User login
Metastatic Primary Extramammary Paget Disease: A Case Series
Metastatic Primary Extramammary Paget Disease: A Case Series
Extramammary Paget disease (EMPD) is a rare cutaneous malignancy typically seen in apocrine-rich areas, including the axillae and anogenital region. It presents as a slow-growing, erythematous patch or plaque that commonly is misdiagnosed as an infectious or inflammatory condition.1,2 Primary EMPD occurs as a intraepithelial neoplasm, whereas secondary EMPD occurs due to epidermotropic metastases or direct extension of an underlying adenocarcinoma into the skin.1 Most commonly, primary EMPD occurs in situ; however, when present, dermal invasion and metastases from the skin are associated with poorer outcomes.3 Given the rarity of metastatic disease, existing literature is limited to case reports and case series.
We present 2 patients with metastatic primary EMPD who had evidence of invasion on initial biopsy and died secondary to metastatic EMPD. We conducted a comprehensive review of the literature for invasive and metastatic EMPD to highlight key clinicopathologic features, treatment considerations, and the potential for rapid disease progression in cases of invasive EMPD.
Case Series
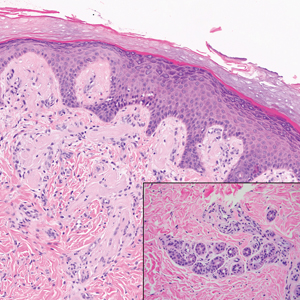
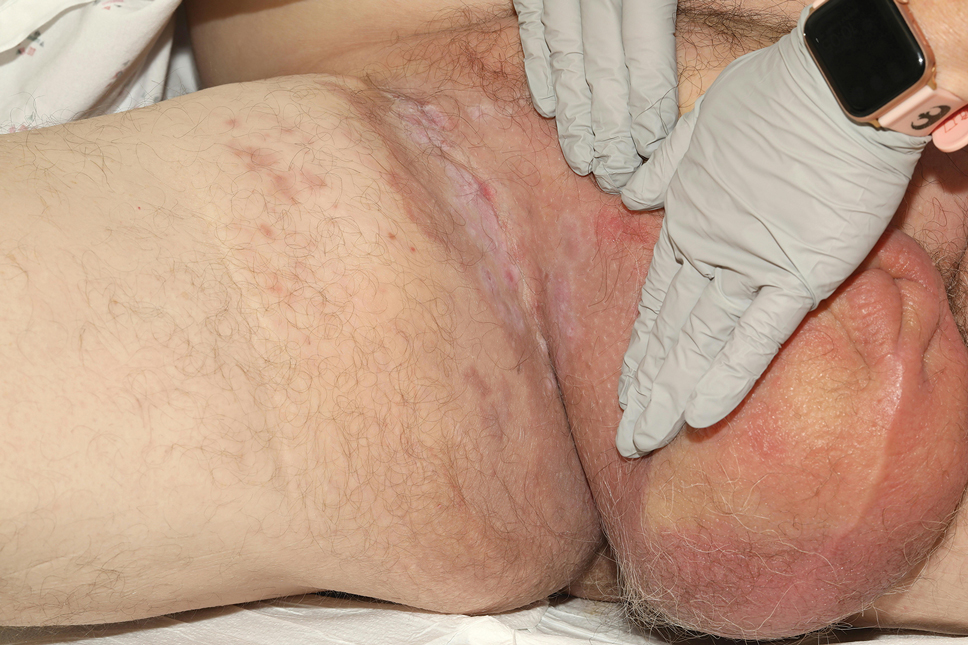
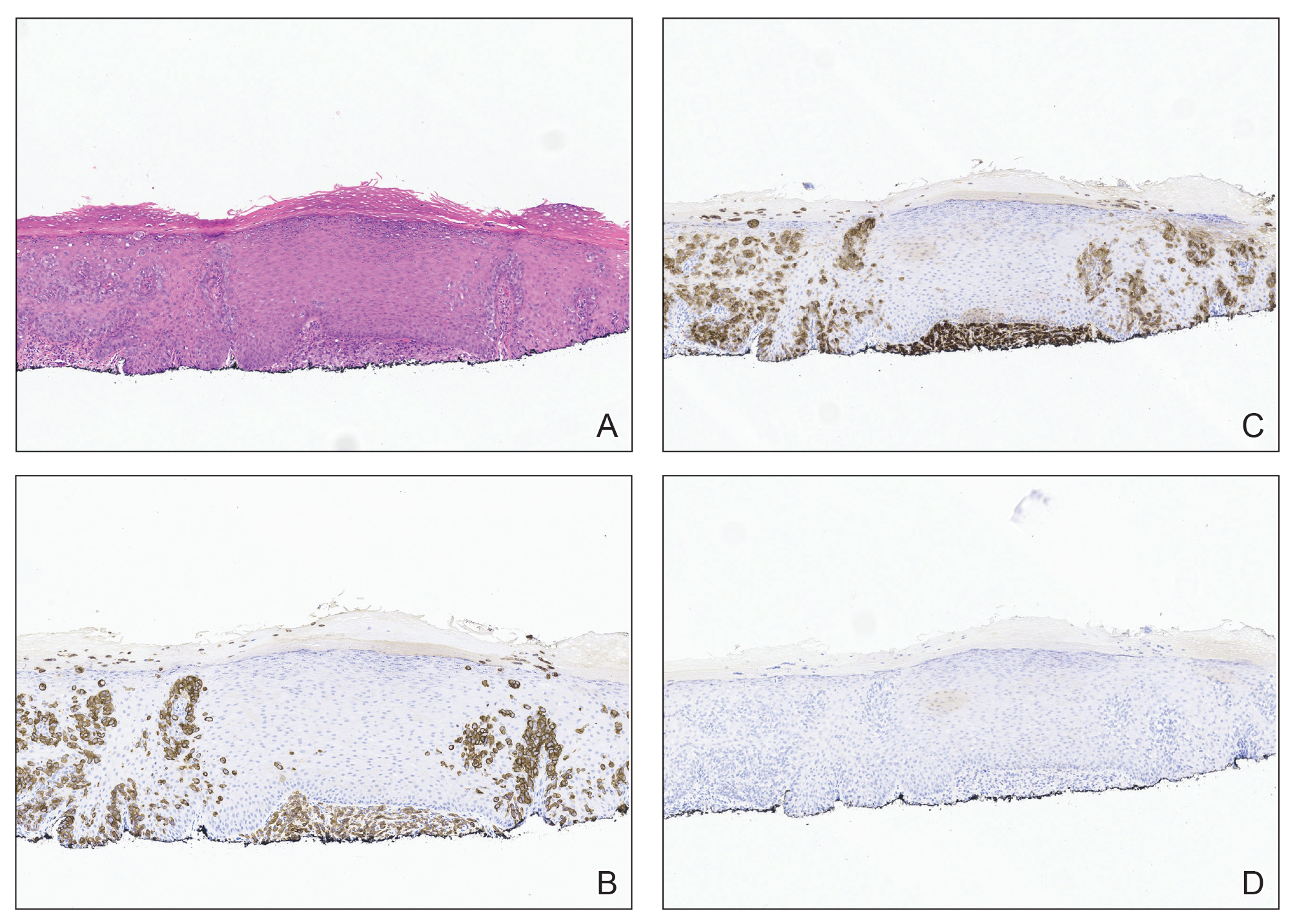
Patient 1—A 68-year-old White man with a history of breast cancer (in remission) presented to our clinic for further management of biopsy-proven scrotal EMPD. Prior to biopsy, he described a 6-month history of worsening scrotal rash treated with topical antifungals, oral antibiotics, and topical steroids due to presumed diagnosis of intertrigo, cellulitis, and dermatitis, respectively. Clinical examination showed indurated, erythematous, ulcerated plaques involving the bilateral groin, genitalia, and perineum (Figure 1). Skin biopsy confirmed a diagnosis of EMPD with both dermal and lymphovascular invasion. An immunohistochemical profile was positive for CK7 and carcinoembryonic antigen (CEA) and negative for CK20 (Figure 2).
At presentation, the patient had palpable lymphadenopathy and scrotal edema concerning for inguinal and iliac lymph node metastases. Workup for an underlying adenocarcinoma included computed tomography (CT) of the chest, abdomen, and pelvis; urologic consultation with cystoscopy; and a screening colonoscopy. The CT scan revealed multiple enlarged inguinal and external iliac lymph nodes. Fine-needle aspiration revealed CK7- and CEA-positive neoplastic cells consistent with metastatic EMPD. The patient was treated with 6 cycles of carboplatin-paclitaxel, palliative radiation therapy, and pembrolizumab with minimal response to treatment and development of osteolytic vertebral lesions concerning for disease progression. He died 1 year after the initial diagnosis secondary to the disease.
Patient 2—A 79-year-old White man presented for further management of an outside diagnosis of superficially invasive primary EMPD of the bilateral inguinal folds and scrotum that had been present for 5 months prior to biopsy and diagnosis. Clinical examination at initial presentation revealed erythematous patches of the bilateral inguinal folds and scrotum, as well as an erythematous scaling plaque in the right axilla. There was no palpable clinical lymphadenopathy. Biopsy of the axilla and groin were both consistent with invasive EMPD with positive staining for CK7 and negative staining for CK20 and CDX2. Workup for underlying adenocarcinoma with whole-body positron emission tomography/CT, mammography, esophagogastroduodenoscopy, serum CEA, colonoscopy, and cystoscopy were all negative for a metastatic adenocarcinoma. There was no imaging or clinical evidence of lymphadenopathy. Complete circumferential peripheral and deep-margin assessment was performed in a staged manner on both sites, and negative margins were obtained.
Surveillance imaging 6 months after surgery revealed suspicious hepatic lesions. Fine-needle aspiration of the hepatic lesions demonstrated positive staining for CK7 and negative staining for CK20, CDX2, prostate-specific antigen, and thyroid transcription factor 1, consistent with metastatic EMPD. Oncology recommended carboplatin and docetaxel or docetaxel monotherapy chemotherapy. The patient was further managed by an outside oncologist due to ease of travel but died secondary to the disease 15 months following the initial diagnosis.
Comment
Extramammary Paget disease is an uncommon cutaneous malignancy that manifests as pruritic erythematous plaques within apocrine-rich areas such as the genitalia, axillae, or anal region. It most commonly occurs in patients older than 65 years, with White women and Asian men being affected at disproportionately higher rates.1,4 Delay in diagnosis is common, as EMPD can mimic other benign inflammatory or infectious conditions, including contact dermatitis, seborrheic dermatitis, tinea, candidiasis, and eczema.1
Metastatic and multifocal cases of primary EMPD are especially rare. According to a search of PubMed articles indexed for MEDLINE published through December 2023 using the terms extramammary Paget disease, EMPD, neoplasm metastasis, invasive extramammary, and neoplasm invasiveness, we identified 5040 cases of invasive EMPD and 477 cases of metastatic EMPD.5-37 Of the reports that disclosed patient demographic information, 3627 patients were female 1410 were male, and the mean age was 67 years. Sites of metastases included regional lymph nodes, liver, lungs, cervix, bladder, bone, brain, skin, kidney, and adrenal glands
Workup for EMPD—The initial steps for workup of EMPD include a thorough physical examination and lymph node assessment. A skin biopsy also should be performed for patients presenting with refractory, pruritic, and eczematous rashes in apocrine-rich areas to evaluate for EMPD.1 Characterization of large and complex tumors is better achieved through multiple biopsies with particular focus on nodular or thickened areas, as these may indicate invasive disease.2 Primary EMPD is characterized by pagetoid cells with abundant pale cytoplasm proliferating in a single-cell or nested pattern within the epidermis or dermis in invasive disease and often is accompanied by dermal lymphocytic inflammation.1 Immunohistochemistry demonstrates positive staining for CEA, CK7, and CK8, with negative staining for indicators of secondary EMPD including CK20 and CDX2.1,2
As part of the workup, it is critical to distinguish between primary disease and secondary EMPD.1 Beyond skin and clinical lymph node examination, additional workup should be based on age-appropriate and location-directed malignant neoplasm screenings, including colonoscopy, cystoscopy, prostate examination, mammography, and Papanicolaou test. Advanced imaging such as CT, positron emission tomography, or magnetic resonance imaging can be used to assess for metastatic disease if internal malignant neoplasms are present on initial screening or clinical lymphadenopathy is identified.2 Additionally, it can be helpful in the evaluation for nodal disease in cases of invasive EMPD.
The likelihood of associated underlying carcinomas varies depending on the site of involvement.38,39 For example, vulvar involvement constitutes approximately 65% of EMPD cases, with 11% to 20% of cases being associated with underlying gastrointestinal or genitourinary carcinomas. Involvement of the male genitalia, as in our 2 patients, is rare, accounting for approximately 14% of cases, 11% of which are associated with prostate, testicular, and bladder carcinoma. Perianal involvement comprises 20% of EMPD cases and has the greatest risk for underlying malignancy with an incidence of 33% to 86%, the majority of which are rectal or tubo-ovarian cancers.38,39 Consideration of the frequency and types of underlying carcinoma of respective sites of involvement can be helpful when ruling out secondary EMPD.
In both of our patients, palpable lymphadenopathy at the time of original diagnosis and histologic invasive disease on initial biopsy warranted thorough imaging and laboratory workup; there was no evidence of primary malignancy. Given the absence of an underlying carcinoma, both patients were classified as having metastatic primary EMPD.
Assessment of lymphadenopathy is an essential aspect of disease workup, as it is associated with a statistically higher rate of lymph node metastases. A study by Fujisawa et al20 demonstrated that 80% of patients with lymphadenopathy had regional metastases compared to only 15% of patients without clinical lymphadenopathy. The presence of invasive disease also has been shown to correspond with lymph node metastases.40 Ogata et al40 showed that 0% of cases with in situ EMPD had a positive sentinel lymph node biopsy (SLNB) compared to 4% and 43% in cases that showed evidence of microinvasion and dermal invasion, respectively. Lymph node metastases are associated with poor prognosis, with increasingly worse prognosis when there are multiple lymph nodes affected.41 In our case series, patient 1 had lymphadenopathy and both patients had invasive EMPD; they both later developed metastases and died.
Lymphadenopathy should be further investigated with imaging and biopsy or fine-needle aspiration.42 Recent expert consensus guidelines recommended this method of investigation over routine use of SLNB, as there is no evidence that a positive SLNB affects treatment that changes disease-specific survival.2
Treatment of EMPD—Surgical excision of the primary lesion is the first-line treatment of EMPD,1,2 which can be performed by wide local excision; however, studies have demonstrated higher recurrence-free survival with margin-controlled surgery (complete circumferential peripheral and deep margin assessment) or Mohs micrographic surgery (MMS), especially with CK7 immunostaining.2,37,43 The literature on MMS of invasive EMPD is sparse, accounting for 57 patients.25,37,44 Other reports describe management with surgical excision, wide local excision, regional resection, or vulvectomy, in addition to lymph node dissection, radiation therapy (RT), and/or chemotherapy.1-36,39,43-46 Despite the improved outcomes with MMS, the predominance of other surgical approaches in our search suggests that MMS may be currently underutilized for the treatment of invasive or locally advanced EMPD.
Among patients with unresectable disease or distant metastases, management includes RT with curative intent, chemotherapy, or a combination of both.1,2 In our review, 267 cases were treated using RT and 77 with chemotherapy. Radiation therapy is an effective therapeutic option with a reported response rate of 62% to 100% and can be employed as either primary or adjuvant treatment.3 For patients with lymph node metastasis the combination of RT and lymph node dissection has been shown to have improved outcomes compared to lymph node dissection alone, with 1 study showing a 5-year survival of 75% for patients who received adjuvant RT compared to 0% for lymph node dissection alone.45
There are currently no consensus guidelines on the best chemotherapeutic regimen for metastatic EMPD. Several regimens have been reported, including docetaxel monotherapy; low-dose 5-fluorouracil and cisplatin; combination chemotherapy FECOM (5-fluorouracil, epirubicin, carboplatin, vincristine, mitomycin); or combination therapy with cisplatin, epirubicin, and paclitaxel.1
Prognosis of Metastatic EMPD—Because invasive and metastatic EMPD is rare, its natural history is hard to predict. Poor prognosis is associated with nodule formation, tumor thickness, perianal or vaginal involvement, lymphovascular invasion, nodal metastasis, and distant metastasis. The 5-year survival for metastatic EMPD has been reported to be less than 10%.46 Our cases underscore the poor prognostic risk associated with metastatic EMPD.
For all cases of EMPD, close follow-up is warranted. Guidelines recommend physical examination with lymph node assessment every 3 to 6 months for 3 years and every 6 to 12 months for the subsequent 5 years.2 Specific recommendations for follow-up in invasive disease have not yet been described, though the 20% probability of developing an internal malignancy within 5 years after diagnosis and poor prognostic outcomes associated with invasive and metastatic disease support the need for close monitoring.2
Conclusion
Although in situ EMPD often is a slow-growing tumor with good prognosis, invasive disease has high potential to behave aggressively with high morbidity and mortality. Increased awareness and prompt identification of invasive EMPD, expedited comprehensive workup, and early multidisciplinary management might impact patient outcomes.
Acknowledgment—The authors would like to thank Ellen Aaronson, MLIS, AHIP (Mayo Clinic Libraries [Jacksonville, FL]), for creating and conducting the narrative literature search in the MEDLINE database.
- Hashimoto H, Ito T. Current management and treatment of extramammary Paget’s disease. Curr Treat Options Oncol. 2022;23:818-830. doi:10.1007/s11864-021-00923-3
- Kibbi N, Owen JL, Worley B, et al. Evidence-based clinical practice guidelines for extramammary Paget disease. JAMA Oncol. 2022;8:618-628. doi:10.1001/jamaoncol.2021.7148
- Morris CR, Hurst EA. Extramammary Paget’s disease: a review of the literature part II: treatment and prognosis. Dermatol Surg. 2020;46:305-311. doi:10.1097/DSS.0000000000002240
- Merritt BG, Degesys CA, Brodland DG. Extramammary Paget disease. Dermatol Clin. 2019;37:261-267. doi:10.1016/j.det.2019.02.002
- Aroche Gutierrez LL, Holloway SB, Donthi D, et al. Docetaxel treatment for widely metastatic invasive vulvar extramammary Paget’s disease with multifocal bone metastasis. Gynecol Oncol Rep. 2022;45:101114. doi:10.1016/j.gore.2022.101114
- Ueda M, Omori M, Sakai A. Invasive extramammary Paget’s disease with lymph node metastases and high-grade B-cell lymphoma. An Bras Dermatol. 2023;98:414-418. doi:10.1016/j.abd.2022.04.012
- Rathore R, Yadav D, Agarwal S, et al. Primary extra mammary Paget’s disease of vulva, with apocrine adenocarcinoma, signet ring cell differentiation and distant metastasis. J Family Reprod Health. 2020;14:276-280. doi:10.18502/jfrh.v14i4.5213
- Kawahara Y, Umeda Y, Yamaguchi B, et al. Long-term resolution of invasive extramammary Paget’s disease with multiple regional lymph node metastases solely with regional lymph node dissection. J Dermatol. 2021;48:E452-E453. doi:10.1111/1346-8138.16007
- Hanyu T, Fujitani S, Ito A, et al. Brain metastasis from extramammary Paget’s disease. Nagoya J Med Sci. 2020;82:791-798. doi:10.18999/nagjms.82.4.791
- Waki Y, Nobeyama Y, Ogawa T, et al. Case of extramammary Paget’s disease causing pulmonary tumor embolism. J Dermatol. 2020;47:E133-E134. doi:10.1111/1346-8138.15267
- Li ZG, Qin XJ. Extensive invasive extramammary Paget disease evaluated by F-18 FDG PET/CT: a case report. Medicine (Baltimore). 2015;94:E371. doi:10.1097/MD.0000000000000371
- Kato N, Matsue K, Sotodate A, et al. Extramammary Paget’s disease with distant skin metastasis. J Dermatol. 1996;23:408-414. doi:10.1111/j.1346-8138.1996.tb04043.x
- Hosomi M, Miyake O, Matsumiya K, et al. Extramammary Paget’s disease with a large mass in male genitalia: a case report. Article in Japanese. Hinyokika Kiyo. 1989;35:1981-1984.
- Hardy LE, Baxter L, Wan K, et al. Invasive cervical adenocarcinoma arising from extension of recurrent vulval Paget’s disease. BMJ Case Rep. 2020;13e232424. doi:10.1136/bcr-2019-232424
- Onaiwu CO, Ramirez PT, Kamat A, et al. Invasive extramammary Paget’s disease of the bladder diagnosed 18 years after noninvasive extramammary Paget’s disease of the vulva. Gynecol Oncol Case Rep. 2014;8:27-29. doi:10.1016/j.gynor.2014.03.004
- Yao H, Xie M, Fu S, et al. Survival analysis of patients with invasive extramammary Paget disease: implications of anatomic sites. BMC Cancer. 2018;18:403. doi:10.1186/s12885-018-4257-1
- Kato H, Watanabe S, Kariya K, et al. Efficacy of low-dose 5-fluorouracil/cisplatin therapy for invasive extramammary Paget’s disease. J Dermatol. 2018;45:560-563. doi:10.1111/1346-8138.14247
- Yoshino K, Fujisawa Y, Kiyohara Y, et al. Usefulness of docetaxel as first-line chemotherapy for metastatic extramammary Paget’s disease. J Dermatol. 2016;43:633-637. doi:10.1111/1346-8138.13200
- Shu B, Shen XX, Chen P, et al. Primary invasive extramammary Paget disease on penoscrotum: a clinicopathological analysis of 41 cases. Hum Pathol. 2016;47:70-77. doi:10.1016/j.humpath.2015.09.005References
- Fujisawa Y, Yoshino K, Kiyohara Y, et al. The role of sentinel lymph node biopsy in the management of invasive extramammary Paget’s disease: multi-center, retrospective study of 151 patients. J Dermatol Sci. 2015;79:38-42. doi:10.1016/j.jdermsci.2015.03.014
- Dai B, Kong YY, Chang K, et al. Primary invasive carcinoma associated with penoscrotal extramammary Paget’s disease: a clinicopathological analysis of 56 cases. BJU Int. 2015;115:153-160. doi:10.1111/bju.12776
- Shiomi T, Noguchi T, Nakayama H, et al. Clinicopathological study of invasive extramammary Paget’s disease: subgroup comparison according to invasion depth. J Eur Acad Dermatol Venereol. 2013;27:589-592. doi:10.1111/j.1468-3083.2012.04489.x
- Hatta N, Morita R, Yamada M, et al. Sentinel lymph node biopsy in patients with extramammary Paget’s disease. Dermatol Surg. 2004;30:1329-1334. doi:10.1111/j.1524-4725.2004.30377.x
- Karam A, Dorigo O. Treatment outcomes in a large cohort of patients with invasive extramammary Paget’s disease. Gynecol Oncol. 2012;125:346-351. doi:10.1016/j.ygyno.2012.01.032
- Guo L, Liu X, Li H, et al. Clinicopathological features of extramammary Paget’s disease: a report of 75 cases. Article in Chinese. Zhonghua Yi Xue Za Zhi. 2015;95:1751-1754.
- Kilts TP, Long B, Glasgow AE, et al. Invasive vulvar extramammary Paget’s disease in the United States. Gynecol Oncol. 2020;157:649-655. doi:10.1016/j.ygyno.2020.03.018
- Kusatake K, Harada Y, Mizumoto K, et al. Usefulness of sentinel lymph node biopsy for the detection of metastasis in the early stage of extramammary Paget’s disease. Eur J Dermatol. 2015;25:156-161. doi:10.1684/ejd.2015.2534
- Jeong BK, Kim KR. Invasive extramammary Paget disease of the vulva with signet ring cell morphology in a patient with signet ring cell carcinoma of the stomach: report of a case. Int J Gynecol Pathol. 2018;37:147-151. doi:10.1097/PGP.0000000000000405
- Pagnanelli M, De Nardi P, Martella S, et al. Local excision of a mucinous adenocarcinoma of the anal margin (extramammary Paget’s disease) and reconstruction with a bilateral V-Y flap. Case Rep Surg. 2019;2019:9073982. doi:10.1155/2019/9073982
- Sopracordevole F, Di Giuseppe J, De Piero G, et al. Surgical treatment of Paget disease of the vulva: prognostic significance of stromal invasion and surgical margin status. J Low Genit Tract Dis. 2016;20:184-188. doi:10.1097/LGT.0000000000000191
- Evans AT, Neven P. Invasive adenocarcinoma arising in extramammary Paget’s disease of the vulva. Histopathology. 1991;18:355-360. doi:10.1111/j.1365-2559.1991.tb00857.x
- Kitano A, Izumi M, Tamura K, et al. Brain metastasis from cutaneous squamous cell carcinoma coexistent with extramammary Paget’s disease: a case report. Pathol Int. 2019;69:619-625. doi:10.1111/pin.12846
- Miracco C, Francini E, Torre P, et al. Extramammary invasive Paget’s disease and apocrine angiomatous hamartoma: an unusual association. Eur J Dermatol. 2018;28:853-855. doi:10.1684/ejd.2018.3438
- Kambayashi Y, Fujimura T, Ohuchi K, et al. Advanced invasive extramammary Paget’s disease concomitant with cecal cancer possessing rare variant of TP53 single nucleotide polymorphism. Case Rep Oncol. 2019;12:855-860. doi:10.1159/000504339
- Fujimura T, Furudate S, Kambayashi Y, et al. Potential use of bisphosphonates in invasive extramammary Paget’s disease: an immunohistochemical investigation. Clin Dev Immunol. 2013;2013:164982. doi:10.1155/2013/164982
- Kawamura H, Ogata K, Miura H, et al. Patellar metastases. A report of two cases. Int Orthop. 1993;17:57-59. doi:10.1007/BF00195227
- Damavandy AA, Terushkin V, Zitelli JA, et al. Intraoperative immunostaining for cytokeratin-7 during Mohs micrographic surgery demonstrates low local recurrence rates in extramammary Paget’s disease. Dermatol Surg. 2018;44:354-364. doi:10.1097/DSS.0000000000001355
- Morris CR, Hurst EA. Extramammary Paget disease: a review of the literature-part I: history, epidemiology, pathogenesis, presentation, histopathology, and diagnostic work-up. Dermatol Surg. 2020;46:151-158. doi:10.1097/DSS.0000000000002064
- Simonds RM, Segal RJ, Sharma A. Extramammary Paget’s disease: a review of the literature. Int J Dermatol. 2019;58:871-879. doi:10.1111/ijd.14328
- Ogata D, Kiyohara Y, Yoshikawa S, et al. Usefulness of sentinel lymph node biopsy for prognostic prediction in extramammary Paget’s disease. Eur J Dermatol. 2016;26:254-259. doi:10.1684/ejd.2016.2744
- Ohara K, Fujisawa Y, Yoshino K, et al. A proposal for a TNM staging system for extramammary Paget disease: retrospective analysis of 301 patients with invasive primary tumors. J Dermatol Sci. 2016;83:234-239. doi:10.1016/j.jdermsci.2016.06.004
- Fujisawa Y, Yoshino K, Kiyohara Y, et al. The role of sentinel lymph node biopsy in the management of invasive extramammary Paget’s disease: multi-center, retrospective study of 151 patients. J Dermatol Sci. 2015;79:38-42. doi:10.1016/j.jdermsci.2015.03.014
- Kim SJ, Thompson AK, Zubair AS, et al. Surgical treatment and outcomes of patients with extramammary Paget disease: a cohort study. Dermatol Surg. 2017;43:708-714. doi:10.1097/DSS.0000000000001051
- Wollina U. Extensive invasive extramammary Paget’s disease: surgical treatment. J Cutan Aesthet Surg. 2013;6:41-44. doi:10.4103/0974-2077.110098
- Tsutsui K, Takahashi A, Muto Y, et al. Outcomes of lymph node dissection in the treatment of extramammary Paget’s disease: a single-institution study. J Dermatol. 2020;47:512-517. doi:10.1111/1346-8138.15285
- Guercio BJ, Iyer G, Kidwai WZ, et al. Treatment of metastatic extramammary Paget disease with combination ipilimumab and nivolumab: a case report. Case Rep Oncol. 2021;14:430-438. doi:10.1159/000514345
Extramammary Paget disease (EMPD) is a rare cutaneous malignancy typically seen in apocrine-rich areas, including the axillae and anogenital region. It presents as a slow-growing, erythematous patch or plaque that commonly is misdiagnosed as an infectious or inflammatory condition.1,2 Primary EMPD occurs as a intraepithelial neoplasm, whereas secondary EMPD occurs due to epidermotropic metastases or direct extension of an underlying adenocarcinoma into the skin.1 Most commonly, primary EMPD occurs in situ; however, when present, dermal invasion and metastases from the skin are associated with poorer outcomes.3 Given the rarity of metastatic disease, existing literature is limited to case reports and case series.
We present 2 patients with metastatic primary EMPD who had evidence of invasion on initial biopsy and died secondary to metastatic EMPD. We conducted a comprehensive review of the literature for invasive and metastatic EMPD to highlight key clinicopathologic features, treatment considerations, and the potential for rapid disease progression in cases of invasive EMPD.
Case Series
Patient 1—A 68-year-old White man with a history of breast cancer (in remission) presented to our clinic for further management of biopsy-proven scrotal EMPD. Prior to biopsy, he described a 6-month history of worsening scrotal rash treated with topical antifungals, oral antibiotics, and topical steroids due to presumed diagnosis of intertrigo, cellulitis, and dermatitis, respectively. Clinical examination showed indurated, erythematous, ulcerated plaques involving the bilateral groin, genitalia, and perineum (Figure 1). Skin biopsy confirmed a diagnosis of EMPD with both dermal and lymphovascular invasion. An immunohistochemical profile was positive for CK7 and carcinoembryonic antigen (CEA) and negative for CK20 (Figure 2).
At presentation, the patient had palpable lymphadenopathy and scrotal edema concerning for inguinal and iliac lymph node metastases. Workup for an underlying adenocarcinoma included computed tomography (CT) of the chest, abdomen, and pelvis; urologic consultation with cystoscopy; and a screening colonoscopy. The CT scan revealed multiple enlarged inguinal and external iliac lymph nodes. Fine-needle aspiration revealed CK7- and CEA-positive neoplastic cells consistent with metastatic EMPD. The patient was treated with 6 cycles of carboplatin-paclitaxel, palliative radiation therapy, and pembrolizumab with minimal response to treatment and development of osteolytic vertebral lesions concerning for disease progression. He died 1 year after the initial diagnosis secondary to the disease.
Patient 2—A 79-year-old White man presented for further management of an outside diagnosis of superficially invasive primary EMPD of the bilateral inguinal folds and scrotum that had been present for 5 months prior to biopsy and diagnosis. Clinical examination at initial presentation revealed erythematous patches of the bilateral inguinal folds and scrotum, as well as an erythematous scaling plaque in the right axilla. There was no palpable clinical lymphadenopathy. Biopsy of the axilla and groin were both consistent with invasive EMPD with positive staining for CK7 and negative staining for CK20 and CDX2. Workup for underlying adenocarcinoma with whole-body positron emission tomography/CT, mammography, esophagogastroduodenoscopy, serum CEA, colonoscopy, and cystoscopy were all negative for a metastatic adenocarcinoma. There was no imaging or clinical evidence of lymphadenopathy. Complete circumferential peripheral and deep-margin assessment was performed in a staged manner on both sites, and negative margins were obtained.
Surveillance imaging 6 months after surgery revealed suspicious hepatic lesions. Fine-needle aspiration of the hepatic lesions demonstrated positive staining for CK7 and negative staining for CK20, CDX2, prostate-specific antigen, and thyroid transcription factor 1, consistent with metastatic EMPD. Oncology recommended carboplatin and docetaxel or docetaxel monotherapy chemotherapy. The patient was further managed by an outside oncologist due to ease of travel but died secondary to the disease 15 months following the initial diagnosis.
Comment
Extramammary Paget disease is an uncommon cutaneous malignancy that manifests as pruritic erythematous plaques within apocrine-rich areas such as the genitalia, axillae, or anal region. It most commonly occurs in patients older than 65 years, with White women and Asian men being affected at disproportionately higher rates.1,4 Delay in diagnosis is common, as EMPD can mimic other benign inflammatory or infectious conditions, including contact dermatitis, seborrheic dermatitis, tinea, candidiasis, and eczema.1
Metastatic and multifocal cases of primary EMPD are especially rare. According to a search of PubMed articles indexed for MEDLINE published through December 2023 using the terms extramammary Paget disease, EMPD, neoplasm metastasis, invasive extramammary, and neoplasm invasiveness, we identified 5040 cases of invasive EMPD and 477 cases of metastatic EMPD.5-37 Of the reports that disclosed patient demographic information, 3627 patients were female 1410 were male, and the mean age was 67 years. Sites of metastases included regional lymph nodes, liver, lungs, cervix, bladder, bone, brain, skin, kidney, and adrenal glands
Workup for EMPD—The initial steps for workup of EMPD include a thorough physical examination and lymph node assessment. A skin biopsy also should be performed for patients presenting with refractory, pruritic, and eczematous rashes in apocrine-rich areas to evaluate for EMPD.1 Characterization of large and complex tumors is better achieved through multiple biopsies with particular focus on nodular or thickened areas, as these may indicate invasive disease.2 Primary EMPD is characterized by pagetoid cells with abundant pale cytoplasm proliferating in a single-cell or nested pattern within the epidermis or dermis in invasive disease and often is accompanied by dermal lymphocytic inflammation.1 Immunohistochemistry demonstrates positive staining for CEA, CK7, and CK8, with negative staining for indicators of secondary EMPD including CK20 and CDX2.1,2
As part of the workup, it is critical to distinguish between primary disease and secondary EMPD.1 Beyond skin and clinical lymph node examination, additional workup should be based on age-appropriate and location-directed malignant neoplasm screenings, including colonoscopy, cystoscopy, prostate examination, mammography, and Papanicolaou test. Advanced imaging such as CT, positron emission tomography, or magnetic resonance imaging can be used to assess for metastatic disease if internal malignant neoplasms are present on initial screening or clinical lymphadenopathy is identified.2 Additionally, it can be helpful in the evaluation for nodal disease in cases of invasive EMPD.
The likelihood of associated underlying carcinomas varies depending on the site of involvement.38,39 For example, vulvar involvement constitutes approximately 65% of EMPD cases, with 11% to 20% of cases being associated with underlying gastrointestinal or genitourinary carcinomas. Involvement of the male genitalia, as in our 2 patients, is rare, accounting for approximately 14% of cases, 11% of which are associated with prostate, testicular, and bladder carcinoma. Perianal involvement comprises 20% of EMPD cases and has the greatest risk for underlying malignancy with an incidence of 33% to 86%, the majority of which are rectal or tubo-ovarian cancers.38,39 Consideration of the frequency and types of underlying carcinoma of respective sites of involvement can be helpful when ruling out secondary EMPD.
In both of our patients, palpable lymphadenopathy at the time of original diagnosis and histologic invasive disease on initial biopsy warranted thorough imaging and laboratory workup; there was no evidence of primary malignancy. Given the absence of an underlying carcinoma, both patients were classified as having metastatic primary EMPD.
Assessment of lymphadenopathy is an essential aspect of disease workup, as it is associated with a statistically higher rate of lymph node metastases. A study by Fujisawa et al20 demonstrated that 80% of patients with lymphadenopathy had regional metastases compared to only 15% of patients without clinical lymphadenopathy. The presence of invasive disease also has been shown to correspond with lymph node metastases.40 Ogata et al40 showed that 0% of cases with in situ EMPD had a positive sentinel lymph node biopsy (SLNB) compared to 4% and 43% in cases that showed evidence of microinvasion and dermal invasion, respectively. Lymph node metastases are associated with poor prognosis, with increasingly worse prognosis when there are multiple lymph nodes affected.41 In our case series, patient 1 had lymphadenopathy and both patients had invasive EMPD; they both later developed metastases and died.
Lymphadenopathy should be further investigated with imaging and biopsy or fine-needle aspiration.42 Recent expert consensus guidelines recommended this method of investigation over routine use of SLNB, as there is no evidence that a positive SLNB affects treatment that changes disease-specific survival.2
Treatment of EMPD—Surgical excision of the primary lesion is the first-line treatment of EMPD,1,2 which can be performed by wide local excision; however, studies have demonstrated higher recurrence-free survival with margin-controlled surgery (complete circumferential peripheral and deep margin assessment) or Mohs micrographic surgery (MMS), especially with CK7 immunostaining.2,37,43 The literature on MMS of invasive EMPD is sparse, accounting for 57 patients.25,37,44 Other reports describe management with surgical excision, wide local excision, regional resection, or vulvectomy, in addition to lymph node dissection, radiation therapy (RT), and/or chemotherapy.1-36,39,43-46 Despite the improved outcomes with MMS, the predominance of other surgical approaches in our search suggests that MMS may be currently underutilized for the treatment of invasive or locally advanced EMPD.
Among patients with unresectable disease or distant metastases, management includes RT with curative intent, chemotherapy, or a combination of both.1,2 In our review, 267 cases were treated using RT and 77 with chemotherapy. Radiation therapy is an effective therapeutic option with a reported response rate of 62% to 100% and can be employed as either primary or adjuvant treatment.3 For patients with lymph node metastasis the combination of RT and lymph node dissection has been shown to have improved outcomes compared to lymph node dissection alone, with 1 study showing a 5-year survival of 75% for patients who received adjuvant RT compared to 0% for lymph node dissection alone.45
There are currently no consensus guidelines on the best chemotherapeutic regimen for metastatic EMPD. Several regimens have been reported, including docetaxel monotherapy; low-dose 5-fluorouracil and cisplatin; combination chemotherapy FECOM (5-fluorouracil, epirubicin, carboplatin, vincristine, mitomycin); or combination therapy with cisplatin, epirubicin, and paclitaxel.1
Prognosis of Metastatic EMPD—Because invasive and metastatic EMPD is rare, its natural history is hard to predict. Poor prognosis is associated with nodule formation, tumor thickness, perianal or vaginal involvement, lymphovascular invasion, nodal metastasis, and distant metastasis. The 5-year survival for metastatic EMPD has been reported to be less than 10%.46 Our cases underscore the poor prognostic risk associated with metastatic EMPD.
For all cases of EMPD, close follow-up is warranted. Guidelines recommend physical examination with lymph node assessment every 3 to 6 months for 3 years and every 6 to 12 months for the subsequent 5 years.2 Specific recommendations for follow-up in invasive disease have not yet been described, though the 20% probability of developing an internal malignancy within 5 years after diagnosis and poor prognostic outcomes associated with invasive and metastatic disease support the need for close monitoring.2
Conclusion
Although in situ EMPD often is a slow-growing tumor with good prognosis, invasive disease has high potential to behave aggressively with high morbidity and mortality. Increased awareness and prompt identification of invasive EMPD, expedited comprehensive workup, and early multidisciplinary management might impact patient outcomes.
Acknowledgment—The authors would like to thank Ellen Aaronson, MLIS, AHIP (Mayo Clinic Libraries [Jacksonville, FL]), for creating and conducting the narrative literature search in the MEDLINE database.
Extramammary Paget disease (EMPD) is a rare cutaneous malignancy typically seen in apocrine-rich areas, including the axillae and anogenital region. It presents as a slow-growing, erythematous patch or plaque that commonly is misdiagnosed as an infectious or inflammatory condition.1,2 Primary EMPD occurs as a intraepithelial neoplasm, whereas secondary EMPD occurs due to epidermotropic metastases or direct extension of an underlying adenocarcinoma into the skin.1 Most commonly, primary EMPD occurs in situ; however, when present, dermal invasion and metastases from the skin are associated with poorer outcomes.3 Given the rarity of metastatic disease, existing literature is limited to case reports and case series.
We present 2 patients with metastatic primary EMPD who had evidence of invasion on initial biopsy and died secondary to metastatic EMPD. We conducted a comprehensive review of the literature for invasive and metastatic EMPD to highlight key clinicopathologic features, treatment considerations, and the potential for rapid disease progression in cases of invasive EMPD.
Case Series
Patient 1—A 68-year-old White man with a history of breast cancer (in remission) presented to our clinic for further management of biopsy-proven scrotal EMPD. Prior to biopsy, he described a 6-month history of worsening scrotal rash treated with topical antifungals, oral antibiotics, and topical steroids due to presumed diagnosis of intertrigo, cellulitis, and dermatitis, respectively. Clinical examination showed indurated, erythematous, ulcerated plaques involving the bilateral groin, genitalia, and perineum (Figure 1). Skin biopsy confirmed a diagnosis of EMPD with both dermal and lymphovascular invasion. An immunohistochemical profile was positive for CK7 and carcinoembryonic antigen (CEA) and negative for CK20 (Figure 2).
At presentation, the patient had palpable lymphadenopathy and scrotal edema concerning for inguinal and iliac lymph node metastases. Workup for an underlying adenocarcinoma included computed tomography (CT) of the chest, abdomen, and pelvis; urologic consultation with cystoscopy; and a screening colonoscopy. The CT scan revealed multiple enlarged inguinal and external iliac lymph nodes. Fine-needle aspiration revealed CK7- and CEA-positive neoplastic cells consistent with metastatic EMPD. The patient was treated with 6 cycles of carboplatin-paclitaxel, palliative radiation therapy, and pembrolizumab with minimal response to treatment and development of osteolytic vertebral lesions concerning for disease progression. He died 1 year after the initial diagnosis secondary to the disease.
Patient 2—A 79-year-old White man presented for further management of an outside diagnosis of superficially invasive primary EMPD of the bilateral inguinal folds and scrotum that had been present for 5 months prior to biopsy and diagnosis. Clinical examination at initial presentation revealed erythematous patches of the bilateral inguinal folds and scrotum, as well as an erythematous scaling plaque in the right axilla. There was no palpable clinical lymphadenopathy. Biopsy of the axilla and groin were both consistent with invasive EMPD with positive staining for CK7 and negative staining for CK20 and CDX2. Workup for underlying adenocarcinoma with whole-body positron emission tomography/CT, mammography, esophagogastroduodenoscopy, serum CEA, colonoscopy, and cystoscopy were all negative for a metastatic adenocarcinoma. There was no imaging or clinical evidence of lymphadenopathy. Complete circumferential peripheral and deep-margin assessment was performed in a staged manner on both sites, and negative margins were obtained.
Surveillance imaging 6 months after surgery revealed suspicious hepatic lesions. Fine-needle aspiration of the hepatic lesions demonstrated positive staining for CK7 and negative staining for CK20, CDX2, prostate-specific antigen, and thyroid transcription factor 1, consistent with metastatic EMPD. Oncology recommended carboplatin and docetaxel or docetaxel monotherapy chemotherapy. The patient was further managed by an outside oncologist due to ease of travel but died secondary to the disease 15 months following the initial diagnosis.
Comment
Extramammary Paget disease is an uncommon cutaneous malignancy that manifests as pruritic erythematous plaques within apocrine-rich areas such as the genitalia, axillae, or anal region. It most commonly occurs in patients older than 65 years, with White women and Asian men being affected at disproportionately higher rates.1,4 Delay in diagnosis is common, as EMPD can mimic other benign inflammatory or infectious conditions, including contact dermatitis, seborrheic dermatitis, tinea, candidiasis, and eczema.1
Metastatic and multifocal cases of primary EMPD are especially rare. According to a search of PubMed articles indexed for MEDLINE published through December 2023 using the terms extramammary Paget disease, EMPD, neoplasm metastasis, invasive extramammary, and neoplasm invasiveness, we identified 5040 cases of invasive EMPD and 477 cases of metastatic EMPD.5-37 Of the reports that disclosed patient demographic information, 3627 patients were female 1410 were male, and the mean age was 67 years. Sites of metastases included regional lymph nodes, liver, lungs, cervix, bladder, bone, brain, skin, kidney, and adrenal glands
Workup for EMPD—The initial steps for workup of EMPD include a thorough physical examination and lymph node assessment. A skin biopsy also should be performed for patients presenting with refractory, pruritic, and eczematous rashes in apocrine-rich areas to evaluate for EMPD.1 Characterization of large and complex tumors is better achieved through multiple biopsies with particular focus on nodular or thickened areas, as these may indicate invasive disease.2 Primary EMPD is characterized by pagetoid cells with abundant pale cytoplasm proliferating in a single-cell or nested pattern within the epidermis or dermis in invasive disease and often is accompanied by dermal lymphocytic inflammation.1 Immunohistochemistry demonstrates positive staining for CEA, CK7, and CK8, with negative staining for indicators of secondary EMPD including CK20 and CDX2.1,2
As part of the workup, it is critical to distinguish between primary disease and secondary EMPD.1 Beyond skin and clinical lymph node examination, additional workup should be based on age-appropriate and location-directed malignant neoplasm screenings, including colonoscopy, cystoscopy, prostate examination, mammography, and Papanicolaou test. Advanced imaging such as CT, positron emission tomography, or magnetic resonance imaging can be used to assess for metastatic disease if internal malignant neoplasms are present on initial screening or clinical lymphadenopathy is identified.2 Additionally, it can be helpful in the evaluation for nodal disease in cases of invasive EMPD.
The likelihood of associated underlying carcinomas varies depending on the site of involvement.38,39 For example, vulvar involvement constitutes approximately 65% of EMPD cases, with 11% to 20% of cases being associated with underlying gastrointestinal or genitourinary carcinomas. Involvement of the male genitalia, as in our 2 patients, is rare, accounting for approximately 14% of cases, 11% of which are associated with prostate, testicular, and bladder carcinoma. Perianal involvement comprises 20% of EMPD cases and has the greatest risk for underlying malignancy with an incidence of 33% to 86%, the majority of which are rectal or tubo-ovarian cancers.38,39 Consideration of the frequency and types of underlying carcinoma of respective sites of involvement can be helpful when ruling out secondary EMPD.
In both of our patients, palpable lymphadenopathy at the time of original diagnosis and histologic invasive disease on initial biopsy warranted thorough imaging and laboratory workup; there was no evidence of primary malignancy. Given the absence of an underlying carcinoma, both patients were classified as having metastatic primary EMPD.
Assessment of lymphadenopathy is an essential aspect of disease workup, as it is associated with a statistically higher rate of lymph node metastases. A study by Fujisawa et al20 demonstrated that 80% of patients with lymphadenopathy had regional metastases compared to only 15% of patients without clinical lymphadenopathy. The presence of invasive disease also has been shown to correspond with lymph node metastases.40 Ogata et al40 showed that 0% of cases with in situ EMPD had a positive sentinel lymph node biopsy (SLNB) compared to 4% and 43% in cases that showed evidence of microinvasion and dermal invasion, respectively. Lymph node metastases are associated with poor prognosis, with increasingly worse prognosis when there are multiple lymph nodes affected.41 In our case series, patient 1 had lymphadenopathy and both patients had invasive EMPD; they both later developed metastases and died.
Lymphadenopathy should be further investigated with imaging and biopsy or fine-needle aspiration.42 Recent expert consensus guidelines recommended this method of investigation over routine use of SLNB, as there is no evidence that a positive SLNB affects treatment that changes disease-specific survival.2
Treatment of EMPD—Surgical excision of the primary lesion is the first-line treatment of EMPD,1,2 which can be performed by wide local excision; however, studies have demonstrated higher recurrence-free survival with margin-controlled surgery (complete circumferential peripheral and deep margin assessment) or Mohs micrographic surgery (MMS), especially with CK7 immunostaining.2,37,43 The literature on MMS of invasive EMPD is sparse, accounting for 57 patients.25,37,44 Other reports describe management with surgical excision, wide local excision, regional resection, or vulvectomy, in addition to lymph node dissection, radiation therapy (RT), and/or chemotherapy.1-36,39,43-46 Despite the improved outcomes with MMS, the predominance of other surgical approaches in our search suggests that MMS may be currently underutilized for the treatment of invasive or locally advanced EMPD.
Among patients with unresectable disease or distant metastases, management includes RT with curative intent, chemotherapy, or a combination of both.1,2 In our review, 267 cases were treated using RT and 77 with chemotherapy. Radiation therapy is an effective therapeutic option with a reported response rate of 62% to 100% and can be employed as either primary or adjuvant treatment.3 For patients with lymph node metastasis the combination of RT and lymph node dissection has been shown to have improved outcomes compared to lymph node dissection alone, with 1 study showing a 5-year survival of 75% for patients who received adjuvant RT compared to 0% for lymph node dissection alone.45
There are currently no consensus guidelines on the best chemotherapeutic regimen for metastatic EMPD. Several regimens have been reported, including docetaxel monotherapy; low-dose 5-fluorouracil and cisplatin; combination chemotherapy FECOM (5-fluorouracil, epirubicin, carboplatin, vincristine, mitomycin); or combination therapy with cisplatin, epirubicin, and paclitaxel.1
Prognosis of Metastatic EMPD—Because invasive and metastatic EMPD is rare, its natural history is hard to predict. Poor prognosis is associated with nodule formation, tumor thickness, perianal or vaginal involvement, lymphovascular invasion, nodal metastasis, and distant metastasis. The 5-year survival for metastatic EMPD has been reported to be less than 10%.46 Our cases underscore the poor prognostic risk associated with metastatic EMPD.
For all cases of EMPD, close follow-up is warranted. Guidelines recommend physical examination with lymph node assessment every 3 to 6 months for 3 years and every 6 to 12 months for the subsequent 5 years.2 Specific recommendations for follow-up in invasive disease have not yet been described, though the 20% probability of developing an internal malignancy within 5 years after diagnosis and poor prognostic outcomes associated with invasive and metastatic disease support the need for close monitoring.2
Conclusion
Although in situ EMPD often is a slow-growing tumor with good prognosis, invasive disease has high potential to behave aggressively with high morbidity and mortality. Increased awareness and prompt identification of invasive EMPD, expedited comprehensive workup, and early multidisciplinary management might impact patient outcomes.
Acknowledgment—The authors would like to thank Ellen Aaronson, MLIS, AHIP (Mayo Clinic Libraries [Jacksonville, FL]), for creating and conducting the narrative literature search in the MEDLINE database.
- Hashimoto H, Ito T. Current management and treatment of extramammary Paget’s disease. Curr Treat Options Oncol. 2022;23:818-830. doi:10.1007/s11864-021-00923-3
- Kibbi N, Owen JL, Worley B, et al. Evidence-based clinical practice guidelines for extramammary Paget disease. JAMA Oncol. 2022;8:618-628. doi:10.1001/jamaoncol.2021.7148
- Morris CR, Hurst EA. Extramammary Paget’s disease: a review of the literature part II: treatment and prognosis. Dermatol Surg. 2020;46:305-311. doi:10.1097/DSS.0000000000002240
- Merritt BG, Degesys CA, Brodland DG. Extramammary Paget disease. Dermatol Clin. 2019;37:261-267. doi:10.1016/j.det.2019.02.002
- Aroche Gutierrez LL, Holloway SB, Donthi D, et al. Docetaxel treatment for widely metastatic invasive vulvar extramammary Paget’s disease with multifocal bone metastasis. Gynecol Oncol Rep. 2022;45:101114. doi:10.1016/j.gore.2022.101114
- Ueda M, Omori M, Sakai A. Invasive extramammary Paget’s disease with lymph node metastases and high-grade B-cell lymphoma. An Bras Dermatol. 2023;98:414-418. doi:10.1016/j.abd.2022.04.012
- Rathore R, Yadav D, Agarwal S, et al. Primary extra mammary Paget’s disease of vulva, with apocrine adenocarcinoma, signet ring cell differentiation and distant metastasis. J Family Reprod Health. 2020;14:276-280. doi:10.18502/jfrh.v14i4.5213
- Kawahara Y, Umeda Y, Yamaguchi B, et al. Long-term resolution of invasive extramammary Paget’s disease with multiple regional lymph node metastases solely with regional lymph node dissection. J Dermatol. 2021;48:E452-E453. doi:10.1111/1346-8138.16007
- Hanyu T, Fujitani S, Ito A, et al. Brain metastasis from extramammary Paget’s disease. Nagoya J Med Sci. 2020;82:791-798. doi:10.18999/nagjms.82.4.791
- Waki Y, Nobeyama Y, Ogawa T, et al. Case of extramammary Paget’s disease causing pulmonary tumor embolism. J Dermatol. 2020;47:E133-E134. doi:10.1111/1346-8138.15267
- Li ZG, Qin XJ. Extensive invasive extramammary Paget disease evaluated by F-18 FDG PET/CT: a case report. Medicine (Baltimore). 2015;94:E371. doi:10.1097/MD.0000000000000371
- Kato N, Matsue K, Sotodate A, et al. Extramammary Paget’s disease with distant skin metastasis. J Dermatol. 1996;23:408-414. doi:10.1111/j.1346-8138.1996.tb04043.x
- Hosomi M, Miyake O, Matsumiya K, et al. Extramammary Paget’s disease with a large mass in male genitalia: a case report. Article in Japanese. Hinyokika Kiyo. 1989;35:1981-1984.
- Hardy LE, Baxter L, Wan K, et al. Invasive cervical adenocarcinoma arising from extension of recurrent vulval Paget’s disease. BMJ Case Rep. 2020;13e232424. doi:10.1136/bcr-2019-232424
- Onaiwu CO, Ramirez PT, Kamat A, et al. Invasive extramammary Paget’s disease of the bladder diagnosed 18 years after noninvasive extramammary Paget’s disease of the vulva. Gynecol Oncol Case Rep. 2014;8:27-29. doi:10.1016/j.gynor.2014.03.004
- Yao H, Xie M, Fu S, et al. Survival analysis of patients with invasive extramammary Paget disease: implications of anatomic sites. BMC Cancer. 2018;18:403. doi:10.1186/s12885-018-4257-1
- Kato H, Watanabe S, Kariya K, et al. Efficacy of low-dose 5-fluorouracil/cisplatin therapy for invasive extramammary Paget’s disease. J Dermatol. 2018;45:560-563. doi:10.1111/1346-8138.14247
- Yoshino K, Fujisawa Y, Kiyohara Y, et al. Usefulness of docetaxel as first-line chemotherapy for metastatic extramammary Paget’s disease. J Dermatol. 2016;43:633-637. doi:10.1111/1346-8138.13200
- Shu B, Shen XX, Chen P, et al. Primary invasive extramammary Paget disease on penoscrotum: a clinicopathological analysis of 41 cases. Hum Pathol. 2016;47:70-77. doi:10.1016/j.humpath.2015.09.005References
- Fujisawa Y, Yoshino K, Kiyohara Y, et al. The role of sentinel lymph node biopsy in the management of invasive extramammary Paget’s disease: multi-center, retrospective study of 151 patients. J Dermatol Sci. 2015;79:38-42. doi:10.1016/j.jdermsci.2015.03.014
- Dai B, Kong YY, Chang K, et al. Primary invasive carcinoma associated with penoscrotal extramammary Paget’s disease: a clinicopathological analysis of 56 cases. BJU Int. 2015;115:153-160. doi:10.1111/bju.12776
- Shiomi T, Noguchi T, Nakayama H, et al. Clinicopathological study of invasive extramammary Paget’s disease: subgroup comparison according to invasion depth. J Eur Acad Dermatol Venereol. 2013;27:589-592. doi:10.1111/j.1468-3083.2012.04489.x
- Hatta N, Morita R, Yamada M, et al. Sentinel lymph node biopsy in patients with extramammary Paget’s disease. Dermatol Surg. 2004;30:1329-1334. doi:10.1111/j.1524-4725.2004.30377.x
- Karam A, Dorigo O. Treatment outcomes in a large cohort of patients with invasive extramammary Paget’s disease. Gynecol Oncol. 2012;125:346-351. doi:10.1016/j.ygyno.2012.01.032
- Guo L, Liu X, Li H, et al. Clinicopathological features of extramammary Paget’s disease: a report of 75 cases. Article in Chinese. Zhonghua Yi Xue Za Zhi. 2015;95:1751-1754.
- Kilts TP, Long B, Glasgow AE, et al. Invasive vulvar extramammary Paget’s disease in the United States. Gynecol Oncol. 2020;157:649-655. doi:10.1016/j.ygyno.2020.03.018
- Kusatake K, Harada Y, Mizumoto K, et al. Usefulness of sentinel lymph node biopsy for the detection of metastasis in the early stage of extramammary Paget’s disease. Eur J Dermatol. 2015;25:156-161. doi:10.1684/ejd.2015.2534
- Jeong BK, Kim KR. Invasive extramammary Paget disease of the vulva with signet ring cell morphology in a patient with signet ring cell carcinoma of the stomach: report of a case. Int J Gynecol Pathol. 2018;37:147-151. doi:10.1097/PGP.0000000000000405
- Pagnanelli M, De Nardi P, Martella S, et al. Local excision of a mucinous adenocarcinoma of the anal margin (extramammary Paget’s disease) and reconstruction with a bilateral V-Y flap. Case Rep Surg. 2019;2019:9073982. doi:10.1155/2019/9073982
- Sopracordevole F, Di Giuseppe J, De Piero G, et al. Surgical treatment of Paget disease of the vulva: prognostic significance of stromal invasion and surgical margin status. J Low Genit Tract Dis. 2016;20:184-188. doi:10.1097/LGT.0000000000000191
- Evans AT, Neven P. Invasive adenocarcinoma arising in extramammary Paget’s disease of the vulva. Histopathology. 1991;18:355-360. doi:10.1111/j.1365-2559.1991.tb00857.x
- Kitano A, Izumi M, Tamura K, et al. Brain metastasis from cutaneous squamous cell carcinoma coexistent with extramammary Paget’s disease: a case report. Pathol Int. 2019;69:619-625. doi:10.1111/pin.12846
- Miracco C, Francini E, Torre P, et al. Extramammary invasive Paget’s disease and apocrine angiomatous hamartoma: an unusual association. Eur J Dermatol. 2018;28:853-855. doi:10.1684/ejd.2018.3438
- Kambayashi Y, Fujimura T, Ohuchi K, et al. Advanced invasive extramammary Paget’s disease concomitant with cecal cancer possessing rare variant of TP53 single nucleotide polymorphism. Case Rep Oncol. 2019;12:855-860. doi:10.1159/000504339
- Fujimura T, Furudate S, Kambayashi Y, et al. Potential use of bisphosphonates in invasive extramammary Paget’s disease: an immunohistochemical investigation. Clin Dev Immunol. 2013;2013:164982. doi:10.1155/2013/164982
- Kawamura H, Ogata K, Miura H, et al. Patellar metastases. A report of two cases. Int Orthop. 1993;17:57-59. doi:10.1007/BF00195227
- Damavandy AA, Terushkin V, Zitelli JA, et al. Intraoperative immunostaining for cytokeratin-7 during Mohs micrographic surgery demonstrates low local recurrence rates in extramammary Paget’s disease. Dermatol Surg. 2018;44:354-364. doi:10.1097/DSS.0000000000001355
- Morris CR, Hurst EA. Extramammary Paget disease: a review of the literature-part I: history, epidemiology, pathogenesis, presentation, histopathology, and diagnostic work-up. Dermatol Surg. 2020;46:151-158. doi:10.1097/DSS.0000000000002064
- Simonds RM, Segal RJ, Sharma A. Extramammary Paget’s disease: a review of the literature. Int J Dermatol. 2019;58:871-879. doi:10.1111/ijd.14328
- Ogata D, Kiyohara Y, Yoshikawa S, et al. Usefulness of sentinel lymph node biopsy for prognostic prediction in extramammary Paget’s disease. Eur J Dermatol. 2016;26:254-259. doi:10.1684/ejd.2016.2744
- Ohara K, Fujisawa Y, Yoshino K, et al. A proposal for a TNM staging system for extramammary Paget disease: retrospective analysis of 301 patients with invasive primary tumors. J Dermatol Sci. 2016;83:234-239. doi:10.1016/j.jdermsci.2016.06.004
- Fujisawa Y, Yoshino K, Kiyohara Y, et al. The role of sentinel lymph node biopsy in the management of invasive extramammary Paget’s disease: multi-center, retrospective study of 151 patients. J Dermatol Sci. 2015;79:38-42. doi:10.1016/j.jdermsci.2015.03.014
- Kim SJ, Thompson AK, Zubair AS, et al. Surgical treatment and outcomes of patients with extramammary Paget disease: a cohort study. Dermatol Surg. 2017;43:708-714. doi:10.1097/DSS.0000000000001051
- Wollina U. Extensive invasive extramammary Paget’s disease: surgical treatment. J Cutan Aesthet Surg. 2013;6:41-44. doi:10.4103/0974-2077.110098
- Tsutsui K, Takahashi A, Muto Y, et al. Outcomes of lymph node dissection in the treatment of extramammary Paget’s disease: a single-institution study. J Dermatol. 2020;47:512-517. doi:10.1111/1346-8138.15285
- Guercio BJ, Iyer G, Kidwai WZ, et al. Treatment of metastatic extramammary Paget disease with combination ipilimumab and nivolumab: a case report. Case Rep Oncol. 2021;14:430-438. doi:10.1159/000514345
- Hashimoto H, Ito T. Current management and treatment of extramammary Paget’s disease. Curr Treat Options Oncol. 2022;23:818-830. doi:10.1007/s11864-021-00923-3
- Kibbi N, Owen JL, Worley B, et al. Evidence-based clinical practice guidelines for extramammary Paget disease. JAMA Oncol. 2022;8:618-628. doi:10.1001/jamaoncol.2021.7148
- Morris CR, Hurst EA. Extramammary Paget’s disease: a review of the literature part II: treatment and prognosis. Dermatol Surg. 2020;46:305-311. doi:10.1097/DSS.0000000000002240
- Merritt BG, Degesys CA, Brodland DG. Extramammary Paget disease. Dermatol Clin. 2019;37:261-267. doi:10.1016/j.det.2019.02.002
- Aroche Gutierrez LL, Holloway SB, Donthi D, et al. Docetaxel treatment for widely metastatic invasive vulvar extramammary Paget’s disease with multifocal bone metastasis. Gynecol Oncol Rep. 2022;45:101114. doi:10.1016/j.gore.2022.101114
- Ueda M, Omori M, Sakai A. Invasive extramammary Paget’s disease with lymph node metastases and high-grade B-cell lymphoma. An Bras Dermatol. 2023;98:414-418. doi:10.1016/j.abd.2022.04.012
- Rathore R, Yadav D, Agarwal S, et al. Primary extra mammary Paget’s disease of vulva, with apocrine adenocarcinoma, signet ring cell differentiation and distant metastasis. J Family Reprod Health. 2020;14:276-280. doi:10.18502/jfrh.v14i4.5213
- Kawahara Y, Umeda Y, Yamaguchi B, et al. Long-term resolution of invasive extramammary Paget’s disease with multiple regional lymph node metastases solely with regional lymph node dissection. J Dermatol. 2021;48:E452-E453. doi:10.1111/1346-8138.16007
- Hanyu T, Fujitani S, Ito A, et al. Brain metastasis from extramammary Paget’s disease. Nagoya J Med Sci. 2020;82:791-798. doi:10.18999/nagjms.82.4.791
- Waki Y, Nobeyama Y, Ogawa T, et al. Case of extramammary Paget’s disease causing pulmonary tumor embolism. J Dermatol. 2020;47:E133-E134. doi:10.1111/1346-8138.15267
- Li ZG, Qin XJ. Extensive invasive extramammary Paget disease evaluated by F-18 FDG PET/CT: a case report. Medicine (Baltimore). 2015;94:E371. doi:10.1097/MD.0000000000000371
- Kato N, Matsue K, Sotodate A, et al. Extramammary Paget’s disease with distant skin metastasis. J Dermatol. 1996;23:408-414. doi:10.1111/j.1346-8138.1996.tb04043.x
- Hosomi M, Miyake O, Matsumiya K, et al. Extramammary Paget’s disease with a large mass in male genitalia: a case report. Article in Japanese. Hinyokika Kiyo. 1989;35:1981-1984.
- Hardy LE, Baxter L, Wan K, et al. Invasive cervical adenocarcinoma arising from extension of recurrent vulval Paget’s disease. BMJ Case Rep. 2020;13e232424. doi:10.1136/bcr-2019-232424
- Onaiwu CO, Ramirez PT, Kamat A, et al. Invasive extramammary Paget’s disease of the bladder diagnosed 18 years after noninvasive extramammary Paget’s disease of the vulva. Gynecol Oncol Case Rep. 2014;8:27-29. doi:10.1016/j.gynor.2014.03.004
- Yao H, Xie M, Fu S, et al. Survival analysis of patients with invasive extramammary Paget disease: implications of anatomic sites. BMC Cancer. 2018;18:403. doi:10.1186/s12885-018-4257-1
- Kato H, Watanabe S, Kariya K, et al. Efficacy of low-dose 5-fluorouracil/cisplatin therapy for invasive extramammary Paget’s disease. J Dermatol. 2018;45:560-563. doi:10.1111/1346-8138.14247
- Yoshino K, Fujisawa Y, Kiyohara Y, et al. Usefulness of docetaxel as first-line chemotherapy for metastatic extramammary Paget’s disease. J Dermatol. 2016;43:633-637. doi:10.1111/1346-8138.13200
- Shu B, Shen XX, Chen P, et al. Primary invasive extramammary Paget disease on penoscrotum: a clinicopathological analysis of 41 cases. Hum Pathol. 2016;47:70-77. doi:10.1016/j.humpath.2015.09.005References
- Fujisawa Y, Yoshino K, Kiyohara Y, et al. The role of sentinel lymph node biopsy in the management of invasive extramammary Paget’s disease: multi-center, retrospective study of 151 patients. J Dermatol Sci. 2015;79:38-42. doi:10.1016/j.jdermsci.2015.03.014
- Dai B, Kong YY, Chang K, et al. Primary invasive carcinoma associated with penoscrotal extramammary Paget’s disease: a clinicopathological analysis of 56 cases. BJU Int. 2015;115:153-160. doi:10.1111/bju.12776
- Shiomi T, Noguchi T, Nakayama H, et al. Clinicopathological study of invasive extramammary Paget’s disease: subgroup comparison according to invasion depth. J Eur Acad Dermatol Venereol. 2013;27:589-592. doi:10.1111/j.1468-3083.2012.04489.x
- Hatta N, Morita R, Yamada M, et al. Sentinel lymph node biopsy in patients with extramammary Paget’s disease. Dermatol Surg. 2004;30:1329-1334. doi:10.1111/j.1524-4725.2004.30377.x
- Karam A, Dorigo O. Treatment outcomes in a large cohort of patients with invasive extramammary Paget’s disease. Gynecol Oncol. 2012;125:346-351. doi:10.1016/j.ygyno.2012.01.032
- Guo L, Liu X, Li H, et al. Clinicopathological features of extramammary Paget’s disease: a report of 75 cases. Article in Chinese. Zhonghua Yi Xue Za Zhi. 2015;95:1751-1754.
- Kilts TP, Long B, Glasgow AE, et al. Invasive vulvar extramammary Paget’s disease in the United States. Gynecol Oncol. 2020;157:649-655. doi:10.1016/j.ygyno.2020.03.018
- Kusatake K, Harada Y, Mizumoto K, et al. Usefulness of sentinel lymph node biopsy for the detection of metastasis in the early stage of extramammary Paget’s disease. Eur J Dermatol. 2015;25:156-161. doi:10.1684/ejd.2015.2534
- Jeong BK, Kim KR. Invasive extramammary Paget disease of the vulva with signet ring cell morphology in a patient with signet ring cell carcinoma of the stomach: report of a case. Int J Gynecol Pathol. 2018;37:147-151. doi:10.1097/PGP.0000000000000405
- Pagnanelli M, De Nardi P, Martella S, et al. Local excision of a mucinous adenocarcinoma of the anal margin (extramammary Paget’s disease) and reconstruction with a bilateral V-Y flap. Case Rep Surg. 2019;2019:9073982. doi:10.1155/2019/9073982
- Sopracordevole F, Di Giuseppe J, De Piero G, et al. Surgical treatment of Paget disease of the vulva: prognostic significance of stromal invasion and surgical margin status. J Low Genit Tract Dis. 2016;20:184-188. doi:10.1097/LGT.0000000000000191
- Evans AT, Neven P. Invasive adenocarcinoma arising in extramammary Paget’s disease of the vulva. Histopathology. 1991;18:355-360. doi:10.1111/j.1365-2559.1991.tb00857.x
- Kitano A, Izumi M, Tamura K, et al. Brain metastasis from cutaneous squamous cell carcinoma coexistent with extramammary Paget’s disease: a case report. Pathol Int. 2019;69:619-625. doi:10.1111/pin.12846
- Miracco C, Francini E, Torre P, et al. Extramammary invasive Paget’s disease and apocrine angiomatous hamartoma: an unusual association. Eur J Dermatol. 2018;28:853-855. doi:10.1684/ejd.2018.3438
- Kambayashi Y, Fujimura T, Ohuchi K, et al. Advanced invasive extramammary Paget’s disease concomitant with cecal cancer possessing rare variant of TP53 single nucleotide polymorphism. Case Rep Oncol. 2019;12:855-860. doi:10.1159/000504339
- Fujimura T, Furudate S, Kambayashi Y, et al. Potential use of bisphosphonates in invasive extramammary Paget’s disease: an immunohistochemical investigation. Clin Dev Immunol. 2013;2013:164982. doi:10.1155/2013/164982
- Kawamura H, Ogata K, Miura H, et al. Patellar metastases. A report of two cases. Int Orthop. 1993;17:57-59. doi:10.1007/BF00195227
- Damavandy AA, Terushkin V, Zitelli JA, et al. Intraoperative immunostaining for cytokeratin-7 during Mohs micrographic surgery demonstrates low local recurrence rates in extramammary Paget’s disease. Dermatol Surg. 2018;44:354-364. doi:10.1097/DSS.0000000000001355
- Morris CR, Hurst EA. Extramammary Paget disease: a review of the literature-part I: history, epidemiology, pathogenesis, presentation, histopathology, and diagnostic work-up. Dermatol Surg. 2020;46:151-158. doi:10.1097/DSS.0000000000002064
- Simonds RM, Segal RJ, Sharma A. Extramammary Paget’s disease: a review of the literature. Int J Dermatol. 2019;58:871-879. doi:10.1111/ijd.14328
- Ogata D, Kiyohara Y, Yoshikawa S, et al. Usefulness of sentinel lymph node biopsy for prognostic prediction in extramammary Paget’s disease. Eur J Dermatol. 2016;26:254-259. doi:10.1684/ejd.2016.2744
- Ohara K, Fujisawa Y, Yoshino K, et al. A proposal for a TNM staging system for extramammary Paget disease: retrospective analysis of 301 patients with invasive primary tumors. J Dermatol Sci. 2016;83:234-239. doi:10.1016/j.jdermsci.2016.06.004
- Fujisawa Y, Yoshino K, Kiyohara Y, et al. The role of sentinel lymph node biopsy in the management of invasive extramammary Paget’s disease: multi-center, retrospective study of 151 patients. J Dermatol Sci. 2015;79:38-42. doi:10.1016/j.jdermsci.2015.03.014
- Kim SJ, Thompson AK, Zubair AS, et al. Surgical treatment and outcomes of patients with extramammary Paget disease: a cohort study. Dermatol Surg. 2017;43:708-714. doi:10.1097/DSS.0000000000001051
- Wollina U. Extensive invasive extramammary Paget’s disease: surgical treatment. J Cutan Aesthet Surg. 2013;6:41-44. doi:10.4103/0974-2077.110098
- Tsutsui K, Takahashi A, Muto Y, et al. Outcomes of lymph node dissection in the treatment of extramammary Paget’s disease: a single-institution study. J Dermatol. 2020;47:512-517. doi:10.1111/1346-8138.15285
- Guercio BJ, Iyer G, Kidwai WZ, et al. Treatment of metastatic extramammary Paget disease with combination ipilimumab and nivolumab: a case report. Case Rep Oncol. 2021;14:430-438. doi:10.1159/000514345
Metastatic Primary Extramammary Paget Disease: A Case Series
Metastatic Primary Extramammary Paget Disease: A Case Series
Practice Points
- Invasive primary extramammary Paget disease has a higher risk for lymph node metastasis.
- Consider extramammary Paget disease in patients presenting with erythematous pruritic plaques in apocrine-rich areas that fail to respond to topical steroids or antifungals.
- Prompt diagnosis can expedite comprehensive malignancy work-up and multidisciplinary management, potentially impacting patient outcomes.

Evaluation of Micrographic Surgery and Dermatologic Oncology Fellowship Program Websites
To the Editor:
Micrographic surgery and dermatologic oncology (MSDO) is a highly competitive subspecialty fellowship in dermatology. Prospective applicants often depend on the Internet to obtain pertinent information about fellowship programs to navigate the application process. An up-to-date and comprehensive fellowship website has the potential to be advantageous for both applicants and programs—applicants can more readily identify programs that align with their goals and values, and programs can effectively attract compatible applicants. These advantages are increasingly relevant with the virtual application process that has become essential considering the COVID-19 pandemic. At the height of the COVID-19 pandemic in 2020, we sought to evaluate the comprehensiveness of the content of Accreditation Council for Graduate Medical Education (ACGME) MSDO fellowship program websites to identify possible areas for improvement.
We obtained a list of all ACGME MSDO fellowships from the ACGME website (https://www.acgme.org/) and verified it against the list of MSDO programs in FREIDA, the American Medical Association residency and fellowship database (https://freida.ama-assn.org/). All programs without a website were excluded from further analysis. All data collection from currently accessible fellowship websites and evaluation occurred in April 2020.
The remaining MSDO fellowship program websites were evaluated using 25 criteria distributed among 5 domains: education/research, clinical training, program information, application process, and incentives. These criteria were determined based on earlier studies that similarly evaluated the website content of fellowship programs with inclusion of information that was considered valuable in the appraisal of fellowship programs.1,2 Criteria were further refined by direct consideration of relevance and importance to MSDO fellowship applicants (eg, inclusion of case volume, exclusion of call schedule).
Each criterion was independently assessed by 2 investigators (J.Y.C. and S.J.E.S.). A third investigator (J.R.P.) then independently evaluated those 2 assessments for agreement. Where disagreement was discovered, the third evaluator (J.R.P.) provided a final appraisal. Cohen’s kappa (κ) was conducted to evaluate for concordance between the 2 primary website evaluators. We found there to be substantial agreement between the reviewers within the education/research (κ [SD]=0.772 [0.077]), clinical training (κ [SD]=0.740 [0.051]), application process (κ [SD]=0.726 [0.103]), and incentives domains (κ [SD]=0.730 [0.110]). There was moderate agreement (κ [SD]=0.603 [0.128]) between the reviewers within the program information domain.
We identified 77 active MSDO fellowship programs. Sixty of those 77 programs (77.9%) had a dedicated fellowship website that was readily accessible. Most programs that had a dedicated fellowship website had a core or affiliated residency program (49/60 [81.7%]).
Websites that we evaluated fulfilled a mean (SD) of 9.37 (4.17) of the 25 identified criteria. Only 13 of 60 (21.7%) websites fulfilled more than 50% of evaluated criteria.
There was no statistical difference in the number of criteria fulfilled based on whether the fellowship program had a core or affiliated residency program.
Upon reviewing website accessibility directly from FREIDA, only 5 of 60 programs (8.3%) provided applicants with a link directly to their fellowship page (Table). Most programs (41 [68.3%]) provided a link to the dermatology department website, not to the specific fellowship program page, thus requiring a multistep process to find the fellowship-specific page. The remaining programs had an inaccessible (4 [6.7%]) or absent (10 [16.7%]) link on FREIDA, though a fellowship website could be identified by an Internet search of the program name.
")
The domain most fulfilled was program information with an average of 51.1% of programs satisfying the criteria, whereas the incentives domain was least fulfilled with an average of only 20.8% of programs satisfying the criteria. Across the various criteria, websites more often included a description of the program (58 [96.6%]), mentioned accreditation (53 [88.3%]), and provided case descriptions (48 [80.0%]). They less often reported information regarding a fellow’s call responsibility (3 [5%]); evaluation criteria (5 [8.3%]); and rotation schedule or options (6 [10.0%]).
The highest number of criteria fulfilled by a single program was 19 (76%). The lowest number of criteria met was 2 (8%). These findings suggest a large variation in comprehensiveness across fellowship websites.
Our research suggests that many current MSDO fellowship programs have room to maximize the information provided to applicants through their websites, which is particularly relevant following the COVID-19 pandemic, as the value of providing comprehensive and transparent information through an online platform is greater than ever. Given the ongoing desire to limit travel, virtual methods for navigating the application process have been readily used, including online videoconferencing for interviews and virtual program visits. This scenario has placed applicants in a challenging situation—their ability to directly evaluate their compatibility with a given program has been limited.3
Earlier studies that analyzed rheumatology fellowship recruitment during the COVID-19 pandemic found that programs may have more difficulty highlighting the strengths of their institution (eg, clinical facilities, professional opportunities, educational environment).4 An updated and comprehensive fellowship website was recommended4 as a key part in facing these new challenges. On the other hand, given the large number of applicants each year for fellowship positions in any given program, we acknowledge the potential benefit programs may obtain from limiting electronic information that is readily accessible to all applicants, as doing so may encourage applicants to communicate directly with a program and allow programs to identify candidates who are more interested.
In light of the movement to a more virtual-friendly and technology-driven fellowship application process, we identified 25 content areas that fellowships may want to include on their websites so that potential applicants can be well informed about the program before submitting an application and scheduling an interview. Efforts to improve accessibility and maximize the content of these websites may help programs attract compatible candidates, improve transparency, and guide applicants throughout the application process.
- Lu F, Vijayasarathi A, Murray N, et al. Evaluation of pediatric radiology fellowship website content in USA and Canada. Curr Prob Diagn Radiol. 2021;50:151-155. doi:10.1067/j.cpradiol.2020.01.007
- Cantrell CK, Bergstresser SL, Schuh AC, et al. Accessibility and content of abdominal transplant fellowship program websites in the United States. J Surg Res. 2018;232:271-274. doi:10.1016/j.jss.2018.06.052
- Nesemeier BR, Lebo NL, Schmalbach CE, et al. Impact of the COVID-19 global pandemic on the otolaryngology fellowship application process. Otolaryngol Head Neck Surg. 2020;163:712-713. doi:10.1177/0194599820934370
- Kilian A, Dua AB, Bolster MB, et al. Rheumatology fellowship recruitment in 2020: benefits, challenges, and adaptations. Arthritis Care Res (Hoboken). 2021;73:459-461. doi:10.1002/acr.24445
To the Editor:
Micrographic surgery and dermatologic oncology (MSDO) is a highly competitive subspecialty fellowship in dermatology. Prospective applicants often depend on the Internet to obtain pertinent information about fellowship programs to navigate the application process. An up-to-date and comprehensive fellowship website has the potential to be advantageous for both applicants and programs—applicants can more readily identify programs that align with their goals and values, and programs can effectively attract compatible applicants. These advantages are increasingly relevant with the virtual application process that has become essential considering the COVID-19 pandemic. At the height of the COVID-19 pandemic in 2020, we sought to evaluate the comprehensiveness of the content of Accreditation Council for Graduate Medical Education (ACGME) MSDO fellowship program websites to identify possible areas for improvement.
We obtained a list of all ACGME MSDO fellowships from the ACGME website (https://www.acgme.org/) and verified it against the list of MSDO programs in FREIDA, the American Medical Association residency and fellowship database (https://freida.ama-assn.org/). All programs without a website were excluded from further analysis. All data collection from currently accessible fellowship websites and evaluation occurred in April 2020.
The remaining MSDO fellowship program websites were evaluated using 25 criteria distributed among 5 domains: education/research, clinical training, program information, application process, and incentives. These criteria were determined based on earlier studies that similarly evaluated the website content of fellowship programs with inclusion of information that was considered valuable in the appraisal of fellowship programs.1,2 Criteria were further refined by direct consideration of relevance and importance to MSDO fellowship applicants (eg, inclusion of case volume, exclusion of call schedule).
Each criterion was independently assessed by 2 investigators (J.Y.C. and S.J.E.S.). A third investigator (J.R.P.) then independently evaluated those 2 assessments for agreement. Where disagreement was discovered, the third evaluator (J.R.P.) provided a final appraisal. Cohen’s kappa (κ) was conducted to evaluate for concordance between the 2 primary website evaluators. We found there to be substantial agreement between the reviewers within the education/research (κ [SD]=0.772 [0.077]), clinical training (κ [SD]=0.740 [0.051]), application process (κ [SD]=0.726 [0.103]), and incentives domains (κ [SD]=0.730 [0.110]). There was moderate agreement (κ [SD]=0.603 [0.128]) between the reviewers within the program information domain.
We identified 77 active MSDO fellowship programs. Sixty of those 77 programs (77.9%) had a dedicated fellowship website that was readily accessible. Most programs that had a dedicated fellowship website had a core or affiliated residency program (49/60 [81.7%]).
Websites that we evaluated fulfilled a mean (SD) of 9.37 (4.17) of the 25 identified criteria. Only 13 of 60 (21.7%) websites fulfilled more than 50% of evaluated criteria.
There was no statistical difference in the number of criteria fulfilled based on whether the fellowship program had a core or affiliated residency program.
Upon reviewing website accessibility directly from FREIDA, only 5 of 60 programs (8.3%) provided applicants with a link directly to their fellowship page (Table). Most programs (41 [68.3%]) provided a link to the dermatology department website, not to the specific fellowship program page, thus requiring a multistep process to find the fellowship-specific page. The remaining programs had an inaccessible (4 [6.7%]) or absent (10 [16.7%]) link on FREIDA, though a fellowship website could be identified by an Internet search of the program name.
The domain most fulfilled was program information with an average of 51.1% of programs satisfying the criteria, whereas the incentives domain was least fulfilled with an average of only 20.8% of programs satisfying the criteria. Across the various criteria, websites more often included a description of the program (58 [96.6%]), mentioned accreditation (53 [88.3%]), and provided case descriptions (48 [80.0%]). They less often reported information regarding a fellow’s call responsibility (3 [5%]); evaluation criteria (5 [8.3%]); and rotation schedule or options (6 [10.0%]).
The highest number of criteria fulfilled by a single program was 19 (76%). The lowest number of criteria met was 2 (8%). These findings suggest a large variation in comprehensiveness across fellowship websites.
Our research suggests that many current MSDO fellowship programs have room to maximize the information provided to applicants through their websites, which is particularly relevant following the COVID-19 pandemic, as the value of providing comprehensive and transparent information through an online platform is greater than ever. Given the ongoing desire to limit travel, virtual methods for navigating the application process have been readily used, including online videoconferencing for interviews and virtual program visits. This scenario has placed applicants in a challenging situation—their ability to directly evaluate their compatibility with a given program has been limited.3
Earlier studies that analyzed rheumatology fellowship recruitment during the COVID-19 pandemic found that programs may have more difficulty highlighting the strengths of their institution (eg, clinical facilities, professional opportunities, educational environment).4 An updated and comprehensive fellowship website was recommended4 as a key part in facing these new challenges. On the other hand, given the large number of applicants each year for fellowship positions in any given program, we acknowledge the potential benefit programs may obtain from limiting electronic information that is readily accessible to all applicants, as doing so may encourage applicants to communicate directly with a program and allow programs to identify candidates who are more interested.
In light of the movement to a more virtual-friendly and technology-driven fellowship application process, we identified 25 content areas that fellowships may want to include on their websites so that potential applicants can be well informed about the program before submitting an application and scheduling an interview. Efforts to improve accessibility and maximize the content of these websites may help programs attract compatible candidates, improve transparency, and guide applicants throughout the application process.
To the Editor:
Micrographic surgery and dermatologic oncology (MSDO) is a highly competitive subspecialty fellowship in dermatology. Prospective applicants often depend on the Internet to obtain pertinent information about fellowship programs to navigate the application process. An up-to-date and comprehensive fellowship website has the potential to be advantageous for both applicants and programs—applicants can more readily identify programs that align with their goals and values, and programs can effectively attract compatible applicants. These advantages are increasingly relevant with the virtual application process that has become essential considering the COVID-19 pandemic. At the height of the COVID-19 pandemic in 2020, we sought to evaluate the comprehensiveness of the content of Accreditation Council for Graduate Medical Education (ACGME) MSDO fellowship program websites to identify possible areas for improvement.
We obtained a list of all ACGME MSDO fellowships from the ACGME website (https://www.acgme.org/) and verified it against the list of MSDO programs in FREIDA, the American Medical Association residency and fellowship database (https://freida.ama-assn.org/). All programs without a website were excluded from further analysis. All data collection from currently accessible fellowship websites and evaluation occurred in April 2020.
The remaining MSDO fellowship program websites were evaluated using 25 criteria distributed among 5 domains: education/research, clinical training, program information, application process, and incentives. These criteria were determined based on earlier studies that similarly evaluated the website content of fellowship programs with inclusion of information that was considered valuable in the appraisal of fellowship programs.1,2 Criteria were further refined by direct consideration of relevance and importance to MSDO fellowship applicants (eg, inclusion of case volume, exclusion of call schedule).
Each criterion was independently assessed by 2 investigators (J.Y.C. and S.J.E.S.). A third investigator (J.R.P.) then independently evaluated those 2 assessments for agreement. Where disagreement was discovered, the third evaluator (J.R.P.) provided a final appraisal. Cohen’s kappa (κ) was conducted to evaluate for concordance between the 2 primary website evaluators. We found there to be substantial agreement between the reviewers within the education/research (κ [SD]=0.772 [0.077]), clinical training (κ [SD]=0.740 [0.051]), application process (κ [SD]=0.726 [0.103]), and incentives domains (κ [SD]=0.730 [0.110]). There was moderate agreement (κ [SD]=0.603 [0.128]) between the reviewers within the program information domain.
We identified 77 active MSDO fellowship programs. Sixty of those 77 programs (77.9%) had a dedicated fellowship website that was readily accessible. Most programs that had a dedicated fellowship website had a core or affiliated residency program (49/60 [81.7%]).
Websites that we evaluated fulfilled a mean (SD) of 9.37 (4.17) of the 25 identified criteria. Only 13 of 60 (21.7%) websites fulfilled more than 50% of evaluated criteria.
There was no statistical difference in the number of criteria fulfilled based on whether the fellowship program had a core or affiliated residency program.
Upon reviewing website accessibility directly from FREIDA, only 5 of 60 programs (8.3%) provided applicants with a link directly to their fellowship page (Table). Most programs (41 [68.3%]) provided a link to the dermatology department website, not to the specific fellowship program page, thus requiring a multistep process to find the fellowship-specific page. The remaining programs had an inaccessible (4 [6.7%]) or absent (10 [16.7%]) link on FREIDA, though a fellowship website could be identified by an Internet search of the program name.
The domain most fulfilled was program information with an average of 51.1% of programs satisfying the criteria, whereas the incentives domain was least fulfilled with an average of only 20.8% of programs satisfying the criteria. Across the various criteria, websites more often included a description of the program (58 [96.6%]), mentioned accreditation (53 [88.3%]), and provided case descriptions (48 [80.0%]). They less often reported information regarding a fellow’s call responsibility (3 [5%]); evaluation criteria (5 [8.3%]); and rotation schedule or options (6 [10.0%]).
The highest number of criteria fulfilled by a single program was 19 (76%). The lowest number of criteria met was 2 (8%). These findings suggest a large variation in comprehensiveness across fellowship websites.
Our research suggests that many current MSDO fellowship programs have room to maximize the information provided to applicants through their websites, which is particularly relevant following the COVID-19 pandemic, as the value of providing comprehensive and transparent information through an online platform is greater than ever. Given the ongoing desire to limit travel, virtual methods for navigating the application process have been readily used, including online videoconferencing for interviews and virtual program visits. This scenario has placed applicants in a challenging situation—their ability to directly evaluate their compatibility with a given program has been limited.3
Earlier studies that analyzed rheumatology fellowship recruitment during the COVID-19 pandemic found that programs may have more difficulty highlighting the strengths of their institution (eg, clinical facilities, professional opportunities, educational environment).4 An updated and comprehensive fellowship website was recommended4 as a key part in facing these new challenges. On the other hand, given the large number of applicants each year for fellowship positions in any given program, we acknowledge the potential benefit programs may obtain from limiting electronic information that is readily accessible to all applicants, as doing so may encourage applicants to communicate directly with a program and allow programs to identify candidates who are more interested.
In light of the movement to a more virtual-friendly and technology-driven fellowship application process, we identified 25 content areas that fellowships may want to include on their websites so that potential applicants can be well informed about the program before submitting an application and scheduling an interview. Efforts to improve accessibility and maximize the content of these websites may help programs attract compatible candidates, improve transparency, and guide applicants throughout the application process.
- Lu F, Vijayasarathi A, Murray N, et al. Evaluation of pediatric radiology fellowship website content in USA and Canada. Curr Prob Diagn Radiol. 2021;50:151-155. doi:10.1067/j.cpradiol.2020.01.007
- Cantrell CK, Bergstresser SL, Schuh AC, et al. Accessibility and content of abdominal transplant fellowship program websites in the United States. J Surg Res. 2018;232:271-274. doi:10.1016/j.jss.2018.06.052
- Nesemeier BR, Lebo NL, Schmalbach CE, et al. Impact of the COVID-19 global pandemic on the otolaryngology fellowship application process. Otolaryngol Head Neck Surg. 2020;163:712-713. doi:10.1177/0194599820934370
- Kilian A, Dua AB, Bolster MB, et al. Rheumatology fellowship recruitment in 2020: benefits, challenges, and adaptations. Arthritis Care Res (Hoboken). 2021;73:459-461. doi:10.1002/acr.24445
- Lu F, Vijayasarathi A, Murray N, et al. Evaluation of pediatric radiology fellowship website content in USA and Canada. Curr Prob Diagn Radiol. 2021;50:151-155. doi:10.1067/j.cpradiol.2020.01.007
- Cantrell CK, Bergstresser SL, Schuh AC, et al. Accessibility and content of abdominal transplant fellowship program websites in the United States. J Surg Res. 2018;232:271-274. doi:10.1016/j.jss.2018.06.052
- Nesemeier BR, Lebo NL, Schmalbach CE, et al. Impact of the COVID-19 global pandemic on the otolaryngology fellowship application process. Otolaryngol Head Neck Surg. 2020;163:712-713. doi:10.1177/0194599820934370
- Kilian A, Dua AB, Bolster MB, et al. Rheumatology fellowship recruitment in 2020: benefits, challenges, and adaptations. Arthritis Care Res (Hoboken). 2021;73:459-461. doi:10.1002/acr.24445
Practice Points
- With the COVID-19 pandemic and the movement to a virtual fellowship application process, fellowship program websites that are comprehensive and accessible may help programs attract compatible candidates, improve transparency, and guide applicants through the application process.
- There is variation in the content of current micrographic surgery and dermatologic oncology fellowship program websites and areas upon which programs may seek to augment their website content to better reflect program strengths while attracting competitive candidates best suited for their program.
Primary Effusion Lymphoma: An Infiltrative Plaque in a Patient With HIV
To the Editor:
A 47-year-old man presented to the dermatology service with an asymptomatic plaque on the right thigh of 2 months’ duration. He had a medical history of HIV and Kaposi sarcoma as well as a recently relapsed primary effusion lymphoma (PEL) subsequent to an allogeneic bone marrow transplant. He initially was diagnosed with PEL 3 years prior to the current presentation during a workup for fever and weight loss. Imaging at the time demonstrated a bladder mass, which was biopsied and demonstrated PEL. Further imaging demonstrated both sinus and bone marrow involvement. Prior to dermatologic consultation, he had been treated with 6 cycles of etoposide, prednisolone, vincristine, cyclophosphamide, and doxorubicin (EPOCH); 6 cycles of brentuximab; 4 cycles of rituximab with gemcitabine and oxaliplatin; and 2 cycles of ifosfamide, carboplatin, and etoposide. Despite these therapies, he had 3 relapses, and oncology determined the need for a matched unrelated donor allogeneic stem cell transplant for his PEL.

At the time of dermatology consultation, the patient was being managed on daratumumab and bortezomib. Physical examination revealed an infiltrative plaque on the right inferomedial thigh measuring approximately 6.0 cm (largest dimension) with a small amount of peripheral scale (Figure 1). An ultrasound revealed notable subcutaneous tissue edema and increased vascularity without a discrete mass or fluid collection. A 4-mm punch biopsy demonstrated a dense infiltrate comprised of collections of histiocytes admixed with scattered plasma cells and mature lymphoid aggregates. Additionally, rare enlarged plasmablastic cells with scant basophilic cytoplasm and slightly irregular nuclear contours were visualized (Figure 2A). Immunohistochemistry was positive for CD3 with a normal CD4:CD8 ratio, CD68-highlighted histiocytes within the lymphoid aggregates, and human herpesvirus 8 (HHV-8)(or Kaposi sarcoma–associated herpesvirus) demonstrated stippled nuclear staining within the scattered large cells (Figure 2B). Epstein-Barr virus–encoded RNA staining was negative, though the area of interest was lost on deeper sectioning of the tissue block. The histopathologic findings were consistent with cutaneous extracavitary PEL. Shortly after this diagnosis, he died from disease complications.
. B, Stippled staining of scattered large cells also was noted (HHV-8, original magnification ×400).")
Primary effusion lymphoma is an aggressive non-Hodgkin B-cell lymphoma that was first described by Knowles et al1 in 1989. Primary effusion lymphoma occurs exclusively in the setting of HHV-8 infection and typically is associated with chronic immunosuppression related to HIV/AIDS. Cases that are negative for HIV-1 are rare but have been reported in organ transplant recipients and elderly men from areas with a high prevalence of HHV-8 infections. Most HIV-associated cases show concurrent Epstein-Barr virus infection, though the pathogenic meaning of this co-infection remains unclear.2,3
Primary effusion lymphoma classically presents as an isolated effusion of malignant lymphoid cells within body cavities in the absence of solid tumor masses. The pleural, peritoneal, and pericardial spaces most commonly are involved. Extracavitary PEL, a rare variant, may present as a solid mass without effusion. In general, extracavitary tumors may occur in the setting of de novo malignancy or recurrent PEL.4 Cutaneous manifestations associated with extracavitary PEL are rare; 4 cases have been described in which skin lesions were the heralding sign of the disease.3 Interestingly, despite obligatory underlying HHV-8 infection, a review by Pielasinski et al3 noted only 2 patients with cutaneous PEL who had prior or concurrent Kaposi sarcoma. This heterogeneity in HHV-8–related phenotypes may be related to differences in microRNA expression, but further study is needed.5
The diagnosis of PEL relies on histologic, immunophenotypic, and molecular analysis of the affected tissue. The malignant cells typically are large with round to irregular nuclei. These cells may demonstrate a variety of appearances, including anaplastic, plasmablastic, and immunoblastic morphologies.6,7 The immunophenotype displays CD45 positivity and markers of lymphocyte activation (CD30, CD38, CD71), while typical B-cell (CD19, CD20, CD79a) and T-cell (CD3, CD4, CD8) markers often are absent.6-8 Human herpesvirus 8 detection by polymerase chain reaction testing of the peripheral blood or by immunohistochemistry staining of the affected tissue is required for diagnosis.6,7 Epstein-Barr virus infection may be detected via in situ hybridization, though it is not required for diagnosis.
The overall prognosis for PEL is poor; Brimo et al6 reported a median survival of less than 6 months, and Guillet et al9 reported 5-year overall survival (OS) for PEL vs extracavitary PEL to be 43% vs 39%. Another review noted variation in survival contingent on the number of body cavities involved; patients with a single body cavity involved experienced a median OS of 18 months, whereas patients with multiple involved cavities experienced a median OS of 4 months,7 possibly due to the limited study of treatment regimens or disease aggressiveness. Even in cases of successful initial treatment, relapse within 6 to 8 months is common. Extracavitary PEL may have improved disease-free survival relative to classic PEL, though the data were less clear for OS.9 Limitations of the Guillet et al9 study included a small sample size, the impossibility to randomize to disease type, and loss of power on the log-rank test for OS in the setting of possible nonproportional hazards (crossing survival curves). Overall, prognostic differences between the groups may be challenging to ascertain until further data are obtained.
As with many HIV-associated neoplasms, antiretroviral treatment (ART) for HIV-positive patients affords a better prognosis when used in addition to therapy directed at malignancy.7 The general approach is for concurrent ART with systemic therapies such as rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone for the rare CD20+ cases, and cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) or dose-adjusted EPOCH therapy in the more common CD20− PEL cases. Narkhede et al7 suggested avoidance of methotrexate in patients with effusions because of increased toxicity, but it is unclear if this recommendation is applicable in extracavitary PEL patients without an effusion. Additionally, second-line treatment modalities include radiation for solid PEL masses, HHV-8–targeted antivirals, and stem cell transplantation, though evidence is limited. Of note, there is a phase I-II trial (ClinicalTrials.gov identifier NCT02911142) ongoing for treatment-naïve PEL patients involving the experimental treatment DA-EPOCH-R plus lenalidomide, but the trial is ongoing.10
We report a case of cutaneous PEL in a patient with a history of Kaposi sarcoma. The patient’s deterioration and ultimate death despite initial treatment with EPOCH and bone marrow transplantation followed by final management with daratumumab and bortezomib confirm other reports that PEL has a poor prognosis and that optimal treatments are not well delineated for these patients. In general, the current approach is to utilize ART for HIV-positive patients and to then implement chemotherapy such as CHOP. Without continued research and careful planning of treatments, data will remain limited on how best to serve patients with PEL.
- Knowles DM, Inghirami G, Ubriaco A, et al. Molecular genetic analysis of three AIDS-associated neoplasms of uncertain lineage demonstrates their B-cell derivation and the possible pathogenetic role of the Epstein-Barr virus. Blood. 1989;73:792-799.
- Kugasia IAR, Kumar A, Khatri A, et al. Primary effusion lymphoma of the pleural space: report of a rare complication of cardiac transplant with review of the literature. Transpl Infect Dis. 2019;21:E13005.
- Pielasinski U, Santonja C, Rodriguez-Pinilla SM, et al. Extracavitary primary effusion lymphoma presenting as a cutaneous tumor: a case report and literature review. J Cutan Pathol. 2014;41:745-753.
- Boulanger E, Meignin V, Afonso PV, et al. Extracavitary tumor after primary effusion lymphoma: relapse or second distinct lymphoma? Haematologica. 2007;92:1275-1276.
- Goncalves PH, Uldrick TS, Yarchoan R. HIV-associated Kaposi sarcoma and related diseases. AIDS. 2017;31:1903-1916.
- Brimo F, Michel RP, Khetani K, et al. Primary effusion lymphoma: a series of 4 cases and review of the literature with emphasis on cytomorphologic and immunocytochemical differential diagnosis. Cancer. 2007;111:224-233.
- Narkhede M, Arora S, Ujjani C. Primary effusion lymphoma: current perspectives. Onco Targets Ther. 2018;11:3747-3754.
- Chen YB, Rahemtullah A, Hochberg E. Primary effusion lymphoma. Oncologist. 2007;12:569-576.
- Guillet S, Gerard L, Meignin V, et al. Classic and extracavitary primary effusion lymphoma in 51 HIV-infected patients from a single institution. Am J Hematol. 2016;91:233-237.
To the Editor:
A 47-year-old man presented to the dermatology service with an asymptomatic plaque on the right thigh of 2 months’ duration. He had a medical history of HIV and Kaposi sarcoma as well as a recently relapsed primary effusion lymphoma (PEL) subsequent to an allogeneic bone marrow transplant. He initially was diagnosed with PEL 3 years prior to the current presentation during a workup for fever and weight loss. Imaging at the time demonstrated a bladder mass, which was biopsied and demonstrated PEL. Further imaging demonstrated both sinus and bone marrow involvement. Prior to dermatologic consultation, he had been treated with 6 cycles of etoposide, prednisolone, vincristine, cyclophosphamide, and doxorubicin (EPOCH); 6 cycles of brentuximab; 4 cycles of rituximab with gemcitabine and oxaliplatin; and 2 cycles of ifosfamide, carboplatin, and etoposide. Despite these therapies, he had 3 relapses, and oncology determined the need for a matched unrelated donor allogeneic stem cell transplant for his PEL.
At the time of dermatology consultation, the patient was being managed on daratumumab and bortezomib. Physical examination revealed an infiltrative plaque on the right inferomedial thigh measuring approximately 6.0 cm (largest dimension) with a small amount of peripheral scale (Figure 1). An ultrasound revealed notable subcutaneous tissue edema and increased vascularity without a discrete mass or fluid collection. A 4-mm punch biopsy demonstrated a dense infiltrate comprised of collections of histiocytes admixed with scattered plasma cells and mature lymphoid aggregates. Additionally, rare enlarged plasmablastic cells with scant basophilic cytoplasm and slightly irregular nuclear contours were visualized (Figure 2A). Immunohistochemistry was positive for CD3 with a normal CD4:CD8 ratio, CD68-highlighted histiocytes within the lymphoid aggregates, and human herpesvirus 8 (HHV-8)(or Kaposi sarcoma–associated herpesvirus) demonstrated stippled nuclear staining within the scattered large cells (Figure 2B). Epstein-Barr virus–encoded RNA staining was negative, though the area of interest was lost on deeper sectioning of the tissue block. The histopathologic findings were consistent with cutaneous extracavitary PEL. Shortly after this diagnosis, he died from disease complications.
Primary effusion lymphoma is an aggressive non-Hodgkin B-cell lymphoma that was first described by Knowles et al1 in 1989. Primary effusion lymphoma occurs exclusively in the setting of HHV-8 infection and typically is associated with chronic immunosuppression related to HIV/AIDS. Cases that are negative for HIV-1 are rare but have been reported in organ transplant recipients and elderly men from areas with a high prevalence of HHV-8 infections. Most HIV-associated cases show concurrent Epstein-Barr virus infection, though the pathogenic meaning of this co-infection remains unclear.2,3
Primary effusion lymphoma classically presents as an isolated effusion of malignant lymphoid cells within body cavities in the absence of solid tumor masses. The pleural, peritoneal, and pericardial spaces most commonly are involved. Extracavitary PEL, a rare variant, may present as a solid mass without effusion. In general, extracavitary tumors may occur in the setting of de novo malignancy or recurrent PEL.4 Cutaneous manifestations associated with extracavitary PEL are rare; 4 cases have been described in which skin lesions were the heralding sign of the disease.3 Interestingly, despite obligatory underlying HHV-8 infection, a review by Pielasinski et al3 noted only 2 patients with cutaneous PEL who had prior or concurrent Kaposi sarcoma. This heterogeneity in HHV-8–related phenotypes may be related to differences in microRNA expression, but further study is needed.5
The diagnosis of PEL relies on histologic, immunophenotypic, and molecular analysis of the affected tissue. The malignant cells typically are large with round to irregular nuclei. These cells may demonstrate a variety of appearances, including anaplastic, plasmablastic, and immunoblastic morphologies.6,7 The immunophenotype displays CD45 positivity and markers of lymphocyte activation (CD30, CD38, CD71), while typical B-cell (CD19, CD20, CD79a) and T-cell (CD3, CD4, CD8) markers often are absent.6-8 Human herpesvirus 8 detection by polymerase chain reaction testing of the peripheral blood or by immunohistochemistry staining of the affected tissue is required for diagnosis.6,7 Epstein-Barr virus infection may be detected via in situ hybridization, though it is not required for diagnosis.
The overall prognosis for PEL is poor; Brimo et al6 reported a median survival of less than 6 months, and Guillet et al9 reported 5-year overall survival (OS) for PEL vs extracavitary PEL to be 43% vs 39%. Another review noted variation in survival contingent on the number of body cavities involved; patients with a single body cavity involved experienced a median OS of 18 months, whereas patients with multiple involved cavities experienced a median OS of 4 months,7 possibly due to the limited study of treatment regimens or disease aggressiveness. Even in cases of successful initial treatment, relapse within 6 to 8 months is common. Extracavitary PEL may have improved disease-free survival relative to classic PEL, though the data were less clear for OS.9 Limitations of the Guillet et al9 study included a small sample size, the impossibility to randomize to disease type, and loss of power on the log-rank test for OS in the setting of possible nonproportional hazards (crossing survival curves). Overall, prognostic differences between the groups may be challenging to ascertain until further data are obtained.
As with many HIV-associated neoplasms, antiretroviral treatment (ART) for HIV-positive patients affords a better prognosis when used in addition to therapy directed at malignancy.7 The general approach is for concurrent ART with systemic therapies such as rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone for the rare CD20+ cases, and cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) or dose-adjusted EPOCH therapy in the more common CD20− PEL cases. Narkhede et al7 suggested avoidance of methotrexate in patients with effusions because of increased toxicity, but it is unclear if this recommendation is applicable in extracavitary PEL patients without an effusion. Additionally, second-line treatment modalities include radiation for solid PEL masses, HHV-8–targeted antivirals, and stem cell transplantation, though evidence is limited. Of note, there is a phase I-II trial (ClinicalTrials.gov identifier NCT02911142) ongoing for treatment-naïve PEL patients involving the experimental treatment DA-EPOCH-R plus lenalidomide, but the trial is ongoing.10
We report a case of cutaneous PEL in a patient with a history of Kaposi sarcoma. The patient’s deterioration and ultimate death despite initial treatment with EPOCH and bone marrow transplantation followed by final management with daratumumab and bortezomib confirm other reports that PEL has a poor prognosis and that optimal treatments are not well delineated for these patients. In general, the current approach is to utilize ART for HIV-positive patients and to then implement chemotherapy such as CHOP. Without continued research and careful planning of treatments, data will remain limited on how best to serve patients with PEL.
To the Editor:
A 47-year-old man presented to the dermatology service with an asymptomatic plaque on the right thigh of 2 months’ duration. He had a medical history of HIV and Kaposi sarcoma as well as a recently relapsed primary effusion lymphoma (PEL) subsequent to an allogeneic bone marrow transplant. He initially was diagnosed with PEL 3 years prior to the current presentation during a workup for fever and weight loss. Imaging at the time demonstrated a bladder mass, which was biopsied and demonstrated PEL. Further imaging demonstrated both sinus and bone marrow involvement. Prior to dermatologic consultation, he had been treated with 6 cycles of etoposide, prednisolone, vincristine, cyclophosphamide, and doxorubicin (EPOCH); 6 cycles of brentuximab; 4 cycles of rituximab with gemcitabine and oxaliplatin; and 2 cycles of ifosfamide, carboplatin, and etoposide. Despite these therapies, he had 3 relapses, and oncology determined the need for a matched unrelated donor allogeneic stem cell transplant for his PEL.
At the time of dermatology consultation, the patient was being managed on daratumumab and bortezomib. Physical examination revealed an infiltrative plaque on the right inferomedial thigh measuring approximately 6.0 cm (largest dimension) with a small amount of peripheral scale (Figure 1). An ultrasound revealed notable subcutaneous tissue edema and increased vascularity without a discrete mass or fluid collection. A 4-mm punch biopsy demonstrated a dense infiltrate comprised of collections of histiocytes admixed with scattered plasma cells and mature lymphoid aggregates. Additionally, rare enlarged plasmablastic cells with scant basophilic cytoplasm and slightly irregular nuclear contours were visualized (Figure 2A). Immunohistochemistry was positive for CD3 with a normal CD4:CD8 ratio, CD68-highlighted histiocytes within the lymphoid aggregates, and human herpesvirus 8 (HHV-8)(or Kaposi sarcoma–associated herpesvirus) demonstrated stippled nuclear staining within the scattered large cells (Figure 2B). Epstein-Barr virus–encoded RNA staining was negative, though the area of interest was lost on deeper sectioning of the tissue block. The histopathologic findings were consistent with cutaneous extracavitary PEL. Shortly after this diagnosis, he died from disease complications.
Primary effusion lymphoma is an aggressive non-Hodgkin B-cell lymphoma that was first described by Knowles et al1 in 1989. Primary effusion lymphoma occurs exclusively in the setting of HHV-8 infection and typically is associated with chronic immunosuppression related to HIV/AIDS. Cases that are negative for HIV-1 are rare but have been reported in organ transplant recipients and elderly men from areas with a high prevalence of HHV-8 infections. Most HIV-associated cases show concurrent Epstein-Barr virus infection, though the pathogenic meaning of this co-infection remains unclear.2,3
Primary effusion lymphoma classically presents as an isolated effusion of malignant lymphoid cells within body cavities in the absence of solid tumor masses. The pleural, peritoneal, and pericardial spaces most commonly are involved. Extracavitary PEL, a rare variant, may present as a solid mass without effusion. In general, extracavitary tumors may occur in the setting of de novo malignancy or recurrent PEL.4 Cutaneous manifestations associated with extracavitary PEL are rare; 4 cases have been described in which skin lesions were the heralding sign of the disease.3 Interestingly, despite obligatory underlying HHV-8 infection, a review by Pielasinski et al3 noted only 2 patients with cutaneous PEL who had prior or concurrent Kaposi sarcoma. This heterogeneity in HHV-8–related phenotypes may be related to differences in microRNA expression, but further study is needed.5
The diagnosis of PEL relies on histologic, immunophenotypic, and molecular analysis of the affected tissue. The malignant cells typically are large with round to irregular nuclei. These cells may demonstrate a variety of appearances, including anaplastic, plasmablastic, and immunoblastic morphologies.6,7 The immunophenotype displays CD45 positivity and markers of lymphocyte activation (CD30, CD38, CD71), while typical B-cell (CD19, CD20, CD79a) and T-cell (CD3, CD4, CD8) markers often are absent.6-8 Human herpesvirus 8 detection by polymerase chain reaction testing of the peripheral blood or by immunohistochemistry staining of the affected tissue is required for diagnosis.6,7 Epstein-Barr virus infection may be detected via in situ hybridization, though it is not required for diagnosis.
The overall prognosis for PEL is poor; Brimo et al6 reported a median survival of less than 6 months, and Guillet et al9 reported 5-year overall survival (OS) for PEL vs extracavitary PEL to be 43% vs 39%. Another review noted variation in survival contingent on the number of body cavities involved; patients with a single body cavity involved experienced a median OS of 18 months, whereas patients with multiple involved cavities experienced a median OS of 4 months,7 possibly due to the limited study of treatment regimens or disease aggressiveness. Even in cases of successful initial treatment, relapse within 6 to 8 months is common. Extracavitary PEL may have improved disease-free survival relative to classic PEL, though the data were less clear for OS.9 Limitations of the Guillet et al9 study included a small sample size, the impossibility to randomize to disease type, and loss of power on the log-rank test for OS in the setting of possible nonproportional hazards (crossing survival curves). Overall, prognostic differences between the groups may be challenging to ascertain until further data are obtained.
As with many HIV-associated neoplasms, antiretroviral treatment (ART) for HIV-positive patients affords a better prognosis when used in addition to therapy directed at malignancy.7 The general approach is for concurrent ART with systemic therapies such as rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone for the rare CD20+ cases, and cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) or dose-adjusted EPOCH therapy in the more common CD20− PEL cases. Narkhede et al7 suggested avoidance of methotrexate in patients with effusions because of increased toxicity, but it is unclear if this recommendation is applicable in extracavitary PEL patients without an effusion. Additionally, second-line treatment modalities include radiation for solid PEL masses, HHV-8–targeted antivirals, and stem cell transplantation, though evidence is limited. Of note, there is a phase I-II trial (ClinicalTrials.gov identifier NCT02911142) ongoing for treatment-naïve PEL patients involving the experimental treatment DA-EPOCH-R plus lenalidomide, but the trial is ongoing.10
We report a case of cutaneous PEL in a patient with a history of Kaposi sarcoma. The patient’s deterioration and ultimate death despite initial treatment with EPOCH and bone marrow transplantation followed by final management with daratumumab and bortezomib confirm other reports that PEL has a poor prognosis and that optimal treatments are not well delineated for these patients. In general, the current approach is to utilize ART for HIV-positive patients and to then implement chemotherapy such as CHOP. Without continued research and careful planning of treatments, data will remain limited on how best to serve patients with PEL.
- Knowles DM, Inghirami G, Ubriaco A, et al. Molecular genetic analysis of three AIDS-associated neoplasms of uncertain lineage demonstrates their B-cell derivation and the possible pathogenetic role of the Epstein-Barr virus. Blood. 1989;73:792-799.
- Kugasia IAR, Kumar A, Khatri A, et al. Primary effusion lymphoma of the pleural space: report of a rare complication of cardiac transplant with review of the literature. Transpl Infect Dis. 2019;21:E13005.
- Pielasinski U, Santonja C, Rodriguez-Pinilla SM, et al. Extracavitary primary effusion lymphoma presenting as a cutaneous tumor: a case report and literature review. J Cutan Pathol. 2014;41:745-753.
- Boulanger E, Meignin V, Afonso PV, et al. Extracavitary tumor after primary effusion lymphoma: relapse or second distinct lymphoma? Haematologica. 2007;92:1275-1276.
- Goncalves PH, Uldrick TS, Yarchoan R. HIV-associated Kaposi sarcoma and related diseases. AIDS. 2017;31:1903-1916.
- Brimo F, Michel RP, Khetani K, et al. Primary effusion lymphoma: a series of 4 cases and review of the literature with emphasis on cytomorphologic and immunocytochemical differential diagnosis. Cancer. 2007;111:224-233.
- Narkhede M, Arora S, Ujjani C. Primary effusion lymphoma: current perspectives. Onco Targets Ther. 2018;11:3747-3754.
- Chen YB, Rahemtullah A, Hochberg E. Primary effusion lymphoma. Oncologist. 2007;12:569-576.
- Guillet S, Gerard L, Meignin V, et al. Classic and extracavitary primary effusion lymphoma in 51 HIV-infected patients from a single institution. Am J Hematol. 2016;91:233-237.
- Knowles DM, Inghirami G, Ubriaco A, et al. Molecular genetic analysis of three AIDS-associated neoplasms of uncertain lineage demonstrates their B-cell derivation and the possible pathogenetic role of the Epstein-Barr virus. Blood. 1989;73:792-799.
- Kugasia IAR, Kumar A, Khatri A, et al. Primary effusion lymphoma of the pleural space: report of a rare complication of cardiac transplant with review of the literature. Transpl Infect Dis. 2019;21:E13005.
- Pielasinski U, Santonja C, Rodriguez-Pinilla SM, et al. Extracavitary primary effusion lymphoma presenting as a cutaneous tumor: a case report and literature review. J Cutan Pathol. 2014;41:745-753.
- Boulanger E, Meignin V, Afonso PV, et al. Extracavitary tumor after primary effusion lymphoma: relapse or second distinct lymphoma? Haematologica. 2007;92:1275-1276.
- Goncalves PH, Uldrick TS, Yarchoan R. HIV-associated Kaposi sarcoma and related diseases. AIDS. 2017;31:1903-1916.
- Brimo F, Michel RP, Khetani K, et al. Primary effusion lymphoma: a series of 4 cases and review of the literature with emphasis on cytomorphologic and immunocytochemical differential diagnosis. Cancer. 2007;111:224-233.
- Narkhede M, Arora S, Ujjani C. Primary effusion lymphoma: current perspectives. Onco Targets Ther. 2018;11:3747-3754.
- Chen YB, Rahemtullah A, Hochberg E. Primary effusion lymphoma. Oncologist. 2007;12:569-576.
- Guillet S, Gerard L, Meignin V, et al. Classic and extracavitary primary effusion lymphoma in 51 HIV-infected patients from a single institution. Am J Hematol. 2016;91:233-237.
Practice Points
- Extracavitary primary effusion lymphoma is an aggressive non-Hodgkin B-cell lymphoma that occurs solely in the presence of human herpesvirus 8 infection and typically is associated with HIV/AIDS.
- Diagnosis necessitates a thorough workup and correlation of histologic, molecular, and immunophenotypic analysis.
- Antiretroviral therapy in HIV-positive patients and intensive chemotherapy regimens are the current recommended treatments. Despite newer targeted agents, the prognosis remains poor.

Depressed Shiny Scars and Crusted Erosions
The Diagnosis: Erythropoietic Protoporphyria
Erythropoietic protoporphyria (EPP) is an autosomal-recessive photodermatosis that results from loss of activity of ferrochelatase, the last enzyme in the heme biosynthetic pathway.1 Erythropoietic protoporphyria normally involves sun-exposed areas of the body. Skin that is exposed to sunlight develops intense burning and stinging pain followed by erythema, edema, crusting, and petechiae that develops into waxy scarring over time. In contrast to other porphyrias, blistering generally is not seen.2 Accurate diagnosis often can be delayed by a decade or more following symptom onset due to the prominence of subjective pain as the presenting sign.
The histologic appearance of EPP differs depending on the chronicity of lesions. Biopsies of acute lesions show vacuolization of epidermal cells with intercellular edema, vacuolization and cytolysis of endothelial cells in superficial blood vessels, and focal red blood cell extravasation.3,4 A largely neutrophilic inflammatory infiltrate can be present.5 Hyaline cuffing develops over time in and around vessels in the papillary and superficial reticular dermis with notable sparing of adnexal structures. The perivascular deposits are strongly periodic acid-Schiff (PAS) positive and diastase resistant (Figure 1). Direct immunofluorescence shows mainly IgG and some IgM, fibrinogen, and C3 outlining characteristic donut-shaped blood vessels in the papillary dermis.6 The prominent thickness of the perivascular hyaline material depositions and the absence of subepidermal blistering can help differentiate EPP from porphyria cutanea tarda (PCT) and pseudoporphyria.6,7 When the deposition is extensive and involves the surrounding dermis, EPP can mimic colloid milium. Additional histologic differential diagnoses of EPP include other dermal depositional diseases such as lipoid proteinosis and amyloidosis.
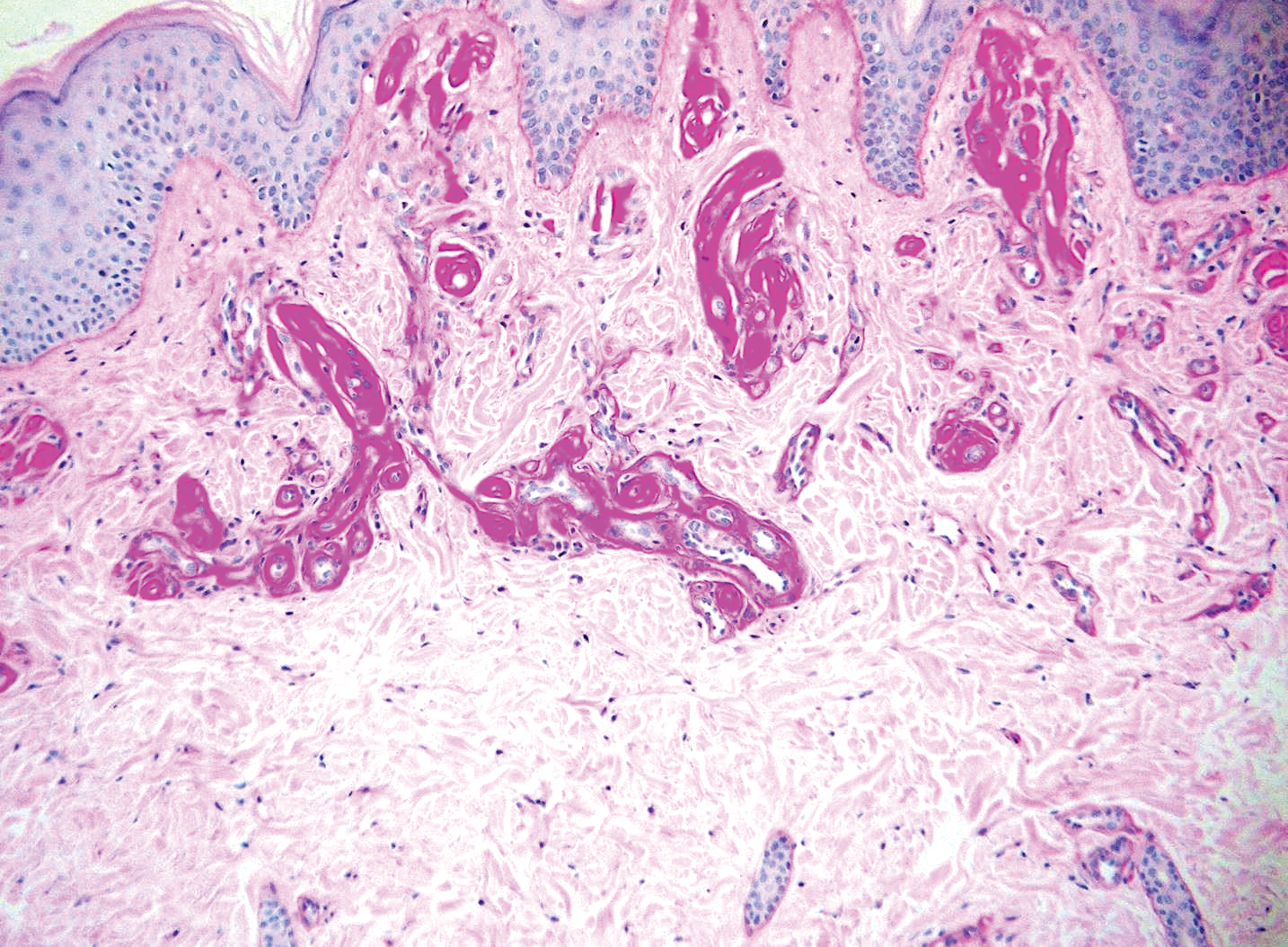
Lipoid proteinosis is an autosomal-recessive multisystem genodermatosis caused by mutations in extracellular matrix gene 1, ECM1. The first clinical sign can be a hoarse cry in infancy due to infiltration of vocal cords.3 Development of papulonodular lesions along the eyelids can result in a string-of-beads appearance called moniliform blepharosis, which is pathognomonic for lipoid proteinosis.6 With chronicity, the involved skin can become yellow, waxy, and thickened, particularly in the flexures or areas of trauma. Histologically, the dermis in lipoid proteinosis becomes diffusely thickened due to deposition of PAS-positive eosinophilic hyaline material that stains weakly with Congo red and thioflavin T.6 Early lesions demonstrate pale pink, hyalinelike thickening of the papillary dermal capillaries. Chronic lesions reveal an acanthotic epidermis, occasional papillomatosis with overlying hyperkeratosis, and a thickened dermis where diffuse thick bundles of pink hyaline deposits are oriented perpendicularly to the dermoepidermal junction.1,6 Lipoid proteinosis can be differentiated from EPP by the involvement of adnexal structures such as hair follicles, sebaceous glands, and arrector pili muscles (Figure 2), as opposed to EPP where adnexal structures are spared.1 Additionally, depositions in lipoid proteinosis are centered around both superficial and deep vessels with an onion skin-like pattern, while EPP involves mainly superficial vessels with more mild and focal hyalinization.
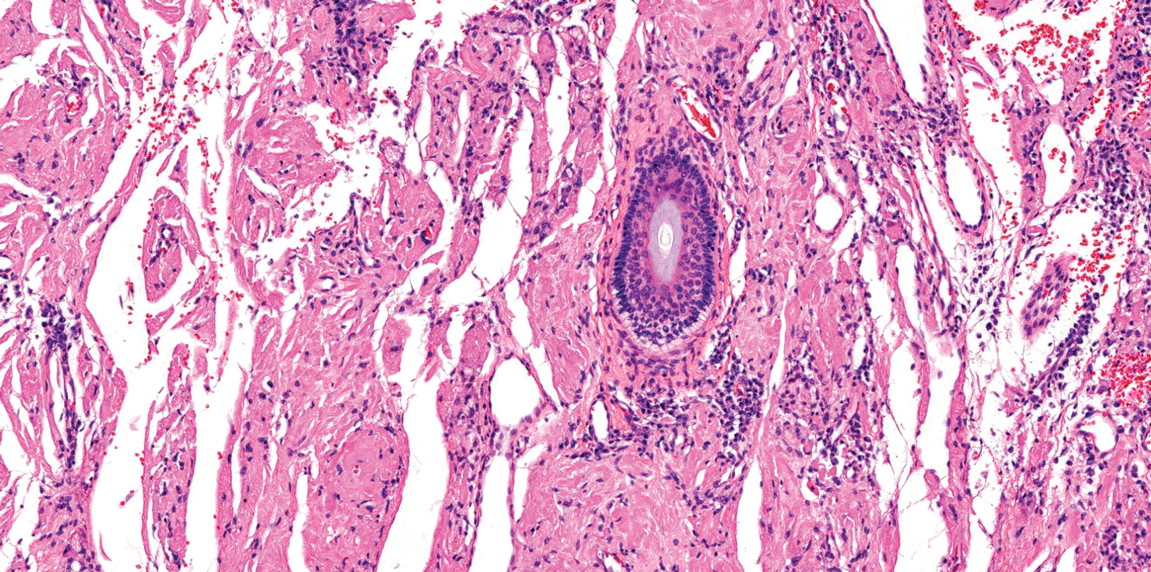
Juvenile colloid milium (JCM) is a rare condition that presents before puberty with discrete, yellow-brown, translucent papules predominantly located on the cheeks and nose and around the mouth. A gelatinous material can be expressed after puncturing a lesion.6 Gingival deposits and ligneous conjunctivitis also can be present. On histopathology, JCM shows degeneration of epidermal keratinocytes that form colloid bodies within the superficial dermis following apoptosis.6 Hematoxylin and eosin staining shows amorphous, fissured, pale pink deposits completely filling and expanding the superficial to mid dermis with clefting and no inflammation (Figure 3). Spindle-shaped fibroblasts may be seen within the lines of colloid fissuring and dispersed throughout the deposits.1 Histologically, JCM can be differentiated from EPP because deposits in EPP are distributed around and within superficial blood vessel walls, causing prominent vascular thickening not seen in JCM.6 The adult variant of colloid milium also can be distinguished from EPP by the presence of solar elastosis, which is absent in EPP due to a history of sun avoidance.3,7
Lichen amyloidosis presents with highly pruritic, red-brown, hyperkeratotic papules that commonly are found on the anterior lower legs and extensor forearms.1 The calves, ankles, dorsal aspects of the feet, thighs, and trunk also may be affected. Excoriations, lichenification, and nodular prurigo-like lesions due to chronic scratching can be present.6 Lichen amyloidosis is characterized by large, pink, amorphous deposits in the papillary dermis with epidermal acanthosis, hypergranulosis, and hyperkeratosis (Figure 4).6 Perivascular deposits are not a feature of primary cutaneous localized amyloid lesions.6 The diagnosis can be confirmed with Congo red staining under polarized light, which classically demonstrates apple green birefringence.1 For cases of amyloid that are not detected by Congo red or are not clear-cut, direct immunofluorescence and immunohistochemistry can be used as adjuncts for diagnosis. Amyloid deposits fluoresce positively for immunoglobulins or complements, particularly IgM and C3,8 and immunohistochemistry confirms the presence of keratin epitopes in deposits.9
Porphyria cutanea tarda can appear histologically similar to EPP. Caterpillar bodies, or linearly arranged eosinophilic PAS-positive globules in the epidermis overlying subepidermal bullae, are a diagnostic histopathologic finding in both PCT and EPP but are seen in less than half of both cases.7,10 Compared to EPP, the perivascular deposits in PCT typically are less pronounced and limited to the vessel wall with smaller hyaline cuffs (Figure 5).7 Additionally, solar elastosis can be seen in PCT lesions but not in EPP, as patients with PCT tend to be older and have increased cumulative sun damage.
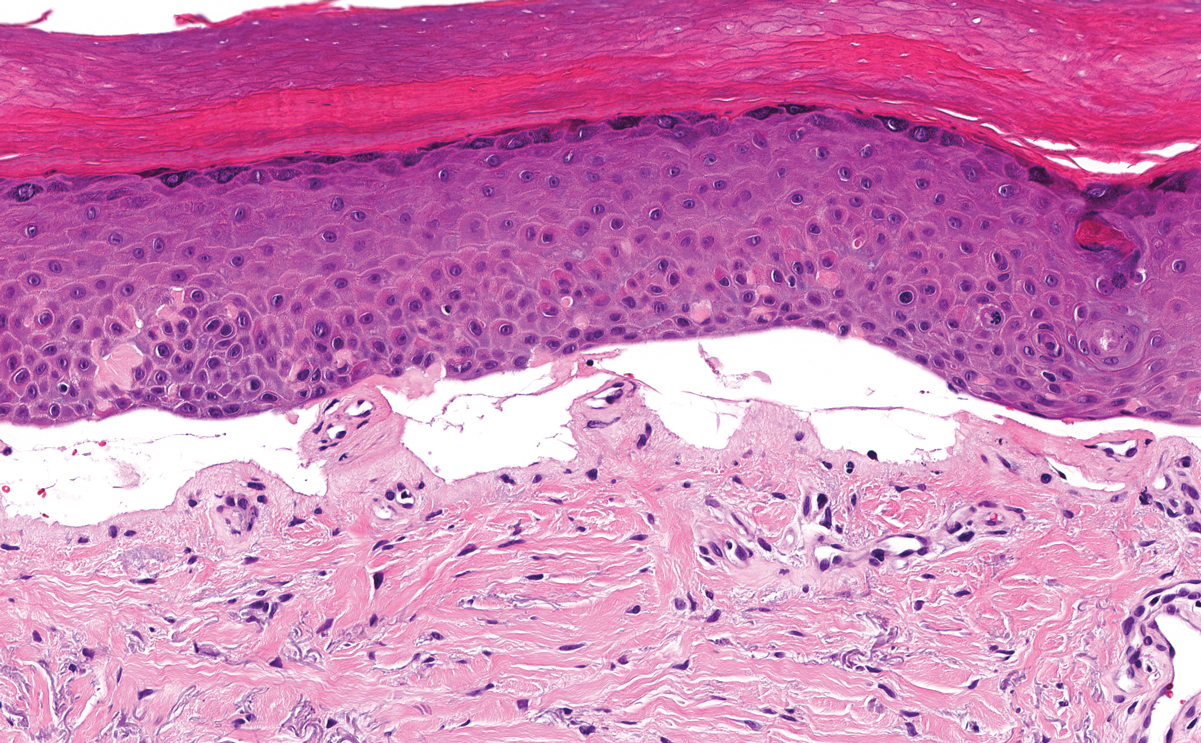
- Touart DM, Sau P. Cutaneous deposition diseases. part I. J Am Acad Dermatol. 1998;39(2, pt 1):149-171; quiz 172-144.
- Lim HW. Pathogenesis of photosensitivity in the cutaneous porphyrias. J Invest Dermatol. 2005;124:xvi-xvii.
- In: Alikhan A, Hocker TLH, eds. Review of Dermatology. China: Elsevier; 2017.
- Horner ME, Alikhan A, Tintle S, et al. Cutaneous porphyrias part I: epidemiology, pathogenesis, presentation, diagnosis, and histopathology. Int J Dermatol. 2013;52:1464-1480.
- Michaels BD, Del Rosso JQ, Mobini N, et al. Erythropoietic protoporphyria: a case report and literature review. J Clin Aesthet Dermatol. 2010;3:44-48.
- Calonje E, Brenn T, Lazar A, et al, eds. McKee's Pathology of the Skin. 4th ed. China: Elsevier Saunders; 2012.
- Patterson JW. Weedon's Skin Pathology. 4th ed. China: Elsevier Limited; 2016.
- MacDonald DM, Black MM, Ramnarain N. Immunofluorescence studies in primary localized cutaneous amyloidosis. Br J Dermatol. 1977;96:635-641.
- Ortiz-Romero PL, Ballestin-Carcavilla C, Lopez-Estebaranz JL, et al. Clinicopathologic and immunohistochemical studies on lichen amyloidosis and macular amyloidosis. Arch Dermatol. 1994;130:1559-1560.
- Raso DS, Greene WB, Maize JC, et al. Caterpillar bodies of porphyria cutanea tarda ultrastructurally represent a unique arrangement of colloid and basement membrane bodies. Am J Dermatopathol. 1996;18:24-29.
The Diagnosis: Erythropoietic Protoporphyria
Erythropoietic protoporphyria (EPP) is an autosomal-recessive photodermatosis that results from loss of activity of ferrochelatase, the last enzyme in the heme biosynthetic pathway.1 Erythropoietic protoporphyria normally involves sun-exposed areas of the body. Skin that is exposed to sunlight develops intense burning and stinging pain followed by erythema, edema, crusting, and petechiae that develops into waxy scarring over time. In contrast to other porphyrias, blistering generally is not seen.2 Accurate diagnosis often can be delayed by a decade or more following symptom onset due to the prominence of subjective pain as the presenting sign.
The histologic appearance of EPP differs depending on the chronicity of lesions. Biopsies of acute lesions show vacuolization of epidermal cells with intercellular edema, vacuolization and cytolysis of endothelial cells in superficial blood vessels, and focal red blood cell extravasation.3,4 A largely neutrophilic inflammatory infiltrate can be present.5 Hyaline cuffing develops over time in and around vessels in the papillary and superficial reticular dermis with notable sparing of adnexal structures. The perivascular deposits are strongly periodic acid-Schiff (PAS) positive and diastase resistant (Figure 1). Direct immunofluorescence shows mainly IgG and some IgM, fibrinogen, and C3 outlining characteristic donut-shaped blood vessels in the papillary dermis.6 The prominent thickness of the perivascular hyaline material depositions and the absence of subepidermal blistering can help differentiate EPP from porphyria cutanea tarda (PCT) and pseudoporphyria.6,7 When the deposition is extensive and involves the surrounding dermis, EPP can mimic colloid milium. Additional histologic differential diagnoses of EPP include other dermal depositional diseases such as lipoid proteinosis and amyloidosis.
Lipoid proteinosis is an autosomal-recessive multisystem genodermatosis caused by mutations in extracellular matrix gene 1, ECM1. The first clinical sign can be a hoarse cry in infancy due to infiltration of vocal cords.3 Development of papulonodular lesions along the eyelids can result in a string-of-beads appearance called moniliform blepharosis, which is pathognomonic for lipoid proteinosis.6 With chronicity, the involved skin can become yellow, waxy, and thickened, particularly in the flexures or areas of trauma. Histologically, the dermis in lipoid proteinosis becomes diffusely thickened due to deposition of PAS-positive eosinophilic hyaline material that stains weakly with Congo red and thioflavin T.6 Early lesions demonstrate pale pink, hyalinelike thickening of the papillary dermal capillaries. Chronic lesions reveal an acanthotic epidermis, occasional papillomatosis with overlying hyperkeratosis, and a thickened dermis where diffuse thick bundles of pink hyaline deposits are oriented perpendicularly to the dermoepidermal junction.1,6 Lipoid proteinosis can be differentiated from EPP by the involvement of adnexal structures such as hair follicles, sebaceous glands, and arrector pili muscles (Figure 2), as opposed to EPP where adnexal structures are spared.1 Additionally, depositions in lipoid proteinosis are centered around both superficial and deep vessels with an onion skin-like pattern, while EPP involves mainly superficial vessels with more mild and focal hyalinization.
Juvenile colloid milium (JCM) is a rare condition that presents before puberty with discrete, yellow-brown, translucent papules predominantly located on the cheeks and nose and around the mouth. A gelatinous material can be expressed after puncturing a lesion.6 Gingival deposits and ligneous conjunctivitis also can be present. On histopathology, JCM shows degeneration of epidermal keratinocytes that form colloid bodies within the superficial dermis following apoptosis.6 Hematoxylin and eosin staining shows amorphous, fissured, pale pink deposits completely filling and expanding the superficial to mid dermis with clefting and no inflammation (Figure 3). Spindle-shaped fibroblasts may be seen within the lines of colloid fissuring and dispersed throughout the deposits.1 Histologically, JCM can be differentiated from EPP because deposits in EPP are distributed around and within superficial blood vessel walls, causing prominent vascular thickening not seen in JCM.6 The adult variant of colloid milium also can be distinguished from EPP by the presence of solar elastosis, which is absent in EPP due to a history of sun avoidance.3,7
Lichen amyloidosis presents with highly pruritic, red-brown, hyperkeratotic papules that commonly are found on the anterior lower legs and extensor forearms.1 The calves, ankles, dorsal aspects of the feet, thighs, and trunk also may be affected. Excoriations, lichenification, and nodular prurigo-like lesions due to chronic scratching can be present.6 Lichen amyloidosis is characterized by large, pink, amorphous deposits in the papillary dermis with epidermal acanthosis, hypergranulosis, and hyperkeratosis (Figure 4).6 Perivascular deposits are not a feature of primary cutaneous localized amyloid lesions.6 The diagnosis can be confirmed with Congo red staining under polarized light, which classically demonstrates apple green birefringence.1 For cases of amyloid that are not detected by Congo red or are not clear-cut, direct immunofluorescence and immunohistochemistry can be used as adjuncts for diagnosis. Amyloid deposits fluoresce positively for immunoglobulins or complements, particularly IgM and C3,8 and immunohistochemistry confirms the presence of keratin epitopes in deposits.9
Porphyria cutanea tarda can appear histologically similar to EPP. Caterpillar bodies, or linearly arranged eosinophilic PAS-positive globules in the epidermis overlying subepidermal bullae, are a diagnostic histopathologic finding in both PCT and EPP but are seen in less than half of both cases.7,10 Compared to EPP, the perivascular deposits in PCT typically are less pronounced and limited to the vessel wall with smaller hyaline cuffs (Figure 5).7 Additionally, solar elastosis can be seen in PCT lesions but not in EPP, as patients with PCT tend to be older and have increased cumulative sun damage.
The Diagnosis: Erythropoietic Protoporphyria
Erythropoietic protoporphyria (EPP) is an autosomal-recessive photodermatosis that results from loss of activity of ferrochelatase, the last enzyme in the heme biosynthetic pathway.1 Erythropoietic protoporphyria normally involves sun-exposed areas of the body. Skin that is exposed to sunlight develops intense burning and stinging pain followed by erythema, edema, crusting, and petechiae that develops into waxy scarring over time. In contrast to other porphyrias, blistering generally is not seen.2 Accurate diagnosis often can be delayed by a decade or more following symptom onset due to the prominence of subjective pain as the presenting sign.
The histologic appearance of EPP differs depending on the chronicity of lesions. Biopsies of acute lesions show vacuolization of epidermal cells with intercellular edema, vacuolization and cytolysis of endothelial cells in superficial blood vessels, and focal red blood cell extravasation.3,4 A largely neutrophilic inflammatory infiltrate can be present.5 Hyaline cuffing develops over time in and around vessels in the papillary and superficial reticular dermis with notable sparing of adnexal structures. The perivascular deposits are strongly periodic acid-Schiff (PAS) positive and diastase resistant (Figure 1). Direct immunofluorescence shows mainly IgG and some IgM, fibrinogen, and C3 outlining characteristic donut-shaped blood vessels in the papillary dermis.6 The prominent thickness of the perivascular hyaline material depositions and the absence of subepidermal blistering can help differentiate EPP from porphyria cutanea tarda (PCT) and pseudoporphyria.6,7 When the deposition is extensive and involves the surrounding dermis, EPP can mimic colloid milium. Additional histologic differential diagnoses of EPP include other dermal depositional diseases such as lipoid proteinosis and amyloidosis.
Lipoid proteinosis is an autosomal-recessive multisystem genodermatosis caused by mutations in extracellular matrix gene 1, ECM1. The first clinical sign can be a hoarse cry in infancy due to infiltration of vocal cords.3 Development of papulonodular lesions along the eyelids can result in a string-of-beads appearance called moniliform blepharosis, which is pathognomonic for lipoid proteinosis.6 With chronicity, the involved skin can become yellow, waxy, and thickened, particularly in the flexures or areas of trauma. Histologically, the dermis in lipoid proteinosis becomes diffusely thickened due to deposition of PAS-positive eosinophilic hyaline material that stains weakly with Congo red and thioflavin T.6 Early lesions demonstrate pale pink, hyalinelike thickening of the papillary dermal capillaries. Chronic lesions reveal an acanthotic epidermis, occasional papillomatosis with overlying hyperkeratosis, and a thickened dermis where diffuse thick bundles of pink hyaline deposits are oriented perpendicularly to the dermoepidermal junction.1,6 Lipoid proteinosis can be differentiated from EPP by the involvement of adnexal structures such as hair follicles, sebaceous glands, and arrector pili muscles (Figure 2), as opposed to EPP where adnexal structures are spared.1 Additionally, depositions in lipoid proteinosis are centered around both superficial and deep vessels with an onion skin-like pattern, while EPP involves mainly superficial vessels with more mild and focal hyalinization.
Juvenile colloid milium (JCM) is a rare condition that presents before puberty with discrete, yellow-brown, translucent papules predominantly located on the cheeks and nose and around the mouth. A gelatinous material can be expressed after puncturing a lesion.6 Gingival deposits and ligneous conjunctivitis also can be present. On histopathology, JCM shows degeneration of epidermal keratinocytes that form colloid bodies within the superficial dermis following apoptosis.6 Hematoxylin and eosin staining shows amorphous, fissured, pale pink deposits completely filling and expanding the superficial to mid dermis with clefting and no inflammation (Figure 3). Spindle-shaped fibroblasts may be seen within the lines of colloid fissuring and dispersed throughout the deposits.1 Histologically, JCM can be differentiated from EPP because deposits in EPP are distributed around and within superficial blood vessel walls, causing prominent vascular thickening not seen in JCM.6 The adult variant of colloid milium also can be distinguished from EPP by the presence of solar elastosis, which is absent in EPP due to a history of sun avoidance.3,7
Lichen amyloidosis presents with highly pruritic, red-brown, hyperkeratotic papules that commonly are found on the anterior lower legs and extensor forearms.1 The calves, ankles, dorsal aspects of the feet, thighs, and trunk also may be affected. Excoriations, lichenification, and nodular prurigo-like lesions due to chronic scratching can be present.6 Lichen amyloidosis is characterized by large, pink, amorphous deposits in the papillary dermis with epidermal acanthosis, hypergranulosis, and hyperkeratosis (Figure 4).6 Perivascular deposits are not a feature of primary cutaneous localized amyloid lesions.6 The diagnosis can be confirmed with Congo red staining under polarized light, which classically demonstrates apple green birefringence.1 For cases of amyloid that are not detected by Congo red or are not clear-cut, direct immunofluorescence and immunohistochemistry can be used as adjuncts for diagnosis. Amyloid deposits fluoresce positively for immunoglobulins or complements, particularly IgM and C3,8 and immunohistochemistry confirms the presence of keratin epitopes in deposits.9
Porphyria cutanea tarda can appear histologically similar to EPP. Caterpillar bodies, or linearly arranged eosinophilic PAS-positive globules in the epidermis overlying subepidermal bullae, are a diagnostic histopathologic finding in both PCT and EPP but are seen in less than half of both cases.7,10 Compared to EPP, the perivascular deposits in PCT typically are less pronounced and limited to the vessel wall with smaller hyaline cuffs (Figure 5).7 Additionally, solar elastosis can be seen in PCT lesions but not in EPP, as patients with PCT tend to be older and have increased cumulative sun damage.
- Touart DM, Sau P. Cutaneous deposition diseases. part I. J Am Acad Dermatol. 1998;39(2, pt 1):149-171; quiz 172-144.
- Lim HW. Pathogenesis of photosensitivity in the cutaneous porphyrias. J Invest Dermatol. 2005;124:xvi-xvii.
- In: Alikhan A, Hocker TLH, eds. Review of Dermatology. China: Elsevier; 2017.
- Horner ME, Alikhan A, Tintle S, et al. Cutaneous porphyrias part I: epidemiology, pathogenesis, presentation, diagnosis, and histopathology. Int J Dermatol. 2013;52:1464-1480.
- Michaels BD, Del Rosso JQ, Mobini N, et al. Erythropoietic protoporphyria: a case report and literature review. J Clin Aesthet Dermatol. 2010;3:44-48.
- Calonje E, Brenn T, Lazar A, et al, eds. McKee's Pathology of the Skin. 4th ed. China: Elsevier Saunders; 2012.
- Patterson JW. Weedon's Skin Pathology. 4th ed. China: Elsevier Limited; 2016.
- MacDonald DM, Black MM, Ramnarain N. Immunofluorescence studies in primary localized cutaneous amyloidosis. Br J Dermatol. 1977;96:635-641.
- Ortiz-Romero PL, Ballestin-Carcavilla C, Lopez-Estebaranz JL, et al. Clinicopathologic and immunohistochemical studies on lichen amyloidosis and macular amyloidosis. Arch Dermatol. 1994;130:1559-1560.
- Raso DS, Greene WB, Maize JC, et al. Caterpillar bodies of porphyria cutanea tarda ultrastructurally represent a unique arrangement of colloid and basement membrane bodies. Am J Dermatopathol. 1996;18:24-29.
- Touart DM, Sau P. Cutaneous deposition diseases. part I. J Am Acad Dermatol. 1998;39(2, pt 1):149-171; quiz 172-144.
- Lim HW. Pathogenesis of photosensitivity in the cutaneous porphyrias. J Invest Dermatol. 2005;124:xvi-xvii.
- In: Alikhan A, Hocker TLH, eds. Review of Dermatology. China: Elsevier; 2017.
- Horner ME, Alikhan A, Tintle S, et al. Cutaneous porphyrias part I: epidemiology, pathogenesis, presentation, diagnosis, and histopathology. Int J Dermatol. 2013;52:1464-1480.
- Michaels BD, Del Rosso JQ, Mobini N, et al. Erythropoietic protoporphyria: a case report and literature review. J Clin Aesthet Dermatol. 2010;3:44-48.
- Calonje E, Brenn T, Lazar A, et al, eds. McKee's Pathology of the Skin. 4th ed. China: Elsevier Saunders; 2012.
- Patterson JW. Weedon's Skin Pathology. 4th ed. China: Elsevier Limited; 2016.
- MacDonald DM, Black MM, Ramnarain N. Immunofluorescence studies in primary localized cutaneous amyloidosis. Br J Dermatol. 1977;96:635-641.
- Ortiz-Romero PL, Ballestin-Carcavilla C, Lopez-Estebaranz JL, et al. Clinicopathologic and immunohistochemical studies on lichen amyloidosis and macular amyloidosis. Arch Dermatol. 1994;130:1559-1560.
- Raso DS, Greene WB, Maize JC, et al. Caterpillar bodies of porphyria cutanea tarda ultrastructurally represent a unique arrangement of colloid and basement membrane bodies. Am J Dermatopathol. 1996;18:24-29.
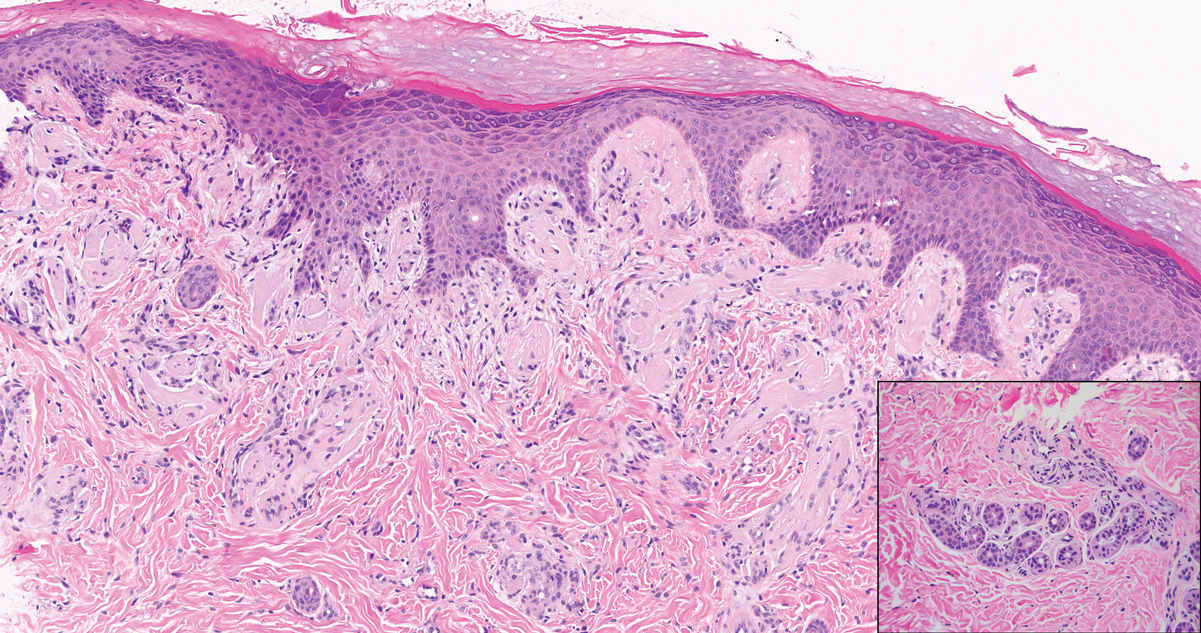
A 9-year-old girl presented with unexplained burning pain on the face, hands, and feet of 3 years' duration. Physical examination showed depressed shiny scars and crusted erosions on the dorsal aspect of the nose, arms, hands, and fingers. A 3-mm punch biopsy specimen was obtained from the right hand.