User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Antidepressant failure in adolescents
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Preventing drinking relapse in patients with alcoholic liver disease
Alcohol use disorder (AUD) is a mosaic of psychiatric and medical symptoms. Alcoholic liver disease (ALD) in its acute and chronic forms is a common clinical consequence of long-standing AUD. Patients with ALD require specialized care from professionals in addiction, gastroenterology, and psychiatry. However, medical specialists treating ALD might not regularly consider medications to treat AUD because of their limited experience with the drugs or the lack of studies in patients with significant liver disease.1 Similarly, psychiatrists might be reticent to prescribe medications for AUD, fearing that liver disease will be made worse or that they will cause other medical complications. As a result, patients with ALD might not receive care that could help treat their AUD (Box).

Given the high worldwide prevalence and morbidity of ALD,2 general and subspecialized psychiatrists routinely evaluate patients with AUD in and out of the hospital. This article aims to equip a psychiatrist with:
• a practical understanding of the natural history and categorization of ALD
• basic skills to detect symptoms of ALD
• preparation to collaborate with medical colleagues in multidisciplinary management of co-occurring AUD and ALD
• a summary of the pharmacotherapeutics of AUD, with emphasis on patients with clinically apparent ALD.
Categorization and clinical features
Alcoholic liver damage encompasses a spectrum of disorders, including alcoholic fatty liver, acute alcohol hepatitis (AH), and cirrhosis following varying durations and patterns of alcohol use. Manifestations of ALD vary from asymptomatic fatty liver with minimal liver enzyme elevation to severe acute AH with jaundice, coagulopathy, and high short-term mortality (Table 1). Symptoms seen in patients with AH include fever, abdominal pain, anorexia, jaundice, leukocytosis, and coagulopathy.3
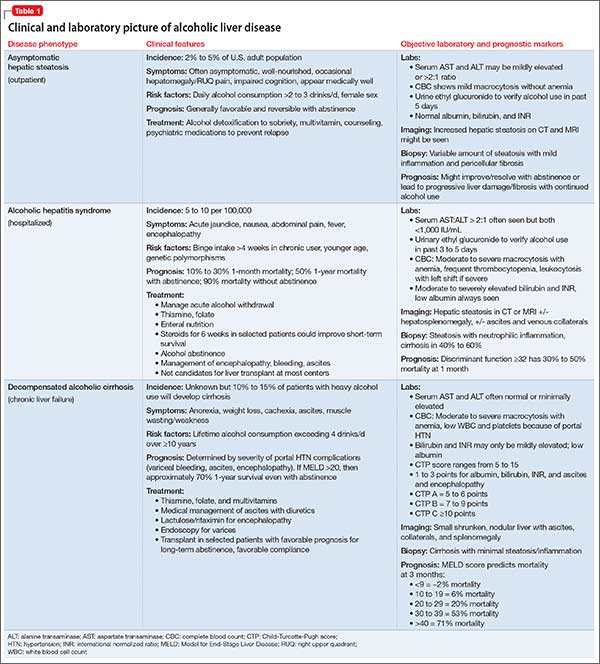
Patients with chronic ALD often develop cirrhosis, persistent elevation of the serum aminotransferase level (even after prolonged alcohol abstinence), signs of portal hypertension (ascites, encephalopathy, variceal bleeding), and profound malnutrition. The survival of ALD patients with chronic liver failure is predicted in part by a Model for End-Stage Liver Disease (MELD) score that incorporates their serum total bilirubin level, creatinine level, and international normalized ratio. The MELD score, which ranges from 6 to 40, also is used to gauge the need for liver transplantation; most patients who have a MELD score >15 benefit from transplant. To definitively determine the severity of ALD, a liver biopsy is required but usually is not performed in clinical practice.
All patients who drink heavily or suffer with AUD are at risk of developing AH; women and binge drinkers are particularly vulnerable.4 Liver dysfunction and malnutrition in ALD patients compromise the immune system, increasing the risk of infection. Patients hospitalized with AH have a 10% to 30% risk of inpatient mortality; their 1- and 2-month post-discharge survival is 50% to 65%, largely determined by whether the patient can maintain sobriety.5 Psychiatrists’ contribution to ALD treatment therefore has the potential to save lives.
Screening and detection of ALD
Because of the high mortality associated with AH and cirrhosis, symptom recognition and collaborative medical and psychiatric management are critical (Table 2). A psychiatrist evaluating a jaundiced patient who continues to drink should arrange urgent medical evaluation. While gathering a history, mental health providers might hear a patient refer to symptoms of gastrointestinal bleeding (vomiting blood, bloody or dark stool), painful abdominal distension, fevers, or confusion that should prompt a referral to a gastroenterologist or the emergency department. Testing for urinary ethyl glucuronide—a direct metabolite of ethanol that can be detected for as long as 90 hours after ethanol ingestion—is useful in detecting alcohol use in the past 4 or 5 days.

Medical management of ALD
Corticosteroids are a mainstay in pharmacotherapy for severe AH. There is evidence for improved outcomes in patients with severe AH treated with prednisolone for 4 to 6 weeks.5 Prognostic models such as the Maddrey’s Discriminant Function, Lille Model, and the MELD score help determine the need for steroid use and identify high-risk patients. Patients with active infection or bleeding are not a candidate for steroid treatment. An experienced gastroenterologist or hepatologist should initiate medical intervention after thorough evaluation.
Liver transplantation. A select group of patients with refractory liver failure are considered for liver transplantation. Although transplant programs differ in their criteria for organ listing, many require patients to demonstrate at least 6 months of verified abstinence from alcohol and illicit drugs as well as adherence to a formal AUD treatment and rehabilitation plan. The patient’s psychological health and prognosis for sustained sobriety are central to candidacy for organ listing, which highlights the key role of psychiatrists.
Further considerations. Thiamine and folate often are given to patients with ALD. Abdominal imaging and screening for HIV and viral hepatitis—identified in 10% to 20% of ALD patients—is routine. Alcohol abstinence remains central to survival because relapse increases the risk of recurrent, severe liver disease. Regrettably, many physical symptoms of liver disease, such as portal hypertension, ascites, and jaundice, can take months to improve with abstinence.
Treating AUD in patients with ALD
Successful treatment is multifaceted and includes more than just medications. Initial management often includes addressing alcohol withdrawal in dependent patients.6
Behavioral interventions are effective and indispensable components in preventing relapse,7 including a written relapse prevention plan that formally outlines the patient’s commitment to change, identifies triggers, and outlines a discrete plan of action. Primary psychiatric pathology, including depression and anxiety, often are comorbid with AUD; concurrent treatment of these disorders could improve patient outcomes.8
Benzodiazepines often are used during acute alcohol withdrawal. They should not be used for relapse prevention in ALD because of their additive interactions with alcohol, cognitive and psychomotor side effects, and abuse potential.9,10 Many of these drugs are cleared by the liver and generally are not recommended for use in patients with ALD.
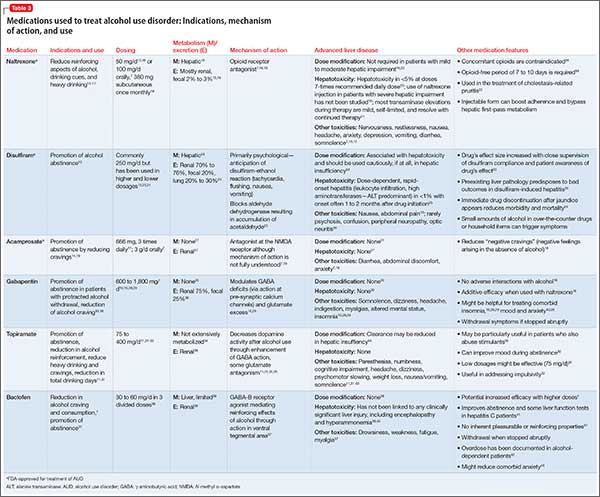
Other agents, further considerations. Drug trials in AUD largely have been conducted in small, heterogeneous populations and revealed modest and, at times, conflicting drug effect sizes.6,11,12 The placebo effect among the AUD population is pronounced.6,7,13 Despite these caveats, several agents have been studied and validated by the FDA to treat AUD. Additional agents with promising pilot data are being investigated. Table 31,7,10,11,13-43 summarizes drugs used to treat AUD—those with and without FDA approval—with a focus on how they might be used in patients with ALD. Of note, several of these agents do not rely on the liver for metabolism or excretion.
There is no agreed-upon algorithm or safety profile to guide a prescriber’s decision making about drug or dosage choices when treating AUD in patients with ALD. Because liver function can vary among patients as well as during an individual patient’s disease course, treatment decisions should be made on a clinical, collaborative, and case-by-case basis.
That being said, the AUD treatment literature suggests that specific drugs might be more useful in patients with varying severity of disease and during different phases of recovery:
• Acamprosate has been found to be effective in supporting abstinence in sober patients.14,44
• Naltrexone has been shown to be useful in patients with severe alcohol cravings. By modulating alcohol’s rewarding effects, naltrexone also reduces heavy alcohol consumption in patients who are drinking.14,15,44
• Disulfiram generally is not recommended for use in patients with clinically apparent hepatic insufficiency, such as decompensated cirrhosis or preexisting jaundice.
Although alcohol abstinence remains the treatment goal and a requirement for liver transplant, providers must recognize that some patients might not be able to maintain long-term sobriety. Therefore, harm reduction models are important companions to abstinence-only models of AUD treatment.45 The array of behavioral, pharmacological, and philosophical approaches to AUD treatment underlines the need for an individualized approach to relapse prevention.
Collaboration between medicine and psychiatry
When AUD and ALD are comorbid, psychiatrists might worry about making the patient’s medical condition worse by prescribing additional psychoactive medications—particularly ones that are cleared by the liver. Remember that AUD confers a substantial mortality rate that is more than 3 times that of the general population, along with severe medical46 and psychosocial31 effects. Although prescribers must remain vigilant for adverse drug effects, medications easily can be blamed for what might be the natural progression and symptoms of AUD in patients with ALD.26 This erroneous conclusion can lead to premature medication discontinuation and under-treatment of AUD.
In the end, keeping the patient sober and mentally well might be more beneficial than eliminating the burden of any medication side effects. Collaborative medical and psychiatric management of ALD patients can ensure that clinicians properly weigh the risks, benefits, and duration of treatment unique to each patient.
Starting AUD treatment promptly after alcohol relapse is essential and entails a multidisciplinary effort between medicine and psychiatry, both in and out of the hospital. Because the relapsing, ill ALD patient most often will be admitted to a medical specialist, AUD might not receive enough attention during the medical admission. Psychiatrists can help in initiating AUD treatment in the acute medical setting, which has been shown to improve the outpatient course.6 For medically stable ALD patients admitted for inpatient psychiatric care or presenting a clinic, the mental health clinician should be aware of key laboratory and physical exam findings.
Bottom Line
Patients with alcoholic liver disease (ALD) require collaborative care from specialists in addiction, gastroenterology, and psychiatry. Psychiatrists have a role in identifying signs of ALD, prescribing medication to treat alcohol use disorder, and encouraging abstinence. There is some evidence supporting specific medications for varying severity of disease and different phases of recovery. Pharmacotherapy decisions should be made case by case.
Related Resources
• Khan A, Tansel A, White DL, et al. Efficacy of psychosocial interventions in inducing and maintaining alcohol abstinence in patients with chronic liver disease: a systematic review [published online August 6, 2015]. Clin Gastroenterol Hepatol. doi: 10.1016/j.cgh.2015.07.047.
• Vuittonet CL, Halse M, Leggio L, et al. Pharmacotherapy for alcoholic patients with alcoholic liver disease. Am J Health Syst Pharm. 2014;71(15):1265-1276.
Drug Brand Names
Acamprosate • Campral
Baclofen • Lioresal
Disulfiram • Antabuse
Gabapentin • Neurontin
Naltrexone • ReVia, Vivitrol
Pentoxifylline • Trental
Prednisolone • Prelone
Rifaximin • Xifaxan
Topiramate • Topamax
Disclosures
Dr. Winder and Dr. Mellinger report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products. Dr. Fontana receives research funding from Bristol Myers Squibb, Gilead, and Janssen and consults for the Chronic Liver Disease Foundation.
1. Gache P, Hadengue A. Baclofen improves abstinence in alcoholic cirrhosis: still better to come? J Hepatol. 2008;49(6):1083-1085.
2. Rehm J, Mathers C, Popova S, et al. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet. 2009;373(9682):2223-2233.
3. Singal AK, Kamath PS, Gores GJ, et al. Alcoholic hepatitis: current challenges and future directions. Clin Gastroenterol Hepatol. 2014;12(4):555-564; quiz e31-32.
4. Becker U, Deis A, Sørensen TI, et al. Prediction of risk of liver disease by alcohol intake, sex, and age: a prospective population study. Hepatology. 1996;23(5):1025-1029.
5. Mathurin P, Lucey MR. Management of alcoholic hepatitis. J Hepatol. 2012;56(suppl 1):S39-S45.
6. Mann K, Lemenager T, Hoffmann S, et al; PREDICT Study Team. Results of a double-blind, placebo-controlled pharmacotherapy trial in alcoholism conducted in Germany and comparison with the US COMBINE study. Addict Biol. 2013;18(6):937-946.
7. Anton RF, O’Malley SS, Ciraulo DA, et al; COMBINE Study Research Group. Combined pharmacotherapies and behavioral interventions for alcohol dependence: the COMBINE study: a randomized controlled trial. JAMA. 2006;295(17):2003-2017.
8. Kranzler HR, Rosenthal RN. Dual diagnosis: alcoholism and co-morbid psychiatric disorders. Am J Addict. 2003;12(suppl 1):S26-S40.
9. Book SW, Myrick H. Novel anticonvulsants in the treatment of alcoholism. Expert Opin Investig Drugs. 2005;14(4):371-376.
10. Furieri FA, Nakamura-Palacios EM. Gabapentin reduces alcohol consumption and craving: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2007;68(11):1691-1700.
11. Blodgett JC, Del Re AC, Maisel NC, et al. A meta-analysis of topiramate’s effects for individuals with alcohol use disorders. Alcohol Clin Exp Res. 2014;38(6):1481-1488.
12. Krystal JH, Cramer JA, Krol WF, et al; Veterans Affairs Naltrexone Cooperative Study 425 Group. Naltrexone in the treatment of alcohol dependence. N Engl J Med. 2001;345(24):1734-1739.
13. Petrakis IL, Poling J, Levinson C, et al; VA New England VISN I MIRECC Study Group. Naltrexone and disulfiram in patients with alcohol dependence and comorbid psychiatric disorders. Biol Psychiatry. 2005;57(10):1128-1137.
14. Maisel NC, Blodgett JC, Wilbourne PL, et al. Meta-analysis of naltrexone and acamprosate for treating alcohol use disorders: when are these medications most helpful? Addiction. 2013;108(2):275-293.
15. Pettinati HM, O’Brien CP, Rabinowitz AR, et al. The status of naltrexone in the treatment of alcohol dependence: specific effects on heavy drinking. J Clin Psychopharmacol. 2006;26(6):610-625.
16. Anton RF, Myrick H, Wright TM, et al. Gabapentin combined with naltrexone for the treatment of alcohol dependence. Am J Psychiatry. 2011;168(7):709-717.
17. Srisurapanont M, Jarusuraisin N. Opioid antagonists for alcohol dependence. Cochrane Database Syst Rev. 2005(1):CD001867.
18. Naltrexone. 2014. http://www.micromedexsolutions.com. Accessed January 31, 2015.
19. Soyka M, Chick J. Use of acamprosate and opioid antagonists in the treatment of alcohol dependence: a European perspective. Am J Addict. 2003;12(suppl 1):S69-S80.
20. Turncliff RZ, Dunbar JL, Dong Q, et al. Pharmacokinetics of long-acting naltrexone in subjects with mild to moderate hepatic impairment. J Clin Pharmacol. 2005;45(11):1259-1267.
21. United States National Library of Medicine. Naltrexone. http://livertox.nlm.nih.gov/Naltrexone.htm. Updated September 30, 2015. Accessed November 10, 2015.
22. Terg R, Coronel E, Sordá J, et al. Efficacy and safety of oral naltrexone treatment for pruritus of cholestasis, a crossover, double blind, placebo-controlled study. J Hepatol. 2002;37(6):717-722.
23. Skinner MD, Lahmek P, Pham H, et al. Disulfiram efficacy in the treatment of alcohol dependence: a meta-analysis [published online February 10, 2014]. PLoS One. 2014;9(2):e87366. doi: 10.1371/journal.pone.0087366.
24. Disulfiram. 2014. http://www.micromedexsolutions.com. Accessed January 31, 2015.
25. Björnsson E, Nordlinder H, Olsson R. Clinical characteristics and prognostic markers in disulfiram-induced liver injury. J Hepatol. 2006;44(4):791-797.
26. Chick J. Safety issues concerning the use of disulfiram in treating alcohol dependence. Drug Saf. 1999;20(5):427-435.
27. Campral [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2012.
28. Brower KJ, Myra Kim H, Strobbe S, et al. A randomized double-blind pilot trial of gabapentin versus placebo to treat alcohol dependence and comorbid insomnia. Alcohol Clin Exp Res. 2008;32(8):1429-1438.
29. Mason BJ, Quello S, Goodell V, et al. Gabapentin treatment for alcohol dependence: a randomized clinical trial. JAMA Intern Med. 2014;174(1):70-77.
30. Neurontin [package insert]. New York, NY: Pfizer; 2015.
31. Johnson BA, Ait-Daoud N, Akhtar FZ, et al. Oral topiramate reduces the consequences of drinking and improves the quality of life of alcohol-dependent individuals: a randomized controlled trial. Arch Gen Psychiatry. 2004;61(9):905-912.
32. Paparrigopoulos T, Tzavellas E, Karaiskos D, et al. Treatment of alcohol dependence with low-dose topiramate: an open-label controlled study. BMC Psychiatry. 2011;11:41.
33. Rubio G, Ponce G, Jiménez-Arriero MA, et al. Effects of topiramate in the treatment of alcohol dependence. Pharmacopsychiatry. 2004;37(1):37-40.
34. Topamax [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2009.
35. De Sousa AA, De Sousa J, Kapoor H. An open randomized trial comparing disulfiram and topiramate in the treatment of alcohol dependence. J Subst Abuse Treat. 2008;34(4):460-463.
36. Kampman KM, Pettinati HM, Lynch KG, et al. A double-blind, placebo-controlled trial of topiramate for the treatment of comorbid cocaine and alcohol dependence. Drug Alcohol Depend. 2013;133(1):94-99.
37. Addolorato G, Leggio L, Ferrulli A, et al. Dose-response effect of baclofen in reducing daily alcohol intake in alcohol dependence: secondary analysis of a randomized, double-blind, placebo-controlled trial. Alcohol Alcohol. 2011;46(3):312-317.
38. Balcofen [package insert]. Concord, NC: McKesson Packing Services; 2013.
39. United States National Library of Medicine. Baclofen. 2015. http://livertox.nlm.nih.gov/Baclofen.htm. Accessed November 7, 2015.
40. Addolorato G, Leggio L, Ferrulli A, et al. Effectiveness and safety of baclofen for maintenance of alcohol abstinence in alcohol-dependent patients with liver cirrhosis: randomised, double-blind controlled study. Lancet. 2007;370(9603):1915-1922.
41. Leggio L, Ferrulli A, Zambon A, et al. Baclofen promotes alcohol abstinence in alcohol dependent cirrhotic patients with hepatitis C virus (HCV) infection. Addict Behav. 2012;37(4):561-564.
42. Franchitto N, Pelissier F, Lauque D, et al. Self-intoxication with baclofen in alcohol-dependent patients with co-existing psychiatric illness: an emergency department case series. Alcohol Alcohol. 2014;49(1):79-83.
43. Brennan JL, Leung JG, Gagliardi JP, et al. Clinical effectiveness of baclofen for the treatment of alcohol dependence: a review. Clin Pharmacol. 2013;5:99-107.
44. Rösner S, Leucht S, Lehert P, et al. Acamprosate supports abstinence, naltrexone prevents excessive drinking: evidence from a meta-analysis with unreported outcomes. J Psychopharmacol. 2008;22(1):11-23.
45. Marlatt GA, Witkiewitz K. Harm reduction approaches to alcohol use: health promotion, prevention, and treatment. Addict Behav. 2002;27(6):867-886.
46. O’Shea RS, Dasarathy S, McCullough AJ. Alcoholic liver disease. Am J Gastroenterol. 2010;105(1):14-32; quiz 33.
Alcohol use disorder (AUD) is a mosaic of psychiatric and medical symptoms. Alcoholic liver disease (ALD) in its acute and chronic forms is a common clinical consequence of long-standing AUD. Patients with ALD require specialized care from professionals in addiction, gastroenterology, and psychiatry. However, medical specialists treating ALD might not regularly consider medications to treat AUD because of their limited experience with the drugs or the lack of studies in patients with significant liver disease.1 Similarly, psychiatrists might be reticent to prescribe medications for AUD, fearing that liver disease will be made worse or that they will cause other medical complications. As a result, patients with ALD might not receive care that could help treat their AUD (Box).
Given the high worldwide prevalence and morbidity of ALD,2 general and subspecialized psychiatrists routinely evaluate patients with AUD in and out of the hospital. This article aims to equip a psychiatrist with:
• a practical understanding of the natural history and categorization of ALD
• basic skills to detect symptoms of ALD
• preparation to collaborate with medical colleagues in multidisciplinary management of co-occurring AUD and ALD
• a summary of the pharmacotherapeutics of AUD, with emphasis on patients with clinically apparent ALD.
Categorization and clinical features
Alcoholic liver damage encompasses a spectrum of disorders, including alcoholic fatty liver, acute alcohol hepatitis (AH), and cirrhosis following varying durations and patterns of alcohol use. Manifestations of ALD vary from asymptomatic fatty liver with minimal liver enzyme elevation to severe acute AH with jaundice, coagulopathy, and high short-term mortality (Table 1). Symptoms seen in patients with AH include fever, abdominal pain, anorexia, jaundice, leukocytosis, and coagulopathy.3
Patients with chronic ALD often develop cirrhosis, persistent elevation of the serum aminotransferase level (even after prolonged alcohol abstinence), signs of portal hypertension (ascites, encephalopathy, variceal bleeding), and profound malnutrition. The survival of ALD patients with chronic liver failure is predicted in part by a Model for End-Stage Liver Disease (MELD) score that incorporates their serum total bilirubin level, creatinine level, and international normalized ratio. The MELD score, which ranges from 6 to 40, also is used to gauge the need for liver transplantation; most patients who have a MELD score >15 benefit from transplant. To definitively determine the severity of ALD, a liver biopsy is required but usually is not performed in clinical practice.
All patients who drink heavily or suffer with AUD are at risk of developing AH; women and binge drinkers are particularly vulnerable.4 Liver dysfunction and malnutrition in ALD patients compromise the immune system, increasing the risk of infection. Patients hospitalized with AH have a 10% to 30% risk of inpatient mortality; their 1- and 2-month post-discharge survival is 50% to 65%, largely determined by whether the patient can maintain sobriety.5 Psychiatrists’ contribution to ALD treatment therefore has the potential to save lives.
Screening and detection of ALD
Because of the high mortality associated with AH and cirrhosis, symptom recognition and collaborative medical and psychiatric management are critical (Table 2). A psychiatrist evaluating a jaundiced patient who continues to drink should arrange urgent medical evaluation. While gathering a history, mental health providers might hear a patient refer to symptoms of gastrointestinal bleeding (vomiting blood, bloody or dark stool), painful abdominal distension, fevers, or confusion that should prompt a referral to a gastroenterologist or the emergency department. Testing for urinary ethyl glucuronide—a direct metabolite of ethanol that can be detected for as long as 90 hours after ethanol ingestion—is useful in detecting alcohol use in the past 4 or 5 days.
Medical management of ALD
Corticosteroids are a mainstay in pharmacotherapy for severe AH. There is evidence for improved outcomes in patients with severe AH treated with prednisolone for 4 to 6 weeks.5 Prognostic models such as the Maddrey’s Discriminant Function, Lille Model, and the MELD score help determine the need for steroid use and identify high-risk patients. Patients with active infection or bleeding are not a candidate for steroid treatment. An experienced gastroenterologist or hepatologist should initiate medical intervention after thorough evaluation.
Liver transplantation. A select group of patients with refractory liver failure are considered for liver transplantation. Although transplant programs differ in their criteria for organ listing, many require patients to demonstrate at least 6 months of verified abstinence from alcohol and illicit drugs as well as adherence to a formal AUD treatment and rehabilitation plan. The patient’s psychological health and prognosis for sustained sobriety are central to candidacy for organ listing, which highlights the key role of psychiatrists.
Further considerations. Thiamine and folate often are given to patients with ALD. Abdominal imaging and screening for HIV and viral hepatitis—identified in 10% to 20% of ALD patients—is routine. Alcohol abstinence remains central to survival because relapse increases the risk of recurrent, severe liver disease. Regrettably, many physical symptoms of liver disease, such as portal hypertension, ascites, and jaundice, can take months to improve with abstinence.
Treating AUD in patients with ALD
Successful treatment is multifaceted and includes more than just medications. Initial management often includes addressing alcohol withdrawal in dependent patients.6
Behavioral interventions are effective and indispensable components in preventing relapse,7 including a written relapse prevention plan that formally outlines the patient’s commitment to change, identifies triggers, and outlines a discrete plan of action. Primary psychiatric pathology, including depression and anxiety, often are comorbid with AUD; concurrent treatment of these disorders could improve patient outcomes.8
Benzodiazepines often are used during acute alcohol withdrawal. They should not be used for relapse prevention in ALD because of their additive interactions with alcohol, cognitive and psychomotor side effects, and abuse potential.9,10 Many of these drugs are cleared by the liver and generally are not recommended for use in patients with ALD.
Other agents, further considerations. Drug trials in AUD largely have been conducted in small, heterogeneous populations and revealed modest and, at times, conflicting drug effect sizes.6,11,12 The placebo effect among the AUD population is pronounced.6,7,13 Despite these caveats, several agents have been studied and validated by the FDA to treat AUD. Additional agents with promising pilot data are being investigated. Table 31,7,10,11,13-43 summarizes drugs used to treat AUD—those with and without FDA approval—with a focus on how they might be used in patients with ALD. Of note, several of these agents do not rely on the liver for metabolism or excretion.
There is no agreed-upon algorithm or safety profile to guide a prescriber’s decision making about drug or dosage choices when treating AUD in patients with ALD. Because liver function can vary among patients as well as during an individual patient’s disease course, treatment decisions should be made on a clinical, collaborative, and case-by-case basis.
That being said, the AUD treatment literature suggests that specific drugs might be more useful in patients with varying severity of disease and during different phases of recovery:
• Acamprosate has been found to be effective in supporting abstinence in sober patients.14,44
• Naltrexone has been shown to be useful in patients with severe alcohol cravings. By modulating alcohol’s rewarding effects, naltrexone also reduces heavy alcohol consumption in patients who are drinking.14,15,44
• Disulfiram generally is not recommended for use in patients with clinically apparent hepatic insufficiency, such as decompensated cirrhosis or preexisting jaundice.
Although alcohol abstinence remains the treatment goal and a requirement for liver transplant, providers must recognize that some patients might not be able to maintain long-term sobriety. Therefore, harm reduction models are important companions to abstinence-only models of AUD treatment.45 The array of behavioral, pharmacological, and philosophical approaches to AUD treatment underlines the need for an individualized approach to relapse prevention.
Collaboration between medicine and psychiatry
When AUD and ALD are comorbid, psychiatrists might worry about making the patient’s medical condition worse by prescribing additional psychoactive medications—particularly ones that are cleared by the liver. Remember that AUD confers a substantial mortality rate that is more than 3 times that of the general population, along with severe medical46 and psychosocial31 effects. Although prescribers must remain vigilant for adverse drug effects, medications easily can be blamed for what might be the natural progression and symptoms of AUD in patients with ALD.26 This erroneous conclusion can lead to premature medication discontinuation and under-treatment of AUD.
In the end, keeping the patient sober and mentally well might be more beneficial than eliminating the burden of any medication side effects. Collaborative medical and psychiatric management of ALD patients can ensure that clinicians properly weigh the risks, benefits, and duration of treatment unique to each patient.
Starting AUD treatment promptly after alcohol relapse is essential and entails a multidisciplinary effort between medicine and psychiatry, both in and out of the hospital. Because the relapsing, ill ALD patient most often will be admitted to a medical specialist, AUD might not receive enough attention during the medical admission. Psychiatrists can help in initiating AUD treatment in the acute medical setting, which has been shown to improve the outpatient course.6 For medically stable ALD patients admitted for inpatient psychiatric care or presenting a clinic, the mental health clinician should be aware of key laboratory and physical exam findings.
Bottom Line
Patients with alcoholic liver disease (ALD) require collaborative care from specialists in addiction, gastroenterology, and psychiatry. Psychiatrists have a role in identifying signs of ALD, prescribing medication to treat alcohol use disorder, and encouraging abstinence. There is some evidence supporting specific medications for varying severity of disease and different phases of recovery. Pharmacotherapy decisions should be made case by case.
Related Resources
• Khan A, Tansel A, White DL, et al. Efficacy of psychosocial interventions in inducing and maintaining alcohol abstinence in patients with chronic liver disease: a systematic review [published online August 6, 2015]. Clin Gastroenterol Hepatol. doi: 10.1016/j.cgh.2015.07.047.
• Vuittonet CL, Halse M, Leggio L, et al. Pharmacotherapy for alcoholic patients with alcoholic liver disease. Am J Health Syst Pharm. 2014;71(15):1265-1276.
Drug Brand Names
Acamprosate • Campral
Baclofen • Lioresal
Disulfiram • Antabuse
Gabapentin • Neurontin
Naltrexone • ReVia, Vivitrol
Pentoxifylline • Trental
Prednisolone • Prelone
Rifaximin • Xifaxan
Topiramate • Topamax
Disclosures
Dr. Winder and Dr. Mellinger report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products. Dr. Fontana receives research funding from Bristol Myers Squibb, Gilead, and Janssen and consults for the Chronic Liver Disease Foundation.
Alcohol use disorder (AUD) is a mosaic of psychiatric and medical symptoms. Alcoholic liver disease (ALD) in its acute and chronic forms is a common clinical consequence of long-standing AUD. Patients with ALD require specialized care from professionals in addiction, gastroenterology, and psychiatry. However, medical specialists treating ALD might not regularly consider medications to treat AUD because of their limited experience with the drugs or the lack of studies in patients with significant liver disease.1 Similarly, psychiatrists might be reticent to prescribe medications for AUD, fearing that liver disease will be made worse or that they will cause other medical complications. As a result, patients with ALD might not receive care that could help treat their AUD (Box).
Given the high worldwide prevalence and morbidity of ALD,2 general and subspecialized psychiatrists routinely evaluate patients with AUD in and out of the hospital. This article aims to equip a psychiatrist with:
• a practical understanding of the natural history and categorization of ALD
• basic skills to detect symptoms of ALD
• preparation to collaborate with medical colleagues in multidisciplinary management of co-occurring AUD and ALD
• a summary of the pharmacotherapeutics of AUD, with emphasis on patients with clinically apparent ALD.
Categorization and clinical features
Alcoholic liver damage encompasses a spectrum of disorders, including alcoholic fatty liver, acute alcohol hepatitis (AH), and cirrhosis following varying durations and patterns of alcohol use. Manifestations of ALD vary from asymptomatic fatty liver with minimal liver enzyme elevation to severe acute AH with jaundice, coagulopathy, and high short-term mortality (Table 1). Symptoms seen in patients with AH include fever, abdominal pain, anorexia, jaundice, leukocytosis, and coagulopathy.3
Patients with chronic ALD often develop cirrhosis, persistent elevation of the serum aminotransferase level (even after prolonged alcohol abstinence), signs of portal hypertension (ascites, encephalopathy, variceal bleeding), and profound malnutrition. The survival of ALD patients with chronic liver failure is predicted in part by a Model for End-Stage Liver Disease (MELD) score that incorporates their serum total bilirubin level, creatinine level, and international normalized ratio. The MELD score, which ranges from 6 to 40, also is used to gauge the need for liver transplantation; most patients who have a MELD score >15 benefit from transplant. To definitively determine the severity of ALD, a liver biopsy is required but usually is not performed in clinical practice.
All patients who drink heavily or suffer with AUD are at risk of developing AH; women and binge drinkers are particularly vulnerable.4 Liver dysfunction and malnutrition in ALD patients compromise the immune system, increasing the risk of infection. Patients hospitalized with AH have a 10% to 30% risk of inpatient mortality; their 1- and 2-month post-discharge survival is 50% to 65%, largely determined by whether the patient can maintain sobriety.5 Psychiatrists’ contribution to ALD treatment therefore has the potential to save lives.
Screening and detection of ALD
Because of the high mortality associated with AH and cirrhosis, symptom recognition and collaborative medical and psychiatric management are critical (Table 2). A psychiatrist evaluating a jaundiced patient who continues to drink should arrange urgent medical evaluation. While gathering a history, mental health providers might hear a patient refer to symptoms of gastrointestinal bleeding (vomiting blood, bloody or dark stool), painful abdominal distension, fevers, or confusion that should prompt a referral to a gastroenterologist or the emergency department. Testing for urinary ethyl glucuronide—a direct metabolite of ethanol that can be detected for as long as 90 hours after ethanol ingestion—is useful in detecting alcohol use in the past 4 or 5 days.
Medical management of ALD
Corticosteroids are a mainstay in pharmacotherapy for severe AH. There is evidence for improved outcomes in patients with severe AH treated with prednisolone for 4 to 6 weeks.5 Prognostic models such as the Maddrey’s Discriminant Function, Lille Model, and the MELD score help determine the need for steroid use and identify high-risk patients. Patients with active infection or bleeding are not a candidate for steroid treatment. An experienced gastroenterologist or hepatologist should initiate medical intervention after thorough evaluation.
Liver transplantation. A select group of patients with refractory liver failure are considered for liver transplantation. Although transplant programs differ in their criteria for organ listing, many require patients to demonstrate at least 6 months of verified abstinence from alcohol and illicit drugs as well as adherence to a formal AUD treatment and rehabilitation plan. The patient’s psychological health and prognosis for sustained sobriety are central to candidacy for organ listing, which highlights the key role of psychiatrists.
Further considerations. Thiamine and folate often are given to patients with ALD. Abdominal imaging and screening for HIV and viral hepatitis—identified in 10% to 20% of ALD patients—is routine. Alcohol abstinence remains central to survival because relapse increases the risk of recurrent, severe liver disease. Regrettably, many physical symptoms of liver disease, such as portal hypertension, ascites, and jaundice, can take months to improve with abstinence.
Treating AUD in patients with ALD
Successful treatment is multifaceted and includes more than just medications. Initial management often includes addressing alcohol withdrawal in dependent patients.6
Behavioral interventions are effective and indispensable components in preventing relapse,7 including a written relapse prevention plan that formally outlines the patient’s commitment to change, identifies triggers, and outlines a discrete plan of action. Primary psychiatric pathology, including depression and anxiety, often are comorbid with AUD; concurrent treatment of these disorders could improve patient outcomes.8
Benzodiazepines often are used during acute alcohol withdrawal. They should not be used for relapse prevention in ALD because of their additive interactions with alcohol, cognitive and psychomotor side effects, and abuse potential.9,10 Many of these drugs are cleared by the liver and generally are not recommended for use in patients with ALD.
Other agents, further considerations. Drug trials in AUD largely have been conducted in small, heterogeneous populations and revealed modest and, at times, conflicting drug effect sizes.6,11,12 The placebo effect among the AUD population is pronounced.6,7,13 Despite these caveats, several agents have been studied and validated by the FDA to treat AUD. Additional agents with promising pilot data are being investigated. Table 31,7,10,11,13-43 summarizes drugs used to treat AUD—those with and without FDA approval—with a focus on how they might be used in patients with ALD. Of note, several of these agents do not rely on the liver for metabolism or excretion.
There is no agreed-upon algorithm or safety profile to guide a prescriber’s decision making about drug or dosage choices when treating AUD in patients with ALD. Because liver function can vary among patients as well as during an individual patient’s disease course, treatment decisions should be made on a clinical, collaborative, and case-by-case basis.
That being said, the AUD treatment literature suggests that specific drugs might be more useful in patients with varying severity of disease and during different phases of recovery:
• Acamprosate has been found to be effective in supporting abstinence in sober patients.14,44
• Naltrexone has been shown to be useful in patients with severe alcohol cravings. By modulating alcohol’s rewarding effects, naltrexone also reduces heavy alcohol consumption in patients who are drinking.14,15,44
• Disulfiram generally is not recommended for use in patients with clinically apparent hepatic insufficiency, such as decompensated cirrhosis or preexisting jaundice.
Although alcohol abstinence remains the treatment goal and a requirement for liver transplant, providers must recognize that some patients might not be able to maintain long-term sobriety. Therefore, harm reduction models are important companions to abstinence-only models of AUD treatment.45 The array of behavioral, pharmacological, and philosophical approaches to AUD treatment underlines the need for an individualized approach to relapse prevention.
Collaboration between medicine and psychiatry
When AUD and ALD are comorbid, psychiatrists might worry about making the patient’s medical condition worse by prescribing additional psychoactive medications—particularly ones that are cleared by the liver. Remember that AUD confers a substantial mortality rate that is more than 3 times that of the general population, along with severe medical46 and psychosocial31 effects. Although prescribers must remain vigilant for adverse drug effects, medications easily can be blamed for what might be the natural progression and symptoms of AUD in patients with ALD.26 This erroneous conclusion can lead to premature medication discontinuation and under-treatment of AUD.
In the end, keeping the patient sober and mentally well might be more beneficial than eliminating the burden of any medication side effects. Collaborative medical and psychiatric management of ALD patients can ensure that clinicians properly weigh the risks, benefits, and duration of treatment unique to each patient.
Starting AUD treatment promptly after alcohol relapse is essential and entails a multidisciplinary effort between medicine and psychiatry, both in and out of the hospital. Because the relapsing, ill ALD patient most often will be admitted to a medical specialist, AUD might not receive enough attention during the medical admission. Psychiatrists can help in initiating AUD treatment in the acute medical setting, which has been shown to improve the outpatient course.6 For medically stable ALD patients admitted for inpatient psychiatric care or presenting a clinic, the mental health clinician should be aware of key laboratory and physical exam findings.
Bottom Line
Patients with alcoholic liver disease (ALD) require collaborative care from specialists in addiction, gastroenterology, and psychiatry. Psychiatrists have a role in identifying signs of ALD, prescribing medication to treat alcohol use disorder, and encouraging abstinence. There is some evidence supporting specific medications for varying severity of disease and different phases of recovery. Pharmacotherapy decisions should be made case by case.
Related Resources
• Khan A, Tansel A, White DL, et al. Efficacy of psychosocial interventions in inducing and maintaining alcohol abstinence in patients with chronic liver disease: a systematic review [published online August 6, 2015]. Clin Gastroenterol Hepatol. doi: 10.1016/j.cgh.2015.07.047.
• Vuittonet CL, Halse M, Leggio L, et al. Pharmacotherapy for alcoholic patients with alcoholic liver disease. Am J Health Syst Pharm. 2014;71(15):1265-1276.
Drug Brand Names
Acamprosate • Campral
Baclofen • Lioresal
Disulfiram • Antabuse
Gabapentin • Neurontin
Naltrexone • ReVia, Vivitrol
Pentoxifylline • Trental
Prednisolone • Prelone
Rifaximin • Xifaxan
Topiramate • Topamax
Disclosures
Dr. Winder and Dr. Mellinger report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products. Dr. Fontana receives research funding from Bristol Myers Squibb, Gilead, and Janssen and consults for the Chronic Liver Disease Foundation.
1. Gache P, Hadengue A. Baclofen improves abstinence in alcoholic cirrhosis: still better to come? J Hepatol. 2008;49(6):1083-1085.
2. Rehm J, Mathers C, Popova S, et al. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet. 2009;373(9682):2223-2233.
3. Singal AK, Kamath PS, Gores GJ, et al. Alcoholic hepatitis: current challenges and future directions. Clin Gastroenterol Hepatol. 2014;12(4):555-564; quiz e31-32.
4. Becker U, Deis A, Sørensen TI, et al. Prediction of risk of liver disease by alcohol intake, sex, and age: a prospective population study. Hepatology. 1996;23(5):1025-1029.
5. Mathurin P, Lucey MR. Management of alcoholic hepatitis. J Hepatol. 2012;56(suppl 1):S39-S45.
6. Mann K, Lemenager T, Hoffmann S, et al; PREDICT Study Team. Results of a double-blind, placebo-controlled pharmacotherapy trial in alcoholism conducted in Germany and comparison with the US COMBINE study. Addict Biol. 2013;18(6):937-946.
7. Anton RF, O’Malley SS, Ciraulo DA, et al; COMBINE Study Research Group. Combined pharmacotherapies and behavioral interventions for alcohol dependence: the COMBINE study: a randomized controlled trial. JAMA. 2006;295(17):2003-2017.
8. Kranzler HR, Rosenthal RN. Dual diagnosis: alcoholism and co-morbid psychiatric disorders. Am J Addict. 2003;12(suppl 1):S26-S40.
9. Book SW, Myrick H. Novel anticonvulsants in the treatment of alcoholism. Expert Opin Investig Drugs. 2005;14(4):371-376.
10. Furieri FA, Nakamura-Palacios EM. Gabapentin reduces alcohol consumption and craving: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2007;68(11):1691-1700.
11. Blodgett JC, Del Re AC, Maisel NC, et al. A meta-analysis of topiramate’s effects for individuals with alcohol use disorders. Alcohol Clin Exp Res. 2014;38(6):1481-1488.
12. Krystal JH, Cramer JA, Krol WF, et al; Veterans Affairs Naltrexone Cooperative Study 425 Group. Naltrexone in the treatment of alcohol dependence. N Engl J Med. 2001;345(24):1734-1739.
13. Petrakis IL, Poling J, Levinson C, et al; VA New England VISN I MIRECC Study Group. Naltrexone and disulfiram in patients with alcohol dependence and comorbid psychiatric disorders. Biol Psychiatry. 2005;57(10):1128-1137.
14. Maisel NC, Blodgett JC, Wilbourne PL, et al. Meta-analysis of naltrexone and acamprosate for treating alcohol use disorders: when are these medications most helpful? Addiction. 2013;108(2):275-293.
15. Pettinati HM, O’Brien CP, Rabinowitz AR, et al. The status of naltrexone in the treatment of alcohol dependence: specific effects on heavy drinking. J Clin Psychopharmacol. 2006;26(6):610-625.
16. Anton RF, Myrick H, Wright TM, et al. Gabapentin combined with naltrexone for the treatment of alcohol dependence. Am J Psychiatry. 2011;168(7):709-717.
17. Srisurapanont M, Jarusuraisin N. Opioid antagonists for alcohol dependence. Cochrane Database Syst Rev. 2005(1):CD001867.
18. Naltrexone. 2014. http://www.micromedexsolutions.com. Accessed January 31, 2015.
19. Soyka M, Chick J. Use of acamprosate and opioid antagonists in the treatment of alcohol dependence: a European perspective. Am J Addict. 2003;12(suppl 1):S69-S80.
20. Turncliff RZ, Dunbar JL, Dong Q, et al. Pharmacokinetics of long-acting naltrexone in subjects with mild to moderate hepatic impairment. J Clin Pharmacol. 2005;45(11):1259-1267.
21. United States National Library of Medicine. Naltrexone. http://livertox.nlm.nih.gov/Naltrexone.htm. Updated September 30, 2015. Accessed November 10, 2015.
22. Terg R, Coronel E, Sordá J, et al. Efficacy and safety of oral naltrexone treatment for pruritus of cholestasis, a crossover, double blind, placebo-controlled study. J Hepatol. 2002;37(6):717-722.
23. Skinner MD, Lahmek P, Pham H, et al. Disulfiram efficacy in the treatment of alcohol dependence: a meta-analysis [published online February 10, 2014]. PLoS One. 2014;9(2):e87366. doi: 10.1371/journal.pone.0087366.
24. Disulfiram. 2014. http://www.micromedexsolutions.com. Accessed January 31, 2015.
25. Björnsson E, Nordlinder H, Olsson R. Clinical characteristics and prognostic markers in disulfiram-induced liver injury. J Hepatol. 2006;44(4):791-797.
26. Chick J. Safety issues concerning the use of disulfiram in treating alcohol dependence. Drug Saf. 1999;20(5):427-435.
27. Campral [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2012.
28. Brower KJ, Myra Kim H, Strobbe S, et al. A randomized double-blind pilot trial of gabapentin versus placebo to treat alcohol dependence and comorbid insomnia. Alcohol Clin Exp Res. 2008;32(8):1429-1438.
29. Mason BJ, Quello S, Goodell V, et al. Gabapentin treatment for alcohol dependence: a randomized clinical trial. JAMA Intern Med. 2014;174(1):70-77.
30. Neurontin [package insert]. New York, NY: Pfizer; 2015.
31. Johnson BA, Ait-Daoud N, Akhtar FZ, et al. Oral topiramate reduces the consequences of drinking and improves the quality of life of alcohol-dependent individuals: a randomized controlled trial. Arch Gen Psychiatry. 2004;61(9):905-912.
32. Paparrigopoulos T, Tzavellas E, Karaiskos D, et al. Treatment of alcohol dependence with low-dose topiramate: an open-label controlled study. BMC Psychiatry. 2011;11:41.
33. Rubio G, Ponce G, Jiménez-Arriero MA, et al. Effects of topiramate in the treatment of alcohol dependence. Pharmacopsychiatry. 2004;37(1):37-40.
34. Topamax [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2009.
35. De Sousa AA, De Sousa J, Kapoor H. An open randomized trial comparing disulfiram and topiramate in the treatment of alcohol dependence. J Subst Abuse Treat. 2008;34(4):460-463.
36. Kampman KM, Pettinati HM, Lynch KG, et al. A double-blind, placebo-controlled trial of topiramate for the treatment of comorbid cocaine and alcohol dependence. Drug Alcohol Depend. 2013;133(1):94-99.
37. Addolorato G, Leggio L, Ferrulli A, et al. Dose-response effect of baclofen in reducing daily alcohol intake in alcohol dependence: secondary analysis of a randomized, double-blind, placebo-controlled trial. Alcohol Alcohol. 2011;46(3):312-317.
38. Balcofen [package insert]. Concord, NC: McKesson Packing Services; 2013.
39. United States National Library of Medicine. Baclofen. 2015. http://livertox.nlm.nih.gov/Baclofen.htm. Accessed November 7, 2015.
40. Addolorato G, Leggio L, Ferrulli A, et al. Effectiveness and safety of baclofen for maintenance of alcohol abstinence in alcohol-dependent patients with liver cirrhosis: randomised, double-blind controlled study. Lancet. 2007;370(9603):1915-1922.
41. Leggio L, Ferrulli A, Zambon A, et al. Baclofen promotes alcohol abstinence in alcohol dependent cirrhotic patients with hepatitis C virus (HCV) infection. Addict Behav. 2012;37(4):561-564.
42. Franchitto N, Pelissier F, Lauque D, et al. Self-intoxication with baclofen in alcohol-dependent patients with co-existing psychiatric illness: an emergency department case series. Alcohol Alcohol. 2014;49(1):79-83.
43. Brennan JL, Leung JG, Gagliardi JP, et al. Clinical effectiveness of baclofen for the treatment of alcohol dependence: a review. Clin Pharmacol. 2013;5:99-107.
44. Rösner S, Leucht S, Lehert P, et al. Acamprosate supports abstinence, naltrexone prevents excessive drinking: evidence from a meta-analysis with unreported outcomes. J Psychopharmacol. 2008;22(1):11-23.
45. Marlatt GA, Witkiewitz K. Harm reduction approaches to alcohol use: health promotion, prevention, and treatment. Addict Behav. 2002;27(6):867-886.
46. O’Shea RS, Dasarathy S, McCullough AJ. Alcoholic liver disease. Am J Gastroenterol. 2010;105(1):14-32; quiz 33.
1. Gache P, Hadengue A. Baclofen improves abstinence in alcoholic cirrhosis: still better to come? J Hepatol. 2008;49(6):1083-1085.
2. Rehm J, Mathers C, Popova S, et al. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet. 2009;373(9682):2223-2233.
3. Singal AK, Kamath PS, Gores GJ, et al. Alcoholic hepatitis: current challenges and future directions. Clin Gastroenterol Hepatol. 2014;12(4):555-564; quiz e31-32.
4. Becker U, Deis A, Sørensen TI, et al. Prediction of risk of liver disease by alcohol intake, sex, and age: a prospective population study. Hepatology. 1996;23(5):1025-1029.
5. Mathurin P, Lucey MR. Management of alcoholic hepatitis. J Hepatol. 2012;56(suppl 1):S39-S45.
6. Mann K, Lemenager T, Hoffmann S, et al; PREDICT Study Team. Results of a double-blind, placebo-controlled pharmacotherapy trial in alcoholism conducted in Germany and comparison with the US COMBINE study. Addict Biol. 2013;18(6):937-946.
7. Anton RF, O’Malley SS, Ciraulo DA, et al; COMBINE Study Research Group. Combined pharmacotherapies and behavioral interventions for alcohol dependence: the COMBINE study: a randomized controlled trial. JAMA. 2006;295(17):2003-2017.
8. Kranzler HR, Rosenthal RN. Dual diagnosis: alcoholism and co-morbid psychiatric disorders. Am J Addict. 2003;12(suppl 1):S26-S40.
9. Book SW, Myrick H. Novel anticonvulsants in the treatment of alcoholism. Expert Opin Investig Drugs. 2005;14(4):371-376.
10. Furieri FA, Nakamura-Palacios EM. Gabapentin reduces alcohol consumption and craving: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2007;68(11):1691-1700.
11. Blodgett JC, Del Re AC, Maisel NC, et al. A meta-analysis of topiramate’s effects for individuals with alcohol use disorders. Alcohol Clin Exp Res. 2014;38(6):1481-1488.
12. Krystal JH, Cramer JA, Krol WF, et al; Veterans Affairs Naltrexone Cooperative Study 425 Group. Naltrexone in the treatment of alcohol dependence. N Engl J Med. 2001;345(24):1734-1739.
13. Petrakis IL, Poling J, Levinson C, et al; VA New England VISN I MIRECC Study Group. Naltrexone and disulfiram in patients with alcohol dependence and comorbid psychiatric disorders. Biol Psychiatry. 2005;57(10):1128-1137.
14. Maisel NC, Blodgett JC, Wilbourne PL, et al. Meta-analysis of naltrexone and acamprosate for treating alcohol use disorders: when are these medications most helpful? Addiction. 2013;108(2):275-293.
15. Pettinati HM, O’Brien CP, Rabinowitz AR, et al. The status of naltrexone in the treatment of alcohol dependence: specific effects on heavy drinking. J Clin Psychopharmacol. 2006;26(6):610-625.
16. Anton RF, Myrick H, Wright TM, et al. Gabapentin combined with naltrexone for the treatment of alcohol dependence. Am J Psychiatry. 2011;168(7):709-717.
17. Srisurapanont M, Jarusuraisin N. Opioid antagonists for alcohol dependence. Cochrane Database Syst Rev. 2005(1):CD001867.
18. Naltrexone. 2014. http://www.micromedexsolutions.com. Accessed January 31, 2015.
19. Soyka M, Chick J. Use of acamprosate and opioid antagonists in the treatment of alcohol dependence: a European perspective. Am J Addict. 2003;12(suppl 1):S69-S80.
20. Turncliff RZ, Dunbar JL, Dong Q, et al. Pharmacokinetics of long-acting naltrexone in subjects with mild to moderate hepatic impairment. J Clin Pharmacol. 2005;45(11):1259-1267.
21. United States National Library of Medicine. Naltrexone. http://livertox.nlm.nih.gov/Naltrexone.htm. Updated September 30, 2015. Accessed November 10, 2015.
22. Terg R, Coronel E, Sordá J, et al. Efficacy and safety of oral naltrexone treatment for pruritus of cholestasis, a crossover, double blind, placebo-controlled study. J Hepatol. 2002;37(6):717-722.
23. Skinner MD, Lahmek P, Pham H, et al. Disulfiram efficacy in the treatment of alcohol dependence: a meta-analysis [published online February 10, 2014]. PLoS One. 2014;9(2):e87366. doi: 10.1371/journal.pone.0087366.
24. Disulfiram. 2014. http://www.micromedexsolutions.com. Accessed January 31, 2015.
25. Björnsson E, Nordlinder H, Olsson R. Clinical characteristics and prognostic markers in disulfiram-induced liver injury. J Hepatol. 2006;44(4):791-797.
26. Chick J. Safety issues concerning the use of disulfiram in treating alcohol dependence. Drug Saf. 1999;20(5):427-435.
27. Campral [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2012.
28. Brower KJ, Myra Kim H, Strobbe S, et al. A randomized double-blind pilot trial of gabapentin versus placebo to treat alcohol dependence and comorbid insomnia. Alcohol Clin Exp Res. 2008;32(8):1429-1438.
29. Mason BJ, Quello S, Goodell V, et al. Gabapentin treatment for alcohol dependence: a randomized clinical trial. JAMA Intern Med. 2014;174(1):70-77.
30. Neurontin [package insert]. New York, NY: Pfizer; 2015.
31. Johnson BA, Ait-Daoud N, Akhtar FZ, et al. Oral topiramate reduces the consequences of drinking and improves the quality of life of alcohol-dependent individuals: a randomized controlled trial. Arch Gen Psychiatry. 2004;61(9):905-912.
32. Paparrigopoulos T, Tzavellas E, Karaiskos D, et al. Treatment of alcohol dependence with low-dose topiramate: an open-label controlled study. BMC Psychiatry. 2011;11:41.
33. Rubio G, Ponce G, Jiménez-Arriero MA, et al. Effects of topiramate in the treatment of alcohol dependence. Pharmacopsychiatry. 2004;37(1):37-40.
34. Topamax [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2009.
35. De Sousa AA, De Sousa J, Kapoor H. An open randomized trial comparing disulfiram and topiramate in the treatment of alcohol dependence. J Subst Abuse Treat. 2008;34(4):460-463.
36. Kampman KM, Pettinati HM, Lynch KG, et al. A double-blind, placebo-controlled trial of topiramate for the treatment of comorbid cocaine and alcohol dependence. Drug Alcohol Depend. 2013;133(1):94-99.
37. Addolorato G, Leggio L, Ferrulli A, et al. Dose-response effect of baclofen in reducing daily alcohol intake in alcohol dependence: secondary analysis of a randomized, double-blind, placebo-controlled trial. Alcohol Alcohol. 2011;46(3):312-317.
38. Balcofen [package insert]. Concord, NC: McKesson Packing Services; 2013.
39. United States National Library of Medicine. Baclofen. 2015. http://livertox.nlm.nih.gov/Baclofen.htm. Accessed November 7, 2015.
40. Addolorato G, Leggio L, Ferrulli A, et al. Effectiveness and safety of baclofen for maintenance of alcohol abstinence in alcohol-dependent patients with liver cirrhosis: randomised, double-blind controlled study. Lancet. 2007;370(9603):1915-1922.
41. Leggio L, Ferrulli A, Zambon A, et al. Baclofen promotes alcohol abstinence in alcohol dependent cirrhotic patients with hepatitis C virus (HCV) infection. Addict Behav. 2012;37(4):561-564.
42. Franchitto N, Pelissier F, Lauque D, et al. Self-intoxication with baclofen in alcohol-dependent patients with co-existing psychiatric illness: an emergency department case series. Alcohol Alcohol. 2014;49(1):79-83.
43. Brennan JL, Leung JG, Gagliardi JP, et al. Clinical effectiveness of baclofen for the treatment of alcohol dependence: a review. Clin Pharmacol. 2013;5:99-107.
44. Rösner S, Leucht S, Lehert P, et al. Acamprosate supports abstinence, naltrexone prevents excessive drinking: evidence from a meta-analysis with unreported outcomes. J Psychopharmacol. 2008;22(1):11-23.
45. Marlatt GA, Witkiewitz K. Harm reduction approaches to alcohol use: health promotion, prevention, and treatment. Addict Behav. 2002;27(6):867-886.
46. O’Shea RS, Dasarathy S, McCullough AJ. Alcoholic liver disease. Am J Gastroenterol. 2010;105(1):14-32; quiz 33.

Think beyond prazosin when treating nightmares in PTSD
Nightmares are a common feature of posttraumatic stress disorder (PTSD) that could lead to fatigue, impaired concentration, and poor work performance. The α-1 antagonist prazosin decreases noradrenergic hyperactivity and reduces nightmares; however, it can cause adverse effects, be contraindicated, or provide no benefit to some patients. Consider these alternative medications to reduce nightmares in PTSD.
Alpha-2 agonists
Clonidine and guanfacine are α-2 agonists, used to treat attention-deficit/hyperactivity disorder and high blood pressure, that decrease noradrenergic activity, and either medication might be preferable to prazosin because they are more likely to cause sedation. A review and a case series showed that many patients—some with comorbid traumatic brain injury—reported fewer nightmares after taking 0.2 to 0.6 mg of clonidine.1,2 Guanfacine might be more beneficial because it has a longer half-life; 2 mg of guanfacine eliminated nightmares in 1 patient.3 However, in a double-blind placebo-controlled study and an extension study, guanfacine did not reduce nightmares or other PTSD symptoms.4,5
Initiate 0.1 mg of clonidine at bedtime, and titrate to efficacy or to 0.6 mg. Similarly, initiate guanfacine at 1 mg, and titrate to efficacy or to 4 mg. Monitor for hypotension, excess sedation, dry mouth, and rebound hypertension.
Cyproheptadine
Used to treat serotonin syndrome, cyproheptadine’s antagonism of serotonin 2A receptors has varying efficacy for reducing nightmares. Some patients have reported a decrease in nightmares at dosages ranging from 4 to 24 mg.1,6 Other studies found no reduction in nightmares or diminished quality of sleep.1,7
Initiate cyproheptadine at 4 mg/d, titrate every 2 or 3 days, and monitor for sedation, confusion, or reduced efficacy of concurrent serotonergic medications. Cyproheptadine might be preferable for its sedating effect and potential to reduce sexual adverse effects from serotonergic medications.
Topiramate
Topiramate is approved for treatment of epilepsy and migraine headache. At 75 to 100 mg/d in a clinical trial, topiramate partially or completely suppressed nightmares.8 Start with 25 mg/d, titrate to efficacy, and monitor for anorexia, paresthesias, and cognitive impairment. Topiramate might be better than prazosin for patients without renal impairment who want sedation, weight loss, or reduced irritability.
Gabapentin
Gabapentin is approved to treat seizures and postherpetic neuralgia and also is used to treat neuropathic pain. When 300 to 3,600 mg/d (mean dosage, 1,300 mg/d) of gabapentin was added to medication regimens, most patients reported decreased frequency or intensity of nightmares.9 Monitor patients for sedation, dizziness, mood changes, and weight gain. Gabapentin might be an option for patients without renal impairment who have comorbid pain, insomnia, or anxiety.
Are these reasonable alternatives?
Despite small sample sizes in published studies and few randomized trials, clonidine, guanfacine, cyproheptadine, topiramate, and gabapentin are reasonable alternatives to prazosin for reducing nightmares in patients with PTSD.
Disclosure
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Aurora RN, Zak RS, Auerbach SH, et al; Standards of Practice Committee; American Academy of Sleep Medicine. Best practice guide for the treatment of nightmare disorder in adults. J Clin Sleep Med. 2010;6(4):389-401.
2. Alao A, Selvarajah J, Razi S. The use of clonidine in the treatment of nightmares among patients with co-morbid PTSD and traumatic brain injury. Int J Psychiatry Med. 2012;44(2):165-169.
3. Horrigan JP, Barnhill LJ. The suppression of nightmares with guanfacine. J Clin Psychiatry. 1996;57(8):371.
4. Davis LL, Ward C, Rasmusson A, et al. A placebo-controlled trial of guanfacine for the treatment of posttraumatic stress disorder in veterans. Psychopharmacol Bull. 2008;41(1):8-18.
5. Neylan TC, Lenoci M, Samuelson KW, et al. No improvement of posttraumatic stress disorder symptoms with guanfacine treatment. Am J Psychiatry. 2006;163(12):2186-2188.
6. Harsch HH. Cyproheptadine for recurrent nightmares. Am J Psychiatry. 1986;143(11):1491-1492.
7. Jacobs-Rebhun S, Schnurr PP, Friedman MJ, et al. Posttraumatic stress disorder and sleep difficulty. Am J Psychiatry. 2000;157(9):1525-1526.
8. Berlant J, van Kammen DP. Open-label topiramate as primary or adjunctive therapy in chronic civilian posttraumatic stress disorder: a preliminary report. J Clin Psychiatry. 2002;63(1):15-20.
9. Hamner MB, Brodrick PS, Labbate LA. Gabapentin in PTSD: a retrospective, clinical series of adjunctive therapy. Ann Clin Psychiatry. 2001;13(3):141-146.
Nightmares are a common feature of posttraumatic stress disorder (PTSD) that could lead to fatigue, impaired concentration, and poor work performance. The α-1 antagonist prazosin decreases noradrenergic hyperactivity and reduces nightmares; however, it can cause adverse effects, be contraindicated, or provide no benefit to some patients. Consider these alternative medications to reduce nightmares in PTSD.
Alpha-2 agonists
Clonidine and guanfacine are α-2 agonists, used to treat attention-deficit/hyperactivity disorder and high blood pressure, that decrease noradrenergic activity, and either medication might be preferable to prazosin because they are more likely to cause sedation. A review and a case series showed that many patients—some with comorbid traumatic brain injury—reported fewer nightmares after taking 0.2 to 0.6 mg of clonidine.1,2 Guanfacine might be more beneficial because it has a longer half-life; 2 mg of guanfacine eliminated nightmares in 1 patient.3 However, in a double-blind placebo-controlled study and an extension study, guanfacine did not reduce nightmares or other PTSD symptoms.4,5
Initiate 0.1 mg of clonidine at bedtime, and titrate to efficacy or to 0.6 mg. Similarly, initiate guanfacine at 1 mg, and titrate to efficacy or to 4 mg. Monitor for hypotension, excess sedation, dry mouth, and rebound hypertension.
Cyproheptadine
Used to treat serotonin syndrome, cyproheptadine’s antagonism of serotonin 2A receptors has varying efficacy for reducing nightmares. Some patients have reported a decrease in nightmares at dosages ranging from 4 to 24 mg.1,6 Other studies found no reduction in nightmares or diminished quality of sleep.1,7
Initiate cyproheptadine at 4 mg/d, titrate every 2 or 3 days, and monitor for sedation, confusion, or reduced efficacy of concurrent serotonergic medications. Cyproheptadine might be preferable for its sedating effect and potential to reduce sexual adverse effects from serotonergic medications.
Topiramate
Topiramate is approved for treatment of epilepsy and migraine headache. At 75 to 100 mg/d in a clinical trial, topiramate partially or completely suppressed nightmares.8 Start with 25 mg/d, titrate to efficacy, and monitor for anorexia, paresthesias, and cognitive impairment. Topiramate might be better than prazosin for patients without renal impairment who want sedation, weight loss, or reduced irritability.
Gabapentin
Gabapentin is approved to treat seizures and postherpetic neuralgia and also is used to treat neuropathic pain. When 300 to 3,600 mg/d (mean dosage, 1,300 mg/d) of gabapentin was added to medication regimens, most patients reported decreased frequency or intensity of nightmares.9 Monitor patients for sedation, dizziness, mood changes, and weight gain. Gabapentin might be an option for patients without renal impairment who have comorbid pain, insomnia, or anxiety.
Are these reasonable alternatives?
Despite small sample sizes in published studies and few randomized trials, clonidine, guanfacine, cyproheptadine, topiramate, and gabapentin are reasonable alternatives to prazosin for reducing nightmares in patients with PTSD.
Disclosure
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Nightmares are a common feature of posttraumatic stress disorder (PTSD) that could lead to fatigue, impaired concentration, and poor work performance. The α-1 antagonist prazosin decreases noradrenergic hyperactivity and reduces nightmares; however, it can cause adverse effects, be contraindicated, or provide no benefit to some patients. Consider these alternative medications to reduce nightmares in PTSD.
Alpha-2 agonists
Clonidine and guanfacine are α-2 agonists, used to treat attention-deficit/hyperactivity disorder and high blood pressure, that decrease noradrenergic activity, and either medication might be preferable to prazosin because they are more likely to cause sedation. A review and a case series showed that many patients—some with comorbid traumatic brain injury—reported fewer nightmares after taking 0.2 to 0.6 mg of clonidine.1,2 Guanfacine might be more beneficial because it has a longer half-life; 2 mg of guanfacine eliminated nightmares in 1 patient.3 However, in a double-blind placebo-controlled study and an extension study, guanfacine did not reduce nightmares or other PTSD symptoms.4,5
Initiate 0.1 mg of clonidine at bedtime, and titrate to efficacy or to 0.6 mg. Similarly, initiate guanfacine at 1 mg, and titrate to efficacy or to 4 mg. Monitor for hypotension, excess sedation, dry mouth, and rebound hypertension.
Cyproheptadine
Used to treat serotonin syndrome, cyproheptadine’s antagonism of serotonin 2A receptors has varying efficacy for reducing nightmares. Some patients have reported a decrease in nightmares at dosages ranging from 4 to 24 mg.1,6 Other studies found no reduction in nightmares or diminished quality of sleep.1,7
Initiate cyproheptadine at 4 mg/d, titrate every 2 or 3 days, and monitor for sedation, confusion, or reduced efficacy of concurrent serotonergic medications. Cyproheptadine might be preferable for its sedating effect and potential to reduce sexual adverse effects from serotonergic medications.
Topiramate
Topiramate is approved for treatment of epilepsy and migraine headache. At 75 to 100 mg/d in a clinical trial, topiramate partially or completely suppressed nightmares.8 Start with 25 mg/d, titrate to efficacy, and monitor for anorexia, paresthesias, and cognitive impairment. Topiramate might be better than prazosin for patients without renal impairment who want sedation, weight loss, or reduced irritability.
Gabapentin
Gabapentin is approved to treat seizures and postherpetic neuralgia and also is used to treat neuropathic pain. When 300 to 3,600 mg/d (mean dosage, 1,300 mg/d) of gabapentin was added to medication regimens, most patients reported decreased frequency or intensity of nightmares.9 Monitor patients for sedation, dizziness, mood changes, and weight gain. Gabapentin might be an option for patients without renal impairment who have comorbid pain, insomnia, or anxiety.
Are these reasonable alternatives?
Despite small sample sizes in published studies and few randomized trials, clonidine, guanfacine, cyproheptadine, topiramate, and gabapentin are reasonable alternatives to prazosin for reducing nightmares in patients with PTSD.
Disclosure
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Aurora RN, Zak RS, Auerbach SH, et al; Standards of Practice Committee; American Academy of Sleep Medicine. Best practice guide for the treatment of nightmare disorder in adults. J Clin Sleep Med. 2010;6(4):389-401.
2. Alao A, Selvarajah J, Razi S. The use of clonidine in the treatment of nightmares among patients with co-morbid PTSD and traumatic brain injury. Int J Psychiatry Med. 2012;44(2):165-169.
3. Horrigan JP, Barnhill LJ. The suppression of nightmares with guanfacine. J Clin Psychiatry. 1996;57(8):371.
4. Davis LL, Ward C, Rasmusson A, et al. A placebo-controlled trial of guanfacine for the treatment of posttraumatic stress disorder in veterans. Psychopharmacol Bull. 2008;41(1):8-18.
5. Neylan TC, Lenoci M, Samuelson KW, et al. No improvement of posttraumatic stress disorder symptoms with guanfacine treatment. Am J Psychiatry. 2006;163(12):2186-2188.
6. Harsch HH. Cyproheptadine for recurrent nightmares. Am J Psychiatry. 1986;143(11):1491-1492.
7. Jacobs-Rebhun S, Schnurr PP, Friedman MJ, et al. Posttraumatic stress disorder and sleep difficulty. Am J Psychiatry. 2000;157(9):1525-1526.
8. Berlant J, van Kammen DP. Open-label topiramate as primary or adjunctive therapy in chronic civilian posttraumatic stress disorder: a preliminary report. J Clin Psychiatry. 2002;63(1):15-20.
9. Hamner MB, Brodrick PS, Labbate LA. Gabapentin in PTSD: a retrospective, clinical series of adjunctive therapy. Ann Clin Psychiatry. 2001;13(3):141-146.
1. Aurora RN, Zak RS, Auerbach SH, et al; Standards of Practice Committee; American Academy of Sleep Medicine. Best practice guide for the treatment of nightmare disorder in adults. J Clin Sleep Med. 2010;6(4):389-401.
2. Alao A, Selvarajah J, Razi S. The use of clonidine in the treatment of nightmares among patients with co-morbid PTSD and traumatic brain injury. Int J Psychiatry Med. 2012;44(2):165-169.
3. Horrigan JP, Barnhill LJ. The suppression of nightmares with guanfacine. J Clin Psychiatry. 1996;57(8):371.
4. Davis LL, Ward C, Rasmusson A, et al. A placebo-controlled trial of guanfacine for the treatment of posttraumatic stress disorder in veterans. Psychopharmacol Bull. 2008;41(1):8-18.
5. Neylan TC, Lenoci M, Samuelson KW, et al. No improvement of posttraumatic stress disorder symptoms with guanfacine treatment. Am J Psychiatry. 2006;163(12):2186-2188.
6. Harsch HH. Cyproheptadine for recurrent nightmares. Am J Psychiatry. 1986;143(11):1491-1492.
7. Jacobs-Rebhun S, Schnurr PP, Friedman MJ, et al. Posttraumatic stress disorder and sleep difficulty. Am J Psychiatry. 2000;157(9):1525-1526.
8. Berlant J, van Kammen DP. Open-label topiramate as primary or adjunctive therapy in chronic civilian posttraumatic stress disorder: a preliminary report. J Clin Psychiatry. 2002;63(1):15-20.
9. Hamner MB, Brodrick PS, Labbate LA. Gabapentin in PTSD: a retrospective, clinical series of adjunctive therapy. Ann Clin Psychiatry. 2001;13(3):141-146.
What to do when adolescents with ADHD self-medicate with bath salts
Designer drugs are rapidly making inroads with young people, primarily because of easier access, lower overall cost, and nebulous legality. These drugs are made as variants of illicit drugs or new formulations and sold as “research chemicals” and labeled as “not for human consumption,” which allows them to fall outside existing laws. The ingredients typically are not detected in a urine drug screen.
Notoriously addictive, these designer drugs, such as bath salts, are known to incorporate synthetic cathinones—namely, methylone, mephedrone or methylenedioxypyrovalerone (MDPV). The stimulant, amphetamine-like effects of bath salts make the drug attractive to adolescents with attention-deficit/hyperactivity disorder (ADHD).
Why do teens gravitate toward bath salts?
Adolescents with undiagnosed ADHD might self-medicate with drugs that are suited for addressing restlessness, intrapsychic turmoil, and other symptoms of ADHD. In 2 case studies, using the self-medication hypothesis, people with ADHD were more likely to seek cocaine by means of “self-selection.”1 These drug-seeking behaviors often led to cocaine dependence, even when other substances, such as alcohol or Cannabis, were available.
Methylphenidate and other ADHD pharmacotherapies influence the nucleus accumbens in a manner similar to that of cocaine. These findings suggest that adolescents with ADHD and cocaine dependence might respond to therapeutic interventions that substitute cocaine with psychostimulants.1
Bath salts fall within the same spectrum of psychostimulant agents as methylphenidate and cocaine. MDPV approximates the effect of methylphenidate at low doses, and cocaine at higher doses. It often is marketed under the name “Ivory Wave” and could be confused with cocaine. Self-administration of MDPV can induce psychoactive effects that help alleviate ADHD symptoms; adolescents might continue to experience enhanced concentration and overall performance.2 Also, because of the low cost of “legal” bath salts, they are an appealing alternative to cocaine for self-medication.
Managing the sequelae of bath salt intoxication
Bath salts may produce sympathomimetic effects greater than cocaine, which require a proactive approach to symptom management. A medley of unknown ingredients in bath salt preparations makes it difficult for clinicians to gauge the pharmacological impact on individual patients; therefore, therapeutic interventions are on a case-by-case basis. However, emergencies concerning amphetamines and amphetamine analogues and derivatives often have similar presentations.
Cardiovascular effects. MDPV-specific urine and blood tests conducted on patients admitted to the emergency room showed a 10-fold increase in overall dopamine levels compared with those who took cocaine. As a sympathomimetic, high doses of dopamine are responsible for raising blood pressure and could lead to the development of pronounced cardiovascular effects.3,4
Agitation. Clinicians generally are advised to treat agitation before providing a more comprehensive assessment of symptoms. Endotracheal intubation often is a required for adequate control of agitation. Bath salt-induced agitation often is treated with IV benzodiazepines.4,5 Monitor patients for excessive sedation or new-onset “paradoxical agitation” as a function of ongoing benzo-diazepine therapy. Clinicians also may choose to co-administer an antipsychotic with benzodiazepines, although the practice is not universally encouraged for agitation control.
Mephedrone produces a delirious state in conjunction with psychotic symptoms. Antipsychotic therapy has been suggested for addressing ongoing agitation.6
Tachycardia. Symptomatic treatment of tachycardia involves beta blockers, such as labetalol. Nitroglycerine has evidence of efficacy for chest pain associated with cocaine intoxication; however, it is unclear whether it is effective for similar drugs of abuse.4
Multi-organ collapse caused by MDPV necessitates aggressive intervention, including prompt dialysis. Carefully evaluate the patient for the presence of organ-specific insults and initiate supportive measures accordingly. Pronounced agitation with hyperthermia might portend severely compromised renal, hepatic, and/or cardiac function in MDPV users.7 Those who present with MDPV intoxication and concomitant renal injury seem to benefit from hemodialysis.8 Repeat intoxication events may yield a presentation of acute renal injury replete with metabolic derangements, including metabolic acidosis, hyperuricemia, and rhabdomyolysis.9 Thorough patient assessments and interventions are useful in determining long-term outcomes, including issues pertaining to mortality.
Confronting an epidemic
Adolescents are quickly adopting designer drugs as a readily accessible form of recreational “legal highs.”10 Public awareness and educational initiatives can bring to light the dangers of these substances that exert powerful and, sometimes, unpredictable psychoactive effects on the user.
Self-mutilation and suicidal ideation also have been documented among those who ingested bath salts. These reports appear to be escalating across Europe and the United States. On a national level, U.S. poison centers have reported an almost 20-fold increase in calls regarding bath salts between 2010 and 2011.5 It is of utmost importance for clinicians and emergency personnel to familiarize themselves with the sympathomimetic toxidrome and management for bath salt consumption.
1. Mariani JJ, Khantzian EJ, Levin FR. The self-medication hypothesis and psychostimulant treatment of cocaine dependence: an update. Am J Addict. 2014;23(2):189-193.
2. Deluca P, Schifano F, Davey Z, et al. MDPV Report: Psychonaut Web Mapping Research Project. https://catbull.com/alamut/Bibliothek/PsychonautMDPVreport. pdf. Updated June 8, 2010. Accessed October 27, 2015.
3. National Institute on Drug Abuse. What are bath salts? http://teens.drugabuse.gov/drug-facts/bath-salts. Updated October 23, 2015. Accessed October 27, 2015.
4. Richards JR, Derlet RW, Albertson TE, et al. Methamphetamine, “bath salts,” and other amphetamine-related derivatives. Enliven: Toxicology and Allied Clinical Pharmacology. 2014;1(1):1-15.
5. Olives TD, Orozco BS, Stellpflug SJ. Bath salts: the ivory wave of trouble. West J Emerg Med. 2012;13(1):58-62.
6. Kasick DP, McKnight CA, Klisovic E. “Bath salt” ingestion leading to severe intoxication delirium: two cases and a brief review of the emergence of mephedrone use. Am J Drug Alcohol Abuse. 2012;38(2):176-180.
7. Borek HA, Holstege CP. Hyperthermia and multiorgan failure after abuse of “bath salts” containing 3,4-methylenedioxypyrovalerone. Ann Emerg Med. 2012;60(1):103-105.
8. Regunath H, Ariyamuthu VK, Dalal P, et al. Bath salt intoxication causing acute kidney injury requiring hemodialysis. Hemodial Int. 2012;16(suppl 1):S47-S49.
9. Adebamiro A, Perazella MA. Recurrent acute kidney injury following bath salts intoxication. Am J Kidney Dis. 2012;59(2):273-275.
10. Federation of American Societies for Experimental Biology. New designer drug, ‘bath salts,’ may confer additional risk for adolescents. EurekAlert. http://www.eurekalert.org/ pub_releases/2013-04/foas-ndd041813.php. Published April 23, 2013. Accessed November 10, 2015.
Designer drugs are rapidly making inroads with young people, primarily because of easier access, lower overall cost, and nebulous legality. These drugs are made as variants of illicit drugs or new formulations and sold as “research chemicals” and labeled as “not for human consumption,” which allows them to fall outside existing laws. The ingredients typically are not detected in a urine drug screen.
Notoriously addictive, these designer drugs, such as bath salts, are known to incorporate synthetic cathinones—namely, methylone, mephedrone or methylenedioxypyrovalerone (MDPV). The stimulant, amphetamine-like effects of bath salts make the drug attractive to adolescents with attention-deficit/hyperactivity disorder (ADHD).
Why do teens gravitate toward bath salts?
Adolescents with undiagnosed ADHD might self-medicate with drugs that are suited for addressing restlessness, intrapsychic turmoil, and other symptoms of ADHD. In 2 case studies, using the self-medication hypothesis, people with ADHD were more likely to seek cocaine by means of “self-selection.”1 These drug-seeking behaviors often led to cocaine dependence, even when other substances, such as alcohol or Cannabis, were available.
Methylphenidate and other ADHD pharmacotherapies influence the nucleus accumbens in a manner similar to that of cocaine. These findings suggest that adolescents with ADHD and cocaine dependence might respond to therapeutic interventions that substitute cocaine with psychostimulants.1
Bath salts fall within the same spectrum of psychostimulant agents as methylphenidate and cocaine. MDPV approximates the effect of methylphenidate at low doses, and cocaine at higher doses. It often is marketed under the name “Ivory Wave” and could be confused with cocaine. Self-administration of MDPV can induce psychoactive effects that help alleviate ADHD symptoms; adolescents might continue to experience enhanced concentration and overall performance.2 Also, because of the low cost of “legal” bath salts, they are an appealing alternative to cocaine for self-medication.
Managing the sequelae of bath salt intoxication
Bath salts may produce sympathomimetic effects greater than cocaine, which require a proactive approach to symptom management. A medley of unknown ingredients in bath salt preparations makes it difficult for clinicians to gauge the pharmacological impact on individual patients; therefore, therapeutic interventions are on a case-by-case basis. However, emergencies concerning amphetamines and amphetamine analogues and derivatives often have similar presentations.
Cardiovascular effects. MDPV-specific urine and blood tests conducted on patients admitted to the emergency room showed a 10-fold increase in overall dopamine levels compared with those who took cocaine. As a sympathomimetic, high doses of dopamine are responsible for raising blood pressure and could lead to the development of pronounced cardiovascular effects.3,4
Agitation. Clinicians generally are advised to treat agitation before providing a more comprehensive assessment of symptoms. Endotracheal intubation often is a required for adequate control of agitation. Bath salt-induced agitation often is treated with IV benzodiazepines.4,5 Monitor patients for excessive sedation or new-onset “paradoxical agitation” as a function of ongoing benzo-diazepine therapy. Clinicians also may choose to co-administer an antipsychotic with benzodiazepines, although the practice is not universally encouraged for agitation control.
Mephedrone produces a delirious state in conjunction with psychotic symptoms. Antipsychotic therapy has been suggested for addressing ongoing agitation.6
Tachycardia. Symptomatic treatment of tachycardia involves beta blockers, such as labetalol. Nitroglycerine has evidence of efficacy for chest pain associated with cocaine intoxication; however, it is unclear whether it is effective for similar drugs of abuse.4
Multi-organ collapse caused by MDPV necessitates aggressive intervention, including prompt dialysis. Carefully evaluate the patient for the presence of organ-specific insults and initiate supportive measures accordingly. Pronounced agitation with hyperthermia might portend severely compromised renal, hepatic, and/or cardiac function in MDPV users.7 Those who present with MDPV intoxication and concomitant renal injury seem to benefit from hemodialysis.8 Repeat intoxication events may yield a presentation of acute renal injury replete with metabolic derangements, including metabolic acidosis, hyperuricemia, and rhabdomyolysis.9 Thorough patient assessments and interventions are useful in determining long-term outcomes, including issues pertaining to mortality.
Confronting an epidemic
Adolescents are quickly adopting designer drugs as a readily accessible form of recreational “legal highs.”10 Public awareness and educational initiatives can bring to light the dangers of these substances that exert powerful and, sometimes, unpredictable psychoactive effects on the user.
Self-mutilation and suicidal ideation also have been documented among those who ingested bath salts. These reports appear to be escalating across Europe and the United States. On a national level, U.S. poison centers have reported an almost 20-fold increase in calls regarding bath salts between 2010 and 2011.5 It is of utmost importance for clinicians and emergency personnel to familiarize themselves with the sympathomimetic toxidrome and management for bath salt consumption.
Designer drugs are rapidly making inroads with young people, primarily because of easier access, lower overall cost, and nebulous legality. These drugs are made as variants of illicit drugs or new formulations and sold as “research chemicals” and labeled as “not for human consumption,” which allows them to fall outside existing laws. The ingredients typically are not detected in a urine drug screen.
Notoriously addictive, these designer drugs, such as bath salts, are known to incorporate synthetic cathinones—namely, methylone, mephedrone or methylenedioxypyrovalerone (MDPV). The stimulant, amphetamine-like effects of bath salts make the drug attractive to adolescents with attention-deficit/hyperactivity disorder (ADHD).
Why do teens gravitate toward bath salts?
Adolescents with undiagnosed ADHD might self-medicate with drugs that are suited for addressing restlessness, intrapsychic turmoil, and other symptoms of ADHD. In 2 case studies, using the self-medication hypothesis, people with ADHD were more likely to seek cocaine by means of “self-selection.”1 These drug-seeking behaviors often led to cocaine dependence, even when other substances, such as alcohol or Cannabis, were available.
Methylphenidate and other ADHD pharmacotherapies influence the nucleus accumbens in a manner similar to that of cocaine. These findings suggest that adolescents with ADHD and cocaine dependence might respond to therapeutic interventions that substitute cocaine with psychostimulants.1
Bath salts fall within the same spectrum of psychostimulant agents as methylphenidate and cocaine. MDPV approximates the effect of methylphenidate at low doses, and cocaine at higher doses. It often is marketed under the name “Ivory Wave” and could be confused with cocaine. Self-administration of MDPV can induce psychoactive effects that help alleviate ADHD symptoms; adolescents might continue to experience enhanced concentration and overall performance.2 Also, because of the low cost of “legal” bath salts, they are an appealing alternative to cocaine for self-medication.
Managing the sequelae of bath salt intoxication
Bath salts may produce sympathomimetic effects greater than cocaine, which require a proactive approach to symptom management. A medley of unknown ingredients in bath salt preparations makes it difficult for clinicians to gauge the pharmacological impact on individual patients; therefore, therapeutic interventions are on a case-by-case basis. However, emergencies concerning amphetamines and amphetamine analogues and derivatives often have similar presentations.
Cardiovascular effects. MDPV-specific urine and blood tests conducted on patients admitted to the emergency room showed a 10-fold increase in overall dopamine levels compared with those who took cocaine. As a sympathomimetic, high doses of dopamine are responsible for raising blood pressure and could lead to the development of pronounced cardiovascular effects.3,4
Agitation. Clinicians generally are advised to treat agitation before providing a more comprehensive assessment of symptoms. Endotracheal intubation often is a required for adequate control of agitation. Bath salt-induced agitation often is treated with IV benzodiazepines.4,5 Monitor patients for excessive sedation or new-onset “paradoxical agitation” as a function of ongoing benzo-diazepine therapy. Clinicians also may choose to co-administer an antipsychotic with benzodiazepines, although the practice is not universally encouraged for agitation control.
Mephedrone produces a delirious state in conjunction with psychotic symptoms. Antipsychotic therapy has been suggested for addressing ongoing agitation.6
Tachycardia. Symptomatic treatment of tachycardia involves beta blockers, such as labetalol. Nitroglycerine has evidence of efficacy for chest pain associated with cocaine intoxication; however, it is unclear whether it is effective for similar drugs of abuse.4
Multi-organ collapse caused by MDPV necessitates aggressive intervention, including prompt dialysis. Carefully evaluate the patient for the presence of organ-specific insults and initiate supportive measures accordingly. Pronounced agitation with hyperthermia might portend severely compromised renal, hepatic, and/or cardiac function in MDPV users.7 Those who present with MDPV intoxication and concomitant renal injury seem to benefit from hemodialysis.8 Repeat intoxication events may yield a presentation of acute renal injury replete with metabolic derangements, including metabolic acidosis, hyperuricemia, and rhabdomyolysis.9 Thorough patient assessments and interventions are useful in determining long-term outcomes, including issues pertaining to mortality.
Confronting an epidemic
Adolescents are quickly adopting designer drugs as a readily accessible form of recreational “legal highs.”10 Public awareness and educational initiatives can bring to light the dangers of these substances that exert powerful and, sometimes, unpredictable psychoactive effects on the user.
Self-mutilation and suicidal ideation also have been documented among those who ingested bath salts. These reports appear to be escalating across Europe and the United States. On a national level, U.S. poison centers have reported an almost 20-fold increase in calls regarding bath salts between 2010 and 2011.5 It is of utmost importance for clinicians and emergency personnel to familiarize themselves with the sympathomimetic toxidrome and management for bath salt consumption.
1. Mariani JJ, Khantzian EJ, Levin FR. The self-medication hypothesis and psychostimulant treatment of cocaine dependence: an update. Am J Addict. 2014;23(2):189-193.
2. Deluca P, Schifano F, Davey Z, et al. MDPV Report: Psychonaut Web Mapping Research Project. https://catbull.com/alamut/Bibliothek/PsychonautMDPVreport. pdf. Updated June 8, 2010. Accessed October 27, 2015.
3. National Institute on Drug Abuse. What are bath salts? http://teens.drugabuse.gov/drug-facts/bath-salts. Updated October 23, 2015. Accessed October 27, 2015.
4. Richards JR, Derlet RW, Albertson TE, et al. Methamphetamine, “bath salts,” and other amphetamine-related derivatives. Enliven: Toxicology and Allied Clinical Pharmacology. 2014;1(1):1-15.
5. Olives TD, Orozco BS, Stellpflug SJ. Bath salts: the ivory wave of trouble. West J Emerg Med. 2012;13(1):58-62.
6. Kasick DP, McKnight CA, Klisovic E. “Bath salt” ingestion leading to severe intoxication delirium: two cases and a brief review of the emergence of mephedrone use. Am J Drug Alcohol Abuse. 2012;38(2):176-180.
7. Borek HA, Holstege CP. Hyperthermia and multiorgan failure after abuse of “bath salts” containing 3,4-methylenedioxypyrovalerone. Ann Emerg Med. 2012;60(1):103-105.
8. Regunath H, Ariyamuthu VK, Dalal P, et al. Bath salt intoxication causing acute kidney injury requiring hemodialysis. Hemodial Int. 2012;16(suppl 1):S47-S49.
9. Adebamiro A, Perazella MA. Recurrent acute kidney injury following bath salts intoxication. Am J Kidney Dis. 2012;59(2):273-275.
10. Federation of American Societies for Experimental Biology. New designer drug, ‘bath salts,’ may confer additional risk for adolescents. EurekAlert. http://www.eurekalert.org/ pub_releases/2013-04/foas-ndd041813.php. Published April 23, 2013. Accessed November 10, 2015.
1. Mariani JJ, Khantzian EJ, Levin FR. The self-medication hypothesis and psychostimulant treatment of cocaine dependence: an update. Am J Addict. 2014;23(2):189-193.
2. Deluca P, Schifano F, Davey Z, et al. MDPV Report: Psychonaut Web Mapping Research Project. https://catbull.com/alamut/Bibliothek/PsychonautMDPVreport. pdf. Updated June 8, 2010. Accessed October 27, 2015.
3. National Institute on Drug Abuse. What are bath salts? http://teens.drugabuse.gov/drug-facts/bath-salts. Updated October 23, 2015. Accessed October 27, 2015.
4. Richards JR, Derlet RW, Albertson TE, et al. Methamphetamine, “bath salts,” and other amphetamine-related derivatives. Enliven: Toxicology and Allied Clinical Pharmacology. 2014;1(1):1-15.
5. Olives TD, Orozco BS, Stellpflug SJ. Bath salts: the ivory wave of trouble. West J Emerg Med. 2012;13(1):58-62.
6. Kasick DP, McKnight CA, Klisovic E. “Bath salt” ingestion leading to severe intoxication delirium: two cases and a brief review of the emergence of mephedrone use. Am J Drug Alcohol Abuse. 2012;38(2):176-180.
7. Borek HA, Holstege CP. Hyperthermia and multiorgan failure after abuse of “bath salts” containing 3,4-methylenedioxypyrovalerone. Ann Emerg Med. 2012;60(1):103-105.
8. Regunath H, Ariyamuthu VK, Dalal P, et al. Bath salt intoxication causing acute kidney injury requiring hemodialysis. Hemodial Int. 2012;16(suppl 1):S47-S49.
9. Adebamiro A, Perazella MA. Recurrent acute kidney injury following bath salts intoxication. Am J Kidney Dis. 2012;59(2):273-275.
10. Federation of American Societies for Experimental Biology. New designer drug, ‘bath salts,’ may confer additional risk for adolescents. EurekAlert. http://www.eurekalert.org/ pub_releases/2013-04/foas-ndd041813.php. Published April 23, 2013. Accessed November 10, 2015.
To blog or not to blog? That is the marketing question
Few methods can build your practice and reputation as well as blogging— nor can they give you as much grief. Your opinions can become known to a wide audience; you might influence public thinking or behavior; and you might become associated with a particular expertise at almost no financial cost. Yet, having regular deadlines to produce creative content can be stressful, and the time required to do it well has its own cost.
What is it?
“Blog” is the collapsed expression of “Web log.” Blogging is posting your thoughts on a Web site for colleagues or consumers, or both, to read. Typically, a blog is written as if you were writing a newspaper column; word count varies, from 250 to 1,000 words. Alternative formats are auditory (podcasts) or visual (vlog) but those media require greater technical proficiency and take more time to produce.
Whether you decide to write or record your blog entry, be guided by this advice:
• The subject matter can be anything you choose, but will be easiest to write when what you write about is based on your expertise.
• The format can be stream of consciousness,essay, or bulleted lists or slides; the latter is the most common and often follows a how-to or list format (eg, “Top [number] strategies to XYZ” or “[Number] of things you didn’t know about ABC”).
• End the blog with a cliffhanger or a call-to-action statement that invites readers to comment (especially if you then comment on their comments), to help drive interest.
• Generate material at a consistent interval (eg, once a week or twice a month), so your readers can look forward to your soliloquies on a regular basis.
Your professional Web site can serve as a venue for your blog. Using a WordPressa-based site, for example, offers a user-friendly way to compose your dispatch, add formatting (headers, bullets, color, images, etc.) as you see fit, and then publish it. It requires little technical expertise and adds no extra expense to your Web site. Alternatively, you might wish to contact editors at magazines or blog aggregators with story ideas, and let them handle the logistics if your content is appealing to them.
aWordPress is a Web site creation and management tool.
Spreading the word
There is much you can do to publicize your blog.
• Take advantage of social media. Build up your contacts on LinkedIn and follow other bloggers and large news sites on Twitter. Often, recipients will respond in-kind. Then, for each new piece, post or tweet it in these accounts.
• Offer an e-mail subscription so that readers can easily follow you (by means of a free WordPress plug-in, for example).
• Be found in search engines, such as Google, by writing high-quality, original content. Don’t force certain keywords into your article in the hopes that search engines find them—doing so tends to make writing more robotic and can lower your page rank.
Successful strategies
Regularly setting time aside so that the process is enjoyable and not onerously deadline-driven lends satisfaction to the experience and comes through in the quality of the composition. To save time, consider dictating your thoughts to your computer or phone, then outsource transcription.
Don’t overlook the bounty of material in your day-to-day life: stories from sessions; discoveries from your own reading or the latest news; and lectures you give. All of these can serve as inspiration and material for posts. Jot down these moments in a notebook as soon as they come up, or else the memory will likely slip away.
Just as with other forms of social media, be mindful of appropriate boundaries. Do not disclose identifying patient information; even revealing facets of your life might not be appropriate for current or future patients to have access to. On the other hand, it might be therapeutic for them to know select personal information, such as how you have handled past dilemmas, that reveals you are a real person (a “whole object” in psychoanalytic terms), and that models meaningful thoughts or deeds.
You’ll find your voice, in time
Getting started with blogging often is the toughest part. Finding the right format, material, and routine will take time. Eventually, you will find your blogging voice, and will value the unique opportunity to brand your practice and yourself, provide valuable content to your readers, and find an outlet for artistic expression.
Disclosure
Dr. Braslow is the founder of Luminello.com.
Few methods can build your practice and reputation as well as blogging— nor can they give you as much grief. Your opinions can become known to a wide audience; you might influence public thinking or behavior; and you might become associated with a particular expertise at almost no financial cost. Yet, having regular deadlines to produce creative content can be stressful, and the time required to do it well has its own cost.
What is it?
“Blog” is the collapsed expression of “Web log.” Blogging is posting your thoughts on a Web site for colleagues or consumers, or both, to read. Typically, a blog is written as if you were writing a newspaper column; word count varies, from 250 to 1,000 words. Alternative formats are auditory (podcasts) or visual (vlog) but those media require greater technical proficiency and take more time to produce.
Whether you decide to write or record your blog entry, be guided by this advice:
• The subject matter can be anything you choose, but will be easiest to write when what you write about is based on your expertise.
• The format can be stream of consciousness,essay, or bulleted lists or slides; the latter is the most common and often follows a how-to or list format (eg, “Top [number] strategies to XYZ” or “[Number] of things you didn’t know about ABC”).
• End the blog with a cliffhanger or a call-to-action statement that invites readers to comment (especially if you then comment on their comments), to help drive interest.
• Generate material at a consistent interval (eg, once a week or twice a month), so your readers can look forward to your soliloquies on a regular basis.
Your professional Web site can serve as a venue for your blog. Using a WordPressa-based site, for example, offers a user-friendly way to compose your dispatch, add formatting (headers, bullets, color, images, etc.) as you see fit, and then publish it. It requires little technical expertise and adds no extra expense to your Web site. Alternatively, you might wish to contact editors at magazines or blog aggregators with story ideas, and let them handle the logistics if your content is appealing to them.
aWordPress is a Web site creation and management tool.
Spreading the word
There is much you can do to publicize your blog.
• Take advantage of social media. Build up your contacts on LinkedIn and follow other bloggers and large news sites on Twitter. Often, recipients will respond in-kind. Then, for each new piece, post or tweet it in these accounts.
• Offer an e-mail subscription so that readers can easily follow you (by means of a free WordPress plug-in, for example).
• Be found in search engines, such as Google, by writing high-quality, original content. Don’t force certain keywords into your article in the hopes that search engines find them—doing so tends to make writing more robotic and can lower your page rank.
Successful strategies
Regularly setting time aside so that the process is enjoyable and not onerously deadline-driven lends satisfaction to the experience and comes through in the quality of the composition. To save time, consider dictating your thoughts to your computer or phone, then outsource transcription.
Don’t overlook the bounty of material in your day-to-day life: stories from sessions; discoveries from your own reading or the latest news; and lectures you give. All of these can serve as inspiration and material for posts. Jot down these moments in a notebook as soon as they come up, or else the memory will likely slip away.
Just as with other forms of social media, be mindful of appropriate boundaries. Do not disclose identifying patient information; even revealing facets of your life might not be appropriate for current or future patients to have access to. On the other hand, it might be therapeutic for them to know select personal information, such as how you have handled past dilemmas, that reveals you are a real person (a “whole object” in psychoanalytic terms), and that models meaningful thoughts or deeds.
You’ll find your voice, in time
Getting started with blogging often is the toughest part. Finding the right format, material, and routine will take time. Eventually, you will find your blogging voice, and will value the unique opportunity to brand your practice and yourself, provide valuable content to your readers, and find an outlet for artistic expression.
Disclosure
Dr. Braslow is the founder of Luminello.com.
Few methods can build your practice and reputation as well as blogging— nor can they give you as much grief. Your opinions can become known to a wide audience; you might influence public thinking or behavior; and you might become associated with a particular expertise at almost no financial cost. Yet, having regular deadlines to produce creative content can be stressful, and the time required to do it well has its own cost.
What is it?
“Blog” is the collapsed expression of “Web log.” Blogging is posting your thoughts on a Web site for colleagues or consumers, or both, to read. Typically, a blog is written as if you were writing a newspaper column; word count varies, from 250 to 1,000 words. Alternative formats are auditory (podcasts) or visual (vlog) but those media require greater technical proficiency and take more time to produce.
Whether you decide to write or record your blog entry, be guided by this advice:
• The subject matter can be anything you choose, but will be easiest to write when what you write about is based on your expertise.
• The format can be stream of consciousness,essay, or bulleted lists or slides; the latter is the most common and often follows a how-to or list format (eg, “Top [number] strategies to XYZ” or “[Number] of things you didn’t know about ABC”).
• End the blog with a cliffhanger or a call-to-action statement that invites readers to comment (especially if you then comment on their comments), to help drive interest.
• Generate material at a consistent interval (eg, once a week or twice a month), so your readers can look forward to your soliloquies on a regular basis.
Your professional Web site can serve as a venue for your blog. Using a WordPressa-based site, for example, offers a user-friendly way to compose your dispatch, add formatting (headers, bullets, color, images, etc.) as you see fit, and then publish it. It requires little technical expertise and adds no extra expense to your Web site. Alternatively, you might wish to contact editors at magazines or blog aggregators with story ideas, and let them handle the logistics if your content is appealing to them.
aWordPress is a Web site creation and management tool.
Spreading the word
There is much you can do to publicize your blog.
• Take advantage of social media. Build up your contacts on LinkedIn and follow other bloggers and large news sites on Twitter. Often, recipients will respond in-kind. Then, for each new piece, post or tweet it in these accounts.
• Offer an e-mail subscription so that readers can easily follow you (by means of a free WordPress plug-in, for example).
• Be found in search engines, such as Google, by writing high-quality, original content. Don’t force certain keywords into your article in the hopes that search engines find them—doing so tends to make writing more robotic and can lower your page rank.
Successful strategies
Regularly setting time aside so that the process is enjoyable and not onerously deadline-driven lends satisfaction to the experience and comes through in the quality of the composition. To save time, consider dictating your thoughts to your computer or phone, then outsource transcription.
Don’t overlook the bounty of material in your day-to-day life: stories from sessions; discoveries from your own reading or the latest news; and lectures you give. All of these can serve as inspiration and material for posts. Jot down these moments in a notebook as soon as they come up, or else the memory will likely slip away.
Just as with other forms of social media, be mindful of appropriate boundaries. Do not disclose identifying patient information; even revealing facets of your life might not be appropriate for current or future patients to have access to. On the other hand, it might be therapeutic for them to know select personal information, such as how you have handled past dilemmas, that reveals you are a real person (a “whole object” in psychoanalytic terms), and that models meaningful thoughts or deeds.
You’ll find your voice, in time
Getting started with blogging often is the toughest part. Finding the right format, material, and routine will take time. Eventually, you will find your blogging voice, and will value the unique opportunity to brand your practice and yourself, provide valuable content to your readers, and find an outlet for artistic expression.
Disclosure
Dr. Braslow is the founder of Luminello.com.
30 Years of psychiatry, with no fundamental progress
It is helpful, when evaluating any scientific proposal, to first examine underlying assumptions. In the editorial outlining 10 foundations of advanced care for psychiatrists (Do you practice sophisticated psychiatry? 10 Proposed foundations of care, Current Psychiatry, From the Editor, August 2015, p. 12-13), there are a number of assumptions that warrant discussion. I want to discuss just one of those assumptions: that psychiatric practice is better now than it was 30 years ago.
I argue that psychiatric care in America, as a whole, is much worse now than when I finished training in 1990, primarily because of changes in healthcare reimbursement. When I was in training, it was common for patients to stay in the hospital for a month or longer. One reason for the longer stays was because it takes weeks for anti depressants and antipsychotics to reveal their effectiveness. If, after 10 days, no improvement was seen, there was time to change medications.
With a hospital stay now averaging less than a week, today’s inpatient psychiatrists must rely on faith that the first treatment will work; the patient must hope that he (she) does not fall through the cracks after discharge; and the outpatient psychiatrist, short on time and psychotherapeutic training, might feel more like an assembly line worker than a professional.
Longer stays also meant more intense and extended periods of psychotherapy and psychosocial support than is possible now. It was common, when I was in training, for the biological camp to dismiss the benefits of psychotherapy therapy. After several decades of research, that position is no longer tenable—yet hospital stays remain impractically short.
If all of the medications developed since 1990 disappeared, I believe I could be as effective a psychiatrist as I am today. The simple, sad fact is that there have been no major psychopharmacotherapeutic advances in the past 30 years. Yes, there have been changes in side-effect profiles and improvements around the edges, so to speak, but no fundamental changes in effectiveness since the introduction of clozapine.
What about advances in treating negative symptoms of schizophrenia, you ask? I have not been impressed.
The worst change in psychiatric practice in the last 30 years, and the one that threatens to undermine our profession the most, is relinquishing psychotherapy as a major component of our practice. I do not mean cognitive-behavioral therapy (CBT), although that paint-by-the-numbers set of tools is better than nothing. I mean psychoanalytic psychotherapy, because it is the only comprehensive developmental theory of the mind and its pathology. There is no CBT of development, for example.
To practice and research most effectively, we need good theories of both mind and brain. For all our advances, we are no closer to explaining the mind through a brain-based theory than when Sigmund Freud tried with his Project for a Scientific Psychology.
Ours is, by far, the hardest, most intellectually challenging medical profession. We must be masters of neuroscience to bring forth a future when something such as a Project for a Scientific Psychology will be possible; we must do the best by our patients with available somatic treatments; but we also must be masters at understanding human emotional development and the intricacies of relationships and how these influence the function and epigenetics of our brains. We must reinforce our understanding of the mind through doing psychotherapy with a significant fraction of our patients to further that understanding and its associated skills—or we risk becoming assembly line psychiatrists.
Over the past 25 years of practice, I have learned that the 2 most important tools we have are time and our evolutionarily determined empathic capacity to enter another human’s subjective world. The first session should not be about ordering medical tests and gathering family history; it should be about establishing a relationship. It is on the strength of that relationship that the success of understanding and treatment, whether somatic or psychological, rests.
Thirty years ago, we had all the time we needed. That was, in some ways, a Golden Age of psychiatry. Imagine if we could bring together the greater availability of time of that era with the increasing biological and psychoanalytic understanding of the present. Then, we might have a new Golden Age. For that to happen, we must fight to change the reimbursement system. The quickest way—maybe the only way—to do that is to stop accepting reimbursement from healthcare insurance companies.
Jule P. Miller III, MD
Private Practice
Biloxi, Mississippi
Dr. Nasrallah responds
Dr. Miller is correct: Insurance has defiled psychiatric care, both inpatient and outpatient, including minimizing hospital days of care and reducing psychiatric visits to med-checks with no time to provide even rudimentary psychotherapy—psychodynamic or otherwise. I believe that the optimal medical model of psychiatric care must incorporate empathic capacity, which establishes the indispensable therapeutic alliance with each patient we see.
Dr. Miller is harsh on the lack of psychiatric advances since completing training 25 years ago. I have been in academia throughout those years and I am thrilled by how much neuroscience we have discovered that is directly related to disorders of affect, thought, cognition, and behavior. The challenge continues to be in translating those discoveries into practical therapeutic interventions, which proves the point that psychiatry is the most challenging medical specialty, consistent with the brain (and its mind) being, by far, the most complex organ.
We cannot abdicate insurance reimbursement except for our well-to-do patients. Patients with severe and disabling mental disorders, who need our services the most, cannot afford to pay for care, and depend on Medicare and Medicaid, whose modest reimbursement would impoverish non-salaried psychiatrists. Because of the severe shortage of psychiatrists, who are the only specialists who can competently manage neurobiological disorders such as schizophrenia, bipolar disorder, and severe depression, I believe we should deploy our resources for those vulnerable patients and leave the worried well and walking wounded to other mental health professionals.
Henry A. Nasrallah, MD
Professor and Chairman
Department of Psychiatry
Saint Louis University School of Medicine
St. Louis, Missouri
It is helpful, when evaluating any scientific proposal, to first examine underlying assumptions. In the editorial outlining 10 foundations of advanced care for psychiatrists (Do you practice sophisticated psychiatry? 10 Proposed foundations of care, Current Psychiatry, From the Editor, August 2015, p. 12-13), there are a number of assumptions that warrant discussion. I want to discuss just one of those assumptions: that psychiatric practice is better now than it was 30 years ago.
I argue that psychiatric care in America, as a whole, is much worse now than when I finished training in 1990, primarily because of changes in healthcare reimbursement. When I was in training, it was common for patients to stay in the hospital for a month or longer. One reason for the longer stays was because it takes weeks for anti depressants and antipsychotics to reveal their effectiveness. If, after 10 days, no improvement was seen, there was time to change medications.
With a hospital stay now averaging less than a week, today’s inpatient psychiatrists must rely on faith that the first treatment will work; the patient must hope that he (she) does not fall through the cracks after discharge; and the outpatient psychiatrist, short on time and psychotherapeutic training, might feel more like an assembly line worker than a professional.
Longer stays also meant more intense and extended periods of psychotherapy and psychosocial support than is possible now. It was common, when I was in training, for the biological camp to dismiss the benefits of psychotherapy therapy. After several decades of research, that position is no longer tenable—yet hospital stays remain impractically short.
If all of the medications developed since 1990 disappeared, I believe I could be as effective a psychiatrist as I am today. The simple, sad fact is that there have been no major psychopharmacotherapeutic advances in the past 30 years. Yes, there have been changes in side-effect profiles and improvements around the edges, so to speak, but no fundamental changes in effectiveness since the introduction of clozapine.
What about advances in treating negative symptoms of schizophrenia, you ask? I have not been impressed.
The worst change in psychiatric practice in the last 30 years, and the one that threatens to undermine our profession the most, is relinquishing psychotherapy as a major component of our practice. I do not mean cognitive-behavioral therapy (CBT), although that paint-by-the-numbers set of tools is better than nothing. I mean psychoanalytic psychotherapy, because it is the only comprehensive developmental theory of the mind and its pathology. There is no CBT of development, for example.
To practice and research most effectively, we need good theories of both mind and brain. For all our advances, we are no closer to explaining the mind through a brain-based theory than when Sigmund Freud tried with his Project for a Scientific Psychology.
Ours is, by far, the hardest, most intellectually challenging medical profession. We must be masters of neuroscience to bring forth a future when something such as a Project for a Scientific Psychology will be possible; we must do the best by our patients with available somatic treatments; but we also must be masters at understanding human emotional development and the intricacies of relationships and how these influence the function and epigenetics of our brains. We must reinforce our understanding of the mind through doing psychotherapy with a significant fraction of our patients to further that understanding and its associated skills—or we risk becoming assembly line psychiatrists.
Over the past 25 years of practice, I have learned that the 2 most important tools we have are time and our evolutionarily determined empathic capacity to enter another human’s subjective world. The first session should not be about ordering medical tests and gathering family history; it should be about establishing a relationship. It is on the strength of that relationship that the success of understanding and treatment, whether somatic or psychological, rests.
Thirty years ago, we had all the time we needed. That was, in some ways, a Golden Age of psychiatry. Imagine if we could bring together the greater availability of time of that era with the increasing biological and psychoanalytic understanding of the present. Then, we might have a new Golden Age. For that to happen, we must fight to change the reimbursement system. The quickest way—maybe the only way—to do that is to stop accepting reimbursement from healthcare insurance companies.
Jule P. Miller III, MD
Private Practice
Biloxi, Mississippi
Dr. Nasrallah responds
Dr. Miller is correct: Insurance has defiled psychiatric care, both inpatient and outpatient, including minimizing hospital days of care and reducing psychiatric visits to med-checks with no time to provide even rudimentary psychotherapy—psychodynamic or otherwise. I believe that the optimal medical model of psychiatric care must incorporate empathic capacity, which establishes the indispensable therapeutic alliance with each patient we see.
Dr. Miller is harsh on the lack of psychiatric advances since completing training 25 years ago. I have been in academia throughout those years and I am thrilled by how much neuroscience we have discovered that is directly related to disorders of affect, thought, cognition, and behavior. The challenge continues to be in translating those discoveries into practical therapeutic interventions, which proves the point that psychiatry is the most challenging medical specialty, consistent with the brain (and its mind) being, by far, the most complex organ.
We cannot abdicate insurance reimbursement except for our well-to-do patients. Patients with severe and disabling mental disorders, who need our services the most, cannot afford to pay for care, and depend on Medicare and Medicaid, whose modest reimbursement would impoverish non-salaried psychiatrists. Because of the severe shortage of psychiatrists, who are the only specialists who can competently manage neurobiological disorders such as schizophrenia, bipolar disorder, and severe depression, I believe we should deploy our resources for those vulnerable patients and leave the worried well and walking wounded to other mental health professionals.
Henry A. Nasrallah, MD
Professor and Chairman
Department of Psychiatry
Saint Louis University School of Medicine
St. Louis, Missouri
It is helpful, when evaluating any scientific proposal, to first examine underlying assumptions. In the editorial outlining 10 foundations of advanced care for psychiatrists (Do you practice sophisticated psychiatry? 10 Proposed foundations of care, Current Psychiatry, From the Editor, August 2015, p. 12-13), there are a number of assumptions that warrant discussion. I want to discuss just one of those assumptions: that psychiatric practice is better now than it was 30 years ago.
I argue that psychiatric care in America, as a whole, is much worse now than when I finished training in 1990, primarily because of changes in healthcare reimbursement. When I was in training, it was common for patients to stay in the hospital for a month or longer. One reason for the longer stays was because it takes weeks for anti depressants and antipsychotics to reveal their effectiveness. If, after 10 days, no improvement was seen, there was time to change medications.
With a hospital stay now averaging less than a week, today’s inpatient psychiatrists must rely on faith that the first treatment will work; the patient must hope that he (she) does not fall through the cracks after discharge; and the outpatient psychiatrist, short on time and psychotherapeutic training, might feel more like an assembly line worker than a professional.
Longer stays also meant more intense and extended periods of psychotherapy and psychosocial support than is possible now. It was common, when I was in training, for the biological camp to dismiss the benefits of psychotherapy therapy. After several decades of research, that position is no longer tenable—yet hospital stays remain impractically short.
If all of the medications developed since 1990 disappeared, I believe I could be as effective a psychiatrist as I am today. The simple, sad fact is that there have been no major psychopharmacotherapeutic advances in the past 30 years. Yes, there have been changes in side-effect profiles and improvements around the edges, so to speak, but no fundamental changes in effectiveness since the introduction of clozapine.
What about advances in treating negative symptoms of schizophrenia, you ask? I have not been impressed.
The worst change in psychiatric practice in the last 30 years, and the one that threatens to undermine our profession the most, is relinquishing psychotherapy as a major component of our practice. I do not mean cognitive-behavioral therapy (CBT), although that paint-by-the-numbers set of tools is better than nothing. I mean psychoanalytic psychotherapy, because it is the only comprehensive developmental theory of the mind and its pathology. There is no CBT of development, for example.
To practice and research most effectively, we need good theories of both mind and brain. For all our advances, we are no closer to explaining the mind through a brain-based theory than when Sigmund Freud tried with his Project for a Scientific Psychology.
Ours is, by far, the hardest, most intellectually challenging medical profession. We must be masters of neuroscience to bring forth a future when something such as a Project for a Scientific Psychology will be possible; we must do the best by our patients with available somatic treatments; but we also must be masters at understanding human emotional development and the intricacies of relationships and how these influence the function and epigenetics of our brains. We must reinforce our understanding of the mind through doing psychotherapy with a significant fraction of our patients to further that understanding and its associated skills—or we risk becoming assembly line psychiatrists.
Over the past 25 years of practice, I have learned that the 2 most important tools we have are time and our evolutionarily determined empathic capacity to enter another human’s subjective world. The first session should not be about ordering medical tests and gathering family history; it should be about establishing a relationship. It is on the strength of that relationship that the success of understanding and treatment, whether somatic or psychological, rests.
Thirty years ago, we had all the time we needed. That was, in some ways, a Golden Age of psychiatry. Imagine if we could bring together the greater availability of time of that era with the increasing biological and psychoanalytic understanding of the present. Then, we might have a new Golden Age. For that to happen, we must fight to change the reimbursement system. The quickest way—maybe the only way—to do that is to stop accepting reimbursement from healthcare insurance companies.
Jule P. Miller III, MD
Private Practice
Biloxi, Mississippi
Dr. Nasrallah responds
Dr. Miller is correct: Insurance has defiled psychiatric care, both inpatient and outpatient, including minimizing hospital days of care and reducing psychiatric visits to med-checks with no time to provide even rudimentary psychotherapy—psychodynamic or otherwise. I believe that the optimal medical model of psychiatric care must incorporate empathic capacity, which establishes the indispensable therapeutic alliance with each patient we see.
Dr. Miller is harsh on the lack of psychiatric advances since completing training 25 years ago. I have been in academia throughout those years and I am thrilled by how much neuroscience we have discovered that is directly related to disorders of affect, thought, cognition, and behavior. The challenge continues to be in translating those discoveries into practical therapeutic interventions, which proves the point that psychiatry is the most challenging medical specialty, consistent with the brain (and its mind) being, by far, the most complex organ.
We cannot abdicate insurance reimbursement except for our well-to-do patients. Patients with severe and disabling mental disorders, who need our services the most, cannot afford to pay for care, and depend on Medicare and Medicaid, whose modest reimbursement would impoverish non-salaried psychiatrists. Because of the severe shortage of psychiatrists, who are the only specialists who can competently manage neurobiological disorders such as schizophrenia, bipolar disorder, and severe depression, I believe we should deploy our resources for those vulnerable patients and leave the worried well and walking wounded to other mental health professionals.
Henry A. Nasrallah, MD
Professor and Chairman
Department of Psychiatry
Saint Louis University School of Medicine
St. Louis, Missouri
Awareness and management of obstetrical complications of depression
When a patient who has a preexisting medical illness seeks prenatal care, the obstetrician asks herself (himself) 2 questions:
• What impact will the illness have on the pregnancy?
• What impact will the pregnancy have on the illness?
Depression is both a pregnancy-associated and pregnancy-independent illness, which, in the setting of a pregnant woman who has a depressive disorder, makes these questions particularly difficult to answer. In such a case, coordination of care with a mental health provider is essential.
Awareness of the obstetrical complications associated with depression during pregnancy, as well as their implications for the future health of the mother–infant dyad, is important for the entire care team. This article reviews the associations and interconnectedness of depression with complications of pregnancy, childbirth, and the neonatal period.
Diagnosis of depression during prenatal care
The American College of Obstetricians and Gynecologists (ACOG) states that evidence is insufficient to support a recommendation for universal screening for depression among prenatal patients, although such screening should be considered.1 There is considerable variability among obstetrical providers regarding the practice of depression screening; tools to be used if such screening is done; and screening frequency through the pregnancy.
Discernment of depression is difficult. Many somatic symptoms of depression overlap with common prenatal complaints and, consequentially, can be overlooked. Among a sample of 700 pregnant women, for example, 56% complained of lack of energy; 19%, of insomnia; and 19%, of appetite changes.2 Weight change, of course, is universal.
The 10-question self-rating Edinburgh Postnatal Depression Scale has been validated for use during pregnancy and postnatally. This screening instrument can be helpful for differentiating purely physical complaints from mental distress due to depressive symptoms.2,3
When an obstetrical provider suspects a depressive disorder, or one has been diagnosed, she (he) faces the problem of what to do with that information. Women of low socioeconomic status and victims of domestic violence are at increased risk of depression during pregnancy, but barriers to appropriate referral can seem nearly insurmountable because they lack insurance and social support.4-9
In addition, within the setting of numerous tasks that need attending during the relatively short prenatal period, it is common for women newly given a diagnosis of depression to fail to follow up on a referral to a mental health provider.
Although most providers will “check in” with a depressed or at-risk patient at each prenatal visit about her mood, any effort at follow-up can be overshadowed by tangible physical concerns, such as preterm contractions, fetal growth restriction, and coordination of routine testing that has been delayed because of scant prenatal care. All these physical concerns and circumstances of care are associated with maternal depression, as we will discuss.
Preterm labor and birth
Preterm labor is defined as uterine contractions that lead to cervical change before 37 weeks gestational age. Preterm labor increases the risk of preterm birth; preterm labor precedes 50% of preterm births. Preterm birth is the leading cause of neonatal mortality in the United States, and rates of morbidity and mortality increase as gestational age decreases.10 Common neonatal complications related to prematurity are shown in the Figure.11
Women who suffer from depression have an increased risk of preterm labor and preterm birth, as many studies of treated and untreated depressed pregnant women have shown.12-20 The causative mechanism is unknown; it has been proposed that the increase in maternal cortisol production associated with depression and distress triggers overproduction of placental cortisol releasing hormone, which is thought to be involved in initiation of parturition.21,22 Depression also is associated with other risk factors for preterm birth, such as low socioeconomic status, substance use, and smoking.
Intrauterine growth restriction
Women who have depression during pregnancy have an increased risk of intrauterine growth restriction (IUGR), which leads to delivery of an infant who is small for gestational age (SGA) or of low birth weight (LBW) (weighing <2,500 g at birth), or both.23 Again, the basis of the association between depression and IUGR and SGA is unknown; it is theorized that increased levels of cortisol and catecholamines associated with maternal distress might, by increasing blood pressure and inducing vasoconstriction, cause placental hypoperfusion.24,25
It also is possible that the association of depression with other risk factors for IUGR, such as smoking, substance use, obesity, and poor prenatal care, puts the infants of depressed women at risk of growth restriction.26 Several large-scale studies showed that the association between LBW and depression is lost when smoking and substance use are accounted for; other studies, however, found a persistent association in untreated depressed women when smokers, substance users, and drinkers were excluded.17,26,27
IUGR infants are at increased risk of iatrogenic prematurity and stillbirth. Fetuses that weigh <10th percentile for their gestational age are delivered no later than 40 weeks; delivery can be indicated as early as 32 weeks, depending on the results of other antenatal tests. Women who have a growth-restricted infant have a higher risk of cesarean delivery because growth-restricted infants often have less reserve and poorer tolerance of labor.
Preeclampsia and eclampsia
Preeclampsia is defined as blood pressure >140/90 mm HG on at least 2 occasions, with proteinuria, that occurs later than the twentieth week of pregnancy in women who did not have hypertension or renal dysfunction at baseline. Preeclampsia is a progressive disease that can cause severe maternal morbidity, including renal failure, stroke, hepatic rupture, pulmonary edema, and heart failure.
Eclampsia refers to onset of seizures in the setting of preeclampsia. These 2 hypertensive disorders are the third leading world wide cause of maternal mortality.28
Depressed women have an elevated risk of preeclampsia. The association between preeclampsia and depression might be caused by the presence of increased levels of inflammatory mediators29,30; other comorbidities, such as increased body mass index, also might be involved, but the risk for preeclampsia in depressed women still is increased after controlling for obesity.31
The presence of preeclampsia is responsible for a high percentage of iatrogenic preterm births, because the cure for the disorder is delivery—even at early or previable gestational age. Complication rates for mother and infant are high.
The presence of preeclampsia is a significant risk factor for intrauterine fetal demise. Treating the mother after delivery involves administration of IV magnesium for 24 hours; often, the mother is separated from her infant for a day after birth.
Impact on prenatal care
Depression increases odds that women will have fewer prenatal visits.32 During pregnancy, women typically initiate prenatal care during the first trimester, when pregnancy-dating ultrasonography and early screening tests for chromosomal abnormalities are performed. Prenatal visits occur monthly until the third trimester, then every 2 weeks between 32 and 36 weeks’ gestation, increasing to weekly after 36 weeks’ gestation.
The increased number of visits in late pregnancy allows for early detection and treatment of hypertensive disorders; assesses fetal well-being; and decreases the risks of morbidity and mortality for mother and fetus.33 Because women who suffer from depression are at increased risk of an array of adverse pregnancy outcomes, the importance of regular and timely prenatal care cannot be understated.
In addition, the prenatal visit gives the obstetrician the opportunity to connect women with other specialists for management of any unmet medical needs. One study showed that, when women have adequate prenatal care (measured by the number of visits), the association between preterm birth and self-reported maternal depression was eliminated.34
Substance use
Substance use and depression often co-exist.35,36 Unlike screening for depression, screening for substance use is universal during prenatal care. Studies have shown that women who screen positive for depression are at higher risk of a number of comorbidities, including substance use.37,38 Conversely, women who use substances are more likely to screen positive for depression.
Evidence suggests that best practice might be to screen for depression in any woman who has a positive drug screen, if a provider is not routinely screening their general patient population.39 Substance use in pregnancy is associated with a number of poor outcomes, including placental abruption (cocaine use); dysmorphic facies and congenital anomalies (alcohol); and neonatal abstinence syndrome (heroin).
Antidepressants in pregnancy
A full discussion of the risks and benefits associated with pharmacotherapy for depression in pregnancy is beyond the scope of this article. Generally, antidepressant use is fraught with concerns over teratogenicity and adverse fetal outcomes. Although ACOG states that (1) pharmacotherapy for depression should be individualized and (2) most selective serotonin reuptake inhibitors (SSRIs) are not considered major teratogenic agents, many obstetricians and patients feel uncomfortable using these medications in pregnancy.40 Often, pre-pregnancy antidepressants are discontinued in the first trimester; one large population-based study found that only 0.9% of women who had depression filled their antidepressant prescription consistently throughout their pregnancy.41
It is unclear whether antidepressant use in pregnancy contributes to the risk of preterm birth seen in women who have depression. In a large population-based study, use of antidepressants in the second trimester was associated with preterm delivery but severe depression was not.18 A recent meta-analysis revealed an increased risk of preterm birth in women who used an antidepressant, compared with healthy women and untreated depressed women.42
Research limits, unanswered questions. Regrettably, it is difficult to untangle risk factors for preterm birth among depressed women without randomized controlled studies that are not ethically feasible. It cannot be said with certainty whether antidepressant pharmacotherapy is associated with a higher risk of preterm birth than depression alone.
Likewise, it is difficult to clarify the extent to which antidepressants contribute to infant growth restriction, if at all. Two recent meta-analyses concluded that exposure to antidepressants is associated with a statistically significant risk of LBW.42,43 However, increased severity of depressive symptoms generally is associated with exposure to antidepressants during pregnancy, and a randomized controlled trial is, again, impossible to conduct for ethical reasons.
Whereas a plausible biological mechanism associating IUGR, SGA, and LBW with depression exists, the same cannot be said for antidepressants. In one study, exposure to maternal depression altered the expression of certain placental genes but exposure to SSRIs did not cause further changes. This suggests that, on a cellular level, placental function might differ in depressed women.44 Although antidepressants do cross the placenta, it remains to be seen whether fetal growth is impacted as a result. One study found decreased fetal head circumference in infants who had been exposed to antidepressants during pregnancy, but no increased risk for having a SGA or LWB infant.45
Obstetrical management and mental health implications
Treated or not, women who suffer depression are a high-risk group when it comes to preterm birth and a host of other pregnancy comorbidities. Women with serious complications of pregnancy often are hospitalized for observation, and can undergo a prolonged stay when close proximity to medical services or a surgical suite is required.
For example, hospitalization until delivery is the standard of care for women who have preterm premature rupture of membranes or preeclampsia before 34 weeks’ gestation. Prolonged inpatient admissions and associated restriction of activity is profoundly deleterious on mood, with depression and anxiety significantly correlated with length of stay.46,47 Given the associations between depression and preterm birth, it might be reasonable to consider screening antenatal inpatients at risk of preterm birth for depression on a regular basis, so that treatment can be initiated if needed.
Depression during pregnancy is relatively common; an estimated 12.7% of pregnant women are affected at some time between conception and birth.48 Not only does depression appear to have deleterious effects on pregnancy outcomes, it also plays a pivotal role in the qualitative experience of pregnancy for the mother.
Bottom Line
Awareness of obstetrical complications associated with depression in pregnancy is important for the entire care team, including the psychiatrist and obstetrician. Depression not only appears to have deleterious effects on pregnancy outcomes, it also plays a pivotal role in the qualitative experience of pregnancy for the mother. Antidepressant use generally is fraught with concerns over teratogenicity and adverse fetal outcomes.
Related Resources
• Freeman MP. Some SSRIs are better than others for pregnant women (audio interview). Current Psychiatry. 2014;13(7). http://www.currentpsychiatry.com/specialty-focus/practice-trends/article/some-ssris-are-better-thanothers-for-pregnant-women/e3adb4704e25492f3e15331fc1cc058d.html.
• Freeman MP, Joffe H, Cohen LS. Postpartum depression: Help patients find the right treatment. Current Psychiatry. 2012;11(11):14-16,19-21.
Disclosures
Dr. Habecker reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Freeman is a member of the advisory board of JDS Therapeutics, Sunovion Pharmaceuticals, Inc., and Takeda Pharmaceutical Co. She receives research grant support from Takeda Pharmaceutical Co.
1. American College of Obstetricians and Gynecologists. Committee on Obstetric Practice. Committee opinion no. 630. 2015;125:1268-1271.
2. Apter G, Devouche E, Garez V, et al. Pregnancy, somatic complaints and depression: a French population-based study. Eur J Obstet Gynecol Reprod Biol. 2013;171(1):35-39.
3. Murray D, Cox JL. Screening for depression during pregnancy with the Edinburgh Depression Scale (EDDS). J Reprod Infant Psychol. 1990;8(2):99-107.
4. Gotlib IH, Whiffen VE, Mount JH, et al. Prevalence rates and demographic characteristics associated with depression in pregnancy and the postpartum. J Consult Clin Psychol. 1989;57(2):269-274.
5. Melville JL, Gavin A, Guo Y, et al. Depressive disorders during pregnancy: prevalence and risk factors in a large urban sample. Obstet Gynecol. 2010;116(5):1064-1070.
6. Leddy M, Haaga D, Gray J, et al. Postpartum mental health screening and diagnosis by obstetrician-gynecologists. J Psychosom Obstet Gynaecol. 2011;32(1):27-34.
7. McFarlane J, Maddoux J, Cesario S, et al. Effect of abuse during pregnancy on maternal and child safety and functioning for 24 months after delivery. Obstet Gynecol. 2014;123(4):839-847.
8. Vesga-López O, Bianco C, Keyes K, et al. Psychiatric disorders in pregnant and postpartum women in the United States. Arch Gen Psychiatry. 2008;65(7):805-815.
9. Farr SL, Bitsko RH, Hayes DK, et al. Mental health and access to services among US women of reproductive age. Am J Obstet Gynecol. 2010;203(6):542.e1-e542.e9. doi: 10.1016/j.ajog.2010.07.007.
10. Committee on Practice Bulletins—Obstetrics; The American College of Obstetricians and Gynecologists. Practice bulletin no. 130: prediction and prevention of preterm birth. Obstet Gynecol. 2012;120(4):964-973.
11. Stoll BJ, Hansen NI, Bell EF, et al; Eunice Kennedy Shriver National Institute of Child Health and Human Development Neonatal Research Network. Neonatal outcomes of extremely preterm infants from the NICHD Neonatal Research Network. Pediatrics. 2010;126(3):443-456.
12. Steer RA, Scholl TO, Hediger ML, et al. Self-reported depression and negative pregnancy outcomes. J Clin Epidemiol. 1992;45(10):1093-1099.
13. Goldenberg RL, Cliver SP, Mulvihill FX, et al. Medical, psychosocial, and behavioral risk factors do not explain the increased risk for low birth weight among black women. Am J Obstet Gynecol. 1996;175(5):1317-1324.
14. Orr ST, James SA, Blackmore Prince C. Maternal prenatal depressive symptoms and spontaneous preterm births among African-American women in Baltimore, Maryland. Am J Epidemiol. 2002;156(9):797-802.
15. Dayan J, Creveuil C, Marks MN, et al. Prenatal depression, prenatal anxiety, and spontaneous preterm birth: a prospective cohort study among women with early and regular care. Psychosom Med. 2006;68(6):938-946.
16. Goedhart G, Snijders AC, Hesselink AE, et al. Maternal depressive symptoms in relation to perinatal mortality and morbidity: results from a large multiethnic cohort study. Psychosom Med. 2010;72(8):769-776.
17. Grote NK, Bridge JA, Gavin AR, et al. A meta-analysis of depression during pregnancy and the risk of preterm birth, low birth weight, and intrauterine growth restriction. Arch Gen Psychiatry. 2010;67(10):1012-1024.
18. Hayes RM, Wu P, Shelton RC, et al. Maternal antidepressant use and adverse outcomes: a cohort study of 228,876 pregnancies [published online April 30, 2012]. Am J Obstet Gynecol. 2012;207(1):49.e1-49.e9. doi: 10.1016/j. ajog.2012.04.028.
19. McDonagh MS, Matthews A, Phillipi C, et al. Depression drug treatment outcomes in pregnancy and the postpartum period: a systematic review and meta-analysis. Obstet Gynecol. 2014;124(3):526-534.
20. Sahingöz M, Yuksel G, Karsidag C, et al. Birth weight and preterm birth in babies of pregnant women with major depression in relation to treatment with antidepressants. J Clin Psychopharmacol. 2014;34(2):226-229.
When a patient who has a preexisting medical illness seeks prenatal care, the obstetrician asks herself (himself) 2 questions:
• What impact will the illness have on the pregnancy?
• What impact will the pregnancy have on the illness?
Depression is both a pregnancy-associated and pregnancy-independent illness, which, in the setting of a pregnant woman who has a depressive disorder, makes these questions particularly difficult to answer. In such a case, coordination of care with a mental health provider is essential.
Awareness of the obstetrical complications associated with depression during pregnancy, as well as their implications for the future health of the mother–infant dyad, is important for the entire care team. This article reviews the associations and interconnectedness of depression with complications of pregnancy, childbirth, and the neonatal period.
Diagnosis of depression during prenatal care
The American College of Obstetricians and Gynecologists (ACOG) states that evidence is insufficient to support a recommendation for universal screening for depression among prenatal patients, although such screening should be considered.1 There is considerable variability among obstetrical providers regarding the practice of depression screening; tools to be used if such screening is done; and screening frequency through the pregnancy.
Discernment of depression is difficult. Many somatic symptoms of depression overlap with common prenatal complaints and, consequentially, can be overlooked. Among a sample of 700 pregnant women, for example, 56% complained of lack of energy; 19%, of insomnia; and 19%, of appetite changes.2 Weight change, of course, is universal.
The 10-question self-rating Edinburgh Postnatal Depression Scale has been validated for use during pregnancy and postnatally. This screening instrument can be helpful for differentiating purely physical complaints from mental distress due to depressive symptoms.2,3
When an obstetrical provider suspects a depressive disorder, or one has been diagnosed, she (he) faces the problem of what to do with that information. Women of low socioeconomic status and victims of domestic violence are at increased risk of depression during pregnancy, but barriers to appropriate referral can seem nearly insurmountable because they lack insurance and social support.4-9
In addition, within the setting of numerous tasks that need attending during the relatively short prenatal period, it is common for women newly given a diagnosis of depression to fail to follow up on a referral to a mental health provider.
Although most providers will “check in” with a depressed or at-risk patient at each prenatal visit about her mood, any effort at follow-up can be overshadowed by tangible physical concerns, such as preterm contractions, fetal growth restriction, and coordination of routine testing that has been delayed because of scant prenatal care. All these physical concerns and circumstances of care are associated with maternal depression, as we will discuss.
Preterm labor and birth
Preterm labor is defined as uterine contractions that lead to cervical change before 37 weeks gestational age. Preterm labor increases the risk of preterm birth; preterm labor precedes 50% of preterm births. Preterm birth is the leading cause of neonatal mortality in the United States, and rates of morbidity and mortality increase as gestational age decreases.10 Common neonatal complications related to prematurity are shown in the Figure.11
Women who suffer from depression have an increased risk of preterm labor and preterm birth, as many studies of treated and untreated depressed pregnant women have shown.12-20 The causative mechanism is unknown; it has been proposed that the increase in maternal cortisol production associated with depression and distress triggers overproduction of placental cortisol releasing hormone, which is thought to be involved in initiation of parturition.21,22 Depression also is associated with other risk factors for preterm birth, such as low socioeconomic status, substance use, and smoking.
Intrauterine growth restriction
Women who have depression during pregnancy have an increased risk of intrauterine growth restriction (IUGR), which leads to delivery of an infant who is small for gestational age (SGA) or of low birth weight (LBW) (weighing <2,500 g at birth), or both.23 Again, the basis of the association between depression and IUGR and SGA is unknown; it is theorized that increased levels of cortisol and catecholamines associated with maternal distress might, by increasing blood pressure and inducing vasoconstriction, cause placental hypoperfusion.24,25
It also is possible that the association of depression with other risk factors for IUGR, such as smoking, substance use, obesity, and poor prenatal care, puts the infants of depressed women at risk of growth restriction.26 Several large-scale studies showed that the association between LBW and depression is lost when smoking and substance use are accounted for; other studies, however, found a persistent association in untreated depressed women when smokers, substance users, and drinkers were excluded.17,26,27
IUGR infants are at increased risk of iatrogenic prematurity and stillbirth. Fetuses that weigh <10th percentile for their gestational age are delivered no later than 40 weeks; delivery can be indicated as early as 32 weeks, depending on the results of other antenatal tests. Women who have a growth-restricted infant have a higher risk of cesarean delivery because growth-restricted infants often have less reserve and poorer tolerance of labor.
Preeclampsia and eclampsia
Preeclampsia is defined as blood pressure >140/90 mm HG on at least 2 occasions, with proteinuria, that occurs later than the twentieth week of pregnancy in women who did not have hypertension or renal dysfunction at baseline. Preeclampsia is a progressive disease that can cause severe maternal morbidity, including renal failure, stroke, hepatic rupture, pulmonary edema, and heart failure.
Eclampsia refers to onset of seizures in the setting of preeclampsia. These 2 hypertensive disorders are the third leading world wide cause of maternal mortality.28
Depressed women have an elevated risk of preeclampsia. The association between preeclampsia and depression might be caused by the presence of increased levels of inflammatory mediators29,30; other comorbidities, such as increased body mass index, also might be involved, but the risk for preeclampsia in depressed women still is increased after controlling for obesity.31
The presence of preeclampsia is responsible for a high percentage of iatrogenic preterm births, because the cure for the disorder is delivery—even at early or previable gestational age. Complication rates for mother and infant are high.
The presence of preeclampsia is a significant risk factor for intrauterine fetal demise. Treating the mother after delivery involves administration of IV magnesium for 24 hours; often, the mother is separated from her infant for a day after birth.
Impact on prenatal care
Depression increases odds that women will have fewer prenatal visits.32 During pregnancy, women typically initiate prenatal care during the first trimester, when pregnancy-dating ultrasonography and early screening tests for chromosomal abnormalities are performed. Prenatal visits occur monthly until the third trimester, then every 2 weeks between 32 and 36 weeks’ gestation, increasing to weekly after 36 weeks’ gestation.
The increased number of visits in late pregnancy allows for early detection and treatment of hypertensive disorders; assesses fetal well-being; and decreases the risks of morbidity and mortality for mother and fetus.33 Because women who suffer from depression are at increased risk of an array of adverse pregnancy outcomes, the importance of regular and timely prenatal care cannot be understated.
In addition, the prenatal visit gives the obstetrician the opportunity to connect women with other specialists for management of any unmet medical needs. One study showed that, when women have adequate prenatal care (measured by the number of visits), the association between preterm birth and self-reported maternal depression was eliminated.34
Substance use
Substance use and depression often co-exist.35,36 Unlike screening for depression, screening for substance use is universal during prenatal care. Studies have shown that women who screen positive for depression are at higher risk of a number of comorbidities, including substance use.37,38 Conversely, women who use substances are more likely to screen positive for depression.
Evidence suggests that best practice might be to screen for depression in any woman who has a positive drug screen, if a provider is not routinely screening their general patient population.39 Substance use in pregnancy is associated with a number of poor outcomes, including placental abruption (cocaine use); dysmorphic facies and congenital anomalies (alcohol); and neonatal abstinence syndrome (heroin).
Antidepressants in pregnancy
A full discussion of the risks and benefits associated with pharmacotherapy for depression in pregnancy is beyond the scope of this article. Generally, antidepressant use is fraught with concerns over teratogenicity and adverse fetal outcomes. Although ACOG states that (1) pharmacotherapy for depression should be individualized and (2) most selective serotonin reuptake inhibitors (SSRIs) are not considered major teratogenic agents, many obstetricians and patients feel uncomfortable using these medications in pregnancy.40 Often, pre-pregnancy antidepressants are discontinued in the first trimester; one large population-based study found that only 0.9% of women who had depression filled their antidepressant prescription consistently throughout their pregnancy.41
It is unclear whether antidepressant use in pregnancy contributes to the risk of preterm birth seen in women who have depression. In a large population-based study, use of antidepressants in the second trimester was associated with preterm delivery but severe depression was not.18 A recent meta-analysis revealed an increased risk of preterm birth in women who used an antidepressant, compared with healthy women and untreated depressed women.42
Research limits, unanswered questions. Regrettably, it is difficult to untangle risk factors for preterm birth among depressed women without randomized controlled studies that are not ethically feasible. It cannot be said with certainty whether antidepressant pharmacotherapy is associated with a higher risk of preterm birth than depression alone.
Likewise, it is difficult to clarify the extent to which antidepressants contribute to infant growth restriction, if at all. Two recent meta-analyses concluded that exposure to antidepressants is associated with a statistically significant risk of LBW.42,43 However, increased severity of depressive symptoms generally is associated with exposure to antidepressants during pregnancy, and a randomized controlled trial is, again, impossible to conduct for ethical reasons.
Whereas a plausible biological mechanism associating IUGR, SGA, and LBW with depression exists, the same cannot be said for antidepressants. In one study, exposure to maternal depression altered the expression of certain placental genes but exposure to SSRIs did not cause further changes. This suggests that, on a cellular level, placental function might differ in depressed women.44 Although antidepressants do cross the placenta, it remains to be seen whether fetal growth is impacted as a result. One study found decreased fetal head circumference in infants who had been exposed to antidepressants during pregnancy, but no increased risk for having a SGA or LWB infant.45
Obstetrical management and mental health implications
Treated or not, women who suffer depression are a high-risk group when it comes to preterm birth and a host of other pregnancy comorbidities. Women with serious complications of pregnancy often are hospitalized for observation, and can undergo a prolonged stay when close proximity to medical services or a surgical suite is required.
For example, hospitalization until delivery is the standard of care for women who have preterm premature rupture of membranes or preeclampsia before 34 weeks’ gestation. Prolonged inpatient admissions and associated restriction of activity is profoundly deleterious on mood, with depression and anxiety significantly correlated with length of stay.46,47 Given the associations between depression and preterm birth, it might be reasonable to consider screening antenatal inpatients at risk of preterm birth for depression on a regular basis, so that treatment can be initiated if needed.
Depression during pregnancy is relatively common; an estimated 12.7% of pregnant women are affected at some time between conception and birth.48 Not only does depression appear to have deleterious effects on pregnancy outcomes, it also plays a pivotal role in the qualitative experience of pregnancy for the mother.
Bottom Line
Awareness of obstetrical complications associated with depression in pregnancy is important for the entire care team, including the psychiatrist and obstetrician. Depression not only appears to have deleterious effects on pregnancy outcomes, it also plays a pivotal role in the qualitative experience of pregnancy for the mother. Antidepressant use generally is fraught with concerns over teratogenicity and adverse fetal outcomes.
Related Resources
• Freeman MP. Some SSRIs are better than others for pregnant women (audio interview). Current Psychiatry. 2014;13(7). http://www.currentpsychiatry.com/specialty-focus/practice-trends/article/some-ssris-are-better-thanothers-for-pregnant-women/e3adb4704e25492f3e15331fc1cc058d.html.
• Freeman MP, Joffe H, Cohen LS. Postpartum depression: Help patients find the right treatment. Current Psychiatry. 2012;11(11):14-16,19-21.
Disclosures
Dr. Habecker reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Freeman is a member of the advisory board of JDS Therapeutics, Sunovion Pharmaceuticals, Inc., and Takeda Pharmaceutical Co. She receives research grant support from Takeda Pharmaceutical Co.
When a patient who has a preexisting medical illness seeks prenatal care, the obstetrician asks herself (himself) 2 questions:
• What impact will the illness have on the pregnancy?
• What impact will the pregnancy have on the illness?
Depression is both a pregnancy-associated and pregnancy-independent illness, which, in the setting of a pregnant woman who has a depressive disorder, makes these questions particularly difficult to answer. In such a case, coordination of care with a mental health provider is essential.
Awareness of the obstetrical complications associated with depression during pregnancy, as well as their implications for the future health of the mother–infant dyad, is important for the entire care team. This article reviews the associations and interconnectedness of depression with complications of pregnancy, childbirth, and the neonatal period.
Diagnosis of depression during prenatal care
The American College of Obstetricians and Gynecologists (ACOG) states that evidence is insufficient to support a recommendation for universal screening for depression among prenatal patients, although such screening should be considered.1 There is considerable variability among obstetrical providers regarding the practice of depression screening; tools to be used if such screening is done; and screening frequency through the pregnancy.
Discernment of depression is difficult. Many somatic symptoms of depression overlap with common prenatal complaints and, consequentially, can be overlooked. Among a sample of 700 pregnant women, for example, 56% complained of lack of energy; 19%, of insomnia; and 19%, of appetite changes.2 Weight change, of course, is universal.
The 10-question self-rating Edinburgh Postnatal Depression Scale has been validated for use during pregnancy and postnatally. This screening instrument can be helpful for differentiating purely physical complaints from mental distress due to depressive symptoms.2,3
When an obstetrical provider suspects a depressive disorder, or one has been diagnosed, she (he) faces the problem of what to do with that information. Women of low socioeconomic status and victims of domestic violence are at increased risk of depression during pregnancy, but barriers to appropriate referral can seem nearly insurmountable because they lack insurance and social support.4-9
In addition, within the setting of numerous tasks that need attending during the relatively short prenatal period, it is common for women newly given a diagnosis of depression to fail to follow up on a referral to a mental health provider.
Although most providers will “check in” with a depressed or at-risk patient at each prenatal visit about her mood, any effort at follow-up can be overshadowed by tangible physical concerns, such as preterm contractions, fetal growth restriction, and coordination of routine testing that has been delayed because of scant prenatal care. All these physical concerns and circumstances of care are associated with maternal depression, as we will discuss.
Preterm labor and birth
Preterm labor is defined as uterine contractions that lead to cervical change before 37 weeks gestational age. Preterm labor increases the risk of preterm birth; preterm labor precedes 50% of preterm births. Preterm birth is the leading cause of neonatal mortality in the United States, and rates of morbidity and mortality increase as gestational age decreases.10 Common neonatal complications related to prematurity are shown in the Figure.11
Women who suffer from depression have an increased risk of preterm labor and preterm birth, as many studies of treated and untreated depressed pregnant women have shown.12-20 The causative mechanism is unknown; it has been proposed that the increase in maternal cortisol production associated with depression and distress triggers overproduction of placental cortisol releasing hormone, which is thought to be involved in initiation of parturition.21,22 Depression also is associated with other risk factors for preterm birth, such as low socioeconomic status, substance use, and smoking.
Intrauterine growth restriction
Women who have depression during pregnancy have an increased risk of intrauterine growth restriction (IUGR), which leads to delivery of an infant who is small for gestational age (SGA) or of low birth weight (LBW) (weighing <2,500 g at birth), or both.23 Again, the basis of the association between depression and IUGR and SGA is unknown; it is theorized that increased levels of cortisol and catecholamines associated with maternal distress might, by increasing blood pressure and inducing vasoconstriction, cause placental hypoperfusion.24,25
It also is possible that the association of depression with other risk factors for IUGR, such as smoking, substance use, obesity, and poor prenatal care, puts the infants of depressed women at risk of growth restriction.26 Several large-scale studies showed that the association between LBW and depression is lost when smoking and substance use are accounted for; other studies, however, found a persistent association in untreated depressed women when smokers, substance users, and drinkers were excluded.17,26,27
IUGR infants are at increased risk of iatrogenic prematurity and stillbirth. Fetuses that weigh <10th percentile for their gestational age are delivered no later than 40 weeks; delivery can be indicated as early as 32 weeks, depending on the results of other antenatal tests. Women who have a growth-restricted infant have a higher risk of cesarean delivery because growth-restricted infants often have less reserve and poorer tolerance of labor.
Preeclampsia and eclampsia
Preeclampsia is defined as blood pressure >140/90 mm HG on at least 2 occasions, with proteinuria, that occurs later than the twentieth week of pregnancy in women who did not have hypertension or renal dysfunction at baseline. Preeclampsia is a progressive disease that can cause severe maternal morbidity, including renal failure, stroke, hepatic rupture, pulmonary edema, and heart failure.
Eclampsia refers to onset of seizures in the setting of preeclampsia. These 2 hypertensive disorders are the third leading world wide cause of maternal mortality.28
Depressed women have an elevated risk of preeclampsia. The association between preeclampsia and depression might be caused by the presence of increased levels of inflammatory mediators29,30; other comorbidities, such as increased body mass index, also might be involved, but the risk for preeclampsia in depressed women still is increased after controlling for obesity.31
The presence of preeclampsia is responsible for a high percentage of iatrogenic preterm births, because the cure for the disorder is delivery—even at early or previable gestational age. Complication rates for mother and infant are high.
The presence of preeclampsia is a significant risk factor for intrauterine fetal demise. Treating the mother after delivery involves administration of IV magnesium for 24 hours; often, the mother is separated from her infant for a day after birth.
Impact on prenatal care
Depression increases odds that women will have fewer prenatal visits.32 During pregnancy, women typically initiate prenatal care during the first trimester, when pregnancy-dating ultrasonography and early screening tests for chromosomal abnormalities are performed. Prenatal visits occur monthly until the third trimester, then every 2 weeks between 32 and 36 weeks’ gestation, increasing to weekly after 36 weeks’ gestation.
The increased number of visits in late pregnancy allows for early detection and treatment of hypertensive disorders; assesses fetal well-being; and decreases the risks of morbidity and mortality for mother and fetus.33 Because women who suffer from depression are at increased risk of an array of adverse pregnancy outcomes, the importance of regular and timely prenatal care cannot be understated.
In addition, the prenatal visit gives the obstetrician the opportunity to connect women with other specialists for management of any unmet medical needs. One study showed that, when women have adequate prenatal care (measured by the number of visits), the association between preterm birth and self-reported maternal depression was eliminated.34
Substance use
Substance use and depression often co-exist.35,36 Unlike screening for depression, screening for substance use is universal during prenatal care. Studies have shown that women who screen positive for depression are at higher risk of a number of comorbidities, including substance use.37,38 Conversely, women who use substances are more likely to screen positive for depression.
Evidence suggests that best practice might be to screen for depression in any woman who has a positive drug screen, if a provider is not routinely screening their general patient population.39 Substance use in pregnancy is associated with a number of poor outcomes, including placental abruption (cocaine use); dysmorphic facies and congenital anomalies (alcohol); and neonatal abstinence syndrome (heroin).
Antidepressants in pregnancy
A full discussion of the risks and benefits associated with pharmacotherapy for depression in pregnancy is beyond the scope of this article. Generally, antidepressant use is fraught with concerns over teratogenicity and adverse fetal outcomes. Although ACOG states that (1) pharmacotherapy for depression should be individualized and (2) most selective serotonin reuptake inhibitors (SSRIs) are not considered major teratogenic agents, many obstetricians and patients feel uncomfortable using these medications in pregnancy.40 Often, pre-pregnancy antidepressants are discontinued in the first trimester; one large population-based study found that only 0.9% of women who had depression filled their antidepressant prescription consistently throughout their pregnancy.41
It is unclear whether antidepressant use in pregnancy contributes to the risk of preterm birth seen in women who have depression. In a large population-based study, use of antidepressants in the second trimester was associated with preterm delivery but severe depression was not.18 A recent meta-analysis revealed an increased risk of preterm birth in women who used an antidepressant, compared with healthy women and untreated depressed women.42
Research limits, unanswered questions. Regrettably, it is difficult to untangle risk factors for preterm birth among depressed women without randomized controlled studies that are not ethically feasible. It cannot be said with certainty whether antidepressant pharmacotherapy is associated with a higher risk of preterm birth than depression alone.
Likewise, it is difficult to clarify the extent to which antidepressants contribute to infant growth restriction, if at all. Two recent meta-analyses concluded that exposure to antidepressants is associated with a statistically significant risk of LBW.42,43 However, increased severity of depressive symptoms generally is associated with exposure to antidepressants during pregnancy, and a randomized controlled trial is, again, impossible to conduct for ethical reasons.
Whereas a plausible biological mechanism associating IUGR, SGA, and LBW with depression exists, the same cannot be said for antidepressants. In one study, exposure to maternal depression altered the expression of certain placental genes but exposure to SSRIs did not cause further changes. This suggests that, on a cellular level, placental function might differ in depressed women.44 Although antidepressants do cross the placenta, it remains to be seen whether fetal growth is impacted as a result. One study found decreased fetal head circumference in infants who had been exposed to antidepressants during pregnancy, but no increased risk for having a SGA or LWB infant.45
Obstetrical management and mental health implications
Treated or not, women who suffer depression are a high-risk group when it comes to preterm birth and a host of other pregnancy comorbidities. Women with serious complications of pregnancy often are hospitalized for observation, and can undergo a prolonged stay when close proximity to medical services or a surgical suite is required.
For example, hospitalization until delivery is the standard of care for women who have preterm premature rupture of membranes or preeclampsia before 34 weeks’ gestation. Prolonged inpatient admissions and associated restriction of activity is profoundly deleterious on mood, with depression and anxiety significantly correlated with length of stay.46,47 Given the associations between depression and preterm birth, it might be reasonable to consider screening antenatal inpatients at risk of preterm birth for depression on a regular basis, so that treatment can be initiated if needed.
Depression during pregnancy is relatively common; an estimated 12.7% of pregnant women are affected at some time between conception and birth.48 Not only does depression appear to have deleterious effects on pregnancy outcomes, it also plays a pivotal role in the qualitative experience of pregnancy for the mother.
Bottom Line
Awareness of obstetrical complications associated with depression in pregnancy is important for the entire care team, including the psychiatrist and obstetrician. Depression not only appears to have deleterious effects on pregnancy outcomes, it also plays a pivotal role in the qualitative experience of pregnancy for the mother. Antidepressant use generally is fraught with concerns over teratogenicity and adverse fetal outcomes.
Related Resources
• Freeman MP. Some SSRIs are better than others for pregnant women (audio interview). Current Psychiatry. 2014;13(7). http://www.currentpsychiatry.com/specialty-focus/practice-trends/article/some-ssris-are-better-thanothers-for-pregnant-women/e3adb4704e25492f3e15331fc1cc058d.html.
• Freeman MP, Joffe H, Cohen LS. Postpartum depression: Help patients find the right treatment. Current Psychiatry. 2012;11(11):14-16,19-21.
Disclosures
Dr. Habecker reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Freeman is a member of the advisory board of JDS Therapeutics, Sunovion Pharmaceuticals, Inc., and Takeda Pharmaceutical Co. She receives research grant support from Takeda Pharmaceutical Co.
1. American College of Obstetricians and Gynecologists. Committee on Obstetric Practice. Committee opinion no. 630. 2015;125:1268-1271.
2. Apter G, Devouche E, Garez V, et al. Pregnancy, somatic complaints and depression: a French population-based study. Eur J Obstet Gynecol Reprod Biol. 2013;171(1):35-39.
3. Murray D, Cox JL. Screening for depression during pregnancy with the Edinburgh Depression Scale (EDDS). J Reprod Infant Psychol. 1990;8(2):99-107.
4. Gotlib IH, Whiffen VE, Mount JH, et al. Prevalence rates and demographic characteristics associated with depression in pregnancy and the postpartum. J Consult Clin Psychol. 1989;57(2):269-274.
5. Melville JL, Gavin A, Guo Y, et al. Depressive disorders during pregnancy: prevalence and risk factors in a large urban sample. Obstet Gynecol. 2010;116(5):1064-1070.
6. Leddy M, Haaga D, Gray J, et al. Postpartum mental health screening and diagnosis by obstetrician-gynecologists. J Psychosom Obstet Gynaecol. 2011;32(1):27-34.
7. McFarlane J, Maddoux J, Cesario S, et al. Effect of abuse during pregnancy on maternal and child safety and functioning for 24 months after delivery. Obstet Gynecol. 2014;123(4):839-847.
8. Vesga-López O, Bianco C, Keyes K, et al. Psychiatric disorders in pregnant and postpartum women in the United States. Arch Gen Psychiatry. 2008;65(7):805-815.
9. Farr SL, Bitsko RH, Hayes DK, et al. Mental health and access to services among US women of reproductive age. Am J Obstet Gynecol. 2010;203(6):542.e1-e542.e9. doi: 10.1016/j.ajog.2010.07.007.
10. Committee on Practice Bulletins—Obstetrics; The American College of Obstetricians and Gynecologists. Practice bulletin no. 130: prediction and prevention of preterm birth. Obstet Gynecol. 2012;120(4):964-973.
11. Stoll BJ, Hansen NI, Bell EF, et al; Eunice Kennedy Shriver National Institute of Child Health and Human Development Neonatal Research Network. Neonatal outcomes of extremely preterm infants from the NICHD Neonatal Research Network. Pediatrics. 2010;126(3):443-456.
12. Steer RA, Scholl TO, Hediger ML, et al. Self-reported depression and negative pregnancy outcomes. J Clin Epidemiol. 1992;45(10):1093-1099.
13. Goldenberg RL, Cliver SP, Mulvihill FX, et al. Medical, psychosocial, and behavioral risk factors do not explain the increased risk for low birth weight among black women. Am J Obstet Gynecol. 1996;175(5):1317-1324.
14. Orr ST, James SA, Blackmore Prince C. Maternal prenatal depressive symptoms and spontaneous preterm births among African-American women in Baltimore, Maryland. Am J Epidemiol. 2002;156(9):797-802.
15. Dayan J, Creveuil C, Marks MN, et al. Prenatal depression, prenatal anxiety, and spontaneous preterm birth: a prospective cohort study among women with early and regular care. Psychosom Med. 2006;68(6):938-946.
16. Goedhart G, Snijders AC, Hesselink AE, et al. Maternal depressive symptoms in relation to perinatal mortality and morbidity: results from a large multiethnic cohort study. Psychosom Med. 2010;72(8):769-776.
17. Grote NK, Bridge JA, Gavin AR, et al. A meta-analysis of depression during pregnancy and the risk of preterm birth, low birth weight, and intrauterine growth restriction. Arch Gen Psychiatry. 2010;67(10):1012-1024.
18. Hayes RM, Wu P, Shelton RC, et al. Maternal antidepressant use and adverse outcomes: a cohort study of 228,876 pregnancies [published online April 30, 2012]. Am J Obstet Gynecol. 2012;207(1):49.e1-49.e9. doi: 10.1016/j. ajog.2012.04.028.
19. McDonagh MS, Matthews A, Phillipi C, et al. Depression drug treatment outcomes in pregnancy and the postpartum period: a systematic review and meta-analysis. Obstet Gynecol. 2014;124(3):526-534.
20. Sahingöz M, Yuksel G, Karsidag C, et al. Birth weight and preterm birth in babies of pregnant women with major depression in relation to treatment with antidepressants. J Clin Psychopharmacol. 2014;34(2):226-229.
1. American College of Obstetricians and Gynecologists. Committee on Obstetric Practice. Committee opinion no. 630. 2015;125:1268-1271.
2. Apter G, Devouche E, Garez V, et al. Pregnancy, somatic complaints and depression: a French population-based study. Eur J Obstet Gynecol Reprod Biol. 2013;171(1):35-39.
3. Murray D, Cox JL. Screening for depression during pregnancy with the Edinburgh Depression Scale (EDDS). J Reprod Infant Psychol. 1990;8(2):99-107.
4. Gotlib IH, Whiffen VE, Mount JH, et al. Prevalence rates and demographic characteristics associated with depression in pregnancy and the postpartum. J Consult Clin Psychol. 1989;57(2):269-274.
5. Melville JL, Gavin A, Guo Y, et al. Depressive disorders during pregnancy: prevalence and risk factors in a large urban sample. Obstet Gynecol. 2010;116(5):1064-1070.
6. Leddy M, Haaga D, Gray J, et al. Postpartum mental health screening and diagnosis by obstetrician-gynecologists. J Psychosom Obstet Gynaecol. 2011;32(1):27-34.
7. McFarlane J, Maddoux J, Cesario S, et al. Effect of abuse during pregnancy on maternal and child safety and functioning for 24 months after delivery. Obstet Gynecol. 2014;123(4):839-847.
8. Vesga-López O, Bianco C, Keyes K, et al. Psychiatric disorders in pregnant and postpartum women in the United States. Arch Gen Psychiatry. 2008;65(7):805-815.
9. Farr SL, Bitsko RH, Hayes DK, et al. Mental health and access to services among US women of reproductive age. Am J Obstet Gynecol. 2010;203(6):542.e1-e542.e9. doi: 10.1016/j.ajog.2010.07.007.
10. Committee on Practice Bulletins—Obstetrics; The American College of Obstetricians and Gynecologists. Practice bulletin no. 130: prediction and prevention of preterm birth. Obstet Gynecol. 2012;120(4):964-973.
11. Stoll BJ, Hansen NI, Bell EF, et al; Eunice Kennedy Shriver National Institute of Child Health and Human Development Neonatal Research Network. Neonatal outcomes of extremely preterm infants from the NICHD Neonatal Research Network. Pediatrics. 2010;126(3):443-456.
12. Steer RA, Scholl TO, Hediger ML, et al. Self-reported depression and negative pregnancy outcomes. J Clin Epidemiol. 1992;45(10):1093-1099.
13. Goldenberg RL, Cliver SP, Mulvihill FX, et al. Medical, psychosocial, and behavioral risk factors do not explain the increased risk for low birth weight among black women. Am J Obstet Gynecol. 1996;175(5):1317-1324.
14. Orr ST, James SA, Blackmore Prince C. Maternal prenatal depressive symptoms and spontaneous preterm births among African-American women in Baltimore, Maryland. Am J Epidemiol. 2002;156(9):797-802.
15. Dayan J, Creveuil C, Marks MN, et al. Prenatal depression, prenatal anxiety, and spontaneous preterm birth: a prospective cohort study among women with early and regular care. Psychosom Med. 2006;68(6):938-946.
16. Goedhart G, Snijders AC, Hesselink AE, et al. Maternal depressive symptoms in relation to perinatal mortality and morbidity: results from a large multiethnic cohort study. Psychosom Med. 2010;72(8):769-776.
17. Grote NK, Bridge JA, Gavin AR, et al. A meta-analysis of depression during pregnancy and the risk of preterm birth, low birth weight, and intrauterine growth restriction. Arch Gen Psychiatry. 2010;67(10):1012-1024.
18. Hayes RM, Wu P, Shelton RC, et al. Maternal antidepressant use and adverse outcomes: a cohort study of 228,876 pregnancies [published online April 30, 2012]. Am J Obstet Gynecol. 2012;207(1):49.e1-49.e9. doi: 10.1016/j. ajog.2012.04.028.
19. McDonagh MS, Matthews A, Phillipi C, et al. Depression drug treatment outcomes in pregnancy and the postpartum period: a systematic review and meta-analysis. Obstet Gynecol. 2014;124(3):526-534.
20. Sahingöz M, Yuksel G, Karsidag C, et al. Birth weight and preterm birth in babies of pregnant women with major depression in relation to treatment with antidepressants. J Clin Psychopharmacol. 2014;34(2):226-229.

Malignant catatonia and aphasia follow multiple-drug overdose
CASE Improvement, then decline
Ms. M, age 37, is brought to the hospital after her husband found her at home, after an unknown duration of impaired consciousness. Her husband reports that Ms. M had normal cognitive functioning before this event, with no difficulty completing activities of daily living. Ms. M’s medical and psychiatric histories are notable for type 2 diabetes mellitus, unspecified bipolar disorder, and opioid, cocaine, and alcohol use disorders. Her medications include paroxetine, 40 mg/d, and gabapentin, 1,200 mg/d.
First admission. Poor inspiratory effort and oxygen saturation of 70% leads to emergent intubation. Serum laboratory studies reveal a white blood cell (WBC) count at 10,900/μL and creatinine phosphokinase level of 25,000 U/L. Urine drug screen is positive for tetrahydrocannabinol, cocaine, and opioids.
Ms. M is admitted to the ICU for management of rhabdomyolysis and multi-organ system failure, including acute hypoxic kidney injury.
By hospital Day 7, the tube is extubated with no recorded physical neurologic deficits. Mental status exam is normal, except for impaired memory of events surrounding the admission. Ms. M is discharged home with a recommendation for outpatient follow-up.
2 Weeks later. Ms. M is brought to the emergency department after a progressive decrease in social interaction, limited oral intake, decline in activities of daily living, and urinary incontinence. Results from laboratory studies are within normal limits; brain MRI is negative; EEG shows generalized moderate slowing.
During psychiatric evaluation, Ms. M is mute and staring continuously. Examination reveals oppositional paratonia (gegenhalten), catalepsy, prominent negativism, and waxy flexibility, all suggestive of catatonia. IV lorazepam is initiated at 1 mg every 8 hours, titrated to 2 mg, 3 times a day.
Ms. M is transferred to a psychiatric hospital for further treatment of catatonia.
Second admission. Evaluation with the Bush-Francis Catatonia Rating Scale supported a diagnosis of catatonia, with the presence of >3 features from the 14-item screen and a score of 16 on the 23-item rating scale.1 After titrating lorazepam to 9 mg/d with minimal therapeutic impact, the psychiatry team consults the electroconvulsive therapy (ECT) service, who deems Ms. M to be an appropriate candidate and petitions for court-ordered ECT.
On hospital Day 8, Ms. M has a fever of 104°F, tachycardia at 180 beats per minute, increased rigidity, and a WBC count of 17,800/μL. She is transferred to the ICU, with a presumptive diagnosis of malignant catatonia.
The medical evaluation, including general laboratory studies, EEG, and spinal fluid analysis, is unremarkable. Because of vital sign instability, 2 ECT treatments are completed in the general hospital before Ms. M resumes psychiatric inpatient care.
By the tenth ECT treatment, Ms. M is no longer febrile and experiences no further autonomic instability or psychomotor features of catatonia. Despite these improvements, she is noted to have persistent word-finding difficulty.
Which test would you order as the next step in your work up?
a) EEG
b) lumbar puncture
c) MRI
d) CT
The authors’ observations
In approximately 25% of cases, catatonia is caused by a general medical condition2; as such, a comprehensive medical workup is vital for assessment and management of catatonic patients. In Ms. M’s case, we considered several medical causes, including nutritional deficiency, infection, a toxin, renal or hepatic impairment, hypothyroidism, seizure, and stroke. Evaluation included measurement of thyroid-stimulating hormone, vitamin B12, and folic acid levels; urinalysis and urine drug screen; chest radiography; lumbar puncture; neuroimaging; and EEG (Table 1).
Several conditions in the differential diagnosis were noteworthy. Ms. M’s severe and sudden neurologic decline, along with a positive urine drug screen for substances of abuse, raised concern about overdose leading to toxic encephalopathy or hypoxic brain injury. Ms. M’s oxygen saturation when she was found was moderately hypoxic at 70%, which is not a level associated with hypoxic brain damage.
We also considered posterior reversible encephalopathy syndrome (PRES), which presents variably with nausea, visual impairment, disturbance in consciousness, seizures, and focal neurologic signs.3 Although 67% to 80% of patients with PRES also have acute hypertension, blood pressure elevation is not necessary for the diagnosis.4 Similar to toxic leukoencephalopathy, PRES is diagnosed by brain MRI, with classic signs of posterior white-matter edema.
Case reports also describe an uncommon demyelinating syndrome, delayed post-hypoxic leukoencephalopathy (DPHL), which develops several weeks or months after a cerebral anoxic insult.5 In Ms. M’s case, brain MRI performed during her second medical hospitalization, 7 days after the initial neuropsychiatric decline, was unremarkable. Using this result to rule out DPHL would have been premature because pathognomonic abnormalities can appear as long as 40 days after the anoxic insult. Given our differential diagnosis, we ordered a repeat MRI.
Etiology and pathophysiology
First described in 1979, DPHL is rare, posing diagnostic challenges for clinical providers.6 Although the exact incidence of DPHL is unknown, the precipitating event typically involves cerebral anoxia, which can occur through carbon monoxide (CO) poisoning, strangulation, cardiac arrest, respiratory failure, and overdose from sedatives and narcotics (Table 2).7 DPHL was first observed in a small percentage (2.75%) of patients suffering from CO poisoning.8,9 Progression of the disease generally includes a period of unconsciousness, then a lucid interval that can last 2 to 40 days, followed by the abrupt onset of neuropsychiatric symptoms.10 The specific pathophysiologic mechanism is unknown, but has been hypothesized to involve inferior compensatory response to decreased oxygenation in the white matter.

Diagnosis and clinical features
DPHL can be divided into 2 clinical variations: parkinsonism and akinetic mutism. The former consists of conventional parkinsonian features along with agitation, apathy, hallucinations, dystonic posturing, and odd behaviors. The latter variant presents with apathy, minimal response to pain, functional bowel and bladder incontinence, mutism, and, at times, inappropriate laughter or tearfulness.5 Both variants share similar features with hypokinetic forms of catatonia.
DPHL is a diagnosis of exclusion. A careful history is critical to establish the possibility of a recent anoxic event. MRI findings, including hyperintensities in the cerebral white matter on T2-based sequencing, are suggestive of the disease. A choline peak on magnetic resonance spectroscopy also might be present in patients with DPHL, although it is not specific to the diagnosis.
Early reports of DPHL suggested an associated deficiency of arylsulfatase A, an enzyme required in the modulation of myelin; however, more recent case reports are conflicting.11 Familial mutations in the gene for arylsulfatase A also result in metachromatic leukodystrophy, and adult onset can present with psychiatric symptoms, including delusions and hallucinations.12
Treatment and prognosis
The treatment of DPHL consists primarily of supportive care and rehabilitation with physical, occupational, and speech therapy.11 With these measures, most patients improve after 3 to 6 months; however, a large percentage sustain some long-term cognitive deficit, the most prevalent symptom being frontal executive dysfunction.5
OUTCOME Supportive care
A second MRI shows diffuse hyperintensities in the white matter that spare the cerebellum and brainstem (Figure). This finding is pathognomonic for DPHL.
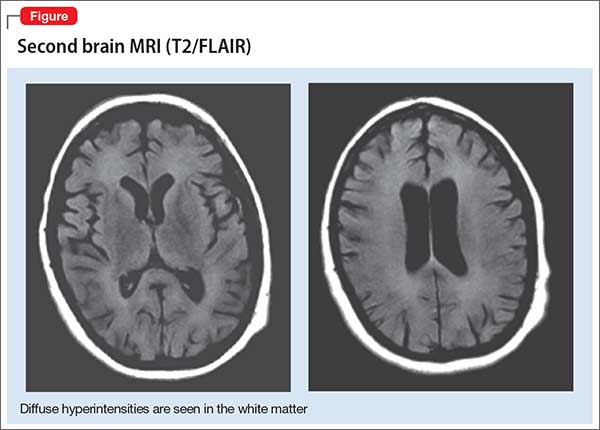
ECT is discontinued because there is no evidence to support ECT-associated improvement in DPHL. Moreover, ECT might worsen the clinical course through increased stress and metabolic demand on the brain.13
Because the primary treatment of DPHL is early rehabilitation, we consider that Ms. M would benefit most from increased supportive care and therapy. She is discharged to a brain injury rehabilitation facility, where metoprolol is prescribed for mild tachycardia, along with thiamine and vitamins B12 and D. Physical, occupational, and speech therapy are continued.
Approximately 3 weeks after admission to the rehabilitation program, Ms. M is discharged home. Although she improves in overall activities of daily living, she continues to experience moderate communication deficits and occasional external distractibility.
Bottom Line
Although delayed post-hypoxic leukoencephalopathy is considered rare, consider it in the differential diagnosis when a patient has a recent history of an anoxic event followed by the abrupt onset of neuropsychiatric symptoms. Keep in mind that the condition can be missed if an MRI is obtained too early, and the clinical signs can mimic hypokinetic catatonia.
Related Resources
• Meyer MA. Delayed post-hypoxic leukoencephalopathy: case report with a review of disease pathophysiology. Neurol Int. 2013;5(3):e13. doi: 10.4081/ni.2013.e13.
• Aljarallah S, Al-Hussain F. Acute fatal posthypoxic leukoencephalopathy following benzodiazepine overdose: a case report and review of the literature. BMC Neurol. 2015;15:69.
Drug Brand Names
Gabapentin • Neurontin
Lorazepam • Ativan
Metoprolol • Lopressor
Paroxetine • Paxil
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of com
1. Bush G, Fink M, Petrides G, et al. Catatonia. I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
2. Azzam PN, Gopalan P. Prototypes of catatonia: diagnostic and therapeutic challenges in the general hospital. Psychosomatics. 2013;54(1):88-93.
3. Tormoehlen LM. Toxic leukoencephalopathies. Neurol Clin. 2011;29(3):591-605
4. Legriel S, Pico F, Azoulay E. Understanding posterior reversible encephalopathy syndrome. In: Vincent JL, ed. Annual update in intensive care and emergency medicine. Heidelberg, Germany: Springer Berlin Heidelberg; 2011:631-653.
5. Schprecher D, Mehta L. The syndrome of delayed post-hypoxic leukoencephalopathy. NeuroRehabilitation. 2010;26(1):65-72.
6. Wallace IR, Dynan C, Esmonde T. One confused patient, many confused physicians: a case of delayed post-hypoxic leucoencephalopathy. QJM. 2010;103(3):193-194.
7. Lou M, Jing CH, Selim MH, et al. Delayed substantia nigra damage and leukoencephalopathy after hypoxic-ischemic injury. J Neurol Sci. 2009;277(1-2):147-149.
8. Choi IS. Delayed neurologic sequelae in carbon monoxide intoxication. Arch Neurol. 1983;40(7):433-435.
9. Molloy S, Soh C, Williams TL. Reversible delayed posthypoxic leukoencephalopathy. AJNR Am J Neuroradiol. 2006;27(8):1763-1765.
10. Shprecher DR, Flanigan KM, Smith AG, et al. Clinical and diagnostic features of delayed hypoxic leukoencephalopathy. J Neuropsychiatry Clin Neurosci. 2008;20(4):473-477.
11. Lee BH, Lyketsos CG. Delayed post-hypoxic leukoencephalopathy. Psychosomatics. 2001;42(6):530-533.
12. Hyde TM, Ziegler JC, Weinberger DR. Psychiatric disturbances in metachromatic leukodystrophy. Insights into the neurobiology of psychosis. Arch Neurol. 1992;49(4):401-406.
13. Quinn DK, Abbott CC. Catatonia after cerebral hypoxia: do the usual treatments apply? Psychosomatics. 2014;55(6):525-535.
CASE Improvement, then decline
Ms. M, age 37, is brought to the hospital after her husband found her at home, after an unknown duration of impaired consciousness. Her husband reports that Ms. M had normal cognitive functioning before this event, with no difficulty completing activities of daily living. Ms. M’s medical and psychiatric histories are notable for type 2 diabetes mellitus, unspecified bipolar disorder, and opioid, cocaine, and alcohol use disorders. Her medications include paroxetine, 40 mg/d, and gabapentin, 1,200 mg/d.
First admission. Poor inspiratory effort and oxygen saturation of 70% leads to emergent intubation. Serum laboratory studies reveal a white blood cell (WBC) count at 10,900/μL and creatinine phosphokinase level of 25,000 U/L. Urine drug screen is positive for tetrahydrocannabinol, cocaine, and opioids.
Ms. M is admitted to the ICU for management of rhabdomyolysis and multi-organ system failure, including acute hypoxic kidney injury.
By hospital Day 7, the tube is extubated with no recorded physical neurologic deficits. Mental status exam is normal, except for impaired memory of events surrounding the admission. Ms. M is discharged home with a recommendation for outpatient follow-up.
2 Weeks later. Ms. M is brought to the emergency department after a progressive decrease in social interaction, limited oral intake, decline in activities of daily living, and urinary incontinence. Results from laboratory studies are within normal limits; brain MRI is negative; EEG shows generalized moderate slowing.
During psychiatric evaluation, Ms. M is mute and staring continuously. Examination reveals oppositional paratonia (gegenhalten), catalepsy, prominent negativism, and waxy flexibility, all suggestive of catatonia. IV lorazepam is initiated at 1 mg every 8 hours, titrated to 2 mg, 3 times a day.
Ms. M is transferred to a psychiatric hospital for further treatment of catatonia.
Second admission. Evaluation with the Bush-Francis Catatonia Rating Scale supported a diagnosis of catatonia, with the presence of >3 features from the 14-item screen and a score of 16 on the 23-item rating scale.1 After titrating lorazepam to 9 mg/d with minimal therapeutic impact, the psychiatry team consults the electroconvulsive therapy (ECT) service, who deems Ms. M to be an appropriate candidate and petitions for court-ordered ECT.
On hospital Day 8, Ms. M has a fever of 104°F, tachycardia at 180 beats per minute, increased rigidity, and a WBC count of 17,800/μL. She is transferred to the ICU, with a presumptive diagnosis of malignant catatonia.
The medical evaluation, including general laboratory studies, EEG, and spinal fluid analysis, is unremarkable. Because of vital sign instability, 2 ECT treatments are completed in the general hospital before Ms. M resumes psychiatric inpatient care.
By the tenth ECT treatment, Ms. M is no longer febrile and experiences no further autonomic instability or psychomotor features of catatonia. Despite these improvements, she is noted to have persistent word-finding difficulty.
Which test would you order as the next step in your work up?
a) EEG
b) lumbar puncture
c) MRI
d) CT
The authors’ observations
In approximately 25% of cases, catatonia is caused by a general medical condition2; as such, a comprehensive medical workup is vital for assessment and management of catatonic patients. In Ms. M’s case, we considered several medical causes, including nutritional deficiency, infection, a toxin, renal or hepatic impairment, hypothyroidism, seizure, and stroke. Evaluation included measurement of thyroid-stimulating hormone, vitamin B12, and folic acid levels; urinalysis and urine drug screen; chest radiography; lumbar puncture; neuroimaging; and EEG (Table 1).
Several conditions in the differential diagnosis were noteworthy. Ms. M’s severe and sudden neurologic decline, along with a positive urine drug screen for substances of abuse, raised concern about overdose leading to toxic encephalopathy or hypoxic brain injury. Ms. M’s oxygen saturation when she was found was moderately hypoxic at 70%, which is not a level associated with hypoxic brain damage.
We also considered posterior reversible encephalopathy syndrome (PRES), which presents variably with nausea, visual impairment, disturbance in consciousness, seizures, and focal neurologic signs.3 Although 67% to 80% of patients with PRES also have acute hypertension, blood pressure elevation is not necessary for the diagnosis.4 Similar to toxic leukoencephalopathy, PRES is diagnosed by brain MRI, with classic signs of posterior white-matter edema.
Case reports also describe an uncommon demyelinating syndrome, delayed post-hypoxic leukoencephalopathy (DPHL), which develops several weeks or months after a cerebral anoxic insult.5 In Ms. M’s case, brain MRI performed during her second medical hospitalization, 7 days after the initial neuropsychiatric decline, was unremarkable. Using this result to rule out DPHL would have been premature because pathognomonic abnormalities can appear as long as 40 days after the anoxic insult. Given our differential diagnosis, we ordered a repeat MRI.
Etiology and pathophysiology
First described in 1979, DPHL is rare, posing diagnostic challenges for clinical providers.6 Although the exact incidence of DPHL is unknown, the precipitating event typically involves cerebral anoxia, which can occur through carbon monoxide (CO) poisoning, strangulation, cardiac arrest, respiratory failure, and overdose from sedatives and narcotics (Table 2).7 DPHL was first observed in a small percentage (2.75%) of patients suffering from CO poisoning.8,9 Progression of the disease generally includes a period of unconsciousness, then a lucid interval that can last 2 to 40 days, followed by the abrupt onset of neuropsychiatric symptoms.10 The specific pathophysiologic mechanism is unknown, but has been hypothesized to involve inferior compensatory response to decreased oxygenation in the white matter.
Diagnosis and clinical features
DPHL can be divided into 2 clinical variations: parkinsonism and akinetic mutism. The former consists of conventional parkinsonian features along with agitation, apathy, hallucinations, dystonic posturing, and odd behaviors. The latter variant presents with apathy, minimal response to pain, functional bowel and bladder incontinence, mutism, and, at times, inappropriate laughter or tearfulness.5 Both variants share similar features with hypokinetic forms of catatonia.
DPHL is a diagnosis of exclusion. A careful history is critical to establish the possibility of a recent anoxic event. MRI findings, including hyperintensities in the cerebral white matter on T2-based sequencing, are suggestive of the disease. A choline peak on magnetic resonance spectroscopy also might be present in patients with DPHL, although it is not specific to the diagnosis.
Early reports of DPHL suggested an associated deficiency of arylsulfatase A, an enzyme required in the modulation of myelin; however, more recent case reports are conflicting.11 Familial mutations in the gene for arylsulfatase A also result in metachromatic leukodystrophy, and adult onset can present with psychiatric symptoms, including delusions and hallucinations.12
Treatment and prognosis
The treatment of DPHL consists primarily of supportive care and rehabilitation with physical, occupational, and speech therapy.11 With these measures, most patients improve after 3 to 6 months; however, a large percentage sustain some long-term cognitive deficit, the most prevalent symptom being frontal executive dysfunction.5
OUTCOME Supportive care
A second MRI shows diffuse hyperintensities in the white matter that spare the cerebellum and brainstem (Figure). This finding is pathognomonic for DPHL.
ECT is discontinued because there is no evidence to support ECT-associated improvement in DPHL. Moreover, ECT might worsen the clinical course through increased stress and metabolic demand on the brain.13
Because the primary treatment of DPHL is early rehabilitation, we consider that Ms. M would benefit most from increased supportive care and therapy. She is discharged to a brain injury rehabilitation facility, where metoprolol is prescribed for mild tachycardia, along with thiamine and vitamins B12 and D. Physical, occupational, and speech therapy are continued.
Approximately 3 weeks after admission to the rehabilitation program, Ms. M is discharged home. Although she improves in overall activities of daily living, she continues to experience moderate communication deficits and occasional external distractibility.
Bottom Line
Although delayed post-hypoxic leukoencephalopathy is considered rare, consider it in the differential diagnosis when a patient has a recent history of an anoxic event followed by the abrupt onset of neuropsychiatric symptoms. Keep in mind that the condition can be missed if an MRI is obtained too early, and the clinical signs can mimic hypokinetic catatonia.
Related Resources
• Meyer MA. Delayed post-hypoxic leukoencephalopathy: case report with a review of disease pathophysiology. Neurol Int. 2013;5(3):e13. doi: 10.4081/ni.2013.e13.
• Aljarallah S, Al-Hussain F. Acute fatal posthypoxic leukoencephalopathy following benzodiazepine overdose: a case report and review of the literature. BMC Neurol. 2015;15:69.
Drug Brand Names
Gabapentin • Neurontin
Lorazepam • Ativan
Metoprolol • Lopressor
Paroxetine • Paxil
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of com
CASE Improvement, then decline
Ms. M, age 37, is brought to the hospital after her husband found her at home, after an unknown duration of impaired consciousness. Her husband reports that Ms. M had normal cognitive functioning before this event, with no difficulty completing activities of daily living. Ms. M’s medical and psychiatric histories are notable for type 2 diabetes mellitus, unspecified bipolar disorder, and opioid, cocaine, and alcohol use disorders. Her medications include paroxetine, 40 mg/d, and gabapentin, 1,200 mg/d.
First admission. Poor inspiratory effort and oxygen saturation of 70% leads to emergent intubation. Serum laboratory studies reveal a white blood cell (WBC) count at 10,900/μL and creatinine phosphokinase level of 25,000 U/L. Urine drug screen is positive for tetrahydrocannabinol, cocaine, and opioids.
Ms. M is admitted to the ICU for management of rhabdomyolysis and multi-organ system failure, including acute hypoxic kidney injury.
By hospital Day 7, the tube is extubated with no recorded physical neurologic deficits. Mental status exam is normal, except for impaired memory of events surrounding the admission. Ms. M is discharged home with a recommendation for outpatient follow-up.
2 Weeks later. Ms. M is brought to the emergency department after a progressive decrease in social interaction, limited oral intake, decline in activities of daily living, and urinary incontinence. Results from laboratory studies are within normal limits; brain MRI is negative; EEG shows generalized moderate slowing.
During psychiatric evaluation, Ms. M is mute and staring continuously. Examination reveals oppositional paratonia (gegenhalten), catalepsy, prominent negativism, and waxy flexibility, all suggestive of catatonia. IV lorazepam is initiated at 1 mg every 8 hours, titrated to 2 mg, 3 times a day.
Ms. M is transferred to a psychiatric hospital for further treatment of catatonia.
Second admission. Evaluation with the Bush-Francis Catatonia Rating Scale supported a diagnosis of catatonia, with the presence of >3 features from the 14-item screen and a score of 16 on the 23-item rating scale.1 After titrating lorazepam to 9 mg/d with minimal therapeutic impact, the psychiatry team consults the electroconvulsive therapy (ECT) service, who deems Ms. M to be an appropriate candidate and petitions for court-ordered ECT.
On hospital Day 8, Ms. M has a fever of 104°F, tachycardia at 180 beats per minute, increased rigidity, and a WBC count of 17,800/μL. She is transferred to the ICU, with a presumptive diagnosis of malignant catatonia.
The medical evaluation, including general laboratory studies, EEG, and spinal fluid analysis, is unremarkable. Because of vital sign instability, 2 ECT treatments are completed in the general hospital before Ms. M resumes psychiatric inpatient care.
By the tenth ECT treatment, Ms. M is no longer febrile and experiences no further autonomic instability or psychomotor features of catatonia. Despite these improvements, she is noted to have persistent word-finding difficulty.
Which test would you order as the next step in your work up?
a) EEG
b) lumbar puncture
c) MRI
d) CT
The authors’ observations
In approximately 25% of cases, catatonia is caused by a general medical condition2; as such, a comprehensive medical workup is vital for assessment and management of catatonic patients. In Ms. M’s case, we considered several medical causes, including nutritional deficiency, infection, a toxin, renal or hepatic impairment, hypothyroidism, seizure, and stroke. Evaluation included measurement of thyroid-stimulating hormone, vitamin B12, and folic acid levels; urinalysis and urine drug screen; chest radiography; lumbar puncture; neuroimaging; and EEG (Table 1).
Several conditions in the differential diagnosis were noteworthy. Ms. M’s severe and sudden neurologic decline, along with a positive urine drug screen for substances of abuse, raised concern about overdose leading to toxic encephalopathy or hypoxic brain injury. Ms. M’s oxygen saturation when she was found was moderately hypoxic at 70%, which is not a level associated with hypoxic brain damage.
We also considered posterior reversible encephalopathy syndrome (PRES), which presents variably with nausea, visual impairment, disturbance in consciousness, seizures, and focal neurologic signs.3 Although 67% to 80% of patients with PRES also have acute hypertension, blood pressure elevation is not necessary for the diagnosis.4 Similar to toxic leukoencephalopathy, PRES is diagnosed by brain MRI, with classic signs of posterior white-matter edema.
Case reports also describe an uncommon demyelinating syndrome, delayed post-hypoxic leukoencephalopathy (DPHL), which develops several weeks or months after a cerebral anoxic insult.5 In Ms. M’s case, brain MRI performed during her second medical hospitalization, 7 days after the initial neuropsychiatric decline, was unremarkable. Using this result to rule out DPHL would have been premature because pathognomonic abnormalities can appear as long as 40 days after the anoxic insult. Given our differential diagnosis, we ordered a repeat MRI.
Etiology and pathophysiology
First described in 1979, DPHL is rare, posing diagnostic challenges for clinical providers.6 Although the exact incidence of DPHL is unknown, the precipitating event typically involves cerebral anoxia, which can occur through carbon monoxide (CO) poisoning, strangulation, cardiac arrest, respiratory failure, and overdose from sedatives and narcotics (Table 2).7 DPHL was first observed in a small percentage (2.75%) of patients suffering from CO poisoning.8,9 Progression of the disease generally includes a period of unconsciousness, then a lucid interval that can last 2 to 40 days, followed by the abrupt onset of neuropsychiatric symptoms.10 The specific pathophysiologic mechanism is unknown, but has been hypothesized to involve inferior compensatory response to decreased oxygenation in the white matter.
Diagnosis and clinical features
DPHL can be divided into 2 clinical variations: parkinsonism and akinetic mutism. The former consists of conventional parkinsonian features along with agitation, apathy, hallucinations, dystonic posturing, and odd behaviors. The latter variant presents with apathy, minimal response to pain, functional bowel and bladder incontinence, mutism, and, at times, inappropriate laughter or tearfulness.5 Both variants share similar features with hypokinetic forms of catatonia.
DPHL is a diagnosis of exclusion. A careful history is critical to establish the possibility of a recent anoxic event. MRI findings, including hyperintensities in the cerebral white matter on T2-based sequencing, are suggestive of the disease. A choline peak on magnetic resonance spectroscopy also might be present in patients with DPHL, although it is not specific to the diagnosis.
Early reports of DPHL suggested an associated deficiency of arylsulfatase A, an enzyme required in the modulation of myelin; however, more recent case reports are conflicting.11 Familial mutations in the gene for arylsulfatase A also result in metachromatic leukodystrophy, and adult onset can present with psychiatric symptoms, including delusions and hallucinations.12
Treatment and prognosis
The treatment of DPHL consists primarily of supportive care and rehabilitation with physical, occupational, and speech therapy.11 With these measures, most patients improve after 3 to 6 months; however, a large percentage sustain some long-term cognitive deficit, the most prevalent symptom being frontal executive dysfunction.5
OUTCOME Supportive care
A second MRI shows diffuse hyperintensities in the white matter that spare the cerebellum and brainstem (Figure). This finding is pathognomonic for DPHL.
ECT is discontinued because there is no evidence to support ECT-associated improvement in DPHL. Moreover, ECT might worsen the clinical course through increased stress and metabolic demand on the brain.13
Because the primary treatment of DPHL is early rehabilitation, we consider that Ms. M would benefit most from increased supportive care and therapy. She is discharged to a brain injury rehabilitation facility, where metoprolol is prescribed for mild tachycardia, along with thiamine and vitamins B12 and D. Physical, occupational, and speech therapy are continued.
Approximately 3 weeks after admission to the rehabilitation program, Ms. M is discharged home. Although she improves in overall activities of daily living, she continues to experience moderate communication deficits and occasional external distractibility.
Bottom Line
Although delayed post-hypoxic leukoencephalopathy is considered rare, consider it in the differential diagnosis when a patient has a recent history of an anoxic event followed by the abrupt onset of neuropsychiatric symptoms. Keep in mind that the condition can be missed if an MRI is obtained too early, and the clinical signs can mimic hypokinetic catatonia.
Related Resources
• Meyer MA. Delayed post-hypoxic leukoencephalopathy: case report with a review of disease pathophysiology. Neurol Int. 2013;5(3):e13. doi: 10.4081/ni.2013.e13.
• Aljarallah S, Al-Hussain F. Acute fatal posthypoxic leukoencephalopathy following benzodiazepine overdose: a case report and review of the literature. BMC Neurol. 2015;15:69.
Drug Brand Names
Gabapentin • Neurontin
Lorazepam • Ativan
Metoprolol • Lopressor
Paroxetine • Paxil
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of com
1. Bush G, Fink M, Petrides G, et al. Catatonia. I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
2. Azzam PN, Gopalan P. Prototypes of catatonia: diagnostic and therapeutic challenges in the general hospital. Psychosomatics. 2013;54(1):88-93.
3. Tormoehlen LM. Toxic leukoencephalopathies. Neurol Clin. 2011;29(3):591-605
4. Legriel S, Pico F, Azoulay E. Understanding posterior reversible encephalopathy syndrome. In: Vincent JL, ed. Annual update in intensive care and emergency medicine. Heidelberg, Germany: Springer Berlin Heidelberg; 2011:631-653.
5. Schprecher D, Mehta L. The syndrome of delayed post-hypoxic leukoencephalopathy. NeuroRehabilitation. 2010;26(1):65-72.
6. Wallace IR, Dynan C, Esmonde T. One confused patient, many confused physicians: a case of delayed post-hypoxic leucoencephalopathy. QJM. 2010;103(3):193-194.
7. Lou M, Jing CH, Selim MH, et al. Delayed substantia nigra damage and leukoencephalopathy after hypoxic-ischemic injury. J Neurol Sci. 2009;277(1-2):147-149.
8. Choi IS. Delayed neurologic sequelae in carbon monoxide intoxication. Arch Neurol. 1983;40(7):433-435.
9. Molloy S, Soh C, Williams TL. Reversible delayed posthypoxic leukoencephalopathy. AJNR Am J Neuroradiol. 2006;27(8):1763-1765.
10. Shprecher DR, Flanigan KM, Smith AG, et al. Clinical and diagnostic features of delayed hypoxic leukoencephalopathy. J Neuropsychiatry Clin Neurosci. 2008;20(4):473-477.
11. Lee BH, Lyketsos CG. Delayed post-hypoxic leukoencephalopathy. Psychosomatics. 2001;42(6):530-533.
12. Hyde TM, Ziegler JC, Weinberger DR. Psychiatric disturbances in metachromatic leukodystrophy. Insights into the neurobiology of psychosis. Arch Neurol. 1992;49(4):401-406.
13. Quinn DK, Abbott CC. Catatonia after cerebral hypoxia: do the usual treatments apply? Psychosomatics. 2014;55(6):525-535.
1. Bush G, Fink M, Petrides G, et al. Catatonia. I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
2. Azzam PN, Gopalan P. Prototypes of catatonia: diagnostic and therapeutic challenges in the general hospital. Psychosomatics. 2013;54(1):88-93.
3. Tormoehlen LM. Toxic leukoencephalopathies. Neurol Clin. 2011;29(3):591-605
4. Legriel S, Pico F, Azoulay E. Understanding posterior reversible encephalopathy syndrome. In: Vincent JL, ed. Annual update in intensive care and emergency medicine. Heidelberg, Germany: Springer Berlin Heidelberg; 2011:631-653.
5. Schprecher D, Mehta L. The syndrome of delayed post-hypoxic leukoencephalopathy. NeuroRehabilitation. 2010;26(1):65-72.
6. Wallace IR, Dynan C, Esmonde T. One confused patient, many confused physicians: a case of delayed post-hypoxic leucoencephalopathy. QJM. 2010;103(3):193-194.
7. Lou M, Jing CH, Selim MH, et al. Delayed substantia nigra damage and leukoencephalopathy after hypoxic-ischemic injury. J Neurol Sci. 2009;277(1-2):147-149.
8. Choi IS. Delayed neurologic sequelae in carbon monoxide intoxication. Arch Neurol. 1983;40(7):433-435.
9. Molloy S, Soh C, Williams TL. Reversible delayed posthypoxic leukoencephalopathy. AJNR Am J Neuroradiol. 2006;27(8):1763-1765.
10. Shprecher DR, Flanigan KM, Smith AG, et al. Clinical and diagnostic features of delayed hypoxic leukoencephalopathy. J Neuropsychiatry Clin Neurosci. 2008;20(4):473-477.
11. Lee BH, Lyketsos CG. Delayed post-hypoxic leukoencephalopathy. Psychosomatics. 2001;42(6):530-533.
12. Hyde TM, Ziegler JC, Weinberger DR. Psychiatric disturbances in metachromatic leukodystrophy. Insights into the neurobiology of psychosis. Arch Neurol. 1992;49(4):401-406.
13. Quinn DK, Abbott CC. Catatonia after cerebral hypoxia: do the usual treatments apply? Psychosomatics. 2014;55(6):525-535.
A decade after the CATIE study, the focus has shifted from effectiveness to neuroprotection
This past September, exactly 10 years after publication of the primary findings of the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study1—namely, that effectiveness (defined as all-cause discontinuation) was not different across first-generation antipsychotics (FGAs) and second generation antipsychotics (SGAs)— a new meta-analysis by Vita et al2 of differences in cortical gray-matter change between those 2 classes of antipsychotics offers a reminder: The clinical focus of the CATIE study overlooked important neurobiological and neuroprotective differences between FGAs and SGAs.
How drastically 1 decade can change the scientific perspective! Vita et al’s meta-analysis and meta-regression encompassed all 18 MRI studies of cortical gray matter in patients with schizophrenia.2 Earlier studies (published between 1983 and 2014) had lumped together patients who were receiving an FGA and those receiving an SGA, and authors reported overall reduction in cortical gray matter with prolonged antipsychotic treatment.
Remarkable findings emerge
When Vita et al2 analyzed FGA- and SGA-treated patients separately, however, they found a significant reduction in cortical gray matter in the FGA group but not in the SGA group. In fact, while higher daily dosages of FGAs were associated with greater reduction in cortical gray matter, higher dosages of SGAs were associated with lower cortical gray matter reduction and, in some samples, with an increase in volume of cortical gray matter.
The researchers hypothesized that the differential effects of FGAs and SGAs might be attributable to the neurotoxicity of typical FGAs and the neuroprotective effect of atypical SGAs.
Hindsight
The key neurobiological difference between FGAs and SGAs reported by Vita et al2 was not addressed in the CATIE study, leading, at that time, to a rush to judgment that all antipsychotics are the same. This conclusion emboldened managed-care organizations to mandate use of older (and cheaper) generic FGAs instead of newer (and more expensive) SGAs— most of which have become available as generic equivalents since the CATIE study was completed.
Investigators in the CATIE study— of which I was one—cannot be blamed for not focusing on neurotoxicity and neuroprotection; those data were not on the psychiatry’s radar when the CATIE study was designed in 1998. The major focus was on whether SGAs (new on the scene in the late 1990s) were more efficacious, safe, and tolerable (that is, more effective) than FGAs.
In fact, the first study reporting that SGAs stimulated neurogenesis (in animals) was published in 2002,3 when the CATIE study was more than half complete. Research into the neuroprotective properties of SGAs then grew rapidly. In fact, the principal investigator of the CATIE study conducted a head-to-head comparison of FGA haloperidol and SGA olanzapine in a sample of first-episode schizophrenia patients4; over 1 year of follow-up, it was determined that patients in the haloperidol-treated group exhibited significant brain volume loss on MRI but those in the olanzapine-treated group did not. This study was published in 2005—the same year the CATIE study was published!
SGAs offer neuroprotection
Over the past decade, the neuroprotective effects of SGAs5 and the neurotoxic effects of FGAs6 have been studied intensively, revealing that SGAs have multiple neuroprotective effects. These effects include:
• stimulation of the production of new brain cells (neurons and glia), known as neurogenesis5,7,8
• an increase in neurotrophic factors, such as nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF),9 which are found at a significantly low level in patients with psychosis10
• reversal of phencyclidine (PCP)-induced changes in gene expression11
• neuroprotection against ischemic stroke12-14
• reversal of PCP-induced loss of dendritic spines in the frontal cortex15
• prevention of oligodendrocyte damage caused by interferon gamma-stimulated microglia16,17
• reversal of loss of dendritic spines in the prefrontal cortex induced by dopamine depletion18
• an anti-inflammatory effect19,20
• protection against β-amyloid and hydrogen peroxide-induced cell death21
• protection against prefrontal cortical neuronal damage caused by dizocilpine (MK-801)22
• reversal of a PCP-induced decrease in the glutathione level and alteration of antioxidant defenses23
• protection of cortical neurons from glutamate neurotoxicity.24
One reason why SGAs are neuroprotective, but FGAs are not, can be attributed to their receptor profiles. FGAs block dopamine D2 receptors far more than serotonin 2A receptors, whereas SGAs do the opposite: They block 5-HT2A receptors 500% to 1,000% more than they block D2 receptors. This difference is associated in turn with a different neurobiological and neuroprotective profiles, such as a decrease or an increase in BDNF.25,26
Neither similar nor interchangeable
Since publication of the findings of the CATIE study, the primary investigator has proposed that neuroprotection can be a therapeutic strategy to prevent neurodegeneration and neurodeterioration associated with schizophrenia.27 Given the preponderance of data showing that SGAs have numerous neuroprotective properties but FGAs have many neurotoxic effects,6 the message to psychiatric practitioners, a decade after the CATIE study, is that the 2 generations of antipsychotic agents are not really similar or interchangeable. They might have similar clinical effectiveness but they exert very different neurobiological effects.
The proof of the pudding is in the eating: Despite the findings of the CATIE study, the vast majority of psychiatrists would prefer to treat their own family members with an SGA, not an FGA, if the need for antipsychotic medication arises.
1. Lieberman JA, Stroup TS, McEvoy JP, et al; Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209-1223.
2. Vita A, De Peri L, Deste G, et al. The effect of antipsychotic treatment on cortical gray matter changes in schizophrenia: does the class matter? A meta-analysis and meta-regression of longitudinal magnetic resonance imaging studies. Biol Psychiatry. 2015;78(6):403-412.
3. Wakade CG, Mahadik SP, Waller JL, et al. Atypical neuroleptics stimulate neurogenesis in adult rat brain. J Neurosci Res. 2002;69(1):72-79.
4. Lieberman JA, Tollefson GD, Charles C, et al; HGDH Study Group. Antipsychotic drug effects on brain morphology in first-episode psychosis. Arch Gen Psychiatry. 2005;62(4):361-370.
5. Nasrallah HA. Impaired neuroplasticity in schizophrenia and the neuro-regenerative effects of atypical antipsychotics. Medscape Psychiatry. http://www.medscape.org/viewarticle/569521. Published January 31, 2008. Accessed November 10, 2015.
6. Nasrallah HA. Haloperidol clearly is neurotoxic. Should it be banned? Current Psychiatry. 2012;12(7):7-8.
7. Nandra KS, Agius M. The differences between typical and atypical antipsychotics: the effects on neurogenesis. Psychiatr Danub. 2012;24(suppl 1):S95-S99.
8. Nasrallah HA, Hopkins T, Pixley SK, et al. Differential effects of antipsychotic and antidepressant drugs on neurogenic region in rats. Brain Res. 2010;354:23-29.
9. Pillai A, Tery AV, Mahadik SP. Differential effects of long-term treatment with typical and atypical antipsychotics on NGF and BNDF levels in rat striatum and hippocampus. Schizophr Res. 2006;82(1):95-106.
10. Buckley PF, Pillai A, Evans D, et al. Brain derived neurotropic factor in first-episode psychosis. Schizophr Res. 2007;91(1-3):1-5.
11. Martin MV, Mimics K, Nisenbaum LK, et al. Olanzapine reversed brain gene expression changes induced by phencyclidines treatment in non-human primates. Mol Neuropsychiatry. 2015;1(2):82-93.
12. Yan BC, Park JH, Ahn JH, et al. Neuroprotection of posttreatment with risperidone, an atypical antipsychotic drug, in rat and gerbil models of ischemic stroke and the maintenance of antioxidants in a gerbil model of ischemic stroke. J Neurosci Res. 2014;92(6):795-807.
13. Yulug B, Yildiz A, Güzel O, et al. Risperidone attenuates brain damage after focal cerebral ischemia in vivo. Brain Res Bull. 2006;69(6):656-659.
14. Yulug B, Yildiz A, Hüdaoglu O, et al. Olanzapine attenuates brain damage after focal cerebral ischemia in vivo. Brain Res Bull. 2006;71(1-3):296-300.
15. Elsworth JD, Morrow BA. Hajszan T, et al. Phencyclidine-induced loss of asymmetric spine synapses in rodent prefrontal cortex is reversed by acute and chronic treatment with olanzapine. Neuropsychopharmacology. 2001;36(10):2054-2061.
16. Seki Y, Kato TA, Monji A, et al. Pretreatment of aripiprazole and minocycline, but not haloperidol, suppresses oligodendrocyte damage from interferon-y-stimulated microglia in co-culture model. Schizophr Res. 2013;151(1-3):20-28.
17. Bian Q, Kato T, Monji A, et al. The effect of atypical antipsychotics, perospirone, ziprasidone and quetiapine on microglial activation induced by interferon-gamma. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(1):42-48.
18. Wang HD, Deutch AY. Dopamine depletion of the prefrontal cortex induces dendritic spine loss: reversal by atypical antipsychotic drug treatment. Neuropsychopharmacology. 2008;33(6):1276-1286.
19. Miller BJ, Buckley P, Seabolt W, et al. Meta-analysis of cytokine alternations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry. 2011;70(7):663-671.
20. Nasrallah HA. Beyond dopamine: The ‘other’ effects of antipsychotics. Current Psychiatry. 2013;12(6):8-9.
21. Yang MC, Lung FW. Neuroprotection of paliperidone on SH-SY5Y cells against β-amyloid peptide(25-35), N-methyl-4-phenylpyridinium ion, and hydrogen peroxide-induced cell death. Psychopharmacology (Berl). 2011;217(3):397-410.
22. Peng L, Zhu D, Feng X, et al. Paliperidone protects prefrontal cortical neurons from damages caused by MK-801 via Akt1/GSK3β _signaling pathway. Schizophr Res. 2013;147(1):14-23.23.
Stojkovic´ T, Radonjic´ NV, Velimirovic´ M, et al. Risperidone reverses phencyclidine induced decrease in glutathione levels and alternations of antioxidant defense in rat brain. Prog Neuropsychopharmacol Biol Psychiatry. 2012;39(1):192-199.
24. Koprivica V, Regardie K, Wolff C, et al. Aripiprazole protects cortical neurons from glutamate toxicity. Eur J Pharmacol. 2011;651(1-3):73-76.
25. Vaidya VA, Marek GJ, Aghajanian GK, et al. 5-HT2A receptor-mediated regulation of brain-derived neurotrophic factor mRNA in the hippocampus and the neocortex. J Neurosci. 1997;17(8):2785-2795.
26. Meridith GE, Switzer RC 3rd, Napier TC. Short-term, D2 receptor blockade induces synaptic degeneration, reduces levels of tyrosine hydroxylase and brain-derived neurotrophic factor, and enhances D2-mediated firing in the ventral pallidum. Brain Res. 2004;995(1):14-22.
27. Lieberman JA, Perkins DO, Jarskog LF. Neuroprotection: a therapeutic strategy to prevent deterioration associated with schizophrenia. CNS Spectr. 2007;12(suppl 4):1-13; quiz 14.
This past September, exactly 10 years after publication of the primary findings of the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study1—namely, that effectiveness (defined as all-cause discontinuation) was not different across first-generation antipsychotics (FGAs) and second generation antipsychotics (SGAs)— a new meta-analysis by Vita et al2 of differences in cortical gray-matter change between those 2 classes of antipsychotics offers a reminder: The clinical focus of the CATIE study overlooked important neurobiological and neuroprotective differences between FGAs and SGAs.
How drastically 1 decade can change the scientific perspective! Vita et al’s meta-analysis and meta-regression encompassed all 18 MRI studies of cortical gray matter in patients with schizophrenia.2 Earlier studies (published between 1983 and 2014) had lumped together patients who were receiving an FGA and those receiving an SGA, and authors reported overall reduction in cortical gray matter with prolonged antipsychotic treatment.
Remarkable findings emerge
When Vita et al2 analyzed FGA- and SGA-treated patients separately, however, they found a significant reduction in cortical gray matter in the FGA group but not in the SGA group. In fact, while higher daily dosages of FGAs were associated with greater reduction in cortical gray matter, higher dosages of SGAs were associated with lower cortical gray matter reduction and, in some samples, with an increase in volume of cortical gray matter.
The researchers hypothesized that the differential effects of FGAs and SGAs might be attributable to the neurotoxicity of typical FGAs and the neuroprotective effect of atypical SGAs.
Hindsight
The key neurobiological difference between FGAs and SGAs reported by Vita et al2 was not addressed in the CATIE study, leading, at that time, to a rush to judgment that all antipsychotics are the same. This conclusion emboldened managed-care organizations to mandate use of older (and cheaper) generic FGAs instead of newer (and more expensive) SGAs— most of which have become available as generic equivalents since the CATIE study was completed.
Investigators in the CATIE study— of which I was one—cannot be blamed for not focusing on neurotoxicity and neuroprotection; those data were not on the psychiatry’s radar when the CATIE study was designed in 1998. The major focus was on whether SGAs (new on the scene in the late 1990s) were more efficacious, safe, and tolerable (that is, more effective) than FGAs.
In fact, the first study reporting that SGAs stimulated neurogenesis (in animals) was published in 2002,3 when the CATIE study was more than half complete. Research into the neuroprotective properties of SGAs then grew rapidly. In fact, the principal investigator of the CATIE study conducted a head-to-head comparison of FGA haloperidol and SGA olanzapine in a sample of first-episode schizophrenia patients4; over 1 year of follow-up, it was determined that patients in the haloperidol-treated group exhibited significant brain volume loss on MRI but those in the olanzapine-treated group did not. This study was published in 2005—the same year the CATIE study was published!
SGAs offer neuroprotection
Over the past decade, the neuroprotective effects of SGAs5 and the neurotoxic effects of FGAs6 have been studied intensively, revealing that SGAs have multiple neuroprotective effects. These effects include:
• stimulation of the production of new brain cells (neurons and glia), known as neurogenesis5,7,8
• an increase in neurotrophic factors, such as nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF),9 which are found at a significantly low level in patients with psychosis10
• reversal of phencyclidine (PCP)-induced changes in gene expression11
• neuroprotection against ischemic stroke12-14
• reversal of PCP-induced loss of dendritic spines in the frontal cortex15
• prevention of oligodendrocyte damage caused by interferon gamma-stimulated microglia16,17
• reversal of loss of dendritic spines in the prefrontal cortex induced by dopamine depletion18
• an anti-inflammatory effect19,20
• protection against β-amyloid and hydrogen peroxide-induced cell death21
• protection against prefrontal cortical neuronal damage caused by dizocilpine (MK-801)22
• reversal of a PCP-induced decrease in the glutathione level and alteration of antioxidant defenses23
• protection of cortical neurons from glutamate neurotoxicity.24
One reason why SGAs are neuroprotective, but FGAs are not, can be attributed to their receptor profiles. FGAs block dopamine D2 receptors far more than serotonin 2A receptors, whereas SGAs do the opposite: They block 5-HT2A receptors 500% to 1,000% more than they block D2 receptors. This difference is associated in turn with a different neurobiological and neuroprotective profiles, such as a decrease or an increase in BDNF.25,26
Neither similar nor interchangeable
Since publication of the findings of the CATIE study, the primary investigator has proposed that neuroprotection can be a therapeutic strategy to prevent neurodegeneration and neurodeterioration associated with schizophrenia.27 Given the preponderance of data showing that SGAs have numerous neuroprotective properties but FGAs have many neurotoxic effects,6 the message to psychiatric practitioners, a decade after the CATIE study, is that the 2 generations of antipsychotic agents are not really similar or interchangeable. They might have similar clinical effectiveness but they exert very different neurobiological effects.
The proof of the pudding is in the eating: Despite the findings of the CATIE study, the vast majority of psychiatrists would prefer to treat their own family members with an SGA, not an FGA, if the need for antipsychotic medication arises.
This past September, exactly 10 years after publication of the primary findings of the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study1—namely, that effectiveness (defined as all-cause discontinuation) was not different across first-generation antipsychotics (FGAs) and second generation antipsychotics (SGAs)— a new meta-analysis by Vita et al2 of differences in cortical gray-matter change between those 2 classes of antipsychotics offers a reminder: The clinical focus of the CATIE study overlooked important neurobiological and neuroprotective differences between FGAs and SGAs.
How drastically 1 decade can change the scientific perspective! Vita et al’s meta-analysis and meta-regression encompassed all 18 MRI studies of cortical gray matter in patients with schizophrenia.2 Earlier studies (published between 1983 and 2014) had lumped together patients who were receiving an FGA and those receiving an SGA, and authors reported overall reduction in cortical gray matter with prolonged antipsychotic treatment.
Remarkable findings emerge
When Vita et al2 analyzed FGA- and SGA-treated patients separately, however, they found a significant reduction in cortical gray matter in the FGA group but not in the SGA group. In fact, while higher daily dosages of FGAs were associated with greater reduction in cortical gray matter, higher dosages of SGAs were associated with lower cortical gray matter reduction and, in some samples, with an increase in volume of cortical gray matter.
The researchers hypothesized that the differential effects of FGAs and SGAs might be attributable to the neurotoxicity of typical FGAs and the neuroprotective effect of atypical SGAs.
Hindsight
The key neurobiological difference between FGAs and SGAs reported by Vita et al2 was not addressed in the CATIE study, leading, at that time, to a rush to judgment that all antipsychotics are the same. This conclusion emboldened managed-care organizations to mandate use of older (and cheaper) generic FGAs instead of newer (and more expensive) SGAs— most of which have become available as generic equivalents since the CATIE study was completed.
Investigators in the CATIE study— of which I was one—cannot be blamed for not focusing on neurotoxicity and neuroprotection; those data were not on the psychiatry’s radar when the CATIE study was designed in 1998. The major focus was on whether SGAs (new on the scene in the late 1990s) were more efficacious, safe, and tolerable (that is, more effective) than FGAs.
In fact, the first study reporting that SGAs stimulated neurogenesis (in animals) was published in 2002,3 when the CATIE study was more than half complete. Research into the neuroprotective properties of SGAs then grew rapidly. In fact, the principal investigator of the CATIE study conducted a head-to-head comparison of FGA haloperidol and SGA olanzapine in a sample of first-episode schizophrenia patients4; over 1 year of follow-up, it was determined that patients in the haloperidol-treated group exhibited significant brain volume loss on MRI but those in the olanzapine-treated group did not. This study was published in 2005—the same year the CATIE study was published!
SGAs offer neuroprotection
Over the past decade, the neuroprotective effects of SGAs5 and the neurotoxic effects of FGAs6 have been studied intensively, revealing that SGAs have multiple neuroprotective effects. These effects include:
• stimulation of the production of new brain cells (neurons and glia), known as neurogenesis5,7,8
• an increase in neurotrophic factors, such as nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF),9 which are found at a significantly low level in patients with psychosis10
• reversal of phencyclidine (PCP)-induced changes in gene expression11
• neuroprotection against ischemic stroke12-14
• reversal of PCP-induced loss of dendritic spines in the frontal cortex15
• prevention of oligodendrocyte damage caused by interferon gamma-stimulated microglia16,17
• reversal of loss of dendritic spines in the prefrontal cortex induced by dopamine depletion18
• an anti-inflammatory effect19,20
• protection against β-amyloid and hydrogen peroxide-induced cell death21
• protection against prefrontal cortical neuronal damage caused by dizocilpine (MK-801)22
• reversal of a PCP-induced decrease in the glutathione level and alteration of antioxidant defenses23
• protection of cortical neurons from glutamate neurotoxicity.24
One reason why SGAs are neuroprotective, but FGAs are not, can be attributed to their receptor profiles. FGAs block dopamine D2 receptors far more than serotonin 2A receptors, whereas SGAs do the opposite: They block 5-HT2A receptors 500% to 1,000% more than they block D2 receptors. This difference is associated in turn with a different neurobiological and neuroprotective profiles, such as a decrease or an increase in BDNF.25,26
Neither similar nor interchangeable
Since publication of the findings of the CATIE study, the primary investigator has proposed that neuroprotection can be a therapeutic strategy to prevent neurodegeneration and neurodeterioration associated with schizophrenia.27 Given the preponderance of data showing that SGAs have numerous neuroprotective properties but FGAs have many neurotoxic effects,6 the message to psychiatric practitioners, a decade after the CATIE study, is that the 2 generations of antipsychotic agents are not really similar or interchangeable. They might have similar clinical effectiveness but they exert very different neurobiological effects.
The proof of the pudding is in the eating: Despite the findings of the CATIE study, the vast majority of psychiatrists would prefer to treat their own family members with an SGA, not an FGA, if the need for antipsychotic medication arises.
1. Lieberman JA, Stroup TS, McEvoy JP, et al; Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209-1223.
2. Vita A, De Peri L, Deste G, et al. The effect of antipsychotic treatment on cortical gray matter changes in schizophrenia: does the class matter? A meta-analysis and meta-regression of longitudinal magnetic resonance imaging studies. Biol Psychiatry. 2015;78(6):403-412.
3. Wakade CG, Mahadik SP, Waller JL, et al. Atypical neuroleptics stimulate neurogenesis in adult rat brain. J Neurosci Res. 2002;69(1):72-79.
4. Lieberman JA, Tollefson GD, Charles C, et al; HGDH Study Group. Antipsychotic drug effects on brain morphology in first-episode psychosis. Arch Gen Psychiatry. 2005;62(4):361-370.
5. Nasrallah HA. Impaired neuroplasticity in schizophrenia and the neuro-regenerative effects of atypical antipsychotics. Medscape Psychiatry. http://www.medscape.org/viewarticle/569521. Published January 31, 2008. Accessed November 10, 2015.
6. Nasrallah HA. Haloperidol clearly is neurotoxic. Should it be banned? Current Psychiatry. 2012;12(7):7-8.
7. Nandra KS, Agius M. The differences between typical and atypical antipsychotics: the effects on neurogenesis. Psychiatr Danub. 2012;24(suppl 1):S95-S99.
8. Nasrallah HA, Hopkins T, Pixley SK, et al. Differential effects of antipsychotic and antidepressant drugs on neurogenic region in rats. Brain Res. 2010;354:23-29.
9. Pillai A, Tery AV, Mahadik SP. Differential effects of long-term treatment with typical and atypical antipsychotics on NGF and BNDF levels in rat striatum and hippocampus. Schizophr Res. 2006;82(1):95-106.
10. Buckley PF, Pillai A, Evans D, et al. Brain derived neurotropic factor in first-episode psychosis. Schizophr Res. 2007;91(1-3):1-5.
11. Martin MV, Mimics K, Nisenbaum LK, et al. Olanzapine reversed brain gene expression changes induced by phencyclidines treatment in non-human primates. Mol Neuropsychiatry. 2015;1(2):82-93.
12. Yan BC, Park JH, Ahn JH, et al. Neuroprotection of posttreatment with risperidone, an atypical antipsychotic drug, in rat and gerbil models of ischemic stroke and the maintenance of antioxidants in a gerbil model of ischemic stroke. J Neurosci Res. 2014;92(6):795-807.
13. Yulug B, Yildiz A, Güzel O, et al. Risperidone attenuates brain damage after focal cerebral ischemia in vivo. Brain Res Bull. 2006;69(6):656-659.
14. Yulug B, Yildiz A, Hüdaoglu O, et al. Olanzapine attenuates brain damage after focal cerebral ischemia in vivo. Brain Res Bull. 2006;71(1-3):296-300.
15. Elsworth JD, Morrow BA. Hajszan T, et al. Phencyclidine-induced loss of asymmetric spine synapses in rodent prefrontal cortex is reversed by acute and chronic treatment with olanzapine. Neuropsychopharmacology. 2001;36(10):2054-2061.
16. Seki Y, Kato TA, Monji A, et al. Pretreatment of aripiprazole and minocycline, but not haloperidol, suppresses oligodendrocyte damage from interferon-y-stimulated microglia in co-culture model. Schizophr Res. 2013;151(1-3):20-28.
17. Bian Q, Kato T, Monji A, et al. The effect of atypical antipsychotics, perospirone, ziprasidone and quetiapine on microglial activation induced by interferon-gamma. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(1):42-48.
18. Wang HD, Deutch AY. Dopamine depletion of the prefrontal cortex induces dendritic spine loss: reversal by atypical antipsychotic drug treatment. Neuropsychopharmacology. 2008;33(6):1276-1286.
19. Miller BJ, Buckley P, Seabolt W, et al. Meta-analysis of cytokine alternations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry. 2011;70(7):663-671.
20. Nasrallah HA. Beyond dopamine: The ‘other’ effects of antipsychotics. Current Psychiatry. 2013;12(6):8-9.
21. Yang MC, Lung FW. Neuroprotection of paliperidone on SH-SY5Y cells against β-amyloid peptide(25-35), N-methyl-4-phenylpyridinium ion, and hydrogen peroxide-induced cell death. Psychopharmacology (Berl). 2011;217(3):397-410.
22. Peng L, Zhu D, Feng X, et al. Paliperidone protects prefrontal cortical neurons from damages caused by MK-801 via Akt1/GSK3β _signaling pathway. Schizophr Res. 2013;147(1):14-23.23.
Stojkovic´ T, Radonjic´ NV, Velimirovic´ M, et al. Risperidone reverses phencyclidine induced decrease in glutathione levels and alternations of antioxidant defense in rat brain. Prog Neuropsychopharmacol Biol Psychiatry. 2012;39(1):192-199.
24. Koprivica V, Regardie K, Wolff C, et al. Aripiprazole protects cortical neurons from glutamate toxicity. Eur J Pharmacol. 2011;651(1-3):73-76.
25. Vaidya VA, Marek GJ, Aghajanian GK, et al. 5-HT2A receptor-mediated regulation of brain-derived neurotrophic factor mRNA in the hippocampus and the neocortex. J Neurosci. 1997;17(8):2785-2795.
26. Meridith GE, Switzer RC 3rd, Napier TC. Short-term, D2 receptor blockade induces synaptic degeneration, reduces levels of tyrosine hydroxylase and brain-derived neurotrophic factor, and enhances D2-mediated firing in the ventral pallidum. Brain Res. 2004;995(1):14-22.
27. Lieberman JA, Perkins DO, Jarskog LF. Neuroprotection: a therapeutic strategy to prevent deterioration associated with schizophrenia. CNS Spectr. 2007;12(suppl 4):1-13; quiz 14.
1. Lieberman JA, Stroup TS, McEvoy JP, et al; Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209-1223.
2. Vita A, De Peri L, Deste G, et al. The effect of antipsychotic treatment on cortical gray matter changes in schizophrenia: does the class matter? A meta-analysis and meta-regression of longitudinal magnetic resonance imaging studies. Biol Psychiatry. 2015;78(6):403-412.
3. Wakade CG, Mahadik SP, Waller JL, et al. Atypical neuroleptics stimulate neurogenesis in adult rat brain. J Neurosci Res. 2002;69(1):72-79.
4. Lieberman JA, Tollefson GD, Charles C, et al; HGDH Study Group. Antipsychotic drug effects on brain morphology in first-episode psychosis. Arch Gen Psychiatry. 2005;62(4):361-370.
5. Nasrallah HA. Impaired neuroplasticity in schizophrenia and the neuro-regenerative effects of atypical antipsychotics. Medscape Psychiatry. http://www.medscape.org/viewarticle/569521. Published January 31, 2008. Accessed November 10, 2015.
6. Nasrallah HA. Haloperidol clearly is neurotoxic. Should it be banned? Current Psychiatry. 2012;12(7):7-8.
7. Nandra KS, Agius M. The differences between typical and atypical antipsychotics: the effects on neurogenesis. Psychiatr Danub. 2012;24(suppl 1):S95-S99.
8. Nasrallah HA, Hopkins T, Pixley SK, et al. Differential effects of antipsychotic and antidepressant drugs on neurogenic region in rats. Brain Res. 2010;354:23-29.
9. Pillai A, Tery AV, Mahadik SP. Differential effects of long-term treatment with typical and atypical antipsychotics on NGF and BNDF levels in rat striatum and hippocampus. Schizophr Res. 2006;82(1):95-106.
10. Buckley PF, Pillai A, Evans D, et al. Brain derived neurotropic factor in first-episode psychosis. Schizophr Res. 2007;91(1-3):1-5.
11. Martin MV, Mimics K, Nisenbaum LK, et al. Olanzapine reversed brain gene expression changes induced by phencyclidines treatment in non-human primates. Mol Neuropsychiatry. 2015;1(2):82-93.
12. Yan BC, Park JH, Ahn JH, et al. Neuroprotection of posttreatment with risperidone, an atypical antipsychotic drug, in rat and gerbil models of ischemic stroke and the maintenance of antioxidants in a gerbil model of ischemic stroke. J Neurosci Res. 2014;92(6):795-807.
13. Yulug B, Yildiz A, Güzel O, et al. Risperidone attenuates brain damage after focal cerebral ischemia in vivo. Brain Res Bull. 2006;69(6):656-659.
14. Yulug B, Yildiz A, Hüdaoglu O, et al. Olanzapine attenuates brain damage after focal cerebral ischemia in vivo. Brain Res Bull. 2006;71(1-3):296-300.
15. Elsworth JD, Morrow BA. Hajszan T, et al. Phencyclidine-induced loss of asymmetric spine synapses in rodent prefrontal cortex is reversed by acute and chronic treatment with olanzapine. Neuropsychopharmacology. 2001;36(10):2054-2061.
16. Seki Y, Kato TA, Monji A, et al. Pretreatment of aripiprazole and minocycline, but not haloperidol, suppresses oligodendrocyte damage from interferon-y-stimulated microglia in co-culture model. Schizophr Res. 2013;151(1-3):20-28.
17. Bian Q, Kato T, Monji A, et al. The effect of atypical antipsychotics, perospirone, ziprasidone and quetiapine on microglial activation induced by interferon-gamma. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(1):42-48.
18. Wang HD, Deutch AY. Dopamine depletion of the prefrontal cortex induces dendritic spine loss: reversal by atypical antipsychotic drug treatment. Neuropsychopharmacology. 2008;33(6):1276-1286.
19. Miller BJ, Buckley P, Seabolt W, et al. Meta-analysis of cytokine alternations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry. 2011;70(7):663-671.
20. Nasrallah HA. Beyond dopamine: The ‘other’ effects of antipsychotics. Current Psychiatry. 2013;12(6):8-9.
21. Yang MC, Lung FW. Neuroprotection of paliperidone on SH-SY5Y cells against β-amyloid peptide(25-35), N-methyl-4-phenylpyridinium ion, and hydrogen peroxide-induced cell death. Psychopharmacology (Berl). 2011;217(3):397-410.
22. Peng L, Zhu D, Feng X, et al. Paliperidone protects prefrontal cortical neurons from damages caused by MK-801 via Akt1/GSK3β _signaling pathway. Schizophr Res. 2013;147(1):14-23.23.
Stojkovic´ T, Radonjic´ NV, Velimirovic´ M, et al. Risperidone reverses phencyclidine induced decrease in glutathione levels and alternations of antioxidant defense in rat brain. Prog Neuropsychopharmacol Biol Psychiatry. 2012;39(1):192-199.
24. Koprivica V, Regardie K, Wolff C, et al. Aripiprazole protects cortical neurons from glutamate toxicity. Eur J Pharmacol. 2011;651(1-3):73-76.
25. Vaidya VA, Marek GJ, Aghajanian GK, et al. 5-HT2A receptor-mediated regulation of brain-derived neurotrophic factor mRNA in the hippocampus and the neocortex. J Neurosci. 1997;17(8):2785-2795.
26. Meridith GE, Switzer RC 3rd, Napier TC. Short-term, D2 receptor blockade induces synaptic degeneration, reduces levels of tyrosine hydroxylase and brain-derived neurotrophic factor, and enhances D2-mediated firing in the ventral pallidum. Brain Res. 2004;995(1):14-22.
27. Lieberman JA, Perkins DO, Jarskog LF. Neuroprotection: a therapeutic strategy to prevent deterioration associated with schizophrenia. CNS Spectr. 2007;12(suppl 4):1-13; quiz 14.
‘You’ve been served’: What to do if you receive a subpoena
Dear Dr. Mossman,
Psychiatrists should not reveal what their patients say except to avert a threat to health or safety or to report abuse. So, how can psychiatrists be subpoenaed to provide information for a trial? If I receive a subpoena, how can I comply without violating patient privacy? If I have to go to court, can I “plead the Fifth”?
Submitted by “Dr. S”
Physicians who are served with a subpoena feel upset for the reason Dr. S described: Complying with a subpoena seems to violate the obligation to protect patients’ privacy. But physicians can’t “plead the Fifth” under these circumstances, because the Fifth Amendment to the U.S. Constitution only bars forcing someone to give self-incriminating testimony.1
If you receive a subpoena for information gleaned during patient care, you should not ignore it. Failing to respond might place you in contempt of court and subject you to a fine or even jail time. Yet simply complying could have legal and professional implications, too.
Often, a psychiatrist who receives a subpoena should seek an attorney’s advice on how to best respond. But understanding what subpoenas are and how they work might let you feel less anxious as you go through the process of responding. With this goal in mind, this article covers:
• what a subpoena is and isn’t
• 2 types of privacy obligations
• legal options
• avoiding potential embarrassment (Box).2

What is a subpoena?
All citizens have a legal obligation to furnish courts with the information needed to decide legal issues.3 Statutes and legal rules dictate how such material comes to court.
Issuing a subpoena (from the Latin sub poena, “under penalty”) is one way of obtaining information needed for a legal proceeding. A subpoena ad testificandum directs the recipient to appear at a legal proceeding and provide testimony. A subpoena duces tecum (“you shall bring with you”) directs the recipient to produce specific records or to appear at a legal proceeding with the records.
Usually, subpoenas are issued by attorneys or court clerks, not by judges. As such, they are not court orders. If you receive a subpoena, you should make a timely response of some sort. But, ultimately, you might not have to release the information. Although physicians have to follow the same rules as other citizens, courts recognize that doctors also have professional obligations to their patients.
Confidentiality: Your reason to hesitate
Receiving a subpoena doesn’t change your obligation to protect your patient’s confidentiality. From the law’s standpoint, patient confidentiality is a function of the rules that govern use of information in legal proceedings.
The Privacy Rule4 that arose from the Health Insurance Portability and Accountability Act (HIPAA) of 19965 provides guidance on acceptable responses to subpoenas by “covered entities,” which includes most physicians’ practices. HIPAA permits disclosure of the minimum amount of personal information needed to fulfill the demands of a subpoena. The Table6,7 explains HIPAA’s rules about specific responses to subpoenas, depending on their source.
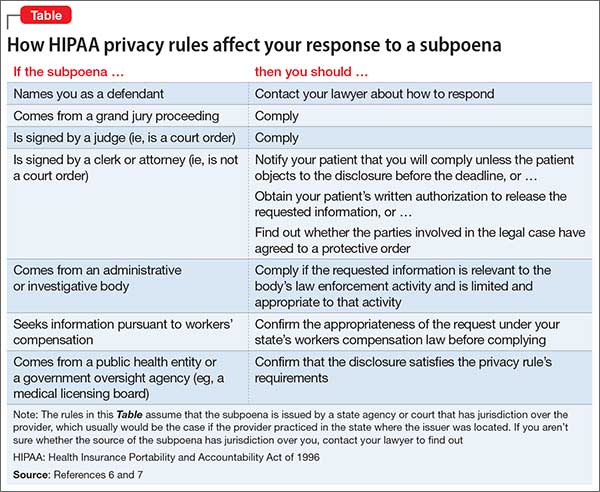
Many states have patient privacy laws that are stricter than HIPAA rules. If you practice in one of those states, you have to follow the more stringent rule.8 For example, Ohio law does not let subpoenaed providers tender medical records for use in a grand jury proceeding without a release signed by the patient, although HIPAA would allow this (Table).6,7 Out of concern that “giving law enforcement unbridled access to medical records could discourage patients from seeking medical treatment,” Ohio protects patient records more than HIPAA does.9 New York State’s privilege rules also are stricter than HIPAA10 and contain specific provisions about releasing certain types of information (eg, HIV status11). State courts expect physicians to follow their laws about patient privacy and to consult attorneys to make sure that releasing information is done properly.12
Releasing information improperly could become grounds for legal action against you, even if you released the information in response to a subpoena. Legal action could take the form of a lawsuit for breach of confidentiality, a HIPAA-based complaint, a complaint to the state medical board, or all 3 of these.
Must you turn over information?
Before you testify or turn over documents, you need to verify that the legal and ethical requirements for the disclosure are met—as you would for any release of patient information. You can do this by obtaining your patient’s formal, written consent for the disclosure. Before you accept the patient’s agreement, however, you might—and in most cases should— consider discussing how the disclosure could affect the patient’s well-being or your treatment relationship.
If the patient will not agree to the disclosure, the patient or the patient’s attorney can seek to have the subpoena modified or quashed (declared void). One tactic for doing this is by asserting doctor–patient privilege, a legal doctrine codified in most state’s laws. The privilege recognizes that, because privacy is important in medical care, stopping clinical information from automatically coming in court serves an important social purpose.13
The doctor–patient privilege belongs to the litigant—here, your patient—not to you, so your patient has to raise the objection to releasing information.14 Also, the privilege is not absolute. If having the clinical information is necessary, the judge may issue a court order denying the patient’s motion to quash. Unless the judge later modifies or vacates the order, you risk being found in contempt of court if you still refuse to turn over documents demanded by the subpoena.
Fact witness or expert witness?
If the subpoena demands your testimony, the issuing party might want you to serve as a fact witness or expert witness. Persons with relevant personal knowledge to a legal proceeding can serve as fact witnesses, and testify about things they did or perceived.15 For example, a psychiatrist serving as a fact witness could recount having heard or seen a patient talking aloud as if arguing with someone when no real interlocutor was present.
A witness whom the court deems an “expert” by virtue of special knowledge, skill, experience, training, or education may offer opinions based on specific sets of facts. Courts hear such testimony when the expert’s specialized knowledge will help the jury understand the evidence or reach a verdict in a case.16 To return to the example above: a psychiatric expert witness might tell jurors that the patient’s “arguing” was evidence that she was hallucinating and suffered from schizophrenia.
If you receive a subpoena to testify about someone you have treated, you should notify the issuing party that you will provide fact testimony if required to do so. You cannot be compelled to serve as an expert witness, however. In many situations, attempting to provide objective expert testimony about one’s own patient could create unresolvable conflicts between the obligation to tell the truth and your obligation to serve your patient’s interests.17
If the subpoena requests deposition testimony about a patient, you probably will be able to schedule the deposition at a time that is convenient for you and the attorneys involved. Yet you should not agree to be deposed unless (a) you have received the patient’s authorization, (b) a court has ordered you to testify despite the patient’s objection, or (c) your attorney (whom you have consulted about the situation) has advised you that providing testimony is appropriate.
If you are called as a fact witness for a trial, the attorney or court that has subpoenaed you often will try to schedule things to minimize the time taken away from your other duties. Once in court, you can ask the judge (on the record) whether you must answer if you are asked questions about a patient who has not previously authorized you to release treatment information. A judge’s explicit command to respond absolves you of any further ethical obligation to withhold confidential information about the patient’s care.
Bottom Line
If you receive a subpoena for records or testimony, obtaining the patient’s written authorization should allow you to release the information without violating confidentiality obligations. If your patient won’t agree to the release, if turning over information might adversely affect the patient, or if you’re not sure what to do, seek advice from an attorney who knows about medical privacy rules. That way, you can be sure you are meeting all legal and professional standards that apply.
Disclosure
The author reports no financial relationship with any company whose products are mentioned in this article or with any manufacturers of competing products.
1. Kastigar v United States, 406 US 441 (1972).
2. Barsky AE. Clinicians in court, second edition: A guide to subpoenas, depositions, testifying, and everything else you need to know. New York, NY: Guilford Press; 2012.
3. United States v Bryan, 339 US 323 (1950).
4. 45 CFR §164.50.
5. 45 CFR Parts 160 and 164.
6. 45 CFR §164.512.
7. Stanger K. HIPAA: responding to subpoenas, orders, and administrative demands. http://www.hhhealthlawblog.com/2013/10/hipaa-responding-to-subpoenas-orders-and-administrative-demands.html. Published October 9, 2013. Accessed September 22, 2015.
8. Zwerling AL, de Harder WA, Tarpey CM. To disclose or not to disclose, that is the question: issues to consider before responding to a subpoena. J Am Coll Radiol. 2012;9(4):279-281.
9. Turk v Oilier, 732 F Supp 2d 758 (ND Ohio 2010).
10. In re Antonia E, 16 Misc 3d 637 (Fam Ct Queens County 2007).
11. NY PBH Law §2785.
12. Crescenzo v Crane, 796 A2d 283 (NJ Super Ct App Div 2002).
13. In re Bruendl’s Will, 102 Wis 45, 78 NW 169 (1899).
14. In re Lifschutz, 2 Cal 3d 415, 85 Cal Rptr 829, 467 P2d 557 (1970).
15. Broun KS, Dix GE, Imwinkelried EJ, et al. McCormick on evidence. 7th ed. St. Paul, MN: West Group; 2013.
16. Fed Evid R 702.
17. Strasburger LH, Gutheil TG, Brodsky A. On wearing two hats: role conflict in serving as both psychotherapist and expert witness. Am J Psychiatry. 1997;154:448-456.
Dear Dr. Mossman,
Psychiatrists should not reveal what their patients say except to avert a threat to health or safety or to report abuse. So, how can psychiatrists be subpoenaed to provide information for a trial? If I receive a subpoena, how can I comply without violating patient privacy? If I have to go to court, can I “plead the Fifth”?
Submitted by “Dr. S”
Physicians who are served with a subpoena feel upset for the reason Dr. S described: Complying with a subpoena seems to violate the obligation to protect patients’ privacy. But physicians can’t “plead the Fifth” under these circumstances, because the Fifth Amendment to the U.S. Constitution only bars forcing someone to give self-incriminating testimony.1
If you receive a subpoena for information gleaned during patient care, you should not ignore it. Failing to respond might place you in contempt of court and subject you to a fine or even jail time. Yet simply complying could have legal and professional implications, too.
Often, a psychiatrist who receives a subpoena should seek an attorney’s advice on how to best respond. But understanding what subpoenas are and how they work might let you feel less anxious as you go through the process of responding. With this goal in mind, this article covers:
• what a subpoena is and isn’t
• 2 types of privacy obligations
• legal options
• avoiding potential embarrassment (Box).2
What is a subpoena?
All citizens have a legal obligation to furnish courts with the information needed to decide legal issues.3 Statutes and legal rules dictate how such material comes to court.
Issuing a subpoena (from the Latin sub poena, “under penalty”) is one way of obtaining information needed for a legal proceeding. A subpoena ad testificandum directs the recipient to appear at a legal proceeding and provide testimony. A subpoena duces tecum (“you shall bring with you”) directs the recipient to produce specific records or to appear at a legal proceeding with the records.
Usually, subpoenas are issued by attorneys or court clerks, not by judges. As such, they are not court orders. If you receive a subpoena, you should make a timely response of some sort. But, ultimately, you might not have to release the information. Although physicians have to follow the same rules as other citizens, courts recognize that doctors also have professional obligations to their patients.
Confidentiality: Your reason to hesitate
Receiving a subpoena doesn’t change your obligation to protect your patient’s confidentiality. From the law’s standpoint, patient confidentiality is a function of the rules that govern use of information in legal proceedings.
The Privacy Rule4 that arose from the Health Insurance Portability and Accountability Act (HIPAA) of 19965 provides guidance on acceptable responses to subpoenas by “covered entities,” which includes most physicians’ practices. HIPAA permits disclosure of the minimum amount of personal information needed to fulfill the demands of a subpoena. The Table6,7 explains HIPAA’s rules about specific responses to subpoenas, depending on their source.
Many states have patient privacy laws that are stricter than HIPAA rules. If you practice in one of those states, you have to follow the more stringent rule.8 For example, Ohio law does not let subpoenaed providers tender medical records for use in a grand jury proceeding without a release signed by the patient, although HIPAA would allow this (Table).6,7 Out of concern that “giving law enforcement unbridled access to medical records could discourage patients from seeking medical treatment,” Ohio protects patient records more than HIPAA does.9 New York State’s privilege rules also are stricter than HIPAA10 and contain specific provisions about releasing certain types of information (eg, HIV status11). State courts expect physicians to follow their laws about patient privacy and to consult attorneys to make sure that releasing information is done properly.12
Releasing information improperly could become grounds for legal action against you, even if you released the information in response to a subpoena. Legal action could take the form of a lawsuit for breach of confidentiality, a HIPAA-based complaint, a complaint to the state medical board, or all 3 of these.
Must you turn over information?
Before you testify or turn over documents, you need to verify that the legal and ethical requirements for the disclosure are met—as you would for any release of patient information. You can do this by obtaining your patient’s formal, written consent for the disclosure. Before you accept the patient’s agreement, however, you might—and in most cases should— consider discussing how the disclosure could affect the patient’s well-being or your treatment relationship.
If the patient will not agree to the disclosure, the patient or the patient’s attorney can seek to have the subpoena modified or quashed (declared void). One tactic for doing this is by asserting doctor–patient privilege, a legal doctrine codified in most state’s laws. The privilege recognizes that, because privacy is important in medical care, stopping clinical information from automatically coming in court serves an important social purpose.13
The doctor–patient privilege belongs to the litigant—here, your patient—not to you, so your patient has to raise the objection to releasing information.14 Also, the privilege is not absolute. If having the clinical information is necessary, the judge may issue a court order denying the patient’s motion to quash. Unless the judge later modifies or vacates the order, you risk being found in contempt of court if you still refuse to turn over documents demanded by the subpoena.
Fact witness or expert witness?
If the subpoena demands your testimony, the issuing party might want you to serve as a fact witness or expert witness. Persons with relevant personal knowledge to a legal proceeding can serve as fact witnesses, and testify about things they did or perceived.15 For example, a psychiatrist serving as a fact witness could recount having heard or seen a patient talking aloud as if arguing with someone when no real interlocutor was present.
A witness whom the court deems an “expert” by virtue of special knowledge, skill, experience, training, or education may offer opinions based on specific sets of facts. Courts hear such testimony when the expert’s specialized knowledge will help the jury understand the evidence or reach a verdict in a case.16 To return to the example above: a psychiatric expert witness might tell jurors that the patient’s “arguing” was evidence that she was hallucinating and suffered from schizophrenia.
If you receive a subpoena to testify about someone you have treated, you should notify the issuing party that you will provide fact testimony if required to do so. You cannot be compelled to serve as an expert witness, however. In many situations, attempting to provide objective expert testimony about one’s own patient could create unresolvable conflicts between the obligation to tell the truth and your obligation to serve your patient’s interests.17
If the subpoena requests deposition testimony about a patient, you probably will be able to schedule the deposition at a time that is convenient for you and the attorneys involved. Yet you should not agree to be deposed unless (a) you have received the patient’s authorization, (b) a court has ordered you to testify despite the patient’s objection, or (c) your attorney (whom you have consulted about the situation) has advised you that providing testimony is appropriate.
If you are called as a fact witness for a trial, the attorney or court that has subpoenaed you often will try to schedule things to minimize the time taken away from your other duties. Once in court, you can ask the judge (on the record) whether you must answer if you are asked questions about a patient who has not previously authorized you to release treatment information. A judge’s explicit command to respond absolves you of any further ethical obligation to withhold confidential information about the patient’s care.
Bottom Line
If you receive a subpoena for records or testimony, obtaining the patient’s written authorization should allow you to release the information without violating confidentiality obligations. If your patient won’t agree to the release, if turning over information might adversely affect the patient, or if you’re not sure what to do, seek advice from an attorney who knows about medical privacy rules. That way, you can be sure you are meeting all legal and professional standards that apply.
Disclosure
The author reports no financial relationship with any company whose products are mentioned in this article or with any manufacturers of competing products.
Dear Dr. Mossman,
Psychiatrists should not reveal what their patients say except to avert a threat to health or safety or to report abuse. So, how can psychiatrists be subpoenaed to provide information for a trial? If I receive a subpoena, how can I comply without violating patient privacy? If I have to go to court, can I “plead the Fifth”?
Submitted by “Dr. S”
Physicians who are served with a subpoena feel upset for the reason Dr. S described: Complying with a subpoena seems to violate the obligation to protect patients’ privacy. But physicians can’t “plead the Fifth” under these circumstances, because the Fifth Amendment to the U.S. Constitution only bars forcing someone to give self-incriminating testimony.1
If you receive a subpoena for information gleaned during patient care, you should not ignore it. Failing to respond might place you in contempt of court and subject you to a fine or even jail time. Yet simply complying could have legal and professional implications, too.
Often, a psychiatrist who receives a subpoena should seek an attorney’s advice on how to best respond. But understanding what subpoenas are and how they work might let you feel less anxious as you go through the process of responding. With this goal in mind, this article covers:
• what a subpoena is and isn’t
• 2 types of privacy obligations
• legal options
• avoiding potential embarrassment (Box).2
What is a subpoena?
All citizens have a legal obligation to furnish courts with the information needed to decide legal issues.3 Statutes and legal rules dictate how such material comes to court.
Issuing a subpoena (from the Latin sub poena, “under penalty”) is one way of obtaining information needed for a legal proceeding. A subpoena ad testificandum directs the recipient to appear at a legal proceeding and provide testimony. A subpoena duces tecum (“you shall bring with you”) directs the recipient to produce specific records or to appear at a legal proceeding with the records.
Usually, subpoenas are issued by attorneys or court clerks, not by judges. As such, they are not court orders. If you receive a subpoena, you should make a timely response of some sort. But, ultimately, you might not have to release the information. Although physicians have to follow the same rules as other citizens, courts recognize that doctors also have professional obligations to their patients.
Confidentiality: Your reason to hesitate
Receiving a subpoena doesn’t change your obligation to protect your patient’s confidentiality. From the law’s standpoint, patient confidentiality is a function of the rules that govern use of information in legal proceedings.
The Privacy Rule4 that arose from the Health Insurance Portability and Accountability Act (HIPAA) of 19965 provides guidance on acceptable responses to subpoenas by “covered entities,” which includes most physicians’ practices. HIPAA permits disclosure of the minimum amount of personal information needed to fulfill the demands of a subpoena. The Table6,7 explains HIPAA’s rules about specific responses to subpoenas, depending on their source.
Many states have patient privacy laws that are stricter than HIPAA rules. If you practice in one of those states, you have to follow the more stringent rule.8 For example, Ohio law does not let subpoenaed providers tender medical records for use in a grand jury proceeding without a release signed by the patient, although HIPAA would allow this (Table).6,7 Out of concern that “giving law enforcement unbridled access to medical records could discourage patients from seeking medical treatment,” Ohio protects patient records more than HIPAA does.9 New York State’s privilege rules also are stricter than HIPAA10 and contain specific provisions about releasing certain types of information (eg, HIV status11). State courts expect physicians to follow their laws about patient privacy and to consult attorneys to make sure that releasing information is done properly.12
Releasing information improperly could become grounds for legal action against you, even if you released the information in response to a subpoena. Legal action could take the form of a lawsuit for breach of confidentiality, a HIPAA-based complaint, a complaint to the state medical board, or all 3 of these.
Must you turn over information?
Before you testify or turn over documents, you need to verify that the legal and ethical requirements for the disclosure are met—as you would for any release of patient information. You can do this by obtaining your patient’s formal, written consent for the disclosure. Before you accept the patient’s agreement, however, you might—and in most cases should— consider discussing how the disclosure could affect the patient’s well-being or your treatment relationship.
If the patient will not agree to the disclosure, the patient or the patient’s attorney can seek to have the subpoena modified or quashed (declared void). One tactic for doing this is by asserting doctor–patient privilege, a legal doctrine codified in most state’s laws. The privilege recognizes that, because privacy is important in medical care, stopping clinical information from automatically coming in court serves an important social purpose.13
The doctor–patient privilege belongs to the litigant—here, your patient—not to you, so your patient has to raise the objection to releasing information.14 Also, the privilege is not absolute. If having the clinical information is necessary, the judge may issue a court order denying the patient’s motion to quash. Unless the judge later modifies or vacates the order, you risk being found in contempt of court if you still refuse to turn over documents demanded by the subpoena.
Fact witness or expert witness?
If the subpoena demands your testimony, the issuing party might want you to serve as a fact witness or expert witness. Persons with relevant personal knowledge to a legal proceeding can serve as fact witnesses, and testify about things they did or perceived.15 For example, a psychiatrist serving as a fact witness could recount having heard or seen a patient talking aloud as if arguing with someone when no real interlocutor was present.
A witness whom the court deems an “expert” by virtue of special knowledge, skill, experience, training, or education may offer opinions based on specific sets of facts. Courts hear such testimony when the expert’s specialized knowledge will help the jury understand the evidence or reach a verdict in a case.16 To return to the example above: a psychiatric expert witness might tell jurors that the patient’s “arguing” was evidence that she was hallucinating and suffered from schizophrenia.
If you receive a subpoena to testify about someone you have treated, you should notify the issuing party that you will provide fact testimony if required to do so. You cannot be compelled to serve as an expert witness, however. In many situations, attempting to provide objective expert testimony about one’s own patient could create unresolvable conflicts between the obligation to tell the truth and your obligation to serve your patient’s interests.17
If the subpoena requests deposition testimony about a patient, you probably will be able to schedule the deposition at a time that is convenient for you and the attorneys involved. Yet you should not agree to be deposed unless (a) you have received the patient’s authorization, (b) a court has ordered you to testify despite the patient’s objection, or (c) your attorney (whom you have consulted about the situation) has advised you that providing testimony is appropriate.
If you are called as a fact witness for a trial, the attorney or court that has subpoenaed you often will try to schedule things to minimize the time taken away from your other duties. Once in court, you can ask the judge (on the record) whether you must answer if you are asked questions about a patient who has not previously authorized you to release treatment information. A judge’s explicit command to respond absolves you of any further ethical obligation to withhold confidential information about the patient’s care.
Bottom Line
If you receive a subpoena for records or testimony, obtaining the patient’s written authorization should allow you to release the information without violating confidentiality obligations. If your patient won’t agree to the release, if turning over information might adversely affect the patient, or if you’re not sure what to do, seek advice from an attorney who knows about medical privacy rules. That way, you can be sure you are meeting all legal and professional standards that apply.
Disclosure
The author reports no financial relationship with any company whose products are mentioned in this article or with any manufacturers of competing products.
1. Kastigar v United States, 406 US 441 (1972).
2. Barsky AE. Clinicians in court, second edition: A guide to subpoenas, depositions, testifying, and everything else you need to know. New York, NY: Guilford Press; 2012.
3. United States v Bryan, 339 US 323 (1950).
4. 45 CFR §164.50.
5. 45 CFR Parts 160 and 164.
6. 45 CFR §164.512.
7. Stanger K. HIPAA: responding to subpoenas, orders, and administrative demands. http://www.hhhealthlawblog.com/2013/10/hipaa-responding-to-subpoenas-orders-and-administrative-demands.html. Published October 9, 2013. Accessed September 22, 2015.
8. Zwerling AL, de Harder WA, Tarpey CM. To disclose or not to disclose, that is the question: issues to consider before responding to a subpoena. J Am Coll Radiol. 2012;9(4):279-281.
9. Turk v Oilier, 732 F Supp 2d 758 (ND Ohio 2010).
10. In re Antonia E, 16 Misc 3d 637 (Fam Ct Queens County 2007).
11. NY PBH Law §2785.
12. Crescenzo v Crane, 796 A2d 283 (NJ Super Ct App Div 2002).
13. In re Bruendl’s Will, 102 Wis 45, 78 NW 169 (1899).
14. In re Lifschutz, 2 Cal 3d 415, 85 Cal Rptr 829, 467 P2d 557 (1970).
15. Broun KS, Dix GE, Imwinkelried EJ, et al. McCormick on evidence. 7th ed. St. Paul, MN: West Group; 2013.
16. Fed Evid R 702.
17. Strasburger LH, Gutheil TG, Brodsky A. On wearing two hats: role conflict in serving as both psychotherapist and expert witness. Am J Psychiatry. 1997;154:448-456.
1. Kastigar v United States, 406 US 441 (1972).
2. Barsky AE. Clinicians in court, second edition: A guide to subpoenas, depositions, testifying, and everything else you need to know. New York, NY: Guilford Press; 2012.
3. United States v Bryan, 339 US 323 (1950).
4. 45 CFR §164.50.
5. 45 CFR Parts 160 and 164.
6. 45 CFR §164.512.
7. Stanger K. HIPAA: responding to subpoenas, orders, and administrative demands. http://www.hhhealthlawblog.com/2013/10/hipaa-responding-to-subpoenas-orders-and-administrative-demands.html. Published October 9, 2013. Accessed September 22, 2015.
8. Zwerling AL, de Harder WA, Tarpey CM. To disclose or not to disclose, that is the question: issues to consider before responding to a subpoena. J Am Coll Radiol. 2012;9(4):279-281.
9. Turk v Oilier, 732 F Supp 2d 758 (ND Ohio 2010).
10. In re Antonia E, 16 Misc 3d 637 (Fam Ct Queens County 2007).
11. NY PBH Law §2785.
12. Crescenzo v Crane, 796 A2d 283 (NJ Super Ct App Div 2002).
13. In re Bruendl’s Will, 102 Wis 45, 78 NW 169 (1899).
14. In re Lifschutz, 2 Cal 3d 415, 85 Cal Rptr 829, 467 P2d 557 (1970).
15. Broun KS, Dix GE, Imwinkelried EJ, et al. McCormick on evidence. 7th ed. St. Paul, MN: West Group; 2013.
16. Fed Evid R 702.
17. Strasburger LH, Gutheil TG, Brodsky A. On wearing two hats: role conflict in serving as both psychotherapist and expert witness. Am J Psychiatry. 1997;154:448-456.