User login
Postirradiation Pseudosclerodermatous Panniculitis: A Rare Complication of Megavoltage External Beam Radiotherapy
To the Editor:
Postirradiation pseudosclerodermatous panniculitis (PIPP) is a rarely reported complication of megavoltage external beam radiotherapy that was first identified in 1993 by Winkelmann et al.1 The condition presents as an erythematous or hyperpigmented indurated plaque at a site of prior radiotherapy. Lesions caused by PIPP most commonly arise several months after treatment, although they may emerge up to 17 years following exposure.2 Herein, we report a rare case of a patient with PIPP occurring on the leg who previously had been treated for Kaposi sarcoma.
An 84-year-old woman presented with a tender plaque on the right lower leg of 2 months’ duration. Her medical history was remarkable for Kaposi sarcoma, with multiple sites on the body treated with megavoltage external beam radiotherapy during the prior 4 years. The most recent treatment occurred 8 months prior to presentation, at which time she had undergone radiotherapy for lesions on the posterior lower right leg. Physical examination demonstrated a hyperpigmented and indurated plaque at the treatment site (Figure 1). Skin biopsy results showed a mildly sclerotic dermis with atypical radiation fibroblasts scattered interstitially between collagen bundles, and a lobular panniculitis with degenerated adipocytes and foamy histiocytes (Figure 2). Hyalinized dermal vessels also were present. Based on the constellation of these biopsy findings, a diagnosis of PIPP was made.

The diagnosis of PIPP is challenging and invariably requires histologic examination. Clinically, the differential diagnosis includes cutaneous metastasis of the primary neoplasm, cellulitis, lipodermatosclerosis, morphea, and chronic radiation dermatitis.

Histologically, PIPP is characterized by a lobular panniculitis without vasculitis. Typical findings include the presence of centrilobular necrotic adipocytes along with a foamy histiocytic infiltrate containing lipophagic granulomas at the periphery of the fat lobules. Septal thickening and sclerosis around fat lobules also have been described, and dermal changes associated with chronic radiation dermatitis, such as papillary dermal sclerosis, endothelial swelling, vascular hyaline arteriosclerosis, and atypical star-shaped radiation fibroblasts, may be present.2 Features of radiation-induced vasculopathy commonly are seen, although the appearance of these features varies over time. Intimal injury and mural thrombosis can develop within 5 years of radiation therapy, fibrosis of the vessel wall can occur within 10 years of radiation therapy, and atherosclerosis and periarterial fibrosis can appear within 20 years of radiation therapy.2,3 The histologic findings in our patient showed characteristic dermal findings seen in radiation dermatitis in addition to a lobular panniculitis with foamy histiocytes and mild vessel damage.
In contrast, lipodermatosclerosis is a septal and lobular panniculitis with septal fibrosis. Membranocystic fat necrosis is present, characterized by fat microcysts lined by feathery eosinophilic material. Stasis changes in the dermis and epidermis are accompanied by a mild perivascular lymphocytic infiltrate.
Patients with traumatic panniculitis, which also may enter the clinical differential diagnosis of PIPP, often demonstrate nonspecific histologic changes. Early lesions show a perivascular infiltrate of lymphocytes and macrophages. Evolving lesions show variably sized fat microcysts surrounded by histiocytes, in addition to possible calcifications and a foreign-body giant cell reaction. A fibrous capsule may develop, surrounding the fat necrosis to form a mobile encapsulated lipoma. Late lesions frequently demonstrate lipomembranous changes and calcium deposits.4
To date, nearly all cases of PIPP in the literature have been described in breast cancer patients.1,2,5,6 However, Sandoval et al7 reported a case of PIPP occurring in the leg of a patient after radiotherapy for a soft tissue sarcoma. Similar to our patient, this patient presented with a painful, dully erythematous, indurated plaque, although her symptoms arose 5 years after radiotherapy.
Megavoltage external beam radiotherapy has become a widely used modality in the treatment of various cancers. As such, PIPP may represent an underdiagnosed condition with potential cases remaining unidentified when the clinical differential diagnosis does not lead to biopsy. Effective therapies have yet to be widely reported, and our patient failed to experience notable improvement with either topical or intralesional corticosteroids. Further studies are needed in order to address this knowledge gap.
- Winkelmann RK, Grado GL, Quimby SR, et al. Pseudosclerodermatous panniculitis after irradiation: an unusual complication of megavoltage treatment of breast carcinoma. Mayo Clin Proc. 1993;68:122-127.
- Pielasinski U, Machan S, Camacho D, et al. Postirradiation pseudosclerodermatous panniculitis: three new cases with additional histopathologic features supporting the radiotherapy etiology. Am J Dermatopathol. 2013;35:129-134.
- Butler MJ, Lane RH, Webster JH. Irradiation injury to large arteries. Br J Surg. 1980;67:341-343. Moreno A, Marcoval J, Peyri J. Traumatic panniculitis. Dermatol Clin. 2008;26:481-483.
- Shirsat HS, Walsh NM, McDonald LJ, et al. Postirradiation pseudosclerodermatous panniculitis with involvement of breast parenchyma: a dramatic example of a rare entity and a pitfall in diagnosis. J Cutan Pathol. 2016;43:444-450.
- Carrasco L, Moreno C, Pastor MA, et al. Postirradiation pseudosclerodermatous panniculitis. Am J Dermatopathol. 2001;23:283-287.
- Sandoval M, Giesen L, Cataldo K, et al. Postirradiation pseudosclerodermatous panniculitis of the leg: report of a case and review of the literature. Am J Dermatopathol. 2015;37:587-589.
To the Editor:
Postirradiation pseudosclerodermatous panniculitis (PIPP) is a rarely reported complication of megavoltage external beam radiotherapy that was first identified in 1993 by Winkelmann et al.1 The condition presents as an erythematous or hyperpigmented indurated plaque at a site of prior radiotherapy. Lesions caused by PIPP most commonly arise several months after treatment, although they may emerge up to 17 years following exposure.2 Herein, we report a rare case of a patient with PIPP occurring on the leg who previously had been treated for Kaposi sarcoma.
An 84-year-old woman presented with a tender plaque on the right lower leg of 2 months’ duration. Her medical history was remarkable for Kaposi sarcoma, with multiple sites on the body treated with megavoltage external beam radiotherapy during the prior 4 years. The most recent treatment occurred 8 months prior to presentation, at which time she had undergone radiotherapy for lesions on the posterior lower right leg. Physical examination demonstrated a hyperpigmented and indurated plaque at the treatment site (Figure 1). Skin biopsy results showed a mildly sclerotic dermis with atypical radiation fibroblasts scattered interstitially between collagen bundles, and a lobular panniculitis with degenerated adipocytes and foamy histiocytes (Figure 2). Hyalinized dermal vessels also were present. Based on the constellation of these biopsy findings, a diagnosis of PIPP was made.
The diagnosis of PIPP is challenging and invariably requires histologic examination. Clinically, the differential diagnosis includes cutaneous metastasis of the primary neoplasm, cellulitis, lipodermatosclerosis, morphea, and chronic radiation dermatitis.
Histologically, PIPP is characterized by a lobular panniculitis without vasculitis. Typical findings include the presence of centrilobular necrotic adipocytes along with a foamy histiocytic infiltrate containing lipophagic granulomas at the periphery of the fat lobules. Septal thickening and sclerosis around fat lobules also have been described, and dermal changes associated with chronic radiation dermatitis, such as papillary dermal sclerosis, endothelial swelling, vascular hyaline arteriosclerosis, and atypical star-shaped radiation fibroblasts, may be present.2 Features of radiation-induced vasculopathy commonly are seen, although the appearance of these features varies over time. Intimal injury and mural thrombosis can develop within 5 years of radiation therapy, fibrosis of the vessel wall can occur within 10 years of radiation therapy, and atherosclerosis and periarterial fibrosis can appear within 20 years of radiation therapy.2,3 The histologic findings in our patient showed characteristic dermal findings seen in radiation dermatitis in addition to a lobular panniculitis with foamy histiocytes and mild vessel damage.
In contrast, lipodermatosclerosis is a septal and lobular panniculitis with septal fibrosis. Membranocystic fat necrosis is present, characterized by fat microcysts lined by feathery eosinophilic material. Stasis changes in the dermis and epidermis are accompanied by a mild perivascular lymphocytic infiltrate.
Patients with traumatic panniculitis, which also may enter the clinical differential diagnosis of PIPP, often demonstrate nonspecific histologic changes. Early lesions show a perivascular infiltrate of lymphocytes and macrophages. Evolving lesions show variably sized fat microcysts surrounded by histiocytes, in addition to possible calcifications and a foreign-body giant cell reaction. A fibrous capsule may develop, surrounding the fat necrosis to form a mobile encapsulated lipoma. Late lesions frequently demonstrate lipomembranous changes and calcium deposits.4
To date, nearly all cases of PIPP in the literature have been described in breast cancer patients.1,2,5,6 However, Sandoval et al7 reported a case of PIPP occurring in the leg of a patient after radiotherapy for a soft tissue sarcoma. Similar to our patient, this patient presented with a painful, dully erythematous, indurated plaque, although her symptoms arose 5 years after radiotherapy.
Megavoltage external beam radiotherapy has become a widely used modality in the treatment of various cancers. As such, PIPP may represent an underdiagnosed condition with potential cases remaining unidentified when the clinical differential diagnosis does not lead to biopsy. Effective therapies have yet to be widely reported, and our patient failed to experience notable improvement with either topical or intralesional corticosteroids. Further studies are needed in order to address this knowledge gap.
To the Editor:
Postirradiation pseudosclerodermatous panniculitis (PIPP) is a rarely reported complication of megavoltage external beam radiotherapy that was first identified in 1993 by Winkelmann et al.1 The condition presents as an erythematous or hyperpigmented indurated plaque at a site of prior radiotherapy. Lesions caused by PIPP most commonly arise several months after treatment, although they may emerge up to 17 years following exposure.2 Herein, we report a rare case of a patient with PIPP occurring on the leg who previously had been treated for Kaposi sarcoma.
An 84-year-old woman presented with a tender plaque on the right lower leg of 2 months’ duration. Her medical history was remarkable for Kaposi sarcoma, with multiple sites on the body treated with megavoltage external beam radiotherapy during the prior 4 years. The most recent treatment occurred 8 months prior to presentation, at which time she had undergone radiotherapy for lesions on the posterior lower right leg. Physical examination demonstrated a hyperpigmented and indurated plaque at the treatment site (Figure 1). Skin biopsy results showed a mildly sclerotic dermis with atypical radiation fibroblasts scattered interstitially between collagen bundles, and a lobular panniculitis with degenerated adipocytes and foamy histiocytes (Figure 2). Hyalinized dermal vessels also were present. Based on the constellation of these biopsy findings, a diagnosis of PIPP was made.
The diagnosis of PIPP is challenging and invariably requires histologic examination. Clinically, the differential diagnosis includes cutaneous metastasis of the primary neoplasm, cellulitis, lipodermatosclerosis, morphea, and chronic radiation dermatitis.
Histologically, PIPP is characterized by a lobular panniculitis without vasculitis. Typical findings include the presence of centrilobular necrotic adipocytes along with a foamy histiocytic infiltrate containing lipophagic granulomas at the periphery of the fat lobules. Septal thickening and sclerosis around fat lobules also have been described, and dermal changes associated with chronic radiation dermatitis, such as papillary dermal sclerosis, endothelial swelling, vascular hyaline arteriosclerosis, and atypical star-shaped radiation fibroblasts, may be present.2 Features of radiation-induced vasculopathy commonly are seen, although the appearance of these features varies over time. Intimal injury and mural thrombosis can develop within 5 years of radiation therapy, fibrosis of the vessel wall can occur within 10 years of radiation therapy, and atherosclerosis and periarterial fibrosis can appear within 20 years of radiation therapy.2,3 The histologic findings in our patient showed characteristic dermal findings seen in radiation dermatitis in addition to a lobular panniculitis with foamy histiocytes and mild vessel damage.
In contrast, lipodermatosclerosis is a septal and lobular panniculitis with septal fibrosis. Membranocystic fat necrosis is present, characterized by fat microcysts lined by feathery eosinophilic material. Stasis changes in the dermis and epidermis are accompanied by a mild perivascular lymphocytic infiltrate.
Patients with traumatic panniculitis, which also may enter the clinical differential diagnosis of PIPP, often demonstrate nonspecific histologic changes. Early lesions show a perivascular infiltrate of lymphocytes and macrophages. Evolving lesions show variably sized fat microcysts surrounded by histiocytes, in addition to possible calcifications and a foreign-body giant cell reaction. A fibrous capsule may develop, surrounding the fat necrosis to form a mobile encapsulated lipoma. Late lesions frequently demonstrate lipomembranous changes and calcium deposits.4
To date, nearly all cases of PIPP in the literature have been described in breast cancer patients.1,2,5,6 However, Sandoval et al7 reported a case of PIPP occurring in the leg of a patient after radiotherapy for a soft tissue sarcoma. Similar to our patient, this patient presented with a painful, dully erythematous, indurated plaque, although her symptoms arose 5 years after radiotherapy.
Megavoltage external beam radiotherapy has become a widely used modality in the treatment of various cancers. As such, PIPP may represent an underdiagnosed condition with potential cases remaining unidentified when the clinical differential diagnosis does not lead to biopsy. Effective therapies have yet to be widely reported, and our patient failed to experience notable improvement with either topical or intralesional corticosteroids. Further studies are needed in order to address this knowledge gap.
- Winkelmann RK, Grado GL, Quimby SR, et al. Pseudosclerodermatous panniculitis after irradiation: an unusual complication of megavoltage treatment of breast carcinoma. Mayo Clin Proc. 1993;68:122-127.
- Pielasinski U, Machan S, Camacho D, et al. Postirradiation pseudosclerodermatous panniculitis: three new cases with additional histopathologic features supporting the radiotherapy etiology. Am J Dermatopathol. 2013;35:129-134.
- Butler MJ, Lane RH, Webster JH. Irradiation injury to large arteries. Br J Surg. 1980;67:341-343. Moreno A, Marcoval J, Peyri J. Traumatic panniculitis. Dermatol Clin. 2008;26:481-483.
- Shirsat HS, Walsh NM, McDonald LJ, et al. Postirradiation pseudosclerodermatous panniculitis with involvement of breast parenchyma: a dramatic example of a rare entity and a pitfall in diagnosis. J Cutan Pathol. 2016;43:444-450.
- Carrasco L, Moreno C, Pastor MA, et al. Postirradiation pseudosclerodermatous panniculitis. Am J Dermatopathol. 2001;23:283-287.
- Sandoval M, Giesen L, Cataldo K, et al. Postirradiation pseudosclerodermatous panniculitis of the leg: report of a case and review of the literature. Am J Dermatopathol. 2015;37:587-589.
- Winkelmann RK, Grado GL, Quimby SR, et al. Pseudosclerodermatous panniculitis after irradiation: an unusual complication of megavoltage treatment of breast carcinoma. Mayo Clin Proc. 1993;68:122-127.
- Pielasinski U, Machan S, Camacho D, et al. Postirradiation pseudosclerodermatous panniculitis: three new cases with additional histopathologic features supporting the radiotherapy etiology. Am J Dermatopathol. 2013;35:129-134.
- Butler MJ, Lane RH, Webster JH. Irradiation injury to large arteries. Br J Surg. 1980;67:341-343. Moreno A, Marcoval J, Peyri J. Traumatic panniculitis. Dermatol Clin. 2008;26:481-483.
- Shirsat HS, Walsh NM, McDonald LJ, et al. Postirradiation pseudosclerodermatous panniculitis with involvement of breast parenchyma: a dramatic example of a rare entity and a pitfall in diagnosis. J Cutan Pathol. 2016;43:444-450.
- Carrasco L, Moreno C, Pastor MA, et al. Postirradiation pseudosclerodermatous panniculitis. Am J Dermatopathol. 2001;23:283-287.
- Sandoval M, Giesen L, Cataldo K, et al. Postirradiation pseudosclerodermatous panniculitis of the leg: report of a case and review of the literature. Am J Dermatopathol. 2015;37:587-589.
Practice Points
- Postirradiation pseudosclerodermatous panniculitis presents as an erythematous or indurated plaque at a site of prior radiotherapy.
- This rare entity may be underreported and requires biopsy for accurate diagnosis.

DRESS Syndrome Due to Cefdinir Mimicking Superinfected Eczema in a Pediatric Patient
To the Editor:
Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, or drug-induced hypersensitivity syndrome, is a serious and potentially fatal multiorgan drug hypersensitivity reaction. Drug reaction with eosinophilia and systemic symptoms syndrome shares many clinical features with viral exanthems and may be difficult to diagnose in the setting of atopic dermatitis (AD) in which children may have baseline eosinophilia from an atopic diathesis. The cutaneous exanthema also may be variable in presentation, further complicating diagnosis.1,2
A 3-year-old boy with AD since infancy and a history of anaphylaxis to peanuts presented to the emergency department with reported fever, rash, sore throat, and decreased oral intake. Ten days prior, the patient was treated for cellulitis of the left foot with a 7-day course of cefdinir with complete resolution of symptoms. Four days prior to admission, the patient started developing “bumps” on the face and fevers. He was seen at an outside facility, where a rapid test for Streptococcus was negative, and the patient was treated with ibuprofen and fluids for a presumed viral exanthem. The rash subsequently spread to involve the trunk and extremities. On the day of admission, the patient had a positive rapid test for Streptococcus and was referred to the emergency department with concern for superinfected eczema and eczema herpeticum. The patient recently traveled to Puerto Rico, where he had contact with an aunt with active herpes zoster but no other sick contacts. The patient’s immunizations were reported to be up-to-date.
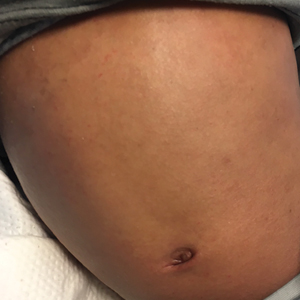
Physical examination revealed the patient was afebrile but irritable and had erythematous crusted papules and patches on the face, arms, and legs, as well as erythematous dry patches on the chest, abdomen, and back (Figure). There were no conjunctival erythematous or oral erosions. The patient was admitted to the hospital for presumed superinfected AD and possible eczema herpeticum. He was started on intravenous clindamycin and acyclovir.

The following day, the patient had new facial edema and fever (temperature, 102.8 °F [39.36 °C]) in addition to palpable mobile cervical, axillary, and inguinal lymphadenopathy. He also was noted to have notably worsening eosinophilia from 1288 (14%) to 2570 (29.2%) cells/µL (reference range, 0%–5%) and new-onset transaminitis. Herpes and varicella-zoster direct fluorescent antibody tests, culture, and serum polymerase chain reaction were all negative, and acyclovir was discontinued. Repeat laboratory tests 12 hours later showed a continued uptrend in transaminitis. Serologies for acute and chronic cytomegalovirus; Epstein-Barr virus; and hepatitis A, B, and C were all nonreactive. The patient was started on intravenous methylprednisolone 1 mg/kg daily for suspected DRESS syndrome likely due to cefdinir.
The patient’s eosinophilia completely resolved (from approximately 2600 to 100 cells/µL) after 1 dose of steroids, and his transaminitis trended down over the next few days. He remained afebrile for the remainder of his admission, and his facial swelling and rash continued to improve. Bacterial culture from the skin grew oxacillin-susceptible Staphylococcus aureus and group A Streptococcus pyogenes. A blood culture was negative. The patient was discharged home to complete a 10-day course of clindamycin and was given topical steroids for the eczema. He continued on oral prednisolone 1 mg/kg daily for 10 days, after which the dose was tapered down for a total 1-month course of systemic corticosteroids. At 1-month follow-up after completing the course of steroids, he was doing well with normal hepatic enzyme levels and no recurrence of fever, facial edema, or rash. He continues to be followed for management of the AD.
Drug reaction with eosinophilia and systemic symptoms syndrome is a serious systemic adverse drug reaction, with high morbidity and even mortality, estimated at 10% in the adult population, though more specific pediatric mortality data are not available.1,2 The exact pathogenesis of DRESS syndrome has not been elucidated. Certain human leukocyte antigen class I alleles are predisposed to the development of DRESS syndrome, but there has not been a human leukocyte antigen subtype identified with beta-lactam–associated DRESS syndrome. Some studies have demonstrated a reactivation of human herpesvirus 6, human herpesvirus 7, and Epstein-Barr virus.3 One study involving 40 patients with DRESS syndrome identified viremia in 76% (29/38) of patients and identified CD8+ T-cell populations directed toward viral epitopes.3 Finally, DRESS syndrome may be related to the slow detoxification and elimination of intermediary products of offending medications that serve as an immunogenic stimulus for the inflammatory cascade.2
In adults, DRESS syndrome was first identified in association with phenytoin, but more recently other drugs have been identified, including other aromatic anticonvulsants (ie, lamotrigine, phenobarbital, carbamazepine), allopurinol, sulfonamides, antiretrovirals (particularly abacavir), and minocycline.2 In a 3-year pediatric prospective study, 11 cases of DRESS syndrome were identified: 4 cases due to lamotrigine, and 3 caused by penicillins.4 The trigger in our patient’s case was the beta-lactam, third-generation cephalosporin cefdinir, and his symptoms developed within 6 days of starting the medication. Many articles report that beta-lactams are a rare cause of DRESS syndrome, with only a handful of cases reported.1,5,6
The diagnosis of DRESS syndrome often can be delayed, as children present acutely febrile and toxic appearing. Unlike many adverse drug reactions, DRESS syndrome does not show rapid resolution with withdrawal of the causative agent, further complicating the diagnosis. The typical onset of DRESS syndrome generally ranges from 2 to 6 weeks after the initiation of the offending drug; however, faster onset of symptoms, similar to our case, has been noted in antibiotic-triggered cases. In the prospective pediatric series by Sasidharanpillai et al,4 the average time to onset among 3 antibiotic-triggered DRESS cases was 5.8 days vs 23.9 days among the 4 cases of lamotrigine-associated DRESS syndrome.
Our patient demonstrated the classic features of DRESS syndrome, including fever, rash, lymphadenopathy, facial edema, peripheral eosinophilia, atypical lymphocytosis, and hepatitis. Based on the proposed RegiSCAR scoring system, our patient was classified as a “definite” case of DRESS syndrome.1,7 Other hematologic findings in DRESS syndrome may include thrombocytopenia and anemia. The liver is the most commonly affected internal organ in DRESS syndrome, with pneumonitis, carditis, and nephritis reported less frequently.1 The pattern of liver injury in our patient was mixed (hepatocellular and cholestatic), the second most common pattern in patients with DRESS syndrome (the cholestatic pattern is most common).8
The exanthem of DRESS syndrome can vary in morphology, with up to 7% of patients reported to have eczemalike lesions in the multinational prospective RegiSCAR study.1 Other entities in the differential diagnosis for our patient included Kawasaki disease, where conjunctivitis and strawberry tongue are classically present, as well as erythrodermic AD, where internal organ involvement is not common.2 Our patient’s exanthem initially was considered to be a flare of AD with superimposed bacterial infection and possible eczema herpeticum. Although bacterial cultures did grow Staphylococcus and Streptococcus, viral studies were all negative, and this alone would not have explained the facial edema, rapidly rising eosinophil count, and transaminitis. The dramatic drop in his eosinophil count and decrease in hepatic enzymes after 1 dose of intravenous methylprednisolone also supported the diagnosis of DRESS syndrome.
Treatment recommendations remain largely anecdotal. Early systemic steroids generally are accepted as the first line of therapy, with a slow taper. Although the average required duration of systemic steroids in 1 series of adults was reported at 50.1 days,9 the duration was shorter (21–35 days) in a series of pediatric patients.4 Our patient’s clinical symptoms and laboratory values normalized after completing a 1-month steroid taper. Other therapies have been tried for recalcitrant cases, including intravenous immunoglobulin, plasmapheresis, rituximab, and valganciclovir.2
Early clinical recognition of the signs and symptoms of DRESS syndrome in the setting of a new medication can decrease morbidity and mortality. Although DRESS syndrome in pediatric patients presents with many similar clinical features as in adults, it may be a greater diagnostic challenge. As in adult cases, timely administration of systemic corticosteroids and tapering based on clinical signs and symptoms can lead to resolution of the hypersensitivity syndrome.
- Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. results from the prospective RegiSCAR study. Br J Dermatol. 2013;169:1071-1080.
- Fernando SL. Drug-reaction eosinophilia and systemic symptoms and drug-induced hypersensitivity syndrome. Australas J Dermatol. 2014;55:15-23.
- Picard D, Janela B, Descamps V, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): a multiorgan antiviral T cell response. Sci Transl Med. 2010;2:46ra62.
- Sasidharanpillai S, Sabitha S, Riyaz N, et al. Drug reaction with eosinophilia and systemic symptoms in children: a prospective study. Pediatr Dermatol. 2016;33:E162-E165.
- Aouam K, Chaabane A, Toumi A, et al. Drug rash with eosinophilia and systemic symptoms (DRESS) probably induced by cefotaxime: a report of two cases. Clin Med Res. 2012;10:32-35.
- Guleria VS, Dhillon M, Gill S, et al. Ceftriaxone induced drug rash with eosinophilia and systemic symptoms. J Res Pharm Pract. 2014;3:72-74.
- Kardaun SH, Sidoroff A, Valeyrie-Allanore L, et al. Variability in the clinical pattern of cutaneous side-effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br J Dermatol. 2007;156:609-611.
- Lin IC, Yang HC, Strong C, et al. Liver injury in patients with DRESS: a clinical study of 72 cases. J Am Acad Dermatol. 2015;72:984-991.
- Ang CC, Wang YS, Yoosuff EL, et al. Retrospective analysis of drug-induced hypersensitivity syndrome: a study of 27 patients. J Am Acad Dermatol. 2010;63:219-227.
To the Editor:
Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, or drug-induced hypersensitivity syndrome, is a serious and potentially fatal multiorgan drug hypersensitivity reaction. Drug reaction with eosinophilia and systemic symptoms syndrome shares many clinical features with viral exanthems and may be difficult to diagnose in the setting of atopic dermatitis (AD) in which children may have baseline eosinophilia from an atopic diathesis. The cutaneous exanthema also may be variable in presentation, further complicating diagnosis.1,2
A 3-year-old boy with AD since infancy and a history of anaphylaxis to peanuts presented to the emergency department with reported fever, rash, sore throat, and decreased oral intake. Ten days prior, the patient was treated for cellulitis of the left foot with a 7-day course of cefdinir with complete resolution of symptoms. Four days prior to admission, the patient started developing “bumps” on the face and fevers. He was seen at an outside facility, where a rapid test for Streptococcus was negative, and the patient was treated with ibuprofen and fluids for a presumed viral exanthem. The rash subsequently spread to involve the trunk and extremities. On the day of admission, the patient had a positive rapid test for Streptococcus and was referred to the emergency department with concern for superinfected eczema and eczema herpeticum. The patient recently traveled to Puerto Rico, where he had contact with an aunt with active herpes zoster but no other sick contacts. The patient’s immunizations were reported to be up-to-date.
Physical examination revealed the patient was afebrile but irritable and had erythematous crusted papules and patches on the face, arms, and legs, as well as erythematous dry patches on the chest, abdomen, and back (Figure). There were no conjunctival erythematous or oral erosions. The patient was admitted to the hospital for presumed superinfected AD and possible eczema herpeticum. He was started on intravenous clindamycin and acyclovir.
The following day, the patient had new facial edema and fever (temperature, 102.8 °F [39.36 °C]) in addition to palpable mobile cervical, axillary, and inguinal lymphadenopathy. He also was noted to have notably worsening eosinophilia from 1288 (14%) to 2570 (29.2%) cells/µL (reference range, 0%–5%) and new-onset transaminitis. Herpes and varicella-zoster direct fluorescent antibody tests, culture, and serum polymerase chain reaction were all negative, and acyclovir was discontinued. Repeat laboratory tests 12 hours later showed a continued uptrend in transaminitis. Serologies for acute and chronic cytomegalovirus; Epstein-Barr virus; and hepatitis A, B, and C were all nonreactive. The patient was started on intravenous methylprednisolone 1 mg/kg daily for suspected DRESS syndrome likely due to cefdinir.
The patient’s eosinophilia completely resolved (from approximately 2600 to 100 cells/µL) after 1 dose of steroids, and his transaminitis trended down over the next few days. He remained afebrile for the remainder of his admission, and his facial swelling and rash continued to improve. Bacterial culture from the skin grew oxacillin-susceptible Staphylococcus aureus and group A Streptococcus pyogenes. A blood culture was negative. The patient was discharged home to complete a 10-day course of clindamycin and was given topical steroids for the eczema. He continued on oral prednisolone 1 mg/kg daily for 10 days, after which the dose was tapered down for a total 1-month course of systemic corticosteroids. At 1-month follow-up after completing the course of steroids, he was doing well with normal hepatic enzyme levels and no recurrence of fever, facial edema, or rash. He continues to be followed for management of the AD.
Drug reaction with eosinophilia and systemic symptoms syndrome is a serious systemic adverse drug reaction, with high morbidity and even mortality, estimated at 10% in the adult population, though more specific pediatric mortality data are not available.1,2 The exact pathogenesis of DRESS syndrome has not been elucidated. Certain human leukocyte antigen class I alleles are predisposed to the development of DRESS syndrome, but there has not been a human leukocyte antigen subtype identified with beta-lactam–associated DRESS syndrome. Some studies have demonstrated a reactivation of human herpesvirus 6, human herpesvirus 7, and Epstein-Barr virus.3 One study involving 40 patients with DRESS syndrome identified viremia in 76% (29/38) of patients and identified CD8+ T-cell populations directed toward viral epitopes.3 Finally, DRESS syndrome may be related to the slow detoxification and elimination of intermediary products of offending medications that serve as an immunogenic stimulus for the inflammatory cascade.2
In adults, DRESS syndrome was first identified in association with phenytoin, but more recently other drugs have been identified, including other aromatic anticonvulsants (ie, lamotrigine, phenobarbital, carbamazepine), allopurinol, sulfonamides, antiretrovirals (particularly abacavir), and minocycline.2 In a 3-year pediatric prospective study, 11 cases of DRESS syndrome were identified: 4 cases due to lamotrigine, and 3 caused by penicillins.4 The trigger in our patient’s case was the beta-lactam, third-generation cephalosporin cefdinir, and his symptoms developed within 6 days of starting the medication. Many articles report that beta-lactams are a rare cause of DRESS syndrome, with only a handful of cases reported.1,5,6
The diagnosis of DRESS syndrome often can be delayed, as children present acutely febrile and toxic appearing. Unlike many adverse drug reactions, DRESS syndrome does not show rapid resolution with withdrawal of the causative agent, further complicating the diagnosis. The typical onset of DRESS syndrome generally ranges from 2 to 6 weeks after the initiation of the offending drug; however, faster onset of symptoms, similar to our case, has been noted in antibiotic-triggered cases. In the prospective pediatric series by Sasidharanpillai et al,4 the average time to onset among 3 antibiotic-triggered DRESS cases was 5.8 days vs 23.9 days among the 4 cases of lamotrigine-associated DRESS syndrome.
Our patient demonstrated the classic features of DRESS syndrome, including fever, rash, lymphadenopathy, facial edema, peripheral eosinophilia, atypical lymphocytosis, and hepatitis. Based on the proposed RegiSCAR scoring system, our patient was classified as a “definite” case of DRESS syndrome.1,7 Other hematologic findings in DRESS syndrome may include thrombocytopenia and anemia. The liver is the most commonly affected internal organ in DRESS syndrome, with pneumonitis, carditis, and nephritis reported less frequently.1 The pattern of liver injury in our patient was mixed (hepatocellular and cholestatic), the second most common pattern in patients with DRESS syndrome (the cholestatic pattern is most common).8
The exanthem of DRESS syndrome can vary in morphology, with up to 7% of patients reported to have eczemalike lesions in the multinational prospective RegiSCAR study.1 Other entities in the differential diagnosis for our patient included Kawasaki disease, where conjunctivitis and strawberry tongue are classically present, as well as erythrodermic AD, where internal organ involvement is not common.2 Our patient’s exanthem initially was considered to be a flare of AD with superimposed bacterial infection and possible eczema herpeticum. Although bacterial cultures did grow Staphylococcus and Streptococcus, viral studies were all negative, and this alone would not have explained the facial edema, rapidly rising eosinophil count, and transaminitis. The dramatic drop in his eosinophil count and decrease in hepatic enzymes after 1 dose of intravenous methylprednisolone also supported the diagnosis of DRESS syndrome.
Treatment recommendations remain largely anecdotal. Early systemic steroids generally are accepted as the first line of therapy, with a slow taper. Although the average required duration of systemic steroids in 1 series of adults was reported at 50.1 days,9 the duration was shorter (21–35 days) in a series of pediatric patients.4 Our patient’s clinical symptoms and laboratory values normalized after completing a 1-month steroid taper. Other therapies have been tried for recalcitrant cases, including intravenous immunoglobulin, plasmapheresis, rituximab, and valganciclovir.2
Early clinical recognition of the signs and symptoms of DRESS syndrome in the setting of a new medication can decrease morbidity and mortality. Although DRESS syndrome in pediatric patients presents with many similar clinical features as in adults, it may be a greater diagnostic challenge. As in adult cases, timely administration of systemic corticosteroids and tapering based on clinical signs and symptoms can lead to resolution of the hypersensitivity syndrome.
To the Editor:
Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, or drug-induced hypersensitivity syndrome, is a serious and potentially fatal multiorgan drug hypersensitivity reaction. Drug reaction with eosinophilia and systemic symptoms syndrome shares many clinical features with viral exanthems and may be difficult to diagnose in the setting of atopic dermatitis (AD) in which children may have baseline eosinophilia from an atopic diathesis. The cutaneous exanthema also may be variable in presentation, further complicating diagnosis.1,2
A 3-year-old boy with AD since infancy and a history of anaphylaxis to peanuts presented to the emergency department with reported fever, rash, sore throat, and decreased oral intake. Ten days prior, the patient was treated for cellulitis of the left foot with a 7-day course of cefdinir with complete resolution of symptoms. Four days prior to admission, the patient started developing “bumps” on the face and fevers. He was seen at an outside facility, where a rapid test for Streptococcus was negative, and the patient was treated with ibuprofen and fluids for a presumed viral exanthem. The rash subsequently spread to involve the trunk and extremities. On the day of admission, the patient had a positive rapid test for Streptococcus and was referred to the emergency department with concern for superinfected eczema and eczema herpeticum. The patient recently traveled to Puerto Rico, where he had contact with an aunt with active herpes zoster but no other sick contacts. The patient’s immunizations were reported to be up-to-date.
Physical examination revealed the patient was afebrile but irritable and had erythematous crusted papules and patches on the face, arms, and legs, as well as erythematous dry patches on the chest, abdomen, and back (Figure). There were no conjunctival erythematous or oral erosions. The patient was admitted to the hospital for presumed superinfected AD and possible eczema herpeticum. He was started on intravenous clindamycin and acyclovir.
The following day, the patient had new facial edema and fever (temperature, 102.8 °F [39.36 °C]) in addition to palpable mobile cervical, axillary, and inguinal lymphadenopathy. He also was noted to have notably worsening eosinophilia from 1288 (14%) to 2570 (29.2%) cells/µL (reference range, 0%–5%) and new-onset transaminitis. Herpes and varicella-zoster direct fluorescent antibody tests, culture, and serum polymerase chain reaction were all negative, and acyclovir was discontinued. Repeat laboratory tests 12 hours later showed a continued uptrend in transaminitis. Serologies for acute and chronic cytomegalovirus; Epstein-Barr virus; and hepatitis A, B, and C were all nonreactive. The patient was started on intravenous methylprednisolone 1 mg/kg daily for suspected DRESS syndrome likely due to cefdinir.
The patient’s eosinophilia completely resolved (from approximately 2600 to 100 cells/µL) after 1 dose of steroids, and his transaminitis trended down over the next few days. He remained afebrile for the remainder of his admission, and his facial swelling and rash continued to improve. Bacterial culture from the skin grew oxacillin-susceptible Staphylococcus aureus and group A Streptococcus pyogenes. A blood culture was negative. The patient was discharged home to complete a 10-day course of clindamycin and was given topical steroids for the eczema. He continued on oral prednisolone 1 mg/kg daily for 10 days, after which the dose was tapered down for a total 1-month course of systemic corticosteroids. At 1-month follow-up after completing the course of steroids, he was doing well with normal hepatic enzyme levels and no recurrence of fever, facial edema, or rash. He continues to be followed for management of the AD.
Drug reaction with eosinophilia and systemic symptoms syndrome is a serious systemic adverse drug reaction, with high morbidity and even mortality, estimated at 10% in the adult population, though more specific pediatric mortality data are not available.1,2 The exact pathogenesis of DRESS syndrome has not been elucidated. Certain human leukocyte antigen class I alleles are predisposed to the development of DRESS syndrome, but there has not been a human leukocyte antigen subtype identified with beta-lactam–associated DRESS syndrome. Some studies have demonstrated a reactivation of human herpesvirus 6, human herpesvirus 7, and Epstein-Barr virus.3 One study involving 40 patients with DRESS syndrome identified viremia in 76% (29/38) of patients and identified CD8+ T-cell populations directed toward viral epitopes.3 Finally, DRESS syndrome may be related to the slow detoxification and elimination of intermediary products of offending medications that serve as an immunogenic stimulus for the inflammatory cascade.2
In adults, DRESS syndrome was first identified in association with phenytoin, but more recently other drugs have been identified, including other aromatic anticonvulsants (ie, lamotrigine, phenobarbital, carbamazepine), allopurinol, sulfonamides, antiretrovirals (particularly abacavir), and minocycline.2 In a 3-year pediatric prospective study, 11 cases of DRESS syndrome were identified: 4 cases due to lamotrigine, and 3 caused by penicillins.4 The trigger in our patient’s case was the beta-lactam, third-generation cephalosporin cefdinir, and his symptoms developed within 6 days of starting the medication. Many articles report that beta-lactams are a rare cause of DRESS syndrome, with only a handful of cases reported.1,5,6
The diagnosis of DRESS syndrome often can be delayed, as children present acutely febrile and toxic appearing. Unlike many adverse drug reactions, DRESS syndrome does not show rapid resolution with withdrawal of the causative agent, further complicating the diagnosis. The typical onset of DRESS syndrome generally ranges from 2 to 6 weeks after the initiation of the offending drug; however, faster onset of symptoms, similar to our case, has been noted in antibiotic-triggered cases. In the prospective pediatric series by Sasidharanpillai et al,4 the average time to onset among 3 antibiotic-triggered DRESS cases was 5.8 days vs 23.9 days among the 4 cases of lamotrigine-associated DRESS syndrome.
Our patient demonstrated the classic features of DRESS syndrome, including fever, rash, lymphadenopathy, facial edema, peripheral eosinophilia, atypical lymphocytosis, and hepatitis. Based on the proposed RegiSCAR scoring system, our patient was classified as a “definite” case of DRESS syndrome.1,7 Other hematologic findings in DRESS syndrome may include thrombocytopenia and anemia. The liver is the most commonly affected internal organ in DRESS syndrome, with pneumonitis, carditis, and nephritis reported less frequently.1 The pattern of liver injury in our patient was mixed (hepatocellular and cholestatic), the second most common pattern in patients with DRESS syndrome (the cholestatic pattern is most common).8
The exanthem of DRESS syndrome can vary in morphology, with up to 7% of patients reported to have eczemalike lesions in the multinational prospective RegiSCAR study.1 Other entities in the differential diagnosis for our patient included Kawasaki disease, where conjunctivitis and strawberry tongue are classically present, as well as erythrodermic AD, where internal organ involvement is not common.2 Our patient’s exanthem initially was considered to be a flare of AD with superimposed bacterial infection and possible eczema herpeticum. Although bacterial cultures did grow Staphylococcus and Streptococcus, viral studies were all negative, and this alone would not have explained the facial edema, rapidly rising eosinophil count, and transaminitis. The dramatic drop in his eosinophil count and decrease in hepatic enzymes after 1 dose of intravenous methylprednisolone also supported the diagnosis of DRESS syndrome.
Treatment recommendations remain largely anecdotal. Early systemic steroids generally are accepted as the first line of therapy, with a slow taper. Although the average required duration of systemic steroids in 1 series of adults was reported at 50.1 days,9 the duration was shorter (21–35 days) in a series of pediatric patients.4 Our patient’s clinical symptoms and laboratory values normalized after completing a 1-month steroid taper. Other therapies have been tried for recalcitrant cases, including intravenous immunoglobulin, plasmapheresis, rituximab, and valganciclovir.2
Early clinical recognition of the signs and symptoms of DRESS syndrome in the setting of a new medication can decrease morbidity and mortality. Although DRESS syndrome in pediatric patients presents with many similar clinical features as in adults, it may be a greater diagnostic challenge. As in adult cases, timely administration of systemic corticosteroids and tapering based on clinical signs and symptoms can lead to resolution of the hypersensitivity syndrome.
- Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. results from the prospective RegiSCAR study. Br J Dermatol. 2013;169:1071-1080.
- Fernando SL. Drug-reaction eosinophilia and systemic symptoms and drug-induced hypersensitivity syndrome. Australas J Dermatol. 2014;55:15-23.
- Picard D, Janela B, Descamps V, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): a multiorgan antiviral T cell response. Sci Transl Med. 2010;2:46ra62.
- Sasidharanpillai S, Sabitha S, Riyaz N, et al. Drug reaction with eosinophilia and systemic symptoms in children: a prospective study. Pediatr Dermatol. 2016;33:E162-E165.
- Aouam K, Chaabane A, Toumi A, et al. Drug rash with eosinophilia and systemic symptoms (DRESS) probably induced by cefotaxime: a report of two cases. Clin Med Res. 2012;10:32-35.
- Guleria VS, Dhillon M, Gill S, et al. Ceftriaxone induced drug rash with eosinophilia and systemic symptoms. J Res Pharm Pract. 2014;3:72-74.
- Kardaun SH, Sidoroff A, Valeyrie-Allanore L, et al. Variability in the clinical pattern of cutaneous side-effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br J Dermatol. 2007;156:609-611.
- Lin IC, Yang HC, Strong C, et al. Liver injury in patients with DRESS: a clinical study of 72 cases. J Am Acad Dermatol. 2015;72:984-991.
- Ang CC, Wang YS, Yoosuff EL, et al. Retrospective analysis of drug-induced hypersensitivity syndrome: a study of 27 patients. J Am Acad Dermatol. 2010;63:219-227.
- Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. results from the prospective RegiSCAR study. Br J Dermatol. 2013;169:1071-1080.
- Fernando SL. Drug-reaction eosinophilia and systemic symptoms and drug-induced hypersensitivity syndrome. Australas J Dermatol. 2014;55:15-23.
- Picard D, Janela B, Descamps V, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): a multiorgan antiviral T cell response. Sci Transl Med. 2010;2:46ra62.
- Sasidharanpillai S, Sabitha S, Riyaz N, et al. Drug reaction with eosinophilia and systemic symptoms in children: a prospective study. Pediatr Dermatol. 2016;33:E162-E165.
- Aouam K, Chaabane A, Toumi A, et al. Drug rash with eosinophilia and systemic symptoms (DRESS) probably induced by cefotaxime: a report of two cases. Clin Med Res. 2012;10:32-35.
- Guleria VS, Dhillon M, Gill S, et al. Ceftriaxone induced drug rash with eosinophilia and systemic symptoms. J Res Pharm Pract. 2014;3:72-74.
- Kardaun SH, Sidoroff A, Valeyrie-Allanore L, et al. Variability in the clinical pattern of cutaneous side-effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br J Dermatol. 2007;156:609-611.
- Lin IC, Yang HC, Strong C, et al. Liver injury in patients with DRESS: a clinical study of 72 cases. J Am Acad Dermatol. 2015;72:984-991.
- Ang CC, Wang YS, Yoosuff EL, et al. Retrospective analysis of drug-induced hypersensitivity syndrome: a study of 27 patients. J Am Acad Dermatol. 2010;63:219-227.
Practice Points
- Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome shares many clinical features with viral exanthems and may be difficult to diagnose in the setting of atopic dermatitis in which children may have baseline eosinophilia from an atopic diathesis.
- Early clinical recognition of the signs and symptoms of DRESS syndrome in the setting of a new medication can decrease morbidity and mortality.