroma (turban tumor). J Invest Dermatol. 1962;39:577-595.
2. Tellechea O, Reis JP, Ilheu O, et al. Dermal cylindroma. an immunohistochemical study of thirteen cases. Am J Dermatopathol. 1995;17:260-265.
3. Biggs PJ, Wooster R, Ford D, et al. Familial cylindromatosis (turban tumour syndrome) gene localised to chromosome 16q12-q13: evidence for its role as a tumour suppressor gene. Nat Genet. 1995;11:441-443.
4. Bignell GR, Warren W, Seal S, et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet. 2000;25:160-165.
5. Bowen S, Gill M, Lee DA, et al. Mutations in the CYLD gene in Brooke-Spiegler syndrome, familial cylindromatosis, and multiple familial trichoepithelioma: lack of genotype-phenotype correlation. J Invest Dermatol. 2005;124:919-920.
6. Gerretsen AL, van der Putte SC, Deenstra W, et al. Cutaneous cylindroma with malignant transformation. Cancer. 1993;72:1618-1623.
The Diagnosis: Dermal Cylindroma
Microsopic evaluation of a tangential biopsy revealed findings of a dermal process consisting of well-circumscribed islands of pale and darker blue cells with little cytoplasm outlined by a hyaline basement membrane (Figure). These cellular islands were arranged in a jigsawlike configuration. These findings were thought to be consistent with a diagnosis of cylindroma.
|
Cylindromas are benign appendageal neoplasms with a somewhat controversial histogenesis. Munger and colleagues1 investigated the pattern of acid mucopolysaccharide secretion by these tumors in association with prosecretory vacuoles in proximity to the Golgi apparatus, which led to their impression that cylindromas most resemble eccrine rather than apocrine sweat glands. Other researchers, however, have concluded that cylindromas are of apocrine derivation.2
Clinically, cylindromas appear most often in 2 settings: isolated or as a manifestation of one of several inherited familial syndromes. One such syndrome is familial cylindromatosis, a rare autosomal-dominant disorder in which affected individuals develop multiple cylindromas, usually on the head and neck. The merging of multiple lesions gives rise to the often-employed term turban tumor.3 This syndrome has been linked to mutations in the cylindromatosis gene, CYLD.4 Brooke-Spiegler syndrome also has been associated with the development of multiple cylindromas. Similar to familial cylindromatosis, it is inherited in an autosomal-dominant fashion. Brooke-Spiegler syndrome is typified by the appearance of multiple cylindromas, trichoepitheliomas, and less commonly spiradenomas. Mutations in the CYLD gene also have been linked to Brooke-Spiegler syndrome in some cases.5
Although considered a benign entity, in rare cases cylindromas have shown evidence of malignant transformation to cylindrocarcinoma. This more aggressive tumor may occur in the setting of isolated cylindromas or more commonly in individuals with numerous lesions, as with both familial cylindromatosis and Brooke-Spiegler syndrome. These lesions may appear to grow rapidly, ulcerate, or bleed, traits that are not associated with their benign counterparts.
Diagnosis of cylindromas rests on histopathologic confirmation, which demonstrates well-defined dermal islands of epithelial cells comprised of dark- and pale-staining nuclei. These tumor islands are surrounded by a hyaline basement membrane and often take on the appearance of a jigsaw puzzle. Cylindrocarcinomas exhibit greater cellular pleomorphism and higher mitotic rates.
Dermal cylindromas require no further treatment but can be electively excised, while treatment of cylindrocarcinoma with excision is curative.6 Definitive excision was offered to our patient, but she declined treatment.
The Diagnosis: Dermal Cylindroma
Microsopic evaluation of a tangential biopsy revealed findings of a dermal process consisting of well-circumscribed islands of pale and darker blue cells with little cytoplasm outlined by a hyaline basement membrane (Figure). These cellular islands were arranged in a jigsawlike configuration. These findings were thought to be consistent with a diagnosis of cylindroma.
|
Cylindromas are benign appendageal neoplasms with a somewhat controversial histogenesis. Munger and colleagues1 investigated the pattern of acid mucopolysaccharide secretion by these tumors in association with prosecretory vacuoles in proximity to the Golgi apparatus, which led to their impression that cylindromas most resemble eccrine rather than apocrine sweat glands. Other researchers, however, have concluded that cylindromas are of apocrine derivation.2
Clinically, cylindromas appear most often in 2 settings: isolated or as a manifestation of one of several inherited familial syndromes. One such syndrome is familial cylindromatosis, a rare autosomal-dominant disorder in which affected individuals develop multiple cylindromas, usually on the head and neck. The merging of multiple lesions gives rise to the often-employed term turban tumor.3 This syndrome has been linked to mutations in the cylindromatosis gene, CYLD.4 Brooke-Spiegler syndrome also has been associated with the development of multiple cylindromas. Similar to familial cylindromatosis, it is inherited in an autosomal-dominant fashion. Brooke-Spiegler syndrome is typified by the appearance of multiple cylindromas, trichoepitheliomas, and less commonly spiradenomas. Mutations in the CYLD gene also have been linked to Brooke-Spiegler syndrome in some cases.5
Although considered a benign entity, in rare cases cylindromas have shown evidence of malignant transformation to cylindrocarcinoma. This more aggressive tumor may occur in the setting of isolated cylindromas or more commonly in individuals with numerous lesions, as with both familial cylindromatosis and Brooke-Spiegler syndrome. These lesions may appear to grow rapidly, ulcerate, or bleed, traits that are not associated with their benign counterparts.
Diagnosis of cylindromas rests on histopathologic confirmation, which demonstrates well-defined dermal islands of epithelial cells comprised of dark- and pale-staining nuclei. These tumor islands are surrounded by a hyaline basement membrane and often take on the appearance of a jigsaw puzzle. Cylindrocarcinomas exhibit greater cellular pleomorphism and higher mitotic rates.
Dermal cylindromas require no further treatment but can be electively excised, while treatment of cylindrocarcinoma with excision is curative.6 Definitive excision was offered to our patient, but she declined treatment.
1. Munger BL, Graham JH, Helwig EB. Ultrastructure and histochemical characteristics of dermal eccrine cylindroma (turban tumor). J Invest Dermatol. 1962;39:577-595.
2. Tellechea O, Reis JP, Ilheu O, et al. Dermal cylindroma. an immunohistochemical study of thirteen cases. Am J Dermatopathol. 1995;17:260-265.
3. Biggs PJ, Wooster R, Ford D, et al. Familial cylindromatosis (turban tumour syndrome) gene localised to chromosome 16q12-q13: evidence for its role as a tumour suppressor gene. Nat Genet. 1995;11:441-443.
4. Bignell GR, Warren W, Seal S, et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet. 2000;25:160-165.
5. Bowen S, Gill M, Lee DA, et al. Mutations in the CYLD gene in Brooke-Spiegler syndrome, familial cylindromatosis, and multiple familial trichoepithelioma: lack of genotype-phenotype correlation. J Invest Dermatol. 2005;124:919-920.
6. Gerretsen AL, van der Putte SC, Deenstra W, et al. Cutaneous cylindroma with malignant transformation. Cancer. 1993;72:1618-1623.
1. Munger BL, Graham JH, Helwig EB. Ultrastructure and histochemical characteristics of dermal eccrine cylindroma (turban tumor). J Invest Dermatol. 1962;39:577-595.
2. Tellechea O, Reis JP, Ilheu O, et al. Dermal cylindroma. an immunohistochemical study of thirteen cases. Am J Dermatopathol. 1995;17:260-265.
3. Biggs PJ, Wooster R, Ford D, et al. Familial cylindromatosis (turban tumour syndrome) gene localised to chromosome 16q12-q13: evidence for its role as a tumour suppressor gene. Nat Genet. 1995;11:441-443.
4. Bignell GR, Warren W, Seal S, et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet. 2000;25:160-165.
5. Bowen S, Gill M, Lee DA, et al. Mutations in the CYLD gene in Brooke-Spiegler syndrome, familial cylindromatosis, and multiple familial trichoepithelioma: lack of genotype-phenotype correlation. J Invest Dermatol. 2005;124:919-920.
6. Gerretsen AL, van der Putte SC, Deenstra W, et al. Cutaneous cylindroma with malignant transformation. Cancer. 1993;72:1618-1623.
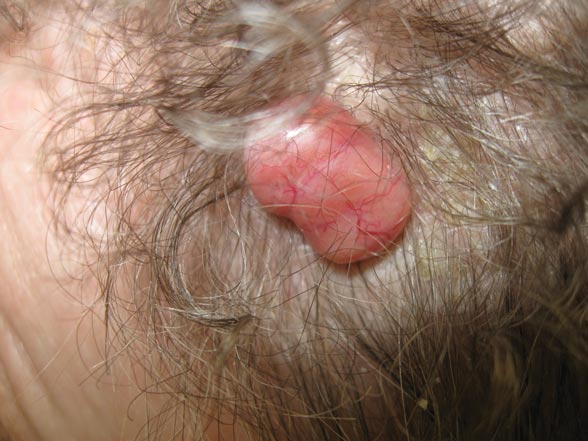
A 79-year-old woman presented with a lesion on the left side of the scalp of several years’ duration that had slowly increased in size. Despite its growth, the lesion remained asymptomatic. Physical examination revealed an exophytic, lobular-appearing nodule on the left side of the temporoparietal scalp, measuring 1.5 cm in size.

Hyperpigmented patches on the neck, shoulder, and back
During a routine examination, a 17-year-old boy was noted to have unilateral hyperpigmented patches on his left neck, shoulder, and back. The lesions had been present since birth, had not changed in size or color, and were asymptomatic. His mother had noted an increase in the size of his left jaw starting at age 1.

The hyperpigmented patches had irregular borders (Figure 1). The skin was otherwise clear. Laboratory testing from 5 years earlier showed normal levels of dehydroepiandrosterone, prolactin, parathyroid hormone, thyroxine, and thyroid-stimulating hormone. Computed tomography showed a “ground-glass” appearance of the bones at the base of the skull, consistent with polyostotic fibrous dysplasia (Figure 2).
Q: Which is the most likely diagnosis?
- Neurofibromatosis
- Congenital melanocytic nevus
- McCune-Albright syndrome
- Tuberous sclerosis

A: This patient had typical features of McCune-Albright syndrome (or Albright syndrome), the classic triad of fibrous dysplasia of bone, large unilateral café-au-lait macules or patches, and precocious puberty or other endocrinopathy.1 The syndrome is rare, with an estimated prevalence of 1/100,000 to 1/1,000,000.2 It results from somatic mutation of the GNAS gene (chromosome 20q13) during embryonic development, which causes constitutive activation of intracellular cyclic adenosine monophosphate (cAMP) signaling and cellular dysplasia.3
THE DIAGNOSIS IS CLINICAL
McCune-Albright syndrome is a clinical diagnosis based on the presence of at least two features of the classic triad.1,4
Other conditions, such as neurofibromatosis, also cause café-au-lait macules in children; but the lesions of McCune-Albright syndrome are fewer in number, larger, and darker and may follow Blaschko lines, with a linear or segmental configuration.5 McCune-Albright lesions tend to have jagged, “coast-of-Maine” borders, as opposed to the smoother “coast-of-California” borders of the lesions of neurofibromatosis.1
Nevertheless, because café-au-lait macules of McCune-Albright syndrome are sometimes indistinguishable from those of neurofibromatosis, the endocrine and skeletal manifestations are essential to making the diagnosis.5
SIGNS OF GENETIC MOSAICISM
The somatic (postzygotic) nature of the GNAS mutation means that patients have normal and abnormal cell lines, ie, mosaicism. Therefore, the extent of disease depends on the precise stage in development during which the mutation occurred. This determines which tissues contain mutated cells and the proportion and distribution of affected cells at these loci.1,4
In addition, differential sensitivity to cAMP signaling between cell types and tissue-specific imprinting of GNAS may contribute to the phenotypic variation seen in McCune-Albright syndrome.4 This means that the clinical features often vary, and the classic clinical triad is not always present.6
The most common clinical features are fibrous dysplasia, which occurs in 46% to 98% of patients, and café-au-lait macules, which occur in 53.1% to 92.5% of patients.1,2 Fibrous dysplasia is typically polyostotic, ie, it involves multiple skeletal sites, with the proximal femur and skull base being the most common.6 It presents as bone pain, asymmetry, or pathologic fracture (or a combination of these) and shows a characteristic “ground-glass” appearance on computed tomography (Figure 2).1 Café-au-lait lesions present at birth or shortly thereafter are often unappreciated as a potential presenting sign.1 These hyperpigmented lesions are typically large and unilateral, often favoring the forehead, nuchal area, sacrum, and buttocks.5.6
Precocious puberty is the most common endocrinopathy in McCune-Albright syndrome, seen in 64% to 79% of cases, and is more common in girls than in boys.2 Other associated endocrinopathies include hyperthyroidism (20% to 30%), excess growth hormone, renal phosphate wasting, and Cushing syndrome.1,6
SCREEN FOR OTHER MANIFESTATIONS
McCune-Albright syndrome can involve a broad spectrum of tissues. Therefore, once the diagnosis is made, the patient should be thoroughly evaluated for other manifestations.1 The evaluation may include imaging studies and biochemical testing and may necessitate referral to an endocrinologist, a radiologist, and an orthopedic surgeon.
Young girls with premature vaginal bleeding or recurrent follicular cysts should always be evaluated for McCune-Albright syndrome, since ovarian enlargement can be mistaken for an ovarian tumor.7 Likewise, adults with isolated fibrous dysplasia or large unilateral café-au-lait macules should also be evaluated, since patients with McCune-Albright syndrome have a normal life span and so may present later in life.4,6
TREATMENT
Drug treatment of this syndrome aims to block the effects of prolonged exposure of end-organs to sex steroids. Since precocious puberty of McCune-Alright syndrome is typically peripheral in origin, it is unresponsive to gonadotropin-releasing hormone agonist drugs; instead, aromatase inhibitors (testolactone) with antiestrogens (tamoxifen) may be used in girls, or antiandrogens (spironolactone) in boys.8
Unfortunately, despite our advanced mechanistic understanding of this disease, medical management remains challenging, with poor long-term efficacy and few studies on long-term outcomes, such as skeletal growth.
GENETIC TESTING HAS LIMITED VALUE
Although genetic testing for GNAS mutations is available, the mosaic nature of McCune-Albright syndrome makes the detection of mutant alleles in affected tissues and circulating cells exceedingly difficult.1,4 These constraints, coupled with high costs, have limited the clinical utility of genetic testing at present. In addition, the lack of a known genotype-phenotype correlation in this syndrome limits the value of genetic testing.1 In the future, improvements in molecular techniques may make genetic testing more useful in the diagnosis and management of McCune-Albright syndrome, especially if clinically relevant genotype-phenotype correlates are identified.4 At this time, although genetic testing is not a standard of care, genetic counseling should be offered to all patients with this syndrome.
- Dumitrescu CE, Collins MT. McCune-Albright syndrome. Orphanet J Rare Dis 2008; 3:12.
- Aycan Z, Önder A, Çetinkaya S. Eight-year follow-up of a girl with McCune-Albright syndrome. J Clin Res Pediatr Endocrinol 2011; 3:40–42.
- Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med 1991; 325:1688–1695.
- Lietman SA, Schwindinger WF, Levine MA. Genetic and molecular aspects of McCune-Albright syndrome. Pediatr Endocrinol Rev 2007; 4(suppl 4):380–385.
- Bolognia JL, Jorizzo JL, Rapini RP, editors. Dermatology. 2nd ed. St. Louis, MO: Mosby/Elsevier; 2008.
- Spitz JL, editor. Genodermatoses: A Clinical Guide to Genetic Skin Disorders. Philadelphia: Lippincott Williams & Wilkins; 2005.
- Nabhan ZM, West KW, Eugster EA. Oophorectomy in McCune-Albright syndrome: a case of mistaken identity. J Pediatr Surg 2007; 42:1578–1583.
- Haddad N, Eugster E. An update on the treatment of precocious puberty in McCune-Albright syndrome and testotoxicosis. J Pediatr Endocrinol Metab 2007; 20:653–661.
During a routine examination, a 17-year-old boy was noted to have unilateral hyperpigmented patches on his left neck, shoulder, and back. The lesions had been present since birth, had not changed in size or color, and were asymptomatic. His mother had noted an increase in the size of his left jaw starting at age 1.
The hyperpigmented patches had irregular borders (Figure 1). The skin was otherwise clear. Laboratory testing from 5 years earlier showed normal levels of dehydroepiandrosterone, prolactin, parathyroid hormone, thyroxine, and thyroid-stimulating hormone. Computed tomography showed a “ground-glass” appearance of the bones at the base of the skull, consistent with polyostotic fibrous dysplasia (Figure 2).
Q: Which is the most likely diagnosis?
- Neurofibromatosis
- Congenital melanocytic nevus
- McCune-Albright syndrome
- Tuberous sclerosis
A: This patient had typical features of McCune-Albright syndrome (or Albright syndrome), the classic triad of fibrous dysplasia of bone, large unilateral café-au-lait macules or patches, and precocious puberty or other endocrinopathy.1 The syndrome is rare, with an estimated prevalence of 1/100,000 to 1/1,000,000.2 It results from somatic mutation of the GNAS gene (chromosome 20q13) during embryonic development, which causes constitutive activation of intracellular cyclic adenosine monophosphate (cAMP) signaling and cellular dysplasia.3
THE DIAGNOSIS IS CLINICAL
McCune-Albright syndrome is a clinical diagnosis based on the presence of at least two features of the classic triad.1,4
Other conditions, such as neurofibromatosis, also cause café-au-lait macules in children; but the lesions of McCune-Albright syndrome are fewer in number, larger, and darker and may follow Blaschko lines, with a linear or segmental configuration.5 McCune-Albright lesions tend to have jagged, “coast-of-Maine” borders, as opposed to the smoother “coast-of-California” borders of the lesions of neurofibromatosis.1
Nevertheless, because café-au-lait macules of McCune-Albright syndrome are sometimes indistinguishable from those of neurofibromatosis, the endocrine and skeletal manifestations are essential to making the diagnosis.5
SIGNS OF GENETIC MOSAICISM
The somatic (postzygotic) nature of the GNAS mutation means that patients have normal and abnormal cell lines, ie, mosaicism. Therefore, the extent of disease depends on the precise stage in development during which the mutation occurred. This determines which tissues contain mutated cells and the proportion and distribution of affected cells at these loci.1,4
In addition, differential sensitivity to cAMP signaling between cell types and tissue-specific imprinting of GNAS may contribute to the phenotypic variation seen in McCune-Albright syndrome.4 This means that the clinical features often vary, and the classic clinical triad is not always present.6
The most common clinical features are fibrous dysplasia, which occurs in 46% to 98% of patients, and café-au-lait macules, which occur in 53.1% to 92.5% of patients.1,2 Fibrous dysplasia is typically polyostotic, ie, it involves multiple skeletal sites, with the proximal femur and skull base being the most common.6 It presents as bone pain, asymmetry, or pathologic fracture (or a combination of these) and shows a characteristic “ground-glass” appearance on computed tomography (Figure 2).1 Café-au-lait lesions present at birth or shortly thereafter are often unappreciated as a potential presenting sign.1 These hyperpigmented lesions are typically large and unilateral, often favoring the forehead, nuchal area, sacrum, and buttocks.5.6
Precocious puberty is the most common endocrinopathy in McCune-Albright syndrome, seen in 64% to 79% of cases, and is more common in girls than in boys.2 Other associated endocrinopathies include hyperthyroidism (20% to 30%), excess growth hormone, renal phosphate wasting, and Cushing syndrome.1,6
SCREEN FOR OTHER MANIFESTATIONS
McCune-Albright syndrome can involve a broad spectrum of tissues. Therefore, once the diagnosis is made, the patient should be thoroughly evaluated for other manifestations.1 The evaluation may include imaging studies and biochemical testing and may necessitate referral to an endocrinologist, a radiologist, and an orthopedic surgeon.
Young girls with premature vaginal bleeding or recurrent follicular cysts should always be evaluated for McCune-Albright syndrome, since ovarian enlargement can be mistaken for an ovarian tumor.7 Likewise, adults with isolated fibrous dysplasia or large unilateral café-au-lait macules should also be evaluated, since patients with McCune-Albright syndrome have a normal life span and so may present later in life.4,6
TREATMENT
Drug treatment of this syndrome aims to block the effects of prolonged exposure of end-organs to sex steroids. Since precocious puberty of McCune-Alright syndrome is typically peripheral in origin, it is unresponsive to gonadotropin-releasing hormone agonist drugs; instead, aromatase inhibitors (testolactone) with antiestrogens (tamoxifen) may be used in girls, or antiandrogens (spironolactone) in boys.8
Unfortunately, despite our advanced mechanistic understanding of this disease, medical management remains challenging, with poor long-term efficacy and few studies on long-term outcomes, such as skeletal growth.
GENETIC TESTING HAS LIMITED VALUE
Although genetic testing for GNAS mutations is available, the mosaic nature of McCune-Albright syndrome makes the detection of mutant alleles in affected tissues and circulating cells exceedingly difficult.1,4 These constraints, coupled with high costs, have limited the clinical utility of genetic testing at present. In addition, the lack of a known genotype-phenotype correlation in this syndrome limits the value of genetic testing.1 In the future, improvements in molecular techniques may make genetic testing more useful in the diagnosis and management of McCune-Albright syndrome, especially if clinically relevant genotype-phenotype correlates are identified.4 At this time, although genetic testing is not a standard of care, genetic counseling should be offered to all patients with this syndrome.
During a routine examination, a 17-year-old boy was noted to have unilateral hyperpigmented patches on his left neck, shoulder, and back. The lesions had been present since birth, had not changed in size or color, and were asymptomatic. His mother had noted an increase in the size of his left jaw starting at age 1.
The hyperpigmented patches had irregular borders (Figure 1). The skin was otherwise clear. Laboratory testing from 5 years earlier showed normal levels of dehydroepiandrosterone, prolactin, parathyroid hormone, thyroxine, and thyroid-stimulating hormone. Computed tomography showed a “ground-glass” appearance of the bones at the base of the skull, consistent with polyostotic fibrous dysplasia (Figure 2).
Q: Which is the most likely diagnosis?
- Neurofibromatosis
- Congenital melanocytic nevus
- McCune-Albright syndrome
- Tuberous sclerosis
A: This patient had typical features of McCune-Albright syndrome (or Albright syndrome), the classic triad of fibrous dysplasia of bone, large unilateral café-au-lait macules or patches, and precocious puberty or other endocrinopathy.1 The syndrome is rare, with an estimated prevalence of 1/100,000 to 1/1,000,000.2 It results from somatic mutation of the GNAS gene (chromosome 20q13) during embryonic development, which causes constitutive activation of intracellular cyclic adenosine monophosphate (cAMP) signaling and cellular dysplasia.3
THE DIAGNOSIS IS CLINICAL
McCune-Albright syndrome is a clinical diagnosis based on the presence of at least two features of the classic triad.1,4
Other conditions, such as neurofibromatosis, also cause café-au-lait macules in children; but the lesions of McCune-Albright syndrome are fewer in number, larger, and darker and may follow Blaschko lines, with a linear or segmental configuration.5 McCune-Albright lesions tend to have jagged, “coast-of-Maine” borders, as opposed to the smoother “coast-of-California” borders of the lesions of neurofibromatosis.1
Nevertheless, because café-au-lait macules of McCune-Albright syndrome are sometimes indistinguishable from those of neurofibromatosis, the endocrine and skeletal manifestations are essential to making the diagnosis.5
SIGNS OF GENETIC MOSAICISM
The somatic (postzygotic) nature of the GNAS mutation means that patients have normal and abnormal cell lines, ie, mosaicism. Therefore, the extent of disease depends on the precise stage in development during which the mutation occurred. This determines which tissues contain mutated cells and the proportion and distribution of affected cells at these loci.1,4
In addition, differential sensitivity to cAMP signaling between cell types and tissue-specific imprinting of GNAS may contribute to the phenotypic variation seen in McCune-Albright syndrome.4 This means that the clinical features often vary, and the classic clinical triad is not always present.6
The most common clinical features are fibrous dysplasia, which occurs in 46% to 98% of patients, and café-au-lait macules, which occur in 53.1% to 92.5% of patients.1,2 Fibrous dysplasia is typically polyostotic, ie, it involves multiple skeletal sites, with the proximal femur and skull base being the most common.6 It presents as bone pain, asymmetry, or pathologic fracture (or a combination of these) and shows a characteristic “ground-glass” appearance on computed tomography (Figure 2).1 Café-au-lait lesions present at birth or shortly thereafter are often unappreciated as a potential presenting sign.1 These hyperpigmented lesions are typically large and unilateral, often favoring the forehead, nuchal area, sacrum, and buttocks.5.6
Precocious puberty is the most common endocrinopathy in McCune-Albright syndrome, seen in 64% to 79% of cases, and is more common in girls than in boys.2 Other associated endocrinopathies include hyperthyroidism (20% to 30%), excess growth hormone, renal phosphate wasting, and Cushing syndrome.1,6
SCREEN FOR OTHER MANIFESTATIONS
McCune-Albright syndrome can involve a broad spectrum of tissues. Therefore, once the diagnosis is made, the patient should be thoroughly evaluated for other manifestations.1 The evaluation may include imaging studies and biochemical testing and may necessitate referral to an endocrinologist, a radiologist, and an orthopedic surgeon.
Young girls with premature vaginal bleeding or recurrent follicular cysts should always be evaluated for McCune-Albright syndrome, since ovarian enlargement can be mistaken for an ovarian tumor.7 Likewise, adults with isolated fibrous dysplasia or large unilateral café-au-lait macules should also be evaluated, since patients with McCune-Albright syndrome have a normal life span and so may present later in life.4,6
TREATMENT
Drug treatment of this syndrome aims to block the effects of prolonged exposure of end-organs to sex steroids. Since precocious puberty of McCune-Alright syndrome is typically peripheral in origin, it is unresponsive to gonadotropin-releasing hormone agonist drugs; instead, aromatase inhibitors (testolactone) with antiestrogens (tamoxifen) may be used in girls, or antiandrogens (spironolactone) in boys.8
Unfortunately, despite our advanced mechanistic understanding of this disease, medical management remains challenging, with poor long-term efficacy and few studies on long-term outcomes, such as skeletal growth.
GENETIC TESTING HAS LIMITED VALUE
Although genetic testing for GNAS mutations is available, the mosaic nature of McCune-Albright syndrome makes the detection of mutant alleles in affected tissues and circulating cells exceedingly difficult.1,4 These constraints, coupled with high costs, have limited the clinical utility of genetic testing at present. In addition, the lack of a known genotype-phenotype correlation in this syndrome limits the value of genetic testing.1 In the future, improvements in molecular techniques may make genetic testing more useful in the diagnosis and management of McCune-Albright syndrome, especially if clinically relevant genotype-phenotype correlates are identified.4 At this time, although genetic testing is not a standard of care, genetic counseling should be offered to all patients with this syndrome.
- Dumitrescu CE, Collins MT. McCune-Albright syndrome. Orphanet J Rare Dis 2008; 3:12.
- Aycan Z, Önder A, Çetinkaya S. Eight-year follow-up of a girl with McCune-Albright syndrome. J Clin Res Pediatr Endocrinol 2011; 3:40–42.
- Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med 1991; 325:1688–1695.
- Lietman SA, Schwindinger WF, Levine MA. Genetic and molecular aspects of McCune-Albright syndrome. Pediatr Endocrinol Rev 2007; 4(suppl 4):380–385.
- Bolognia JL, Jorizzo JL, Rapini RP, editors. Dermatology. 2nd ed. St. Louis, MO: Mosby/Elsevier; 2008.
- Spitz JL, editor. Genodermatoses: A Clinical Guide to Genetic Skin Disorders. Philadelphia: Lippincott Williams & Wilkins; 2005.
- Nabhan ZM, West KW, Eugster EA. Oophorectomy in McCune-Albright syndrome: a case of mistaken identity. J Pediatr Surg 2007; 42:1578–1583.
- Haddad N, Eugster E. An update on the treatment of precocious puberty in McCune-Albright syndrome and testotoxicosis. J Pediatr Endocrinol Metab 2007; 20:653–661.
- Dumitrescu CE, Collins MT. McCune-Albright syndrome. Orphanet J Rare Dis 2008; 3:12.
- Aycan Z, Önder A, Çetinkaya S. Eight-year follow-up of a girl with McCune-Albright syndrome. J Clin Res Pediatr Endocrinol 2011; 3:40–42.
- Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med 1991; 325:1688–1695.
- Lietman SA, Schwindinger WF, Levine MA. Genetic and molecular aspects of McCune-Albright syndrome. Pediatr Endocrinol Rev 2007; 4(suppl 4):380–385.
- Bolognia JL, Jorizzo JL, Rapini RP, editors. Dermatology. 2nd ed. St. Louis, MO: Mosby/Elsevier; 2008.
- Spitz JL, editor. Genodermatoses: A Clinical Guide to Genetic Skin Disorders. Philadelphia: Lippincott Williams & Wilkins; 2005.
- Nabhan ZM, West KW, Eugster EA. Oophorectomy in McCune-Albright syndrome: a case of mistaken identity. J Pediatr Surg 2007; 42:1578–1583.
- Haddad N, Eugster E. An update on the treatment of precocious puberty in McCune-Albright syndrome and testotoxicosis. J Pediatr Endocrinol Metab 2007; 20:653–661.
