User login
Orlistat-Induced Bullous Leukocytoclastic Vasculitis
The Appearance of Pili Annulati Following Alopecia Areata
Thrombogenic Vasculopathy With Diffuse Neutrophilic Inflammation: A Histologic Manifestation of a Tick Bite
Localized Argyria After Exposure to Aerosolized Solder
Localized argyria is a rare disorder that occurs less frequently than generalized argyria. The pathogenesis involves direct implantation of silver in the skin or, more rarely, percutaneous absorption of silver salts via the eccrine glands. The silver salts are released into the surrounding tissues. Occupational exposure is the most common cause of localized argyria and occurs most frequently in miners, photographic laboratory workers, and jewelers. Round or oval well-demarcated blue-gray macules typically are seen. Generalized argyria most often results from systemic treatment with drugs that contain silver salts or from inhalation of silver particulates in the workplace.1 Generalized argyria usually presents with blue-gray discoloration of the skin, including non—sun-exposed skin, the lips, tongue, mucous membranes, lunulae, and sclera. Permanent diffuse blue-gray pigmentary change of the skin is observed with generalized argyria.
Case Report
A 58-year-old man presented with an ashen color to his face that had progressed over several years. The patient denied taking any medications and had no significant past medical history. The results of a complete blood count, chemistry panel, liver function test, and hepatitis panel were within reference range. His serum silver level also was within reference range at 11 ng/mL (reference range, 0—14 ng/mL). The results of a physical examination revealed diffuse blue-gray pigmentation distributed over the face and neck with a sharp line of demarcation at the collar (Figure 1). There was no photoaccentuation and no involvement of the nails, mucous membrane, or sclera. For the past 20 years, the patient worked as an electronics technician soldering silver-containing wire in the construction of electronic devices. He wore gloves, long-sleeved shirts, and pants but no protective mask.
The biopsy specimen revealed small, black, refractile granules within the membrane propria of the eccrine glands (Figure 2). Additional biopsy specimens were taken from the healthy skin of the right upper back and revealed no silver granules. These histologic features were consistent with argyria.
Comment
Localized argyria is a rare disorder that presents with asymptomatic blue-gray macules.2 The lesions may be large and ill defined or sharply demarcated, resembling blue nevi.1 The most common cause of localized argyria is occupational exposure; small silver particles enter the skin by mechanical impregnation of workers involved in silver mining, silver refining, silverware and metal alloy manufacturing, and photographic processing.3 Localized argyria also has been attributed to surgical and dental procedures, silver earrings, and acupuncture needles.4-6 Additionally, localized argyria may be caused by percutaneous absorption of silver salts via the eccrine glands, which most likely occurred in this case. Generalized argyria most commonly results from long-term systemic use of silver-containing nose drops or colloidal silver—containing dietary supplements,7 homemade silver solution,8 and ingested or topical silver nitrate.9 Inhalation of silver-bearing dust in industries such as silver refining or metal grinding also may cause generalized argyria. Corneal argyrosis associated with silver soldering has been previously reported10,11; full cutaneous examination was not described in these patients. The presentation of generalized argyria typically begins with gray-brown staining of the gums that progresses to involve the skin diffusely. The mucocutaneous findings in argyria are the results of elevated serum silver levels, which lead to dermal and mucosal deposition of the metal. Histopathology evaluations reveal black-silver granules around the eccrine glands, in the walls of blood vessels, and along elastic fibers. The granules occasionally are found in the arrector pili muscles, perineural tissue, and around collagen fibrils. The slate gray, metallic, or blue-gray pigmentation seen in argyria may be clinically apparent after a few months but usually takes years to develop and depends on the degree of exposure.12,13 In some patients, the entire skin may acquire a slate blue—gray color. Hyperpigmentation is most apparent in sun-exposed areas of the skin, especially the forehead, nose, and hands. Although pigmentary changes occur primarily in sun-exposed sites, the granules are deposited evenly throughout the skin. Light causes silver-containing compounds complexed with proteins in the skin to be reduced to elemental silver, similar to the process of developing photographs.14 In addition, the silver stimulates melanocyte tyrosinase activity, which results in an increase in melanin production.15 The sclerae, nail beds, and mucous membranes also may become hyperpigmented. Viscera, including the spleen, liver, and gut, tend to show a blue discoloration that is evident during abdominal surgery or at postmortem examination. Our patient is unique because he presented with the diffuse pigmentary changes that would be seen with generalized argyria, but the pigmentary changes were limited to his face and neck. Other features suggestive of generalized argyria, such as sclerae and nail changes or silver impregnation in non—sun-exposed skin, were not present. The presence of silver granules in the eccrine glands of only exposed skin favors a diagnosis of localized argyria because of percutaneous silver absorption via the eccrine glands. A careful history is necessary in the diagnosis of argyria, with inquiries about possible occupational and environmental exposure and the use of dietary supplements containing colloidal silver protein. Habitual use of silver-based nose drops may produce pigmentation that is most apparent on the nose and nail lunulae. Scar-localized argyria may occur secondary to the use of silver sulfadiazine cream.16 Other causes of diffuse blue-gray pigmentation include medications (eg, phenothiazines, antimalarials, amiodarone, minocycline), heavy metal exposure (eg, mercury, bismuth, arsenic, gold, lead), hemochromatosis, ochronosis, cyanosis, polycythemia vera, and diffuse melanosis in metastatic melanoma.17 The average human body contains approximately 1 mg of silver.18 Serum silver has a reference range of 0 to 14 ng/mL. The smallest amount of silver reported to produce generalized argyria in humans ranges from 5 to 40 g.19 Although the amount of silver in argyria usually results in no serious effects on human health, a few cases of notable clinical symptoms and signs have been documented. Some of the complications of systemic toxic effects of silver include gastrointestinal tract catarrh, tissue wasting, uremia, albuminuria, fatty degeneration of the liver, hemorrhage, and idiopathic thrombocytopenia.20 The treatment of both localized and generalized argyria is difficult. Hydroquinone, depigmenting creams, and dermabrasion are not successful.9 Selenium and sulfur have been shown to have favorable modifying effects on the metabolism and toxicity of silver by forming complexes with the silver. The Q-switched double-frequency Nd:YAG laser, which has been used in the treatment of tattoos, also may be effective in the treatment of localized argyria.21 Unfortunately, no completely satisfactory treatment modality exists and some pigmentation remains permanent. However, sunscreens and opaque cosmetics may be helpful in masking discoloration and preventing further pigmentary darkening.17 To our knowledge, this is the first presented case of localized argyria secondary to aerosolized silver. This case emphasizes the need for skin protection in individuals with occupational exposure to aerosolized solder.
- Gettler AO, Rhoads CP, Weiss S. A contribution to the pathology of generalized argyria with a discussion of the state of silver in the human body. Am J Pathol. 1927;3:631-652.
- Espinel ML, Ferrando L, Diaz JF. Asymptomatic blue nevus-like macule. Arch Dermatol. 1996;132:459-464.
- Kapur N, Landon G, Yu RC. Localized argyria in an antique restorer. Br J Dermatol. 2001;144:191-193.
- McGinnis JP Jr, Greer JL, Daniels DS. Amalgam tattoo. J Am Dent Assoc. 1985;110:52-54.
- Shall L, Stevens A, Millard LG. An unusual case of acquired localized argyria. Br J Dermatol. 1990;123:403-407.
- Tanita Y, Kato T, Hanada K, et al. Blue macules of localized argyria caused by implanted acupuncture needles. Arch Dermatol. 1985;121:1550-1552.
- Wadhera A, Fung M. Systemic argyria associated with ingestion of colloidal silver. Dermatol Online J. 2005;11:12.
- Brandt D, Park B, Hoag M, et al. Argyria secondary to ingestion of homemade silver solution. J Am Acad Dermatol. 2005;53(2 suppl 1):S105-S107.
- Marshall JP, Schneider RP. Systemic argyria secondary to topical silver nitrate. Arch Dermatol. 1977;113:1077-1079.
- Scroggs MW, Lewis JS, Proia AD. Corneal argyrosis associated with silver soldering. Cornea. 1992;11:164-169.
- Sanchez-Huerta V, De Wit-Carter G, Hernandez-Quintela E, et al. Occupational corneal argyrosis in art silver solderers. Cornea. 2003;22:604-611.
- Greene RM, Su WPD. Argyria. Am Fam Pract. 1987;36:151-154.
- Peterson WC. Argyria. Minn Med. 1968;51:533-534.
- Shelley WB, Shelley ED, Burmeister V. Argyria: the intradermal photograph, a manifestation of passive photosensitivity. J Am Acad Dermatol. 1987;16:211-217.
- Buckley WR, Terhaar CJ. The skin as an excretory organ in argyria. Trans St Johns Hosp Dermatol Soc. 1973;59:39-44.
- Fisher NM, Marsh E, Lazova R. Scar-localized argyria secondary to silver sulfadiazine cream. J Am Acad Dermatol. 2003;49:730-732.
- Tanner LS, Gross DJ. Generalized argyria. Cutis. 1990;45:237-239.
- Venugopal B, Luckey TD. Metal toxicity in mammals. In: Venugopal B, Luckey TD, eds. Chemical Toxicology of Metals and Metalloids. Vol 2. New York, NY: Academic Press; 1978:32-36.
- Bleehen SS, Gould DJ, Harrington CI, et al. Occupational argyria; light and electron microscopic studies and X-ray microanalysis. Br J Dermatol. 1981;104:19-26.
- Sato S, Sueki H, Nishijima A. Two unusual cases of argyria: the application of an improved tissue processing method for x-ray microanalysis of selenium and sulphur in silver-laden granules. Br J Dermatol. 1999;140:158-163.
- Robinson-Bostom L, Pomerantz D, Wilkel C, et al. Localized argyria with pseudo-ochronosis. J Am
Localized argyria is a rare disorder that occurs less frequently than generalized argyria. The pathogenesis involves direct implantation of silver in the skin or, more rarely, percutaneous absorption of silver salts via the eccrine glands. The silver salts are released into the surrounding tissues. Occupational exposure is the most common cause of localized argyria and occurs most frequently in miners, photographic laboratory workers, and jewelers. Round or oval well-demarcated blue-gray macules typically are seen. Generalized argyria most often results from systemic treatment with drugs that contain silver salts or from inhalation of silver particulates in the workplace.1 Generalized argyria usually presents with blue-gray discoloration of the skin, including non—sun-exposed skin, the lips, tongue, mucous membranes, lunulae, and sclera. Permanent diffuse blue-gray pigmentary change of the skin is observed with generalized argyria.
Case Report
A 58-year-old man presented with an ashen color to his face that had progressed over several years. The patient denied taking any medications and had no significant past medical history. The results of a complete blood count, chemistry panel, liver function test, and hepatitis panel were within reference range. His serum silver level also was within reference range at 11 ng/mL (reference range, 0—14 ng/mL). The results of a physical examination revealed diffuse blue-gray pigmentation distributed over the face and neck with a sharp line of demarcation at the collar (Figure 1). There was no photoaccentuation and no involvement of the nails, mucous membrane, or sclera. For the past 20 years, the patient worked as an electronics technician soldering silver-containing wire in the construction of electronic devices. He wore gloves, long-sleeved shirts, and pants but no protective mask.
The biopsy specimen revealed small, black, refractile granules within the membrane propria of the eccrine glands (Figure 2). Additional biopsy specimens were taken from the healthy skin of the right upper back and revealed no silver granules. These histologic features were consistent with argyria.
Comment
Localized argyria is a rare disorder that presents with asymptomatic blue-gray macules.2 The lesions may be large and ill defined or sharply demarcated, resembling blue nevi.1 The most common cause of localized argyria is occupational exposure; small silver particles enter the skin by mechanical impregnation of workers involved in silver mining, silver refining, silverware and metal alloy manufacturing, and photographic processing.3 Localized argyria also has been attributed to surgical and dental procedures, silver earrings, and acupuncture needles.4-6 Additionally, localized argyria may be caused by percutaneous absorption of silver salts via the eccrine glands, which most likely occurred in this case. Generalized argyria most commonly results from long-term systemic use of silver-containing nose drops or colloidal silver—containing dietary supplements,7 homemade silver solution,8 and ingested or topical silver nitrate.9 Inhalation of silver-bearing dust in industries such as silver refining or metal grinding also may cause generalized argyria. Corneal argyrosis associated with silver soldering has been previously reported10,11; full cutaneous examination was not described in these patients. The presentation of generalized argyria typically begins with gray-brown staining of the gums that progresses to involve the skin diffusely. The mucocutaneous findings in argyria are the results of elevated serum silver levels, which lead to dermal and mucosal deposition of the metal. Histopathology evaluations reveal black-silver granules around the eccrine glands, in the walls of blood vessels, and along elastic fibers. The granules occasionally are found in the arrector pili muscles, perineural tissue, and around collagen fibrils. The slate gray, metallic, or blue-gray pigmentation seen in argyria may be clinically apparent after a few months but usually takes years to develop and depends on the degree of exposure.12,13 In some patients, the entire skin may acquire a slate blue—gray color. Hyperpigmentation is most apparent in sun-exposed areas of the skin, especially the forehead, nose, and hands. Although pigmentary changes occur primarily in sun-exposed sites, the granules are deposited evenly throughout the skin. Light causes silver-containing compounds complexed with proteins in the skin to be reduced to elemental silver, similar to the process of developing photographs.14 In addition, the silver stimulates melanocyte tyrosinase activity, which results in an increase in melanin production.15 The sclerae, nail beds, and mucous membranes also may become hyperpigmented. Viscera, including the spleen, liver, and gut, tend to show a blue discoloration that is evident during abdominal surgery or at postmortem examination. Our patient is unique because he presented with the diffuse pigmentary changes that would be seen with generalized argyria, but the pigmentary changes were limited to his face and neck. Other features suggestive of generalized argyria, such as sclerae and nail changes or silver impregnation in non—sun-exposed skin, were not present. The presence of silver granules in the eccrine glands of only exposed skin favors a diagnosis of localized argyria because of percutaneous silver absorption via the eccrine glands. A careful history is necessary in the diagnosis of argyria, with inquiries about possible occupational and environmental exposure and the use of dietary supplements containing colloidal silver protein. Habitual use of silver-based nose drops may produce pigmentation that is most apparent on the nose and nail lunulae. Scar-localized argyria may occur secondary to the use of silver sulfadiazine cream.16 Other causes of diffuse blue-gray pigmentation include medications (eg, phenothiazines, antimalarials, amiodarone, minocycline), heavy metal exposure (eg, mercury, bismuth, arsenic, gold, lead), hemochromatosis, ochronosis, cyanosis, polycythemia vera, and diffuse melanosis in metastatic melanoma.17 The average human body contains approximately 1 mg of silver.18 Serum silver has a reference range of 0 to 14 ng/mL. The smallest amount of silver reported to produce generalized argyria in humans ranges from 5 to 40 g.19 Although the amount of silver in argyria usually results in no serious effects on human health, a few cases of notable clinical symptoms and signs have been documented. Some of the complications of systemic toxic effects of silver include gastrointestinal tract catarrh, tissue wasting, uremia, albuminuria, fatty degeneration of the liver, hemorrhage, and idiopathic thrombocytopenia.20 The treatment of both localized and generalized argyria is difficult. Hydroquinone, depigmenting creams, and dermabrasion are not successful.9 Selenium and sulfur have been shown to have favorable modifying effects on the metabolism and toxicity of silver by forming complexes with the silver. The Q-switched double-frequency Nd:YAG laser, which has been used in the treatment of tattoos, also may be effective in the treatment of localized argyria.21 Unfortunately, no completely satisfactory treatment modality exists and some pigmentation remains permanent. However, sunscreens and opaque cosmetics may be helpful in masking discoloration and preventing further pigmentary darkening.17 To our knowledge, this is the first presented case of localized argyria secondary to aerosolized silver. This case emphasizes the need for skin protection in individuals with occupational exposure to aerosolized solder.
Localized argyria is a rare disorder that occurs less frequently than generalized argyria. The pathogenesis involves direct implantation of silver in the skin or, more rarely, percutaneous absorption of silver salts via the eccrine glands. The silver salts are released into the surrounding tissues. Occupational exposure is the most common cause of localized argyria and occurs most frequently in miners, photographic laboratory workers, and jewelers. Round or oval well-demarcated blue-gray macules typically are seen. Generalized argyria most often results from systemic treatment with drugs that contain silver salts or from inhalation of silver particulates in the workplace.1 Generalized argyria usually presents with blue-gray discoloration of the skin, including non—sun-exposed skin, the lips, tongue, mucous membranes, lunulae, and sclera. Permanent diffuse blue-gray pigmentary change of the skin is observed with generalized argyria.
Case Report
A 58-year-old man presented with an ashen color to his face that had progressed over several years. The patient denied taking any medications and had no significant past medical history. The results of a complete blood count, chemistry panel, liver function test, and hepatitis panel were within reference range. His serum silver level also was within reference range at 11 ng/mL (reference range, 0—14 ng/mL). The results of a physical examination revealed diffuse blue-gray pigmentation distributed over the face and neck with a sharp line of demarcation at the collar (Figure 1). There was no photoaccentuation and no involvement of the nails, mucous membrane, or sclera. For the past 20 years, the patient worked as an electronics technician soldering silver-containing wire in the construction of electronic devices. He wore gloves, long-sleeved shirts, and pants but no protective mask.
The biopsy specimen revealed small, black, refractile granules within the membrane propria of the eccrine glands (Figure 2). Additional biopsy specimens were taken from the healthy skin of the right upper back and revealed no silver granules. These histologic features were consistent with argyria.
Comment
Localized argyria is a rare disorder that presents with asymptomatic blue-gray macules.2 The lesions may be large and ill defined or sharply demarcated, resembling blue nevi.1 The most common cause of localized argyria is occupational exposure; small silver particles enter the skin by mechanical impregnation of workers involved in silver mining, silver refining, silverware and metal alloy manufacturing, and photographic processing.3 Localized argyria also has been attributed to surgical and dental procedures, silver earrings, and acupuncture needles.4-6 Additionally, localized argyria may be caused by percutaneous absorption of silver salts via the eccrine glands, which most likely occurred in this case. Generalized argyria most commonly results from long-term systemic use of silver-containing nose drops or colloidal silver—containing dietary supplements,7 homemade silver solution,8 and ingested or topical silver nitrate.9 Inhalation of silver-bearing dust in industries such as silver refining or metal grinding also may cause generalized argyria. Corneal argyrosis associated with silver soldering has been previously reported10,11; full cutaneous examination was not described in these patients. The presentation of generalized argyria typically begins with gray-brown staining of the gums that progresses to involve the skin diffusely. The mucocutaneous findings in argyria are the results of elevated serum silver levels, which lead to dermal and mucosal deposition of the metal. Histopathology evaluations reveal black-silver granules around the eccrine glands, in the walls of blood vessels, and along elastic fibers. The granules occasionally are found in the arrector pili muscles, perineural tissue, and around collagen fibrils. The slate gray, metallic, or blue-gray pigmentation seen in argyria may be clinically apparent after a few months but usually takes years to develop and depends on the degree of exposure.12,13 In some patients, the entire skin may acquire a slate blue—gray color. Hyperpigmentation is most apparent in sun-exposed areas of the skin, especially the forehead, nose, and hands. Although pigmentary changes occur primarily in sun-exposed sites, the granules are deposited evenly throughout the skin. Light causes silver-containing compounds complexed with proteins in the skin to be reduced to elemental silver, similar to the process of developing photographs.14 In addition, the silver stimulates melanocyte tyrosinase activity, which results in an increase in melanin production.15 The sclerae, nail beds, and mucous membranes also may become hyperpigmented. Viscera, including the spleen, liver, and gut, tend to show a blue discoloration that is evident during abdominal surgery or at postmortem examination. Our patient is unique because he presented with the diffuse pigmentary changes that would be seen with generalized argyria, but the pigmentary changes were limited to his face and neck. Other features suggestive of generalized argyria, such as sclerae and nail changes or silver impregnation in non—sun-exposed skin, were not present. The presence of silver granules in the eccrine glands of only exposed skin favors a diagnosis of localized argyria because of percutaneous silver absorption via the eccrine glands. A careful history is necessary in the diagnosis of argyria, with inquiries about possible occupational and environmental exposure and the use of dietary supplements containing colloidal silver protein. Habitual use of silver-based nose drops may produce pigmentation that is most apparent on the nose and nail lunulae. Scar-localized argyria may occur secondary to the use of silver sulfadiazine cream.16 Other causes of diffuse blue-gray pigmentation include medications (eg, phenothiazines, antimalarials, amiodarone, minocycline), heavy metal exposure (eg, mercury, bismuth, arsenic, gold, lead), hemochromatosis, ochronosis, cyanosis, polycythemia vera, and diffuse melanosis in metastatic melanoma.17 The average human body contains approximately 1 mg of silver.18 Serum silver has a reference range of 0 to 14 ng/mL. The smallest amount of silver reported to produce generalized argyria in humans ranges from 5 to 40 g.19 Although the amount of silver in argyria usually results in no serious effects on human health, a few cases of notable clinical symptoms and signs have been documented. Some of the complications of systemic toxic effects of silver include gastrointestinal tract catarrh, tissue wasting, uremia, albuminuria, fatty degeneration of the liver, hemorrhage, and idiopathic thrombocytopenia.20 The treatment of both localized and generalized argyria is difficult. Hydroquinone, depigmenting creams, and dermabrasion are not successful.9 Selenium and sulfur have been shown to have favorable modifying effects on the metabolism and toxicity of silver by forming complexes with the silver. The Q-switched double-frequency Nd:YAG laser, which has been used in the treatment of tattoos, also may be effective in the treatment of localized argyria.21 Unfortunately, no completely satisfactory treatment modality exists and some pigmentation remains permanent. However, sunscreens and opaque cosmetics may be helpful in masking discoloration and preventing further pigmentary darkening.17 To our knowledge, this is the first presented case of localized argyria secondary to aerosolized silver. This case emphasizes the need for skin protection in individuals with occupational exposure to aerosolized solder.
- Gettler AO, Rhoads CP, Weiss S. A contribution to the pathology of generalized argyria with a discussion of the state of silver in the human body. Am J Pathol. 1927;3:631-652.
- Espinel ML, Ferrando L, Diaz JF. Asymptomatic blue nevus-like macule. Arch Dermatol. 1996;132:459-464.
- Kapur N, Landon G, Yu RC. Localized argyria in an antique restorer. Br J Dermatol. 2001;144:191-193.
- McGinnis JP Jr, Greer JL, Daniels DS. Amalgam tattoo. J Am Dent Assoc. 1985;110:52-54.
- Shall L, Stevens A, Millard LG. An unusual case of acquired localized argyria. Br J Dermatol. 1990;123:403-407.
- Tanita Y, Kato T, Hanada K, et al. Blue macules of localized argyria caused by implanted acupuncture needles. Arch Dermatol. 1985;121:1550-1552.
- Wadhera A, Fung M. Systemic argyria associated with ingestion of colloidal silver. Dermatol Online J. 2005;11:12.
- Brandt D, Park B, Hoag M, et al. Argyria secondary to ingestion of homemade silver solution. J Am Acad Dermatol. 2005;53(2 suppl 1):S105-S107.
- Marshall JP, Schneider RP. Systemic argyria secondary to topical silver nitrate. Arch Dermatol. 1977;113:1077-1079.
- Scroggs MW, Lewis JS, Proia AD. Corneal argyrosis associated with silver soldering. Cornea. 1992;11:164-169.
- Sanchez-Huerta V, De Wit-Carter G, Hernandez-Quintela E, et al. Occupational corneal argyrosis in art silver solderers. Cornea. 2003;22:604-611.
- Greene RM, Su WPD. Argyria. Am Fam Pract. 1987;36:151-154.
- Peterson WC. Argyria. Minn Med. 1968;51:533-534.
- Shelley WB, Shelley ED, Burmeister V. Argyria: the intradermal photograph, a manifestation of passive photosensitivity. J Am Acad Dermatol. 1987;16:211-217.
- Buckley WR, Terhaar CJ. The skin as an excretory organ in argyria. Trans St Johns Hosp Dermatol Soc. 1973;59:39-44.
- Fisher NM, Marsh E, Lazova R. Scar-localized argyria secondary to silver sulfadiazine cream. J Am Acad Dermatol. 2003;49:730-732.
- Tanner LS, Gross DJ. Generalized argyria. Cutis. 1990;45:237-239.
- Venugopal B, Luckey TD. Metal toxicity in mammals. In: Venugopal B, Luckey TD, eds. Chemical Toxicology of Metals and Metalloids. Vol 2. New York, NY: Academic Press; 1978:32-36.
- Bleehen SS, Gould DJ, Harrington CI, et al. Occupational argyria; light and electron microscopic studies and X-ray microanalysis. Br J Dermatol. 1981;104:19-26.
- Sato S, Sueki H, Nishijima A. Two unusual cases of argyria: the application of an improved tissue processing method for x-ray microanalysis of selenium and sulphur in silver-laden granules. Br J Dermatol. 1999;140:158-163.
- Robinson-Bostom L, Pomerantz D, Wilkel C, et al. Localized argyria with pseudo-ochronosis. J Am
- Gettler AO, Rhoads CP, Weiss S. A contribution to the pathology of generalized argyria with a discussion of the state of silver in the human body. Am J Pathol. 1927;3:631-652.
- Espinel ML, Ferrando L, Diaz JF. Asymptomatic blue nevus-like macule. Arch Dermatol. 1996;132:459-464.
- Kapur N, Landon G, Yu RC. Localized argyria in an antique restorer. Br J Dermatol. 2001;144:191-193.
- McGinnis JP Jr, Greer JL, Daniels DS. Amalgam tattoo. J Am Dent Assoc. 1985;110:52-54.
- Shall L, Stevens A, Millard LG. An unusual case of acquired localized argyria. Br J Dermatol. 1990;123:403-407.
- Tanita Y, Kato T, Hanada K, et al. Blue macules of localized argyria caused by implanted acupuncture needles. Arch Dermatol. 1985;121:1550-1552.
- Wadhera A, Fung M. Systemic argyria associated with ingestion of colloidal silver. Dermatol Online J. 2005;11:12.
- Brandt D, Park B, Hoag M, et al. Argyria secondary to ingestion of homemade silver solution. J Am Acad Dermatol. 2005;53(2 suppl 1):S105-S107.
- Marshall JP, Schneider RP. Systemic argyria secondary to topical silver nitrate. Arch Dermatol. 1977;113:1077-1079.
- Scroggs MW, Lewis JS, Proia AD. Corneal argyrosis associated with silver soldering. Cornea. 1992;11:164-169.
- Sanchez-Huerta V, De Wit-Carter G, Hernandez-Quintela E, et al. Occupational corneal argyrosis in art silver solderers. Cornea. 2003;22:604-611.
- Greene RM, Su WPD. Argyria. Am Fam Pract. 1987;36:151-154.
- Peterson WC. Argyria. Minn Med. 1968;51:533-534.
- Shelley WB, Shelley ED, Burmeister V. Argyria: the intradermal photograph, a manifestation of passive photosensitivity. J Am Acad Dermatol. 1987;16:211-217.
- Buckley WR, Terhaar CJ. The skin as an excretory organ in argyria. Trans St Johns Hosp Dermatol Soc. 1973;59:39-44.
- Fisher NM, Marsh E, Lazova R. Scar-localized argyria secondary to silver sulfadiazine cream. J Am Acad Dermatol. 2003;49:730-732.
- Tanner LS, Gross DJ. Generalized argyria. Cutis. 1990;45:237-239.
- Venugopal B, Luckey TD. Metal toxicity in mammals. In: Venugopal B, Luckey TD, eds. Chemical Toxicology of Metals and Metalloids. Vol 2. New York, NY: Academic Press; 1978:32-36.
- Bleehen SS, Gould DJ, Harrington CI, et al. Occupational argyria; light and electron microscopic studies and X-ray microanalysis. Br J Dermatol. 1981;104:19-26.
- Sato S, Sueki H, Nishijima A. Two unusual cases of argyria: the application of an improved tissue processing method for x-ray microanalysis of selenium and sulphur in silver-laden granules. Br J Dermatol. 1999;140:158-163.
- Robinson-Bostom L, Pomerantz D, Wilkel C, et al. Localized argyria with pseudo-ochronosis. J Am
Bednar Tumor: Treatment With Mohs Micrographic Surgery
Multinucleated Atypia of the Vulva
α1-Antitrypsin Deficiency Panniculitis
α1-Antitrypsin deficiency panniculitis (A1ADP) is a rare form of panniculitis that affects children and adults. Clinical and histologic features, precipitating actors, and treatments are discussed.
α1-Antitrypsin, a serine protease inhibitor synthesized in the liver, regulates the action of proteolytic enzymes including trypsin, collagenase, elastase, factor VIII, chymotrypsin, and kallikrein. A deficiency of this protein is hypothesized to lead to
inadequate inhibition of proteases released by neutrophils and monocytes, which in turn results in unchecked inflammation and tissue necrosis.1 The systemic sequelae of this phenomenon include panacinar emphysema, hepatitis, cirrhosis, hemorrhagic diathesis, and panniculitis.2 Panniculitis most commonly occurs in patients with the severe homozygous deficiency PiZZ phenotype, resulting in serum α1-antitrypsin levels that are 10% of normal. The lesions may mimic cellulitis and are most frequently found on the trunk and proximal extremities. Characteristic microscopic features include neutrophils between collagen bundles in
reticular dermis, septal panniculitis with liquefactive necrosis, and collagenolysis with large areas of normal fat lobules adjacent to necrotic fat.3
Case Report
After falling down the stairs at home, a previously healthy 40-year-old woman presented to the emergency department with a tender edematous 25-cm hematoma on the right lateral thigh. The patient was treated empirically for an infected hematoma; use of oral antibiotics resulted in partial improvement. Subsequently, atraumatic indurated plaques and nodules developed on the
extremities; these plaques and nodules were minimally responsive to oral and intravenous antibiotics. The hematoma was incised and drained. Culture results were negative for bacterial and fungal growth. Serous fluid drained from a left-buttock nodule that had spontaneously ulcerated. The patient was admitted to our hospital for further diagnosis and management.
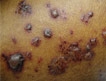
The patient was comfortable and nontoxic. Her temperature was 99.8ºF, and her heart rate was 88 bpm. Distributed over the right axilla, the medial area of the left elbow, the left ankle, the distal area of the right tibia, and the lateral area of the right thigh were multiple, deep, red, tender, indurated, 4- to 25-cm nodules and plaques (Figure 1). On the left buttock was an ulcerated 5-cm nodule.
A biopsy of the right axillary lesion was performed. Results of histologic examination showed normal epidermis and dermis. Mixed lobular and septal panniculitis included normal fat lobules adjacent to necrotic fat lobules (Figure 2). The infiltrate was composed of lymphocytes, histiocytes, and pools of neutrophils with suppuration, liquefactive necrosis, and collagenolysis (Figure 3). Neutrophils were splayed between collagen bundles (Figure 4). Granulomata were not evident, and there was no evidence of vasculitis. Periodic acid–Schiff, Gram, and acid-fast bacillus stains were negative for microorganisms. Refractile material was not evident under polariscopic examination. The microscopic differential diagnosis included infections, factitial panniculitis, subcutaneous Sweet syndrome, pancreatic fat necrosis, and α1-antitrypsin deficiency panniculitis (A1ADP).
Levels of antineutrophil cytoplasmic antibodies (p-ANCA, c-ANCA), C3, C4, and CH50 were normal. Cryoglobulins were not detected. Complete blood cell count and amylase and lipase levels were normal. Erythrocyte sedimentation rate was
elevated (52 mm/h).
Serum α1-antitrypsin level was 37.0 mg/dL (reference range, 84–218 mg/dL) with a ZZ phenotype. Results of a chest radiograph, liver function tests, and pulmonary function tests were normal. Given the clinical and pathologic findings and the results from the genetic α1 phenotyping, α1-antitrypsin deficiency was diagnosed. The patient, treated with dapsone, improved dramatically.
Comment
α1-Antitrypsin, a polypeptide glycoprotein synthesized by hepatocytes, inhibits collagenase, elastase, factor VIII, chymotrypsin, and kallikrein.1 α1-Antitrypsin is an acute-phase reactant that increases in serum concentration with stress from illness or trauma. Protease activation in the absence of α1-antitrypsin may trigger a cascade of inflammatory events that ultimately damage the tissues they are
meant to protect.2 Speculation is that absence of α1-antitrypsin allows inflammation to continue unabated and thus leads to panniculitis.
α1-Antitrypsin deficiency most frequently causes severe and rapidly progressive panacinar emphysema. This deficiency also is associated with hepatitis, cirrhosis,
vasculitis, acquired angioedema, Marshall syndrome, and severe psoriasis.4 Recently, α1-antitrypsin deficiency was used as a model for conformational
diseases (including liver cirrhosis) and neurodegenerative disorders (including Alzheimer disease and spongiform encephalopathies).5
Serum concentration of α1-antitrypsin is determined by inheritance of autosomal codominant alleles—M, S, and Z being the most common.6 Most cases of A1ADP occur in individuals with a severe homozygous deficiency (ZZ phenotype).3
Ninety-five percent of the US population shares the normal protease inhibitor MM phenotype designated type M. SZ heterozygotes have one third of the normal inhibitor level and a relatively low risk of developing emphysema.6 Prevalence of
the SZ phenotype ranges from 1 in 180 to 1 in 2500 individuals, depending on geographic location.7 Type Z, an α1-antitrypsin variant, differs from the M protein by a single amino acid substitution (lysine for glutamic acid).1 This substitution
results in a changed conformation leading to inhibition of α1-antitrypsin release from hepatocytes and decreased serum levels in patients with the protease inhibitor ZZ phenotype. Homozygous deficiency occurs in about 1 in 2500 individuals; heterozygous deficiency occurs in about 1 in 50.
The case reported here demonstrates many important features of A1ADP. First, our patient linked the onset of her symptoms to her fall down the stairs and her resulting injury of the right lower extremity. Another reviewer found that 6 of
18 cases of A1ADP were precipitated by trauma,8 and an investigator reported the case of a patient who had subclinical 1-antitrypsin deficiency and who developed panniculitis after trauma induced by cryosurgery.9
Second, our case demonstrates the typical clinical characteristics of A1ADP, including location on the proximal area of the lower extremities and axilla
and drainage of serous fluid. Lesions of this disease begin as tender, erythematous, indurated subcutaneous nodules that may be widely disseminated on the trunk or extremities. These lesions spontaneously ulcerate and drain oily, serosanguineous fluid.1 As reported in a review, 16 patients developed such lesions predominantly on the trunk and proximal area of the extremities.8
Third, our patient was diagnosed with a secondarily infected hematoma and cellulitis; repeated trials of antibiotics failed. Antibacterial treatment is completely ineffective in the management of A1ADP. Failure of multiple trials of oral antibiotics
and intravenous antibiotics exemplifies the difficulties encountered in making the diagnosis of A1ADP.
Integral to the diagnosis of our patient’s condition was the right axillary skin biopsy and laboratory evaluation of serum α1-antitrypsin level. Foci of fat necrosis adjacent to large areas of normal fat and acute lobular panniculitis with a large number of neutrophils are characteristic findings.
Other histologic findings should be addressed. Geller and Su3 described the earliest histopathologic findings of A1ADP as splaying of neutrophils between collagen bundles in the reticular dermis. Degeneration of collagen within the dermis, progressive dermal necrosis, and subsequent involvement of fibrous septa and subcutaneous fat are additional features of A1ADP. Therefore, histologic examination is an important diagnostic tool.
The cornerstone of the diagnosis in our patient’s case was the finding of a low level of α1-antitrypsin (37.0 mg/dL) and P1 typing of ZZ. Given the availability of the assay and the prolonged diagnostic challenge marked by multiple unsuccessful trials of antibiotics, running the assay earlier in the workup may be beneficial in cases with a high index of suspicion.
Treatment of A1ADP should include avoidance of trauma and surgical debridements—frequent precipitating factors of panniculitis.10 Dapsone, seemingly the treatment of choice, has been anecdotally effective in a number of cases of A1ADP.
For homozygous patients who have severe forms of the disease and who present with severe emphysema and liver failure, supplemental infusion of exogenous α1 protease inhibitor concentrate has been suggested as the most important therapeutic possibility.9
A1ADP is difficult to diagnose but should be considered when a patient with recurrent painful indurated plaques presents after sustaining a localized trauma.
α1-Antitrypsin deficiency panniculitis (A1ADP) is a rare form of panniculitis that affects children and adults. Clinical and histologic features, precipitating actors, and treatments are discussed.
α1-Antitrypsin, a serine protease inhibitor synthesized in the liver, regulates the action of proteolytic enzymes including trypsin, collagenase, elastase, factor VIII, chymotrypsin, and kallikrein. A deficiency of this protein is hypothesized to lead to
inadequate inhibition of proteases released by neutrophils and monocytes, which in turn results in unchecked inflammation and tissue necrosis.1 The systemic sequelae of this phenomenon include panacinar emphysema, hepatitis, cirrhosis, hemorrhagic diathesis, and panniculitis.2 Panniculitis most commonly occurs in patients with the severe homozygous deficiency PiZZ phenotype, resulting in serum α1-antitrypsin levels that are 10% of normal. The lesions may mimic cellulitis and are most frequently found on the trunk and proximal extremities. Characteristic microscopic features include neutrophils between collagen bundles in
reticular dermis, septal panniculitis with liquefactive necrosis, and collagenolysis with large areas of normal fat lobules adjacent to necrotic fat.3
Case Report
After falling down the stairs at home, a previously healthy 40-year-old woman presented to the emergency department with a tender edematous 25-cm hematoma on the right lateral thigh. The patient was treated empirically for an infected hematoma; use of oral antibiotics resulted in partial improvement. Subsequently, atraumatic indurated plaques and nodules developed on the
extremities; these plaques and nodules were minimally responsive to oral and intravenous antibiotics. The hematoma was incised and drained. Culture results were negative for bacterial and fungal growth. Serous fluid drained from a left-buttock nodule that had spontaneously ulcerated. The patient was admitted to our hospital for further diagnosis and management.
The patient was comfortable and nontoxic. Her temperature was 99.8ºF, and her heart rate was 88 bpm. Distributed over the right axilla, the medial area of the left elbow, the left ankle, the distal area of the right tibia, and the lateral area of the right thigh were multiple, deep, red, tender, indurated, 4- to 25-cm nodules and plaques (Figure 1). On the left buttock was an ulcerated 5-cm nodule.
A biopsy of the right axillary lesion was performed. Results of histologic examination showed normal epidermis and dermis. Mixed lobular and septal panniculitis included normal fat lobules adjacent to necrotic fat lobules (Figure 2). The infiltrate was composed of lymphocytes, histiocytes, and pools of neutrophils with suppuration, liquefactive necrosis, and collagenolysis (Figure 3). Neutrophils were splayed between collagen bundles (Figure 4). Granulomata were not evident, and there was no evidence of vasculitis. Periodic acid–Schiff, Gram, and acid-fast bacillus stains were negative for microorganisms. Refractile material was not evident under polariscopic examination. The microscopic differential diagnosis included infections, factitial panniculitis, subcutaneous Sweet syndrome, pancreatic fat necrosis, and α1-antitrypsin deficiency panniculitis (A1ADP).
Levels of antineutrophil cytoplasmic antibodies (p-ANCA, c-ANCA), C3, C4, and CH50 were normal. Cryoglobulins were not detected. Complete blood cell count and amylase and lipase levels were normal. Erythrocyte sedimentation rate was
elevated (52 mm/h).
Serum α1-antitrypsin level was 37.0 mg/dL (reference range, 84–218 mg/dL) with a ZZ phenotype. Results of a chest radiograph, liver function tests, and pulmonary function tests were normal. Given the clinical and pathologic findings and the results from the genetic α1 phenotyping, α1-antitrypsin deficiency was diagnosed. The patient, treated with dapsone, improved dramatically.
Comment
α1-Antitrypsin, a polypeptide glycoprotein synthesized by hepatocytes, inhibits collagenase, elastase, factor VIII, chymotrypsin, and kallikrein.1 α1-Antitrypsin is an acute-phase reactant that increases in serum concentration with stress from illness or trauma. Protease activation in the absence of α1-antitrypsin may trigger a cascade of inflammatory events that ultimately damage the tissues they are
meant to protect.2 Speculation is that absence of α1-antitrypsin allows inflammation to continue unabated and thus leads to panniculitis.
α1-Antitrypsin deficiency most frequently causes severe and rapidly progressive panacinar emphysema. This deficiency also is associated with hepatitis, cirrhosis,
vasculitis, acquired angioedema, Marshall syndrome, and severe psoriasis.4 Recently, α1-antitrypsin deficiency was used as a model for conformational
diseases (including liver cirrhosis) and neurodegenerative disorders (including Alzheimer disease and spongiform encephalopathies).5
Serum concentration of α1-antitrypsin is determined by inheritance of autosomal codominant alleles—M, S, and Z being the most common.6 Most cases of A1ADP occur in individuals with a severe homozygous deficiency (ZZ phenotype).3
Ninety-five percent of the US population shares the normal protease inhibitor MM phenotype designated type M. SZ heterozygotes have one third of the normal inhibitor level and a relatively low risk of developing emphysema.6 Prevalence of
the SZ phenotype ranges from 1 in 180 to 1 in 2500 individuals, depending on geographic location.7 Type Z, an α1-antitrypsin variant, differs from the M protein by a single amino acid substitution (lysine for glutamic acid).1 This substitution
results in a changed conformation leading to inhibition of α1-antitrypsin release from hepatocytes and decreased serum levels in patients with the protease inhibitor ZZ phenotype. Homozygous deficiency occurs in about 1 in 2500 individuals; heterozygous deficiency occurs in about 1 in 50.
The case reported here demonstrates many important features of A1ADP. First, our patient linked the onset of her symptoms to her fall down the stairs and her resulting injury of the right lower extremity. Another reviewer found that 6 of
18 cases of A1ADP were precipitated by trauma,8 and an investigator reported the case of a patient who had subclinical 1-antitrypsin deficiency and who developed panniculitis after trauma induced by cryosurgery.9
Second, our case demonstrates the typical clinical characteristics of A1ADP, including location on the proximal area of the lower extremities and axilla
and drainage of serous fluid. Lesions of this disease begin as tender, erythematous, indurated subcutaneous nodules that may be widely disseminated on the trunk or extremities. These lesions spontaneously ulcerate and drain oily, serosanguineous fluid.1 As reported in a review, 16 patients developed such lesions predominantly on the trunk and proximal area of the extremities.8
Third, our patient was diagnosed with a secondarily infected hematoma and cellulitis; repeated trials of antibiotics failed. Antibacterial treatment is completely ineffective in the management of A1ADP. Failure of multiple trials of oral antibiotics
and intravenous antibiotics exemplifies the difficulties encountered in making the diagnosis of A1ADP.
Integral to the diagnosis of our patient’s condition was the right axillary skin biopsy and laboratory evaluation of serum α1-antitrypsin level. Foci of fat necrosis adjacent to large areas of normal fat and acute lobular panniculitis with a large number of neutrophils are characteristic findings.
Other histologic findings should be addressed. Geller and Su3 described the earliest histopathologic findings of A1ADP as splaying of neutrophils between collagen bundles in the reticular dermis. Degeneration of collagen within the dermis, progressive dermal necrosis, and subsequent involvement of fibrous septa and subcutaneous fat are additional features of A1ADP. Therefore, histologic examination is an important diagnostic tool.
The cornerstone of the diagnosis in our patient’s case was the finding of a low level of α1-antitrypsin (37.0 mg/dL) and P1 typing of ZZ. Given the availability of the assay and the prolonged diagnostic challenge marked by multiple unsuccessful trials of antibiotics, running the assay earlier in the workup may be beneficial in cases with a high index of suspicion.
Treatment of A1ADP should include avoidance of trauma and surgical debridements—frequent precipitating factors of panniculitis.10 Dapsone, seemingly the treatment of choice, has been anecdotally effective in a number of cases of A1ADP.
For homozygous patients who have severe forms of the disease and who present with severe emphysema and liver failure, supplemental infusion of exogenous α1 protease inhibitor concentrate has been suggested as the most important therapeutic possibility.9
A1ADP is difficult to diagnose but should be considered when a patient with recurrent painful indurated plaques presents after sustaining a localized trauma.
α1-Antitrypsin deficiency panniculitis (A1ADP) is a rare form of panniculitis that affects children and adults. Clinical and histologic features, precipitating actors, and treatments are discussed.
α1-Antitrypsin, a serine protease inhibitor synthesized in the liver, regulates the action of proteolytic enzymes including trypsin, collagenase, elastase, factor VIII, chymotrypsin, and kallikrein. A deficiency of this protein is hypothesized to lead to
inadequate inhibition of proteases released by neutrophils and monocytes, which in turn results in unchecked inflammation and tissue necrosis.1 The systemic sequelae of this phenomenon include panacinar emphysema, hepatitis, cirrhosis, hemorrhagic diathesis, and panniculitis.2 Panniculitis most commonly occurs in patients with the severe homozygous deficiency PiZZ phenotype, resulting in serum α1-antitrypsin levels that are 10% of normal. The lesions may mimic cellulitis and are most frequently found on the trunk and proximal extremities. Characteristic microscopic features include neutrophils between collagen bundles in
reticular dermis, septal panniculitis with liquefactive necrosis, and collagenolysis with large areas of normal fat lobules adjacent to necrotic fat.3
Case Report
After falling down the stairs at home, a previously healthy 40-year-old woman presented to the emergency department with a tender edematous 25-cm hematoma on the right lateral thigh. The patient was treated empirically for an infected hematoma; use of oral antibiotics resulted in partial improvement. Subsequently, atraumatic indurated plaques and nodules developed on the
extremities; these plaques and nodules were minimally responsive to oral and intravenous antibiotics. The hematoma was incised and drained. Culture results were negative for bacterial and fungal growth. Serous fluid drained from a left-buttock nodule that had spontaneously ulcerated. The patient was admitted to our hospital for further diagnosis and management.
The patient was comfortable and nontoxic. Her temperature was 99.8ºF, and her heart rate was 88 bpm. Distributed over the right axilla, the medial area of the left elbow, the left ankle, the distal area of the right tibia, and the lateral area of the right thigh were multiple, deep, red, tender, indurated, 4- to 25-cm nodules and plaques (Figure 1). On the left buttock was an ulcerated 5-cm nodule.
A biopsy of the right axillary lesion was performed. Results of histologic examination showed normal epidermis and dermis. Mixed lobular and septal panniculitis included normal fat lobules adjacent to necrotic fat lobules (Figure 2). The infiltrate was composed of lymphocytes, histiocytes, and pools of neutrophils with suppuration, liquefactive necrosis, and collagenolysis (Figure 3). Neutrophils were splayed between collagen bundles (Figure 4). Granulomata were not evident, and there was no evidence of vasculitis. Periodic acid–Schiff, Gram, and acid-fast bacillus stains were negative for microorganisms. Refractile material was not evident under polariscopic examination. The microscopic differential diagnosis included infections, factitial panniculitis, subcutaneous Sweet syndrome, pancreatic fat necrosis, and α1-antitrypsin deficiency panniculitis (A1ADP).
Levels of antineutrophil cytoplasmic antibodies (p-ANCA, c-ANCA), C3, C4, and CH50 were normal. Cryoglobulins were not detected. Complete blood cell count and amylase and lipase levels were normal. Erythrocyte sedimentation rate was
elevated (52 mm/h).
Serum α1-antitrypsin level was 37.0 mg/dL (reference range, 84–218 mg/dL) with a ZZ phenotype. Results of a chest radiograph, liver function tests, and pulmonary function tests were normal. Given the clinical and pathologic findings and the results from the genetic α1 phenotyping, α1-antitrypsin deficiency was diagnosed. The patient, treated with dapsone, improved dramatically.
Comment
α1-Antitrypsin, a polypeptide glycoprotein synthesized by hepatocytes, inhibits collagenase, elastase, factor VIII, chymotrypsin, and kallikrein.1 α1-Antitrypsin is an acute-phase reactant that increases in serum concentration with stress from illness or trauma. Protease activation in the absence of α1-antitrypsin may trigger a cascade of inflammatory events that ultimately damage the tissues they are
meant to protect.2 Speculation is that absence of α1-antitrypsin allows inflammation to continue unabated and thus leads to panniculitis.
α1-Antitrypsin deficiency most frequently causes severe and rapidly progressive panacinar emphysema. This deficiency also is associated with hepatitis, cirrhosis,
vasculitis, acquired angioedema, Marshall syndrome, and severe psoriasis.4 Recently, α1-antitrypsin deficiency was used as a model for conformational
diseases (including liver cirrhosis) and neurodegenerative disorders (including Alzheimer disease and spongiform encephalopathies).5
Serum concentration of α1-antitrypsin is determined by inheritance of autosomal codominant alleles—M, S, and Z being the most common.6 Most cases of A1ADP occur in individuals with a severe homozygous deficiency (ZZ phenotype).3
Ninety-five percent of the US population shares the normal protease inhibitor MM phenotype designated type M. SZ heterozygotes have one third of the normal inhibitor level and a relatively low risk of developing emphysema.6 Prevalence of
the SZ phenotype ranges from 1 in 180 to 1 in 2500 individuals, depending on geographic location.7 Type Z, an α1-antitrypsin variant, differs from the M protein by a single amino acid substitution (lysine for glutamic acid).1 This substitution
results in a changed conformation leading to inhibition of α1-antitrypsin release from hepatocytes and decreased serum levels in patients with the protease inhibitor ZZ phenotype. Homozygous deficiency occurs in about 1 in 2500 individuals; heterozygous deficiency occurs in about 1 in 50.
The case reported here demonstrates many important features of A1ADP. First, our patient linked the onset of her symptoms to her fall down the stairs and her resulting injury of the right lower extremity. Another reviewer found that 6 of
18 cases of A1ADP were precipitated by trauma,8 and an investigator reported the case of a patient who had subclinical 1-antitrypsin deficiency and who developed panniculitis after trauma induced by cryosurgery.9
Second, our case demonstrates the typical clinical characteristics of A1ADP, including location on the proximal area of the lower extremities and axilla
and drainage of serous fluid. Lesions of this disease begin as tender, erythematous, indurated subcutaneous nodules that may be widely disseminated on the trunk or extremities. These lesions spontaneously ulcerate and drain oily, serosanguineous fluid.1 As reported in a review, 16 patients developed such lesions predominantly on the trunk and proximal area of the extremities.8
Third, our patient was diagnosed with a secondarily infected hematoma and cellulitis; repeated trials of antibiotics failed. Antibacterial treatment is completely ineffective in the management of A1ADP. Failure of multiple trials of oral antibiotics
and intravenous antibiotics exemplifies the difficulties encountered in making the diagnosis of A1ADP.
Integral to the diagnosis of our patient’s condition was the right axillary skin biopsy and laboratory evaluation of serum α1-antitrypsin level. Foci of fat necrosis adjacent to large areas of normal fat and acute lobular panniculitis with a large number of neutrophils are characteristic findings.
Other histologic findings should be addressed. Geller and Su3 described the earliest histopathologic findings of A1ADP as splaying of neutrophils between collagen bundles in the reticular dermis. Degeneration of collagen within the dermis, progressive dermal necrosis, and subsequent involvement of fibrous septa and subcutaneous fat are additional features of A1ADP. Therefore, histologic examination is an important diagnostic tool.
The cornerstone of the diagnosis in our patient’s case was the finding of a low level of α1-antitrypsin (37.0 mg/dL) and P1 typing of ZZ. Given the availability of the assay and the prolonged diagnostic challenge marked by multiple unsuccessful trials of antibiotics, running the assay earlier in the workup may be beneficial in cases with a high index of suspicion.
Treatment of A1ADP should include avoidance of trauma and surgical debridements—frequent precipitating factors of panniculitis.10 Dapsone, seemingly the treatment of choice, has been anecdotally effective in a number of cases of A1ADP.
For homozygous patients who have severe forms of the disease and who present with severe emphysema and liver failure, supplemental infusion of exogenous α1 protease inhibitor concentrate has been suggested as the most important therapeutic possibility.9
A1ADP is difficult to diagnose but should be considered when a patient with recurrent painful indurated plaques presents after sustaining a localized trauma.