User login
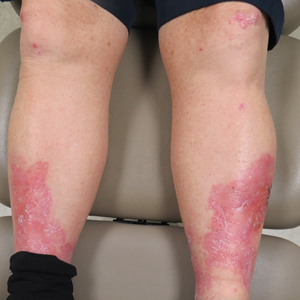
Erythema, Blisters, and Scars on the Elbows, Knees, and Legs
The Diagnosis: Epidermolysis Bullosa Acquisita
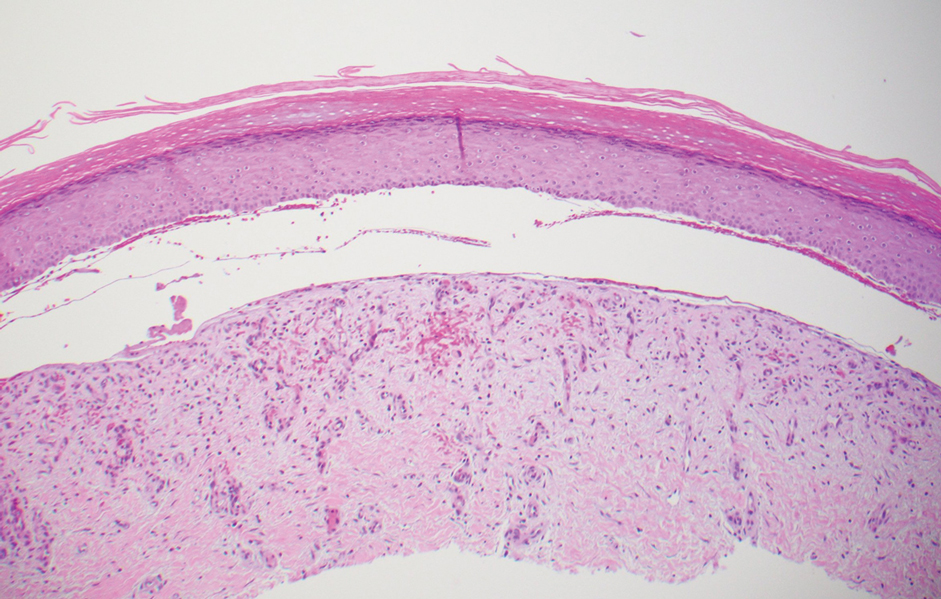
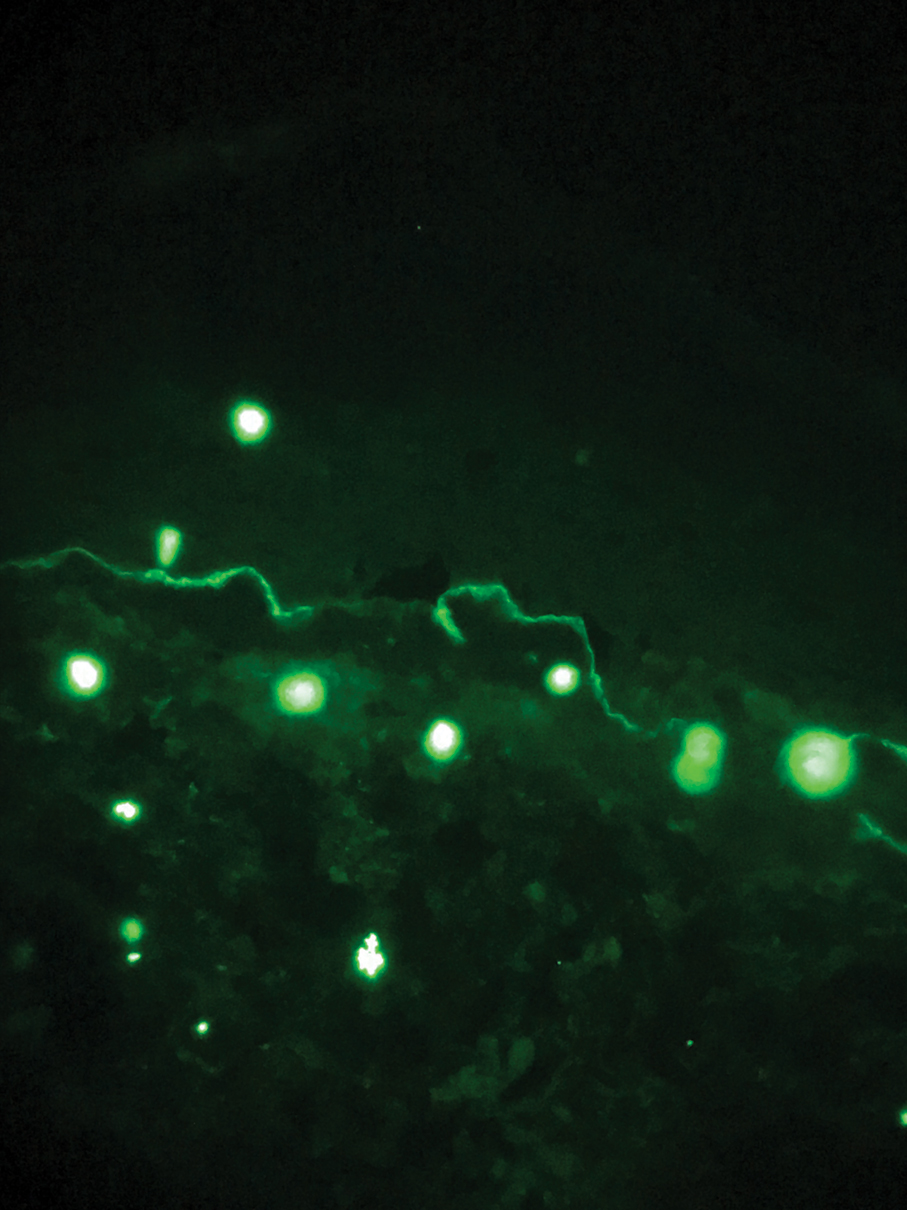
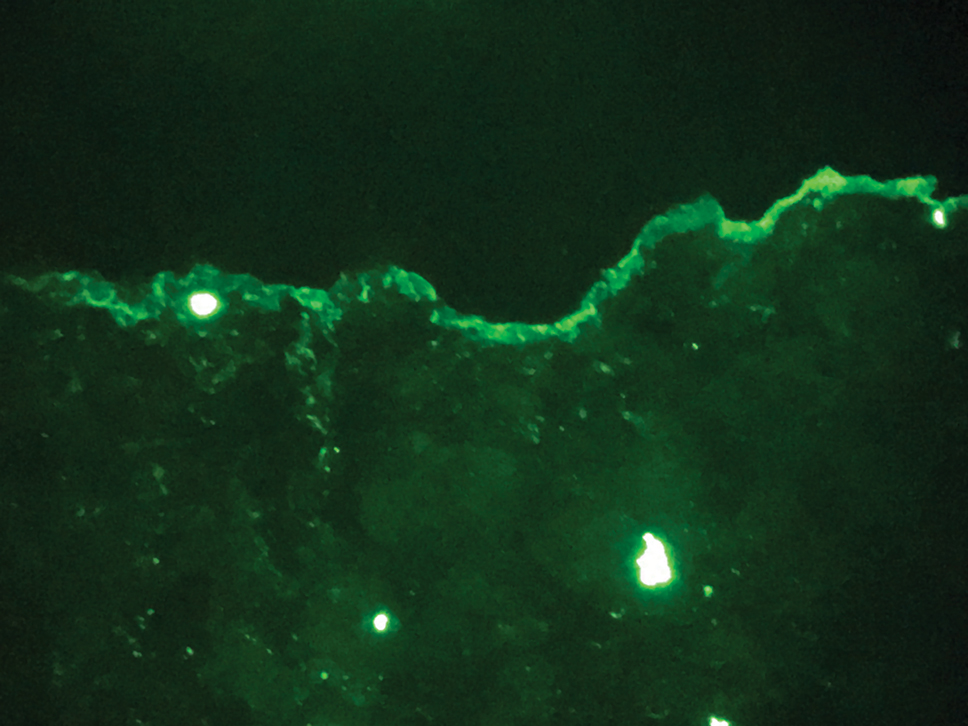
The diagnosis of epidermolysis bullosa acquisita (EBA) was made based on the clinical and pathologic findings. A blistering disorder that resolves with milia is characteristic of EBA. Hematoxylin and eosin staining demonstrated a pauci-inflammatory separation between the epidermis and dermis (Figure 1). Direct immunofluorescence studies showed linear IgG deposition along the basement membrane zone while C3 was negative (Figure 2). Salt-split skin was essential, as it revealed IgG deposition to the floor of the split (Figure 3), a pattern seen in EBA and not bullous pemphigoid (BP).1
Epidermolysis bullosa acquisita is an acquired autoimmune bullous disorder that results from antibodies to type VII collagen, an anchoring fibril that attaches the lamina densa to the dermis. The epidemiology and etiology of the trigger that leads to antibody production are not well known, but an association between EBA and inflammatory bowel disease has been described.2 Although this disease may present in childhood, EBA most commonly is a disorder seen in adults and the elderly. A classic noninflammatory mechanobullous form as well as an inflammatory BP-like form are the most commonly encountered presentations. Light microscopy demonstrates subepidermal cleavage without acantholysis. In the inflammatory BP-like subtype, an inflammatory infiltrate may be present. Direct immunofluorescence is remarkable for a linear band of IgG deposits along the basement membrane zone, with or without C3 deposition in a similar pattern.1
Bullous pemphigoid is within the differential of EBA. It can be difficult to differentiate clinically, especially when a patient has the BP-like variant of EBA because, as the name implies, it mimics BP. Patients with BP often will report a pruritic patch that will then develop into an urticarial plaque. Scarring and milia rarely are seen in BP but can be observed in the multiple presentations of EBA. Hematoxylin and eosin staining and direct immunofluorescence may be almost identical, and differentiating between the 2 disorders can be a challenge. Immunodeposition in EBA occurs in a U-shaped, serrated pattern, while the pattern in BP is N-shaped and serrated.3 Although the U-shaped, serrated pattern is relatively specific, it is not always easy to interpret and requires a high-quality biopsy specimen, which can be difficult to discern with certainty in suboptimal preparations. Another way to differentiate between the 2 entities is to utilize the salt-split skin technique, as performed in our patient. With salt-split skin, the biopsy is placed into a solution of 1 mol/L sodium chloride and incubated at 4 °C (39 °F) for 18 to 24 hours. A blister is then produced at the level of the lamina lucida, which allows for the staining of immunoreactants to occur either above or below that split (commonly referred to as staining on the roof or floor of the blister cavity). With EBA, there is immunoreactant deposition on the floor of the blister, while the opposite occurs in BP.4
Epidermolysis bullosa simplex is the most common type of epidermolysis bullosa, with keratin genes KRT5 and KRT14 as frequent mutations. Patients develop blisters, vesicles, bullae, and milia on traumatized areas of the body such as the hands, elbows, knees, and feet. This disease presents early in childhood. Histology exhibits a cell-poor subepidermal blister.5 With porphyria cutanea tarda, reduced activity of uroporphyrinogen decarboxylase, a major enzyme in the heme synthesis pathway, leads to blisters with erosions and milia on sun-exposed areas of the body. Histologic evaluation reveals a subepidermal pauci-inflammatory vesicle with festooning of the dermal papillae and amphophilic basement membrane within the epidermis. Direct immunofluorescence of porphyria cutanea tarda demonstrates IgM and C3 in the vessels.6 Sweet syndrome is a neutrophilic dermatosis that presents as erythematous, edematous, hot, and tender plaques along with fever and leukocytosis. It is associated with myeloproliferative disorders. Biopsy demonstrates papillary dermal edema along with diffuse neutrophilic infiltrate.7
Numerous medications have been recommended for the treatment of EBA, ranging from steroids to steroid-sparing drugs such as colchicine and dapsone.8,9 Our patient was educated on physical precautions and was started on dapsone alone due to comorbid diabetes mellitus and renal disease. Within a few weeks of initiating dapsone, he observed a reduction in erythema, and within months he experienced a decrease in blister eruption frequency.
- Vorobyev A, Ludwig RJ, Schmidt E. Clinical features and diagnosis of epidermolysis bullosa acquisita. Expert Rev Clin Immunol. 2017;13:157-169.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-230.
- Vodegel RM, Jonkman MF, Pas HH, et al. U-serrated immunodeposition pattern differentiates type VII collagen targeting bullous diseases from other subepidermal bullous autoimmune diseases. Br J Dermatol. 2004;151:112-118.
- Gardner KM, Crawford RI. Distinguishing epidermolysis bullosa acquisita from bullous pemphigoid without direct immunofluorescence. J Cutan Med Surg. 2018;22:22-24.
- Sprecher E. Epidermolysis bullosa simplex. Dermatol Clin. 2010;28:23-32.
- Maynard B, Peters MS. Histologic and immunofluorescence study of cutaneous porphyrias. J Cutan Pathol. 1992;19:40-47.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018:79:987-1006.
- Kirtschig G, Murrell D, Wojnarowska F, et al. Interventions for mucous membrane pemphigoid and epidermolysis bullosa acquisita. Cochrane Database Syst Rev. 2003;1:CD004056
- Gürcan HM, Ahmed AR. Current concepts in the treatment of epidermolysis bullosa acquisita. Expert Opin Pharmacother. 2011;12:1259-1268.
The Diagnosis: Epidermolysis Bullosa Acquisita
The diagnosis of epidermolysis bullosa acquisita (EBA) was made based on the clinical and pathologic findings. A blistering disorder that resolves with milia is characteristic of EBA. Hematoxylin and eosin staining demonstrated a pauci-inflammatory separation between the epidermis and dermis (Figure 1). Direct immunofluorescence studies showed linear IgG deposition along the basement membrane zone while C3 was negative (Figure 2). Salt-split skin was essential, as it revealed IgG deposition to the floor of the split (Figure 3), a pattern seen in EBA and not bullous pemphigoid (BP).1
Epidermolysis bullosa acquisita is an acquired autoimmune bullous disorder that results from antibodies to type VII collagen, an anchoring fibril that attaches the lamina densa to the dermis. The epidemiology and etiology of the trigger that leads to antibody production are not well known, but an association between EBA and inflammatory bowel disease has been described.2 Although this disease may present in childhood, EBA most commonly is a disorder seen in adults and the elderly. A classic noninflammatory mechanobullous form as well as an inflammatory BP-like form are the most commonly encountered presentations. Light microscopy demonstrates subepidermal cleavage without acantholysis. In the inflammatory BP-like subtype, an inflammatory infiltrate may be present. Direct immunofluorescence is remarkable for a linear band of IgG deposits along the basement membrane zone, with or without C3 deposition in a similar pattern.1
Bullous pemphigoid is within the differential of EBA. It can be difficult to differentiate clinically, especially when a patient has the BP-like variant of EBA because, as the name implies, it mimics BP. Patients with BP often will report a pruritic patch that will then develop into an urticarial plaque. Scarring and milia rarely are seen in BP but can be observed in the multiple presentations of EBA. Hematoxylin and eosin staining and direct immunofluorescence may be almost identical, and differentiating between the 2 disorders can be a challenge. Immunodeposition in EBA occurs in a U-shaped, serrated pattern, while the pattern in BP is N-shaped and serrated.3 Although the U-shaped, serrated pattern is relatively specific, it is not always easy to interpret and requires a high-quality biopsy specimen, which can be difficult to discern with certainty in suboptimal preparations. Another way to differentiate between the 2 entities is to utilize the salt-split skin technique, as performed in our patient. With salt-split skin, the biopsy is placed into a solution of 1 mol/L sodium chloride and incubated at 4 °C (39 °F) for 18 to 24 hours. A blister is then produced at the level of the lamina lucida, which allows for the staining of immunoreactants to occur either above or below that split (commonly referred to as staining on the roof or floor of the blister cavity). With EBA, there is immunoreactant deposition on the floor of the blister, while the opposite occurs in BP.4
Epidermolysis bullosa simplex is the most common type of epidermolysis bullosa, with keratin genes KRT5 and KRT14 as frequent mutations. Patients develop blisters, vesicles, bullae, and milia on traumatized areas of the body such as the hands, elbows, knees, and feet. This disease presents early in childhood. Histology exhibits a cell-poor subepidermal blister.5 With porphyria cutanea tarda, reduced activity of uroporphyrinogen decarboxylase, a major enzyme in the heme synthesis pathway, leads to blisters with erosions and milia on sun-exposed areas of the body. Histologic evaluation reveals a subepidermal pauci-inflammatory vesicle with festooning of the dermal papillae and amphophilic basement membrane within the epidermis. Direct immunofluorescence of porphyria cutanea tarda demonstrates IgM and C3 in the vessels.6 Sweet syndrome is a neutrophilic dermatosis that presents as erythematous, edematous, hot, and tender plaques along with fever and leukocytosis. It is associated with myeloproliferative disorders. Biopsy demonstrates papillary dermal edema along with diffuse neutrophilic infiltrate.7
Numerous medications have been recommended for the treatment of EBA, ranging from steroids to steroid-sparing drugs such as colchicine and dapsone.8,9 Our patient was educated on physical precautions and was started on dapsone alone due to comorbid diabetes mellitus and renal disease. Within a few weeks of initiating dapsone, he observed a reduction in erythema, and within months he experienced a decrease in blister eruption frequency.
The Diagnosis: Epidermolysis Bullosa Acquisita
The diagnosis of epidermolysis bullosa acquisita (EBA) was made based on the clinical and pathologic findings. A blistering disorder that resolves with milia is characteristic of EBA. Hematoxylin and eosin staining demonstrated a pauci-inflammatory separation between the epidermis and dermis (Figure 1). Direct immunofluorescence studies showed linear IgG deposition along the basement membrane zone while C3 was negative (Figure 2). Salt-split skin was essential, as it revealed IgG deposition to the floor of the split (Figure 3), a pattern seen in EBA and not bullous pemphigoid (BP).1
Epidermolysis bullosa acquisita is an acquired autoimmune bullous disorder that results from antibodies to type VII collagen, an anchoring fibril that attaches the lamina densa to the dermis. The epidemiology and etiology of the trigger that leads to antibody production are not well known, but an association between EBA and inflammatory bowel disease has been described.2 Although this disease may present in childhood, EBA most commonly is a disorder seen in adults and the elderly. A classic noninflammatory mechanobullous form as well as an inflammatory BP-like form are the most commonly encountered presentations. Light microscopy demonstrates subepidermal cleavage without acantholysis. In the inflammatory BP-like subtype, an inflammatory infiltrate may be present. Direct immunofluorescence is remarkable for a linear band of IgG deposits along the basement membrane zone, with or without C3 deposition in a similar pattern.1
Bullous pemphigoid is within the differential of EBA. It can be difficult to differentiate clinically, especially when a patient has the BP-like variant of EBA because, as the name implies, it mimics BP. Patients with BP often will report a pruritic patch that will then develop into an urticarial plaque. Scarring and milia rarely are seen in BP but can be observed in the multiple presentations of EBA. Hematoxylin and eosin staining and direct immunofluorescence may be almost identical, and differentiating between the 2 disorders can be a challenge. Immunodeposition in EBA occurs in a U-shaped, serrated pattern, while the pattern in BP is N-shaped and serrated.3 Although the U-shaped, serrated pattern is relatively specific, it is not always easy to interpret and requires a high-quality biopsy specimen, which can be difficult to discern with certainty in suboptimal preparations. Another way to differentiate between the 2 entities is to utilize the salt-split skin technique, as performed in our patient. With salt-split skin, the biopsy is placed into a solution of 1 mol/L sodium chloride and incubated at 4 °C (39 °F) for 18 to 24 hours. A blister is then produced at the level of the lamina lucida, which allows for the staining of immunoreactants to occur either above or below that split (commonly referred to as staining on the roof or floor of the blister cavity). With EBA, there is immunoreactant deposition on the floor of the blister, while the opposite occurs in BP.4
Epidermolysis bullosa simplex is the most common type of epidermolysis bullosa, with keratin genes KRT5 and KRT14 as frequent mutations. Patients develop blisters, vesicles, bullae, and milia on traumatized areas of the body such as the hands, elbows, knees, and feet. This disease presents early in childhood. Histology exhibits a cell-poor subepidermal blister.5 With porphyria cutanea tarda, reduced activity of uroporphyrinogen decarboxylase, a major enzyme in the heme synthesis pathway, leads to blisters with erosions and milia on sun-exposed areas of the body. Histologic evaluation reveals a subepidermal pauci-inflammatory vesicle with festooning of the dermal papillae and amphophilic basement membrane within the epidermis. Direct immunofluorescence of porphyria cutanea tarda demonstrates IgM and C3 in the vessels.6 Sweet syndrome is a neutrophilic dermatosis that presents as erythematous, edematous, hot, and tender plaques along with fever and leukocytosis. It is associated with myeloproliferative disorders. Biopsy demonstrates papillary dermal edema along with diffuse neutrophilic infiltrate.7
Numerous medications have been recommended for the treatment of EBA, ranging from steroids to steroid-sparing drugs such as colchicine and dapsone.8,9 Our patient was educated on physical precautions and was started on dapsone alone due to comorbid diabetes mellitus and renal disease. Within a few weeks of initiating dapsone, he observed a reduction in erythema, and within months he experienced a decrease in blister eruption frequency.
- Vorobyev A, Ludwig RJ, Schmidt E. Clinical features and diagnosis of epidermolysis bullosa acquisita. Expert Rev Clin Immunol. 2017;13:157-169.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-230.
- Vodegel RM, Jonkman MF, Pas HH, et al. U-serrated immunodeposition pattern differentiates type VII collagen targeting bullous diseases from other subepidermal bullous autoimmune diseases. Br J Dermatol. 2004;151:112-118.
- Gardner KM, Crawford RI. Distinguishing epidermolysis bullosa acquisita from bullous pemphigoid without direct immunofluorescence. J Cutan Med Surg. 2018;22:22-24.
- Sprecher E. Epidermolysis bullosa simplex. Dermatol Clin. 2010;28:23-32.
- Maynard B, Peters MS. Histologic and immunofluorescence study of cutaneous porphyrias. J Cutan Pathol. 1992;19:40-47.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018:79:987-1006.
- Kirtschig G, Murrell D, Wojnarowska F, et al. Interventions for mucous membrane pemphigoid and epidermolysis bullosa acquisita. Cochrane Database Syst Rev. 2003;1:CD004056
- Gürcan HM, Ahmed AR. Current concepts in the treatment of epidermolysis bullosa acquisita. Expert Opin Pharmacother. 2011;12:1259-1268.
- Vorobyev A, Ludwig RJ, Schmidt E. Clinical features and diagnosis of epidermolysis bullosa acquisita. Expert Rev Clin Immunol. 2017;13:157-169.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-230.
- Vodegel RM, Jonkman MF, Pas HH, et al. U-serrated immunodeposition pattern differentiates type VII collagen targeting bullous diseases from other subepidermal bullous autoimmune diseases. Br J Dermatol. 2004;151:112-118.
- Gardner KM, Crawford RI. Distinguishing epidermolysis bullosa acquisita from bullous pemphigoid without direct immunofluorescence. J Cutan Med Surg. 2018;22:22-24.
- Sprecher E. Epidermolysis bullosa simplex. Dermatol Clin. 2010;28:23-32.
- Maynard B, Peters MS. Histologic and immunofluorescence study of cutaneous porphyrias. J Cutan Pathol. 1992;19:40-47.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018:79:987-1006.
- Kirtschig G, Murrell D, Wojnarowska F, et al. Interventions for mucous membrane pemphigoid and epidermolysis bullosa acquisita. Cochrane Database Syst Rev. 2003;1:CD004056
- Gürcan HM, Ahmed AR. Current concepts in the treatment of epidermolysis bullosa acquisita. Expert Opin Pharmacother. 2011;12:1259-1268.
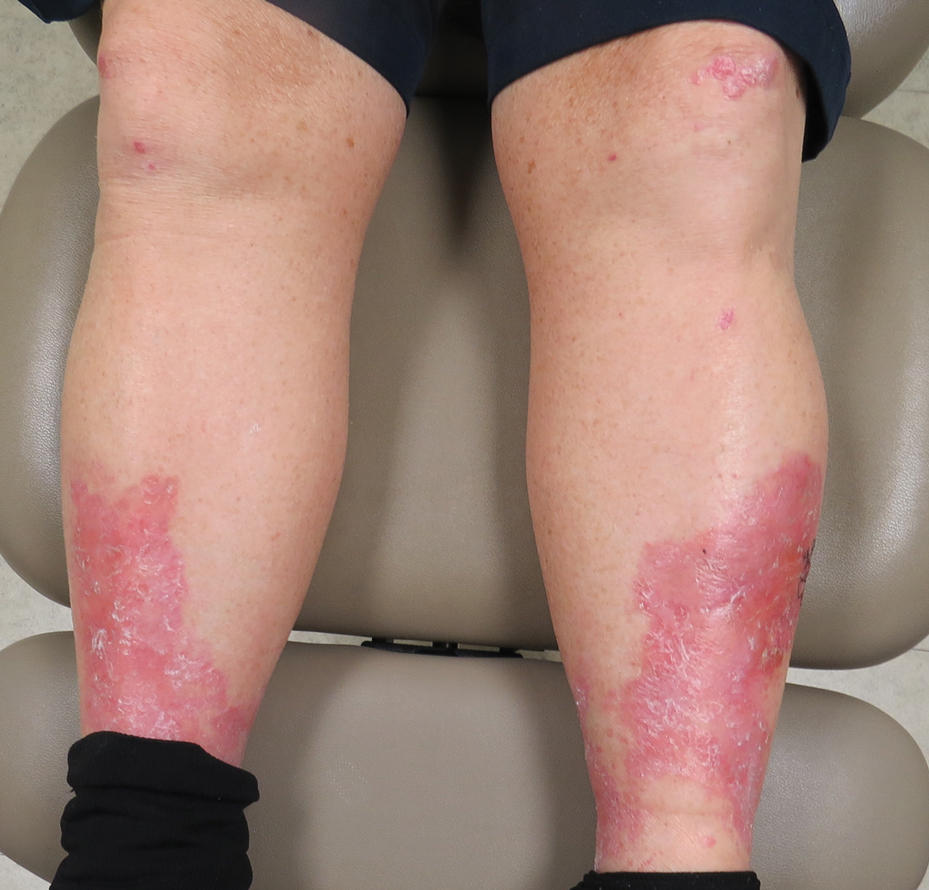
A 69-year-old man presented with an asymptomatic rash on the extensor surfaces of 2 years' duration. He reported recurrent blisters that would then scar over. The lesions did not occur in relation to any known trauma. The patient's medical history revealed dialysis-dependent end-stage renal disease secondary to type 2 diabetes mellitus. His medications were noncontributory, and there was no family history of blistering disorders. He had tried triamcinolone cream without any improvement. Physical examination was remarkable for erythematous blisters and bullae with scales and milia on the elbows, knees, and lower legs. The oral mucosa was unremarkable. Shave biopsies of the skin for direct immunofluorescence and salt-split skin studies were obtained.