User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Can combining triptans with SSRIs or SNRIs cause serotonin syndrome?
In 2006, the FDA issued a warning of the risk of potentially fatal serotonin syndrome when 5-hydroxytryptamine receptor agonist antimigraine medications (triptans) and selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRI) are coprescribed.1 As a result, most drug interaction programs trigger a serotonin syndrome warning when triptans are prescribed with an SSRI or SNRI.2 However, many patients with depression or anxiety also suffer from migraines and require treatment with both triptans and an SSRI or SNRI.3,4 Kalaydjian et al4 found the incidence of major depression and generalized anxiety disorder were approximately 3 times greater in patients with migraines than in those without migraines. Should we avoid coprescribing triptans and SSRIs or SNRIs?
What is serotonin syndrome?
Serotonin syndrome is an adverse drug reaction that results from excessive serotonin stimulation. There are 2 sets of validated diagnostic criteria: the Sternbach Criteria and the Hunter Serotonin Toxicity Criteria; the latter is considered more stringent.3,5-7 Symptoms of serotonin syndrome include mental status changes, autonomic hyperactivity, and neuromuscular changes such as muscle rigidity.5-7 Typical manifestations of serotonin syndrome on physical exam include spontaneous and/or inducible clonus, agitation, diaphoresis, tremor, hyperreflexia, hypertonia, and temperature >38°C.6 In severe cases, serotonin syndrome can lead to seizures, coma, and death. Management includes supportive treatment, discontinuing the offending agents, controlling agitation with medications such as benzodiazepines, and possibly administering cyproheptadine, a 5HT2A antagonist.8 Most cases resolve within 24 hours of discontinuing the offending agents or appropriate treatment.5
What did the FDA say?
The 2006 FDA warning initially was based on 27 reports of serotonin syndrome in patients receiving triptans and SSRIs or SNRIs; this was later expanded to include 29 patients.1,9 No patients died but 13 required hospitalization and 2 had life-threatening symptoms. However, most cases lacked data necessary to diagnose serotonin syndrome.9 Further, reviews of the available clinical information have suggested that in some cases, clinicians did not rule out other disorders as required by diagnostic criteria, while others were viral in nature or resolved despite ongoing treatment with the presumed offending agents.9-11
Some clinicians met the FDA’s assessment with skepticism. Only 10 of the 29 cases met the Sternbach criteria of serotonin syndrome and none met the more rigorous Hunter criteria. Additionally, the theoretical basis has been questioned.9-11 Available evidence indicates that serotonin syndrome requires activation of 5HT2A receptors and a possible limited role of 5HT1A.9-12 However, triptans are agonists at the 5HT1B/1D/1F receptor subtypes, with weak affinity for 5HT1A receptors and no activity at the 5HT2 receptors.13,14 Additionally, triptan medications are used as needed, not as standing treatments, with parameters limiting the maximum dose, dosing interval, and frequency of use. In clinical practice, it appears that these dosing guidelines are being followed: Tepper et al15 found the typical female patient experiences 1 to 2 migraines per month; on average, patients use 1.2 to 1.8 triptan tablets per month.
Our opinion
We believe it is reasonable to coprescribe SSRIs or SNRIs with triptans because:
- data indicate that many patients are treated with a combination of triptans and SSRIs or SNRIs but the number of reported cases of serotonin syndrome is extremely limited
- the nature of serotonin syndrome cases reported in the literature is questionable
- the interaction is biologically implausible
- triptans remain in the body for a limited time
- triptans are used infrequently.5-11
This view is supported by the most recent American Headache Society position paper,11 which states that inadequate data are available to assess the risk but current evidence does not support limiting use of triptans with SSRIs and SNRIs.
How we deal with the warning in clinical practice. In practice we are alerted to this interaction by notification in our e-prescribing systems, by pharmacists calling with concerns about dispensing an SSRI or SNRI for a patient already receiving a triptan, and during patient visits that involve prescribing an SSRI or SNRI.
Although it is relatively easy to override a drug interaction warning in our e-prescribing system, we discuss the issue with pharmacists and patients. We provide information about the signs and symptoms of serotonin syndrome and its potential dangerousness. We note that serotonin syndrome is a theoretical concern, but highly unlikely with this combination of medications because of their pharmacologic properties. We explain the parameters of triptan use, recommend that our patients use triptans for migraines when needed, and reassure patients we are available to answer questions. When a patient uses triptans more than twice monthly, we consider discussing this usage with the patient and the treating physician.
Related Resource
- Sclar DA, Robison LM, Castillo LV, et al. Concomitant use of triptan, and SSRI or SNRI after the US Food and Drug Administration alert on serotonin syndrome. Headache. 2012. www.headachejournal.org/SpringboardWebApp/userfiles/headache/file/sclar.pdf.
Drug Brand Name
- Cyproheptadine • Perinctin
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. U.S. Food and Drug Administration. Public health advisory—combined use of 5-hydroxytryptamine receptor agonists (triptans), selective serotonin reuptake inhibitors (SSRIs) or selective serotonin/norepinephrine reuptake inhibitors (SNRIs) may result in life-threatening serotonin syndrome. http://1.usa.gov/U0A0V4. Published July 19, 2006. Accessed September 18, 2012.
2. Kogut SJ. Do triptan antimigraine medications interact with SSRI/SNRI antidepressants? What does your decision support system say? J Manag Care Pharm. 2011;17(7):547-551.
3. Tepper SJ. Serotonin syndrome: SSRIs SNRIs, triptans, and current clinical practice. Headache. 2012;52(2):195-197.
4. Kalaydjian A, Merikangas K. Physical and mental comorbidity of headache in a nationally representative sample of US adults. Psychosom Med. 2008;70(7):773-780.
5. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352(11):1112-1120.
6. Sternbach H. The serotonin syndrome. Am J Psychiatry. 1991;148(6):705-713.
7. Dunkley EJ, Isbister GK, Sibbritt D, et al. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM. 2003;96(9):635-642.
8. Ables AZ, Nagubilli R. Prevention recognition, and management of serotonin syndrome. Am Fam Physician. 2010;81(9):1139-1142.
9. Evans RW. The FDA alert on serotonin syndrome with combined use of SSRIs or SNRIs and triptans: an analysis of the 29 case reports. MedGenMed. 2007;9(3):48.-
10. Gillman PK. Triptans serotonin agonists, and serotonin syndrome (serotonin toxicity): a review. Headache. 2010;50(2):264-272.
11. Evans RW, Tepper SJ, Shapiro RE, et al. The FDA alert on serotonin syndrome with use of triptans combined with selective serotonin reuptake inhibitors or selective serotonin-norepinephrine reuptake inhibitors: American Headache Society position paper. Headache. 2010;50(6):1089-1099.
12. Ahn AH, Basbaum AI. Where do triptans act in the treatment of migraine? Pain. 2005;115(1-2):1-4.
13. Pediatric & Neonatal Lexi-Drugs. Hudson, OH: Lexi-Comp, Inc.; 2011.
14. Sclar DA, Robison LM, Castillo LV, et al. Concomitant use of triptan, and SSRI or SNRI after the US Food and Drug Administration alert on serotonin syndrome. Headache. 2012;52(2):198-203.
15. Tepper S, Allen C, Sanders D, et al. Coprescription of triptans with potentially interacting medications: a cohort study involving 240,268 patients. Headache. 2003;43(1):44-48.
In 2006, the FDA issued a warning of the risk of potentially fatal serotonin syndrome when 5-hydroxytryptamine receptor agonist antimigraine medications (triptans) and selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRI) are coprescribed.1 As a result, most drug interaction programs trigger a serotonin syndrome warning when triptans are prescribed with an SSRI or SNRI.2 However, many patients with depression or anxiety also suffer from migraines and require treatment with both triptans and an SSRI or SNRI.3,4 Kalaydjian et al4 found the incidence of major depression and generalized anxiety disorder were approximately 3 times greater in patients with migraines than in those without migraines. Should we avoid coprescribing triptans and SSRIs or SNRIs?
What is serotonin syndrome?
Serotonin syndrome is an adverse drug reaction that results from excessive serotonin stimulation. There are 2 sets of validated diagnostic criteria: the Sternbach Criteria and the Hunter Serotonin Toxicity Criteria; the latter is considered more stringent.3,5-7 Symptoms of serotonin syndrome include mental status changes, autonomic hyperactivity, and neuromuscular changes such as muscle rigidity.5-7 Typical manifestations of serotonin syndrome on physical exam include spontaneous and/or inducible clonus, agitation, diaphoresis, tremor, hyperreflexia, hypertonia, and temperature >38°C.6 In severe cases, serotonin syndrome can lead to seizures, coma, and death. Management includes supportive treatment, discontinuing the offending agents, controlling agitation with medications such as benzodiazepines, and possibly administering cyproheptadine, a 5HT2A antagonist.8 Most cases resolve within 24 hours of discontinuing the offending agents or appropriate treatment.5
What did the FDA say?
The 2006 FDA warning initially was based on 27 reports of serotonin syndrome in patients receiving triptans and SSRIs or SNRIs; this was later expanded to include 29 patients.1,9 No patients died but 13 required hospitalization and 2 had life-threatening symptoms. However, most cases lacked data necessary to diagnose serotonin syndrome.9 Further, reviews of the available clinical information have suggested that in some cases, clinicians did not rule out other disorders as required by diagnostic criteria, while others were viral in nature or resolved despite ongoing treatment with the presumed offending agents.9-11
Some clinicians met the FDA’s assessment with skepticism. Only 10 of the 29 cases met the Sternbach criteria of serotonin syndrome and none met the more rigorous Hunter criteria. Additionally, the theoretical basis has been questioned.9-11 Available evidence indicates that serotonin syndrome requires activation of 5HT2A receptors and a possible limited role of 5HT1A.9-12 However, triptans are agonists at the 5HT1B/1D/1F receptor subtypes, with weak affinity for 5HT1A receptors and no activity at the 5HT2 receptors.13,14 Additionally, triptan medications are used as needed, not as standing treatments, with parameters limiting the maximum dose, dosing interval, and frequency of use. In clinical practice, it appears that these dosing guidelines are being followed: Tepper et al15 found the typical female patient experiences 1 to 2 migraines per month; on average, patients use 1.2 to 1.8 triptan tablets per month.
Our opinion
We believe it is reasonable to coprescribe SSRIs or SNRIs with triptans because:
- data indicate that many patients are treated with a combination of triptans and SSRIs or SNRIs but the number of reported cases of serotonin syndrome is extremely limited
- the nature of serotonin syndrome cases reported in the literature is questionable
- the interaction is biologically implausible
- triptans remain in the body for a limited time
- triptans are used infrequently.5-11
This view is supported by the most recent American Headache Society position paper,11 which states that inadequate data are available to assess the risk but current evidence does not support limiting use of triptans with SSRIs and SNRIs.
How we deal with the warning in clinical practice. In practice we are alerted to this interaction by notification in our e-prescribing systems, by pharmacists calling with concerns about dispensing an SSRI or SNRI for a patient already receiving a triptan, and during patient visits that involve prescribing an SSRI or SNRI.
Although it is relatively easy to override a drug interaction warning in our e-prescribing system, we discuss the issue with pharmacists and patients. We provide information about the signs and symptoms of serotonin syndrome and its potential dangerousness. We note that serotonin syndrome is a theoretical concern, but highly unlikely with this combination of medications because of their pharmacologic properties. We explain the parameters of triptan use, recommend that our patients use triptans for migraines when needed, and reassure patients we are available to answer questions. When a patient uses triptans more than twice monthly, we consider discussing this usage with the patient and the treating physician.
Related Resource
- Sclar DA, Robison LM, Castillo LV, et al. Concomitant use of triptan, and SSRI or SNRI after the US Food and Drug Administration alert on serotonin syndrome. Headache. 2012. www.headachejournal.org/SpringboardWebApp/userfiles/headache/file/sclar.pdf.
Drug Brand Name
- Cyproheptadine • Perinctin
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
In 2006, the FDA issued a warning of the risk of potentially fatal serotonin syndrome when 5-hydroxytryptamine receptor agonist antimigraine medications (triptans) and selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRI) are coprescribed.1 As a result, most drug interaction programs trigger a serotonin syndrome warning when triptans are prescribed with an SSRI or SNRI.2 However, many patients with depression or anxiety also suffer from migraines and require treatment with both triptans and an SSRI or SNRI.3,4 Kalaydjian et al4 found the incidence of major depression and generalized anxiety disorder were approximately 3 times greater in patients with migraines than in those without migraines. Should we avoid coprescribing triptans and SSRIs or SNRIs?
What is serotonin syndrome?
Serotonin syndrome is an adverse drug reaction that results from excessive serotonin stimulation. There are 2 sets of validated diagnostic criteria: the Sternbach Criteria and the Hunter Serotonin Toxicity Criteria; the latter is considered more stringent.3,5-7 Symptoms of serotonin syndrome include mental status changes, autonomic hyperactivity, and neuromuscular changes such as muscle rigidity.5-7 Typical manifestations of serotonin syndrome on physical exam include spontaneous and/or inducible clonus, agitation, diaphoresis, tremor, hyperreflexia, hypertonia, and temperature >38°C.6 In severe cases, serotonin syndrome can lead to seizures, coma, and death. Management includes supportive treatment, discontinuing the offending agents, controlling agitation with medications such as benzodiazepines, and possibly administering cyproheptadine, a 5HT2A antagonist.8 Most cases resolve within 24 hours of discontinuing the offending agents or appropriate treatment.5
What did the FDA say?
The 2006 FDA warning initially was based on 27 reports of serotonin syndrome in patients receiving triptans and SSRIs or SNRIs; this was later expanded to include 29 patients.1,9 No patients died but 13 required hospitalization and 2 had life-threatening symptoms. However, most cases lacked data necessary to diagnose serotonin syndrome.9 Further, reviews of the available clinical information have suggested that in some cases, clinicians did not rule out other disorders as required by diagnostic criteria, while others were viral in nature or resolved despite ongoing treatment with the presumed offending agents.9-11
Some clinicians met the FDA’s assessment with skepticism. Only 10 of the 29 cases met the Sternbach criteria of serotonin syndrome and none met the more rigorous Hunter criteria. Additionally, the theoretical basis has been questioned.9-11 Available evidence indicates that serotonin syndrome requires activation of 5HT2A receptors and a possible limited role of 5HT1A.9-12 However, triptans are agonists at the 5HT1B/1D/1F receptor subtypes, with weak affinity for 5HT1A receptors and no activity at the 5HT2 receptors.13,14 Additionally, triptan medications are used as needed, not as standing treatments, with parameters limiting the maximum dose, dosing interval, and frequency of use. In clinical practice, it appears that these dosing guidelines are being followed: Tepper et al15 found the typical female patient experiences 1 to 2 migraines per month; on average, patients use 1.2 to 1.8 triptan tablets per month.
Our opinion
We believe it is reasonable to coprescribe SSRIs or SNRIs with triptans because:
- data indicate that many patients are treated with a combination of triptans and SSRIs or SNRIs but the number of reported cases of serotonin syndrome is extremely limited
- the nature of serotonin syndrome cases reported in the literature is questionable
- the interaction is biologically implausible
- triptans remain in the body for a limited time
- triptans are used infrequently.5-11
This view is supported by the most recent American Headache Society position paper,11 which states that inadequate data are available to assess the risk but current evidence does not support limiting use of triptans with SSRIs and SNRIs.
How we deal with the warning in clinical practice. In practice we are alerted to this interaction by notification in our e-prescribing systems, by pharmacists calling with concerns about dispensing an SSRI or SNRI for a patient already receiving a triptan, and during patient visits that involve prescribing an SSRI or SNRI.
Although it is relatively easy to override a drug interaction warning in our e-prescribing system, we discuss the issue with pharmacists and patients. We provide information about the signs and symptoms of serotonin syndrome and its potential dangerousness. We note that serotonin syndrome is a theoretical concern, but highly unlikely with this combination of medications because of their pharmacologic properties. We explain the parameters of triptan use, recommend that our patients use triptans for migraines when needed, and reassure patients we are available to answer questions. When a patient uses triptans more than twice monthly, we consider discussing this usage with the patient and the treating physician.
Related Resource
- Sclar DA, Robison LM, Castillo LV, et al. Concomitant use of triptan, and SSRI or SNRI after the US Food and Drug Administration alert on serotonin syndrome. Headache. 2012. www.headachejournal.org/SpringboardWebApp/userfiles/headache/file/sclar.pdf.
Drug Brand Name
- Cyproheptadine • Perinctin
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. U.S. Food and Drug Administration. Public health advisory—combined use of 5-hydroxytryptamine receptor agonists (triptans), selective serotonin reuptake inhibitors (SSRIs) or selective serotonin/norepinephrine reuptake inhibitors (SNRIs) may result in life-threatening serotonin syndrome. http://1.usa.gov/U0A0V4. Published July 19, 2006. Accessed September 18, 2012.
2. Kogut SJ. Do triptan antimigraine medications interact with SSRI/SNRI antidepressants? What does your decision support system say? J Manag Care Pharm. 2011;17(7):547-551.
3. Tepper SJ. Serotonin syndrome: SSRIs SNRIs, triptans, and current clinical practice. Headache. 2012;52(2):195-197.
4. Kalaydjian A, Merikangas K. Physical and mental comorbidity of headache in a nationally representative sample of US adults. Psychosom Med. 2008;70(7):773-780.
5. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352(11):1112-1120.
6. Sternbach H. The serotonin syndrome. Am J Psychiatry. 1991;148(6):705-713.
7. Dunkley EJ, Isbister GK, Sibbritt D, et al. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM. 2003;96(9):635-642.
8. Ables AZ, Nagubilli R. Prevention recognition, and management of serotonin syndrome. Am Fam Physician. 2010;81(9):1139-1142.
9. Evans RW. The FDA alert on serotonin syndrome with combined use of SSRIs or SNRIs and triptans: an analysis of the 29 case reports. MedGenMed. 2007;9(3):48.-
10. Gillman PK. Triptans serotonin agonists, and serotonin syndrome (serotonin toxicity): a review. Headache. 2010;50(2):264-272.
11. Evans RW, Tepper SJ, Shapiro RE, et al. The FDA alert on serotonin syndrome with use of triptans combined with selective serotonin reuptake inhibitors or selective serotonin-norepinephrine reuptake inhibitors: American Headache Society position paper. Headache. 2010;50(6):1089-1099.
12. Ahn AH, Basbaum AI. Where do triptans act in the treatment of migraine? Pain. 2005;115(1-2):1-4.
13. Pediatric & Neonatal Lexi-Drugs. Hudson, OH: Lexi-Comp, Inc.; 2011.
14. Sclar DA, Robison LM, Castillo LV, et al. Concomitant use of triptan, and SSRI or SNRI after the US Food and Drug Administration alert on serotonin syndrome. Headache. 2012;52(2):198-203.
15. Tepper S, Allen C, Sanders D, et al. Coprescription of triptans with potentially interacting medications: a cohort study involving 240,268 patients. Headache. 2003;43(1):44-48.
1. U.S. Food and Drug Administration. Public health advisory—combined use of 5-hydroxytryptamine receptor agonists (triptans), selective serotonin reuptake inhibitors (SSRIs) or selective serotonin/norepinephrine reuptake inhibitors (SNRIs) may result in life-threatening serotonin syndrome. http://1.usa.gov/U0A0V4. Published July 19, 2006. Accessed September 18, 2012.
2. Kogut SJ. Do triptan antimigraine medications interact with SSRI/SNRI antidepressants? What does your decision support system say? J Manag Care Pharm. 2011;17(7):547-551.
3. Tepper SJ. Serotonin syndrome: SSRIs SNRIs, triptans, and current clinical practice. Headache. 2012;52(2):195-197.
4. Kalaydjian A, Merikangas K. Physical and mental comorbidity of headache in a nationally representative sample of US adults. Psychosom Med. 2008;70(7):773-780.
5. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352(11):1112-1120.
6. Sternbach H. The serotonin syndrome. Am J Psychiatry. 1991;148(6):705-713.
7. Dunkley EJ, Isbister GK, Sibbritt D, et al. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM. 2003;96(9):635-642.
8. Ables AZ, Nagubilli R. Prevention recognition, and management of serotonin syndrome. Am Fam Physician. 2010;81(9):1139-1142.
9. Evans RW. The FDA alert on serotonin syndrome with combined use of SSRIs or SNRIs and triptans: an analysis of the 29 case reports. MedGenMed. 2007;9(3):48.-
10. Gillman PK. Triptans serotonin agonists, and serotonin syndrome (serotonin toxicity): a review. Headache. 2010;50(2):264-272.
11. Evans RW, Tepper SJ, Shapiro RE, et al. The FDA alert on serotonin syndrome with use of triptans combined with selective serotonin reuptake inhibitors or selective serotonin-norepinephrine reuptake inhibitors: American Headache Society position paper. Headache. 2010;50(6):1089-1099.
12. Ahn AH, Basbaum AI. Where do triptans act in the treatment of migraine? Pain. 2005;115(1-2):1-4.
13. Pediatric & Neonatal Lexi-Drugs. Hudson, OH: Lexi-Comp, Inc.; 2011.
14. Sclar DA, Robison LM, Castillo LV, et al. Concomitant use of triptan, and SSRI or SNRI after the US Food and Drug Administration alert on serotonin syndrome. Headache. 2012;52(2):198-203.
15. Tepper S, Allen C, Sanders D, et al. Coprescription of triptans with potentially interacting medications: a cohort study involving 240,268 patients. Headache. 2003;43(1):44-48.
The link between schizophrenia and diabetes
Discuss this article at www.facebook.com/CurrentPsychiatry
Although diabetes and schizophrenia are common companions, it is unclear how this association should influence our practice. What do we need to know about diabetes, and what are the key intervention points for psychiatrists?
This article is informed by my experience monitoring >1,000 patients with schizophrenia in a large urban mental health facility using an electronic metabolic monitoring system and consulting on hundreds of individuals with comorbid schizophrenia and diabetes in a mental health metabolic clinic.
A significant link
The association between schizophrenia and diabetes has been recognized for more than a century.1 The prevalence of diabetes is increased 2- to 3-fold in patients with schizophrenia.2,3 This relationship is specific to type 2 diabetes mellitus (T2DM); type 1 diabetes mellitus, an autoimmune disease, is less common in patients with schizophrenia.4 Factors that contribute to comorbidity between schizophrenia and T2DM include:
- illness susceptibility: the mechanisms remain unclear but include the thrifty phenotype hypothesis,5 autonomic hyperactivity,6 and potential cellular and genetic links7,8
- lifestyle: diet, physical inactivity, and cigarette smoking9-12
- antipsychotic use13
- social health determinants, such as income, housing, and food insecurity.14
When evaluating a patient’s risk for a cardiac event, we consider having a diabetes diagnosis equivalent to having had a myocardial infarction.15 Likely, the high prevalence of T2DM among schizophrenia patients and challenges in managing diabetes and prediabetes underlies these patients’ reduced life expectancy.16 Self-care, a cornerstone of diabetes management, is challenging for patients with schizophrenia because of deficits in executive functioning, working memory, and motivation, coupled with negative symptoms and social and economic disadvantages that often accompany schizophrenia.
How diabetes impacts practice
What psychiatrists need to know. Insulin resistance—reduced biologic effectiveness of insulin—is the precursor of T2DM. Insulin is required to move glucose from the blood into cells. Weight gain, particularly abdominal adiposity, is the principal driver of insulin resistance. The body responds by producing more insulin (hyperinsulinemia) to maintain glucose homeostasis. Hyperinsulinemia underlies metabolic syndrome, an important risk marker for developing T2DM. Diabetes usually develops after many years when the pancreas fails to compensate for insulin resistance.
In most cases the development of diabetes in patients with schizophrenia follows this course. Weight gain, a consequence of lifestyle factors as well as antipsychotics and other psychotropics that promote obesity, leads to progressive insulin resistance. Consequently, metabolic syndrome is twice as prevalent among patients with schizophrenia compared with matched controls.17,18
Occasionally patients develop T2DM within a few weeks or months of starting antipsychotic treatment—usually with clozapine or olanzapine—before they gain weight, which suggests a second mechanism may be involved. Animal studies have documented rapid development of insulin resistance after a single subcutaneous injection of antipsychotics that have high metabolic liability, possibly through a direct effect on insulin signaling.19 This phenomenon has been difficult to demonstrate in humans.20
Managing diabetes is complex and ideally involves a range of health practitioners who work with patients to provide education, promote self-care behaviors, and direct complex health care. These services are outside the scope of psychiatric practice, but given the functional deficits in seriously mentally ill patients, it is important to have an overview of diabetes care (Table 3).
In addition to diagnosing diabetes, psychiatrists should be able to identify patients at risk for developing diabetes and initiate prevention strategies. Interventions are focused on lifestyle—weight reduction, increased physical activity, diet, and smoking cessation—as well as pharmacologic strategies such as metformin.
What patients need to know. Similar to schizophrenia, a diabetes diagnosis may be difficult for patients to accept. Initially, a patient may have no manifestations or symptoms. However, untreated diabetes has serious long-term health consequences—including blindness, amputations, kidney disease, and early death from heart attacks.
Patients should actively participate in treatment that involves learning about the illness, making lifestyle changes, working on self-care, and keeping regular medical appointments. Three components of lifestyle change must be addressed:
- Diet: counseling with a dietician or other health professional to reduce or stabilize body weight and make changes in diet quality, portion size, and meal frequency to improve glucose control and reduce long-term diabetes complications
- Physical activity: increasing physical activity, initially by walking daily, to benefit glucose control and weight maintenance
- Smoking: reducing or stopping cigarette smoking to improve glucose control and reduce diabetes complications.
American Diabetes Association diagnostic criteria for diabetes
| Testa | Threshold | Qualifier |
|---|---|---|
| A1C, or | ≥6.5% | Lab NGSP certified, standardized DCCT assay |
| Fasting glucose, or | ≥126 mg/dL | No caloric intake for at least 8 hours |
| 2-hour glucose, or | ≥200 mg/dL | After 75 g of anhydrous glucose |
| Random glucose | ≥200 mg/dL | Plus classic hyperglycemic symptoms or crisis |
| aResults should be confirmed by repeat testing DCCT: Diabetes Control and Complications Trial; NGSP: National Glycohemoglobin Standardization Program Source: References 21-23 | ||
Signs and symptoms of diabetes and diabetic ketoacidosis
| Diabetes |
| Frequent urination |
| Excessive thirst |
| Extreme hunger |
| Unusual weight loss |
| Increased fatigue |
| Irritability |
| Blurry vision |
| Diabetic ketoacidosisa |
| Thirst or very dry mouth |
| Constantly feeling tired |
| Dry or flushed skin |
| Nausea, vomiting, or abdominal pain |
| Difficulty breathing (short, deep breaths) |
| Fruity odor on breath |
| Difficulty paying attention or confusion |
| aVomiting is a sign of escalation Source: References 24,25 |
Components of diabetes care
| Self-care tasks | Tests/annual assessments |
|---|---|
| Self-monitoring of glucose | A1C (2 to 4 times/year) |
| Medication management | Urinary microalbumin |
| Meal planning | Fasting lipids |
| Exercise | Blood pressure |
| Smoking cessation | Dilated eye exam |
| Foot self-examination and foot care | Foot exam |
| Stress management | General health and cardiovascular exam |
Managing patients at risk for diabetes
| Prediabetes21 | Management |
|---|---|
| Impaired fasting glucose (100 to 125 mg/dL) | Weight reduction (7%) Activity (150 minutes/week) At least yearly glucose monitoring |
| Impaired glucose tolerance (2-hour plasma glucose: 140 to 199 mg/dL) | |
| Prediabetic A1C (5.7% to 6.4%) | |
| Metabolic syndrome (any 3)26 | Management |
| Waist circumferencea (men >40 inches; women >35 inches) | Weight reduction Reduce consumption of refined carbohydrates Exercise Focused interventions for individual criteria |
| Fasting triglycerides (≥150 mg/dL) | |
| Fasting high-density lipoprotein cholesterol (men | |
| Fasting glucose (≥100 mg/dL or taking medication) | |
| Blood pressure (≥130/85 mm Hg or taking medication) | |
| aWaist circumference guidelines are ethnicity specific. The International Diabetes Federation27 has published specific cutoffs for those of Asian background (men: ≥90 cm [35 inches] and women: ≥80 cm [31 inches]) | |
Metabolic monitoring
Metabolic monitoring is the key to keeping patients with schizophrenia well. Treating metabolic conditions falls outside of psychiatric practice; however, many argue that mental health clinicians should monitor basic metabolic parameters during antipsychotic treatment and advocate medical interventions when indicated because:
- most antipsychotics are associated with weight gain and metabolic side effects
- patients with schizophrenia have cognitive deficits that impact health maintenance
- mental health providers often are the primary health care contacts for patients with serious mental illness.
- identify treatable medical conditions such as diabetes, dyslipidemia, and hypertension when treatment delay or no treatment has consequences
- identify individuals with prediabetes and metabolic syndrome for targeted prevention
- determine the association between antipsychotic treatment and metabolic disturbance to evaluate the risk of treatment vs antipsychotic switching.
How to intervene
To switch or not to switch? For many psychiatrists, deciding whether or when to switch from a high or intermediate metabolic liability antipsychotic to one with low metabolic liability is difficult. Clinicians must balance potential metabolic benefits against the risk of psychotic decompensation and side effects. Ultimately, patients and their families make the decision, taking into account information provided to them. For medical-legal purposes, document the discussion of potential risks and benefits. These are difficult decisions and there are no clear guidelines. In my clinical experience, the following issues need to be considered:
- The antipsychotics that many clinicians consider to be the most effective—clozapine and olanzapine—also have the greatest metabolic liability and risk for emergent T2DM.
- Patients who are stable and in psychotic remission may risk a relapse of their illness if switched.
- The clearest indication for switching is when a patient who does not have diabetes develops the condition shortly after starting an antipsychotic. This scenario is rare, but evidence suggests that diabetes may resolve or reverse with an antipsychotic switch.33
- In patients who gain weight while taking a high- or intermediate-liability antipsychotic and are able to tolerate a switch to a low-liability antipsychotic, the effect size of weight reduction can be large and may result in a patient returning to their pretreatment weight.
- To reduce relapse risk, patients switching antipsychotics should be closely monitored at least weekly for ≥1 month. A plateau cross-taper—building the new antipsychotic up to therapeutic levels before gradually reducing the first antipsychotic—may be safer than abrupt discontinuation or standard cross-titration.
- Switching from one high or intermediate liability antipsychotic to another (eg, olanzapine to quetiapine or risperidone) often provides little if any metabolic benefit on body weight or diabetes control.
- Established diabetes (type 1 or type 2) should not be a contraindication to antipsychotic treatment, including clozapine, if clinically warranted. Monitor metabolic parameters more closely for 6 to 12 months after the switch. In most cases, patients experience limited, if any, metabolic consequences. If so, diabetes medication can be adjusted.
- Patients who have experienced significant weight gain on an atypical antipsychotic often do not gain more weight when switched to clozapine. A patient may reach a “ceiling” in terms of weight gain and medication-related metabolic effects.
Data from metabolic monitoring informs the decision to switch and metabolic consequences of switching. Conducting monitoring at baseline, when starting an antipsychotic, when switching to a high-liability agent, 3 months after the switch, and then annually provides data needed to consider switching or initiating medical and behavioral or lifestyle interventions.
Facilitate early diabetes treatment. Clinicians who are most closely involved in caring for patients with schizophrenia often are best situated to screen for diabetes. I have found that without a close working relationship with my patients’ primary care practitioners, patients may experience a long delay in receiving care. After your patient is diagnosed with diabetes, establish a relationship with diabetes treatment providers and work with your patient to ensure they engage in diabetes care.
Contribute to diabetes chronic disease management. Mental health practitioners can complement diabetes care in patients with serious mental illness by:
- navigating the health system and negotiating for service on patients’ behalf
- promoting positive relationships among diabetes and mental health treatment teams
- evaluating and treating depression that may be comorbid with diabetes
- assessing treatment capacity, self-care deficits, cognitive functioning, psychotic symptoms, negative symptoms, etc., that impact diabetes self-care and collaborating with diabetes care providers to support patients.
Start with a low-liability agent
Patients who are early in the course of psychotic illness are most susceptible to the metabolic effects of antipsychotics.13 The average weight gain observed with olanzapine was 34 lbs at 2 years in first episode psychosis patients (mean age 24 ± 4.9).34 Metabolic consequences with medium-liability second-generation antipsychotics—such as quetiapine and risperidone—are extreme, particularly in children, adolescents, and young adults (age 35,36 Although frank diabetes is uncommon in early psychosis because patients are, to a certain extent, protected by insulin compensation—increased insulin secretion maintains glucose levels within a therapeutic range—diabetes risk is increased, and hyperinsulinemia and hypertriglyceridemia are early markers of metabolic strain. Also, response to initial antipsychotic treatment—possibly independent of the agent selected—is robust in early psychosis.37
Related Resources
- American Diabetes Association. www.diabetes.org.
- International Diabetes Federation. www.idf.org.
- Framingham Heart Study. Coronary heart disease (10-year risk). www.framinghamheartstudy.org/risk/coronary.html.
- National Cholesterol Education Program. Risk assessment tool for estimating your 10-year risk of having a heart attack. http://hp2010.nhlbihin.net/atpiii/calculator.asp.
- Clozapine • Clozaril
- Metformin • Glucophage
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
Dr. Cohn is a speaker for Pfizer Canada.
1. Kohen D. Diabetes mellitus and schizophrenia: historical perspective. Br J Psychiatry Suppl. 2004;47:S64-S66.
2. Dixon L, Weiden P, Delahanty J, et al. Prevalence and correlates of diabetes in national schizophrenia samples. Schizophr Bull. 2000;26(4):903-912.
3. De Hert M, van Winkel R, Van Eyck D, et al. Prevalence of diabetes, metabolic syndrome and metabolic abnormalities in schizophrenia over the course of the illness: a cross-sectional study. Clin Pract Epidemol Ment Health. 2006;2:14.-
4. Juvonen H, Reunanen A, Haukka J, et al. Incidence of schizophrenia in a nationwide cohort of patients with type 1 diabetes mellitus. Arch Gen Psychiatry. 2007;64(8):894-899.
5. Hales CN, Barker DJ. The thrifty phenotype hypothesis. Br Med Bull. 2001;60:5-20.
6. Ryan MC, Sharifi N, Condren R, et al. Evidence of basal pituitary-adrenal overactivity in first episode, drug naive patients with schizophrenia. Psychoneuroendocrinology. 2004;29(8):1065-1070.
7. Odawara M, Isaka M, Tada K, et al. Diabetes mellitus associated with mitochondrial myopathy and schizophrenia: a possible link between diabetes mellitus and schizophrenia. Diabet Med. 1997;14(6):503.-
8. Siuta MA, Robertson SD, Kocalis H, et al. Dysregulation of the norepinephrine transporter sustains cortical hypodopaminergia and schizophrenia-like behaviors in neuronal rictor null mice. PLoS Biol. 2010;8(6):e1000393.-
9. Strassnig M, Brar JS, Ganguli R. Nutritional assessment of patients with schizophrenia: a preliminary study. Schizophr Bull. 2003;29(2):393-397.
10. Daumit GL, Goldberg RW, Anthony C, et al. Physical activity patterns in adults with severe mental illness. J Nerv Ment Dis. 2005;193(10):641-646.
11. Ussher M, Stanbury L, Cheeseman V, et al. Physical activity p and perceived barriers to activity among persons with severe mental illness in the United Kingdom. Psychiatr Serv. 2007;58(3):405-408.
12. Cho NH, Chan JC, Jang HC, et al. Cigarette smoking is an independent risk factor for type 2 diabetes: a four-year community-based prospective study. Clin Endocrinol (Oxf). 2009;71(5):679-685.
13. Newcomer JW. Second-generation (atypical) antipsychotics and metabolic effects: a comprehensive literature review. CNS Drugs. 2005;19(suppl 1):1-93.
14. Yu VL, Raphael D. Identifying and addressing the social determinants of the incidence and successful management of type 2 diabetes mellitus in Canada. Can J Public Health. 2004;95(5):366-368.
15. Barr EL, Zimmet PZ, Welborn TA, et al. Risk of cardiovascular and all-cause mortality in individuals with diabetes mellitus, impaired fasting glucose, and impaired glucose tolerance: the Australian Diabetes, Obesity, and Lifestyle Study (AusDiab). Circulation. 2007;116(2):151-157.
16. Colton CW, Manderscheid RW. Congruencies in increased mortality rates years of potential life lost, and causes of death among public mental health clients in eight states. Prev Chronic Dis. 2006;3(2):A42.-
17. Cohn T, Prud’homme D, Streiner D, et al. Characterizing coronary heart disease risk in chronic schizophrenia: high prevalence of the metabolic syndrome. Can J Psychiatry. 2004;49(11):753-760.
18. Meyer JM, Stahl SM. The metabolic syndrome and schizophrenia. Acta Psychiatr Scand. 2009;119(1):4-14.
19. Chintoh AF, Mann SW, Lam L, et al. Insulin resistance and decreased glucose-stimulated insulin secretion after acute olanzapine administration. J Clin Psychopharmacol. 2008;28(5):494-499.
20. Hahn MK, Arenovich T, Wolever T, et al. Single dose administration of olanzapine: effects on glucose metabolism, endocrine and inflammatory markers in healthy volunteers. Poster presented at: Schizophrenia International Research Society 3rd Biennial Conference; April 14-18, 2012; Florence, Italy.
21. American Diabetes Association. Standards of medical care in diabetes—2012. Diabetes Care. 2012;35(suppl 1):S11-S63.
22. Little RR. Glycated hemoglobin standardization—National Glycohemoglobin Standardization Program (NGSP) perspective. Clin Chem Lab Med. 2003;41(9):1191-1198.
23. Keen H. The Diabetes Control and Complications Trial (DCCT). Health Trends. 1994;26(2):41-43.
24. American Diabetes Association. Symptoms. http://www.diabetes.org/diabetes-basics/symptoms. Accessed August 27 2012.
25. American Diabetes Association. Ketoacidosis (DKA). http://www.diabetes.org/living-with-diabetes/complications/ketoacidosis-dka.html. Accessed August 27 2012.
26. Grundy SM, Cleeman JI, Daniels SR, et al. American Heart Association; National Heart, Lung, and Blood Institute. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112(17):2735-2752.
27. Alberti KG, Zimmet P, Shaw J. Metabolic syndrome—a new world-wide definition. A consensus statement from the International Diabetes Federation. Diabet Med. 2006;23(5):469-480.
28. Rodbard HW, Blonde L, Braithwaite SS, et al. AACE Diabetes Mellitus Clinical Practice Guidelines Task Force. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the management of diabetes mellitus. Endocr Pract. 2007;13(suppl 1):1-68.
29. Cohn TA, Sernyak MJ. Metabolic monitoring for patients treated with antipsychotic medications. Can J Psychiatry. 2006;51(8):492-501.
30. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists; North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
31. Newcomer JW, Nasrallah HA, Loebel AD. The Atypical Antipsychotic Therapy and Metabolic Issues National Survey: practice patterns and knowledge of psychiatrists. J Clin Psychopharmacol. 2004;24(5 suppl 1):S1-S6.
32. Khoury A, Sproule BA, Cohn TA. Development and implementation of the Metabolic Health Monitor at the Centre for Addiction and Mental Health. Poster presented at: BC Psychopharmacology Conference; February 15-16 2008; Vancouver, British Columbia, Canada.
33. Koller EA, Doraiswamy PM. Olanzapine-associated diabetes mellitus. Pharmacotherapy. 2002;22(7):841-852.
34. Zipursky RB, Gu H, Green AI, et al. Course and predictors of weight gain in people with first-episode psychosis treated with olanzapine or haloperidol. Br J Psychiatry. 2005;187:537-543.
35. Correll CU, Carlson HE. Endocrine and metabolic adverse effects of psychotropic medications in children and adolescents. J Am Acad Child Adolesc Psychiatry. 2006;45(7):771-791.
36. Correll CU, Manu P, Olshanskiy V, et al. Cardiometabolic risk of second-generation antipsychotic medications during first-time use in children and adolescents. JAMA. 2009;302(16):1765-1773.
37. Nicol G, Newcomer J. Review: children and adolescents with schizophrenia spectrum disorders respond to antipsychotics but are susceptible to adverse events. Evid Based Ment Health. 2008;11(3):81.-
Discuss this article at www.facebook.com/CurrentPsychiatry
Although diabetes and schizophrenia are common companions, it is unclear how this association should influence our practice. What do we need to know about diabetes, and what are the key intervention points for psychiatrists?
This article is informed by my experience monitoring >1,000 patients with schizophrenia in a large urban mental health facility using an electronic metabolic monitoring system and consulting on hundreds of individuals with comorbid schizophrenia and diabetes in a mental health metabolic clinic.
A significant link
The association between schizophrenia and diabetes has been recognized for more than a century.1 The prevalence of diabetes is increased 2- to 3-fold in patients with schizophrenia.2,3 This relationship is specific to type 2 diabetes mellitus (T2DM); type 1 diabetes mellitus, an autoimmune disease, is less common in patients with schizophrenia.4 Factors that contribute to comorbidity between schizophrenia and T2DM include:
- illness susceptibility: the mechanisms remain unclear but include the thrifty phenotype hypothesis,5 autonomic hyperactivity,6 and potential cellular and genetic links7,8
- lifestyle: diet, physical inactivity, and cigarette smoking9-12
- antipsychotic use13
- social health determinants, such as income, housing, and food insecurity.14
When evaluating a patient’s risk for a cardiac event, we consider having a diabetes diagnosis equivalent to having had a myocardial infarction.15 Likely, the high prevalence of T2DM among schizophrenia patients and challenges in managing diabetes and prediabetes underlies these patients’ reduced life expectancy.16 Self-care, a cornerstone of diabetes management, is challenging for patients with schizophrenia because of deficits in executive functioning, working memory, and motivation, coupled with negative symptoms and social and economic disadvantages that often accompany schizophrenia.
How diabetes impacts practice
What psychiatrists need to know. Insulin resistance—reduced biologic effectiveness of insulin—is the precursor of T2DM. Insulin is required to move glucose from the blood into cells. Weight gain, particularly abdominal adiposity, is the principal driver of insulin resistance. The body responds by producing more insulin (hyperinsulinemia) to maintain glucose homeostasis. Hyperinsulinemia underlies metabolic syndrome, an important risk marker for developing T2DM. Diabetes usually develops after many years when the pancreas fails to compensate for insulin resistance.
In most cases the development of diabetes in patients with schizophrenia follows this course. Weight gain, a consequence of lifestyle factors as well as antipsychotics and other psychotropics that promote obesity, leads to progressive insulin resistance. Consequently, metabolic syndrome is twice as prevalent among patients with schizophrenia compared with matched controls.17,18
Occasionally patients develop T2DM within a few weeks or months of starting antipsychotic treatment—usually with clozapine or olanzapine—before they gain weight, which suggests a second mechanism may be involved. Animal studies have documented rapid development of insulin resistance after a single subcutaneous injection of antipsychotics that have high metabolic liability, possibly through a direct effect on insulin signaling.19 This phenomenon has been difficult to demonstrate in humans.20
Managing diabetes is complex and ideally involves a range of health practitioners who work with patients to provide education, promote self-care behaviors, and direct complex health care. These services are outside the scope of psychiatric practice, but given the functional deficits in seriously mentally ill patients, it is important to have an overview of diabetes care (Table 3).
In addition to diagnosing diabetes, psychiatrists should be able to identify patients at risk for developing diabetes and initiate prevention strategies. Interventions are focused on lifestyle—weight reduction, increased physical activity, diet, and smoking cessation—as well as pharmacologic strategies such as metformin.
What patients need to know. Similar to schizophrenia, a diabetes diagnosis may be difficult for patients to accept. Initially, a patient may have no manifestations or symptoms. However, untreated diabetes has serious long-term health consequences—including blindness, amputations, kidney disease, and early death from heart attacks.
Patients should actively participate in treatment that involves learning about the illness, making lifestyle changes, working on self-care, and keeping regular medical appointments. Three components of lifestyle change must be addressed:
- Diet: counseling with a dietician or other health professional to reduce or stabilize body weight and make changes in diet quality, portion size, and meal frequency to improve glucose control and reduce long-term diabetes complications
- Physical activity: increasing physical activity, initially by walking daily, to benefit glucose control and weight maintenance
- Smoking: reducing or stopping cigarette smoking to improve glucose control and reduce diabetes complications.
American Diabetes Association diagnostic criteria for diabetes
| Testa | Threshold | Qualifier |
|---|---|---|
| A1C, or | ≥6.5% | Lab NGSP certified, standardized DCCT assay |
| Fasting glucose, or | ≥126 mg/dL | No caloric intake for at least 8 hours |
| 2-hour glucose, or | ≥200 mg/dL | After 75 g of anhydrous glucose |
| Random glucose | ≥200 mg/dL | Plus classic hyperglycemic symptoms or crisis |
| aResults should be confirmed by repeat testing DCCT: Diabetes Control and Complications Trial; NGSP: National Glycohemoglobin Standardization Program Source: References 21-23 | ||
Signs and symptoms of diabetes and diabetic ketoacidosis
| Diabetes |
| Frequent urination |
| Excessive thirst |
| Extreme hunger |
| Unusual weight loss |
| Increased fatigue |
| Irritability |
| Blurry vision |
| Diabetic ketoacidosisa |
| Thirst or very dry mouth |
| Constantly feeling tired |
| Dry or flushed skin |
| Nausea, vomiting, or abdominal pain |
| Difficulty breathing (short, deep breaths) |
| Fruity odor on breath |
| Difficulty paying attention or confusion |
| aVomiting is a sign of escalation Source: References 24,25 |
Components of diabetes care
| Self-care tasks | Tests/annual assessments |
|---|---|
| Self-monitoring of glucose | A1C (2 to 4 times/year) |
| Medication management | Urinary microalbumin |
| Meal planning | Fasting lipids |
| Exercise | Blood pressure |
| Smoking cessation | Dilated eye exam |
| Foot self-examination and foot care | Foot exam |
| Stress management | General health and cardiovascular exam |
Managing patients at risk for diabetes
| Prediabetes21 | Management |
|---|---|
| Impaired fasting glucose (100 to 125 mg/dL) | Weight reduction (7%) Activity (150 minutes/week) At least yearly glucose monitoring |
| Impaired glucose tolerance (2-hour plasma glucose: 140 to 199 mg/dL) | |
| Prediabetic A1C (5.7% to 6.4%) | |
| Metabolic syndrome (any 3)26 | Management |
| Waist circumferencea (men >40 inches; women >35 inches) | Weight reduction Reduce consumption of refined carbohydrates Exercise Focused interventions for individual criteria |
| Fasting triglycerides (≥150 mg/dL) | |
| Fasting high-density lipoprotein cholesterol (men | |
| Fasting glucose (≥100 mg/dL or taking medication) | |
| Blood pressure (≥130/85 mm Hg or taking medication) | |
| aWaist circumference guidelines are ethnicity specific. The International Diabetes Federation27 has published specific cutoffs for those of Asian background (men: ≥90 cm [35 inches] and women: ≥80 cm [31 inches]) | |
Metabolic monitoring
Metabolic monitoring is the key to keeping patients with schizophrenia well. Treating metabolic conditions falls outside of psychiatric practice; however, many argue that mental health clinicians should monitor basic metabolic parameters during antipsychotic treatment and advocate medical interventions when indicated because:
- most antipsychotics are associated with weight gain and metabolic side effects
- patients with schizophrenia have cognitive deficits that impact health maintenance
- mental health providers often are the primary health care contacts for patients with serious mental illness.
- identify treatable medical conditions such as diabetes, dyslipidemia, and hypertension when treatment delay or no treatment has consequences
- identify individuals with prediabetes and metabolic syndrome for targeted prevention
- determine the association between antipsychotic treatment and metabolic disturbance to evaluate the risk of treatment vs antipsychotic switching.
How to intervene
To switch or not to switch? For many psychiatrists, deciding whether or when to switch from a high or intermediate metabolic liability antipsychotic to one with low metabolic liability is difficult. Clinicians must balance potential metabolic benefits against the risk of psychotic decompensation and side effects. Ultimately, patients and their families make the decision, taking into account information provided to them. For medical-legal purposes, document the discussion of potential risks and benefits. These are difficult decisions and there are no clear guidelines. In my clinical experience, the following issues need to be considered:
- The antipsychotics that many clinicians consider to be the most effective—clozapine and olanzapine—also have the greatest metabolic liability and risk for emergent T2DM.
- Patients who are stable and in psychotic remission may risk a relapse of their illness if switched.
- The clearest indication for switching is when a patient who does not have diabetes develops the condition shortly after starting an antipsychotic. This scenario is rare, but evidence suggests that diabetes may resolve or reverse with an antipsychotic switch.33
- In patients who gain weight while taking a high- or intermediate-liability antipsychotic and are able to tolerate a switch to a low-liability antipsychotic, the effect size of weight reduction can be large and may result in a patient returning to their pretreatment weight.
- To reduce relapse risk, patients switching antipsychotics should be closely monitored at least weekly for ≥1 month. A plateau cross-taper—building the new antipsychotic up to therapeutic levels before gradually reducing the first antipsychotic—may be safer than abrupt discontinuation or standard cross-titration.
- Switching from one high or intermediate liability antipsychotic to another (eg, olanzapine to quetiapine or risperidone) often provides little if any metabolic benefit on body weight or diabetes control.
- Established diabetes (type 1 or type 2) should not be a contraindication to antipsychotic treatment, including clozapine, if clinically warranted. Monitor metabolic parameters more closely for 6 to 12 months after the switch. In most cases, patients experience limited, if any, metabolic consequences. If so, diabetes medication can be adjusted.
- Patients who have experienced significant weight gain on an atypical antipsychotic often do not gain more weight when switched to clozapine. A patient may reach a “ceiling” in terms of weight gain and medication-related metabolic effects.
Data from metabolic monitoring informs the decision to switch and metabolic consequences of switching. Conducting monitoring at baseline, when starting an antipsychotic, when switching to a high-liability agent, 3 months after the switch, and then annually provides data needed to consider switching or initiating medical and behavioral or lifestyle interventions.
Facilitate early diabetes treatment. Clinicians who are most closely involved in caring for patients with schizophrenia often are best situated to screen for diabetes. I have found that without a close working relationship with my patients’ primary care practitioners, patients may experience a long delay in receiving care. After your patient is diagnosed with diabetes, establish a relationship with diabetes treatment providers and work with your patient to ensure they engage in diabetes care.
Contribute to diabetes chronic disease management. Mental health practitioners can complement diabetes care in patients with serious mental illness by:
- navigating the health system and negotiating for service on patients’ behalf
- promoting positive relationships among diabetes and mental health treatment teams
- evaluating and treating depression that may be comorbid with diabetes
- assessing treatment capacity, self-care deficits, cognitive functioning, psychotic symptoms, negative symptoms, etc., that impact diabetes self-care and collaborating with diabetes care providers to support patients.
Start with a low-liability agent
Patients who are early in the course of psychotic illness are most susceptible to the metabolic effects of antipsychotics.13 The average weight gain observed with olanzapine was 34 lbs at 2 years in first episode psychosis patients (mean age 24 ± 4.9).34 Metabolic consequences with medium-liability second-generation antipsychotics—such as quetiapine and risperidone—are extreme, particularly in children, adolescents, and young adults (age 35,36 Although frank diabetes is uncommon in early psychosis because patients are, to a certain extent, protected by insulin compensation—increased insulin secretion maintains glucose levels within a therapeutic range—diabetes risk is increased, and hyperinsulinemia and hypertriglyceridemia are early markers of metabolic strain. Also, response to initial antipsychotic treatment—possibly independent of the agent selected—is robust in early psychosis.37
Related Resources
- American Diabetes Association. www.diabetes.org.
- International Diabetes Federation. www.idf.org.
- Framingham Heart Study. Coronary heart disease (10-year risk). www.framinghamheartstudy.org/risk/coronary.html.
- National Cholesterol Education Program. Risk assessment tool for estimating your 10-year risk of having a heart attack. http://hp2010.nhlbihin.net/atpiii/calculator.asp.
- Clozapine • Clozaril
- Metformin • Glucophage
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
Dr. Cohn is a speaker for Pfizer Canada.
Discuss this article at www.facebook.com/CurrentPsychiatry
Although diabetes and schizophrenia are common companions, it is unclear how this association should influence our practice. What do we need to know about diabetes, and what are the key intervention points for psychiatrists?
This article is informed by my experience monitoring >1,000 patients with schizophrenia in a large urban mental health facility using an electronic metabolic monitoring system and consulting on hundreds of individuals with comorbid schizophrenia and diabetes in a mental health metabolic clinic.
A significant link
The association between schizophrenia and diabetes has been recognized for more than a century.1 The prevalence of diabetes is increased 2- to 3-fold in patients with schizophrenia.2,3 This relationship is specific to type 2 diabetes mellitus (T2DM); type 1 diabetes mellitus, an autoimmune disease, is less common in patients with schizophrenia.4 Factors that contribute to comorbidity between schizophrenia and T2DM include:
- illness susceptibility: the mechanisms remain unclear but include the thrifty phenotype hypothesis,5 autonomic hyperactivity,6 and potential cellular and genetic links7,8
- lifestyle: diet, physical inactivity, and cigarette smoking9-12
- antipsychotic use13
- social health determinants, such as income, housing, and food insecurity.14
When evaluating a patient’s risk for a cardiac event, we consider having a diabetes diagnosis equivalent to having had a myocardial infarction.15 Likely, the high prevalence of T2DM among schizophrenia patients and challenges in managing diabetes and prediabetes underlies these patients’ reduced life expectancy.16 Self-care, a cornerstone of diabetes management, is challenging for patients with schizophrenia because of deficits in executive functioning, working memory, and motivation, coupled with negative symptoms and social and economic disadvantages that often accompany schizophrenia.
How diabetes impacts practice
What psychiatrists need to know. Insulin resistance—reduced biologic effectiveness of insulin—is the precursor of T2DM. Insulin is required to move glucose from the blood into cells. Weight gain, particularly abdominal adiposity, is the principal driver of insulin resistance. The body responds by producing more insulin (hyperinsulinemia) to maintain glucose homeostasis. Hyperinsulinemia underlies metabolic syndrome, an important risk marker for developing T2DM. Diabetes usually develops after many years when the pancreas fails to compensate for insulin resistance.
In most cases the development of diabetes in patients with schizophrenia follows this course. Weight gain, a consequence of lifestyle factors as well as antipsychotics and other psychotropics that promote obesity, leads to progressive insulin resistance. Consequently, metabolic syndrome is twice as prevalent among patients with schizophrenia compared with matched controls.17,18
Occasionally patients develop T2DM within a few weeks or months of starting antipsychotic treatment—usually with clozapine or olanzapine—before they gain weight, which suggests a second mechanism may be involved. Animal studies have documented rapid development of insulin resistance after a single subcutaneous injection of antipsychotics that have high metabolic liability, possibly through a direct effect on insulin signaling.19 This phenomenon has been difficult to demonstrate in humans.20
Managing diabetes is complex and ideally involves a range of health practitioners who work with patients to provide education, promote self-care behaviors, and direct complex health care. These services are outside the scope of psychiatric practice, but given the functional deficits in seriously mentally ill patients, it is important to have an overview of diabetes care (Table 3).
In addition to diagnosing diabetes, psychiatrists should be able to identify patients at risk for developing diabetes and initiate prevention strategies. Interventions are focused on lifestyle—weight reduction, increased physical activity, diet, and smoking cessation—as well as pharmacologic strategies such as metformin.
What patients need to know. Similar to schizophrenia, a diabetes diagnosis may be difficult for patients to accept. Initially, a patient may have no manifestations or symptoms. However, untreated diabetes has serious long-term health consequences—including blindness, amputations, kidney disease, and early death from heart attacks.
Patients should actively participate in treatment that involves learning about the illness, making lifestyle changes, working on self-care, and keeping regular medical appointments. Three components of lifestyle change must be addressed:
- Diet: counseling with a dietician or other health professional to reduce or stabilize body weight and make changes in diet quality, portion size, and meal frequency to improve glucose control and reduce long-term diabetes complications
- Physical activity: increasing physical activity, initially by walking daily, to benefit glucose control and weight maintenance
- Smoking: reducing or stopping cigarette smoking to improve glucose control and reduce diabetes complications.
American Diabetes Association diagnostic criteria for diabetes
| Testa | Threshold | Qualifier |
|---|---|---|
| A1C, or | ≥6.5% | Lab NGSP certified, standardized DCCT assay |
| Fasting glucose, or | ≥126 mg/dL | No caloric intake for at least 8 hours |
| 2-hour glucose, or | ≥200 mg/dL | After 75 g of anhydrous glucose |
| Random glucose | ≥200 mg/dL | Plus classic hyperglycemic symptoms or crisis |
| aResults should be confirmed by repeat testing DCCT: Diabetes Control and Complications Trial; NGSP: National Glycohemoglobin Standardization Program Source: References 21-23 | ||
Signs and symptoms of diabetes and diabetic ketoacidosis
| Diabetes |
| Frequent urination |
| Excessive thirst |
| Extreme hunger |
| Unusual weight loss |
| Increased fatigue |
| Irritability |
| Blurry vision |
| Diabetic ketoacidosisa |
| Thirst or very dry mouth |
| Constantly feeling tired |
| Dry or flushed skin |
| Nausea, vomiting, or abdominal pain |
| Difficulty breathing (short, deep breaths) |
| Fruity odor on breath |
| Difficulty paying attention or confusion |
| aVomiting is a sign of escalation Source: References 24,25 |
Components of diabetes care
| Self-care tasks | Tests/annual assessments |
|---|---|
| Self-monitoring of glucose | A1C (2 to 4 times/year) |
| Medication management | Urinary microalbumin |
| Meal planning | Fasting lipids |
| Exercise | Blood pressure |
| Smoking cessation | Dilated eye exam |
| Foot self-examination and foot care | Foot exam |
| Stress management | General health and cardiovascular exam |
Managing patients at risk for diabetes
| Prediabetes21 | Management |
|---|---|
| Impaired fasting glucose (100 to 125 mg/dL) | Weight reduction (7%) Activity (150 minutes/week) At least yearly glucose monitoring |
| Impaired glucose tolerance (2-hour plasma glucose: 140 to 199 mg/dL) | |
| Prediabetic A1C (5.7% to 6.4%) | |
| Metabolic syndrome (any 3)26 | Management |
| Waist circumferencea (men >40 inches; women >35 inches) | Weight reduction Reduce consumption of refined carbohydrates Exercise Focused interventions for individual criteria |
| Fasting triglycerides (≥150 mg/dL) | |
| Fasting high-density lipoprotein cholesterol (men | |
| Fasting glucose (≥100 mg/dL or taking medication) | |
| Blood pressure (≥130/85 mm Hg or taking medication) | |
| aWaist circumference guidelines are ethnicity specific. The International Diabetes Federation27 has published specific cutoffs for those of Asian background (men: ≥90 cm [35 inches] and women: ≥80 cm [31 inches]) | |
Metabolic monitoring
Metabolic monitoring is the key to keeping patients with schizophrenia well. Treating metabolic conditions falls outside of psychiatric practice; however, many argue that mental health clinicians should monitor basic metabolic parameters during antipsychotic treatment and advocate medical interventions when indicated because:
- most antipsychotics are associated with weight gain and metabolic side effects
- patients with schizophrenia have cognitive deficits that impact health maintenance
- mental health providers often are the primary health care contacts for patients with serious mental illness.
- identify treatable medical conditions such as diabetes, dyslipidemia, and hypertension when treatment delay or no treatment has consequences
- identify individuals with prediabetes and metabolic syndrome for targeted prevention
- determine the association between antipsychotic treatment and metabolic disturbance to evaluate the risk of treatment vs antipsychotic switching.
How to intervene
To switch or not to switch? For many psychiatrists, deciding whether or when to switch from a high or intermediate metabolic liability antipsychotic to one with low metabolic liability is difficult. Clinicians must balance potential metabolic benefits against the risk of psychotic decompensation and side effects. Ultimately, patients and their families make the decision, taking into account information provided to them. For medical-legal purposes, document the discussion of potential risks and benefits. These are difficult decisions and there are no clear guidelines. In my clinical experience, the following issues need to be considered:
- The antipsychotics that many clinicians consider to be the most effective—clozapine and olanzapine—also have the greatest metabolic liability and risk for emergent T2DM.
- Patients who are stable and in psychotic remission may risk a relapse of their illness if switched.
- The clearest indication for switching is when a patient who does not have diabetes develops the condition shortly after starting an antipsychotic. This scenario is rare, but evidence suggests that diabetes may resolve or reverse with an antipsychotic switch.33
- In patients who gain weight while taking a high- or intermediate-liability antipsychotic and are able to tolerate a switch to a low-liability antipsychotic, the effect size of weight reduction can be large and may result in a patient returning to their pretreatment weight.
- To reduce relapse risk, patients switching antipsychotics should be closely monitored at least weekly for ≥1 month. A plateau cross-taper—building the new antipsychotic up to therapeutic levels before gradually reducing the first antipsychotic—may be safer than abrupt discontinuation or standard cross-titration.
- Switching from one high or intermediate liability antipsychotic to another (eg, olanzapine to quetiapine or risperidone) often provides little if any metabolic benefit on body weight or diabetes control.
- Established diabetes (type 1 or type 2) should not be a contraindication to antipsychotic treatment, including clozapine, if clinically warranted. Monitor metabolic parameters more closely for 6 to 12 months after the switch. In most cases, patients experience limited, if any, metabolic consequences. If so, diabetes medication can be adjusted.
- Patients who have experienced significant weight gain on an atypical antipsychotic often do not gain more weight when switched to clozapine. A patient may reach a “ceiling” in terms of weight gain and medication-related metabolic effects.
Data from metabolic monitoring informs the decision to switch and metabolic consequences of switching. Conducting monitoring at baseline, when starting an antipsychotic, when switching to a high-liability agent, 3 months after the switch, and then annually provides data needed to consider switching or initiating medical and behavioral or lifestyle interventions.
Facilitate early diabetes treatment. Clinicians who are most closely involved in caring for patients with schizophrenia often are best situated to screen for diabetes. I have found that without a close working relationship with my patients’ primary care practitioners, patients may experience a long delay in receiving care. After your patient is diagnosed with diabetes, establish a relationship with diabetes treatment providers and work with your patient to ensure they engage in diabetes care.
Contribute to diabetes chronic disease management. Mental health practitioners can complement diabetes care in patients with serious mental illness by:
- navigating the health system and negotiating for service on patients’ behalf
- promoting positive relationships among diabetes and mental health treatment teams
- evaluating and treating depression that may be comorbid with diabetes
- assessing treatment capacity, self-care deficits, cognitive functioning, psychotic symptoms, negative symptoms, etc., that impact diabetes self-care and collaborating with diabetes care providers to support patients.
Start with a low-liability agent
Patients who are early in the course of psychotic illness are most susceptible to the metabolic effects of antipsychotics.13 The average weight gain observed with olanzapine was 34 lbs at 2 years in first episode psychosis patients (mean age 24 ± 4.9).34 Metabolic consequences with medium-liability second-generation antipsychotics—such as quetiapine and risperidone—are extreme, particularly in children, adolescents, and young adults (age 35,36 Although frank diabetes is uncommon in early psychosis because patients are, to a certain extent, protected by insulin compensation—increased insulin secretion maintains glucose levels within a therapeutic range—diabetes risk is increased, and hyperinsulinemia and hypertriglyceridemia are early markers of metabolic strain. Also, response to initial antipsychotic treatment—possibly independent of the agent selected—is robust in early psychosis.37
Related Resources
- American Diabetes Association. www.diabetes.org.
- International Diabetes Federation. www.idf.org.
- Framingham Heart Study. Coronary heart disease (10-year risk). www.framinghamheartstudy.org/risk/coronary.html.
- National Cholesterol Education Program. Risk assessment tool for estimating your 10-year risk of having a heart attack. http://hp2010.nhlbihin.net/atpiii/calculator.asp.
- Clozapine • Clozaril
- Metformin • Glucophage
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
Dr. Cohn is a speaker for Pfizer Canada.
1. Kohen D. Diabetes mellitus and schizophrenia: historical perspective. Br J Psychiatry Suppl. 2004;47:S64-S66.
2. Dixon L, Weiden P, Delahanty J, et al. Prevalence and correlates of diabetes in national schizophrenia samples. Schizophr Bull. 2000;26(4):903-912.
3. De Hert M, van Winkel R, Van Eyck D, et al. Prevalence of diabetes, metabolic syndrome and metabolic abnormalities in schizophrenia over the course of the illness: a cross-sectional study. Clin Pract Epidemol Ment Health. 2006;2:14.-
4. Juvonen H, Reunanen A, Haukka J, et al. Incidence of schizophrenia in a nationwide cohort of patients with type 1 diabetes mellitus. Arch Gen Psychiatry. 2007;64(8):894-899.
5. Hales CN, Barker DJ. The thrifty phenotype hypothesis. Br Med Bull. 2001;60:5-20.
6. Ryan MC, Sharifi N, Condren R, et al. Evidence of basal pituitary-adrenal overactivity in first episode, drug naive patients with schizophrenia. Psychoneuroendocrinology. 2004;29(8):1065-1070.
7. Odawara M, Isaka M, Tada K, et al. Diabetes mellitus associated with mitochondrial myopathy and schizophrenia: a possible link between diabetes mellitus and schizophrenia. Diabet Med. 1997;14(6):503.-
8. Siuta MA, Robertson SD, Kocalis H, et al. Dysregulation of the norepinephrine transporter sustains cortical hypodopaminergia and schizophrenia-like behaviors in neuronal rictor null mice. PLoS Biol. 2010;8(6):e1000393.-
9. Strassnig M, Brar JS, Ganguli R. Nutritional assessment of patients with schizophrenia: a preliminary study. Schizophr Bull. 2003;29(2):393-397.
10. Daumit GL, Goldberg RW, Anthony C, et al. Physical activity patterns in adults with severe mental illness. J Nerv Ment Dis. 2005;193(10):641-646.
11. Ussher M, Stanbury L, Cheeseman V, et al. Physical activity p and perceived barriers to activity among persons with severe mental illness in the United Kingdom. Psychiatr Serv. 2007;58(3):405-408.
12. Cho NH, Chan JC, Jang HC, et al. Cigarette smoking is an independent risk factor for type 2 diabetes: a four-year community-based prospective study. Clin Endocrinol (Oxf). 2009;71(5):679-685.
13. Newcomer JW. Second-generation (atypical) antipsychotics and metabolic effects: a comprehensive literature review. CNS Drugs. 2005;19(suppl 1):1-93.
14. Yu VL, Raphael D. Identifying and addressing the social determinants of the incidence and successful management of type 2 diabetes mellitus in Canada. Can J Public Health. 2004;95(5):366-368.
15. Barr EL, Zimmet PZ, Welborn TA, et al. Risk of cardiovascular and all-cause mortality in individuals with diabetes mellitus, impaired fasting glucose, and impaired glucose tolerance: the Australian Diabetes, Obesity, and Lifestyle Study (AusDiab). Circulation. 2007;116(2):151-157.
16. Colton CW, Manderscheid RW. Congruencies in increased mortality rates years of potential life lost, and causes of death among public mental health clients in eight states. Prev Chronic Dis. 2006;3(2):A42.-
17. Cohn T, Prud’homme D, Streiner D, et al. Characterizing coronary heart disease risk in chronic schizophrenia: high prevalence of the metabolic syndrome. Can J Psychiatry. 2004;49(11):753-760.
18. Meyer JM, Stahl SM. The metabolic syndrome and schizophrenia. Acta Psychiatr Scand. 2009;119(1):4-14.
19. Chintoh AF, Mann SW, Lam L, et al. Insulin resistance and decreased glucose-stimulated insulin secretion after acute olanzapine administration. J Clin Psychopharmacol. 2008;28(5):494-499.
20. Hahn MK, Arenovich T, Wolever T, et al. Single dose administration of olanzapine: effects on glucose metabolism, endocrine and inflammatory markers in healthy volunteers. Poster presented at: Schizophrenia International Research Society 3rd Biennial Conference; April 14-18, 2012; Florence, Italy.
21. American Diabetes Association. Standards of medical care in diabetes—2012. Diabetes Care. 2012;35(suppl 1):S11-S63.
22. Little RR. Glycated hemoglobin standardization—National Glycohemoglobin Standardization Program (NGSP) perspective. Clin Chem Lab Med. 2003;41(9):1191-1198.
23. Keen H. The Diabetes Control and Complications Trial (DCCT). Health Trends. 1994;26(2):41-43.
24. American Diabetes Association. Symptoms. http://www.diabetes.org/diabetes-basics/symptoms. Accessed August 27 2012.
25. American Diabetes Association. Ketoacidosis (DKA). http://www.diabetes.org/living-with-diabetes/complications/ketoacidosis-dka.html. Accessed August 27 2012.
26. Grundy SM, Cleeman JI, Daniels SR, et al. American Heart Association; National Heart, Lung, and Blood Institute. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112(17):2735-2752.
27. Alberti KG, Zimmet P, Shaw J. Metabolic syndrome—a new world-wide definition. A consensus statement from the International Diabetes Federation. Diabet Med. 2006;23(5):469-480.
28. Rodbard HW, Blonde L, Braithwaite SS, et al. AACE Diabetes Mellitus Clinical Practice Guidelines Task Force. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the management of diabetes mellitus. Endocr Pract. 2007;13(suppl 1):1-68.
29. Cohn TA, Sernyak MJ. Metabolic monitoring for patients treated with antipsychotic medications. Can J Psychiatry. 2006;51(8):492-501.
30. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists; North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
31. Newcomer JW, Nasrallah HA, Loebel AD. The Atypical Antipsychotic Therapy and Metabolic Issues National Survey: practice patterns and knowledge of psychiatrists. J Clin Psychopharmacol. 2004;24(5 suppl 1):S1-S6.
32. Khoury A, Sproule BA, Cohn TA. Development and implementation of the Metabolic Health Monitor at the Centre for Addiction and Mental Health. Poster presented at: BC Psychopharmacology Conference; February 15-16 2008; Vancouver, British Columbia, Canada.
33. Koller EA, Doraiswamy PM. Olanzapine-associated diabetes mellitus. Pharmacotherapy. 2002;22(7):841-852.
34. Zipursky RB, Gu H, Green AI, et al. Course and predictors of weight gain in people with first-episode psychosis treated with olanzapine or haloperidol. Br J Psychiatry. 2005;187:537-543.
35. Correll CU, Carlson HE. Endocrine and metabolic adverse effects of psychotropic medications in children and adolescents. J Am Acad Child Adolesc Psychiatry. 2006;45(7):771-791.
36. Correll CU, Manu P, Olshanskiy V, et al. Cardiometabolic risk of second-generation antipsychotic medications during first-time use in children and adolescents. JAMA. 2009;302(16):1765-1773.
37. Nicol G, Newcomer J. Review: children and adolescents with schizophrenia spectrum disorders respond to antipsychotics but are susceptible to adverse events. Evid Based Ment Health. 2008;11(3):81.-
1. Kohen D. Diabetes mellitus and schizophrenia: historical perspective. Br J Psychiatry Suppl. 2004;47:S64-S66.
2. Dixon L, Weiden P, Delahanty J, et al. Prevalence and correlates of diabetes in national schizophrenia samples. Schizophr Bull. 2000;26(4):903-912.
3. De Hert M, van Winkel R, Van Eyck D, et al. Prevalence of diabetes, metabolic syndrome and metabolic abnormalities in schizophrenia over the course of the illness: a cross-sectional study. Clin Pract Epidemol Ment Health. 2006;2:14.-
4. Juvonen H, Reunanen A, Haukka J, et al. Incidence of schizophrenia in a nationwide cohort of patients with type 1 diabetes mellitus. Arch Gen Psychiatry. 2007;64(8):894-899.
5. Hales CN, Barker DJ. The thrifty phenotype hypothesis. Br Med Bull. 2001;60:5-20.
6. Ryan MC, Sharifi N, Condren R, et al. Evidence of basal pituitary-adrenal overactivity in first episode, drug naive patients with schizophrenia. Psychoneuroendocrinology. 2004;29(8):1065-1070.
7. Odawara M, Isaka M, Tada K, et al. Diabetes mellitus associated with mitochondrial myopathy and schizophrenia: a possible link between diabetes mellitus and schizophrenia. Diabet Med. 1997;14(6):503.-
8. Siuta MA, Robertson SD, Kocalis H, et al. Dysregulation of the norepinephrine transporter sustains cortical hypodopaminergia and schizophrenia-like behaviors in neuronal rictor null mice. PLoS Biol. 2010;8(6):e1000393.-
9. Strassnig M, Brar JS, Ganguli R. Nutritional assessment of patients with schizophrenia: a preliminary study. Schizophr Bull. 2003;29(2):393-397.
10. Daumit GL, Goldberg RW, Anthony C, et al. Physical activity patterns in adults with severe mental illness. J Nerv Ment Dis. 2005;193(10):641-646.
11. Ussher M, Stanbury L, Cheeseman V, et al. Physical activity p and perceived barriers to activity among persons with severe mental illness in the United Kingdom. Psychiatr Serv. 2007;58(3):405-408.
12. Cho NH, Chan JC, Jang HC, et al. Cigarette smoking is an independent risk factor for type 2 diabetes: a four-year community-based prospective study. Clin Endocrinol (Oxf). 2009;71(5):679-685.
13. Newcomer JW. Second-generation (atypical) antipsychotics and metabolic effects: a comprehensive literature review. CNS Drugs. 2005;19(suppl 1):1-93.
14. Yu VL, Raphael D. Identifying and addressing the social determinants of the incidence and successful management of type 2 diabetes mellitus in Canada. Can J Public Health. 2004;95(5):366-368.
15. Barr EL, Zimmet PZ, Welborn TA, et al. Risk of cardiovascular and all-cause mortality in individuals with diabetes mellitus, impaired fasting glucose, and impaired glucose tolerance: the Australian Diabetes, Obesity, and Lifestyle Study (AusDiab). Circulation. 2007;116(2):151-157.
16. Colton CW, Manderscheid RW. Congruencies in increased mortality rates years of potential life lost, and causes of death among public mental health clients in eight states. Prev Chronic Dis. 2006;3(2):A42.-
17. Cohn T, Prud’homme D, Streiner D, et al. Characterizing coronary heart disease risk in chronic schizophrenia: high prevalence of the metabolic syndrome. Can J Psychiatry. 2004;49(11):753-760.
18. Meyer JM, Stahl SM. The metabolic syndrome and schizophrenia. Acta Psychiatr Scand. 2009;119(1):4-14.
19. Chintoh AF, Mann SW, Lam L, et al. Insulin resistance and decreased glucose-stimulated insulin secretion after acute olanzapine administration. J Clin Psychopharmacol. 2008;28(5):494-499.
20. Hahn MK, Arenovich T, Wolever T, et al. Single dose administration of olanzapine: effects on glucose metabolism, endocrine and inflammatory markers in healthy volunteers. Poster presented at: Schizophrenia International Research Society 3rd Biennial Conference; April 14-18, 2012; Florence, Italy.
21. American Diabetes Association. Standards of medical care in diabetes—2012. Diabetes Care. 2012;35(suppl 1):S11-S63.
22. Little RR. Glycated hemoglobin standardization—National Glycohemoglobin Standardization Program (NGSP) perspective. Clin Chem Lab Med. 2003;41(9):1191-1198.
23. Keen H. The Diabetes Control and Complications Trial (DCCT). Health Trends. 1994;26(2):41-43.
24. American Diabetes Association. Symptoms. http://www.diabetes.org/diabetes-basics/symptoms. Accessed August 27 2012.
25. American Diabetes Association. Ketoacidosis (DKA). http://www.diabetes.org/living-with-diabetes/complications/ketoacidosis-dka.html. Accessed August 27 2012.
26. Grundy SM, Cleeman JI, Daniels SR, et al. American Heart Association; National Heart, Lung, and Blood Institute. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112(17):2735-2752.
27. Alberti KG, Zimmet P, Shaw J. Metabolic syndrome—a new world-wide definition. A consensus statement from the International Diabetes Federation. Diabet Med. 2006;23(5):469-480.
28. Rodbard HW, Blonde L, Braithwaite SS, et al. AACE Diabetes Mellitus Clinical Practice Guidelines Task Force. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the management of diabetes mellitus. Endocr Pract. 2007;13(suppl 1):1-68.
29. Cohn TA, Sernyak MJ. Metabolic monitoring for patients treated with antipsychotic medications. Can J Psychiatry. 2006;51(8):492-501.
30. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists; North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
31. Newcomer JW, Nasrallah HA, Loebel AD. The Atypical Antipsychotic Therapy and Metabolic Issues National Survey: practice patterns and knowledge of psychiatrists. J Clin Psychopharmacol. 2004;24(5 suppl 1):S1-S6.
32. Khoury A, Sproule BA, Cohn TA. Development and implementation of the Metabolic Health Monitor at the Centre for Addiction and Mental Health. Poster presented at: BC Psychopharmacology Conference; February 15-16 2008; Vancouver, British Columbia, Canada.
33. Koller EA, Doraiswamy PM. Olanzapine-associated diabetes mellitus. Pharmacotherapy. 2002;22(7):841-852.
34. Zipursky RB, Gu H, Green AI, et al. Course and predictors of weight gain in people with first-episode psychosis treated with olanzapine or haloperidol. Br J Psychiatry. 2005;187:537-543.
35. Correll CU, Carlson HE. Endocrine and metabolic adverse effects of psychotropic medications in children and adolescents. J Am Acad Child Adolesc Psychiatry. 2006;45(7):771-791.
36. Correll CU, Manu P, Olshanskiy V, et al. Cardiometabolic risk of second-generation antipsychotic medications during first-time use in children and adolescents. JAMA. 2009;302(16):1765-1773.
37. Nicol G, Newcomer J. Review: children and adolescents with schizophrenia spectrum disorders respond to antipsychotics but are susceptible to adverse events. Evid Based Ment Health. 2008;11(3):81.-
Primum non nocere
Dr. Nasrallah’s June editorial (“Innovative approaches to treatment-resistant depression,” From the Editor, Current Psychiatry, June 2012, p. 4-5; http://bit.ly/1GM92oV) and the authors’ response to a letter on treating resistant depression (Current Psychiatry, June 2012, p. 19; http://bit.ly/1QU1sOR) remind us of depression’s complexity and the wide range of treatments available. I question whether in our zeal to help our patients we have forgotten the bedrock principle of medicine: Primum non nocere (First, do no harm).
Do we as psychiatrists make this principle a staple of our daily practice? Do we ask, “Which treatment modality offers the greatest likelihood of restoring wellness with the least risk of harm?” or do we restrict such inquiry to the confines of pharmacotherapy, considering only which medicine is least harmful? Is it not common practice to prescribe atypical antipsychotics to patients who have failed antidepressants? Do we offer modalities such as transcranial magnetic stimulation (TMS) before introducing atypical antipsychotics? If we keep our oath to abstain from doing harm, should we offer TMS before atypicals?
Some have argued that the high cost of TMS is reason not to offer it. But what is the cost of developing type 2 diabetes mellitus? Should we put patients at risk for such a disorder without giving them the option to choose a modality that doesn’t confer such risk?
The language used in Drs. Desseilles, Fava, Mischoulon, and Freeman’s “Comments & Controversies” response suggesting TMS was in the “same vein” as vagus nerve stimulation and deep brain stimulation concerned me. Such a comment—hopefully inadvertently—suggests a failure to recognize our oath to first, do no harm. Do the authors really believe that such invasive procedures confer no greater risk of harm than TMS? Are such modalities in the same vein as TMS, or do they take us to a new level of treatment risk and complexity?
Although no evidence suggests TMS is a panacea that successfully treats all patients with treatment-resistant depression, we can say with great confidence it is the safest of all somatic treatments and confers the least risk of harm. Because no evidence demonstrates that any other somatic treatment provides greater efficacy, do our ethics not require us to offer TMS as part of informed consent, before starting atypical antipsychotics, which carry a risk of metabolic syndrome, type 2 diabetes mellitus, sexual dysfunction, parkinsonism, and a host of other potentially life-altering problems and complications?
Timothy R. Jennings, MD, FAPA
President, TMS Chattanooga
Adjunct Faculty
University of Tennessee College of Medicine
Chattanooga, TN
Dr. Nasrallah responds
Dr. Jennings is correct in reminding us that above all, physicians must do no harm. However, there are certain other principles in medicine: 1) the lack of treatment for severe illness can result in serious harm, and 2) there always is a side-effect burden with any treatment. The risk-benefit considerations are complicated when dealing with chronically suffering, disabled, or suicidal patients with refractory depression.
Bold new interventions must be developed for such desperate cases at the cost of side effects, which must not be unacceptably severe. That’s why controlled studies to prove the usefulness of a new therapy are conducted in a few hundred patients so that millions of others can benefit from a new treatment mechanism. That’s how medical science advances, always balancing risks and benefits. It’s up to the clinician to determine which intervention is the best and least harmful for each patient. However, what may be considered an effective treatment may quickly be discarded when better and less harmful treatments are found, such as abandoning prefrontal lobotomy for aggressive psychotic patients shortly after chlorpromazine was discovered.
The authors respond
We thank Dr. Jennings for his comments and appreciate his concern to offer patients pharmacologic or nonpharmacologic treatments with the least amount of side effects. We also recognize the diversity of clinical situations, which, according to factors such as the degree of depression severity, the therapeutic choice of the patient, and the availability of treatments, lead the clinician to suggest an antidepressant treatment that is the most efficient and least harmful as possible.
By proposing that the Massachusetts General Hospital Antidepressant Treatment Response Questionnaire consider the diversity of antidepressant treatments that various clinical situations have necessitated, we by no means encourage the use of antidepressant treatments that are inefficient and harmful to the patient. We look to provide clinicians with tools that take into consideration the therapeutic interventions available for treatment-resistant depression. In the same manner, we have not established a hierarchy of pharmacologic options, because we hope clinicians will identify the multiple treatments that the patient needs. If it is essential to “abstain from doing harm,” we must not forget that if healing is an ideal objective, we often only “treat” our patients with the best available methods.
As Dr. Jennings suggests, the least harmful treatments often are those that target depression’s physiopathology with the highest degree of specificity. In the same vein, neuromodulation treatments target the different neurobiologic mechanisms underlying depression. However, response predictors of TMS include age, degrees of treatment resistance, and the absence of comorbid anxiety or psychotic symptoms.Innovative approaches to treatment-resistant depression,” From the Editor, Current Psychiatry, June 2012, p. 4-5; http://bit.ly/FTE612). This so-called side effect may be the central antidepressant effect, because short-term memory loss may be a central effect in any seizure therapy for depression.
John S. Kafka, MD
Private Practice
Emeritus Professor of Psychiatry and Behavioral Sciences
George Washington University School of Medicine and Health Sciences
Washington, DC
Dr. Nasrallah responds
Memory loss with regular and heavy ketamine use or with a course of bilateral electroconvulsive therapy (ECT) is widely regarded as an undesirable side effect, not a therapeutic effect or mechanism. The side effects of short-term ketamine use in refractory depression studies included dissociation and unusual beliefs—such as conspiracy theories—as well as full-fledged delusions.
Both ketamine and ECT increase brain-derived neurotrophic factor (BDNF), which has been found to significantly decline in depression. The BDNF deficit is emerging as the leading mechanism of antidepressant therapy, both pharmacologic and non-pharmacologic, in both animal models and clinical populations.
Dr. Nasrallah’s June editorial (“Innovative approaches to treatment-resistant depression,” From the Editor, Current Psychiatry, June 2012, p. 4-5; http://bit.ly/1GM92oV) and the authors’ response to a letter on treating resistant depression (Current Psychiatry, June 2012, p. 19; http://bit.ly/1QU1sOR) remind us of depression’s complexity and the wide range of treatments available. I question whether in our zeal to help our patients we have forgotten the bedrock principle of medicine: Primum non nocere (First, do no harm).
Do we as psychiatrists make this principle a staple of our daily practice? Do we ask, “Which treatment modality offers the greatest likelihood of restoring wellness with the least risk of harm?” or do we restrict such inquiry to the confines of pharmacotherapy, considering only which medicine is least harmful? Is it not common practice to prescribe atypical antipsychotics to patients who have failed antidepressants? Do we offer modalities such as transcranial magnetic stimulation (TMS) before introducing atypical antipsychotics? If we keep our oath to abstain from doing harm, should we offer TMS before atypicals?
Some have argued that the high cost of TMS is reason not to offer it. But what is the cost of developing type 2 diabetes mellitus? Should we put patients at risk for such a disorder without giving them the option to choose a modality that doesn’t confer such risk?
The language used in Drs. Desseilles, Fava, Mischoulon, and Freeman’s “Comments & Controversies” response suggesting TMS was in the “same vein” as vagus nerve stimulation and deep brain stimulation concerned me. Such a comment—hopefully inadvertently—suggests a failure to recognize our oath to first, do no harm. Do the authors really believe that such invasive procedures confer no greater risk of harm than TMS? Are such modalities in the same vein as TMS, or do they take us to a new level of treatment risk and complexity?
Although no evidence suggests TMS is a panacea that successfully treats all patients with treatment-resistant depression, we can say with great confidence it is the safest of all somatic treatments and confers the least risk of harm. Because no evidence demonstrates that any other somatic treatment provides greater efficacy, do our ethics not require us to offer TMS as part of informed consent, before starting atypical antipsychotics, which carry a risk of metabolic syndrome, type 2 diabetes mellitus, sexual dysfunction, parkinsonism, and a host of other potentially life-altering problems and complications?
Timothy R. Jennings, MD, FAPA
President, TMS Chattanooga
Adjunct Faculty
University of Tennessee College of Medicine
Chattanooga, TN
Dr. Nasrallah responds
Dr. Jennings is correct in reminding us that above all, physicians must do no harm. However, there are certain other principles in medicine: 1) the lack of treatment for severe illness can result in serious harm, and 2) there always is a side-effect burden with any treatment. The risk-benefit considerations are complicated when dealing with chronically suffering, disabled, or suicidal patients with refractory depression.
Bold new interventions must be developed for such desperate cases at the cost of side effects, which must not be unacceptably severe. That’s why controlled studies to prove the usefulness of a new therapy are conducted in a few hundred patients so that millions of others can benefit from a new treatment mechanism. That’s how medical science advances, always balancing risks and benefits. It’s up to the clinician to determine which intervention is the best and least harmful for each patient. However, what may be considered an effective treatment may quickly be discarded when better and less harmful treatments are found, such as abandoning prefrontal lobotomy for aggressive psychotic patients shortly after chlorpromazine was discovered.
The authors respond
We thank Dr. Jennings for his comments and appreciate his concern to offer patients pharmacologic or nonpharmacologic treatments with the least amount of side effects. We also recognize the diversity of clinical situations, which, according to factors such as the degree of depression severity, the therapeutic choice of the patient, and the availability of treatments, lead the clinician to suggest an antidepressant treatment that is the most efficient and least harmful as possible.
By proposing that the Massachusetts General Hospital Antidepressant Treatment Response Questionnaire consider the diversity of antidepressant treatments that various clinical situations have necessitated, we by no means encourage the use of antidepressant treatments that are inefficient and harmful to the patient. We look to provide clinicians with tools that take into consideration the therapeutic interventions available for treatment-resistant depression. In the same manner, we have not established a hierarchy of pharmacologic options, because we hope clinicians will identify the multiple treatments that the patient needs. If it is essential to “abstain from doing harm,” we must not forget that if healing is an ideal objective, we often only “treat” our patients with the best available methods.
As Dr. Jennings suggests, the least harmful treatments often are those that target depression’s physiopathology with the highest degree of specificity. In the same vein, neuromodulation treatments target the different neurobiologic mechanisms underlying depression. However, response predictors of TMS include age, degrees of treatment resistance, and the absence of comorbid anxiety or psychotic symptoms.Innovative approaches to treatment-resistant depression,” From the Editor, Current Psychiatry, June 2012, p. 4-5; http://bit.ly/FTE612). This so-called side effect may be the central antidepressant effect, because short-term memory loss may be a central effect in any seizure therapy for depression.
John S. Kafka, MD
Private Practice
Emeritus Professor of Psychiatry and Behavioral Sciences
George Washington University School of Medicine and Health Sciences
Washington, DC
Dr. Nasrallah responds
Memory loss with regular and heavy ketamine use or with a course of bilateral electroconvulsive therapy (ECT) is widely regarded as an undesirable side effect, not a therapeutic effect or mechanism. The side effects of short-term ketamine use in refractory depression studies included dissociation and unusual beliefs—such as conspiracy theories—as well as full-fledged delusions.
Both ketamine and ECT increase brain-derived neurotrophic factor (BDNF), which has been found to significantly decline in depression. The BDNF deficit is emerging as the leading mechanism of antidepressant therapy, both pharmacologic and non-pharmacologic, in both animal models and clinical populations.
Dr. Nasrallah’s June editorial (“Innovative approaches to treatment-resistant depression,” From the Editor, Current Psychiatry, June 2012, p. 4-5; http://bit.ly/1GM92oV) and the authors’ response to a letter on treating resistant depression (Current Psychiatry, June 2012, p. 19; http://bit.ly/1QU1sOR) remind us of depression’s complexity and the wide range of treatments available. I question whether in our zeal to help our patients we have forgotten the bedrock principle of medicine: Primum non nocere (First, do no harm).
Do we as psychiatrists make this principle a staple of our daily practice? Do we ask, “Which treatment modality offers the greatest likelihood of restoring wellness with the least risk of harm?” or do we restrict such inquiry to the confines of pharmacotherapy, considering only which medicine is least harmful? Is it not common practice to prescribe atypical antipsychotics to patients who have failed antidepressants? Do we offer modalities such as transcranial magnetic stimulation (TMS) before introducing atypical antipsychotics? If we keep our oath to abstain from doing harm, should we offer TMS before atypicals?
Some have argued that the high cost of TMS is reason not to offer it. But what is the cost of developing type 2 diabetes mellitus? Should we put patients at risk for such a disorder without giving them the option to choose a modality that doesn’t confer such risk?
The language used in Drs. Desseilles, Fava, Mischoulon, and Freeman’s “Comments & Controversies” response suggesting TMS was in the “same vein” as vagus nerve stimulation and deep brain stimulation concerned me. Such a comment—hopefully inadvertently—suggests a failure to recognize our oath to first, do no harm. Do the authors really believe that such invasive procedures confer no greater risk of harm than TMS? Are such modalities in the same vein as TMS, or do they take us to a new level of treatment risk and complexity?
Although no evidence suggests TMS is a panacea that successfully treats all patients with treatment-resistant depression, we can say with great confidence it is the safest of all somatic treatments and confers the least risk of harm. Because no evidence demonstrates that any other somatic treatment provides greater efficacy, do our ethics not require us to offer TMS as part of informed consent, before starting atypical antipsychotics, which carry a risk of metabolic syndrome, type 2 diabetes mellitus, sexual dysfunction, parkinsonism, and a host of other potentially life-altering problems and complications?
Timothy R. Jennings, MD, FAPA
President, TMS Chattanooga
Adjunct Faculty
University of Tennessee College of Medicine
Chattanooga, TN
Dr. Nasrallah responds
Dr. Jennings is correct in reminding us that above all, physicians must do no harm. However, there are certain other principles in medicine: 1) the lack of treatment for severe illness can result in serious harm, and 2) there always is a side-effect burden with any treatment. The risk-benefit considerations are complicated when dealing with chronically suffering, disabled, or suicidal patients with refractory depression.
Bold new interventions must be developed for such desperate cases at the cost of side effects, which must not be unacceptably severe. That’s why controlled studies to prove the usefulness of a new therapy are conducted in a few hundred patients so that millions of others can benefit from a new treatment mechanism. That’s how medical science advances, always balancing risks and benefits. It’s up to the clinician to determine which intervention is the best and least harmful for each patient. However, what may be considered an effective treatment may quickly be discarded when better and less harmful treatments are found, such as abandoning prefrontal lobotomy for aggressive psychotic patients shortly after chlorpromazine was discovered.
The authors respond
We thank Dr. Jennings for his comments and appreciate his concern to offer patients pharmacologic or nonpharmacologic treatments with the least amount of side effects. We also recognize the diversity of clinical situations, which, according to factors such as the degree of depression severity, the therapeutic choice of the patient, and the availability of treatments, lead the clinician to suggest an antidepressant treatment that is the most efficient and least harmful as possible.
By proposing that the Massachusetts General Hospital Antidepressant Treatment Response Questionnaire consider the diversity of antidepressant treatments that various clinical situations have necessitated, we by no means encourage the use of antidepressant treatments that are inefficient and harmful to the patient. We look to provide clinicians with tools that take into consideration the therapeutic interventions available for treatment-resistant depression. In the same manner, we have not established a hierarchy of pharmacologic options, because we hope clinicians will identify the multiple treatments that the patient needs. If it is essential to “abstain from doing harm,” we must not forget that if healing is an ideal objective, we often only “treat” our patients with the best available methods.
As Dr. Jennings suggests, the least harmful treatments often are those that target depression’s physiopathology with the highest degree of specificity. In the same vein, neuromodulation treatments target the different neurobiologic mechanisms underlying depression. However, response predictors of TMS include age, degrees of treatment resistance, and the absence of comorbid anxiety or psychotic symptoms.Innovative approaches to treatment-resistant depression,” From the Editor, Current Psychiatry, June 2012, p. 4-5; http://bit.ly/FTE612). This so-called side effect may be the central antidepressant effect, because short-term memory loss may be a central effect in any seizure therapy for depression.
John S. Kafka, MD
Private Practice
Emeritus Professor of Psychiatry and Behavioral Sciences
George Washington University School of Medicine and Health Sciences
Washington, DC
Dr. Nasrallah responds
Memory loss with regular and heavy ketamine use or with a course of bilateral electroconvulsive therapy (ECT) is widely regarded as an undesirable side effect, not a therapeutic effect or mechanism. The side effects of short-term ketamine use in refractory depression studies included dissociation and unusual beliefs—such as conspiracy theories—as well as full-fledged delusions.
Both ketamine and ECT increase brain-derived neurotrophic factor (BDNF), which has been found to significantly decline in depression. The BDNF deficit is emerging as the leading mechanism of antidepressant therapy, both pharmacologic and non-pharmacologic, in both animal models and clinical populations.
Sleep disturbances in older adults
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Pediatric anxiety
Epilepsy or something else?
CASE: Seizure-like symptoms
Ms. T, age 20, is brought to the emergency room (ER) by her father because she refuses to eat and drink, is unable to function at home, lies in bed all day, and does not attend to her activities of daily living (ADLs). Ms. T lives with her family, is not enrolled in school, and is unemployed. In the ER she initially is uncooperative and mute and then suddenly becomes agitated and has a seizure-like episode characterized by jerking of her trunk followed by random, asymmetrical movements of her legs and arms, closing both eyes, weeping, foaming at the mouth, moaning, and marked unresponsiveness. The episode lasts for >5 minutes.
The authors’ observations
Based on Ms. T’s presentation, the medical team considered acute epileptic seizures. Asymmetrical jerking of the body may be seen in frontal lobe epilepsy or seizures of the supplementary sensorimotor area. Frontal lobe epilepsy can present with bilateral asynchronous motor activity with consciousness during the event and a lack of postictal confusion.1 Seizures of the supplementary sensorimotor area—also known as the secondary motor area—are particularly problematic because typically they present with bilateral asymmetric tonic posturing followed by a few clonic movements, intact consciousness, and rarely postictal confusion. Adding to the diagnostic uncertainty, some “soft signs” thought to indicate PNES (eg, pelvic thrusting, crying) are common with frontal lobe epilepsy.1,2
PNES are episodes of altered movement, sensation, or experience that may be mistaken for epileptic seizures but are not a consequence of abnormal cortical discharges. Instead they are caused by physiological or psychological factors.3 Behaviors or signs that strongly suggest PNES include:
- gradual onset or termination
- pseudosleep, when the patient appears to be asleep but electroencephalography (EEG) findings indicate he or she is awake
- discontinuous (stop-and-go), irregular, or asynchronous (out-of-phase) activity—including side-to-side head movement, pelvic thrusting, and opisthotonic posturing—stuttering, and weeping4
- eye closure.5
Ms. T’s father said his daughter had been hospitalized several times for episodes characterized by pelvic thrusting, stuttering, and pseudosleep, which raised the possibility of PNES. Definitive diagnosis of PNES comes from video EEG when a patient is observed having typical seizures without accompanying EEG abnormalities.6
EVALUATION: Inconclusive data
Ms. T is admitted to the medical unit to rule out a seizure disorder. Physical examination is unremarkable and laboratory tests are within normal limits. The neurology service requests a head MRI, which is inconclusive. Inpatient video EEG with 24-hour monitoring does not indicate acute epileptic seizures. Ms. T’s father says that she has experienced many paroxysmal motor episodes and all neurologic tests, exams, and labs have failed to find a cause for these episodes. She did not receive any antiepileptic medications. A psychiatric consult is requested to clarify the diagnosis. Ms. T is transferred to an inpatient psychiatric unit for further evaluation and management.
The authors’ observations
Fleisher et al7 suggested that traumatic events may lead to presentations similar to PNES. Because Ms. T was molested by a family friend as a child, we considered posttraumatic stress disorder (PTSD) in the differential diagnosis, although she has not reported symptoms of intrusive recollections, avoidance, numbing, or hyperarousal.
We also considered conversion disorder and dissociative disorder. Patients with conversion disorder have ≥1 symptoms or signs that affect voluntary motor or sensory function that cannot be explained by a neurologic or general medical condition.8 Dissociative disorder is a disruption in usually integrated functions of consciousness, memory, identity, or perception of the environment.8 The presentation of patients with PNES may resemble that of patients with dissociative disorder.8 In a study of 45 adult PNES patients, Bowman et al8 found that PNES often are comorbid with other psychiatric disorders, including somatoform disorders (89%), dissociative disorders (91%), affective disorders (64%), personality disorders (62%), PTSD (49%), and other anxiety disorders (47%).
TREATMENT: Managing aggression
In the psychiatric unit, Ms. T initially is irritable and disorganized with poor oral intake and regressed behavior; she often is found in the fetal position, crying and talking in a childish manner. Throughout her admission, she receives several anxiolytics and antipsychotics—including lorazepam, up to 6 mg/d, clonazepam, up to 3 mg/d, haloperidol, up to 10 mg/d, and quetiapine, up to 200 mg/d—to help manage her aggressive behaviors after her seizure-like episodes. Further evaluation reveals that Ms. T has no psychotic symptoms, overt delusions, or perceptual disturbances and her thought process is coherent and clear. She has no history of substance abuse. Her ability to perform ADLs improves within a few days. She complains of depressed mood and engages in head banging, which requires close observation.
Ms. T has a history of mood and behavioral problems since early childhood characterized by episodic dysphoric mood, anxiety, and agitation. She has had trials of several antidepressants, including sertraline, fluoxetine, venlafaxine, and escitalopram, and anxiolytics, including lorazepam, clonazepam, and alprazolam. Her outpatient psychiatrist describes a history of physical and sexual abuse starting at age 7. At age 9, after her mother died from breast cancer, Ms. T and her siblings were moved to foster care, where she was physically abused by the staff. She remained in foster care until age 18.
The authors’ observations
PNES pose a diagnostic and therapeutic challenge. Many PNES patients seek medical attention for their seizures. PNES patients misdiagnosed as having epilepsy have a worse prognosis because they do not receive appropriate treatment9 and may experience side effects if antiepileptics are prescribed.10 Finally, the financial burden of medical care can be significant. Ms. T had several hospitalizations, including extensive neurologic workup, intensive care unit admissions for intubation, and use of antiepileptics with almost no benefit.
Psychosocial assessments of PNES patients have revealed that sexual abuse, family conflicts, and death of a family member often play an important role.11 It is possible that as a result of childhood trauma, Ms. T exhibited a regressed and primitive defense mechanism to deal with the trauma. PNES usually are considered when a patient presents with:
- absence of therapeutic response to antiepileptics
- loss of response (therapeutic failure) to antiepileptics
- paradoxical response to antiepileptics (worsening or unexpected responses)
- atypical, multiple, or inconsistent seizures
- seizures that occur soon after emotional stress.12
We concluded Ms. T had PNES because of the unusual presentations of her seizures, negative video EEG findings, failure to respond to antiepileptics, lack of risk factors for epilepsy, and aggressive behaviors before or after the seizures ( Table ).4,10,11,13 Diagnosing PNES early allows clinicians to focus on appropriate treatment modalities (eg, psychotherapy, antidepressants), prevents costly neurologic workups and treatments (eg, routine EEGs, trials of several antiepileptics), and provides patients with diagnostic assurance.10
Table
Characteristics of psychogenic nonepileptic seizures
| Characteristic | Comment |
|---|---|
| Duration | May be prolonged |
| Timing | Usually occur only during the day |
| Physical harm | Rare |
| Tongue biting | Rare |
| Urinary incontinence | Rare |
| Motor activity | Prolonged |
| Cyanosis | No |
| Postictal confusion | Rare |
| Related to medication changes | No |
| Interictal EEG | Normal |
| Ictal EEG | Normal |
| Presence of secondary gain | Common |
| EEG: electroencephalography Source: References 4,10,11,13 | |
3 components of treatment
Presenting the PNES diagnosis to the patient. The neurologist and the psychiatrist should convey to the patient that they see the symptoms as “real” and not “all in your head.”14
Withdrawing antiepileptic medications. Antiepileptic medication withdrawal is recommended when a thorough diagnostic workup shows no evidence of epileptic seizures.15 Oto et al16 reported 49% of PNES patients became seizure-free 12 months after discontinuing antiepileptics.
Psychotherapy and pharmacotherapy. Open-label studies of psychological treatments for PNES have demonstrated that a cognitive-behavioral therapy-based approach and brief augmented psychodynamic interpersonal therapy could reduce seizures.17 In a pilot, randomized, placebo-controlled trial, PNES patients who received flexibly dosed sertraline reported a 45% reduction in seizures compared with an 8% increase in the placebo group.18 Similar improvements in seizure frequency have been reported in PNES patients with anxiety or depression treated with venlafaxine.19
OUTCOME: Support, improvement
During the next several days, Ms. T has random episodes of seizures with foaming of the mouth and unresponsiveness. These episodes last from 5 to 30 minutes and require transfer to the ER. After each episode, Ms. T is medically cleared and sent back to the psychiatric unit. The neurologist recommends avoiding antiepileptics. Ms. T responds well to the structured inpatient setting and supportive psychotherapy. Her episodes decrease and her mood becomes more stable. She refrains from self-injurious behaviors and is discharged home with outpatient follow-up.
Related Resource
- Marsh P, Benbadis S, Fernandez F. Psychogenic nonepileptic seizures: ways to win over skeptical patients. Current Psychiatry. 2008;7(1):21-35.
Drug Brand Names
- Alprazolam • Xanax
- Clonazepam • Klonopin
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Lorazepam • Ativan
- Quetiapine • Seroquel
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Kellinghaus C, Lüders HO. Frontal lobe epilepsy. Epileptic Disord. 2004;6(4):223-239.
2. Kanner AM, Morris HH, Lüders H, et al. Supplementary motor seizures mimicking pseudoseizures: some clinical differences. Neurology. 1990;40(9):1404-1407.
3. Hall-Patch L, Brown R, House A, et al. Acceptability and effectiveness of a strategy for the communication of the diagnosis of psychogenic nonepileptic seizures. Epilepsia. 2010;51(1):70-78.
4. Reuber M, Elger CE. Psychogenic nonepileptic seizures: review and update. Epilepsy Behav. 2003;4(3):205-216.
5. Chung SS, Gerber P, Kirlin KA. Ictal eye closure is a reliable indicator for psychogenic nonepileptic seizures. Neurology. 2006;66(11):1730-1731.
6. Mostacci B, Bisulli F, Alvisi L, et al. Ictal characteristics of psychogenic nonepileptic seizures: what we have learned from video/EEG recordings—a literature review. Epilepsy Behav. 2011;22(2):144-153.
7. Fleisher W, Staley D, Krawetz P, et al. Comparative study of trauma-related phenomena in subjects with pseudoseizures and subjects with epilepsy. Am J Psychiatry. 2002;159(4):660-663.
8. Bowman ES, Markand ON. Psychodynamics and psychiatric diagnoses of pseudoseizure subjects. Am J Psychiatry. 1996;153(1):57-63.
9. Benbadis SR. The EEG in nonepileptic seizures. J Clin Neurophysiol. 2006;23(4):340-352.
10. Brown RJ, Syed TU, Benbadis S, et al. Psychogenic nonepileptic seizures. Epilepsy Behav. 2011;22(1):85-93.
11. Bodde NM, Brooks JL, Baker GA, et al. Psychogenic non-epileptic seizures—definition, etiology, treatment and prognostic issues: a critical review. Seizure. 2009;18(8):543-553.
12. Alsaadi TM, Marquez AV. Psychogenic nonepileptic seizures. Am Fam Physician. 2005;72(5):849-856.
13. Bradley WG, Daroff RB, Fenichel GM, et al. eds. Neurology in clinical practice: principles of diagnosis and management. 4th ed. Philadelphia, PA: Butterworth Heinemann; 2004:19-20, 1971–1972.
14. Harden CL, Ferrando SJ. Delivering the diagnosis of psychogenic pseudoseizures: should the neurologist or the psychiatrist be responsible? Epilepsy Behav. 2001;2(6):519-523.
15. Oto M, Espie CA, Duncan R. An exploratory randomized controlled trial of immediate versus delayed withdrawal of antiepileptic drugs in patients with psychogenic nonepileptic attacks (PNEAs). Epilepsia. 2010;51(10):1994-1999.
16. Oto M, Espie C, Pelosi A, et al. The safety of antiepileptic drug withdrawal in patients with non-epileptic seizures. J Neurol Neurosurg Psychiatry. 2005;76(12):1682-1685.
17. Goldstein LH, Mellers JD. Recent developments in our understanding of the semiology and treatment of psychogenic nonepileptic seizures. Curr Neurol Neurosci Rep. 2012;12(4):436-444.
18. LaFrance WC, Jr, Keitner GI, Papandonatos GD, et al. Pilot pharmacologic randomized controlled trial for psychogenic nonepileptic seizures. Neurology. 2010;75(13):1166-1173.
19. Pintor L, Baillés E, Matrai S, et al. Efficiency of venlafaxine in patients with psychogenic nonepileptic seizures and anxiety and/or depressive disorders. J Neuropsychiatry Clin Neurosci. 2010;22(4):401-408.
CASE: Seizure-like symptoms
Ms. T, age 20, is brought to the emergency room (ER) by her father because she refuses to eat and drink, is unable to function at home, lies in bed all day, and does not attend to her activities of daily living (ADLs). Ms. T lives with her family, is not enrolled in school, and is unemployed. In the ER she initially is uncooperative and mute and then suddenly becomes agitated and has a seizure-like episode characterized by jerking of her trunk followed by random, asymmetrical movements of her legs and arms, closing both eyes, weeping, foaming at the mouth, moaning, and marked unresponsiveness. The episode lasts for >5 minutes.
The authors’ observations
Based on Ms. T’s presentation, the medical team considered acute epileptic seizures. Asymmetrical jerking of the body may be seen in frontal lobe epilepsy or seizures of the supplementary sensorimotor area. Frontal lobe epilepsy can present with bilateral asynchronous motor activity with consciousness during the event and a lack of postictal confusion.1 Seizures of the supplementary sensorimotor area—also known as the secondary motor area—are particularly problematic because typically they present with bilateral asymmetric tonic posturing followed by a few clonic movements, intact consciousness, and rarely postictal confusion. Adding to the diagnostic uncertainty, some “soft signs” thought to indicate PNES (eg, pelvic thrusting, crying) are common with frontal lobe epilepsy.1,2
PNES are episodes of altered movement, sensation, or experience that may be mistaken for epileptic seizures but are not a consequence of abnormal cortical discharges. Instead they are caused by physiological or psychological factors.3 Behaviors or signs that strongly suggest PNES include:
- gradual onset or termination
- pseudosleep, when the patient appears to be asleep but electroencephalography (EEG) findings indicate he or she is awake
- discontinuous (stop-and-go), irregular, or asynchronous (out-of-phase) activity—including side-to-side head movement, pelvic thrusting, and opisthotonic posturing—stuttering, and weeping4
- eye closure.5
Ms. T’s father said his daughter had been hospitalized several times for episodes characterized by pelvic thrusting, stuttering, and pseudosleep, which raised the possibility of PNES. Definitive diagnosis of PNES comes from video EEG when a patient is observed having typical seizures without accompanying EEG abnormalities.6
EVALUATION: Inconclusive data
Ms. T is admitted to the medical unit to rule out a seizure disorder. Physical examination is unremarkable and laboratory tests are within normal limits. The neurology service requests a head MRI, which is inconclusive. Inpatient video EEG with 24-hour monitoring does not indicate acute epileptic seizures. Ms. T’s father says that she has experienced many paroxysmal motor episodes and all neurologic tests, exams, and labs have failed to find a cause for these episodes. She did not receive any antiepileptic medications. A psychiatric consult is requested to clarify the diagnosis. Ms. T is transferred to an inpatient psychiatric unit for further evaluation and management.
The authors’ observations
Fleisher et al7 suggested that traumatic events may lead to presentations similar to PNES. Because Ms. T was molested by a family friend as a child, we considered posttraumatic stress disorder (PTSD) in the differential diagnosis, although she has not reported symptoms of intrusive recollections, avoidance, numbing, or hyperarousal.
We also considered conversion disorder and dissociative disorder. Patients with conversion disorder have ≥1 symptoms or signs that affect voluntary motor or sensory function that cannot be explained by a neurologic or general medical condition.8 Dissociative disorder is a disruption in usually integrated functions of consciousness, memory, identity, or perception of the environment.8 The presentation of patients with PNES may resemble that of patients with dissociative disorder.8 In a study of 45 adult PNES patients, Bowman et al8 found that PNES often are comorbid with other psychiatric disorders, including somatoform disorders (89%), dissociative disorders (91%), affective disorders (64%), personality disorders (62%), PTSD (49%), and other anxiety disorders (47%).
TREATMENT: Managing aggression
In the psychiatric unit, Ms. T initially is irritable and disorganized with poor oral intake and regressed behavior; she often is found in the fetal position, crying and talking in a childish manner. Throughout her admission, she receives several anxiolytics and antipsychotics—including lorazepam, up to 6 mg/d, clonazepam, up to 3 mg/d, haloperidol, up to 10 mg/d, and quetiapine, up to 200 mg/d—to help manage her aggressive behaviors after her seizure-like episodes. Further evaluation reveals that Ms. T has no psychotic symptoms, overt delusions, or perceptual disturbances and her thought process is coherent and clear. She has no history of substance abuse. Her ability to perform ADLs improves within a few days. She complains of depressed mood and engages in head banging, which requires close observation.
Ms. T has a history of mood and behavioral problems since early childhood characterized by episodic dysphoric mood, anxiety, and agitation. She has had trials of several antidepressants, including sertraline, fluoxetine, venlafaxine, and escitalopram, and anxiolytics, including lorazepam, clonazepam, and alprazolam. Her outpatient psychiatrist describes a history of physical and sexual abuse starting at age 7. At age 9, after her mother died from breast cancer, Ms. T and her siblings were moved to foster care, where she was physically abused by the staff. She remained in foster care until age 18.
The authors’ observations
PNES pose a diagnostic and therapeutic challenge. Many PNES patients seek medical attention for their seizures. PNES patients misdiagnosed as having epilepsy have a worse prognosis because they do not receive appropriate treatment9 and may experience side effects if antiepileptics are prescribed.10 Finally, the financial burden of medical care can be significant. Ms. T had several hospitalizations, including extensive neurologic workup, intensive care unit admissions for intubation, and use of antiepileptics with almost no benefit.
Psychosocial assessments of PNES patients have revealed that sexual abuse, family conflicts, and death of a family member often play an important role.11 It is possible that as a result of childhood trauma, Ms. T exhibited a regressed and primitive defense mechanism to deal with the trauma. PNES usually are considered when a patient presents with:
- absence of therapeutic response to antiepileptics
- loss of response (therapeutic failure) to antiepileptics
- paradoxical response to antiepileptics (worsening or unexpected responses)
- atypical, multiple, or inconsistent seizures
- seizures that occur soon after emotional stress.12
We concluded Ms. T had PNES because of the unusual presentations of her seizures, negative video EEG findings, failure to respond to antiepileptics, lack of risk factors for epilepsy, and aggressive behaviors before or after the seizures ( Table ).4,10,11,13 Diagnosing PNES early allows clinicians to focus on appropriate treatment modalities (eg, psychotherapy, antidepressants), prevents costly neurologic workups and treatments (eg, routine EEGs, trials of several antiepileptics), and provides patients with diagnostic assurance.10
Table
Characteristics of psychogenic nonepileptic seizures
| Characteristic | Comment |
|---|---|
| Duration | May be prolonged |
| Timing | Usually occur only during the day |
| Physical harm | Rare |
| Tongue biting | Rare |
| Urinary incontinence | Rare |
| Motor activity | Prolonged |
| Cyanosis | No |
| Postictal confusion | Rare |
| Related to medication changes | No |
| Interictal EEG | Normal |
| Ictal EEG | Normal |
| Presence of secondary gain | Common |
| EEG: electroencephalography Source: References 4,10,11,13 | |
3 components of treatment
Presenting the PNES diagnosis to the patient. The neurologist and the psychiatrist should convey to the patient that they see the symptoms as “real” and not “all in your head.”14
Withdrawing antiepileptic medications. Antiepileptic medication withdrawal is recommended when a thorough diagnostic workup shows no evidence of epileptic seizures.15 Oto et al16 reported 49% of PNES patients became seizure-free 12 months after discontinuing antiepileptics.
Psychotherapy and pharmacotherapy. Open-label studies of psychological treatments for PNES have demonstrated that a cognitive-behavioral therapy-based approach and brief augmented psychodynamic interpersonal therapy could reduce seizures.17 In a pilot, randomized, placebo-controlled trial, PNES patients who received flexibly dosed sertraline reported a 45% reduction in seizures compared with an 8% increase in the placebo group.18 Similar improvements in seizure frequency have been reported in PNES patients with anxiety or depression treated with venlafaxine.19
OUTCOME: Support, improvement
During the next several days, Ms. T has random episodes of seizures with foaming of the mouth and unresponsiveness. These episodes last from 5 to 30 minutes and require transfer to the ER. After each episode, Ms. T is medically cleared and sent back to the psychiatric unit. The neurologist recommends avoiding antiepileptics. Ms. T responds well to the structured inpatient setting and supportive psychotherapy. Her episodes decrease and her mood becomes more stable. She refrains from self-injurious behaviors and is discharged home with outpatient follow-up.
Related Resource
- Marsh P, Benbadis S, Fernandez F. Psychogenic nonepileptic seizures: ways to win over skeptical patients. Current Psychiatry. 2008;7(1):21-35.
Drug Brand Names
- Alprazolam • Xanax
- Clonazepam • Klonopin
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Lorazepam • Ativan
- Quetiapine • Seroquel
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE: Seizure-like symptoms
Ms. T, age 20, is brought to the emergency room (ER) by her father because she refuses to eat and drink, is unable to function at home, lies in bed all day, and does not attend to her activities of daily living (ADLs). Ms. T lives with her family, is not enrolled in school, and is unemployed. In the ER she initially is uncooperative and mute and then suddenly becomes agitated and has a seizure-like episode characterized by jerking of her trunk followed by random, asymmetrical movements of her legs and arms, closing both eyes, weeping, foaming at the mouth, moaning, and marked unresponsiveness. The episode lasts for >5 minutes.
The authors’ observations
Based on Ms. T’s presentation, the medical team considered acute epileptic seizures. Asymmetrical jerking of the body may be seen in frontal lobe epilepsy or seizures of the supplementary sensorimotor area. Frontal lobe epilepsy can present with bilateral asynchronous motor activity with consciousness during the event and a lack of postictal confusion.1 Seizures of the supplementary sensorimotor area—also known as the secondary motor area—are particularly problematic because typically they present with bilateral asymmetric tonic posturing followed by a few clonic movements, intact consciousness, and rarely postictal confusion. Adding to the diagnostic uncertainty, some “soft signs” thought to indicate PNES (eg, pelvic thrusting, crying) are common with frontal lobe epilepsy.1,2
PNES are episodes of altered movement, sensation, or experience that may be mistaken for epileptic seizures but are not a consequence of abnormal cortical discharges. Instead they are caused by physiological or psychological factors.3 Behaviors or signs that strongly suggest PNES include:
- gradual onset or termination
- pseudosleep, when the patient appears to be asleep but electroencephalography (EEG) findings indicate he or she is awake
- discontinuous (stop-and-go), irregular, or asynchronous (out-of-phase) activity—including side-to-side head movement, pelvic thrusting, and opisthotonic posturing—stuttering, and weeping4
- eye closure.5
Ms. T’s father said his daughter had been hospitalized several times for episodes characterized by pelvic thrusting, stuttering, and pseudosleep, which raised the possibility of PNES. Definitive diagnosis of PNES comes from video EEG when a patient is observed having typical seizures without accompanying EEG abnormalities.6
EVALUATION: Inconclusive data
Ms. T is admitted to the medical unit to rule out a seizure disorder. Physical examination is unremarkable and laboratory tests are within normal limits. The neurology service requests a head MRI, which is inconclusive. Inpatient video EEG with 24-hour monitoring does not indicate acute epileptic seizures. Ms. T’s father says that she has experienced many paroxysmal motor episodes and all neurologic tests, exams, and labs have failed to find a cause for these episodes. She did not receive any antiepileptic medications. A psychiatric consult is requested to clarify the diagnosis. Ms. T is transferred to an inpatient psychiatric unit for further evaluation and management.
The authors’ observations
Fleisher et al7 suggested that traumatic events may lead to presentations similar to PNES. Because Ms. T was molested by a family friend as a child, we considered posttraumatic stress disorder (PTSD) in the differential diagnosis, although she has not reported symptoms of intrusive recollections, avoidance, numbing, or hyperarousal.
We also considered conversion disorder and dissociative disorder. Patients with conversion disorder have ≥1 symptoms or signs that affect voluntary motor or sensory function that cannot be explained by a neurologic or general medical condition.8 Dissociative disorder is a disruption in usually integrated functions of consciousness, memory, identity, or perception of the environment.8 The presentation of patients with PNES may resemble that of patients with dissociative disorder.8 In a study of 45 adult PNES patients, Bowman et al8 found that PNES often are comorbid with other psychiatric disorders, including somatoform disorders (89%), dissociative disorders (91%), affective disorders (64%), personality disorders (62%), PTSD (49%), and other anxiety disorders (47%).
TREATMENT: Managing aggression
In the psychiatric unit, Ms. T initially is irritable and disorganized with poor oral intake and regressed behavior; she often is found in the fetal position, crying and talking in a childish manner. Throughout her admission, she receives several anxiolytics and antipsychotics—including lorazepam, up to 6 mg/d, clonazepam, up to 3 mg/d, haloperidol, up to 10 mg/d, and quetiapine, up to 200 mg/d—to help manage her aggressive behaviors after her seizure-like episodes. Further evaluation reveals that Ms. T has no psychotic symptoms, overt delusions, or perceptual disturbances and her thought process is coherent and clear. She has no history of substance abuse. Her ability to perform ADLs improves within a few days. She complains of depressed mood and engages in head banging, which requires close observation.
Ms. T has a history of mood and behavioral problems since early childhood characterized by episodic dysphoric mood, anxiety, and agitation. She has had trials of several antidepressants, including sertraline, fluoxetine, venlafaxine, and escitalopram, and anxiolytics, including lorazepam, clonazepam, and alprazolam. Her outpatient psychiatrist describes a history of physical and sexual abuse starting at age 7. At age 9, after her mother died from breast cancer, Ms. T and her siblings were moved to foster care, where she was physically abused by the staff. She remained in foster care until age 18.
The authors’ observations
PNES pose a diagnostic and therapeutic challenge. Many PNES patients seek medical attention for their seizures. PNES patients misdiagnosed as having epilepsy have a worse prognosis because they do not receive appropriate treatment9 and may experience side effects if antiepileptics are prescribed.10 Finally, the financial burden of medical care can be significant. Ms. T had several hospitalizations, including extensive neurologic workup, intensive care unit admissions for intubation, and use of antiepileptics with almost no benefit.
Psychosocial assessments of PNES patients have revealed that sexual abuse, family conflicts, and death of a family member often play an important role.11 It is possible that as a result of childhood trauma, Ms. T exhibited a regressed and primitive defense mechanism to deal with the trauma. PNES usually are considered when a patient presents with:
- absence of therapeutic response to antiepileptics
- loss of response (therapeutic failure) to antiepileptics
- paradoxical response to antiepileptics (worsening or unexpected responses)
- atypical, multiple, or inconsistent seizures
- seizures that occur soon after emotional stress.12
We concluded Ms. T had PNES because of the unusual presentations of her seizures, negative video EEG findings, failure to respond to antiepileptics, lack of risk factors for epilepsy, and aggressive behaviors before or after the seizures ( Table ).4,10,11,13 Diagnosing PNES early allows clinicians to focus on appropriate treatment modalities (eg, psychotherapy, antidepressants), prevents costly neurologic workups and treatments (eg, routine EEGs, trials of several antiepileptics), and provides patients with diagnostic assurance.10
Table
Characteristics of psychogenic nonepileptic seizures
| Characteristic | Comment |
|---|---|
| Duration | May be prolonged |
| Timing | Usually occur only during the day |
| Physical harm | Rare |
| Tongue biting | Rare |
| Urinary incontinence | Rare |
| Motor activity | Prolonged |
| Cyanosis | No |
| Postictal confusion | Rare |
| Related to medication changes | No |
| Interictal EEG | Normal |
| Ictal EEG | Normal |
| Presence of secondary gain | Common |
| EEG: electroencephalography Source: References 4,10,11,13 | |
3 components of treatment
Presenting the PNES diagnosis to the patient. The neurologist and the psychiatrist should convey to the patient that they see the symptoms as “real” and not “all in your head.”14
Withdrawing antiepileptic medications. Antiepileptic medication withdrawal is recommended when a thorough diagnostic workup shows no evidence of epileptic seizures.15 Oto et al16 reported 49% of PNES patients became seizure-free 12 months after discontinuing antiepileptics.
Psychotherapy and pharmacotherapy. Open-label studies of psychological treatments for PNES have demonstrated that a cognitive-behavioral therapy-based approach and brief augmented psychodynamic interpersonal therapy could reduce seizures.17 In a pilot, randomized, placebo-controlled trial, PNES patients who received flexibly dosed sertraline reported a 45% reduction in seizures compared with an 8% increase in the placebo group.18 Similar improvements in seizure frequency have been reported in PNES patients with anxiety or depression treated with venlafaxine.19
OUTCOME: Support, improvement
During the next several days, Ms. T has random episodes of seizures with foaming of the mouth and unresponsiveness. These episodes last from 5 to 30 minutes and require transfer to the ER. After each episode, Ms. T is medically cleared and sent back to the psychiatric unit. The neurologist recommends avoiding antiepileptics. Ms. T responds well to the structured inpatient setting and supportive psychotherapy. Her episodes decrease and her mood becomes more stable. She refrains from self-injurious behaviors and is discharged home with outpatient follow-up.
Related Resource
- Marsh P, Benbadis S, Fernandez F. Psychogenic nonepileptic seizures: ways to win over skeptical patients. Current Psychiatry. 2008;7(1):21-35.
Drug Brand Names
- Alprazolam • Xanax
- Clonazepam • Klonopin
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Lorazepam • Ativan
- Quetiapine • Seroquel
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Kellinghaus C, Lüders HO. Frontal lobe epilepsy. Epileptic Disord. 2004;6(4):223-239.
2. Kanner AM, Morris HH, Lüders H, et al. Supplementary motor seizures mimicking pseudoseizures: some clinical differences. Neurology. 1990;40(9):1404-1407.
3. Hall-Patch L, Brown R, House A, et al. Acceptability and effectiveness of a strategy for the communication of the diagnosis of psychogenic nonepileptic seizures. Epilepsia. 2010;51(1):70-78.
4. Reuber M, Elger CE. Psychogenic nonepileptic seizures: review and update. Epilepsy Behav. 2003;4(3):205-216.
5. Chung SS, Gerber P, Kirlin KA. Ictal eye closure is a reliable indicator for psychogenic nonepileptic seizures. Neurology. 2006;66(11):1730-1731.
6. Mostacci B, Bisulli F, Alvisi L, et al. Ictal characteristics of psychogenic nonepileptic seizures: what we have learned from video/EEG recordings—a literature review. Epilepsy Behav. 2011;22(2):144-153.
7. Fleisher W, Staley D, Krawetz P, et al. Comparative study of trauma-related phenomena in subjects with pseudoseizures and subjects with epilepsy. Am J Psychiatry. 2002;159(4):660-663.
8. Bowman ES, Markand ON. Psychodynamics and psychiatric diagnoses of pseudoseizure subjects. Am J Psychiatry. 1996;153(1):57-63.
9. Benbadis SR. The EEG in nonepileptic seizures. J Clin Neurophysiol. 2006;23(4):340-352.
10. Brown RJ, Syed TU, Benbadis S, et al. Psychogenic nonepileptic seizures. Epilepsy Behav. 2011;22(1):85-93.
11. Bodde NM, Brooks JL, Baker GA, et al. Psychogenic non-epileptic seizures—definition, etiology, treatment and prognostic issues: a critical review. Seizure. 2009;18(8):543-553.
12. Alsaadi TM, Marquez AV. Psychogenic nonepileptic seizures. Am Fam Physician. 2005;72(5):849-856.
13. Bradley WG, Daroff RB, Fenichel GM, et al. eds. Neurology in clinical practice: principles of diagnosis and management. 4th ed. Philadelphia, PA: Butterworth Heinemann; 2004:19-20, 1971–1972.
14. Harden CL, Ferrando SJ. Delivering the diagnosis of psychogenic pseudoseizures: should the neurologist or the psychiatrist be responsible? Epilepsy Behav. 2001;2(6):519-523.
15. Oto M, Espie CA, Duncan R. An exploratory randomized controlled trial of immediate versus delayed withdrawal of antiepileptic drugs in patients with psychogenic nonepileptic attacks (PNEAs). Epilepsia. 2010;51(10):1994-1999.
16. Oto M, Espie C, Pelosi A, et al. The safety of antiepileptic drug withdrawal in patients with non-epileptic seizures. J Neurol Neurosurg Psychiatry. 2005;76(12):1682-1685.
17. Goldstein LH, Mellers JD. Recent developments in our understanding of the semiology and treatment of psychogenic nonepileptic seizures. Curr Neurol Neurosci Rep. 2012;12(4):436-444.
18. LaFrance WC, Jr, Keitner GI, Papandonatos GD, et al. Pilot pharmacologic randomized controlled trial for psychogenic nonepileptic seizures. Neurology. 2010;75(13):1166-1173.
19. Pintor L, Baillés E, Matrai S, et al. Efficiency of venlafaxine in patients with psychogenic nonepileptic seizures and anxiety and/or depressive disorders. J Neuropsychiatry Clin Neurosci. 2010;22(4):401-408.
1. Kellinghaus C, Lüders HO. Frontal lobe epilepsy. Epileptic Disord. 2004;6(4):223-239.
2. Kanner AM, Morris HH, Lüders H, et al. Supplementary motor seizures mimicking pseudoseizures: some clinical differences. Neurology. 1990;40(9):1404-1407.
3. Hall-Patch L, Brown R, House A, et al. Acceptability and effectiveness of a strategy for the communication of the diagnosis of psychogenic nonepileptic seizures. Epilepsia. 2010;51(1):70-78.
4. Reuber M, Elger CE. Psychogenic nonepileptic seizures: review and update. Epilepsy Behav. 2003;4(3):205-216.
5. Chung SS, Gerber P, Kirlin KA. Ictal eye closure is a reliable indicator for psychogenic nonepileptic seizures. Neurology. 2006;66(11):1730-1731.
6. Mostacci B, Bisulli F, Alvisi L, et al. Ictal characteristics of psychogenic nonepileptic seizures: what we have learned from video/EEG recordings—a literature review. Epilepsy Behav. 2011;22(2):144-153.
7. Fleisher W, Staley D, Krawetz P, et al. Comparative study of trauma-related phenomena in subjects with pseudoseizures and subjects with epilepsy. Am J Psychiatry. 2002;159(4):660-663.
8. Bowman ES, Markand ON. Psychodynamics and psychiatric diagnoses of pseudoseizure subjects. Am J Psychiatry. 1996;153(1):57-63.
9. Benbadis SR. The EEG in nonepileptic seizures. J Clin Neurophysiol. 2006;23(4):340-352.
10. Brown RJ, Syed TU, Benbadis S, et al. Psychogenic nonepileptic seizures. Epilepsy Behav. 2011;22(1):85-93.
11. Bodde NM, Brooks JL, Baker GA, et al. Psychogenic non-epileptic seizures—definition, etiology, treatment and prognostic issues: a critical review. Seizure. 2009;18(8):543-553.
12. Alsaadi TM, Marquez AV. Psychogenic nonepileptic seizures. Am Fam Physician. 2005;72(5):849-856.
13. Bradley WG, Daroff RB, Fenichel GM, et al. eds. Neurology in clinical practice: principles of diagnosis and management. 4th ed. Philadelphia, PA: Butterworth Heinemann; 2004:19-20, 1971–1972.
14. Harden CL, Ferrando SJ. Delivering the diagnosis of psychogenic pseudoseizures: should the neurologist or the psychiatrist be responsible? Epilepsy Behav. 2001;2(6):519-523.
15. Oto M, Espie CA, Duncan R. An exploratory randomized controlled trial of immediate versus delayed withdrawal of antiepileptic drugs in patients with psychogenic nonepileptic attacks (PNEAs). Epilepsia. 2010;51(10):1994-1999.
16. Oto M, Espie C, Pelosi A, et al. The safety of antiepileptic drug withdrawal in patients with non-epileptic seizures. J Neurol Neurosurg Psychiatry. 2005;76(12):1682-1685.
17. Goldstein LH, Mellers JD. Recent developments in our understanding of the semiology and treatment of psychogenic nonepileptic seizures. Curr Neurol Neurosci Rep. 2012;12(4):436-444.
18. LaFrance WC, Jr, Keitner GI, Papandonatos GD, et al. Pilot pharmacologic randomized controlled trial for psychogenic nonepileptic seizures. Neurology. 2010;75(13):1166-1173.
19. Pintor L, Baillés E, Matrai S, et al. Efficiency of venlafaxine in patients with psychogenic nonepileptic seizures and anxiety and/or depressive disorders. J Neuropsychiatry Clin Neurosci. 2010;22(4):401-408.
TMS for depression
I want to correct an error in a recent letter regarding the use of transcranial magnetic stimulation (TMS) in treatment-resistant depression (TRD) (Comments & Controversies, Current Psychiatry, June 2012, p. 19; http://bit.ly/1QU1sOR). The letter writers assert that a March 2012 article in Current Psychiatry should have included TMS as a treatment for TRD because it is “FDA-approved for TRD.” TMS is FDA-approved for the treatment of unipolar depression in patients who failed to respond to a single antidepressant trial. There are various definitions of TRD, but failing to respond to 1 antidepressant trial would not satisfy criteria for any such definition. A pivotal study of TMS, upon which the FDA approval was based, found that patients who had failed to respond to a single antidepressant were significantly more likely to respond to TMS than those who failed 2 to 4 antidepressant trials (P = .021).1 Therefore, TMS is of questionable utility in treating TRD.
Brian Feldman, MD
Boca Raton Psychiatric Group
Boca Raton, FL
The authors respond
We appreciate Dr. Feldman noting our technical error. He is correct that TMS is FDA-approved for treating unipolar depression in individuals who had failed 1 adequate antidepressant trial during their current episode. However, Dr. Feldman is not correct that by no definition would this constitute TRD. For example, Fava1 stated TRD “typically refers to inadequate response to at least one antidepressant trial of adequate doses and duration.”
Based on the research literature, we also disagree with Dr. Feldman’s assertion of “questionable utility” of TMS in TRD. As part of research presented to the FDA, O’Reardon et al2 characterized the study sample as having failed an average of 1.6 adequate antidepressant treatment trial, with approximately one-half having failed ≥2 treatments in their current episode. Connolly et al3 described results in treating 100 consecutive depressed patients with TMS as equivalent to research findings. Most patients had >1 failed adequate antidepressant trial in their current episode. The Agency for Healthcare Research and Quality concluded that evidence supported use of TMS.4 Overall, the panel concluded that there is a substantial and well-replicated body of evidence that TMS is beneficial compared with controls in severity of symptoms, response rate, and remission rate. In a head-to-head comparison with electroconvulsive therapy, TMS was equally effective. TMS is a valuable addition to the therapeutic armamentarium that can help patients early in an illness not fall into a treatment-resistant state, and can offer another chance for those who have.
Gordon Baumbacher MD
Caroline Mulder, MD
Private Practice
Corte Madera, CA
Richard Bermudes, MD
Private Practice
Sacramento, CA
Jennifer Beck, MD
Private Practice
Santa Rosa, CA
I want to correct an error in a recent letter regarding the use of transcranial magnetic stimulation (TMS) in treatment-resistant depression (TRD) (Comments & Controversies, Current Psychiatry, June 2012, p. 19; http://bit.ly/1QU1sOR). The letter writers assert that a March 2012 article in Current Psychiatry should have included TMS as a treatment for TRD because it is “FDA-approved for TRD.” TMS is FDA-approved for the treatment of unipolar depression in patients who failed to respond to a single antidepressant trial. There are various definitions of TRD, but failing to respond to 1 antidepressant trial would not satisfy criteria for any such definition. A pivotal study of TMS, upon which the FDA approval was based, found that patients who had failed to respond to a single antidepressant were significantly more likely to respond to TMS than those who failed 2 to 4 antidepressant trials (P = .021).1 Therefore, TMS is of questionable utility in treating TRD.
Brian Feldman, MD
Boca Raton Psychiatric Group
Boca Raton, FL
The authors respond
We appreciate Dr. Feldman noting our technical error. He is correct that TMS is FDA-approved for treating unipolar depression in individuals who had failed 1 adequate antidepressant trial during their current episode. However, Dr. Feldman is not correct that by no definition would this constitute TRD. For example, Fava1 stated TRD “typically refers to inadequate response to at least one antidepressant trial of adequate doses and duration.”
Based on the research literature, we also disagree with Dr. Feldman’s assertion of “questionable utility” of TMS in TRD. As part of research presented to the FDA, O’Reardon et al2 characterized the study sample as having failed an average of 1.6 adequate antidepressant treatment trial, with approximately one-half having failed ≥2 treatments in their current episode. Connolly et al3 described results in treating 100 consecutive depressed patients with TMS as equivalent to research findings. Most patients had >1 failed adequate antidepressant trial in their current episode. The Agency for Healthcare Research and Quality concluded that evidence supported use of TMS.4 Overall, the panel concluded that there is a substantial and well-replicated body of evidence that TMS is beneficial compared with controls in severity of symptoms, response rate, and remission rate. In a head-to-head comparison with electroconvulsive therapy, TMS was equally effective. TMS is a valuable addition to the therapeutic armamentarium that can help patients early in an illness not fall into a treatment-resistant state, and can offer another chance for those who have.
Gordon Baumbacher MD
Caroline Mulder, MD
Private Practice
Corte Madera, CA
Richard Bermudes, MD
Private Practice
Sacramento, CA
Jennifer Beck, MD
Private Practice
Santa Rosa, CA
I want to correct an error in a recent letter regarding the use of transcranial magnetic stimulation (TMS) in treatment-resistant depression (TRD) (Comments & Controversies, Current Psychiatry, June 2012, p. 19; http://bit.ly/1QU1sOR). The letter writers assert that a March 2012 article in Current Psychiatry should have included TMS as a treatment for TRD because it is “FDA-approved for TRD.” TMS is FDA-approved for the treatment of unipolar depression in patients who failed to respond to a single antidepressant trial. There are various definitions of TRD, but failing to respond to 1 antidepressant trial would not satisfy criteria for any such definition. A pivotal study of TMS, upon which the FDA approval was based, found that patients who had failed to respond to a single antidepressant were significantly more likely to respond to TMS than those who failed 2 to 4 antidepressant trials (P = .021).1 Therefore, TMS is of questionable utility in treating TRD.
Brian Feldman, MD
Boca Raton Psychiatric Group
Boca Raton, FL
The authors respond
We appreciate Dr. Feldman noting our technical error. He is correct that TMS is FDA-approved for treating unipolar depression in individuals who had failed 1 adequate antidepressant trial during their current episode. However, Dr. Feldman is not correct that by no definition would this constitute TRD. For example, Fava1 stated TRD “typically refers to inadequate response to at least one antidepressant trial of adequate doses and duration.”
Based on the research literature, we also disagree with Dr. Feldman’s assertion of “questionable utility” of TMS in TRD. As part of research presented to the FDA, O’Reardon et al2 characterized the study sample as having failed an average of 1.6 adequate antidepressant treatment trial, with approximately one-half having failed ≥2 treatments in their current episode. Connolly et al3 described results in treating 100 consecutive depressed patients with TMS as equivalent to research findings. Most patients had >1 failed adequate antidepressant trial in their current episode. The Agency for Healthcare Research and Quality concluded that evidence supported use of TMS.4 Overall, the panel concluded that there is a substantial and well-replicated body of evidence that TMS is beneficial compared with controls in severity of symptoms, response rate, and remission rate. In a head-to-head comparison with electroconvulsive therapy, TMS was equally effective. TMS is a valuable addition to the therapeutic armamentarium that can help patients early in an illness not fall into a treatment-resistant state, and can offer another chance for those who have.
Gordon Baumbacher MD
Caroline Mulder, MD
Private Practice
Corte Madera, CA
Richard Bermudes, MD
Private Practice
Sacramento, CA
Jennifer Beck, MD
Private Practice
Santa Rosa, CA
The dark side of the human brain
On July 20 of every year, I recall the transcendent wonder I felt on that day in 1969 when the entire world and I were glued to our television sets, witnessing one of the greatest achievements in the history of the human race: American astronauts landing on the moon and beaming pictures of their extraordinary celestial expedition to the billions of earthlings sitting on the edge of their seats.
Every year on July 20, I ponder the supreme and brilliant abilities of the human brain that transformed walking on the moon from an absurd fantasy to a thrilling reality. After becoming a psychiatrist, trained to observe everything through the prism of mind and brain, the moon landing represented the zenith achievement of the divinely evolved human neocortex, especially the prefrontal lobe of President John F. Kennedy, who established that lofty goal, and the advanced brains of thousands of NASA scientists and engineers, who set out to fulfill that towering expectation in less than a decade. That’s why every year on July 20, my abiding faith in the limitless capacities of the human brain to do great things is confirmed and reinforced.
However, this year was different as July 20 took on a much darker meaning. The tragic shooting in Aurora, CO that killed 12 people and wounded 58 others—perpetrated by the bizarre machinations of a graduate school student who had been receiving psychiatric treatment when he suddenly dropped out—rudely reminded me how, like the moon, the human brain has a dark side. A healthy brain that can envision, plan, and execute a magnificent moon landing is capable, when perturbed, of carrying out a dreadfully heinous crime. So henceforth, July 20 always will remind me of the zenith vs nadir dichotomy of the human mind potential in health and disease.
As a longtime academic psychiatrist, I have repeatedly witnessed the tragic disability my patients suffer when the frontal lobe—the most advanced component of their brains— becomes seriously impaired, rendering them dysfunctional. Unless driven by an intense delusion or command hallucinations, patients with schizophrenia rarely commit a murder. However, the media often has linked psychosis with violent crime, whether in movies1 or in news reporting, which has led to public misconception that every person suffering from psychosis is a potential mass murderer. The truth is that the vast majority of murders are committed by nonpsychotic criminals with severe antisocial traits.
Because of the shocking killings in Aurora, July 20 may become an annual reminder that perpetuates the unfair and ignorant notion that mental illness always is associated with horrendous crimes. That would further darken the discriminatory stigma of mental illness and may foster a hatred of and aversion to mentally ill individuals, the vast majority of whom are law-abiding citizens. Even the legally mandated “not guilty by reason of insanity” defense is being described as a spurious “excuse” and is under assault, which may lead to death sentences for medically ill persons whose actions are triggered by a severe brain pathology that impairs judgment and distorts reality testing of what is right or wrong.
The devastated families of the victims also are very much on my mind. For them, July 20 will become a dark anniversary of how their loved ones—most of whom were in the prime of life—lost theirs lives while watching a movie called The Dark Knight Rises. Their annual grief on July 20 will for many years reopen the deep wounds inflicted on their souls.
In the future, on July 20 I will no longer be merely inspired by the human brain’s potential for achievements. I now also will remember the potential for despicable and murderous actions when a sickened human brain and its convoluted mind go terribly awry. As an academic investigator dedicated to finding a cure for schizophrenia and related disorders, I regard the tragic events in Aurora on July 20, 2012 as another call to escalate the commitment of researchers to restore normalcy to the blighted brains of those afflicted by this cruel disease, or, better yet, to ultimately discover how to prevent schizophrenia from developing. Only then will the darkness of severe mental illness finally ebb and vanish.
Editor's Note: This editorial was written before Neil Armstrong, the first man to walk on the moon, died on August 25, 2012.
1. Owen PR. Portrayals of schizophrenia by entertainment media: a content analysis of contemporary movies. Psychiatr Serv. 2012;63(7):655-659.

Henry A. Nasrallah, MD
Editor-in-Chief
To comment on this editorial or other topics of interest, visit http://www.facebook.com/CurrentPsychiatry, or click on the “Send Letters” link.
Henry A. Nasrallah, MD
Editor-in-Chief
To comment on this editorial or other topics of interest, visit http://www.facebook.com/CurrentPsychiatry, or click on the “Send Letters” link.
Henry A. Nasrallah, MD
Editor-in-Chief
To comment on this editorial or other topics of interest, visit http://www.facebook.com/CurrentPsychiatry, or click on the “Send Letters” link.
On July 20 of every year, I recall the transcendent wonder I felt on that day in 1969 when the entire world and I were glued to our television sets, witnessing one of the greatest achievements in the history of the human race: American astronauts landing on the moon and beaming pictures of their extraordinary celestial expedition to the billions of earthlings sitting on the edge of their seats.
Every year on July 20, I ponder the supreme and brilliant abilities of the human brain that transformed walking on the moon from an absurd fantasy to a thrilling reality. After becoming a psychiatrist, trained to observe everything through the prism of mind and brain, the moon landing represented the zenith achievement of the divinely evolved human neocortex, especially the prefrontal lobe of President John F. Kennedy, who established that lofty goal, and the advanced brains of thousands of NASA scientists and engineers, who set out to fulfill that towering expectation in less than a decade. That’s why every year on July 20, my abiding faith in the limitless capacities of the human brain to do great things is confirmed and reinforced.
However, this year was different as July 20 took on a much darker meaning. The tragic shooting in Aurora, CO that killed 12 people and wounded 58 others—perpetrated by the bizarre machinations of a graduate school student who had been receiving psychiatric treatment when he suddenly dropped out—rudely reminded me how, like the moon, the human brain has a dark side. A healthy brain that can envision, plan, and execute a magnificent moon landing is capable, when perturbed, of carrying out a dreadfully heinous crime. So henceforth, July 20 always will remind me of the zenith vs nadir dichotomy of the human mind potential in health and disease.
As a longtime academic psychiatrist, I have repeatedly witnessed the tragic disability my patients suffer when the frontal lobe—the most advanced component of their brains— becomes seriously impaired, rendering them dysfunctional. Unless driven by an intense delusion or command hallucinations, patients with schizophrenia rarely commit a murder. However, the media often has linked psychosis with violent crime, whether in movies1 or in news reporting, which has led to public misconception that every person suffering from psychosis is a potential mass murderer. The truth is that the vast majority of murders are committed by nonpsychotic criminals with severe antisocial traits.
Because of the shocking killings in Aurora, July 20 may become an annual reminder that perpetuates the unfair and ignorant notion that mental illness always is associated with horrendous crimes. That would further darken the discriminatory stigma of mental illness and may foster a hatred of and aversion to mentally ill individuals, the vast majority of whom are law-abiding citizens. Even the legally mandated “not guilty by reason of insanity” defense is being described as a spurious “excuse” and is under assault, which may lead to death sentences for medically ill persons whose actions are triggered by a severe brain pathology that impairs judgment and distorts reality testing of what is right or wrong.
The devastated families of the victims also are very much on my mind. For them, July 20 will become a dark anniversary of how their loved ones—most of whom were in the prime of life—lost theirs lives while watching a movie called The Dark Knight Rises. Their annual grief on July 20 will for many years reopen the deep wounds inflicted on their souls.
In the future, on July 20 I will no longer be merely inspired by the human brain’s potential for achievements. I now also will remember the potential for despicable and murderous actions when a sickened human brain and its convoluted mind go terribly awry. As an academic investigator dedicated to finding a cure for schizophrenia and related disorders, I regard the tragic events in Aurora on July 20, 2012 as another call to escalate the commitment of researchers to restore normalcy to the blighted brains of those afflicted by this cruel disease, or, better yet, to ultimately discover how to prevent schizophrenia from developing. Only then will the darkness of severe mental illness finally ebb and vanish.
Editor's Note: This editorial was written before Neil Armstrong, the first man to walk on the moon, died on August 25, 2012.
On July 20 of every year, I recall the transcendent wonder I felt on that day in 1969 when the entire world and I were glued to our television sets, witnessing one of the greatest achievements in the history of the human race: American astronauts landing on the moon and beaming pictures of their extraordinary celestial expedition to the billions of earthlings sitting on the edge of their seats.
Every year on July 20, I ponder the supreme and brilliant abilities of the human brain that transformed walking on the moon from an absurd fantasy to a thrilling reality. After becoming a psychiatrist, trained to observe everything through the prism of mind and brain, the moon landing represented the zenith achievement of the divinely evolved human neocortex, especially the prefrontal lobe of President John F. Kennedy, who established that lofty goal, and the advanced brains of thousands of NASA scientists and engineers, who set out to fulfill that towering expectation in less than a decade. That’s why every year on July 20, my abiding faith in the limitless capacities of the human brain to do great things is confirmed and reinforced.
However, this year was different as July 20 took on a much darker meaning. The tragic shooting in Aurora, CO that killed 12 people and wounded 58 others—perpetrated by the bizarre machinations of a graduate school student who had been receiving psychiatric treatment when he suddenly dropped out—rudely reminded me how, like the moon, the human brain has a dark side. A healthy brain that can envision, plan, and execute a magnificent moon landing is capable, when perturbed, of carrying out a dreadfully heinous crime. So henceforth, July 20 always will remind me of the zenith vs nadir dichotomy of the human mind potential in health and disease.
As a longtime academic psychiatrist, I have repeatedly witnessed the tragic disability my patients suffer when the frontal lobe—the most advanced component of their brains— becomes seriously impaired, rendering them dysfunctional. Unless driven by an intense delusion or command hallucinations, patients with schizophrenia rarely commit a murder. However, the media often has linked psychosis with violent crime, whether in movies1 or in news reporting, which has led to public misconception that every person suffering from psychosis is a potential mass murderer. The truth is that the vast majority of murders are committed by nonpsychotic criminals with severe antisocial traits.
Because of the shocking killings in Aurora, July 20 may become an annual reminder that perpetuates the unfair and ignorant notion that mental illness always is associated with horrendous crimes. That would further darken the discriminatory stigma of mental illness and may foster a hatred of and aversion to mentally ill individuals, the vast majority of whom are law-abiding citizens. Even the legally mandated “not guilty by reason of insanity” defense is being described as a spurious “excuse” and is under assault, which may lead to death sentences for medically ill persons whose actions are triggered by a severe brain pathology that impairs judgment and distorts reality testing of what is right or wrong.
The devastated families of the victims also are very much on my mind. For them, July 20 will become a dark anniversary of how their loved ones—most of whom were in the prime of life—lost theirs lives while watching a movie called The Dark Knight Rises. Their annual grief on July 20 will for many years reopen the deep wounds inflicted on their souls.
In the future, on July 20 I will no longer be merely inspired by the human brain’s potential for achievements. I now also will remember the potential for despicable and murderous actions when a sickened human brain and its convoluted mind go terribly awry. As an academic investigator dedicated to finding a cure for schizophrenia and related disorders, I regard the tragic events in Aurora on July 20, 2012 as another call to escalate the commitment of researchers to restore normalcy to the blighted brains of those afflicted by this cruel disease, or, better yet, to ultimately discover how to prevent schizophrenia from developing. Only then will the darkness of severe mental illness finally ebb and vanish.
Editor's Note: This editorial was written before Neil Armstrong, the first man to walk on the moon, died on August 25, 2012.
1. Owen PR. Portrayals of schizophrenia by entertainment media: a content analysis of contemporary movies. Psychiatr Serv. 2012;63(7):655-659.
1. Owen PR. Portrayals of schizophrenia by entertainment media: a content analysis of contemporary movies. Psychiatr Serv. 2012;63(7):655-659.
An evidence-based approach to treating pediatric anxiety disorders
Anxiety disorders are remarkably common among pediatric patients1,2 and are associated with significant morbidity3 and increased risk of suicidality in adolescents.4,5 Effective diagnosis and treatment of pediatric anxiety disorders are critical for reducing psychosocial morbidity,3,6 suicidality, and the risk of secondary mood disorders.7
This article summarizes open-label studies and randomized controlled trials (RCTs) of selective serotonin reuptake inhibitors (SSRIs), selective serotonin-norepinephrine reuptake inhibitors, atypical anxiolytics, and benzodiazepines in children and adolescents with generalized anxiety disorder (GAD), social phobia, separation anxiety disorder, and panic disorder. Although we focus on psychopharmacologic treatments, the best outcomes generally are observed with multimodal treatments that combine psychotherapy and pharmacotherapy.
Generalized anxiety disorder
Researchers have evaluated SSRIs, benzodiazepines, and buspirone in pediatric patients with GAD. In a double-blind, placebo-controlled trial of 22 patients age 5 to 17, sertraline, 50 mg/d, was associated with improvement in Hamilton Anxiety Rating Scale (HAM-A), Clinical Global Impression-Severity (CGI-S), and Clinical Global Impression-Improvement (CGI-I) scores over 9 weeks.8 The Child-Adolescent Anxiety Multimodal Study compared cognitive-behavioral therapy (CBT) to sertraline or sertraline plus CBT in 488 patients age 7 to 17, 78% of whom had GAD.9 Sertraline monotherapy was superior to placebo and not statistically different from CBT, while combination treatment was superior to both monotherapy conditions in improving CGI score. In both trials, sertraline was well tolerated.
One study evaluated fluoxetine, 5 to 40 mg/d, or CBT in 14 youths with GAD; both treatments improved symptoms.10 In a study of 320 GAD patients age 6 to 17, venlafaxine extended-release (XR) initiated at 37.5 mg/d was associated with improved HAM-A scores.11 In general, venlafaxine was well tolerated; adverse effects included increased blood pressure, asthenia, pain, anorexia, somnolence, weight loss, and possibly treatment-emergent suicidal ideation.
Two RCTs of buspirone, 15 to 60 mg/d, that evaluated 559 children and adolescents age 6 to 17 with GAD did not observe significant differences between buspirone and placebo.12 By contrast, 2 open-label studies of youths with anxiety suggested improvement associated with buspirone.12 Treatment-emergent adverse events included nausea, stomachache, and headache.
Clinical trials of benzodiazepines in anxious children and adolescents have yielded mixed results. A 4-week, open-label trial of alprazolam, 0.5 mg to 1.5 mg/d, in 12 adolescents with overanxious disorder—the DSM-III forerunner of GAD—found improvements in anxiety, depression, psychomotor excitation, and hyperactivity, but patients experienced sedation, activation, headache, and nausea.13 However, a double-blind RCT in 30 youths age 8 to 16 found no statistically significant difference between alprazolam and placebo.14 Alprazolam generally was well tolerated; fatigue and dry mouth were reported, but no withdrawal symptoms. Additionally, benzodiazepine use may be associated with tolerance and—in young children—disinhibition.
Social phobia
Researchers have evaluated paroxetine, citalopram, fluoxetine, and venlafaxine for treating social phobia in pediatric patients. In an RCT, 78% of paroxetine-treated patients with social phobia responded compared with 38% for placebo over 16 weeks. Adverse events—including withdrawal symptoms—were twice as likely in patients who received paroxetine. Additionally, 4 paroxetine patients exhibited suicidal ideation vs 0 patients who received placebo.15
In an RCT of 293 children and adolescents age 8 to 17 with social phobia, venlafaxine XR was initiated at 37.5 mg/d and titrated to 112.5 mg/d, 150 mg/d, or 225 mg/d, depending on body weight.16 The venlafaxine group experienced significantly improved anxiety symptoms and the medication generally was well tolerated, although 3 venlafaxine-treated patients developed suicidal ideation compared with 0 in the placebo group.
An RCT compared Social Effectiveness Therapy for Children (SET-C) and fluoxetine, 10 to 40 mg/d, for 139 patients age 7 to 17 with social phobia.17 SET-C is a CBT for children and adolescents that focuses on increasing interpersonal skills and becoming more comfortable in social situations; it involves psychoeducation, social skills training, and exposure exercises. At endpoint, 53% of patients in the SET-C group no longer met diagnostic criteria for social phobia. Fluoxetine was well tolerated; no severe adverse events were reported.
In an open-label study of sertraline (mean dose = 123 mg/d) for 14 young persons with social phobia, 36% of patients responded and 29% partially responded at 8 weeks.18 Adverse events generally were mild and included nausea, diarrhea, and headache. In a 12-week study, 12 pediatric patients with social phobia received citalopram, 10 to 40 mg/d, and eight 15-minute counseling sessions. At endpoint, clinicians rated 83% of patients as much improved or very much improved. The medication generally was well tolerated.19
Separation anxiety disorder
In a 4-week, double-blind crossover pilot study, researchers randomly assigned 15 children age 7 to 13 with separation anxiety disorder to clonazepam, up to 2 mg/d, or placebo.20 There was no significant difference in CGI-I score between clonazepam and placebo. Side effects—including drowsiness, irritability and “oppositional behavior”—were more frequent in patients treated with clonazepam.
Panic disorder
Only 2 open-label studies of SSRIs have been conducted in pediatric patients with panic disorder. The first evaluated the effectiveness and tolerability of fluoxetine, sertraline, or paroxetine over 6 months in 12 patients; 67% no longer met criteria for panic disorder at endpoint.21 In this study, benzodiazepines—including clonazepam and lorazepam—were used in 67% of patients at the start of SSRI treatment. The authors suggested this strategy may be clinically useful for patients with panic disorder.
In the second study, Fairbanks et al22 examined the use of fluoxetine for 6 to 9 weeks in 16 outpatients with mixed anxiety disorders who did not respond to psychotherapy. Patients age ≤12 were given 5 to 40 mg/d and those age ≥13 received 5 to 80 mg/d. Fluoxetine was associated with clinically significant improvement in 3 of the 5 patients who had panic disorder. Although overall fluoxetine was well tolerated, drowsiness, dyssomnia, decreased appetite, nausea, and abdominal pain were the most common side effects. Fluoxetine was not associated with suicidal ideation.
Mixed anxiety disorders
Most trials of pediatric anxiety have evaluated patients with “mixed anxiety disorders” because GAD, social phobia, and separation anxiety disorder are highly comorbid and share diagnostic features (Figure 1).9 An RCT of fluvoxamine, up to 300 mg/d, in 128 pediatric patients with ≥1 anxiety disorders found significant differences in CGI-I and endpoint Pediatric Anxiety Rating Scale (PARS) scores.23 Fluvoxamine was well tolerated but associated with increased motor activity and abdominal discomfort compared with placebo.
Two open-label trials of pediatric patients with mixed anxiety disorders suggested fluoxetine may be beneficial. Fairbanks et al22 documented clinical improvement in 10 of 10 patients with separation anxiety disorder, 8 of 10 with social phobia, 4 of 6 with specific phobia, 3 of 5 with panic disorder, and 1 of 7 with GAD. Birmaher et al24 evaluated 21 pediatric patients with overanxious disorder, social phobia, or separation anxiety who had not responded to psychotherapy and were not depressed; all patients received flexibly-dosed fluoxetine for up to 10 months. Fluoxetine was well tolerated and 81% of patients improved.
Finally, in a 12-week RCT of 74 patients age 7 to 17 with GAD, separation anxiety disorder, and/or social phobia, fluoxetine, 10 to 20 mg/d, was associated with improved scores on the Screen for Anxiety Related Emotional Disorders, PARS, CGI-I, CGI-S, and Children’s Global Assessment Scale.25 A follow-up open-label trial suggested that maintenance treatment is associated with sustained improvement.26
Figure 1: The pediatric anxiety disorders triad: Comorbidity is common
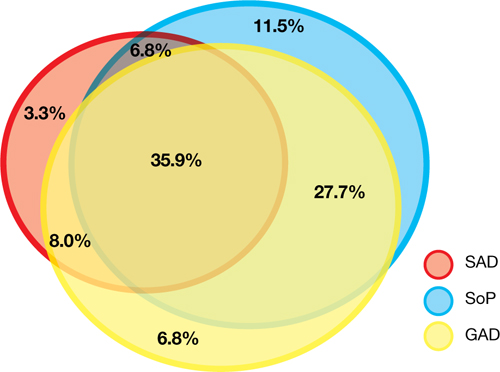
In the Child-Adolescent Multimodal Treatment Study, GAD was the most common disorder; however, GAD, SAD, and SoP were highly comorbid
GAD: generalized anxiety disorder; SAD: separation anxiety disorder; SoP: social phobia
Source: Reference 9
Anxiety disorders with ADHD
Anxiety disorders often are comorbid with attention-deficit/hyperactivity disorder (ADHD). An RCT of patients age 8 to 17 with ADHD and comorbid anxiety found that atomoxetine was associated with improved PARS scores and ADHD symptoms.27 The target dose was 1.2 mg/kg/d. Atomoxetine was well-tolerated; decreased appetite was the only significant adverse event in the treatment group vs placebo.
Multimodal treatment
Although this article reviews evidence for psychopharmacologic treatments, psychotherapeutic treatment of young patients with anxiety disorders has seen significant advances.28 Most psychotherapy studies have evaluated the efficacy of CBT,29-31 although there is evidence for psychodynamic therapy and interpersonal therapy.32 The American Academy of Child & Adolescent Psychiatry recommends a multimodal treatment approach because combination treatment appears to be more effective than monotherapy.8,28,33 Also, clinicians who treat pediatric patients who have an anxiety disorder should evaluate the family’s role on anxiety symptoms and may consider family therapy.
Treatment considerations
Evidence supports the efficacy of sertraline, citalopram, paroxetine, fluvoxamine, fluoxetine, and venlafaxine for treating children and adolescents with anxiety disorders (Figure 2).8,9,11,15,16,23,25 Some practitioners suggest using differing dosing strategies for pediatric anxiety disorders compared with those used to treat adults (Table).34 When considering SSRIs for children and adolescents, keep in mind the “black-box” warning regarding suicidality in these patients. Carefully monitor patients for treatment-emergent suicidality and routinely reassess for the presence and severity of suicidal ideation and suicide risk.
Figure 2: Number needed to treat for SSRIs and SNRIs in pediatric anxiety disorders

GAD: generalized anxiety disorder; RUPP: Research Unit on Pediatric Psychopharmacology; SAD: separation anxiety disorder; SNRI: serotonin-norepinephrine reuptake inhibitor; SoP: social phobia; SSRI: selective serotonin reuptake inhibitorTable
Practical dosing of SSRIs and SNRIs in pediatric patients with anxietya
| Medication | Initial child dose (age <12; mg/d) | Initial adolescent dose (age 12 to 17; mg/d) | Target dose (mg/d) |
|---|---|---|---|
| Citalopram | 5 to 10 | 10 | 20 to 40 |
| Escitalopram | 2.5 to 5 | 5 to 10 | 10 to 20 |
| Fluoxetineb | 10 | 20 | 20 to 40 (children), 40 to 60 (adolescents) |
| Paroxetineb | 5 to 10 | 10 | 20 |
| Sertralinec | 10 to 12.5 | 25 | 150 |
| Venlafaxine | 37.5 | 37.5 | 150 |
| aGeneralized anxiety disorder, social phobia, and separation anxiety disorder bMay consider cytochrome P450 genotyping for 2D6, which may suggest an alternate dosing strategy cSertraline is available in a liquid formulation (20 mg/mL) SNRI: serotonin-norepinephrine reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor Source: Adapted from reference 34 | |||
Related Resources
- Connolly SD, Bernstein GA; Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
- Anxiety and Depression Association of America. www.adaa.org.
- American Academy of Child & Adolescent Psychiatry. www.aacap.org.
Drug Brand Names
- Alprazolam • Xanax
- Atomoxetine • Strattera
- Buspirone • BuSpar
- Citalopram • Celexa
- Clonazepam • Klonopin
- Fluoxetine • Prozac
- Fluvoxamine • Luvox, Luvox CR
- Lorazepam • Ativan
- Paroxetine • Paxil, Paxil CR
- Sertraline • Zoloft
- Venlafaxine • Effexor, Effexor XR
Disclosures
Dr. Strawn has received research support from the American Academy of Child & Adolescent Psychiatry, Eli Lilly and Company, and Shire, and is an employee of the University of Cincinnati, Cincinnati, OH.
Dr. McReynolds was employed by Eli Lilly and Company from 1997 to 2005.
1. Beesdo K, Knappe S, Pine DS. Anxiety and anxiety disorders in children and adolescents: developmental issues and implications for DSM-V. Psychiatr Clin North Am. 2009;32(3):483-524.
2. Beesdo K, Pine DS, Lieb R, et al. Incidence and risk patterns of anxiety and depressive disorders and categorization of generalized anxiety disorder. Arch Gen Psychiatry. 2010;67(1):47-57.
3. Ialongo N, Edelsohn G, Werthamer-Larsson L, et al. The significance of self-reported anxious symptoms in first grade children: prediction to anxious symptoms and adaptive functioning in fifth grade. J Child Psychol Psychiatry. 1995;36(3):427-437.
4. Foley DL, Goldston DB, Costello EJ, et al. Proximal psychiatric risk factors for suicidality in youth: the Great Smoky Mountains Study. Arch Gen Psychiatry. 2006;63(9):1017-1024.
5. Jacobson CM, Muehlenkamp JJ, Miller AL, et al. Psychiatric impairment among adolescents engaging in different types of deliberate self-harm. J Clin Child Adolesc Psychol. 2008;37(2):363-375.
6. Ialongo N, Edelsohn G, Werthamer-Larsson L, et al. The significance of self-reported anxious symptoms in first-grade children. J Abnorm Child Psychol. 1994;22(4):441-455.
7. Pine DS, Cohen P, Gurley D, et al. The risk for early-adulthood anxiety and depressive disorders in adolescents with anxiety and depressive disorders. Arch Gen Psychiatry. 1998;55(1):56-64.
8. Rynn MA, Siqueland L, Rickels K. Placebo-controlled trial of sertraline in the treatment of children with generalized anxiety disorders. Am J Psychiatry. 2001;158(12):2008-2014.
9. Walkup JT, Albano AM, Piacentini J, et al. Cognitive behavioral therapy, sertraline, or a combination in childhood anxiety. N Engl J Med. 2008;359(26):2753-2766.
10. Maslowsky J, Mogg K, Bradley BP, et al. A preliminary investigation of neural correlates of treatment in adolescents with generalized anxiety disorder. J Child Adolesc Psychopharmacol. 2010;20(2):105-111.
11. Rynn MA, Riddle MA, Yeung PP, et al. Efficacy and safety of extended-release venlafaxine in the treatment of generalized anxiety disorder in children and adolescents: two placebo-controlled trials. Am J Psychiatry. 2007;164(2):290-300.
12. BuSpar [package insert] Princeton NJ: Bristol-Myers Squibb; 2010.
13. Simeon JG, Ferguson HB. Alprazolam effects in children with anxiety disorders. Can J Psychiatry. 1987;32(7):570-574.
14. Simeon JG, Ferguson HB, Knott V, et al. Clinical, cognitive, and neurophysiological effects of alprazolam in children and adolescents with overanxious and avoidant disorders. J Am Acad Child Adolesc Psychiatry. 1992;31(1):29-33.
15. Wagner KD, Berard R, Stein MB, et al. A multicenter, randomized, double-blind, placebo-controlled trial of paroxetine in children and adolescents with social anxiety disorder. Arch Gen Psychiatry. 2004;61(11):1153-1162.
16. March JS, Entusah AR, Rynn M, et al. A randomized controlled trial of venlafaxine ER versus placebo in pediatric social anxiety disorder. Biol Psychiatry. 2007;62(10):1149-1154.
17. Beidel DC, Turner SM, Sallee FR, et al. SET-C versus fluoxetine in the treatment of childhood social phobia. J Am Acad Child Adolesc Psychiatry. 2007;46(12):1622-1632.
18. Compton SN, Grant PJ, Chrisman AK, et al. Sertraline in children and adolescents with social anxiety disorder: an open trial. J Am Acad Child Adolesc Psychiatry. 2001;40(5):564-571.
19. Chavira DA, Stein MB. Combined psychoeducation and treatment with selective serotonin reuptake inhibitors for youth with generalized social anxiety disorder. J Child Adolesc Psychopharmacol. 2002;12(1):47-54.
20. Graae F, Milner J, Rizzotto L, et al. Clonazepam in childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 1994;33(3):372-376.
21. Renaud J, Birmaher B, Wassick SC, et al. Use of selective serotonin reuptake inhibitors for the treatment of childhood panic disorder: a pilot study. J Child Adolesc Psychopharmacol. 1999;9(2):73-83.
22. Fairbanks JM, Pine DS, Tancer NK, et al. Open fluoxetine treatment of mixed anxiety disorders in children and adolescents. J Child Adolesc Psychopharmacol. 1997;7(1):17-29.
23. The Research Unit on Pediatric Psychopharmacology Anxiety Study Group. Fluvoxamine for the treatment of anxiety disorders in children and adolescents. N Engl J Med. 2001;344(17):1279-1285.
24. Birmaher B, Waterman GS, Ryan N, et al. Fluoxetine for childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 1994;33(7):993-999.
25. Birmaher B, Axelson DA, Monk K, et al. Fluoxetine for the treatment of childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2003;42(4):415-423.
26. Clark DB, Birmaher B, Axelson D, et al. Fluoxetine for the treatment of childhood anxiety disorders: open-label, long-term extension to a controlled trial. J Am Acad Child Adolesc Psychiatry. 2005;44(12):1263-1270.
27. Geller D, Donnelly C, Lopez F, et al. Atomoxetine treatment for pediatric patients with attention-deficit/hyperactivity disorder with comorbid anxiety disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(9):1119-1127.
28. Connolly SD, Bernstein GA. Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
29. Kendall PC. Treating anxiety disorders in children: results of a randomized clinical trial. J Consult Clin Psychol. 1994;62(1):100-110.
30. Kendall PC, Flannery-Schroeder E, Panichelli-Mindel SM, et al. Therapy for youths with anxiety disorders: a second randomized clinical trial. J Consult Clin Psychol. 1997;65(3):366-380.
31. Reynolds S, Wilson C, Austin J, et al. Effects of psychotherapy for anxiety in children and adolescents: a meta-analytic review. Clin Psychol Rev. 2012;32(4):251-262.
32. Strawn JR, Wehry AM, DelBello MP, et al. Establishing the neurobiologic basis of treatment in children and adolescents with generalized anxiety disorder. Depress Anxiety. 2012;29(4):328-339.
33. Ginsburg GS, Kendall PC, Sakolsky D, et al. Remission after acute treatment in children and adolescents with anxiety disorders: findings from the CAMS. J Consult Clin Psychol. 2011;79(6):806-813.
34. Findling RL, Kowatch RA. How (not) to dose antidepressants and antipsychotics for children. Current Psychiatry. 2007;6(6):79-83.
Anxiety disorders are remarkably common among pediatric patients1,2 and are associated with significant morbidity3 and increased risk of suicidality in adolescents.4,5 Effective diagnosis and treatment of pediatric anxiety disorders are critical for reducing psychosocial morbidity,3,6 suicidality, and the risk of secondary mood disorders.7
This article summarizes open-label studies and randomized controlled trials (RCTs) of selective serotonin reuptake inhibitors (SSRIs), selective serotonin-norepinephrine reuptake inhibitors, atypical anxiolytics, and benzodiazepines in children and adolescents with generalized anxiety disorder (GAD), social phobia, separation anxiety disorder, and panic disorder. Although we focus on psychopharmacologic treatments, the best outcomes generally are observed with multimodal treatments that combine psychotherapy and pharmacotherapy.
Generalized anxiety disorder
Researchers have evaluated SSRIs, benzodiazepines, and buspirone in pediatric patients with GAD. In a double-blind, placebo-controlled trial of 22 patients age 5 to 17, sertraline, 50 mg/d, was associated with improvement in Hamilton Anxiety Rating Scale (HAM-A), Clinical Global Impression-Severity (CGI-S), and Clinical Global Impression-Improvement (CGI-I) scores over 9 weeks.8 The Child-Adolescent Anxiety Multimodal Study compared cognitive-behavioral therapy (CBT) to sertraline or sertraline plus CBT in 488 patients age 7 to 17, 78% of whom had GAD.9 Sertraline monotherapy was superior to placebo and not statistically different from CBT, while combination treatment was superior to both monotherapy conditions in improving CGI score. In both trials, sertraline was well tolerated.
One study evaluated fluoxetine, 5 to 40 mg/d, or CBT in 14 youths with GAD; both treatments improved symptoms.10 In a study of 320 GAD patients age 6 to 17, venlafaxine extended-release (XR) initiated at 37.5 mg/d was associated with improved HAM-A scores.11 In general, venlafaxine was well tolerated; adverse effects included increased blood pressure, asthenia, pain, anorexia, somnolence, weight loss, and possibly treatment-emergent suicidal ideation.
Two RCTs of buspirone, 15 to 60 mg/d, that evaluated 559 children and adolescents age 6 to 17 with GAD did not observe significant differences between buspirone and placebo.12 By contrast, 2 open-label studies of youths with anxiety suggested improvement associated with buspirone.12 Treatment-emergent adverse events included nausea, stomachache, and headache.
Clinical trials of benzodiazepines in anxious children and adolescents have yielded mixed results. A 4-week, open-label trial of alprazolam, 0.5 mg to 1.5 mg/d, in 12 adolescents with overanxious disorder—the DSM-III forerunner of GAD—found improvements in anxiety, depression, psychomotor excitation, and hyperactivity, but patients experienced sedation, activation, headache, and nausea.13 However, a double-blind RCT in 30 youths age 8 to 16 found no statistically significant difference between alprazolam and placebo.14 Alprazolam generally was well tolerated; fatigue and dry mouth were reported, but no withdrawal symptoms. Additionally, benzodiazepine use may be associated with tolerance and—in young children—disinhibition.
Social phobia
Researchers have evaluated paroxetine, citalopram, fluoxetine, and venlafaxine for treating social phobia in pediatric patients. In an RCT, 78% of paroxetine-treated patients with social phobia responded compared with 38% for placebo over 16 weeks. Adverse events—including withdrawal symptoms—were twice as likely in patients who received paroxetine. Additionally, 4 paroxetine patients exhibited suicidal ideation vs 0 patients who received placebo.15
In an RCT of 293 children and adolescents age 8 to 17 with social phobia, venlafaxine XR was initiated at 37.5 mg/d and titrated to 112.5 mg/d, 150 mg/d, or 225 mg/d, depending on body weight.16 The venlafaxine group experienced significantly improved anxiety symptoms and the medication generally was well tolerated, although 3 venlafaxine-treated patients developed suicidal ideation compared with 0 in the placebo group.
An RCT compared Social Effectiveness Therapy for Children (SET-C) and fluoxetine, 10 to 40 mg/d, for 139 patients age 7 to 17 with social phobia.17 SET-C is a CBT for children and adolescents that focuses on increasing interpersonal skills and becoming more comfortable in social situations; it involves psychoeducation, social skills training, and exposure exercises. At endpoint, 53% of patients in the SET-C group no longer met diagnostic criteria for social phobia. Fluoxetine was well tolerated; no severe adverse events were reported.
In an open-label study of sertraline (mean dose = 123 mg/d) for 14 young persons with social phobia, 36% of patients responded and 29% partially responded at 8 weeks.18 Adverse events generally were mild and included nausea, diarrhea, and headache. In a 12-week study, 12 pediatric patients with social phobia received citalopram, 10 to 40 mg/d, and eight 15-minute counseling sessions. At endpoint, clinicians rated 83% of patients as much improved or very much improved. The medication generally was well tolerated.19
Separation anxiety disorder
In a 4-week, double-blind crossover pilot study, researchers randomly assigned 15 children age 7 to 13 with separation anxiety disorder to clonazepam, up to 2 mg/d, or placebo.20 There was no significant difference in CGI-I score between clonazepam and placebo. Side effects—including drowsiness, irritability and “oppositional behavior”—were more frequent in patients treated with clonazepam.
Panic disorder
Only 2 open-label studies of SSRIs have been conducted in pediatric patients with panic disorder. The first evaluated the effectiveness and tolerability of fluoxetine, sertraline, or paroxetine over 6 months in 12 patients; 67% no longer met criteria for panic disorder at endpoint.21 In this study, benzodiazepines—including clonazepam and lorazepam—were used in 67% of patients at the start of SSRI treatment. The authors suggested this strategy may be clinically useful for patients with panic disorder.
In the second study, Fairbanks et al22 examined the use of fluoxetine for 6 to 9 weeks in 16 outpatients with mixed anxiety disorders who did not respond to psychotherapy. Patients age ≤12 were given 5 to 40 mg/d and those age ≥13 received 5 to 80 mg/d. Fluoxetine was associated with clinically significant improvement in 3 of the 5 patients who had panic disorder. Although overall fluoxetine was well tolerated, drowsiness, dyssomnia, decreased appetite, nausea, and abdominal pain were the most common side effects. Fluoxetine was not associated with suicidal ideation.
Mixed anxiety disorders
Most trials of pediatric anxiety have evaluated patients with “mixed anxiety disorders” because GAD, social phobia, and separation anxiety disorder are highly comorbid and share diagnostic features (Figure 1).9 An RCT of fluvoxamine, up to 300 mg/d, in 128 pediatric patients with ≥1 anxiety disorders found significant differences in CGI-I and endpoint Pediatric Anxiety Rating Scale (PARS) scores.23 Fluvoxamine was well tolerated but associated with increased motor activity and abdominal discomfort compared with placebo.
Two open-label trials of pediatric patients with mixed anxiety disorders suggested fluoxetine may be beneficial. Fairbanks et al22 documented clinical improvement in 10 of 10 patients with separation anxiety disorder, 8 of 10 with social phobia, 4 of 6 with specific phobia, 3 of 5 with panic disorder, and 1 of 7 with GAD. Birmaher et al24 evaluated 21 pediatric patients with overanxious disorder, social phobia, or separation anxiety who had not responded to psychotherapy and were not depressed; all patients received flexibly-dosed fluoxetine for up to 10 months. Fluoxetine was well tolerated and 81% of patients improved.
Finally, in a 12-week RCT of 74 patients age 7 to 17 with GAD, separation anxiety disorder, and/or social phobia, fluoxetine, 10 to 20 mg/d, was associated with improved scores on the Screen for Anxiety Related Emotional Disorders, PARS, CGI-I, CGI-S, and Children’s Global Assessment Scale.25 A follow-up open-label trial suggested that maintenance treatment is associated with sustained improvement.26
Figure 1: The pediatric anxiety disorders triad: Comorbidity is common
In the Child-Adolescent Multimodal Treatment Study, GAD was the most common disorder; however, GAD, SAD, and SoP were highly comorbid
GAD: generalized anxiety disorder; SAD: separation anxiety disorder; SoP: social phobia
Source: Reference 9
Anxiety disorders with ADHD
Anxiety disorders often are comorbid with attention-deficit/hyperactivity disorder (ADHD). An RCT of patients age 8 to 17 with ADHD and comorbid anxiety found that atomoxetine was associated with improved PARS scores and ADHD symptoms.27 The target dose was 1.2 mg/kg/d. Atomoxetine was well-tolerated; decreased appetite was the only significant adverse event in the treatment group vs placebo.
Multimodal treatment
Although this article reviews evidence for psychopharmacologic treatments, psychotherapeutic treatment of young patients with anxiety disorders has seen significant advances.28 Most psychotherapy studies have evaluated the efficacy of CBT,29-31 although there is evidence for psychodynamic therapy and interpersonal therapy.32 The American Academy of Child & Adolescent Psychiatry recommends a multimodal treatment approach because combination treatment appears to be more effective than monotherapy.8,28,33 Also, clinicians who treat pediatric patients who have an anxiety disorder should evaluate the family’s role on anxiety symptoms and may consider family therapy.
Treatment considerations
Evidence supports the efficacy of sertraline, citalopram, paroxetine, fluvoxamine, fluoxetine, and venlafaxine for treating children and adolescents with anxiety disorders (Figure 2).8,9,11,15,16,23,25 Some practitioners suggest using differing dosing strategies for pediatric anxiety disorders compared with those used to treat adults (Table).34 When considering SSRIs for children and adolescents, keep in mind the “black-box” warning regarding suicidality in these patients. Carefully monitor patients for treatment-emergent suicidality and routinely reassess for the presence and severity of suicidal ideation and suicide risk.
Figure 2: Number needed to treat for SSRIs and SNRIs in pediatric anxiety disorders
GAD: generalized anxiety disorder; RUPP: Research Unit on Pediatric Psychopharmacology; SAD: separation anxiety disorder; SNRI: serotonin-norepinephrine reuptake inhibitor; SoP: social phobia; SSRI: selective serotonin reuptake inhibitorTable
Practical dosing of SSRIs and SNRIs in pediatric patients with anxietya
| Medication | Initial child dose (age <12; mg/d) | Initial adolescent dose (age 12 to 17; mg/d) | Target dose (mg/d) |
|---|---|---|---|
| Citalopram | 5 to 10 | 10 | 20 to 40 |
| Escitalopram | 2.5 to 5 | 5 to 10 | 10 to 20 |
| Fluoxetineb | 10 | 20 | 20 to 40 (children), 40 to 60 (adolescents) |
| Paroxetineb | 5 to 10 | 10 | 20 |
| Sertralinec | 10 to 12.5 | 25 | 150 |
| Venlafaxine | 37.5 | 37.5 | 150 |
| aGeneralized anxiety disorder, social phobia, and separation anxiety disorder bMay consider cytochrome P450 genotyping for 2D6, which may suggest an alternate dosing strategy cSertraline is available in a liquid formulation (20 mg/mL) SNRI: serotonin-norepinephrine reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor Source: Adapted from reference 34 | |||
Related Resources
- Connolly SD, Bernstein GA; Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
- Anxiety and Depression Association of America. www.adaa.org.
- American Academy of Child & Adolescent Psychiatry. www.aacap.org.
Drug Brand Names
- Alprazolam • Xanax
- Atomoxetine • Strattera
- Buspirone • BuSpar
- Citalopram • Celexa
- Clonazepam • Klonopin
- Fluoxetine • Prozac
- Fluvoxamine • Luvox, Luvox CR
- Lorazepam • Ativan
- Paroxetine • Paxil, Paxil CR
- Sertraline • Zoloft
- Venlafaxine • Effexor, Effexor XR
Disclosures
Dr. Strawn has received research support from the American Academy of Child & Adolescent Psychiatry, Eli Lilly and Company, and Shire, and is an employee of the University of Cincinnati, Cincinnati, OH.
Dr. McReynolds was employed by Eli Lilly and Company from 1997 to 2005.
Anxiety disorders are remarkably common among pediatric patients1,2 and are associated with significant morbidity3 and increased risk of suicidality in adolescents.4,5 Effective diagnosis and treatment of pediatric anxiety disorders are critical for reducing psychosocial morbidity,3,6 suicidality, and the risk of secondary mood disorders.7
This article summarizes open-label studies and randomized controlled trials (RCTs) of selective serotonin reuptake inhibitors (SSRIs), selective serotonin-norepinephrine reuptake inhibitors, atypical anxiolytics, and benzodiazepines in children and adolescents with generalized anxiety disorder (GAD), social phobia, separation anxiety disorder, and panic disorder. Although we focus on psychopharmacologic treatments, the best outcomes generally are observed with multimodal treatments that combine psychotherapy and pharmacotherapy.
Generalized anxiety disorder
Researchers have evaluated SSRIs, benzodiazepines, and buspirone in pediatric patients with GAD. In a double-blind, placebo-controlled trial of 22 patients age 5 to 17, sertraline, 50 mg/d, was associated with improvement in Hamilton Anxiety Rating Scale (HAM-A), Clinical Global Impression-Severity (CGI-S), and Clinical Global Impression-Improvement (CGI-I) scores over 9 weeks.8 The Child-Adolescent Anxiety Multimodal Study compared cognitive-behavioral therapy (CBT) to sertraline or sertraline plus CBT in 488 patients age 7 to 17, 78% of whom had GAD.9 Sertraline monotherapy was superior to placebo and not statistically different from CBT, while combination treatment was superior to both monotherapy conditions in improving CGI score. In both trials, sertraline was well tolerated.
One study evaluated fluoxetine, 5 to 40 mg/d, or CBT in 14 youths with GAD; both treatments improved symptoms.10 In a study of 320 GAD patients age 6 to 17, venlafaxine extended-release (XR) initiated at 37.5 mg/d was associated with improved HAM-A scores.11 In general, venlafaxine was well tolerated; adverse effects included increased blood pressure, asthenia, pain, anorexia, somnolence, weight loss, and possibly treatment-emergent suicidal ideation.
Two RCTs of buspirone, 15 to 60 mg/d, that evaluated 559 children and adolescents age 6 to 17 with GAD did not observe significant differences between buspirone and placebo.12 By contrast, 2 open-label studies of youths with anxiety suggested improvement associated with buspirone.12 Treatment-emergent adverse events included nausea, stomachache, and headache.
Clinical trials of benzodiazepines in anxious children and adolescents have yielded mixed results. A 4-week, open-label trial of alprazolam, 0.5 mg to 1.5 mg/d, in 12 adolescents with overanxious disorder—the DSM-III forerunner of GAD—found improvements in anxiety, depression, psychomotor excitation, and hyperactivity, but patients experienced sedation, activation, headache, and nausea.13 However, a double-blind RCT in 30 youths age 8 to 16 found no statistically significant difference between alprazolam and placebo.14 Alprazolam generally was well tolerated; fatigue and dry mouth were reported, but no withdrawal symptoms. Additionally, benzodiazepine use may be associated with tolerance and—in young children—disinhibition.
Social phobia
Researchers have evaluated paroxetine, citalopram, fluoxetine, and venlafaxine for treating social phobia in pediatric patients. In an RCT, 78% of paroxetine-treated patients with social phobia responded compared with 38% for placebo over 16 weeks. Adverse events—including withdrawal symptoms—were twice as likely in patients who received paroxetine. Additionally, 4 paroxetine patients exhibited suicidal ideation vs 0 patients who received placebo.15
In an RCT of 293 children and adolescents age 8 to 17 with social phobia, venlafaxine XR was initiated at 37.5 mg/d and titrated to 112.5 mg/d, 150 mg/d, or 225 mg/d, depending on body weight.16 The venlafaxine group experienced significantly improved anxiety symptoms and the medication generally was well tolerated, although 3 venlafaxine-treated patients developed suicidal ideation compared with 0 in the placebo group.
An RCT compared Social Effectiveness Therapy for Children (SET-C) and fluoxetine, 10 to 40 mg/d, for 139 patients age 7 to 17 with social phobia.17 SET-C is a CBT for children and adolescents that focuses on increasing interpersonal skills and becoming more comfortable in social situations; it involves psychoeducation, social skills training, and exposure exercises. At endpoint, 53% of patients in the SET-C group no longer met diagnostic criteria for social phobia. Fluoxetine was well tolerated; no severe adverse events were reported.
In an open-label study of sertraline (mean dose = 123 mg/d) for 14 young persons with social phobia, 36% of patients responded and 29% partially responded at 8 weeks.18 Adverse events generally were mild and included nausea, diarrhea, and headache. In a 12-week study, 12 pediatric patients with social phobia received citalopram, 10 to 40 mg/d, and eight 15-minute counseling sessions. At endpoint, clinicians rated 83% of patients as much improved or very much improved. The medication generally was well tolerated.19
Separation anxiety disorder
In a 4-week, double-blind crossover pilot study, researchers randomly assigned 15 children age 7 to 13 with separation anxiety disorder to clonazepam, up to 2 mg/d, or placebo.20 There was no significant difference in CGI-I score between clonazepam and placebo. Side effects—including drowsiness, irritability and “oppositional behavior”—were more frequent in patients treated with clonazepam.
Panic disorder
Only 2 open-label studies of SSRIs have been conducted in pediatric patients with panic disorder. The first evaluated the effectiveness and tolerability of fluoxetine, sertraline, or paroxetine over 6 months in 12 patients; 67% no longer met criteria for panic disorder at endpoint.21 In this study, benzodiazepines—including clonazepam and lorazepam—were used in 67% of patients at the start of SSRI treatment. The authors suggested this strategy may be clinically useful for patients with panic disorder.
In the second study, Fairbanks et al22 examined the use of fluoxetine for 6 to 9 weeks in 16 outpatients with mixed anxiety disorders who did not respond to psychotherapy. Patients age ≤12 were given 5 to 40 mg/d and those age ≥13 received 5 to 80 mg/d. Fluoxetine was associated with clinically significant improvement in 3 of the 5 patients who had panic disorder. Although overall fluoxetine was well tolerated, drowsiness, dyssomnia, decreased appetite, nausea, and abdominal pain were the most common side effects. Fluoxetine was not associated with suicidal ideation.
Mixed anxiety disorders
Most trials of pediatric anxiety have evaluated patients with “mixed anxiety disorders” because GAD, social phobia, and separation anxiety disorder are highly comorbid and share diagnostic features (Figure 1).9 An RCT of fluvoxamine, up to 300 mg/d, in 128 pediatric patients with ≥1 anxiety disorders found significant differences in CGI-I and endpoint Pediatric Anxiety Rating Scale (PARS) scores.23 Fluvoxamine was well tolerated but associated with increased motor activity and abdominal discomfort compared with placebo.
Two open-label trials of pediatric patients with mixed anxiety disorders suggested fluoxetine may be beneficial. Fairbanks et al22 documented clinical improvement in 10 of 10 patients with separation anxiety disorder, 8 of 10 with social phobia, 4 of 6 with specific phobia, 3 of 5 with panic disorder, and 1 of 7 with GAD. Birmaher et al24 evaluated 21 pediatric patients with overanxious disorder, social phobia, or separation anxiety who had not responded to psychotherapy and were not depressed; all patients received flexibly-dosed fluoxetine for up to 10 months. Fluoxetine was well tolerated and 81% of patients improved.
Finally, in a 12-week RCT of 74 patients age 7 to 17 with GAD, separation anxiety disorder, and/or social phobia, fluoxetine, 10 to 20 mg/d, was associated with improved scores on the Screen for Anxiety Related Emotional Disorders, PARS, CGI-I, CGI-S, and Children’s Global Assessment Scale.25 A follow-up open-label trial suggested that maintenance treatment is associated with sustained improvement.26
Figure 1: The pediatric anxiety disorders triad: Comorbidity is common
In the Child-Adolescent Multimodal Treatment Study, GAD was the most common disorder; however, GAD, SAD, and SoP were highly comorbid
GAD: generalized anxiety disorder; SAD: separation anxiety disorder; SoP: social phobia
Source: Reference 9
Anxiety disorders with ADHD
Anxiety disorders often are comorbid with attention-deficit/hyperactivity disorder (ADHD). An RCT of patients age 8 to 17 with ADHD and comorbid anxiety found that atomoxetine was associated with improved PARS scores and ADHD symptoms.27 The target dose was 1.2 mg/kg/d. Atomoxetine was well-tolerated; decreased appetite was the only significant adverse event in the treatment group vs placebo.
Multimodal treatment
Although this article reviews evidence for psychopharmacologic treatments, psychotherapeutic treatment of young patients with anxiety disorders has seen significant advances.28 Most psychotherapy studies have evaluated the efficacy of CBT,29-31 although there is evidence for psychodynamic therapy and interpersonal therapy.32 The American Academy of Child & Adolescent Psychiatry recommends a multimodal treatment approach because combination treatment appears to be more effective than monotherapy.8,28,33 Also, clinicians who treat pediatric patients who have an anxiety disorder should evaluate the family’s role on anxiety symptoms and may consider family therapy.
Treatment considerations
Evidence supports the efficacy of sertraline, citalopram, paroxetine, fluvoxamine, fluoxetine, and venlafaxine for treating children and adolescents with anxiety disorders (Figure 2).8,9,11,15,16,23,25 Some practitioners suggest using differing dosing strategies for pediatric anxiety disorders compared with those used to treat adults (Table).34 When considering SSRIs for children and adolescents, keep in mind the “black-box” warning regarding suicidality in these patients. Carefully monitor patients for treatment-emergent suicidality and routinely reassess for the presence and severity of suicidal ideation and suicide risk.
Figure 2: Number needed to treat for SSRIs and SNRIs in pediatric anxiety disorders
GAD: generalized anxiety disorder; RUPP: Research Unit on Pediatric Psychopharmacology; SAD: separation anxiety disorder; SNRI: serotonin-norepinephrine reuptake inhibitor; SoP: social phobia; SSRI: selective serotonin reuptake inhibitorTable
Practical dosing of SSRIs and SNRIs in pediatric patients with anxietya
| Medication | Initial child dose (age <12; mg/d) | Initial adolescent dose (age 12 to 17; mg/d) | Target dose (mg/d) |
|---|---|---|---|
| Citalopram | 5 to 10 | 10 | 20 to 40 |
| Escitalopram | 2.5 to 5 | 5 to 10 | 10 to 20 |
| Fluoxetineb | 10 | 20 | 20 to 40 (children), 40 to 60 (adolescents) |
| Paroxetineb | 5 to 10 | 10 | 20 |
| Sertralinec | 10 to 12.5 | 25 | 150 |
| Venlafaxine | 37.5 | 37.5 | 150 |
| aGeneralized anxiety disorder, social phobia, and separation anxiety disorder bMay consider cytochrome P450 genotyping for 2D6, which may suggest an alternate dosing strategy cSertraline is available in a liquid formulation (20 mg/mL) SNRI: serotonin-norepinephrine reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor Source: Adapted from reference 34 | |||
Related Resources
- Connolly SD, Bernstein GA; Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
- Anxiety and Depression Association of America. www.adaa.org.
- American Academy of Child & Adolescent Psychiatry. www.aacap.org.
Drug Brand Names
- Alprazolam • Xanax
- Atomoxetine • Strattera
- Buspirone • BuSpar
- Citalopram • Celexa
- Clonazepam • Klonopin
- Fluoxetine • Prozac
- Fluvoxamine • Luvox, Luvox CR
- Lorazepam • Ativan
- Paroxetine • Paxil, Paxil CR
- Sertraline • Zoloft
- Venlafaxine • Effexor, Effexor XR
Disclosures
Dr. Strawn has received research support from the American Academy of Child & Adolescent Psychiatry, Eli Lilly and Company, and Shire, and is an employee of the University of Cincinnati, Cincinnati, OH.
Dr. McReynolds was employed by Eli Lilly and Company from 1997 to 2005.
1. Beesdo K, Knappe S, Pine DS. Anxiety and anxiety disorders in children and adolescents: developmental issues and implications for DSM-V. Psychiatr Clin North Am. 2009;32(3):483-524.
2. Beesdo K, Pine DS, Lieb R, et al. Incidence and risk patterns of anxiety and depressive disorders and categorization of generalized anxiety disorder. Arch Gen Psychiatry. 2010;67(1):47-57.
3. Ialongo N, Edelsohn G, Werthamer-Larsson L, et al. The significance of self-reported anxious symptoms in first grade children: prediction to anxious symptoms and adaptive functioning in fifth grade. J Child Psychol Psychiatry. 1995;36(3):427-437.
4. Foley DL, Goldston DB, Costello EJ, et al. Proximal psychiatric risk factors for suicidality in youth: the Great Smoky Mountains Study. Arch Gen Psychiatry. 2006;63(9):1017-1024.
5. Jacobson CM, Muehlenkamp JJ, Miller AL, et al. Psychiatric impairment among adolescents engaging in different types of deliberate self-harm. J Clin Child Adolesc Psychol. 2008;37(2):363-375.
6. Ialongo N, Edelsohn G, Werthamer-Larsson L, et al. The significance of self-reported anxious symptoms in first-grade children. J Abnorm Child Psychol. 1994;22(4):441-455.
7. Pine DS, Cohen P, Gurley D, et al. The risk for early-adulthood anxiety and depressive disorders in adolescents with anxiety and depressive disorders. Arch Gen Psychiatry. 1998;55(1):56-64.
8. Rynn MA, Siqueland L, Rickels K. Placebo-controlled trial of sertraline in the treatment of children with generalized anxiety disorders. Am J Psychiatry. 2001;158(12):2008-2014.
9. Walkup JT, Albano AM, Piacentini J, et al. Cognitive behavioral therapy, sertraline, or a combination in childhood anxiety. N Engl J Med. 2008;359(26):2753-2766.
10. Maslowsky J, Mogg K, Bradley BP, et al. A preliminary investigation of neural correlates of treatment in adolescents with generalized anxiety disorder. J Child Adolesc Psychopharmacol. 2010;20(2):105-111.
11. Rynn MA, Riddle MA, Yeung PP, et al. Efficacy and safety of extended-release venlafaxine in the treatment of generalized anxiety disorder in children and adolescents: two placebo-controlled trials. Am J Psychiatry. 2007;164(2):290-300.
12. BuSpar [package insert] Princeton NJ: Bristol-Myers Squibb; 2010.
13. Simeon JG, Ferguson HB. Alprazolam effects in children with anxiety disorders. Can J Psychiatry. 1987;32(7):570-574.
14. Simeon JG, Ferguson HB, Knott V, et al. Clinical, cognitive, and neurophysiological effects of alprazolam in children and adolescents with overanxious and avoidant disorders. J Am Acad Child Adolesc Psychiatry. 1992;31(1):29-33.
15. Wagner KD, Berard R, Stein MB, et al. A multicenter, randomized, double-blind, placebo-controlled trial of paroxetine in children and adolescents with social anxiety disorder. Arch Gen Psychiatry. 2004;61(11):1153-1162.
16. March JS, Entusah AR, Rynn M, et al. A randomized controlled trial of venlafaxine ER versus placebo in pediatric social anxiety disorder. Biol Psychiatry. 2007;62(10):1149-1154.
17. Beidel DC, Turner SM, Sallee FR, et al. SET-C versus fluoxetine in the treatment of childhood social phobia. J Am Acad Child Adolesc Psychiatry. 2007;46(12):1622-1632.
18. Compton SN, Grant PJ, Chrisman AK, et al. Sertraline in children and adolescents with social anxiety disorder: an open trial. J Am Acad Child Adolesc Psychiatry. 2001;40(5):564-571.
19. Chavira DA, Stein MB. Combined psychoeducation and treatment with selective serotonin reuptake inhibitors for youth with generalized social anxiety disorder. J Child Adolesc Psychopharmacol. 2002;12(1):47-54.
20. Graae F, Milner J, Rizzotto L, et al. Clonazepam in childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 1994;33(3):372-376.
21. Renaud J, Birmaher B, Wassick SC, et al. Use of selective serotonin reuptake inhibitors for the treatment of childhood panic disorder: a pilot study. J Child Adolesc Psychopharmacol. 1999;9(2):73-83.
22. Fairbanks JM, Pine DS, Tancer NK, et al. Open fluoxetine treatment of mixed anxiety disorders in children and adolescents. J Child Adolesc Psychopharmacol. 1997;7(1):17-29.
23. The Research Unit on Pediatric Psychopharmacology Anxiety Study Group. Fluvoxamine for the treatment of anxiety disorders in children and adolescents. N Engl J Med. 2001;344(17):1279-1285.
24. Birmaher B, Waterman GS, Ryan N, et al. Fluoxetine for childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 1994;33(7):993-999.
25. Birmaher B, Axelson DA, Monk K, et al. Fluoxetine for the treatment of childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2003;42(4):415-423.
26. Clark DB, Birmaher B, Axelson D, et al. Fluoxetine for the treatment of childhood anxiety disorders: open-label, long-term extension to a controlled trial. J Am Acad Child Adolesc Psychiatry. 2005;44(12):1263-1270.
27. Geller D, Donnelly C, Lopez F, et al. Atomoxetine treatment for pediatric patients with attention-deficit/hyperactivity disorder with comorbid anxiety disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(9):1119-1127.
28. Connolly SD, Bernstein GA. Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
29. Kendall PC. Treating anxiety disorders in children: results of a randomized clinical trial. J Consult Clin Psychol. 1994;62(1):100-110.
30. Kendall PC, Flannery-Schroeder E, Panichelli-Mindel SM, et al. Therapy for youths with anxiety disorders: a second randomized clinical trial. J Consult Clin Psychol. 1997;65(3):366-380.
31. Reynolds S, Wilson C, Austin J, et al. Effects of psychotherapy for anxiety in children and adolescents: a meta-analytic review. Clin Psychol Rev. 2012;32(4):251-262.
32. Strawn JR, Wehry AM, DelBello MP, et al. Establishing the neurobiologic basis of treatment in children and adolescents with generalized anxiety disorder. Depress Anxiety. 2012;29(4):328-339.
33. Ginsburg GS, Kendall PC, Sakolsky D, et al. Remission after acute treatment in children and adolescents with anxiety disorders: findings from the CAMS. J Consult Clin Psychol. 2011;79(6):806-813.
34. Findling RL, Kowatch RA. How (not) to dose antidepressants and antipsychotics for children. Current Psychiatry. 2007;6(6):79-83.
1. Beesdo K, Knappe S, Pine DS. Anxiety and anxiety disorders in children and adolescents: developmental issues and implications for DSM-V. Psychiatr Clin North Am. 2009;32(3):483-524.
2. Beesdo K, Pine DS, Lieb R, et al. Incidence and risk patterns of anxiety and depressive disorders and categorization of generalized anxiety disorder. Arch Gen Psychiatry. 2010;67(1):47-57.
3. Ialongo N, Edelsohn G, Werthamer-Larsson L, et al. The significance of self-reported anxious symptoms in first grade children: prediction to anxious symptoms and adaptive functioning in fifth grade. J Child Psychol Psychiatry. 1995;36(3):427-437.
4. Foley DL, Goldston DB, Costello EJ, et al. Proximal psychiatric risk factors for suicidality in youth: the Great Smoky Mountains Study. Arch Gen Psychiatry. 2006;63(9):1017-1024.
5. Jacobson CM, Muehlenkamp JJ, Miller AL, et al. Psychiatric impairment among adolescents engaging in different types of deliberate self-harm. J Clin Child Adolesc Psychol. 2008;37(2):363-375.
6. Ialongo N, Edelsohn G, Werthamer-Larsson L, et al. The significance of self-reported anxious symptoms in first-grade children. J Abnorm Child Psychol. 1994;22(4):441-455.
7. Pine DS, Cohen P, Gurley D, et al. The risk for early-adulthood anxiety and depressive disorders in adolescents with anxiety and depressive disorders. Arch Gen Psychiatry. 1998;55(1):56-64.
8. Rynn MA, Siqueland L, Rickels K. Placebo-controlled trial of sertraline in the treatment of children with generalized anxiety disorders. Am J Psychiatry. 2001;158(12):2008-2014.
9. Walkup JT, Albano AM, Piacentini J, et al. Cognitive behavioral therapy, sertraline, or a combination in childhood anxiety. N Engl J Med. 2008;359(26):2753-2766.
10. Maslowsky J, Mogg K, Bradley BP, et al. A preliminary investigation of neural correlates of treatment in adolescents with generalized anxiety disorder. J Child Adolesc Psychopharmacol. 2010;20(2):105-111.
11. Rynn MA, Riddle MA, Yeung PP, et al. Efficacy and safety of extended-release venlafaxine in the treatment of generalized anxiety disorder in children and adolescents: two placebo-controlled trials. Am J Psychiatry. 2007;164(2):290-300.
12. BuSpar [package insert] Princeton NJ: Bristol-Myers Squibb; 2010.
13. Simeon JG, Ferguson HB. Alprazolam effects in children with anxiety disorders. Can J Psychiatry. 1987;32(7):570-574.
14. Simeon JG, Ferguson HB, Knott V, et al. Clinical, cognitive, and neurophysiological effects of alprazolam in children and adolescents with overanxious and avoidant disorders. J Am Acad Child Adolesc Psychiatry. 1992;31(1):29-33.
15. Wagner KD, Berard R, Stein MB, et al. A multicenter, randomized, double-blind, placebo-controlled trial of paroxetine in children and adolescents with social anxiety disorder. Arch Gen Psychiatry. 2004;61(11):1153-1162.
16. March JS, Entusah AR, Rynn M, et al. A randomized controlled trial of venlafaxine ER versus placebo in pediatric social anxiety disorder. Biol Psychiatry. 2007;62(10):1149-1154.
17. Beidel DC, Turner SM, Sallee FR, et al. SET-C versus fluoxetine in the treatment of childhood social phobia. J Am Acad Child Adolesc Psychiatry. 2007;46(12):1622-1632.
18. Compton SN, Grant PJ, Chrisman AK, et al. Sertraline in children and adolescents with social anxiety disorder: an open trial. J Am Acad Child Adolesc Psychiatry. 2001;40(5):564-571.
19. Chavira DA, Stein MB. Combined psychoeducation and treatment with selective serotonin reuptake inhibitors for youth with generalized social anxiety disorder. J Child Adolesc Psychopharmacol. 2002;12(1):47-54.
20. Graae F, Milner J, Rizzotto L, et al. Clonazepam in childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 1994;33(3):372-376.
21. Renaud J, Birmaher B, Wassick SC, et al. Use of selective serotonin reuptake inhibitors for the treatment of childhood panic disorder: a pilot study. J Child Adolesc Psychopharmacol. 1999;9(2):73-83.
22. Fairbanks JM, Pine DS, Tancer NK, et al. Open fluoxetine treatment of mixed anxiety disorders in children and adolescents. J Child Adolesc Psychopharmacol. 1997;7(1):17-29.
23. The Research Unit on Pediatric Psychopharmacology Anxiety Study Group. Fluvoxamine for the treatment of anxiety disorders in children and adolescents. N Engl J Med. 2001;344(17):1279-1285.
24. Birmaher B, Waterman GS, Ryan N, et al. Fluoxetine for childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 1994;33(7):993-999.
25. Birmaher B, Axelson DA, Monk K, et al. Fluoxetine for the treatment of childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2003;42(4):415-423.
26. Clark DB, Birmaher B, Axelson D, et al. Fluoxetine for the treatment of childhood anxiety disorders: open-label, long-term extension to a controlled trial. J Am Acad Child Adolesc Psychiatry. 2005;44(12):1263-1270.
27. Geller D, Donnelly C, Lopez F, et al. Atomoxetine treatment for pediatric patients with attention-deficit/hyperactivity disorder with comorbid anxiety disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(9):1119-1127.
28. Connolly SD, Bernstein GA. Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
29. Kendall PC. Treating anxiety disorders in children: results of a randomized clinical trial. J Consult Clin Psychol. 1994;62(1):100-110.
30. Kendall PC, Flannery-Schroeder E, Panichelli-Mindel SM, et al. Therapy for youths with anxiety disorders: a second randomized clinical trial. J Consult Clin Psychol. 1997;65(3):366-380.
31. Reynolds S, Wilson C, Austin J, et al. Effects of psychotherapy for anxiety in children and adolescents: a meta-analytic review. Clin Psychol Rev. 2012;32(4):251-262.
32. Strawn JR, Wehry AM, DelBello MP, et al. Establishing the neurobiologic basis of treatment in children and adolescents with generalized anxiety disorder. Depress Anxiety. 2012;29(4):328-339.
33. Ginsburg GS, Kendall PC, Sakolsky D, et al. Remission after acute treatment in children and adolescents with anxiety disorders: findings from the CAMS. J Consult Clin Psychol. 2011;79(6):806-813.
34. Findling RL, Kowatch RA. How (not) to dose antidepressants and antipsychotics for children. Current Psychiatry. 2007;6(6):79-83.
Reducing CYP450 drug interactions caused by antidepressants
Most psychiatrists are aware that some antidepressants can cause clinically significant drug interactions, especially through the cytochrome P450 (CYP450) hepatic enzyme system. Antidepressants’ potential for drug interactions is especially important for patients who take >1 other medication, including cardiovascular agents.1
Unfortunately, drug interactions can be difficult to remember and are commonly missed. One strategy to help remember a list of antidepressants with a relatively low potential for CYP450 drug interactions is to use the mnemonic Various Medicines Definitely Commingle Very Easily (VMDCVE) to recall venlafaxine, mirtazapine, desvenlafaxine,2 citalopram, vilazodone,3 and escitalopram. The order in which these medications are listed does not indicate a preference for any of the 6 antidepressants. Bupropion and duloxetine are not included in this list because they are moderately potent inhibitors of the 2D6 isoenzyme.4,5
A few caveats
There are some important caveats in using this mnemonic:
- None of these antidepressants is completely devoid of effects on the CYP450 system. However, compared with the antidepressants included in this mnemonic, fluoxetine, paroxetine, fluvoxamine, duloxetine, bupropion, and nefazodone are more likely to have clinically significant effects on CYP450.4,5
- Although sertraline has a lower potential for CYP450-mediated drug interactions at low doses, it is not included in this mnemonic because it may have greater effects on 2D6 inhibition in some patients, especially at higher doses, such as ≥150 mg/d.5 Also, sertraline may significantly increase lamotrigine levels through a different mechanism: inhibition of uridine 5’-diphosphate glucuronosyltransferase 1A4.4
- Antidepressants also may be the substrates for CYP450 drug interactions caused by other medications.
- This mnemonic refers only to CYP450-mediated drug interactions. Antidepressants included in this mnemonic may have a high potential for drug interactions mediated by displacement from carrier proteins— eg, with digoxin or warfarin.
- Pharmacodynamic drug interactions also are possible—eg, serotonin syndrome as a result of combining a selective serotonin reuptake inhibitor with another serotonergic medication.
To remain vigilant for drug-drug interactions, routinely use a drug interaction software, in addition to this mnemonic.
Disclosure
Dr. Mago receives grant/research, support from Bristol-Myers Squibb, Eli Lilly and Company, and NARSAD.
1. Williams S, Wynn G, Cozza K, et al. Cardiovascular medications. Psychosomatics. 2007;48(6):537-547.
2. Nichols AI, Tourian KA, Tse SY, et al. Desvenlafaxine for major depressive disorder: incremental clinical benefits from a second-generation serotonin-norepinephrine reuptake inhibitor. Expert Opin Drug Metab Toxicol. 2010;6(12):1565-1574.
3. Laughren TP, Gobburu J, Temple RJ, et al. Vilazodone: clinical basis for the US Food and Drug Administration’s approval of a new antidepressant. J Clin Psychiatry. 2011;72(9):1166-1173.
4. Sandson NB, Armstrong SC, Cozza KL. An overview of psychotropic drug-drug interactions. Psychosomatics. 2005;46(5):464-494.
5. Spina E, Santoro V, D’Arrigo C. Clinically relevant pharmacokinetic drug interactions with second-generation antidepressants: an update. Clin Ther. 2008;30(7):1206-1227.
Most psychiatrists are aware that some antidepressants can cause clinically significant drug interactions, especially through the cytochrome P450 (CYP450) hepatic enzyme system. Antidepressants’ potential for drug interactions is especially important for patients who take >1 other medication, including cardiovascular agents.1
Unfortunately, drug interactions can be difficult to remember and are commonly missed. One strategy to help remember a list of antidepressants with a relatively low potential for CYP450 drug interactions is to use the mnemonic Various Medicines Definitely Commingle Very Easily (VMDCVE) to recall venlafaxine, mirtazapine, desvenlafaxine,2 citalopram, vilazodone,3 and escitalopram. The order in which these medications are listed does not indicate a preference for any of the 6 antidepressants. Bupropion and duloxetine are not included in this list because they are moderately potent inhibitors of the 2D6 isoenzyme.4,5
A few caveats
There are some important caveats in using this mnemonic:
- None of these antidepressants is completely devoid of effects on the CYP450 system. However, compared with the antidepressants included in this mnemonic, fluoxetine, paroxetine, fluvoxamine, duloxetine, bupropion, and nefazodone are more likely to have clinically significant effects on CYP450.4,5
- Although sertraline has a lower potential for CYP450-mediated drug interactions at low doses, it is not included in this mnemonic because it may have greater effects on 2D6 inhibition in some patients, especially at higher doses, such as ≥150 mg/d.5 Also, sertraline may significantly increase lamotrigine levels through a different mechanism: inhibition of uridine 5’-diphosphate glucuronosyltransferase 1A4.4
- Antidepressants also may be the substrates for CYP450 drug interactions caused by other medications.
- This mnemonic refers only to CYP450-mediated drug interactions. Antidepressants included in this mnemonic may have a high potential for drug interactions mediated by displacement from carrier proteins— eg, with digoxin or warfarin.
- Pharmacodynamic drug interactions also are possible—eg, serotonin syndrome as a result of combining a selective serotonin reuptake inhibitor with another serotonergic medication.
To remain vigilant for drug-drug interactions, routinely use a drug interaction software, in addition to this mnemonic.
Disclosure
Dr. Mago receives grant/research, support from Bristol-Myers Squibb, Eli Lilly and Company, and NARSAD.
Most psychiatrists are aware that some antidepressants can cause clinically significant drug interactions, especially through the cytochrome P450 (CYP450) hepatic enzyme system. Antidepressants’ potential for drug interactions is especially important for patients who take >1 other medication, including cardiovascular agents.1
Unfortunately, drug interactions can be difficult to remember and are commonly missed. One strategy to help remember a list of antidepressants with a relatively low potential for CYP450 drug interactions is to use the mnemonic Various Medicines Definitely Commingle Very Easily (VMDCVE) to recall venlafaxine, mirtazapine, desvenlafaxine,2 citalopram, vilazodone,3 and escitalopram. The order in which these medications are listed does not indicate a preference for any of the 6 antidepressants. Bupropion and duloxetine are not included in this list because they are moderately potent inhibitors of the 2D6 isoenzyme.4,5
A few caveats
There are some important caveats in using this mnemonic:
- None of these antidepressants is completely devoid of effects on the CYP450 system. However, compared with the antidepressants included in this mnemonic, fluoxetine, paroxetine, fluvoxamine, duloxetine, bupropion, and nefazodone are more likely to have clinically significant effects on CYP450.4,5
- Although sertraline has a lower potential for CYP450-mediated drug interactions at low doses, it is not included in this mnemonic because it may have greater effects on 2D6 inhibition in some patients, especially at higher doses, such as ≥150 mg/d.5 Also, sertraline may significantly increase lamotrigine levels through a different mechanism: inhibition of uridine 5’-diphosphate glucuronosyltransferase 1A4.4
- Antidepressants also may be the substrates for CYP450 drug interactions caused by other medications.
- This mnemonic refers only to CYP450-mediated drug interactions. Antidepressants included in this mnemonic may have a high potential for drug interactions mediated by displacement from carrier proteins— eg, with digoxin or warfarin.
- Pharmacodynamic drug interactions also are possible—eg, serotonin syndrome as a result of combining a selective serotonin reuptake inhibitor with another serotonergic medication.
To remain vigilant for drug-drug interactions, routinely use a drug interaction software, in addition to this mnemonic.
Disclosure
Dr. Mago receives grant/research, support from Bristol-Myers Squibb, Eli Lilly and Company, and NARSAD.
1. Williams S, Wynn G, Cozza K, et al. Cardiovascular medications. Psychosomatics. 2007;48(6):537-547.
2. Nichols AI, Tourian KA, Tse SY, et al. Desvenlafaxine for major depressive disorder: incremental clinical benefits from a second-generation serotonin-norepinephrine reuptake inhibitor. Expert Opin Drug Metab Toxicol. 2010;6(12):1565-1574.
3. Laughren TP, Gobburu J, Temple RJ, et al. Vilazodone: clinical basis for the US Food and Drug Administration’s approval of a new antidepressant. J Clin Psychiatry. 2011;72(9):1166-1173.
4. Sandson NB, Armstrong SC, Cozza KL. An overview of psychotropic drug-drug interactions. Psychosomatics. 2005;46(5):464-494.
5. Spina E, Santoro V, D’Arrigo C. Clinically relevant pharmacokinetic drug interactions with second-generation antidepressants: an update. Clin Ther. 2008;30(7):1206-1227.
1. Williams S, Wynn G, Cozza K, et al. Cardiovascular medications. Psychosomatics. 2007;48(6):537-547.
2. Nichols AI, Tourian KA, Tse SY, et al. Desvenlafaxine for major depressive disorder: incremental clinical benefits from a second-generation serotonin-norepinephrine reuptake inhibitor. Expert Opin Drug Metab Toxicol. 2010;6(12):1565-1574.
3. Laughren TP, Gobburu J, Temple RJ, et al. Vilazodone: clinical basis for the US Food and Drug Administration’s approval of a new antidepressant. J Clin Psychiatry. 2011;72(9):1166-1173.
4. Sandson NB, Armstrong SC, Cozza KL. An overview of psychotropic drug-drug interactions. Psychosomatics. 2005;46(5):464-494.
5. Spina E, Santoro V, D’Arrigo C. Clinically relevant pharmacokinetic drug interactions with second-generation antidepressants: an update. Clin Ther. 2008;30(7):1206-1227.