User login
To the Editor:
Langerhans cell histiocytosis (LCH) is a rare inflammatory neoplasia caused by accumulation of clonal Langerhans cells in 1 or more organs. The clinical spectrum is diverse, ranging from mild, single-organ involvement that may resolve spontaneously to severe progressive multisystem disease that can be fatal. It is most prevalent in children, affecting an estimated 4 to 5 children for every 1 million annually, with male predominance.1 The pathogenesis is driven by activating mutations in the mitogen-activated protein kinase pathway, with the BRAF V600E mutation detected in most LCH patients, resulting in proliferation of pathologic Langerhans cells and dysregulated expression of inflammatory cytokines in LCH lesions.2 A biopsy of lesional tissue is required for definitive diagnosis. Histopathology reveals a mixed inflammatory infiltrate and characteristic mononuclear cells with reniform nuclei that are positive for CD1a and CD207 proteins on immunohistochemical staining.3
Langerhans cell histiocytosis is categorized by the extent of organ involvement. It commonly affects the bones, skin, pituitary gland, liver, lungs, bone marrow, and lymph nodes.4 Single-system LCH involves a single organ with unifocal or multifocal lesions; multisystem LCH involves 2 or more organs and has a worse prognosis if risk organs (eg, liver, spleen, bone marrow) are involved.4
Skin lesions are reported in more than half of LCH cases and are the most common initial manifestation in patients younger than 2 years.4 Cutaneous findings are highly variable, which poses a diagnostic challenge. Common morphologies include erythematous papules, pustules, papulovesicles, scaly plaques, erosions, and petechiae. Lesions can be solitary or widespread and favor the trunk, head, and face.4 We describe an atypical case of hypopigmented cutaneous LCH and review the literature on this morphology in patients with skin of color.
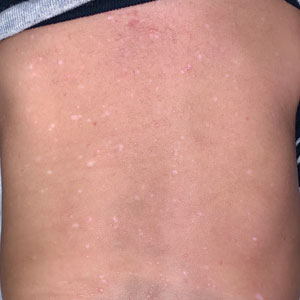
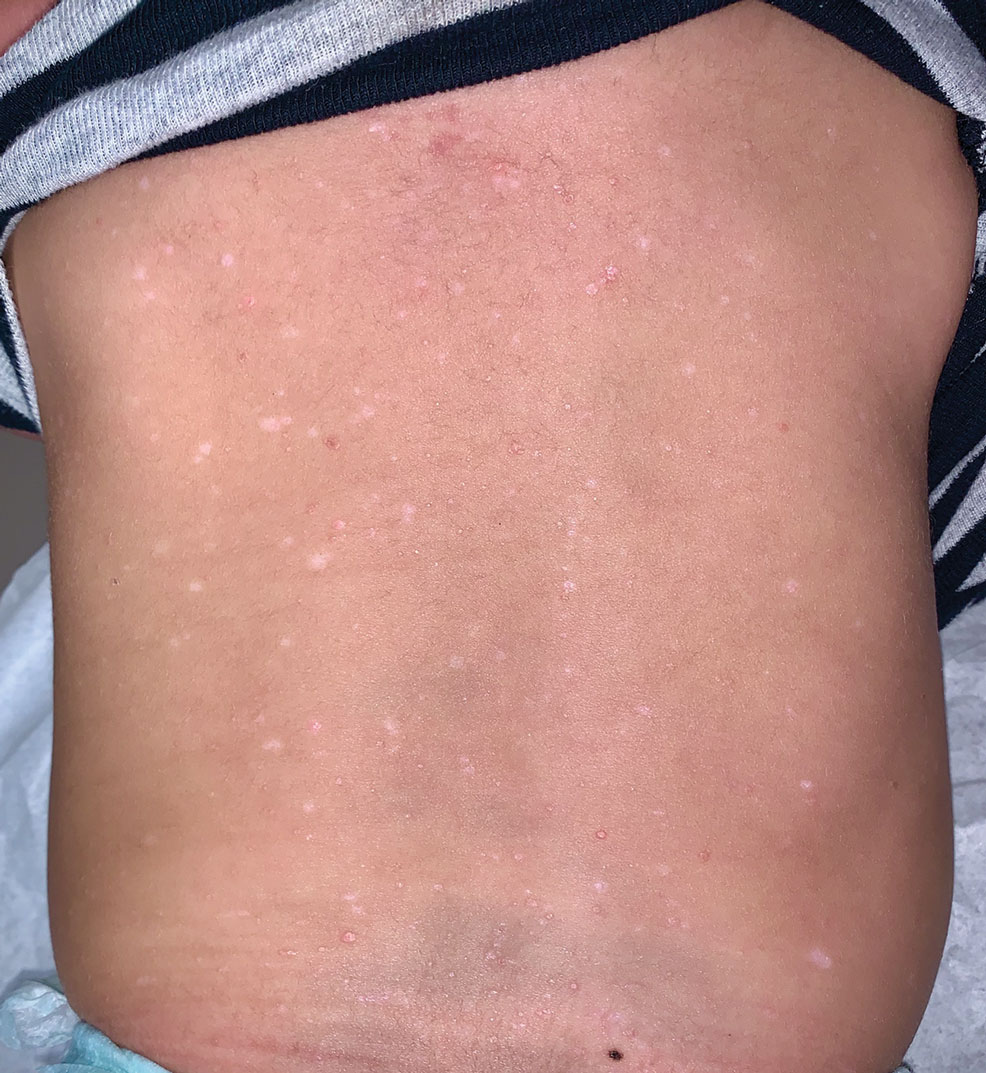
A 7-month-old Hispanic male infant who was otherwise healthy presented with numerous hypopigmented macules and pink papules on the trunk and groin that had progressed since birth. A review of systems was unremarkable. Physical examination revealed 1- to 3-mm, discrete, hypopigmented macules intermixed with 1- to 2-mm pearly pink papules scattered on the back, chest, abdomen, and inguinal folds (Figure 1). Some lesions appeared koebnerized; however, the parents denied a history of scratching or trauma.
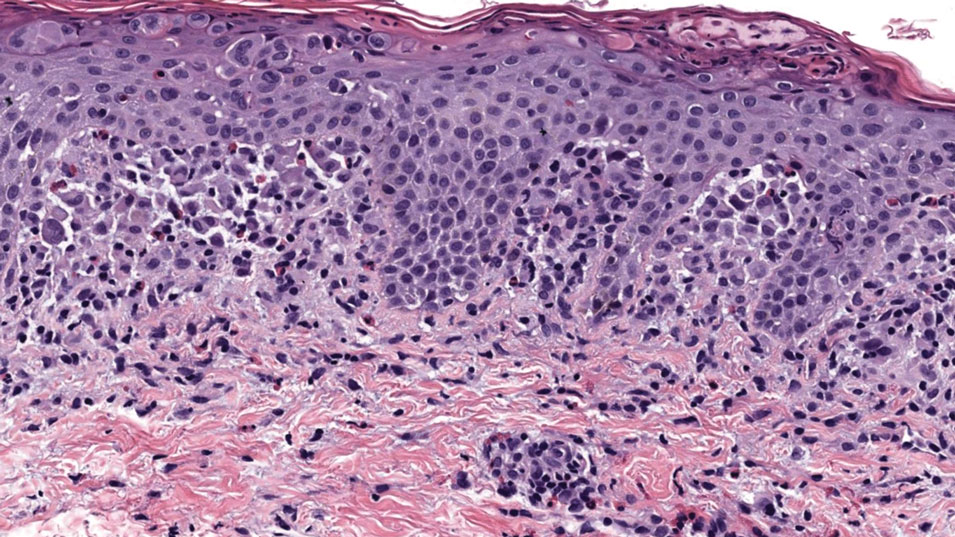
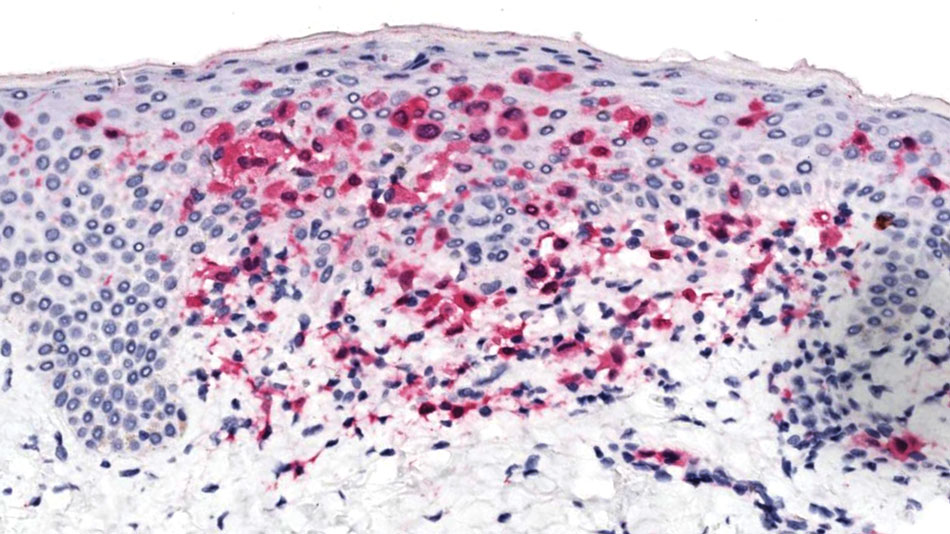
Histopathology of a lesion in the inguinal fold showed aggregates of mononuclear cells with reniform nuclei and abundant amphophilic cytoplasm in the papillary dermis, with focal extension into the epidermis. Scattered eosinophils and multinucleated giant cells were present in the dermal inflammatory infiltrate (Figure 2). Immunohistochemical staining was positive for CD1a (Figure 3) and S-100 protein (Figure 4). Although epidermal Langerhans cell collections also can be seen in allergic contact dermatitis,5 predominant involvement of the papillary dermis and the presence of multinucleated giant cells are characteristic of LCH.4 Given these findings, which were consistent with LCH, the dermatopathology deemed BRAF V600E immunostaining unnecessary for diagnostic purposes.
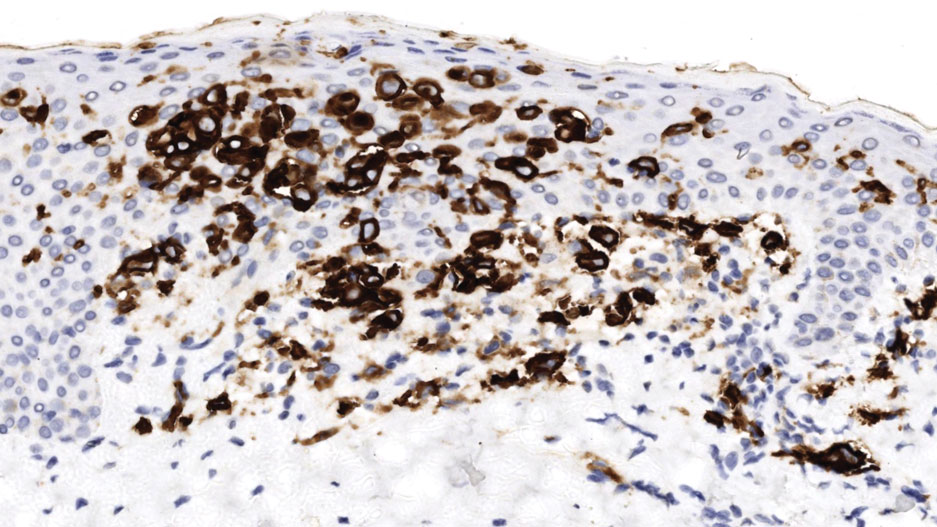
The patient was referred to the hematology and oncology department to undergo thorough evaluation for extracutaneous involvement. The workup included a complete blood cell count, liver function testing, electrolyte assessment, skeletal survey, chest radiography, and ultrasonography of the liver and spleen. All results were negative, suggesting a diagnosis of single-system cutaneous LCH.
Three months later, the patient presented to dermatology with spontaneous regression of all skin lesions. Continued follow-up—every 6 months for 5 years—was recommended to monitor for disease recurrence or progression to multisystem disease.
Cutaneous LCH is a clinically heterogeneous disease with the potential for multisystem involvement and long-term sequelae; therefore, timely diagnosis is paramount to optimize outcomes. However, delayed diagnosis is common because of the spectrum of skin findings that can mimic common pediatric dermatoses, such as seborrheic dermatitis, atopic dermatitis, and diaper dermatitis.4 In one study, the median time from onset of skin lesions to diagnostic biopsy was longer than 3 months (maximum, 5 years).6 Our patient was referred to dermatology 7 months after onset of hypopigmented macules, a rarely reported cutaneous manifestation of LCH.
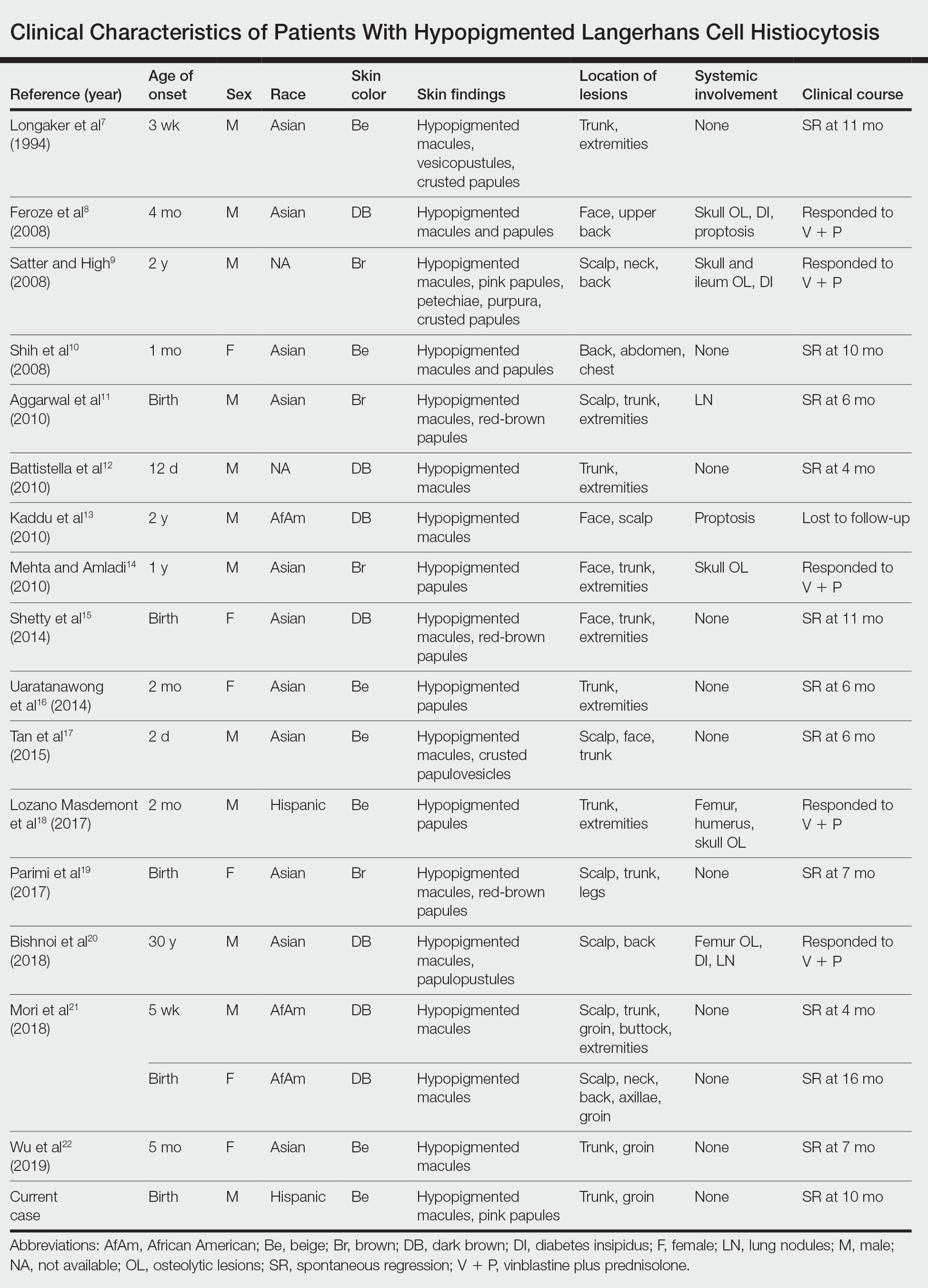
A PubMed search of articles indexed for MEDLINE from 1994 to 2019 using the terms Langerhans cell histiocytotis and hypopigmented yielded 17 cases of LCH presenting as hypopigmented skin lesions (Table).7-22 All cases occurred in patients with skin of color (ie, patients of Asian, Hispanic, or African descent). Hypopigmented macules were the only cutaneous manifestation in 10 (59%) cases. Lesions most commonly were distributed on the trunk (16/17 [94%]) and extremities (8/17 [47%]). The median age of onset was 1 month; 76% (13/17) of patients developed skin lesions before 1 year of age, indicating that this morphology may be more common in newborns. In most patients, the diagnosis was single-system cutaneous LCH; they exhibited spontaneous regression by 8 months of age on average, suggesting that this variant may be associated with a better prognosis. Mori and colleagues21 hypothesized that hypopigmented lesions may represent the resolving stage of active LCH based on histopathologic findings of dermal pallor and fibrosis in a hypopigmented LCH lesion. However, systemic involvement was reported in 7 cases of hypopigmented LCH, highlighting the importance of assessing for multisystem disease regardless of cutaneous morphology.21Langerhans cell histiocytosis should be considered in the differential diagnosis when evaluating hypopigmented skin eruptions in infants with darker skin types. Prompt diagnosis of this atypical variant requires a higher index of suspicion because of its rarity and the polymorphic nature of cutaneous LCH. This morphology may go undiagnosed in the setting of mild or spontaneously resolving disease; notwithstanding, accurate diagnosis and longitudinal surveillance are necessary given the potential for progressive systemic involvement.
1. Guyot-Goubin A, Donadieu J, Barkaoui M, et al. Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000–2004. Pediatr Blood Cancer. 2008;51:71-75. doi:10.1002/pbc.21498
2. Badalian-Very G, Vergilio J-A, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919-1923. doi:10.1182/blood-2010-04-279083
3. Haupt R, Minkov M, Astigarraga I, et al; Euro Histio Network. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work‐up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-184. doi:10.1002/pbc.24367
4. Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: history, classification, pathobiology, clinical manifestations, and prognosis. J Am Acad Dermatol. 2018;78:1035-1044. doi:10.1016/j.jaad.2017.05.059
5. Rosa G, Fernandez AP, Vij A, et al. Langerhans cell collections, but not eosinophils, are clues to a diagnosis of allergic contact dermatitis in appropriate skin biopsies. J Cutan Pathol. 2016;43:498-504. doi:10.1111/cup.12707
6. Simko SJ, Garmezy B, Abhyankar H, et al. Differentiating skin-limited and multisystem Langerhans cell histiocytosis. J Pediatr. 2014;165:990-996. doi:10.1016/j.jpeds.2014.07.063
7. Longaker MA, Frieden IJ, LeBoit PE, et al. Congenital “self-healing” Langerhans cell histiocytosis: the need for long-term follow-up. J Am Acad Dermatol. 1994;31(5, pt 2):910-916. doi:10.1016/s0190-9622(94)70258-6
8. Feroze K, Unni M, Jayasree MG, et al. Langerhans cell histiocytosis presenting with hypopigmented macules. Indian J Dermatol Venereol Leprol. 2008;74:670-672. doi:10.4103/0378-6323.45128
9. Satter EK, High WA. Langerhans cell histiocytosis: a case report and summary of the current recommendations of the Histiocyte Society. Dermatol Online J. 2008;14:3.
10. Chang SL, Shih IH, Kuo TT, et al. Congenital self-healing reticulohistiocytosis presenting as hypopigmented macules and papules in a neonate. Dermatologica Sinica 2008;26:80-84.
11. Aggarwal V, Seth A, Jain M, et al. Congenital Langerhans cell histiocytosis with skin and lung involvement: spontaneous regression. Indian J Pediatr. 2010;77:811-812.
12. Battistella M, Fraitag S, Teillac DH, et al. Neonatal and early infantile cutaneous Langerhans cell histiocytosis: comparison of self-regressive and non-self-regressive forms. Arch Dermatol. 2010;146:149-156. doi:10.1001/archdermatol.2009.360
13. Kaddu S, Mulyowa G, Kovarik C. Hypopigmented scaly, scalp and facial lesions and disfiguring exopthalmus. Clin Exp Dermatol. 2010;3:E52-E53. doi:10.1111/j.1365-2230.2009.03336.x
14. Mehta B, Amladi S. Langerhans cell histiocytosis presenting as hypopigmented papules. Pediatr Dermatol. 2010;27:215-217. doi:10.1111/j.1525-1470.2010.01104.x
15. Shetty S, Monappa V, Pai K, et al. Congenital self-healing reticulohistiocytosis: a case report. Our Dermatol Online. 2014;5:264-266.
16. Uaratanawong R, Kootiratrakarn T, Sudtikoonaseth P, et al. Congenital self-healing reticulohistiocytosis presented with multiple hypopigmented flat-topped papules: a case report and review of literatures. J Med Assoc Thai. 2014;97:993-997.
17. Tan Q, Gan LQ, Wang H. Congenital self-healing Langerhans cell histiocytosis in a male neonate. Indian J Dermatol Venereol Leprol. 2015;81:75-77. doi:10.4103/0378-6323.148587
18. Lozano Masdemont B, Gómez‐Recuero Muñoz L, Villanueva Álvarez‐Santullano A, et al. Langerhans cell histiocytosis mimicking lichen nitidus with bone involvement. Australas J Dermatol. 2017;58:231-233. doi:10.1111/ajd.12467
19. Parimi LR, You J, Hong L, et al. Congenital self-healing reticulohistiocytosis with spontaneous regression. An Bras Dermatol. 2017;92:553-555. doi:10.1590/abd1806-4841.20175432
20. Bishnoi A, De D, Khullar G, et al. Hypopigmented and acneiform lesions: an unusual initial presentation of adult-onset multisystem Langerhans cell histiocytosis. Indian J Dermatol Venereol Leprol. 2018;84:621-626. doi:10.4103/ijdvl.IJDVL_639_17
21. Mori S, Adar T, Kazlouskaya V, et al. Cutaneous Langerhans cell histiocytosis presenting with hypopigmented lesions: report of two cases and review of literature. Pediatr Dermatol. 2018;35:502-506. doi:10.1111/pde.13509
22. Wu X, Huang J, Jiang L, et al. Congenital self‐healing reticulohistiocytosis with BRAF V600E mutation in an infant. Clin Exp Dermatol. 2019;44:647-650. doi:10.1111/ced.13880
To the Editor:
Langerhans cell histiocytosis (LCH) is a rare inflammatory neoplasia caused by accumulation of clonal Langerhans cells in 1 or more organs. The clinical spectrum is diverse, ranging from mild, single-organ involvement that may resolve spontaneously to severe progressive multisystem disease that can be fatal. It is most prevalent in children, affecting an estimated 4 to 5 children for every 1 million annually, with male predominance.1 The pathogenesis is driven by activating mutations in the mitogen-activated protein kinase pathway, with the BRAF V600E mutation detected in most LCH patients, resulting in proliferation of pathologic Langerhans cells and dysregulated expression of inflammatory cytokines in LCH lesions.2 A biopsy of lesional tissue is required for definitive diagnosis. Histopathology reveals a mixed inflammatory infiltrate and characteristic mononuclear cells with reniform nuclei that are positive for CD1a and CD207 proteins on immunohistochemical staining.3
Langerhans cell histiocytosis is categorized by the extent of organ involvement. It commonly affects the bones, skin, pituitary gland, liver, lungs, bone marrow, and lymph nodes.4 Single-system LCH involves a single organ with unifocal or multifocal lesions; multisystem LCH involves 2 or more organs and has a worse prognosis if risk organs (eg, liver, spleen, bone marrow) are involved.4
Skin lesions are reported in more than half of LCH cases and are the most common initial manifestation in patients younger than 2 years.4 Cutaneous findings are highly variable, which poses a diagnostic challenge. Common morphologies include erythematous papules, pustules, papulovesicles, scaly plaques, erosions, and petechiae. Lesions can be solitary or widespread and favor the trunk, head, and face.4 We describe an atypical case of hypopigmented cutaneous LCH and review the literature on this morphology in patients with skin of color.
A 7-month-old Hispanic male infant who was otherwise healthy presented with numerous hypopigmented macules and pink papules on the trunk and groin that had progressed since birth. A review of systems was unremarkable. Physical examination revealed 1- to 3-mm, discrete, hypopigmented macules intermixed with 1- to 2-mm pearly pink papules scattered on the back, chest, abdomen, and inguinal folds (Figure 1). Some lesions appeared koebnerized; however, the parents denied a history of scratching or trauma.
Histopathology of a lesion in the inguinal fold showed aggregates of mononuclear cells with reniform nuclei and abundant amphophilic cytoplasm in the papillary dermis, with focal extension into the epidermis. Scattered eosinophils and multinucleated giant cells were present in the dermal inflammatory infiltrate (Figure 2). Immunohistochemical staining was positive for CD1a (Figure 3) and S-100 protein (Figure 4). Although epidermal Langerhans cell collections also can be seen in allergic contact dermatitis,5 predominant involvement of the papillary dermis and the presence of multinucleated giant cells are characteristic of LCH.4 Given these findings, which were consistent with LCH, the dermatopathology deemed BRAF V600E immunostaining unnecessary for diagnostic purposes.
The patient was referred to the hematology and oncology department to undergo thorough evaluation for extracutaneous involvement. The workup included a complete blood cell count, liver function testing, electrolyte assessment, skeletal survey, chest radiography, and ultrasonography of the liver and spleen. All results were negative, suggesting a diagnosis of single-system cutaneous LCH.
Three months later, the patient presented to dermatology with spontaneous regression of all skin lesions. Continued follow-up—every 6 months for 5 years—was recommended to monitor for disease recurrence or progression to multisystem disease.
Cutaneous LCH is a clinically heterogeneous disease with the potential for multisystem involvement and long-term sequelae; therefore, timely diagnosis is paramount to optimize outcomes. However, delayed diagnosis is common because of the spectrum of skin findings that can mimic common pediatric dermatoses, such as seborrheic dermatitis, atopic dermatitis, and diaper dermatitis.4 In one study, the median time from onset of skin lesions to diagnostic biopsy was longer than 3 months (maximum, 5 years).6 Our patient was referred to dermatology 7 months after onset of hypopigmented macules, a rarely reported cutaneous manifestation of LCH.
A PubMed search of articles indexed for MEDLINE from 1994 to 2019 using the terms Langerhans cell histiocytotis and hypopigmented yielded 17 cases of LCH presenting as hypopigmented skin lesions (Table).7-22 All cases occurred in patients with skin of color (ie, patients of Asian, Hispanic, or African descent). Hypopigmented macules were the only cutaneous manifestation in 10 (59%) cases. Lesions most commonly were distributed on the trunk (16/17 [94%]) and extremities (8/17 [47%]). The median age of onset was 1 month; 76% (13/17) of patients developed skin lesions before 1 year of age, indicating that this morphology may be more common in newborns. In most patients, the diagnosis was single-system cutaneous LCH; they exhibited spontaneous regression by 8 months of age on average, suggesting that this variant may be associated with a better prognosis. Mori and colleagues21 hypothesized that hypopigmented lesions may represent the resolving stage of active LCH based on histopathologic findings of dermal pallor and fibrosis in a hypopigmented LCH lesion. However, systemic involvement was reported in 7 cases of hypopigmented LCH, highlighting the importance of assessing for multisystem disease regardless of cutaneous morphology.21Langerhans cell histiocytosis should be considered in the differential diagnosis when evaluating hypopigmented skin eruptions in infants with darker skin types. Prompt diagnosis of this atypical variant requires a higher index of suspicion because of its rarity and the polymorphic nature of cutaneous LCH. This morphology may go undiagnosed in the setting of mild or spontaneously resolving disease; notwithstanding, accurate diagnosis and longitudinal surveillance are necessary given the potential for progressive systemic involvement.
To the Editor:
Langerhans cell histiocytosis (LCH) is a rare inflammatory neoplasia caused by accumulation of clonal Langerhans cells in 1 or more organs. The clinical spectrum is diverse, ranging from mild, single-organ involvement that may resolve spontaneously to severe progressive multisystem disease that can be fatal. It is most prevalent in children, affecting an estimated 4 to 5 children for every 1 million annually, with male predominance.1 The pathogenesis is driven by activating mutations in the mitogen-activated protein kinase pathway, with the BRAF V600E mutation detected in most LCH patients, resulting in proliferation of pathologic Langerhans cells and dysregulated expression of inflammatory cytokines in LCH lesions.2 A biopsy of lesional tissue is required for definitive diagnosis. Histopathology reveals a mixed inflammatory infiltrate and characteristic mononuclear cells with reniform nuclei that are positive for CD1a and CD207 proteins on immunohistochemical staining.3
Langerhans cell histiocytosis is categorized by the extent of organ involvement. It commonly affects the bones, skin, pituitary gland, liver, lungs, bone marrow, and lymph nodes.4 Single-system LCH involves a single organ with unifocal or multifocal lesions; multisystem LCH involves 2 or more organs and has a worse prognosis if risk organs (eg, liver, spleen, bone marrow) are involved.4
Skin lesions are reported in more than half of LCH cases and are the most common initial manifestation in patients younger than 2 years.4 Cutaneous findings are highly variable, which poses a diagnostic challenge. Common morphologies include erythematous papules, pustules, papulovesicles, scaly plaques, erosions, and petechiae. Lesions can be solitary or widespread and favor the trunk, head, and face.4 We describe an atypical case of hypopigmented cutaneous LCH and review the literature on this morphology in patients with skin of color.
A 7-month-old Hispanic male infant who was otherwise healthy presented with numerous hypopigmented macules and pink papules on the trunk and groin that had progressed since birth. A review of systems was unremarkable. Physical examination revealed 1- to 3-mm, discrete, hypopigmented macules intermixed with 1- to 2-mm pearly pink papules scattered on the back, chest, abdomen, and inguinal folds (Figure 1). Some lesions appeared koebnerized; however, the parents denied a history of scratching or trauma.
Histopathology of a lesion in the inguinal fold showed aggregates of mononuclear cells with reniform nuclei and abundant amphophilic cytoplasm in the papillary dermis, with focal extension into the epidermis. Scattered eosinophils and multinucleated giant cells were present in the dermal inflammatory infiltrate (Figure 2). Immunohistochemical staining was positive for CD1a (Figure 3) and S-100 protein (Figure 4). Although epidermal Langerhans cell collections also can be seen in allergic contact dermatitis,5 predominant involvement of the papillary dermis and the presence of multinucleated giant cells are characteristic of LCH.4 Given these findings, which were consistent with LCH, the dermatopathology deemed BRAF V600E immunostaining unnecessary for diagnostic purposes.
The patient was referred to the hematology and oncology department to undergo thorough evaluation for extracutaneous involvement. The workup included a complete blood cell count, liver function testing, electrolyte assessment, skeletal survey, chest radiography, and ultrasonography of the liver and spleen. All results were negative, suggesting a diagnosis of single-system cutaneous LCH.
Three months later, the patient presented to dermatology with spontaneous regression of all skin lesions. Continued follow-up—every 6 months for 5 years—was recommended to monitor for disease recurrence or progression to multisystem disease.
Cutaneous LCH is a clinically heterogeneous disease with the potential for multisystem involvement and long-term sequelae; therefore, timely diagnosis is paramount to optimize outcomes. However, delayed diagnosis is common because of the spectrum of skin findings that can mimic common pediatric dermatoses, such as seborrheic dermatitis, atopic dermatitis, and diaper dermatitis.4 In one study, the median time from onset of skin lesions to diagnostic biopsy was longer than 3 months (maximum, 5 years).6 Our patient was referred to dermatology 7 months after onset of hypopigmented macules, a rarely reported cutaneous manifestation of LCH.
A PubMed search of articles indexed for MEDLINE from 1994 to 2019 using the terms Langerhans cell histiocytotis and hypopigmented yielded 17 cases of LCH presenting as hypopigmented skin lesions (Table).7-22 All cases occurred in patients with skin of color (ie, patients of Asian, Hispanic, or African descent). Hypopigmented macules were the only cutaneous manifestation in 10 (59%) cases. Lesions most commonly were distributed on the trunk (16/17 [94%]) and extremities (8/17 [47%]). The median age of onset was 1 month; 76% (13/17) of patients developed skin lesions before 1 year of age, indicating that this morphology may be more common in newborns. In most patients, the diagnosis was single-system cutaneous LCH; they exhibited spontaneous regression by 8 months of age on average, suggesting that this variant may be associated with a better prognosis. Mori and colleagues21 hypothesized that hypopigmented lesions may represent the resolving stage of active LCH based on histopathologic findings of dermal pallor and fibrosis in a hypopigmented LCH lesion. However, systemic involvement was reported in 7 cases of hypopigmented LCH, highlighting the importance of assessing for multisystem disease regardless of cutaneous morphology.21Langerhans cell histiocytosis should be considered in the differential diagnosis when evaluating hypopigmented skin eruptions in infants with darker skin types. Prompt diagnosis of this atypical variant requires a higher index of suspicion because of its rarity and the polymorphic nature of cutaneous LCH. This morphology may go undiagnosed in the setting of mild or spontaneously resolving disease; notwithstanding, accurate diagnosis and longitudinal surveillance are necessary given the potential for progressive systemic involvement.
1. Guyot-Goubin A, Donadieu J, Barkaoui M, et al. Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000–2004. Pediatr Blood Cancer. 2008;51:71-75. doi:10.1002/pbc.21498
2. Badalian-Very G, Vergilio J-A, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919-1923. doi:10.1182/blood-2010-04-279083
3. Haupt R, Minkov M, Astigarraga I, et al; Euro Histio Network. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work‐up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-184. doi:10.1002/pbc.24367
4. Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: history, classification, pathobiology, clinical manifestations, and prognosis. J Am Acad Dermatol. 2018;78:1035-1044. doi:10.1016/j.jaad.2017.05.059
5. Rosa G, Fernandez AP, Vij A, et al. Langerhans cell collections, but not eosinophils, are clues to a diagnosis of allergic contact dermatitis in appropriate skin biopsies. J Cutan Pathol. 2016;43:498-504. doi:10.1111/cup.12707
6. Simko SJ, Garmezy B, Abhyankar H, et al. Differentiating skin-limited and multisystem Langerhans cell histiocytosis. J Pediatr. 2014;165:990-996. doi:10.1016/j.jpeds.2014.07.063
7. Longaker MA, Frieden IJ, LeBoit PE, et al. Congenital “self-healing” Langerhans cell histiocytosis: the need for long-term follow-up. J Am Acad Dermatol. 1994;31(5, pt 2):910-916. doi:10.1016/s0190-9622(94)70258-6
8. Feroze K, Unni M, Jayasree MG, et al. Langerhans cell histiocytosis presenting with hypopigmented macules. Indian J Dermatol Venereol Leprol. 2008;74:670-672. doi:10.4103/0378-6323.45128
9. Satter EK, High WA. Langerhans cell histiocytosis: a case report and summary of the current recommendations of the Histiocyte Society. Dermatol Online J. 2008;14:3.
10. Chang SL, Shih IH, Kuo TT, et al. Congenital self-healing reticulohistiocytosis presenting as hypopigmented macules and papules in a neonate. Dermatologica Sinica 2008;26:80-84.
11. Aggarwal V, Seth A, Jain M, et al. Congenital Langerhans cell histiocytosis with skin and lung involvement: spontaneous regression. Indian J Pediatr. 2010;77:811-812.
12. Battistella M, Fraitag S, Teillac DH, et al. Neonatal and early infantile cutaneous Langerhans cell histiocytosis: comparison of self-regressive and non-self-regressive forms. Arch Dermatol. 2010;146:149-156. doi:10.1001/archdermatol.2009.360
13. Kaddu S, Mulyowa G, Kovarik C. Hypopigmented scaly, scalp and facial lesions and disfiguring exopthalmus. Clin Exp Dermatol. 2010;3:E52-E53. doi:10.1111/j.1365-2230.2009.03336.x
14. Mehta B, Amladi S. Langerhans cell histiocytosis presenting as hypopigmented papules. Pediatr Dermatol. 2010;27:215-217. doi:10.1111/j.1525-1470.2010.01104.x
15. Shetty S, Monappa V, Pai K, et al. Congenital self-healing reticulohistiocytosis: a case report. Our Dermatol Online. 2014;5:264-266.
16. Uaratanawong R, Kootiratrakarn T, Sudtikoonaseth P, et al. Congenital self-healing reticulohistiocytosis presented with multiple hypopigmented flat-topped papules: a case report and review of literatures. J Med Assoc Thai. 2014;97:993-997.
17. Tan Q, Gan LQ, Wang H. Congenital self-healing Langerhans cell histiocytosis in a male neonate. Indian J Dermatol Venereol Leprol. 2015;81:75-77. doi:10.4103/0378-6323.148587
18. Lozano Masdemont B, Gómez‐Recuero Muñoz L, Villanueva Álvarez‐Santullano A, et al. Langerhans cell histiocytosis mimicking lichen nitidus with bone involvement. Australas J Dermatol. 2017;58:231-233. doi:10.1111/ajd.12467
19. Parimi LR, You J, Hong L, et al. Congenital self-healing reticulohistiocytosis with spontaneous regression. An Bras Dermatol. 2017;92:553-555. doi:10.1590/abd1806-4841.20175432
20. Bishnoi A, De D, Khullar G, et al. Hypopigmented and acneiform lesions: an unusual initial presentation of adult-onset multisystem Langerhans cell histiocytosis. Indian J Dermatol Venereol Leprol. 2018;84:621-626. doi:10.4103/ijdvl.IJDVL_639_17
21. Mori S, Adar T, Kazlouskaya V, et al. Cutaneous Langerhans cell histiocytosis presenting with hypopigmented lesions: report of two cases and review of literature. Pediatr Dermatol. 2018;35:502-506. doi:10.1111/pde.13509
22. Wu X, Huang J, Jiang L, et al. Congenital self‐healing reticulohistiocytosis with BRAF V600E mutation in an infant. Clin Exp Dermatol. 2019;44:647-650. doi:10.1111/ced.13880
1. Guyot-Goubin A, Donadieu J, Barkaoui M, et al. Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000–2004. Pediatr Blood Cancer. 2008;51:71-75. doi:10.1002/pbc.21498
2. Badalian-Very G, Vergilio J-A, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919-1923. doi:10.1182/blood-2010-04-279083
3. Haupt R, Minkov M, Astigarraga I, et al; Euro Histio Network. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work‐up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-184. doi:10.1002/pbc.24367
4. Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: history, classification, pathobiology, clinical manifestations, and prognosis. J Am Acad Dermatol. 2018;78:1035-1044. doi:10.1016/j.jaad.2017.05.059
5. Rosa G, Fernandez AP, Vij A, et al. Langerhans cell collections, but not eosinophils, are clues to a diagnosis of allergic contact dermatitis in appropriate skin biopsies. J Cutan Pathol. 2016;43:498-504. doi:10.1111/cup.12707
6. Simko SJ, Garmezy B, Abhyankar H, et al. Differentiating skin-limited and multisystem Langerhans cell histiocytosis. J Pediatr. 2014;165:990-996. doi:10.1016/j.jpeds.2014.07.063
7. Longaker MA, Frieden IJ, LeBoit PE, et al. Congenital “self-healing” Langerhans cell histiocytosis: the need for long-term follow-up. J Am Acad Dermatol. 1994;31(5, pt 2):910-916. doi:10.1016/s0190-9622(94)70258-6
8. Feroze K, Unni M, Jayasree MG, et al. Langerhans cell histiocytosis presenting with hypopigmented macules. Indian J Dermatol Venereol Leprol. 2008;74:670-672. doi:10.4103/0378-6323.45128
9. Satter EK, High WA. Langerhans cell histiocytosis: a case report and summary of the current recommendations of the Histiocyte Society. Dermatol Online J. 2008;14:3.
10. Chang SL, Shih IH, Kuo TT, et al. Congenital self-healing reticulohistiocytosis presenting as hypopigmented macules and papules in a neonate. Dermatologica Sinica 2008;26:80-84.
11. Aggarwal V, Seth A, Jain M, et al. Congenital Langerhans cell histiocytosis with skin and lung involvement: spontaneous regression. Indian J Pediatr. 2010;77:811-812.
12. Battistella M, Fraitag S, Teillac DH, et al. Neonatal and early infantile cutaneous Langerhans cell histiocytosis: comparison of self-regressive and non-self-regressive forms. Arch Dermatol. 2010;146:149-156. doi:10.1001/archdermatol.2009.360
13. Kaddu S, Mulyowa G, Kovarik C. Hypopigmented scaly, scalp and facial lesions and disfiguring exopthalmus. Clin Exp Dermatol. 2010;3:E52-E53. doi:10.1111/j.1365-2230.2009.03336.x
14. Mehta B, Amladi S. Langerhans cell histiocytosis presenting as hypopigmented papules. Pediatr Dermatol. 2010;27:215-217. doi:10.1111/j.1525-1470.2010.01104.x
15. Shetty S, Monappa V, Pai K, et al. Congenital self-healing reticulohistiocytosis: a case report. Our Dermatol Online. 2014;5:264-266.
16. Uaratanawong R, Kootiratrakarn T, Sudtikoonaseth P, et al. Congenital self-healing reticulohistiocytosis presented with multiple hypopigmented flat-topped papules: a case report and review of literatures. J Med Assoc Thai. 2014;97:993-997.
17. Tan Q, Gan LQ, Wang H. Congenital self-healing Langerhans cell histiocytosis in a male neonate. Indian J Dermatol Venereol Leprol. 2015;81:75-77. doi:10.4103/0378-6323.148587
18. Lozano Masdemont B, Gómez‐Recuero Muñoz L, Villanueva Álvarez‐Santullano A, et al. Langerhans cell histiocytosis mimicking lichen nitidus with bone involvement. Australas J Dermatol. 2017;58:231-233. doi:10.1111/ajd.12467
19. Parimi LR, You J, Hong L, et al. Congenital self-healing reticulohistiocytosis with spontaneous regression. An Bras Dermatol. 2017;92:553-555. doi:10.1590/abd1806-4841.20175432
20. Bishnoi A, De D, Khullar G, et al. Hypopigmented and acneiform lesions: an unusual initial presentation of adult-onset multisystem Langerhans cell histiocytosis. Indian J Dermatol Venereol Leprol. 2018;84:621-626. doi:10.4103/ijdvl.IJDVL_639_17
21. Mori S, Adar T, Kazlouskaya V, et al. Cutaneous Langerhans cell histiocytosis presenting with hypopigmented lesions: report of two cases and review of literature. Pediatr Dermatol. 2018;35:502-506. doi:10.1111/pde.13509
22. Wu X, Huang J, Jiang L, et al. Congenital self‐healing reticulohistiocytosis with BRAF V600E mutation in an infant. Clin Exp Dermatol. 2019;44:647-650. doi:10.1111/ced.13880
Practice Points
- Dermatologists should be aware of the hypopigmented variant of cutaneous Langerhans cell histiocytosis (LCH), which has been reported exclusively in patients with skin of color.
- Langerhans cell histiocytosis should be included in the differential diagnosis of hypopigmented macules, which may be the only cutaneous manifestation or may coincide with typical lesions of LCH.
- Hypopigmented cutaneous LCH may be more common in newborns and associated with a better prognosis.