User login
The Diagnosis: Erythropoietic Protoporphyria
Erythropoietic protoporphyria (EPP) is an autosomal-recessive photodermatosis that results from loss of activity of ferrochelatase, the last enzyme in the heme biosynthetic pathway.1 Erythropoietic protoporphyria normally involves sun-exposed areas of the body. Skin that is exposed to sunlight develops intense burning and stinging pain followed by erythema, edema, crusting, and petechiae that develops into waxy scarring over time. In contrast to other porphyrias, blistering generally is not seen.2 Accurate diagnosis often can be delayed by a decade or more following symptom onset due to the prominence of subjective pain as the presenting sign.
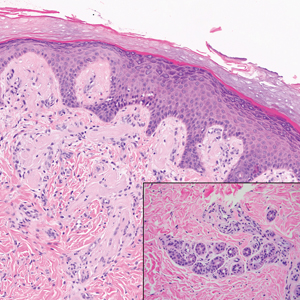
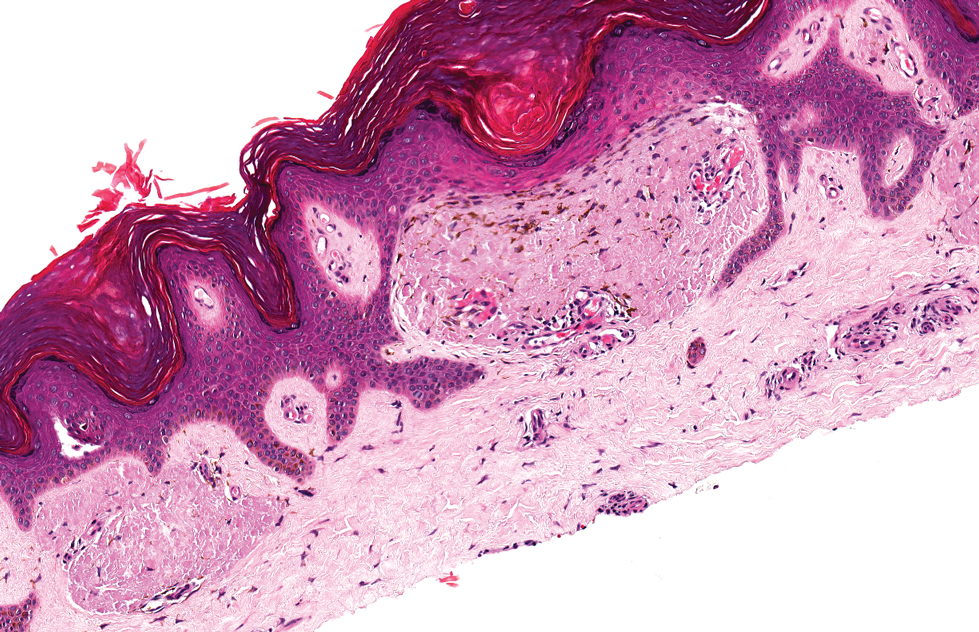
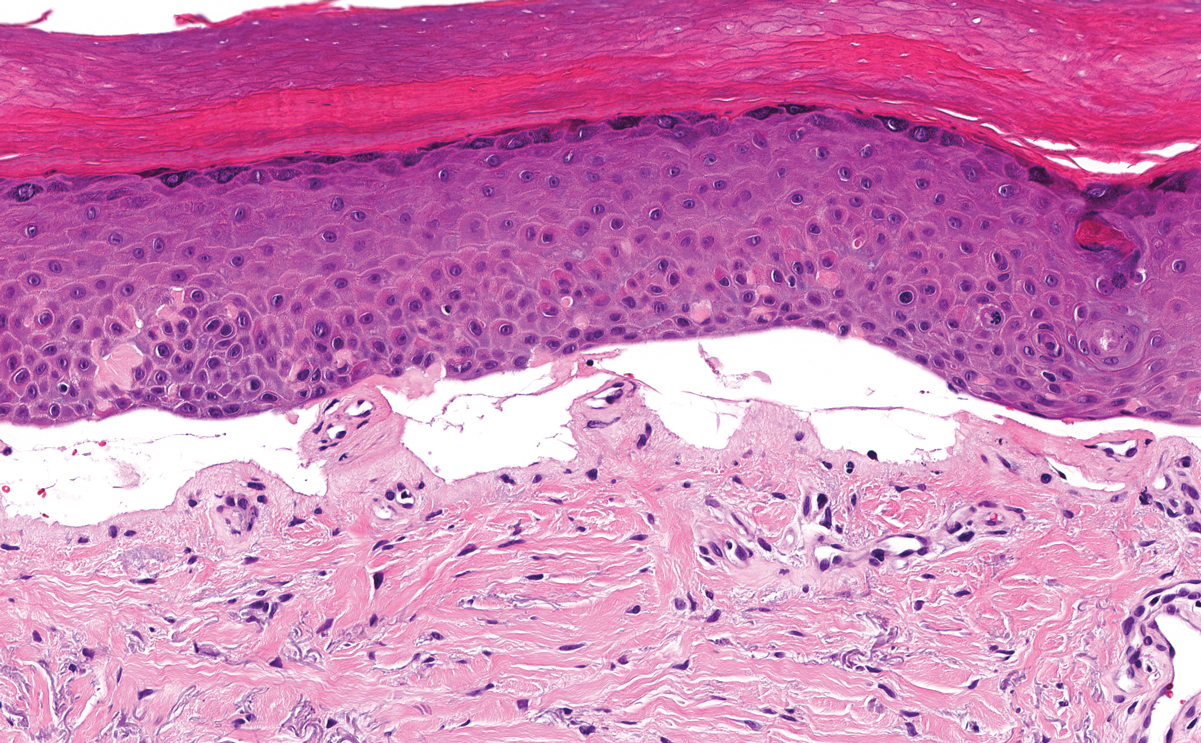
The histologic appearance of EPP differs depending on the chronicity of lesions. Biopsies of acute lesions show vacuolization of epidermal cells with intercellular edema, vacuolization and cytolysis of endothelial cells in superficial blood vessels, and focal red blood cell extravasation.3,4 A largely neutrophilic inflammatory infiltrate can be present.5 Hyaline cuffing develops over time in and around vessels in the papillary and superficial reticular dermis with notable sparing of adnexal structures. The perivascular deposits are strongly periodic acid-Schiff (PAS) positive and diastase resistant (Figure 1). Direct immunofluorescence shows mainly IgG and some IgM, fibrinogen, and C3 outlining characteristic donut-shaped blood vessels in the papillary dermis.6 The prominent thickness of the perivascular hyaline material depositions and the absence of subepidermal blistering can help differentiate EPP from porphyria cutanea tarda (PCT) and pseudoporphyria.6,7 When the deposition is extensive and involves the surrounding dermis, EPP can mimic colloid milium. Additional histologic differential diagnoses of EPP include other dermal depositional diseases such as lipoid proteinosis and amyloidosis.
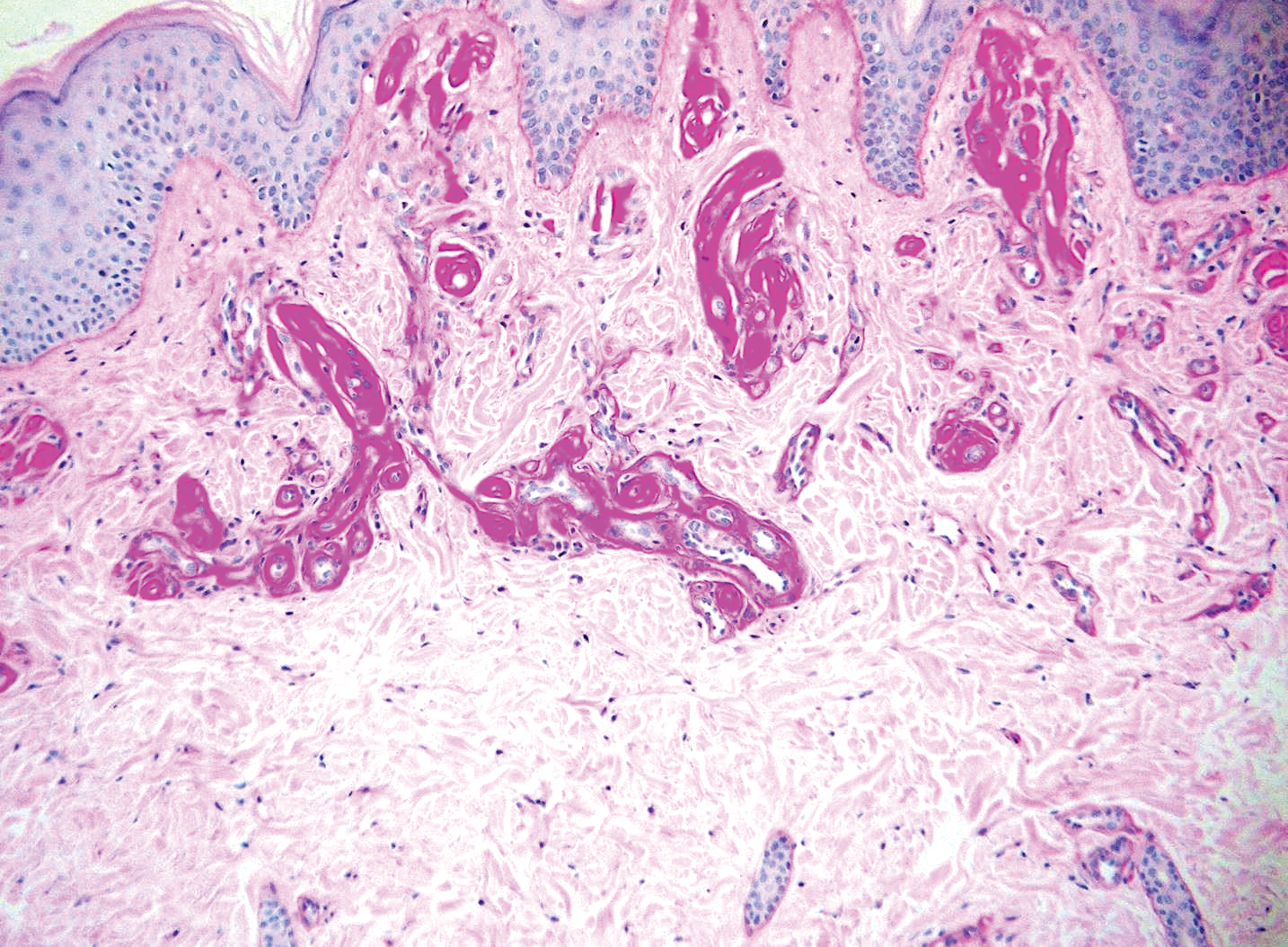
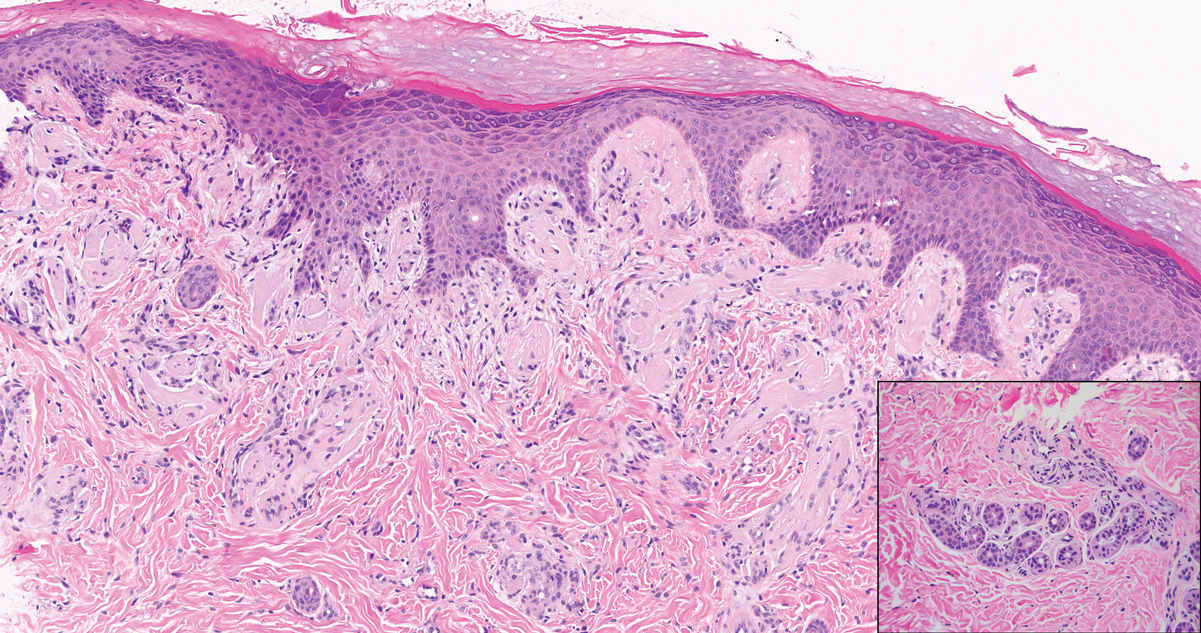
Lipoid proteinosis is an autosomal-recessive multisystem genodermatosis caused by mutations in extracellular matrix gene 1, ECM1. The first clinical sign can be a hoarse cry in infancy due to infiltration of vocal cords.3 Development of papulonodular lesions along the eyelids can result in a string-of-beads appearance called moniliform blepharosis, which is pathognomonic for lipoid proteinosis.6 With chronicity, the involved skin can become yellow, waxy, and thickened, particularly in the flexures or areas of trauma. Histologically, the dermis in lipoid proteinosis becomes diffusely thickened due to deposition of PAS-positive eosinophilic hyaline material that stains weakly with Congo red and thioflavin T.6 Early lesions demonstrate pale pink, hyalinelike thickening of the papillary dermal capillaries. Chronic lesions reveal an acanthotic epidermis, occasional papillomatosis with overlying hyperkeratosis, and a thickened dermis where diffuse thick bundles of pink hyaline deposits are oriented perpendicularly to the dermoepidermal junction.1,6 Lipoid proteinosis can be differentiated from EPP by the involvement of adnexal structures such as hair follicles, sebaceous glands, and arrector pili muscles (Figure 2), as opposed to EPP where adnexal structures are spared.1 Additionally, depositions in lipoid proteinosis are centered around both superficial and deep vessels with an onion skin-like pattern, while EPP involves mainly superficial vessels with more mild and focal hyalinization.
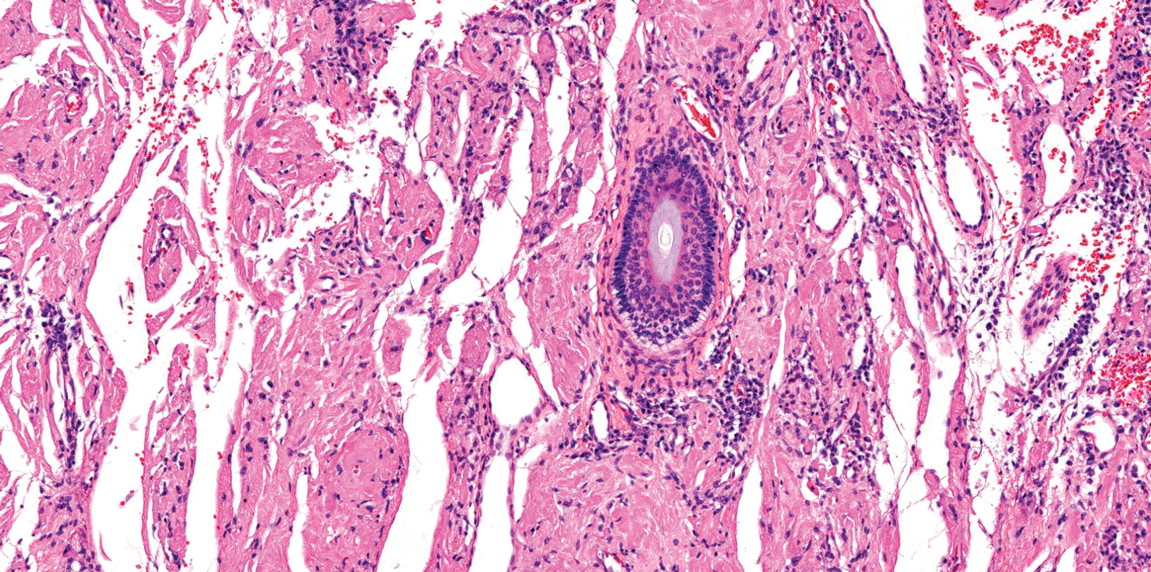
Juvenile colloid milium (JCM) is a rare condition that presents before puberty with discrete, yellow-brown, translucent papules predominantly located on the cheeks and nose and around the mouth. A gelatinous material can be expressed after puncturing a lesion.6 Gingival deposits and ligneous conjunctivitis also can be present. On histopathology, JCM shows degeneration of epidermal keratinocytes that form colloid bodies within the superficial dermis following apoptosis.6 Hematoxylin and eosin staining shows amorphous, fissured, pale pink deposits completely filling and expanding the superficial to mid dermis with clefting and no inflammation (Figure 3). Spindle-shaped fibroblasts may be seen within the lines of colloid fissuring and dispersed throughout the deposits.1 Histologically, JCM can be differentiated from EPP because deposits in EPP are distributed around and within superficial blood vessel walls, causing prominent vascular thickening not seen in JCM.6 The adult variant of colloid milium also can be distinguished from EPP by the presence of solar elastosis, which is absent in EPP due to a history of sun avoidance.3,7
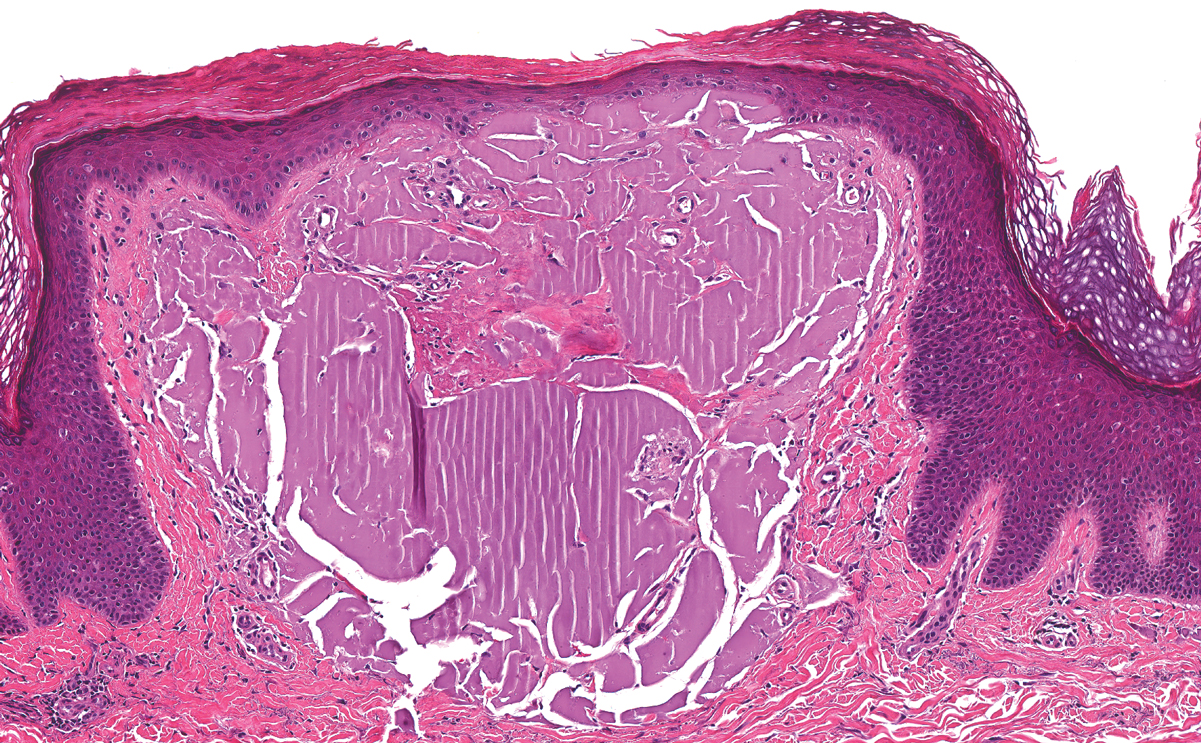
Lichen amyloidosis presents with highly pruritic, red-brown, hyperkeratotic papules that commonly are found on the anterior lower legs and extensor forearms.1 The calves, ankles, dorsal aspects of the feet, thighs, and trunk also may be affected. Excoriations, lichenification, and nodular prurigo-like lesions due to chronic scratching can be present.6 Lichen amyloidosis is characterized by large, pink, amorphous deposits in the papillary dermis with epidermal acanthosis, hypergranulosis, and hyperkeratosis (Figure 4).6 Perivascular deposits are not a feature of primary cutaneous localized amyloid lesions.6 The diagnosis can be confirmed with Congo red staining under polarized light, which classically demonstrates apple green birefringence.1 For cases of amyloid that are not detected by Congo red or are not clear-cut, direct immunofluorescence and immunohistochemistry can be used as adjuncts for diagnosis. Amyloid deposits fluoresce positively for immunoglobulins or complements, particularly IgM and C3,8 and immunohistochemistry confirms the presence of keratin epitopes in deposits.9
Porphyria cutanea tarda can appear histologically similar to EPP. Caterpillar bodies, or linearly arranged eosinophilic PAS-positive globules in the epidermis overlying subepidermal bullae, are a diagnostic histopathologic finding in both PCT and EPP but are seen in less than half of both cases.7,10 Compared to EPP, the perivascular deposits in PCT typically are less pronounced and limited to the vessel wall with smaller hyaline cuffs (Figure 5).7 Additionally, solar elastosis can be seen in PCT lesions but not in EPP, as patients with PCT tend to be older and have increased cumulative sun damage.
- Touart DM, Sau P. Cutaneous deposition diseases. part I. J Am Acad Dermatol. 1998;39(2, pt 1):149-171; quiz 172-144.
- Lim HW. Pathogenesis of photosensitivity in the cutaneous porphyrias. J Invest Dermatol. 2005;124:xvi-xvii.
- In: Alikhan A, Hocker TLH, eds. Review of Dermatology. China: Elsevier; 2017.
- Horner ME, Alikhan A, Tintle S, et al. Cutaneous porphyrias part I: epidemiology, pathogenesis, presentation, diagnosis, and histopathology. Int J Dermatol. 2013;52:1464-1480.
- Michaels BD, Del Rosso JQ, Mobini N, et al. Erythropoietic protoporphyria: a case report and literature review. J Clin Aesthet Dermatol. 2010;3:44-48.
- Calonje E, Brenn T, Lazar A, et al, eds. McKee's Pathology of the Skin. 4th ed. China: Elsevier Saunders; 2012.
- Patterson JW. Weedon's Skin Pathology. 4th ed. China: Elsevier Limited; 2016.
- MacDonald DM, Black MM, Ramnarain N. Immunofluorescence studies in primary localized cutaneous amyloidosis. Br J Dermatol. 1977;96:635-641.
- Ortiz-Romero PL, Ballestin-Carcavilla C, Lopez-Estebaranz JL, et al. Clinicopathologic and immunohistochemical studies on lichen amyloidosis and macular amyloidosis. Arch Dermatol. 1994;130:1559-1560.
- Raso DS, Greene WB, Maize JC, et al. Caterpillar bodies of porphyria cutanea tarda ultrastructurally represent a unique arrangement of colloid and basement membrane bodies. Am J Dermatopathol. 1996;18:24-29.
The Diagnosis: Erythropoietic Protoporphyria
Erythropoietic protoporphyria (EPP) is an autosomal-recessive photodermatosis that results from loss of activity of ferrochelatase, the last enzyme in the heme biosynthetic pathway.1 Erythropoietic protoporphyria normally involves sun-exposed areas of the body. Skin that is exposed to sunlight develops intense burning and stinging pain followed by erythema, edema, crusting, and petechiae that develops into waxy scarring over time. In contrast to other porphyrias, blistering generally is not seen.2 Accurate diagnosis often can be delayed by a decade or more following symptom onset due to the prominence of subjective pain as the presenting sign.
The histologic appearance of EPP differs depending on the chronicity of lesions. Biopsies of acute lesions show vacuolization of epidermal cells with intercellular edema, vacuolization and cytolysis of endothelial cells in superficial blood vessels, and focal red blood cell extravasation.3,4 A largely neutrophilic inflammatory infiltrate can be present.5 Hyaline cuffing develops over time in and around vessels in the papillary and superficial reticular dermis with notable sparing of adnexal structures. The perivascular deposits are strongly periodic acid-Schiff (PAS) positive and diastase resistant (Figure 1). Direct immunofluorescence shows mainly IgG and some IgM, fibrinogen, and C3 outlining characteristic donut-shaped blood vessels in the papillary dermis.6 The prominent thickness of the perivascular hyaline material depositions and the absence of subepidermal blistering can help differentiate EPP from porphyria cutanea tarda (PCT) and pseudoporphyria.6,7 When the deposition is extensive and involves the surrounding dermis, EPP can mimic colloid milium. Additional histologic differential diagnoses of EPP include other dermal depositional diseases such as lipoid proteinosis and amyloidosis.
Lipoid proteinosis is an autosomal-recessive multisystem genodermatosis caused by mutations in extracellular matrix gene 1, ECM1. The first clinical sign can be a hoarse cry in infancy due to infiltration of vocal cords.3 Development of papulonodular lesions along the eyelids can result in a string-of-beads appearance called moniliform blepharosis, which is pathognomonic for lipoid proteinosis.6 With chronicity, the involved skin can become yellow, waxy, and thickened, particularly in the flexures or areas of trauma. Histologically, the dermis in lipoid proteinosis becomes diffusely thickened due to deposition of PAS-positive eosinophilic hyaline material that stains weakly with Congo red and thioflavin T.6 Early lesions demonstrate pale pink, hyalinelike thickening of the papillary dermal capillaries. Chronic lesions reveal an acanthotic epidermis, occasional papillomatosis with overlying hyperkeratosis, and a thickened dermis where diffuse thick bundles of pink hyaline deposits are oriented perpendicularly to the dermoepidermal junction.1,6 Lipoid proteinosis can be differentiated from EPP by the involvement of adnexal structures such as hair follicles, sebaceous glands, and arrector pili muscles (Figure 2), as opposed to EPP where adnexal structures are spared.1 Additionally, depositions in lipoid proteinosis are centered around both superficial and deep vessels with an onion skin-like pattern, while EPP involves mainly superficial vessels with more mild and focal hyalinization.
Juvenile colloid milium (JCM) is a rare condition that presents before puberty with discrete, yellow-brown, translucent papules predominantly located on the cheeks and nose and around the mouth. A gelatinous material can be expressed after puncturing a lesion.6 Gingival deposits and ligneous conjunctivitis also can be present. On histopathology, JCM shows degeneration of epidermal keratinocytes that form colloid bodies within the superficial dermis following apoptosis.6 Hematoxylin and eosin staining shows amorphous, fissured, pale pink deposits completely filling and expanding the superficial to mid dermis with clefting and no inflammation (Figure 3). Spindle-shaped fibroblasts may be seen within the lines of colloid fissuring and dispersed throughout the deposits.1 Histologically, JCM can be differentiated from EPP because deposits in EPP are distributed around and within superficial blood vessel walls, causing prominent vascular thickening not seen in JCM.6 The adult variant of colloid milium also can be distinguished from EPP by the presence of solar elastosis, which is absent in EPP due to a history of sun avoidance.3,7
Lichen amyloidosis presents with highly pruritic, red-brown, hyperkeratotic papules that commonly are found on the anterior lower legs and extensor forearms.1 The calves, ankles, dorsal aspects of the feet, thighs, and trunk also may be affected. Excoriations, lichenification, and nodular prurigo-like lesions due to chronic scratching can be present.6 Lichen amyloidosis is characterized by large, pink, amorphous deposits in the papillary dermis with epidermal acanthosis, hypergranulosis, and hyperkeratosis (Figure 4).6 Perivascular deposits are not a feature of primary cutaneous localized amyloid lesions.6 The diagnosis can be confirmed with Congo red staining under polarized light, which classically demonstrates apple green birefringence.1 For cases of amyloid that are not detected by Congo red or are not clear-cut, direct immunofluorescence and immunohistochemistry can be used as adjuncts for diagnosis. Amyloid deposits fluoresce positively for immunoglobulins or complements, particularly IgM and C3,8 and immunohistochemistry confirms the presence of keratin epitopes in deposits.9
Porphyria cutanea tarda can appear histologically similar to EPP. Caterpillar bodies, or linearly arranged eosinophilic PAS-positive globules in the epidermis overlying subepidermal bullae, are a diagnostic histopathologic finding in both PCT and EPP but are seen in less than half of both cases.7,10 Compared to EPP, the perivascular deposits in PCT typically are less pronounced and limited to the vessel wall with smaller hyaline cuffs (Figure 5).7 Additionally, solar elastosis can be seen in PCT lesions but not in EPP, as patients with PCT tend to be older and have increased cumulative sun damage.
The Diagnosis: Erythropoietic Protoporphyria
Erythropoietic protoporphyria (EPP) is an autosomal-recessive photodermatosis that results from loss of activity of ferrochelatase, the last enzyme in the heme biosynthetic pathway.1 Erythropoietic protoporphyria normally involves sun-exposed areas of the body. Skin that is exposed to sunlight develops intense burning and stinging pain followed by erythema, edema, crusting, and petechiae that develops into waxy scarring over time. In contrast to other porphyrias, blistering generally is not seen.2 Accurate diagnosis often can be delayed by a decade or more following symptom onset due to the prominence of subjective pain as the presenting sign.
The histologic appearance of EPP differs depending on the chronicity of lesions. Biopsies of acute lesions show vacuolization of epidermal cells with intercellular edema, vacuolization and cytolysis of endothelial cells in superficial blood vessels, and focal red blood cell extravasation.3,4 A largely neutrophilic inflammatory infiltrate can be present.5 Hyaline cuffing develops over time in and around vessels in the papillary and superficial reticular dermis with notable sparing of adnexal structures. The perivascular deposits are strongly periodic acid-Schiff (PAS) positive and diastase resistant (Figure 1). Direct immunofluorescence shows mainly IgG and some IgM, fibrinogen, and C3 outlining characteristic donut-shaped blood vessels in the papillary dermis.6 The prominent thickness of the perivascular hyaline material depositions and the absence of subepidermal blistering can help differentiate EPP from porphyria cutanea tarda (PCT) and pseudoporphyria.6,7 When the deposition is extensive and involves the surrounding dermis, EPP can mimic colloid milium. Additional histologic differential diagnoses of EPP include other dermal depositional diseases such as lipoid proteinosis and amyloidosis.
Lipoid proteinosis is an autosomal-recessive multisystem genodermatosis caused by mutations in extracellular matrix gene 1, ECM1. The first clinical sign can be a hoarse cry in infancy due to infiltration of vocal cords.3 Development of papulonodular lesions along the eyelids can result in a string-of-beads appearance called moniliform blepharosis, which is pathognomonic for lipoid proteinosis.6 With chronicity, the involved skin can become yellow, waxy, and thickened, particularly in the flexures or areas of trauma. Histologically, the dermis in lipoid proteinosis becomes diffusely thickened due to deposition of PAS-positive eosinophilic hyaline material that stains weakly with Congo red and thioflavin T.6 Early lesions demonstrate pale pink, hyalinelike thickening of the papillary dermal capillaries. Chronic lesions reveal an acanthotic epidermis, occasional papillomatosis with overlying hyperkeratosis, and a thickened dermis where diffuse thick bundles of pink hyaline deposits are oriented perpendicularly to the dermoepidermal junction.1,6 Lipoid proteinosis can be differentiated from EPP by the involvement of adnexal structures such as hair follicles, sebaceous glands, and arrector pili muscles (Figure 2), as opposed to EPP where adnexal structures are spared.1 Additionally, depositions in lipoid proteinosis are centered around both superficial and deep vessels with an onion skin-like pattern, while EPP involves mainly superficial vessels with more mild and focal hyalinization.
Juvenile colloid milium (JCM) is a rare condition that presents before puberty with discrete, yellow-brown, translucent papules predominantly located on the cheeks and nose and around the mouth. A gelatinous material can be expressed after puncturing a lesion.6 Gingival deposits and ligneous conjunctivitis also can be present. On histopathology, JCM shows degeneration of epidermal keratinocytes that form colloid bodies within the superficial dermis following apoptosis.6 Hematoxylin and eosin staining shows amorphous, fissured, pale pink deposits completely filling and expanding the superficial to mid dermis with clefting and no inflammation (Figure 3). Spindle-shaped fibroblasts may be seen within the lines of colloid fissuring and dispersed throughout the deposits.1 Histologically, JCM can be differentiated from EPP because deposits in EPP are distributed around and within superficial blood vessel walls, causing prominent vascular thickening not seen in JCM.6 The adult variant of colloid milium also can be distinguished from EPP by the presence of solar elastosis, which is absent in EPP due to a history of sun avoidance.3,7
Lichen amyloidosis presents with highly pruritic, red-brown, hyperkeratotic papules that commonly are found on the anterior lower legs and extensor forearms.1 The calves, ankles, dorsal aspects of the feet, thighs, and trunk also may be affected. Excoriations, lichenification, and nodular prurigo-like lesions due to chronic scratching can be present.6 Lichen amyloidosis is characterized by large, pink, amorphous deposits in the papillary dermis with epidermal acanthosis, hypergranulosis, and hyperkeratosis (Figure 4).6 Perivascular deposits are not a feature of primary cutaneous localized amyloid lesions.6 The diagnosis can be confirmed with Congo red staining under polarized light, which classically demonstrates apple green birefringence.1 For cases of amyloid that are not detected by Congo red or are not clear-cut, direct immunofluorescence and immunohistochemistry can be used as adjuncts for diagnosis. Amyloid deposits fluoresce positively for immunoglobulins or complements, particularly IgM and C3,8 and immunohistochemistry confirms the presence of keratin epitopes in deposits.9
Porphyria cutanea tarda can appear histologically similar to EPP. Caterpillar bodies, or linearly arranged eosinophilic PAS-positive globules in the epidermis overlying subepidermal bullae, are a diagnostic histopathologic finding in both PCT and EPP but are seen in less than half of both cases.7,10 Compared to EPP, the perivascular deposits in PCT typically are less pronounced and limited to the vessel wall with smaller hyaline cuffs (Figure 5).7 Additionally, solar elastosis can be seen in PCT lesions but not in EPP, as patients with PCT tend to be older and have increased cumulative sun damage.
- Touart DM, Sau P. Cutaneous deposition diseases. part I. J Am Acad Dermatol. 1998;39(2, pt 1):149-171; quiz 172-144.
- Lim HW. Pathogenesis of photosensitivity in the cutaneous porphyrias. J Invest Dermatol. 2005;124:xvi-xvii.
- In: Alikhan A, Hocker TLH, eds. Review of Dermatology. China: Elsevier; 2017.
- Horner ME, Alikhan A, Tintle S, et al. Cutaneous porphyrias part I: epidemiology, pathogenesis, presentation, diagnosis, and histopathology. Int J Dermatol. 2013;52:1464-1480.
- Michaels BD, Del Rosso JQ, Mobini N, et al. Erythropoietic protoporphyria: a case report and literature review. J Clin Aesthet Dermatol. 2010;3:44-48.
- Calonje E, Brenn T, Lazar A, et al, eds. McKee's Pathology of the Skin. 4th ed. China: Elsevier Saunders; 2012.
- Patterson JW. Weedon's Skin Pathology. 4th ed. China: Elsevier Limited; 2016.
- MacDonald DM, Black MM, Ramnarain N. Immunofluorescence studies in primary localized cutaneous amyloidosis. Br J Dermatol. 1977;96:635-641.
- Ortiz-Romero PL, Ballestin-Carcavilla C, Lopez-Estebaranz JL, et al. Clinicopathologic and immunohistochemical studies on lichen amyloidosis and macular amyloidosis. Arch Dermatol. 1994;130:1559-1560.
- Raso DS, Greene WB, Maize JC, et al. Caterpillar bodies of porphyria cutanea tarda ultrastructurally represent a unique arrangement of colloid and basement membrane bodies. Am J Dermatopathol. 1996;18:24-29.
- Touart DM, Sau P. Cutaneous deposition diseases. part I. J Am Acad Dermatol. 1998;39(2, pt 1):149-171; quiz 172-144.
- Lim HW. Pathogenesis of photosensitivity in the cutaneous porphyrias. J Invest Dermatol. 2005;124:xvi-xvii.
- In: Alikhan A, Hocker TLH, eds. Review of Dermatology. China: Elsevier; 2017.
- Horner ME, Alikhan A, Tintle S, et al. Cutaneous porphyrias part I: epidemiology, pathogenesis, presentation, diagnosis, and histopathology. Int J Dermatol. 2013;52:1464-1480.
- Michaels BD, Del Rosso JQ, Mobini N, et al. Erythropoietic protoporphyria: a case report and literature review. J Clin Aesthet Dermatol. 2010;3:44-48.
- Calonje E, Brenn T, Lazar A, et al, eds. McKee's Pathology of the Skin. 4th ed. China: Elsevier Saunders; 2012.
- Patterson JW. Weedon's Skin Pathology. 4th ed. China: Elsevier Limited; 2016.
- MacDonald DM, Black MM, Ramnarain N. Immunofluorescence studies in primary localized cutaneous amyloidosis. Br J Dermatol. 1977;96:635-641.
- Ortiz-Romero PL, Ballestin-Carcavilla C, Lopez-Estebaranz JL, et al. Clinicopathologic and immunohistochemical studies on lichen amyloidosis and macular amyloidosis. Arch Dermatol. 1994;130:1559-1560.
- Raso DS, Greene WB, Maize JC, et al. Caterpillar bodies of porphyria cutanea tarda ultrastructurally represent a unique arrangement of colloid and basement membrane bodies. Am J Dermatopathol. 1996;18:24-29.
A 9-year-old girl presented with unexplained burning pain on the face, hands, and feet of 3 years' duration. Physical examination showed depressed shiny scars and crusted erosions on the dorsal aspect of the nose, arms, hands, and fingers. A 3-mm punch biopsy specimen was obtained from the right hand.