User login
Atrophic Lesions in a Pregnant Woman
The Diagnosis: Degos Disease
The pathophysiology of Degos disease (malignant atrophic papulosis) is unknown.1 Histopathology demonstrates a wedge-shaped area of dermal necrosis with edema and mucin deposition extending from the papillary dermis to the deep reticular dermis. Occluded vessels, thrombosis, and perivascular lymphocytic infiltrates also may be seen, particularly at the dermal subcutaneous junction and at the periphery of the wedge-shaped infarction. The vascular damage that occurs may be the result of vasculitis, coagulopathy, or endothelial cell dysfunction.1
Patients typically present with small, round, erythematous papules that eventually develop atrophic porcelain white centers and telangiectatic rims. These lesions most commonly occur on the trunk and arms. In the benign form of atrophic papulosis, only the skin is involved; however, systemic involvement of the gastrointestinal tract and central nervous system can occur, resulting in bowel perforation and stroke, respectively.1 Although there is no definitive treatment of Degos disease, successful therapy with aspirin or dipyridamole has been reported.1 Eculizumab, a monoclonal antibody that binds C5, and treprostinil, a prostacyclin analog, are emerging treatment options.2,3 The differential diagnosis of Degos disease may include granuloma annulare, guttate extragenital lichen sclerosus, livedoid vasculopathy, and lymphomatoid papulosis.
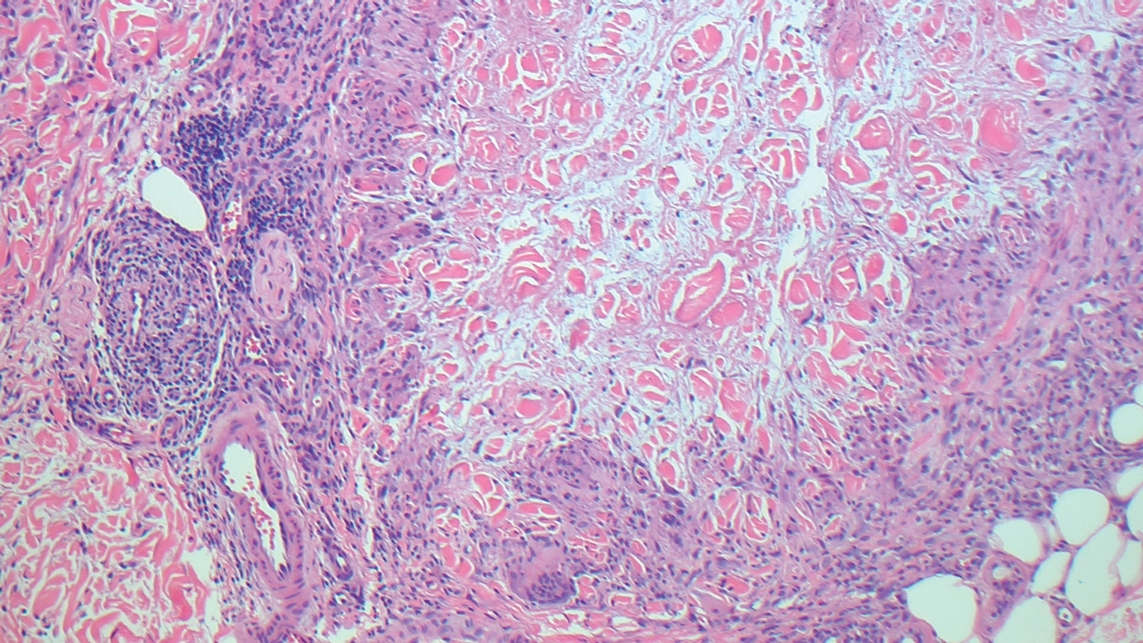
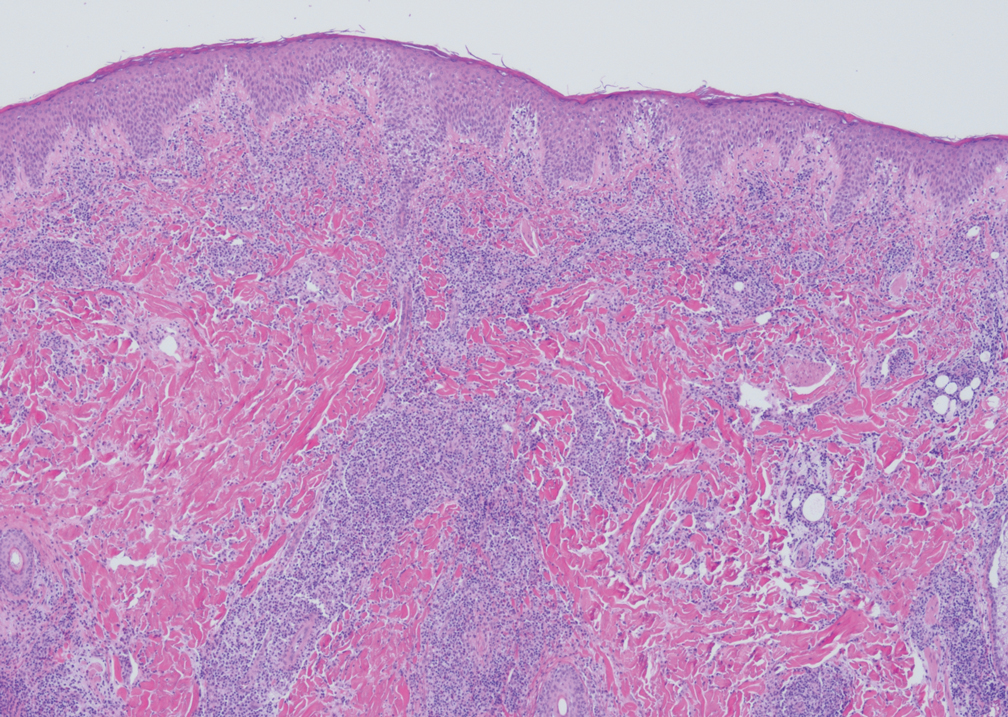
Granuloma annulare may clinically mimic the erythematous papules seen in early Degos disease, and histopathology can be used to distinguish between these two disease processes. Localized granuloma annulare is the most common variant and clinically presents as pink papules and plaques in an annular configuration.4 Histopathology demonstrates an unremarkable epidermis; however, the dermis contains degenerated collagen surrounded by palisading histiocytes as well as lymphocytes. Similar to Degos disease, increased mucin is seen within these areas of degeneration, but occluded vessels and thrombosis typically are not seen (Figure 1).4,5
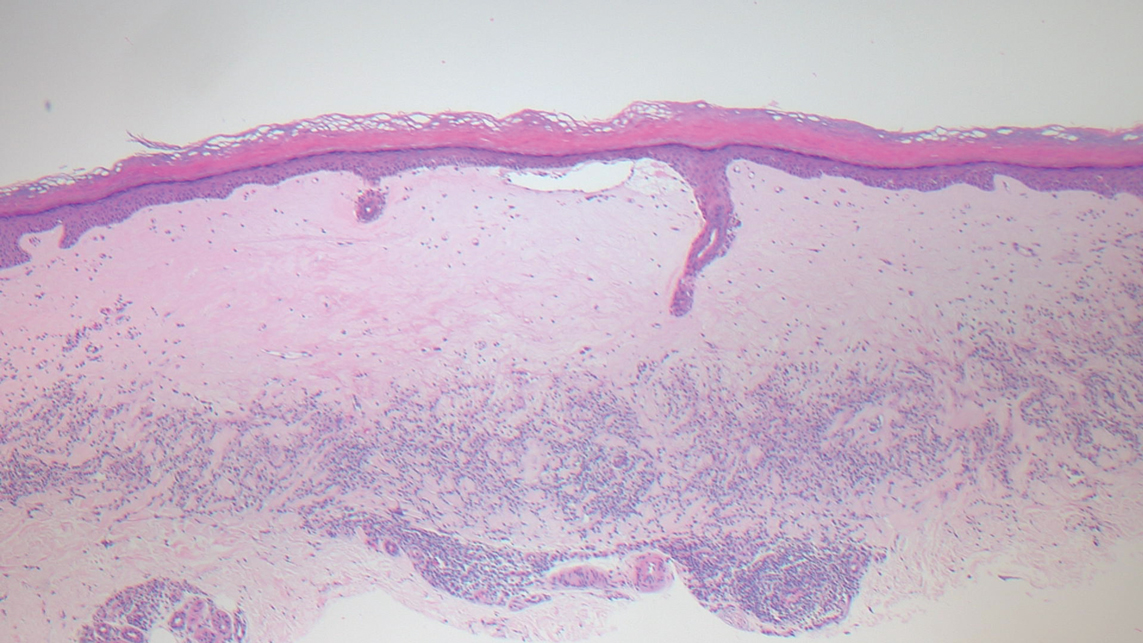
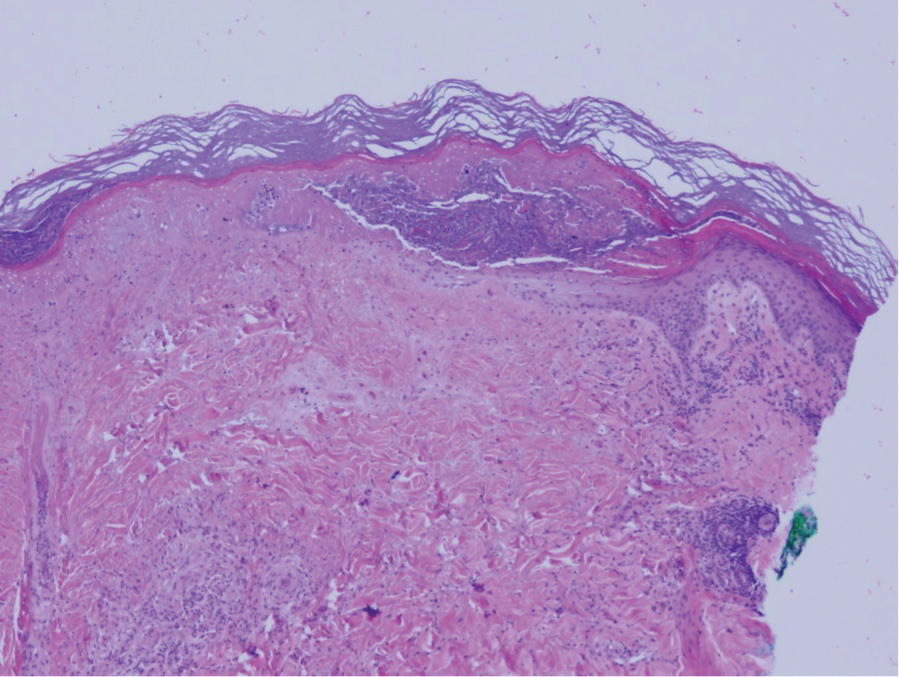
Guttate extragenital lichen sclerosus initially presents as polygonal, bluish white papules that coalesce into plaques.6 Over time, these lesions become more atrophic and may mimic Degos disease but appear differently on histopathology. Histopathology of lichen sclerosus classically demonstrates atrophy of the epidermis with loss of the rete ridges and vacuolar surface changes. Homogenization of the superficial/papillary dermis with an underlying bandlike lymphocytic infiltrate also is seen (Figure 2).6
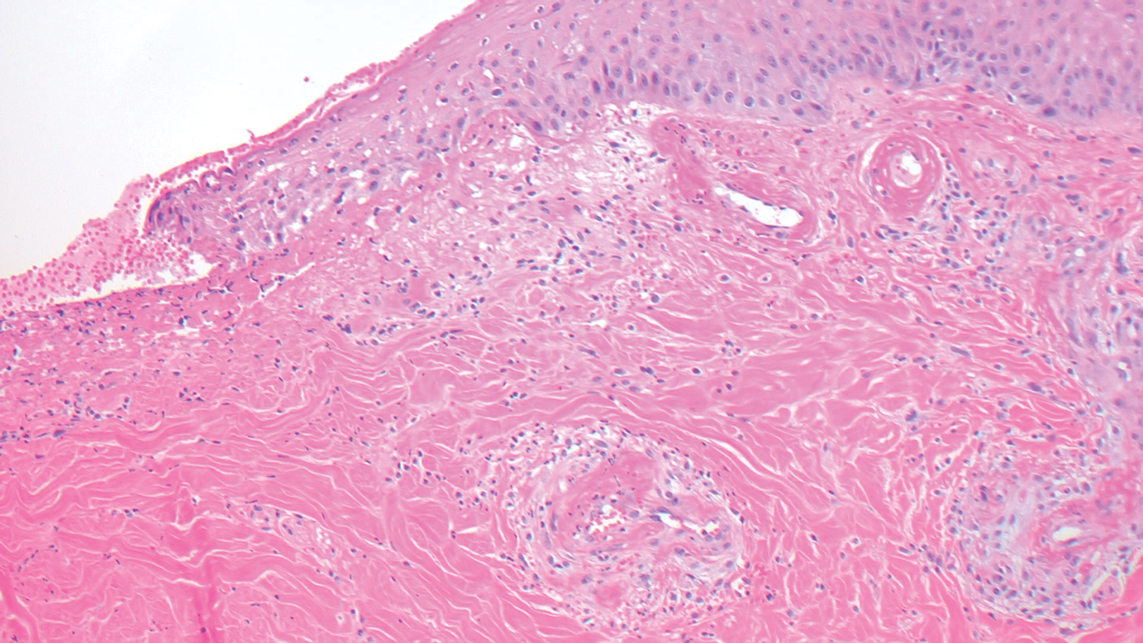
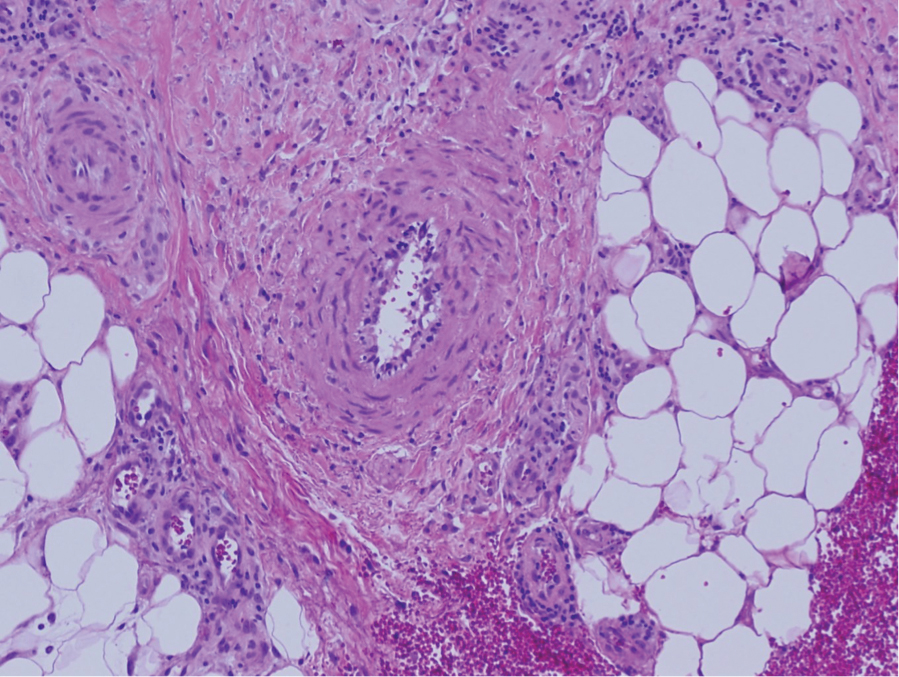
Livedoid vasculopathy is characterized by chronic recurrent ulceration of the legs secondary to thrombosis and subsequent ischemia. In the initial phase of this disease, livedo reticularis is seen followed by the development of ulcerations. As these ulcerations heal, they leave behind porcelain white scars referred to as atrophie blanche.7 The areas of scarring in livedoid vasculopathy are broad and angulated, differentiating them from the small, round, porcelain white macules in end-stage Degos disease. Histopathology demonstrates thrombosis and fibrin occlusion of the upper and mid dermal vessels. Very minimal perivascular infiltrate typically is seen, but when it is present, the infiltrate mostly is lymphocytic. Hyalinization of the vessel walls also is seen, particularly in the atrophie blanche stage (Figure 3).7
Lymphomatoid papulosis classically presents with pruritic red papules that often spontaneously involute. After resolution of the primary lesions, atrophic varioliform scars may be left behind that can resemble Degos disease.8 Classically, there are 5 histopathologic subtypes: A, B, C, D, and E. Type A is the most common type of lymphomatoid papulosis, and histopathology demonstrates a dermal lymphocytic infiltrate that consists of cells arranged in small clusters. Numerous medium- to large-sized atypical lymphocytes with prominent nucleoli and abundant cytoplasm are seen, and mitotic figures are common (Figure 4).8
Our case was particularly interesting because the patient was 2 to 3 weeks pregnant. Degos disease in pregnancy appears to be quite exceptional. A PubMed search of articles indexed for MEDLINE using the terms Degos disease and pregnancy revealed only 4 other cases reported in the literature.9-12 With the exception of a single case that was complicated by severe abdominal pain requiring labor induction, the other reported cases resulted in uncomplicated pregnancies.9-12 Conversely, our patient's pregnancy was complicated by gestational hypertension and fetal hydrops requiring a preterm cesarean delivery. Furthermore, the infant had multiple complications, which were attributed to both placental insufficiency and a coagulopathic state.
Our patient also was found to have a heterozygous factor V Leiden mutation on workup. A PubMed search using the terms factor V Leiden mutation and Degos disease revealed 2 other cases of factor V Leiden mutation-associated Degos disease.13,14 The importance of factor V Leiden mutations in patients with Degos disease currently is unclear.
- Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Köhlmeier-Degos disease)--a review. Orphanet J Rare Dis. 2013;8:10.
- Oliver B, Boehm M, Rosing DR, et al. Diffuse atrophic papules and plaques, intermittent abdominal pain, paresthesias, and cardiac abnormalities in a 55-year-old woman. J Am Acad Dermatol. 2016;75:1274-1277.
- Magro CM, Wang X, Garrett-Bakelman F, et al. The effects of eculizumab on the pathology of malignant atrophic papulosis. Orphanet J Rare Dis. 2013;8:185.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Tronnier M, Mitteldorf C. Histologic features of granulomatous skin diseases. part 1: non-infectious granulomatous disorders. J Dtsch Dermatol Ges. 2015;13:211-216.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus: an update. Am J Clin Dermatol. 2013;14:27-47.
- Vasudevan B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478‐488.
- Martinez-Cabriales SA, Walsh S, Sade S, et al. Lymphomatoid papulosis: an update and review. J Eur Acad Dermatol Venereol. 2020;34:59-73.
- Moulin G, Barrut D, Franc MP, et al. Familial Degos' atrophic papulosis (mother-daughter). Ann Dermatol Venereol. 1984;111:149-155.
- Bogenrieder T, Kuske M, Landthaler M, et al. Benign Degos' disease developing during pregnancy and followed for 10 years. Acta Derm Venereol. 2002;82:284-287.
- Sharma S, Brennan B, Naden R, et al. A case of Degos disease in pregnancy. Obstet Med. 2016;9:167-168.
- Zhao Q, Zhang S, Dong A. An unusual case of abdominal pain. Gastroenterology. 2018;154:E1-E2.
- Darwich E, Guilabert A, Mascaró JM Jr, et al. Dermoscopic description of a patient with thrombocythemia and factor V Leiden mutation-associated Degos' disease. Int J Dermatol. 2011;50:604-606.
- Hohwy T, Jensen MG, Tøttrup A, et al. A fatal case of malignant atrophic papulosis (Degos' disease) in a man with factor V Leiden mutation and lupus anticoagulant. Acta Derm Venereol. 2006;86:245-247.
The Diagnosis: Degos Disease
The pathophysiology of Degos disease (malignant atrophic papulosis) is unknown.1 Histopathology demonstrates a wedge-shaped area of dermal necrosis with edema and mucin deposition extending from the papillary dermis to the deep reticular dermis. Occluded vessels, thrombosis, and perivascular lymphocytic infiltrates also may be seen, particularly at the dermal subcutaneous junction and at the periphery of the wedge-shaped infarction. The vascular damage that occurs may be the result of vasculitis, coagulopathy, or endothelial cell dysfunction.1
Patients typically present with small, round, erythematous papules that eventually develop atrophic porcelain white centers and telangiectatic rims. These lesions most commonly occur on the trunk and arms. In the benign form of atrophic papulosis, only the skin is involved; however, systemic involvement of the gastrointestinal tract and central nervous system can occur, resulting in bowel perforation and stroke, respectively.1 Although there is no definitive treatment of Degos disease, successful therapy with aspirin or dipyridamole has been reported.1 Eculizumab, a monoclonal antibody that binds C5, and treprostinil, a prostacyclin analog, are emerging treatment options.2,3 The differential diagnosis of Degos disease may include granuloma annulare, guttate extragenital lichen sclerosus, livedoid vasculopathy, and lymphomatoid papulosis.
Granuloma annulare may clinically mimic the erythematous papules seen in early Degos disease, and histopathology can be used to distinguish between these two disease processes. Localized granuloma annulare is the most common variant and clinically presents as pink papules and plaques in an annular configuration.4 Histopathology demonstrates an unremarkable epidermis; however, the dermis contains degenerated collagen surrounded by palisading histiocytes as well as lymphocytes. Similar to Degos disease, increased mucin is seen within these areas of degeneration, but occluded vessels and thrombosis typically are not seen (Figure 1).4,5
Guttate extragenital lichen sclerosus initially presents as polygonal, bluish white papules that coalesce into plaques.6 Over time, these lesions become more atrophic and may mimic Degos disease but appear differently on histopathology. Histopathology of lichen sclerosus classically demonstrates atrophy of the epidermis with loss of the rete ridges and vacuolar surface changes. Homogenization of the superficial/papillary dermis with an underlying bandlike lymphocytic infiltrate also is seen (Figure 2).6
Livedoid vasculopathy is characterized by chronic recurrent ulceration of the legs secondary to thrombosis and subsequent ischemia. In the initial phase of this disease, livedo reticularis is seen followed by the development of ulcerations. As these ulcerations heal, they leave behind porcelain white scars referred to as atrophie blanche.7 The areas of scarring in livedoid vasculopathy are broad and angulated, differentiating them from the small, round, porcelain white macules in end-stage Degos disease. Histopathology demonstrates thrombosis and fibrin occlusion of the upper and mid dermal vessels. Very minimal perivascular infiltrate typically is seen, but when it is present, the infiltrate mostly is lymphocytic. Hyalinization of the vessel walls also is seen, particularly in the atrophie blanche stage (Figure 3).7
Lymphomatoid papulosis classically presents with pruritic red papules that often spontaneously involute. After resolution of the primary lesions, atrophic varioliform scars may be left behind that can resemble Degos disease.8 Classically, there are 5 histopathologic subtypes: A, B, C, D, and E. Type A is the most common type of lymphomatoid papulosis, and histopathology demonstrates a dermal lymphocytic infiltrate that consists of cells arranged in small clusters. Numerous medium- to large-sized atypical lymphocytes with prominent nucleoli and abundant cytoplasm are seen, and mitotic figures are common (Figure 4).8
Our case was particularly interesting because the patient was 2 to 3 weeks pregnant. Degos disease in pregnancy appears to be quite exceptional. A PubMed search of articles indexed for MEDLINE using the terms Degos disease and pregnancy revealed only 4 other cases reported in the literature.9-12 With the exception of a single case that was complicated by severe abdominal pain requiring labor induction, the other reported cases resulted in uncomplicated pregnancies.9-12 Conversely, our patient's pregnancy was complicated by gestational hypertension and fetal hydrops requiring a preterm cesarean delivery. Furthermore, the infant had multiple complications, which were attributed to both placental insufficiency and a coagulopathic state.
Our patient also was found to have a heterozygous factor V Leiden mutation on workup. A PubMed search using the terms factor V Leiden mutation and Degos disease revealed 2 other cases of factor V Leiden mutation-associated Degos disease.13,14 The importance of factor V Leiden mutations in patients with Degos disease currently is unclear.
The Diagnosis: Degos Disease
The pathophysiology of Degos disease (malignant atrophic papulosis) is unknown.1 Histopathology demonstrates a wedge-shaped area of dermal necrosis with edema and mucin deposition extending from the papillary dermis to the deep reticular dermis. Occluded vessels, thrombosis, and perivascular lymphocytic infiltrates also may be seen, particularly at the dermal subcutaneous junction and at the periphery of the wedge-shaped infarction. The vascular damage that occurs may be the result of vasculitis, coagulopathy, or endothelial cell dysfunction.1
Patients typically present with small, round, erythematous papules that eventually develop atrophic porcelain white centers and telangiectatic rims. These lesions most commonly occur on the trunk and arms. In the benign form of atrophic papulosis, only the skin is involved; however, systemic involvement of the gastrointestinal tract and central nervous system can occur, resulting in bowel perforation and stroke, respectively.1 Although there is no definitive treatment of Degos disease, successful therapy with aspirin or dipyridamole has been reported.1 Eculizumab, a monoclonal antibody that binds C5, and treprostinil, a prostacyclin analog, are emerging treatment options.2,3 The differential diagnosis of Degos disease may include granuloma annulare, guttate extragenital lichen sclerosus, livedoid vasculopathy, and lymphomatoid papulosis.
Granuloma annulare may clinically mimic the erythematous papules seen in early Degos disease, and histopathology can be used to distinguish between these two disease processes. Localized granuloma annulare is the most common variant and clinically presents as pink papules and plaques in an annular configuration.4 Histopathology demonstrates an unremarkable epidermis; however, the dermis contains degenerated collagen surrounded by palisading histiocytes as well as lymphocytes. Similar to Degos disease, increased mucin is seen within these areas of degeneration, but occluded vessels and thrombosis typically are not seen (Figure 1).4,5
Guttate extragenital lichen sclerosus initially presents as polygonal, bluish white papules that coalesce into plaques.6 Over time, these lesions become more atrophic and may mimic Degos disease but appear differently on histopathology. Histopathology of lichen sclerosus classically demonstrates atrophy of the epidermis with loss of the rete ridges and vacuolar surface changes. Homogenization of the superficial/papillary dermis with an underlying bandlike lymphocytic infiltrate also is seen (Figure 2).6
Livedoid vasculopathy is characterized by chronic recurrent ulceration of the legs secondary to thrombosis and subsequent ischemia. In the initial phase of this disease, livedo reticularis is seen followed by the development of ulcerations. As these ulcerations heal, they leave behind porcelain white scars referred to as atrophie blanche.7 The areas of scarring in livedoid vasculopathy are broad and angulated, differentiating them from the small, round, porcelain white macules in end-stage Degos disease. Histopathology demonstrates thrombosis and fibrin occlusion of the upper and mid dermal vessels. Very minimal perivascular infiltrate typically is seen, but when it is present, the infiltrate mostly is lymphocytic. Hyalinization of the vessel walls also is seen, particularly in the atrophie blanche stage (Figure 3).7
Lymphomatoid papulosis classically presents with pruritic red papules that often spontaneously involute. After resolution of the primary lesions, atrophic varioliform scars may be left behind that can resemble Degos disease.8 Classically, there are 5 histopathologic subtypes: A, B, C, D, and E. Type A is the most common type of lymphomatoid papulosis, and histopathology demonstrates a dermal lymphocytic infiltrate that consists of cells arranged in small clusters. Numerous medium- to large-sized atypical lymphocytes with prominent nucleoli and abundant cytoplasm are seen, and mitotic figures are common (Figure 4).8
Our case was particularly interesting because the patient was 2 to 3 weeks pregnant. Degos disease in pregnancy appears to be quite exceptional. A PubMed search of articles indexed for MEDLINE using the terms Degos disease and pregnancy revealed only 4 other cases reported in the literature.9-12 With the exception of a single case that was complicated by severe abdominal pain requiring labor induction, the other reported cases resulted in uncomplicated pregnancies.9-12 Conversely, our patient's pregnancy was complicated by gestational hypertension and fetal hydrops requiring a preterm cesarean delivery. Furthermore, the infant had multiple complications, which were attributed to both placental insufficiency and a coagulopathic state.
Our patient also was found to have a heterozygous factor V Leiden mutation on workup. A PubMed search using the terms factor V Leiden mutation and Degos disease revealed 2 other cases of factor V Leiden mutation-associated Degos disease.13,14 The importance of factor V Leiden mutations in patients with Degos disease currently is unclear.
- Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Köhlmeier-Degos disease)--a review. Orphanet J Rare Dis. 2013;8:10.
- Oliver B, Boehm M, Rosing DR, et al. Diffuse atrophic papules and plaques, intermittent abdominal pain, paresthesias, and cardiac abnormalities in a 55-year-old woman. J Am Acad Dermatol. 2016;75:1274-1277.
- Magro CM, Wang X, Garrett-Bakelman F, et al. The effects of eculizumab on the pathology of malignant atrophic papulosis. Orphanet J Rare Dis. 2013;8:185.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Tronnier M, Mitteldorf C. Histologic features of granulomatous skin diseases. part 1: non-infectious granulomatous disorders. J Dtsch Dermatol Ges. 2015;13:211-216.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus: an update. Am J Clin Dermatol. 2013;14:27-47.
- Vasudevan B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478‐488.
- Martinez-Cabriales SA, Walsh S, Sade S, et al. Lymphomatoid papulosis: an update and review. J Eur Acad Dermatol Venereol. 2020;34:59-73.
- Moulin G, Barrut D, Franc MP, et al. Familial Degos' atrophic papulosis (mother-daughter). Ann Dermatol Venereol. 1984;111:149-155.
- Bogenrieder T, Kuske M, Landthaler M, et al. Benign Degos' disease developing during pregnancy and followed for 10 years. Acta Derm Venereol. 2002;82:284-287.
- Sharma S, Brennan B, Naden R, et al. A case of Degos disease in pregnancy. Obstet Med. 2016;9:167-168.
- Zhao Q, Zhang S, Dong A. An unusual case of abdominal pain. Gastroenterology. 2018;154:E1-E2.
- Darwich E, Guilabert A, Mascaró JM Jr, et al. Dermoscopic description of a patient with thrombocythemia and factor V Leiden mutation-associated Degos' disease. Int J Dermatol. 2011;50:604-606.
- Hohwy T, Jensen MG, Tøttrup A, et al. A fatal case of malignant atrophic papulosis (Degos' disease) in a man with factor V Leiden mutation and lupus anticoagulant. Acta Derm Venereol. 2006;86:245-247.
- Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Köhlmeier-Degos disease)--a review. Orphanet J Rare Dis. 2013;8:10.
- Oliver B, Boehm M, Rosing DR, et al. Diffuse atrophic papules and plaques, intermittent abdominal pain, paresthesias, and cardiac abnormalities in a 55-year-old woman. J Am Acad Dermatol. 2016;75:1274-1277.
- Magro CM, Wang X, Garrett-Bakelman F, et al. The effects of eculizumab on the pathology of malignant atrophic papulosis. Orphanet J Rare Dis. 2013;8:185.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Tronnier M, Mitteldorf C. Histologic features of granulomatous skin diseases. part 1: non-infectious granulomatous disorders. J Dtsch Dermatol Ges. 2015;13:211-216.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus: an update. Am J Clin Dermatol. 2013;14:27-47.
- Vasudevan B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478‐488.
- Martinez-Cabriales SA, Walsh S, Sade S, et al. Lymphomatoid papulosis: an update and review. J Eur Acad Dermatol Venereol. 2020;34:59-73.
- Moulin G, Barrut D, Franc MP, et al. Familial Degos' atrophic papulosis (mother-daughter). Ann Dermatol Venereol. 1984;111:149-155.
- Bogenrieder T, Kuske M, Landthaler M, et al. Benign Degos' disease developing during pregnancy and followed for 10 years. Acta Derm Venereol. 2002;82:284-287.
- Sharma S, Brennan B, Naden R, et al. A case of Degos disease in pregnancy. Obstet Med. 2016;9:167-168.
- Zhao Q, Zhang S, Dong A. An unusual case of abdominal pain. Gastroenterology. 2018;154:E1-E2.
- Darwich E, Guilabert A, Mascaró JM Jr, et al. Dermoscopic description of a patient with thrombocythemia and factor V Leiden mutation-associated Degos' disease. Int J Dermatol. 2011;50:604-606.
- Hohwy T, Jensen MG, Tøttrup A, et al. A fatal case of malignant atrophic papulosis (Degos' disease) in a man with factor V Leiden mutation and lupus anticoagulant. Acta Derm Venereol. 2006;86:245-247.
A 36-year-old pregnant woman presented with painful erythematous papules on the palms and fingers of 2 months’ duration. Similar lesions developed on the thighs and feet several weeks later. Two tender macules with central areas of porcelain white scarring rimmed by telangiectases on the right foot also were present. A punch biopsy of these lesions demonstrated a wedge-shaped area of ischemic necrosis associated with dermal mucin without associated necrobiosis. Fibrin thrombi were seen within several small dermal vessels and were associated with a perivascular lymphocytic infiltrate. Endotheliitis was observed within a deep dermal vessel. Laboratory workup including syphilis IgG, antinuclear antibodies, extractable nuclear antigen antibodies, anti–double-stranded DNA, antistreptolysin O antibodies, Russell viper venom time, cryoglobulin, hepatitis screening, perinuclear antineutrophil cytoplasmic antibodies (ANCA), and cytoplasmic ANCA was unremarkable. Hypercoagulable studies including prothrombin gene mutation, factor V Leiden, plasminogen, proteins C and S, antithrombin III, homocysteine, and antiphospholipid IgM and IgG antibodies were notable only for heterozygosity for factor V Leiden.