User login
Diabetes management: More than just cardiovascular risk?
Diabetes mellitus and its management have become the center of controversy in recent years. More emphasis is being placed on the potential for adverse cardiovascular outcomes with more aggressive glycemic control as well as on the potential for adverse cardiovascular events with newer antidiabetic therapies, and less on the importance of glycemic control, particularly early in the disease course.
Although it is important to take new data into consideration when managing diabetes, it appears that the results of recent clinical trials are being misinterpreted and incorrectly applied to the wrong patient populations, and in the process, the results of older landmark clinical trials are being neglected. Inadequate glycemic control not only plays a role in cardiovascular risk, it also remains the leading cause of blindness, kidney failure, and nontraumatic lower-limb amputations in the United States.1
Although we need to recognize the potential for adverse cardiovascular outcomes with diabetes and its management, we cannot lose sight of the big picture—ie, that inadequate glycemic control confers both microvascular and macrovascular risk, and that the available data show that restoring near-euglycemia in patients with diabetes considerably reduces the risk of microvascular and macrovascular complications.
Several recently published clinical trials—the Action to Control Cardiovascular Risk in Diabetes (ACCORD),2 the Veterans Affairs Diabetes Trial (VADT),3 and the Action in Diabetes and Vascular Disease (ADVANCE)4—failed to demonstrate improved cardiovascular outcomes with improved glycemic control. However, we should not take this to mean that glycemic control is unimportant.
In this article, we will discuss why the results of these recent clinical trials are not valid for the general population of patients with diabetes. We will review evidence from landmark clinical trials that clearly demonstrates that better glycemic control reduces both microvascular and macrovascular complications of diabetes (the “glucose hypothesis”). We contend that excellent glycemic control clearly decreases the microvascular complications of diabetes, and that results from long-term follow-up studies in both type 1 and type 2 diabetes show reduced rates of heart attack and stroke in patients treated intensively earlier in the course of their disease.5,6
EVIDENCE FOR THE GLUCOSE HYPOTHESIS
Diabetes Control and Complications Trial
The first major trial demonstrating that improved glycemic control provides benefit was the Diabetes Control and Complications Trial (DCCT).7 This study enrolled 1,441 patients with insulin-dependent diabetes mellitus, 726 of whom had no retinopathy at baseline (the primary-prevention cohort) and 715 of whom had mild retinopathy (the secondary-intervention cohort).
Patients were randomly assigned to intensive therapy (three or more insulin injections per day or an insulin pump) or to conventional therapy with one or two daily insulin injections. They were followed for a mean of 6.5 years, and the appearance and progression of retinopathy and other complications were assessed regularly.
During the study, the hemoglobin A1c level averaged 9% in the control group and 7% in the intensively treated group. The cumulative incidence of retinopathy was defined as a change of three steps or more on fundus photography that was sustained over a 6-month period.
Effect on retinopathy. At study completion, the cumulative incidence of retinopathy in the intensive-therapy group was approximately 50% less than in the conventional-therapy group. Intensive therapy reduced the adjusted mean risk of retinopathy by 76% (95% confidence interval [CI] 62%–85%) in the primary-prevention cohort. In the secondary-prevention cohort, intensive therapy reduced the average risk of progression by 54% (95% CI 39%–66%). Intensive therapy reduced the adjusted risk of proliferative or severe nonproliferative retinopathy by 47% (P = .011) and that of treatment with photocoagulation by 56% (P = .002).
Effect on nephropathy. Intensive therapy reduced the mean adjusted risk of microalbuminuria by 34% (P = .04) in the primary-prevention cohort and by 43% (P = .001) in the secondary-intervention cohort. The risk of macroalbuminuria was reduced by 56% (P = .01) in the secondary-intervention cohort.
Effect on neuropathy. In the patients in the primary-prevention cohort who did not have neuropathy at baseline, intensive therapy reduced the incidence of neuropathy at 5 years by 69% (to 3%, vs 10% in the conventional-therapy group; P = .006). Similarly, in the secondary-intervention cohort, intensive therapy reduced the incidence of clinical neuropathy at 5 years by 57% (to 7%, vs 16%; P < .001).
Effect on macrovascular events. In the initial trial, a nonsignificant 41% reduction in combined cardiovascular and peripheral vascular disease events was observed.
DCCT long-term follow-up
After DCCT concluded, the control and treatment groups’ hemoglobin A1c levels converged to approximately 8%. The two groups were then followed to determine the long-term effects of their prior separation of glycemic levels on micro- and macrovascular out comes.5 More than 90% of the original DCCT patients were followed for a mean of 17 years.

Intensive treatment reduced the risk of any cardiovascular disease event by 42% (95% CI 9%–63%; P = .02) and the risk of nonfatal myocardial infarction, stroke, or death from cardiovascular disease by 57% (95% CI 12%– 79%; P = .02). This result was observed despite separation of glucose control in the two groups only for the first 6.5 years. This beneficial effect of intensive early glycemic control has been termed metabolic memory.
United Kingdom Prospective Diabetes Study
A second major trial, the United Kingdom Prospective Diabetes Study (UKPDS),8 assessed the effect of excellent diabetes control on diabetes complications in patients with type 2 diabetes. A total of 3,867 patients newly diagnosed with type 2 diabetes, median age 54, who after 3 months of diet treatment had mean fasting plasma glucose concentrations of 110 to 270 mg/dL, were randomly assigned to an intensive policy (with a sulfonylurea or insulin or, if overweight, metformin) or a conventional policy with diet. The aim in the intensive group was a fasting plasma glucose less than 108 mg/dL. In the conventional group, the aim was the best achievable fasting plasma glucose with diet alone; drugs were added only if there were hyperglycemic symptoms or a fasting plasma glucose greater than 270 mg/dL.
Over 10 years, the median hemoglobin A1c level was 7.0% (interquartile range 6.2%–8.2%) in the intensive group compared with 7.9% (6.9%–8.8%) in the conventional group. Compared with the conventional group, the risk of any diabetes-related end point was 12% lower in the intensive group (95% CI 1%–21%, P = .029), the risk of any diabetes-related death was 10% lower (−11% to 27%, P = .34), and the rate of all-cause mortality was 6% lower (−10% to 20%, P = .44). Most of the reduction in risk of any diabetes-related end point was from a 25% risk reduction (95% CI 7%–40%, P = .0099) in microvascular end points, including the need for retinal photocoagulation.
UKPDS long-term follow-up
In 2008, Holman et al published the results of long-term follow-up of patients included in the UKPDS.6 In posttrial monitoring, 3,277 patients were asked to attend annual UKPDS clinics for 5 years, but no attempts were made to maintain their previously assigned therapies. Annual questionnaires were used to follow patients who were unable to attend the clinics, and all patients in years 6 to 10 were assessed through questionnaires.
Between-group differences in hemoglobin A1c levels were lost after the first year. However, in the sulfonylurea-insulin group, relative reductions in risk persisted at 10 years for any diabetes-related end point (9%, P = .04) and microvascular disease (24%, P = .001), while risk reductions for myocardial infarction (15%, P = .01) and death from any cause (13%, P = .007) emerged over time as more events occurred. In the metformin group, significant risk reductions persisted for any diabetes-related end point (21%, P = .01), myocardial infarction (33%, P = .005), and death from any cause (27%, P = .002).
The long-term follow-up to the UKPDS, like the long-term follow-up to the DCCT, demonstrated metabolic memory: that is, despite an early loss of glycemic differences after completion of the trial, a continued reduction in microvascular risk and an emergent risk reduction for myocardial infarction and death from any cause were observed.
These long-term randomized prospective trials in patients with type 1 and type 2 diabetes clearly show that the glucose hypothesis is in fact correct: intensive glucose control lowers the risk of both microvascular and macrovascular complications of diabetes.
IS THERE DISCORDANCE BETWEEN OLDER AND MORE RECENT TRIALS?
If the results of these older landmark clinical trials are true, why did the more recent clinical trials fail to show cardiovascular benefit with stricter glycemic control, and in one trial2 demonstrate the potential for harm? (ACCORD2 found an increased death rate in patients who received intensive therapy, targeting a hemoglobin A1c below 6.0%.)
The answer lies in the populations studied. ACCORD,2 VADT,3 and ADVANCE4 were performed in older patients with prior cardiac events or with several risk factors for cardiovascular events. The study populations were picked to increase the number of cardiac events in a short time frame. Therefore, extrapolating the results of these studies to the younger population of patients with diabetes, most of whom have yet to develop macrovascular disease, is inappropriate.
The available evidence suggests that early aggressive management of diabetes reduces the risk of macrovascular disease, but that this benefit is delayed. In the UKPDS and DCCT trials, it took 10 to 17 years to show cardiac benefit in younger patients.
The results of ACCORD,2 VADT,3 and ADVANCE4 are important when considered in the correct clinical context. Two of these trials did demonstrate some microvascular benefit as a result of better glycemic control in older patients, many of whom had longstanding diabetes. These studies suggest that, in patients who already have established cardiovascular disease or have several risk factors for cardiovascular events, a less-strict glycemic target may be warranted.
These trials should not be interpreted as saying that glycemic control is unimportant in older patients at higher risk. Rather, they suggest that an individualized approach to diabetes management, supported by the most recent American Diabetes Association guidelines,9 is more appropriate.
Physicians may reasonably suggest a stricter A1c goal (ie, < 6.5%) in certain patients if it can be achieved without significant hypoglycemia. Stricter glycemic targets would seem appropriate in patients recently diagnosed with diabetes, those who have a long life expectancy, and those who have not yet developed significant cardiovascular disease.9
However, in patients who already have developed advanced microvascular and macrovascular complications, who have long-standing diabetes, who have a history of severe hypoglycemia (or hypoglycemia unawareness), or who have a limited life expectancy or numerous adverse comorbidities, a less strict glycemic target (hemoglobin A1c < 8%) may be more appropriate.9
CARDIOVASCULAR RISK, HYPOGLYCEMIA, AND ATTAINING GLYCEMIC TARGETS
Metformin, in the absence of contraindications or intolerability, is generally the recommended first-line therapy to manage glycemia in patients with type 2 diabetes mellitus.10,11 However, there are only limited data to direct clinicians as to which antidiabetic medication to use if further therapy is required to obtain glycemic control.
Much of the cardiovascular and mortality risk associated with aggressive diabetes management (ie, lower A1c targets) is related to hypoglycemia. Thus, antidiabetic therapies that pose no risk or only a low risk of hypoglycemia should be chosen, particularly in older patients and in those with known cardiovascular disease. This may allow for better glycemic control without the risk of hypoglycemia and adverse cardiovascular outcomes.
However, in practice, clinicians continue to use a sulfonylurea as the second-line agent. Although sulfonylureas may appear to be a great option because of their low cost, they are associated with a higher risk of hypoglycemic episodes than other classes of diabetes drugs. We need to consider the frequency and cost of hypoglycemic episodes and the potential morbidity associated with them, because these episodes are a barrier to our efforts to achieve better glycemic control.
Budnitz et al12 reported that from 2007 through 2009, in US adults age 65 and older, insulins were implicated in 13.9% of hospitalizations related to adverse drug events, and oral hypoglycemic agents (ie, insulin secretagogues) in 10.7%.
Quilliam et al13 reported that hypoglycemia resulted in a mean cost of $17,564 for an inpatient admission, $1,387 for an emergency department visit, and $394 for an outpatient visit. Thus, the cost savings associated with prescribing a sulfonylurea vs one of the newer oral antidiabetic agents that do not increase the risk of hypoglycemia (unless used concurrently with insulin or an insulin secretagogue) can quickly be eroded by severe hypoglycemic episodes requiring medical care.
Moreover, once patients start to experience hypoglycemic episodes, they are very reluctant, as are their physicians, to intensify therapy, even if it is indicated by their elevated A1c.
There are now seven classes of oral antidiabetic therapies other than insulin secretagogues (ie, other than sulfonylureas and the meglitinides nateglinide and repaglinide), as well as a few noninsulin injectable therapies (glucagon-like peptide-1 agonists), that are not associated with hypoglycemia. We believe these agents should be tried before prescribing an agent that carries the risk of hypoglycemia (ie, sulfonylureas).
If agents that do not cause hypoglycemia are used, more-aggressive glycemic targets may be achieved safely. The ACCORD study,2 which included patients at high cardiovascular risk and aimed at an aggressive glycemic target of 6%, may have yielded much different results had agents that carry a high risk of hypoglycemia been excluded.
Of importance, cardiovascular risk is also influenced by the common comorbidities seen in patients with diabetes, such as hypertension and hypercholesterolemia. Intensive, multifactorial interventions that address not only glycemic control but also blood pressure and lipids and that include low-dose aspirin therapy have been shown to lower the risk of death from cardiovascular causes and the risk of cardiovascular events.14 Likewise, smoking cessation is very important in reducing cardiovascular risk, especially in patients with diabetes.15
CLINICAL TRIALS IN CONTEXT
In conclusion, there is more to diabetes management than cardiovascular complications. Clearly, improved glycemic control decreases the risk of retinopathy, nephropathy, and neuropathy in patients with type 1 and type 2 diabetes. The DCCT and UKPDS extension studies further found that excellent glycemic control decreases rates of cardiac events.
The best way to treat diabetes may be different in otherwise healthy younger patients who have yet to develop significant complications than it is in older patients known to have cardiovascular disease or several risk factors for cardiovascular events. The available evidence suggests it would be reasonable to aim for stricter glycemic targets in the younger patients and less stringent targets in the older patients, particularly in those with long-standing diabetes who have already developed significant micro- and macrovascular complications.
We should interpret clinical trials within their narrow clinical context, emphasizing the actual population of patients included in the study, so as to avoid the inappropriate extrapolation of the results to all.
- Centers for Disease Control and Prevention. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, 2011. www.cdc.gov/diabetes/pubs/pdf/ndfs_2011.pdf. Accessed October 7, 2014.
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008; 358:2545–2559.
- Duckworth W, Abraira C, Moritz T, et al; VADT Investigators. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 2009; 360:129–139.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008; 358:2560–2572.
- Nathan DM, Cleary PA, Backlund JY, et al; Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 2005; 353:2643–2653.
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359:1577–1589.
- The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 1993; 329:977–986.
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998; 352:837–853.
- American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care 2013; 36(Suppl 1):S11–S66.
- Inzucchi SE, Bergenstal RM, Buse JB, et al; American Diabetes Association (ADA); European Association for the Study of Diabetes (EASD). Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2012; 35:1364–1379.
- Garber AJ, Abrahamson MJ, Barzilay JI, et al; American Association of Clinical Endocrinologists. AACE comprehensive diabetes management algorithm 2013. Endocr Pract 2013; 19:327–336.
- Budnitz DS, Lovegrove MC, Shehab N, Richards CL. Emergency hospitalizations for adverse drug events in older Americans. N Engl J Med 2011; 365:2002–2012.
- Quilliam BJ, Simeone JC, Ozbay AB, Kogut SJ. The incidence and costs of hypoglycemia in type 2 diabetes. Am J Manag Care 2011; 17:673–680.
- Gaede P, Lund-Andersen H, Parving HH, Pedersen O. Effect of a multifactorial intervention on mortality in type 2 diabetes. N Engl J Med 2008; 358:580–591.
- Chaturvedi N, Stevens L, Fuller JH. Which features of smoking determine mortality risk in former cigarette smokers with diabetes? The World Health Organization Multinational Study Group. Diabetes Care 1997; 20:1266–1272.
Diabetes mellitus and its management have become the center of controversy in recent years. More emphasis is being placed on the potential for adverse cardiovascular outcomes with more aggressive glycemic control as well as on the potential for adverse cardiovascular events with newer antidiabetic therapies, and less on the importance of glycemic control, particularly early in the disease course.
Although it is important to take new data into consideration when managing diabetes, it appears that the results of recent clinical trials are being misinterpreted and incorrectly applied to the wrong patient populations, and in the process, the results of older landmark clinical trials are being neglected. Inadequate glycemic control not only plays a role in cardiovascular risk, it also remains the leading cause of blindness, kidney failure, and nontraumatic lower-limb amputations in the United States.1
Although we need to recognize the potential for adverse cardiovascular outcomes with diabetes and its management, we cannot lose sight of the big picture—ie, that inadequate glycemic control confers both microvascular and macrovascular risk, and that the available data show that restoring near-euglycemia in patients with diabetes considerably reduces the risk of microvascular and macrovascular complications.
Several recently published clinical trials—the Action to Control Cardiovascular Risk in Diabetes (ACCORD),2 the Veterans Affairs Diabetes Trial (VADT),3 and the Action in Diabetes and Vascular Disease (ADVANCE)4—failed to demonstrate improved cardiovascular outcomes with improved glycemic control. However, we should not take this to mean that glycemic control is unimportant.
In this article, we will discuss why the results of these recent clinical trials are not valid for the general population of patients with diabetes. We will review evidence from landmark clinical trials that clearly demonstrates that better glycemic control reduces both microvascular and macrovascular complications of diabetes (the “glucose hypothesis”). We contend that excellent glycemic control clearly decreases the microvascular complications of diabetes, and that results from long-term follow-up studies in both type 1 and type 2 diabetes show reduced rates of heart attack and stroke in patients treated intensively earlier in the course of their disease.5,6
EVIDENCE FOR THE GLUCOSE HYPOTHESIS
Diabetes Control and Complications Trial
The first major trial demonstrating that improved glycemic control provides benefit was the Diabetes Control and Complications Trial (DCCT).7 This study enrolled 1,441 patients with insulin-dependent diabetes mellitus, 726 of whom had no retinopathy at baseline (the primary-prevention cohort) and 715 of whom had mild retinopathy (the secondary-intervention cohort).
Patients were randomly assigned to intensive therapy (three or more insulin injections per day or an insulin pump) or to conventional therapy with one or two daily insulin injections. They were followed for a mean of 6.5 years, and the appearance and progression of retinopathy and other complications were assessed regularly.
During the study, the hemoglobin A1c level averaged 9% in the control group and 7% in the intensively treated group. The cumulative incidence of retinopathy was defined as a change of three steps or more on fundus photography that was sustained over a 6-month period.
Effect on retinopathy. At study completion, the cumulative incidence of retinopathy in the intensive-therapy group was approximately 50% less than in the conventional-therapy group. Intensive therapy reduced the adjusted mean risk of retinopathy by 76% (95% confidence interval [CI] 62%–85%) in the primary-prevention cohort. In the secondary-prevention cohort, intensive therapy reduced the average risk of progression by 54% (95% CI 39%–66%). Intensive therapy reduced the adjusted risk of proliferative or severe nonproliferative retinopathy by 47% (P = .011) and that of treatment with photocoagulation by 56% (P = .002).
Effect on nephropathy. Intensive therapy reduced the mean adjusted risk of microalbuminuria by 34% (P = .04) in the primary-prevention cohort and by 43% (P = .001) in the secondary-intervention cohort. The risk of macroalbuminuria was reduced by 56% (P = .01) in the secondary-intervention cohort.
Effect on neuropathy. In the patients in the primary-prevention cohort who did not have neuropathy at baseline, intensive therapy reduced the incidence of neuropathy at 5 years by 69% (to 3%, vs 10% in the conventional-therapy group; P = .006). Similarly, in the secondary-intervention cohort, intensive therapy reduced the incidence of clinical neuropathy at 5 years by 57% (to 7%, vs 16%; P < .001).
Effect on macrovascular events. In the initial trial, a nonsignificant 41% reduction in combined cardiovascular and peripheral vascular disease events was observed.
DCCT long-term follow-up
After DCCT concluded, the control and treatment groups’ hemoglobin A1c levels converged to approximately 8%. The two groups were then followed to determine the long-term effects of their prior separation of glycemic levels on micro- and macrovascular out comes.5 More than 90% of the original DCCT patients were followed for a mean of 17 years.
Intensive treatment reduced the risk of any cardiovascular disease event by 42% (95% CI 9%–63%; P = .02) and the risk of nonfatal myocardial infarction, stroke, or death from cardiovascular disease by 57% (95% CI 12%– 79%; P = .02). This result was observed despite separation of glucose control in the two groups only for the first 6.5 years. This beneficial effect of intensive early glycemic control has been termed metabolic memory.
United Kingdom Prospective Diabetes Study
A second major trial, the United Kingdom Prospective Diabetes Study (UKPDS),8 assessed the effect of excellent diabetes control on diabetes complications in patients with type 2 diabetes. A total of 3,867 patients newly diagnosed with type 2 diabetes, median age 54, who after 3 months of diet treatment had mean fasting plasma glucose concentrations of 110 to 270 mg/dL, were randomly assigned to an intensive policy (with a sulfonylurea or insulin or, if overweight, metformin) or a conventional policy with diet. The aim in the intensive group was a fasting plasma glucose less than 108 mg/dL. In the conventional group, the aim was the best achievable fasting plasma glucose with diet alone; drugs were added only if there were hyperglycemic symptoms or a fasting plasma glucose greater than 270 mg/dL.
Over 10 years, the median hemoglobin A1c level was 7.0% (interquartile range 6.2%–8.2%) in the intensive group compared with 7.9% (6.9%–8.8%) in the conventional group. Compared with the conventional group, the risk of any diabetes-related end point was 12% lower in the intensive group (95% CI 1%–21%, P = .029), the risk of any diabetes-related death was 10% lower (−11% to 27%, P = .34), and the rate of all-cause mortality was 6% lower (−10% to 20%, P = .44). Most of the reduction in risk of any diabetes-related end point was from a 25% risk reduction (95% CI 7%–40%, P = .0099) in microvascular end points, including the need for retinal photocoagulation.
UKPDS long-term follow-up
In 2008, Holman et al published the results of long-term follow-up of patients included in the UKPDS.6 In posttrial monitoring, 3,277 patients were asked to attend annual UKPDS clinics for 5 years, but no attempts were made to maintain their previously assigned therapies. Annual questionnaires were used to follow patients who were unable to attend the clinics, and all patients in years 6 to 10 were assessed through questionnaires.
Between-group differences in hemoglobin A1c levels were lost after the first year. However, in the sulfonylurea-insulin group, relative reductions in risk persisted at 10 years for any diabetes-related end point (9%, P = .04) and microvascular disease (24%, P = .001), while risk reductions for myocardial infarction (15%, P = .01) and death from any cause (13%, P = .007) emerged over time as more events occurred. In the metformin group, significant risk reductions persisted for any diabetes-related end point (21%, P = .01), myocardial infarction (33%, P = .005), and death from any cause (27%, P = .002).
The long-term follow-up to the UKPDS, like the long-term follow-up to the DCCT, demonstrated metabolic memory: that is, despite an early loss of glycemic differences after completion of the trial, a continued reduction in microvascular risk and an emergent risk reduction for myocardial infarction and death from any cause were observed.
These long-term randomized prospective trials in patients with type 1 and type 2 diabetes clearly show that the glucose hypothesis is in fact correct: intensive glucose control lowers the risk of both microvascular and macrovascular complications of diabetes.
IS THERE DISCORDANCE BETWEEN OLDER AND MORE RECENT TRIALS?
If the results of these older landmark clinical trials are true, why did the more recent clinical trials fail to show cardiovascular benefit with stricter glycemic control, and in one trial2 demonstrate the potential for harm? (ACCORD2 found an increased death rate in patients who received intensive therapy, targeting a hemoglobin A1c below 6.0%.)
The answer lies in the populations studied. ACCORD,2 VADT,3 and ADVANCE4 were performed in older patients with prior cardiac events or with several risk factors for cardiovascular events. The study populations were picked to increase the number of cardiac events in a short time frame. Therefore, extrapolating the results of these studies to the younger population of patients with diabetes, most of whom have yet to develop macrovascular disease, is inappropriate.
The available evidence suggests that early aggressive management of diabetes reduces the risk of macrovascular disease, but that this benefit is delayed. In the UKPDS and DCCT trials, it took 10 to 17 years to show cardiac benefit in younger patients.
The results of ACCORD,2 VADT,3 and ADVANCE4 are important when considered in the correct clinical context. Two of these trials did demonstrate some microvascular benefit as a result of better glycemic control in older patients, many of whom had longstanding diabetes. These studies suggest that, in patients who already have established cardiovascular disease or have several risk factors for cardiovascular events, a less-strict glycemic target may be warranted.
These trials should not be interpreted as saying that glycemic control is unimportant in older patients at higher risk. Rather, they suggest that an individualized approach to diabetes management, supported by the most recent American Diabetes Association guidelines,9 is more appropriate.
Physicians may reasonably suggest a stricter A1c goal (ie, < 6.5%) in certain patients if it can be achieved without significant hypoglycemia. Stricter glycemic targets would seem appropriate in patients recently diagnosed with diabetes, those who have a long life expectancy, and those who have not yet developed significant cardiovascular disease.9
However, in patients who already have developed advanced microvascular and macrovascular complications, who have long-standing diabetes, who have a history of severe hypoglycemia (or hypoglycemia unawareness), or who have a limited life expectancy or numerous adverse comorbidities, a less strict glycemic target (hemoglobin A1c < 8%) may be more appropriate.9
CARDIOVASCULAR RISK, HYPOGLYCEMIA, AND ATTAINING GLYCEMIC TARGETS
Metformin, in the absence of contraindications or intolerability, is generally the recommended first-line therapy to manage glycemia in patients with type 2 diabetes mellitus.10,11 However, there are only limited data to direct clinicians as to which antidiabetic medication to use if further therapy is required to obtain glycemic control.
Much of the cardiovascular and mortality risk associated with aggressive diabetes management (ie, lower A1c targets) is related to hypoglycemia. Thus, antidiabetic therapies that pose no risk or only a low risk of hypoglycemia should be chosen, particularly in older patients and in those with known cardiovascular disease. This may allow for better glycemic control without the risk of hypoglycemia and adverse cardiovascular outcomes.
However, in practice, clinicians continue to use a sulfonylurea as the second-line agent. Although sulfonylureas may appear to be a great option because of their low cost, they are associated with a higher risk of hypoglycemic episodes than other classes of diabetes drugs. We need to consider the frequency and cost of hypoglycemic episodes and the potential morbidity associated with them, because these episodes are a barrier to our efforts to achieve better glycemic control.
Budnitz et al12 reported that from 2007 through 2009, in US adults age 65 and older, insulins were implicated in 13.9% of hospitalizations related to adverse drug events, and oral hypoglycemic agents (ie, insulin secretagogues) in 10.7%.
Quilliam et al13 reported that hypoglycemia resulted in a mean cost of $17,564 for an inpatient admission, $1,387 for an emergency department visit, and $394 for an outpatient visit. Thus, the cost savings associated with prescribing a sulfonylurea vs one of the newer oral antidiabetic agents that do not increase the risk of hypoglycemia (unless used concurrently with insulin or an insulin secretagogue) can quickly be eroded by severe hypoglycemic episodes requiring medical care.
Moreover, once patients start to experience hypoglycemic episodes, they are very reluctant, as are their physicians, to intensify therapy, even if it is indicated by their elevated A1c.
There are now seven classes of oral antidiabetic therapies other than insulin secretagogues (ie, other than sulfonylureas and the meglitinides nateglinide and repaglinide), as well as a few noninsulin injectable therapies (glucagon-like peptide-1 agonists), that are not associated with hypoglycemia. We believe these agents should be tried before prescribing an agent that carries the risk of hypoglycemia (ie, sulfonylureas).
If agents that do not cause hypoglycemia are used, more-aggressive glycemic targets may be achieved safely. The ACCORD study,2 which included patients at high cardiovascular risk and aimed at an aggressive glycemic target of 6%, may have yielded much different results had agents that carry a high risk of hypoglycemia been excluded.
Of importance, cardiovascular risk is also influenced by the common comorbidities seen in patients with diabetes, such as hypertension and hypercholesterolemia. Intensive, multifactorial interventions that address not only glycemic control but also blood pressure and lipids and that include low-dose aspirin therapy have been shown to lower the risk of death from cardiovascular causes and the risk of cardiovascular events.14 Likewise, smoking cessation is very important in reducing cardiovascular risk, especially in patients with diabetes.15
CLINICAL TRIALS IN CONTEXT
In conclusion, there is more to diabetes management than cardiovascular complications. Clearly, improved glycemic control decreases the risk of retinopathy, nephropathy, and neuropathy in patients with type 1 and type 2 diabetes. The DCCT and UKPDS extension studies further found that excellent glycemic control decreases rates of cardiac events.
The best way to treat diabetes may be different in otherwise healthy younger patients who have yet to develop significant complications than it is in older patients known to have cardiovascular disease or several risk factors for cardiovascular events. The available evidence suggests it would be reasonable to aim for stricter glycemic targets in the younger patients and less stringent targets in the older patients, particularly in those with long-standing diabetes who have already developed significant micro- and macrovascular complications.
We should interpret clinical trials within their narrow clinical context, emphasizing the actual population of patients included in the study, so as to avoid the inappropriate extrapolation of the results to all.
Diabetes mellitus and its management have become the center of controversy in recent years. More emphasis is being placed on the potential for adverse cardiovascular outcomes with more aggressive glycemic control as well as on the potential for adverse cardiovascular events with newer antidiabetic therapies, and less on the importance of glycemic control, particularly early in the disease course.
Although it is important to take new data into consideration when managing diabetes, it appears that the results of recent clinical trials are being misinterpreted and incorrectly applied to the wrong patient populations, and in the process, the results of older landmark clinical trials are being neglected. Inadequate glycemic control not only plays a role in cardiovascular risk, it also remains the leading cause of blindness, kidney failure, and nontraumatic lower-limb amputations in the United States.1
Although we need to recognize the potential for adverse cardiovascular outcomes with diabetes and its management, we cannot lose sight of the big picture—ie, that inadequate glycemic control confers both microvascular and macrovascular risk, and that the available data show that restoring near-euglycemia in patients with diabetes considerably reduces the risk of microvascular and macrovascular complications.
Several recently published clinical trials—the Action to Control Cardiovascular Risk in Diabetes (ACCORD),2 the Veterans Affairs Diabetes Trial (VADT),3 and the Action in Diabetes and Vascular Disease (ADVANCE)4—failed to demonstrate improved cardiovascular outcomes with improved glycemic control. However, we should not take this to mean that glycemic control is unimportant.
In this article, we will discuss why the results of these recent clinical trials are not valid for the general population of patients with diabetes. We will review evidence from landmark clinical trials that clearly demonstrates that better glycemic control reduces both microvascular and macrovascular complications of diabetes (the “glucose hypothesis”). We contend that excellent glycemic control clearly decreases the microvascular complications of diabetes, and that results from long-term follow-up studies in both type 1 and type 2 diabetes show reduced rates of heart attack and stroke in patients treated intensively earlier in the course of their disease.5,6
EVIDENCE FOR THE GLUCOSE HYPOTHESIS
Diabetes Control and Complications Trial
The first major trial demonstrating that improved glycemic control provides benefit was the Diabetes Control and Complications Trial (DCCT).7 This study enrolled 1,441 patients with insulin-dependent diabetes mellitus, 726 of whom had no retinopathy at baseline (the primary-prevention cohort) and 715 of whom had mild retinopathy (the secondary-intervention cohort).
Patients were randomly assigned to intensive therapy (three or more insulin injections per day or an insulin pump) or to conventional therapy with one or two daily insulin injections. They were followed for a mean of 6.5 years, and the appearance and progression of retinopathy and other complications were assessed regularly.
During the study, the hemoglobin A1c level averaged 9% in the control group and 7% in the intensively treated group. The cumulative incidence of retinopathy was defined as a change of three steps or more on fundus photography that was sustained over a 6-month period.
Effect on retinopathy. At study completion, the cumulative incidence of retinopathy in the intensive-therapy group was approximately 50% less than in the conventional-therapy group. Intensive therapy reduced the adjusted mean risk of retinopathy by 76% (95% confidence interval [CI] 62%–85%) in the primary-prevention cohort. In the secondary-prevention cohort, intensive therapy reduced the average risk of progression by 54% (95% CI 39%–66%). Intensive therapy reduced the adjusted risk of proliferative or severe nonproliferative retinopathy by 47% (P = .011) and that of treatment with photocoagulation by 56% (P = .002).
Effect on nephropathy. Intensive therapy reduced the mean adjusted risk of microalbuminuria by 34% (P = .04) in the primary-prevention cohort and by 43% (P = .001) in the secondary-intervention cohort. The risk of macroalbuminuria was reduced by 56% (P = .01) in the secondary-intervention cohort.
Effect on neuropathy. In the patients in the primary-prevention cohort who did not have neuropathy at baseline, intensive therapy reduced the incidence of neuropathy at 5 years by 69% (to 3%, vs 10% in the conventional-therapy group; P = .006). Similarly, in the secondary-intervention cohort, intensive therapy reduced the incidence of clinical neuropathy at 5 years by 57% (to 7%, vs 16%; P < .001).
Effect on macrovascular events. In the initial trial, a nonsignificant 41% reduction in combined cardiovascular and peripheral vascular disease events was observed.
DCCT long-term follow-up
After DCCT concluded, the control and treatment groups’ hemoglobin A1c levels converged to approximately 8%. The two groups were then followed to determine the long-term effects of their prior separation of glycemic levels on micro- and macrovascular out comes.5 More than 90% of the original DCCT patients were followed for a mean of 17 years.
Intensive treatment reduced the risk of any cardiovascular disease event by 42% (95% CI 9%–63%; P = .02) and the risk of nonfatal myocardial infarction, stroke, or death from cardiovascular disease by 57% (95% CI 12%– 79%; P = .02). This result was observed despite separation of glucose control in the two groups only for the first 6.5 years. This beneficial effect of intensive early glycemic control has been termed metabolic memory.
United Kingdom Prospective Diabetes Study
A second major trial, the United Kingdom Prospective Diabetes Study (UKPDS),8 assessed the effect of excellent diabetes control on diabetes complications in patients with type 2 diabetes. A total of 3,867 patients newly diagnosed with type 2 diabetes, median age 54, who after 3 months of diet treatment had mean fasting plasma glucose concentrations of 110 to 270 mg/dL, were randomly assigned to an intensive policy (with a sulfonylurea or insulin or, if overweight, metformin) or a conventional policy with diet. The aim in the intensive group was a fasting plasma glucose less than 108 mg/dL. In the conventional group, the aim was the best achievable fasting plasma glucose with diet alone; drugs were added only if there were hyperglycemic symptoms or a fasting plasma glucose greater than 270 mg/dL.
Over 10 years, the median hemoglobin A1c level was 7.0% (interquartile range 6.2%–8.2%) in the intensive group compared with 7.9% (6.9%–8.8%) in the conventional group. Compared with the conventional group, the risk of any diabetes-related end point was 12% lower in the intensive group (95% CI 1%–21%, P = .029), the risk of any diabetes-related death was 10% lower (−11% to 27%, P = .34), and the rate of all-cause mortality was 6% lower (−10% to 20%, P = .44). Most of the reduction in risk of any diabetes-related end point was from a 25% risk reduction (95% CI 7%–40%, P = .0099) in microvascular end points, including the need for retinal photocoagulation.
UKPDS long-term follow-up
In 2008, Holman et al published the results of long-term follow-up of patients included in the UKPDS.6 In posttrial monitoring, 3,277 patients were asked to attend annual UKPDS clinics for 5 years, but no attempts were made to maintain their previously assigned therapies. Annual questionnaires were used to follow patients who were unable to attend the clinics, and all patients in years 6 to 10 were assessed through questionnaires.
Between-group differences in hemoglobin A1c levels were lost after the first year. However, in the sulfonylurea-insulin group, relative reductions in risk persisted at 10 years for any diabetes-related end point (9%, P = .04) and microvascular disease (24%, P = .001), while risk reductions for myocardial infarction (15%, P = .01) and death from any cause (13%, P = .007) emerged over time as more events occurred. In the metformin group, significant risk reductions persisted for any diabetes-related end point (21%, P = .01), myocardial infarction (33%, P = .005), and death from any cause (27%, P = .002).
The long-term follow-up to the UKPDS, like the long-term follow-up to the DCCT, demonstrated metabolic memory: that is, despite an early loss of glycemic differences after completion of the trial, a continued reduction in microvascular risk and an emergent risk reduction for myocardial infarction and death from any cause were observed.
These long-term randomized prospective trials in patients with type 1 and type 2 diabetes clearly show that the glucose hypothesis is in fact correct: intensive glucose control lowers the risk of both microvascular and macrovascular complications of diabetes.
IS THERE DISCORDANCE BETWEEN OLDER AND MORE RECENT TRIALS?
If the results of these older landmark clinical trials are true, why did the more recent clinical trials fail to show cardiovascular benefit with stricter glycemic control, and in one trial2 demonstrate the potential for harm? (ACCORD2 found an increased death rate in patients who received intensive therapy, targeting a hemoglobin A1c below 6.0%.)
The answer lies in the populations studied. ACCORD,2 VADT,3 and ADVANCE4 were performed in older patients with prior cardiac events or with several risk factors for cardiovascular events. The study populations were picked to increase the number of cardiac events in a short time frame. Therefore, extrapolating the results of these studies to the younger population of patients with diabetes, most of whom have yet to develop macrovascular disease, is inappropriate.
The available evidence suggests that early aggressive management of diabetes reduces the risk of macrovascular disease, but that this benefit is delayed. In the UKPDS and DCCT trials, it took 10 to 17 years to show cardiac benefit in younger patients.
The results of ACCORD,2 VADT,3 and ADVANCE4 are important when considered in the correct clinical context. Two of these trials did demonstrate some microvascular benefit as a result of better glycemic control in older patients, many of whom had longstanding diabetes. These studies suggest that, in patients who already have established cardiovascular disease or have several risk factors for cardiovascular events, a less-strict glycemic target may be warranted.
These trials should not be interpreted as saying that glycemic control is unimportant in older patients at higher risk. Rather, they suggest that an individualized approach to diabetes management, supported by the most recent American Diabetes Association guidelines,9 is more appropriate.
Physicians may reasonably suggest a stricter A1c goal (ie, < 6.5%) in certain patients if it can be achieved without significant hypoglycemia. Stricter glycemic targets would seem appropriate in patients recently diagnosed with diabetes, those who have a long life expectancy, and those who have not yet developed significant cardiovascular disease.9
However, in patients who already have developed advanced microvascular and macrovascular complications, who have long-standing diabetes, who have a history of severe hypoglycemia (or hypoglycemia unawareness), or who have a limited life expectancy or numerous adverse comorbidities, a less strict glycemic target (hemoglobin A1c < 8%) may be more appropriate.9
CARDIOVASCULAR RISK, HYPOGLYCEMIA, AND ATTAINING GLYCEMIC TARGETS
Metformin, in the absence of contraindications or intolerability, is generally the recommended first-line therapy to manage glycemia in patients with type 2 diabetes mellitus.10,11 However, there are only limited data to direct clinicians as to which antidiabetic medication to use if further therapy is required to obtain glycemic control.
Much of the cardiovascular and mortality risk associated with aggressive diabetes management (ie, lower A1c targets) is related to hypoglycemia. Thus, antidiabetic therapies that pose no risk or only a low risk of hypoglycemia should be chosen, particularly in older patients and in those with known cardiovascular disease. This may allow for better glycemic control without the risk of hypoglycemia and adverse cardiovascular outcomes.
However, in practice, clinicians continue to use a sulfonylurea as the second-line agent. Although sulfonylureas may appear to be a great option because of their low cost, they are associated with a higher risk of hypoglycemic episodes than other classes of diabetes drugs. We need to consider the frequency and cost of hypoglycemic episodes and the potential morbidity associated with them, because these episodes are a barrier to our efforts to achieve better glycemic control.
Budnitz et al12 reported that from 2007 through 2009, in US adults age 65 and older, insulins were implicated in 13.9% of hospitalizations related to adverse drug events, and oral hypoglycemic agents (ie, insulin secretagogues) in 10.7%.
Quilliam et al13 reported that hypoglycemia resulted in a mean cost of $17,564 for an inpatient admission, $1,387 for an emergency department visit, and $394 for an outpatient visit. Thus, the cost savings associated with prescribing a sulfonylurea vs one of the newer oral antidiabetic agents that do not increase the risk of hypoglycemia (unless used concurrently with insulin or an insulin secretagogue) can quickly be eroded by severe hypoglycemic episodes requiring medical care.
Moreover, once patients start to experience hypoglycemic episodes, they are very reluctant, as are their physicians, to intensify therapy, even if it is indicated by their elevated A1c.
There are now seven classes of oral antidiabetic therapies other than insulin secretagogues (ie, other than sulfonylureas and the meglitinides nateglinide and repaglinide), as well as a few noninsulin injectable therapies (glucagon-like peptide-1 agonists), that are not associated with hypoglycemia. We believe these agents should be tried before prescribing an agent that carries the risk of hypoglycemia (ie, sulfonylureas).
If agents that do not cause hypoglycemia are used, more-aggressive glycemic targets may be achieved safely. The ACCORD study,2 which included patients at high cardiovascular risk and aimed at an aggressive glycemic target of 6%, may have yielded much different results had agents that carry a high risk of hypoglycemia been excluded.
Of importance, cardiovascular risk is also influenced by the common comorbidities seen in patients with diabetes, such as hypertension and hypercholesterolemia. Intensive, multifactorial interventions that address not only glycemic control but also blood pressure and lipids and that include low-dose aspirin therapy have been shown to lower the risk of death from cardiovascular causes and the risk of cardiovascular events.14 Likewise, smoking cessation is very important in reducing cardiovascular risk, especially in patients with diabetes.15
CLINICAL TRIALS IN CONTEXT
In conclusion, there is more to diabetes management than cardiovascular complications. Clearly, improved glycemic control decreases the risk of retinopathy, nephropathy, and neuropathy in patients with type 1 and type 2 diabetes. The DCCT and UKPDS extension studies further found that excellent glycemic control decreases rates of cardiac events.
The best way to treat diabetes may be different in otherwise healthy younger patients who have yet to develop significant complications than it is in older patients known to have cardiovascular disease or several risk factors for cardiovascular events. The available evidence suggests it would be reasonable to aim for stricter glycemic targets in the younger patients and less stringent targets in the older patients, particularly in those with long-standing diabetes who have already developed significant micro- and macrovascular complications.
We should interpret clinical trials within their narrow clinical context, emphasizing the actual population of patients included in the study, so as to avoid the inappropriate extrapolation of the results to all.
- Centers for Disease Control and Prevention. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, 2011. www.cdc.gov/diabetes/pubs/pdf/ndfs_2011.pdf. Accessed October 7, 2014.
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008; 358:2545–2559.
- Duckworth W, Abraira C, Moritz T, et al; VADT Investigators. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 2009; 360:129–139.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008; 358:2560–2572.
- Nathan DM, Cleary PA, Backlund JY, et al; Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 2005; 353:2643–2653.
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359:1577–1589.
- The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 1993; 329:977–986.
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998; 352:837–853.
- American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care 2013; 36(Suppl 1):S11–S66.
- Inzucchi SE, Bergenstal RM, Buse JB, et al; American Diabetes Association (ADA); European Association for the Study of Diabetes (EASD). Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2012; 35:1364–1379.
- Garber AJ, Abrahamson MJ, Barzilay JI, et al; American Association of Clinical Endocrinologists. AACE comprehensive diabetes management algorithm 2013. Endocr Pract 2013; 19:327–336.
- Budnitz DS, Lovegrove MC, Shehab N, Richards CL. Emergency hospitalizations for adverse drug events in older Americans. N Engl J Med 2011; 365:2002–2012.
- Quilliam BJ, Simeone JC, Ozbay AB, Kogut SJ. The incidence and costs of hypoglycemia in type 2 diabetes. Am J Manag Care 2011; 17:673–680.
- Gaede P, Lund-Andersen H, Parving HH, Pedersen O. Effect of a multifactorial intervention on mortality in type 2 diabetes. N Engl J Med 2008; 358:580–591.
- Chaturvedi N, Stevens L, Fuller JH. Which features of smoking determine mortality risk in former cigarette smokers with diabetes? The World Health Organization Multinational Study Group. Diabetes Care 1997; 20:1266–1272.
- Centers for Disease Control and Prevention. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, 2011. www.cdc.gov/diabetes/pubs/pdf/ndfs_2011.pdf. Accessed October 7, 2014.
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008; 358:2545–2559.
- Duckworth W, Abraira C, Moritz T, et al; VADT Investigators. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 2009; 360:129–139.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008; 358:2560–2572.
- Nathan DM, Cleary PA, Backlund JY, et al; Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 2005; 353:2643–2653.
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359:1577–1589.
- The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 1993; 329:977–986.
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998; 352:837–853.
- American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care 2013; 36(Suppl 1):S11–S66.
- Inzucchi SE, Bergenstal RM, Buse JB, et al; American Diabetes Association (ADA); European Association for the Study of Diabetes (EASD). Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2012; 35:1364–1379.
- Garber AJ, Abrahamson MJ, Barzilay JI, et al; American Association of Clinical Endocrinologists. AACE comprehensive diabetes management algorithm 2013. Endocr Pract 2013; 19:327–336.
- Budnitz DS, Lovegrove MC, Shehab N, Richards CL. Emergency hospitalizations for adverse drug events in older Americans. N Engl J Med 2011; 365:2002–2012.
- Quilliam BJ, Simeone JC, Ozbay AB, Kogut SJ. The incidence and costs of hypoglycemia in type 2 diabetes. Am J Manag Care 2011; 17:673–680.
- Gaede P, Lund-Andersen H, Parving HH, Pedersen O. Effect of a multifactorial intervention on mortality in type 2 diabetes. N Engl J Med 2008; 358:580–591.
- Chaturvedi N, Stevens L, Fuller JH. Which features of smoking determine mortality risk in former cigarette smokers with diabetes? The World Health Organization Multinational Study Group. Diabetes Care 1997; 20:1266–1272.
Male hypogonadism: More than just a low testosterone
Editor’s note: This article on the differential diagnosis of hypogonadism in men is the first of two articles. The second, to be published next month, focuses on the appropriate use of testosterone therapy.
A 54-year-old man is referred for evaluation of low testosterone. He had seen his primary care physician for complaints of diminished libido and erectile dysfunction for the past year and worsening fatigue over the past few years. He has not been formally diagnosed with any medical condition. His serum testosterone level is 180 ng/dL (reference range 249–836 ng/dL).
On physical examination, he is obese (body mass index 31 kg/m2) with a normal-appearing male body habitus, no gynecomastia, and normal testicles and prostate gland.
How should this patient be evaluated?
LOW TESTOSTERONE HAS MANY CAUSES
Male hypogonadism, ie, failure of the testes to produce adequate amounts of androgen or sperm, has become a common clinical finding, particularly in the older population. This is more likely the result of an increase in awareness and detection of the disorder by physicians rather than a true increase in prevalence.
The finding of a low serum testosterone value needs to be confirmed and thoroughly evaluated before starting treatment. It is important to determine whether the cause is a primary (hypergonadotropic) testicular disorder or secondary to a hypothalamic-pituitary process (hypogonadotropic or normogonadotropic).
THE HYPOTHALAMIC-PITUITARY-GONADAL AXIS
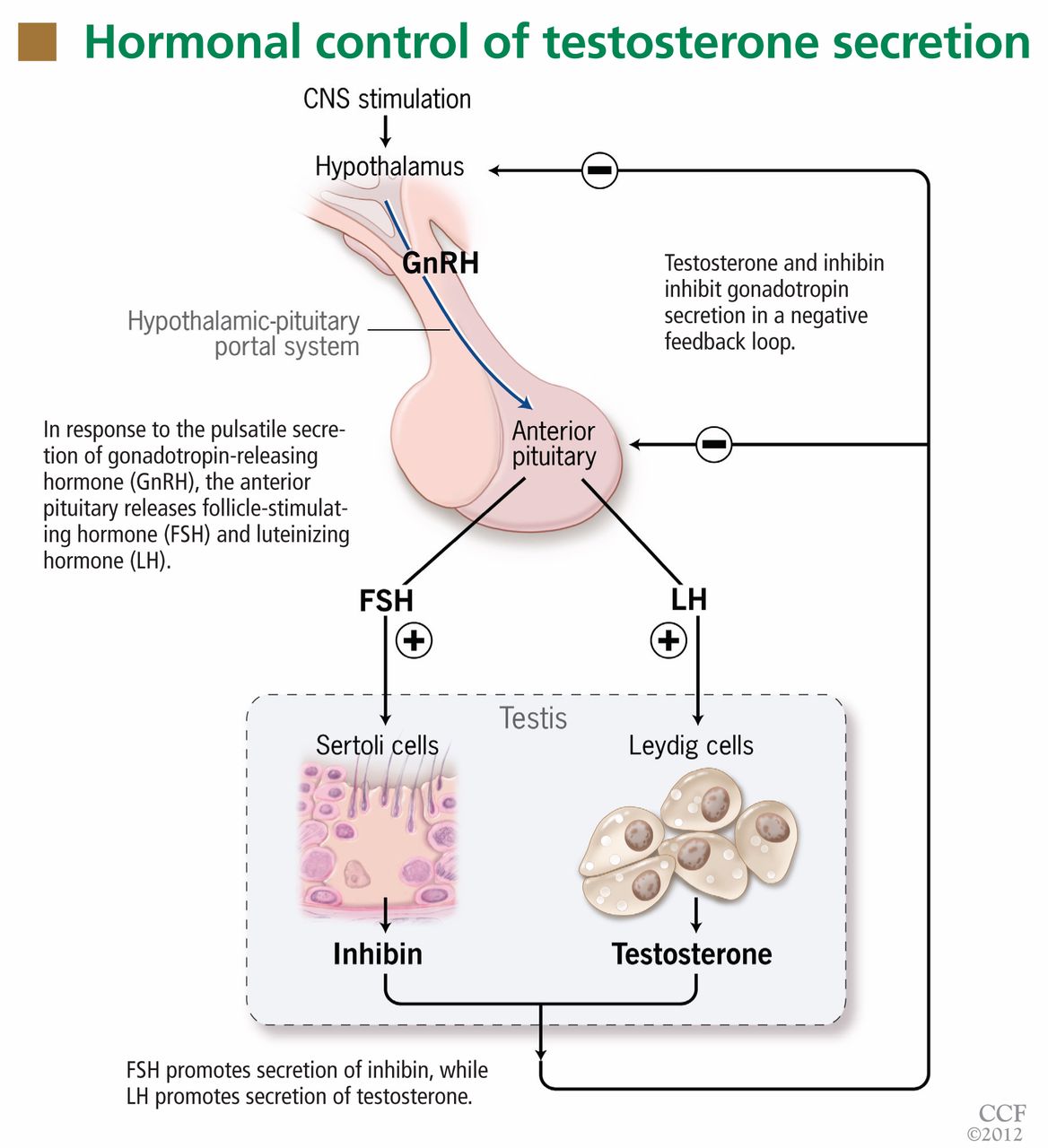
Testosterone production is under the control of luteinizing hormone (LH), whereas sperm production is under the control of follicle-stimulating hormone (FSH) (Figure 1). Both of these pituitary hormones are regulated by the pulsatile secretion of hypothalamic gonadotropin-releasing hormone (GnRH).
Testosterone (produced by Leydig cells) and inhibin B (produced by Sertoli cells within the seminiferous tubules) result in negative feedback inhibition of gonadotropin (LH and FSH) secretion. Testosterone and estradiol (produced by aromatization of testosterone) act at both pituitary and hypothalamic sites and are the principal regulators of LH secretion.1,2 Inhibin B is the major regulator of FSH secretion in men,3 but steroid feedback also occurs.2,4
TO FOLLOW UP A LOW TESTOSTERONE, CONFIRM THE VALUE NEAR 8 am
If a testosterone value is found to be low, it is important to determine the time that the sample was obtained. Serum testosterone levels follow a diurnal rhythm, at least in younger men, with values near 8 am being, on average, 30% higher than the trough levels later in the day.5–7 The timing of the diurnal variation may be different in night-shift workers, who may require assessment at a more appropriate time of the day (ie, upon awakening).
Another factor affecting testosterone levels is the patient’s health status at the time of testing. Values obtained in the hospital during an acute illness should be repeated once the event has resolved, as testosterone values decrease considerably in this setting.8 Even in outpatients, particularly in men over age 60, one must be sure that the low testosterone level was not obtained during a period of decompensation of one of the many comorbidities seen in these patients, such as coronary artery disease, congestive heart failure, or uncontrolled diabetes.
If an 8 am testosterone value is low, it is reasonable to obtain at least one confirmatory testosterone level on another day, near 8 am, in the next few weeks, when the patient is in good health. Confirming the testosterone level is important, particularly since commercially available testosterone assays are not well standardized and some are frankly unreliable.9,10 A repeat confirmatory level should always be performed by a reliable reference laboratory. If the testosterone level is still low, further evaluation is warranted.
TOTAL TESTOSTERONE VS BIOAVAILABLE TESTOSTERONE VS FREE TESTOSTERONE
Of the total circulating testosterone, 60% is bound to sex hormone-binding globulin (SHBG), 38% is bound to albumin, and only 2% is free. All of these fractions can be measured to assess for testosterone deficiency.
Free testosterone is the biologically active form of this hormone and, thus, the free testosterone level is considered to be a better representation of the true testosterone status. However, some clinicians believe that bioavailable testosterone (testosterone loosely bound to albumin + free testosterone) is a better reflection of the true level of the active hormone than the level of free testosterone alone.
There are situations in which the total testosterone level is low but bioavailable or free testosterone levels are normal. The level of total testosterone is affected by alterations in the levels of SHBG and albumin. A reduction in the level of SHBG can result in low total serum testosterone levels in patients with obesity or type 2 diabetes (states of insulin resistance), and also in cachexia, malnutrition, advanced cirrhosis, acromegaly, hypothyroidism, and nephrotic syndrome. SHBG can also be low in patients taking glucocorticoids, progestins, or androgenic steroids.11 In these settings, checking the level of free testosterone (the active hormone), bioavailable testosterone, or both, by a reliable reference laboratory, may be more appropriate.9,10
But regardless of which measurement is chosen, all testosterone levels—especially bioavailable and free testosterone values—should be interpreted with caution if they are not measured at a reliable reference laboratory.9,10 Interested readers may wish to see the US Centers for Disease Control and Prevention (CDC) Hormone Standardization Program Web site (www.cdc.gov/labstandards/hs.html) for more details, including a list of CDC-certified laboratories.
CLINICAL FEATURES OF LOW TESTOSTERONE
A history of erectile dysfunction, decreased libido, and fatigue may be seen in patients with low testosterone. However, one must realize that these symptoms—as well as others reported by men with low testosterone, such as depression, difficulty concentrating, irritability, and insomnia—are nonspecific and may be related to other medical conditions.12
Likewise, physical findings such as muscle weakness, reduced body hair, and altered fat distribution (abdominal obesity) are seen in men with low testosterone, but also in those with a number of other medical conditions.
Additional features suggest specific disorders, eg, anosmia in Kallmann syndrome; eunuchoid body habitus, gynecomastia, and small testes in Klinefelter syndrome.
Men with low testosterone may have low bone mineral density or anemia, or both.
Careful examination of the breasts for gynecomastia and the testes for size, consistency, and masses (testicular tumors) helps in formulating a differential diagnosis and in appropriately directing subsequent laboratory evaluation and diagnostic imaging.
LOW TESTOSTERONE: PRIMARY VS SECONDARY
A history of testicular trauma, systemic chemotherapy, or mumps orchitis should direct the physician’s attention to a testicular etiology. On the other hand, darkened or tanned skin (suggesting hemochromatosis), galactorrhea (suggesting hyperprolactinemia), or visual field deficits (suggesting a sellar mass) should direct the physician’s attention toward a pituitary-hypothalamic process.

Once the low testosterone value has been confirmed at least one time near 8 am, one should obtain LH and FSH values to help direct further evaluation in deciphering the etiology (Figure 2). Elevated (hypergonadotropic) values indicate a testicular disorder (primary hypogonadism), whereas low (hypogonadotropic) or normal (normogonadotropic) values point to a pituitary-hypothalamic process (secondary hypogonadism). It should be emphasized that, in the setting of a low testosterone level, LH and FSH values within the normal range are “inappropriately normal” so that further investigation is required.
This evaluation should also include serum prolactin, thyroid-stimulating hormone (TSH, also known as thyrotropin), free thyroxine (T4), and ferritin levels, the latter because hemochromatosis (iron overload) can cause both primary and secondary hypogonadism. If at any time in the evaluation the laboratory results suggest secondary hypogonadism, a full assessment of pituitary function should be undertaken.
Semen analysis is usually reserved for patients presenting with the primary complaint of infertility.
PRIMARY HYPOGONADISM
The patient should be carefully questioned about the age at which his problems began, about pubertal development, and about fertility. Causes of primary hypogonadism include:
- Karyotype abnormalities—Klinefelter syndrome (47, XXY syndrome) is the most common
- Toxin exposure, chemotherapy
- Congenital defects—anorchia, cryptorchidism13
- Orchitis (mumps, autoimmune)
- Testicular trauma or infarction
- Hemochromatosis
- Medications that inhibit androgen biosynthesis, eg, ketoconazole (Nizoral)14
- Increase in temperature of the testicular environment (due to varicocele or a large panniculus).
SECONDARY HYPOGONADISM
Causes of secondary hypogonadism include the following:
Congenital disorders
These disorders are usually diagnosed in childhood or adolescence, often after the patient is brought to the physician because of short stature or pubertal delay.
- Kallmann syndrome (anosmia and GnRH deficiency)15
- GnRH receptor mutation and deficiency16
- Genetic mutations associated with pituitary hormone deficiencies, eg, PROP-1 mutation.17
Acquired disorders that suppress gonadotrophs
Drugs. Long-term therapy with common medications such as opioids or corticosteroids can result in secondary hypogonadism.18–20 Others are GnRH analogues such as leuprolide (Lupron), which are used in treating advanced prostate cancer. The hypogonadism is usually transient and resolves after stopping the offending agent.
Obesity and related conditions such as obstructive sleep apnea, insulin resistance, and type 2 diabetes mellitus are associated with low testosterone levels.21 Treatment should be directed at these underlying conditions and should include lifestyle measures such as weight loss and exercise, rather than simple prescribing of testosterone supplementation, as these efforts may provide multiple health benefits in addition to raising testosterone levels.22
Insulin resistance. In the setting of obesity, the total testosterone level may be low but the bioavailable and free testosterone (active hormone) levels may be normal. This is due to the effect of hyperinsulinemia on the liver, which results in a reduction in SHBG production.23 Low levels of both total and free testosterone can be seen in morbid obesity,24 but the cause remains unclear.
Type 2 diabetes mellitus. Testosterone levels have been reported to be lower in obese men who have diabetes than in those with obesity alone.24 This decrement, comparable in magnitude to that seen with other chronic diseases, suggests that low testosterone may simply be a marker of poor health.22,25,26
Sleep apnea. Disturbances in the sleep cycle, regardless of the underlying cause, can result in decreases in serum testosterone levels. Often, correcting the underlying sleep disturbance can result in a normalization of serum testosterone levels.27,28 A caveat about testosterone therapy: a thorough evaluation for sleep apnea should be undertaken in patients at high risk, since testosterone replacement therapy can adversely affect ventilatory drive and induce or worsen obstructive sleep apnea.29
Aging. Most reports have shown an agerelated decline in both total and free serum testosterone levels (commonly referred to as “andropause”), particularly in men over 60 years of age. There also appears to be a loss of circadian rhythm,30 although not all reports agree.6 It appears that factors such as functional status and overall health may play a more important role in the pathophysiology of hypogonadism in men of advanced age than age alone.
Hemochromatosis. Iron overload, regardless of the cause, can result in hypogonadism via deposition of iron in the hypothalamus, pituitary, or testes. Hereditary hemochromatosis is a common autosomal recessive disease characterized by increased iron absorption. Although both primary and secondary hypogonadism can occur with long-standing iron overload, the latter is much more common.31 Some cases of hypogonadism have been reported to reverse with iron depletion therapy.32
Hyperprolactinemia. Recognized causes of hyperprolactinemia in men include medications (dopamine antagonists, antipsychotics, metoclopramide [Reglan]), pituitary adenomas (microadenomas < 10 mm, macroadenomas ≥ 10 mm), lactotroph hyperfunction (stalk compression interrupting or reducing the tonic suppression of prolactin secretion by dopamine), hypothyroidism, stress, chronic renal failure, cirrhosis, chest wall injury (trauma), and active herpes zoster. The ensuing hypogonadism may be due to the compressive effect of a sellar mass or the direct effect of the prolactin elevation alone, since prolactin disrupts the pulsatile release of GnRH from the hypothalamus,33 required for normal LH and FSH secretion.
Estrogen excess can be either exogenous (from exposure to estrogen-containing contraceptives and creams) or endogenous (from testicular34,35 or very rare adrenal36 estrogen-secreting tumors). Of note, some cases of testicular neoplasms may be detectable only with ultrasonography. Computed tomography may be performed if an adrenal lesion is suspected.
Anabolic steroid abuse. Exposure to anabolic steroids, deliberately or inadvertently, can result in secondary hypogonadism and testicular atrophy, both of which may persist for years after stopping the anabolic agents. If you suspect anabolic steroid abuse, a urine anabolic steroid screen can be obtained.
Anorexia nervosa is far less common in men than in women.37,38 Elements in the history that suggest this disorder include excessive exercise and a low body mass index. Chronic malnutrition (cachexia), regardless of the cause, can result in secondary hypogonadism.
Acute illness (gonadotroph sick syndrome). Hypogonadism is a relatively common finding in any critical illness (analogous to euthyroid sick syndrome with respect to the hypothalamic-pituitary-thyroid axis).8 Testosterone levels are invariably low, so that assessment of testosterone status is not recommended in this setting. The low testosterone phase is usually transient and resolves with resolution or improvement of the underlying medical condition, such as sepsis or myocardial infarction.
HIV. Human immunodeficiency virus (HIV) infection can result in primary or secondary hypogonadism. It can occur with active HIV infection, in patients in whom control of viral replication has been achieved with highly active antiretroviral therapy, and even in patients who have normalized CD4+ cell counts.39 Hypogonadism in HIV patients is multifactorial and may be related to weight loss, opportunistic infections of the pituitary-hypothalamus or testes, or medications such as opioids (licit or illicit), ganciclovir (Cytovene), ketoconazole, the appetite stimulant megestrol (Megace), or cyclophosphamide (Cytoxan). Testosterone replacement therapy does not adversely affect the HIV disease process and in fact may help to avoid complications.
Chronic medical conditions such as cirrhosis, renal failure, and rheumatoid arthritis commonly result in hypogonadism, the pathogenesis of which may involve dysfunction at all levels of the hypothalamic-pituitary-go-nadal axis.40–45 Hypogonadism in the setting of chronic disease is multifactorial, being due not only to the metabolic disturbances seen with these illnesses (uremia in renal failure, elevated circulating estrogens in liver cirrhosis), but also to recurrent acute illness and hospitalization for infection in these immuno-compromised hosts, either from the underlying medical condition or as a result of medications (corticosteroids).
Alcohol abuse. Alcohol can have adverse effects at all levels of the hypothalamic-pituitary-gonadal axis, resulting in low serum testosterone and reduced spermatogenesis.46
Severe chronic primary hypothyroidism, manifested by an extreme elevation of serum thyroid-stimulating hormone (TSH), can result in hypopituitarism. Pituitary function usually recovers with restoration of euthyroidism.47,48
Pubertal delay. Depending on the age of presentation, differentiating pubertal delay from permanent hypogonadotropic hypogonadism can be challenging.
Acquired disorders that damage gonadotrophs
- Sellar mass or cyst—pituitary adenoma, craniopharyngioma, Rathke cleft cyst, meningioma
- Infiltrative lesion—lymphocytic hypophysitis, Langerhans cell histiocytosis, hemochromatosis, sarcoidosis, infection
- Metastatic lesion
- Trauma (head injury)
- Radiation exposure
- Surgery
- Stalk severance
- Pituitary apoplexy.
See Table 1 for a summary of the causes of male hypogonadism.
WHEN IS MRI INDICATED IN EVALUATING SECONDARY HYPOGONADISM?

The yield of pituitary-hypothalamic imaging in older men with secondary hypogonadism is fairly low in the absence of other pituitary hormone abnormalities and deficiencies. There are limited data regarding appropriate criteria for performing hypothalamic-pituitary imaging studies. However, a patient who has multiple anterior pituitary abnormalities on laboratory evaluation should undergo dedicated hypothalamic-pituitary magnetic resonance imaging (MRI).
The Endocrine Society Clinical Practice Guidelines11 recommend that MRI be performed to exclude a pituitary or hypothalamic tumor or infiltrative disease if the patient has severe secondary hypogonadism (serum testosterone < 150 ng/dL), panhypopituitarism, persistent hyperprolactinemia, or symptoms or signs of tumor mass effect such as headache, visual impairment, or a visual field defect.
WHO SHOULD UNDERGO ASSESSMENT OF TESTOSTERONE STATUS?
Screening for androgen deficiency in the asymptomatic general population is not recommended.11 The nonspecific nature of many of the signs and symptoms of androgen deficiency makes it difficult to give concrete recommendations as to who should have testosterone levels measured. Clinicians should consider testing if there is evidence of certain clinical disorders that are associated with low testosterone levels (see earlier discussion on the specific causes of primary and secondary hypogonadism).
When a male patient complains of erectile dysfunction, the investigation should include an assessment of serum testosterone. However, if a man who has a constellation of nonspecific symptoms asks for his testosterone level to be assessed (which is common, given the aggressive marketing of testosterone replacement by the pharmaceutical industry), we would recommend a basic evaluation that includes a comprehensive metabolic panel, complete blood count, and TSH level. Further testing should be determined by the history and physical examination. If no obvious explanation has been found for the patient’s symptoms at that point, assessment of serum testosterone may be warranted. More often than not the patient’s weight and limited physical activity are the driving forces behind the nonspecific symptoms, and counseling a patient on a life-style change can provide much benefit if the patient follows through with the physician’s recommendations.
Men whom we believe should not undergo assessment for testosterone deficiency are those who are acutely ill and hospitalized and those who are severely obese and are complaining of fatigue. Testosterone levels should be assessed only after the acute illness has resolved and, in a severely obese patient with fatigue, only after a thorough evaluation for sleep apnea has been undertaken.
TREAT THE UNDERLYING CAUSE, IF ONE CAN BE FOUND
If the evaluation of low testosterone leads to the diagnosis of a clear underlying condition that is amenable to treatment, such as prolactin elevation or sleep apnea, then treatment should be directed at the underlying cause, with subsequent monitoring of the patient’s symptoms and response in serum testosterone levels. In general, the use of dopamine agonist therapy in the management of hyperprolactinemia and, in cases of panhypopituitarism, of replacement therapy with levothyroxine (Synthroid), hydrocortisone, and possibly growth hormone and desmopressin (DDAVP), fall best under the purview of an endocrinologist. A caveat: serum TSH cannot be used to monitor levothyroxine replacement therapy in cases of secondary hypothyroidism. The clinical picture and serum free T4 and free T3 levels are used instead.
In the absence of a correctable (or immediately correctable) cause, testosterone supplementation can be initiated on an individualized basis in select patients who have clinical signs and symptoms of androgen deficiency if the benefits of treatment appear to outweigh the potential risks, and only after a thorough discussion with the patient.11 The Endocrine Society recommends against offering testosterone therapy to all older men with low testosterone.11
INFERTILITY
In men presenting with low serum testosterone, semen analysis is not routine. It is usually reserved for patients presenting with the primary complaint of infertility.
If an endocrine disorder such as prolactin elevation or hypothyroidism is the suspected cause of infertility, the patient should be referred to an endocrinologist for further evaluation and management. Treatment of male infertility should be directed at the underlying cause, but often requires exogenous human chorionic gonadotropin, FSH, GnRH (via a pulsatile pump), and possibly sperm harvesting from the testis with subsequent in vitro fertilization with intracytoplasmic sperm injection. It is critical that the partner be included in the evaluation of infertility.
These patients should be referred to a urologic or fertility center specializing in the diagnosis and treatment of infertility. For further information regarding male infertility, patients can be directed to www.fertilitylifelines.com.
CASE CONCLUDED
The patient’s low serum testosterone was confirmed on subsequent measurements at 8 am, with levels of 128 and 182 ng/dL (reference range 249–836). Other laboratory values:
- LH 1.4 mIU/mL (reference range 1.2–8.6)
- FSH 2.7 mIU/mL (1.3–9.9 mIU/mL)
(Both of these values are inappropriately normal in the setting of the low testosterone.)
- TSH 248 μIU/mL (0.4–5.5)
- Prolactin 24.6 ng/mL (1.6–18.8).
The patient was started on levothyroxine replacement therapy and after 3 months was noted to be euthyroid (TSH 1.8 μIU/mL) and to have a normal serum prolactin level. Testosterone levels (8 am) at this time were 350 ng/dL and 420 ng/dL.
Therefore, the cause of this patient’s hypogonadism was severe hypothyroidism and associated mild hyperprolactinemia. This case shows that a thorough evaluation is warranted before initiating testosterone therapy.
- Pitteloud N, Dwyer AA, DeCruz S, et al. Inhibition of luteinizing hormone secretion by testosterone in men requires aromatization for its pituitary but not its hypo-thalamic effects: evidence from the tandem study of normal and gonadotropin-releasing hormone-deficient men. J Clin Endocrinol Metab 2008; 93:784–791.
- Hayes FJ, DeCruz S, Seminara SB, Boepple PA, Crowley WF. Differential regulation of gonadotropin secretion by testosterone in the human male: absence of a negative feedback effect of testosterone on follicle-stimulating hormone secretion. J Clin Endocrinol Metab 2001; 86:53–58.
- Hayes FJ, Pitteloud N, DeCruz S, Crowley WF, Boepple PA. Importance of inhibin B in the regulation of FSH secretion in the human male. J Clin Endocrinol Metab 2001; 86:5541–5546.
- Pitteloud N, Dwyer AA, DeCruz S, et al. The relative role of gonadal sex steroids and gonadotropin-releasing hormone pulse frequency in the regulation of follicle-stimulating hormone secretion in men. J Clin Endocrinol Metab 2008; 93:2686–2692.
- Cooke RR, McIntosh JE, McIntosh RP. Circadian variation in serum free and non-SHBG-bound testosterone in normal men: measurements, and simulation using a mass action model. Clin Endocrinol (Oxf) 1993; 39:163–171.
- Diver MJ, Imtiaz KE, Ahmad AM, Vora JP, Fraser WD. Diurnal rhythms of serum total, free and bioavailable testosterone and of SHBG in middle-aged men compared with those in young men. Clin Endocrinol (Oxf) 2003; 58:710–717.
- Clair P, Claustrat B, Jordan D, Dechaud H, Sassolas G. Daily variations of plasma sex hormone-binding globulin binding capacity, testosterone and luteinizing hormone concentrations in healthy rested adult males. Horm Res 1985; 21:220–223.
- Woolf PD, Hamill RW, McDonald JV, Lee LA, Kelly M. Transient hypogonadotropic hypogonadism caused by critical illness. J Clin Endocrinol Metab 1985; 60:444–450.
- Rosner W, Auchus RJ, Azziz R, Sluss PM, Raff H. Position statement: utility, limitations, and pitfalls in measuring testosterone: an Endocrine Society position statement. J Clin Endocrinol Metab 2007; 92:405–413.
- Rosner W, Vesper H, et al; Endocrine Society; American Association for Clinical Chemistry; American Association of Clinical Endocrinologists; et al. Toward excellence in testosterone testing: a consensus statement. J Clin Endocrinol Metab 2010; 95:4542–4548.
- Bhasin S, Cunningham GR, Hayes FJ, et al; Task Force, Endocrine Society. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2010; 95:2536–2559.
- Wu FC, Tajar A, Beynon JM, et al; EMAS Group. Identification of late-onset hypogonadism in middle-aged and elderly men. N Engl J Med 2010; 363:123–135.
- Farrer JH, Sikka SC, Xie HW, Constantinide D, Rajfer J. Impaired testosterone biosynthesis in cryptorchidism. Fertil Steril 1985; 44:125–132.
- Sikka SC, Swerdloff RS, Rajfer J. In vitro inhibition of testosterone biosynthesis by ketoconazole. Endocrinology 1985; 116:1920–1925.
- Pallais JC, Au M, Pitteloud N, Seminara S, Crowley WF Jr. Kallmann syndrome. In:Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, eds. GeneReviews™ (Internet). Seattle, WA: University of Washington; 1993.
- Chevrier L, Guimiot F, de Roux N. GnRH receptor mutations in isolated gonadotropic deficiency. Mol Cell Endocrinol 2011; 346:21–28.
- Romero CJ, Pine-Twaddell E, Radovick S. Novel mutations associated with combined pituitary hormone deficiency. J Mol Endocrinol 2011; 46:R93–R102.
- Colameco S, Coren JS, Ciervo CA. Continuous opioid treatment for chronic noncancer pain: a time for moderation in prescribing. Postgrad Med 2009; 121:61–66.
- Fraser LA, Morrison D, Morley-Forster P, et al. Oral opioids for chronic non-cancer pain: higher prevalence of hypogonadism in men than in women. Exp Clin Endocrinol Diabetes 2009; 117:38–43.
- Morrison D, Capewell S, Reynolds SP, et al. Testosterone levels during systemic and inhaled corticosteroid therapy. Respir Med 1994; 88:659–663.
- Mah PM, Wittert GA. Obesity and testicular function. Mol Cell Endocrinol 2010; 316:180–186.
- Grossmann M. Low testosterone in men with type 2 diabetes: significance and treatment. J Clin Endocrinol Metab 2011; 96:2341–2353.
- Gascón F, Valle M, Martos R, et al. Sex hormone-binding globulin as a marker for hyperinsulinemia and/or insulin resistance in obese children. Eur J Endocrinol 2000; 143:85–89.
- Dhindsa S, Miller MG, McWhirter CL, et al. Testosterone concentrations in diabetic and nondiabetic obese men. Diabetes Care 2010; 33:1186–1192.
- Grossmann M, Gianatti EJ, Zajac JD. Testosterone and type 2 diabetes. Curr Opin Endocrinol Diabetes Obes 2010; 17:247–256.
- Andersson B, Mårin P, Lissner L, Vermeulen A, Björntorp P. Testosterone concentrations in women and men with NIDDM. Diabetes Care 1994; 17:405–411.
- Santamaria JD, Prior JC, Fleetham JA. Reversible reproductive dysfunction in men with obstructive sleep apnoea. Clin Endocrinol (Oxf) 1988; 28:461–470.
- Grunstein RR, Handelsman DJ, Lawrence SJ, Blackwell C, Caterson ID, Sullivan CE. Neuroendocrine dysfunction in sleep apnea: reversal by continuous positive airways pressure therapy. J Clin Endocrinol Metab 1989; 68:352–358.
- Matsumoto AM, Sandblom RE, Schoene RB, et al. Testosterone replacement in hypogonadal men: effects on obstructive sleep apnoea, respiratory drives, and sleep. Clin Endocrinol (Oxf) 1985; 22:713–721.
- Bremner WJ, Vitiello MV, Prinz PN. Loss of circadian rhythmicity in blood testosterone levels with aging in normal men. J Clin Endocrinol Metab 1983; 56:1278–1281.
- McDermott JH, Walsh CH. Hypogonadism in hereditary hemochromatosis. J Clin Endocrinol Metab 2005; 90:2451–2455.
- Kelly TM, Edwards CQ, Meikle AW, Kushner JP. Hypogonadism in hemochromatosis: reversal with iron depletion. Ann Intern Med 1984; 101:629–632.
- Milenkovic L, D’Angelo G, Kelly PA, Weiner RI. Inhibition of gonadotropin hormone-releasing hormone release by prolactin from GT1 neuronal cell lines through prolactin receptors. Proc Natl Acad Sci U S A 1994; 91:1244–1247.
- Valensi P, Coussieu C, Kemeny JL, Attali JR, Amouroux J, Sebaoun J. Endocrine investigations in two cases of feminizing Leydig cell tumour. Acta Endocrinol (Copenh) 1987; 115:365–372.
- Young S, Gooneratne S, Straus FH, Zeller WP, Bulun SE, Rosenthal IM. Feminizing Sertoli cell tumors in boys with Peutz-Jeghers syndrome. Am J Surg Pathol 1995; 19:50–58.
- Zayed A, Stock JL, Liepman MK, Wollin M, Longcope C. Feminization as a result of both peripheral conversion of androgens and direct estrogen production from an adrenocortical carcinoma. J Endocrinol Invest 1994; 17:275–278.
- Russ MJ, Ackerman SH, Barakat R, Levy B. Hypogonadotropic hypogonadism and delayed puberty in a man with anorexia nervosa. Psychosomatics 1986; 27:737–739.
- Rigotti NA, Neer RM, Jameson L. Osteopenia and bone fractures in a man with anorexia nervosa and hypogonadism. JAMA 1986; 256:385–388.
- Cohan GR. HIV-associated hypogonadism. AIDS Read 2006; 16:341–345,348,352–354.
- Handelsman DJ, Strasser S, McDonald JA, Conway AJ, McCaughan GW. Hypothalamic-pituitary-testicular function in end-stage nonalcoholic liver disease before and after liver transplantation. Clin Endocrinol (Oxf) 1995; 43:331–337.
- Lim VS, Fang VS. Gonadal dysfunction in uremic men. A study of the hypothalamo-pituitary-testicular axis before and after renal transplantation. Am J Med 1975; 58:655–662.
- Handelsman DJ, Dong Q. Hypothalamo-pituitary gonadal axis in chronic renal failure. Endocrinol Metab Clin North Am 1993; 22:145–161.
- Handelsman DJ, Spaliviero JA, Turtle JR. Hypothalamic-pituitary function in experimental uremic hypogonadism. Endocrinology 1985; 117:1984–1995.
- Tengstrand B, Carlström K, Hafström I. Bioavailable testosterone in men with rheumatoid arthritis-high frequency of hypogonadism. Rheumatology (Oxford) 2002; 41:285–289.
- Tengstrand B, Carlström K, Hafström I. Gonadal hormones in men with rheumatoid arthritis--from onset through 2 years. J Rheumatol 2009; 36:887–892.
- Emanuele MA, Emanuele NV. Alcohol’s effects on male reproduction. Alcohol Health Res World 1998; 22:195–201.
- Meikle AW. The interrelationships between thyroid dysfunction and hypogonadism in men and boys. Thyroid 2004; 14( suppl 1):S17–S25.
- Vagenakis AG, Dole K, Braverman LE. Pituitary enlargement, pituitary failure, and primary hypothyroidism. Ann Intern Med 1976; 85:195–198.
Editor’s note: This article on the differential diagnosis of hypogonadism in men is the first of two articles. The second, to be published next month, focuses on the appropriate use of testosterone therapy.
A 54-year-old man is referred for evaluation of low testosterone. He had seen his primary care physician for complaints of diminished libido and erectile dysfunction for the past year and worsening fatigue over the past few years. He has not been formally diagnosed with any medical condition. His serum testosterone level is 180 ng/dL (reference range 249–836 ng/dL).
On physical examination, he is obese (body mass index 31 kg/m2) with a normal-appearing male body habitus, no gynecomastia, and normal testicles and prostate gland.
How should this patient be evaluated?
LOW TESTOSTERONE HAS MANY CAUSES
Male hypogonadism, ie, failure of the testes to produce adequate amounts of androgen or sperm, has become a common clinical finding, particularly in the older population. This is more likely the result of an increase in awareness and detection of the disorder by physicians rather than a true increase in prevalence.
The finding of a low serum testosterone value needs to be confirmed and thoroughly evaluated before starting treatment. It is important to determine whether the cause is a primary (hypergonadotropic) testicular disorder or secondary to a hypothalamic-pituitary process (hypogonadotropic or normogonadotropic).
THE HYPOTHALAMIC-PITUITARY-GONADAL AXIS
Testosterone production is under the control of luteinizing hormone (LH), whereas sperm production is under the control of follicle-stimulating hormone (FSH) (Figure 1). Both of these pituitary hormones are regulated by the pulsatile secretion of hypothalamic gonadotropin-releasing hormone (GnRH).
Testosterone (produced by Leydig cells) and inhibin B (produced by Sertoli cells within the seminiferous tubules) result in negative feedback inhibition of gonadotropin (LH and FSH) secretion. Testosterone and estradiol (produced by aromatization of testosterone) act at both pituitary and hypothalamic sites and are the principal regulators of LH secretion.1,2 Inhibin B is the major regulator of FSH secretion in men,3 but steroid feedback also occurs.2,4
TO FOLLOW UP A LOW TESTOSTERONE, CONFIRM THE VALUE NEAR 8 am
If a testosterone value is found to be low, it is important to determine the time that the sample was obtained. Serum testosterone levels follow a diurnal rhythm, at least in younger men, with values near 8 am being, on average, 30% higher than the trough levels later in the day.5–7 The timing of the diurnal variation may be different in night-shift workers, who may require assessment at a more appropriate time of the day (ie, upon awakening).
Another factor affecting testosterone levels is the patient’s health status at the time of testing. Values obtained in the hospital during an acute illness should be repeated once the event has resolved, as testosterone values decrease considerably in this setting.8 Even in outpatients, particularly in men over age 60, one must be sure that the low testosterone level was not obtained during a period of decompensation of one of the many comorbidities seen in these patients, such as coronary artery disease, congestive heart failure, or uncontrolled diabetes.
If an 8 am testosterone value is low, it is reasonable to obtain at least one confirmatory testosterone level on another day, near 8 am, in the next few weeks, when the patient is in good health. Confirming the testosterone level is important, particularly since commercially available testosterone assays are not well standardized and some are frankly unreliable.9,10 A repeat confirmatory level should always be performed by a reliable reference laboratory. If the testosterone level is still low, further evaluation is warranted.
TOTAL TESTOSTERONE VS BIOAVAILABLE TESTOSTERONE VS FREE TESTOSTERONE
Of the total circulating testosterone, 60% is bound to sex hormone-binding globulin (SHBG), 38% is bound to albumin, and only 2% is free. All of these fractions can be measured to assess for testosterone deficiency.
Free testosterone is the biologically active form of this hormone and, thus, the free testosterone level is considered to be a better representation of the true testosterone status. However, some clinicians believe that bioavailable testosterone (testosterone loosely bound to albumin + free testosterone) is a better reflection of the true level of the active hormone than the level of free testosterone alone.
There are situations in which the total testosterone level is low but bioavailable or free testosterone levels are normal. The level of total testosterone is affected by alterations in the levels of SHBG and albumin. A reduction in the level of SHBG can result in low total serum testosterone levels in patients with obesity or type 2 diabetes (states of insulin resistance), and also in cachexia, malnutrition, advanced cirrhosis, acromegaly, hypothyroidism, and nephrotic syndrome. SHBG can also be low in patients taking glucocorticoids, progestins, or androgenic steroids.11 In these settings, checking the level of free testosterone (the active hormone), bioavailable testosterone, or both, by a reliable reference laboratory, may be more appropriate.9,10
But regardless of which measurement is chosen, all testosterone levels—especially bioavailable and free testosterone values—should be interpreted with caution if they are not measured at a reliable reference laboratory.9,10 Interested readers may wish to see the US Centers for Disease Control and Prevention (CDC) Hormone Standardization Program Web site (www.cdc.gov/labstandards/hs.html) for more details, including a list of CDC-certified laboratories.
CLINICAL FEATURES OF LOW TESTOSTERONE
A history of erectile dysfunction, decreased libido, and fatigue may be seen in patients with low testosterone. However, one must realize that these symptoms—as well as others reported by men with low testosterone, such as depression, difficulty concentrating, irritability, and insomnia—are nonspecific and may be related to other medical conditions.12
Likewise, physical findings such as muscle weakness, reduced body hair, and altered fat distribution (abdominal obesity) are seen in men with low testosterone, but also in those with a number of other medical conditions.
Additional features suggest specific disorders, eg, anosmia in Kallmann syndrome; eunuchoid body habitus, gynecomastia, and small testes in Klinefelter syndrome.
Men with low testosterone may have low bone mineral density or anemia, or both.
Careful examination of the breasts for gynecomastia and the testes for size, consistency, and masses (testicular tumors) helps in formulating a differential diagnosis and in appropriately directing subsequent laboratory evaluation and diagnostic imaging.
LOW TESTOSTERONE: PRIMARY VS SECONDARY
A history of testicular trauma, systemic chemotherapy, or mumps orchitis should direct the physician’s attention to a testicular etiology. On the other hand, darkened or tanned skin (suggesting hemochromatosis), galactorrhea (suggesting hyperprolactinemia), or visual field deficits (suggesting a sellar mass) should direct the physician’s attention toward a pituitary-hypothalamic process.
Once the low testosterone value has been confirmed at least one time near 8 am, one should obtain LH and FSH values to help direct further evaluation in deciphering the etiology (Figure 2). Elevated (hypergonadotropic) values indicate a testicular disorder (primary hypogonadism), whereas low (hypogonadotropic) or normal (normogonadotropic) values point to a pituitary-hypothalamic process (secondary hypogonadism). It should be emphasized that, in the setting of a low testosterone level, LH and FSH values within the normal range are “inappropriately normal” so that further investigation is required.
This evaluation should also include serum prolactin, thyroid-stimulating hormone (TSH, also known as thyrotropin), free thyroxine (T4), and ferritin levels, the latter because hemochromatosis (iron overload) can cause both primary and secondary hypogonadism. If at any time in the evaluation the laboratory results suggest secondary hypogonadism, a full assessment of pituitary function should be undertaken.
Semen analysis is usually reserved for patients presenting with the primary complaint of infertility.
PRIMARY HYPOGONADISM
The patient should be carefully questioned about the age at which his problems began, about pubertal development, and about fertility. Causes of primary hypogonadism include:
- Karyotype abnormalities—Klinefelter syndrome (47, XXY syndrome) is the most common
- Toxin exposure, chemotherapy
- Congenital defects—anorchia, cryptorchidism13
- Orchitis (mumps, autoimmune)
- Testicular trauma or infarction
- Hemochromatosis
- Medications that inhibit androgen biosynthesis, eg, ketoconazole (Nizoral)14
- Increase in temperature of the testicular environment (due to varicocele or a large panniculus).
SECONDARY HYPOGONADISM
Causes of secondary hypogonadism include the following:
Congenital disorders
These disorders are usually diagnosed in childhood or adolescence, often after the patient is brought to the physician because of short stature or pubertal delay.
- Kallmann syndrome (anosmia and GnRH deficiency)15
- GnRH receptor mutation and deficiency16
- Genetic mutations associated with pituitary hormone deficiencies, eg, PROP-1 mutation.17
Acquired disorders that suppress gonadotrophs
Drugs. Long-term therapy with common medications such as opioids or corticosteroids can result in secondary hypogonadism.18–20 Others are GnRH analogues such as leuprolide (Lupron), which are used in treating advanced prostate cancer. The hypogonadism is usually transient and resolves after stopping the offending agent.
Obesity and related conditions such as obstructive sleep apnea, insulin resistance, and type 2 diabetes mellitus are associated with low testosterone levels.21 Treatment should be directed at these underlying conditions and should include lifestyle measures such as weight loss and exercise, rather than simple prescribing of testosterone supplementation, as these efforts may provide multiple health benefits in addition to raising testosterone levels.22
Insulin resistance. In the setting of obesity, the total testosterone level may be low but the bioavailable and free testosterone (active hormone) levels may be normal. This is due to the effect of hyperinsulinemia on the liver, which results in a reduction in SHBG production.23 Low levels of both total and free testosterone can be seen in morbid obesity,24 but the cause remains unclear.
Type 2 diabetes mellitus. Testosterone levels have been reported to be lower in obese men who have diabetes than in those with obesity alone.24 This decrement, comparable in magnitude to that seen with other chronic diseases, suggests that low testosterone may simply be a marker of poor health.22,25,26
Sleep apnea. Disturbances in the sleep cycle, regardless of the underlying cause, can result in decreases in serum testosterone levels. Often, correcting the underlying sleep disturbance can result in a normalization of serum testosterone levels.27,28 A caveat about testosterone therapy: a thorough evaluation for sleep apnea should be undertaken in patients at high risk, since testosterone replacement therapy can adversely affect ventilatory drive and induce or worsen obstructive sleep apnea.29
Aging. Most reports have shown an agerelated decline in both total and free serum testosterone levels (commonly referred to as “andropause”), particularly in men over 60 years of age. There also appears to be a loss of circadian rhythm,30 although not all reports agree.6 It appears that factors such as functional status and overall health may play a more important role in the pathophysiology of hypogonadism in men of advanced age than age alone.
Hemochromatosis. Iron overload, regardless of the cause, can result in hypogonadism via deposition of iron in the hypothalamus, pituitary, or testes. Hereditary hemochromatosis is a common autosomal recessive disease characterized by increased iron absorption. Although both primary and secondary hypogonadism can occur with long-standing iron overload, the latter is much more common.31 Some cases of hypogonadism have been reported to reverse with iron depletion therapy.32
Hyperprolactinemia. Recognized causes of hyperprolactinemia in men include medications (dopamine antagonists, antipsychotics, metoclopramide [Reglan]), pituitary adenomas (microadenomas < 10 mm, macroadenomas ≥ 10 mm), lactotroph hyperfunction (stalk compression interrupting or reducing the tonic suppression of prolactin secretion by dopamine), hypothyroidism, stress, chronic renal failure, cirrhosis, chest wall injury (trauma), and active herpes zoster. The ensuing hypogonadism may be due to the compressive effect of a sellar mass or the direct effect of the prolactin elevation alone, since prolactin disrupts the pulsatile release of GnRH from the hypothalamus,33 required for normal LH and FSH secretion.
Estrogen excess can be either exogenous (from exposure to estrogen-containing contraceptives and creams) or endogenous (from testicular34,35 or very rare adrenal36 estrogen-secreting tumors). Of note, some cases of testicular neoplasms may be detectable only with ultrasonography. Computed tomography may be performed if an adrenal lesion is suspected.
Anabolic steroid abuse. Exposure to anabolic steroids, deliberately or inadvertently, can result in secondary hypogonadism and testicular atrophy, both of which may persist for years after stopping the anabolic agents. If you suspect anabolic steroid abuse, a urine anabolic steroid screen can be obtained.
Anorexia nervosa is far less common in men than in women.37,38 Elements in the history that suggest this disorder include excessive exercise and a low body mass index. Chronic malnutrition (cachexia), regardless of the cause, can result in secondary hypogonadism.
Acute illness (gonadotroph sick syndrome). Hypogonadism is a relatively common finding in any critical illness (analogous to euthyroid sick syndrome with respect to the hypothalamic-pituitary-thyroid axis).8 Testosterone levels are invariably low, so that assessment of testosterone status is not recommended in this setting. The low testosterone phase is usually transient and resolves with resolution or improvement of the underlying medical condition, such as sepsis or myocardial infarction.
HIV. Human immunodeficiency virus (HIV) infection can result in primary or secondary hypogonadism. It can occur with active HIV infection, in patients in whom control of viral replication has been achieved with highly active antiretroviral therapy, and even in patients who have normalized CD4+ cell counts.39 Hypogonadism in HIV patients is multifactorial and may be related to weight loss, opportunistic infections of the pituitary-hypothalamus or testes, or medications such as opioids (licit or illicit), ganciclovir (Cytovene), ketoconazole, the appetite stimulant megestrol (Megace), or cyclophosphamide (Cytoxan). Testosterone replacement therapy does not adversely affect the HIV disease process and in fact may help to avoid complications.
Chronic medical conditions such as cirrhosis, renal failure, and rheumatoid arthritis commonly result in hypogonadism, the pathogenesis of which may involve dysfunction at all levels of the hypothalamic-pituitary-go-nadal axis.40–45 Hypogonadism in the setting of chronic disease is multifactorial, being due not only to the metabolic disturbances seen with these illnesses (uremia in renal failure, elevated circulating estrogens in liver cirrhosis), but also to recurrent acute illness and hospitalization for infection in these immuno-compromised hosts, either from the underlying medical condition or as a result of medications (corticosteroids).
Alcohol abuse. Alcohol can have adverse effects at all levels of the hypothalamic-pituitary-gonadal axis, resulting in low serum testosterone and reduced spermatogenesis.46
Severe chronic primary hypothyroidism, manifested by an extreme elevation of serum thyroid-stimulating hormone (TSH), can result in hypopituitarism. Pituitary function usually recovers with restoration of euthyroidism.47,48
Pubertal delay. Depending on the age of presentation, differentiating pubertal delay from permanent hypogonadotropic hypogonadism can be challenging.
Acquired disorders that damage gonadotrophs
- Sellar mass or cyst—pituitary adenoma, craniopharyngioma, Rathke cleft cyst, meningioma
- Infiltrative lesion—lymphocytic hypophysitis, Langerhans cell histiocytosis, hemochromatosis, sarcoidosis, infection
- Metastatic lesion
- Trauma (head injury)
- Radiation exposure
- Surgery
- Stalk severance
- Pituitary apoplexy.
See Table 1 for a summary of the causes of male hypogonadism.
WHEN IS MRI INDICATED IN EVALUATING SECONDARY HYPOGONADISM?
The yield of pituitary-hypothalamic imaging in older men with secondary hypogonadism is fairly low in the absence of other pituitary hormone abnormalities and deficiencies. There are limited data regarding appropriate criteria for performing hypothalamic-pituitary imaging studies. However, a patient who has multiple anterior pituitary abnormalities on laboratory evaluation should undergo dedicated hypothalamic-pituitary magnetic resonance imaging (MRI).
The Endocrine Society Clinical Practice Guidelines11 recommend that MRI be performed to exclude a pituitary or hypothalamic tumor or infiltrative disease if the patient has severe secondary hypogonadism (serum testosterone < 150 ng/dL), panhypopituitarism, persistent hyperprolactinemia, or symptoms or signs of tumor mass effect such as headache, visual impairment, or a visual field defect.
WHO SHOULD UNDERGO ASSESSMENT OF TESTOSTERONE STATUS?
Screening for androgen deficiency in the asymptomatic general population is not recommended.11 The nonspecific nature of many of the signs and symptoms of androgen deficiency makes it difficult to give concrete recommendations as to who should have testosterone levels measured. Clinicians should consider testing if there is evidence of certain clinical disorders that are associated with low testosterone levels (see earlier discussion on the specific causes of primary and secondary hypogonadism).
When a male patient complains of erectile dysfunction, the investigation should include an assessment of serum testosterone. However, if a man who has a constellation of nonspecific symptoms asks for his testosterone level to be assessed (which is common, given the aggressive marketing of testosterone replacement by the pharmaceutical industry), we would recommend a basic evaluation that includes a comprehensive metabolic panel, complete blood count, and TSH level. Further testing should be determined by the history and physical examination. If no obvious explanation has been found for the patient’s symptoms at that point, assessment of serum testosterone may be warranted. More often than not the patient’s weight and limited physical activity are the driving forces behind the nonspecific symptoms, and counseling a patient on a life-style change can provide much benefit if the patient follows through with the physician’s recommendations.
Men whom we believe should not undergo assessment for testosterone deficiency are those who are acutely ill and hospitalized and those who are severely obese and are complaining of fatigue. Testosterone levels should be assessed only after the acute illness has resolved and, in a severely obese patient with fatigue, only after a thorough evaluation for sleep apnea has been undertaken.
TREAT THE UNDERLYING CAUSE, IF ONE CAN BE FOUND
If the evaluation of low testosterone leads to the diagnosis of a clear underlying condition that is amenable to treatment, such as prolactin elevation or sleep apnea, then treatment should be directed at the underlying cause, with subsequent monitoring of the patient’s symptoms and response in serum testosterone levels. In general, the use of dopamine agonist therapy in the management of hyperprolactinemia and, in cases of panhypopituitarism, of replacement therapy with levothyroxine (Synthroid), hydrocortisone, and possibly growth hormone and desmopressin (DDAVP), fall best under the purview of an endocrinologist. A caveat: serum TSH cannot be used to monitor levothyroxine replacement therapy in cases of secondary hypothyroidism. The clinical picture and serum free T4 and free T3 levels are used instead.
In the absence of a correctable (or immediately correctable) cause, testosterone supplementation can be initiated on an individualized basis in select patients who have clinical signs and symptoms of androgen deficiency if the benefits of treatment appear to outweigh the potential risks, and only after a thorough discussion with the patient.11 The Endocrine Society recommends against offering testosterone therapy to all older men with low testosterone.11
INFERTILITY
In men presenting with low serum testosterone, semen analysis is not routine. It is usually reserved for patients presenting with the primary complaint of infertility.
If an endocrine disorder such as prolactin elevation or hypothyroidism is the suspected cause of infertility, the patient should be referred to an endocrinologist for further evaluation and management. Treatment of male infertility should be directed at the underlying cause, but often requires exogenous human chorionic gonadotropin, FSH, GnRH (via a pulsatile pump), and possibly sperm harvesting from the testis with subsequent in vitro fertilization with intracytoplasmic sperm injection. It is critical that the partner be included in the evaluation of infertility.
These patients should be referred to a urologic or fertility center specializing in the diagnosis and treatment of infertility. For further information regarding male infertility, patients can be directed to www.fertilitylifelines.com.
CASE CONCLUDED
The patient’s low serum testosterone was confirmed on subsequent measurements at 8 am, with levels of 128 and 182 ng/dL (reference range 249–836). Other laboratory values:
- LH 1.4 mIU/mL (reference range 1.2–8.6)
- FSH 2.7 mIU/mL (1.3–9.9 mIU/mL)
(Both of these values are inappropriately normal in the setting of the low testosterone.)
- TSH 248 μIU/mL (0.4–5.5)
- Prolactin 24.6 ng/mL (1.6–18.8).
The patient was started on levothyroxine replacement therapy and after 3 months was noted to be euthyroid (TSH 1.8 μIU/mL) and to have a normal serum prolactin level. Testosterone levels (8 am) at this time were 350 ng/dL and 420 ng/dL.
Therefore, the cause of this patient’s hypogonadism was severe hypothyroidism and associated mild hyperprolactinemia. This case shows that a thorough evaluation is warranted before initiating testosterone therapy.
Editor’s note: This article on the differential diagnosis of hypogonadism in men is the first of two articles. The second, to be published next month, focuses on the appropriate use of testosterone therapy.
A 54-year-old man is referred for evaluation of low testosterone. He had seen his primary care physician for complaints of diminished libido and erectile dysfunction for the past year and worsening fatigue over the past few years. He has not been formally diagnosed with any medical condition. His serum testosterone level is 180 ng/dL (reference range 249–836 ng/dL).
On physical examination, he is obese (body mass index 31 kg/m2) with a normal-appearing male body habitus, no gynecomastia, and normal testicles and prostate gland.
How should this patient be evaluated?
LOW TESTOSTERONE HAS MANY CAUSES
Male hypogonadism, ie, failure of the testes to produce adequate amounts of androgen or sperm, has become a common clinical finding, particularly in the older population. This is more likely the result of an increase in awareness and detection of the disorder by physicians rather than a true increase in prevalence.
The finding of a low serum testosterone value needs to be confirmed and thoroughly evaluated before starting treatment. It is important to determine whether the cause is a primary (hypergonadotropic) testicular disorder or secondary to a hypothalamic-pituitary process (hypogonadotropic or normogonadotropic).
THE HYPOTHALAMIC-PITUITARY-GONADAL AXIS
Testosterone production is under the control of luteinizing hormone (LH), whereas sperm production is under the control of follicle-stimulating hormone (FSH) (Figure 1). Both of these pituitary hormones are regulated by the pulsatile secretion of hypothalamic gonadotropin-releasing hormone (GnRH).
Testosterone (produced by Leydig cells) and inhibin B (produced by Sertoli cells within the seminiferous tubules) result in negative feedback inhibition of gonadotropin (LH and FSH) secretion. Testosterone and estradiol (produced by aromatization of testosterone) act at both pituitary and hypothalamic sites and are the principal regulators of LH secretion.1,2 Inhibin B is the major regulator of FSH secretion in men,3 but steroid feedback also occurs.2,4
TO FOLLOW UP A LOW TESTOSTERONE, CONFIRM THE VALUE NEAR 8 am
If a testosterone value is found to be low, it is important to determine the time that the sample was obtained. Serum testosterone levels follow a diurnal rhythm, at least in younger men, with values near 8 am being, on average, 30% higher than the trough levels later in the day.5–7 The timing of the diurnal variation may be different in night-shift workers, who may require assessment at a more appropriate time of the day (ie, upon awakening).
Another factor affecting testosterone levels is the patient’s health status at the time of testing. Values obtained in the hospital during an acute illness should be repeated once the event has resolved, as testosterone values decrease considerably in this setting.8 Even in outpatients, particularly in men over age 60, one must be sure that the low testosterone level was not obtained during a period of decompensation of one of the many comorbidities seen in these patients, such as coronary artery disease, congestive heart failure, or uncontrolled diabetes.
If an 8 am testosterone value is low, it is reasonable to obtain at least one confirmatory testosterone level on another day, near 8 am, in the next few weeks, when the patient is in good health. Confirming the testosterone level is important, particularly since commercially available testosterone assays are not well standardized and some are frankly unreliable.9,10 A repeat confirmatory level should always be performed by a reliable reference laboratory. If the testosterone level is still low, further evaluation is warranted.
TOTAL TESTOSTERONE VS BIOAVAILABLE TESTOSTERONE VS FREE TESTOSTERONE
Of the total circulating testosterone, 60% is bound to sex hormone-binding globulin (SHBG), 38% is bound to albumin, and only 2% is free. All of these fractions can be measured to assess for testosterone deficiency.
Free testosterone is the biologically active form of this hormone and, thus, the free testosterone level is considered to be a better representation of the true testosterone status. However, some clinicians believe that bioavailable testosterone (testosterone loosely bound to albumin + free testosterone) is a better reflection of the true level of the active hormone than the level of free testosterone alone.
There are situations in which the total testosterone level is low but bioavailable or free testosterone levels are normal. The level of total testosterone is affected by alterations in the levels of SHBG and albumin. A reduction in the level of SHBG can result in low total serum testosterone levels in patients with obesity or type 2 diabetes (states of insulin resistance), and also in cachexia, malnutrition, advanced cirrhosis, acromegaly, hypothyroidism, and nephrotic syndrome. SHBG can also be low in patients taking glucocorticoids, progestins, or androgenic steroids.11 In these settings, checking the level of free testosterone (the active hormone), bioavailable testosterone, or both, by a reliable reference laboratory, may be more appropriate.9,10
But regardless of which measurement is chosen, all testosterone levels—especially bioavailable and free testosterone values—should be interpreted with caution if they are not measured at a reliable reference laboratory.9,10 Interested readers may wish to see the US Centers for Disease Control and Prevention (CDC) Hormone Standardization Program Web site (www.cdc.gov/labstandards/hs.html) for more details, including a list of CDC-certified laboratories.
CLINICAL FEATURES OF LOW TESTOSTERONE
A history of erectile dysfunction, decreased libido, and fatigue may be seen in patients with low testosterone. However, one must realize that these symptoms—as well as others reported by men with low testosterone, such as depression, difficulty concentrating, irritability, and insomnia—are nonspecific and may be related to other medical conditions.12
Likewise, physical findings such as muscle weakness, reduced body hair, and altered fat distribution (abdominal obesity) are seen in men with low testosterone, but also in those with a number of other medical conditions.
Additional features suggest specific disorders, eg, anosmia in Kallmann syndrome; eunuchoid body habitus, gynecomastia, and small testes in Klinefelter syndrome.
Men with low testosterone may have low bone mineral density or anemia, or both.
Careful examination of the breasts for gynecomastia and the testes for size, consistency, and masses (testicular tumors) helps in formulating a differential diagnosis and in appropriately directing subsequent laboratory evaluation and diagnostic imaging.
LOW TESTOSTERONE: PRIMARY VS SECONDARY
A history of testicular trauma, systemic chemotherapy, or mumps orchitis should direct the physician’s attention to a testicular etiology. On the other hand, darkened or tanned skin (suggesting hemochromatosis), galactorrhea (suggesting hyperprolactinemia), or visual field deficits (suggesting a sellar mass) should direct the physician’s attention toward a pituitary-hypothalamic process.
Once the low testosterone value has been confirmed at least one time near 8 am, one should obtain LH and FSH values to help direct further evaluation in deciphering the etiology (Figure 2). Elevated (hypergonadotropic) values indicate a testicular disorder (primary hypogonadism), whereas low (hypogonadotropic) or normal (normogonadotropic) values point to a pituitary-hypothalamic process (secondary hypogonadism). It should be emphasized that, in the setting of a low testosterone level, LH and FSH values within the normal range are “inappropriately normal” so that further investigation is required.
This evaluation should also include serum prolactin, thyroid-stimulating hormone (TSH, also known as thyrotropin), free thyroxine (T4), and ferritin levels, the latter because hemochromatosis (iron overload) can cause both primary and secondary hypogonadism. If at any time in the evaluation the laboratory results suggest secondary hypogonadism, a full assessment of pituitary function should be undertaken.
Semen analysis is usually reserved for patients presenting with the primary complaint of infertility.
PRIMARY HYPOGONADISM
The patient should be carefully questioned about the age at which his problems began, about pubertal development, and about fertility. Causes of primary hypogonadism include:
- Karyotype abnormalities—Klinefelter syndrome (47, XXY syndrome) is the most common
- Toxin exposure, chemotherapy
- Congenital defects—anorchia, cryptorchidism13
- Orchitis (mumps, autoimmune)
- Testicular trauma or infarction
- Hemochromatosis
- Medications that inhibit androgen biosynthesis, eg, ketoconazole (Nizoral)14
- Increase in temperature of the testicular environment (due to varicocele or a large panniculus).
SECONDARY HYPOGONADISM
Causes of secondary hypogonadism include the following:
Congenital disorders
These disorders are usually diagnosed in childhood or adolescence, often after the patient is brought to the physician because of short stature or pubertal delay.
- Kallmann syndrome (anosmia and GnRH deficiency)15
- GnRH receptor mutation and deficiency16
- Genetic mutations associated with pituitary hormone deficiencies, eg, PROP-1 mutation.17
Acquired disorders that suppress gonadotrophs
Drugs. Long-term therapy with common medications such as opioids or corticosteroids can result in secondary hypogonadism.18–20 Others are GnRH analogues such as leuprolide (Lupron), which are used in treating advanced prostate cancer. The hypogonadism is usually transient and resolves after stopping the offending agent.
Obesity and related conditions such as obstructive sleep apnea, insulin resistance, and type 2 diabetes mellitus are associated with low testosterone levels.21 Treatment should be directed at these underlying conditions and should include lifestyle measures such as weight loss and exercise, rather than simple prescribing of testosterone supplementation, as these efforts may provide multiple health benefits in addition to raising testosterone levels.22
Insulin resistance. In the setting of obesity, the total testosterone level may be low but the bioavailable and free testosterone (active hormone) levels may be normal. This is due to the effect of hyperinsulinemia on the liver, which results in a reduction in SHBG production.23 Low levels of both total and free testosterone can be seen in morbid obesity,24 but the cause remains unclear.
Type 2 diabetes mellitus. Testosterone levels have been reported to be lower in obese men who have diabetes than in those with obesity alone.24 This decrement, comparable in magnitude to that seen with other chronic diseases, suggests that low testosterone may simply be a marker of poor health.22,25,26
Sleep apnea. Disturbances in the sleep cycle, regardless of the underlying cause, can result in decreases in serum testosterone levels. Often, correcting the underlying sleep disturbance can result in a normalization of serum testosterone levels.27,28 A caveat about testosterone therapy: a thorough evaluation for sleep apnea should be undertaken in patients at high risk, since testosterone replacement therapy can adversely affect ventilatory drive and induce or worsen obstructive sleep apnea.29
Aging. Most reports have shown an agerelated decline in both total and free serum testosterone levels (commonly referred to as “andropause”), particularly in men over 60 years of age. There also appears to be a loss of circadian rhythm,30 although not all reports agree.6 It appears that factors such as functional status and overall health may play a more important role in the pathophysiology of hypogonadism in men of advanced age than age alone.
Hemochromatosis. Iron overload, regardless of the cause, can result in hypogonadism via deposition of iron in the hypothalamus, pituitary, or testes. Hereditary hemochromatosis is a common autosomal recessive disease characterized by increased iron absorption. Although both primary and secondary hypogonadism can occur with long-standing iron overload, the latter is much more common.31 Some cases of hypogonadism have been reported to reverse with iron depletion therapy.32
Hyperprolactinemia. Recognized causes of hyperprolactinemia in men include medications (dopamine antagonists, antipsychotics, metoclopramide [Reglan]), pituitary adenomas (microadenomas < 10 mm, macroadenomas ≥ 10 mm), lactotroph hyperfunction (stalk compression interrupting or reducing the tonic suppression of prolactin secretion by dopamine), hypothyroidism, stress, chronic renal failure, cirrhosis, chest wall injury (trauma), and active herpes zoster. The ensuing hypogonadism may be due to the compressive effect of a sellar mass or the direct effect of the prolactin elevation alone, since prolactin disrupts the pulsatile release of GnRH from the hypothalamus,33 required for normal LH and FSH secretion.
Estrogen excess can be either exogenous (from exposure to estrogen-containing contraceptives and creams) or endogenous (from testicular34,35 or very rare adrenal36 estrogen-secreting tumors). Of note, some cases of testicular neoplasms may be detectable only with ultrasonography. Computed tomography may be performed if an adrenal lesion is suspected.
Anabolic steroid abuse. Exposure to anabolic steroids, deliberately or inadvertently, can result in secondary hypogonadism and testicular atrophy, both of which may persist for years after stopping the anabolic agents. If you suspect anabolic steroid abuse, a urine anabolic steroid screen can be obtained.
Anorexia nervosa is far less common in men than in women.37,38 Elements in the history that suggest this disorder include excessive exercise and a low body mass index. Chronic malnutrition (cachexia), regardless of the cause, can result in secondary hypogonadism.
Acute illness (gonadotroph sick syndrome). Hypogonadism is a relatively common finding in any critical illness (analogous to euthyroid sick syndrome with respect to the hypothalamic-pituitary-thyroid axis).8 Testosterone levels are invariably low, so that assessment of testosterone status is not recommended in this setting. The low testosterone phase is usually transient and resolves with resolution or improvement of the underlying medical condition, such as sepsis or myocardial infarction.
HIV. Human immunodeficiency virus (HIV) infection can result in primary or secondary hypogonadism. It can occur with active HIV infection, in patients in whom control of viral replication has been achieved with highly active antiretroviral therapy, and even in patients who have normalized CD4+ cell counts.39 Hypogonadism in HIV patients is multifactorial and may be related to weight loss, opportunistic infections of the pituitary-hypothalamus or testes, or medications such as opioids (licit or illicit), ganciclovir (Cytovene), ketoconazole, the appetite stimulant megestrol (Megace), or cyclophosphamide (Cytoxan). Testosterone replacement therapy does not adversely affect the HIV disease process and in fact may help to avoid complications.
Chronic medical conditions such as cirrhosis, renal failure, and rheumatoid arthritis commonly result in hypogonadism, the pathogenesis of which may involve dysfunction at all levels of the hypothalamic-pituitary-go-nadal axis.40–45 Hypogonadism in the setting of chronic disease is multifactorial, being due not only to the metabolic disturbances seen with these illnesses (uremia in renal failure, elevated circulating estrogens in liver cirrhosis), but also to recurrent acute illness and hospitalization for infection in these immuno-compromised hosts, either from the underlying medical condition or as a result of medications (corticosteroids).
Alcohol abuse. Alcohol can have adverse effects at all levels of the hypothalamic-pituitary-gonadal axis, resulting in low serum testosterone and reduced spermatogenesis.46
Severe chronic primary hypothyroidism, manifested by an extreme elevation of serum thyroid-stimulating hormone (TSH), can result in hypopituitarism. Pituitary function usually recovers with restoration of euthyroidism.47,48
Pubertal delay. Depending on the age of presentation, differentiating pubertal delay from permanent hypogonadotropic hypogonadism can be challenging.
Acquired disorders that damage gonadotrophs
- Sellar mass or cyst—pituitary adenoma, craniopharyngioma, Rathke cleft cyst, meningioma
- Infiltrative lesion—lymphocytic hypophysitis, Langerhans cell histiocytosis, hemochromatosis, sarcoidosis, infection
- Metastatic lesion
- Trauma (head injury)
- Radiation exposure
- Surgery
- Stalk severance
- Pituitary apoplexy.
See Table 1 for a summary of the causes of male hypogonadism.
WHEN IS MRI INDICATED IN EVALUATING SECONDARY HYPOGONADISM?
The yield of pituitary-hypothalamic imaging in older men with secondary hypogonadism is fairly low in the absence of other pituitary hormone abnormalities and deficiencies. There are limited data regarding appropriate criteria for performing hypothalamic-pituitary imaging studies. However, a patient who has multiple anterior pituitary abnormalities on laboratory evaluation should undergo dedicated hypothalamic-pituitary magnetic resonance imaging (MRI).
The Endocrine Society Clinical Practice Guidelines11 recommend that MRI be performed to exclude a pituitary or hypothalamic tumor or infiltrative disease if the patient has severe secondary hypogonadism (serum testosterone < 150 ng/dL), panhypopituitarism, persistent hyperprolactinemia, or symptoms or signs of tumor mass effect such as headache, visual impairment, or a visual field defect.
WHO SHOULD UNDERGO ASSESSMENT OF TESTOSTERONE STATUS?
Screening for androgen deficiency in the asymptomatic general population is not recommended.11 The nonspecific nature of many of the signs and symptoms of androgen deficiency makes it difficult to give concrete recommendations as to who should have testosterone levels measured. Clinicians should consider testing if there is evidence of certain clinical disorders that are associated with low testosterone levels (see earlier discussion on the specific causes of primary and secondary hypogonadism).
When a male patient complains of erectile dysfunction, the investigation should include an assessment of serum testosterone. However, if a man who has a constellation of nonspecific symptoms asks for his testosterone level to be assessed (which is common, given the aggressive marketing of testosterone replacement by the pharmaceutical industry), we would recommend a basic evaluation that includes a comprehensive metabolic panel, complete blood count, and TSH level. Further testing should be determined by the history and physical examination. If no obvious explanation has been found for the patient’s symptoms at that point, assessment of serum testosterone may be warranted. More often than not the patient’s weight and limited physical activity are the driving forces behind the nonspecific symptoms, and counseling a patient on a life-style change can provide much benefit if the patient follows through with the physician’s recommendations.
Men whom we believe should not undergo assessment for testosterone deficiency are those who are acutely ill and hospitalized and those who are severely obese and are complaining of fatigue. Testosterone levels should be assessed only after the acute illness has resolved and, in a severely obese patient with fatigue, only after a thorough evaluation for sleep apnea has been undertaken.
TREAT THE UNDERLYING CAUSE, IF ONE CAN BE FOUND
If the evaluation of low testosterone leads to the diagnosis of a clear underlying condition that is amenable to treatment, such as prolactin elevation or sleep apnea, then treatment should be directed at the underlying cause, with subsequent monitoring of the patient’s symptoms and response in serum testosterone levels. In general, the use of dopamine agonist therapy in the management of hyperprolactinemia and, in cases of panhypopituitarism, of replacement therapy with levothyroxine (Synthroid), hydrocortisone, and possibly growth hormone and desmopressin (DDAVP), fall best under the purview of an endocrinologist. A caveat: serum TSH cannot be used to monitor levothyroxine replacement therapy in cases of secondary hypothyroidism. The clinical picture and serum free T4 and free T3 levels are used instead.
In the absence of a correctable (or immediately correctable) cause, testosterone supplementation can be initiated on an individualized basis in select patients who have clinical signs and symptoms of androgen deficiency if the benefits of treatment appear to outweigh the potential risks, and only after a thorough discussion with the patient.11 The Endocrine Society recommends against offering testosterone therapy to all older men with low testosterone.11
INFERTILITY
In men presenting with low serum testosterone, semen analysis is not routine. It is usually reserved for patients presenting with the primary complaint of infertility.
If an endocrine disorder such as prolactin elevation or hypothyroidism is the suspected cause of infertility, the patient should be referred to an endocrinologist for further evaluation and management. Treatment of male infertility should be directed at the underlying cause, but often requires exogenous human chorionic gonadotropin, FSH, GnRH (via a pulsatile pump), and possibly sperm harvesting from the testis with subsequent in vitro fertilization with intracytoplasmic sperm injection. It is critical that the partner be included in the evaluation of infertility.
These patients should be referred to a urologic or fertility center specializing in the diagnosis and treatment of infertility. For further information regarding male infertility, patients can be directed to www.fertilitylifelines.com.
CASE CONCLUDED
The patient’s low serum testosterone was confirmed on subsequent measurements at 8 am, with levels of 128 and 182 ng/dL (reference range 249–836). Other laboratory values:
- LH 1.4 mIU/mL (reference range 1.2–8.6)
- FSH 2.7 mIU/mL (1.3–9.9 mIU/mL)
(Both of these values are inappropriately normal in the setting of the low testosterone.)
- TSH 248 μIU/mL (0.4–5.5)
- Prolactin 24.6 ng/mL (1.6–18.8).
The patient was started on levothyroxine replacement therapy and after 3 months was noted to be euthyroid (TSH 1.8 μIU/mL) and to have a normal serum prolactin level. Testosterone levels (8 am) at this time were 350 ng/dL and 420 ng/dL.
Therefore, the cause of this patient’s hypogonadism was severe hypothyroidism and associated mild hyperprolactinemia. This case shows that a thorough evaluation is warranted before initiating testosterone therapy.
- Pitteloud N, Dwyer AA, DeCruz S, et al. Inhibition of luteinizing hormone secretion by testosterone in men requires aromatization for its pituitary but not its hypo-thalamic effects: evidence from the tandem study of normal and gonadotropin-releasing hormone-deficient men. J Clin Endocrinol Metab 2008; 93:784–791.
- Hayes FJ, DeCruz S, Seminara SB, Boepple PA, Crowley WF. Differential regulation of gonadotropin secretion by testosterone in the human male: absence of a negative feedback effect of testosterone on follicle-stimulating hormone secretion. J Clin Endocrinol Metab 2001; 86:53–58.
- Hayes FJ, Pitteloud N, DeCruz S, Crowley WF, Boepple PA. Importance of inhibin B in the regulation of FSH secretion in the human male. J Clin Endocrinol Metab 2001; 86:5541–5546.
- Pitteloud N, Dwyer AA, DeCruz S, et al. The relative role of gonadal sex steroids and gonadotropin-releasing hormone pulse frequency in the regulation of follicle-stimulating hormone secretion in men. J Clin Endocrinol Metab 2008; 93:2686–2692.
- Cooke RR, McIntosh JE, McIntosh RP. Circadian variation in serum free and non-SHBG-bound testosterone in normal men: measurements, and simulation using a mass action model. Clin Endocrinol (Oxf) 1993; 39:163–171.
- Diver MJ, Imtiaz KE, Ahmad AM, Vora JP, Fraser WD. Diurnal rhythms of serum total, free and bioavailable testosterone and of SHBG in middle-aged men compared with those in young men. Clin Endocrinol (Oxf) 2003; 58:710–717.
- Clair P, Claustrat B, Jordan D, Dechaud H, Sassolas G. Daily variations of plasma sex hormone-binding globulin binding capacity, testosterone and luteinizing hormone concentrations in healthy rested adult males. Horm Res 1985; 21:220–223.
- Woolf PD, Hamill RW, McDonald JV, Lee LA, Kelly M. Transient hypogonadotropic hypogonadism caused by critical illness. J Clin Endocrinol Metab 1985; 60:444–450.
- Rosner W, Auchus RJ, Azziz R, Sluss PM, Raff H. Position statement: utility, limitations, and pitfalls in measuring testosterone: an Endocrine Society position statement. J Clin Endocrinol Metab 2007; 92:405–413.
- Rosner W, Vesper H, et al; Endocrine Society; American Association for Clinical Chemistry; American Association of Clinical Endocrinologists; et al. Toward excellence in testosterone testing: a consensus statement. J Clin Endocrinol Metab 2010; 95:4542–4548.
- Bhasin S, Cunningham GR, Hayes FJ, et al; Task Force, Endocrine Society. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2010; 95:2536–2559.
- Wu FC, Tajar A, Beynon JM, et al; EMAS Group. Identification of late-onset hypogonadism in middle-aged and elderly men. N Engl J Med 2010; 363:123–135.
- Farrer JH, Sikka SC, Xie HW, Constantinide D, Rajfer J. Impaired testosterone biosynthesis in cryptorchidism. Fertil Steril 1985; 44:125–132.
- Sikka SC, Swerdloff RS, Rajfer J. In vitro inhibition of testosterone biosynthesis by ketoconazole. Endocrinology 1985; 116:1920–1925.
- Pallais JC, Au M, Pitteloud N, Seminara S, Crowley WF Jr. Kallmann syndrome. In:Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, eds. GeneReviews™ (Internet). Seattle, WA: University of Washington; 1993.
- Chevrier L, Guimiot F, de Roux N. GnRH receptor mutations in isolated gonadotropic deficiency. Mol Cell Endocrinol 2011; 346:21–28.
- Romero CJ, Pine-Twaddell E, Radovick S. Novel mutations associated with combined pituitary hormone deficiency. J Mol Endocrinol 2011; 46:R93–R102.
- Colameco S, Coren JS, Ciervo CA. Continuous opioid treatment for chronic noncancer pain: a time for moderation in prescribing. Postgrad Med 2009; 121:61–66.
- Fraser LA, Morrison D, Morley-Forster P, et al. Oral opioids for chronic non-cancer pain: higher prevalence of hypogonadism in men than in women. Exp Clin Endocrinol Diabetes 2009; 117:38–43.
- Morrison D, Capewell S, Reynolds SP, et al. Testosterone levels during systemic and inhaled corticosteroid therapy. Respir Med 1994; 88:659–663.
- Mah PM, Wittert GA. Obesity and testicular function. Mol Cell Endocrinol 2010; 316:180–186.
- Grossmann M. Low testosterone in men with type 2 diabetes: significance and treatment. J Clin Endocrinol Metab 2011; 96:2341–2353.
- Gascón F, Valle M, Martos R, et al. Sex hormone-binding globulin as a marker for hyperinsulinemia and/or insulin resistance in obese children. Eur J Endocrinol 2000; 143:85–89.
- Dhindsa S, Miller MG, McWhirter CL, et al. Testosterone concentrations in diabetic and nondiabetic obese men. Diabetes Care 2010; 33:1186–1192.
- Grossmann M, Gianatti EJ, Zajac JD. Testosterone and type 2 diabetes. Curr Opin Endocrinol Diabetes Obes 2010; 17:247–256.
- Andersson B, Mårin P, Lissner L, Vermeulen A, Björntorp P. Testosterone concentrations in women and men with NIDDM. Diabetes Care 1994; 17:405–411.
- Santamaria JD, Prior JC, Fleetham JA. Reversible reproductive dysfunction in men with obstructive sleep apnoea. Clin Endocrinol (Oxf) 1988; 28:461–470.
- Grunstein RR, Handelsman DJ, Lawrence SJ, Blackwell C, Caterson ID, Sullivan CE. Neuroendocrine dysfunction in sleep apnea: reversal by continuous positive airways pressure therapy. J Clin Endocrinol Metab 1989; 68:352–358.
- Matsumoto AM, Sandblom RE, Schoene RB, et al. Testosterone replacement in hypogonadal men: effects on obstructive sleep apnoea, respiratory drives, and sleep. Clin Endocrinol (Oxf) 1985; 22:713–721.
- Bremner WJ, Vitiello MV, Prinz PN. Loss of circadian rhythmicity in blood testosterone levels with aging in normal men. J Clin Endocrinol Metab 1983; 56:1278–1281.
- McDermott JH, Walsh CH. Hypogonadism in hereditary hemochromatosis. J Clin Endocrinol Metab 2005; 90:2451–2455.
- Kelly TM, Edwards CQ, Meikle AW, Kushner JP. Hypogonadism in hemochromatosis: reversal with iron depletion. Ann Intern Med 1984; 101:629–632.
- Milenkovic L, D’Angelo G, Kelly PA, Weiner RI. Inhibition of gonadotropin hormone-releasing hormone release by prolactin from GT1 neuronal cell lines through prolactin receptors. Proc Natl Acad Sci U S A 1994; 91:1244–1247.
- Valensi P, Coussieu C, Kemeny JL, Attali JR, Amouroux J, Sebaoun J. Endocrine investigations in two cases of feminizing Leydig cell tumour. Acta Endocrinol (Copenh) 1987; 115:365–372.
- Young S, Gooneratne S, Straus FH, Zeller WP, Bulun SE, Rosenthal IM. Feminizing Sertoli cell tumors in boys with Peutz-Jeghers syndrome. Am J Surg Pathol 1995; 19:50–58.
- Zayed A, Stock JL, Liepman MK, Wollin M, Longcope C. Feminization as a result of both peripheral conversion of androgens and direct estrogen production from an adrenocortical carcinoma. J Endocrinol Invest 1994; 17:275–278.
- Russ MJ, Ackerman SH, Barakat R, Levy B. Hypogonadotropic hypogonadism and delayed puberty in a man with anorexia nervosa. Psychosomatics 1986; 27:737–739.
- Rigotti NA, Neer RM, Jameson L. Osteopenia and bone fractures in a man with anorexia nervosa and hypogonadism. JAMA 1986; 256:385–388.
- Cohan GR. HIV-associated hypogonadism. AIDS Read 2006; 16:341–345,348,352–354.
- Handelsman DJ, Strasser S, McDonald JA, Conway AJ, McCaughan GW. Hypothalamic-pituitary-testicular function in end-stage nonalcoholic liver disease before and after liver transplantation. Clin Endocrinol (Oxf) 1995; 43:331–337.
- Lim VS, Fang VS. Gonadal dysfunction in uremic men. A study of the hypothalamo-pituitary-testicular axis before and after renal transplantation. Am J Med 1975; 58:655–662.
- Handelsman DJ, Dong Q. Hypothalamo-pituitary gonadal axis in chronic renal failure. Endocrinol Metab Clin North Am 1993; 22:145–161.
- Handelsman DJ, Spaliviero JA, Turtle JR. Hypothalamic-pituitary function in experimental uremic hypogonadism. Endocrinology 1985; 117:1984–1995.
- Tengstrand B, Carlström K, Hafström I. Bioavailable testosterone in men with rheumatoid arthritis-high frequency of hypogonadism. Rheumatology (Oxford) 2002; 41:285–289.
- Tengstrand B, Carlström K, Hafström I. Gonadal hormones in men with rheumatoid arthritis--from onset through 2 years. J Rheumatol 2009; 36:887–892.
- Emanuele MA, Emanuele NV. Alcohol’s effects on male reproduction. Alcohol Health Res World 1998; 22:195–201.
- Meikle AW. The interrelationships between thyroid dysfunction and hypogonadism in men and boys. Thyroid 2004; 14( suppl 1):S17–S25.
- Vagenakis AG, Dole K, Braverman LE. Pituitary enlargement, pituitary failure, and primary hypothyroidism. Ann Intern Med 1976; 85:195–198.
- Pitteloud N, Dwyer AA, DeCruz S, et al. Inhibition of luteinizing hormone secretion by testosterone in men requires aromatization for its pituitary but not its hypo-thalamic effects: evidence from the tandem study of normal and gonadotropin-releasing hormone-deficient men. J Clin Endocrinol Metab 2008; 93:784–791.
- Hayes FJ, DeCruz S, Seminara SB, Boepple PA, Crowley WF. Differential regulation of gonadotropin secretion by testosterone in the human male: absence of a negative feedback effect of testosterone on follicle-stimulating hormone secretion. J Clin Endocrinol Metab 2001; 86:53–58.
- Hayes FJ, Pitteloud N, DeCruz S, Crowley WF, Boepple PA. Importance of inhibin B in the regulation of FSH secretion in the human male. J Clin Endocrinol Metab 2001; 86:5541–5546.
- Pitteloud N, Dwyer AA, DeCruz S, et al. The relative role of gonadal sex steroids and gonadotropin-releasing hormone pulse frequency in the regulation of follicle-stimulating hormone secretion in men. J Clin Endocrinol Metab 2008; 93:2686–2692.
- Cooke RR, McIntosh JE, McIntosh RP. Circadian variation in serum free and non-SHBG-bound testosterone in normal men: measurements, and simulation using a mass action model. Clin Endocrinol (Oxf) 1993; 39:163–171.
- Diver MJ, Imtiaz KE, Ahmad AM, Vora JP, Fraser WD. Diurnal rhythms of serum total, free and bioavailable testosterone and of SHBG in middle-aged men compared with those in young men. Clin Endocrinol (Oxf) 2003; 58:710–717.
- Clair P, Claustrat B, Jordan D, Dechaud H, Sassolas G. Daily variations of plasma sex hormone-binding globulin binding capacity, testosterone and luteinizing hormone concentrations in healthy rested adult males. Horm Res 1985; 21:220–223.
- Woolf PD, Hamill RW, McDonald JV, Lee LA, Kelly M. Transient hypogonadotropic hypogonadism caused by critical illness. J Clin Endocrinol Metab 1985; 60:444–450.
- Rosner W, Auchus RJ, Azziz R, Sluss PM, Raff H. Position statement: utility, limitations, and pitfalls in measuring testosterone: an Endocrine Society position statement. J Clin Endocrinol Metab 2007; 92:405–413.
- Rosner W, Vesper H, et al; Endocrine Society; American Association for Clinical Chemistry; American Association of Clinical Endocrinologists; et al. Toward excellence in testosterone testing: a consensus statement. J Clin Endocrinol Metab 2010; 95:4542–4548.
- Bhasin S, Cunningham GR, Hayes FJ, et al; Task Force, Endocrine Society. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2010; 95:2536–2559.
- Wu FC, Tajar A, Beynon JM, et al; EMAS Group. Identification of late-onset hypogonadism in middle-aged and elderly men. N Engl J Med 2010; 363:123–135.
- Farrer JH, Sikka SC, Xie HW, Constantinide D, Rajfer J. Impaired testosterone biosynthesis in cryptorchidism. Fertil Steril 1985; 44:125–132.
- Sikka SC, Swerdloff RS, Rajfer J. In vitro inhibition of testosterone biosynthesis by ketoconazole. Endocrinology 1985; 116:1920–1925.
- Pallais JC, Au M, Pitteloud N, Seminara S, Crowley WF Jr. Kallmann syndrome. In:Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, eds. GeneReviews™ (Internet). Seattle, WA: University of Washington; 1993.
- Chevrier L, Guimiot F, de Roux N. GnRH receptor mutations in isolated gonadotropic deficiency. Mol Cell Endocrinol 2011; 346:21–28.
- Romero CJ, Pine-Twaddell E, Radovick S. Novel mutations associated with combined pituitary hormone deficiency. J Mol Endocrinol 2011; 46:R93–R102.
- Colameco S, Coren JS, Ciervo CA. Continuous opioid treatment for chronic noncancer pain: a time for moderation in prescribing. Postgrad Med 2009; 121:61–66.
- Fraser LA, Morrison D, Morley-Forster P, et al. Oral opioids for chronic non-cancer pain: higher prevalence of hypogonadism in men than in women. Exp Clin Endocrinol Diabetes 2009; 117:38–43.
- Morrison D, Capewell S, Reynolds SP, et al. Testosterone levels during systemic and inhaled corticosteroid therapy. Respir Med 1994; 88:659–663.
- Mah PM, Wittert GA. Obesity and testicular function. Mol Cell Endocrinol 2010; 316:180–186.
- Grossmann M. Low testosterone in men with type 2 diabetes: significance and treatment. J Clin Endocrinol Metab 2011; 96:2341–2353.
- Gascón F, Valle M, Martos R, et al. Sex hormone-binding globulin as a marker for hyperinsulinemia and/or insulin resistance in obese children. Eur J Endocrinol 2000; 143:85–89.
- Dhindsa S, Miller MG, McWhirter CL, et al. Testosterone concentrations in diabetic and nondiabetic obese men. Diabetes Care 2010; 33:1186–1192.
- Grossmann M, Gianatti EJ, Zajac JD. Testosterone and type 2 diabetes. Curr Opin Endocrinol Diabetes Obes 2010; 17:247–256.
- Andersson B, Mårin P, Lissner L, Vermeulen A, Björntorp P. Testosterone concentrations in women and men with NIDDM. Diabetes Care 1994; 17:405–411.
- Santamaria JD, Prior JC, Fleetham JA. Reversible reproductive dysfunction in men with obstructive sleep apnoea. Clin Endocrinol (Oxf) 1988; 28:461–470.
- Grunstein RR, Handelsman DJ, Lawrence SJ, Blackwell C, Caterson ID, Sullivan CE. Neuroendocrine dysfunction in sleep apnea: reversal by continuous positive airways pressure therapy. J Clin Endocrinol Metab 1989; 68:352–358.
- Matsumoto AM, Sandblom RE, Schoene RB, et al. Testosterone replacement in hypogonadal men: effects on obstructive sleep apnoea, respiratory drives, and sleep. Clin Endocrinol (Oxf) 1985; 22:713–721.
- Bremner WJ, Vitiello MV, Prinz PN. Loss of circadian rhythmicity in blood testosterone levels with aging in normal men. J Clin Endocrinol Metab 1983; 56:1278–1281.
- McDermott JH, Walsh CH. Hypogonadism in hereditary hemochromatosis. J Clin Endocrinol Metab 2005; 90:2451–2455.
- Kelly TM, Edwards CQ, Meikle AW, Kushner JP. Hypogonadism in hemochromatosis: reversal with iron depletion. Ann Intern Med 1984; 101:629–632.
- Milenkovic L, D’Angelo G, Kelly PA, Weiner RI. Inhibition of gonadotropin hormone-releasing hormone release by prolactin from GT1 neuronal cell lines through prolactin receptors. Proc Natl Acad Sci U S A 1994; 91:1244–1247.
- Valensi P, Coussieu C, Kemeny JL, Attali JR, Amouroux J, Sebaoun J. Endocrine investigations in two cases of feminizing Leydig cell tumour. Acta Endocrinol (Copenh) 1987; 115:365–372.
- Young S, Gooneratne S, Straus FH, Zeller WP, Bulun SE, Rosenthal IM. Feminizing Sertoli cell tumors in boys with Peutz-Jeghers syndrome. Am J Surg Pathol 1995; 19:50–58.
- Zayed A, Stock JL, Liepman MK, Wollin M, Longcope C. Feminization as a result of both peripheral conversion of androgens and direct estrogen production from an adrenocortical carcinoma. J Endocrinol Invest 1994; 17:275–278.
- Russ MJ, Ackerman SH, Barakat R, Levy B. Hypogonadotropic hypogonadism and delayed puberty in a man with anorexia nervosa. Psychosomatics 1986; 27:737–739.
- Rigotti NA, Neer RM, Jameson L. Osteopenia and bone fractures in a man with anorexia nervosa and hypogonadism. JAMA 1986; 256:385–388.
- Cohan GR. HIV-associated hypogonadism. AIDS Read 2006; 16:341–345,348,352–354.
- Handelsman DJ, Strasser S, McDonald JA, Conway AJ, McCaughan GW. Hypothalamic-pituitary-testicular function in end-stage nonalcoholic liver disease before and after liver transplantation. Clin Endocrinol (Oxf) 1995; 43:331–337.
- Lim VS, Fang VS. Gonadal dysfunction in uremic men. A study of the hypothalamo-pituitary-testicular axis before and after renal transplantation. Am J Med 1975; 58:655–662.
- Handelsman DJ, Dong Q. Hypothalamo-pituitary gonadal axis in chronic renal failure. Endocrinol Metab Clin North Am 1993; 22:145–161.
- Handelsman DJ, Spaliviero JA, Turtle JR. Hypothalamic-pituitary function in experimental uremic hypogonadism. Endocrinology 1985; 117:1984–1995.
- Tengstrand B, Carlström K, Hafström I. Bioavailable testosterone in men with rheumatoid arthritis-high frequency of hypogonadism. Rheumatology (Oxford) 2002; 41:285–289.
- Tengstrand B, Carlström K, Hafström I. Gonadal hormones in men with rheumatoid arthritis--from onset through 2 years. J Rheumatol 2009; 36:887–892.
- Emanuele MA, Emanuele NV. Alcohol’s effects on male reproduction. Alcohol Health Res World 1998; 22:195–201.
- Meikle AW. The interrelationships between thyroid dysfunction and hypogonadism in men and boys. Thyroid 2004; 14( suppl 1):S17–S25.
- Vagenakis AG, Dole K, Braverman LE. Pituitary enlargement, pituitary failure, and primary hypothyroidism. Ann Intern Med 1976; 85:195–198.
KEY POINTS
- Blood samples for testosterone measurements should be drawn near 8 am.
- A low serum testosterone value should always be confirmed by a reliable reference laboratory.
- The definition of a low testosterone level varies from laboratory to laboratory. In general, values less than 200 or 250 ng/dL are considered low, and values between 250 and 350 ng/dL may be considered borderline low.
- If testosterone is low, determine if the cause is primary (testicular) or secondary (hypothalamic-pituitary).
- Acute illness and treatment with opioids, anabolic steroids, or corticosteroids can result in transient hypogonadism.