User login
Rubbery Nodule on the Face of an Infant
The Diagnosis: Juvenile Xanthogranuloma
Juvenile xanthogranuloma (JXG) was first described in 1905 by Adamson1 as solitary or multiple plaquiform or nodular lesions that are yellow to yellowish brown. In 1954, Helwig and Hackney2 coined the term juvenile xanthogranuloma to define this histiocytic cutaneous granulomatous tumor.
Juvenile xanthogranuloma is a rare dermatologic disorder that may be present at birth and primarily affects infants and young children. The benign lesions generally occur in the first 4 years of life, with a median age of onset of 2 years.3 Lesions range in size from millimeters to several centimeters in diameter.4 The skin of the head and neck is the most commonly involved site in JXG. The most frequent noncutaneous site of JXG involvement is the eye, particularly the iris, accounting for 0.4% of cases.5,6 Extracutaneous sites such as the heart, liver, lungs, spleen, oral cavity, and brain also may be involved.4
Most children with JXG are asymptomatic. Skin lesions present as well-demarcated, rubbery, tan-orange papules or nodules. They usually are solitary, and multiple nodules can increase the risk for extracutaneous involvement.4 A case series of patients with neurofibromatosis type 1 showed 14 of 77 (18%) patients examined in the first year of life presented with JXG or other non–Langerhans cell histiocytosis.7 The adult form of cutaneous xanthogranuloma often presents with severe bronchial asthma.8
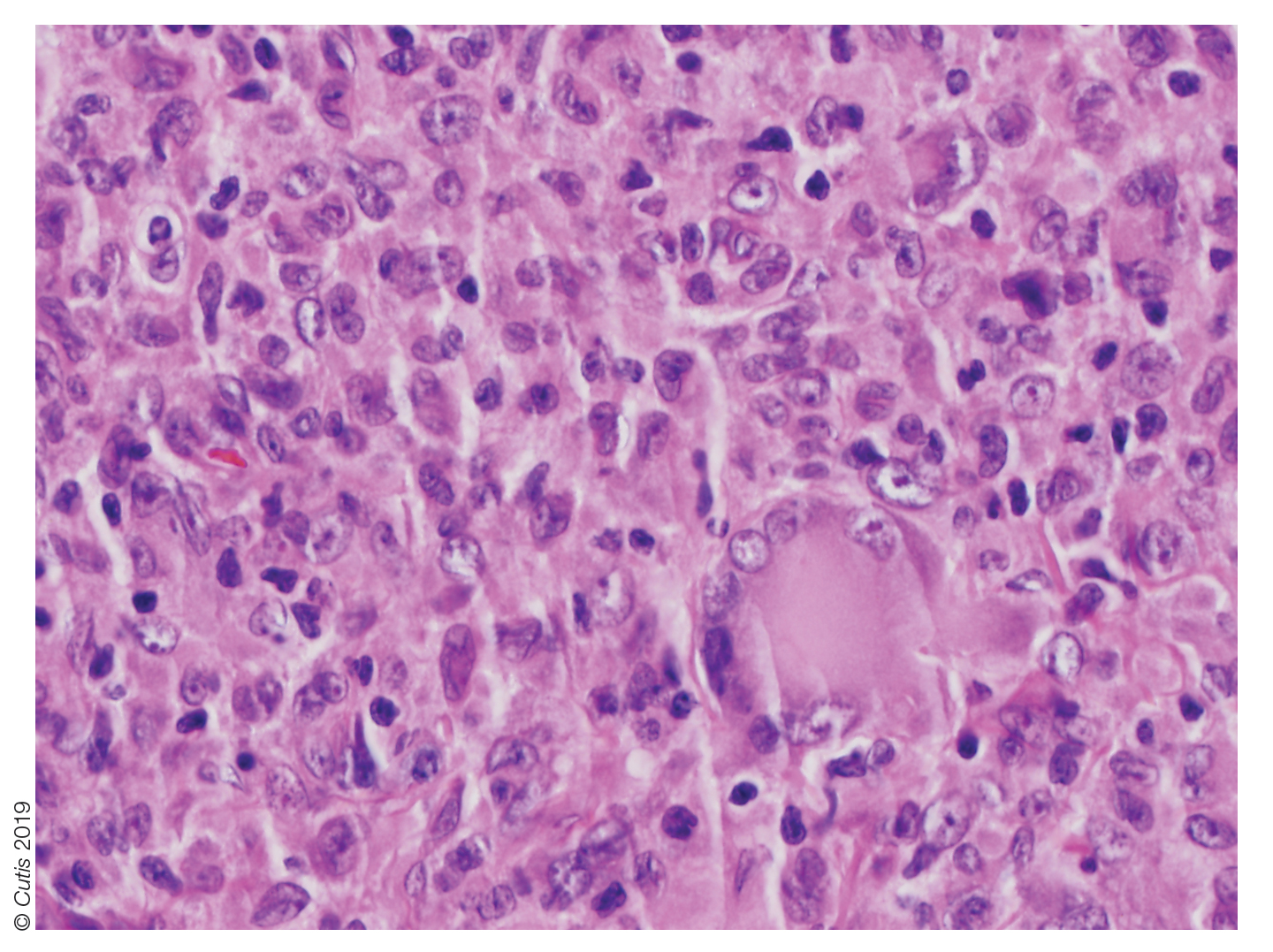
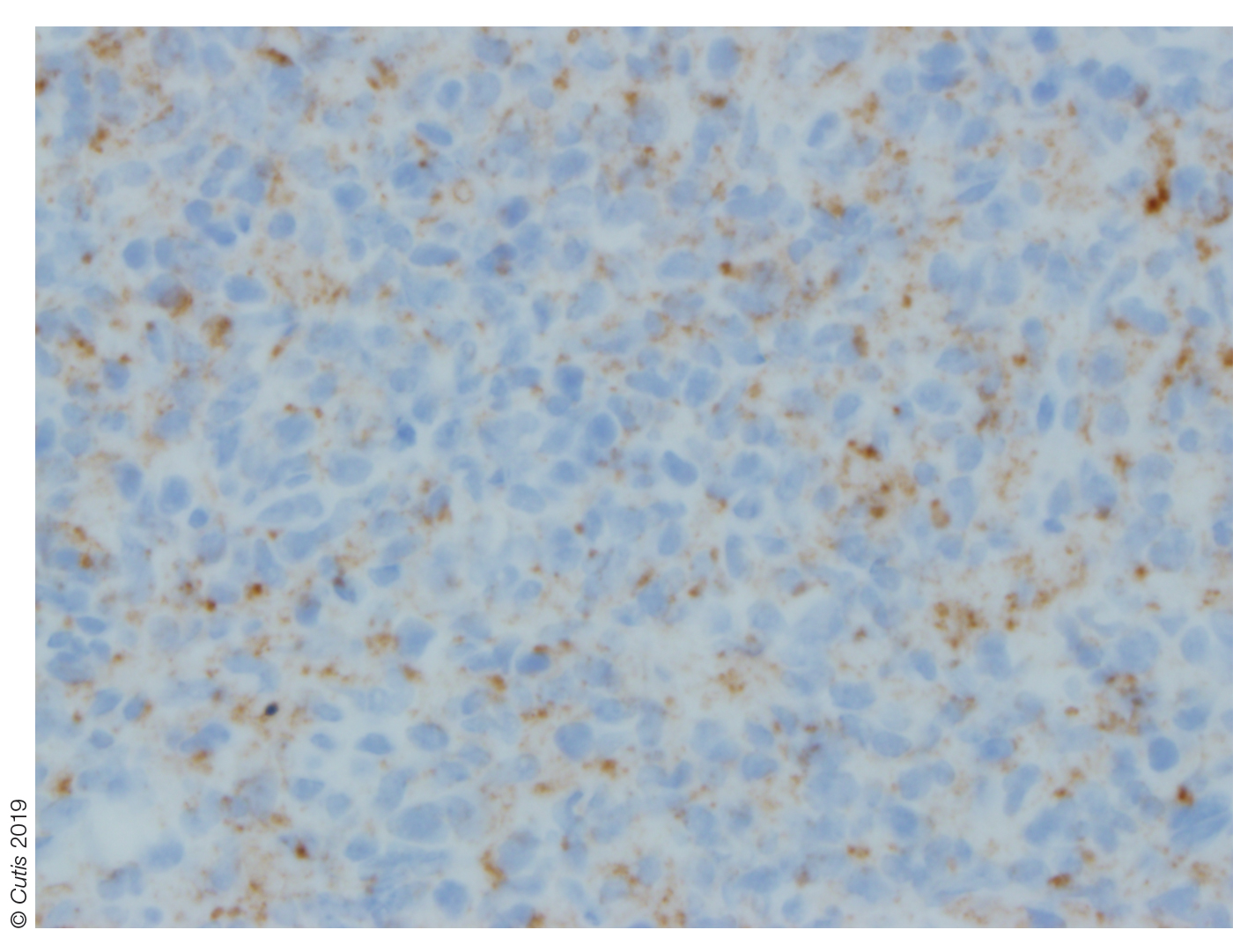
Histopathologic examination of a biopsy of the lesion typically demonstrates well-circumscribed nodules with dense infiltrates of polyhedral histiocytes with vaculoles.3-7 In 85% of cases, Touton giant cells are present.4,9 A prominent vascular network often is present, which was observed in our patient’s biopsy (Figure 1). Immunohistochemistry typically is positive for CD14, CD68, CD163, fascin, and factor XIIIa.4,10 Classically, the cells are negative for S-100 and CD1a, which differentiates these lesions from Langerhans cell histiocytosis.4-7,10 Our patient demonstrated scattered S-100–positive cells representing background dendritic cells and macrophages (Figure 2). The remainder of the clinical, morphologic, and immunophenotypic findings were consistent with non–Langerhans cell histiocytosis, specifically JXG. 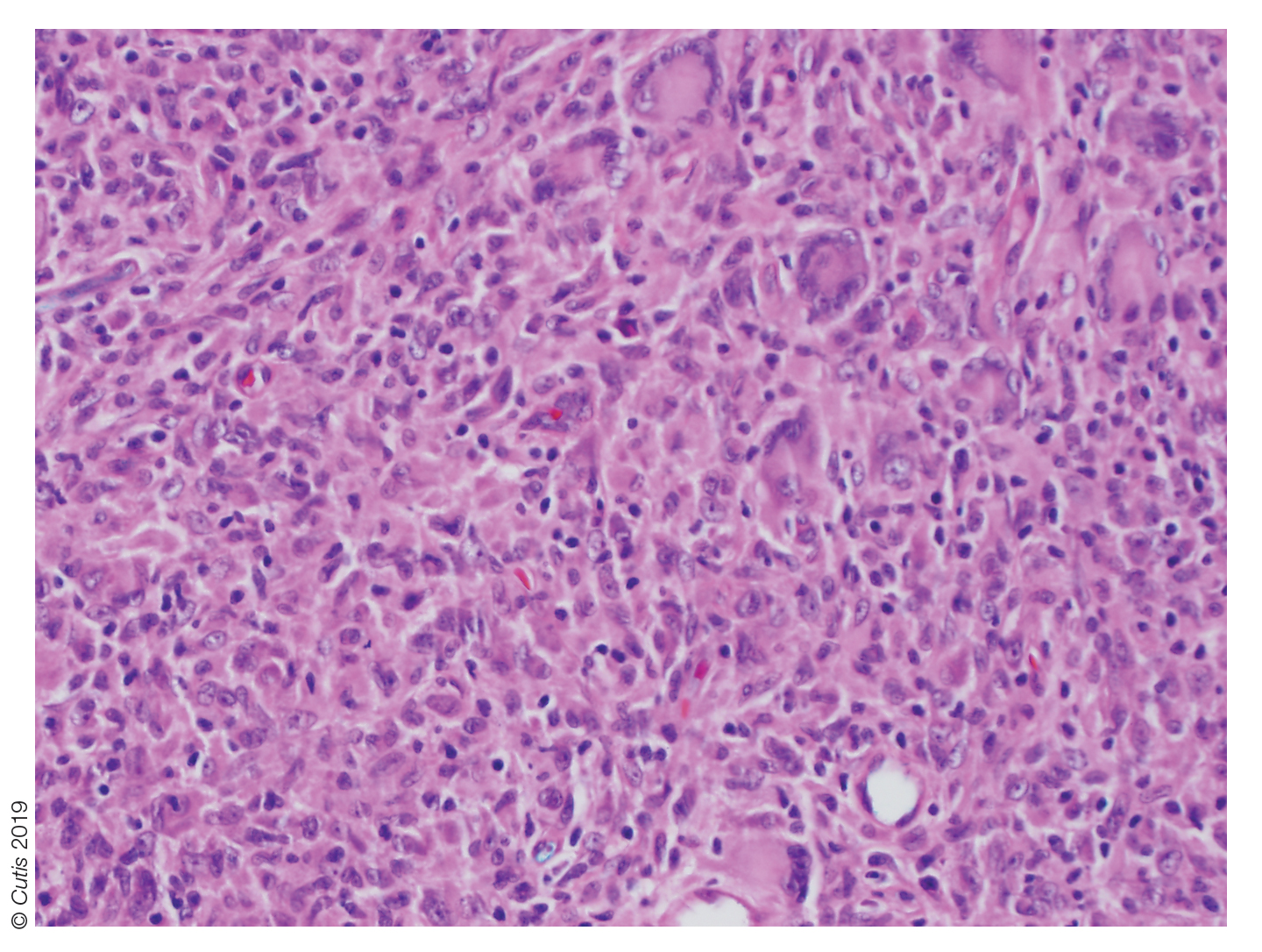
Biopsy should be performed in all cases, and basic laboratory test results such as a complete blood cell count and basic metabolic panel also are appropriate. Routine referral of all patients with cutaneous JXG for ophthalmologic evaluation is not recommended.11 Most patients with ocular involvement present with acute ocular concerns; asymptomatic eye involvement is rare. It is reasonable to consider referral to ophthalmology for patients younger than 2 years who present with multiple lesions, as they may have a higher risk for ocular involvement.11
Juvenile xanthogranuloma usually is a benign disorder with management involving observation, as the lesions typically involute spontaneously.3-7,9,10,12 Systemic or intralesional corticosteroids may be used for treatment in lesions that do not resolve. Ocular JXG refractory to steroid treatment has been managed in several cases with intravitreal bevacizumab.13 Additionally, surgical excision can be considered if malignancy is suspected, the lesion does not resolve with observation or steroid treatment, or the lesion is located near vital structures.4-7,9-13
Spitz nevus presents as a single dome-shaped papule, but histology shows a symmetrical proliferation of spindle and epithelioid cells. Trachoma can present in and around the eye as a follicular hypertrophy but most commonly is seen in the conjunctiva. Dermoid cysts present as solitary subcutaneous cystic lesions; histology demonstrates a lining of keratinizing squamous epithelium with the presence of pilosebaceous structures. Dermatofibroma appears as a tan to reddish-brown papule in an area of prior minor trauma; pathology demonstrates an acanthotic epidermis with an underlying zone of normal papillary dermis and unencapsulated lesions with spindle cells overlapping in fascicles and whorls at the periphery.
1. Adamson HG. Society intelligence: the Dermatological Society of
London. Br J Dermatol. 1905;17:222-223.
2. Helwig E, Hackney VC. Juvenile xanthogranuloma (nevoxanthoendothelioma).
Am J Pathol. 1954;30:625-626.
3. Farrugia EJ, Stephen AP, Raza SA. Juvenile xanthogranuloma of
temporal bone—a case report. J Laryngol Otol. 1997;111:63-65.
4. Cypel TK, Zuker RM. Juvenile xanthogranuloma: case report and review
of the literature. Can J Plast Surg. 2008;16:175-177.
5. Chang MW, Frieden IJ, Good W. The risk of intraocular juvenile
xanthogranuloma: survey of current practices and assessment of risk.
J Am Acad Dermatol. 1996;34:445-449.
6. Zimmerman LE. Ocular lesions of juvenile xanthogranuloma.
nevoxanthoendothelioma. Am J Ophthalmol. 1965;60:1011-1035.
7. Cambiaghi S, Restano L, Caputo R. Juvenile xanthogranuloma associated
with neurofibromatosis 1: 14 patients without evidence of hematologic
malignancies. Pediatr Dermatol. 2004;21:97-101.
8. Stover DG, Alapati S, Regueira O, et al. Treatment of juvenile xanthogranuloma.
Pediatr Blood Cancer. 2008;51:130-133.
9. Dehner LP. Juvenile xanthogranulomas in the first two decades of life. a
clinicopathologic study of 174 cases with cutaneous and extracutaneous
manifestations. Am J Surg Pathol. 2003;27:579-593.
10. Weitzman S, Jaffe R. Uncommon histiocytic disorders: the non-
Langerhans cell histiocytoses. Pediatr Blood Cancer. 2005;45:256-264.
11. Chang MW, Frieden IJ, Good W. The risk of intraocular juvenile
xanthogranuloma: survey of current practices and assessment of risk.
J Am Acad Dermatol. 1996;34:445.
12. Eggli KD, Caro P, Quioque T, et al. Juvenile xanthogranuloma: non-X
histiocytosis with systemic involvement. Pediatr Radiol. 1992;22:374-376.
13. Ashkenazy N, Henry CR, Abbey AM, et al. Successful treatment of juvenile
xanthogranuloma using bevacizumab. J AAPOS. 2014;18:295-297.
The Diagnosis: Juvenile Xanthogranuloma
Juvenile xanthogranuloma (JXG) was first described in 1905 by Adamson1 as solitary or multiple plaquiform or nodular lesions that are yellow to yellowish brown. In 1954, Helwig and Hackney2 coined the term juvenile xanthogranuloma to define this histiocytic cutaneous granulomatous tumor.
Juvenile xanthogranuloma is a rare dermatologic disorder that may be present at birth and primarily affects infants and young children. The benign lesions generally occur in the first 4 years of life, with a median age of onset of 2 years.3 Lesions range in size from millimeters to several centimeters in diameter.4 The skin of the head and neck is the most commonly involved site in JXG. The most frequent noncutaneous site of JXG involvement is the eye, particularly the iris, accounting for 0.4% of cases.5,6 Extracutaneous sites such as the heart, liver, lungs, spleen, oral cavity, and brain also may be involved.4
Most children with JXG are asymptomatic. Skin lesions present as well-demarcated, rubbery, tan-orange papules or nodules. They usually are solitary, and multiple nodules can increase the risk for extracutaneous involvement.4 A case series of patients with neurofibromatosis type 1 showed 14 of 77 (18%) patients examined in the first year of life presented with JXG or other non–Langerhans cell histiocytosis.7 The adult form of cutaneous xanthogranuloma often presents with severe bronchial asthma.8
Histopathologic examination of a biopsy of the lesion typically demonstrates well-circumscribed nodules with dense infiltrates of polyhedral histiocytes with vaculoles.3-7 In 85% of cases, Touton giant cells are present.4,9 A prominent vascular network often is present, which was observed in our patient’s biopsy (Figure 1). Immunohistochemistry typically is positive for CD14, CD68, CD163, fascin, and factor XIIIa.4,10 Classically, the cells are negative for S-100 and CD1a, which differentiates these lesions from Langerhans cell histiocytosis.4-7,10 Our patient demonstrated scattered S-100–positive cells representing background dendritic cells and macrophages (Figure 2). The remainder of the clinical, morphologic, and immunophenotypic findings were consistent with non–Langerhans cell histiocytosis, specifically JXG.
Biopsy should be performed in all cases, and basic laboratory test results such as a complete blood cell count and basic metabolic panel also are appropriate. Routine referral of all patients with cutaneous JXG for ophthalmologic evaluation is not recommended.11 Most patients with ocular involvement present with acute ocular concerns; asymptomatic eye involvement is rare. It is reasonable to consider referral to ophthalmology for patients younger than 2 years who present with multiple lesions, as they may have a higher risk for ocular involvement.11
Juvenile xanthogranuloma usually is a benign disorder with management involving observation, as the lesions typically involute spontaneously.3-7,9,10,12 Systemic or intralesional corticosteroids may be used for treatment in lesions that do not resolve. Ocular JXG refractory to steroid treatment has been managed in several cases with intravitreal bevacizumab.13 Additionally, surgical excision can be considered if malignancy is suspected, the lesion does not resolve with observation or steroid treatment, or the lesion is located near vital structures.4-7,9-13
Spitz nevus presents as a single dome-shaped papule, but histology shows a symmetrical proliferation of spindle and epithelioid cells. Trachoma can present in and around the eye as a follicular hypertrophy but most commonly is seen in the conjunctiva. Dermoid cysts present as solitary subcutaneous cystic lesions; histology demonstrates a lining of keratinizing squamous epithelium with the presence of pilosebaceous structures. Dermatofibroma appears as a tan to reddish-brown papule in an area of prior minor trauma; pathology demonstrates an acanthotic epidermis with an underlying zone of normal papillary dermis and unencapsulated lesions with spindle cells overlapping in fascicles and whorls at the periphery.
The Diagnosis: Juvenile Xanthogranuloma
Juvenile xanthogranuloma (JXG) was first described in 1905 by Adamson1 as solitary or multiple plaquiform or nodular lesions that are yellow to yellowish brown. In 1954, Helwig and Hackney2 coined the term juvenile xanthogranuloma to define this histiocytic cutaneous granulomatous tumor.
Juvenile xanthogranuloma is a rare dermatologic disorder that may be present at birth and primarily affects infants and young children. The benign lesions generally occur in the first 4 years of life, with a median age of onset of 2 years.3 Lesions range in size from millimeters to several centimeters in diameter.4 The skin of the head and neck is the most commonly involved site in JXG. The most frequent noncutaneous site of JXG involvement is the eye, particularly the iris, accounting for 0.4% of cases.5,6 Extracutaneous sites such as the heart, liver, lungs, spleen, oral cavity, and brain also may be involved.4
Most children with JXG are asymptomatic. Skin lesions present as well-demarcated, rubbery, tan-orange papules or nodules. They usually are solitary, and multiple nodules can increase the risk for extracutaneous involvement.4 A case series of patients with neurofibromatosis type 1 showed 14 of 77 (18%) patients examined in the first year of life presented with JXG or other non–Langerhans cell histiocytosis.7 The adult form of cutaneous xanthogranuloma often presents with severe bronchial asthma.8
Histopathologic examination of a biopsy of the lesion typically demonstrates well-circumscribed nodules with dense infiltrates of polyhedral histiocytes with vaculoles.3-7 In 85% of cases, Touton giant cells are present.4,9 A prominent vascular network often is present, which was observed in our patient’s biopsy (Figure 1). Immunohistochemistry typically is positive for CD14, CD68, CD163, fascin, and factor XIIIa.4,10 Classically, the cells are negative for S-100 and CD1a, which differentiates these lesions from Langerhans cell histiocytosis.4-7,10 Our patient demonstrated scattered S-100–positive cells representing background dendritic cells and macrophages (Figure 2). The remainder of the clinical, morphologic, and immunophenotypic findings were consistent with non–Langerhans cell histiocytosis, specifically JXG.
Biopsy should be performed in all cases, and basic laboratory test results such as a complete blood cell count and basic metabolic panel also are appropriate. Routine referral of all patients with cutaneous JXG for ophthalmologic evaluation is not recommended.11 Most patients with ocular involvement present with acute ocular concerns; asymptomatic eye involvement is rare. It is reasonable to consider referral to ophthalmology for patients younger than 2 years who present with multiple lesions, as they may have a higher risk for ocular involvement.11
Juvenile xanthogranuloma usually is a benign disorder with management involving observation, as the lesions typically involute spontaneously.3-7,9,10,12 Systemic or intralesional corticosteroids may be used for treatment in lesions that do not resolve. Ocular JXG refractory to steroid treatment has been managed in several cases with intravitreal bevacizumab.13 Additionally, surgical excision can be considered if malignancy is suspected, the lesion does not resolve with observation or steroid treatment, or the lesion is located near vital structures.4-7,9-13
Spitz nevus presents as a single dome-shaped papule, but histology shows a symmetrical proliferation of spindle and epithelioid cells. Trachoma can present in and around the eye as a follicular hypertrophy but most commonly is seen in the conjunctiva. Dermoid cysts present as solitary subcutaneous cystic lesions; histology demonstrates a lining of keratinizing squamous epithelium with the presence of pilosebaceous structures. Dermatofibroma appears as a tan to reddish-brown papule in an area of prior minor trauma; pathology demonstrates an acanthotic epidermis with an underlying zone of normal papillary dermis and unencapsulated lesions with spindle cells overlapping in fascicles and whorls at the periphery.
1. Adamson HG. Society intelligence: the Dermatological Society of
London. Br J Dermatol. 1905;17:222-223.
2. Helwig E, Hackney VC. Juvenile xanthogranuloma (nevoxanthoendothelioma).
Am J Pathol. 1954;30:625-626.
3. Farrugia EJ, Stephen AP, Raza SA. Juvenile xanthogranuloma of
temporal bone—a case report. J Laryngol Otol. 1997;111:63-65.
4. Cypel TK, Zuker RM. Juvenile xanthogranuloma: case report and review
of the literature. Can J Plast Surg. 2008;16:175-177.
5. Chang MW, Frieden IJ, Good W. The risk of intraocular juvenile
xanthogranuloma: survey of current practices and assessment of risk.
J Am Acad Dermatol. 1996;34:445-449.
6. Zimmerman LE. Ocular lesions of juvenile xanthogranuloma.
nevoxanthoendothelioma. Am J Ophthalmol. 1965;60:1011-1035.
7. Cambiaghi S, Restano L, Caputo R. Juvenile xanthogranuloma associated
with neurofibromatosis 1: 14 patients without evidence of hematologic
malignancies. Pediatr Dermatol. 2004;21:97-101.
8. Stover DG, Alapati S, Regueira O, et al. Treatment of juvenile xanthogranuloma.
Pediatr Blood Cancer. 2008;51:130-133.
9. Dehner LP. Juvenile xanthogranulomas in the first two decades of life. a
clinicopathologic study of 174 cases with cutaneous and extracutaneous
manifestations. Am J Surg Pathol. 2003;27:579-593.
10. Weitzman S, Jaffe R. Uncommon histiocytic disorders: the non-
Langerhans cell histiocytoses. Pediatr Blood Cancer. 2005;45:256-264.
11. Chang MW, Frieden IJ, Good W. The risk of intraocular juvenile
xanthogranuloma: survey of current practices and assessment of risk.
J Am Acad Dermatol. 1996;34:445.
12. Eggli KD, Caro P, Quioque T, et al. Juvenile xanthogranuloma: non-X
histiocytosis with systemic involvement. Pediatr Radiol. 1992;22:374-376.
13. Ashkenazy N, Henry CR, Abbey AM, et al. Successful treatment of juvenile
xanthogranuloma using bevacizumab. J AAPOS. 2014;18:295-297.
1. Adamson HG. Society intelligence: the Dermatological Society of
London. Br J Dermatol. 1905;17:222-223.
2. Helwig E, Hackney VC. Juvenile xanthogranuloma (nevoxanthoendothelioma).
Am J Pathol. 1954;30:625-626.
3. Farrugia EJ, Stephen AP, Raza SA. Juvenile xanthogranuloma of
temporal bone—a case report. J Laryngol Otol. 1997;111:63-65.
4. Cypel TK, Zuker RM. Juvenile xanthogranuloma: case report and review
of the literature. Can J Plast Surg. 2008;16:175-177.
5. Chang MW, Frieden IJ, Good W. The risk of intraocular juvenile
xanthogranuloma: survey of current practices and assessment of risk.
J Am Acad Dermatol. 1996;34:445-449.
6. Zimmerman LE. Ocular lesions of juvenile xanthogranuloma.
nevoxanthoendothelioma. Am J Ophthalmol. 1965;60:1011-1035.
7. Cambiaghi S, Restano L, Caputo R. Juvenile xanthogranuloma associated
with neurofibromatosis 1: 14 patients without evidence of hematologic
malignancies. Pediatr Dermatol. 2004;21:97-101.
8. Stover DG, Alapati S, Regueira O, et al. Treatment of juvenile xanthogranuloma.
Pediatr Blood Cancer. 2008;51:130-133.
9. Dehner LP. Juvenile xanthogranulomas in the first two decades of life. a
clinicopathologic study of 174 cases with cutaneous and extracutaneous
manifestations. Am J Surg Pathol. 2003;27:579-593.
10. Weitzman S, Jaffe R. Uncommon histiocytic disorders: the non-
Langerhans cell histiocytoses. Pediatr Blood Cancer. 2005;45:256-264.
11. Chang MW, Frieden IJ, Good W. The risk of intraocular juvenile
xanthogranuloma: survey of current practices and assessment of risk.
J Am Acad Dermatol. 1996;34:445.
12. Eggli KD, Caro P, Quioque T, et al. Juvenile xanthogranuloma: non-X
histiocytosis with systemic involvement. Pediatr Radiol. 1992;22:374-376.
13. Ashkenazy N, Henry CR, Abbey AM, et al. Successful treatment of juvenile
xanthogranuloma using bevacizumab. J AAPOS. 2014;18:295-297.
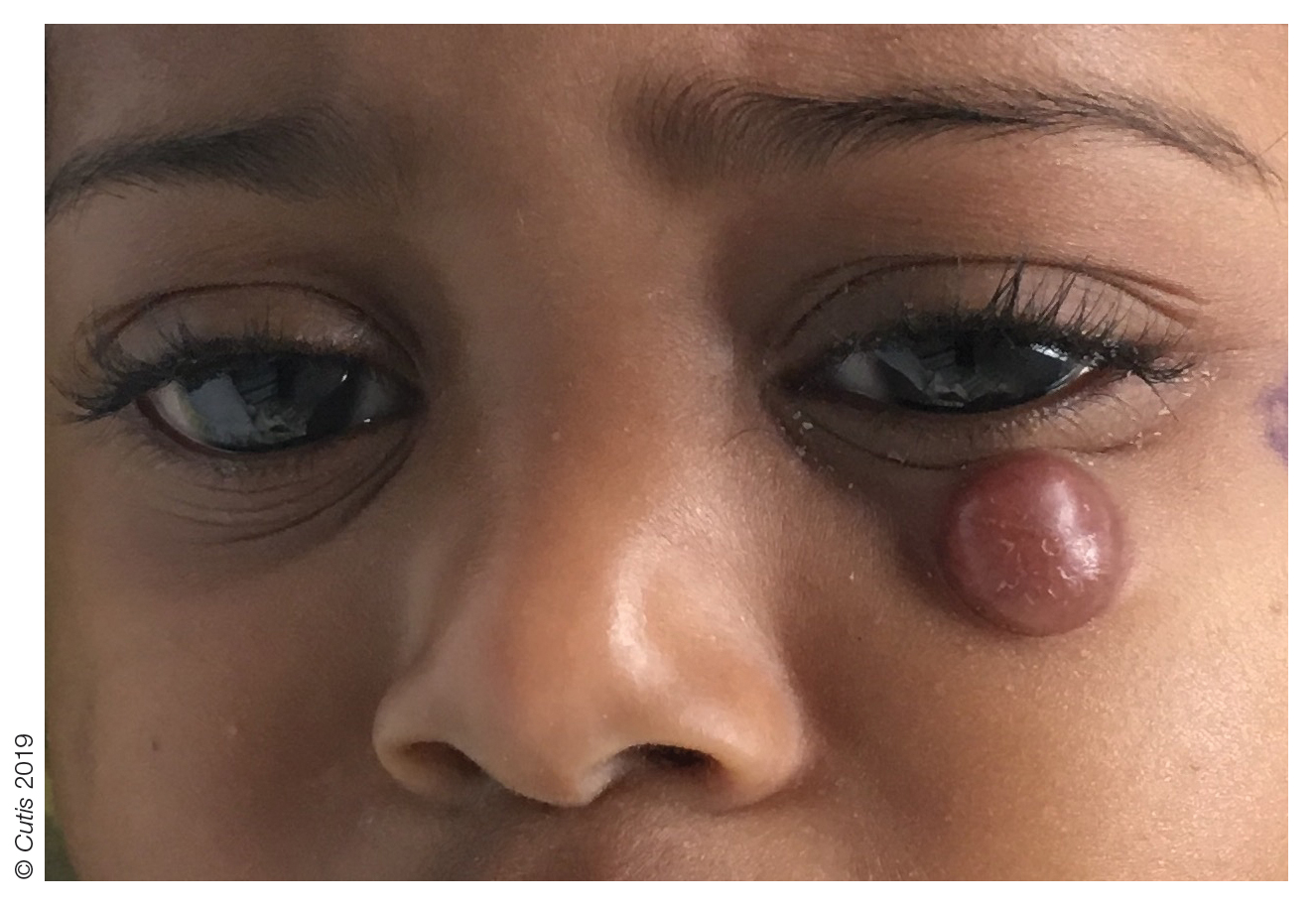
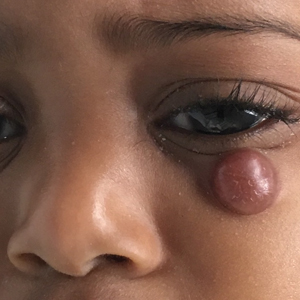
A 10-month-old girl presented with a facial nodule of 7 months’ duration that started as a small lesion. On physical examination, a single 10×10-mm, nontender, well-circumscribed, firm, freely mobile nodule was observed in the left infraorbital area. The patient was born full term at 37 weeks’ gestation via spontaneous vaginal delivery and had no other notable findings on physical examination. Excision was performed by an oculoplastic surgeon. Pathology revealed a relatively well-circumscribed, diffuse, dermal infiltrate of cells arranged in short fascicles and a storiform pattern. The cells had abundant clear to amphophilic cytoplasm, ovoid to reniform nuclei with vesicular chromatin and focal grooves, and diffuse CD68+ immunoreactivity, as well as scattered S-100–positive cells. The patient did well with the excision and no new lesions have developed.