User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Should you attend a plaintiff’s deposition?
A psychiatrist named in a malpractice suit may doubt the need to attend the deposition of the plaintiff and his hired experts. In a time when many psychiatrists are handling busy private practices, you may be tempted to skip the plaintiff’s deposition because often the law does not require you to attend.
However, based on our forensic psychiatry, expert witness, and risk management experience, we highly recommend that psychiatrists involved in a malpractice lawsuit attend plaintiff’s depositions for several reasons.
Counteract countertransference
One of the most often encountered ingredients of a malpractice lawsuit is negative transference toward the defendant psychiatrist and/or negative countertransference by the psychiatrist toward the plaintiff.1 Attending the deposition gives you the opportunity to identify and analyze these reactions, consider how they could impair your objectivity, and allow you and your attorney to put together the best defense.2
Identify errors
Listening to depositions lets you identify errors, misunderstandings, misinterpretations, or distortions of facts in the plaintiff’s allegations or his experts’ testimony and supply your attorney fuel for an effective cross-examination.2–4
Make your presence known
Sometimes your physical presence during the deposition might prevent any misstatements or false allegations.
Assess your case
Attending a plaintiff’s deposition is an excellent opportunity to see and assess the case as a whole while deepening your and your attorney’s understanding of the malpractice lawsuit.2,5
Discuss your decision to attend the deposition with your attorney. If you choose not to attend, be sure to carefully read the transcripts of all depositions and use this information when preparing for your deposition.
Although attending the plaintiff’s deposition is optional, your presence during a malpractice trial is mandatory. Your expertise is most valuable when your attorney cross-examines witnesses. Your presence in the courtroom is necessary to establish your credibility, professionalism, and personality. In most trials jurors will assess the defendant psychiatrist, and not showing up could damage your case.
1. Malmquist CP, Notman MT. Psychiatrist-patient boundary issues following treatment termination. Am J Psychiatry. 2001;158(7):1010-1018.
2. Meadow W. Evidence-based expert testimony. Clin Perinatol. 2005;32(1):251-275, ix.
3. Bettman JW. A lexicon for the expert witness and defendant. Surv Ophthalmol. 1988;32(6):433-434.
4. Critelli N. Head injury—cervical strain—carpal tunnel syndrome—a videotaped evidence deposition of plaintiff’s neurosurgeon—direct and cross-examination. Med Trial Tech Q. 1982;29(1):114-136.
5. Epstein JI. Pathologists and the judicial process: how to avoid it. Am J Surg Pathol. 2001;25(4):527-537.
A psychiatrist named in a malpractice suit may doubt the need to attend the deposition of the plaintiff and his hired experts. In a time when many psychiatrists are handling busy private practices, you may be tempted to skip the plaintiff’s deposition because often the law does not require you to attend.
However, based on our forensic psychiatry, expert witness, and risk management experience, we highly recommend that psychiatrists involved in a malpractice lawsuit attend plaintiff’s depositions for several reasons.
Counteract countertransference
One of the most often encountered ingredients of a malpractice lawsuit is negative transference toward the defendant psychiatrist and/or negative countertransference by the psychiatrist toward the plaintiff.1 Attending the deposition gives you the opportunity to identify and analyze these reactions, consider how they could impair your objectivity, and allow you and your attorney to put together the best defense.2
Identify errors
Listening to depositions lets you identify errors, misunderstandings, misinterpretations, or distortions of facts in the plaintiff’s allegations or his experts’ testimony and supply your attorney fuel for an effective cross-examination.2–4
Make your presence known
Sometimes your physical presence during the deposition might prevent any misstatements or false allegations.
Assess your case
Attending a plaintiff’s deposition is an excellent opportunity to see and assess the case as a whole while deepening your and your attorney’s understanding of the malpractice lawsuit.2,5
Discuss your decision to attend the deposition with your attorney. If you choose not to attend, be sure to carefully read the transcripts of all depositions and use this information when preparing for your deposition.
Although attending the plaintiff’s deposition is optional, your presence during a malpractice trial is mandatory. Your expertise is most valuable when your attorney cross-examines witnesses. Your presence in the courtroom is necessary to establish your credibility, professionalism, and personality. In most trials jurors will assess the defendant psychiatrist, and not showing up could damage your case.
A psychiatrist named in a malpractice suit may doubt the need to attend the deposition of the plaintiff and his hired experts. In a time when many psychiatrists are handling busy private practices, you may be tempted to skip the plaintiff’s deposition because often the law does not require you to attend.
However, based on our forensic psychiatry, expert witness, and risk management experience, we highly recommend that psychiatrists involved in a malpractice lawsuit attend plaintiff’s depositions for several reasons.
Counteract countertransference
One of the most often encountered ingredients of a malpractice lawsuit is negative transference toward the defendant psychiatrist and/or negative countertransference by the psychiatrist toward the plaintiff.1 Attending the deposition gives you the opportunity to identify and analyze these reactions, consider how they could impair your objectivity, and allow you and your attorney to put together the best defense.2
Identify errors
Listening to depositions lets you identify errors, misunderstandings, misinterpretations, or distortions of facts in the plaintiff’s allegations or his experts’ testimony and supply your attorney fuel for an effective cross-examination.2–4
Make your presence known
Sometimes your physical presence during the deposition might prevent any misstatements or false allegations.
Assess your case
Attending a plaintiff’s deposition is an excellent opportunity to see and assess the case as a whole while deepening your and your attorney’s understanding of the malpractice lawsuit.2,5
Discuss your decision to attend the deposition with your attorney. If you choose not to attend, be sure to carefully read the transcripts of all depositions and use this information when preparing for your deposition.
Although attending the plaintiff’s deposition is optional, your presence during a malpractice trial is mandatory. Your expertise is most valuable when your attorney cross-examines witnesses. Your presence in the courtroom is necessary to establish your credibility, professionalism, and personality. In most trials jurors will assess the defendant psychiatrist, and not showing up could damage your case.
1. Malmquist CP, Notman MT. Psychiatrist-patient boundary issues following treatment termination. Am J Psychiatry. 2001;158(7):1010-1018.
2. Meadow W. Evidence-based expert testimony. Clin Perinatol. 2005;32(1):251-275, ix.
3. Bettman JW. A lexicon for the expert witness and defendant. Surv Ophthalmol. 1988;32(6):433-434.
4. Critelli N. Head injury—cervical strain—carpal tunnel syndrome—a videotaped evidence deposition of plaintiff’s neurosurgeon—direct and cross-examination. Med Trial Tech Q. 1982;29(1):114-136.
5. Epstein JI. Pathologists and the judicial process: how to avoid it. Am J Surg Pathol. 2001;25(4):527-537.
1. Malmquist CP, Notman MT. Psychiatrist-patient boundary issues following treatment termination. Am J Psychiatry. 2001;158(7):1010-1018.
2. Meadow W. Evidence-based expert testimony. Clin Perinatol. 2005;32(1):251-275, ix.
3. Bettman JW. A lexicon for the expert witness and defendant. Surv Ophthalmol. 1988;32(6):433-434.
4. Critelli N. Head injury—cervical strain—carpal tunnel syndrome—a videotaped evidence deposition of plaintiff’s neurosurgeon—direct and cross-examination. Med Trial Tech Q. 1982;29(1):114-136.
5. Epstein JI. Pathologists and the judicial process: how to avoid it. Am J Surg Pathol. 2001;25(4):527-537.
Immunization update: How to protect your at-risk patients
Principal Source: Advisory Committee on Immunization Practices. Recommended adult immunization schedule: United States, October 2007-September 2008. Ann Intern Med. 2007;147:725-729.—Discussant: Daniel Goldsmith, MD
Dr. Goldsmith is associate director, internal medicine residency program, Capital Health System, Trenton, NJ.
- Recommend hepatitis A and B vaccination for psychiatric patients who abuse substances or engage in high-risk sexual behaviors.
- Tobacco use and subsequent chronic pulmonary disease—common among psychiatric populations—is an indication for annual influenza vaccination.
- All women ≤26 are eligible to receive the human papilloma virus vaccine.
- Immunity testing is required for hepatitis A and B and varicella vaccines.
Psychiatric patients who use drugs, alcohol, or tobacco and those who engage in high-risk sexual behaviors can be protected from acquiring viral infections such as hepatitis A and B, influenza, and human papillomavirus (HPV). The recently updated adult immunization schedule (Table)1,2 from the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices (ACIP) gives mental health professionals the opportunity to recognize risk and refer patients for vaccinations as part of preventive care. (see Related Resources) for a link to the complete CDC vaccination recommendations.
Hepatitis A and B. Substance abuse and high-risk sexual behaviors contribute to the high prevalence of comorbid alcohol-related liver disease and viral hepatitis among psychiatric patients. Individuals with chronic liver disease—regardless of its cause—should be screened for hepatitis A and B infection and, if negative, offered the appropriate vaccination series. Acute viral hepatitis in patients with pre-existing chronic hepatitis from any cause, such as alcohol abuse or hepatitis C, is associated with severe hepatic dysfunction and liver failure.3
Hepatitis B screening and vaccination is recommended for psychiatric populations that include clients of substance abuse treatment centers and institutions and daycare facilities for developmental disabilities, as well as IV drug users. Anyone who uses illegal drugs—injectable or noninjectable—should be vaccinated for hepatitis A.
Only individuals susceptible to hepatitis A and B should be vaccinated.3 To determine immune status for hepatitis B, serum tests for hepatitis B surface antigen (HepBsAg), hepatitis B surface antibody IgG (HepBsAb), and hepatitis B core antibody IgG (HepBcAb) are necessary to differentiate among patients who:
- are susceptible to infection
- had an infection that cleared
- have chronic active infection
- already have been vaccinated.
The hepatitis A IgG serum test determines a patient’s hepatitis A immune status. A negative result shows no previous infection, and the patient is eligible for vaccination. A positive result indicates previous infection meaning vaccination has no benefit.3
Influenza. Tobacco use and subsequent chronic pulmonary disease is an indication for annual influenza vaccination.4 Most individuals will receive the injectable, inactivated vaccine. The intranasal, live attenuated vaccine is reserved for nonpregnant adults age ≤49 without high-risk medical conditions and who are not in close contact with immunocompromised persons.1
Pneumococcal and influenza vaccinations also are recommended for persons with chronic liver disease.1 Consider recommending influenza, pneumococcal, varicella, and hepatitis B vaccinations for psychiatric patients in long-term care facilities.1
HPV. Cervical cancer is highly associated with HPV, a sexually transmitted organism, and the HPV vaccine can effectively prevent infection and subsequent neoplasia. All women age ≤26 are eligible for the vaccine. Women with evidence of HPV infection—such as abnormal Pap smear, genital warts, or a positive HPV DNA test—are still eligible to receive the HPV vaccine because several viral strains cause disease.1 Because mental health providers may treat otherwise medically healthy young people, including those who engage in high-risk behaviors, psychiatrists have an opportunity to refer for vaccinations individuals who may not consistently utilize primary care.
Immunity screening considerations. Vaccines for HPV, influenza, and pneumonia are recommended regardless of evidence of immunity or prior infection. Hepatitis A and B vaccines require a history of never having had the illness or laboratory evidence of a lack of immunity.
Table
Updated CDC adult vaccination recommendations
| Vaccination | Changes in recommendations |
|---|---|
| Human papilloma virus | New recombinant vaccine (2007): quadrivalent, 3-dose series Indicated for all women age ≤26 years |
| Herpes zoster | New live attenuated vaccine (2006): single dose Indicated for immunocompetent adults age ≥60 |
| Acellular pertussis | New vaccine (2006) may substitute for tetanus and diphtheria booster |
| Influenza | New indications: patients with aspiration risk or pregnancy during flu season |
| Hepatitis B | Broader wording: all sexually-active persons not in long-term mutually monogamous relationships |
| Mumps, measles, and rubella | New recommendation: second dose for health care workers as result of recent mumps outbreaks |
| Varicella | Broader indication: all immunocompetent adults without immunity to varicella |
| Various live attenuated vaccines | Recommendations for HIV-infected individuals split by CD4+ T lymphocyte count of |
| HIV: human immunodeficiency virus; CD4: cluster of differentiation 4; CDC: Centers for Disease Control and Prevention | |
| Source: Reference 1,2 | |
- Immunization Action Coalition: Vaccine information for health care professionals. www.immunize.org.
- Centers for Disease Control and Prevention. Adult immunization schedule. www.cdc.gov/vaccines/recs/schedules/adult-schedule.htm.
Disclosure
Dr. Goldsmith reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Advisory Committee on Immunization Practices. Recommended adult immunization schedule: United States, October 2007-September 2008. Ann Intern Med. 2007;147:725-729.
2. Poland GA, Schaffner W. Adult immunization guidelines: a patient safety and quality-of-care issue. Ann Intern Med. 2007;147:735-737.
3. Lau DT, Hewlett AT. Screening for hepatitis A and B antibodies in patients with chronic liver disease. Am J Med. 2005;118(suppl 10A):28S-33S.
4. Arcavi L, Benowitz NL. Cigarette smoking and infection. Arch Intern Med. 2004;164(20):2206-2216.
Principal Source: Advisory Committee on Immunization Practices. Recommended adult immunization schedule: United States, October 2007-September 2008. Ann Intern Med. 2007;147:725-729.—Discussant: Daniel Goldsmith, MD
Dr. Goldsmith is associate director, internal medicine residency program, Capital Health System, Trenton, NJ.
- Recommend hepatitis A and B vaccination for psychiatric patients who abuse substances or engage in high-risk sexual behaviors.
- Tobacco use and subsequent chronic pulmonary disease—common among psychiatric populations—is an indication for annual influenza vaccination.
- All women ≤26 are eligible to receive the human papilloma virus vaccine.
- Immunity testing is required for hepatitis A and B and varicella vaccines.
Psychiatric patients who use drugs, alcohol, or tobacco and those who engage in high-risk sexual behaviors can be protected from acquiring viral infections such as hepatitis A and B, influenza, and human papillomavirus (HPV). The recently updated adult immunization schedule (Table)1,2 from the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices (ACIP) gives mental health professionals the opportunity to recognize risk and refer patients for vaccinations as part of preventive care. (see Related Resources) for a link to the complete CDC vaccination recommendations.
Hepatitis A and B. Substance abuse and high-risk sexual behaviors contribute to the high prevalence of comorbid alcohol-related liver disease and viral hepatitis among psychiatric patients. Individuals with chronic liver disease—regardless of its cause—should be screened for hepatitis A and B infection and, if negative, offered the appropriate vaccination series. Acute viral hepatitis in patients with pre-existing chronic hepatitis from any cause, such as alcohol abuse or hepatitis C, is associated with severe hepatic dysfunction and liver failure.3
Hepatitis B screening and vaccination is recommended for psychiatric populations that include clients of substance abuse treatment centers and institutions and daycare facilities for developmental disabilities, as well as IV drug users. Anyone who uses illegal drugs—injectable or noninjectable—should be vaccinated for hepatitis A.
Only individuals susceptible to hepatitis A and B should be vaccinated.3 To determine immune status for hepatitis B, serum tests for hepatitis B surface antigen (HepBsAg), hepatitis B surface antibody IgG (HepBsAb), and hepatitis B core antibody IgG (HepBcAb) are necessary to differentiate among patients who:
- are susceptible to infection
- had an infection that cleared
- have chronic active infection
- already have been vaccinated.
The hepatitis A IgG serum test determines a patient’s hepatitis A immune status. A negative result shows no previous infection, and the patient is eligible for vaccination. A positive result indicates previous infection meaning vaccination has no benefit.3
Influenza. Tobacco use and subsequent chronic pulmonary disease is an indication for annual influenza vaccination.4 Most individuals will receive the injectable, inactivated vaccine. The intranasal, live attenuated vaccine is reserved for nonpregnant adults age ≤49 without high-risk medical conditions and who are not in close contact with immunocompromised persons.1
Pneumococcal and influenza vaccinations also are recommended for persons with chronic liver disease.1 Consider recommending influenza, pneumococcal, varicella, and hepatitis B vaccinations for psychiatric patients in long-term care facilities.1
HPV. Cervical cancer is highly associated with HPV, a sexually transmitted organism, and the HPV vaccine can effectively prevent infection and subsequent neoplasia. All women age ≤26 are eligible for the vaccine. Women with evidence of HPV infection—such as abnormal Pap smear, genital warts, or a positive HPV DNA test—are still eligible to receive the HPV vaccine because several viral strains cause disease.1 Because mental health providers may treat otherwise medically healthy young people, including those who engage in high-risk behaviors, psychiatrists have an opportunity to refer for vaccinations individuals who may not consistently utilize primary care.
Immunity screening considerations. Vaccines for HPV, influenza, and pneumonia are recommended regardless of evidence of immunity or prior infection. Hepatitis A and B vaccines require a history of never having had the illness or laboratory evidence of a lack of immunity.
Table
Updated CDC adult vaccination recommendations
| Vaccination | Changes in recommendations |
|---|---|
| Human papilloma virus | New recombinant vaccine (2007): quadrivalent, 3-dose series Indicated for all women age ≤26 years |
| Herpes zoster | New live attenuated vaccine (2006): single dose Indicated for immunocompetent adults age ≥60 |
| Acellular pertussis | New vaccine (2006) may substitute for tetanus and diphtheria booster |
| Influenza | New indications: patients with aspiration risk or pregnancy during flu season |
| Hepatitis B | Broader wording: all sexually-active persons not in long-term mutually monogamous relationships |
| Mumps, measles, and rubella | New recommendation: second dose for health care workers as result of recent mumps outbreaks |
| Varicella | Broader indication: all immunocompetent adults without immunity to varicella |
| Various live attenuated vaccines | Recommendations for HIV-infected individuals split by CD4+ T lymphocyte count of |
| HIV: human immunodeficiency virus; CD4: cluster of differentiation 4; CDC: Centers for Disease Control and Prevention | |
| Source: Reference 1,2 | |
- Immunization Action Coalition: Vaccine information for health care professionals. www.immunize.org.
- Centers for Disease Control and Prevention. Adult immunization schedule. www.cdc.gov/vaccines/recs/schedules/adult-schedule.htm.
Disclosure
Dr. Goldsmith reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Principal Source: Advisory Committee on Immunization Practices. Recommended adult immunization schedule: United States, October 2007-September 2008. Ann Intern Med. 2007;147:725-729.—Discussant: Daniel Goldsmith, MD
Dr. Goldsmith is associate director, internal medicine residency program, Capital Health System, Trenton, NJ.
- Recommend hepatitis A and B vaccination for psychiatric patients who abuse substances or engage in high-risk sexual behaviors.
- Tobacco use and subsequent chronic pulmonary disease—common among psychiatric populations—is an indication for annual influenza vaccination.
- All women ≤26 are eligible to receive the human papilloma virus vaccine.
- Immunity testing is required for hepatitis A and B and varicella vaccines.
Psychiatric patients who use drugs, alcohol, or tobacco and those who engage in high-risk sexual behaviors can be protected from acquiring viral infections such as hepatitis A and B, influenza, and human papillomavirus (HPV). The recently updated adult immunization schedule (Table)1,2 from the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices (ACIP) gives mental health professionals the opportunity to recognize risk and refer patients for vaccinations as part of preventive care. (see Related Resources) for a link to the complete CDC vaccination recommendations.
Hepatitis A and B. Substance abuse and high-risk sexual behaviors contribute to the high prevalence of comorbid alcohol-related liver disease and viral hepatitis among psychiatric patients. Individuals with chronic liver disease—regardless of its cause—should be screened for hepatitis A and B infection and, if negative, offered the appropriate vaccination series. Acute viral hepatitis in patients with pre-existing chronic hepatitis from any cause, such as alcohol abuse or hepatitis C, is associated with severe hepatic dysfunction and liver failure.3
Hepatitis B screening and vaccination is recommended for psychiatric populations that include clients of substance abuse treatment centers and institutions and daycare facilities for developmental disabilities, as well as IV drug users. Anyone who uses illegal drugs—injectable or noninjectable—should be vaccinated for hepatitis A.
Only individuals susceptible to hepatitis A and B should be vaccinated.3 To determine immune status for hepatitis B, serum tests for hepatitis B surface antigen (HepBsAg), hepatitis B surface antibody IgG (HepBsAb), and hepatitis B core antibody IgG (HepBcAb) are necessary to differentiate among patients who:
- are susceptible to infection
- had an infection that cleared
- have chronic active infection
- already have been vaccinated.
The hepatitis A IgG serum test determines a patient’s hepatitis A immune status. A negative result shows no previous infection, and the patient is eligible for vaccination. A positive result indicates previous infection meaning vaccination has no benefit.3
Influenza. Tobacco use and subsequent chronic pulmonary disease is an indication for annual influenza vaccination.4 Most individuals will receive the injectable, inactivated vaccine. The intranasal, live attenuated vaccine is reserved for nonpregnant adults age ≤49 without high-risk medical conditions and who are not in close contact with immunocompromised persons.1
Pneumococcal and influenza vaccinations also are recommended for persons with chronic liver disease.1 Consider recommending influenza, pneumococcal, varicella, and hepatitis B vaccinations for psychiatric patients in long-term care facilities.1
HPV. Cervical cancer is highly associated with HPV, a sexually transmitted organism, and the HPV vaccine can effectively prevent infection and subsequent neoplasia. All women age ≤26 are eligible for the vaccine. Women with evidence of HPV infection—such as abnormal Pap smear, genital warts, or a positive HPV DNA test—are still eligible to receive the HPV vaccine because several viral strains cause disease.1 Because mental health providers may treat otherwise medically healthy young people, including those who engage in high-risk behaviors, psychiatrists have an opportunity to refer for vaccinations individuals who may not consistently utilize primary care.
Immunity screening considerations. Vaccines for HPV, influenza, and pneumonia are recommended regardless of evidence of immunity or prior infection. Hepatitis A and B vaccines require a history of never having had the illness or laboratory evidence of a lack of immunity.
Table
Updated CDC adult vaccination recommendations
| Vaccination | Changes in recommendations |
|---|---|
| Human papilloma virus | New recombinant vaccine (2007): quadrivalent, 3-dose series Indicated for all women age ≤26 years |
| Herpes zoster | New live attenuated vaccine (2006): single dose Indicated for immunocompetent adults age ≥60 |
| Acellular pertussis | New vaccine (2006) may substitute for tetanus and diphtheria booster |
| Influenza | New indications: patients with aspiration risk or pregnancy during flu season |
| Hepatitis B | Broader wording: all sexually-active persons not in long-term mutually monogamous relationships |
| Mumps, measles, and rubella | New recommendation: second dose for health care workers as result of recent mumps outbreaks |
| Varicella | Broader indication: all immunocompetent adults without immunity to varicella |
| Various live attenuated vaccines | Recommendations for HIV-infected individuals split by CD4+ T lymphocyte count of |
| HIV: human immunodeficiency virus; CD4: cluster of differentiation 4; CDC: Centers for Disease Control and Prevention | |
| Source: Reference 1,2 | |
- Immunization Action Coalition: Vaccine information for health care professionals. www.immunize.org.
- Centers for Disease Control and Prevention. Adult immunization schedule. www.cdc.gov/vaccines/recs/schedules/adult-schedule.htm.
Disclosure
Dr. Goldsmith reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Advisory Committee on Immunization Practices. Recommended adult immunization schedule: United States, October 2007-September 2008. Ann Intern Med. 2007;147:725-729.
2. Poland GA, Schaffner W. Adult immunization guidelines: a patient safety and quality-of-care issue. Ann Intern Med. 2007;147:735-737.
3. Lau DT, Hewlett AT. Screening for hepatitis A and B antibodies in patients with chronic liver disease. Am J Med. 2005;118(suppl 10A):28S-33S.
4. Arcavi L, Benowitz NL. Cigarette smoking and infection. Arch Intern Med. 2004;164(20):2206-2216.
1. Advisory Committee on Immunization Practices. Recommended adult immunization schedule: United States, October 2007-September 2008. Ann Intern Med. 2007;147:725-729.
2. Poland GA, Schaffner W. Adult immunization guidelines: a patient safety and quality-of-care issue. Ann Intern Med. 2007;147:735-737.
3. Lau DT, Hewlett AT. Screening for hepatitis A and B antibodies in patients with chronic liver disease. Am J Med. 2005;118(suppl 10A):28S-33S.
4. Arcavi L, Benowitz NL. Cigarette smoking and infection. Arch Intern Med. 2004;164(20):2206-2216.
Treat dementia holistically
The authors of “Antipsychotics in dementia: Beyond ‘black-box’ warnings” (Current Psychiatry, June 2008) comment that the list of drugs being taken by Mrs. B is revealing for reasons that, unfortunately, are not rare. If the reasons were rare, this likely would be a much shorter article. Despite repeatedly being catheterized, Mrs. B has bladder distention. She also has fecal impaction. She is said to be getting one-to-one care, so why isn’t staff aware of “input/output” issues? If they were aware, did they communicate this to the treating psychiatrist? It is not surprising that Mrs. B became agitated.
More disturbing is that the care facility obtained informed consent at admission. This is not so much an issue of authority as of having a family member or proxy decision-maker in the loop with a “big picture” perspective.
There may be instances when atypical antipsychotic drugs are indicated. However, my sense is that these drugs have the effect of lowering the volume on a TV set; it’s still turned on.
Samuel Lyons
Alexandria, VA
To comment on articles in this issue or other topics, send letters in care of Erica Vonderheid, Current Psychiatry, 110 Summit Avenue, Montvale, NJ 07645, [email protected] or click here.
The authors of “Antipsychotics in dementia: Beyond ‘black-box’ warnings” (Current Psychiatry, June 2008) comment that the list of drugs being taken by Mrs. B is revealing for reasons that, unfortunately, are not rare. If the reasons were rare, this likely would be a much shorter article. Despite repeatedly being catheterized, Mrs. B has bladder distention. She also has fecal impaction. She is said to be getting one-to-one care, so why isn’t staff aware of “input/output” issues? If they were aware, did they communicate this to the treating psychiatrist? It is not surprising that Mrs. B became agitated.
More disturbing is that the care facility obtained informed consent at admission. This is not so much an issue of authority as of having a family member or proxy decision-maker in the loop with a “big picture” perspective.
There may be instances when atypical antipsychotic drugs are indicated. However, my sense is that these drugs have the effect of lowering the volume on a TV set; it’s still turned on.
Samuel Lyons
Alexandria, VA
The authors of “Antipsychotics in dementia: Beyond ‘black-box’ warnings” (Current Psychiatry, June 2008) comment that the list of drugs being taken by Mrs. B is revealing for reasons that, unfortunately, are not rare. If the reasons were rare, this likely would be a much shorter article. Despite repeatedly being catheterized, Mrs. B has bladder distention. She also has fecal impaction. She is said to be getting one-to-one care, so why isn’t staff aware of “input/output” issues? If they were aware, did they communicate this to the treating psychiatrist? It is not surprising that Mrs. B became agitated.
More disturbing is that the care facility obtained informed consent at admission. This is not so much an issue of authority as of having a family member or proxy decision-maker in the loop with a “big picture” perspective.
There may be instances when atypical antipsychotic drugs are indicated. However, my sense is that these drugs have the effect of lowering the volume on a TV set; it’s still turned on.
Samuel Lyons
Alexandria, VA
To comment on articles in this issue or other topics, send letters in care of Erica Vonderheid, Current Psychiatry, 110 Summit Avenue, Montvale, NJ 07645, [email protected] or click here.
To comment on articles in this issue or other topics, send letters in care of Erica Vonderheid, Current Psychiatry, 110 Summit Avenue, Montvale, NJ 07645, [email protected] or click here.
Mnemonic possession
I was surprised to see my TRAUMA mnemonic featured in the article “Mnemonics in a mnutshell: 32 aids to psychiatric diagnosis” (Current Psychiatry, October 2008) and attributed to the article by Dr. Khouzam. I created my first mnemonic—TRAMA—for posttraumatic stress disorder (PTSD) in 1992. When the clinically significant distress/impaired functioning criterion was included in DSM-IV, I added the “U” for “unable.” I first presented a copyrighted version of TRAUMA in 1994 during a lecture to medical students at Columbia University. Since then I have presented it on numerous occasions.
The authors also did not include my mnemonics for subcriteria corresponding to the 3 symptom clusters of PTSD:
R3D2 (think Star Wars and add an “R”) stands for:
- Recollections, Recurring, and Reactivity (physiological) in response to cues of the traumatic event
- Dreams (distressing) and Distress (psychological).
AFRAID equals:
- Avoid thoughts, feelings, conversations, people, places, or activities associated with the trauma
- Foreshortened future
- Recall (inability to)
- Affect (restricted)
- Interest (diminished)
- Detachment.
SCARE represents:
- Sleep (difficulty falling or staying)
- Concentration (difficulty)
- Anger (outbursts or irritability)
- Really vigilant
- Exaggerated startle response.
Finally, to accurately quote DSM-IV or DSM-IV-TR, the symptoms you listed in the TRAUMA mnemonic need to persist for “more than 1 month” instead of a “month or more.”
Joseph C. Napoli, MD, DFAPA
Assistant clinical professor of psychiatry
Columbia University
New York, NY
Drs. Caplan and Stern Respond
Although we made a good-faith effort to find the original sources of mnemonics included in our article, we were aware that lore and oral history might not allow us to properly cite the contributions of these innovators. In this case, because to the best of our memory neither of the authors have attended a lecture by Dr. Napoli, our awareness of the TRAUMA mnemonic originated from the cited published article.
Since publication of our article, we also have heard from William Falk, MD, at Massachusetts General Hospital informing us that he created DIG FAST to help remember the criteria for mania. We offer apologies to those ingenious clinicians for not our citing their role in the genesis of these mnemonics, and we are grateful that they have enhanced the accuracy of the information we provided.
We also could say: Some Oversights Shall Occasionally Result in Remorse and Yearning (SO SORRY).
Jason P. Caplan, MD
Assistant clinical professor of psychiatry
Creighton University School of Medicine
Omaha, NE
Theodore A. Stern, MD
Professor of psychiatry
Harvard Medical School
Boston, MA
To comment on articles in this issue or other topics, send letters in care of Erica Vonderheid, Current Psychiatry, 110 Summit Avenue, Montvale, NJ 07645, [email protected] or click here.
I was surprised to see my TRAUMA mnemonic featured in the article “Mnemonics in a mnutshell: 32 aids to psychiatric diagnosis” (Current Psychiatry, October 2008) and attributed to the article by Dr. Khouzam. I created my first mnemonic—TRAMA—for posttraumatic stress disorder (PTSD) in 1992. When the clinically significant distress/impaired functioning criterion was included in DSM-IV, I added the “U” for “unable.” I first presented a copyrighted version of TRAUMA in 1994 during a lecture to medical students at Columbia University. Since then I have presented it on numerous occasions.
The authors also did not include my mnemonics for subcriteria corresponding to the 3 symptom clusters of PTSD:
R3D2 (think Star Wars and add an “R”) stands for:
- Recollections, Recurring, and Reactivity (physiological) in response to cues of the traumatic event
- Dreams (distressing) and Distress (psychological).
AFRAID equals:
- Avoid thoughts, feelings, conversations, people, places, or activities associated with the trauma
- Foreshortened future
- Recall (inability to)
- Affect (restricted)
- Interest (diminished)
- Detachment.
SCARE represents:
- Sleep (difficulty falling or staying)
- Concentration (difficulty)
- Anger (outbursts or irritability)
- Really vigilant
- Exaggerated startle response.
Finally, to accurately quote DSM-IV or DSM-IV-TR, the symptoms you listed in the TRAUMA mnemonic need to persist for “more than 1 month” instead of a “month or more.”
Joseph C. Napoli, MD, DFAPA
Assistant clinical professor of psychiatry
Columbia University
New York, NY
Drs. Caplan and Stern Respond
Although we made a good-faith effort to find the original sources of mnemonics included in our article, we were aware that lore and oral history might not allow us to properly cite the contributions of these innovators. In this case, because to the best of our memory neither of the authors have attended a lecture by Dr. Napoli, our awareness of the TRAUMA mnemonic originated from the cited published article.
Since publication of our article, we also have heard from William Falk, MD, at Massachusetts General Hospital informing us that he created DIG FAST to help remember the criteria for mania. We offer apologies to those ingenious clinicians for not our citing their role in the genesis of these mnemonics, and we are grateful that they have enhanced the accuracy of the information we provided.
We also could say: Some Oversights Shall Occasionally Result in Remorse and Yearning (SO SORRY).
Jason P. Caplan, MD
Assistant clinical professor of psychiatry
Creighton University School of Medicine
Omaha, NE
Theodore A. Stern, MD
Professor of psychiatry
Harvard Medical School
Boston, MA
I was surprised to see my TRAUMA mnemonic featured in the article “Mnemonics in a mnutshell: 32 aids to psychiatric diagnosis” (Current Psychiatry, October 2008) and attributed to the article by Dr. Khouzam. I created my first mnemonic—TRAMA—for posttraumatic stress disorder (PTSD) in 1992. When the clinically significant distress/impaired functioning criterion was included in DSM-IV, I added the “U” for “unable.” I first presented a copyrighted version of TRAUMA in 1994 during a lecture to medical students at Columbia University. Since then I have presented it on numerous occasions.
The authors also did not include my mnemonics for subcriteria corresponding to the 3 symptom clusters of PTSD:
R3D2 (think Star Wars and add an “R”) stands for:
- Recollections, Recurring, and Reactivity (physiological) in response to cues of the traumatic event
- Dreams (distressing) and Distress (psychological).
AFRAID equals:
- Avoid thoughts, feelings, conversations, people, places, or activities associated with the trauma
- Foreshortened future
- Recall (inability to)
- Affect (restricted)
- Interest (diminished)
- Detachment.
SCARE represents:
- Sleep (difficulty falling or staying)
- Concentration (difficulty)
- Anger (outbursts or irritability)
- Really vigilant
- Exaggerated startle response.
Finally, to accurately quote DSM-IV or DSM-IV-TR, the symptoms you listed in the TRAUMA mnemonic need to persist for “more than 1 month” instead of a “month or more.”
Joseph C. Napoli, MD, DFAPA
Assistant clinical professor of psychiatry
Columbia University
New York, NY
Drs. Caplan and Stern Respond
Although we made a good-faith effort to find the original sources of mnemonics included in our article, we were aware that lore and oral history might not allow us to properly cite the contributions of these innovators. In this case, because to the best of our memory neither of the authors have attended a lecture by Dr. Napoli, our awareness of the TRAUMA mnemonic originated from the cited published article.
Since publication of our article, we also have heard from William Falk, MD, at Massachusetts General Hospital informing us that he created DIG FAST to help remember the criteria for mania. We offer apologies to those ingenious clinicians for not our citing their role in the genesis of these mnemonics, and we are grateful that they have enhanced the accuracy of the information we provided.
We also could say: Some Oversights Shall Occasionally Result in Remorse and Yearning (SO SORRY).
Jason P. Caplan, MD
Assistant clinical professor of psychiatry
Creighton University School of Medicine
Omaha, NE
Theodore A. Stern, MD
Professor of psychiatry
Harvard Medical School
Boston, MA
To comment on articles in this issue or other topics, send letters in care of Erica Vonderheid, Current Psychiatry, 110 Summit Avenue, Montvale, NJ 07645, [email protected] or click here.
To comment on articles in this issue or other topics, send letters in care of Erica Vonderheid, Current Psychiatry, 110 Summit Avenue, Montvale, NJ 07645, [email protected] or click here.
A pacemaker patient’s electrical dilemma
CASE: Relapsing depression
Mrs. A, age 41, presents with worsening depression and suicidal ideation with a plan to take an overdose of her medications. She describes herself as “tense, anxious, and worrying all the time.” She reports worsening mood, loss of interest in previously pleasurable activities, lack of energy and drive, and difficulties performing routine household tasks. She also endorses a combination of initial and middle insomnia. According to her husband, the patient has been slow in movement and speech and has not been taking adequate care of herself.
Mrs. A denies auditory or visual hallucinations, thought insertion, thought withdrawal, thought broadcast, ideas of reference, or paranoid ideation. She also denies recent or past symptoms of mania or hypomania.
Mrs. A has a history of alcohol abuse and major depressive disorder. For her first depressive episode 5 years ago, she was treated with paroxetine, 20 to 80 mg/d, with good results. Following a depressive relapse, she was switched to fluoxetine, 80 mg/d, which improved her depressive symptoms. Approximately 2 years later, she experienced another depressive relapse that resulted in hospitalization. During hospitalization and subsequent outpatient visits, she was treated with citalopram, 20 mg/d, ziprasidone, 80 mg bid, and lorazepam, 1 mg tid. Her depressive symptoms were in partial remission for 2 years until her current relapse.
Her medical history includes syncope of unexplained origin, for which she received an implanted cardiac pacemaker 3 years ago. She takes sertraline, 150 mg/d, methylphenidate, 15 mg/d, and trazodone, 200 mg at night. Laboratory testing is unremarkable.
On mental status examination, Mrs. A’s mood is sad and her affect constricted. Her speech is fluent but slow, and she speaks only when spoken to. We note that Mrs. A has thought blocking but no hallucinations or delusions. She is alert and oriented, but her attention and concentration are impaired. Her insight is fair, and judgment is poor.
The authors’ observations
Somatic therapy for severe major depressive disorders has been limited principally to pharmacotherapy. Despite the availability of effective antidepressants and aggressive treatment, for many patients—such as Mrs. A—the course of depression is characterized by relapse, recurrence, and chronicity.1,2
Because Mrs. A has treatment-refractory depression, we decide to treat her with ECT. ECT has few contraindications and typically is well tolerated. It commonly is used to treat depression in patients with cardiac conditions and generally is quite safe in this population.3,4
ECT in patients with cardiac pacemakers in situ theoretically presents an increased risk of complications, however.5 Specific concerns of administering ECT to pacemaker patients include electrical interference from ECT stimulus and pacemaker sensing of:
- myopotentials that originate from succinylcholine-induced fasciculation (muscular twitching of contiguous groups of muscle fibers)
- muscle contractions that result in incomplete muscle paralysis
- dysrhythmias during the seizure.
Skeletal muscle can generate significant electrical potentials that are well within the sensing capabilities of most newer pulse generators. This happens most frequently in some dual-chamber pacemakers that can automatically perform mode switching or adapt their sensing and pacing thresholds to new situations, which might make them more sensitive to interference by ECT.
Similar concerns apply to administering ECT to patients receiving vagus nerve stimulation (VNS) therapy, as both VNS pulse generators and cardiac pacemakers are battery-powered, electrical signal-producing mechanisms housed in a metal case. The safety of concurrent ECT and VNS therapy is unknown (Box).6,7
Although vagus nerve stimulation (VNS) and electroconvulsive therapy (ECT) are not mutually exclusive, the safety of concurrent use of these 2 therapies is uncertain.6 The manufacturer of the VNS device recommends turning off the VNS pulse generator before administering ECT. In at least 1 case report, however, ECT was administered safely without the VNS pulse generator turned off.7
No case reports describe the safety of VNS in patients with an implanted device such as a pacemaker or automatic cardioverter defibrillator. According to the manufacturer, the VNS system may affect the operation of other devices. For VNS patients who require an implantable pacemaker, defibrillator therapy, or other types of stimulators, the VNS manufacturer advises careful programming of each system and implanting the 2 stimulators at least 10 centimeters (4 inches) apart to avoid communication interference.
What the evidence says
In evidence-based medicine, we tend to say: “In God we trust; all the others have to bring their data.” Unfortunately, it is difficult to conduct a trial of patients with multiple medical issues. Based on anecdotal reports, it appears that ECT use in patients with an implanted cardiac device such as a pacemaker or automatic internal cardioverter-defibrillator (AICD) generally is safe.8-12
One case report describes successful administration of ECT in a treatment-refractory depressed patient with an AICD. The AICD was deactivated during ECT and re-activated immediately upon completion of each treatment. The case report’s authors concluded that the presence of an AICD should not be a contraindication to ECT.13
A chart review of 3 patients with ICDs who received concurrent ECT found treatment was generally uneventful.12 One patient developed tachycardia with a rate-dependent left bundle branch block and hypotension in the recovery room, which responded promptly to esmolol. She did not experience similar events after subsequent ECT treatments.
Minimizing risk
In the absence of controlled data about the use of ECT in patients with implanted cardiac devices, crucial therapeutic decisions depend on the physician’s skill and judgment. Risk strategies can minimize complications (Algorithm).12 An internist or cardiologist experienced in pacemaker management should conduct a device interrogation—evaluating thresholds, lead impedance, and battery voltage and reviewing histograms, mode switch episodes, and stored electrograms—before the first ECT session and after the final one.
Most modern implantable pacemakers work in the synchronous (demand), rate-adaptive mode. In a patient in whom non-cardiac electrical signals cause bradycardia or asystole during ECT, the pacemaker can be reprogrammed to be less sensitive by placing a magnet over the pulse generator, which converts the pacemaker to an asynchronous (fixed), non-sensing mode. It is important to keep in mind that magnet application will not “turn off” a pacemaker; although each pacemaker is programmed to respond to a magnet in a specific fashion, the main response is asynchronous pacing.
Careful cardiac monitoring during ECT is essential (Table). The cardiologist or internist should be available during the first few ECT sessions to monitor for potential pacemaker interference or malfunction. This physician should be familiar with the pacemaker model and type of lead system so he or she can deactivate, reactivate, or reprogram the device.
Algorithm
Reducing risk when administering ECT to cardiac pacemaker patients
| Step 1 | |
| Evaluate the patient to ensure medical suitability for ECT and associated anesthesia | |
| Step 2 | ↓ |
| Conduct pacemaker interrogation (evaluating thresholds, lead impedance, and battery voltage and reviewing histograms, mode switch episodes, and stored electrograms) prior to first ECT treatment and after completion of full ECT course | |
| Step 3 | ↓ |
| Perform cardiac monitoring during and immediately after administering ECT | |
| Step 4 | ↓ |
| Have a magnet available to reprogram the pacemaker in the event of pacemaker inhibition or symptomatic bradycardia during ECT | |
| Step 5 | ↓ |
| Check that all monitoring devices are properly grounded, insulate the patient’s stretcher, and ensure that the patient does not touch anyone who is in contact with the ground during presentation of the ECT electrical stimulus | |
| ECT: electroconvulsive therapy | |
| Source: Reference 12 | |
Guidelines for monitoring cardiac pacemaker patients during ECT
| Use multilead ECG monitoring |
| Have equipment available to rapidly obtain central access (if vasoactive medications or transvenous pacing is needed) |
| Assess the plethysmography tracing of the pulse oximeter (a useful surrogate if the patient experiences dysrhythmias) |
| Have ready an external defibrillator |
TREATMENT: Successful ECT
We seek a medical consultation before initiating ECT. An internist performs device interrogation before the first ECT treatment and is present in the ECT treatment suite to ensure proper pacemaker conversion and to monitor for cardiac complications. The internist conducts another device interrogation after the acute series of ECT treatments.
Mrs. A tolerates the ECT sessions without cardiac complications. Her depressive symptoms respond well to 12 ECT sessions. She is more interactive and reports better attention and concentration. Although Mrs. A still has middle and initial insomnia, she denies thoughts of harming herself or anyone else.
Related resources
- Yarlagadda C. Pacemaker failure. www.emedicine.com/med/TOPIC1704.HTM.
- Atracurium • Tracrium
- Citalopram • Celexa
- Esmolol • Brevibloc
- Fluoxetine • Prozac
- Lorazepam • Ativan
- Methylphenidate • Ritalin, Concerta, others
- Nortriptyline • Aventyl, Pamelor, others
- Paroxetine • Paxil
- Sertraline • Zoloft
- Succinylcholine • Anectine
- Trazodone • Desyrel
- Venlafaxine • Effexor
- Ziprasidone • Geodon
Dr. Romanowicz reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Ramaswamy receives research support from Bristol-Myers Squibb, Shire, and Forest Pharmaceuticals and is a consultant to Dainippon Sumitomo Pharma.
1. American Psychiatric Association Committee on ECT. The practice of electroconvulsive therapy: recommendations for treatment, training, and privileging. 2nd ed. Washington, D.C: American Psychiatric Association; 2001.
2. Russell JC, Rasmussen KG, O’Connor MK, et al. Long-term maintenance ECT: a retrospective review of efficacy and cognitive outcome. J ECT. 2003;19(1):4-9.
3. Alexopoulos GS, Shamoian CJ, Lucas J, et al. Medical problems of geriatric psychiatric patients and younger controls during electroconvulsive therapy. J Am Geriatr Soc. 1984;32(9):651-654.
4. Rasmussen KG, Rummans TA, Richardson JR. Electroconvulsive therapy in the medically ill. Psychiatric Clin North Am. 2002;25:177-193.
5. MacPherson RD, Loo CK, Barrett N. Electroconvulsive therapy in patients with cardiac pacemakers. Anaesth Intensive Care. 2006;34(4):470-474.
6. Burke MJ, Husain MM. Concomitant use of vagus nerve stimulation and electroconvulsive therapy for treatment-resistant depression. J ECT. 2006;22(3):218-222.
7. Husain MM, Montgomery JH, Fernandes P, et al. Safety of vagus nerve stimulation with ECT. Am J Psychiatry. 2002;159:1243.-
8. Alexopoulos GS, Frances RJ. ECT and cardiac patients with pacemakers. Am J Psychiatry. 1980;137(9):1111-1112.
9. Stone KR, McPherson CA. Assessment and management of patients with pacemakers and implantable cardioverter defibrillators. Crit Care Med. 2004;32(4 suppl):S155-S165.
10. Maisel WH, Sweeney MO, Stevenson WG, et al. Recalls and safety alerts involving pacemakers and implantable cardioverter-defibrillator generators. JAMA. 2001;286(7):793-799.
11. Gibson TC, Leaman DM, Devors J, et al. Pacemaker function in relation to electroconvulsive therapy. Chest. 1973;63(6):1025-1027.
12. Dolenc TJ, Barnes RD, Hayes DL, et al. Electroconvulsive therapy in patients with cardiac pacemakers and implantable cardioverter defibrillators. Pacing Clin Electrophysiol. 2004;27(9):1257-1263.
13. Lapid MI, Rummans TA, Hofmann VE, et al. ECT and automatic internal cardioverter-defibrillator. J ECT. 2001;17(2):146-148.
CASE: Relapsing depression
Mrs. A, age 41, presents with worsening depression and suicidal ideation with a plan to take an overdose of her medications. She describes herself as “tense, anxious, and worrying all the time.” She reports worsening mood, loss of interest in previously pleasurable activities, lack of energy and drive, and difficulties performing routine household tasks. She also endorses a combination of initial and middle insomnia. According to her husband, the patient has been slow in movement and speech and has not been taking adequate care of herself.
Mrs. A denies auditory or visual hallucinations, thought insertion, thought withdrawal, thought broadcast, ideas of reference, or paranoid ideation. She also denies recent or past symptoms of mania or hypomania.
Mrs. A has a history of alcohol abuse and major depressive disorder. For her first depressive episode 5 years ago, she was treated with paroxetine, 20 to 80 mg/d, with good results. Following a depressive relapse, she was switched to fluoxetine, 80 mg/d, which improved her depressive symptoms. Approximately 2 years later, she experienced another depressive relapse that resulted in hospitalization. During hospitalization and subsequent outpatient visits, she was treated with citalopram, 20 mg/d, ziprasidone, 80 mg bid, and lorazepam, 1 mg tid. Her depressive symptoms were in partial remission for 2 years until her current relapse.
Her medical history includes syncope of unexplained origin, for which she received an implanted cardiac pacemaker 3 years ago. She takes sertraline, 150 mg/d, methylphenidate, 15 mg/d, and trazodone, 200 mg at night. Laboratory testing is unremarkable.
On mental status examination, Mrs. A’s mood is sad and her affect constricted. Her speech is fluent but slow, and she speaks only when spoken to. We note that Mrs. A has thought blocking but no hallucinations or delusions. She is alert and oriented, but her attention and concentration are impaired. Her insight is fair, and judgment is poor.
The authors’ observations
Somatic therapy for severe major depressive disorders has been limited principally to pharmacotherapy. Despite the availability of effective antidepressants and aggressive treatment, for many patients—such as Mrs. A—the course of depression is characterized by relapse, recurrence, and chronicity.1,2
Because Mrs. A has treatment-refractory depression, we decide to treat her with ECT. ECT has few contraindications and typically is well tolerated. It commonly is used to treat depression in patients with cardiac conditions and generally is quite safe in this population.3,4
ECT in patients with cardiac pacemakers in situ theoretically presents an increased risk of complications, however.5 Specific concerns of administering ECT to pacemaker patients include electrical interference from ECT stimulus and pacemaker sensing of:
- myopotentials that originate from succinylcholine-induced fasciculation (muscular twitching of contiguous groups of muscle fibers)
- muscle contractions that result in incomplete muscle paralysis
- dysrhythmias during the seizure.
Skeletal muscle can generate significant electrical potentials that are well within the sensing capabilities of most newer pulse generators. This happens most frequently in some dual-chamber pacemakers that can automatically perform mode switching or adapt their sensing and pacing thresholds to new situations, which might make them more sensitive to interference by ECT.
Similar concerns apply to administering ECT to patients receiving vagus nerve stimulation (VNS) therapy, as both VNS pulse generators and cardiac pacemakers are battery-powered, electrical signal-producing mechanisms housed in a metal case. The safety of concurrent ECT and VNS therapy is unknown (Box).6,7
Although vagus nerve stimulation (VNS) and electroconvulsive therapy (ECT) are not mutually exclusive, the safety of concurrent use of these 2 therapies is uncertain.6 The manufacturer of the VNS device recommends turning off the VNS pulse generator before administering ECT. In at least 1 case report, however, ECT was administered safely without the VNS pulse generator turned off.7
No case reports describe the safety of VNS in patients with an implanted device such as a pacemaker or automatic cardioverter defibrillator. According to the manufacturer, the VNS system may affect the operation of other devices. For VNS patients who require an implantable pacemaker, defibrillator therapy, or other types of stimulators, the VNS manufacturer advises careful programming of each system and implanting the 2 stimulators at least 10 centimeters (4 inches) apart to avoid communication interference.
What the evidence says
In evidence-based medicine, we tend to say: “In God we trust; all the others have to bring their data.” Unfortunately, it is difficult to conduct a trial of patients with multiple medical issues. Based on anecdotal reports, it appears that ECT use in patients with an implanted cardiac device such as a pacemaker or automatic internal cardioverter-defibrillator (AICD) generally is safe.8-12
One case report describes successful administration of ECT in a treatment-refractory depressed patient with an AICD. The AICD was deactivated during ECT and re-activated immediately upon completion of each treatment. The case report’s authors concluded that the presence of an AICD should not be a contraindication to ECT.13
A chart review of 3 patients with ICDs who received concurrent ECT found treatment was generally uneventful.12 One patient developed tachycardia with a rate-dependent left bundle branch block and hypotension in the recovery room, which responded promptly to esmolol. She did not experience similar events after subsequent ECT treatments.
Minimizing risk
In the absence of controlled data about the use of ECT in patients with implanted cardiac devices, crucial therapeutic decisions depend on the physician’s skill and judgment. Risk strategies can minimize complications (Algorithm).12 An internist or cardiologist experienced in pacemaker management should conduct a device interrogation—evaluating thresholds, lead impedance, and battery voltage and reviewing histograms, mode switch episodes, and stored electrograms—before the first ECT session and after the final one.
Most modern implantable pacemakers work in the synchronous (demand), rate-adaptive mode. In a patient in whom non-cardiac electrical signals cause bradycardia or asystole during ECT, the pacemaker can be reprogrammed to be less sensitive by placing a magnet over the pulse generator, which converts the pacemaker to an asynchronous (fixed), non-sensing mode. It is important to keep in mind that magnet application will not “turn off” a pacemaker; although each pacemaker is programmed to respond to a magnet in a specific fashion, the main response is asynchronous pacing.
Careful cardiac monitoring during ECT is essential (Table). The cardiologist or internist should be available during the first few ECT sessions to monitor for potential pacemaker interference or malfunction. This physician should be familiar with the pacemaker model and type of lead system so he or she can deactivate, reactivate, or reprogram the device.
Algorithm
Reducing risk when administering ECT to cardiac pacemaker patients
| Step 1 | |
| Evaluate the patient to ensure medical suitability for ECT and associated anesthesia | |
| Step 2 | ↓ |
| Conduct pacemaker interrogation (evaluating thresholds, lead impedance, and battery voltage and reviewing histograms, mode switch episodes, and stored electrograms) prior to first ECT treatment and after completion of full ECT course | |
| Step 3 | ↓ |
| Perform cardiac monitoring during and immediately after administering ECT | |
| Step 4 | ↓ |
| Have a magnet available to reprogram the pacemaker in the event of pacemaker inhibition or symptomatic bradycardia during ECT | |
| Step 5 | ↓ |
| Check that all monitoring devices are properly grounded, insulate the patient’s stretcher, and ensure that the patient does not touch anyone who is in contact with the ground during presentation of the ECT electrical stimulus | |
| ECT: electroconvulsive therapy | |
| Source: Reference 12 | |
Guidelines for monitoring cardiac pacemaker patients during ECT
| Use multilead ECG monitoring |
| Have equipment available to rapidly obtain central access (if vasoactive medications or transvenous pacing is needed) |
| Assess the plethysmography tracing of the pulse oximeter (a useful surrogate if the patient experiences dysrhythmias) |
| Have ready an external defibrillator |
TREATMENT: Successful ECT
We seek a medical consultation before initiating ECT. An internist performs device interrogation before the first ECT treatment and is present in the ECT treatment suite to ensure proper pacemaker conversion and to monitor for cardiac complications. The internist conducts another device interrogation after the acute series of ECT treatments.
Mrs. A tolerates the ECT sessions without cardiac complications. Her depressive symptoms respond well to 12 ECT sessions. She is more interactive and reports better attention and concentration. Although Mrs. A still has middle and initial insomnia, she denies thoughts of harming herself or anyone else.
Related resources
- Yarlagadda C. Pacemaker failure. www.emedicine.com/med/TOPIC1704.HTM.
- Atracurium • Tracrium
- Citalopram • Celexa
- Esmolol • Brevibloc
- Fluoxetine • Prozac
- Lorazepam • Ativan
- Methylphenidate • Ritalin, Concerta, others
- Nortriptyline • Aventyl, Pamelor, others
- Paroxetine • Paxil
- Sertraline • Zoloft
- Succinylcholine • Anectine
- Trazodone • Desyrel
- Venlafaxine • Effexor
- Ziprasidone • Geodon
Dr. Romanowicz reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Ramaswamy receives research support from Bristol-Myers Squibb, Shire, and Forest Pharmaceuticals and is a consultant to Dainippon Sumitomo Pharma.
CASE: Relapsing depression
Mrs. A, age 41, presents with worsening depression and suicidal ideation with a plan to take an overdose of her medications. She describes herself as “tense, anxious, and worrying all the time.” She reports worsening mood, loss of interest in previously pleasurable activities, lack of energy and drive, and difficulties performing routine household tasks. She also endorses a combination of initial and middle insomnia. According to her husband, the patient has been slow in movement and speech and has not been taking adequate care of herself.
Mrs. A denies auditory or visual hallucinations, thought insertion, thought withdrawal, thought broadcast, ideas of reference, or paranoid ideation. She also denies recent or past symptoms of mania or hypomania.
Mrs. A has a history of alcohol abuse and major depressive disorder. For her first depressive episode 5 years ago, she was treated with paroxetine, 20 to 80 mg/d, with good results. Following a depressive relapse, she was switched to fluoxetine, 80 mg/d, which improved her depressive symptoms. Approximately 2 years later, she experienced another depressive relapse that resulted in hospitalization. During hospitalization and subsequent outpatient visits, she was treated with citalopram, 20 mg/d, ziprasidone, 80 mg bid, and lorazepam, 1 mg tid. Her depressive symptoms were in partial remission for 2 years until her current relapse.
Her medical history includes syncope of unexplained origin, for which she received an implanted cardiac pacemaker 3 years ago. She takes sertraline, 150 mg/d, methylphenidate, 15 mg/d, and trazodone, 200 mg at night. Laboratory testing is unremarkable.
On mental status examination, Mrs. A’s mood is sad and her affect constricted. Her speech is fluent but slow, and she speaks only when spoken to. We note that Mrs. A has thought blocking but no hallucinations or delusions. She is alert and oriented, but her attention and concentration are impaired. Her insight is fair, and judgment is poor.
The authors’ observations
Somatic therapy for severe major depressive disorders has been limited principally to pharmacotherapy. Despite the availability of effective antidepressants and aggressive treatment, for many patients—such as Mrs. A—the course of depression is characterized by relapse, recurrence, and chronicity.1,2
Because Mrs. A has treatment-refractory depression, we decide to treat her with ECT. ECT has few contraindications and typically is well tolerated. It commonly is used to treat depression in patients with cardiac conditions and generally is quite safe in this population.3,4
ECT in patients with cardiac pacemakers in situ theoretically presents an increased risk of complications, however.5 Specific concerns of administering ECT to pacemaker patients include electrical interference from ECT stimulus and pacemaker sensing of:
- myopotentials that originate from succinylcholine-induced fasciculation (muscular twitching of contiguous groups of muscle fibers)
- muscle contractions that result in incomplete muscle paralysis
- dysrhythmias during the seizure.
Skeletal muscle can generate significant electrical potentials that are well within the sensing capabilities of most newer pulse generators. This happens most frequently in some dual-chamber pacemakers that can automatically perform mode switching or adapt their sensing and pacing thresholds to new situations, which might make them more sensitive to interference by ECT.
Similar concerns apply to administering ECT to patients receiving vagus nerve stimulation (VNS) therapy, as both VNS pulse generators and cardiac pacemakers are battery-powered, electrical signal-producing mechanisms housed in a metal case. The safety of concurrent ECT and VNS therapy is unknown (Box).6,7
Although vagus nerve stimulation (VNS) and electroconvulsive therapy (ECT) are not mutually exclusive, the safety of concurrent use of these 2 therapies is uncertain.6 The manufacturer of the VNS device recommends turning off the VNS pulse generator before administering ECT. In at least 1 case report, however, ECT was administered safely without the VNS pulse generator turned off.7
No case reports describe the safety of VNS in patients with an implanted device such as a pacemaker or automatic cardioverter defibrillator. According to the manufacturer, the VNS system may affect the operation of other devices. For VNS patients who require an implantable pacemaker, defibrillator therapy, or other types of stimulators, the VNS manufacturer advises careful programming of each system and implanting the 2 stimulators at least 10 centimeters (4 inches) apart to avoid communication interference.
What the evidence says
In evidence-based medicine, we tend to say: “In God we trust; all the others have to bring their data.” Unfortunately, it is difficult to conduct a trial of patients with multiple medical issues. Based on anecdotal reports, it appears that ECT use in patients with an implanted cardiac device such as a pacemaker or automatic internal cardioverter-defibrillator (AICD) generally is safe.8-12
One case report describes successful administration of ECT in a treatment-refractory depressed patient with an AICD. The AICD was deactivated during ECT and re-activated immediately upon completion of each treatment. The case report’s authors concluded that the presence of an AICD should not be a contraindication to ECT.13
A chart review of 3 patients with ICDs who received concurrent ECT found treatment was generally uneventful.12 One patient developed tachycardia with a rate-dependent left bundle branch block and hypotension in the recovery room, which responded promptly to esmolol. She did not experience similar events after subsequent ECT treatments.
Minimizing risk
In the absence of controlled data about the use of ECT in patients with implanted cardiac devices, crucial therapeutic decisions depend on the physician’s skill and judgment. Risk strategies can minimize complications (Algorithm).12 An internist or cardiologist experienced in pacemaker management should conduct a device interrogation—evaluating thresholds, lead impedance, and battery voltage and reviewing histograms, mode switch episodes, and stored electrograms—before the first ECT session and after the final one.
Most modern implantable pacemakers work in the synchronous (demand), rate-adaptive mode. In a patient in whom non-cardiac electrical signals cause bradycardia or asystole during ECT, the pacemaker can be reprogrammed to be less sensitive by placing a magnet over the pulse generator, which converts the pacemaker to an asynchronous (fixed), non-sensing mode. It is important to keep in mind that magnet application will not “turn off” a pacemaker; although each pacemaker is programmed to respond to a magnet in a specific fashion, the main response is asynchronous pacing.
Careful cardiac monitoring during ECT is essential (Table). The cardiologist or internist should be available during the first few ECT sessions to monitor for potential pacemaker interference or malfunction. This physician should be familiar with the pacemaker model and type of lead system so he or she can deactivate, reactivate, or reprogram the device.
Algorithm
Reducing risk when administering ECT to cardiac pacemaker patients
| Step 1 | |
| Evaluate the patient to ensure medical suitability for ECT and associated anesthesia | |
| Step 2 | ↓ |
| Conduct pacemaker interrogation (evaluating thresholds, lead impedance, and battery voltage and reviewing histograms, mode switch episodes, and stored electrograms) prior to first ECT treatment and after completion of full ECT course | |
| Step 3 | ↓ |
| Perform cardiac monitoring during and immediately after administering ECT | |
| Step 4 | ↓ |
| Have a magnet available to reprogram the pacemaker in the event of pacemaker inhibition or symptomatic bradycardia during ECT | |
| Step 5 | ↓ |
| Check that all monitoring devices are properly grounded, insulate the patient’s stretcher, and ensure that the patient does not touch anyone who is in contact with the ground during presentation of the ECT electrical stimulus | |
| ECT: electroconvulsive therapy | |
| Source: Reference 12 | |
Guidelines for monitoring cardiac pacemaker patients during ECT
| Use multilead ECG monitoring |
| Have equipment available to rapidly obtain central access (if vasoactive medications or transvenous pacing is needed) |
| Assess the plethysmography tracing of the pulse oximeter (a useful surrogate if the patient experiences dysrhythmias) |
| Have ready an external defibrillator |
TREATMENT: Successful ECT
We seek a medical consultation before initiating ECT. An internist performs device interrogation before the first ECT treatment and is present in the ECT treatment suite to ensure proper pacemaker conversion and to monitor for cardiac complications. The internist conducts another device interrogation after the acute series of ECT treatments.
Mrs. A tolerates the ECT sessions without cardiac complications. Her depressive symptoms respond well to 12 ECT sessions. She is more interactive and reports better attention and concentration. Although Mrs. A still has middle and initial insomnia, she denies thoughts of harming herself or anyone else.
Related resources
- Yarlagadda C. Pacemaker failure. www.emedicine.com/med/TOPIC1704.HTM.
- Atracurium • Tracrium
- Citalopram • Celexa
- Esmolol • Brevibloc
- Fluoxetine • Prozac
- Lorazepam • Ativan
- Methylphenidate • Ritalin, Concerta, others
- Nortriptyline • Aventyl, Pamelor, others
- Paroxetine • Paxil
- Sertraline • Zoloft
- Succinylcholine • Anectine
- Trazodone • Desyrel
- Venlafaxine • Effexor
- Ziprasidone • Geodon
Dr. Romanowicz reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Ramaswamy receives research support from Bristol-Myers Squibb, Shire, and Forest Pharmaceuticals and is a consultant to Dainippon Sumitomo Pharma.
1. American Psychiatric Association Committee on ECT. The practice of electroconvulsive therapy: recommendations for treatment, training, and privileging. 2nd ed. Washington, D.C: American Psychiatric Association; 2001.
2. Russell JC, Rasmussen KG, O’Connor MK, et al. Long-term maintenance ECT: a retrospective review of efficacy and cognitive outcome. J ECT. 2003;19(1):4-9.
3. Alexopoulos GS, Shamoian CJ, Lucas J, et al. Medical problems of geriatric psychiatric patients and younger controls during electroconvulsive therapy. J Am Geriatr Soc. 1984;32(9):651-654.
4. Rasmussen KG, Rummans TA, Richardson JR. Electroconvulsive therapy in the medically ill. Psychiatric Clin North Am. 2002;25:177-193.
5. MacPherson RD, Loo CK, Barrett N. Electroconvulsive therapy in patients with cardiac pacemakers. Anaesth Intensive Care. 2006;34(4):470-474.
6. Burke MJ, Husain MM. Concomitant use of vagus nerve stimulation and electroconvulsive therapy for treatment-resistant depression. J ECT. 2006;22(3):218-222.
7. Husain MM, Montgomery JH, Fernandes P, et al. Safety of vagus nerve stimulation with ECT. Am J Psychiatry. 2002;159:1243.-
8. Alexopoulos GS, Frances RJ. ECT and cardiac patients with pacemakers. Am J Psychiatry. 1980;137(9):1111-1112.
9. Stone KR, McPherson CA. Assessment and management of patients with pacemakers and implantable cardioverter defibrillators. Crit Care Med. 2004;32(4 suppl):S155-S165.
10. Maisel WH, Sweeney MO, Stevenson WG, et al. Recalls and safety alerts involving pacemakers and implantable cardioverter-defibrillator generators. JAMA. 2001;286(7):793-799.
11. Gibson TC, Leaman DM, Devors J, et al. Pacemaker function in relation to electroconvulsive therapy. Chest. 1973;63(6):1025-1027.
12. Dolenc TJ, Barnes RD, Hayes DL, et al. Electroconvulsive therapy in patients with cardiac pacemakers and implantable cardioverter defibrillators. Pacing Clin Electrophysiol. 2004;27(9):1257-1263.
13. Lapid MI, Rummans TA, Hofmann VE, et al. ECT and automatic internal cardioverter-defibrillator. J ECT. 2001;17(2):146-148.
1. American Psychiatric Association Committee on ECT. The practice of electroconvulsive therapy: recommendations for treatment, training, and privileging. 2nd ed. Washington, D.C: American Psychiatric Association; 2001.
2. Russell JC, Rasmussen KG, O’Connor MK, et al. Long-term maintenance ECT: a retrospective review of efficacy and cognitive outcome. J ECT. 2003;19(1):4-9.
3. Alexopoulos GS, Shamoian CJ, Lucas J, et al. Medical problems of geriatric psychiatric patients and younger controls during electroconvulsive therapy. J Am Geriatr Soc. 1984;32(9):651-654.
4. Rasmussen KG, Rummans TA, Richardson JR. Electroconvulsive therapy in the medically ill. Psychiatric Clin North Am. 2002;25:177-193.
5. MacPherson RD, Loo CK, Barrett N. Electroconvulsive therapy in patients with cardiac pacemakers. Anaesth Intensive Care. 2006;34(4):470-474.
6. Burke MJ, Husain MM. Concomitant use of vagus nerve stimulation and electroconvulsive therapy for treatment-resistant depression. J ECT. 2006;22(3):218-222.
7. Husain MM, Montgomery JH, Fernandes P, et al. Safety of vagus nerve stimulation with ECT. Am J Psychiatry. 2002;159:1243.-
8. Alexopoulos GS, Frances RJ. ECT and cardiac patients with pacemakers. Am J Psychiatry. 1980;137(9):1111-1112.
9. Stone KR, McPherson CA. Assessment and management of patients with pacemakers and implantable cardioverter defibrillators. Crit Care Med. 2004;32(4 suppl):S155-S165.
10. Maisel WH, Sweeney MO, Stevenson WG, et al. Recalls and safety alerts involving pacemakers and implantable cardioverter-defibrillator generators. JAMA. 2001;286(7):793-799.
11. Gibson TC, Leaman DM, Devors J, et al. Pacemaker function in relation to electroconvulsive therapy. Chest. 1973;63(6):1025-1027.
12. Dolenc TJ, Barnes RD, Hayes DL, et al. Electroconvulsive therapy in patients with cardiac pacemakers and implantable cardioverter defibrillators. Pacing Clin Electrophysiol. 2004;27(9):1257-1263.
13. Lapid MI, Rummans TA, Hofmann VE, et al. ECT and automatic internal cardioverter-defibrillator. J ECT. 2001;17(2):146-148.
Loss of enzyme induction: Ups and downs of a hidden drug-drug interaction
Mr. P, age 35 with schizophrenia and seizure disorder, has been maintained on risperidone, 6 mg qhs, and phenytoin, 300 mg qhs. For clinical reasons, the treating neurologist changes the anticonvulsant to divalproex. One week later, Mr. P presents to the emergency room complaining of jaw and neck stiffness.
Ms. K, age 43 with a history of schizoaffective disorder, bipolar type, and erratic medication adherence, is being treated with quetiapine, 600 mg at bedtime, and carbamazepine, 1,000 mg/d. Between appointments she stops taking carbamazepine, believing it is causing her to hear voices from her television. Two weeks later, the manager of Ms. K’s independent living facility tells the psychiatrist that the patient appears excessively sedated and has fallen twice in the past few days.
Mrs. T, a 39-year-old state hospital resident with schizoaffective disorder, bipolar type, has been treated with clozapine, 250 mg bid for 6 months; her most recent trough serum level was 492 ng/mL. She smokes 15 cigarettes/d. Two weeks after the hospital institutes a no-smoking policy, Mrs. T complains of excessive drooling and lightheadedness. Her trough clozapine level is now 875 ng/mL.
Discontinuing a medication that has enzyme-inducing effects presents a hidden problem for patients receiving antipsychotic pharmacotherapy. Certain hepatic enzymes responsible for antipsychotic metabolism—as well those involved in intercellular drug transport—are induced by medication or environmental exposures.1,2 Adding a medication that induces these enzymes to the regimen of a patient receiving antipsychotic therapy can result in markedly reduced serum antipsychotic levels, and discontinuing an inducing agent can result in increased antipsychotic levels.
Drug-drug interactions (DDIs) are a substantial contributor to adverse drug reactions (Box).3-7 Antipsychotic prescribing information highlights potential DDIs from the use of enzyme inhibitors and inducers but identifies only effects caused by adding a second agent. The prescriber remains the sole line of defense for monitoring for DDIs when discontinuing a medication that has inducing or inhibiting effects.
Most psychiatrists are aware that certain medications have clinically significant effects on cytochrome P450 (CYP) activity and of the potential for CYP inhibitors to generate DDIs. Clinicians often are aware of antidepressant medications’ CYP-inhibiting effects, know that levels of other medications will change when discontinuing a potent P450 inhibitor, and understand the need to increase dosages of medications influenced by such agents.8
However, few studies have evaluated the effects of enzyme induction on antipsychotic drug levels,9,10 and the literature rarely discusses changes in serum drug levels after loss of enzyme or drug transport induction.11 If unrecognized, these changes may have significant clinical consequences.
Drug-drug interactions (DDIs) are a common and often preventable cause of morbidity and mortality. National surveillance data showed 700,000 emergency room visits related to adverse drug reactions (ADRs) in the 2 years from January 2004 through 2005.3 ADRs are particularly concerning for psychiatrists managing polypharmacy regimens for patients with severe mental disorders such as schizophrenia.
Literature on DDIs with antipsychotics focuses primarily on kinetic interactions that generate supratherapeutic drug levels.4,5 Because development of side effects is associated with reduced adherence, these kinetic interactions may increase the risk of adverse effects and lead to patients stopping the antipsychotic treatment.6,7
Two induction pathways
The primary mechanism underlying clinically significant DDIs occurs during CYP-mediated phase I metabolism. Molecules undergo oxidative conversion into metabolites that can be conjugated by phase II enzymes, generating more soluble forms that facilitate excretion.
The workhorse of human CYP metabolism is 3A4 (Table 1),12,13 which comprises 30% of hepatic activity and 70% of gut cytochrome activity.14 CYP 1A2 is responsible for 10% to 15% of CYP activity.
Both CYP 3A4 and 1A2 are inducible. A wide variety of medications induce 3A4 activity. The list of 1A2 inducers is shorter; the most common are aryl hydrocarbons from cigarette smoke and proton pump inhibitors.
CYP 2D6 accounts for 20% of hepatic cytochrome activity but is not inducible. CYP 2D6 is well known to psychiatrists because some selective serotonin reuptake inhibitors (SSRIs) and the non-SSRI antidepressant bupropion are potent inhibitors of this enzyme.15,16
P-glycoprotein (PGP) induction. Transmembrane shuttles such as P-glycoprotein (PGP) are an important component of drug disposition. PGP belongs to the family of ATP binding cassette (ABC) transporters that bring molecules across cellular barriers.17,18 It was first described in cancer cells that developed multiple drug resistance (MDR) and is often referred to as MDR1.19 PGP is encoded on human chromosome 7 and expressed in normal tissues, particularly in areas where cells seek to limit drug influx, such as those lining the luminal surface of the small and large intestine and those lining the blood-brain barrier and blood-testis barrier. The expression of PGP in hepatic cells promotes drug clearance by enhancing biliary drug excretion.
PGP is encoded on the same chromosome as CYP 3A4, and these 2 proteins frequently are expressed in the same cells, particularly in the intestinal lining and liver. Moreover, PGP is inducible, and there is substantial overlap between medications that are substrates for—or inducers of—PGP and CYP 3A4 activity. This makes it challenging to determine whether the kinetic effects of a second medication are the result of interference of 3A4, PGP, or both.
Polymorphisms in PGP activity may influence the penetration of psychotropic medications into the CNS. Studies indicate an association between certain PGP polymorphisms and treatment outcomes.17,18
Table 1
What induces CYP 1A2 and 3A4?
| Enzyme | Description | Inducers* |
|---|---|---|
| CYP 1A2 |
| Aryl hydrocarbons (smoking), protonpump inhibitors (omeprazole > lansoprazole > pantoprazole), modafinil, St. John’s wort, chargrilled meat, cruciferous vegetables such as broccoli and cabbage, flavones, protein supplements |
| CYP 3A4 |
| Carbamazepine, phenytoin, phenobarbital, rifampin, oxcarbazepine, efavirenz, glucocorticoids, modafinil, nevirapine, pioglitazone, St. John’s wort |
| * Listed in order from strongest to weakest induction | ||
| CYP: cytochrome P450; PGP: P-glycoprotein | ||
| Source: References 12,13 | ||
Stopping an inducer
In general, inducers of CYP enzymes stimulate gene transcription within hours of exposure; maximum transcriptional activity occurs after 10 to 12 hours of exposure. As transcription increases, the concentration of the CYP mRNA transcript steadily accumulates, as does concentration of CYP protein.
After an inducer is discontinued, transcription returns to basal levels within 18 hours; however, the degradation of CYP proteins is a first-order process, with a half-life of 8 to 30 hours. As a result, the decrease in cellular CYP concentration—and the level of activity—lags behind the decreased synthesis from reduced mRNA levels.
Interactions with antipsychotics
Effects on serum antipsychotic levels caused by discontinuing a CYP or PGP inducer can be predicted from data on decreases in antipsychotic levels following inducer exposure. Except for ziprasidone and paliperidone, most atypical antipsychotics are prone to substantial decreases during concomitant inducer use (Table 2).21
The effect of enzyme inducers on risperidone is particularly interesting. Conversion of risperidone to its active metabolite 9-OH risperidone (paliperidone) occurs primarily via 2D6,22 yet concurrent use of carbamazepine—a potent CYP 3A4 inducer—results in a 50% decrease in the concentration of the active moiety (risperidone plus 9-OH risperidone). This finding and other early investigations suggested that CYP 3A had a role in risperidone metabolism,23,24 but these early studies and case series often involved molecules that had activity at both 3A4 and PGP. Further research clarified that effects on PGP—and not 3A4—are responsible for the changes in risperidone metabolism observed with the use of carbamazepine and other medications.25,26
Induction in case patients: Follow-up. Regardless of whether induction is mediated by ≥1 metabolic pathways, the loss of the inducer will result in serum antipsychotic increases that are proportional to the initial decrease.20 For example, with risperidone, the expected decrease is 50%. Therefore, after Mr. P stopped taking phenytoin, his serum risperidone level would be expected to double, which resulted in extrapyramidal side effects.
Quetiapine clearance is increased 5-fold by inducer exposure, so a clinician treating Ms. K would expect a marked increase in somnolence—and possibly orthostasis—as serum quetiapine levels peak 1 to 2 weeks following carbamazepine discontinuation.
The effects of smoking cessation on serum clozapine levels have been well-documented.1,27 Clinicians should anticipate median increases in serum clozapine levels of 55% after a patient discontinues smoking (aryl hydrocarbon exposure), but changes vary substantially among individuals. Mrs. T’s serum clozapine increased approximately 78%.
Careful clinical monitoring and slow downward adjustment of antipsychotic doses could have prevented the adverse effects these 3 patients experienced after loss of CYP/PGP induction and the consequences those side effects present for future medication adherence. When loss of induction is unplanned—as when Ms. K stopped taking carbamazepine but continued quetiapine—clinicians need to be alert to the fact that the patient was prescribed an inducer and include the loss of induction as a hypothesis for the patient’s somnolence.
Table 2
Effects of CYP/PGP induction on atypical antipsychotics
| Antipsychotic | Metabolic pathways | Effect of induction |
|---|---|---|
| Aripiprazole | 2D6 and 3A4 convert aripiprazole to active metabolite dehydro-aripiprazole | 3A4 induction decreases maximum concentration of aripiprazole and metabolite by 70% |
| Clozapine | Multiple enzymes convert clozapine to N-desmethylclozapine; mean contributions of CYP 1A2, 2C19, 3A4, 2C9, and 2D6 are 30%, 24%, 22%, 12%, and 6%, respectively, with CYP 1A2 predominantly involved at low concentrations | Loss of smoking-related 1A2 induction results in 50% increase in serum levels |
| Olanzapine | Direct glucuronidation or 1A2-mediated oxidation to N-desmethlyolanzapine | Carbamazepine use increases clearance by 50%. Olanzapine concentration:dose ratio is about 5-fold lower in smokers (7.9 +/- 2.6) than in nonsmokers (1.56 +/- 1.1; P |
| Paliperidone | 59% excreted unchanged in urine; phase I metabolism accounts for ≤10% of drug clearance | Unlikely to significantly impact levels, but impact of PGP induction is unknown |
| Quetiapine | 3A4-mediated sulfoxidation to inactive metabolite is primary pathway, but numerous metabolites noted, with 1 active metabolite (norquetiapine) | Phenytoin increases clearance 5-fold |
| Risperidone | 2D6 converts risperidone to active metabolite 9-OH risperidone | In a drug interaction study of risperidone, 6 mg/d for 3 weeks, followed by 3 weeks of carbamazepine, active moiety concentration was decreased by about 50% |
| Ziprasidone | 3A4 (~1/3); aldehyde oxidase (~2/3) | Approximately 35% decrease in ziprasidone exposure by carbamazepine |
| CYP: cytochrome P450; PGP: P-glycoprotein | ||
| Source: Reference 21 | ||
Clinical considerations
In the absence of detailed data on antipsychotic metabolism, clinicians can make intelligent decisions regarding potential DDIs by:
- knowing the extent of induction by common offenders (such as carbamazepine or phenytoin) documented in the medication’s prescribing information or demonstrated through convincing case reports or case series
- memorizing the list of CYP 1A2 and CYP 3A4/PGP inducers.
Patients who may be susceptible to effects from loss of enzyme induction (including smokers receiving olanzapine or clozapine or others taking 3A4/PGP inducers) must be identified, and plans made for dosage adjustments if inducing agents are discontinued for a sufficient time (≥1 week) to result in downregulation of CYP or PGP activity. A slow taper of the antipsychotic over 1 to 2 weeks to the new target dose should compensate for loss of enzyme or PGP induction.
For newer antipsychotic medications with limited data, the proposed discontinuation of an inducer should, at the minimum, prompt a discussion between the psychiatrist and patient regarding the expected increase in serum antipsychotic levels and potential adverse effects that may result. Clinicians also must make every attempt to stay apprised of a patient’s current medications, bearing in mind that another provider may prescribe an inducer. Patients with schizophrenia always should be educated to contact the psychiatrist following any change in medication regimen, placing particular emphasis on the 1 or 2 medications that are known to be implicated in DDIs with the patient’s current antipsychotic.
- Flockhart DA. Drug interactions: Cytochrome P450 drug interaction table. Indiana University School of Medicine. 2007. http://medicine.iupui.edu/flockhart/table.htm.
- Cozza KL, Armstrong SC, Oesterheld JR. Concise guide to drug interaction principles for medical practice: Cytochrome P450s, UGTS, p-glycoproteins. Washington, DC: American Psychiatric Press, Inc; 2003.
- Aripiprazole • Abilify
- Bupropion • Wellbutrin
- Carbamazepine • Carbatrol, Tegretol
- Clozapine • Clozaril
- Divalproex • Depakote
- Efavirenz • Sustiva
- Lansoprazole • Prevacid
- Modafinil • Provigil
- Nevirapine • Viramune
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Oxcarbazepine • Trileptal
- Paliperidone • Invega
- Pantoprazole • Protonix
- Phenobarbital • Barbita, Luminal, others
- Phenytoin • Dilantin
- Pioglitazone • Actos
- Quetiapine • Seroquel
- Rifampin • Rifadin, Rimactane
- Risperidone • Risperdal
- Ziprasidone • Geodon
Dr. Meyer receives grant/research support from the National Institute of Mental Health, Pfizer Inc., and the University of California. He is a consultant to Bristol-Myers Squibb, Organon, Vanda Pharmaceuticals, and Wyeth, and a speaker for AstraZeneca, Bristol-Myers Squibb, Dainippon Sumitomo Pharma, and Pfizer Inc.
Ms. Leckband reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Meyer JM. Individual changes in clozapine levels after smoking cessation: results and a predictive model. J Clin Psychopharmacol. 2001;21:569-574.
2. Wong YW, Yeh C, Thyrum PT. The effects of concomitant phenytoin administration on the steady-state pharmacokinetics of quetiapine. J Clin Psychopharmacol. 2001;21:89-93.
3. Budnitz DS, Pollock DA, Weidenbach KN, et al. National surveillance of emergency department visits for outpatient adverse drug events. JAMA. 2006;296:1858-1866.
4. Prior TI, Baker GB. Interactions between the cytochrome P450 system and the second-generation antipsychotics. J Psychiatry Neurosci. 2003;28:99-112.
5. Spina E, de Leon J. Metabolic drug interactions with newer antipsychotics: a comparative review. Basic Clin Pharmacol Toxicol. 2007;100:4-22.
6. Preskorn SH. Drug-drug interactions: proof of relevance (part II): cause of tolerability problems or noncompliance. J Psychiatr Pract. 2005;11:397-401.
7. Weiden PJ, Mackell JA, McDonnell D. Obesity as a risk factor for antipsychotic noncompliance. Schizophr Res. 2004;66:51-7.
8. Preskorn SH, Flockhart D. 2006 guide to psychiatric drug interactions. Prim Psychiatry. 2006;13:35-64.
9. Spina E, Perucca E. Clinical significance of pharmacokinetic interactions between antiepileptic and psychotropic drugs. Epilepsia. 2002;43(suppl 2):37-44.
10. Meyer JM. Drug-drug interactions with antipsychotics. CNS Spectr. 2007;12:6-9.
11. Takahashi H, Yoshida K, Higuchi H, et al. Development of parkinsonian symptoms after discontinuation of carbamazepine in patients concurrently treated with risperidone: two case reports. Clin Neuropharmacol. 2001;24:358-360.
12. Rendic S. Summary of information on human CYP enzymes: human P450 metabolism data. Drug Metab Rev. 2002;34:83-448.
13. Hong CC, Tang BK, Hammond GL, et al. Cytochrome P450 1A2 (CYP1A2) activity and risk factors for breast cancer: a cross-sectional study. Breast Cancer Res. 2004;6:R352-365.
14. Cozza KL, Armstrong SC, Oesterheld JR. Concise guide to drug interaction principles for medical practice: cytochrome P450s, UGTS, p-glycoproteins. Washington, DC: American Psychiatric Press, Inc; 2003.
15. Kirchheiner J, Seeringer A. Clinical implications of pharmacogenetics of cytochrome P450 drug metabolizing enzymes. Biochim Biophys Acta. 2007;1770:489-494.
16. Kotlyar M, Brauer LH, Tracy TS, et al. Inhibition of CYP2D6 activity by bupropion. J Clin Psychopharmacol. 2005;25:226-229.
17. Uhr M, Tontsch A, Namendorf C, et al. Polymorphisms in the drug transporter gene ABCB1 predict antidepressant treatment response in depression. Neuron. 2008;57:203-209.
18. Bozina N, Kuzman MR, Medved V, et al. Associations between MDR1 gene polymorphisms and schizophrenia and therapeutic response to olanzapine in female schizophrenic patients. J Psychiatr Res. 2008;42:89-97.
19. Kim RB. Drugs as p-glycoprotein substrates, inhibitors, and inducers. Drug Metab Rev. 2002;34:47-54.
20. Hollenberg PF. Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes. Drug Metab Rev. 2002;34:17-35.
21. Physicians’ desk reference. 62nd ed. Montvale, NJ: Thomson Healthcare Inc.; 2007.
22. Heykants J, Huang ML, Mannens G, et al. The pharmacokinetics of risperidone in humans: a summary. J Clin Psychiatry. 1994;55 (suppl):13-7.
23. de Leon J, Bork J. Risperidone and cytochrome P450 3A. J Clin Psychiatry. 1997;58:450.-
24. Lane HY, Chang WH. Risperidone-carbamazepine interactions: is cytochrome P450 3A involved? J Clin Psychiatry. 1998;59:430-431.
25. Ejsing TB, Pedersen AD, Linnet K. P-glycoprotein interaction with risperidone and 9-OH-risperidone studied in vitro, in knock-out mice and in drug-drug interaction experiments. Hum Psychopharmacol. 2005;20:493-500.
26. Cousein E, Barthelemy C, Poullain S, et al. P-glycoprotein and cytochrome P450 3A4 involvement in risperidone transport using an in vitro Caco-2/TC7 model and an in vivo model. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:878-886.
27. Rostami-Hodjegan A, Amin AM, et al. Influence of dose, cigarette smoking, age, sex, and metabolic activity on plasma clozapine concentrations: a predictive model and nomograms to aid clozapine dose adjustment and to assess compliance in individual patients. J Clin Psychopharmacol. 2004;24:70-78.
28. Flockhart DA. Drug interactions: cytochrome P450 drug interaction table. Indiana University School of Medicine. 2007. Available at: http://medicine.iupui.edu/flockhart/table.htm. Accessed October 22, 2008.
Mr. P, age 35 with schizophrenia and seizure disorder, has been maintained on risperidone, 6 mg qhs, and phenytoin, 300 mg qhs. For clinical reasons, the treating neurologist changes the anticonvulsant to divalproex. One week later, Mr. P presents to the emergency room complaining of jaw and neck stiffness.
Ms. K, age 43 with a history of schizoaffective disorder, bipolar type, and erratic medication adherence, is being treated with quetiapine, 600 mg at bedtime, and carbamazepine, 1,000 mg/d. Between appointments she stops taking carbamazepine, believing it is causing her to hear voices from her television. Two weeks later, the manager of Ms. K’s independent living facility tells the psychiatrist that the patient appears excessively sedated and has fallen twice in the past few days.
Mrs. T, a 39-year-old state hospital resident with schizoaffective disorder, bipolar type, has been treated with clozapine, 250 mg bid for 6 months; her most recent trough serum level was 492 ng/mL. She smokes 15 cigarettes/d. Two weeks after the hospital institutes a no-smoking policy, Mrs. T complains of excessive drooling and lightheadedness. Her trough clozapine level is now 875 ng/mL.
Discontinuing a medication that has enzyme-inducing effects presents a hidden problem for patients receiving antipsychotic pharmacotherapy. Certain hepatic enzymes responsible for antipsychotic metabolism—as well those involved in intercellular drug transport—are induced by medication or environmental exposures.1,2 Adding a medication that induces these enzymes to the regimen of a patient receiving antipsychotic therapy can result in markedly reduced serum antipsychotic levels, and discontinuing an inducing agent can result in increased antipsychotic levels.
Drug-drug interactions (DDIs) are a substantial contributor to adverse drug reactions (Box).3-7 Antipsychotic prescribing information highlights potential DDIs from the use of enzyme inhibitors and inducers but identifies only effects caused by adding a second agent. The prescriber remains the sole line of defense for monitoring for DDIs when discontinuing a medication that has inducing or inhibiting effects.
Most psychiatrists are aware that certain medications have clinically significant effects on cytochrome P450 (CYP) activity and of the potential for CYP inhibitors to generate DDIs. Clinicians often are aware of antidepressant medications’ CYP-inhibiting effects, know that levels of other medications will change when discontinuing a potent P450 inhibitor, and understand the need to increase dosages of medications influenced by such agents.8
However, few studies have evaluated the effects of enzyme induction on antipsychotic drug levels,9,10 and the literature rarely discusses changes in serum drug levels after loss of enzyme or drug transport induction.11 If unrecognized, these changes may have significant clinical consequences.
Drug-drug interactions (DDIs) are a common and often preventable cause of morbidity and mortality. National surveillance data showed 700,000 emergency room visits related to adverse drug reactions (ADRs) in the 2 years from January 2004 through 2005.3 ADRs are particularly concerning for psychiatrists managing polypharmacy regimens for patients with severe mental disorders such as schizophrenia.
Literature on DDIs with antipsychotics focuses primarily on kinetic interactions that generate supratherapeutic drug levels.4,5 Because development of side effects is associated with reduced adherence, these kinetic interactions may increase the risk of adverse effects and lead to patients stopping the antipsychotic treatment.6,7
Two induction pathways
The primary mechanism underlying clinically significant DDIs occurs during CYP-mediated phase I metabolism. Molecules undergo oxidative conversion into metabolites that can be conjugated by phase II enzymes, generating more soluble forms that facilitate excretion.
The workhorse of human CYP metabolism is 3A4 (Table 1),12,13 which comprises 30% of hepatic activity and 70% of gut cytochrome activity.14 CYP 1A2 is responsible for 10% to 15% of CYP activity.
Both CYP 3A4 and 1A2 are inducible. A wide variety of medications induce 3A4 activity. The list of 1A2 inducers is shorter; the most common are aryl hydrocarbons from cigarette smoke and proton pump inhibitors.
CYP 2D6 accounts for 20% of hepatic cytochrome activity but is not inducible. CYP 2D6 is well known to psychiatrists because some selective serotonin reuptake inhibitors (SSRIs) and the non-SSRI antidepressant bupropion are potent inhibitors of this enzyme.15,16
P-glycoprotein (PGP) induction. Transmembrane shuttles such as P-glycoprotein (PGP) are an important component of drug disposition. PGP belongs to the family of ATP binding cassette (ABC) transporters that bring molecules across cellular barriers.17,18 It was first described in cancer cells that developed multiple drug resistance (MDR) and is often referred to as MDR1.19 PGP is encoded on human chromosome 7 and expressed in normal tissues, particularly in areas where cells seek to limit drug influx, such as those lining the luminal surface of the small and large intestine and those lining the blood-brain barrier and blood-testis barrier. The expression of PGP in hepatic cells promotes drug clearance by enhancing biliary drug excretion.
PGP is encoded on the same chromosome as CYP 3A4, and these 2 proteins frequently are expressed in the same cells, particularly in the intestinal lining and liver. Moreover, PGP is inducible, and there is substantial overlap between medications that are substrates for—or inducers of—PGP and CYP 3A4 activity. This makes it challenging to determine whether the kinetic effects of a second medication are the result of interference of 3A4, PGP, or both.
Polymorphisms in PGP activity may influence the penetration of psychotropic medications into the CNS. Studies indicate an association between certain PGP polymorphisms and treatment outcomes.17,18
Table 1
What induces CYP 1A2 and 3A4?
| Enzyme | Description | Inducers* |
|---|---|---|
| CYP 1A2 |
| Aryl hydrocarbons (smoking), protonpump inhibitors (omeprazole > lansoprazole > pantoprazole), modafinil, St. John’s wort, chargrilled meat, cruciferous vegetables such as broccoli and cabbage, flavones, protein supplements |
| CYP 3A4 |
| Carbamazepine, phenytoin, phenobarbital, rifampin, oxcarbazepine, efavirenz, glucocorticoids, modafinil, nevirapine, pioglitazone, St. John’s wort |
| * Listed in order from strongest to weakest induction | ||
| CYP: cytochrome P450; PGP: P-glycoprotein | ||
| Source: References 12,13 | ||
Stopping an inducer
In general, inducers of CYP enzymes stimulate gene transcription within hours of exposure; maximum transcriptional activity occurs after 10 to 12 hours of exposure. As transcription increases, the concentration of the CYP mRNA transcript steadily accumulates, as does concentration of CYP protein.
After an inducer is discontinued, transcription returns to basal levels within 18 hours; however, the degradation of CYP proteins is a first-order process, with a half-life of 8 to 30 hours. As a result, the decrease in cellular CYP concentration—and the level of activity—lags behind the decreased synthesis from reduced mRNA levels.
Interactions with antipsychotics
Effects on serum antipsychotic levels caused by discontinuing a CYP or PGP inducer can be predicted from data on decreases in antipsychotic levels following inducer exposure. Except for ziprasidone and paliperidone, most atypical antipsychotics are prone to substantial decreases during concomitant inducer use (Table 2).21
The effect of enzyme inducers on risperidone is particularly interesting. Conversion of risperidone to its active metabolite 9-OH risperidone (paliperidone) occurs primarily via 2D6,22 yet concurrent use of carbamazepine—a potent CYP 3A4 inducer—results in a 50% decrease in the concentration of the active moiety (risperidone plus 9-OH risperidone). This finding and other early investigations suggested that CYP 3A had a role in risperidone metabolism,23,24 but these early studies and case series often involved molecules that had activity at both 3A4 and PGP. Further research clarified that effects on PGP—and not 3A4—are responsible for the changes in risperidone metabolism observed with the use of carbamazepine and other medications.25,26
Induction in case patients: Follow-up. Regardless of whether induction is mediated by ≥1 metabolic pathways, the loss of the inducer will result in serum antipsychotic increases that are proportional to the initial decrease.20 For example, with risperidone, the expected decrease is 50%. Therefore, after Mr. P stopped taking phenytoin, his serum risperidone level would be expected to double, which resulted in extrapyramidal side effects.
Quetiapine clearance is increased 5-fold by inducer exposure, so a clinician treating Ms. K would expect a marked increase in somnolence—and possibly orthostasis—as serum quetiapine levels peak 1 to 2 weeks following carbamazepine discontinuation.
The effects of smoking cessation on serum clozapine levels have been well-documented.1,27 Clinicians should anticipate median increases in serum clozapine levels of 55% after a patient discontinues smoking (aryl hydrocarbon exposure), but changes vary substantially among individuals. Mrs. T’s serum clozapine increased approximately 78%.
Careful clinical monitoring and slow downward adjustment of antipsychotic doses could have prevented the adverse effects these 3 patients experienced after loss of CYP/PGP induction and the consequences those side effects present for future medication adherence. When loss of induction is unplanned—as when Ms. K stopped taking carbamazepine but continued quetiapine—clinicians need to be alert to the fact that the patient was prescribed an inducer and include the loss of induction as a hypothesis for the patient’s somnolence.
Table 2
Effects of CYP/PGP induction on atypical antipsychotics
| Antipsychotic | Metabolic pathways | Effect of induction |
|---|---|---|
| Aripiprazole | 2D6 and 3A4 convert aripiprazole to active metabolite dehydro-aripiprazole | 3A4 induction decreases maximum concentration of aripiprazole and metabolite by 70% |
| Clozapine | Multiple enzymes convert clozapine to N-desmethylclozapine; mean contributions of CYP 1A2, 2C19, 3A4, 2C9, and 2D6 are 30%, 24%, 22%, 12%, and 6%, respectively, with CYP 1A2 predominantly involved at low concentrations | Loss of smoking-related 1A2 induction results in 50% increase in serum levels |
| Olanzapine | Direct glucuronidation or 1A2-mediated oxidation to N-desmethlyolanzapine | Carbamazepine use increases clearance by 50%. Olanzapine concentration:dose ratio is about 5-fold lower in smokers (7.9 +/- 2.6) than in nonsmokers (1.56 +/- 1.1; P |
| Paliperidone | 59% excreted unchanged in urine; phase I metabolism accounts for ≤10% of drug clearance | Unlikely to significantly impact levels, but impact of PGP induction is unknown |
| Quetiapine | 3A4-mediated sulfoxidation to inactive metabolite is primary pathway, but numerous metabolites noted, with 1 active metabolite (norquetiapine) | Phenytoin increases clearance 5-fold |
| Risperidone | 2D6 converts risperidone to active metabolite 9-OH risperidone | In a drug interaction study of risperidone, 6 mg/d for 3 weeks, followed by 3 weeks of carbamazepine, active moiety concentration was decreased by about 50% |
| Ziprasidone | 3A4 (~1/3); aldehyde oxidase (~2/3) | Approximately 35% decrease in ziprasidone exposure by carbamazepine |
| CYP: cytochrome P450; PGP: P-glycoprotein | ||
| Source: Reference 21 | ||
Clinical considerations
In the absence of detailed data on antipsychotic metabolism, clinicians can make intelligent decisions regarding potential DDIs by:
- knowing the extent of induction by common offenders (such as carbamazepine or phenytoin) documented in the medication’s prescribing information or demonstrated through convincing case reports or case series
- memorizing the list of CYP 1A2 and CYP 3A4/PGP inducers.
Patients who may be susceptible to effects from loss of enzyme induction (including smokers receiving olanzapine or clozapine or others taking 3A4/PGP inducers) must be identified, and plans made for dosage adjustments if inducing agents are discontinued for a sufficient time (≥1 week) to result in downregulation of CYP or PGP activity. A slow taper of the antipsychotic over 1 to 2 weeks to the new target dose should compensate for loss of enzyme or PGP induction.
For newer antipsychotic medications with limited data, the proposed discontinuation of an inducer should, at the minimum, prompt a discussion between the psychiatrist and patient regarding the expected increase in serum antipsychotic levels and potential adverse effects that may result. Clinicians also must make every attempt to stay apprised of a patient’s current medications, bearing in mind that another provider may prescribe an inducer. Patients with schizophrenia always should be educated to contact the psychiatrist following any change in medication regimen, placing particular emphasis on the 1 or 2 medications that are known to be implicated in DDIs with the patient’s current antipsychotic.
- Flockhart DA. Drug interactions: Cytochrome P450 drug interaction table. Indiana University School of Medicine. 2007. http://medicine.iupui.edu/flockhart/table.htm.
- Cozza KL, Armstrong SC, Oesterheld JR. Concise guide to drug interaction principles for medical practice: Cytochrome P450s, UGTS, p-glycoproteins. Washington, DC: American Psychiatric Press, Inc; 2003.
- Aripiprazole • Abilify
- Bupropion • Wellbutrin
- Carbamazepine • Carbatrol, Tegretol
- Clozapine • Clozaril
- Divalproex • Depakote
- Efavirenz • Sustiva
- Lansoprazole • Prevacid
- Modafinil • Provigil
- Nevirapine • Viramune
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Oxcarbazepine • Trileptal
- Paliperidone • Invega
- Pantoprazole • Protonix
- Phenobarbital • Barbita, Luminal, others
- Phenytoin • Dilantin
- Pioglitazone • Actos
- Quetiapine • Seroquel
- Rifampin • Rifadin, Rimactane
- Risperidone • Risperdal
- Ziprasidone • Geodon
Dr. Meyer receives grant/research support from the National Institute of Mental Health, Pfizer Inc., and the University of California. He is a consultant to Bristol-Myers Squibb, Organon, Vanda Pharmaceuticals, and Wyeth, and a speaker for AstraZeneca, Bristol-Myers Squibb, Dainippon Sumitomo Pharma, and Pfizer Inc.
Ms. Leckband reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Mr. P, age 35 with schizophrenia and seizure disorder, has been maintained on risperidone, 6 mg qhs, and phenytoin, 300 mg qhs. For clinical reasons, the treating neurologist changes the anticonvulsant to divalproex. One week later, Mr. P presents to the emergency room complaining of jaw and neck stiffness.
Ms. K, age 43 with a history of schizoaffective disorder, bipolar type, and erratic medication adherence, is being treated with quetiapine, 600 mg at bedtime, and carbamazepine, 1,000 mg/d. Between appointments she stops taking carbamazepine, believing it is causing her to hear voices from her television. Two weeks later, the manager of Ms. K’s independent living facility tells the psychiatrist that the patient appears excessively sedated and has fallen twice in the past few days.
Mrs. T, a 39-year-old state hospital resident with schizoaffective disorder, bipolar type, has been treated with clozapine, 250 mg bid for 6 months; her most recent trough serum level was 492 ng/mL. She smokes 15 cigarettes/d. Two weeks after the hospital institutes a no-smoking policy, Mrs. T complains of excessive drooling and lightheadedness. Her trough clozapine level is now 875 ng/mL.
Discontinuing a medication that has enzyme-inducing effects presents a hidden problem for patients receiving antipsychotic pharmacotherapy. Certain hepatic enzymes responsible for antipsychotic metabolism—as well those involved in intercellular drug transport—are induced by medication or environmental exposures.1,2 Adding a medication that induces these enzymes to the regimen of a patient receiving antipsychotic therapy can result in markedly reduced serum antipsychotic levels, and discontinuing an inducing agent can result in increased antipsychotic levels.
Drug-drug interactions (DDIs) are a substantial contributor to adverse drug reactions (Box).3-7 Antipsychotic prescribing information highlights potential DDIs from the use of enzyme inhibitors and inducers but identifies only effects caused by adding a second agent. The prescriber remains the sole line of defense for monitoring for DDIs when discontinuing a medication that has inducing or inhibiting effects.
Most psychiatrists are aware that certain medications have clinically significant effects on cytochrome P450 (CYP) activity and of the potential for CYP inhibitors to generate DDIs. Clinicians often are aware of antidepressant medications’ CYP-inhibiting effects, know that levels of other medications will change when discontinuing a potent P450 inhibitor, and understand the need to increase dosages of medications influenced by such agents.8
However, few studies have evaluated the effects of enzyme induction on antipsychotic drug levels,9,10 and the literature rarely discusses changes in serum drug levels after loss of enzyme or drug transport induction.11 If unrecognized, these changes may have significant clinical consequences.
Drug-drug interactions (DDIs) are a common and often preventable cause of morbidity and mortality. National surveillance data showed 700,000 emergency room visits related to adverse drug reactions (ADRs) in the 2 years from January 2004 through 2005.3 ADRs are particularly concerning for psychiatrists managing polypharmacy regimens for patients with severe mental disorders such as schizophrenia.
Literature on DDIs with antipsychotics focuses primarily on kinetic interactions that generate supratherapeutic drug levels.4,5 Because development of side effects is associated with reduced adherence, these kinetic interactions may increase the risk of adverse effects and lead to patients stopping the antipsychotic treatment.6,7
Two induction pathways
The primary mechanism underlying clinically significant DDIs occurs during CYP-mediated phase I metabolism. Molecules undergo oxidative conversion into metabolites that can be conjugated by phase II enzymes, generating more soluble forms that facilitate excretion.
The workhorse of human CYP metabolism is 3A4 (Table 1),12,13 which comprises 30% of hepatic activity and 70% of gut cytochrome activity.14 CYP 1A2 is responsible for 10% to 15% of CYP activity.
Both CYP 3A4 and 1A2 are inducible. A wide variety of medications induce 3A4 activity. The list of 1A2 inducers is shorter; the most common are aryl hydrocarbons from cigarette smoke and proton pump inhibitors.
CYP 2D6 accounts for 20% of hepatic cytochrome activity but is not inducible. CYP 2D6 is well known to psychiatrists because some selective serotonin reuptake inhibitors (SSRIs) and the non-SSRI antidepressant bupropion are potent inhibitors of this enzyme.15,16
P-glycoprotein (PGP) induction. Transmembrane shuttles such as P-glycoprotein (PGP) are an important component of drug disposition. PGP belongs to the family of ATP binding cassette (ABC) transporters that bring molecules across cellular barriers.17,18 It was first described in cancer cells that developed multiple drug resistance (MDR) and is often referred to as MDR1.19 PGP is encoded on human chromosome 7 and expressed in normal tissues, particularly in areas where cells seek to limit drug influx, such as those lining the luminal surface of the small and large intestine and those lining the blood-brain barrier and blood-testis barrier. The expression of PGP in hepatic cells promotes drug clearance by enhancing biliary drug excretion.
PGP is encoded on the same chromosome as CYP 3A4, and these 2 proteins frequently are expressed in the same cells, particularly in the intestinal lining and liver. Moreover, PGP is inducible, and there is substantial overlap between medications that are substrates for—or inducers of—PGP and CYP 3A4 activity. This makes it challenging to determine whether the kinetic effects of a second medication are the result of interference of 3A4, PGP, or both.
Polymorphisms in PGP activity may influence the penetration of psychotropic medications into the CNS. Studies indicate an association between certain PGP polymorphisms and treatment outcomes.17,18
Table 1
What induces CYP 1A2 and 3A4?
| Enzyme | Description | Inducers* |
|---|---|---|
| CYP 1A2 |
| Aryl hydrocarbons (smoking), protonpump inhibitors (omeprazole > lansoprazole > pantoprazole), modafinil, St. John’s wort, chargrilled meat, cruciferous vegetables such as broccoli and cabbage, flavones, protein supplements |
| CYP 3A4 |
| Carbamazepine, phenytoin, phenobarbital, rifampin, oxcarbazepine, efavirenz, glucocorticoids, modafinil, nevirapine, pioglitazone, St. John’s wort |
| * Listed in order from strongest to weakest induction | ||
| CYP: cytochrome P450; PGP: P-glycoprotein | ||
| Source: References 12,13 | ||
Stopping an inducer
In general, inducers of CYP enzymes stimulate gene transcription within hours of exposure; maximum transcriptional activity occurs after 10 to 12 hours of exposure. As transcription increases, the concentration of the CYP mRNA transcript steadily accumulates, as does concentration of CYP protein.
After an inducer is discontinued, transcription returns to basal levels within 18 hours; however, the degradation of CYP proteins is a first-order process, with a half-life of 8 to 30 hours. As a result, the decrease in cellular CYP concentration—and the level of activity—lags behind the decreased synthesis from reduced mRNA levels.
Interactions with antipsychotics
Effects on serum antipsychotic levels caused by discontinuing a CYP or PGP inducer can be predicted from data on decreases in antipsychotic levels following inducer exposure. Except for ziprasidone and paliperidone, most atypical antipsychotics are prone to substantial decreases during concomitant inducer use (Table 2).21
The effect of enzyme inducers on risperidone is particularly interesting. Conversion of risperidone to its active metabolite 9-OH risperidone (paliperidone) occurs primarily via 2D6,22 yet concurrent use of carbamazepine—a potent CYP 3A4 inducer—results in a 50% decrease in the concentration of the active moiety (risperidone plus 9-OH risperidone). This finding and other early investigations suggested that CYP 3A had a role in risperidone metabolism,23,24 but these early studies and case series often involved molecules that had activity at both 3A4 and PGP. Further research clarified that effects on PGP—and not 3A4—are responsible for the changes in risperidone metabolism observed with the use of carbamazepine and other medications.25,26
Induction in case patients: Follow-up. Regardless of whether induction is mediated by ≥1 metabolic pathways, the loss of the inducer will result in serum antipsychotic increases that are proportional to the initial decrease.20 For example, with risperidone, the expected decrease is 50%. Therefore, after Mr. P stopped taking phenytoin, his serum risperidone level would be expected to double, which resulted in extrapyramidal side effects.
Quetiapine clearance is increased 5-fold by inducer exposure, so a clinician treating Ms. K would expect a marked increase in somnolence—and possibly orthostasis—as serum quetiapine levels peak 1 to 2 weeks following carbamazepine discontinuation.
The effects of smoking cessation on serum clozapine levels have been well-documented.1,27 Clinicians should anticipate median increases in serum clozapine levels of 55% after a patient discontinues smoking (aryl hydrocarbon exposure), but changes vary substantially among individuals. Mrs. T’s serum clozapine increased approximately 78%.
Careful clinical monitoring and slow downward adjustment of antipsychotic doses could have prevented the adverse effects these 3 patients experienced after loss of CYP/PGP induction and the consequences those side effects present for future medication adherence. When loss of induction is unplanned—as when Ms. K stopped taking carbamazepine but continued quetiapine—clinicians need to be alert to the fact that the patient was prescribed an inducer and include the loss of induction as a hypothesis for the patient’s somnolence.
Table 2
Effects of CYP/PGP induction on atypical antipsychotics
| Antipsychotic | Metabolic pathways | Effect of induction |
|---|---|---|
| Aripiprazole | 2D6 and 3A4 convert aripiprazole to active metabolite dehydro-aripiprazole | 3A4 induction decreases maximum concentration of aripiprazole and metabolite by 70% |
| Clozapine | Multiple enzymes convert clozapine to N-desmethylclozapine; mean contributions of CYP 1A2, 2C19, 3A4, 2C9, and 2D6 are 30%, 24%, 22%, 12%, and 6%, respectively, with CYP 1A2 predominantly involved at low concentrations | Loss of smoking-related 1A2 induction results in 50% increase in serum levels |
| Olanzapine | Direct glucuronidation or 1A2-mediated oxidation to N-desmethlyolanzapine | Carbamazepine use increases clearance by 50%. Olanzapine concentration:dose ratio is about 5-fold lower in smokers (7.9 +/- 2.6) than in nonsmokers (1.56 +/- 1.1; P |
| Paliperidone | 59% excreted unchanged in urine; phase I metabolism accounts for ≤10% of drug clearance | Unlikely to significantly impact levels, but impact of PGP induction is unknown |
| Quetiapine | 3A4-mediated sulfoxidation to inactive metabolite is primary pathway, but numerous metabolites noted, with 1 active metabolite (norquetiapine) | Phenytoin increases clearance 5-fold |
| Risperidone | 2D6 converts risperidone to active metabolite 9-OH risperidone | In a drug interaction study of risperidone, 6 mg/d for 3 weeks, followed by 3 weeks of carbamazepine, active moiety concentration was decreased by about 50% |
| Ziprasidone | 3A4 (~1/3); aldehyde oxidase (~2/3) | Approximately 35% decrease in ziprasidone exposure by carbamazepine |
| CYP: cytochrome P450; PGP: P-glycoprotein | ||
| Source: Reference 21 | ||
Clinical considerations
In the absence of detailed data on antipsychotic metabolism, clinicians can make intelligent decisions regarding potential DDIs by:
- knowing the extent of induction by common offenders (such as carbamazepine or phenytoin) documented in the medication’s prescribing information or demonstrated through convincing case reports or case series
- memorizing the list of CYP 1A2 and CYP 3A4/PGP inducers.
Patients who may be susceptible to effects from loss of enzyme induction (including smokers receiving olanzapine or clozapine or others taking 3A4/PGP inducers) must be identified, and plans made for dosage adjustments if inducing agents are discontinued for a sufficient time (≥1 week) to result in downregulation of CYP or PGP activity. A slow taper of the antipsychotic over 1 to 2 weeks to the new target dose should compensate for loss of enzyme or PGP induction.
For newer antipsychotic medications with limited data, the proposed discontinuation of an inducer should, at the minimum, prompt a discussion between the psychiatrist and patient regarding the expected increase in serum antipsychotic levels and potential adverse effects that may result. Clinicians also must make every attempt to stay apprised of a patient’s current medications, bearing in mind that another provider may prescribe an inducer. Patients with schizophrenia always should be educated to contact the psychiatrist following any change in medication regimen, placing particular emphasis on the 1 or 2 medications that are known to be implicated in DDIs with the patient’s current antipsychotic.
- Flockhart DA. Drug interactions: Cytochrome P450 drug interaction table. Indiana University School of Medicine. 2007. http://medicine.iupui.edu/flockhart/table.htm.
- Cozza KL, Armstrong SC, Oesterheld JR. Concise guide to drug interaction principles for medical practice: Cytochrome P450s, UGTS, p-glycoproteins. Washington, DC: American Psychiatric Press, Inc; 2003.
- Aripiprazole • Abilify
- Bupropion • Wellbutrin
- Carbamazepine • Carbatrol, Tegretol
- Clozapine • Clozaril
- Divalproex • Depakote
- Efavirenz • Sustiva
- Lansoprazole • Prevacid
- Modafinil • Provigil
- Nevirapine • Viramune
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Oxcarbazepine • Trileptal
- Paliperidone • Invega
- Pantoprazole • Protonix
- Phenobarbital • Barbita, Luminal, others
- Phenytoin • Dilantin
- Pioglitazone • Actos
- Quetiapine • Seroquel
- Rifampin • Rifadin, Rimactane
- Risperidone • Risperdal
- Ziprasidone • Geodon
Dr. Meyer receives grant/research support from the National Institute of Mental Health, Pfizer Inc., and the University of California. He is a consultant to Bristol-Myers Squibb, Organon, Vanda Pharmaceuticals, and Wyeth, and a speaker for AstraZeneca, Bristol-Myers Squibb, Dainippon Sumitomo Pharma, and Pfizer Inc.
Ms. Leckband reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Meyer JM. Individual changes in clozapine levels after smoking cessation: results and a predictive model. J Clin Psychopharmacol. 2001;21:569-574.
2. Wong YW, Yeh C, Thyrum PT. The effects of concomitant phenytoin administration on the steady-state pharmacokinetics of quetiapine. J Clin Psychopharmacol. 2001;21:89-93.
3. Budnitz DS, Pollock DA, Weidenbach KN, et al. National surveillance of emergency department visits for outpatient adverse drug events. JAMA. 2006;296:1858-1866.
4. Prior TI, Baker GB. Interactions between the cytochrome P450 system and the second-generation antipsychotics. J Psychiatry Neurosci. 2003;28:99-112.
5. Spina E, de Leon J. Metabolic drug interactions with newer antipsychotics: a comparative review. Basic Clin Pharmacol Toxicol. 2007;100:4-22.
6. Preskorn SH. Drug-drug interactions: proof of relevance (part II): cause of tolerability problems or noncompliance. J Psychiatr Pract. 2005;11:397-401.
7. Weiden PJ, Mackell JA, McDonnell D. Obesity as a risk factor for antipsychotic noncompliance. Schizophr Res. 2004;66:51-7.
8. Preskorn SH, Flockhart D. 2006 guide to psychiatric drug interactions. Prim Psychiatry. 2006;13:35-64.
9. Spina E, Perucca E. Clinical significance of pharmacokinetic interactions between antiepileptic and psychotropic drugs. Epilepsia. 2002;43(suppl 2):37-44.
10. Meyer JM. Drug-drug interactions with antipsychotics. CNS Spectr. 2007;12:6-9.
11. Takahashi H, Yoshida K, Higuchi H, et al. Development of parkinsonian symptoms after discontinuation of carbamazepine in patients concurrently treated with risperidone: two case reports. Clin Neuropharmacol. 2001;24:358-360.
12. Rendic S. Summary of information on human CYP enzymes: human P450 metabolism data. Drug Metab Rev. 2002;34:83-448.
13. Hong CC, Tang BK, Hammond GL, et al. Cytochrome P450 1A2 (CYP1A2) activity and risk factors for breast cancer: a cross-sectional study. Breast Cancer Res. 2004;6:R352-365.
14. Cozza KL, Armstrong SC, Oesterheld JR. Concise guide to drug interaction principles for medical practice: cytochrome P450s, UGTS, p-glycoproteins. Washington, DC: American Psychiatric Press, Inc; 2003.
15. Kirchheiner J, Seeringer A. Clinical implications of pharmacogenetics of cytochrome P450 drug metabolizing enzymes. Biochim Biophys Acta. 2007;1770:489-494.
16. Kotlyar M, Brauer LH, Tracy TS, et al. Inhibition of CYP2D6 activity by bupropion. J Clin Psychopharmacol. 2005;25:226-229.
17. Uhr M, Tontsch A, Namendorf C, et al. Polymorphisms in the drug transporter gene ABCB1 predict antidepressant treatment response in depression. Neuron. 2008;57:203-209.
18. Bozina N, Kuzman MR, Medved V, et al. Associations between MDR1 gene polymorphisms and schizophrenia and therapeutic response to olanzapine in female schizophrenic patients. J Psychiatr Res. 2008;42:89-97.
19. Kim RB. Drugs as p-glycoprotein substrates, inhibitors, and inducers. Drug Metab Rev. 2002;34:47-54.
20. Hollenberg PF. Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes. Drug Metab Rev. 2002;34:17-35.
21. Physicians’ desk reference. 62nd ed. Montvale, NJ: Thomson Healthcare Inc.; 2007.
22. Heykants J, Huang ML, Mannens G, et al. The pharmacokinetics of risperidone in humans: a summary. J Clin Psychiatry. 1994;55 (suppl):13-7.
23. de Leon J, Bork J. Risperidone and cytochrome P450 3A. J Clin Psychiatry. 1997;58:450.-
24. Lane HY, Chang WH. Risperidone-carbamazepine interactions: is cytochrome P450 3A involved? J Clin Psychiatry. 1998;59:430-431.
25. Ejsing TB, Pedersen AD, Linnet K. P-glycoprotein interaction with risperidone and 9-OH-risperidone studied in vitro, in knock-out mice and in drug-drug interaction experiments. Hum Psychopharmacol. 2005;20:493-500.
26. Cousein E, Barthelemy C, Poullain S, et al. P-glycoprotein and cytochrome P450 3A4 involvement in risperidone transport using an in vitro Caco-2/TC7 model and an in vivo model. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:878-886.
27. Rostami-Hodjegan A, Amin AM, et al. Influence of dose, cigarette smoking, age, sex, and metabolic activity on plasma clozapine concentrations: a predictive model and nomograms to aid clozapine dose adjustment and to assess compliance in individual patients. J Clin Psychopharmacol. 2004;24:70-78.
28. Flockhart DA. Drug interactions: cytochrome P450 drug interaction table. Indiana University School of Medicine. 2007. Available at: http://medicine.iupui.edu/flockhart/table.htm. Accessed October 22, 2008.
1. Meyer JM. Individual changes in clozapine levels after smoking cessation: results and a predictive model. J Clin Psychopharmacol. 2001;21:569-574.
2. Wong YW, Yeh C, Thyrum PT. The effects of concomitant phenytoin administration on the steady-state pharmacokinetics of quetiapine. J Clin Psychopharmacol. 2001;21:89-93.
3. Budnitz DS, Pollock DA, Weidenbach KN, et al. National surveillance of emergency department visits for outpatient adverse drug events. JAMA. 2006;296:1858-1866.
4. Prior TI, Baker GB. Interactions between the cytochrome P450 system and the second-generation antipsychotics. J Psychiatry Neurosci. 2003;28:99-112.
5. Spina E, de Leon J. Metabolic drug interactions with newer antipsychotics: a comparative review. Basic Clin Pharmacol Toxicol. 2007;100:4-22.
6. Preskorn SH. Drug-drug interactions: proof of relevance (part II): cause of tolerability problems or noncompliance. J Psychiatr Pract. 2005;11:397-401.
7. Weiden PJ, Mackell JA, McDonnell D. Obesity as a risk factor for antipsychotic noncompliance. Schizophr Res. 2004;66:51-7.
8. Preskorn SH, Flockhart D. 2006 guide to psychiatric drug interactions. Prim Psychiatry. 2006;13:35-64.
9. Spina E, Perucca E. Clinical significance of pharmacokinetic interactions between antiepileptic and psychotropic drugs. Epilepsia. 2002;43(suppl 2):37-44.
10. Meyer JM. Drug-drug interactions with antipsychotics. CNS Spectr. 2007;12:6-9.
11. Takahashi H, Yoshida K, Higuchi H, et al. Development of parkinsonian symptoms after discontinuation of carbamazepine in patients concurrently treated with risperidone: two case reports. Clin Neuropharmacol. 2001;24:358-360.
12. Rendic S. Summary of information on human CYP enzymes: human P450 metabolism data. Drug Metab Rev. 2002;34:83-448.
13. Hong CC, Tang BK, Hammond GL, et al. Cytochrome P450 1A2 (CYP1A2) activity and risk factors for breast cancer: a cross-sectional study. Breast Cancer Res. 2004;6:R352-365.
14. Cozza KL, Armstrong SC, Oesterheld JR. Concise guide to drug interaction principles for medical practice: cytochrome P450s, UGTS, p-glycoproteins. Washington, DC: American Psychiatric Press, Inc; 2003.
15. Kirchheiner J, Seeringer A. Clinical implications of pharmacogenetics of cytochrome P450 drug metabolizing enzymes. Biochim Biophys Acta. 2007;1770:489-494.
16. Kotlyar M, Brauer LH, Tracy TS, et al. Inhibition of CYP2D6 activity by bupropion. J Clin Psychopharmacol. 2005;25:226-229.
17. Uhr M, Tontsch A, Namendorf C, et al. Polymorphisms in the drug transporter gene ABCB1 predict antidepressant treatment response in depression. Neuron. 2008;57:203-209.
18. Bozina N, Kuzman MR, Medved V, et al. Associations between MDR1 gene polymorphisms and schizophrenia and therapeutic response to olanzapine in female schizophrenic patients. J Psychiatr Res. 2008;42:89-97.
19. Kim RB. Drugs as p-glycoprotein substrates, inhibitors, and inducers. Drug Metab Rev. 2002;34:47-54.
20. Hollenberg PF. Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes. Drug Metab Rev. 2002;34:17-35.
21. Physicians’ desk reference. 62nd ed. Montvale, NJ: Thomson Healthcare Inc.; 2007.
22. Heykants J, Huang ML, Mannens G, et al. The pharmacokinetics of risperidone in humans: a summary. J Clin Psychiatry. 1994;55 (suppl):13-7.
23. de Leon J, Bork J. Risperidone and cytochrome P450 3A. J Clin Psychiatry. 1997;58:450.-
24. Lane HY, Chang WH. Risperidone-carbamazepine interactions: is cytochrome P450 3A involved? J Clin Psychiatry. 1998;59:430-431.
25. Ejsing TB, Pedersen AD, Linnet K. P-glycoprotein interaction with risperidone and 9-OH-risperidone studied in vitro, in knock-out mice and in drug-drug interaction experiments. Hum Psychopharmacol. 2005;20:493-500.
26. Cousein E, Barthelemy C, Poullain S, et al. P-glycoprotein and cytochrome P450 3A4 involvement in risperidone transport using an in vitro Caco-2/TC7 model and an in vivo model. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:878-886.
27. Rostami-Hodjegan A, Amin AM, et al. Influence of dose, cigarette smoking, age, sex, and metabolic activity on plasma clozapine concentrations: a predictive model and nomograms to aid clozapine dose adjustment and to assess compliance in individual patients. J Clin Psychopharmacol. 2004;24:70-78.
28. Flockhart DA. Drug interactions: cytochrome P450 drug interaction table. Indiana University School of Medicine. 2007. Available at: http://medicine.iupui.edu/flockhart/table.htm. Accessed October 22, 2008.
When Clozapine is not enough: Augment with lamotrigine?
Current antipsychotics are reasonably effective in treating positive symptoms, but they do less to improve the negative and cognitive symptoms1 that contribute to patients’ long-term poor functional capacity and quality of life.2 So what do psychiatrists do in clinical practice to mitigate antipsychotics’ limitations? We augment.
Schizophrenia patients routinely are treated with polypharmacy—often with antidepressants or anticonvulsants—in attempts to improve negative symptoms, aggression, and impulsivity.3 Most adjuncts, however—including divalproex, antidepressants, and lithium—have shown very small, inconsistent, or no effects.4,5 The only agent with a recent meta-analysis supporting its use as augmentation in treatment-resistant schizophrenia is lamotrigine,6 an anticonvulsant approved for use in epilepsy.7
This article examines the evidence supporting off-label use of lamotrigine as an augmenting agent in schizophrenia and explains the rationale, based on lamotrigine’s probable mechanism of action as a stabilizer of glutamate neurotransmission.
Is lamotrigine worth trying?
Some 20% of schizophrenia patients are considered treatment-resistant, with persistent positive symptoms despite having undergone ≥2 adequate antipsychotic trials.8 Evidence suggests clozapine then should be tried,4 but approximately one-half of treatment-resistant patients do not respond to clozapine. Treatment guidelines are limited for these 10% of schizophrenia patients with an inadequate response to available therapies, including clozapine.4
In a meta-analysis of 5 controlled trials in patients with treatment-resistant schizophrenia, adjunctive lamotrigine was shown to significantly reduce Positive and Negative Syndrome Scale (PANSS) total scores, positive symptom subscores, and negative symptom subscores.6 In these trials, lamotrigine was added to various antipsychotics, including clozapine. Based on the results—as outlined below—we suggest:
- In treatment-resistant patients with residual symptoms while taking clozapine, lamotrigine given in dosages ≥200 mg/d could be a first-line adjunct (Figure 1).
- Lamotrigine augmentation also might help patients whose positive symptoms are adequately controlled but who have persistent negative and/or cognitive symptoms.
- Evidence does not support routine use of lamotrigine in patients taking antipsychotics other than clozapine.
Managing side effects. Lamotrigine is generally well tolerated; in the meta-analysis, nausea was the only side effect more common with lamotrigine (9%) than with placebo (3.9%).6 Close follow-up is required, however, as a few case reports have noted worsening positive symptoms when lamotrigine was added to antipsychotics.9,10
Lamotrigine produces a skin rash in approximately 10% of patients; the rash usually is benign but may be severe, including the potentially fatal Stevens-Johnson syndrome.11 In the meta-analysis, rash was no more likely in patients receiving placebo (3%) than those receiving lamotrigine (2.2%), and no serious rashes were reported.6 Even so, lamotrigine needs to be titrated upwards very slowly over weeks, and patients must be able to monitor for rash.
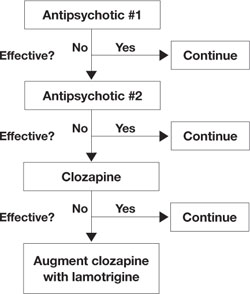
Figure 1 An evidence-based approach to treatment-resistant schizophrenia
Treatment-resistant schizophrenia is defined as residual positive symptoms after ≥2 adequate antipsychotic trials. Evidence supports trying clozapine as the next step.4 When patients show an inadequate response to clozapine, a meta-analysis of 5 controlled trials6 indicates that lamotrigine may be a useful first-line adjunct.
Why consider lamotrigine?
During clinical trials of lamotrigine for epilepsy, patients showed improved mood12 as is seen with other anticonvulsants such as valproate and carbamazepine.13 A series of randomized trials then demonstrated lamotrigine’s effectiveness in treating patients with bipolar I disorder, especially during depressive episodes,14,15 and the FDA approved lamotrigine for maintenance treatment of bipolar I disorder.16 In those early studies, lamotrigine also improved bipolar patients’ quality of life and cognitive function in addition to showing mood-stabilizing properties.12
The glutamate hypothesis. Lamotrigine is an inhibitor of voltage-gated sodium channels and has been shown to inhibit the excessive synaptic release of glutamate.17 Glutamate is the primary excitatory neurotransmitter for at least 60% of neurons in the brain, including all cortical pyramidal neurons. A large body of evidence implicates dysfunctional glutamate signaling in the pathophysiology of schizophrenia.18
For example, phencyclidine (PCP) and ketamine—antagonists of one subtype of glutamate receptor, the N-methyl-D-aspartate (NMDA) receptor—are well known to produce positive psychotic symptoms, negative symptoms, and cognitive dysfunction.19 This led to a long-held hypothesis that schizophrenia is caused by too little glutamate. However, ketamine and PCP also increase the release of glutamate at synapses that then can act on glutamate receptors other than the NMDA receptor, which suggests that too much glutamate also may be involved in schizophrenia.
Too little or too much glutamate? These competing hypotheses could both be at least partially true, suggesting an “inverted-U” pattern of glutamate signaling (Figure 2). Because glutamate is involved in most cortical functions, too little glutamate can cause cognitive and processing deficits such as those seen in schizophrenia. On the other hand, too much glutamate can be toxic to neurons and may be a factor in neurodegeneration, such as in Alzheimer’s disease.20 Indeed, schizophrenia may be associated with gradual neurodegeneration.21
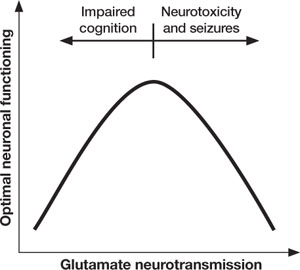
Figure 2 Inverted U-curve may explain dysfunctional glutamate signaling in schizophrenia
Both too little or too much glutamate may play a role in schizophrenia’s pathophysiology. Glutamate, the major excitatory neurotransmitter of the cerebral cortex, is involved in most cognitive functions. Too little (or glutamate inhibition) can impair cognition, whereas too much can lead to seizures, neurotoxicity, and cell death.
Glutamate stabilization?
Because lamotrigine prevents excessive glutamate release at synapses, it stabilizes neuronal membranes by preventing toxicity from too much glutamate without interfering with glutamate’s normal functions.22 Thus, lamotrigine may have potential to maintain optimal glutamate signaling in patients with schizophrenia.
In 16 healthy volunteers, a 300-mg dose of lamotrigine was significantly more effective than placebo in reducing ketamine-induced positive symptoms, as assessed by the Brief Psychiatric Rating Scale positive symptoms subscale (P < .001). Lamotrigine pretreatment also reduced negative symptoms and improved learning and memory.23
More recently, lamotrigine pretreatment was shown to prevent many ketamine-induced changes on functional MRI.24 Few antipsychotics have clinically significant effects on ketamine-induced symptoms—especially in a single dose—although repeated dosing with clozapine attenuates some ketamine-induced effects.25
Given the limitations of available antipsychotics, adding a drug such as lamotrigine—which may modulate and stabilize the glutamate system—could be effective in treatment-resistant schizophrenia.
What is the evidence?
Case reports and open-label case series first showed that lamotrigine augmentation could be effective in treatment-resistant schizophrenia patients receiving clozapine.26–28 One naturalistic case series also included patients receiving olanzapine or risperidone and suggested greater improvement with lamotrigine augmentation in patients on clozapine.26
Controlled trials. In a placebo-controlled trial, Tiihonen et al29 reported significantly lower ratings of positive symptoms—but not negative symptoms—after 38 treatment-resistant schizophrenia patients on clozapine received adjunctive lamotrigine, 200 mg/d, for 14 weeks (Table 1).
A subsequent controlled trial in which Kremer et al30 added lamotrigine, ≤400 mg/d, showed significant improvements in positive and negative symptoms among 31 treatment-resistant schizophrenia patients who completed the 10-week study. Patients were taking conventional and atypical antipsychotics, including clozapine. All groups improved, but the study was not powered to detect differences among the groups.
Table 1
Lamotrigine augmentation: 5 double-blind, placebo-controlled trials
| Trial duration | Patient diagnosis (number) | Antipsychotic(s) | Lamotrigine (mg/d) | Results |
|---|---|---|---|---|
| 14 weeks (Tiihonen et al, 200329) | Treatment-resistant schizophrenia (n=34) | Clozapine | 200 | Significantly reduced psychosis ratings, with no significant improvement in negative symptoms |
| 10 weeks (Kremer et al, 200430) | Treatment-resistant schizophrenia (n=38) | Conventional and atypical, including clozapine | ≤400 | Significant improvements with all antipsychotics, especially clozapine, in positive and negative symptoms* |
| 8 weeks (Akhondzadeh et al, 200531) | Schizophrenia (n=36) | Risperidone | 150 | Significant improvement in negative symptoms and cognition; less improvement in positive symptoms |
| 12 weeks, multicenter (Goff et al, 200732) | Schizophrenia, schizoaffective patients with residual symptoms (n=217+212) | Conventional and atypical, including clozapine | 100 to 400 | No significant improvement in any symptom domain; improved negative symptoms only in study 1 and cognitive symptoms only in study 2 |
| 24 weeks (Zoccali et al, 200733) | Treatment-resistant schizophrenia (n=51) | Clozapine | ≤200 | Significant improvement in positive and negative symptoms as well as some cognitive symptoms |
| * Significance achieved only in study completers, not in the last-observation-carried-forward analysis | ||||
A third trial by Akhondzadeh et al,31 augmenting risperidone with lamotrigine, 150 mg/d, resulted in modest improvements in negative and cognitive symptoms and slight improvement in positive symptoms.
Multicenter trials. Preliminary trials led to 2 randomized, double-blind, multicenter studies. In a total of 429 schizophrenia outpatients with residual psychotic symptoms on atypical antipsychotics, lamotrigine, 100 to 400 mg/d, or placebo was added for 12 weeks.32 The combined results failed to show significant improvement with adjunctive lamotrigine in any symptom domain compared with placebo. One study showed some improved negative symptoms, and the other showed improved cognitive symptoms.
Possible reasons for these negative results were unclear, although:
- a relatively large placebo response, compared with other studies, suggests a “failed” clinical trial
- the small number of patients receiving clozapine in this study suggests that they may have been less treatment-resistant than those enrolled in prior studies.
Meta-analysis. A meta-analysis of data from these 5 randomized, controlled trials found the “positive, negative, and general psychopathology subscale scores as measured with the PANSS … showed significant difference favoring adjuvant lamotrigine” (Table 2).6 As for study limitations, the authors noted that effectiveness data could be usefully analyzed in <70 of the 537 patients from the controlled trials, and “the small mean decrease in scores may not be really clinically relevant.”6 Thus, they said, caution is warranted in translating these results to clinical practice.
One more trial. Since the meta-analysis, an additional placebo-controlled trial has been reported.33 In this 24-week trial, lamotrigine augmentation, ≤200 mg/d, was statistically more effective than placebo in reducing positive and negative symptoms in 51 stable treatment-resistant patients on clozapine. Cognitive function also improved.
Table 2
How symptom scores changed with add-on lamotrigine in the meta-analysis of controlled trials
| PANSS subscales: Individual items scored 1 to 7, with 1=absent and 7=extreme | Change [95% CI]* |
|---|---|
| Positive symptom subscale (max 49) Delusions, conceptual disorganization, hallucinatory behavior, excitement, grandiosity, suspiciousness, hostility | -5.10 [-8.86, -1.34] |
| Negative symptom subscale (max 49) Blunted affect, emotional withdrawal, poor rapport, passive-apathetic social withdrawal, difficulty in abstract thinking, lack of spontaneity and flow of conversation, stereotyped thinking | -5.25 [-7.07, -3.43] |
| General psychopathology subscale (max 112) Somatic concern, anxiety, guilt feelings, tension, mannerisms and posturing, depression, motor retardation, uncooperativeness, unusual thought content, disorientation, poor attention, lack of judgment and insight, disturbance of volition, poor impulse control, preoccupation, active social avoidance | -10.74 [-16.53, -4.96] |
| * See text for limitations of the meta-analysis | |
| CI: confidence interval; PANSS: Positive and Negative Syndrome Scale | |
| Source: Reference 6 | |
Only treatment-resistant patients?
In controlled trials, lamotrigine augmentation has had the greatest effect on positive and negative symptoms in treatment-resistant schizophrenia patients, especially those on clozapine. Could lamotrigine augmentation be of benefit only in treatment-resistant schizophrenia?
Analysis of trial findings. As mentioned, outpatients who comprised the majority of subjects in the 2 large “negative” (or possibly failed) trials32 might have been less treatment-resistant than subjects in the other trials. Lower mean lamotrigine dosages (205 mg/d and 241 mg/d) also were used in the 2 negative trials and in the trial by Akhondzadeh et al (150 mg/d)31—compared with up to 400 mg/d in the trial by Kremer et al.30 This suggests that insufficient dosing might have caused the nonsignificant findings.
Given schizophrenia’s heterogeneity, treatment-resistant patients may represent a subgroup that has greater glutamatergic dysfunction, whereas patients who respond more completely to antipsychotics may have greater dopaminergic dysfunction. Thus, lamotrigine augmentation might be more beneficial in the subset of treatment-resistant patients. Lamotrigine or other glutamate stabilizers have been proposed to act as neuroprotective agents, slowing functional decline in chronic schizophrenia34 (although long-term studies needed to test this hypothesis are unlikely to occur because of cost and time constraints).
Another hypothetical, yet intriguing, explanation for the greater effects of lamotrigine augmentation in patients on clozapine is a pharmacodynamic interaction between these 2 drugs. Clozapine (and possibly olanzapine) have been shown to enhance cortical glutamatergic transmission.25 We propose that clozapine-induced boosting of glutamate in concert with stabilization of the glutamate system by lamotrigine improves neuronal functioning. Clinical trial data regarding lamotrigine augmentation of antipsychotics other than clozapine are needed to determine if the relationship between clozapine and lamotrigine is unique.
Related resources
- Lamotrigine prescribing information and patient handout. www.lamictal.com/bipolar/hcp/prescibing_information. html.
- Augmentation strategies for schizophrenia. IPAP Schizophrenia algorithm flowchart (online interactive version), node 11. www.ipap.org/algorithms.php.
Drug brand names
- Carbamazepine • Carbatrol, Equetro, Tegretol
- Clozapine • Clozaril
- Divalproex • Depakote
- Ketamine • Ketalar
- Lamotrigine • Lamictal
- Olanzapine • Zyprexa
- Risperidone • Risperdal
- Valproate • Depacon, Depakene
Disclosures
Dr. Gray reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Risch receives research support from the National Institute of Mental Health and is a speaker for AstraZeneca and Pfizer Inc.
1. Gray JA, Roth BL. The pipeline and future of drug development in schizophrenia. Mol Psychiatry. 2007;12(10):904-922.
2. Agid Y, Buzsaki G, Diamond DM, et al. How can drug discovery for psychiatric disorders be improved? Nat Rev Drug Discov. 2007;6(3):189-201.
3. Stahl SM, Grady MM. A critical review of atypical antipsychotic utilization: comparing monotherapy with polypharmacy and augmentation. Curr Med Chem. 2004;11(3):313-327.
4. Miller AL, McEvoy SP, Jeste DV, et al. Treatment of chronic schizophrenia. In: Lieberman JA, Stroup TS, Perkins DO, eds. Textbook of schizophrenia. Arlington, VA: American Psychiatric Publishing; 2006:365-381.
5. Miller AL. Combination treatments for schizophrenia. CNS Spectr. 2004;9(9 suppl 9):19-23.
6. Premkumar TS, Pick J. Lamotrigine for schizophrenia. Cochrane Database Syst Rev. 2006;(4):CD005962.-
7. Brodie MJ, Richens A, Yuen AW. Double-blind comparison of lamotrigine and carbamazepine in newly diagnosed epilepsy. UK lamotrigine/carbamazepine monotherapy trial group. Lancet. 1995;345(8948):476-479.
8. Buckley P, Miller A, Olsen J, et al. When symptoms persist: clozapine augmentation strategies. Schizophr Bull. 2001;27(4):615-628.
9. Chan YC, Miller KM, Shaheen N, et al. Worsening of psychotic symptoms in schizophrenia with addition of lamotrigine: a case report. Schizophr Res. 2005;78(2-3):343-345.
10. Konstantakopoulos G, Oulis P, Koulouris GC, et al. Lamotrigine-associated exacerbation of positive symptoms in paranoid schizophrenia. Schizophr Res. 2008;98(1-3):325-326.
11. Messenheimer J, Mullens EL, Giorgi L, et al. Safety review of adult clinical trial experience with lamotrigine. Drug Saf. 1998;18(4):281-296.
12. Smith D, Baker G, Davies G, et al. Outcomes of add-on treatment with lamotrigine in partial epilepsy. Epilepsia. 1993;34(2):312-322.
13. Post RM, Ketter TA, Denicoff K, et al. The place of anticonvulsant therapy in bipolar illness. Psychopharmacology (Berl). 1996;128(2):115-129.
14. Calabrese JR, Bowden CL, Sachs GS, et al. A double-blind placebo-controlled study of lamotrigine monotherapy in outpatients with bipolar I depression. Lamictal 602 study group. J Clin Psychiatry. 1999;60(2):79-88.
15. Calabrese JR, Suppes T, Bowden CL, et al. A double-blind, placebo-controlled, prophylaxis study of lamotrigine in rapid-cycling bipolar disorder. Lamictal 614 study group. J Clin Psychiatry. 2000;61(11):841-850.
16. Bowden CL, Calabrese JR, Sachs G, et al. A placebo-controlled 18-month trial of lamotrigine and lithium maintenance treatment in recently manic or hypomanic patients with bipolar I disorder. Arch Gen Psychiatry. 2003;60(4):392-400.
17. Large CH, Webster EL, Goff DC. The potential role of lamotrigine in schizophrenia. Psychopharmacology (Berl). 2005;181(3):415-436.
18. Goff DC, Coyle JT. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry. 2001;158(9):1367-1377.
19. Javitt DC. Glutamate as a therapeutic target in psychiatric disorders. Mol Psychiatry. 2004;9(11):984-997.
20. Chohan MO, Iqbal K. From tau to toxicity: emerging roles of NMDA receptor in Alzheimer’s disease. J Alzheimers Dis. 2006;10(1):81-87.
21. Konradi C, Heckers S. Molecular aspects of glutamate dysregulation: implications for schizophrenia and its treatment. Pharmacol Ther. 2003;97(2):153-179.
22. Leach MJ, Baxter MG, Critchley MA. Neurochemical and behavioral aspects of lamotrigine. Epilepsia. 1991;32(suppl 2):S4-S8.
23. Anand A, Charney DS, Oren DA, et al. Attenuation of the neuropsychiatric effects of ketamine with lamotrigine: support for hyperglutamatergic effects of n-methyl-D-aspartate receptor antagonists. Arch Gen Psychiatry. 2000;57(3):270-276.
24. Deakin JF, Lees J, McKie S, et al. Glutamate and the neural basis of the subjective effects of ketamine: a pharmaco-magnetic resonance imaging study. Arch Gen Psychiatry. 2008;65(2):154-164.
25. Large CH. Do NMDA receptor antagonist models of schizophrenia predict the clinical efficacy of antipsychotic drugs? J Psychopharmacol. 2007;21(3):283-301.
26. Dursun SM, Deakin JF. Augmenting antipsychotic treatment with lamotrigine or topiramate in patients with treatment-resistant schizophrenia: a naturalistic case-series outcome study. J Psychopharmacol. 2001;15(4):297-301.
27. Dursun SM, McIntosh D, Milliken H. Clozapine plus lamotrigine in treatment-resistant schizophrenia. Arch Gen Psychiatry. 1999;56(10):950.-
28. Saba G, Dumortier G, Kalalou K, et al. Lamotrigine-clozapine combination in refractory schizophrenia: three cases. J Neuropsychiatry Clin Neurosci. 2002;14(1):86.-
29. Tiihonen J, Hallikainen T, Ryynanen OP, et al. Lamotrigine in treatment-resistant schizophrenia: a randomized placebo-controlled crossover trial. Biol Psychiatry. 2003;54(11):1241-1248.
30. Kremer I, Vass A, Gorelik I, et al. Placebo-controlled trial of lamotrigine added to conventional and atypical antipsychotics in schizophrenia. Biol Psychiatry. 2004;56(6):441-446.
31. Akhondzadeh S, Mackinejad K, Ahmadi-Abhari SA, et al. Does the addition of lamotrigine to risperidone improve psychotic symptoms and cognitive impairments in chronic schizophrenia? Therapy. 2005;2(3):399-406.
32. Goff DC, Keefe R, Citrome L, et al. Lamotrigine as add-on therapy in schizophrenia: results of 2 placebo-controlled trials. J Clin Psychopharmacol. 2007;27(6):582-589.
33. Zoccali R, Muscatello MR, Bruno A, et al. The effect of lamotrigine augmentation of clozapine in a sample of treatment-resistant schizophrenic patients: a double-blind, placebo-controlled study. Schizophr Res. 2007;93(1-3):109-116.
34. Lieberman JA, Perkins DO, Jarskog LF. Neuroprotection: a therapeutic strategy to prevent deterioration associated with schizophrenia. CNS Spectr. 2007;12(3 suppl 4):1-13.
Current antipsychotics are reasonably effective in treating positive symptoms, but they do less to improve the negative and cognitive symptoms1 that contribute to patients’ long-term poor functional capacity and quality of life.2 So what do psychiatrists do in clinical practice to mitigate antipsychotics’ limitations? We augment.
Schizophrenia patients routinely are treated with polypharmacy—often with antidepressants or anticonvulsants—in attempts to improve negative symptoms, aggression, and impulsivity.3 Most adjuncts, however—including divalproex, antidepressants, and lithium—have shown very small, inconsistent, or no effects.4,5 The only agent with a recent meta-analysis supporting its use as augmentation in treatment-resistant schizophrenia is lamotrigine,6 an anticonvulsant approved for use in epilepsy.7
This article examines the evidence supporting off-label use of lamotrigine as an augmenting agent in schizophrenia and explains the rationale, based on lamotrigine’s probable mechanism of action as a stabilizer of glutamate neurotransmission.
Is lamotrigine worth trying?
Some 20% of schizophrenia patients are considered treatment-resistant, with persistent positive symptoms despite having undergone ≥2 adequate antipsychotic trials.8 Evidence suggests clozapine then should be tried,4 but approximately one-half of treatment-resistant patients do not respond to clozapine. Treatment guidelines are limited for these 10% of schizophrenia patients with an inadequate response to available therapies, including clozapine.4
In a meta-analysis of 5 controlled trials in patients with treatment-resistant schizophrenia, adjunctive lamotrigine was shown to significantly reduce Positive and Negative Syndrome Scale (PANSS) total scores, positive symptom subscores, and negative symptom subscores.6 In these trials, lamotrigine was added to various antipsychotics, including clozapine. Based on the results—as outlined below—we suggest:
- In treatment-resistant patients with residual symptoms while taking clozapine, lamotrigine given in dosages ≥200 mg/d could be a first-line adjunct (Figure 1).
- Lamotrigine augmentation also might help patients whose positive symptoms are adequately controlled but who have persistent negative and/or cognitive symptoms.
- Evidence does not support routine use of lamotrigine in patients taking antipsychotics other than clozapine.
Managing side effects. Lamotrigine is generally well tolerated; in the meta-analysis, nausea was the only side effect more common with lamotrigine (9%) than with placebo (3.9%).6 Close follow-up is required, however, as a few case reports have noted worsening positive symptoms when lamotrigine was added to antipsychotics.9,10
Lamotrigine produces a skin rash in approximately 10% of patients; the rash usually is benign but may be severe, including the potentially fatal Stevens-Johnson syndrome.11 In the meta-analysis, rash was no more likely in patients receiving placebo (3%) than those receiving lamotrigine (2.2%), and no serious rashes were reported.6 Even so, lamotrigine needs to be titrated upwards very slowly over weeks, and patients must be able to monitor for rash.
Figure 1 An evidence-based approach to treatment-resistant schizophrenia
Treatment-resistant schizophrenia is defined as residual positive symptoms after ≥2 adequate antipsychotic trials. Evidence supports trying clozapine as the next step.4 When patients show an inadequate response to clozapine, a meta-analysis of 5 controlled trials6 indicates that lamotrigine may be a useful first-line adjunct.
Why consider lamotrigine?
During clinical trials of lamotrigine for epilepsy, patients showed improved mood12 as is seen with other anticonvulsants such as valproate and carbamazepine.13 A series of randomized trials then demonstrated lamotrigine’s effectiveness in treating patients with bipolar I disorder, especially during depressive episodes,14,15 and the FDA approved lamotrigine for maintenance treatment of bipolar I disorder.16 In those early studies, lamotrigine also improved bipolar patients’ quality of life and cognitive function in addition to showing mood-stabilizing properties.12
The glutamate hypothesis. Lamotrigine is an inhibitor of voltage-gated sodium channels and has been shown to inhibit the excessive synaptic release of glutamate.17 Glutamate is the primary excitatory neurotransmitter for at least 60% of neurons in the brain, including all cortical pyramidal neurons. A large body of evidence implicates dysfunctional glutamate signaling in the pathophysiology of schizophrenia.18
For example, phencyclidine (PCP) and ketamine—antagonists of one subtype of glutamate receptor, the N-methyl-D-aspartate (NMDA) receptor—are well known to produce positive psychotic symptoms, negative symptoms, and cognitive dysfunction.19 This led to a long-held hypothesis that schizophrenia is caused by too little glutamate. However, ketamine and PCP also increase the release of glutamate at synapses that then can act on glutamate receptors other than the NMDA receptor, which suggests that too much glutamate also may be involved in schizophrenia.
Too little or too much glutamate? These competing hypotheses could both be at least partially true, suggesting an “inverted-U” pattern of glutamate signaling (Figure 2). Because glutamate is involved in most cortical functions, too little glutamate can cause cognitive and processing deficits such as those seen in schizophrenia. On the other hand, too much glutamate can be toxic to neurons and may be a factor in neurodegeneration, such as in Alzheimer’s disease.20 Indeed, schizophrenia may be associated with gradual neurodegeneration.21
Figure 2 Inverted U-curve may explain dysfunctional glutamate signaling in schizophrenia
Both too little or too much glutamate may play a role in schizophrenia’s pathophysiology. Glutamate, the major excitatory neurotransmitter of the cerebral cortex, is involved in most cognitive functions. Too little (or glutamate inhibition) can impair cognition, whereas too much can lead to seizures, neurotoxicity, and cell death.
Glutamate stabilization?
Because lamotrigine prevents excessive glutamate release at synapses, it stabilizes neuronal membranes by preventing toxicity from too much glutamate without interfering with glutamate’s normal functions.22 Thus, lamotrigine may have potential to maintain optimal glutamate signaling in patients with schizophrenia.
In 16 healthy volunteers, a 300-mg dose of lamotrigine was significantly more effective than placebo in reducing ketamine-induced positive symptoms, as assessed by the Brief Psychiatric Rating Scale positive symptoms subscale (P < .001). Lamotrigine pretreatment also reduced negative symptoms and improved learning and memory.23
More recently, lamotrigine pretreatment was shown to prevent many ketamine-induced changes on functional MRI.24 Few antipsychotics have clinically significant effects on ketamine-induced symptoms—especially in a single dose—although repeated dosing with clozapine attenuates some ketamine-induced effects.25
Given the limitations of available antipsychotics, adding a drug such as lamotrigine—which may modulate and stabilize the glutamate system—could be effective in treatment-resistant schizophrenia.
What is the evidence?
Case reports and open-label case series first showed that lamotrigine augmentation could be effective in treatment-resistant schizophrenia patients receiving clozapine.26–28 One naturalistic case series also included patients receiving olanzapine or risperidone and suggested greater improvement with lamotrigine augmentation in patients on clozapine.26
Controlled trials. In a placebo-controlled trial, Tiihonen et al29 reported significantly lower ratings of positive symptoms—but not negative symptoms—after 38 treatment-resistant schizophrenia patients on clozapine received adjunctive lamotrigine, 200 mg/d, for 14 weeks (Table 1).
A subsequent controlled trial in which Kremer et al30 added lamotrigine, ≤400 mg/d, showed significant improvements in positive and negative symptoms among 31 treatment-resistant schizophrenia patients who completed the 10-week study. Patients were taking conventional and atypical antipsychotics, including clozapine. All groups improved, but the study was not powered to detect differences among the groups.
Table 1
Lamotrigine augmentation: 5 double-blind, placebo-controlled trials
| Trial duration | Patient diagnosis (number) | Antipsychotic(s) | Lamotrigine (mg/d) | Results |
|---|---|---|---|---|
| 14 weeks (Tiihonen et al, 200329) | Treatment-resistant schizophrenia (n=34) | Clozapine | 200 | Significantly reduced psychosis ratings, with no significant improvement in negative symptoms |
| 10 weeks (Kremer et al, 200430) | Treatment-resistant schizophrenia (n=38) | Conventional and atypical, including clozapine | ≤400 | Significant improvements with all antipsychotics, especially clozapine, in positive and negative symptoms* |
| 8 weeks (Akhondzadeh et al, 200531) | Schizophrenia (n=36) | Risperidone | 150 | Significant improvement in negative symptoms and cognition; less improvement in positive symptoms |
| 12 weeks, multicenter (Goff et al, 200732) | Schizophrenia, schizoaffective patients with residual symptoms (n=217+212) | Conventional and atypical, including clozapine | 100 to 400 | No significant improvement in any symptom domain; improved negative symptoms only in study 1 and cognitive symptoms only in study 2 |
| 24 weeks (Zoccali et al, 200733) | Treatment-resistant schizophrenia (n=51) | Clozapine | ≤200 | Significant improvement in positive and negative symptoms as well as some cognitive symptoms |
| * Significance achieved only in study completers, not in the last-observation-carried-forward analysis | ||||
A third trial by Akhondzadeh et al,31 augmenting risperidone with lamotrigine, 150 mg/d, resulted in modest improvements in negative and cognitive symptoms and slight improvement in positive symptoms.
Multicenter trials. Preliminary trials led to 2 randomized, double-blind, multicenter studies. In a total of 429 schizophrenia outpatients with residual psychotic symptoms on atypical antipsychotics, lamotrigine, 100 to 400 mg/d, or placebo was added for 12 weeks.32 The combined results failed to show significant improvement with adjunctive lamotrigine in any symptom domain compared with placebo. One study showed some improved negative symptoms, and the other showed improved cognitive symptoms.
Possible reasons for these negative results were unclear, although:
- a relatively large placebo response, compared with other studies, suggests a “failed” clinical trial
- the small number of patients receiving clozapine in this study suggests that they may have been less treatment-resistant than those enrolled in prior studies.
Meta-analysis. A meta-analysis of data from these 5 randomized, controlled trials found the “positive, negative, and general psychopathology subscale scores as measured with the PANSS … showed significant difference favoring adjuvant lamotrigine” (Table 2).6 As for study limitations, the authors noted that effectiveness data could be usefully analyzed in <70 of the 537 patients from the controlled trials, and “the small mean decrease in scores may not be really clinically relevant.”6 Thus, they said, caution is warranted in translating these results to clinical practice.
One more trial. Since the meta-analysis, an additional placebo-controlled trial has been reported.33 In this 24-week trial, lamotrigine augmentation, ≤200 mg/d, was statistically more effective than placebo in reducing positive and negative symptoms in 51 stable treatment-resistant patients on clozapine. Cognitive function also improved.
Table 2
How symptom scores changed with add-on lamotrigine in the meta-analysis of controlled trials
| PANSS subscales: Individual items scored 1 to 7, with 1=absent and 7=extreme | Change [95% CI]* |
|---|---|
| Positive symptom subscale (max 49) Delusions, conceptual disorganization, hallucinatory behavior, excitement, grandiosity, suspiciousness, hostility | -5.10 [-8.86, -1.34] |
| Negative symptom subscale (max 49) Blunted affect, emotional withdrawal, poor rapport, passive-apathetic social withdrawal, difficulty in abstract thinking, lack of spontaneity and flow of conversation, stereotyped thinking | -5.25 [-7.07, -3.43] |
| General psychopathology subscale (max 112) Somatic concern, anxiety, guilt feelings, tension, mannerisms and posturing, depression, motor retardation, uncooperativeness, unusual thought content, disorientation, poor attention, lack of judgment and insight, disturbance of volition, poor impulse control, preoccupation, active social avoidance | -10.74 [-16.53, -4.96] |
| * See text for limitations of the meta-analysis | |
| CI: confidence interval; PANSS: Positive and Negative Syndrome Scale | |
| Source: Reference 6 | |
Only treatment-resistant patients?
In controlled trials, lamotrigine augmentation has had the greatest effect on positive and negative symptoms in treatment-resistant schizophrenia patients, especially those on clozapine. Could lamotrigine augmentation be of benefit only in treatment-resistant schizophrenia?
Analysis of trial findings. As mentioned, outpatients who comprised the majority of subjects in the 2 large “negative” (or possibly failed) trials32 might have been less treatment-resistant than subjects in the other trials. Lower mean lamotrigine dosages (205 mg/d and 241 mg/d) also were used in the 2 negative trials and in the trial by Akhondzadeh et al (150 mg/d)31—compared with up to 400 mg/d in the trial by Kremer et al.30 This suggests that insufficient dosing might have caused the nonsignificant findings.
Given schizophrenia’s heterogeneity, treatment-resistant patients may represent a subgroup that has greater glutamatergic dysfunction, whereas patients who respond more completely to antipsychotics may have greater dopaminergic dysfunction. Thus, lamotrigine augmentation might be more beneficial in the subset of treatment-resistant patients. Lamotrigine or other glutamate stabilizers have been proposed to act as neuroprotective agents, slowing functional decline in chronic schizophrenia34 (although long-term studies needed to test this hypothesis are unlikely to occur because of cost and time constraints).
Another hypothetical, yet intriguing, explanation for the greater effects of lamotrigine augmentation in patients on clozapine is a pharmacodynamic interaction between these 2 drugs. Clozapine (and possibly olanzapine) have been shown to enhance cortical glutamatergic transmission.25 We propose that clozapine-induced boosting of glutamate in concert with stabilization of the glutamate system by lamotrigine improves neuronal functioning. Clinical trial data regarding lamotrigine augmentation of antipsychotics other than clozapine are needed to determine if the relationship between clozapine and lamotrigine is unique.
Related resources
- Lamotrigine prescribing information and patient handout. www.lamictal.com/bipolar/hcp/prescibing_information. html.
- Augmentation strategies for schizophrenia. IPAP Schizophrenia algorithm flowchart (online interactive version), node 11. www.ipap.org/algorithms.php.
Drug brand names
- Carbamazepine • Carbatrol, Equetro, Tegretol
- Clozapine • Clozaril
- Divalproex • Depakote
- Ketamine • Ketalar
- Lamotrigine • Lamictal
- Olanzapine • Zyprexa
- Risperidone • Risperdal
- Valproate • Depacon, Depakene
Disclosures
Dr. Gray reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Risch receives research support from the National Institute of Mental Health and is a speaker for AstraZeneca and Pfizer Inc.
Current antipsychotics are reasonably effective in treating positive symptoms, but they do less to improve the negative and cognitive symptoms1 that contribute to patients’ long-term poor functional capacity and quality of life.2 So what do psychiatrists do in clinical practice to mitigate antipsychotics’ limitations? We augment.
Schizophrenia patients routinely are treated with polypharmacy—often with antidepressants or anticonvulsants—in attempts to improve negative symptoms, aggression, and impulsivity.3 Most adjuncts, however—including divalproex, antidepressants, and lithium—have shown very small, inconsistent, or no effects.4,5 The only agent with a recent meta-analysis supporting its use as augmentation in treatment-resistant schizophrenia is lamotrigine,6 an anticonvulsant approved for use in epilepsy.7
This article examines the evidence supporting off-label use of lamotrigine as an augmenting agent in schizophrenia and explains the rationale, based on lamotrigine’s probable mechanism of action as a stabilizer of glutamate neurotransmission.
Is lamotrigine worth trying?
Some 20% of schizophrenia patients are considered treatment-resistant, with persistent positive symptoms despite having undergone ≥2 adequate antipsychotic trials.8 Evidence suggests clozapine then should be tried,4 but approximately one-half of treatment-resistant patients do not respond to clozapine. Treatment guidelines are limited for these 10% of schizophrenia patients with an inadequate response to available therapies, including clozapine.4
In a meta-analysis of 5 controlled trials in patients with treatment-resistant schizophrenia, adjunctive lamotrigine was shown to significantly reduce Positive and Negative Syndrome Scale (PANSS) total scores, positive symptom subscores, and negative symptom subscores.6 In these trials, lamotrigine was added to various antipsychotics, including clozapine. Based on the results—as outlined below—we suggest:
- In treatment-resistant patients with residual symptoms while taking clozapine, lamotrigine given in dosages ≥200 mg/d could be a first-line adjunct (Figure 1).
- Lamotrigine augmentation also might help patients whose positive symptoms are adequately controlled but who have persistent negative and/or cognitive symptoms.
- Evidence does not support routine use of lamotrigine in patients taking antipsychotics other than clozapine.
Managing side effects. Lamotrigine is generally well tolerated; in the meta-analysis, nausea was the only side effect more common with lamotrigine (9%) than with placebo (3.9%).6 Close follow-up is required, however, as a few case reports have noted worsening positive symptoms when lamotrigine was added to antipsychotics.9,10
Lamotrigine produces a skin rash in approximately 10% of patients; the rash usually is benign but may be severe, including the potentially fatal Stevens-Johnson syndrome.11 In the meta-analysis, rash was no more likely in patients receiving placebo (3%) than those receiving lamotrigine (2.2%), and no serious rashes were reported.6 Even so, lamotrigine needs to be titrated upwards very slowly over weeks, and patients must be able to monitor for rash.
Figure 1 An evidence-based approach to treatment-resistant schizophrenia
Treatment-resistant schizophrenia is defined as residual positive symptoms after ≥2 adequate antipsychotic trials. Evidence supports trying clozapine as the next step.4 When patients show an inadequate response to clozapine, a meta-analysis of 5 controlled trials6 indicates that lamotrigine may be a useful first-line adjunct.
Why consider lamotrigine?
During clinical trials of lamotrigine for epilepsy, patients showed improved mood12 as is seen with other anticonvulsants such as valproate and carbamazepine.13 A series of randomized trials then demonstrated lamotrigine’s effectiveness in treating patients with bipolar I disorder, especially during depressive episodes,14,15 and the FDA approved lamotrigine for maintenance treatment of bipolar I disorder.16 In those early studies, lamotrigine also improved bipolar patients’ quality of life and cognitive function in addition to showing mood-stabilizing properties.12
The glutamate hypothesis. Lamotrigine is an inhibitor of voltage-gated sodium channels and has been shown to inhibit the excessive synaptic release of glutamate.17 Glutamate is the primary excitatory neurotransmitter for at least 60% of neurons in the brain, including all cortical pyramidal neurons. A large body of evidence implicates dysfunctional glutamate signaling in the pathophysiology of schizophrenia.18
For example, phencyclidine (PCP) and ketamine—antagonists of one subtype of glutamate receptor, the N-methyl-D-aspartate (NMDA) receptor—are well known to produce positive psychotic symptoms, negative symptoms, and cognitive dysfunction.19 This led to a long-held hypothesis that schizophrenia is caused by too little glutamate. However, ketamine and PCP also increase the release of glutamate at synapses that then can act on glutamate receptors other than the NMDA receptor, which suggests that too much glutamate also may be involved in schizophrenia.
Too little or too much glutamate? These competing hypotheses could both be at least partially true, suggesting an “inverted-U” pattern of glutamate signaling (Figure 2). Because glutamate is involved in most cortical functions, too little glutamate can cause cognitive and processing deficits such as those seen in schizophrenia. On the other hand, too much glutamate can be toxic to neurons and may be a factor in neurodegeneration, such as in Alzheimer’s disease.20 Indeed, schizophrenia may be associated with gradual neurodegeneration.21
Figure 2 Inverted U-curve may explain dysfunctional glutamate signaling in schizophrenia
Both too little or too much glutamate may play a role in schizophrenia’s pathophysiology. Glutamate, the major excitatory neurotransmitter of the cerebral cortex, is involved in most cognitive functions. Too little (or glutamate inhibition) can impair cognition, whereas too much can lead to seizures, neurotoxicity, and cell death.
Glutamate stabilization?
Because lamotrigine prevents excessive glutamate release at synapses, it stabilizes neuronal membranes by preventing toxicity from too much glutamate without interfering with glutamate’s normal functions.22 Thus, lamotrigine may have potential to maintain optimal glutamate signaling in patients with schizophrenia.
In 16 healthy volunteers, a 300-mg dose of lamotrigine was significantly more effective than placebo in reducing ketamine-induced positive symptoms, as assessed by the Brief Psychiatric Rating Scale positive symptoms subscale (P < .001). Lamotrigine pretreatment also reduced negative symptoms and improved learning and memory.23
More recently, lamotrigine pretreatment was shown to prevent many ketamine-induced changes on functional MRI.24 Few antipsychotics have clinically significant effects on ketamine-induced symptoms—especially in a single dose—although repeated dosing with clozapine attenuates some ketamine-induced effects.25
Given the limitations of available antipsychotics, adding a drug such as lamotrigine—which may modulate and stabilize the glutamate system—could be effective in treatment-resistant schizophrenia.
What is the evidence?
Case reports and open-label case series first showed that lamotrigine augmentation could be effective in treatment-resistant schizophrenia patients receiving clozapine.26–28 One naturalistic case series also included patients receiving olanzapine or risperidone and suggested greater improvement with lamotrigine augmentation in patients on clozapine.26
Controlled trials. In a placebo-controlled trial, Tiihonen et al29 reported significantly lower ratings of positive symptoms—but not negative symptoms—after 38 treatment-resistant schizophrenia patients on clozapine received adjunctive lamotrigine, 200 mg/d, for 14 weeks (Table 1).
A subsequent controlled trial in which Kremer et al30 added lamotrigine, ≤400 mg/d, showed significant improvements in positive and negative symptoms among 31 treatment-resistant schizophrenia patients who completed the 10-week study. Patients were taking conventional and atypical antipsychotics, including clozapine. All groups improved, but the study was not powered to detect differences among the groups.
Table 1
Lamotrigine augmentation: 5 double-blind, placebo-controlled trials
| Trial duration | Patient diagnosis (number) | Antipsychotic(s) | Lamotrigine (mg/d) | Results |
|---|---|---|---|---|
| 14 weeks (Tiihonen et al, 200329) | Treatment-resistant schizophrenia (n=34) | Clozapine | 200 | Significantly reduced psychosis ratings, with no significant improvement in negative symptoms |
| 10 weeks (Kremer et al, 200430) | Treatment-resistant schizophrenia (n=38) | Conventional and atypical, including clozapine | ≤400 | Significant improvements with all antipsychotics, especially clozapine, in positive and negative symptoms* |
| 8 weeks (Akhondzadeh et al, 200531) | Schizophrenia (n=36) | Risperidone | 150 | Significant improvement in negative symptoms and cognition; less improvement in positive symptoms |
| 12 weeks, multicenter (Goff et al, 200732) | Schizophrenia, schizoaffective patients with residual symptoms (n=217+212) | Conventional and atypical, including clozapine | 100 to 400 | No significant improvement in any symptom domain; improved negative symptoms only in study 1 and cognitive symptoms only in study 2 |
| 24 weeks (Zoccali et al, 200733) | Treatment-resistant schizophrenia (n=51) | Clozapine | ≤200 | Significant improvement in positive and negative symptoms as well as some cognitive symptoms |
| * Significance achieved only in study completers, not in the last-observation-carried-forward analysis | ||||
A third trial by Akhondzadeh et al,31 augmenting risperidone with lamotrigine, 150 mg/d, resulted in modest improvements in negative and cognitive symptoms and slight improvement in positive symptoms.
Multicenter trials. Preliminary trials led to 2 randomized, double-blind, multicenter studies. In a total of 429 schizophrenia outpatients with residual psychotic symptoms on atypical antipsychotics, lamotrigine, 100 to 400 mg/d, or placebo was added for 12 weeks.32 The combined results failed to show significant improvement with adjunctive lamotrigine in any symptom domain compared with placebo. One study showed some improved negative symptoms, and the other showed improved cognitive symptoms.
Possible reasons for these negative results were unclear, although:
- a relatively large placebo response, compared with other studies, suggests a “failed” clinical trial
- the small number of patients receiving clozapine in this study suggests that they may have been less treatment-resistant than those enrolled in prior studies.
Meta-analysis. A meta-analysis of data from these 5 randomized, controlled trials found the “positive, negative, and general psychopathology subscale scores as measured with the PANSS … showed significant difference favoring adjuvant lamotrigine” (Table 2).6 As for study limitations, the authors noted that effectiveness data could be usefully analyzed in <70 of the 537 patients from the controlled trials, and “the small mean decrease in scores may not be really clinically relevant.”6 Thus, they said, caution is warranted in translating these results to clinical practice.
One more trial. Since the meta-analysis, an additional placebo-controlled trial has been reported.33 In this 24-week trial, lamotrigine augmentation, ≤200 mg/d, was statistically more effective than placebo in reducing positive and negative symptoms in 51 stable treatment-resistant patients on clozapine. Cognitive function also improved.
Table 2
How symptom scores changed with add-on lamotrigine in the meta-analysis of controlled trials
| PANSS subscales: Individual items scored 1 to 7, with 1=absent and 7=extreme | Change [95% CI]* |
|---|---|
| Positive symptom subscale (max 49) Delusions, conceptual disorganization, hallucinatory behavior, excitement, grandiosity, suspiciousness, hostility | -5.10 [-8.86, -1.34] |
| Negative symptom subscale (max 49) Blunted affect, emotional withdrawal, poor rapport, passive-apathetic social withdrawal, difficulty in abstract thinking, lack of spontaneity and flow of conversation, stereotyped thinking | -5.25 [-7.07, -3.43] |
| General psychopathology subscale (max 112) Somatic concern, anxiety, guilt feelings, tension, mannerisms and posturing, depression, motor retardation, uncooperativeness, unusual thought content, disorientation, poor attention, lack of judgment and insight, disturbance of volition, poor impulse control, preoccupation, active social avoidance | -10.74 [-16.53, -4.96] |
| * See text for limitations of the meta-analysis | |
| CI: confidence interval; PANSS: Positive and Negative Syndrome Scale | |
| Source: Reference 6 | |
Only treatment-resistant patients?
In controlled trials, lamotrigine augmentation has had the greatest effect on positive and negative symptoms in treatment-resistant schizophrenia patients, especially those on clozapine. Could lamotrigine augmentation be of benefit only in treatment-resistant schizophrenia?
Analysis of trial findings. As mentioned, outpatients who comprised the majority of subjects in the 2 large “negative” (or possibly failed) trials32 might have been less treatment-resistant than subjects in the other trials. Lower mean lamotrigine dosages (205 mg/d and 241 mg/d) also were used in the 2 negative trials and in the trial by Akhondzadeh et al (150 mg/d)31—compared with up to 400 mg/d in the trial by Kremer et al.30 This suggests that insufficient dosing might have caused the nonsignificant findings.
Given schizophrenia’s heterogeneity, treatment-resistant patients may represent a subgroup that has greater glutamatergic dysfunction, whereas patients who respond more completely to antipsychotics may have greater dopaminergic dysfunction. Thus, lamotrigine augmentation might be more beneficial in the subset of treatment-resistant patients. Lamotrigine or other glutamate stabilizers have been proposed to act as neuroprotective agents, slowing functional decline in chronic schizophrenia34 (although long-term studies needed to test this hypothesis are unlikely to occur because of cost and time constraints).
Another hypothetical, yet intriguing, explanation for the greater effects of lamotrigine augmentation in patients on clozapine is a pharmacodynamic interaction between these 2 drugs. Clozapine (and possibly olanzapine) have been shown to enhance cortical glutamatergic transmission.25 We propose that clozapine-induced boosting of glutamate in concert with stabilization of the glutamate system by lamotrigine improves neuronal functioning. Clinical trial data regarding lamotrigine augmentation of antipsychotics other than clozapine are needed to determine if the relationship between clozapine and lamotrigine is unique.
Related resources
- Lamotrigine prescribing information and patient handout. www.lamictal.com/bipolar/hcp/prescibing_information. html.
- Augmentation strategies for schizophrenia. IPAP Schizophrenia algorithm flowchart (online interactive version), node 11. www.ipap.org/algorithms.php.
Drug brand names
- Carbamazepine • Carbatrol, Equetro, Tegretol
- Clozapine • Clozaril
- Divalproex • Depakote
- Ketamine • Ketalar
- Lamotrigine • Lamictal
- Olanzapine • Zyprexa
- Risperidone • Risperdal
- Valproate • Depacon, Depakene
Disclosures
Dr. Gray reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Risch receives research support from the National Institute of Mental Health and is a speaker for AstraZeneca and Pfizer Inc.
1. Gray JA, Roth BL. The pipeline and future of drug development in schizophrenia. Mol Psychiatry. 2007;12(10):904-922.
2. Agid Y, Buzsaki G, Diamond DM, et al. How can drug discovery for psychiatric disorders be improved? Nat Rev Drug Discov. 2007;6(3):189-201.
3. Stahl SM, Grady MM. A critical review of atypical antipsychotic utilization: comparing monotherapy with polypharmacy and augmentation. Curr Med Chem. 2004;11(3):313-327.
4. Miller AL, McEvoy SP, Jeste DV, et al. Treatment of chronic schizophrenia. In: Lieberman JA, Stroup TS, Perkins DO, eds. Textbook of schizophrenia. Arlington, VA: American Psychiatric Publishing; 2006:365-381.
5. Miller AL. Combination treatments for schizophrenia. CNS Spectr. 2004;9(9 suppl 9):19-23.
6. Premkumar TS, Pick J. Lamotrigine for schizophrenia. Cochrane Database Syst Rev. 2006;(4):CD005962.-
7. Brodie MJ, Richens A, Yuen AW. Double-blind comparison of lamotrigine and carbamazepine in newly diagnosed epilepsy. UK lamotrigine/carbamazepine monotherapy trial group. Lancet. 1995;345(8948):476-479.
8. Buckley P, Miller A, Olsen J, et al. When symptoms persist: clozapine augmentation strategies. Schizophr Bull. 2001;27(4):615-628.
9. Chan YC, Miller KM, Shaheen N, et al. Worsening of psychotic symptoms in schizophrenia with addition of lamotrigine: a case report. Schizophr Res. 2005;78(2-3):343-345.
10. Konstantakopoulos G, Oulis P, Koulouris GC, et al. Lamotrigine-associated exacerbation of positive symptoms in paranoid schizophrenia. Schizophr Res. 2008;98(1-3):325-326.
11. Messenheimer J, Mullens EL, Giorgi L, et al. Safety review of adult clinical trial experience with lamotrigine. Drug Saf. 1998;18(4):281-296.
12. Smith D, Baker G, Davies G, et al. Outcomes of add-on treatment with lamotrigine in partial epilepsy. Epilepsia. 1993;34(2):312-322.
13. Post RM, Ketter TA, Denicoff K, et al. The place of anticonvulsant therapy in bipolar illness. Psychopharmacology (Berl). 1996;128(2):115-129.
14. Calabrese JR, Bowden CL, Sachs GS, et al. A double-blind placebo-controlled study of lamotrigine monotherapy in outpatients with bipolar I depression. Lamictal 602 study group. J Clin Psychiatry. 1999;60(2):79-88.
15. Calabrese JR, Suppes T, Bowden CL, et al. A double-blind, placebo-controlled, prophylaxis study of lamotrigine in rapid-cycling bipolar disorder. Lamictal 614 study group. J Clin Psychiatry. 2000;61(11):841-850.
16. Bowden CL, Calabrese JR, Sachs G, et al. A placebo-controlled 18-month trial of lamotrigine and lithium maintenance treatment in recently manic or hypomanic patients with bipolar I disorder. Arch Gen Psychiatry. 2003;60(4):392-400.
17. Large CH, Webster EL, Goff DC. The potential role of lamotrigine in schizophrenia. Psychopharmacology (Berl). 2005;181(3):415-436.
18. Goff DC, Coyle JT. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry. 2001;158(9):1367-1377.
19. Javitt DC. Glutamate as a therapeutic target in psychiatric disorders. Mol Psychiatry. 2004;9(11):984-997.
20. Chohan MO, Iqbal K. From tau to toxicity: emerging roles of NMDA receptor in Alzheimer’s disease. J Alzheimers Dis. 2006;10(1):81-87.
21. Konradi C, Heckers S. Molecular aspects of glutamate dysregulation: implications for schizophrenia and its treatment. Pharmacol Ther. 2003;97(2):153-179.
22. Leach MJ, Baxter MG, Critchley MA. Neurochemical and behavioral aspects of lamotrigine. Epilepsia. 1991;32(suppl 2):S4-S8.
23. Anand A, Charney DS, Oren DA, et al. Attenuation of the neuropsychiatric effects of ketamine with lamotrigine: support for hyperglutamatergic effects of n-methyl-D-aspartate receptor antagonists. Arch Gen Psychiatry. 2000;57(3):270-276.
24. Deakin JF, Lees J, McKie S, et al. Glutamate and the neural basis of the subjective effects of ketamine: a pharmaco-magnetic resonance imaging study. Arch Gen Psychiatry. 2008;65(2):154-164.
25. Large CH. Do NMDA receptor antagonist models of schizophrenia predict the clinical efficacy of antipsychotic drugs? J Psychopharmacol. 2007;21(3):283-301.
26. Dursun SM, Deakin JF. Augmenting antipsychotic treatment with lamotrigine or topiramate in patients with treatment-resistant schizophrenia: a naturalistic case-series outcome study. J Psychopharmacol. 2001;15(4):297-301.
27. Dursun SM, McIntosh D, Milliken H. Clozapine plus lamotrigine in treatment-resistant schizophrenia. Arch Gen Psychiatry. 1999;56(10):950.-
28. Saba G, Dumortier G, Kalalou K, et al. Lamotrigine-clozapine combination in refractory schizophrenia: three cases. J Neuropsychiatry Clin Neurosci. 2002;14(1):86.-
29. Tiihonen J, Hallikainen T, Ryynanen OP, et al. Lamotrigine in treatment-resistant schizophrenia: a randomized placebo-controlled crossover trial. Biol Psychiatry. 2003;54(11):1241-1248.
30. Kremer I, Vass A, Gorelik I, et al. Placebo-controlled trial of lamotrigine added to conventional and atypical antipsychotics in schizophrenia. Biol Psychiatry. 2004;56(6):441-446.
31. Akhondzadeh S, Mackinejad K, Ahmadi-Abhari SA, et al. Does the addition of lamotrigine to risperidone improve psychotic symptoms and cognitive impairments in chronic schizophrenia? Therapy. 2005;2(3):399-406.
32. Goff DC, Keefe R, Citrome L, et al. Lamotrigine as add-on therapy in schizophrenia: results of 2 placebo-controlled trials. J Clin Psychopharmacol. 2007;27(6):582-589.
33. Zoccali R, Muscatello MR, Bruno A, et al. The effect of lamotrigine augmentation of clozapine in a sample of treatment-resistant schizophrenic patients: a double-blind, placebo-controlled study. Schizophr Res. 2007;93(1-3):109-116.
34. Lieberman JA, Perkins DO, Jarskog LF. Neuroprotection: a therapeutic strategy to prevent deterioration associated with schizophrenia. CNS Spectr. 2007;12(3 suppl 4):1-13.
1. Gray JA, Roth BL. The pipeline and future of drug development in schizophrenia. Mol Psychiatry. 2007;12(10):904-922.
2. Agid Y, Buzsaki G, Diamond DM, et al. How can drug discovery for psychiatric disorders be improved? Nat Rev Drug Discov. 2007;6(3):189-201.
3. Stahl SM, Grady MM. A critical review of atypical antipsychotic utilization: comparing monotherapy with polypharmacy and augmentation. Curr Med Chem. 2004;11(3):313-327.
4. Miller AL, McEvoy SP, Jeste DV, et al. Treatment of chronic schizophrenia. In: Lieberman JA, Stroup TS, Perkins DO, eds. Textbook of schizophrenia. Arlington, VA: American Psychiatric Publishing; 2006:365-381.
5. Miller AL. Combination treatments for schizophrenia. CNS Spectr. 2004;9(9 suppl 9):19-23.
6. Premkumar TS, Pick J. Lamotrigine for schizophrenia. Cochrane Database Syst Rev. 2006;(4):CD005962.-
7. Brodie MJ, Richens A, Yuen AW. Double-blind comparison of lamotrigine and carbamazepine in newly diagnosed epilepsy. UK lamotrigine/carbamazepine monotherapy trial group. Lancet. 1995;345(8948):476-479.
8. Buckley P, Miller A, Olsen J, et al. When symptoms persist: clozapine augmentation strategies. Schizophr Bull. 2001;27(4):615-628.
9. Chan YC, Miller KM, Shaheen N, et al. Worsening of psychotic symptoms in schizophrenia with addition of lamotrigine: a case report. Schizophr Res. 2005;78(2-3):343-345.
10. Konstantakopoulos G, Oulis P, Koulouris GC, et al. Lamotrigine-associated exacerbation of positive symptoms in paranoid schizophrenia. Schizophr Res. 2008;98(1-3):325-326.
11. Messenheimer J, Mullens EL, Giorgi L, et al. Safety review of adult clinical trial experience with lamotrigine. Drug Saf. 1998;18(4):281-296.
12. Smith D, Baker G, Davies G, et al. Outcomes of add-on treatment with lamotrigine in partial epilepsy. Epilepsia. 1993;34(2):312-322.
13. Post RM, Ketter TA, Denicoff K, et al. The place of anticonvulsant therapy in bipolar illness. Psychopharmacology (Berl). 1996;128(2):115-129.
14. Calabrese JR, Bowden CL, Sachs GS, et al. A double-blind placebo-controlled study of lamotrigine monotherapy in outpatients with bipolar I depression. Lamictal 602 study group. J Clin Psychiatry. 1999;60(2):79-88.
15. Calabrese JR, Suppes T, Bowden CL, et al. A double-blind, placebo-controlled, prophylaxis study of lamotrigine in rapid-cycling bipolar disorder. Lamictal 614 study group. J Clin Psychiatry. 2000;61(11):841-850.
16. Bowden CL, Calabrese JR, Sachs G, et al. A placebo-controlled 18-month trial of lamotrigine and lithium maintenance treatment in recently manic or hypomanic patients with bipolar I disorder. Arch Gen Psychiatry. 2003;60(4):392-400.
17. Large CH, Webster EL, Goff DC. The potential role of lamotrigine in schizophrenia. Psychopharmacology (Berl). 2005;181(3):415-436.
18. Goff DC, Coyle JT. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry. 2001;158(9):1367-1377.
19. Javitt DC. Glutamate as a therapeutic target in psychiatric disorders. Mol Psychiatry. 2004;9(11):984-997.
20. Chohan MO, Iqbal K. From tau to toxicity: emerging roles of NMDA receptor in Alzheimer’s disease. J Alzheimers Dis. 2006;10(1):81-87.
21. Konradi C, Heckers S. Molecular aspects of glutamate dysregulation: implications for schizophrenia and its treatment. Pharmacol Ther. 2003;97(2):153-179.
22. Leach MJ, Baxter MG, Critchley MA. Neurochemical and behavioral aspects of lamotrigine. Epilepsia. 1991;32(suppl 2):S4-S8.
23. Anand A, Charney DS, Oren DA, et al. Attenuation of the neuropsychiatric effects of ketamine with lamotrigine: support for hyperglutamatergic effects of n-methyl-D-aspartate receptor antagonists. Arch Gen Psychiatry. 2000;57(3):270-276.
24. Deakin JF, Lees J, McKie S, et al. Glutamate and the neural basis of the subjective effects of ketamine: a pharmaco-magnetic resonance imaging study. Arch Gen Psychiatry. 2008;65(2):154-164.
25. Large CH. Do NMDA receptor antagonist models of schizophrenia predict the clinical efficacy of antipsychotic drugs? J Psychopharmacol. 2007;21(3):283-301.
26. Dursun SM, Deakin JF. Augmenting antipsychotic treatment with lamotrigine or topiramate in patients with treatment-resistant schizophrenia: a naturalistic case-series outcome study. J Psychopharmacol. 2001;15(4):297-301.
27. Dursun SM, McIntosh D, Milliken H. Clozapine plus lamotrigine in treatment-resistant schizophrenia. Arch Gen Psychiatry. 1999;56(10):950.-
28. Saba G, Dumortier G, Kalalou K, et al. Lamotrigine-clozapine combination in refractory schizophrenia: three cases. J Neuropsychiatry Clin Neurosci. 2002;14(1):86.-
29. Tiihonen J, Hallikainen T, Ryynanen OP, et al. Lamotrigine in treatment-resistant schizophrenia: a randomized placebo-controlled crossover trial. Biol Psychiatry. 2003;54(11):1241-1248.
30. Kremer I, Vass A, Gorelik I, et al. Placebo-controlled trial of lamotrigine added to conventional and atypical antipsychotics in schizophrenia. Biol Psychiatry. 2004;56(6):441-446.
31. Akhondzadeh S, Mackinejad K, Ahmadi-Abhari SA, et al. Does the addition of lamotrigine to risperidone improve psychotic symptoms and cognitive impairments in chronic schizophrenia? Therapy. 2005;2(3):399-406.
32. Goff DC, Keefe R, Citrome L, et al. Lamotrigine as add-on therapy in schizophrenia: results of 2 placebo-controlled trials. J Clin Psychopharmacol. 2007;27(6):582-589.
33. Zoccali R, Muscatello MR, Bruno A, et al. The effect of lamotrigine augmentation of clozapine in a sample of treatment-resistant schizophrenic patients: a double-blind, placebo-controlled study. Schizophr Res. 2007;93(1-3):109-116.
34. Lieberman JA, Perkins DO, Jarskog LF. Neuroprotection: a therapeutic strategy to prevent deterioration associated with schizophrenia. CNS Spectr. 2007;12(3 suppl 4):1-13.
Emerging clues: Is this teen at risk for substance abuse?
Traditionally, clinicians have identified children at high risk for substance abuse disorders (SUDs) by their family history—such as “children of alcoholics.” Advances in etiology research, however, have led to the identification of other risks for SUDs seen during childhood (Table). The clustering of these SUD risk factors—genetic influences, family characteristics, and predictive phenotypes—makes it feasible to identify children and adolescents who are very likely to develop problematic substance use.
Table
Risk factors for substance abuse in children and adolescents
| Genetic predisposition |
| Parental substance use |
| Maltreatment |
| Inadequate supervision |
| Impulsive behavior, inattention, irritability |
| Substance availability |
| Early substance use |
Nature vs nurture
Genetic influences. Heritable risk accounts for a substantial proportion of the variation in SUDs, as multiple genes differentially influence substance initiation, metabolism, and reinforcing properties.1 For example, well-characterized genetic variations determine individual differences in alcohol dehydrogenase and aldehyde dehydrogenase—the enzymes involved in alcohol metabolism—and influence liability to alcohol use disorders (AUDs).2,3 Researchers are exploring ways in which genes might impact SUD risk (Box 1).1,4,5
Genetic influences on substance use may be less important during adolescence than adulthood. In a study of 1,796 male twins’ alcohol, nicotine, and cannabis use from early adolescence to middle adulthood, genetic variations had little or no influence on substance use in early adolescence. The influence of genetic factors gradually increased with age.6
Familial environmental factors, by contrast, were important in early adolescence and gradually decreased in effect with increasing age. During adolescence, the family’s influence on substance use apparently operates more through environmental characteristics than through heritable factors.6
Familial influences. Parents with ongoing SUDs model problematic substance use and create environments of child maltreatment and inadequate supervision.
Child maltreatment. Children of parents with SUDs are more likely to suffer sexual abuse, physical abuse, or neglect.7 The effects of sexual abuse on the child may vary by abuse severity and the child’s gender, developmental stage, and relationship to the perpetrator. Maltreatment may cause the child difficulties in psychological regulation and social development, leading to related psychopathology; these characteristics may contribute to later SUDs.8
Inadequate supervision. Adolescents who report that their parents do not effectively monitor their activities have an increased likelihood of developing SUDs. However, children/adolescents who exhibit difficulties with psychological regulation—such as impulsive behavior and irritability—are difficult to parent, and adolescents with early substance involvement may subvert parental supervision efforts.9,10
Recent investigations have examined genes that might confer risk across substance types. Promising research has focused on:
- genes that influence functional variations in neurotransmitter systems
- gene-environment interactions
- the search for neurobiological endophenotypes—characteristics that cannot be observed by conventional means, such as brain development characteristics that are seen through neuroimaging.1,4,5
Specific molecular-level genetic variations can be measured in individual patients but cannot yet validly quantify risk.
Predictive phenotypes
Predictive phenotypes—measurable individual characteristics that predict SUDs—may be considered risk factors but should not be viewed as causal influences akin to genetic and familial/environmental factors. Rather, predictive phenotypes may reflect propensities that are manifested by specific behaviors and other features according to developmental stage and environmental facilitation.
- specific psychiatric disorders
- specific personality traits that collectively are called psychological dysregulation
- early substance use.11
Recent studies have demonstrated that this clustering of problems—including impulsive behavior, inattention, and negative affect—represents a single continuous dimension termed psychological dysregulation.12 The construct of psychological dysregulation has origins in neuropathology and provides a conceptual link between childhood psychopathological characteristics known to predict SUD and neurobiological deficits.5 Childhood indices of psychological dysregulation—such as the Behavior Rating Inventory of Executive Function (BRIEF)14—complement other risk factors, such as parental SUDs and early substance use, in predicting accelerated substance use and SUDs.15
Neurobiological characteristics. Recent investigations have focused on relationships between variations in normal brain development and differences in psychological regulation.5 Several brain structures thought relevant to the development of psychological regulation—including the prefrontal cortex, limbic structures, and reward circuits—develop during adolescence. Delays or deficits in the development of these structures are called neurodevelopmental dysmaturation.5
Variation in genes that influence these brain areas may interact with environmental influences—including child maltreatment and early substance use—to produce neurodevelopmental dysmaturation that manifests as psychological dysregulation. Thus, genetic and environmental causes are hypothesized to lead to an endophenotype (neurodevelopmental dysmaturation) and developmentally specific phenotypes, such as:
- ADHD in childhood
- CD and accelerated substance use initiation in early adolescence
- SUDs involving alcohol and cannabis in late adolescence.5
Consuming small quantities of alcohol under parental supervision is culturally normative and does not predict problematic drinking.17 On the other hand, regularly consuming “standard drink” quantities of alcohol in late childhood typically occurs in unsupervised settings and predicts adolescent-onset AUDs.18
Problem-focused interview methods—including CAGE, TWEAK, and CRAFFT—have been developed and tested to screen adolescents for AUDs. None has been as consistently successful as the World Health Organization’s Alcohol Use Disorders Identification Test (AUDIT) questionnaire18 (see Related Resources).
Childhood cigarette smoking also predicts accelerated substance use and SUDs.15 Marijuana use predicts both cannabis use disorders and other illicit drug use.1 This observation supports the controversial “gateway hypothesis,” which proposes that marijuana use accelerates the onset of other illicit drug use.15,19,20 An alternate hypothesis proposes that use of marijuana and other illicit drugs is a developmentally specific manifestation of a more general liability for SUDs.1
Identifying those at high risk
Screening for SUD risk factors makes it possible to identify children and adolescents who are very likely to develop problematic substance use. For example, in a study of 560 children age 10 to 12 at recruitment, this author (DBC) identified subjects as high risk if they had 2 parents with SUDs, tobacco or alcohol use by age 12, and high psychological dysregulation as measured by combined assessments of cognitive, emotional, and behavioral regulation. By age 18:
- three-quarters of these adolescents used tobacco daily
- more than one-half had alcohol problems
- nearly one-half had cannabis abuse or dependence.15
Recommendations. Children and adolescents receiving health care services—including primary care, ongoing treatment for chronic conditions, and treatment for psychiatric disorders—should be evaluated for SUD risk. Screening ideally occurs at the initial evaluation or early in the course of treatment. Family history determines genetic risk.
Direct questioning is needed because unstructured evaluations often fail to reveal the presence of important SUD risks.21 Explore possible child maltreatment by questioning the parent and child about physical abuse, sexual abuse, and neglect. Key mental disorders include CD, ADHD, and PTSD. Ask about use of tobacco, alcohol, cannabis, and other drugs. Follow acknowledgement of use with inquiries on frequency, quantity, and problems.
Prevention and early intervention
By identifying characteristics that confer risk for SUDs, you can target these characteristics in prevention and early treatment efforts. These efforts may involve parents as well as children. Many promising approaches have been developed, including universal or selective interventions based on family, school, community, or multi-component approaches.22
Because parental SUDs are a prominent risk factor for children, interventions to reduce or eliminate parental substance use may be helpful, particularly for diminishing childhood psychological dysregulation.23 Early treatment of childhood predictive phenotypes, including CD and ADHD, is another promising approach.12 Community efforts to limit adolescents’ access to addictive substances have met with some success.22
Parents, teachers, and children and adolescents can obtain a wealth of information from the Web sites of the National Institute on Alcohol Abuse and Alcoholism and the National Institute on Drug Abuse (Box 2). The Centers for Disease Control and Prevention offers infomation about preventing smoking (see Related Resources.)
Parents/teachers
The National Institute on Drug Abuse (NIDA) provides a Web site for parents and teachers at www.drugabuse.gov/parent-teacher.html. Parents also can find resources at NIDA for teens at http://teens.drugabuse.gov.
The National Institute on Alcohol Abuse and Alcoholism (NIAAA) provides several free booklets for parents at www.niaaa.nih.gov/Publications/PamphletsBrochuresPosters/English.
My Brain, My Body (www.mybrainmybody.com) is an NIAAA-supported educational tool for teaching middle school students. Developmentally specific NIDA Junior Scientist Programs have been developed for kindergarten through grade 5. The Office of Safe and Drug-Free Schools, part of the U.S. Department of Education, also provides relevant resources.
Children and adolescents
An NIAAA-sponsored educational resource (www.thecoolspot.gov) provides education on alcohol designed for young teens (age 11 to 13). The site includes quizzes, suggestions for resisting peer pressure, and activities that encourage refusing drinking opportunities.
NIDA for Teens (http://teens.drugabuse.gov) provides information and activities designed for those age 11 to 15.
Related resources
- Alcohol Use Disorders Identification Test (AUDIT). World Health Organization. http://whqlibdoc.who.int/hq/2001/WHO_MSD_MSB_01.6a.pdf.
- National Institute on Alcohol Abuse and Alcoholism. www.niaaa.nih.gov.
- National Institute on Drug Abuse. www.nida.nih.gov.
- Centers for Disease Control and Prevention. Youth Tobacco Prevention. www.cdc.gov/tobacco/youth.
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Vanyukov MM, Tarter RE, Kirisci L, et al. Liability to substance use disorders: 1. Common mechanisms and manifestations. Neurosci Biobehav Rev. 2003;27(6):507-515.
2. Karpyak VM, Hall-Flavin DK, Mrazek DA. Can genetics predict risk for alcohol dependence? Current Psychiatry. 2008;7(3):57-73.
3. Kuo PH, Kalsi G, Prescott CA, et al. Association of ADH and ALDH genes with alcohol dependence in the Irish Affected Sib Pair Study of alcohol dependence (IASPSAD) sample. Alcohol Clin Exp Res. 2008;32(5):785-795.
4. Vanyukov MM, Maher BS, Devlin B, et al. The MAOA promoter polymorphism, disruptive behavior disorders, and early onset substance use disorder: gene-environment interaction. Psychiatr Genet. 2007;17(6):323-332.
5. Clark DB, Thatcher DL, Tapert S. Alcohol, psychological dysregulation and adolescent brain development. Alcohol Clin Exp Res. 2008;32(3):375-385.
6. Kendler KS, Schmitt E, Aggen SH, et al. Genetic and environmental influences on alcohol, caffeine, cannabis, and nicotine use from early adolescence to middle adulthood. Arch Gen Psychiatry. 2008;65(6):674-682.
7. Sher KJ, Gershuny BS, Peterson L, et al. The role of childhood stressors in the intergenerational transmission of alcohol use disorders. J Stud Alcohol. 1997;58:414-427.
8. Clark DB, De Bellis MD, Lynch KG, et al. Physical and sexual abuse, depression and alcohol use disorders in adolescents. Drug Alcohol Depend. 2003;69(1):51-60.
9. Clark DB, Thatcher DL, Maisto S. Adolescent neglect and alcohol use disorders in two-parent families. Child Maltreat. 2004;9(4):357-370.
10. Clark DB, Kirisci L, Mezzich A, et al. Parental supervision and alcohol use in early adolescence: developmentally specific interactions. J Dev Behav Pediatr. 2008;29(4):285-292.
11. Clark DB, Kirisci L, Moss HB. Early adolescent gateway drug use in sons of fathers with substance use disorders. Addict Behav. 1998;23:561-566.
12. Clark DB, Winters KC. Measuring risks and outcomes in substance use disorders prevention research. J Consult Clin Psychol. 2002;70(6):1207-1223.
13. Clark DB, Cornelius JR, Wood DS, et al. Psychopathology risk transmission in children of parents with substance use disorders. Am J Psychiatry. 2004;161(4):685-691.
14. Guy SC, Isquith PK, Gioia GA. Behavior rating inventory of executive function—self-report version professional manual. Lutz, FL: Psychological Assessment Resources; 2004.
15. Clark DB, Cornelius JR, Kirisci L, et al. Childhood risk categories for adolescent substance involvement: a general liability typology. Drug Alcohol Depend. 2005;77(1):13-21.
16. Clark DB. The natural history of adolescent alcohol use disorders. Addiction. 2004;99(suppl 2):5-22.
17. Donovan JE, Molina BS. Children’s introduction to alcohol use: sips and tastes. Alcohol Clin Exp Res. 2008;32(1):108-119.
18. Clark DB, Chung T, Martin CS. Alcohol use frequency as a screen for alcohol use disorders in adolescents. Int J Adolesc Med Health. 2006;18(1):181-187.
19. Tarter RE, Kirisci L, Mezzich A, et al. Neurobehavioral disinhibition in childhood predicts early age onset substance use disorder. Am J Psychiatry. 2003;160(6):1078-1085.
20. Tarter RE, Vanyukov M, Kirisci L, et al. Predictors of marijuana use in adolescents before and after licit drug use: examination of the gateway hypothesis. Am J Psychiatry. 2006;163(12):2134-2140.
21. Clark DB, Bukstein OG, Smith MG, et al. Identifying anxiety disorders in adolescents hospitalized for alcohol abuse or dependence. Psychiatr Serv. 1995;46:618-620.
22. Spoth R, Greenberg M, Turrisi R. Preventive interventions addressing underage drinking: state of the evidence and steps toward public health impact. Pediatrics. 2008;121(suppl 4):S311-336.
23. Kelley ML, Fals-Stewart W. Treating paternal drug abuse using Learning Sobriety Together: effects on adolescents versus children. Drug Alcohol Depend. 2008;92(1-3):228-238.
Traditionally, clinicians have identified children at high risk for substance abuse disorders (SUDs) by their family history—such as “children of alcoholics.” Advances in etiology research, however, have led to the identification of other risks for SUDs seen during childhood (Table). The clustering of these SUD risk factors—genetic influences, family characteristics, and predictive phenotypes—makes it feasible to identify children and adolescents who are very likely to develop problematic substance use.
Table
Risk factors for substance abuse in children and adolescents
| Genetic predisposition |
| Parental substance use |
| Maltreatment |
| Inadequate supervision |
| Impulsive behavior, inattention, irritability |
| Substance availability |
| Early substance use |
Nature vs nurture
Genetic influences. Heritable risk accounts for a substantial proportion of the variation in SUDs, as multiple genes differentially influence substance initiation, metabolism, and reinforcing properties.1 For example, well-characterized genetic variations determine individual differences in alcohol dehydrogenase and aldehyde dehydrogenase—the enzymes involved in alcohol metabolism—and influence liability to alcohol use disorders (AUDs).2,3 Researchers are exploring ways in which genes might impact SUD risk (Box 1).1,4,5
Genetic influences on substance use may be less important during adolescence than adulthood. In a study of 1,796 male twins’ alcohol, nicotine, and cannabis use from early adolescence to middle adulthood, genetic variations had little or no influence on substance use in early adolescence. The influence of genetic factors gradually increased with age.6
Familial environmental factors, by contrast, were important in early adolescence and gradually decreased in effect with increasing age. During adolescence, the family’s influence on substance use apparently operates more through environmental characteristics than through heritable factors.6
Familial influences. Parents with ongoing SUDs model problematic substance use and create environments of child maltreatment and inadequate supervision.
Child maltreatment. Children of parents with SUDs are more likely to suffer sexual abuse, physical abuse, or neglect.7 The effects of sexual abuse on the child may vary by abuse severity and the child’s gender, developmental stage, and relationship to the perpetrator. Maltreatment may cause the child difficulties in psychological regulation and social development, leading to related psychopathology; these characteristics may contribute to later SUDs.8
Inadequate supervision. Adolescents who report that their parents do not effectively monitor their activities have an increased likelihood of developing SUDs. However, children/adolescents who exhibit difficulties with psychological regulation—such as impulsive behavior and irritability—are difficult to parent, and adolescents with early substance involvement may subvert parental supervision efforts.9,10
Recent investigations have examined genes that might confer risk across substance types. Promising research has focused on:
- genes that influence functional variations in neurotransmitter systems
- gene-environment interactions
- the search for neurobiological endophenotypes—characteristics that cannot be observed by conventional means, such as brain development characteristics that are seen through neuroimaging.1,4,5
Specific molecular-level genetic variations can be measured in individual patients but cannot yet validly quantify risk.
Predictive phenotypes
Predictive phenotypes—measurable individual characteristics that predict SUDs—may be considered risk factors but should not be viewed as causal influences akin to genetic and familial/environmental factors. Rather, predictive phenotypes may reflect propensities that are manifested by specific behaviors and other features according to developmental stage and environmental facilitation.
- specific psychiatric disorders
- specific personality traits that collectively are called psychological dysregulation
- early substance use.11
Recent studies have demonstrated that this clustering of problems—including impulsive behavior, inattention, and negative affect—represents a single continuous dimension termed psychological dysregulation.12 The construct of psychological dysregulation has origins in neuropathology and provides a conceptual link between childhood psychopathological characteristics known to predict SUD and neurobiological deficits.5 Childhood indices of psychological dysregulation—such as the Behavior Rating Inventory of Executive Function (BRIEF)14—complement other risk factors, such as parental SUDs and early substance use, in predicting accelerated substance use and SUDs.15
Neurobiological characteristics. Recent investigations have focused on relationships between variations in normal brain development and differences in psychological regulation.5 Several brain structures thought relevant to the development of psychological regulation—including the prefrontal cortex, limbic structures, and reward circuits—develop during adolescence. Delays or deficits in the development of these structures are called neurodevelopmental dysmaturation.5
Variation in genes that influence these brain areas may interact with environmental influences—including child maltreatment and early substance use—to produce neurodevelopmental dysmaturation that manifests as psychological dysregulation. Thus, genetic and environmental causes are hypothesized to lead to an endophenotype (neurodevelopmental dysmaturation) and developmentally specific phenotypes, such as:
- ADHD in childhood
- CD and accelerated substance use initiation in early adolescence
- SUDs involving alcohol and cannabis in late adolescence.5
Consuming small quantities of alcohol under parental supervision is culturally normative and does not predict problematic drinking.17 On the other hand, regularly consuming “standard drink” quantities of alcohol in late childhood typically occurs in unsupervised settings and predicts adolescent-onset AUDs.18
Problem-focused interview methods—including CAGE, TWEAK, and CRAFFT—have been developed and tested to screen adolescents for AUDs. None has been as consistently successful as the World Health Organization’s Alcohol Use Disorders Identification Test (AUDIT) questionnaire18 (see Related Resources).
Childhood cigarette smoking also predicts accelerated substance use and SUDs.15 Marijuana use predicts both cannabis use disorders and other illicit drug use.1 This observation supports the controversial “gateway hypothesis,” which proposes that marijuana use accelerates the onset of other illicit drug use.15,19,20 An alternate hypothesis proposes that use of marijuana and other illicit drugs is a developmentally specific manifestation of a more general liability for SUDs.1
Identifying those at high risk
Screening for SUD risk factors makes it possible to identify children and adolescents who are very likely to develop problematic substance use. For example, in a study of 560 children age 10 to 12 at recruitment, this author (DBC) identified subjects as high risk if they had 2 parents with SUDs, tobacco or alcohol use by age 12, and high psychological dysregulation as measured by combined assessments of cognitive, emotional, and behavioral regulation. By age 18:
- three-quarters of these adolescents used tobacco daily
- more than one-half had alcohol problems
- nearly one-half had cannabis abuse or dependence.15
Recommendations. Children and adolescents receiving health care services—including primary care, ongoing treatment for chronic conditions, and treatment for psychiatric disorders—should be evaluated for SUD risk. Screening ideally occurs at the initial evaluation or early in the course of treatment. Family history determines genetic risk.
Direct questioning is needed because unstructured evaluations often fail to reveal the presence of important SUD risks.21 Explore possible child maltreatment by questioning the parent and child about physical abuse, sexual abuse, and neglect. Key mental disorders include CD, ADHD, and PTSD. Ask about use of tobacco, alcohol, cannabis, and other drugs. Follow acknowledgement of use with inquiries on frequency, quantity, and problems.
Prevention and early intervention
By identifying characteristics that confer risk for SUDs, you can target these characteristics in prevention and early treatment efforts. These efforts may involve parents as well as children. Many promising approaches have been developed, including universal or selective interventions based on family, school, community, or multi-component approaches.22
Because parental SUDs are a prominent risk factor for children, interventions to reduce or eliminate parental substance use may be helpful, particularly for diminishing childhood psychological dysregulation.23 Early treatment of childhood predictive phenotypes, including CD and ADHD, is another promising approach.12 Community efforts to limit adolescents’ access to addictive substances have met with some success.22
Parents, teachers, and children and adolescents can obtain a wealth of information from the Web sites of the National Institute on Alcohol Abuse and Alcoholism and the National Institute on Drug Abuse (Box 2). The Centers for Disease Control and Prevention offers infomation about preventing smoking (see Related Resources.)
Parents/teachers
The National Institute on Drug Abuse (NIDA) provides a Web site for parents and teachers at www.drugabuse.gov/parent-teacher.html. Parents also can find resources at NIDA for teens at http://teens.drugabuse.gov.
The National Institute on Alcohol Abuse and Alcoholism (NIAAA) provides several free booklets for parents at www.niaaa.nih.gov/Publications/PamphletsBrochuresPosters/English.
My Brain, My Body (www.mybrainmybody.com) is an NIAAA-supported educational tool for teaching middle school students. Developmentally specific NIDA Junior Scientist Programs have been developed for kindergarten through grade 5. The Office of Safe and Drug-Free Schools, part of the U.S. Department of Education, also provides relevant resources.
Children and adolescents
An NIAAA-sponsored educational resource (www.thecoolspot.gov) provides education on alcohol designed for young teens (age 11 to 13). The site includes quizzes, suggestions for resisting peer pressure, and activities that encourage refusing drinking opportunities.
NIDA for Teens (http://teens.drugabuse.gov) provides information and activities designed for those age 11 to 15.
Related resources
- Alcohol Use Disorders Identification Test (AUDIT). World Health Organization. http://whqlibdoc.who.int/hq/2001/WHO_MSD_MSB_01.6a.pdf.
- National Institute on Alcohol Abuse and Alcoholism. www.niaaa.nih.gov.
- National Institute on Drug Abuse. www.nida.nih.gov.
- Centers for Disease Control and Prevention. Youth Tobacco Prevention. www.cdc.gov/tobacco/youth.
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Traditionally, clinicians have identified children at high risk for substance abuse disorders (SUDs) by their family history—such as “children of alcoholics.” Advances in etiology research, however, have led to the identification of other risks for SUDs seen during childhood (Table). The clustering of these SUD risk factors—genetic influences, family characteristics, and predictive phenotypes—makes it feasible to identify children and adolescents who are very likely to develop problematic substance use.
Table
Risk factors for substance abuse in children and adolescents
| Genetic predisposition |
| Parental substance use |
| Maltreatment |
| Inadequate supervision |
| Impulsive behavior, inattention, irritability |
| Substance availability |
| Early substance use |
Nature vs nurture
Genetic influences. Heritable risk accounts for a substantial proportion of the variation in SUDs, as multiple genes differentially influence substance initiation, metabolism, and reinforcing properties.1 For example, well-characterized genetic variations determine individual differences in alcohol dehydrogenase and aldehyde dehydrogenase—the enzymes involved in alcohol metabolism—and influence liability to alcohol use disorders (AUDs).2,3 Researchers are exploring ways in which genes might impact SUD risk (Box 1).1,4,5
Genetic influences on substance use may be less important during adolescence than adulthood. In a study of 1,796 male twins’ alcohol, nicotine, and cannabis use from early adolescence to middle adulthood, genetic variations had little or no influence on substance use in early adolescence. The influence of genetic factors gradually increased with age.6
Familial environmental factors, by contrast, were important in early adolescence and gradually decreased in effect with increasing age. During adolescence, the family’s influence on substance use apparently operates more through environmental characteristics than through heritable factors.6
Familial influences. Parents with ongoing SUDs model problematic substance use and create environments of child maltreatment and inadequate supervision.
Child maltreatment. Children of parents with SUDs are more likely to suffer sexual abuse, physical abuse, or neglect.7 The effects of sexual abuse on the child may vary by abuse severity and the child’s gender, developmental stage, and relationship to the perpetrator. Maltreatment may cause the child difficulties in psychological regulation and social development, leading to related psychopathology; these characteristics may contribute to later SUDs.8
Inadequate supervision. Adolescents who report that their parents do not effectively monitor their activities have an increased likelihood of developing SUDs. However, children/adolescents who exhibit difficulties with psychological regulation—such as impulsive behavior and irritability—are difficult to parent, and adolescents with early substance involvement may subvert parental supervision efforts.9,10
Recent investigations have examined genes that might confer risk across substance types. Promising research has focused on:
- genes that influence functional variations in neurotransmitter systems
- gene-environment interactions
- the search for neurobiological endophenotypes—characteristics that cannot be observed by conventional means, such as brain development characteristics that are seen through neuroimaging.1,4,5
Specific molecular-level genetic variations can be measured in individual patients but cannot yet validly quantify risk.
Predictive phenotypes
Predictive phenotypes—measurable individual characteristics that predict SUDs—may be considered risk factors but should not be viewed as causal influences akin to genetic and familial/environmental factors. Rather, predictive phenotypes may reflect propensities that are manifested by specific behaviors and other features according to developmental stage and environmental facilitation.
- specific psychiatric disorders
- specific personality traits that collectively are called psychological dysregulation
- early substance use.11
Recent studies have demonstrated that this clustering of problems—including impulsive behavior, inattention, and negative affect—represents a single continuous dimension termed psychological dysregulation.12 The construct of psychological dysregulation has origins in neuropathology and provides a conceptual link between childhood psychopathological characteristics known to predict SUD and neurobiological deficits.5 Childhood indices of psychological dysregulation—such as the Behavior Rating Inventory of Executive Function (BRIEF)14—complement other risk factors, such as parental SUDs and early substance use, in predicting accelerated substance use and SUDs.15
Neurobiological characteristics. Recent investigations have focused on relationships between variations in normal brain development and differences in psychological regulation.5 Several brain structures thought relevant to the development of psychological regulation—including the prefrontal cortex, limbic structures, and reward circuits—develop during adolescence. Delays or deficits in the development of these structures are called neurodevelopmental dysmaturation.5
Variation in genes that influence these brain areas may interact with environmental influences—including child maltreatment and early substance use—to produce neurodevelopmental dysmaturation that manifests as psychological dysregulation. Thus, genetic and environmental causes are hypothesized to lead to an endophenotype (neurodevelopmental dysmaturation) and developmentally specific phenotypes, such as:
- ADHD in childhood
- CD and accelerated substance use initiation in early adolescence
- SUDs involving alcohol and cannabis in late adolescence.5
Consuming small quantities of alcohol under parental supervision is culturally normative and does not predict problematic drinking.17 On the other hand, regularly consuming “standard drink” quantities of alcohol in late childhood typically occurs in unsupervised settings and predicts adolescent-onset AUDs.18
Problem-focused interview methods—including CAGE, TWEAK, and CRAFFT—have been developed and tested to screen adolescents for AUDs. None has been as consistently successful as the World Health Organization’s Alcohol Use Disorders Identification Test (AUDIT) questionnaire18 (see Related Resources).
Childhood cigarette smoking also predicts accelerated substance use and SUDs.15 Marijuana use predicts both cannabis use disorders and other illicit drug use.1 This observation supports the controversial “gateway hypothesis,” which proposes that marijuana use accelerates the onset of other illicit drug use.15,19,20 An alternate hypothesis proposes that use of marijuana and other illicit drugs is a developmentally specific manifestation of a more general liability for SUDs.1
Identifying those at high risk
Screening for SUD risk factors makes it possible to identify children and adolescents who are very likely to develop problematic substance use. For example, in a study of 560 children age 10 to 12 at recruitment, this author (DBC) identified subjects as high risk if they had 2 parents with SUDs, tobacco or alcohol use by age 12, and high psychological dysregulation as measured by combined assessments of cognitive, emotional, and behavioral regulation. By age 18:
- three-quarters of these adolescents used tobacco daily
- more than one-half had alcohol problems
- nearly one-half had cannabis abuse or dependence.15
Recommendations. Children and adolescents receiving health care services—including primary care, ongoing treatment for chronic conditions, and treatment for psychiatric disorders—should be evaluated for SUD risk. Screening ideally occurs at the initial evaluation or early in the course of treatment. Family history determines genetic risk.
Direct questioning is needed because unstructured evaluations often fail to reveal the presence of important SUD risks.21 Explore possible child maltreatment by questioning the parent and child about physical abuse, sexual abuse, and neglect. Key mental disorders include CD, ADHD, and PTSD. Ask about use of tobacco, alcohol, cannabis, and other drugs. Follow acknowledgement of use with inquiries on frequency, quantity, and problems.
Prevention and early intervention
By identifying characteristics that confer risk for SUDs, you can target these characteristics in prevention and early treatment efforts. These efforts may involve parents as well as children. Many promising approaches have been developed, including universal or selective interventions based on family, school, community, or multi-component approaches.22
Because parental SUDs are a prominent risk factor for children, interventions to reduce or eliminate parental substance use may be helpful, particularly for diminishing childhood psychological dysregulation.23 Early treatment of childhood predictive phenotypes, including CD and ADHD, is another promising approach.12 Community efforts to limit adolescents’ access to addictive substances have met with some success.22
Parents, teachers, and children and adolescents can obtain a wealth of information from the Web sites of the National Institute on Alcohol Abuse and Alcoholism and the National Institute on Drug Abuse (Box 2). The Centers for Disease Control and Prevention offers infomation about preventing smoking (see Related Resources.)
Parents/teachers
The National Institute on Drug Abuse (NIDA) provides a Web site for parents and teachers at www.drugabuse.gov/parent-teacher.html. Parents also can find resources at NIDA for teens at http://teens.drugabuse.gov.
The National Institute on Alcohol Abuse and Alcoholism (NIAAA) provides several free booklets for parents at www.niaaa.nih.gov/Publications/PamphletsBrochuresPosters/English.
My Brain, My Body (www.mybrainmybody.com) is an NIAAA-supported educational tool for teaching middle school students. Developmentally specific NIDA Junior Scientist Programs have been developed for kindergarten through grade 5. The Office of Safe and Drug-Free Schools, part of the U.S. Department of Education, also provides relevant resources.
Children and adolescents
An NIAAA-sponsored educational resource (www.thecoolspot.gov) provides education on alcohol designed for young teens (age 11 to 13). The site includes quizzes, suggestions for resisting peer pressure, and activities that encourage refusing drinking opportunities.
NIDA for Teens (http://teens.drugabuse.gov) provides information and activities designed for those age 11 to 15.
Related resources
- Alcohol Use Disorders Identification Test (AUDIT). World Health Organization. http://whqlibdoc.who.int/hq/2001/WHO_MSD_MSB_01.6a.pdf.
- National Institute on Alcohol Abuse and Alcoholism. www.niaaa.nih.gov.
- National Institute on Drug Abuse. www.nida.nih.gov.
- Centers for Disease Control and Prevention. Youth Tobacco Prevention. www.cdc.gov/tobacco/youth.
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Vanyukov MM, Tarter RE, Kirisci L, et al. Liability to substance use disorders: 1. Common mechanisms and manifestations. Neurosci Biobehav Rev. 2003;27(6):507-515.
2. Karpyak VM, Hall-Flavin DK, Mrazek DA. Can genetics predict risk for alcohol dependence? Current Psychiatry. 2008;7(3):57-73.
3. Kuo PH, Kalsi G, Prescott CA, et al. Association of ADH and ALDH genes with alcohol dependence in the Irish Affected Sib Pair Study of alcohol dependence (IASPSAD) sample. Alcohol Clin Exp Res. 2008;32(5):785-795.
4. Vanyukov MM, Maher BS, Devlin B, et al. The MAOA promoter polymorphism, disruptive behavior disorders, and early onset substance use disorder: gene-environment interaction. Psychiatr Genet. 2007;17(6):323-332.
5. Clark DB, Thatcher DL, Tapert S. Alcohol, psychological dysregulation and adolescent brain development. Alcohol Clin Exp Res. 2008;32(3):375-385.
6. Kendler KS, Schmitt E, Aggen SH, et al. Genetic and environmental influences on alcohol, caffeine, cannabis, and nicotine use from early adolescence to middle adulthood. Arch Gen Psychiatry. 2008;65(6):674-682.
7. Sher KJ, Gershuny BS, Peterson L, et al. The role of childhood stressors in the intergenerational transmission of alcohol use disorders. J Stud Alcohol. 1997;58:414-427.
8. Clark DB, De Bellis MD, Lynch KG, et al. Physical and sexual abuse, depression and alcohol use disorders in adolescents. Drug Alcohol Depend. 2003;69(1):51-60.
9. Clark DB, Thatcher DL, Maisto S. Adolescent neglect and alcohol use disorders in two-parent families. Child Maltreat. 2004;9(4):357-370.
10. Clark DB, Kirisci L, Mezzich A, et al. Parental supervision and alcohol use in early adolescence: developmentally specific interactions. J Dev Behav Pediatr. 2008;29(4):285-292.
11. Clark DB, Kirisci L, Moss HB. Early adolescent gateway drug use in sons of fathers with substance use disorders. Addict Behav. 1998;23:561-566.
12. Clark DB, Winters KC. Measuring risks and outcomes in substance use disorders prevention research. J Consult Clin Psychol. 2002;70(6):1207-1223.
13. Clark DB, Cornelius JR, Wood DS, et al. Psychopathology risk transmission in children of parents with substance use disorders. Am J Psychiatry. 2004;161(4):685-691.
14. Guy SC, Isquith PK, Gioia GA. Behavior rating inventory of executive function—self-report version professional manual. Lutz, FL: Psychological Assessment Resources; 2004.
15. Clark DB, Cornelius JR, Kirisci L, et al. Childhood risk categories for adolescent substance involvement: a general liability typology. Drug Alcohol Depend. 2005;77(1):13-21.
16. Clark DB. The natural history of adolescent alcohol use disorders. Addiction. 2004;99(suppl 2):5-22.
17. Donovan JE, Molina BS. Children’s introduction to alcohol use: sips and tastes. Alcohol Clin Exp Res. 2008;32(1):108-119.
18. Clark DB, Chung T, Martin CS. Alcohol use frequency as a screen for alcohol use disorders in adolescents. Int J Adolesc Med Health. 2006;18(1):181-187.
19. Tarter RE, Kirisci L, Mezzich A, et al. Neurobehavioral disinhibition in childhood predicts early age onset substance use disorder. Am J Psychiatry. 2003;160(6):1078-1085.
20. Tarter RE, Vanyukov M, Kirisci L, et al. Predictors of marijuana use in adolescents before and after licit drug use: examination of the gateway hypothesis. Am J Psychiatry. 2006;163(12):2134-2140.
21. Clark DB, Bukstein OG, Smith MG, et al. Identifying anxiety disorders in adolescents hospitalized for alcohol abuse or dependence. Psychiatr Serv. 1995;46:618-620.
22. Spoth R, Greenberg M, Turrisi R. Preventive interventions addressing underage drinking: state of the evidence and steps toward public health impact. Pediatrics. 2008;121(suppl 4):S311-336.
23. Kelley ML, Fals-Stewart W. Treating paternal drug abuse using Learning Sobriety Together: effects on adolescents versus children. Drug Alcohol Depend. 2008;92(1-3):228-238.
1. Vanyukov MM, Tarter RE, Kirisci L, et al. Liability to substance use disorders: 1. Common mechanisms and manifestations. Neurosci Biobehav Rev. 2003;27(6):507-515.
2. Karpyak VM, Hall-Flavin DK, Mrazek DA. Can genetics predict risk for alcohol dependence? Current Psychiatry. 2008;7(3):57-73.
3. Kuo PH, Kalsi G, Prescott CA, et al. Association of ADH and ALDH genes with alcohol dependence in the Irish Affected Sib Pair Study of alcohol dependence (IASPSAD) sample. Alcohol Clin Exp Res. 2008;32(5):785-795.
4. Vanyukov MM, Maher BS, Devlin B, et al. The MAOA promoter polymorphism, disruptive behavior disorders, and early onset substance use disorder: gene-environment interaction. Psychiatr Genet. 2007;17(6):323-332.
5. Clark DB, Thatcher DL, Tapert S. Alcohol, psychological dysregulation and adolescent brain development. Alcohol Clin Exp Res. 2008;32(3):375-385.
6. Kendler KS, Schmitt E, Aggen SH, et al. Genetic and environmental influences on alcohol, caffeine, cannabis, and nicotine use from early adolescence to middle adulthood. Arch Gen Psychiatry. 2008;65(6):674-682.
7. Sher KJ, Gershuny BS, Peterson L, et al. The role of childhood stressors in the intergenerational transmission of alcohol use disorders. J Stud Alcohol. 1997;58:414-427.
8. Clark DB, De Bellis MD, Lynch KG, et al. Physical and sexual abuse, depression and alcohol use disorders in adolescents. Drug Alcohol Depend. 2003;69(1):51-60.
9. Clark DB, Thatcher DL, Maisto S. Adolescent neglect and alcohol use disorders in two-parent families. Child Maltreat. 2004;9(4):357-370.
10. Clark DB, Kirisci L, Mezzich A, et al. Parental supervision and alcohol use in early adolescence: developmentally specific interactions. J Dev Behav Pediatr. 2008;29(4):285-292.
11. Clark DB, Kirisci L, Moss HB. Early adolescent gateway drug use in sons of fathers with substance use disorders. Addict Behav. 1998;23:561-566.
12. Clark DB, Winters KC. Measuring risks and outcomes in substance use disorders prevention research. J Consult Clin Psychol. 2002;70(6):1207-1223.
13. Clark DB, Cornelius JR, Wood DS, et al. Psychopathology risk transmission in children of parents with substance use disorders. Am J Psychiatry. 2004;161(4):685-691.
14. Guy SC, Isquith PK, Gioia GA. Behavior rating inventory of executive function—self-report version professional manual. Lutz, FL: Psychological Assessment Resources; 2004.
15. Clark DB, Cornelius JR, Kirisci L, et al. Childhood risk categories for adolescent substance involvement: a general liability typology. Drug Alcohol Depend. 2005;77(1):13-21.
16. Clark DB. The natural history of adolescent alcohol use disorders. Addiction. 2004;99(suppl 2):5-22.
17. Donovan JE, Molina BS. Children’s introduction to alcohol use: sips and tastes. Alcohol Clin Exp Res. 2008;32(1):108-119.
18. Clark DB, Chung T, Martin CS. Alcohol use frequency as a screen for alcohol use disorders in adolescents. Int J Adolesc Med Health. 2006;18(1):181-187.
19. Tarter RE, Kirisci L, Mezzich A, et al. Neurobehavioral disinhibition in childhood predicts early age onset substance use disorder. Am J Psychiatry. 2003;160(6):1078-1085.
20. Tarter RE, Vanyukov M, Kirisci L, et al. Predictors of marijuana use in adolescents before and after licit drug use: examination of the gateway hypothesis. Am J Psychiatry. 2006;163(12):2134-2140.
21. Clark DB, Bukstein OG, Smith MG, et al. Identifying anxiety disorders in adolescents hospitalized for alcohol abuse or dependence. Psychiatr Serv. 1995;46:618-620.
22. Spoth R, Greenberg M, Turrisi R. Preventive interventions addressing underage drinking: state of the evidence and steps toward public health impact. Pediatrics. 2008;121(suppl 4):S311-336.
23. Kelley ML, Fals-Stewart W. Treating paternal drug abuse using Learning Sobriety Together: effects on adolescents versus children. Drug Alcohol Depend. 2008;92(1-3):228-238.
Transcranial magnetic stimulation for depression
Only 28% to 33% of patients with major depression experience remission after their first antidepressant treatment, according to results of the Sequenced Treatment Alternative to Relieve Depression (STAR*D) trial.1 Therapeutic options include switching to an alternate antidepressant, augmentation with a second antidepressant, psychotherapy, mood stabilizers, or second-generation antipsychotics.
In October 2008, the FDA approved a new option: transcranial magnetic stimulation (NeuroStar TMS Therapy), a neuro-modulation approach indicated for patients with major depressive disorder (MDD) who failed 1 adequate antidepressant trial in the current episode (Table 1).
Table 1
Transcranial magnetic stimulation: Fast facts
| Brand name: NeuroStar TMS Therapy |
| Class: Class II medical device |
| Indication: Treatment of major depressive disorder in adults who failed to achieve satisfactory improvement from 1 prior antidepressant medication at or above the minimal effective dose and duration in the current depressive episode |
| Approval date: October 7, 2008 |
| Availability: Limited number of treatment centers; see www.NeuroStarTMS.com |
| Manufacturer: Neuronetics, Inc. |
| Recommended dose: 75 10-Hz, 4-second trains; 26-second intertrain interval; administered over the left dorsolateral prefrontal cortex; 5 days a week, up to 6 weeks |
How it works
TMS delivers intense intermittent magnetic pulses produced by an electrical charge into a ferromagnetic coil. The intensity of the pulse is similar to that of MRI (1.5 to 2 tesla); however, in MRI the magnetic field is constantly on, whereas in TMS the field is exceptionally brief (milliseconds).
For depression treatment, the coil is usually placed on the scalp over the left dorsolateral prefrontal cortex (DLPFC). Pulses are delivered in a rapid, repetitive train, causing neuronal depolarization in a small area of the cerebral cortex and distal effects in other neurocircuits.
For depression, standard outpatient treatment consists of 5 daily sessions per week for up to 6 weeks. Each session takes approximately 40 minutes, and patients typically return to normal daily activities without difficulty. Initially, NeuroStar TMS will be available in a limited number of treatment centers (see Related Resource).
Intensity of treatment is individualized by adjusting parameters that affect delivery of the magnetic pulses. Motor threshold (MT) is the level of stimulation required to produce movement in a contralateral target muscle, such as the abductor pollicis brevis that causes contraction of the thumb. Once this level is determined, pulses are administered at an intensity relative to the MT (such as 120%). Single TMS pulses are used to find the relevant area of the motor cortex, whereas repetitive pulses are applied over the left DLPFC for therapy.
Frequency of stimulation is measured in cycles per second or hertz (Hz). Stimulation train is the duration during which pulses are administered, and the intertrain interval (ITI) is the time between stimulation trains. Other parameters include site of stimulation and number of treatments per day, week, and course. Recommended treatment levels appear in (Table 2).
Table 2
TMS depression treatment parameters
| Parameter | Definition | Recommended treatment level | |
|---|---|---|---|
| Motor threshold | Level of stimulation required to produce contractions in the contralateral target muscle (abductor pollicis brevis, which causes contraction of the thumb) | 120% | |
| Frequency of stimulation | Measured in cycles per second or hertz (Hz) | 10 Hz | |
| Stimulation train | Duration of the stimulation | 4 seconds | |
| Intertrain interval | Time between stimulation trains | 26 seconds | |
| Site of stimulation | Where in the brain the stimulation will occur | Left dorsolateral prefrontal cortex | |
| Number of treatments | How many times the patient receives stimulation/treatment | 5 days per week for up to 6 weeks | |
| Total stimulation time | Number of stimulations given in a session | 3,000 stimulations per session | |
| TMS: transcranial magnetic stimulation | |||
Efficacy
George et al2 first reported TMS for depression in 1995. Initial small, open-label studies examined a variety of treatment intensities, durations, and stimulation sites. Several sham-controlled studies further refined treatment parameters. These studies generally found TMS efficacious, but questioned the robustness of the clinical effect.
To better assess the antidepressant effect of TMS, studies employed larger samples and more aggressive treatment parameters. Avery et al3 randomized 68 patients to 15 sessions of active or sham TMS over the left DLPFC. Each treatment consisted of 32 10-Hz, 5-second trains at 110% MT with a 25-second ITI. At 1 and 2 weeks after treatment, 31% of subjects in the active treatment group showed a significant decrease in symptoms—defined as ≥50% reduction in Hamilton Depression Rating Scale (HDRS) score—versus 6% in the sham group. In addition, 20% of subjects in the active TMS group achieved remission (defined as HDRS score
The largest trial of TMS monotherapy (N=301) for moderately treatment-resistant major depression was completed in 2007.4 This 3-phase study began with a 4- to 6-week, randomized, double-blind activeversus-sham TMS procedure, followed by 6 weeks of open-label TMS in initial nonresponders. The third phase reintroduced TMS over 6 months as needed to augment maintenance antidepressant medication.
This trial used the most aggressive treatment parameters to date: 75 10-Hz, 4-second trains at 120% MT with a 26-second ITI, delivering 3,000 pulses per treatment over an average of 26 sessions. To maintain an adequate blind, the study utilized sham and active coils with similar appearances, placement, and acoustic properties. The sham coil had an embedded aluminum shield, which limited the magnetic energy reaching the cortex to ≤10% of the active coil. Although there was no assessment of the adequacy of the blind in this trial:
- subjects were naive to TMS in the sham-controlled phase
- TMS operators did not assess efficacy
- TMS operators and subjects did not discuss the treatment experience with the efficacy raters.
Compared with those who received the sham procedure, subjects who received active TMS showed significantly better response rates on the Montgomery-Åsberg Depression Rating Scale (MADRS) at weeks 4 and 6. Similar results were found for the 17- and 24-item HDRS. At 6 weeks, the remission rate (defined as a MADRS score
A post-hoc analysis found that the greatest benefit occurred in patients who had only 1 failed adequate antidepressant trial (effect size=0.83).5
TMS vs ECT. Dowd et al6 summarized 8 published trials that compared TMS with electroconvulsive therapy (ECT) for severe depression:
- 5 reported equivalent efficacy
- 1 found unilateral ECT (UL-ECT) and bilateral ECT (BL-ECT) superior to TMS
- 1 reported UL-ECT superior to TMS
- 1 found UL-ECT plus medication superior to TMS monotherapy in patients with psychosis but comparable in efficacy to TMS in the absence of psychosis.
These results need to be interpreted with caution because of the studies’ diverse designs, nonblinded assessments, and small sample sizes.
Tolerability and safety
The most frequently reported adverse effects of TMS are headache and pain at the site of stimulation. Seizures had been reported in early trials, but the extremely low occurrence has been much lower since Wasserman7 published consensus guidelines on the safe use of TMS in 1996.
Janicak et al8 examined safety data from the 3-phase trial mentioned above, which included >10,000 cumulative treatment sessions. TMS was well-tolerated, with a low discontinuation rate associated with adverse effects: 4.5% in the active treatment group versus 3.4% in the sham TMS procedure group. No deaths, seizures, or cases of treatment-emergent mania occurred. The most commonly reported adverse effects were transient headache and discomfort at the stimulation site. Most patients acclimated to these effects in the first week. No changes were seen in cognitive functioning or auditory thresholds.
As in previous studies, TMS was safely combined with antidepressants in the third phase of this trial; however, patients at risk for seizure or on medications that could lower the seizure threshold were excluded. Thus, risk of seizure may be increased under these conditions. TMS is contraindicated for patients with implanted metallic devices or nonremovable objects in or around the head, except for dental hardware or braces.
- For availability information, contact the manufacturer, Neuronetics, at (877) 6000-7555 or www.NeuroStarTMS.com.
Disclosures
Drs. Dowd, Rado, and Janicak receive research support from and are consultants to Neuronetics, Inc.
Dr. Welch receives research support from Neuronetics, Inc.
1. Trivedi MH, Rush AJ, Wisniewski SR, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 2006;163(1):28-40.
2. George MS, Wassermann EM, Williams WA, et al. Daily repetitive transcranial magnetic stimulation (rTMS) improves mood in depression. Neuroreport 1995;6(14):1853-6.
3. Avery DH, Holtzheimer PE, III, Fawaz W, et al. A controlled study of repetitive transcranial magnetic stimulation in medication-resistant major depression. Biol Psychiatry 2006;59:187-94.
4. O’Reardon JP, Solvason HB, Janicak PG, et al. Efficacy and safety of transcranial magnetic stimulation in the acute treatment of major depression: a multi-site randomized controlled trial. Biol Psychiatry 2007;62:1208-16.
5. Lisanby SH, Husain MM, Rosenquist PB, et al. Daily left prefrontal repetitive transcranial magnetic stimulation in the acute treatment of major depression: clinical predictors of outcome in a multisite, randomized controlled clinical trial. Neuropsychopharmacology Epub 2008 Aug 13.
6. Dowd SM, Janicak PG. Transcranial magnetic stimulation for major depression: part II. Psychopharm Review 2007;42(1):1-8.
7. Wasserman EM. Risk and safety of repetitive transcranial magnetic stimulation: report and suggested guidelines from the International Workshop on the Safety of Repetitive Transcranial Magnetic Stimulation, June 5-7, 1996. Electroencephalogr Clin Neurophysiol 1998;108(1):1-16.
8. Janicak PG, O’Reardon JP, Sampson SM, et al. Transcranial magnetic stimulation in the treatment of major depressive disorder: a comprehensive summary of safety experience from acute exposure, extended exposure, and during reintroduction. J Clin Psychiatry 2008;69:222-33.
Only 28% to 33% of patients with major depression experience remission after their first antidepressant treatment, according to results of the Sequenced Treatment Alternative to Relieve Depression (STAR*D) trial.1 Therapeutic options include switching to an alternate antidepressant, augmentation with a second antidepressant, psychotherapy, mood stabilizers, or second-generation antipsychotics.
In October 2008, the FDA approved a new option: transcranial magnetic stimulation (NeuroStar TMS Therapy), a neuro-modulation approach indicated for patients with major depressive disorder (MDD) who failed 1 adequate antidepressant trial in the current episode (Table 1).
Table 1
Transcranial magnetic stimulation: Fast facts
| Brand name: NeuroStar TMS Therapy |
| Class: Class II medical device |
| Indication: Treatment of major depressive disorder in adults who failed to achieve satisfactory improvement from 1 prior antidepressant medication at or above the minimal effective dose and duration in the current depressive episode |
| Approval date: October 7, 2008 |
| Availability: Limited number of treatment centers; see www.NeuroStarTMS.com |
| Manufacturer: Neuronetics, Inc. |
| Recommended dose: 75 10-Hz, 4-second trains; 26-second intertrain interval; administered over the left dorsolateral prefrontal cortex; 5 days a week, up to 6 weeks |
How it works
TMS delivers intense intermittent magnetic pulses produced by an electrical charge into a ferromagnetic coil. The intensity of the pulse is similar to that of MRI (1.5 to 2 tesla); however, in MRI the magnetic field is constantly on, whereas in TMS the field is exceptionally brief (milliseconds).
For depression treatment, the coil is usually placed on the scalp over the left dorsolateral prefrontal cortex (DLPFC). Pulses are delivered in a rapid, repetitive train, causing neuronal depolarization in a small area of the cerebral cortex and distal effects in other neurocircuits.
For depression, standard outpatient treatment consists of 5 daily sessions per week for up to 6 weeks. Each session takes approximately 40 minutes, and patients typically return to normal daily activities without difficulty. Initially, NeuroStar TMS will be available in a limited number of treatment centers (see Related Resource).
Intensity of treatment is individualized by adjusting parameters that affect delivery of the magnetic pulses. Motor threshold (MT) is the level of stimulation required to produce movement in a contralateral target muscle, such as the abductor pollicis brevis that causes contraction of the thumb. Once this level is determined, pulses are administered at an intensity relative to the MT (such as 120%). Single TMS pulses are used to find the relevant area of the motor cortex, whereas repetitive pulses are applied over the left DLPFC for therapy.
Frequency of stimulation is measured in cycles per second or hertz (Hz). Stimulation train is the duration during which pulses are administered, and the intertrain interval (ITI) is the time between stimulation trains. Other parameters include site of stimulation and number of treatments per day, week, and course. Recommended treatment levels appear in (Table 2).
Table 2
TMS depression treatment parameters
| Parameter | Definition | Recommended treatment level | |
|---|---|---|---|
| Motor threshold | Level of stimulation required to produce contractions in the contralateral target muscle (abductor pollicis brevis, which causes contraction of the thumb) | 120% | |
| Frequency of stimulation | Measured in cycles per second or hertz (Hz) | 10 Hz | |
| Stimulation train | Duration of the stimulation | 4 seconds | |
| Intertrain interval | Time between stimulation trains | 26 seconds | |
| Site of stimulation | Where in the brain the stimulation will occur | Left dorsolateral prefrontal cortex | |
| Number of treatments | How many times the patient receives stimulation/treatment | 5 days per week for up to 6 weeks | |
| Total stimulation time | Number of stimulations given in a session | 3,000 stimulations per session | |
| TMS: transcranial magnetic stimulation | |||
Efficacy
George et al2 first reported TMS for depression in 1995. Initial small, open-label studies examined a variety of treatment intensities, durations, and stimulation sites. Several sham-controlled studies further refined treatment parameters. These studies generally found TMS efficacious, but questioned the robustness of the clinical effect.
To better assess the antidepressant effect of TMS, studies employed larger samples and more aggressive treatment parameters. Avery et al3 randomized 68 patients to 15 sessions of active or sham TMS over the left DLPFC. Each treatment consisted of 32 10-Hz, 5-second trains at 110% MT with a 25-second ITI. At 1 and 2 weeks after treatment, 31% of subjects in the active treatment group showed a significant decrease in symptoms—defined as ≥50% reduction in Hamilton Depression Rating Scale (HDRS) score—versus 6% in the sham group. In addition, 20% of subjects in the active TMS group achieved remission (defined as HDRS score
The largest trial of TMS monotherapy (N=301) for moderately treatment-resistant major depression was completed in 2007.4 This 3-phase study began with a 4- to 6-week, randomized, double-blind activeversus-sham TMS procedure, followed by 6 weeks of open-label TMS in initial nonresponders. The third phase reintroduced TMS over 6 months as needed to augment maintenance antidepressant medication.
This trial used the most aggressive treatment parameters to date: 75 10-Hz, 4-second trains at 120% MT with a 26-second ITI, delivering 3,000 pulses per treatment over an average of 26 sessions. To maintain an adequate blind, the study utilized sham and active coils with similar appearances, placement, and acoustic properties. The sham coil had an embedded aluminum shield, which limited the magnetic energy reaching the cortex to ≤10% of the active coil. Although there was no assessment of the adequacy of the blind in this trial:
- subjects were naive to TMS in the sham-controlled phase
- TMS operators did not assess efficacy
- TMS operators and subjects did not discuss the treatment experience with the efficacy raters.
Compared with those who received the sham procedure, subjects who received active TMS showed significantly better response rates on the Montgomery-Åsberg Depression Rating Scale (MADRS) at weeks 4 and 6. Similar results were found for the 17- and 24-item HDRS. At 6 weeks, the remission rate (defined as a MADRS score
A post-hoc analysis found that the greatest benefit occurred in patients who had only 1 failed adequate antidepressant trial (effect size=0.83).5
TMS vs ECT. Dowd et al6 summarized 8 published trials that compared TMS with electroconvulsive therapy (ECT) for severe depression:
- 5 reported equivalent efficacy
- 1 found unilateral ECT (UL-ECT) and bilateral ECT (BL-ECT) superior to TMS
- 1 reported UL-ECT superior to TMS
- 1 found UL-ECT plus medication superior to TMS monotherapy in patients with psychosis but comparable in efficacy to TMS in the absence of psychosis.
These results need to be interpreted with caution because of the studies’ diverse designs, nonblinded assessments, and small sample sizes.
Tolerability and safety
The most frequently reported adverse effects of TMS are headache and pain at the site of stimulation. Seizures had been reported in early trials, but the extremely low occurrence has been much lower since Wasserman7 published consensus guidelines on the safe use of TMS in 1996.
Janicak et al8 examined safety data from the 3-phase trial mentioned above, which included >10,000 cumulative treatment sessions. TMS was well-tolerated, with a low discontinuation rate associated with adverse effects: 4.5% in the active treatment group versus 3.4% in the sham TMS procedure group. No deaths, seizures, or cases of treatment-emergent mania occurred. The most commonly reported adverse effects were transient headache and discomfort at the stimulation site. Most patients acclimated to these effects in the first week. No changes were seen in cognitive functioning or auditory thresholds.
As in previous studies, TMS was safely combined with antidepressants in the third phase of this trial; however, patients at risk for seizure or on medications that could lower the seizure threshold were excluded. Thus, risk of seizure may be increased under these conditions. TMS is contraindicated for patients with implanted metallic devices or nonremovable objects in or around the head, except for dental hardware or braces.
- For availability information, contact the manufacturer, Neuronetics, at (877) 6000-7555 or www.NeuroStarTMS.com.
Disclosures
Drs. Dowd, Rado, and Janicak receive research support from and are consultants to Neuronetics, Inc.
Dr. Welch receives research support from Neuronetics, Inc.
Only 28% to 33% of patients with major depression experience remission after their first antidepressant treatment, according to results of the Sequenced Treatment Alternative to Relieve Depression (STAR*D) trial.1 Therapeutic options include switching to an alternate antidepressant, augmentation with a second antidepressant, psychotherapy, mood stabilizers, or second-generation antipsychotics.
In October 2008, the FDA approved a new option: transcranial magnetic stimulation (NeuroStar TMS Therapy), a neuro-modulation approach indicated for patients with major depressive disorder (MDD) who failed 1 adequate antidepressant trial in the current episode (Table 1).
Table 1
Transcranial magnetic stimulation: Fast facts
| Brand name: NeuroStar TMS Therapy |
| Class: Class II medical device |
| Indication: Treatment of major depressive disorder in adults who failed to achieve satisfactory improvement from 1 prior antidepressant medication at or above the minimal effective dose and duration in the current depressive episode |
| Approval date: October 7, 2008 |
| Availability: Limited number of treatment centers; see www.NeuroStarTMS.com |
| Manufacturer: Neuronetics, Inc. |
| Recommended dose: 75 10-Hz, 4-second trains; 26-second intertrain interval; administered over the left dorsolateral prefrontal cortex; 5 days a week, up to 6 weeks |
How it works
TMS delivers intense intermittent magnetic pulses produced by an electrical charge into a ferromagnetic coil. The intensity of the pulse is similar to that of MRI (1.5 to 2 tesla); however, in MRI the magnetic field is constantly on, whereas in TMS the field is exceptionally brief (milliseconds).
For depression treatment, the coil is usually placed on the scalp over the left dorsolateral prefrontal cortex (DLPFC). Pulses are delivered in a rapid, repetitive train, causing neuronal depolarization in a small area of the cerebral cortex and distal effects in other neurocircuits.
For depression, standard outpatient treatment consists of 5 daily sessions per week for up to 6 weeks. Each session takes approximately 40 minutes, and patients typically return to normal daily activities without difficulty. Initially, NeuroStar TMS will be available in a limited number of treatment centers (see Related Resource).
Intensity of treatment is individualized by adjusting parameters that affect delivery of the magnetic pulses. Motor threshold (MT) is the level of stimulation required to produce movement in a contralateral target muscle, such as the abductor pollicis brevis that causes contraction of the thumb. Once this level is determined, pulses are administered at an intensity relative to the MT (such as 120%). Single TMS pulses are used to find the relevant area of the motor cortex, whereas repetitive pulses are applied over the left DLPFC for therapy.
Frequency of stimulation is measured in cycles per second or hertz (Hz). Stimulation train is the duration during which pulses are administered, and the intertrain interval (ITI) is the time between stimulation trains. Other parameters include site of stimulation and number of treatments per day, week, and course. Recommended treatment levels appear in (Table 2).
Table 2
TMS depression treatment parameters
| Parameter | Definition | Recommended treatment level | |
|---|---|---|---|
| Motor threshold | Level of stimulation required to produce contractions in the contralateral target muscle (abductor pollicis brevis, which causes contraction of the thumb) | 120% | |
| Frequency of stimulation | Measured in cycles per second or hertz (Hz) | 10 Hz | |
| Stimulation train | Duration of the stimulation | 4 seconds | |
| Intertrain interval | Time between stimulation trains | 26 seconds | |
| Site of stimulation | Where in the brain the stimulation will occur | Left dorsolateral prefrontal cortex | |
| Number of treatments | How many times the patient receives stimulation/treatment | 5 days per week for up to 6 weeks | |
| Total stimulation time | Number of stimulations given in a session | 3,000 stimulations per session | |
| TMS: transcranial magnetic stimulation | |||
Efficacy
George et al2 first reported TMS for depression in 1995. Initial small, open-label studies examined a variety of treatment intensities, durations, and stimulation sites. Several sham-controlled studies further refined treatment parameters. These studies generally found TMS efficacious, but questioned the robustness of the clinical effect.
To better assess the antidepressant effect of TMS, studies employed larger samples and more aggressive treatment parameters. Avery et al3 randomized 68 patients to 15 sessions of active or sham TMS over the left DLPFC. Each treatment consisted of 32 10-Hz, 5-second trains at 110% MT with a 25-second ITI. At 1 and 2 weeks after treatment, 31% of subjects in the active treatment group showed a significant decrease in symptoms—defined as ≥50% reduction in Hamilton Depression Rating Scale (HDRS) score—versus 6% in the sham group. In addition, 20% of subjects in the active TMS group achieved remission (defined as HDRS score
The largest trial of TMS monotherapy (N=301) for moderately treatment-resistant major depression was completed in 2007.4 This 3-phase study began with a 4- to 6-week, randomized, double-blind activeversus-sham TMS procedure, followed by 6 weeks of open-label TMS in initial nonresponders. The third phase reintroduced TMS over 6 months as needed to augment maintenance antidepressant medication.
This trial used the most aggressive treatment parameters to date: 75 10-Hz, 4-second trains at 120% MT with a 26-second ITI, delivering 3,000 pulses per treatment over an average of 26 sessions. To maintain an adequate blind, the study utilized sham and active coils with similar appearances, placement, and acoustic properties. The sham coil had an embedded aluminum shield, which limited the magnetic energy reaching the cortex to ≤10% of the active coil. Although there was no assessment of the adequacy of the blind in this trial:
- subjects were naive to TMS in the sham-controlled phase
- TMS operators did not assess efficacy
- TMS operators and subjects did not discuss the treatment experience with the efficacy raters.
Compared with those who received the sham procedure, subjects who received active TMS showed significantly better response rates on the Montgomery-Åsberg Depression Rating Scale (MADRS) at weeks 4 and 6. Similar results were found for the 17- and 24-item HDRS. At 6 weeks, the remission rate (defined as a MADRS score
A post-hoc analysis found that the greatest benefit occurred in patients who had only 1 failed adequate antidepressant trial (effect size=0.83).5
TMS vs ECT. Dowd et al6 summarized 8 published trials that compared TMS with electroconvulsive therapy (ECT) for severe depression:
- 5 reported equivalent efficacy
- 1 found unilateral ECT (UL-ECT) and bilateral ECT (BL-ECT) superior to TMS
- 1 reported UL-ECT superior to TMS
- 1 found UL-ECT plus medication superior to TMS monotherapy in patients with psychosis but comparable in efficacy to TMS in the absence of psychosis.
These results need to be interpreted with caution because of the studies’ diverse designs, nonblinded assessments, and small sample sizes.
Tolerability and safety
The most frequently reported adverse effects of TMS are headache and pain at the site of stimulation. Seizures had been reported in early trials, but the extremely low occurrence has been much lower since Wasserman7 published consensus guidelines on the safe use of TMS in 1996.
Janicak et al8 examined safety data from the 3-phase trial mentioned above, which included >10,000 cumulative treatment sessions. TMS was well-tolerated, with a low discontinuation rate associated with adverse effects: 4.5% in the active treatment group versus 3.4% in the sham TMS procedure group. No deaths, seizures, or cases of treatment-emergent mania occurred. The most commonly reported adverse effects were transient headache and discomfort at the stimulation site. Most patients acclimated to these effects in the first week. No changes were seen in cognitive functioning or auditory thresholds.
As in previous studies, TMS was safely combined with antidepressants in the third phase of this trial; however, patients at risk for seizure or on medications that could lower the seizure threshold were excluded. Thus, risk of seizure may be increased under these conditions. TMS is contraindicated for patients with implanted metallic devices or nonremovable objects in or around the head, except for dental hardware or braces.
- For availability information, contact the manufacturer, Neuronetics, at (877) 6000-7555 or www.NeuroStarTMS.com.
Disclosures
Drs. Dowd, Rado, and Janicak receive research support from and are consultants to Neuronetics, Inc.
Dr. Welch receives research support from Neuronetics, Inc.
1. Trivedi MH, Rush AJ, Wisniewski SR, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 2006;163(1):28-40.
2. George MS, Wassermann EM, Williams WA, et al. Daily repetitive transcranial magnetic stimulation (rTMS) improves mood in depression. Neuroreport 1995;6(14):1853-6.
3. Avery DH, Holtzheimer PE, III, Fawaz W, et al. A controlled study of repetitive transcranial magnetic stimulation in medication-resistant major depression. Biol Psychiatry 2006;59:187-94.
4. O’Reardon JP, Solvason HB, Janicak PG, et al. Efficacy and safety of transcranial magnetic stimulation in the acute treatment of major depression: a multi-site randomized controlled trial. Biol Psychiatry 2007;62:1208-16.
5. Lisanby SH, Husain MM, Rosenquist PB, et al. Daily left prefrontal repetitive transcranial magnetic stimulation in the acute treatment of major depression: clinical predictors of outcome in a multisite, randomized controlled clinical trial. Neuropsychopharmacology Epub 2008 Aug 13.
6. Dowd SM, Janicak PG. Transcranial magnetic stimulation for major depression: part II. Psychopharm Review 2007;42(1):1-8.
7. Wasserman EM. Risk and safety of repetitive transcranial magnetic stimulation: report and suggested guidelines from the International Workshop on the Safety of Repetitive Transcranial Magnetic Stimulation, June 5-7, 1996. Electroencephalogr Clin Neurophysiol 1998;108(1):1-16.
8. Janicak PG, O’Reardon JP, Sampson SM, et al. Transcranial magnetic stimulation in the treatment of major depressive disorder: a comprehensive summary of safety experience from acute exposure, extended exposure, and during reintroduction. J Clin Psychiatry 2008;69:222-33.
1. Trivedi MH, Rush AJ, Wisniewski SR, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 2006;163(1):28-40.
2. George MS, Wassermann EM, Williams WA, et al. Daily repetitive transcranial magnetic stimulation (rTMS) improves mood in depression. Neuroreport 1995;6(14):1853-6.
3. Avery DH, Holtzheimer PE, III, Fawaz W, et al. A controlled study of repetitive transcranial magnetic stimulation in medication-resistant major depression. Biol Psychiatry 2006;59:187-94.
4. O’Reardon JP, Solvason HB, Janicak PG, et al. Efficacy and safety of transcranial magnetic stimulation in the acute treatment of major depression: a multi-site randomized controlled trial. Biol Psychiatry 2007;62:1208-16.
5. Lisanby SH, Husain MM, Rosenquist PB, et al. Daily left prefrontal repetitive transcranial magnetic stimulation in the acute treatment of major depression: clinical predictors of outcome in a multisite, randomized controlled clinical trial. Neuropsychopharmacology Epub 2008 Aug 13.
6. Dowd SM, Janicak PG. Transcranial magnetic stimulation for major depression: part II. Psychopharm Review 2007;42(1):1-8.
7. Wasserman EM. Risk and safety of repetitive transcranial magnetic stimulation: report and suggested guidelines from the International Workshop on the Safety of Repetitive Transcranial Magnetic Stimulation, June 5-7, 1996. Electroencephalogr Clin Neurophysiol 1998;108(1):1-16.
8. Janicak PG, O’Reardon JP, Sampson SM, et al. Transcranial magnetic stimulation in the treatment of major depressive disorder: a comprehensive summary of safety experience from acute exposure, extended exposure, and during reintroduction. J Clin Psychiatry 2008;69:222-33.
Support patients coping with medical illness
Psychiatrists often are consulted when patients are struggling with the slings and arrows of outrageous medical fortune, to paraphrase Shakespeare. The goal of coping is to bring about relief, reward, quiescence, and equilibrium.1 This definition focuses on the process and does not assume that all of life’s problems can be solved. If your patient seems to be coping poorly, you can help by first identifying the patient’s main coping mode and then increasing his or her repertoire of coping skills.
Emotion-based coping
Are painful psychological experiences such as anxiety or despair interfering with your patient’s ability to cope? Managing emotions with medications, cognitive therapy, or relaxation does not directly address the causes of distress, but it can mitigate psychological paralysis, prevent secondary problems such as alcoholism or demoralization, and allow patients to use executive brain function.
Humor can be effective for managing emotions, but be careful because not all patients can find humor in a painful situation.
Problem-based coping
How well is your patient dealing with the practical aspects of treatment such as keeping doctors’ appointments or going to work when fatigued from chemotherapy? Thinking rationally is difficult when one is overwhelmed by lack of social support or uncontrolled emotions. Ask what your patient sees as the main problem so you can discuss specific, tangible interventions such as child care, transportation, financial assistance, support groups, or informational materials.
Attitudinal-based coping
Adopting an attitude of accepting unavoidable circumstances—which is not the same as passivity—can come from wrestling with the ideas of secular and religious philosophers or spiritual leaders. Show great sensitivity when recommending bibliotherapy or bringing up philosophical ideas, however, so you don’t make your patient feel inadequate or poorly educated. Emotional growth in times of crisis cannot be accelerated. Determine if your patient can find meaning in the illness by asking “Has this illness taught you anything or changed you?”
Successful adaptation to medical adversity and disability requires that a patient use various coping strategies, shifting flexibly between them. Although these 3 coping modes are not necessarily hierarchical, patients who show only emotion-based coping might benefit from being nudged toward problem-based coping. Start by this process by examining practical implications of the illness.
1. Schlozman SC, Groves JE, Weisman AD. Coping with illness and psychotherapy of the medically ill. In: Stern TA, Fricchione GL, Cassem NH, et al. eds. Massachusetts General Hospital handbook of general hospital psychiatry. 5th ed. Philadelphia, PA: Mosby; 2004.
Psychiatrists often are consulted when patients are struggling with the slings and arrows of outrageous medical fortune, to paraphrase Shakespeare. The goal of coping is to bring about relief, reward, quiescence, and equilibrium.1 This definition focuses on the process and does not assume that all of life’s problems can be solved. If your patient seems to be coping poorly, you can help by first identifying the patient’s main coping mode and then increasing his or her repertoire of coping skills.
Emotion-based coping
Are painful psychological experiences such as anxiety or despair interfering with your patient’s ability to cope? Managing emotions with medications, cognitive therapy, or relaxation does not directly address the causes of distress, but it can mitigate psychological paralysis, prevent secondary problems such as alcoholism or demoralization, and allow patients to use executive brain function.
Humor can be effective for managing emotions, but be careful because not all patients can find humor in a painful situation.
Problem-based coping
How well is your patient dealing with the practical aspects of treatment such as keeping doctors’ appointments or going to work when fatigued from chemotherapy? Thinking rationally is difficult when one is overwhelmed by lack of social support or uncontrolled emotions. Ask what your patient sees as the main problem so you can discuss specific, tangible interventions such as child care, transportation, financial assistance, support groups, or informational materials.
Attitudinal-based coping
Adopting an attitude of accepting unavoidable circumstances—which is not the same as passivity—can come from wrestling with the ideas of secular and religious philosophers or spiritual leaders. Show great sensitivity when recommending bibliotherapy or bringing up philosophical ideas, however, so you don’t make your patient feel inadequate or poorly educated. Emotional growth in times of crisis cannot be accelerated. Determine if your patient can find meaning in the illness by asking “Has this illness taught you anything or changed you?”
Successful adaptation to medical adversity and disability requires that a patient use various coping strategies, shifting flexibly between them. Although these 3 coping modes are not necessarily hierarchical, patients who show only emotion-based coping might benefit from being nudged toward problem-based coping. Start by this process by examining practical implications of the illness.
Psychiatrists often are consulted when patients are struggling with the slings and arrows of outrageous medical fortune, to paraphrase Shakespeare. The goal of coping is to bring about relief, reward, quiescence, and equilibrium.1 This definition focuses on the process and does not assume that all of life’s problems can be solved. If your patient seems to be coping poorly, you can help by first identifying the patient’s main coping mode and then increasing his or her repertoire of coping skills.
Emotion-based coping
Are painful psychological experiences such as anxiety or despair interfering with your patient’s ability to cope? Managing emotions with medications, cognitive therapy, or relaxation does not directly address the causes of distress, but it can mitigate psychological paralysis, prevent secondary problems such as alcoholism or demoralization, and allow patients to use executive brain function.
Humor can be effective for managing emotions, but be careful because not all patients can find humor in a painful situation.
Problem-based coping
How well is your patient dealing with the practical aspects of treatment such as keeping doctors’ appointments or going to work when fatigued from chemotherapy? Thinking rationally is difficult when one is overwhelmed by lack of social support or uncontrolled emotions. Ask what your patient sees as the main problem so you can discuss specific, tangible interventions such as child care, transportation, financial assistance, support groups, or informational materials.
Attitudinal-based coping
Adopting an attitude of accepting unavoidable circumstances—which is not the same as passivity—can come from wrestling with the ideas of secular and religious philosophers or spiritual leaders. Show great sensitivity when recommending bibliotherapy or bringing up philosophical ideas, however, so you don’t make your patient feel inadequate or poorly educated. Emotional growth in times of crisis cannot be accelerated. Determine if your patient can find meaning in the illness by asking “Has this illness taught you anything or changed you?”
Successful adaptation to medical adversity and disability requires that a patient use various coping strategies, shifting flexibly between them. Although these 3 coping modes are not necessarily hierarchical, patients who show only emotion-based coping might benefit from being nudged toward problem-based coping. Start by this process by examining practical implications of the illness.
1. Schlozman SC, Groves JE, Weisman AD. Coping with illness and psychotherapy of the medically ill. In: Stern TA, Fricchione GL, Cassem NH, et al. eds. Massachusetts General Hospital handbook of general hospital psychiatry. 5th ed. Philadelphia, PA: Mosby; 2004.
1. Schlozman SC, Groves JE, Weisman AD. Coping with illness and psychotherapy of the medically ill. In: Stern TA, Fricchione GL, Cassem NH, et al. eds. Massachusetts General Hospital handbook of general hospital psychiatry. 5th ed. Philadelphia, PA: Mosby; 2004.