User login
The Diagnosis: Wells Syndrome
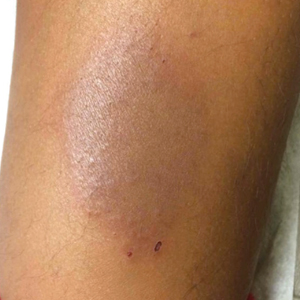
A punch biopsy taken from the perimeter of the lesion demonstrated mild spongiosis overlying a dense nodular to diffuse infiltrate of lymphocytes, neutrophils, and numerous eosinophils, some involving underlying fat lobules (Figure, A and B). In some areas, eosinophilic degeneration of collagen bundles surrounded by a rim of histiocytes, "flame features," were observed (Figure C). The clinical and histological features were consistent with Wells syndrome (WS), also known as eosinophilic cellulitis. Given the localized mild nature of the disease, the patient was started on a midpotency topical corticosteroid.

Wells syndrome is a rare inflammatory condition characterized by clinical polymorphism, suggestive histologic findings, and a recurrent course.1,2 This condition is especially rare in children.3,4 Caputo et al1 described 7 variants in their case series of 19 patients: classic plaque-type variant (the most common clinical presentation in children); annular granuloma-like (the most common clinical presentation in adults); urticarialike; bullous; papulonodular; papulovesicular; and fixed drug eruption-like. Wells syndrome is thought to result from excess production of IL-5 in response to a hypersensitivity reaction to an exogenous or endogenous circulating antigen.3,4 Increased levels of IL-5 enhance eosinophil accumulation in the skin, degranulation, and subsequent tissue destruction.3,4 Reported triggers include insect bites, viral and bacterial infections, drug eruptions, recent vaccination, and paraphenylenediamine in henna tattoos.3-7 Additionally, WS has been reported in the setting of gastrointestinal pathologies, such as celiac disease and ulcerative colitis, and with asthma exacerbations.8,9 However, in half of pediatric cases, no trigger can be identified.7
Clinically, WS presents with pruritic, mildly tender plaques.7 Lesions may be localized or diffuse and range from mild annular or circinate plaques with infiltrated borders to cellulitic-appearing lesions that are occasionally associated with bullae.5,6 Patients often report prodromal symptoms of burning and pruritus.5,6 Lesions rapidly progress over 2 to 3 days, pass through a blue grayish discoloration phase, and gradually resolve over 2 to 8 weeks.5,6,10 Although patients generally heal without scarring, WS lesions have been described to resolve with atrophy and hyperpigmentation resembling morphea.5-7 Additionally, patients typically experience a relapsing remitting course over months to years with eventual spontaneous resolution.1,5 Patients also may experience systemic symptoms including fever, lymphadenopathy, and arthralgia, though they do not develop more widespread systemic manifestations.2,3,7
Diagnosis of WS is based on clinicopathologic correlation. Histopathology of WS lesions demonstrates 3 phases. The acute phase demonstrates edema of the superficial and mid dermis with a dense dermal eosinophilic infiltrate.1,6,10 The subacute granulomatous phase demonstrates flame figures in the dermis.1,2,6,7,10 Flame figures consist of palisading groups of eosinophils and histiocytes around a core of degenerating basophilic collagen bundles associated with major basic protein.1,2,6,7,10 Finally, in the resolution phase, eosinophils gradually disappear while histiocytes and giant cells persist, forming microgranulomas.1,2,10 Notably, no vasculitis is observed and direct immunofluorescence is negative.3,7 Although flame figures are suggestive of WS, they are not pathognomonic and are observed in other conditions including Churg-Strauss syndrome, parasitic and fungal infections, herpes gestationis, bullous pemphigoid, and follicular mucinosis.2,5
Wells syndrome is a self-resolving and benign condition.1,10 Physicians are recommended to gather a complete history including review of medications and vaccinations; a history of insect bites, infections, and asthma; laboratory workup consisting of a complete blood cell count with differential and stool samples for ova and parasites; and a skin biopsy if the diagnosis is unclear.7 Identification and treatment of underlying causes often results in resolution.6 Systemic corticosteroids frequently are used in both adult and pediatric patients, though practitioners should consider alternative treatments when recurrences occur to avoid steroid side effects.3,6 Midpotency topical corticosteroids present a safe alternative to systemic corticosteroids in the pediatric population, especially in cases of localized WS without systemic symptoms.3 Other medications reported in the literature include cyclosporine, dapsone, antimalarial medications, and azathioprine.6 Despite appropriate therapy, patients and physicians should anticipate recurrence over months to years.1,6
- Caputo R, Marzano AV, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
- Smith SM, Kiracofe EA, Clark LN, et al. Idiopathic hypereosinophilic syndrome with cutaneous manifestations and flame figures: a spectrum of eosinophilic dermatoses whose features overlap with Wells' syndrome. Am J Dermatopathol. 2015;37:910-914.
- Gilliam AE, Bruckner AL, Howard RM, et al. Bullous "cellulitis" with eosinophilia: case report and review of Wells' syndrome in childhood. Pediatrics. 2005;116:E149-E155.
- Nacaroglu HT, Celegen M, Karkıner CS, et al. Eosinophilic cellulitis (Wells' syndrome) caused by a temporary henna tattoo. Postepy Dermatol Alergol. 2014;31:322-324.
- Heelan K, Ryan JF, Shear NH, et al. Wells syndrome (eosinophilic cellulitis): proposed diagnostic criteria and a literature review of the drug-induced variant. J Dermatol Case Rep. 2013;7:113-120.
- Sinno H, Lacroix JP, Lee J, et al. Diagnosis and management of eosinophilic cellulitis (Wells' syndrome): a case series and literature review. Can J Plast Surg. 2012;20:91-97.
- Cherng E, McClung AA, Rosenthal HM, et al. Wells' syndrome associated with parvovirus in a 5-year-old boy. Pediatr Dermatol. 2012;29:762-764.
- Eren M, Açikalin M. A case report of Wells' syndrome in a celiac patient. Turk J Gastroenterol. 2010;21:172-174.
- Cruz MJ, Mota A, Baudrier T, et al. Recurrent Wells' syndrome associated with allergic asthma exacerbation. Cutan Ocul Toxicol. 2012;31:154-156.
- Van der Straaten S, Wojciechowski M, Salgado R, et al. Eosinophilic cellulitis or Wells' syndrome in a 6-year-old child. Eur J Pediatr. 2006;165:197-198.
The Diagnosis: Wells Syndrome
A punch biopsy taken from the perimeter of the lesion demonstrated mild spongiosis overlying a dense nodular to diffuse infiltrate of lymphocytes, neutrophils, and numerous eosinophils, some involving underlying fat lobules (Figure, A and B). In some areas, eosinophilic degeneration of collagen bundles surrounded by a rim of histiocytes, "flame features," were observed (Figure C). The clinical and histological features were consistent with Wells syndrome (WS), also known as eosinophilic cellulitis. Given the localized mild nature of the disease, the patient was started on a midpotency topical corticosteroid.
Wells syndrome is a rare inflammatory condition characterized by clinical polymorphism, suggestive histologic findings, and a recurrent course.1,2 This condition is especially rare in children.3,4 Caputo et al1 described 7 variants in their case series of 19 patients: classic plaque-type variant (the most common clinical presentation in children); annular granuloma-like (the most common clinical presentation in adults); urticarialike; bullous; papulonodular; papulovesicular; and fixed drug eruption-like. Wells syndrome is thought to result from excess production of IL-5 in response to a hypersensitivity reaction to an exogenous or endogenous circulating antigen.3,4 Increased levels of IL-5 enhance eosinophil accumulation in the skin, degranulation, and subsequent tissue destruction.3,4 Reported triggers include insect bites, viral and bacterial infections, drug eruptions, recent vaccination, and paraphenylenediamine in henna tattoos.3-7 Additionally, WS has been reported in the setting of gastrointestinal pathologies, such as celiac disease and ulcerative colitis, and with asthma exacerbations.8,9 However, in half of pediatric cases, no trigger can be identified.7
Clinically, WS presents with pruritic, mildly tender plaques.7 Lesions may be localized or diffuse and range from mild annular or circinate plaques with infiltrated borders to cellulitic-appearing lesions that are occasionally associated with bullae.5,6 Patients often report prodromal symptoms of burning and pruritus.5,6 Lesions rapidly progress over 2 to 3 days, pass through a blue grayish discoloration phase, and gradually resolve over 2 to 8 weeks.5,6,10 Although patients generally heal without scarring, WS lesions have been described to resolve with atrophy and hyperpigmentation resembling morphea.5-7 Additionally, patients typically experience a relapsing remitting course over months to years with eventual spontaneous resolution.1,5 Patients also may experience systemic symptoms including fever, lymphadenopathy, and arthralgia, though they do not develop more widespread systemic manifestations.2,3,7
Diagnosis of WS is based on clinicopathologic correlation. Histopathology of WS lesions demonstrates 3 phases. The acute phase demonstrates edema of the superficial and mid dermis with a dense dermal eosinophilic infiltrate.1,6,10 The subacute granulomatous phase demonstrates flame figures in the dermis.1,2,6,7,10 Flame figures consist of palisading groups of eosinophils and histiocytes around a core of degenerating basophilic collagen bundles associated with major basic protein.1,2,6,7,10 Finally, in the resolution phase, eosinophils gradually disappear while histiocytes and giant cells persist, forming microgranulomas.1,2,10 Notably, no vasculitis is observed and direct immunofluorescence is negative.3,7 Although flame figures are suggestive of WS, they are not pathognomonic and are observed in other conditions including Churg-Strauss syndrome, parasitic and fungal infections, herpes gestationis, bullous pemphigoid, and follicular mucinosis.2,5
Wells syndrome is a self-resolving and benign condition.1,10 Physicians are recommended to gather a complete history including review of medications and vaccinations; a history of insect bites, infections, and asthma; laboratory workup consisting of a complete blood cell count with differential and stool samples for ova and parasites; and a skin biopsy if the diagnosis is unclear.7 Identification and treatment of underlying causes often results in resolution.6 Systemic corticosteroids frequently are used in both adult and pediatric patients, though practitioners should consider alternative treatments when recurrences occur to avoid steroid side effects.3,6 Midpotency topical corticosteroids present a safe alternative to systemic corticosteroids in the pediatric population, especially in cases of localized WS without systemic symptoms.3 Other medications reported in the literature include cyclosporine, dapsone, antimalarial medications, and azathioprine.6 Despite appropriate therapy, patients and physicians should anticipate recurrence over months to years.1,6
The Diagnosis: Wells Syndrome
A punch biopsy taken from the perimeter of the lesion demonstrated mild spongiosis overlying a dense nodular to diffuse infiltrate of lymphocytes, neutrophils, and numerous eosinophils, some involving underlying fat lobules (Figure, A and B). In some areas, eosinophilic degeneration of collagen bundles surrounded by a rim of histiocytes, "flame features," were observed (Figure C). The clinical and histological features were consistent with Wells syndrome (WS), also known as eosinophilic cellulitis. Given the localized mild nature of the disease, the patient was started on a midpotency topical corticosteroid.
Wells syndrome is a rare inflammatory condition characterized by clinical polymorphism, suggestive histologic findings, and a recurrent course.1,2 This condition is especially rare in children.3,4 Caputo et al1 described 7 variants in their case series of 19 patients: classic plaque-type variant (the most common clinical presentation in children); annular granuloma-like (the most common clinical presentation in adults); urticarialike; bullous; papulonodular; papulovesicular; and fixed drug eruption-like. Wells syndrome is thought to result from excess production of IL-5 in response to a hypersensitivity reaction to an exogenous or endogenous circulating antigen.3,4 Increased levels of IL-5 enhance eosinophil accumulation in the skin, degranulation, and subsequent tissue destruction.3,4 Reported triggers include insect bites, viral and bacterial infections, drug eruptions, recent vaccination, and paraphenylenediamine in henna tattoos.3-7 Additionally, WS has been reported in the setting of gastrointestinal pathologies, such as celiac disease and ulcerative colitis, and with asthma exacerbations.8,9 However, in half of pediatric cases, no trigger can be identified.7
Clinically, WS presents with pruritic, mildly tender plaques.7 Lesions may be localized or diffuse and range from mild annular or circinate plaques with infiltrated borders to cellulitic-appearing lesions that are occasionally associated with bullae.5,6 Patients often report prodromal symptoms of burning and pruritus.5,6 Lesions rapidly progress over 2 to 3 days, pass through a blue grayish discoloration phase, and gradually resolve over 2 to 8 weeks.5,6,10 Although patients generally heal without scarring, WS lesions have been described to resolve with atrophy and hyperpigmentation resembling morphea.5-7 Additionally, patients typically experience a relapsing remitting course over months to years with eventual spontaneous resolution.1,5 Patients also may experience systemic symptoms including fever, lymphadenopathy, and arthralgia, though they do not develop more widespread systemic manifestations.2,3,7
Diagnosis of WS is based on clinicopathologic correlation. Histopathology of WS lesions demonstrates 3 phases. The acute phase demonstrates edema of the superficial and mid dermis with a dense dermal eosinophilic infiltrate.1,6,10 The subacute granulomatous phase demonstrates flame figures in the dermis.1,2,6,7,10 Flame figures consist of palisading groups of eosinophils and histiocytes around a core of degenerating basophilic collagen bundles associated with major basic protein.1,2,6,7,10 Finally, in the resolution phase, eosinophils gradually disappear while histiocytes and giant cells persist, forming microgranulomas.1,2,10 Notably, no vasculitis is observed and direct immunofluorescence is negative.3,7 Although flame figures are suggestive of WS, they are not pathognomonic and are observed in other conditions including Churg-Strauss syndrome, parasitic and fungal infections, herpes gestationis, bullous pemphigoid, and follicular mucinosis.2,5
Wells syndrome is a self-resolving and benign condition.1,10 Physicians are recommended to gather a complete history including review of medications and vaccinations; a history of insect bites, infections, and asthma; laboratory workup consisting of a complete blood cell count with differential and stool samples for ova and parasites; and a skin biopsy if the diagnosis is unclear.7 Identification and treatment of underlying causes often results in resolution.6 Systemic corticosteroids frequently are used in both adult and pediatric patients, though practitioners should consider alternative treatments when recurrences occur to avoid steroid side effects.3,6 Midpotency topical corticosteroids present a safe alternative to systemic corticosteroids in the pediatric population, especially in cases of localized WS without systemic symptoms.3 Other medications reported in the literature include cyclosporine, dapsone, antimalarial medications, and azathioprine.6 Despite appropriate therapy, patients and physicians should anticipate recurrence over months to years.1,6
- Caputo R, Marzano AV, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
- Smith SM, Kiracofe EA, Clark LN, et al. Idiopathic hypereosinophilic syndrome with cutaneous manifestations and flame figures: a spectrum of eosinophilic dermatoses whose features overlap with Wells' syndrome. Am J Dermatopathol. 2015;37:910-914.
- Gilliam AE, Bruckner AL, Howard RM, et al. Bullous "cellulitis" with eosinophilia: case report and review of Wells' syndrome in childhood. Pediatrics. 2005;116:E149-E155.
- Nacaroglu HT, Celegen M, Karkıner CS, et al. Eosinophilic cellulitis (Wells' syndrome) caused by a temporary henna tattoo. Postepy Dermatol Alergol. 2014;31:322-324.
- Heelan K, Ryan JF, Shear NH, et al. Wells syndrome (eosinophilic cellulitis): proposed diagnostic criteria and a literature review of the drug-induced variant. J Dermatol Case Rep. 2013;7:113-120.
- Sinno H, Lacroix JP, Lee J, et al. Diagnosis and management of eosinophilic cellulitis (Wells' syndrome): a case series and literature review. Can J Plast Surg. 2012;20:91-97.
- Cherng E, McClung AA, Rosenthal HM, et al. Wells' syndrome associated with parvovirus in a 5-year-old boy. Pediatr Dermatol. 2012;29:762-764.
- Eren M, Açikalin M. A case report of Wells' syndrome in a celiac patient. Turk J Gastroenterol. 2010;21:172-174.
- Cruz MJ, Mota A, Baudrier T, et al. Recurrent Wells' syndrome associated with allergic asthma exacerbation. Cutan Ocul Toxicol. 2012;31:154-156.
- Van der Straaten S, Wojciechowski M, Salgado R, et al. Eosinophilic cellulitis or Wells' syndrome in a 6-year-old child. Eur J Pediatr. 2006;165:197-198.
- Caputo R, Marzano AV, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
- Smith SM, Kiracofe EA, Clark LN, et al. Idiopathic hypereosinophilic syndrome with cutaneous manifestations and flame figures: a spectrum of eosinophilic dermatoses whose features overlap with Wells' syndrome. Am J Dermatopathol. 2015;37:910-914.
- Gilliam AE, Bruckner AL, Howard RM, et al. Bullous "cellulitis" with eosinophilia: case report and review of Wells' syndrome in childhood. Pediatrics. 2005;116:E149-E155.
- Nacaroglu HT, Celegen M, Karkıner CS, et al. Eosinophilic cellulitis (Wells' syndrome) caused by a temporary henna tattoo. Postepy Dermatol Alergol. 2014;31:322-324.
- Heelan K, Ryan JF, Shear NH, et al. Wells syndrome (eosinophilic cellulitis): proposed diagnostic criteria and a literature review of the drug-induced variant. J Dermatol Case Rep. 2013;7:113-120.
- Sinno H, Lacroix JP, Lee J, et al. Diagnosis and management of eosinophilic cellulitis (Wells' syndrome): a case series and literature review. Can J Plast Surg. 2012;20:91-97.
- Cherng E, McClung AA, Rosenthal HM, et al. Wells' syndrome associated with parvovirus in a 5-year-old boy. Pediatr Dermatol. 2012;29:762-764.
- Eren M, Açikalin M. A case report of Wells' syndrome in a celiac patient. Turk J Gastroenterol. 2010;21:172-174.
- Cruz MJ, Mota A, Baudrier T, et al. Recurrent Wells' syndrome associated with allergic asthma exacerbation. Cutan Ocul Toxicol. 2012;31:154-156.
- Van der Straaten S, Wojciechowski M, Salgado R, et al. Eosinophilic cellulitis or Wells' syndrome in a 6-year-old child. Eur J Pediatr. 2006;165:197-198.
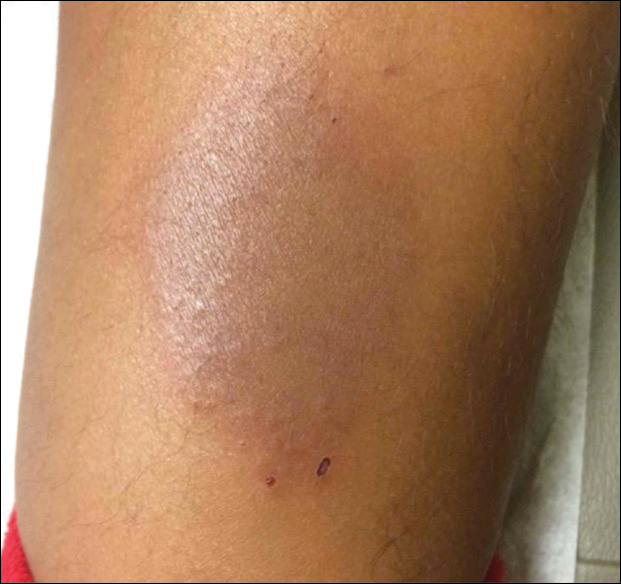
A healthy 7-year-old boy presented with an enlarging hyperpigmented plaque on the anterior aspect of the lower left leg of 2 months' duration. His mother reported onset following a mosquito bite. Clotrimazole was used without improvement. His mother denied recent travel, similar lesions in close contacts, fever, asthma, and arthralgia. Physical examination revealed a 5.2 ×3-cm nonscaly, red-brown, ovoid, thin plaque with a slightly raised border.