User login
Squamoid Eccrine Ductal Carcinoma
Squamoid eccrine ductal carcinoma (SEDC) is an aggressive underrecognized cutaneous malignancy of unknown etiology.1 It is most likely to occur in sun-exposed areas of the body, most commonly the head and neck. Risk factors include male sex, increased age, and chronic immunosuppression.1-4 Current reports suggest that SEDC is likely a high-grade subtype of squamous cell carcinoma (SCC) with a high risk for local recurrence (25%) and metastasis (13%).1,3,5,6 There are as few as 56 cases of SEDC reported in the literature; however, the number of cases may be closer to 100 due to SEDC being classified as either adenosquamous carcinoma of the skin or ductal eccrine carcinoma with squamous differentiation.1
Clinically, SEDC mimics keratinocyte carcinomas. Histologically, SEDC is biphasic, with a superficial portion resembling well-differentiated SCC and a deeply invasive portion having infiltrative irregular cords with ductal differentiation. Perineural invasion (PNI) frequently is present. Multiple connections to the overlying epidermis also can be seen, serving as a subtle clue to the diagnosis on broad superficial specimens.1-3 Due to superficial sampling, approximately 50% of reported cases are misdiagnosed as SCC during the initial biopsy.4 The diagnosis of SEDC often is made during complete excision when deeper tissue is sampled. Establishing an accurate diagnosis is important given the more aggressive nature of SEDC compared with SCC and its proclivity for PNI.1,3,6 The purpose of this review is to increase awareness of this underrecognized entity and describe the histologic findings that help distinguish SEDC from SCC.
Patient Chart Review
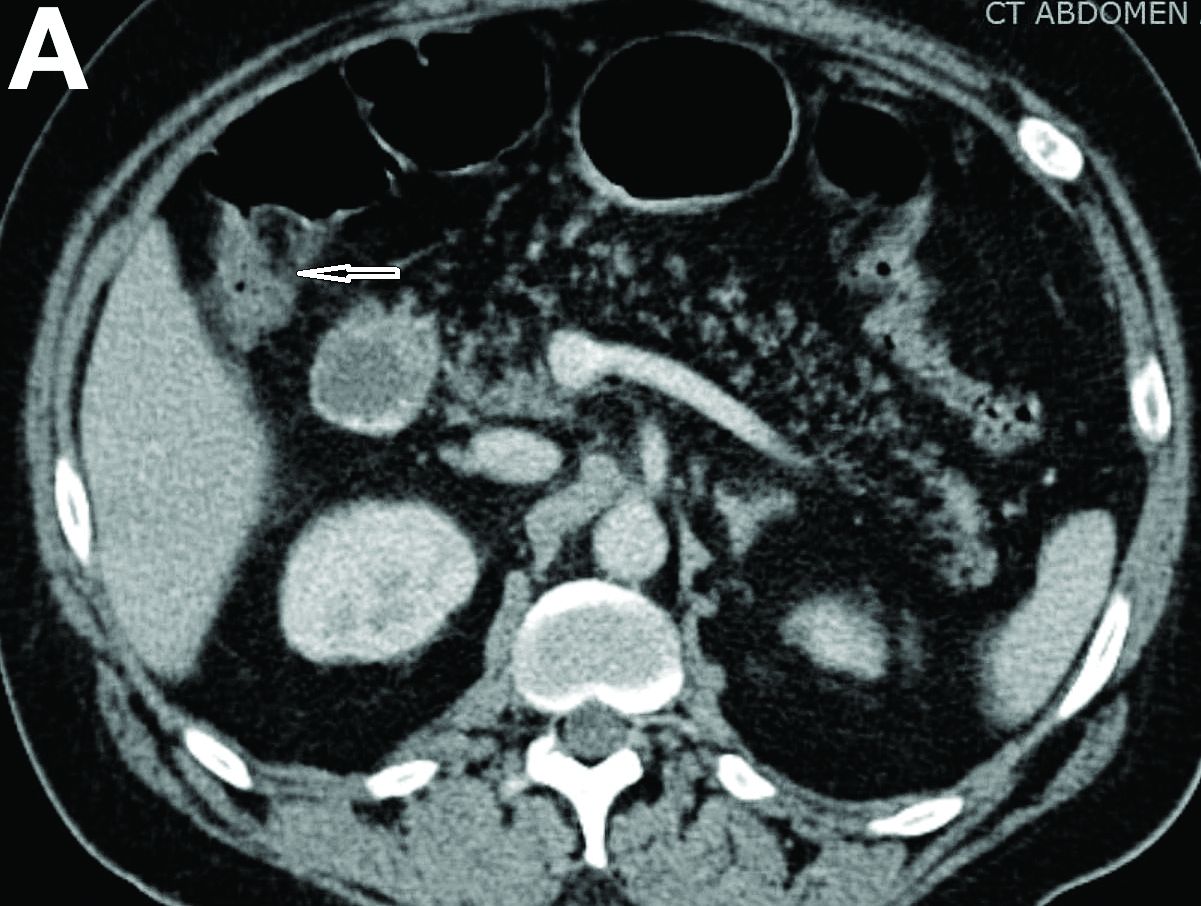
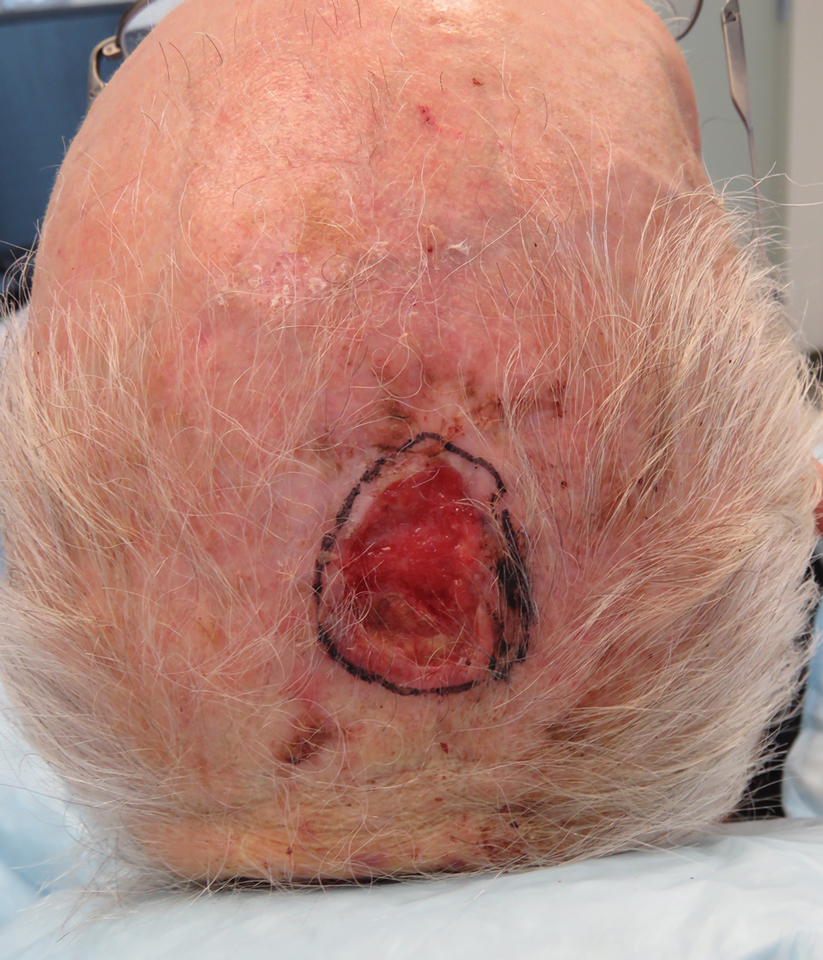
We reviewed chart notes as well as frozen and formalin-fixed paraffin-embedded tissue sections from all 5 patients diagnosed with SEDC at a single institution between November 2018 and May 2020. The mean age of patients was 81 years, and 4 were male. Four of the patients presented for MMS with a preoperative diagnosis of SCC per the original biopsy results. Only 1 patient had a preoperative diagnosis of SEDC. The details of each case are recorded in the Table. All tumors were greater than 2 cm in diameter on initial presentation, were located on the head, and clinically resembled keratinocyte carcinoma with either a nodular or plaquelike appearance (Figure 1).
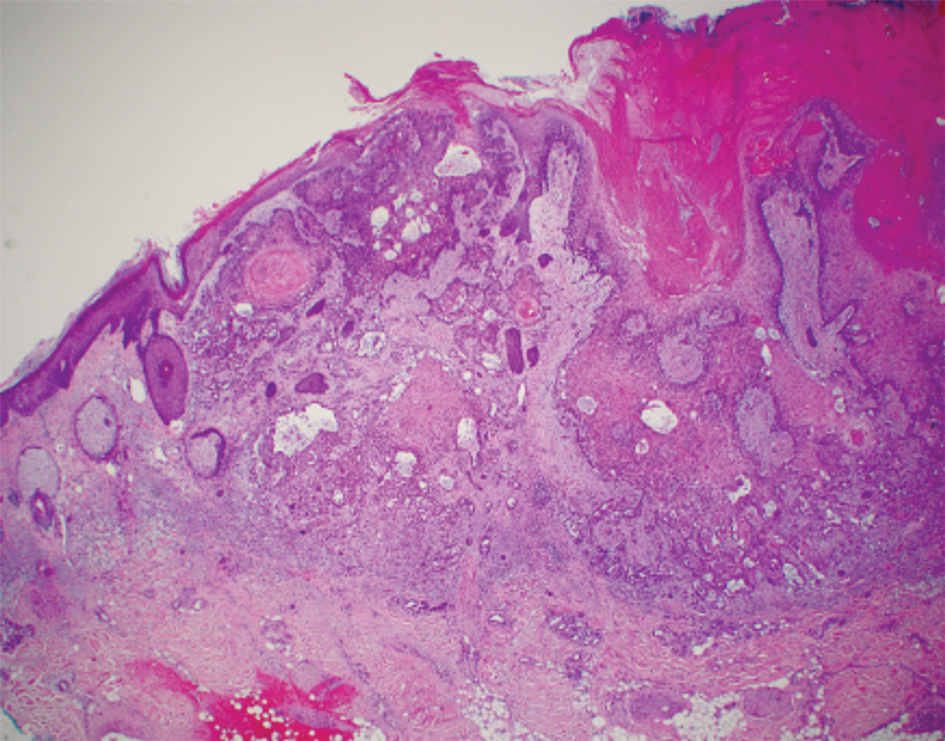
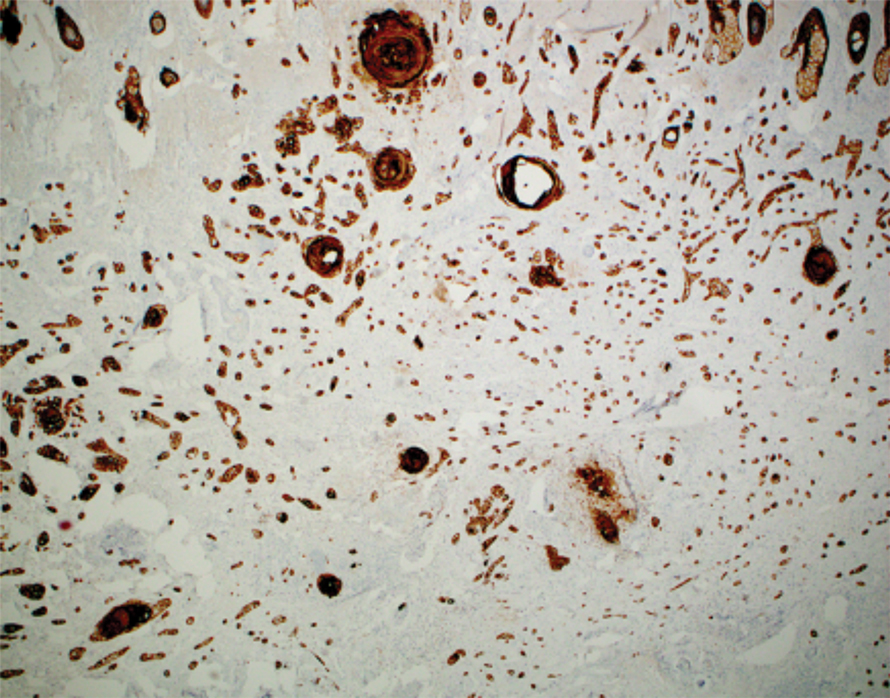
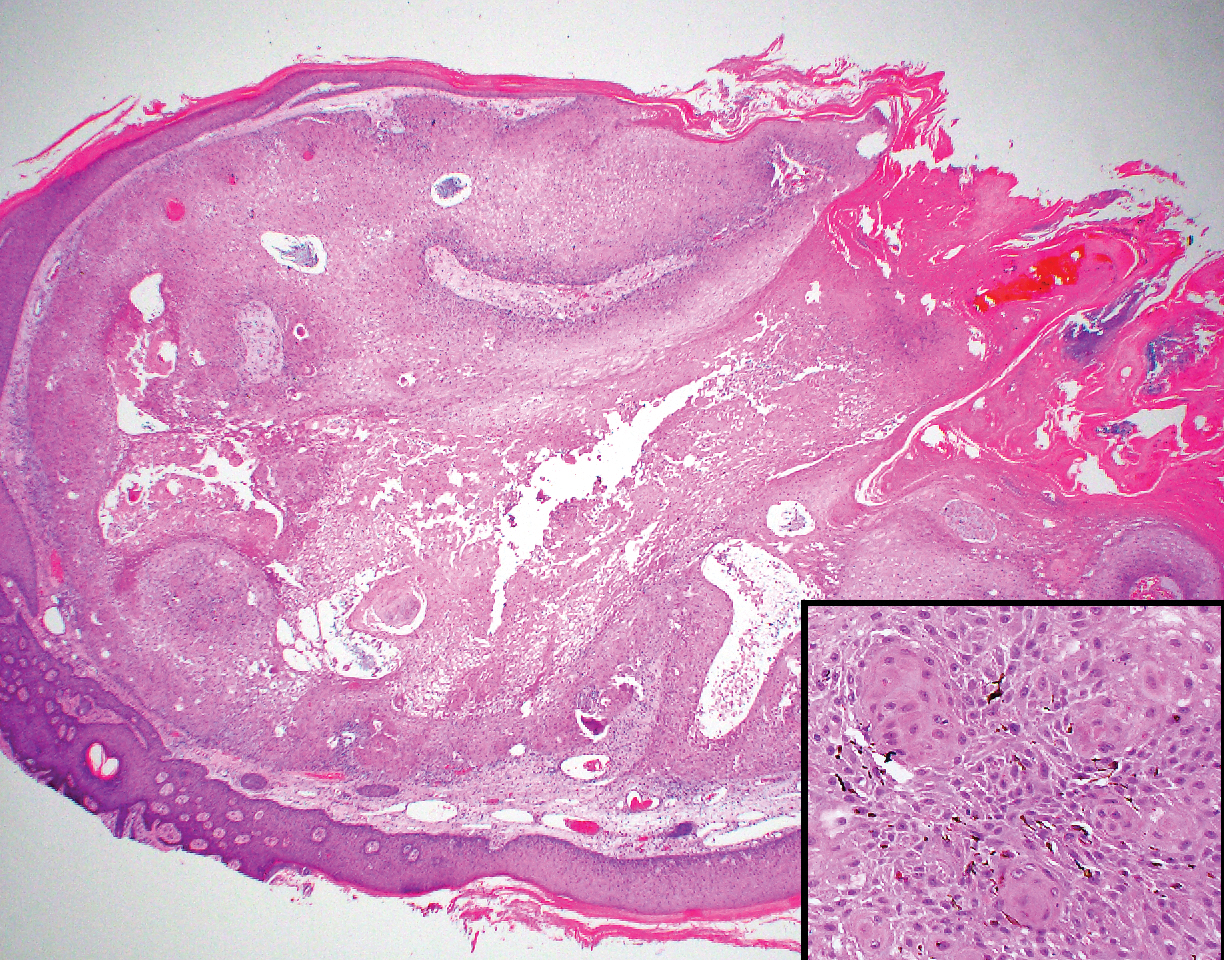
Intraoperative histologic examination of the excised tissue revealed a biphasic pattern consisting of superficial SCC features overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation in all 5 patients (Figures 2–4). Immunohistochemical staining with cytokeratin AE1/AE3 revealed thin strands of carcinoma in the mid to deeper dermis with squamous differentiation and eccrine ductal differentiation (Figure 5), thus confirming the diagnosis in all 5 patients.


The median depth of tumor invasion was 4.1 mm (range, 2.2–5.45 mm). Ulceration was seen in 3 of the patients, and PNI of large-caliber nerves was observed in all 5 patients. A connection with the overlying epidermis was present in all 5 patients. All 5 patients required more than 1 Mohs stage for complete tumor clearance (Table).
In 4 of the patients, nodal imaging performed at the time of diagnosis revealed no evidence of metastasis. Two patients received adjuvant radiation therapy, and none demonstrated evidence of recurrence. The mean follow-up time was 11 months (range, 6.5–18 months) for the 4 cases with available follow-up data (Table).
Literature Review
A PubMed review of the literature using the search term squamoid eccrine ductal carcinoma resulted in 28 articles, 19 of which were included in the review based on inclusion criteria (original articles available in English, in full text, and pertained to SEDC). Our review yielded 56 cases of SEDC.1-19 The mean age of patients with SEDC was 72 years. The number of male and female cases was 52% (29/56) and 48% (27/56), respectively. The most common location of SEDC was on the head or neck (71% [40/56]), followed by the extremities (19% [11/56]). Immunosuppression was noted in 9% (5/56) of cases. Wide local excision was the most commonly employed treatment modality (91% [51/56]), with MMS being used in 4 patients (7%). Adjuvant radiation was reported in 5% (3/56) of cases. Perineural invasion was reported in 34% (19/56) of cases. Recurrence was seen in 23% (13/56) of cases, with a mean time to recurrence of 10.4 months. Metastasis to regional lymph nodes was observed in 13% (7/56) of cases, with 7% (4/56) of those cases having distant metastases.
Comment
Squamoid eccrine ductal carcinoma was successfully treated with MMS in all 5 of the patients we reviewed. Recognition of a distinct biphasic pattern consisting of squamous differentiation superficially with epidermal connection overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation should lead to consideration of this diagnosis. A thorough inspection for PNI also should be performed, as this finding was present in all of 5 cases and in 34% of reported cases in our literature review.
The differential diagnosis for SEDC includes SCC, metastatic adenocarcinoma with squamoid features, and eccrine tumors, including eccrine poroma, microcystic adnexal carcinoma (MAC), and porocarcinoma with squamous differentiation. The combination of histologic features with the immunoexpression profile of carcinoembryonic antigen (CEA), epithelial membrane antigen (EMA), cytokeratin (CK) 5/6, and p63 can effectively exclude the other entities in the differential and confirm the diagnosis of SEDC.1,3,4 While the diagnosis of SEDC relies on the specific histologic features of multiple surface attachments and superficial squamoid changes with deep ductular elements, immunohistochemistry can nonetheless be adjunctive in difficult cases. Positive immunohistochemical staining for CEA and EMA can help to highlight and delineate true glandular elements, whereas CK5/6 highlights the overall contour of the tumor, displaying more clearly the multiple epidermal attachments and the subtle infiltrative nature of the deeper components of invasive cords and ducts. In addition, the combination of CK5/6 and p63 positivity supports the primary cutaneous nature of the lesion rather than metastatic adenocarcinoma.13,20 Other markers of eccrine secretory coils, such as CK7, CAM5.2, and S100, also are sometimes used for confirmation, some of which can aid in distinction from noneccrine sweat gland differentiation, as CK7 and CAM5.2 are negative in both luminal and basal cells of the dermal duct while being positive within the secretory coil, and S100 protein is expressed within eccrine secretory coil but negative within the apocrine sweat glands.2,4,21
The clinical findings from our chart review corroborated those reported in the literature. The mean age of SEDC in the 5 patients we reviewed was 81 years, and all cases presented on the head, consistent with the findings observed in the literature. Although 4 of our cases were male, there may not be a difference in risk based on sex as previously thought.1 Our literature review revealed an almost equivalent percentage of male and female cases, with 52% being male.
Immunosuppression has been associated with an increased risk for SEDC. Our literature review revealed that approximately 9% (5/56) of cases occurred in immunosuppressed individuals. Two of these reported cases were in the setting of underlying chronic lymphocytic leukemia, 2 in individuals with a history of organ transplant, and 1 treated with azathioprine for myasthenia gravis.2,4,10,12,13 Our chart review supported this correlation, as all 5 patients had a medical history potentially consistent with being in an immunocompromised state (Table). Notably, patient 5 represents a unique case of SEDC occurring in the setting of HIV. The patient had HIV for 33 years, with his most recent CD4+ count of 794 mm3 and HIV-1 RNA load of 35 copies/mL. Given that HIV-positive individuals may have more than a 2-fold increased risk of SCC, a greater degree of suspicion for SEDC should be maintained for these patients.22,23
The etiology of SEDC is controversial but is thought to be either an SCC arising from eccrine glands or a variant of eccrine carcinoma with extensive squamoid differentiation.4,6,13,14,17,24 While SEDC certainly appears to share the proclivity for PNI with the malignant eccrine tumor MAC, it is simultaneously quite distinct, demonstrating nuclear pleomorphism and mitotic activity, both of which are lacking in the bland nature of MACs.12,25
The exact prevalence of SEDC is difficult to ascertain because of its frequent misdiagnosis and variable nomenclature used within the literature. Most reported cases of SEDC are mistakenly diagnosed as SCC on the initial shave or punch biopsy because of superficial sampling. This also was the case in 4 of the patients we reviewed. In addition, there are reported cases of SEDC that were referred to by the investigators as cutaneous adenosquamous carcinoma (cASC), among other descriptors, such as ductal eccrine carcinoma with squamous differentiation, adnexal carcinoma with squamous and ductal differentiation, and syringoid eccrine carcinoma.26-32 While the World Health Organization classifies SEDC as a distinct variant of cASC, which is a rare variant of SCC in itself, the 2 can be differentiated. Despite the similar clinical and histologic features shared between cASC and SEDC, the neoplastic aggregates in SEDC exhibit ductal differentiation containing lumina positive for CEA and EMA.4 Overall, we favor the term squamoid eccrine ductal carcinoma, as there has recently been more uniformity for the designation of this disease entity as such.
It is unclear whether the high incidence of local recurrence (23% [13/56]) of SEDC reported in the literature is related to the treatment modality employed (ie, wide local excision) or due to the innate aggressiveness of SEDC.1,3,5 The literature has shown that MMS has lower recurrence rates than other treatments at 5-year follow-up for SCC (3.1%–5%) and eccrine carcinomas (0%–5%).33,34 Although studies assessing tumor behavior or comparing treatment modalities are limited because of the rarity and underrecognition of SEDC, MMS has been used several times for SEDC with only 1 recurrence reported.4,13,17,24 Given that all 5 of the patients we reviewed required more than 1 Mohs stage for complete tumor clearance and none demonstrated evidence of recurrence or metastasis (Table), we recommend MMS as the treatment of choice for SEDC.
Conclusion
Squamoid eccrine ductal carcinoma is a rare but likely underdiagnosed cutaneous tumor of uncertain etiology. Because of its propensity for recurrence and metastasis, excision of SEDC with complete circumferential peripheral and deep margin assessment with close follow-up is recommended.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760.
- Jacob J, Kugelman L. Squamoid eccrine ductal carcinoma. Cutis. 2018;101:378-380, 385.
- Yim S, Lee YH, Chae SW, et al. Squamoid eccrine ductal carcinoma of the ear helix. Clin Case Rep. 2019;7:1409-1411.
- Terushkin E, Leffell DJ, Futoryan T, et al. Squamoid eccrine ductal carcinoma: a case report and review of the literature. Am J Dermatopathol. 2010;32:287-292.
- Jung YH, Jo HJ, Kang MS. Squamoid eccrine ductal carcinoma of the scalp. Korean J Pathol. 2012;46:278-281.
- Saraiva MI, Vieira MA, Portocarrero LK, et al. Squamoid eccrine ductal carcinoma. An Bras Dermatol. 2016;91:799-802.
- Phan K, Kim L, Lim P, et al. A case report of temple squamoid eccrine ductal carcinoma: a diagnostic challenge beneath the tip of the iceberg. Dermatol Ther. 2020;33:E13213.
- McKissack SS, Wohltmann W, Dalton SR, et al. Squamoid eccrine ductal carcinoma: an aggressive mimicker of squamous cell carcinoma. Am J Dermatopathol. 2019;41:140-143.
- Lobo-Jardim MM, Souza BdCE, Kakizaki P, et al. Dermoscopy of squamoid eccrine ductal carcinoma: an aid for early diagnosis. An Bras Dermatol. 2018;93:893-895.
- Chan H, Howard V, Moir D, et al. Squamoid eccrine ductal carcinoma of the scalp. Aust J Dermatol. 2016;57:E117-E119.
- Wang B, Jarell AD, Bingham JL, et al. PET/CT imaging of squamoid eccrine ductal carcinoma. Clin Nucl Med. 2015;40:322-324.
- Frouin E, Vignon-Pennamen MD, Balme B, et al. Anatomoclinical study of 30 cases of sclerosing sweat duct carcinomas (microcystic adnexal carcinoma, syringomatous carcinoma and squamoid eccrine ductal carcinoma). J Eur Acad Dermatol Venereol. 2015;29:1978-1994.
- Clark S, Young A, Piatigorsky E, et al. Mohs micrographic surgery in the setting of squamoid eccrine ductal carcinoma: addressing a diagnostic and therapeutic challenge. J Clin Aesthet Dermatol. 2013;6:33-36.
- Pusiol T, Morichetti D, Zorzi MG, et al. Squamoid eccrine ductal carcinoma: inappropriate diagnosis. Dermatol Surg. 2011;37:1819-1820.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Wasserman DI, Sack J, Gonzalez-Serva A, et al. Sentinel lymph node biopsy for a squamoid eccrine carcinoma with lymphatic invasion. Dermatol Surg. 2007;33:1126-1129.
- Kim YJ, Kim AR, Yu DS. Mohs micrographic surgery for squamoid eccrine ductal carcinoma. Dermatol Surg. 2005;31:1462-1464.
- Herrero J, Monteagudo C, Jorda E, et al. Squamoid eccrine ductal carcinoma. Histopathology. 1998;32:478-480.
- Wong TY, Suster S, Mihm MC. Squamoid eccrine ductal carcinoma. Histopathology. 1997;30:288-293.
- Qureshi HS, Ormsby AH, Lee MW, et al. The diagnostic utility of p63, CK5/6, CK 7, and CK 20 in distinguishing primary cutaneous adnexal neoplasms from metastatic carcinomas. J Cutan Pathol. 2004;31:145-152.
- Dabbs DJ. Diagnostic Immunohistochemistry: Theranostic and Genomic Applications. 4th ed. Elsevier/Saunders; 2014.
- Silverberg MJ, Leyden W, Warton EM, et al. HIV infection status, immunodeficiency, and the incidence of non-melanoma skin cancer. J Natl Cancer Inst. 2013;105:350-360.
- Asgari MM, Ray GT, Quesenberry CP Jr, et al. Association of multiple primary skin cancers with human immunodeficiency virus infection, CD4 count, and viral load. JAMA Dermatol. 2017;153:892-896.
- Tolkachjov SN. Adnexal carcinomas treated with Mohs micrographic surgery: a comprehensive review. Dermatol Surg. 2017;43:1199-1207.
- Kazakov DV. Cutaneous Adnexal Tumors. Wolters Kluwer Health/ Lippincott Williams & Wilkins; 2012.
- Weidner N, Foucar E. Adenosquamous carcinoma of the skin. an aggressive mucin- and gland-forming squamous carcinoma. Arch Dermatol. 1985;121:775-779.
- Banks ER, Cooper PH. Adenosquamous carcinoma of the skin: a report of 10 cases. J Cutan Pathol. 1991;18:227-234.
- Ko CJ, Leffell DJ, McNiff JM. Adenosquamous carcinoma: a report of nine cases with p63 and cytokeratin 5/6 staining. J Cutan Pathol. 2009;36:448-452.
- Patel V, Squires SM, Liu DY, et al. Cutaneous adenosquamous carcinoma: a rare neoplasm with biphasic differentiation. Cutis. 2014;94:231-233.
- Chhibber V, Lyle S, Mahalingam M. Ductal eccrine carcinoma with squamous differentiation: apropos a case. J Cutan Pathol. 2007;34:503-507.
- Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
- Azorín D, López-Ríos F, Ballestín C, et al. Primary cutaneous adenosquamous carcinoma: a case report and review of the literature. J Cutan Pathol. 2001;28:542-545.
- Wildemore JK, Lee JB, Humphreys TR. Mohs surgery for malignant eccrine neoplasms. Dermatol Surg. 2004;30(12 pt 2):1574-1579.
- Garcia-Zuazaga J, Olbricht SM. Cutaneous squamous cell carcinoma. Adv Dermatol. 2008;24:33-57.
Squamoid eccrine ductal carcinoma (SEDC) is an aggressive underrecognized cutaneous malignancy of unknown etiology.1 It is most likely to occur in sun-exposed areas of the body, most commonly the head and neck. Risk factors include male sex, increased age, and chronic immunosuppression.1-4 Current reports suggest that SEDC is likely a high-grade subtype of squamous cell carcinoma (SCC) with a high risk for local recurrence (25%) and metastasis (13%).1,3,5,6 There are as few as 56 cases of SEDC reported in the literature; however, the number of cases may be closer to 100 due to SEDC being classified as either adenosquamous carcinoma of the skin or ductal eccrine carcinoma with squamous differentiation.1
Clinically, SEDC mimics keratinocyte carcinomas. Histologically, SEDC is biphasic, with a superficial portion resembling well-differentiated SCC and a deeply invasive portion having infiltrative irregular cords with ductal differentiation. Perineural invasion (PNI) frequently is present. Multiple connections to the overlying epidermis also can be seen, serving as a subtle clue to the diagnosis on broad superficial specimens.1-3 Due to superficial sampling, approximately 50% of reported cases are misdiagnosed as SCC during the initial biopsy.4 The diagnosis of SEDC often is made during complete excision when deeper tissue is sampled. Establishing an accurate diagnosis is important given the more aggressive nature of SEDC compared with SCC and its proclivity for PNI.1,3,6 The purpose of this review is to increase awareness of this underrecognized entity and describe the histologic findings that help distinguish SEDC from SCC.
Patient Chart Review
We reviewed chart notes as well as frozen and formalin-fixed paraffin-embedded tissue sections from all 5 patients diagnosed with SEDC at a single institution between November 2018 and May 2020. The mean age of patients was 81 years, and 4 were male. Four of the patients presented for MMS with a preoperative diagnosis of SCC per the original biopsy results. Only 1 patient had a preoperative diagnosis of SEDC. The details of each case are recorded in the Table. All tumors were greater than 2 cm in diameter on initial presentation, were located on the head, and clinically resembled keratinocyte carcinoma with either a nodular or plaquelike appearance (Figure 1).
Intraoperative histologic examination of the excised tissue revealed a biphasic pattern consisting of superficial SCC features overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation in all 5 patients (Figures 2–4). Immunohistochemical staining with cytokeratin AE1/AE3 revealed thin strands of carcinoma in the mid to deeper dermis with squamous differentiation and eccrine ductal differentiation (Figure 5), thus confirming the diagnosis in all 5 patients.
The median depth of tumor invasion was 4.1 mm (range, 2.2–5.45 mm). Ulceration was seen in 3 of the patients, and PNI of large-caliber nerves was observed in all 5 patients. A connection with the overlying epidermis was present in all 5 patients. All 5 patients required more than 1 Mohs stage for complete tumor clearance (Table).
In 4 of the patients, nodal imaging performed at the time of diagnosis revealed no evidence of metastasis. Two patients received adjuvant radiation therapy, and none demonstrated evidence of recurrence. The mean follow-up time was 11 months (range, 6.5–18 months) for the 4 cases with available follow-up data (Table).
Literature Review
A PubMed review of the literature using the search term squamoid eccrine ductal carcinoma resulted in 28 articles, 19 of which were included in the review based on inclusion criteria (original articles available in English, in full text, and pertained to SEDC). Our review yielded 56 cases of SEDC.1-19 The mean age of patients with SEDC was 72 years. The number of male and female cases was 52% (29/56) and 48% (27/56), respectively. The most common location of SEDC was on the head or neck (71% [40/56]), followed by the extremities (19% [11/56]). Immunosuppression was noted in 9% (5/56) of cases. Wide local excision was the most commonly employed treatment modality (91% [51/56]), with MMS being used in 4 patients (7%). Adjuvant radiation was reported in 5% (3/56) of cases. Perineural invasion was reported in 34% (19/56) of cases. Recurrence was seen in 23% (13/56) of cases, with a mean time to recurrence of 10.4 months. Metastasis to regional lymph nodes was observed in 13% (7/56) of cases, with 7% (4/56) of those cases having distant metastases.
Comment
Squamoid eccrine ductal carcinoma was successfully treated with MMS in all 5 of the patients we reviewed. Recognition of a distinct biphasic pattern consisting of squamous differentiation superficially with epidermal connection overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation should lead to consideration of this diagnosis. A thorough inspection for PNI also should be performed, as this finding was present in all of 5 cases and in 34% of reported cases in our literature review.
The differential diagnosis for SEDC includes SCC, metastatic adenocarcinoma with squamoid features, and eccrine tumors, including eccrine poroma, microcystic adnexal carcinoma (MAC), and porocarcinoma with squamous differentiation. The combination of histologic features with the immunoexpression profile of carcinoembryonic antigen (CEA), epithelial membrane antigen (EMA), cytokeratin (CK) 5/6, and p63 can effectively exclude the other entities in the differential and confirm the diagnosis of SEDC.1,3,4 While the diagnosis of SEDC relies on the specific histologic features of multiple surface attachments and superficial squamoid changes with deep ductular elements, immunohistochemistry can nonetheless be adjunctive in difficult cases. Positive immunohistochemical staining for CEA and EMA can help to highlight and delineate true glandular elements, whereas CK5/6 highlights the overall contour of the tumor, displaying more clearly the multiple epidermal attachments and the subtle infiltrative nature of the deeper components of invasive cords and ducts. In addition, the combination of CK5/6 and p63 positivity supports the primary cutaneous nature of the lesion rather than metastatic adenocarcinoma.13,20 Other markers of eccrine secretory coils, such as CK7, CAM5.2, and S100, also are sometimes used for confirmation, some of which can aid in distinction from noneccrine sweat gland differentiation, as CK7 and CAM5.2 are negative in both luminal and basal cells of the dermal duct while being positive within the secretory coil, and S100 protein is expressed within eccrine secretory coil but negative within the apocrine sweat glands.2,4,21
The clinical findings from our chart review corroborated those reported in the literature. The mean age of SEDC in the 5 patients we reviewed was 81 years, and all cases presented on the head, consistent with the findings observed in the literature. Although 4 of our cases were male, there may not be a difference in risk based on sex as previously thought.1 Our literature review revealed an almost equivalent percentage of male and female cases, with 52% being male.
Immunosuppression has been associated with an increased risk for SEDC. Our literature review revealed that approximately 9% (5/56) of cases occurred in immunosuppressed individuals. Two of these reported cases were in the setting of underlying chronic lymphocytic leukemia, 2 in individuals with a history of organ transplant, and 1 treated with azathioprine for myasthenia gravis.2,4,10,12,13 Our chart review supported this correlation, as all 5 patients had a medical history potentially consistent with being in an immunocompromised state (Table). Notably, patient 5 represents a unique case of SEDC occurring in the setting of HIV. The patient had HIV for 33 years, with his most recent CD4+ count of 794 mm3 and HIV-1 RNA load of 35 copies/mL. Given that HIV-positive individuals may have more than a 2-fold increased risk of SCC, a greater degree of suspicion for SEDC should be maintained for these patients.22,23
The etiology of SEDC is controversial but is thought to be either an SCC arising from eccrine glands or a variant of eccrine carcinoma with extensive squamoid differentiation.4,6,13,14,17,24 While SEDC certainly appears to share the proclivity for PNI with the malignant eccrine tumor MAC, it is simultaneously quite distinct, demonstrating nuclear pleomorphism and mitotic activity, both of which are lacking in the bland nature of MACs.12,25
The exact prevalence of SEDC is difficult to ascertain because of its frequent misdiagnosis and variable nomenclature used within the literature. Most reported cases of SEDC are mistakenly diagnosed as SCC on the initial shave or punch biopsy because of superficial sampling. This also was the case in 4 of the patients we reviewed. In addition, there are reported cases of SEDC that were referred to by the investigators as cutaneous adenosquamous carcinoma (cASC), among other descriptors, such as ductal eccrine carcinoma with squamous differentiation, adnexal carcinoma with squamous and ductal differentiation, and syringoid eccrine carcinoma.26-32 While the World Health Organization classifies SEDC as a distinct variant of cASC, which is a rare variant of SCC in itself, the 2 can be differentiated. Despite the similar clinical and histologic features shared between cASC and SEDC, the neoplastic aggregates in SEDC exhibit ductal differentiation containing lumina positive for CEA and EMA.4 Overall, we favor the term squamoid eccrine ductal carcinoma, as there has recently been more uniformity for the designation of this disease entity as such.
It is unclear whether the high incidence of local recurrence (23% [13/56]) of SEDC reported in the literature is related to the treatment modality employed (ie, wide local excision) or due to the innate aggressiveness of SEDC.1,3,5 The literature has shown that MMS has lower recurrence rates than other treatments at 5-year follow-up for SCC (3.1%–5%) and eccrine carcinomas (0%–5%).33,34 Although studies assessing tumor behavior or comparing treatment modalities are limited because of the rarity and underrecognition of SEDC, MMS has been used several times for SEDC with only 1 recurrence reported.4,13,17,24 Given that all 5 of the patients we reviewed required more than 1 Mohs stage for complete tumor clearance and none demonstrated evidence of recurrence or metastasis (Table), we recommend MMS as the treatment of choice for SEDC.
Conclusion
Squamoid eccrine ductal carcinoma is a rare but likely underdiagnosed cutaneous tumor of uncertain etiology. Because of its propensity for recurrence and metastasis, excision of SEDC with complete circumferential peripheral and deep margin assessment with close follow-up is recommended.
Squamoid eccrine ductal carcinoma (SEDC) is an aggressive underrecognized cutaneous malignancy of unknown etiology.1 It is most likely to occur in sun-exposed areas of the body, most commonly the head and neck. Risk factors include male sex, increased age, and chronic immunosuppression.1-4 Current reports suggest that SEDC is likely a high-grade subtype of squamous cell carcinoma (SCC) with a high risk for local recurrence (25%) and metastasis (13%).1,3,5,6 There are as few as 56 cases of SEDC reported in the literature; however, the number of cases may be closer to 100 due to SEDC being classified as either adenosquamous carcinoma of the skin or ductal eccrine carcinoma with squamous differentiation.1
Clinically, SEDC mimics keratinocyte carcinomas. Histologically, SEDC is biphasic, with a superficial portion resembling well-differentiated SCC and a deeply invasive portion having infiltrative irregular cords with ductal differentiation. Perineural invasion (PNI) frequently is present. Multiple connections to the overlying epidermis also can be seen, serving as a subtle clue to the diagnosis on broad superficial specimens.1-3 Due to superficial sampling, approximately 50% of reported cases are misdiagnosed as SCC during the initial biopsy.4 The diagnosis of SEDC often is made during complete excision when deeper tissue is sampled. Establishing an accurate diagnosis is important given the more aggressive nature of SEDC compared with SCC and its proclivity for PNI.1,3,6 The purpose of this review is to increase awareness of this underrecognized entity and describe the histologic findings that help distinguish SEDC from SCC.
Patient Chart Review
We reviewed chart notes as well as frozen and formalin-fixed paraffin-embedded tissue sections from all 5 patients diagnosed with SEDC at a single institution between November 2018 and May 2020. The mean age of patients was 81 years, and 4 were male. Four of the patients presented for MMS with a preoperative diagnosis of SCC per the original biopsy results. Only 1 patient had a preoperative diagnosis of SEDC. The details of each case are recorded in the Table. All tumors were greater than 2 cm in diameter on initial presentation, were located on the head, and clinically resembled keratinocyte carcinoma with either a nodular or plaquelike appearance (Figure 1).
Intraoperative histologic examination of the excised tissue revealed a biphasic pattern consisting of superficial SCC features overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation in all 5 patients (Figures 2–4). Immunohistochemical staining with cytokeratin AE1/AE3 revealed thin strands of carcinoma in the mid to deeper dermis with squamous differentiation and eccrine ductal differentiation (Figure 5), thus confirming the diagnosis in all 5 patients.
The median depth of tumor invasion was 4.1 mm (range, 2.2–5.45 mm). Ulceration was seen in 3 of the patients, and PNI of large-caliber nerves was observed in all 5 patients. A connection with the overlying epidermis was present in all 5 patients. All 5 patients required more than 1 Mohs stage for complete tumor clearance (Table).
In 4 of the patients, nodal imaging performed at the time of diagnosis revealed no evidence of metastasis. Two patients received adjuvant radiation therapy, and none demonstrated evidence of recurrence. The mean follow-up time was 11 months (range, 6.5–18 months) for the 4 cases with available follow-up data (Table).
Literature Review
A PubMed review of the literature using the search term squamoid eccrine ductal carcinoma resulted in 28 articles, 19 of which were included in the review based on inclusion criteria (original articles available in English, in full text, and pertained to SEDC). Our review yielded 56 cases of SEDC.1-19 The mean age of patients with SEDC was 72 years. The number of male and female cases was 52% (29/56) and 48% (27/56), respectively. The most common location of SEDC was on the head or neck (71% [40/56]), followed by the extremities (19% [11/56]). Immunosuppression was noted in 9% (5/56) of cases. Wide local excision was the most commonly employed treatment modality (91% [51/56]), with MMS being used in 4 patients (7%). Adjuvant radiation was reported in 5% (3/56) of cases. Perineural invasion was reported in 34% (19/56) of cases. Recurrence was seen in 23% (13/56) of cases, with a mean time to recurrence of 10.4 months. Metastasis to regional lymph nodes was observed in 13% (7/56) of cases, with 7% (4/56) of those cases having distant metastases.
Comment
Squamoid eccrine ductal carcinoma was successfully treated with MMS in all 5 of the patients we reviewed. Recognition of a distinct biphasic pattern consisting of squamous differentiation superficially with epidermal connection overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation should lead to consideration of this diagnosis. A thorough inspection for PNI also should be performed, as this finding was present in all of 5 cases and in 34% of reported cases in our literature review.
The differential diagnosis for SEDC includes SCC, metastatic adenocarcinoma with squamoid features, and eccrine tumors, including eccrine poroma, microcystic adnexal carcinoma (MAC), and porocarcinoma with squamous differentiation. The combination of histologic features with the immunoexpression profile of carcinoembryonic antigen (CEA), epithelial membrane antigen (EMA), cytokeratin (CK) 5/6, and p63 can effectively exclude the other entities in the differential and confirm the diagnosis of SEDC.1,3,4 While the diagnosis of SEDC relies on the specific histologic features of multiple surface attachments and superficial squamoid changes with deep ductular elements, immunohistochemistry can nonetheless be adjunctive in difficult cases. Positive immunohistochemical staining for CEA and EMA can help to highlight and delineate true glandular elements, whereas CK5/6 highlights the overall contour of the tumor, displaying more clearly the multiple epidermal attachments and the subtle infiltrative nature of the deeper components of invasive cords and ducts. In addition, the combination of CK5/6 and p63 positivity supports the primary cutaneous nature of the lesion rather than metastatic adenocarcinoma.13,20 Other markers of eccrine secretory coils, such as CK7, CAM5.2, and S100, also are sometimes used for confirmation, some of which can aid in distinction from noneccrine sweat gland differentiation, as CK7 and CAM5.2 are negative in both luminal and basal cells of the dermal duct while being positive within the secretory coil, and S100 protein is expressed within eccrine secretory coil but negative within the apocrine sweat glands.2,4,21
The clinical findings from our chart review corroborated those reported in the literature. The mean age of SEDC in the 5 patients we reviewed was 81 years, and all cases presented on the head, consistent with the findings observed in the literature. Although 4 of our cases were male, there may not be a difference in risk based on sex as previously thought.1 Our literature review revealed an almost equivalent percentage of male and female cases, with 52% being male.
Immunosuppression has been associated with an increased risk for SEDC. Our literature review revealed that approximately 9% (5/56) of cases occurred in immunosuppressed individuals. Two of these reported cases were in the setting of underlying chronic lymphocytic leukemia, 2 in individuals with a history of organ transplant, and 1 treated with azathioprine for myasthenia gravis.2,4,10,12,13 Our chart review supported this correlation, as all 5 patients had a medical history potentially consistent with being in an immunocompromised state (Table). Notably, patient 5 represents a unique case of SEDC occurring in the setting of HIV. The patient had HIV for 33 years, with his most recent CD4+ count of 794 mm3 and HIV-1 RNA load of 35 copies/mL. Given that HIV-positive individuals may have more than a 2-fold increased risk of SCC, a greater degree of suspicion for SEDC should be maintained for these patients.22,23
The etiology of SEDC is controversial but is thought to be either an SCC arising from eccrine glands or a variant of eccrine carcinoma with extensive squamoid differentiation.4,6,13,14,17,24 While SEDC certainly appears to share the proclivity for PNI with the malignant eccrine tumor MAC, it is simultaneously quite distinct, demonstrating nuclear pleomorphism and mitotic activity, both of which are lacking in the bland nature of MACs.12,25
The exact prevalence of SEDC is difficult to ascertain because of its frequent misdiagnosis and variable nomenclature used within the literature. Most reported cases of SEDC are mistakenly diagnosed as SCC on the initial shave or punch biopsy because of superficial sampling. This also was the case in 4 of the patients we reviewed. In addition, there are reported cases of SEDC that were referred to by the investigators as cutaneous adenosquamous carcinoma (cASC), among other descriptors, such as ductal eccrine carcinoma with squamous differentiation, adnexal carcinoma with squamous and ductal differentiation, and syringoid eccrine carcinoma.26-32 While the World Health Organization classifies SEDC as a distinct variant of cASC, which is a rare variant of SCC in itself, the 2 can be differentiated. Despite the similar clinical and histologic features shared between cASC and SEDC, the neoplastic aggregates in SEDC exhibit ductal differentiation containing lumina positive for CEA and EMA.4 Overall, we favor the term squamoid eccrine ductal carcinoma, as there has recently been more uniformity for the designation of this disease entity as such.
It is unclear whether the high incidence of local recurrence (23% [13/56]) of SEDC reported in the literature is related to the treatment modality employed (ie, wide local excision) or due to the innate aggressiveness of SEDC.1,3,5 The literature has shown that MMS has lower recurrence rates than other treatments at 5-year follow-up for SCC (3.1%–5%) and eccrine carcinomas (0%–5%).33,34 Although studies assessing tumor behavior or comparing treatment modalities are limited because of the rarity and underrecognition of SEDC, MMS has been used several times for SEDC with only 1 recurrence reported.4,13,17,24 Given that all 5 of the patients we reviewed required more than 1 Mohs stage for complete tumor clearance and none demonstrated evidence of recurrence or metastasis (Table), we recommend MMS as the treatment of choice for SEDC.
Conclusion
Squamoid eccrine ductal carcinoma is a rare but likely underdiagnosed cutaneous tumor of uncertain etiology. Because of its propensity for recurrence and metastasis, excision of SEDC with complete circumferential peripheral and deep margin assessment with close follow-up is recommended.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760.
- Jacob J, Kugelman L. Squamoid eccrine ductal carcinoma. Cutis. 2018;101:378-380, 385.
- Yim S, Lee YH, Chae SW, et al. Squamoid eccrine ductal carcinoma of the ear helix. Clin Case Rep. 2019;7:1409-1411.
- Terushkin E, Leffell DJ, Futoryan T, et al. Squamoid eccrine ductal carcinoma: a case report and review of the literature. Am J Dermatopathol. 2010;32:287-292.
- Jung YH, Jo HJ, Kang MS. Squamoid eccrine ductal carcinoma of the scalp. Korean J Pathol. 2012;46:278-281.
- Saraiva MI, Vieira MA, Portocarrero LK, et al. Squamoid eccrine ductal carcinoma. An Bras Dermatol. 2016;91:799-802.
- Phan K, Kim L, Lim P, et al. A case report of temple squamoid eccrine ductal carcinoma: a diagnostic challenge beneath the tip of the iceberg. Dermatol Ther. 2020;33:E13213.
- McKissack SS, Wohltmann W, Dalton SR, et al. Squamoid eccrine ductal carcinoma: an aggressive mimicker of squamous cell carcinoma. Am J Dermatopathol. 2019;41:140-143.
- Lobo-Jardim MM, Souza BdCE, Kakizaki P, et al. Dermoscopy of squamoid eccrine ductal carcinoma: an aid for early diagnosis. An Bras Dermatol. 2018;93:893-895.
- Chan H, Howard V, Moir D, et al. Squamoid eccrine ductal carcinoma of the scalp. Aust J Dermatol. 2016;57:E117-E119.
- Wang B, Jarell AD, Bingham JL, et al. PET/CT imaging of squamoid eccrine ductal carcinoma. Clin Nucl Med. 2015;40:322-324.
- Frouin E, Vignon-Pennamen MD, Balme B, et al. Anatomoclinical study of 30 cases of sclerosing sweat duct carcinomas (microcystic adnexal carcinoma, syringomatous carcinoma and squamoid eccrine ductal carcinoma). J Eur Acad Dermatol Venereol. 2015;29:1978-1994.
- Clark S, Young A, Piatigorsky E, et al. Mohs micrographic surgery in the setting of squamoid eccrine ductal carcinoma: addressing a diagnostic and therapeutic challenge. J Clin Aesthet Dermatol. 2013;6:33-36.
- Pusiol T, Morichetti D, Zorzi MG, et al. Squamoid eccrine ductal carcinoma: inappropriate diagnosis. Dermatol Surg. 2011;37:1819-1820.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Wasserman DI, Sack J, Gonzalez-Serva A, et al. Sentinel lymph node biopsy for a squamoid eccrine carcinoma with lymphatic invasion. Dermatol Surg. 2007;33:1126-1129.
- Kim YJ, Kim AR, Yu DS. Mohs micrographic surgery for squamoid eccrine ductal carcinoma. Dermatol Surg. 2005;31:1462-1464.
- Herrero J, Monteagudo C, Jorda E, et al. Squamoid eccrine ductal carcinoma. Histopathology. 1998;32:478-480.
- Wong TY, Suster S, Mihm MC. Squamoid eccrine ductal carcinoma. Histopathology. 1997;30:288-293.
- Qureshi HS, Ormsby AH, Lee MW, et al. The diagnostic utility of p63, CK5/6, CK 7, and CK 20 in distinguishing primary cutaneous adnexal neoplasms from metastatic carcinomas. J Cutan Pathol. 2004;31:145-152.
- Dabbs DJ. Diagnostic Immunohistochemistry: Theranostic and Genomic Applications. 4th ed. Elsevier/Saunders; 2014.
- Silverberg MJ, Leyden W, Warton EM, et al. HIV infection status, immunodeficiency, and the incidence of non-melanoma skin cancer. J Natl Cancer Inst. 2013;105:350-360.
- Asgari MM, Ray GT, Quesenberry CP Jr, et al. Association of multiple primary skin cancers with human immunodeficiency virus infection, CD4 count, and viral load. JAMA Dermatol. 2017;153:892-896.
- Tolkachjov SN. Adnexal carcinomas treated with Mohs micrographic surgery: a comprehensive review. Dermatol Surg. 2017;43:1199-1207.
- Kazakov DV. Cutaneous Adnexal Tumors. Wolters Kluwer Health/ Lippincott Williams & Wilkins; 2012.
- Weidner N, Foucar E. Adenosquamous carcinoma of the skin. an aggressive mucin- and gland-forming squamous carcinoma. Arch Dermatol. 1985;121:775-779.
- Banks ER, Cooper PH. Adenosquamous carcinoma of the skin: a report of 10 cases. J Cutan Pathol. 1991;18:227-234.
- Ko CJ, Leffell DJ, McNiff JM. Adenosquamous carcinoma: a report of nine cases with p63 and cytokeratin 5/6 staining. J Cutan Pathol. 2009;36:448-452.
- Patel V, Squires SM, Liu DY, et al. Cutaneous adenosquamous carcinoma: a rare neoplasm with biphasic differentiation. Cutis. 2014;94:231-233.
- Chhibber V, Lyle S, Mahalingam M. Ductal eccrine carcinoma with squamous differentiation: apropos a case. J Cutan Pathol. 2007;34:503-507.
- Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
- Azorín D, López-Ríos F, Ballestín C, et al. Primary cutaneous adenosquamous carcinoma: a case report and review of the literature. J Cutan Pathol. 2001;28:542-545.
- Wildemore JK, Lee JB, Humphreys TR. Mohs surgery for malignant eccrine neoplasms. Dermatol Surg. 2004;30(12 pt 2):1574-1579.
- Garcia-Zuazaga J, Olbricht SM. Cutaneous squamous cell carcinoma. Adv Dermatol. 2008;24:33-57.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760.
- Jacob J, Kugelman L. Squamoid eccrine ductal carcinoma. Cutis. 2018;101:378-380, 385.
- Yim S, Lee YH, Chae SW, et al. Squamoid eccrine ductal carcinoma of the ear helix. Clin Case Rep. 2019;7:1409-1411.
- Terushkin E, Leffell DJ, Futoryan T, et al. Squamoid eccrine ductal carcinoma: a case report and review of the literature. Am J Dermatopathol. 2010;32:287-292.
- Jung YH, Jo HJ, Kang MS. Squamoid eccrine ductal carcinoma of the scalp. Korean J Pathol. 2012;46:278-281.
- Saraiva MI, Vieira MA, Portocarrero LK, et al. Squamoid eccrine ductal carcinoma. An Bras Dermatol. 2016;91:799-802.
- Phan K, Kim L, Lim P, et al. A case report of temple squamoid eccrine ductal carcinoma: a diagnostic challenge beneath the tip of the iceberg. Dermatol Ther. 2020;33:E13213.
- McKissack SS, Wohltmann W, Dalton SR, et al. Squamoid eccrine ductal carcinoma: an aggressive mimicker of squamous cell carcinoma. Am J Dermatopathol. 2019;41:140-143.
- Lobo-Jardim MM, Souza BdCE, Kakizaki P, et al. Dermoscopy of squamoid eccrine ductal carcinoma: an aid for early diagnosis. An Bras Dermatol. 2018;93:893-895.
- Chan H, Howard V, Moir D, et al. Squamoid eccrine ductal carcinoma of the scalp. Aust J Dermatol. 2016;57:E117-E119.
- Wang B, Jarell AD, Bingham JL, et al. PET/CT imaging of squamoid eccrine ductal carcinoma. Clin Nucl Med. 2015;40:322-324.
- Frouin E, Vignon-Pennamen MD, Balme B, et al. Anatomoclinical study of 30 cases of sclerosing sweat duct carcinomas (microcystic adnexal carcinoma, syringomatous carcinoma and squamoid eccrine ductal carcinoma). J Eur Acad Dermatol Venereol. 2015;29:1978-1994.
- Clark S, Young A, Piatigorsky E, et al. Mohs micrographic surgery in the setting of squamoid eccrine ductal carcinoma: addressing a diagnostic and therapeutic challenge. J Clin Aesthet Dermatol. 2013;6:33-36.
- Pusiol T, Morichetti D, Zorzi MG, et al. Squamoid eccrine ductal carcinoma: inappropriate diagnosis. Dermatol Surg. 2011;37:1819-1820.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Wasserman DI, Sack J, Gonzalez-Serva A, et al. Sentinel lymph node biopsy for a squamoid eccrine carcinoma with lymphatic invasion. Dermatol Surg. 2007;33:1126-1129.
- Kim YJ, Kim AR, Yu DS. Mohs micrographic surgery for squamoid eccrine ductal carcinoma. Dermatol Surg. 2005;31:1462-1464.
- Herrero J, Monteagudo C, Jorda E, et al. Squamoid eccrine ductal carcinoma. Histopathology. 1998;32:478-480.
- Wong TY, Suster S, Mihm MC. Squamoid eccrine ductal carcinoma. Histopathology. 1997;30:288-293.
- Qureshi HS, Ormsby AH, Lee MW, et al. The diagnostic utility of p63, CK5/6, CK 7, and CK 20 in distinguishing primary cutaneous adnexal neoplasms from metastatic carcinomas. J Cutan Pathol. 2004;31:145-152.
- Dabbs DJ. Diagnostic Immunohistochemistry: Theranostic and Genomic Applications. 4th ed. Elsevier/Saunders; 2014.
- Silverberg MJ, Leyden W, Warton EM, et al. HIV infection status, immunodeficiency, and the incidence of non-melanoma skin cancer. J Natl Cancer Inst. 2013;105:350-360.
- Asgari MM, Ray GT, Quesenberry CP Jr, et al. Association of multiple primary skin cancers with human immunodeficiency virus infection, CD4 count, and viral load. JAMA Dermatol. 2017;153:892-896.
- Tolkachjov SN. Adnexal carcinomas treated with Mohs micrographic surgery: a comprehensive review. Dermatol Surg. 2017;43:1199-1207.
- Kazakov DV. Cutaneous Adnexal Tumors. Wolters Kluwer Health/ Lippincott Williams & Wilkins; 2012.
- Weidner N, Foucar E. Adenosquamous carcinoma of the skin. an aggressive mucin- and gland-forming squamous carcinoma. Arch Dermatol. 1985;121:775-779.
- Banks ER, Cooper PH. Adenosquamous carcinoma of the skin: a report of 10 cases. J Cutan Pathol. 1991;18:227-234.
- Ko CJ, Leffell DJ, McNiff JM. Adenosquamous carcinoma: a report of nine cases with p63 and cytokeratin 5/6 staining. J Cutan Pathol. 2009;36:448-452.
- Patel V, Squires SM, Liu DY, et al. Cutaneous adenosquamous carcinoma: a rare neoplasm with biphasic differentiation. Cutis. 2014;94:231-233.
- Chhibber V, Lyle S, Mahalingam M. Ductal eccrine carcinoma with squamous differentiation: apropos a case. J Cutan Pathol. 2007;34:503-507.
- Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
- Azorín D, López-Ríos F, Ballestín C, et al. Primary cutaneous adenosquamous carcinoma: a case report and review of the literature. J Cutan Pathol. 2001;28:542-545.
- Wildemore JK, Lee JB, Humphreys TR. Mohs surgery for malignant eccrine neoplasms. Dermatol Surg. 2004;30(12 pt 2):1574-1579.
- Garcia-Zuazaga J, Olbricht SM. Cutaneous squamous cell carcinoma. Adv Dermatol. 2008;24:33-57.
PRACTICE POINTS
- Squamoid eccrine ductal carcinoma is an aggressive underrecognized cutaneous malignancy that often is misdiagnosed as squamous cell carcinoma (SCC) during initial biopsy.
- Squamoid eccrine ductal carcinoma has a biphasic histologic appearance with a superficial portion resembling well-differentiated SCC and a deeply invasive portion comprised of infiltrative irregular cords with ductal differentiation.
- Excision with complete circumferential peripheral and deep margin assessment with close follow-up is recommended for these patients because of the high risk for recurrence and metastasis.
Erythema, Blisters, and Scars on the Elbows, Knees, and Legs
The Diagnosis: Epidermolysis Bullosa Acquisita
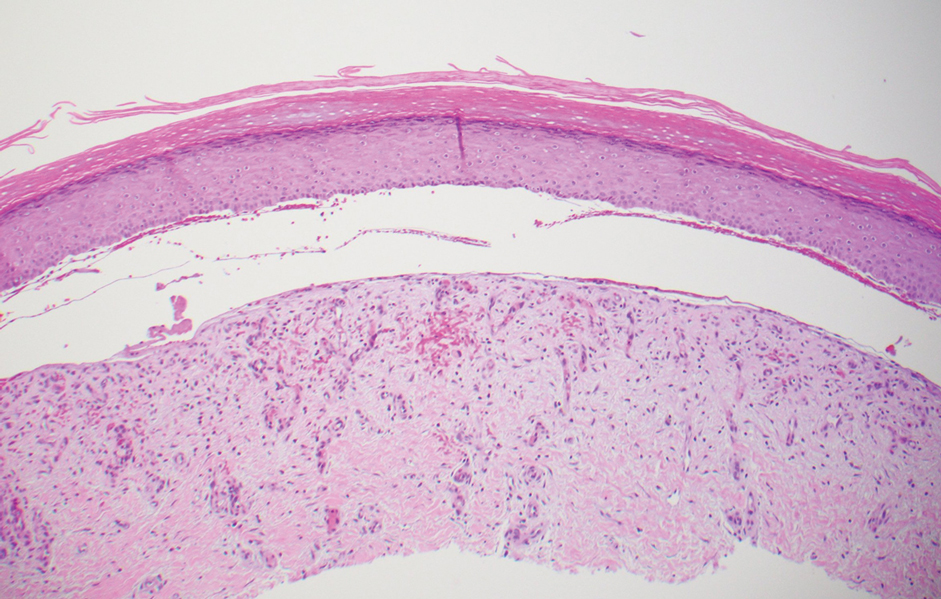
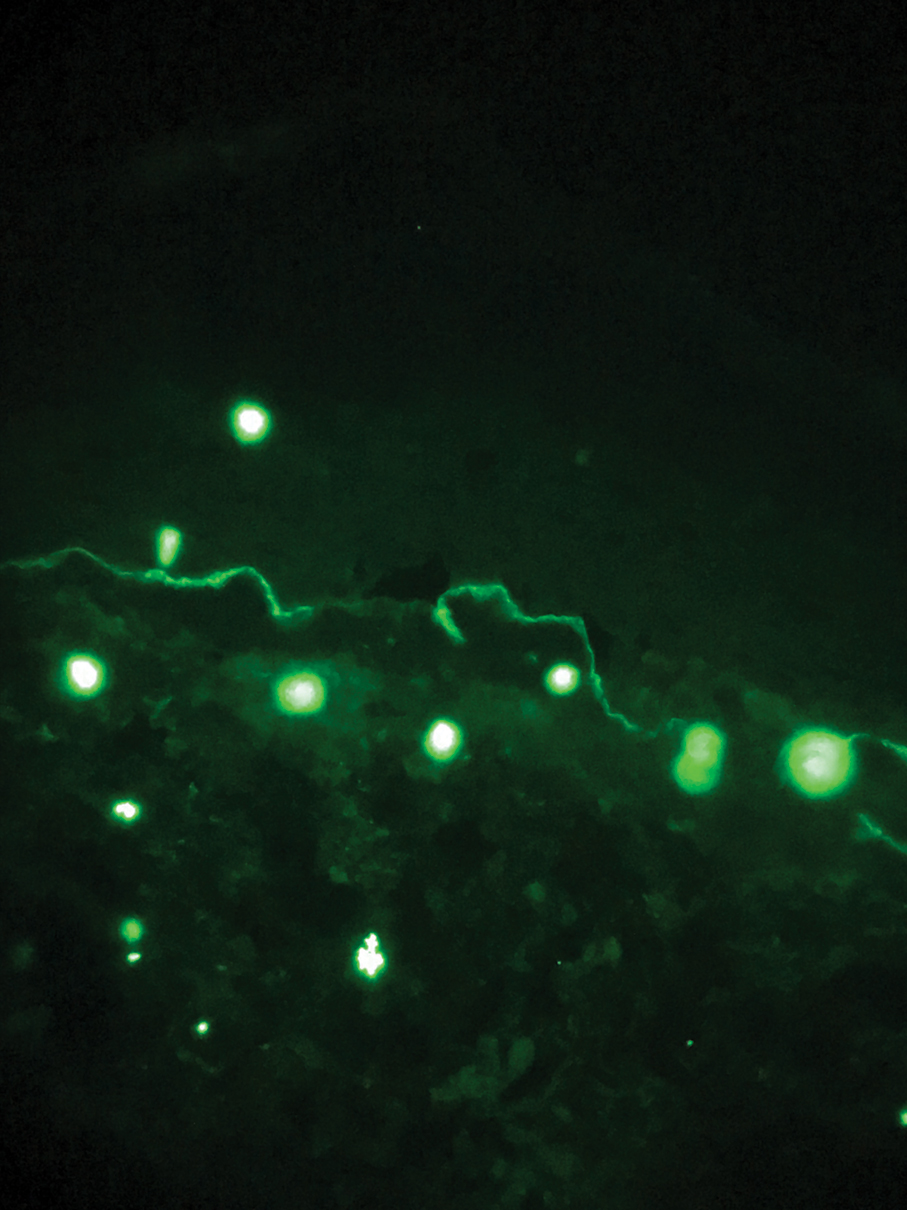
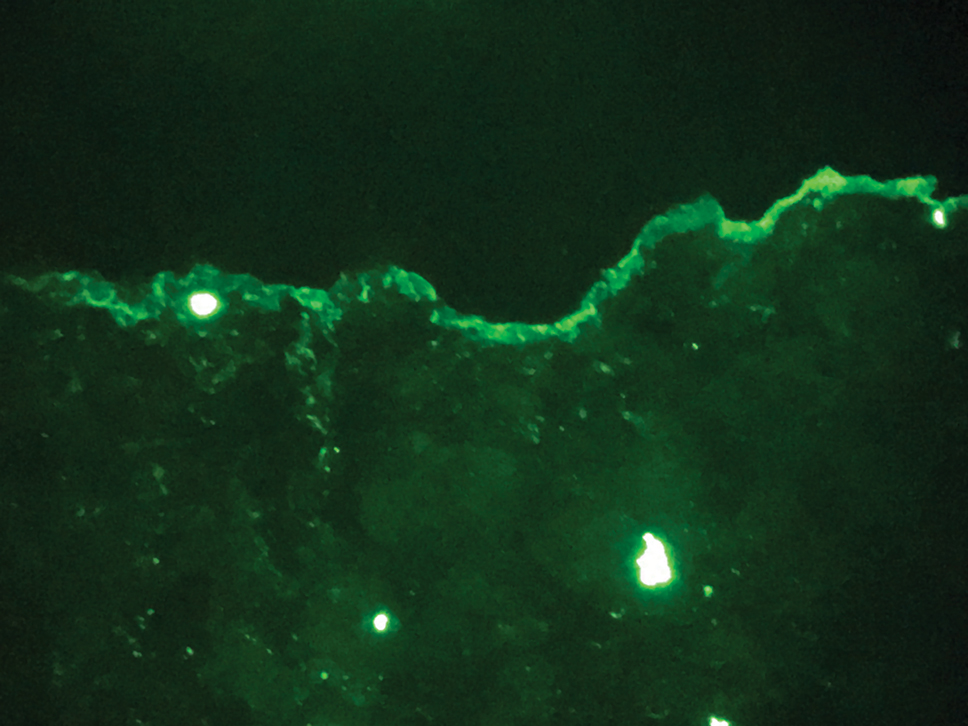
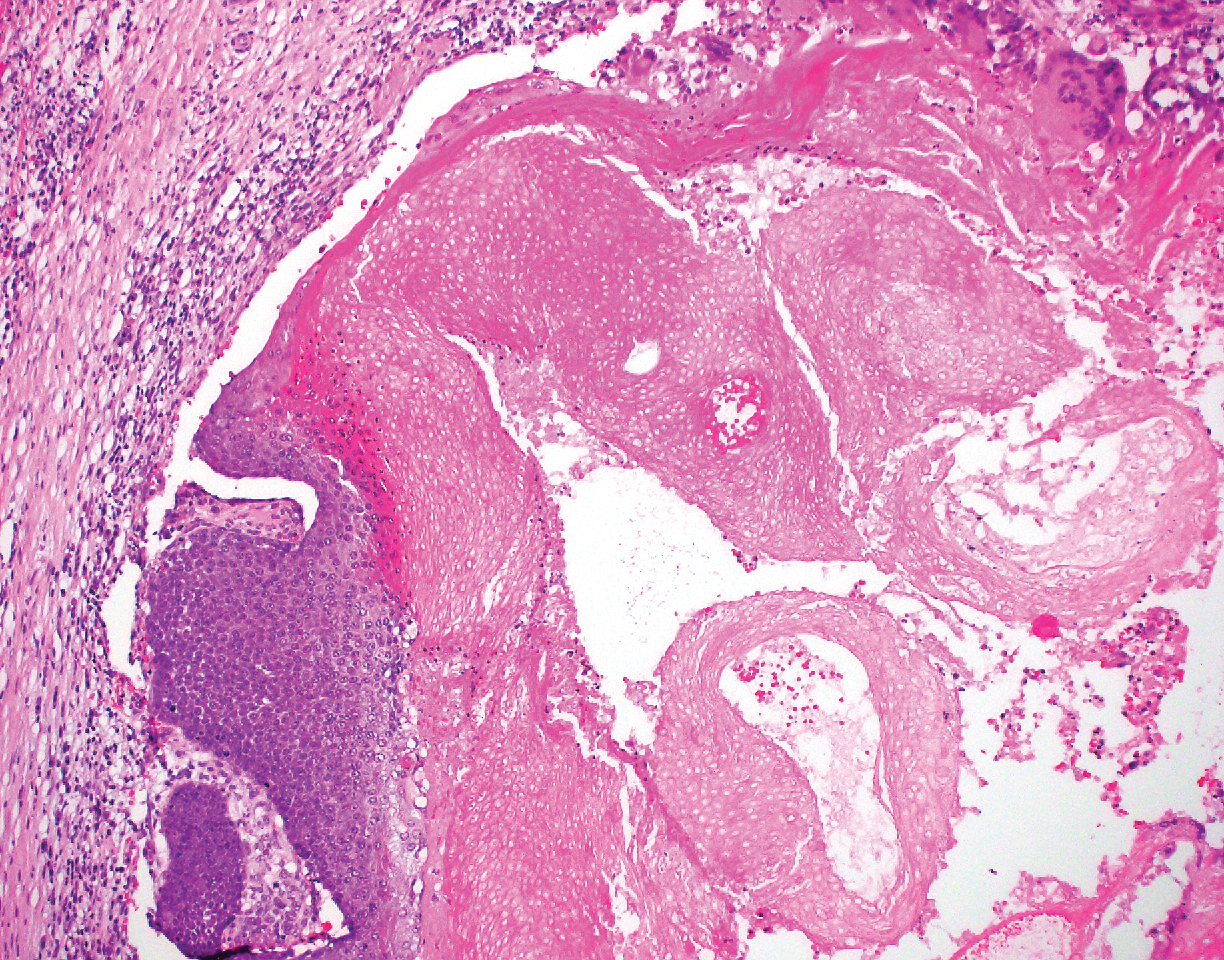
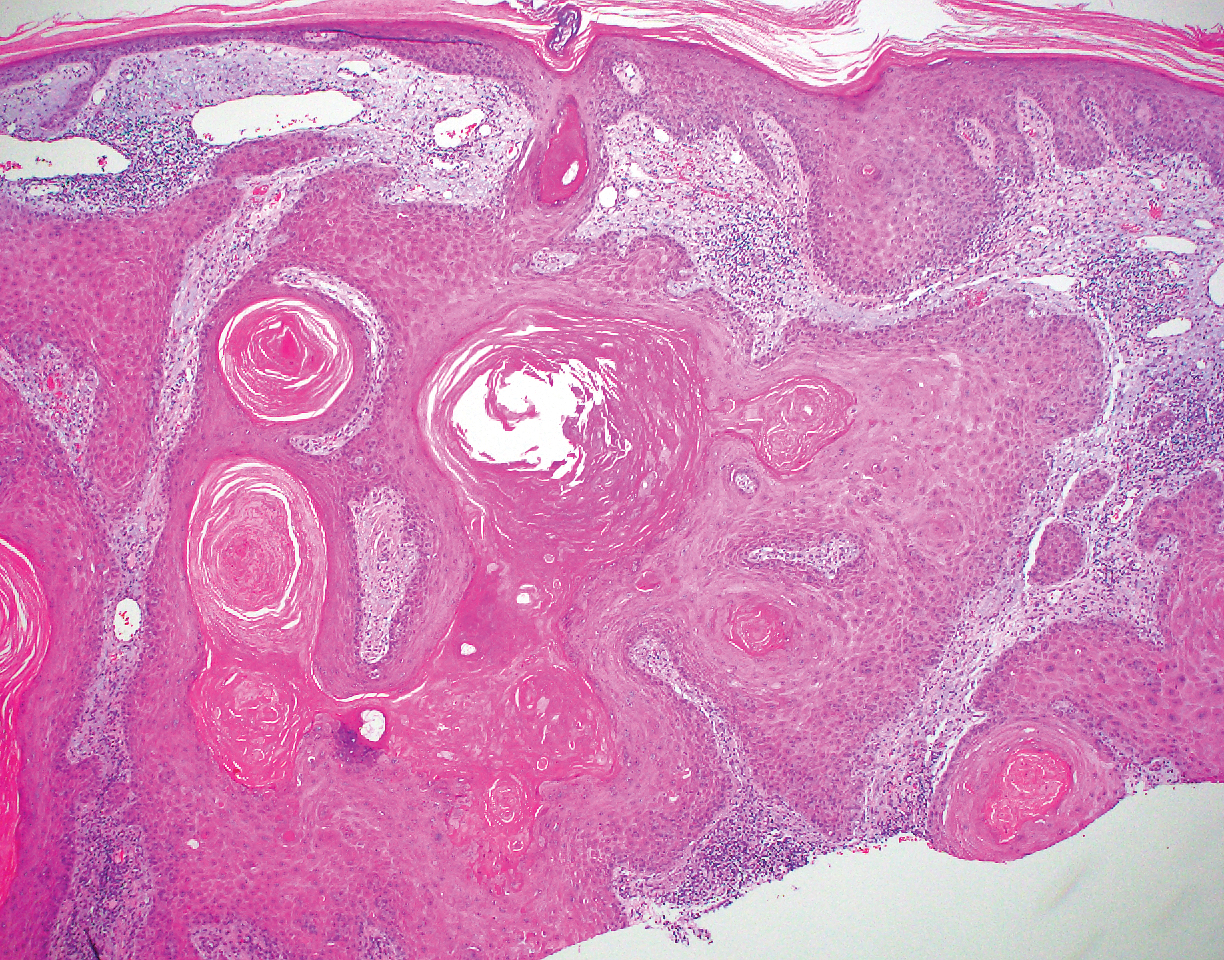
The diagnosis of epidermolysis bullosa acquisita (EBA) was made based on the clinical and pathologic findings. A blistering disorder that resolves with milia is characteristic of EBA. Hematoxylin and eosin staining demonstrated a pauci-inflammatory separation between the epidermis and dermis (Figure 1). Direct immunofluorescence studies showed linear IgG deposition along the basement membrane zone while C3 was negative (Figure 2). Salt-split skin was essential, as it revealed IgG deposition to the floor of the split (Figure 3), a pattern seen in EBA and not bullous pemphigoid (BP).1
Epidermolysis bullosa acquisita is an acquired autoimmune bullous disorder that results from antibodies to type VII collagen, an anchoring fibril that attaches the lamina densa to the dermis. The epidemiology and etiology of the trigger that leads to antibody production are not well known, but an association between EBA and inflammatory bowel disease has been described.2 Although this disease may present in childhood, EBA most commonly is a disorder seen in adults and the elderly. A classic noninflammatory mechanobullous form as well as an inflammatory BP-like form are the most commonly encountered presentations. Light microscopy demonstrates subepidermal cleavage without acantholysis. In the inflammatory BP-like subtype, an inflammatory infiltrate may be present. Direct immunofluorescence is remarkable for a linear band of IgG deposits along the basement membrane zone, with or without C3 deposition in a similar pattern.1
Bullous pemphigoid is within the differential of EBA. It can be difficult to differentiate clinically, especially when a patient has the BP-like variant of EBA because, as the name implies, it mimics BP. Patients with BP often will report a pruritic patch that will then develop into an urticarial plaque. Scarring and milia rarely are seen in BP but can be observed in the multiple presentations of EBA. Hematoxylin and eosin staining and direct immunofluorescence may be almost identical, and differentiating between the 2 disorders can be a challenge. Immunodeposition in EBA occurs in a U-shaped, serrated pattern, while the pattern in BP is N-shaped and serrated.3 Although the U-shaped, serrated pattern is relatively specific, it is not always easy to interpret and requires a high-quality biopsy specimen, which can be difficult to discern with certainty in suboptimal preparations. Another way to differentiate between the 2 entities is to utilize the salt-split skin technique, as performed in our patient. With salt-split skin, the biopsy is placed into a solution of 1 mol/L sodium chloride and incubated at 4 °C (39 °F) for 18 to 24 hours. A blister is then produced at the level of the lamina lucida, which allows for the staining of immunoreactants to occur either above or below that split (commonly referred to as staining on the roof or floor of the blister cavity). With EBA, there is immunoreactant deposition on the floor of the blister, while the opposite occurs in BP.4
Epidermolysis bullosa simplex is the most common type of epidermolysis bullosa, with keratin genes KRT5 and KRT14 as frequent mutations. Patients develop blisters, vesicles, bullae, and milia on traumatized areas of the body such as the hands, elbows, knees, and feet. This disease presents early in childhood. Histology exhibits a cell-poor subepidermal blister.5 With porphyria cutanea tarda, reduced activity of uroporphyrinogen decarboxylase, a major enzyme in the heme synthesis pathway, leads to blisters with erosions and milia on sun-exposed areas of the body. Histologic evaluation reveals a subepidermal pauci-inflammatory vesicle with festooning of the dermal papillae and amphophilic basement membrane within the epidermis. Direct immunofluorescence of porphyria cutanea tarda demonstrates IgM and C3 in the vessels.6 Sweet syndrome is a neutrophilic dermatosis that presents as erythematous, edematous, hot, and tender plaques along with fever and leukocytosis. It is associated with myeloproliferative disorders. Biopsy demonstrates papillary dermal edema along with diffuse neutrophilic infiltrate.7
Numerous medications have been recommended for the treatment of EBA, ranging from steroids to steroid-sparing drugs such as colchicine and dapsone.8,9 Our patient was educated on physical precautions and was started on dapsone alone due to comorbid diabetes mellitus and renal disease. Within a few weeks of initiating dapsone, he observed a reduction in erythema, and within months he experienced a decrease in blister eruption frequency.
- Vorobyev A, Ludwig RJ, Schmidt E. Clinical features and diagnosis of epidermolysis bullosa acquisita. Expert Rev Clin Immunol. 2017;13:157-169.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-230.
- Vodegel RM, Jonkman MF, Pas HH, et al. U-serrated immunodeposition pattern differentiates type VII collagen targeting bullous diseases from other subepidermal bullous autoimmune diseases. Br J Dermatol. 2004;151:112-118.
- Gardner KM, Crawford RI. Distinguishing epidermolysis bullosa acquisita from bullous pemphigoid without direct immunofluorescence. J Cutan Med Surg. 2018;22:22-24.
- Sprecher E. Epidermolysis bullosa simplex. Dermatol Clin. 2010;28:23-32.
- Maynard B, Peters MS. Histologic and immunofluorescence study of cutaneous porphyrias. J Cutan Pathol. 1992;19:40-47.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018:79:987-1006.
- Kirtschig G, Murrell D, Wojnarowska F, et al. Interventions for mucous membrane pemphigoid and epidermolysis bullosa acquisita. Cochrane Database Syst Rev. 2003;1:CD004056
- Gürcan HM, Ahmed AR. Current concepts in the treatment of epidermolysis bullosa acquisita. Expert Opin Pharmacother. 2011;12:1259-1268.
The Diagnosis: Epidermolysis Bullosa Acquisita
The diagnosis of epidermolysis bullosa acquisita (EBA) was made based on the clinical and pathologic findings. A blistering disorder that resolves with milia is characteristic of EBA. Hematoxylin and eosin staining demonstrated a pauci-inflammatory separation between the epidermis and dermis (Figure 1). Direct immunofluorescence studies showed linear IgG deposition along the basement membrane zone while C3 was negative (Figure 2). Salt-split skin was essential, as it revealed IgG deposition to the floor of the split (Figure 3), a pattern seen in EBA and not bullous pemphigoid (BP).1
Epidermolysis bullosa acquisita is an acquired autoimmune bullous disorder that results from antibodies to type VII collagen, an anchoring fibril that attaches the lamina densa to the dermis. The epidemiology and etiology of the trigger that leads to antibody production are not well known, but an association between EBA and inflammatory bowel disease has been described.2 Although this disease may present in childhood, EBA most commonly is a disorder seen in adults and the elderly. A classic noninflammatory mechanobullous form as well as an inflammatory BP-like form are the most commonly encountered presentations. Light microscopy demonstrates subepidermal cleavage without acantholysis. In the inflammatory BP-like subtype, an inflammatory infiltrate may be present. Direct immunofluorescence is remarkable for a linear band of IgG deposits along the basement membrane zone, with or without C3 deposition in a similar pattern.1
Bullous pemphigoid is within the differential of EBA. It can be difficult to differentiate clinically, especially when a patient has the BP-like variant of EBA because, as the name implies, it mimics BP. Patients with BP often will report a pruritic patch that will then develop into an urticarial plaque. Scarring and milia rarely are seen in BP but can be observed in the multiple presentations of EBA. Hematoxylin and eosin staining and direct immunofluorescence may be almost identical, and differentiating between the 2 disorders can be a challenge. Immunodeposition in EBA occurs in a U-shaped, serrated pattern, while the pattern in BP is N-shaped and serrated.3 Although the U-shaped, serrated pattern is relatively specific, it is not always easy to interpret and requires a high-quality biopsy specimen, which can be difficult to discern with certainty in suboptimal preparations. Another way to differentiate between the 2 entities is to utilize the salt-split skin technique, as performed in our patient. With salt-split skin, the biopsy is placed into a solution of 1 mol/L sodium chloride and incubated at 4 °C (39 °F) for 18 to 24 hours. A blister is then produced at the level of the lamina lucida, which allows for the staining of immunoreactants to occur either above or below that split (commonly referred to as staining on the roof or floor of the blister cavity). With EBA, there is immunoreactant deposition on the floor of the blister, while the opposite occurs in BP.4
Epidermolysis bullosa simplex is the most common type of epidermolysis bullosa, with keratin genes KRT5 and KRT14 as frequent mutations. Patients develop blisters, vesicles, bullae, and milia on traumatized areas of the body such as the hands, elbows, knees, and feet. This disease presents early in childhood. Histology exhibits a cell-poor subepidermal blister.5 With porphyria cutanea tarda, reduced activity of uroporphyrinogen decarboxylase, a major enzyme in the heme synthesis pathway, leads to blisters with erosions and milia on sun-exposed areas of the body. Histologic evaluation reveals a subepidermal pauci-inflammatory vesicle with festooning of the dermal papillae and amphophilic basement membrane within the epidermis. Direct immunofluorescence of porphyria cutanea tarda demonstrates IgM and C3 in the vessels.6 Sweet syndrome is a neutrophilic dermatosis that presents as erythematous, edematous, hot, and tender plaques along with fever and leukocytosis. It is associated with myeloproliferative disorders. Biopsy demonstrates papillary dermal edema along with diffuse neutrophilic infiltrate.7
Numerous medications have been recommended for the treatment of EBA, ranging from steroids to steroid-sparing drugs such as colchicine and dapsone.8,9 Our patient was educated on physical precautions and was started on dapsone alone due to comorbid diabetes mellitus and renal disease. Within a few weeks of initiating dapsone, he observed a reduction in erythema, and within months he experienced a decrease in blister eruption frequency.
The Diagnosis: Epidermolysis Bullosa Acquisita
The diagnosis of epidermolysis bullosa acquisita (EBA) was made based on the clinical and pathologic findings. A blistering disorder that resolves with milia is characteristic of EBA. Hematoxylin and eosin staining demonstrated a pauci-inflammatory separation between the epidermis and dermis (Figure 1). Direct immunofluorescence studies showed linear IgG deposition along the basement membrane zone while C3 was negative (Figure 2). Salt-split skin was essential, as it revealed IgG deposition to the floor of the split (Figure 3), a pattern seen in EBA and not bullous pemphigoid (BP).1
Epidermolysis bullosa acquisita is an acquired autoimmune bullous disorder that results from antibodies to type VII collagen, an anchoring fibril that attaches the lamina densa to the dermis. The epidemiology and etiology of the trigger that leads to antibody production are not well known, but an association between EBA and inflammatory bowel disease has been described.2 Although this disease may present in childhood, EBA most commonly is a disorder seen in adults and the elderly. A classic noninflammatory mechanobullous form as well as an inflammatory BP-like form are the most commonly encountered presentations. Light microscopy demonstrates subepidermal cleavage without acantholysis. In the inflammatory BP-like subtype, an inflammatory infiltrate may be present. Direct immunofluorescence is remarkable for a linear band of IgG deposits along the basement membrane zone, with or without C3 deposition in a similar pattern.1
Bullous pemphigoid is within the differential of EBA. It can be difficult to differentiate clinically, especially when a patient has the BP-like variant of EBA because, as the name implies, it mimics BP. Patients with BP often will report a pruritic patch that will then develop into an urticarial plaque. Scarring and milia rarely are seen in BP but can be observed in the multiple presentations of EBA. Hematoxylin and eosin staining and direct immunofluorescence may be almost identical, and differentiating between the 2 disorders can be a challenge. Immunodeposition in EBA occurs in a U-shaped, serrated pattern, while the pattern in BP is N-shaped and serrated.3 Although the U-shaped, serrated pattern is relatively specific, it is not always easy to interpret and requires a high-quality biopsy specimen, which can be difficult to discern with certainty in suboptimal preparations. Another way to differentiate between the 2 entities is to utilize the salt-split skin technique, as performed in our patient. With salt-split skin, the biopsy is placed into a solution of 1 mol/L sodium chloride and incubated at 4 °C (39 °F) for 18 to 24 hours. A blister is then produced at the level of the lamina lucida, which allows for the staining of immunoreactants to occur either above or below that split (commonly referred to as staining on the roof or floor of the blister cavity). With EBA, there is immunoreactant deposition on the floor of the blister, while the opposite occurs in BP.4
Epidermolysis bullosa simplex is the most common type of epidermolysis bullosa, with keratin genes KRT5 and KRT14 as frequent mutations. Patients develop blisters, vesicles, bullae, and milia on traumatized areas of the body such as the hands, elbows, knees, and feet. This disease presents early in childhood. Histology exhibits a cell-poor subepidermal blister.5 With porphyria cutanea tarda, reduced activity of uroporphyrinogen decarboxylase, a major enzyme in the heme synthesis pathway, leads to blisters with erosions and milia on sun-exposed areas of the body. Histologic evaluation reveals a subepidermal pauci-inflammatory vesicle with festooning of the dermal papillae and amphophilic basement membrane within the epidermis. Direct immunofluorescence of porphyria cutanea tarda demonstrates IgM and C3 in the vessels.6 Sweet syndrome is a neutrophilic dermatosis that presents as erythematous, edematous, hot, and tender plaques along with fever and leukocytosis. It is associated with myeloproliferative disorders. Biopsy demonstrates papillary dermal edema along with diffuse neutrophilic infiltrate.7
Numerous medications have been recommended for the treatment of EBA, ranging from steroids to steroid-sparing drugs such as colchicine and dapsone.8,9 Our patient was educated on physical precautions and was started on dapsone alone due to comorbid diabetes mellitus and renal disease. Within a few weeks of initiating dapsone, he observed a reduction in erythema, and within months he experienced a decrease in blister eruption frequency.
- Vorobyev A, Ludwig RJ, Schmidt E. Clinical features and diagnosis of epidermolysis bullosa acquisita. Expert Rev Clin Immunol. 2017;13:157-169.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-230.
- Vodegel RM, Jonkman MF, Pas HH, et al. U-serrated immunodeposition pattern differentiates type VII collagen targeting bullous diseases from other subepidermal bullous autoimmune diseases. Br J Dermatol. 2004;151:112-118.
- Gardner KM, Crawford RI. Distinguishing epidermolysis bullosa acquisita from bullous pemphigoid without direct immunofluorescence. J Cutan Med Surg. 2018;22:22-24.
- Sprecher E. Epidermolysis bullosa simplex. Dermatol Clin. 2010;28:23-32.
- Maynard B, Peters MS. Histologic and immunofluorescence study of cutaneous porphyrias. J Cutan Pathol. 1992;19:40-47.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018:79:987-1006.
- Kirtschig G, Murrell D, Wojnarowska F, et al. Interventions for mucous membrane pemphigoid and epidermolysis bullosa acquisita. Cochrane Database Syst Rev. 2003;1:CD004056
- Gürcan HM, Ahmed AR. Current concepts in the treatment of epidermolysis bullosa acquisita. Expert Opin Pharmacother. 2011;12:1259-1268.
- Vorobyev A, Ludwig RJ, Schmidt E. Clinical features and diagnosis of epidermolysis bullosa acquisita. Expert Rev Clin Immunol. 2017;13:157-169.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-230.
- Vodegel RM, Jonkman MF, Pas HH, et al. U-serrated immunodeposition pattern differentiates type VII collagen targeting bullous diseases from other subepidermal bullous autoimmune diseases. Br J Dermatol. 2004;151:112-118.
- Gardner KM, Crawford RI. Distinguishing epidermolysis bullosa acquisita from bullous pemphigoid without direct immunofluorescence. J Cutan Med Surg. 2018;22:22-24.
- Sprecher E. Epidermolysis bullosa simplex. Dermatol Clin. 2010;28:23-32.
- Maynard B, Peters MS. Histologic and immunofluorescence study of cutaneous porphyrias. J Cutan Pathol. 1992;19:40-47.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018:79:987-1006.
- Kirtschig G, Murrell D, Wojnarowska F, et al. Interventions for mucous membrane pemphigoid and epidermolysis bullosa acquisita. Cochrane Database Syst Rev. 2003;1:CD004056
- Gürcan HM, Ahmed AR. Current concepts in the treatment of epidermolysis bullosa acquisita. Expert Opin Pharmacother. 2011;12:1259-1268.
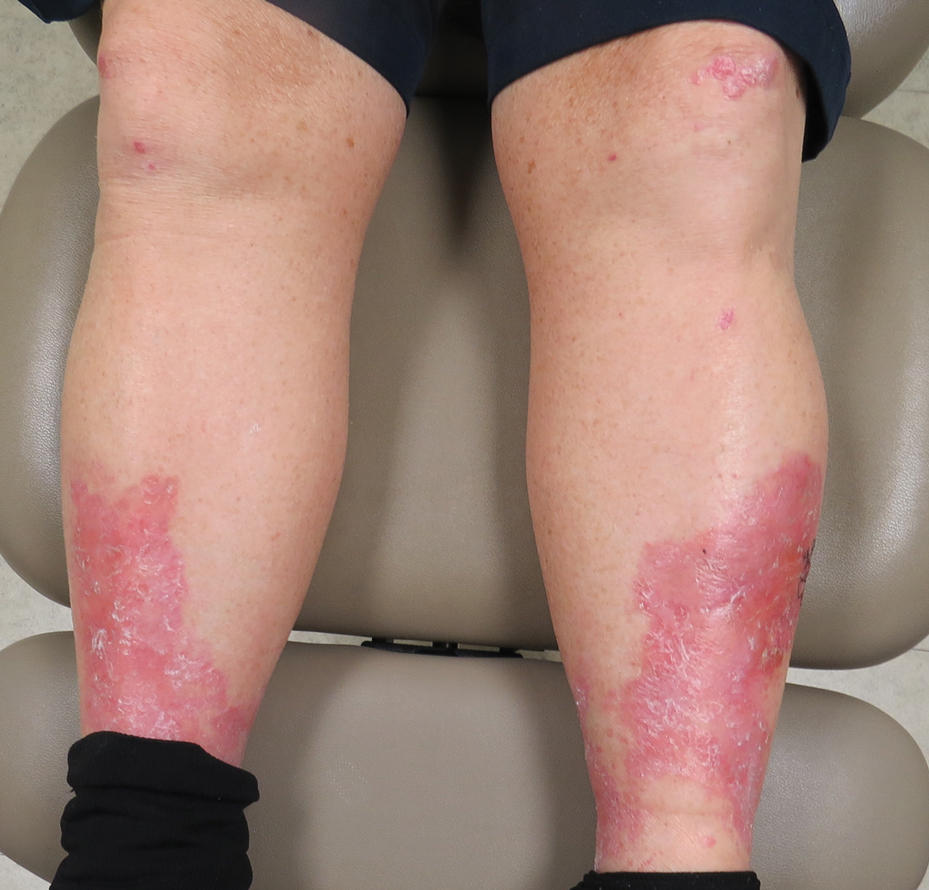
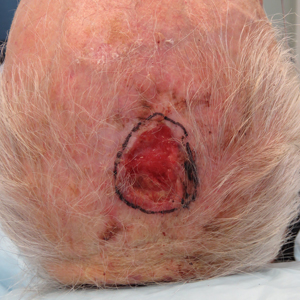
A 69-year-old man presented with an asymptomatic rash on the extensor surfaces of 2 years' duration. He reported recurrent blisters that would then scar over. The lesions did not occur in relation to any known trauma. The patient's medical history revealed dialysis-dependent end-stage renal disease secondary to type 2 diabetes mellitus. His medications were noncontributory, and there was no family history of blistering disorders. He had tried triamcinolone cream without any improvement. Physical examination was remarkable for erythematous blisters and bullae with scales and milia on the elbows, knees, and lower legs. The oral mucosa was unremarkable. Shave biopsies of the skin for direct immunofluorescence and salt-split skin studies were obtained.
Ulcerated Nodule on the Scalp
The Diagnosis: Proliferating Pilar Tumor
Proliferating pilar tumor (PPT), or cyst, is a neoplasm of trichilemmal keratinization first described by Wilson-Jones1 in 1966. Proliferating pilar tumors lie on a spectrum with malignant PPT, which is a rare adnexal neoplasm first described by Saida et al2 in 1983. The incidence of PPT is unknown given the paucity of cases and the possible misdiagnosis as squamous cell carcinoma (SCC). Proliferating pilar tumors tend to present on the head and neck of older females as a multilobular and sometimes ulcerating nodule.3 Although PPT can occur de novo, the majority of cases are thought to develop progressively from a benign pilar cyst. Histopathologically, PPT is characterized by cords and nests of squamous cells that display trichilemmal keratinization (quiz images).
Classification of PPT as benign or malignant is challenging, though criteria have been proposed.3-7 Lesions with minimal infiltration into the surrounding dermis and scant mitosis typically behave in a benign manner, while lesions showing nuclear atypia, atypical mitosis, and irregular infiltration into the surrounding dermis can have up to a 50% locoregional recurrence rate.3 In addition, distinguishing a PPT from an SCC or trichilemmal carcinoma also can be difficult; however, SCC is favored when there is a lack of trichilemmal keratinization or when squamous atypia is present in the adjacent epidermis.8 Trichilemmal carcinoma is a rare tumor that has been questioned as a distinct entity.9-12
Pilomatricoma, also known as calcifying epithelioma of Malherbe, is a benign pilar tumor that presents as a slowly growing nodule on the head or neck area or arms.13,14 Most pilomatricomas develop by the second decade of life. Multiple lesions may be present in association with myotonic dystrophy or Gardner syndrome among other syndromes.15-17 Similar to PPT, pilomatricomas present as large dermal nodules; however, they tend to be circumscribed and have a trabecular network that consists of basophilic cells and eosinophilic keratinized shadow cells (Figure 1).18 Calcification may be seen and bone formation subsequently may occur.19
Most sources now consider keratoacanthoma (KA) as a well-differentiated SCC.20 The typical presentation consists of a rapidly growing erythematous to flesh-colored nodule with a central keratinous plug that develops over a period of weeks. If untreated, KAs may resolve over a period of months and leave a depressed scar. Local destruction can result from KAs, and they have the potential to transform into a more aggressive SCC. Accordingly, most clinicians use tissue destructive methods, excision, or Mohs micrographic surgery for treatment based on location. Histologically, a well-circumscribed proliferation of glassy cytoplasm is noted. A depressed keratin-filled center is surrounded by a lip of epithelium extending over the lesion (Figure 2).20,21 Pseudoepitheliomatous hyperplasia accompanied by hypergranulosis is seen in the center of KAs rather than at the periphery, which is typical of non-KA SCCs. Typical KAs lack acantholysis, a feature suggesting a non-KA type of SCC. Neutrophilic microabscesses and eosinophils commonly are seen in KAs.20,21
Inverted follicular keratosis is a benign tumor that gained traction as its own entity in the 1960s.22 These lesions typically develop from the follicular infundibulum, but some consider them a version of a wart or seborrheic keratosis.23 They generally are flesh-colored nodules on the upper cutaneous lip or face. Treatment usually consists of complete excision. There are many different growth patterns described, but they typically are endophytic tumors with eosinophilic squamous cells in the center and more basophilic cells at the periphery (Figure 3).24 Characteristically, there are squamous eddies throughout the tumor (Figure 3 [inset]). There also may be a scant lymphohistiocytic infiltrate within the dermis surrounding the lesion.
Trichilemmomas are flesh-colored adnexal neoplasms that may present as a solitary lesion or in clusters on the face. They have been reported to occur on all nonglabrous skin sites.25 Multiple lesions may occur in association with Cowden syndrome or with nevus sebaceous.26 A desmoplastic variant of trichilemmomas has been reported.27 Desmoplastic trichilemmomas appear as well-circumscribed tumors of outer root sheath differentiation with lobules extending down into the dermis.28 Vacuolated glycogen-filled keratinocytes are scattered throughout the lesion but are most prominent at the base. At the periphery of the lobules, peripheral palisading of basaloid cells is accompanied by a thickened eosinophilic basement membrane that is periodic acid-Schiff positive. Typical trichilemmomas also can display these features; however, the main differentiating feature of a desmoplastic trichilemmoma is the pink hyalinized stroma separating small islets of basophilic cells (Figure 4). Differentiation from an invasive malignant carcinoma sometimes can be challenging without a focus of typical trichilemmoma or if the biopsy specimen is too superficial.29

Pilar cysts are common tumors that typically arise on the scalp and sometimes are proliferating. Proliferating pilar tumor should be kept on the differential when secondary changes such as ulceration occur in the primary lesion of the scalp. Microscopically, and sometimes clinically, PPT can be difficult to differentiate from other mimickers.
- Wilson-Jones E. Proliferating epidermoid cysts. Arch Dermatol. 1966;94:11-19.
- Saida T, Oohara K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatologica. 1983;166:203-208.
- Ye J, Nappi O, Swanson PE, et al. Proliferating pilar tumours: a clinicopathological study of 76 cases with a proposal for definition of benign and malignant variants. Am J Clin Pathol. 2004;122:566-574.
- Garg PK, Dangi A, Khurana N, et al. Malignant proliferating trichilemmal cyst: a case report with review of literature. Malaysian J Pathol. 2009;31:71-76.
- Herrero J, Monteagudo C, Ruiz A, et al. Malignant proliferating trichilemmal tumors: a histopathological and immunohistochemical study of three cases with DNA ploidy and morphometric evaluation. Histopathology. 1998;33:542-546.
- Haas N, Audring H, Sterry W. Carcinoma arising in a proliferating trichilemmal cyst expresses fetal and trichilemmal hair phenotype. Am J Dermatopathol. 2002;24:340-344.
- Rutty GN, Richman PI, Laing JH. Malignant change in trichilemmal cysts: a study of cell proliferation and DNA content. Histopathology. 1992;21:465-468.
- Brownstein MH, Arluk DJ. Proliferating trichilemmal cyst: a simulant of squamous cell carcinoma. Cancer. 1981;48:1207-1214.
- Misago N, Ackerman AB. Tricholemmal carcinoma? Dermatopathol Pract Concept. 1999;5:205-206.
- Misago N, Narisawa Y. Tricholemmal carcinoma in continuity with trichoblastoma within nevus sebaceous. Am J Dermatopathol. 2002;24:149-155.
- Liang H, Wu H, Giorgadze TA, et al. Podoplanin is a highly sensitive and specific marker to distinguish primary skin adnexal carcinomas from adenocarcinomas metastatic to skin. Am J Surg Pathol. 2007;31:304-310.
- Swanson PE, Marrogi AJ, Williams DJ, et al. Trichilemmal carcinoma: clinicopathologic study of 10 cases. J Cutan Pathol. 1992;19:100-109.
- Mehregan AH. Hair follicle tumors of the skin. J Cutan Pathol. 1985;12:189-195.
- Julian CG, Bowers PW. A clinical review of 209 pilomatricomas. J Am Acad Dermatol. 1998;39:191-195.
- Marrogi AJ, Wick MR, Dehner LP. Pilomatrical neoplasms in children and young adults. Am J Dermatopathol. 1992;14:87-94.
- Berberian BJ, Colonna TM, Battaglia M, et al. Multiple pilomatricomas in association with myotonic dystrophy and a family history of melanoma. J Am Acad Dermatol. 1997;37:268-269.
- Cooper PH, Fechner RE. Pilomatricoma-like changes in the epidermal cysts of Gardner's syndrome. J Am Acad Dermatol. 1983;8:639-644.
- Kaddu S, Soyer HP, Cerroni L, et al. Clinical and histopathologic spectrum of pilomatricomas in adults. Int J Dermatol. 1994;33:705-708.
- Sano Y, Mihara M, Miyamoto T, et al. Simultaneous occurrence of calcification and amyloid deposit in pilomatricoma. Acta Derm Venereol. 1990;70:256-259.
- Schwartz RA. Keratoacanthoma. J Am Acad Dermatol. 1994;30:1-19.
- Kwiek B, Schwartz RA. Keratoacanthoma (KA): an update and review. J Am Acad Dermatol. 2016;74:1220-1233.
- Mehregan AH. Inverted follicular keratosis. Arch Dermatol. 1964;89:117-123.
- Spielvogel RL, Austin C, Ackerman AB. Inverted follicular keratosis is not a specific keratosis but a verruca vulgaris (or seborrheic keratosis) with squamous eddies. Am J Dermatopathol. 1983;5:427-445.
- Mehregan AH. Inverted follicular keratosis is a distinct follicular tumor. Am J Dermatopathol. 1983;5:467-470.
- Brownstein MH. Trichilemmoma. benign follicular tumor or viral wart? Am J Dermatopathol. 1980;2:229-231.
- Brownstein MH. Multiple trichilemmomas in Cowden's syndrome. Arch Dermatol. 1979;115:111.
- Roson E, Gomez Centeno P, Sanchez Aguilar D, et al. Desmoplastic trichilemmoma arising within a nevus sebaceous. Am J Dermatopathol. 1998;20:495-497.
- Tellechea O, Reis JP, Baptista AP. Desmoplastic trichilemmoma. Am J Dermatopathol. 1992;14:107-114.
- Sharma R, Sirohi D, Sengupta P, et al. Desmoplastic trichilemmoma of the facial region mimicking invasive carcinoma. J Maxillofac Oral Surg. 2010;10:71-73.
The Diagnosis: Proliferating Pilar Tumor
Proliferating pilar tumor (PPT), or cyst, is a neoplasm of trichilemmal keratinization first described by Wilson-Jones1 in 1966. Proliferating pilar tumors lie on a spectrum with malignant PPT, which is a rare adnexal neoplasm first described by Saida et al2 in 1983. The incidence of PPT is unknown given the paucity of cases and the possible misdiagnosis as squamous cell carcinoma (SCC). Proliferating pilar tumors tend to present on the head and neck of older females as a multilobular and sometimes ulcerating nodule.3 Although PPT can occur de novo, the majority of cases are thought to develop progressively from a benign pilar cyst. Histopathologically, PPT is characterized by cords and nests of squamous cells that display trichilemmal keratinization (quiz images).
Classification of PPT as benign or malignant is challenging, though criteria have been proposed.3-7 Lesions with minimal infiltration into the surrounding dermis and scant mitosis typically behave in a benign manner, while lesions showing nuclear atypia, atypical mitosis, and irregular infiltration into the surrounding dermis can have up to a 50% locoregional recurrence rate.3 In addition, distinguishing a PPT from an SCC or trichilemmal carcinoma also can be difficult; however, SCC is favored when there is a lack of trichilemmal keratinization or when squamous atypia is present in the adjacent epidermis.8 Trichilemmal carcinoma is a rare tumor that has been questioned as a distinct entity.9-12
Pilomatricoma, also known as calcifying epithelioma of Malherbe, is a benign pilar tumor that presents as a slowly growing nodule on the head or neck area or arms.13,14 Most pilomatricomas develop by the second decade of life. Multiple lesions may be present in association with myotonic dystrophy or Gardner syndrome among other syndromes.15-17 Similar to PPT, pilomatricomas present as large dermal nodules; however, they tend to be circumscribed and have a trabecular network that consists of basophilic cells and eosinophilic keratinized shadow cells (Figure 1).18 Calcification may be seen and bone formation subsequently may occur.19
Most sources now consider keratoacanthoma (KA) as a well-differentiated SCC.20 The typical presentation consists of a rapidly growing erythematous to flesh-colored nodule with a central keratinous plug that develops over a period of weeks. If untreated, KAs may resolve over a period of months and leave a depressed scar. Local destruction can result from KAs, and they have the potential to transform into a more aggressive SCC. Accordingly, most clinicians use tissue destructive methods, excision, or Mohs micrographic surgery for treatment based on location. Histologically, a well-circumscribed proliferation of glassy cytoplasm is noted. A depressed keratin-filled center is surrounded by a lip of epithelium extending over the lesion (Figure 2).20,21 Pseudoepitheliomatous hyperplasia accompanied by hypergranulosis is seen in the center of KAs rather than at the periphery, which is typical of non-KA SCCs. Typical KAs lack acantholysis, a feature suggesting a non-KA type of SCC. Neutrophilic microabscesses and eosinophils commonly are seen in KAs.20,21
Inverted follicular keratosis is a benign tumor that gained traction as its own entity in the 1960s.22 These lesions typically develop from the follicular infundibulum, but some consider them a version of a wart or seborrheic keratosis.23 They generally are flesh-colored nodules on the upper cutaneous lip or face. Treatment usually consists of complete excision. There are many different growth patterns described, but they typically are endophytic tumors with eosinophilic squamous cells in the center and more basophilic cells at the periphery (Figure 3).24 Characteristically, there are squamous eddies throughout the tumor (Figure 3 [inset]). There also may be a scant lymphohistiocytic infiltrate within the dermis surrounding the lesion.
Trichilemmomas are flesh-colored adnexal neoplasms that may present as a solitary lesion or in clusters on the face. They have been reported to occur on all nonglabrous skin sites.25 Multiple lesions may occur in association with Cowden syndrome or with nevus sebaceous.26 A desmoplastic variant of trichilemmomas has been reported.27 Desmoplastic trichilemmomas appear as well-circumscribed tumors of outer root sheath differentiation with lobules extending down into the dermis.28 Vacuolated glycogen-filled keratinocytes are scattered throughout the lesion but are most prominent at the base. At the periphery of the lobules, peripheral palisading of basaloid cells is accompanied by a thickened eosinophilic basement membrane that is periodic acid-Schiff positive. Typical trichilemmomas also can display these features; however, the main differentiating feature of a desmoplastic trichilemmoma is the pink hyalinized stroma separating small islets of basophilic cells (Figure 4). Differentiation from an invasive malignant carcinoma sometimes can be challenging without a focus of typical trichilemmoma or if the biopsy specimen is too superficial.29
Pilar cysts are common tumors that typically arise on the scalp and sometimes are proliferating. Proliferating pilar tumor should be kept on the differential when secondary changes such as ulceration occur in the primary lesion of the scalp. Microscopically, and sometimes clinically, PPT can be difficult to differentiate from other mimickers.
The Diagnosis: Proliferating Pilar Tumor
Proliferating pilar tumor (PPT), or cyst, is a neoplasm of trichilemmal keratinization first described by Wilson-Jones1 in 1966. Proliferating pilar tumors lie on a spectrum with malignant PPT, which is a rare adnexal neoplasm first described by Saida et al2 in 1983. The incidence of PPT is unknown given the paucity of cases and the possible misdiagnosis as squamous cell carcinoma (SCC). Proliferating pilar tumors tend to present on the head and neck of older females as a multilobular and sometimes ulcerating nodule.3 Although PPT can occur de novo, the majority of cases are thought to develop progressively from a benign pilar cyst. Histopathologically, PPT is characterized by cords and nests of squamous cells that display trichilemmal keratinization (quiz images).
Classification of PPT as benign or malignant is challenging, though criteria have been proposed.3-7 Lesions with minimal infiltration into the surrounding dermis and scant mitosis typically behave in a benign manner, while lesions showing nuclear atypia, atypical mitosis, and irregular infiltration into the surrounding dermis can have up to a 50% locoregional recurrence rate.3 In addition, distinguishing a PPT from an SCC or trichilemmal carcinoma also can be difficult; however, SCC is favored when there is a lack of trichilemmal keratinization or when squamous atypia is present in the adjacent epidermis.8 Trichilemmal carcinoma is a rare tumor that has been questioned as a distinct entity.9-12
Pilomatricoma, also known as calcifying epithelioma of Malherbe, is a benign pilar tumor that presents as a slowly growing nodule on the head or neck area or arms.13,14 Most pilomatricomas develop by the second decade of life. Multiple lesions may be present in association with myotonic dystrophy or Gardner syndrome among other syndromes.15-17 Similar to PPT, pilomatricomas present as large dermal nodules; however, they tend to be circumscribed and have a trabecular network that consists of basophilic cells and eosinophilic keratinized shadow cells (Figure 1).18 Calcification may be seen and bone formation subsequently may occur.19
Most sources now consider keratoacanthoma (KA) as a well-differentiated SCC.20 The typical presentation consists of a rapidly growing erythematous to flesh-colored nodule with a central keratinous plug that develops over a period of weeks. If untreated, KAs may resolve over a period of months and leave a depressed scar. Local destruction can result from KAs, and they have the potential to transform into a more aggressive SCC. Accordingly, most clinicians use tissue destructive methods, excision, or Mohs micrographic surgery for treatment based on location. Histologically, a well-circumscribed proliferation of glassy cytoplasm is noted. A depressed keratin-filled center is surrounded by a lip of epithelium extending over the lesion (Figure 2).20,21 Pseudoepitheliomatous hyperplasia accompanied by hypergranulosis is seen in the center of KAs rather than at the periphery, which is typical of non-KA SCCs. Typical KAs lack acantholysis, a feature suggesting a non-KA type of SCC. Neutrophilic microabscesses and eosinophils commonly are seen in KAs.20,21
Inverted follicular keratosis is a benign tumor that gained traction as its own entity in the 1960s.22 These lesions typically develop from the follicular infundibulum, but some consider them a version of a wart or seborrheic keratosis.23 They generally are flesh-colored nodules on the upper cutaneous lip or face. Treatment usually consists of complete excision. There are many different growth patterns described, but they typically are endophytic tumors with eosinophilic squamous cells in the center and more basophilic cells at the periphery (Figure 3).24 Characteristically, there are squamous eddies throughout the tumor (Figure 3 [inset]). There also may be a scant lymphohistiocytic infiltrate within the dermis surrounding the lesion.
Trichilemmomas are flesh-colored adnexal neoplasms that may present as a solitary lesion or in clusters on the face. They have been reported to occur on all nonglabrous skin sites.25 Multiple lesions may occur in association with Cowden syndrome or with nevus sebaceous.26 A desmoplastic variant of trichilemmomas has been reported.27 Desmoplastic trichilemmomas appear as well-circumscribed tumors of outer root sheath differentiation with lobules extending down into the dermis.28 Vacuolated glycogen-filled keratinocytes are scattered throughout the lesion but are most prominent at the base. At the periphery of the lobules, peripheral palisading of basaloid cells is accompanied by a thickened eosinophilic basement membrane that is periodic acid-Schiff positive. Typical trichilemmomas also can display these features; however, the main differentiating feature of a desmoplastic trichilemmoma is the pink hyalinized stroma separating small islets of basophilic cells (Figure 4). Differentiation from an invasive malignant carcinoma sometimes can be challenging without a focus of typical trichilemmoma or if the biopsy specimen is too superficial.29
Pilar cysts are common tumors that typically arise on the scalp and sometimes are proliferating. Proliferating pilar tumor should be kept on the differential when secondary changes such as ulceration occur in the primary lesion of the scalp. Microscopically, and sometimes clinically, PPT can be difficult to differentiate from other mimickers.
- Wilson-Jones E. Proliferating epidermoid cysts. Arch Dermatol. 1966;94:11-19.
- Saida T, Oohara K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatologica. 1983;166:203-208.
- Ye J, Nappi O, Swanson PE, et al. Proliferating pilar tumours: a clinicopathological study of 76 cases with a proposal for definition of benign and malignant variants. Am J Clin Pathol. 2004;122:566-574.
- Garg PK, Dangi A, Khurana N, et al. Malignant proliferating trichilemmal cyst: a case report with review of literature. Malaysian J Pathol. 2009;31:71-76.
- Herrero J, Monteagudo C, Ruiz A, et al. Malignant proliferating trichilemmal tumors: a histopathological and immunohistochemical study of three cases with DNA ploidy and morphometric evaluation. Histopathology. 1998;33:542-546.
- Haas N, Audring H, Sterry W. Carcinoma arising in a proliferating trichilemmal cyst expresses fetal and trichilemmal hair phenotype. Am J Dermatopathol. 2002;24:340-344.
- Rutty GN, Richman PI, Laing JH. Malignant change in trichilemmal cysts: a study of cell proliferation and DNA content. Histopathology. 1992;21:465-468.
- Brownstein MH, Arluk DJ. Proliferating trichilemmal cyst: a simulant of squamous cell carcinoma. Cancer. 1981;48:1207-1214.
- Misago N, Ackerman AB. Tricholemmal carcinoma? Dermatopathol Pract Concept. 1999;5:205-206.
- Misago N, Narisawa Y. Tricholemmal carcinoma in continuity with trichoblastoma within nevus sebaceous. Am J Dermatopathol. 2002;24:149-155.
- Liang H, Wu H, Giorgadze TA, et al. Podoplanin is a highly sensitive and specific marker to distinguish primary skin adnexal carcinomas from adenocarcinomas metastatic to skin. Am J Surg Pathol. 2007;31:304-310.
- Swanson PE, Marrogi AJ, Williams DJ, et al. Trichilemmal carcinoma: clinicopathologic study of 10 cases. J Cutan Pathol. 1992;19:100-109.
- Mehregan AH. Hair follicle tumors of the skin. J Cutan Pathol. 1985;12:189-195.
- Julian CG, Bowers PW. A clinical review of 209 pilomatricomas. J Am Acad Dermatol. 1998;39:191-195.
- Marrogi AJ, Wick MR, Dehner LP. Pilomatrical neoplasms in children and young adults. Am J Dermatopathol. 1992;14:87-94.
- Berberian BJ, Colonna TM, Battaglia M, et al. Multiple pilomatricomas in association with myotonic dystrophy and a family history of melanoma. J Am Acad Dermatol. 1997;37:268-269.
- Cooper PH, Fechner RE. Pilomatricoma-like changes in the epidermal cysts of Gardner's syndrome. J Am Acad Dermatol. 1983;8:639-644.
- Kaddu S, Soyer HP, Cerroni L, et al. Clinical and histopathologic spectrum of pilomatricomas in adults. Int J Dermatol. 1994;33:705-708.
- Sano Y, Mihara M, Miyamoto T, et al. Simultaneous occurrence of calcification and amyloid deposit in pilomatricoma. Acta Derm Venereol. 1990;70:256-259.
- Schwartz RA. Keratoacanthoma. J Am Acad Dermatol. 1994;30:1-19.
- Kwiek B, Schwartz RA. Keratoacanthoma (KA): an update and review. J Am Acad Dermatol. 2016;74:1220-1233.
- Mehregan AH. Inverted follicular keratosis. Arch Dermatol. 1964;89:117-123.
- Spielvogel RL, Austin C, Ackerman AB. Inverted follicular keratosis is not a specific keratosis but a verruca vulgaris (or seborrheic keratosis) with squamous eddies. Am J Dermatopathol. 1983;5:427-445.
- Mehregan AH. Inverted follicular keratosis is a distinct follicular tumor. Am J Dermatopathol. 1983;5:467-470.
- Brownstein MH. Trichilemmoma. benign follicular tumor or viral wart? Am J Dermatopathol. 1980;2:229-231.
- Brownstein MH. Multiple trichilemmomas in Cowden's syndrome. Arch Dermatol. 1979;115:111.
- Roson E, Gomez Centeno P, Sanchez Aguilar D, et al. Desmoplastic trichilemmoma arising within a nevus sebaceous. Am J Dermatopathol. 1998;20:495-497.
- Tellechea O, Reis JP, Baptista AP. Desmoplastic trichilemmoma. Am J Dermatopathol. 1992;14:107-114.
- Sharma R, Sirohi D, Sengupta P, et al. Desmoplastic trichilemmoma of the facial region mimicking invasive carcinoma. J Maxillofac Oral Surg. 2010;10:71-73.
- Wilson-Jones E. Proliferating epidermoid cysts. Arch Dermatol. 1966;94:11-19.
- Saida T, Oohara K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatologica. 1983;166:203-208.
- Ye J, Nappi O, Swanson PE, et al. Proliferating pilar tumours: a clinicopathological study of 76 cases with a proposal for definition of benign and malignant variants. Am J Clin Pathol. 2004;122:566-574.
- Garg PK, Dangi A, Khurana N, et al. Malignant proliferating trichilemmal cyst: a case report with review of literature. Malaysian J Pathol. 2009;31:71-76.
- Herrero J, Monteagudo C, Ruiz A, et al. Malignant proliferating trichilemmal tumors: a histopathological and immunohistochemical study of three cases with DNA ploidy and morphometric evaluation. Histopathology. 1998;33:542-546.
- Haas N, Audring H, Sterry W. Carcinoma arising in a proliferating trichilemmal cyst expresses fetal and trichilemmal hair phenotype. Am J Dermatopathol. 2002;24:340-344.
- Rutty GN, Richman PI, Laing JH. Malignant change in trichilemmal cysts: a study of cell proliferation and DNA content. Histopathology. 1992;21:465-468.
- Brownstein MH, Arluk DJ. Proliferating trichilemmal cyst: a simulant of squamous cell carcinoma. Cancer. 1981;48:1207-1214.
- Misago N, Ackerman AB. Tricholemmal carcinoma? Dermatopathol Pract Concept. 1999;5:205-206.
- Misago N, Narisawa Y. Tricholemmal carcinoma in continuity with trichoblastoma within nevus sebaceous. Am J Dermatopathol. 2002;24:149-155.
- Liang H, Wu H, Giorgadze TA, et al. Podoplanin is a highly sensitive and specific marker to distinguish primary skin adnexal carcinomas from adenocarcinomas metastatic to skin. Am J Surg Pathol. 2007;31:304-310.
- Swanson PE, Marrogi AJ, Williams DJ, et al. Trichilemmal carcinoma: clinicopathologic study of 10 cases. J Cutan Pathol. 1992;19:100-109.
- Mehregan AH. Hair follicle tumors of the skin. J Cutan Pathol. 1985;12:189-195.
- Julian CG, Bowers PW. A clinical review of 209 pilomatricomas. J Am Acad Dermatol. 1998;39:191-195.
- Marrogi AJ, Wick MR, Dehner LP. Pilomatrical neoplasms in children and young adults. Am J Dermatopathol. 1992;14:87-94.
- Berberian BJ, Colonna TM, Battaglia M, et al. Multiple pilomatricomas in association with myotonic dystrophy and a family history of melanoma. J Am Acad Dermatol. 1997;37:268-269.
- Cooper PH, Fechner RE. Pilomatricoma-like changes in the epidermal cysts of Gardner's syndrome. J Am Acad Dermatol. 1983;8:639-644.
- Kaddu S, Soyer HP, Cerroni L, et al. Clinical and histopathologic spectrum of pilomatricomas in adults. Int J Dermatol. 1994;33:705-708.
- Sano Y, Mihara M, Miyamoto T, et al. Simultaneous occurrence of calcification and amyloid deposit in pilomatricoma. Acta Derm Venereol. 1990;70:256-259.
- Schwartz RA. Keratoacanthoma. J Am Acad Dermatol. 1994;30:1-19.
- Kwiek B, Schwartz RA. Keratoacanthoma (KA): an update and review. J Am Acad Dermatol. 2016;74:1220-1233.
- Mehregan AH. Inverted follicular keratosis. Arch Dermatol. 1964;89:117-123.
- Spielvogel RL, Austin C, Ackerman AB. Inverted follicular keratosis is not a specific keratosis but a verruca vulgaris (or seborrheic keratosis) with squamous eddies. Am J Dermatopathol. 1983;5:427-445.
- Mehregan AH. Inverted follicular keratosis is a distinct follicular tumor. Am J Dermatopathol. 1983;5:467-470.
- Brownstein MH. Trichilemmoma. benign follicular tumor or viral wart? Am J Dermatopathol. 1980;2:229-231.
- Brownstein MH. Multiple trichilemmomas in Cowden's syndrome. Arch Dermatol. 1979;115:111.
- Roson E, Gomez Centeno P, Sanchez Aguilar D, et al. Desmoplastic trichilemmoma arising within a nevus sebaceous. Am J Dermatopathol. 1998;20:495-497.
- Tellechea O, Reis JP, Baptista AP. Desmoplastic trichilemmoma. Am J Dermatopathol. 1992;14:107-114.
- Sharma R, Sirohi D, Sengupta P, et al. Desmoplastic trichilemmoma of the facial region mimicking invasive carcinoma. J Maxillofac Oral Surg. 2010;10:71-73.
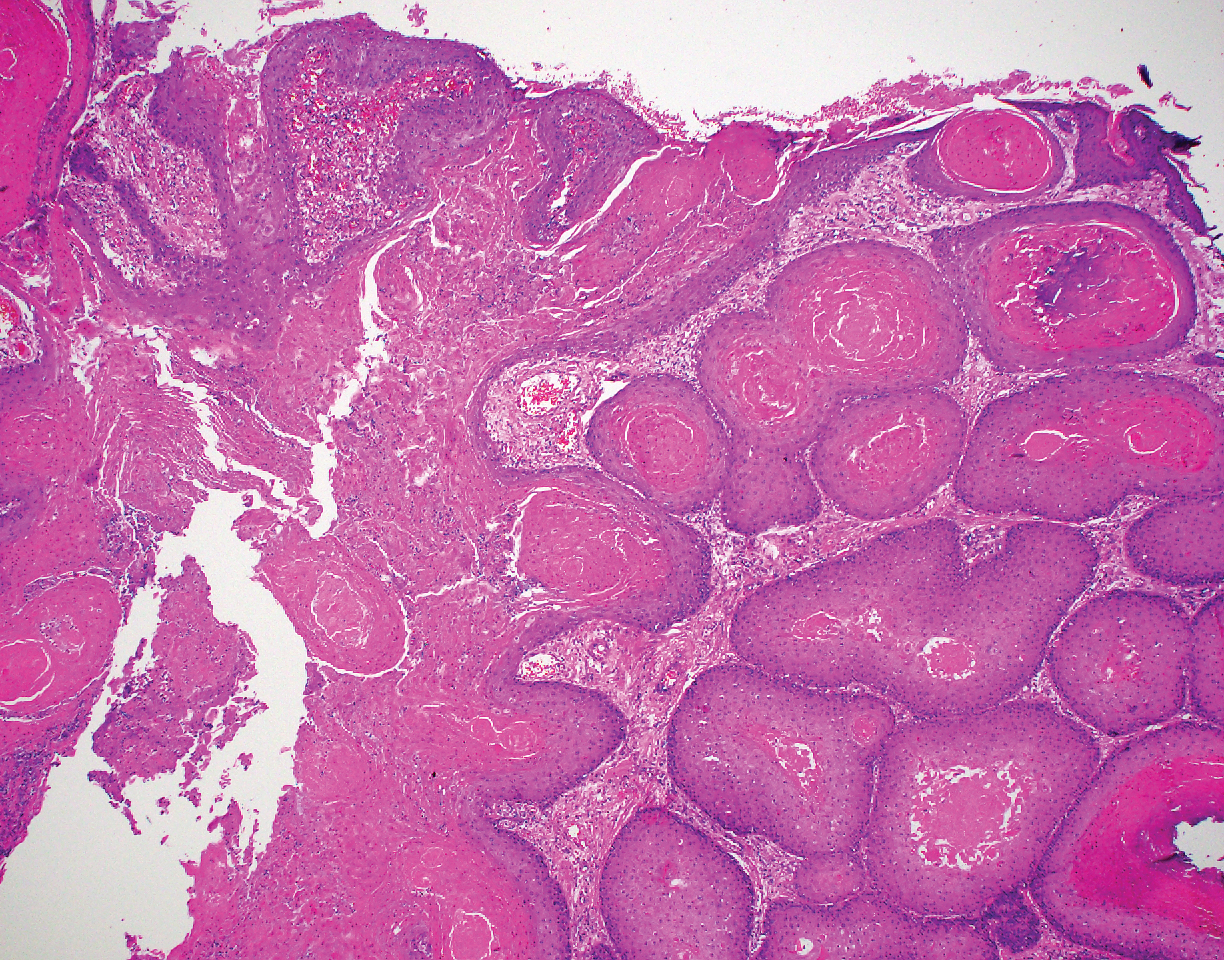
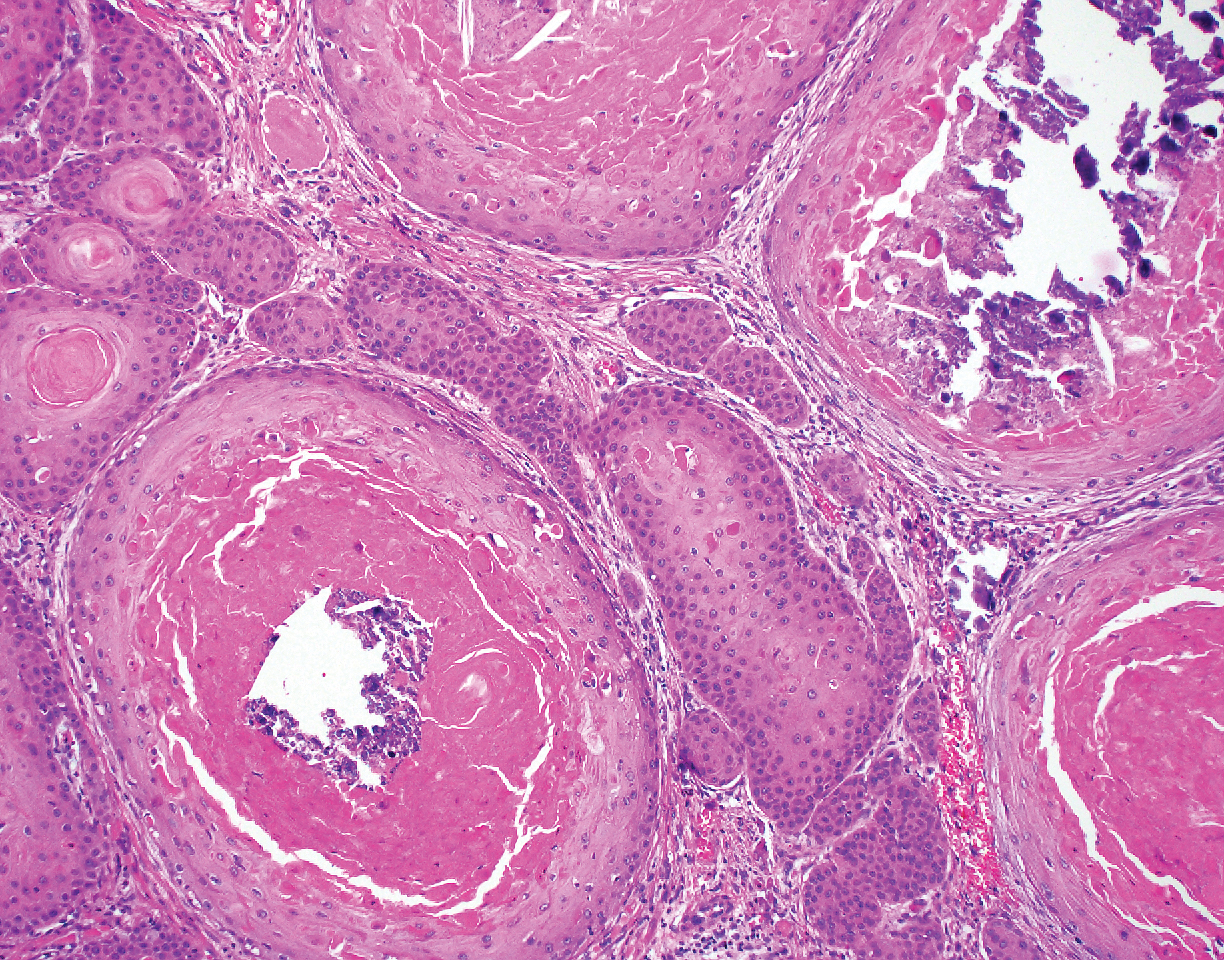
A 66-year-old woman presented to the dermatology clinic with a rapidly enlarging, draining lesion on the scalp. The lesion seemed to enlarge over the last 3 months from a lesion that had been there for years. Physical examination revealed a 2.2-cm ulcerated nodule on the right parietal scalp. A shave biopsy was obtained.
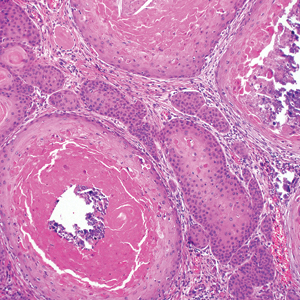
What is your diagnosis? - May 2019
Nonsteroidal anti-inflammatory drug–induced diaphragm disease
Nonsteroidal anti-inflammatory drug (NSAID)–induced diaphragm disease is a rare cause of colonic stricture. To date, only about 50 cases have been reported. Diaphragm-like strictures occur predominantly in the right colon. The most common clinical presentations are obstructive symptoms and gastrointestinal bleeding after taking traditional NSAIDs or cyclo-oxygenase-2 inhibitors for more than 1 year.1 The thin diaphragm strictures are difficult to detect on imaging studies. They are seen during endoscopy or surgery. Concentric strictures in the right colon in the setting of chronic NSAID use are almost diagnostic of colonic diaphragm disease.2 Histopathology demonstrates submucosal fibrosis on resection specimens. Endoscopic biopsies may show lamina propria fibrosis, increased eosinophils with relative paucity of neutrophils, and even crypt distortion. The mechanism is thought to be due to contraction of scar tissue from healing concentric ulceration resulting in diaphragm-like strictures. Discontinuation of NSAIDs is recommended. Surgery is required to relieve obstruction in 75% of reported cases. Some have reported success with endoscopic balloon dilation.3
Using a 15-mm balloon, we performed endoscopic through-the-scope balloon dilation under fluoroscopy (Figures D, E). After dilation, the colonoscopy was completed to the distal ileum. There were two additional proximal concentric colonic strictures that allowed passage of the colonoscope and did not require dilation (Figure F). The patient was advised to stop diclofenac. He had no further gastrointestinal symptoms at the 3-month follow-up visit.
References
1. Wang Y-Z, Sun G, Cai F-C et al. Clinical features, diagnosis, and treatment strategies of gastrointestinal diaphragm disease associated with nonsteroidal anti-inflammatory drugs. Gastroenterol Res Pract. 2016;2016:3679741
2. Püspök A, Kiener HP, Oberhuber G. Clinical, endoscopic, and histologic spectrum of nonsteroidal anti-inflammatory drug-induced lesions in the colon. Dis Colon Rectum. 2000;43:685-91.
3. Smith JA, Pineau BC. Endoscopic therapy of NSAID-induced colonic diaphragm disease: two cases and a review of published reports. Gastrointest Endosc. 2000;52:120-5.
Nonsteroidal anti-inflammatory drug–induced diaphragm disease
Nonsteroidal anti-inflammatory drug (NSAID)–induced diaphragm disease is a rare cause of colonic stricture. To date, only about 50 cases have been reported. Diaphragm-like strictures occur predominantly in the right colon. The most common clinical presentations are obstructive symptoms and gastrointestinal bleeding after taking traditional NSAIDs or cyclo-oxygenase-2 inhibitors for more than 1 year.1 The thin diaphragm strictures are difficult to detect on imaging studies. They are seen during endoscopy or surgery. Concentric strictures in the right colon in the setting of chronic NSAID use are almost diagnostic of colonic diaphragm disease.2 Histopathology demonstrates submucosal fibrosis on resection specimens. Endoscopic biopsies may show lamina propria fibrosis, increased eosinophils with relative paucity of neutrophils, and even crypt distortion. The mechanism is thought to be due to contraction of scar tissue from healing concentric ulceration resulting in diaphragm-like strictures. Discontinuation of NSAIDs is recommended. Surgery is required to relieve obstruction in 75% of reported cases. Some have reported success with endoscopic balloon dilation.3
Using a 15-mm balloon, we performed endoscopic through-the-scope balloon dilation under fluoroscopy (Figures D, E). After dilation, the colonoscopy was completed to the distal ileum. There were two additional proximal concentric colonic strictures that allowed passage of the colonoscope and did not require dilation (Figure F). The patient was advised to stop diclofenac. He had no further gastrointestinal symptoms at the 3-month follow-up visit.
References
1. Wang Y-Z, Sun G, Cai F-C et al. Clinical features, diagnosis, and treatment strategies of gastrointestinal diaphragm disease associated with nonsteroidal anti-inflammatory drugs. Gastroenterol Res Pract. 2016;2016:3679741
2. Püspök A, Kiener HP, Oberhuber G. Clinical, endoscopic, and histologic spectrum of nonsteroidal anti-inflammatory drug-induced lesions in the colon. Dis Colon Rectum. 2000;43:685-91.
3. Smith JA, Pineau BC. Endoscopic therapy of NSAID-induced colonic diaphragm disease: two cases and a review of published reports. Gastrointest Endosc. 2000;52:120-5.
Nonsteroidal anti-inflammatory drug–induced diaphragm disease
Nonsteroidal anti-inflammatory drug (NSAID)–induced diaphragm disease is a rare cause of colonic stricture. To date, only about 50 cases have been reported. Diaphragm-like strictures occur predominantly in the right colon. The most common clinical presentations are obstructive symptoms and gastrointestinal bleeding after taking traditional NSAIDs or cyclo-oxygenase-2 inhibitors for more than 1 year.1 The thin diaphragm strictures are difficult to detect on imaging studies. They are seen during endoscopy or surgery. Concentric strictures in the right colon in the setting of chronic NSAID use are almost diagnostic of colonic diaphragm disease.2 Histopathology demonstrates submucosal fibrosis on resection specimens. Endoscopic biopsies may show lamina propria fibrosis, increased eosinophils with relative paucity of neutrophils, and even crypt distortion. The mechanism is thought to be due to contraction of scar tissue from healing concentric ulceration resulting in diaphragm-like strictures. Discontinuation of NSAIDs is recommended. Surgery is required to relieve obstruction in 75% of reported cases. Some have reported success with endoscopic balloon dilation.3
Using a 15-mm balloon, we performed endoscopic through-the-scope balloon dilation under fluoroscopy (Figures D, E). After dilation, the colonoscopy was completed to the distal ileum. There were two additional proximal concentric colonic strictures that allowed passage of the colonoscope and did not require dilation (Figure F). The patient was advised to stop diclofenac. He had no further gastrointestinal symptoms at the 3-month follow-up visit.
References
1. Wang Y-Z, Sun G, Cai F-C et al. Clinical features, diagnosis, and treatment strategies of gastrointestinal diaphragm disease associated with nonsteroidal anti-inflammatory drugs. Gastroenterol Res Pract. 2016;2016:3679741
2. Püspök A, Kiener HP, Oberhuber G. Clinical, endoscopic, and histologic spectrum of nonsteroidal anti-inflammatory drug-induced lesions in the colon. Dis Colon Rectum. 2000;43:685-91.
3. Smith JA, Pineau BC. Endoscopic therapy of NSAID-induced colonic diaphragm disease: two cases and a review of published reports. Gastrointest Endosc. 2000;52:120-5.
A 57-year-old man presented to our hospital with a week of generalized weakness and abdominal pain.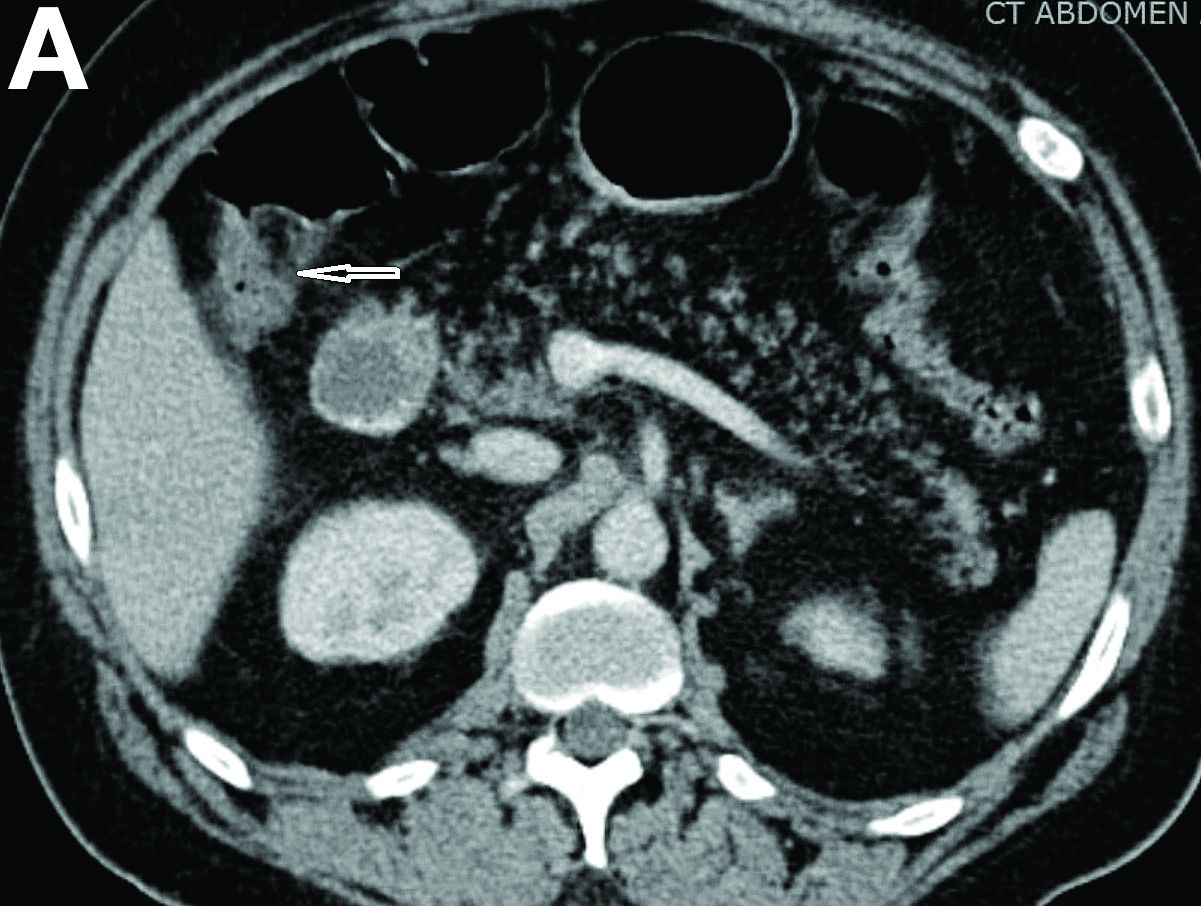
Relevant medications included diclofenac 75 mg twice daily, aspirin 81 mg, and clopidogrel 75 mg/d. Vital signs were normal. Physical examination showed mild diffuse abdominal tenderness.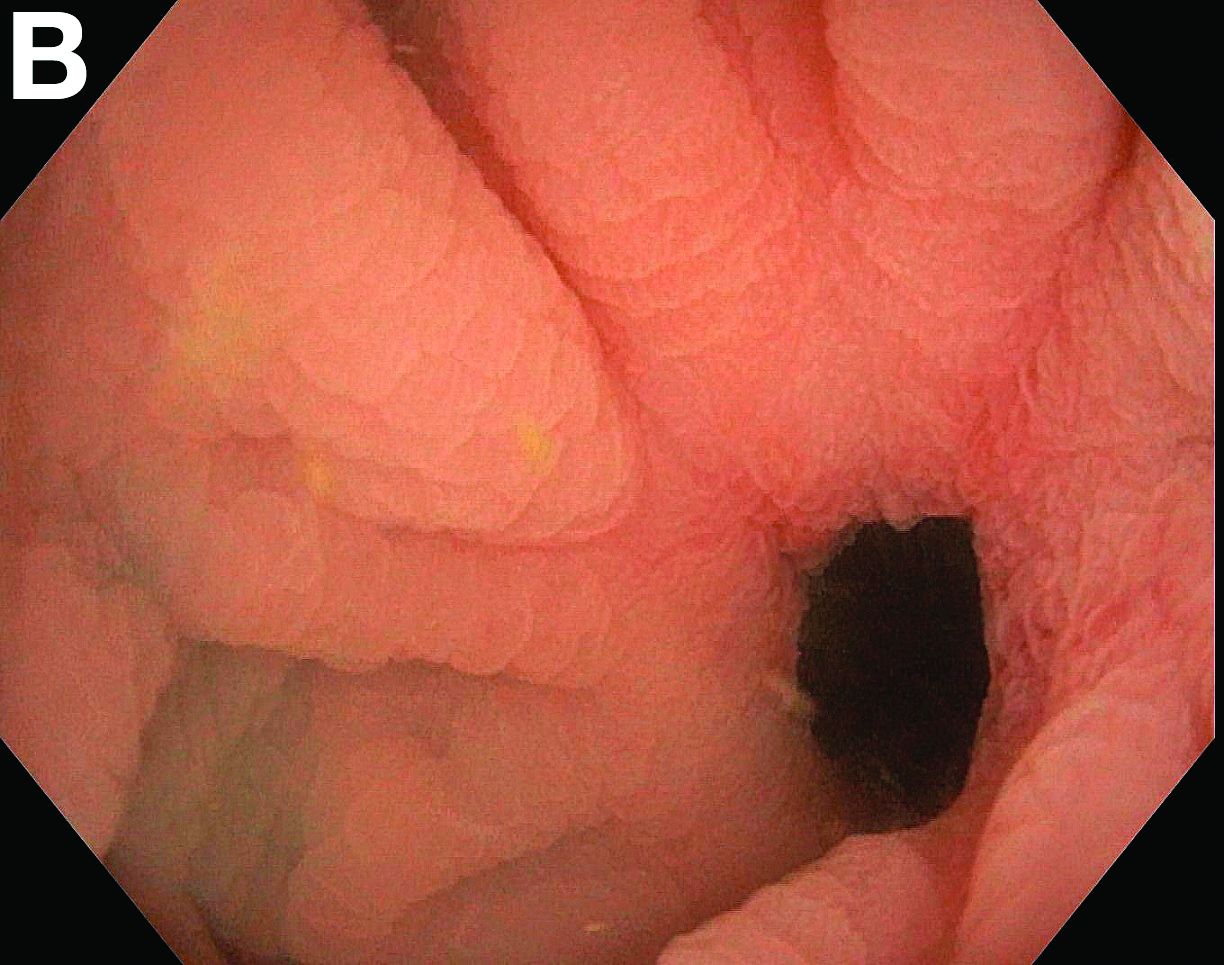
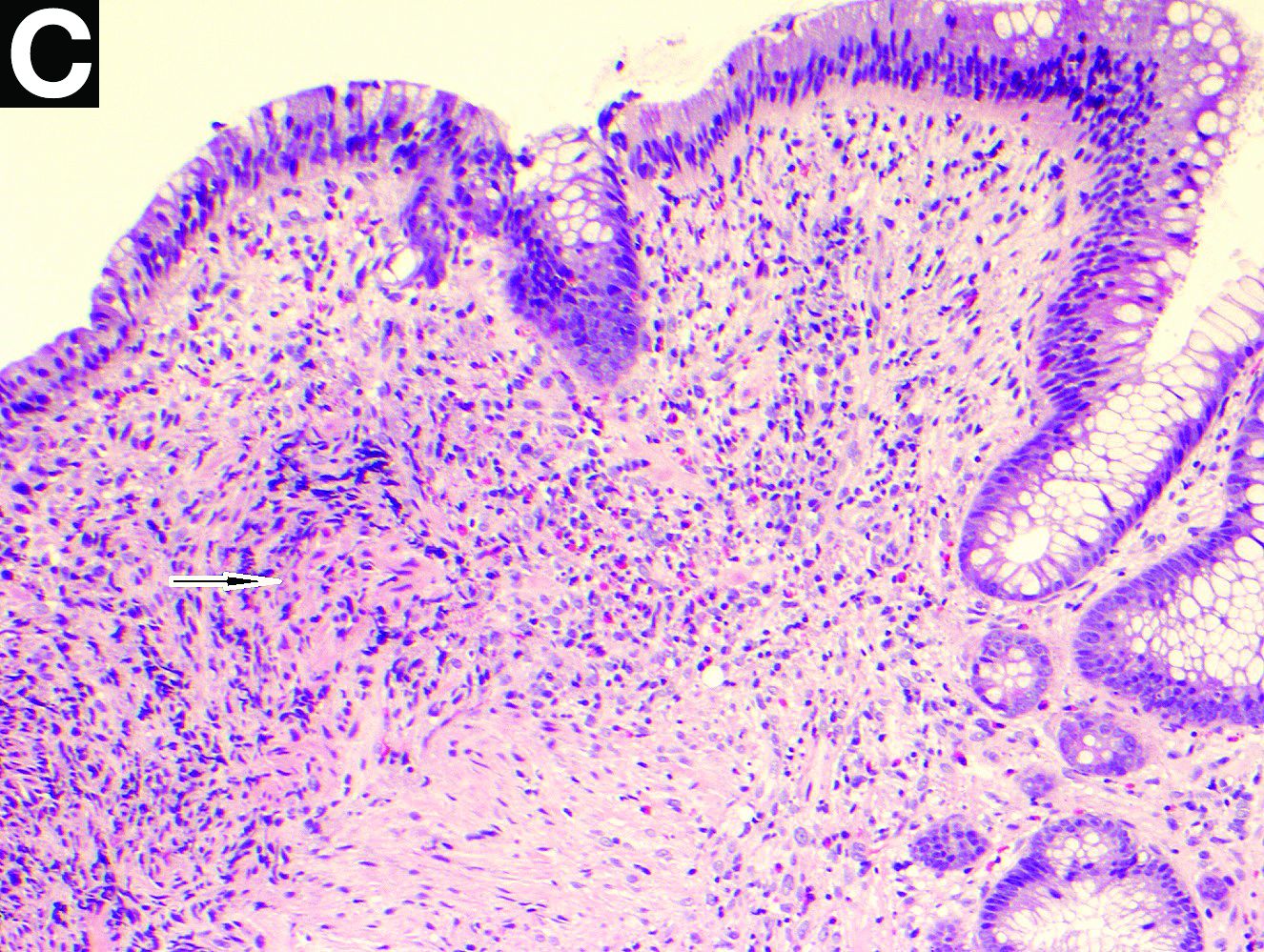
Admission blood work revealed a hemoglobin of 8.8 g/dL, decreased from a baseline hemoglobin of 12 g/dL. The patient did not have overt gastrointestinal bleeding, but tested positive for fecal occult blood. A computed tomography scan demonstrated luminal narrowing at the hepatic flexure without bowel wall thickening or obstruction (Figure A). Esophagogastroduodenoscopy was normal. Colonoscopy revealed a circumferential stricture in the right colon with an estimated diameter of 8 mm (Figure B).
Biopsies of the stricture showed significant lamina propria fibrosis, eosinophilic infiltration, and mild crypt distortion (Figure C).