User login
VIDEO: AGA provides full-service device registry support
SAN FRANCISCO – Taking a device from inception through the Food and Drug Administration approval process, CPT coding, payer coverage, and adoption by physicians can take 10 years or more and millions of dollars. Working with the AGA Center for GI Innovation and Technology to develop a registry and trial design can assist in speeding this process. At the 2015 AGA Tech Summit in San Francisco, Dr. Ashish Atreja, director of medical informatics at the Mount Sinai School of Medicine, New York, and chair of the AGA Registry Oversight Committee, discusses how the AGA can provide full-service device registry support. He also discusses the goals of AGA’s STAR registry and provides a look ahead at upcoming registry initiatives.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN FRANCISCO – Taking a device from inception through the Food and Drug Administration approval process, CPT coding, payer coverage, and adoption by physicians can take 10 years or more and millions of dollars. Working with the AGA Center for GI Innovation and Technology to develop a registry and trial design can assist in speeding this process. At the 2015 AGA Tech Summit in San Francisco, Dr. Ashish Atreja, director of medical informatics at the Mount Sinai School of Medicine, New York, and chair of the AGA Registry Oversight Committee, discusses how the AGA can provide full-service device registry support. He also discusses the goals of AGA’s STAR registry and provides a look ahead at upcoming registry initiatives.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN FRANCISCO – Taking a device from inception through the Food and Drug Administration approval process, CPT coding, payer coverage, and adoption by physicians can take 10 years or more and millions of dollars. Working with the AGA Center for GI Innovation and Technology to develop a registry and trial design can assist in speeding this process. At the 2015 AGA Tech Summit in San Francisco, Dr. Ashish Atreja, director of medical informatics at the Mount Sinai School of Medicine, New York, and chair of the AGA Registry Oversight Committee, discusses how the AGA can provide full-service device registry support. He also discusses the goals of AGA’s STAR registry and provides a look ahead at upcoming registry initiatives.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT THE 2015 AGA TECH SUMMIT

VIDEO: Working with the FDA to get devices to market
SAN FRANCISCO – The AGA Center for GI Innovation and Technology and the Food and Drug Administration are working together in the evaluation of new potential gastroenterological devices and technologies designed to benefit patients. In an interview at the 2015 AGA Tech Summit in San Francisco, Dr. Herbert Lerner, deputy director of the FDA’s Division of Reproductive, Gastro-renal, and Urological Devices, discussed the agency’s willingness to engage openly with experts and innovators with the goal of getting to market new devices and technologies designed to improve patient health.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN FRANCISCO – The AGA Center for GI Innovation and Technology and the Food and Drug Administration are working together in the evaluation of new potential gastroenterological devices and technologies designed to benefit patients. In an interview at the 2015 AGA Tech Summit in San Francisco, Dr. Herbert Lerner, deputy director of the FDA’s Division of Reproductive, Gastro-renal, and Urological Devices, discussed the agency’s willingness to engage openly with experts and innovators with the goal of getting to market new devices and technologies designed to improve patient health.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN FRANCISCO – The AGA Center for GI Innovation and Technology and the Food and Drug Administration are working together in the evaluation of new potential gastroenterological devices and technologies designed to benefit patients. In an interview at the 2015 AGA Tech Summit in San Francisco, Dr. Herbert Lerner, deputy director of the FDA’s Division of Reproductive, Gastro-renal, and Urological Devices, discussed the agency’s willingness to engage openly with experts and innovators with the goal of getting to market new devices and technologies designed to improve patient health.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT THE 2015 AGA TECH SUMMIT

Newly discovered virus led to fatal infection in Kansas man
A newly recognized Thogotovirus species dubbed Bourbon virus has been linked to the death of a previously healthy man in Kansas, according to researchers at the Centers for Disease Control and Prevention.
“It is currently not known how many human infections and disease cases might be attributable to this novel pathogen. On the basis of limited information for our case-patient, health care providers might consider Bourbon virus as a potential infectious etiology in patients in whom fever, leukopenia, and thrombocytopenia develop without a more likely explanation and who have shown negative results for other tick-borne diseases (e.g., ehrlichiosis, anaplasmosis, or Heartland virus disease) or have not responded to doxycycline therapy,” Olga I. Kosoy and her fellow researchers at the CDC wrote (Emerg. Infect. Dis. 2015 May [doi:10.3201/eid2105.150150]).
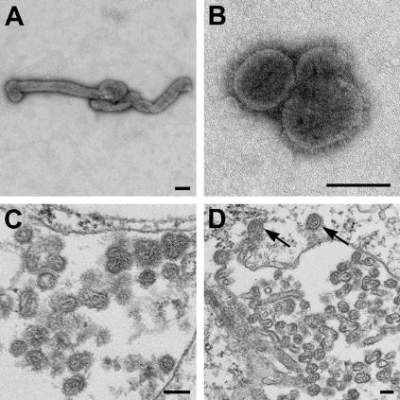
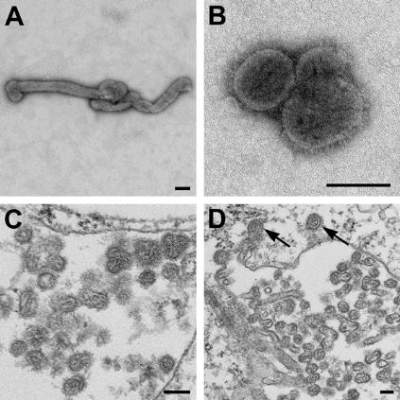
The source case was a man who had a history of tick bite, fever, and fatigue and his lab findings included thrombocytopenia and leukopenia. He was given doxycycline for a presumed tick-borne illness, but his condition did not improve, and he died 11 days later from cardiopulmonary arrest with multiorgan failure.
The patient had been evaluated for a thorough list of potential pathogens. Eliminated potential causes included Rocky Mountain spotted fever, tularemia, Brucella, babesiosis, and Q fever. Also eliminated were a wide variety of fungal pathogens. Evaluations for Cytomegalovirus, Epstein-Barr virus, and Parvovirus showed past infection. Test results for hepatitis B and C viruses, West Nile virus, and HIV were negative. Blood, sputum, and urine bacterial cultures were negative.
At 9 days after the patient’s onset of illness, a whole blood specimen was sent to the CDC for Heartland virus testing. The analysis showed the presence of orthomyxoviral RNA that proved to be a novel Thogotovirus species, which the researchers proposed naming Bourbon virus for the Kansas county where the patient lived.
There have been reports of seven symptomatic human infections associated with Thogotovirus, but those case-patients had meningitis or encephalitis and did not have abnormalities in blood counts.
“The discovery of Bourbon virus, in addition to recent discoveries of tick-associated Heartland and severe fever with thrombocytopenia syndrome viruses, suggests that the public health burden of these pathogens has been underestimated,” the researcher wrote, adding that wider availability of improved methods of detecting pathogens is likely to result in similar discoveries in the future.
A newly recognized Thogotovirus species dubbed Bourbon virus has been linked to the death of a previously healthy man in Kansas, according to researchers at the Centers for Disease Control and Prevention.
“It is currently not known how many human infections and disease cases might be attributable to this novel pathogen. On the basis of limited information for our case-patient, health care providers might consider Bourbon virus as a potential infectious etiology in patients in whom fever, leukopenia, and thrombocytopenia develop without a more likely explanation and who have shown negative results for other tick-borne diseases (e.g., ehrlichiosis, anaplasmosis, or Heartland virus disease) or have not responded to doxycycline therapy,” Olga I. Kosoy and her fellow researchers at the CDC wrote (Emerg. Infect. Dis. 2015 May [doi:10.3201/eid2105.150150]).
The source case was a man who had a history of tick bite, fever, and fatigue and his lab findings included thrombocytopenia and leukopenia. He was given doxycycline for a presumed tick-borne illness, but his condition did not improve, and he died 11 days later from cardiopulmonary arrest with multiorgan failure.
The patient had been evaluated for a thorough list of potential pathogens. Eliminated potential causes included Rocky Mountain spotted fever, tularemia, Brucella, babesiosis, and Q fever. Also eliminated were a wide variety of fungal pathogens. Evaluations for Cytomegalovirus, Epstein-Barr virus, and Parvovirus showed past infection. Test results for hepatitis B and C viruses, West Nile virus, and HIV were negative. Blood, sputum, and urine bacterial cultures were negative.
At 9 days after the patient’s onset of illness, a whole blood specimen was sent to the CDC for Heartland virus testing. The analysis showed the presence of orthomyxoviral RNA that proved to be a novel Thogotovirus species, which the researchers proposed naming Bourbon virus for the Kansas county where the patient lived.
There have been reports of seven symptomatic human infections associated with Thogotovirus, but those case-patients had meningitis or encephalitis and did not have abnormalities in blood counts.
“The discovery of Bourbon virus, in addition to recent discoveries of tick-associated Heartland and severe fever with thrombocytopenia syndrome viruses, suggests that the public health burden of these pathogens has been underestimated,” the researcher wrote, adding that wider availability of improved methods of detecting pathogens is likely to result in similar discoveries in the future.
A newly recognized Thogotovirus species dubbed Bourbon virus has been linked to the death of a previously healthy man in Kansas, according to researchers at the Centers for Disease Control and Prevention.
“It is currently not known how many human infections and disease cases might be attributable to this novel pathogen. On the basis of limited information for our case-patient, health care providers might consider Bourbon virus as a potential infectious etiology in patients in whom fever, leukopenia, and thrombocytopenia develop without a more likely explanation and who have shown negative results for other tick-borne diseases (e.g., ehrlichiosis, anaplasmosis, or Heartland virus disease) or have not responded to doxycycline therapy,” Olga I. Kosoy and her fellow researchers at the CDC wrote (Emerg. Infect. Dis. 2015 May [doi:10.3201/eid2105.150150]).
The source case was a man who had a history of tick bite, fever, and fatigue and his lab findings included thrombocytopenia and leukopenia. He was given doxycycline for a presumed tick-borne illness, but his condition did not improve, and he died 11 days later from cardiopulmonary arrest with multiorgan failure.
The patient had been evaluated for a thorough list of potential pathogens. Eliminated potential causes included Rocky Mountain spotted fever, tularemia, Brucella, babesiosis, and Q fever. Also eliminated were a wide variety of fungal pathogens. Evaluations for Cytomegalovirus, Epstein-Barr virus, and Parvovirus showed past infection. Test results for hepatitis B and C viruses, West Nile virus, and HIV were negative. Blood, sputum, and urine bacterial cultures were negative.
At 9 days after the patient’s onset of illness, a whole blood specimen was sent to the CDC for Heartland virus testing. The analysis showed the presence of orthomyxoviral RNA that proved to be a novel Thogotovirus species, which the researchers proposed naming Bourbon virus for the Kansas county where the patient lived.
There have been reports of seven symptomatic human infections associated with Thogotovirus, but those case-patients had meningitis or encephalitis and did not have abnormalities in blood counts.
“The discovery of Bourbon virus, in addition to recent discoveries of tick-associated Heartland and severe fever with thrombocytopenia syndrome viruses, suggests that the public health burden of these pathogens has been underestimated,” the researcher wrote, adding that wider availability of improved methods of detecting pathogens is likely to result in similar discoveries in the future.
FROM EMERGING INFECTIOUS DISEASES
FDA Approves Mobile Medical App for Real-time Sharing of Glucose Data
The first set of mobile medical apps that permit secure, real-time data sharing by people who have diabetes and use a continuous glucose monitor were approved today by the Food and Drug Administration.
The Dexcom Share Direct Secondary Displays system allows caregivers to a person with diabetes to monitor that individual’s blood sugar levels remotely through an Apple mobile device such as an iPhone. Devices like the Dexcom Share were previously available through open source efforts but were not in compliance with regulatory requirements. The Dexcom Share system is the first legally marketed solution for real-time remote monitoring of a patient’s CGM data, according to a press release from the FDA.
“This innovative technology has been eagerly awaited by the diabetes community, especially caregivers of children with diabetes who want to monitor their glucose levels remotely,” said Alberto Gutierrez, Ph.D., director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health. “Today’s marketing permission paves the way for similar technologies to be marketed in the United States.”
The CGM is worn externally and includes a small, wire-like sensor inserted just under the wearer’s skin. The device monitors and continuously displays an estimate of blood glucose levels as well as the direction and rate of change of these estimates.
The app transmits real-time CGM data to a Web-based storage location. The app of the “follower” can then download the CGM data and display it in real-time.
The FDA has classified the device as low to moderate in risk and exempt from premarket submissions. In the future, manufacturers wishing to market devices like the Dexcom Share system will not need premarket clearance by the FDA prior to marketing. They will still need to register and list their device with the agency, as well as follow other applicable laws and regulations.
“Exempting devices from premarket review is part of the FDA’s effort to ensure these products provide accurate and reliable results while still encouraging the development of devices that meet the needs of people living with diabetes and their caregivers,” said Dr. Gutierrez.
The Dexcom Share system does not replace real-time continuous glucose monitoring or standard home blood glucose monitoring. CGM values alone are not approved to determine dosing of diabetes medications. CGMs must be calibrated by blood glucose meters, and treatment decisions, such as insulin dosing, should be based on readings from a blood glucose meter.The Dexcom Share system is manufactured by Dexcom, Inc., located in San Diego, Calif.
The first set of mobile medical apps that permit secure, real-time data sharing by people who have diabetes and use a continuous glucose monitor were approved today by the Food and Drug Administration.
The Dexcom Share Direct Secondary Displays system allows caregivers to a person with diabetes to monitor that individual’s blood sugar levels remotely through an Apple mobile device such as an iPhone. Devices like the Dexcom Share were previously available through open source efforts but were not in compliance with regulatory requirements. The Dexcom Share system is the first legally marketed solution for real-time remote monitoring of a patient’s CGM data, according to a press release from the FDA.
“This innovative technology has been eagerly awaited by the diabetes community, especially caregivers of children with diabetes who want to monitor their glucose levels remotely,” said Alberto Gutierrez, Ph.D., director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health. “Today’s marketing permission paves the way for similar technologies to be marketed in the United States.”
The CGM is worn externally and includes a small, wire-like sensor inserted just under the wearer’s skin. The device monitors and continuously displays an estimate of blood glucose levels as well as the direction and rate of change of these estimates.
The app transmits real-time CGM data to a Web-based storage location. The app of the “follower” can then download the CGM data and display it in real-time.
The FDA has classified the device as low to moderate in risk and exempt from premarket submissions. In the future, manufacturers wishing to market devices like the Dexcom Share system will not need premarket clearance by the FDA prior to marketing. They will still need to register and list their device with the agency, as well as follow other applicable laws and regulations.
“Exempting devices from premarket review is part of the FDA’s effort to ensure these products provide accurate and reliable results while still encouraging the development of devices that meet the needs of people living with diabetes and their caregivers,” said Dr. Gutierrez.
The Dexcom Share system does not replace real-time continuous glucose monitoring or standard home blood glucose monitoring. CGM values alone are not approved to determine dosing of diabetes medications. CGMs must be calibrated by blood glucose meters, and treatment decisions, such as insulin dosing, should be based on readings from a blood glucose meter.The Dexcom Share system is manufactured by Dexcom, Inc., located in San Diego, Calif.
The first set of mobile medical apps that permit secure, real-time data sharing by people who have diabetes and use a continuous glucose monitor were approved today by the Food and Drug Administration.
The Dexcom Share Direct Secondary Displays system allows caregivers to a person with diabetes to monitor that individual’s blood sugar levels remotely through an Apple mobile device such as an iPhone. Devices like the Dexcom Share were previously available through open source efforts but were not in compliance with regulatory requirements. The Dexcom Share system is the first legally marketed solution for real-time remote monitoring of a patient’s CGM data, according to a press release from the FDA.
“This innovative technology has been eagerly awaited by the diabetes community, especially caregivers of children with diabetes who want to monitor their glucose levels remotely,” said Alberto Gutierrez, Ph.D., director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health. “Today’s marketing permission paves the way for similar technologies to be marketed in the United States.”
The CGM is worn externally and includes a small, wire-like sensor inserted just under the wearer’s skin. The device monitors and continuously displays an estimate of blood glucose levels as well as the direction and rate of change of these estimates.
The app transmits real-time CGM data to a Web-based storage location. The app of the “follower” can then download the CGM data and display it in real-time.
The FDA has classified the device as low to moderate in risk and exempt from premarket submissions. In the future, manufacturers wishing to market devices like the Dexcom Share system will not need premarket clearance by the FDA prior to marketing. They will still need to register and list their device with the agency, as well as follow other applicable laws and regulations.
“Exempting devices from premarket review is part of the FDA’s effort to ensure these products provide accurate and reliable results while still encouraging the development of devices that meet the needs of people living with diabetes and their caregivers,” said Dr. Gutierrez.
The Dexcom Share system does not replace real-time continuous glucose monitoring or standard home blood glucose monitoring. CGM values alone are not approved to determine dosing of diabetes medications. CGMs must be calibrated by blood glucose meters, and treatment decisions, such as insulin dosing, should be based on readings from a blood glucose meter.The Dexcom Share system is manufactured by Dexcom, Inc., located in San Diego, Calif.
FDA approves mobile medical app for real-time sharing of glucose data
The first set of mobile medical apps that permit secure, real-time data sharing by people who have diabetes and use a continuous glucose monitor were approved today by the Food and Drug Administration.
The Dexcom Share Direct Secondary Displays system allows caregivers to a person with diabetes to monitor that individual’s blood sugar levels remotely through an Apple mobile device such as an iPhone. Devices like the Dexcom Share were previously available through open source efforts but were not in compliance with regulatory requirements. The Dexcom Share system is the first legally marketed solution for real-time remote monitoring of a patient’s CGM data, according to a press release from the FDA.
“This innovative technology has been eagerly awaited by the diabetes community, especially caregivers of children with diabetes who want to monitor their glucose levels remotely,” said Alberto Gutierrez, Ph.D., director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health. “Today’s marketing permission paves the way for similar technologies to be marketed in the United States.”
The CGM is worn externally and includes a small, wire-like sensor inserted just under the wearer’s skin. The device monitors and continuously displays an estimate of blood glucose levels as well as the direction and rate of change of these estimates.
The app transmits real-time CGM data to a Web-based storage location. The app of the “follower” can then download the CGM data and display it in real-time.
The FDA has classified the device as low to moderate in risk and exempt from premarket submissions. In the future, manufacturers wishing to market devices like the Dexcom Share system will not need premarket clearance by the FDA prior to marketing. They will still need to register and list their device with the agency, as well as follow other applicable laws and regulations.
“Exempting devices from premarket review is part of the FDA’s effort to ensure these products provide accurate and reliable results while still encouraging the development of devices that meet the needs of people living with diabetes and their caregivers,” said Dr. Gutierrez.
The Dexcom Share system does not replace real-time continuous glucose monitoring or standard home blood glucose monitoring. CGM values alone are not approved to determine dosing of diabetes medications. CGMs must be calibrated by blood glucose meters, and treatment decisions, such as insulin dosing, should be based on readings from a blood glucose meter.The Dexcom Share system is manufactured by Dexcom, Inc., located in San Diego, Calif.
The first set of mobile medical apps that permit secure, real-time data sharing by people who have diabetes and use a continuous glucose monitor were approved today by the Food and Drug Administration.
The Dexcom Share Direct Secondary Displays system allows caregivers to a person with diabetes to monitor that individual’s blood sugar levels remotely through an Apple mobile device such as an iPhone. Devices like the Dexcom Share were previously available through open source efforts but were not in compliance with regulatory requirements. The Dexcom Share system is the first legally marketed solution for real-time remote monitoring of a patient’s CGM data, according to a press release from the FDA.
“This innovative technology has been eagerly awaited by the diabetes community, especially caregivers of children with diabetes who want to monitor their glucose levels remotely,” said Alberto Gutierrez, Ph.D., director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health. “Today’s marketing permission paves the way for similar technologies to be marketed in the United States.”
The CGM is worn externally and includes a small, wire-like sensor inserted just under the wearer’s skin. The device monitors and continuously displays an estimate of blood glucose levels as well as the direction and rate of change of these estimates.
The app transmits real-time CGM data to a Web-based storage location. The app of the “follower” can then download the CGM data and display it in real-time.
The FDA has classified the device as low to moderate in risk and exempt from premarket submissions. In the future, manufacturers wishing to market devices like the Dexcom Share system will not need premarket clearance by the FDA prior to marketing. They will still need to register and list their device with the agency, as well as follow other applicable laws and regulations.
“Exempting devices from premarket review is part of the FDA’s effort to ensure these products provide accurate and reliable results while still encouraging the development of devices that meet the needs of people living with diabetes and their caregivers,” said Dr. Gutierrez.
The Dexcom Share system does not replace real-time continuous glucose monitoring or standard home blood glucose monitoring. CGM values alone are not approved to determine dosing of diabetes medications. CGMs must be calibrated by blood glucose meters, and treatment decisions, such as insulin dosing, should be based on readings from a blood glucose meter.The Dexcom Share system is manufactured by Dexcom, Inc., located in San Diego, Calif.
The first set of mobile medical apps that permit secure, real-time data sharing by people who have diabetes and use a continuous glucose monitor were approved today by the Food and Drug Administration.
The Dexcom Share Direct Secondary Displays system allows caregivers to a person with diabetes to monitor that individual’s blood sugar levels remotely through an Apple mobile device such as an iPhone. Devices like the Dexcom Share were previously available through open source efforts but were not in compliance with regulatory requirements. The Dexcom Share system is the first legally marketed solution for real-time remote monitoring of a patient’s CGM data, according to a press release from the FDA.
“This innovative technology has been eagerly awaited by the diabetes community, especially caregivers of children with diabetes who want to monitor their glucose levels remotely,” said Alberto Gutierrez, Ph.D., director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health. “Today’s marketing permission paves the way for similar technologies to be marketed in the United States.”
The CGM is worn externally and includes a small, wire-like sensor inserted just under the wearer’s skin. The device monitors and continuously displays an estimate of blood glucose levels as well as the direction and rate of change of these estimates.
The app transmits real-time CGM data to a Web-based storage location. The app of the “follower” can then download the CGM data and display it in real-time.
The FDA has classified the device as low to moderate in risk and exempt from premarket submissions. In the future, manufacturers wishing to market devices like the Dexcom Share system will not need premarket clearance by the FDA prior to marketing. They will still need to register and list their device with the agency, as well as follow other applicable laws and regulations.
“Exempting devices from premarket review is part of the FDA’s effort to ensure these products provide accurate and reliable results while still encouraging the development of devices that meet the needs of people living with diabetes and their caregivers,” said Dr. Gutierrez.
The Dexcom Share system does not replace real-time continuous glucose monitoring or standard home blood glucose monitoring. CGM values alone are not approved to determine dosing of diabetes medications. CGMs must be calibrated by blood glucose meters, and treatment decisions, such as insulin dosing, should be based on readings from a blood glucose meter.The Dexcom Share system is manufactured by Dexcom, Inc., located in San Diego, Calif.
Secukinumab tames psoriatic arthritis in FUTURE 2 trial
BOSTON – Secukinumab improved quality of life and symptoms in patients with psoriatic arthritis, regardless of whether they had received prior anti-tumor-necrosis factor therapy or were concurrently receiving methotrexate, Dr. Ian B. McInnes reported at the annual meeting of the American College of Rheumatology.
A 300-mg dose of the investigational drug, which is given subcutaneously, proved to be the most effective dose, Dr. McInnes, of the University of Glasgow, Scotland, said in his presentation of the 24-week results of the FUTURE 2 study. The primary endpoint, a response of at least ACR20, was achieved by 54% of 100 patients given the 300-mg dose and by 51% of 100 patients given the 150-mg dose. The 75-mg dose was far less effective, with a 29% response in 99 patients; the 98 patients given placebo had a 15% response.
At the 150-mg and 300-mg doses, the rates of ACR20 responses were comparable whether or not patients were also taking concomitant methotrexate. Further, the drug’s safety profile was comparable to placebo, Dr. McInnes said.

An ACR50 was achieved by 35% of patients given secukinumab at either 300 mg or 150 mg, and by 18% of those given 75 mg and 7% of those given placebo. About 20% of patients given the higher doses had an ACR70 response, as did 6% of those given 75 mg and 1% of those given placebo.
At the 300-mg dose, secukinumab also resolved dactylitis and enthesitis in approximately half of the affected patients.
Mean improvements in quality of life based on patients’ SF 36 PCS score (Short Form-36 Physical Component Summary) at 24 weeks from baseline were 7.25 in those on the 300-mg dose and 6.39 in those on the 150-mg dose. Those on the 75-mg dose had a 4.38-point mean improvement, whereas patients on placebo had a 1.34-point mean improvement in SF 36 PCS.
No safety signals were noted; adverse events were few and comparable to placebo. Five subjects on the active drug had mild to moderate candidal infections that responded to oral therapy. Neutropenia occurred in one patient in the 300-mg dose group and in one patient in the placebo group, but was transient and patients continued on therapy.
Secukinumab is a fully-human IgG1k monoclonal antibody that selectively targets IL-17A. The drug, manufactured by Novartis, was unanimously recommended for approval by an advisory committee to the Food and Drug Administration. It was administered weekly as a subcutaneous injection for the first 4 weeks of the study, then given again at week 8 and once every 4 weeks thereafter in patients assigned to one of the secukinumab arms of the double-blind, randomized study. Patient assigned to the placebo group were either responders and assigned to receive secukinumab at week 24 and every 4 weeks thereafter or were nonresponders assigned to receive secukinumab at week 16 and every 4 weeks thereafter. Only patients with at least 20% reductions in the number of tender joints or swollen joints continue to receive the drug beyond 1 year.
To be eligible for the study, patients needed to have a diagnosis of active psoriatic arthritis classified by CASPAR criteria and tenderness in at least 3 of 78 joints and swelling of at least 3 of 76 joints. They additionally needed to have an inadequate response to nonsteroidal anti-inflammatory drugs, methotrexate, or anti-TNF therapy
The primary endpoint of the study was ACR20 response at 24 weeks. Secondary endpoints included PASI 75 and PASI 90 responses, change in DAS28-CRP (28-joint Disease Activity Score using C-reactive protein) from baseline, change in SF-36 PCS and HAQ-DI (Health Assessment Questionnaire-Disability Index) from baseline, ACR50 response, the proportion of subjects with dactylitis and enthesitis, and overall safety and tolerability.
As of press time, the FDA was expected to take additional action in January 2015, according to Novartis.
The study was sponsored by Novartis, the maker of secukinumab. Dr. McInnes receives consulting fees from Novartis as well as multiple other pharmaceutical companies.
BOSTON – Secukinumab improved quality of life and symptoms in patients with psoriatic arthritis, regardless of whether they had received prior anti-tumor-necrosis factor therapy or were concurrently receiving methotrexate, Dr. Ian B. McInnes reported at the annual meeting of the American College of Rheumatology.
A 300-mg dose of the investigational drug, which is given subcutaneously, proved to be the most effective dose, Dr. McInnes, of the University of Glasgow, Scotland, said in his presentation of the 24-week results of the FUTURE 2 study. The primary endpoint, a response of at least ACR20, was achieved by 54% of 100 patients given the 300-mg dose and by 51% of 100 patients given the 150-mg dose. The 75-mg dose was far less effective, with a 29% response in 99 patients; the 98 patients given placebo had a 15% response.
At the 150-mg and 300-mg doses, the rates of ACR20 responses were comparable whether or not patients were also taking concomitant methotrexate. Further, the drug’s safety profile was comparable to placebo, Dr. McInnes said.
An ACR50 was achieved by 35% of patients given secukinumab at either 300 mg or 150 mg, and by 18% of those given 75 mg and 7% of those given placebo. About 20% of patients given the higher doses had an ACR70 response, as did 6% of those given 75 mg and 1% of those given placebo.
At the 300-mg dose, secukinumab also resolved dactylitis and enthesitis in approximately half of the affected patients.
Mean improvements in quality of life based on patients’ SF 36 PCS score (Short Form-36 Physical Component Summary) at 24 weeks from baseline were 7.25 in those on the 300-mg dose and 6.39 in those on the 150-mg dose. Those on the 75-mg dose had a 4.38-point mean improvement, whereas patients on placebo had a 1.34-point mean improvement in SF 36 PCS.
No safety signals were noted; adverse events were few and comparable to placebo. Five subjects on the active drug had mild to moderate candidal infections that responded to oral therapy. Neutropenia occurred in one patient in the 300-mg dose group and in one patient in the placebo group, but was transient and patients continued on therapy.
Secukinumab is a fully-human IgG1k monoclonal antibody that selectively targets IL-17A. The drug, manufactured by Novartis, was unanimously recommended for approval by an advisory committee to the Food and Drug Administration. It was administered weekly as a subcutaneous injection for the first 4 weeks of the study, then given again at week 8 and once every 4 weeks thereafter in patients assigned to one of the secukinumab arms of the double-blind, randomized study. Patient assigned to the placebo group were either responders and assigned to receive secukinumab at week 24 and every 4 weeks thereafter or were nonresponders assigned to receive secukinumab at week 16 and every 4 weeks thereafter. Only patients with at least 20% reductions in the number of tender joints or swollen joints continue to receive the drug beyond 1 year.
To be eligible for the study, patients needed to have a diagnosis of active psoriatic arthritis classified by CASPAR criteria and tenderness in at least 3 of 78 joints and swelling of at least 3 of 76 joints. They additionally needed to have an inadequate response to nonsteroidal anti-inflammatory drugs, methotrexate, or anti-TNF therapy
The primary endpoint of the study was ACR20 response at 24 weeks. Secondary endpoints included PASI 75 and PASI 90 responses, change in DAS28-CRP (28-joint Disease Activity Score using C-reactive protein) from baseline, change in SF-36 PCS and HAQ-DI (Health Assessment Questionnaire-Disability Index) from baseline, ACR50 response, the proportion of subjects with dactylitis and enthesitis, and overall safety and tolerability.
As of press time, the FDA was expected to take additional action in January 2015, according to Novartis.
The study was sponsored by Novartis, the maker of secukinumab. Dr. McInnes receives consulting fees from Novartis as well as multiple other pharmaceutical companies.
BOSTON – Secukinumab improved quality of life and symptoms in patients with psoriatic arthritis, regardless of whether they had received prior anti-tumor-necrosis factor therapy or were concurrently receiving methotrexate, Dr. Ian B. McInnes reported at the annual meeting of the American College of Rheumatology.
A 300-mg dose of the investigational drug, which is given subcutaneously, proved to be the most effective dose, Dr. McInnes, of the University of Glasgow, Scotland, said in his presentation of the 24-week results of the FUTURE 2 study. The primary endpoint, a response of at least ACR20, was achieved by 54% of 100 patients given the 300-mg dose and by 51% of 100 patients given the 150-mg dose. The 75-mg dose was far less effective, with a 29% response in 99 patients; the 98 patients given placebo had a 15% response.
At the 150-mg and 300-mg doses, the rates of ACR20 responses were comparable whether or not patients were also taking concomitant methotrexate. Further, the drug’s safety profile was comparable to placebo, Dr. McInnes said.
An ACR50 was achieved by 35% of patients given secukinumab at either 300 mg or 150 mg, and by 18% of those given 75 mg and 7% of those given placebo. About 20% of patients given the higher doses had an ACR70 response, as did 6% of those given 75 mg and 1% of those given placebo.
At the 300-mg dose, secukinumab also resolved dactylitis and enthesitis in approximately half of the affected patients.
Mean improvements in quality of life based on patients’ SF 36 PCS score (Short Form-36 Physical Component Summary) at 24 weeks from baseline were 7.25 in those on the 300-mg dose and 6.39 in those on the 150-mg dose. Those on the 75-mg dose had a 4.38-point mean improvement, whereas patients on placebo had a 1.34-point mean improvement in SF 36 PCS.
No safety signals were noted; adverse events were few and comparable to placebo. Five subjects on the active drug had mild to moderate candidal infections that responded to oral therapy. Neutropenia occurred in one patient in the 300-mg dose group and in one patient in the placebo group, but was transient and patients continued on therapy.
Secukinumab is a fully-human IgG1k monoclonal antibody that selectively targets IL-17A. The drug, manufactured by Novartis, was unanimously recommended for approval by an advisory committee to the Food and Drug Administration. It was administered weekly as a subcutaneous injection for the first 4 weeks of the study, then given again at week 8 and once every 4 weeks thereafter in patients assigned to one of the secukinumab arms of the double-blind, randomized study. Patient assigned to the placebo group were either responders and assigned to receive secukinumab at week 24 and every 4 weeks thereafter or were nonresponders assigned to receive secukinumab at week 16 and every 4 weeks thereafter. Only patients with at least 20% reductions in the number of tender joints or swollen joints continue to receive the drug beyond 1 year.
To be eligible for the study, patients needed to have a diagnosis of active psoriatic arthritis classified by CASPAR criteria and tenderness in at least 3 of 78 joints and swelling of at least 3 of 76 joints. They additionally needed to have an inadequate response to nonsteroidal anti-inflammatory drugs, methotrexate, or anti-TNF therapy
The primary endpoint of the study was ACR20 response at 24 weeks. Secondary endpoints included PASI 75 and PASI 90 responses, change in DAS28-CRP (28-joint Disease Activity Score using C-reactive protein) from baseline, change in SF-36 PCS and HAQ-DI (Health Assessment Questionnaire-Disability Index) from baseline, ACR50 response, the proportion of subjects with dactylitis and enthesitis, and overall safety and tolerability.
As of press time, the FDA was expected to take additional action in January 2015, according to Novartis.
The study was sponsored by Novartis, the maker of secukinumab. Dr. McInnes receives consulting fees from Novartis as well as multiple other pharmaceutical companies.
AT THE ACR ANNUAL MEETING
Key clinical point: Secukinumab could prove to be a major new therapy for psoriatic arthritis.
Major finding: After 24 weeks, a response of at least ACR20 was achieved by 54% of 100 patients given the 300-mg dose of secukinumab.
Data source: The FUTURE 2 study group of nearly 400 patients with psoriatic arthritis who were randomized either to one of three doses of secukinumab or placebo.
Disclosures: The study was sponsored by Novartis, the maker of secukinumab. Dr. McInnes receives consulting fees from Novartis as well as multiple other pharmaceutical companies.

VIDEO: DAGR may prove to be safer alternative to glucocorticoids
BOSTON– In the search for safer alternatives to glucocorticoid therapy, DAGR (PF-0417327) may prove to be a contender. The investigational drug is a selective, high-affinity, dissociated agonist of the glucocorticoid receptor, and appeared to be associated with fewer potential adverse events than are typically seen with glucocorticoids in a phase II trial that compared 8 weeks of the drug against placebo and prednisone.
The findings suggest that DAGR is therapeutically equivalent to prednisone 10 mg once daily, but with side effects comparable with those seen with prednisone 5 mg once daily and with no clinical symptoms of adrenal insufficiency. Dr. Vibeke Strand, a rheumatologist and biopharmaceutical consultant in Portola Valley, Calif., presented data on the drug during the late-breaker session and discussed the implications of the findings at the annual meeting of the American College of Rheumatology.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
BOSTON– In the search for safer alternatives to glucocorticoid therapy, DAGR (PF-0417327) may prove to be a contender. The investigational drug is a selective, high-affinity, dissociated agonist of the glucocorticoid receptor, and appeared to be associated with fewer potential adverse events than are typically seen with glucocorticoids in a phase II trial that compared 8 weeks of the drug against placebo and prednisone.
The findings suggest that DAGR is therapeutically equivalent to prednisone 10 mg once daily, but with side effects comparable with those seen with prednisone 5 mg once daily and with no clinical symptoms of adrenal insufficiency. Dr. Vibeke Strand, a rheumatologist and biopharmaceutical consultant in Portola Valley, Calif., presented data on the drug during the late-breaker session and discussed the implications of the findings at the annual meeting of the American College of Rheumatology.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
BOSTON– In the search for safer alternatives to glucocorticoid therapy, DAGR (PF-0417327) may prove to be a contender. The investigational drug is a selective, high-affinity, dissociated agonist of the glucocorticoid receptor, and appeared to be associated with fewer potential adverse events than are typically seen with glucocorticoids in a phase II trial that compared 8 weeks of the drug against placebo and prednisone.
The findings suggest that DAGR is therapeutically equivalent to prednisone 10 mg once daily, but with side effects comparable with those seen with prednisone 5 mg once daily and with no clinical symptoms of adrenal insufficiency. Dr. Vibeke Strand, a rheumatologist and biopharmaceutical consultant in Portola Valley, Calif., presented data on the drug during the late-breaker session and discussed the implications of the findings at the annual meeting of the American College of Rheumatology.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT THE ACR ANNUAL MEETING

VIDEO: Biomarker may reveal lymphoma risk in Sjögren’s syndrome
BOSTON – Low expression of protein A20 in minor salivary gland tissue appears to be associated with lymphomas in patients with primary Sjögren’s syndrome.
In an interview after his late-breaker presentation at the annual meeting of the American College of Rheumatology, Dr. Svein Joar A. Johnsen of Stavanger (Norway) University Hospital, discussed the implications of his findings and the search for biomarkers of lymphoma risk in these patients.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
BOSTON – Low expression of protein A20 in minor salivary gland tissue appears to be associated with lymphomas in patients with primary Sjögren’s syndrome.
In an interview after his late-breaker presentation at the annual meeting of the American College of Rheumatology, Dr. Svein Joar A. Johnsen of Stavanger (Norway) University Hospital, discussed the implications of his findings and the search for biomarkers of lymphoma risk in these patients.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
BOSTON – Low expression of protein A20 in minor salivary gland tissue appears to be associated with lymphomas in patients with primary Sjögren’s syndrome.
In an interview after his late-breaker presentation at the annual meeting of the American College of Rheumatology, Dr. Svein Joar A. Johnsen of Stavanger (Norway) University Hospital, discussed the implications of his findings and the search for biomarkers of lymphoma risk in these patients.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT ACR 2014

VIDEO: BeST seeks to define best initial therapies for JIA
BOSTON – What is the best initial therapeutic approach to RF-negative polyarticular, oligoarticular, or psoriatic juvenile idiopathic arthritis (JIA)?
The BeST for Kids study is a multicenter, 2-year, randomized trial that is examining the utility of three different treatment arms in disease modifying antirheumatic disease-naive children, aged 2-16 years, with JIA durations of less than 18 months. The study also examines whether inactive disease is a realistic treatment target, if drug-free remissions are possible, and whether restarting medication is effective when relapse occurs.
In our exclusive interview at the annual meeting of the American College of Rheumatology, Dr. Petra C.E. Hissink Muller of Leiden University Medical Center in the Netherlands, discusses the trial’s rationale and initial results from the first 3 months of the study.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
BOSTON – What is the best initial therapeutic approach to RF-negative polyarticular, oligoarticular, or psoriatic juvenile idiopathic arthritis (JIA)?
The BeST for Kids study is a multicenter, 2-year, randomized trial that is examining the utility of three different treatment arms in disease modifying antirheumatic disease-naive children, aged 2-16 years, with JIA durations of less than 18 months. The study also examines whether inactive disease is a realistic treatment target, if drug-free remissions are possible, and whether restarting medication is effective when relapse occurs.
In our exclusive interview at the annual meeting of the American College of Rheumatology, Dr. Petra C.E. Hissink Muller of Leiden University Medical Center in the Netherlands, discusses the trial’s rationale and initial results from the first 3 months of the study.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
BOSTON – What is the best initial therapeutic approach to RF-negative polyarticular, oligoarticular, or psoriatic juvenile idiopathic arthritis (JIA)?
The BeST for Kids study is a multicenter, 2-year, randomized trial that is examining the utility of three different treatment arms in disease modifying antirheumatic disease-naive children, aged 2-16 years, with JIA durations of less than 18 months. The study also examines whether inactive disease is a realistic treatment target, if drug-free remissions are possible, and whether restarting medication is effective when relapse occurs.
In our exclusive interview at the annual meeting of the American College of Rheumatology, Dr. Petra C.E. Hissink Muller of Leiden University Medical Center in the Netherlands, discusses the trial’s rationale and initial results from the first 3 months of the study.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT ACR 2014

VIDEO: Secukinumab rapidly effective against ankylosing spondylitis
BOSTON – Secukinumab showed enduring efficacy in ankylosing spondylitis after 52 weeks of treatment, based on data reported at the annual meeting of the American College of Rheumatology.
The monoclonal antibody, which targets interleukin-17A, is the first drug with demonstrated efficacy against ankylosing spondylitis since the introduction of tumor necrosis factor inhibitors.
In our exclusive video interview, Dr. Dominique Baeten, professor of clinical immunology and rheumatology at the Academic Medical Center of the University of Amsterdam, outlines results from the phase III trial in 371 U.S. and European patients, describes how targeting the IL-17A pathway is uniquely beneficial in AS, and discusses new data from other secukinumab trials in psoriatic arthritis patients.
Secukinumab’s maker, Novartis, sponsored the study. Dr. Baeten has received research grants from Novartis and other drug companies.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
BOSTON – Secukinumab showed enduring efficacy in ankylosing spondylitis after 52 weeks of treatment, based on data reported at the annual meeting of the American College of Rheumatology.
The monoclonal antibody, which targets interleukin-17A, is the first drug with demonstrated efficacy against ankylosing spondylitis since the introduction of tumor necrosis factor inhibitors.
In our exclusive video interview, Dr. Dominique Baeten, professor of clinical immunology and rheumatology at the Academic Medical Center of the University of Amsterdam, outlines results from the phase III trial in 371 U.S. and European patients, describes how targeting the IL-17A pathway is uniquely beneficial in AS, and discusses new data from other secukinumab trials in psoriatic arthritis patients.
Secukinumab’s maker, Novartis, sponsored the study. Dr. Baeten has received research grants from Novartis and other drug companies.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
BOSTON – Secukinumab showed enduring efficacy in ankylosing spondylitis after 52 weeks of treatment, based on data reported at the annual meeting of the American College of Rheumatology.
The monoclonal antibody, which targets interleukin-17A, is the first drug with demonstrated efficacy against ankylosing spondylitis since the introduction of tumor necrosis factor inhibitors.
In our exclusive video interview, Dr. Dominique Baeten, professor of clinical immunology and rheumatology at the Academic Medical Center of the University of Amsterdam, outlines results from the phase III trial in 371 U.S. and European patients, describes how targeting the IL-17A pathway is uniquely beneficial in AS, and discusses new data from other secukinumab trials in psoriatic arthritis patients.
Secukinumab’s maker, Novartis, sponsored the study. Dr. Baeten has received research grants from Novartis and other drug companies.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT THE ACR ANNUAL MEETING
