User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Beware of PHATS in metabolic syndrome
Making time to monitor all five risk factors for metabolic syndrome can be challenging in a busy psychiatric setting. But with higher prevalence in persons with psychiatric disorders and/or taking psychotropics,1 this precursor for type 2 diabetes and cardiovascular disease demands your attention.2 The mnemonic PHATS can help you monitor metabolic syndrome risk factors thoroughly and quickly (Table).
Based on National Cholesterol Education Program (NCEP) criteria,3 patients with three of five PHATS elements have metabolic syndrome. A recent study of patients taking atypical antipsychotics suggests that abdominal obesity and elevated fasting blood glucose might be the most accurate and cost-effective indicators; combining these two factors correctly identified 100% of patients with metabolic syndrome.4 Until additional studies can confirm this finding, use NCEP guidelines—the basis for PHATS.
You can easily monitor for metabolic syndrome with a blood pressure cuff, a tape measure, and periodic blood glucose and lipid profiles. An extra minute or two can help prevent metabolic complications in at-risk patients.
Table
PHATS: 3 of 5 positive criteria indicate metabolic syndrome
| Pressure | >130/85 mm Hg |
| HDL cholesterol | |
| Abdominal obesity | Waist circumference |
| >102 cm in men | |
| >88 cm in women | |
| Triglycerides | ≥150 mg/dL |
| Sugar | Fasting blood glucose |
| ≥110 mg/dL |
1. Casey DE. Dyslipidemia and atypical antipsychotic drugs. J Clin Psychiatry 2004;65(suppl 18):27-35.
2. Gracious BL, Meyer AE. Psychotropic-induced weight gain and potential pharmacologic strategies. Psychiatry 2005;2:36-42.
3. Executive summary of the third report of the national cholesterol education program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults. JAMA 2001;285:2486-97.
4. Straker D, Correll CU, Kramer-Ginsberg E, et al. Cost-effective screening for the metabolic syndrome in patients treated with second-generation antipsychotic medications. Am J Psychiatry 2005;162(6):1217-20.
Dr. Grove, a psychiatrist, practices in Scottsdale, AZ.
Making time to monitor all five risk factors for metabolic syndrome can be challenging in a busy psychiatric setting. But with higher prevalence in persons with psychiatric disorders and/or taking psychotropics,1 this precursor for type 2 diabetes and cardiovascular disease demands your attention.2 The mnemonic PHATS can help you monitor metabolic syndrome risk factors thoroughly and quickly (Table).
Based on National Cholesterol Education Program (NCEP) criteria,3 patients with three of five PHATS elements have metabolic syndrome. A recent study of patients taking atypical antipsychotics suggests that abdominal obesity and elevated fasting blood glucose might be the most accurate and cost-effective indicators; combining these two factors correctly identified 100% of patients with metabolic syndrome.4 Until additional studies can confirm this finding, use NCEP guidelines—the basis for PHATS.
You can easily monitor for metabolic syndrome with a blood pressure cuff, a tape measure, and periodic blood glucose and lipid profiles. An extra minute or two can help prevent metabolic complications in at-risk patients.
Table
PHATS: 3 of 5 positive criteria indicate metabolic syndrome
| Pressure | >130/85 mm Hg |
| HDL cholesterol | |
| Abdominal obesity | Waist circumference |
| >102 cm in men | |
| >88 cm in women | |
| Triglycerides | ≥150 mg/dL |
| Sugar | Fasting blood glucose |
| ≥110 mg/dL |
Making time to monitor all five risk factors for metabolic syndrome can be challenging in a busy psychiatric setting. But with higher prevalence in persons with psychiatric disorders and/or taking psychotropics,1 this precursor for type 2 diabetes and cardiovascular disease demands your attention.2 The mnemonic PHATS can help you monitor metabolic syndrome risk factors thoroughly and quickly (Table).
Based on National Cholesterol Education Program (NCEP) criteria,3 patients with three of five PHATS elements have metabolic syndrome. A recent study of patients taking atypical antipsychotics suggests that abdominal obesity and elevated fasting blood glucose might be the most accurate and cost-effective indicators; combining these two factors correctly identified 100% of patients with metabolic syndrome.4 Until additional studies can confirm this finding, use NCEP guidelines—the basis for PHATS.
You can easily monitor for metabolic syndrome with a blood pressure cuff, a tape measure, and periodic blood glucose and lipid profiles. An extra minute or two can help prevent metabolic complications in at-risk patients.
Table
PHATS: 3 of 5 positive criteria indicate metabolic syndrome
| Pressure | >130/85 mm Hg |
| HDL cholesterol | |
| Abdominal obesity | Waist circumference |
| >102 cm in men | |
| >88 cm in women | |
| Triglycerides | ≥150 mg/dL |
| Sugar | Fasting blood glucose |
| ≥110 mg/dL |
1. Casey DE. Dyslipidemia and atypical antipsychotic drugs. J Clin Psychiatry 2004;65(suppl 18):27-35.
2. Gracious BL, Meyer AE. Psychotropic-induced weight gain and potential pharmacologic strategies. Psychiatry 2005;2:36-42.
3. Executive summary of the third report of the national cholesterol education program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults. JAMA 2001;285:2486-97.
4. Straker D, Correll CU, Kramer-Ginsberg E, et al. Cost-effective screening for the metabolic syndrome in patients treated with second-generation antipsychotic medications. Am J Psychiatry 2005;162(6):1217-20.
Dr. Grove, a psychiatrist, practices in Scottsdale, AZ.
1. Casey DE. Dyslipidemia and atypical antipsychotic drugs. J Clin Psychiatry 2004;65(suppl 18):27-35.
2. Gracious BL, Meyer AE. Psychotropic-induced weight gain and potential pharmacologic strategies. Psychiatry 2005;2:36-42.
3. Executive summary of the third report of the national cholesterol education program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults. JAMA 2001;285:2486-97.
4. Straker D, Correll CU, Kramer-Ginsberg E, et al. Cost-effective screening for the metabolic syndrome in patients treated with second-generation antipsychotic medications. Am J Psychiatry 2005;162(6):1217-20.
Dr. Grove, a psychiatrist, practices in Scottsdale, AZ.
When mixing drugs makes malpractice
Amitriptyline toxicity kills patient
Unknown North Carolina venue
A 26-year-old woman with diabetes saw a psychiatrist to manage her depression. The psychiatrist increased her amitriptyline dosage to 300 mg nightly and over 10 months added:
- alprazolam (unknown dosage, nightly)
- quetiapine (400 mg bid)
- extended-release venlafaxine (225 mg bid)
- and promethazine (100 mg bid).
Several weeks later, the woman was found dead in her home. An autopsy revealed amitriptyline toxicity as the cause of death. The medical examiner noted “a much larger concentration of the metabolite nortriptyline in the liver versus the parent drug,” suggesting a metabolism problem, rather than an overdose, caused the toxic build-up.
The patient’s estate claimed that amitriptyline was cardiotoxic at the prescribed dosage and combined with the other medications used and that the patient was not properly monitored.
- A $2.3 million settlement was reached.
Fatal cardiac arrest after 2 concomitant antidepressants
Gwinnett County (GA) Superior Court
A 40-year-old woman was under a psychiatrist’s care for anxiety and depression. The psychiatrist continued sertraline, which the woman had been taking, and added nortriptyline. Several weeks after the patient began taking the medications together, she had a fatal cardiac arrest.
The patient’s estate argued that:
- toxic levels of the antidepressants caused her death
- sertraline and nortriptyline should not be taken concurrently because one drug inhibits clearance of the other
- the psychiatrist should have monitored the patient to make sure sertraline and nortriptyline levels remained normal.
The medical examiner was unable to say which condition more likely led to the patient’s death.
- The defendant was awarded $3 million. A statutory capitation reduced the award to $1.65 million.
Dr. Grant’s observations
As these cases demonstrate, lawsuits against psychiatrists commonly include allegations of preventable prescribing missteps and drug-drug interactionsOff-label prescribing: 7 steps for safer, more effective treatment”).
When a patient is taking multiple medications, interactions can inhibit drug metabolism and render normal doses excessive.5 When prescribing drugs known to have adverse effects with excessive dosing, such as tricyclic antidepressants and lithium, failing to monitor serum levels could be considered malpractice. In fact, the courts view actions such as prescribing doses that exceed FDA recommendations or failing to monitor levels as prima facie evidence of negligence, requiring the psychiatrist to prove otherwise.
Amitriptyline and nortriptyline have shown cardiac toxicity in overdose,6 and their serum levels increase when used with other antidepressants.7,8 Standard of care dictates serum level monitoring particularly when you use tricyclics:
- in doses higher than recommended by the FDA (300 mg/d for amitriptyline, 150 mg/d for nortriptyline)
- with other drugs that affect their metabolism.9
More than twice as many Americans died from medication errors in 1993 than in 1983, according to a comparative review of U.S. death certificates from that period.2
Between 1985 and 1999, more than 10,000 medication error claims were closed. Patients received payment in 36% of claims, totaling more than $461 million, the Physician Insurers Association of America reported.3
47% of 424 randomly selected visits to a hospital emergency department led to added medication, an analysis found. In 10% of those visits, the new medication added potential for an adverse interaction.2
8% of 1,520 significant adverse drug events were caused by drug-drug interactions, three studies of events occurring between 1976 and 1997 found. Serum levels that could be monitored were done so only 17% of the time. Lawsuits resulted in 13% of the cases with settlements/judgments averaging $3.1 million.4
The following strategies can help you avoid mistakes and malpractice claims.1,11
Clinical practice. Obtain a comprehensive patient history and necessary examinations before prescribing medications. See patients at clinically appropriate intervals.
Ask the patient about other medications he or she is taking, including over-the-counter medications, herbal remedies, and dietary supplements. Remind the patient to report changes in medications or new medications prescribed by another physician.
Put in place a process to obtain appropriate baseline laboratory testing and to ensure that follow-up testing is completed and reviewed. Monitoring lab results becomes particularly important in cases—such as these two—when escalating levels of certain medications can cause adverse effects.
Communicate with the patient’s other physicians about all the medications that are being prescribed to him and about signs, symptoms, and responses to the medications.
Educate yourself by participating in continuing education programs, discussions with colleagues, and through relevant literature. Review drug manufacturer alerts.
Patient education. Educate patients about medication instructions, including the dosage and frequency, ways to identify side effects, and what to do in the event of side effects or a bad reaction. Get informed consent.
Be aware of and inform the patient about potentially lethal side effects of misusing or abusing certain medications. Address the use of street drugs and how they interact with prescription medications; make appropriate treatment assessments and referrals for addiction and dependence issues.
Documentation. Keep thorough records of medications prescribed: dosage, amount, directions for taking them, and other instructions to the patient. Document results of laboratory testing and any decisions you make based on medication serum levels.
When using polypharmacy that increases the risk of adverse interactions, document a clear rationale in patients’ charts.
1. Cash C. A few simple steps can avert medical errors. Psychiatric News 2004;39(3):10.-
2. Kohn LT, Corrigan JM, Donaldson MS, eds. To err is human: Building a safer health system. Committee on Quality of Health Care in America, Institute of Medicine. Washington DC: National Academy Press; 2000.
3. McBride D. Managing risk. Minn Med 2000;83:31-2.
4. Kelly N. Potential risks and prevention, Part 4: reports of significant adverse drug events. Am J Health Syst Pharm 2001;58:1406-12.
5. Armstrong SC, Cozza KL, Benedek DM. Med-psych drug-drug interactions update. Psychosomatics 2002;43:245-7.
6. Thanacoody HK, Thomas SH. Tricyclic antidepressant poisoning: cardiovascular toxicity. Toxicol Rev 2005;24:205-14.
7. Venkatakrishnan K, Greenblatt DJ, von Moltke LL, et al. Five distinct human cytochromes mediate amitriptyline N-demethylation in vitro: dominance of CYP 2C19 and 3A4. J Clin Pharmacol 1998;38:112-21.
8. Venkatakrishnan K, von Moltke LL, Greenblatt DJ. Nortriptyline E-10-hydroxylation in vitro is mediated by human CYP2D6 (high affinity) and CYP3A4 (low affinity): implications for interactions with enzyme-inducing drugs. J Clin Pharmacol 1999;39:567-77.
9. Amsterdam J, Brunswick D, Mendels J. The clinical application of tricyclic antidepressant pharmacokinetics and plasma levels. Am J Psychiatry 1980;137:653-62.
10. Thompson D, Oster G. Use of terfenadine and contraindicated drugs. JAMA 1996;275:1339-41.
11. Simon RI. Litigation hotspots in clinical practice. In: Lifson LE, Simon RI, eds. The mental health practitioner and the law. Cambridge, MA: Harvard University Press; 1998:117-39.
Amitriptyline toxicity kills patient
Unknown North Carolina venue
A 26-year-old woman with diabetes saw a psychiatrist to manage her depression. The psychiatrist increased her amitriptyline dosage to 300 mg nightly and over 10 months added:
- alprazolam (unknown dosage, nightly)
- quetiapine (400 mg bid)
- extended-release venlafaxine (225 mg bid)
- and promethazine (100 mg bid).
Several weeks later, the woman was found dead in her home. An autopsy revealed amitriptyline toxicity as the cause of death. The medical examiner noted “a much larger concentration of the metabolite nortriptyline in the liver versus the parent drug,” suggesting a metabolism problem, rather than an overdose, caused the toxic build-up.
The patient’s estate claimed that amitriptyline was cardiotoxic at the prescribed dosage and combined with the other medications used and that the patient was not properly monitored.
- A $2.3 million settlement was reached.
Fatal cardiac arrest after 2 concomitant antidepressants
Gwinnett County (GA) Superior Court
A 40-year-old woman was under a psychiatrist’s care for anxiety and depression. The psychiatrist continued sertraline, which the woman had been taking, and added nortriptyline. Several weeks after the patient began taking the medications together, she had a fatal cardiac arrest.
The patient’s estate argued that:
- toxic levels of the antidepressants caused her death
- sertraline and nortriptyline should not be taken concurrently because one drug inhibits clearance of the other
- the psychiatrist should have monitored the patient to make sure sertraline and nortriptyline levels remained normal.
The medical examiner was unable to say which condition more likely led to the patient’s death.
- The defendant was awarded $3 million. A statutory capitation reduced the award to $1.65 million.
Dr. Grant’s observations
As these cases demonstrate, lawsuits against psychiatrists commonly include allegations of preventable prescribing missteps and drug-drug interactionsOff-label prescribing: 7 steps for safer, more effective treatment”).
When a patient is taking multiple medications, interactions can inhibit drug metabolism and render normal doses excessive.5 When prescribing drugs known to have adverse effects with excessive dosing, such as tricyclic antidepressants and lithium, failing to monitor serum levels could be considered malpractice. In fact, the courts view actions such as prescribing doses that exceed FDA recommendations or failing to monitor levels as prima facie evidence of negligence, requiring the psychiatrist to prove otherwise.
Amitriptyline and nortriptyline have shown cardiac toxicity in overdose,6 and their serum levels increase when used with other antidepressants.7,8 Standard of care dictates serum level monitoring particularly when you use tricyclics:
- in doses higher than recommended by the FDA (300 mg/d for amitriptyline, 150 mg/d for nortriptyline)
- with other drugs that affect their metabolism.9
More than twice as many Americans died from medication errors in 1993 than in 1983, according to a comparative review of U.S. death certificates from that period.2
Between 1985 and 1999, more than 10,000 medication error claims were closed. Patients received payment in 36% of claims, totaling more than $461 million, the Physician Insurers Association of America reported.3
47% of 424 randomly selected visits to a hospital emergency department led to added medication, an analysis found. In 10% of those visits, the new medication added potential for an adverse interaction.2
8% of 1,520 significant adverse drug events were caused by drug-drug interactions, three studies of events occurring between 1976 and 1997 found. Serum levels that could be monitored were done so only 17% of the time. Lawsuits resulted in 13% of the cases with settlements/judgments averaging $3.1 million.4
The following strategies can help you avoid mistakes and malpractice claims.1,11
Clinical practice. Obtain a comprehensive patient history and necessary examinations before prescribing medications. See patients at clinically appropriate intervals.
Ask the patient about other medications he or she is taking, including over-the-counter medications, herbal remedies, and dietary supplements. Remind the patient to report changes in medications or new medications prescribed by another physician.
Put in place a process to obtain appropriate baseline laboratory testing and to ensure that follow-up testing is completed and reviewed. Monitoring lab results becomes particularly important in cases—such as these two—when escalating levels of certain medications can cause adverse effects.
Communicate with the patient’s other physicians about all the medications that are being prescribed to him and about signs, symptoms, and responses to the medications.
Educate yourself by participating in continuing education programs, discussions with colleagues, and through relevant literature. Review drug manufacturer alerts.
Patient education. Educate patients about medication instructions, including the dosage and frequency, ways to identify side effects, and what to do in the event of side effects or a bad reaction. Get informed consent.
Be aware of and inform the patient about potentially lethal side effects of misusing or abusing certain medications. Address the use of street drugs and how they interact with prescription medications; make appropriate treatment assessments and referrals for addiction and dependence issues.
Documentation. Keep thorough records of medications prescribed: dosage, amount, directions for taking them, and other instructions to the patient. Document results of laboratory testing and any decisions you make based on medication serum levels.
When using polypharmacy that increases the risk of adverse interactions, document a clear rationale in patients’ charts.
Amitriptyline toxicity kills patient
Unknown North Carolina venue
A 26-year-old woman with diabetes saw a psychiatrist to manage her depression. The psychiatrist increased her amitriptyline dosage to 300 mg nightly and over 10 months added:
- alprazolam (unknown dosage, nightly)
- quetiapine (400 mg bid)
- extended-release venlafaxine (225 mg bid)
- and promethazine (100 mg bid).
Several weeks later, the woman was found dead in her home. An autopsy revealed amitriptyline toxicity as the cause of death. The medical examiner noted “a much larger concentration of the metabolite nortriptyline in the liver versus the parent drug,” suggesting a metabolism problem, rather than an overdose, caused the toxic build-up.
The patient’s estate claimed that amitriptyline was cardiotoxic at the prescribed dosage and combined with the other medications used and that the patient was not properly monitored.
- A $2.3 million settlement was reached.
Fatal cardiac arrest after 2 concomitant antidepressants
Gwinnett County (GA) Superior Court
A 40-year-old woman was under a psychiatrist’s care for anxiety and depression. The psychiatrist continued sertraline, which the woman had been taking, and added nortriptyline. Several weeks after the patient began taking the medications together, she had a fatal cardiac arrest.
The patient’s estate argued that:
- toxic levels of the antidepressants caused her death
- sertraline and nortriptyline should not be taken concurrently because one drug inhibits clearance of the other
- the psychiatrist should have monitored the patient to make sure sertraline and nortriptyline levels remained normal.
The medical examiner was unable to say which condition more likely led to the patient’s death.
- The defendant was awarded $3 million. A statutory capitation reduced the award to $1.65 million.
Dr. Grant’s observations
As these cases demonstrate, lawsuits against psychiatrists commonly include allegations of preventable prescribing missteps and drug-drug interactionsOff-label prescribing: 7 steps for safer, more effective treatment”).
When a patient is taking multiple medications, interactions can inhibit drug metabolism and render normal doses excessive.5 When prescribing drugs known to have adverse effects with excessive dosing, such as tricyclic antidepressants and lithium, failing to monitor serum levels could be considered malpractice. In fact, the courts view actions such as prescribing doses that exceed FDA recommendations or failing to monitor levels as prima facie evidence of negligence, requiring the psychiatrist to prove otherwise.
Amitriptyline and nortriptyline have shown cardiac toxicity in overdose,6 and their serum levels increase when used with other antidepressants.7,8 Standard of care dictates serum level monitoring particularly when you use tricyclics:
- in doses higher than recommended by the FDA (300 mg/d for amitriptyline, 150 mg/d for nortriptyline)
- with other drugs that affect their metabolism.9
More than twice as many Americans died from medication errors in 1993 than in 1983, according to a comparative review of U.S. death certificates from that period.2
Between 1985 and 1999, more than 10,000 medication error claims were closed. Patients received payment in 36% of claims, totaling more than $461 million, the Physician Insurers Association of America reported.3
47% of 424 randomly selected visits to a hospital emergency department led to added medication, an analysis found. In 10% of those visits, the new medication added potential for an adverse interaction.2
8% of 1,520 significant adverse drug events were caused by drug-drug interactions, three studies of events occurring between 1976 and 1997 found. Serum levels that could be monitored were done so only 17% of the time. Lawsuits resulted in 13% of the cases with settlements/judgments averaging $3.1 million.4
The following strategies can help you avoid mistakes and malpractice claims.1,11
Clinical practice. Obtain a comprehensive patient history and necessary examinations before prescribing medications. See patients at clinically appropriate intervals.
Ask the patient about other medications he or she is taking, including over-the-counter medications, herbal remedies, and dietary supplements. Remind the patient to report changes in medications or new medications prescribed by another physician.
Put in place a process to obtain appropriate baseline laboratory testing and to ensure that follow-up testing is completed and reviewed. Monitoring lab results becomes particularly important in cases—such as these two—when escalating levels of certain medications can cause adverse effects.
Communicate with the patient’s other physicians about all the medications that are being prescribed to him and about signs, symptoms, and responses to the medications.
Educate yourself by participating in continuing education programs, discussions with colleagues, and through relevant literature. Review drug manufacturer alerts.
Patient education. Educate patients about medication instructions, including the dosage and frequency, ways to identify side effects, and what to do in the event of side effects or a bad reaction. Get informed consent.
Be aware of and inform the patient about potentially lethal side effects of misusing or abusing certain medications. Address the use of street drugs and how they interact with prescription medications; make appropriate treatment assessments and referrals for addiction and dependence issues.
Documentation. Keep thorough records of medications prescribed: dosage, amount, directions for taking them, and other instructions to the patient. Document results of laboratory testing and any decisions you make based on medication serum levels.
When using polypharmacy that increases the risk of adverse interactions, document a clear rationale in patients’ charts.
1. Cash C. A few simple steps can avert medical errors. Psychiatric News 2004;39(3):10.-
2. Kohn LT, Corrigan JM, Donaldson MS, eds. To err is human: Building a safer health system. Committee on Quality of Health Care in America, Institute of Medicine. Washington DC: National Academy Press; 2000.
3. McBride D. Managing risk. Minn Med 2000;83:31-2.
4. Kelly N. Potential risks and prevention, Part 4: reports of significant adverse drug events. Am J Health Syst Pharm 2001;58:1406-12.
5. Armstrong SC, Cozza KL, Benedek DM. Med-psych drug-drug interactions update. Psychosomatics 2002;43:245-7.
6. Thanacoody HK, Thomas SH. Tricyclic antidepressant poisoning: cardiovascular toxicity. Toxicol Rev 2005;24:205-14.
7. Venkatakrishnan K, Greenblatt DJ, von Moltke LL, et al. Five distinct human cytochromes mediate amitriptyline N-demethylation in vitro: dominance of CYP 2C19 and 3A4. J Clin Pharmacol 1998;38:112-21.
8. Venkatakrishnan K, von Moltke LL, Greenblatt DJ. Nortriptyline E-10-hydroxylation in vitro is mediated by human CYP2D6 (high affinity) and CYP3A4 (low affinity): implications for interactions with enzyme-inducing drugs. J Clin Pharmacol 1999;39:567-77.
9. Amsterdam J, Brunswick D, Mendels J. The clinical application of tricyclic antidepressant pharmacokinetics and plasma levels. Am J Psychiatry 1980;137:653-62.
10. Thompson D, Oster G. Use of terfenadine and contraindicated drugs. JAMA 1996;275:1339-41.
11. Simon RI. Litigation hotspots in clinical practice. In: Lifson LE, Simon RI, eds. The mental health practitioner and the law. Cambridge, MA: Harvard University Press; 1998:117-39.
1. Cash C. A few simple steps can avert medical errors. Psychiatric News 2004;39(3):10.-
2. Kohn LT, Corrigan JM, Donaldson MS, eds. To err is human: Building a safer health system. Committee on Quality of Health Care in America, Institute of Medicine. Washington DC: National Academy Press; 2000.
3. McBride D. Managing risk. Minn Med 2000;83:31-2.
4. Kelly N. Potential risks and prevention, Part 4: reports of significant adverse drug events. Am J Health Syst Pharm 2001;58:1406-12.
5. Armstrong SC, Cozza KL, Benedek DM. Med-psych drug-drug interactions update. Psychosomatics 2002;43:245-7.
6. Thanacoody HK, Thomas SH. Tricyclic antidepressant poisoning: cardiovascular toxicity. Toxicol Rev 2005;24:205-14.
7. Venkatakrishnan K, Greenblatt DJ, von Moltke LL, et al. Five distinct human cytochromes mediate amitriptyline N-demethylation in vitro: dominance of CYP 2C19 and 3A4. J Clin Pharmacol 1998;38:112-21.
8. Venkatakrishnan K, von Moltke LL, Greenblatt DJ. Nortriptyline E-10-hydroxylation in vitro is mediated by human CYP2D6 (high affinity) and CYP3A4 (low affinity): implications for interactions with enzyme-inducing drugs. J Clin Pharmacol 1999;39:567-77.
9. Amsterdam J, Brunswick D, Mendels J. The clinical application of tricyclic antidepressant pharmacokinetics and plasma levels. Am J Psychiatry 1980;137:653-62.
10. Thompson D, Oster G. Use of terfenadine and contraindicated drugs. JAMA 1996;275:1339-41.
11. Simon RI. Litigation hotspots in clinical practice. In: Lifson LE, Simon RI, eds. The mental health practitioner and the law. Cambridge, MA: Harvard University Press; 1998:117-39.
Rethinking vitamin E
Vitamin E has been speculated to prevent or treat numerous illnesses. Despite limited efficacy data, many psychiatrists prescribe supplemental vitamin E to prevent or treat dementia and tardive dyskinesia (TD). Findings from several recent meta-analyses, however, question vitamin E’s benefit in certain uses and suggest that it carries some risks.1,2
Mortality risk. In a meta-analysis of 19 clinical trials,2 Miller et al found that vitamin E, 400 IU/d, increased all-cause mortality risk (risk ratio=1.04; P=.035) compared with dosages
- may have pro-oxidant effects at high dosages, which could increase the risk of atherosclerosis
- may lead to withdrawal if used irregularly at high doses
- is an anticoagulant that may increase the risk of hemorrhagic stroke. Vitamin E also can worsen coagulation defects at high dosages and is contraindicated in patients taking coumadin.3
Treating moderate Alzheimer’s disease (AD). Sano et al found little difference in effectiveness between selegiline, 10 mg/d; vitamin E, 2,000 IU/d; concomitant selegiline and vitamin E; or placebo.4 After considering the placebo group’s higher baseline Mini-Mental State Examination scores, the researchers found that primary outcomes (death, institutionalization, lost activities of daily living, progression to severe dementia) were delayed among the treatment groups. Changes in cognitive scores from baseline differed little between the treatment and placebo groups.
Dietary vitamin E consists of various tocopherol forms as well as the alpha-tocopherol usually contained in vitamin E supplements. Increased dietary vitamin E intake may lower the risk of AD7 but probably has different risks and benefits than vitamin E supplementation. For example, Morris et al7 found that increased dietary intake of alpha- and gamma-tocopherols was associated with a reduced AD incidence, whereas beta-tocopherol did not prevent development of AD.
Treating TD. A few small studies have associated vitamin E supplementation with reduced TD symptoms. Soares and McGrath, however, found limited evidence that vitamin E prevents worsening of TD and no evidence that it improves TD symptoms.5 Vitamin E might be most beneficial to patients who have had TD 6
When prescribing vitamin E, be sure to discuss its risks and benefits with patients.
Michael J. Rack, MD
Clinical assistant professor of psychiatry
Sarah Rack, MD
Assistant professor of psychiatry
University of Mississippi Medical Center, Jackson
1. Bjelakovic G, Nikolova D, Simonetti RG, Gluud C. Antioxidant supplements for prevention of gastrointestinal cancers: A systematic review and meta-analysis. Lancet 2004;364:1219-28.
2. Miller ER, 3rd, Pastor-Barriuso R, Dalal D, et al. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med 2005;142:37-46.
3. Berman K, Brodaty H. Tocopherol (vitamin E) in Alzheimer’s disease and other neurodegenerative disorders. CNS Drugs 2004;18:807-25.
4. Sano M, Ernesto C, Thomas RG, et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. N Engl J Med 1997;336:1216-22.
5. Soares KV, McGrath JJ. Vitamin E for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 2001;(4):CD000209.-
6. Lohr JB, Caligiuri MP. A double-blind placebo-controlled study of vitamin E treatment of tardive dyskinesia. J Clin Psychiatry 1996;57:167-73.
7. Morris MC, Evans DA, Tangney CC, et al. Relation of the tocopherol forms to incident Alzheimer disease and to cognitive change. Am J Clin Nutr 2005;81:508-14.
Vitamin E has been speculated to prevent or treat numerous illnesses. Despite limited efficacy data, many psychiatrists prescribe supplemental vitamin E to prevent or treat dementia and tardive dyskinesia (TD). Findings from several recent meta-analyses, however, question vitamin E’s benefit in certain uses and suggest that it carries some risks.1,2
Mortality risk. In a meta-analysis of 19 clinical trials,2 Miller et al found that vitamin E, 400 IU/d, increased all-cause mortality risk (risk ratio=1.04; P=.035) compared with dosages
- may have pro-oxidant effects at high dosages, which could increase the risk of atherosclerosis
- may lead to withdrawal if used irregularly at high doses
- is an anticoagulant that may increase the risk of hemorrhagic stroke. Vitamin E also can worsen coagulation defects at high dosages and is contraindicated in patients taking coumadin.3
Treating moderate Alzheimer’s disease (AD). Sano et al found little difference in effectiveness between selegiline, 10 mg/d; vitamin E, 2,000 IU/d; concomitant selegiline and vitamin E; or placebo.4 After considering the placebo group’s higher baseline Mini-Mental State Examination scores, the researchers found that primary outcomes (death, institutionalization, lost activities of daily living, progression to severe dementia) were delayed among the treatment groups. Changes in cognitive scores from baseline differed little between the treatment and placebo groups.
Dietary vitamin E consists of various tocopherol forms as well as the alpha-tocopherol usually contained in vitamin E supplements. Increased dietary vitamin E intake may lower the risk of AD7 but probably has different risks and benefits than vitamin E supplementation. For example, Morris et al7 found that increased dietary intake of alpha- and gamma-tocopherols was associated with a reduced AD incidence, whereas beta-tocopherol did not prevent development of AD.
Treating TD. A few small studies have associated vitamin E supplementation with reduced TD symptoms. Soares and McGrath, however, found limited evidence that vitamin E prevents worsening of TD and no evidence that it improves TD symptoms.5 Vitamin E might be most beneficial to patients who have had TD 6
When prescribing vitamin E, be sure to discuss its risks and benefits with patients.
Michael J. Rack, MD
Clinical assistant professor of psychiatry
Sarah Rack, MD
Assistant professor of psychiatry
University of Mississippi Medical Center, Jackson
Vitamin E has been speculated to prevent or treat numerous illnesses. Despite limited efficacy data, many psychiatrists prescribe supplemental vitamin E to prevent or treat dementia and tardive dyskinesia (TD). Findings from several recent meta-analyses, however, question vitamin E’s benefit in certain uses and suggest that it carries some risks.1,2
Mortality risk. In a meta-analysis of 19 clinical trials,2 Miller et al found that vitamin E, 400 IU/d, increased all-cause mortality risk (risk ratio=1.04; P=.035) compared with dosages
- may have pro-oxidant effects at high dosages, which could increase the risk of atherosclerosis
- may lead to withdrawal if used irregularly at high doses
- is an anticoagulant that may increase the risk of hemorrhagic stroke. Vitamin E also can worsen coagulation defects at high dosages and is contraindicated in patients taking coumadin.3
Treating moderate Alzheimer’s disease (AD). Sano et al found little difference in effectiveness between selegiline, 10 mg/d; vitamin E, 2,000 IU/d; concomitant selegiline and vitamin E; or placebo.4 After considering the placebo group’s higher baseline Mini-Mental State Examination scores, the researchers found that primary outcomes (death, institutionalization, lost activities of daily living, progression to severe dementia) were delayed among the treatment groups. Changes in cognitive scores from baseline differed little between the treatment and placebo groups.
Dietary vitamin E consists of various tocopherol forms as well as the alpha-tocopherol usually contained in vitamin E supplements. Increased dietary vitamin E intake may lower the risk of AD7 but probably has different risks and benefits than vitamin E supplementation. For example, Morris et al7 found that increased dietary intake of alpha- and gamma-tocopherols was associated with a reduced AD incidence, whereas beta-tocopherol did not prevent development of AD.
Treating TD. A few small studies have associated vitamin E supplementation with reduced TD symptoms. Soares and McGrath, however, found limited evidence that vitamin E prevents worsening of TD and no evidence that it improves TD symptoms.5 Vitamin E might be most beneficial to patients who have had TD 6
When prescribing vitamin E, be sure to discuss its risks and benefits with patients.
Michael J. Rack, MD
Clinical assistant professor of psychiatry
Sarah Rack, MD
Assistant professor of psychiatry
University of Mississippi Medical Center, Jackson
1. Bjelakovic G, Nikolova D, Simonetti RG, Gluud C. Antioxidant supplements for prevention of gastrointestinal cancers: A systematic review and meta-analysis. Lancet 2004;364:1219-28.
2. Miller ER, 3rd, Pastor-Barriuso R, Dalal D, et al. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med 2005;142:37-46.
3. Berman K, Brodaty H. Tocopherol (vitamin E) in Alzheimer’s disease and other neurodegenerative disorders. CNS Drugs 2004;18:807-25.
4. Sano M, Ernesto C, Thomas RG, et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. N Engl J Med 1997;336:1216-22.
5. Soares KV, McGrath JJ. Vitamin E for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 2001;(4):CD000209.-
6. Lohr JB, Caligiuri MP. A double-blind placebo-controlled study of vitamin E treatment of tardive dyskinesia. J Clin Psychiatry 1996;57:167-73.
7. Morris MC, Evans DA, Tangney CC, et al. Relation of the tocopherol forms to incident Alzheimer disease and to cognitive change. Am J Clin Nutr 2005;81:508-14.
1. Bjelakovic G, Nikolova D, Simonetti RG, Gluud C. Antioxidant supplements for prevention of gastrointestinal cancers: A systematic review and meta-analysis. Lancet 2004;364:1219-28.
2. Miller ER, 3rd, Pastor-Barriuso R, Dalal D, et al. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med 2005;142:37-46.
3. Berman K, Brodaty H. Tocopherol (vitamin E) in Alzheimer’s disease and other neurodegenerative disorders. CNS Drugs 2004;18:807-25.
4. Sano M, Ernesto C, Thomas RG, et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. N Engl J Med 1997;336:1216-22.
5. Soares KV, McGrath JJ. Vitamin E for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 2001;(4):CD000209.-
6. Lohr JB, Caligiuri MP. A double-blind placebo-controlled study of vitamin E treatment of tardive dyskinesia. J Clin Psychiatry 1996;57:167-73.
7. Morris MC, Evans DA, Tangney CC, et al. Relation of the tocopherol forms to incident Alzheimer disease and to cognitive change. Am J Clin Nutr 2005;81:508-14.
How 15-minute med checks hurt patient care
I am grateful to Drs. Phil Bohnert and Anne O’Connell for pointing out how the 15-minute medication check is hurting quality of care (Current Psychiatry, January 2006). How can Dr. H. Steven Moffic criticize their work as “a cheap shot?” (Current Psychiatry, March 2006).
On a typical morning, I see 14 patients for 15 minutes each. Some come a couple minutes late, pushing back other appointments so that I am seeing the last patient 15 to 20 minutes later than scheduled. I barely get time for lunch before the afternoon’s first patient arrives.
If Dr. Moffic can handle an “assembly line” of patients, more power to him. For the sake of quality care, I should not be doing that.
Also, let’s say you have one patient in your exam room and another in your waiting room. The patient you are treating announces out of the blue that he or she is suicidal.
How would Dr. Moffic handle that? And how does he deal with calls, pages, patient forms, and other distractions within 15 minutes?
Dr. Moffic’s patients may prefer shorter visits, but over 35 years I have yet to meet a patient who wants to be rushed out the door.
Fuat Ulus, MD
Erie, PA
Dr. Moffic responds
I praised Dr. Bohnert’s and O’Connell’s article as a whole, but was concerned that yet another demonizing of managed care was hurting physician morale.
Time limits might be making practice difficult for Dr. Ulus and many others, but 15-minute medication checks were in place in many community psychiatry settings before managed care and are standard for public-care settings in areas where managed care has yet to make inroads. That said, limited government funding poses the most serious hazard for public medicine.
If Dr. Ulus sees 14 patients nonstop for 15 minutes each—without any no-shows—he must be doing a superb job. No-shows usually provide some cushion for other tasks, including a suicidal patient. I’ve found that patients usually don’t mind waiting or being rescheduled when an emergency occurs. Also, at our multidisciplinary clinic other staff can help where needed. And many patients need less than 15 minutes, allowing more time for other patients and tasks.
If we are stuck with 15-minute med checks because of finances and other system problems, we can make these visits more satisfying and successful. Informing patients of our limits and the reasons behind them will help us build an alliance with them and make the most of our limited time.
Further research could help determine how much clinical time each treatment need requires. Interestingly, no studies have measured psychiatric outcomes after 15-minute checks compared with longer visits.
H. Steven Moffic, MD
Milwaukee, WI
I am grateful to Drs. Phil Bohnert and Anne O’Connell for pointing out how the 15-minute medication check is hurting quality of care (Current Psychiatry, January 2006). How can Dr. H. Steven Moffic criticize their work as “a cheap shot?” (Current Psychiatry, March 2006).
On a typical morning, I see 14 patients for 15 minutes each. Some come a couple minutes late, pushing back other appointments so that I am seeing the last patient 15 to 20 minutes later than scheduled. I barely get time for lunch before the afternoon’s first patient arrives.
If Dr. Moffic can handle an “assembly line” of patients, more power to him. For the sake of quality care, I should not be doing that.
Also, let’s say you have one patient in your exam room and another in your waiting room. The patient you are treating announces out of the blue that he or she is suicidal.
How would Dr. Moffic handle that? And how does he deal with calls, pages, patient forms, and other distractions within 15 minutes?
Dr. Moffic’s patients may prefer shorter visits, but over 35 years I have yet to meet a patient who wants to be rushed out the door.
Fuat Ulus, MD
Erie, PA
Dr. Moffic responds
I praised Dr. Bohnert’s and O’Connell’s article as a whole, but was concerned that yet another demonizing of managed care was hurting physician morale.
Time limits might be making practice difficult for Dr. Ulus and many others, but 15-minute medication checks were in place in many community psychiatry settings before managed care and are standard for public-care settings in areas where managed care has yet to make inroads. That said, limited government funding poses the most serious hazard for public medicine.
If Dr. Ulus sees 14 patients nonstop for 15 minutes each—without any no-shows—he must be doing a superb job. No-shows usually provide some cushion for other tasks, including a suicidal patient. I’ve found that patients usually don’t mind waiting or being rescheduled when an emergency occurs. Also, at our multidisciplinary clinic other staff can help where needed. And many patients need less than 15 minutes, allowing more time for other patients and tasks.
If we are stuck with 15-minute med checks because of finances and other system problems, we can make these visits more satisfying and successful. Informing patients of our limits and the reasons behind them will help us build an alliance with them and make the most of our limited time.
Further research could help determine how much clinical time each treatment need requires. Interestingly, no studies have measured psychiatric outcomes after 15-minute checks compared with longer visits.
H. Steven Moffic, MD
Milwaukee, WI
I am grateful to Drs. Phil Bohnert and Anne O’Connell for pointing out how the 15-minute medication check is hurting quality of care (Current Psychiatry, January 2006). How can Dr. H. Steven Moffic criticize their work as “a cheap shot?” (Current Psychiatry, March 2006).
On a typical morning, I see 14 patients for 15 minutes each. Some come a couple minutes late, pushing back other appointments so that I am seeing the last patient 15 to 20 minutes later than scheduled. I barely get time for lunch before the afternoon’s first patient arrives.
If Dr. Moffic can handle an “assembly line” of patients, more power to him. For the sake of quality care, I should not be doing that.
Also, let’s say you have one patient in your exam room and another in your waiting room. The patient you are treating announces out of the blue that he or she is suicidal.
How would Dr. Moffic handle that? And how does he deal with calls, pages, patient forms, and other distractions within 15 minutes?
Dr. Moffic’s patients may prefer shorter visits, but over 35 years I have yet to meet a patient who wants to be rushed out the door.
Fuat Ulus, MD
Erie, PA
Dr. Moffic responds
I praised Dr. Bohnert’s and O’Connell’s article as a whole, but was concerned that yet another demonizing of managed care was hurting physician morale.
Time limits might be making practice difficult for Dr. Ulus and many others, but 15-minute medication checks were in place in many community psychiatry settings before managed care and are standard for public-care settings in areas where managed care has yet to make inroads. That said, limited government funding poses the most serious hazard for public medicine.
If Dr. Ulus sees 14 patients nonstop for 15 minutes each—without any no-shows—he must be doing a superb job. No-shows usually provide some cushion for other tasks, including a suicidal patient. I’ve found that patients usually don’t mind waiting or being rescheduled when an emergency occurs. Also, at our multidisciplinary clinic other staff can help where needed. And many patients need less than 15 minutes, allowing more time for other patients and tasks.
If we are stuck with 15-minute med checks because of finances and other system problems, we can make these visits more satisfying and successful. Informing patients of our limits and the reasons behind them will help us build an alliance with them and make the most of our limited time.
Further research could help determine how much clinical time each treatment need requires. Interestingly, no studies have measured psychiatric outcomes after 15-minute checks compared with longer visits.
H. Steven Moffic, MD
Milwaukee, WI
Patient need—not finances—should dictate visit length
The article by Drs. Bohnert and O’Connell (Current Psychiatry, January 2006) and Dr. Moffic’s response (Current Psychiatry, March 2006) both miss the point for the average outpatient psychiatrist.
In most cases, isn’t duration of clinical contact the physician’s choice? Basing clinical decisions solely on income is a serious breach of ethics.
Our profession requires us to act in the patient’s best interest, regardless of financial impact to us. No insurance company or governmental body has ever told me how much time to spend with a patient. Our ethics allow us to choose whom we will serve but forbid us from providing substandard care just because we are unhappy with the compensation.
One of my mentors in medical school put it simply: “Just take care of the patient and let the billing take care of itself.” When I no longer can make a living practicing ethical psychiatry, it will be time to find a new career, not a new set of ethics.
Thomas A. Grugle, MD
Richardson, TX
Dr. Bohnert responds
Dr. Grugle makes an excellent point.
I have NEVER limited any medication visit to 15 minutes. I offer each patient a “session” to meet his or her needs, not a time-determined meeting.
Our article’s reference to the stress psychiatrists experience with 15-minute visits was based on literature reports and on discussions with psychiatrists who increasingly feel they have time only for medication checks and must refer psychotherapy to nonpsychiatric therapists.
Phil Bohnert, MD
University of Hawaii, Honolulu
Dr. Moffic responds
I believe Dr. Grugle misinterpreted my attempt to present the most ethical way to conduct brief medication checks.
Section I of AMA Principles of Medical Ethics states an ethical responsibility to provide “competent” (though not ideal) medical service. I believe that competent care generally can be provided with brief medication checks and there are no data to the contrary, but flexibility is needed with each patient.
I appreciate Dr. Grugle’s viewpoint. We can approach this issue from two directions: learn how to do brief med checks as well as possible and (per Section 7 of the principles) advocate for system changes. If we fail to address systemic issues, all of psychiatry will continue to suffer.
H. Steven Moffic, MD
Milwaukee, WI
The article by Drs. Bohnert and O’Connell (Current Psychiatry, January 2006) and Dr. Moffic’s response (Current Psychiatry, March 2006) both miss the point for the average outpatient psychiatrist.
In most cases, isn’t duration of clinical contact the physician’s choice? Basing clinical decisions solely on income is a serious breach of ethics.
Our profession requires us to act in the patient’s best interest, regardless of financial impact to us. No insurance company or governmental body has ever told me how much time to spend with a patient. Our ethics allow us to choose whom we will serve but forbid us from providing substandard care just because we are unhappy with the compensation.
One of my mentors in medical school put it simply: “Just take care of the patient and let the billing take care of itself.” When I no longer can make a living practicing ethical psychiatry, it will be time to find a new career, not a new set of ethics.
Thomas A. Grugle, MD
Richardson, TX
Dr. Bohnert responds
Dr. Grugle makes an excellent point.
I have NEVER limited any medication visit to 15 minutes. I offer each patient a “session” to meet his or her needs, not a time-determined meeting.
Our article’s reference to the stress psychiatrists experience with 15-minute visits was based on literature reports and on discussions with psychiatrists who increasingly feel they have time only for medication checks and must refer psychotherapy to nonpsychiatric therapists.
Phil Bohnert, MD
University of Hawaii, Honolulu
Dr. Moffic responds
I believe Dr. Grugle misinterpreted my attempt to present the most ethical way to conduct brief medication checks.
Section I of AMA Principles of Medical Ethics states an ethical responsibility to provide “competent” (though not ideal) medical service. I believe that competent care generally can be provided with brief medication checks and there are no data to the contrary, but flexibility is needed with each patient.
I appreciate Dr. Grugle’s viewpoint. We can approach this issue from two directions: learn how to do brief med checks as well as possible and (per Section 7 of the principles) advocate for system changes. If we fail to address systemic issues, all of psychiatry will continue to suffer.
H. Steven Moffic, MD
Milwaukee, WI
The article by Drs. Bohnert and O’Connell (Current Psychiatry, January 2006) and Dr. Moffic’s response (Current Psychiatry, March 2006) both miss the point for the average outpatient psychiatrist.
In most cases, isn’t duration of clinical contact the physician’s choice? Basing clinical decisions solely on income is a serious breach of ethics.
Our profession requires us to act in the patient’s best interest, regardless of financial impact to us. No insurance company or governmental body has ever told me how much time to spend with a patient. Our ethics allow us to choose whom we will serve but forbid us from providing substandard care just because we are unhappy with the compensation.
One of my mentors in medical school put it simply: “Just take care of the patient and let the billing take care of itself.” When I no longer can make a living practicing ethical psychiatry, it will be time to find a new career, not a new set of ethics.
Thomas A. Grugle, MD
Richardson, TX
Dr. Bohnert responds
Dr. Grugle makes an excellent point.
I have NEVER limited any medication visit to 15 minutes. I offer each patient a “session” to meet his or her needs, not a time-determined meeting.
Our article’s reference to the stress psychiatrists experience with 15-minute visits was based on literature reports and on discussions with psychiatrists who increasingly feel they have time only for medication checks and must refer psychotherapy to nonpsychiatric therapists.
Phil Bohnert, MD
University of Hawaii, Honolulu
Dr. Moffic responds
I believe Dr. Grugle misinterpreted my attempt to present the most ethical way to conduct brief medication checks.
Section I of AMA Principles of Medical Ethics states an ethical responsibility to provide “competent” (though not ideal) medical service. I believe that competent care generally can be provided with brief medication checks and there are no data to the contrary, but flexibility is needed with each patient.
I appreciate Dr. Grugle’s viewpoint. We can approach this issue from two directions: learn how to do brief med checks as well as possible and (per Section 7 of the principles) advocate for system changes. If we fail to address systemic issues, all of psychiatry will continue to suffer.
H. Steven Moffic, MD
Milwaukee, WI
Depressed and pregnant: Now what?
What do you do when a patient with major depressive disorder who is being successfully maintained on medication wants to become pregnant or has just learned she is pregnant?
If you’re like me, you worry—a lot. If you stop maintenance antidepressants, the chance of depressive relapse is substantial. If you continue the medication and the child has congenital anomalies, the chance of you being blamed is substantial. Either way, you can be sued for malpractice.
Two articles this month address this dilemma: an evidence-based review on use of SSRIs in pregnancy by Caitlin Hasser, MD, Louann Brizendine, MD, and Anna Spielvogel, MD, PhD, from the University of California, San Francisco, and a commentary on the FDA’s paroxetine advisory by Lawson Wulsin, MD, of the University of Cincinnati.
FDA categorizes most drugs we prescribe as pregnancy risk category C, “Risk cannot be ruled out.” Paroxetine has recently been moved to category D, “Evidence of risk to the fetus in human studies.” About 3% of births involve anomalies; a recent study of women who took paroxetine during pregnancy showed a 4% rate. If you treat 30 or more pregnant, depressed patients during your career, odds are that at least one of them will have a child with birth defects, purely by chance.
If we continue antidepressant therapy during pregnancy, the best we can do is study the literature, document that we discussed risks and benefits with the patient, and avoid paroxetine if possible. If we discontinue the medication, the best we can do is document that we discussed risks and benefits with the patient and follow her closely for depressive relapse.
Nobody said this job would be easy.

James Randolph Hillard, MD
Editor-in-Chief
James Randolph Hillard, MD
Editor-in-Chief
James Randolph Hillard, MD
Editor-in-Chief
What do you do when a patient with major depressive disorder who is being successfully maintained on medication wants to become pregnant or has just learned she is pregnant?
If you’re like me, you worry—a lot. If you stop maintenance antidepressants, the chance of depressive relapse is substantial. If you continue the medication and the child has congenital anomalies, the chance of you being blamed is substantial. Either way, you can be sued for malpractice.
Two articles this month address this dilemma: an evidence-based review on use of SSRIs in pregnancy by Caitlin Hasser, MD, Louann Brizendine, MD, and Anna Spielvogel, MD, PhD, from the University of California, San Francisco, and a commentary on the FDA’s paroxetine advisory by Lawson Wulsin, MD, of the University of Cincinnati.
FDA categorizes most drugs we prescribe as pregnancy risk category C, “Risk cannot be ruled out.” Paroxetine has recently been moved to category D, “Evidence of risk to the fetus in human studies.” About 3% of births involve anomalies; a recent study of women who took paroxetine during pregnancy showed a 4% rate. If you treat 30 or more pregnant, depressed patients during your career, odds are that at least one of them will have a child with birth defects, purely by chance.
If we continue antidepressant therapy during pregnancy, the best we can do is study the literature, document that we discussed risks and benefits with the patient, and avoid paroxetine if possible. If we discontinue the medication, the best we can do is document that we discussed risks and benefits with the patient and follow her closely for depressive relapse.
Nobody said this job would be easy.
What do you do when a patient with major depressive disorder who is being successfully maintained on medication wants to become pregnant or has just learned she is pregnant?
If you’re like me, you worry—a lot. If you stop maintenance antidepressants, the chance of depressive relapse is substantial. If you continue the medication and the child has congenital anomalies, the chance of you being blamed is substantial. Either way, you can be sued for malpractice.
Two articles this month address this dilemma: an evidence-based review on use of SSRIs in pregnancy by Caitlin Hasser, MD, Louann Brizendine, MD, and Anna Spielvogel, MD, PhD, from the University of California, San Francisco, and a commentary on the FDA’s paroxetine advisory by Lawson Wulsin, MD, of the University of Cincinnati.
FDA categorizes most drugs we prescribe as pregnancy risk category C, “Risk cannot be ruled out.” Paroxetine has recently been moved to category D, “Evidence of risk to the fetus in human studies.” About 3% of births involve anomalies; a recent study of women who took paroxetine during pregnancy showed a 4% rate. If you treat 30 or more pregnant, depressed patients during your career, odds are that at least one of them will have a child with birth defects, purely by chance.
If we continue antidepressant therapy during pregnancy, the best we can do is study the literature, document that we discussed risks and benefits with the patient, and avoid paroxetine if possible. If we discontinue the medication, the best we can do is document that we discussed risks and benefits with the patient and follow her closely for depressive relapse.
Nobody said this job would be easy.
Paroxetine in pregnancy?
Mrs. J, age 24, has a history of recurrent major depression, for which you have prescribed paroxetine. Newly pregnant, she brings you Internet articles with headlines such as “Depression drugs ‘can raise birth defect risks.’”SSRI use during pregnancy: Do antidepressants’ benefits outweigh the risks?).
FDA recommends avoiding paroxetine in women of child-bearing age. How does this advisory change the way we manage depression in pregnancy? How strong is the evidence supporting it?
SSRIs and birth defect risk
Eight prospective or case-control studies of SSRIs in >5,400 pregnant women have been published since 1993 (Table).3-10 Five included paroxetine.6-10 The studies ranged from small to large, and none showed a significant increase in major malformations with any SSRI. Even in a study of >2,500 women, no single malformation was overrepresented.8
In addition, a recent meta-analysis11 of 7 prospective comparative cohort studies involving 1,774 pregnant women showed no increased risk of major birth defects from exposure to any of the 8 antidepressants studied, including 4 SSRIs used during the first trimester. The review identified no specific malformation or cluster of malformations associated with first-trimester antidepressant use.
Table
8 published studies: No significant increase in birth defects with SSRIs
| Year/location | Authors | Study design | SSRI exposure (# of patients) | Risk of major malformation |
|---|---|---|---|---|
| 1993/USA, Canada | Pastuszak et al3 | Prospective cohort, controlled | Fluoxetine (98) | SSRI: 2% Control: 1.8% (ns) |
| 1996/USA | Chambers et al4 | Prospective cohort, controlled | Fluoxetine (174) | SSRI: 3.4% Control: 2.7% (ns) |
| 1997/worldwide | Goldstein et al5 | Clinical trial | Fluoxetine (28) | SSRI: 3.6% (ns) |
| 1998/USA, Canada, Brazil | Kulin et al6 | Prospective cohort, controlled | Paroxetine (97) | Total SSRI: 4.1% Control: 3.8% (ns) |
| Sertraline (147) | ||||
| Fluvoxamine (26) | ||||
| 1999/Sweden | Ericson et al7 | Case-control | Citalopram (364) | Citalopram: 3.9% Total risk of remaining SSRIs: 3.8% (ns) |
| Paroxetine (118) | ||||
| Sertraline (32) | ||||
| Fluoxetine (15) | ||||
| 2002/USA | Simon et al9 | Case-control | Fluoxetine (129) | Total SSRI: 6.5% Control: 4.9% (ns) |
| Sertraline (32) | ||||
| Paroxetine (28) | ||||
| 2003/USA | Hendrick et al10 | Prospective, uncontrolled | Fluoxetine (13) | Total SSRI: 1.4% (ns) |
| Paroxetine (19) | ||||
| Sertraline (36) | ||||
| 2005/Sweden | Hallberg et al8 | Case-control | Citalopram (1,696) | Citalopram: 3.1% |
| Paroxetine (708) | Paroxetine: 3.4% | |||
| Sertraline (1,067) | Sertraline: 2.0% | |||
| Fluoxetine (574) | Fluoxetine: 3.3% (ns) | |||
| ns: No statistically significant difference | ||||
Evidence cited by FDA
FDA’s advisory (Box) came 3 months after GSK notified health professionals that fetuses exposed to paroxetine during organogenesis may be at increased risk of developing malformations, particularly ventricular septal defect (see Related resources).
What FDA recommends to you and your patients
The FDA is awaiting the final results of the recent studies and accruing additional data related to the use of paroxetine in pregnancy in order to better characterize the risk for congenital malformations associated with paroxetine. In the interim, FDA recommends the following:
Physicians who are caring for women receiving paroxetine should alert them to the potential risk to the fetus if they plan to become pregnant or are currently in their first trimester of pregnancy. Discontinuing paroxetine therapy should be considered for these patients. In individual cases, the benefits of continuing paroxetine may outweigh the potential risk to the fetus. If the decision is made to discontinue paroxetine and switch to another antidepressant or cease antidepressant therapy, paroxetine discontinuation should be undertaken only as directed in the prescribing information. Paroxetine should generally not be initiated in women who are in their first trimester of pregnancy or in women who plan to become pregnant in the near future.
Women who are pregnant, or planning a pregnancy, and currently taking paroxetine should consult with their physician about whether to continue taking it. Women should not stop the drug without discussing the best way to do that with their physician.
Source: Verbatim from FDA advisory, December 2005.
- Major congenital defects occurred in 4% of 527 pregnancies during which women used paroxetine, (adjusted odds ratios [OR], 2.20; 95% CI, 1.34-3.63), compared with 3% prevalence in the general population.
- Cardiovascular malformations occurred at an adjusted rate of 2% (OR, 2.08; 95% CI, 1.03-4.23), compared with 1% in the general population.
- 10 of the 14 cardiovascular malformations were ventricular septal defects.
The GSK investigation was an unpublished retrospective study without peer review when FDA issued its advisory about paroxetine.
Two abstracts. The FDA alert also cited two abstracts that were not peer-reviewed and whose findings were inconsistent with those of GSK.
In the first abstract, a U.S. case-control study showed an increased risk of major malformations in 5,357 infants of women who took SSRIs in the first trimester, compared with 3,366 normal controls.14 Specific birth defects included:
- 161 infants with omphalocele (bowel protrusion through an abdominal wall defect) (OR, 3.0; 95% CI, 1.4-6.1)
- 372 infants with craniosynostosis (deformities caused by premature closure of skull sutures) (OR, 1.8; 95% CI, 1.0-3.2).
In the second abstract, Wogelius et al15 examined a Danish prescription database and found a slightly increased risk for congenital malformation (OR, 1.4; 95% CI, 1.1-1.9) and specifically cardiac malformation (OR, 1.6; 95% CI, 1.0-2.6) with SSRIs during pregnancy. This study included 1,054 women who filled SSRI prescriptions within a window of 30 days before conception to the end of the first trimester. The authors compared these malformation rates with those in 150,908 controls (women who did not fill an SSRI prescription in this period before and during pregnancy).
This cohort study did not report specific data about paroxetine, nor whether the women took the SSRIs they acquired.
Epidemiologic studies. The FDA also cited two unpublished epidemiologic studies.
The first analyzed a Swedish national registry database and found that infants whose mothers received paroxetine in early pregnancy had a 2% risk of cardiac defect, compared with a 1% risk among all registry infants. Although the FDA advisory does not cite a reference, the data may be from the same registry in which previous studies have shown no significant difference in malformations when comparing paroxetine-exposed infants with controls.7,8
The second epidemiologic study used a U.S. insurance claims database and found that infants of women who received paroxetine in early pregnancy had a 1.5% risk of cardiac defect, compared with 1% among infants whose mothers took other antidepressants.
Unfortunately, FDA has refused to release information about these studies beyond what it gave in the advisory, stating simply that the studies are unpublished. Evidence-based medicine can be difficult to practice if clinicians can’t access the evidence to assess its quality.
In clinical practice
Evidence from five peer-reviewed, published studies contradict the FDA advisory on increased risk for congenital cardiac malformations with paroxetine use during pregnancy. So, when prescribing antidepressants for depressed pregnant women, do we rely on the five negative studies or practice defensive medicine and choose SSRIs other than paroxetine?
We have good reasons to question the FDA advisory’s scientific validity, but our patients—and our lawyers—will be more comfortable if we avoid paroxetine in women of child-bearing potential for now. Excluding paroxetine and relying on other SSRIs when necessary to treat major depression during pregnancy is hardly evidence-based medicine, but it’s a legitimate practice of legally-defensive medicine.
This answers our question about how to respond to Mrs. J’s concerns:
- First, she and I would decide if she needs an antidepressant during pregnancy.
- Then, after reviewing with her the FDA warning on paroxetine and discussing its questionable scientific validity, I would recommend that she switch to another SSRI.
- If she chooses to continue paroxetine, I would ask her to sign the note that documents our discussion of the pros and cons of choosing paroxetine instead of alternatives.
- GlaxoSmithKline letter to clinicians about paroxetine during pregnancy, September 2005. www.fda.gov/medwatch/safety/2005/Paxil_dearhcp_letter.pdf
- Food and Drug Administration advisory on paroxetine during pregnancy, December 2005 (full text). www.fda.gov/cder/drug/advisory/paroxetine200512.htm.
- Citalopram • Celexa
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Paroxetine • Paxil, Paxil CR, Pexeva
- Sertraline • Zoloft
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Fleming N. Depression drugs ‘can raise birth defect risk.’ News.telegraph. Available at: www.telegraph.co.uk/news/main.jhtml?xml=/news/2005/09/01/wdep01.xml&sSheet=/news/2005/09/01/ixworld.html. Accessed February 22, 2006.
2. Lamberg L. Risks and benefits key to psychotropic drug use during pregnancy and postpartum period. JAMA 2005;294(13):1604-8.
3. Pastuszak A, Zuber C, et al. Pregnancy outcome following first trimester exposure to fluoxetine. JAMA 1993;269:2246-8.
4. Chambers CD, Johnson KA, Dick LM, et al. Birth outcomes in pregnant women taking fluoxetine. N Engl J Med 1996;335:1010-5.
5. Goldstein DJ, Corbin LA, Sundell KL. Effects of first-trimester fluoxetine exposure on the newborn. Obstet Gynecol 1997;89(5):713-8.
6. Kulin NA, Pastuszak A, Sage SR, et al. Pregnancy outcome following maternal use of the new selective serotonin reuptake inhibitors; a prospective controlled multicenter study. JAMA 1998;279:609-10.
7. Ericson A, Kallen B, Wiholm B. Delivery outcome after the use of antidepressants in early pregnancy. Eur J Clin Pharmacol 1999;55:503-8.
8. Hallberg P, Sjoblom V. The use of selective serotonin reuptake inhibitors during pregnancy and breast-feeding: a review and clinical aspects [review]. J Clin Psychopharmacol 2005;25:59-73.
9. Simon GE, Cunningham ML, Davis RL. Outcomes of prenatal antidepressant exposure. Am J Psychiatry 2002;159:2055-61.
10. Hendrick V, Smith LM, Suri R, et al. Birth outcomes after prenatal exposure to antidepressant medication. Am J Obstet Gynecol 2003;188:812-5.
11. Einarson TR, Einarson A. Newer antidepressants in pregnancy and rates of major malformations: a meta-analysis of prospective comparative studies. Pharmacoepidemiol Drug Saf 2005;14(12):823-7.
12. GlaxoSmithKline study EPIP083. GSK medicine: bupropion and paroxetine. Epidemiology study: preliminary report on bupropion in pregnancy and the occurrence of cardiovascular and major congenital malformation. Available at: http://ctr.gsk.co.uk/summary/paroxetine/epip083.pdf. Accessed February 22, 2006.
13. Honein MA, Paulozzi LJ, Cragan JD, Correa A. Evaluation of selected characteristics of pregnancy drug registries. Teratology 1999;60:356-64.
14. Alwan S, Reefhuis J, Rasmussen S, et al. Maternal use of selective serotonin re-uptake inhibitors and risk for birth defects. Birth Defects Research (Part A). Clin Molecular Teratology 2005;731:291-S143.
15. Wogelius P, Norgaard M, Muff Munk E, et al. Maternal use of selective serotonin reuptake inhibitors and risk of adverse pregnancy outcomes. Pharmacoepidemiol Drug Saf 2005;14:S143.-Available at: www.laegemiddelstyrelsen.dk/db/filarkiv/5611/Pharmacoepidemiology_2005.pdf. Accessed February 22, 2006.
Mrs. J, age 24, has a history of recurrent major depression, for which you have prescribed paroxetine. Newly pregnant, she brings you Internet articles with headlines such as “Depression drugs ‘can raise birth defect risks.’”SSRI use during pregnancy: Do antidepressants’ benefits outweigh the risks?).
FDA recommends avoiding paroxetine in women of child-bearing age. How does this advisory change the way we manage depression in pregnancy? How strong is the evidence supporting it?
SSRIs and birth defect risk
Eight prospective or case-control studies of SSRIs in >5,400 pregnant women have been published since 1993 (Table).3-10 Five included paroxetine.6-10 The studies ranged from small to large, and none showed a significant increase in major malformations with any SSRI. Even in a study of >2,500 women, no single malformation was overrepresented.8
In addition, a recent meta-analysis11 of 7 prospective comparative cohort studies involving 1,774 pregnant women showed no increased risk of major birth defects from exposure to any of the 8 antidepressants studied, including 4 SSRIs used during the first trimester. The review identified no specific malformation or cluster of malformations associated with first-trimester antidepressant use.
Table
8 published studies: No significant increase in birth defects with SSRIs
| Year/location | Authors | Study design | SSRI exposure (# of patients) | Risk of major malformation |
|---|---|---|---|---|
| 1993/USA, Canada | Pastuszak et al3 | Prospective cohort, controlled | Fluoxetine (98) | SSRI: 2% Control: 1.8% (ns) |
| 1996/USA | Chambers et al4 | Prospective cohort, controlled | Fluoxetine (174) | SSRI: 3.4% Control: 2.7% (ns) |
| 1997/worldwide | Goldstein et al5 | Clinical trial | Fluoxetine (28) | SSRI: 3.6% (ns) |
| 1998/USA, Canada, Brazil | Kulin et al6 | Prospective cohort, controlled | Paroxetine (97) | Total SSRI: 4.1% Control: 3.8% (ns) |
| Sertraline (147) | ||||
| Fluvoxamine (26) | ||||
| 1999/Sweden | Ericson et al7 | Case-control | Citalopram (364) | Citalopram: 3.9% Total risk of remaining SSRIs: 3.8% (ns) |
| Paroxetine (118) | ||||
| Sertraline (32) | ||||
| Fluoxetine (15) | ||||
| 2002/USA | Simon et al9 | Case-control | Fluoxetine (129) | Total SSRI: 6.5% Control: 4.9% (ns) |
| Sertraline (32) | ||||
| Paroxetine (28) | ||||
| 2003/USA | Hendrick et al10 | Prospective, uncontrolled | Fluoxetine (13) | Total SSRI: 1.4% (ns) |
| Paroxetine (19) | ||||
| Sertraline (36) | ||||
| 2005/Sweden | Hallberg et al8 | Case-control | Citalopram (1,696) | Citalopram: 3.1% |
| Paroxetine (708) | Paroxetine: 3.4% | |||
| Sertraline (1,067) | Sertraline: 2.0% | |||
| Fluoxetine (574) | Fluoxetine: 3.3% (ns) | |||
| ns: No statistically significant difference | ||||
Evidence cited by FDA
FDA’s advisory (Box) came 3 months after GSK notified health professionals that fetuses exposed to paroxetine during organogenesis may be at increased risk of developing malformations, particularly ventricular septal defect (see Related resources).
What FDA recommends to you and your patients
The FDA is awaiting the final results of the recent studies and accruing additional data related to the use of paroxetine in pregnancy in order to better characterize the risk for congenital malformations associated with paroxetine. In the interim, FDA recommends the following:
Physicians who are caring for women receiving paroxetine should alert them to the potential risk to the fetus if they plan to become pregnant or are currently in their first trimester of pregnancy. Discontinuing paroxetine therapy should be considered for these patients. In individual cases, the benefits of continuing paroxetine may outweigh the potential risk to the fetus. If the decision is made to discontinue paroxetine and switch to another antidepressant or cease antidepressant therapy, paroxetine discontinuation should be undertaken only as directed in the prescribing information. Paroxetine should generally not be initiated in women who are in their first trimester of pregnancy or in women who plan to become pregnant in the near future.
Women who are pregnant, or planning a pregnancy, and currently taking paroxetine should consult with their physician about whether to continue taking it. Women should not stop the drug without discussing the best way to do that with their physician.
Source: Verbatim from FDA advisory, December 2005.
- Major congenital defects occurred in 4% of 527 pregnancies during which women used paroxetine, (adjusted odds ratios [OR], 2.20; 95% CI, 1.34-3.63), compared with 3% prevalence in the general population.
- Cardiovascular malformations occurred at an adjusted rate of 2% (OR, 2.08; 95% CI, 1.03-4.23), compared with 1% in the general population.
- 10 of the 14 cardiovascular malformations were ventricular septal defects.
The GSK investigation was an unpublished retrospective study without peer review when FDA issued its advisory about paroxetine.
Two abstracts. The FDA alert also cited two abstracts that were not peer-reviewed and whose findings were inconsistent with those of GSK.
In the first abstract, a U.S. case-control study showed an increased risk of major malformations in 5,357 infants of women who took SSRIs in the first trimester, compared with 3,366 normal controls.14 Specific birth defects included:
- 161 infants with omphalocele (bowel protrusion through an abdominal wall defect) (OR, 3.0; 95% CI, 1.4-6.1)
- 372 infants with craniosynostosis (deformities caused by premature closure of skull sutures) (OR, 1.8; 95% CI, 1.0-3.2).
In the second abstract, Wogelius et al15 examined a Danish prescription database and found a slightly increased risk for congenital malformation (OR, 1.4; 95% CI, 1.1-1.9) and specifically cardiac malformation (OR, 1.6; 95% CI, 1.0-2.6) with SSRIs during pregnancy. This study included 1,054 women who filled SSRI prescriptions within a window of 30 days before conception to the end of the first trimester. The authors compared these malformation rates with those in 150,908 controls (women who did not fill an SSRI prescription in this period before and during pregnancy).
This cohort study did not report specific data about paroxetine, nor whether the women took the SSRIs they acquired.
Epidemiologic studies. The FDA also cited two unpublished epidemiologic studies.
The first analyzed a Swedish national registry database and found that infants whose mothers received paroxetine in early pregnancy had a 2% risk of cardiac defect, compared with a 1% risk among all registry infants. Although the FDA advisory does not cite a reference, the data may be from the same registry in which previous studies have shown no significant difference in malformations when comparing paroxetine-exposed infants with controls.7,8
The second epidemiologic study used a U.S. insurance claims database and found that infants of women who received paroxetine in early pregnancy had a 1.5% risk of cardiac defect, compared with 1% among infants whose mothers took other antidepressants.
Unfortunately, FDA has refused to release information about these studies beyond what it gave in the advisory, stating simply that the studies are unpublished. Evidence-based medicine can be difficult to practice if clinicians can’t access the evidence to assess its quality.
In clinical practice
Evidence from five peer-reviewed, published studies contradict the FDA advisory on increased risk for congenital cardiac malformations with paroxetine use during pregnancy. So, when prescribing antidepressants for depressed pregnant women, do we rely on the five negative studies or practice defensive medicine and choose SSRIs other than paroxetine?
We have good reasons to question the FDA advisory’s scientific validity, but our patients—and our lawyers—will be more comfortable if we avoid paroxetine in women of child-bearing potential for now. Excluding paroxetine and relying on other SSRIs when necessary to treat major depression during pregnancy is hardly evidence-based medicine, but it’s a legitimate practice of legally-defensive medicine.
This answers our question about how to respond to Mrs. J’s concerns:
- First, she and I would decide if she needs an antidepressant during pregnancy.
- Then, after reviewing with her the FDA warning on paroxetine and discussing its questionable scientific validity, I would recommend that she switch to another SSRI.
- If she chooses to continue paroxetine, I would ask her to sign the note that documents our discussion of the pros and cons of choosing paroxetine instead of alternatives.
- GlaxoSmithKline letter to clinicians about paroxetine during pregnancy, September 2005. www.fda.gov/medwatch/safety/2005/Paxil_dearhcp_letter.pdf
- Food and Drug Administration advisory on paroxetine during pregnancy, December 2005 (full text). www.fda.gov/cder/drug/advisory/paroxetine200512.htm.
- Citalopram • Celexa
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Paroxetine • Paxil, Paxil CR, Pexeva
- Sertraline • Zoloft
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Mrs. J, age 24, has a history of recurrent major depression, for which you have prescribed paroxetine. Newly pregnant, she brings you Internet articles with headlines such as “Depression drugs ‘can raise birth defect risks.’”SSRI use during pregnancy: Do antidepressants’ benefits outweigh the risks?).
FDA recommends avoiding paroxetine in women of child-bearing age. How does this advisory change the way we manage depression in pregnancy? How strong is the evidence supporting it?
SSRIs and birth defect risk
Eight prospective or case-control studies of SSRIs in >5,400 pregnant women have been published since 1993 (Table).3-10 Five included paroxetine.6-10 The studies ranged from small to large, and none showed a significant increase in major malformations with any SSRI. Even in a study of >2,500 women, no single malformation was overrepresented.8
In addition, a recent meta-analysis11 of 7 prospective comparative cohort studies involving 1,774 pregnant women showed no increased risk of major birth defects from exposure to any of the 8 antidepressants studied, including 4 SSRIs used during the first trimester. The review identified no specific malformation or cluster of malformations associated with first-trimester antidepressant use.
Table
8 published studies: No significant increase in birth defects with SSRIs
| Year/location | Authors | Study design | SSRI exposure (# of patients) | Risk of major malformation |
|---|---|---|---|---|
| 1993/USA, Canada | Pastuszak et al3 | Prospective cohort, controlled | Fluoxetine (98) | SSRI: 2% Control: 1.8% (ns) |
| 1996/USA | Chambers et al4 | Prospective cohort, controlled | Fluoxetine (174) | SSRI: 3.4% Control: 2.7% (ns) |
| 1997/worldwide | Goldstein et al5 | Clinical trial | Fluoxetine (28) | SSRI: 3.6% (ns) |
| 1998/USA, Canada, Brazil | Kulin et al6 | Prospective cohort, controlled | Paroxetine (97) | Total SSRI: 4.1% Control: 3.8% (ns) |
| Sertraline (147) | ||||
| Fluvoxamine (26) | ||||
| 1999/Sweden | Ericson et al7 | Case-control | Citalopram (364) | Citalopram: 3.9% Total risk of remaining SSRIs: 3.8% (ns) |
| Paroxetine (118) | ||||
| Sertraline (32) | ||||
| Fluoxetine (15) | ||||
| 2002/USA | Simon et al9 | Case-control | Fluoxetine (129) | Total SSRI: 6.5% Control: 4.9% (ns) |
| Sertraline (32) | ||||
| Paroxetine (28) | ||||
| 2003/USA | Hendrick et al10 | Prospective, uncontrolled | Fluoxetine (13) | Total SSRI: 1.4% (ns) |
| Paroxetine (19) | ||||
| Sertraline (36) | ||||
| 2005/Sweden | Hallberg et al8 | Case-control | Citalopram (1,696) | Citalopram: 3.1% |
| Paroxetine (708) | Paroxetine: 3.4% | |||
| Sertraline (1,067) | Sertraline: 2.0% | |||
| Fluoxetine (574) | Fluoxetine: 3.3% (ns) | |||
| ns: No statistically significant difference | ||||
Evidence cited by FDA
FDA’s advisory (Box) came 3 months after GSK notified health professionals that fetuses exposed to paroxetine during organogenesis may be at increased risk of developing malformations, particularly ventricular septal defect (see Related resources).
What FDA recommends to you and your patients
The FDA is awaiting the final results of the recent studies and accruing additional data related to the use of paroxetine in pregnancy in order to better characterize the risk for congenital malformations associated with paroxetine. In the interim, FDA recommends the following:
Physicians who are caring for women receiving paroxetine should alert them to the potential risk to the fetus if they plan to become pregnant or are currently in their first trimester of pregnancy. Discontinuing paroxetine therapy should be considered for these patients. In individual cases, the benefits of continuing paroxetine may outweigh the potential risk to the fetus. If the decision is made to discontinue paroxetine and switch to another antidepressant or cease antidepressant therapy, paroxetine discontinuation should be undertaken only as directed in the prescribing information. Paroxetine should generally not be initiated in women who are in their first trimester of pregnancy or in women who plan to become pregnant in the near future.
Women who are pregnant, or planning a pregnancy, and currently taking paroxetine should consult with their physician about whether to continue taking it. Women should not stop the drug without discussing the best way to do that with their physician.
Source: Verbatim from FDA advisory, December 2005.
- Major congenital defects occurred in 4% of 527 pregnancies during which women used paroxetine, (adjusted odds ratios [OR], 2.20; 95% CI, 1.34-3.63), compared with 3% prevalence in the general population.
- Cardiovascular malformations occurred at an adjusted rate of 2% (OR, 2.08; 95% CI, 1.03-4.23), compared with 1% in the general population.
- 10 of the 14 cardiovascular malformations were ventricular septal defects.
The GSK investigation was an unpublished retrospective study without peer review when FDA issued its advisory about paroxetine.
Two abstracts. The FDA alert also cited two abstracts that were not peer-reviewed and whose findings were inconsistent with those of GSK.
In the first abstract, a U.S. case-control study showed an increased risk of major malformations in 5,357 infants of women who took SSRIs in the first trimester, compared with 3,366 normal controls.14 Specific birth defects included:
- 161 infants with omphalocele (bowel protrusion through an abdominal wall defect) (OR, 3.0; 95% CI, 1.4-6.1)
- 372 infants with craniosynostosis (deformities caused by premature closure of skull sutures) (OR, 1.8; 95% CI, 1.0-3.2).
In the second abstract, Wogelius et al15 examined a Danish prescription database and found a slightly increased risk for congenital malformation (OR, 1.4; 95% CI, 1.1-1.9) and specifically cardiac malformation (OR, 1.6; 95% CI, 1.0-2.6) with SSRIs during pregnancy. This study included 1,054 women who filled SSRI prescriptions within a window of 30 days before conception to the end of the first trimester. The authors compared these malformation rates with those in 150,908 controls (women who did not fill an SSRI prescription in this period before and during pregnancy).
This cohort study did not report specific data about paroxetine, nor whether the women took the SSRIs they acquired.
Epidemiologic studies. The FDA also cited two unpublished epidemiologic studies.
The first analyzed a Swedish national registry database and found that infants whose mothers received paroxetine in early pregnancy had a 2% risk of cardiac defect, compared with a 1% risk among all registry infants. Although the FDA advisory does not cite a reference, the data may be from the same registry in which previous studies have shown no significant difference in malformations when comparing paroxetine-exposed infants with controls.7,8
The second epidemiologic study used a U.S. insurance claims database and found that infants of women who received paroxetine in early pregnancy had a 1.5% risk of cardiac defect, compared with 1% among infants whose mothers took other antidepressants.
Unfortunately, FDA has refused to release information about these studies beyond what it gave in the advisory, stating simply that the studies are unpublished. Evidence-based medicine can be difficult to practice if clinicians can’t access the evidence to assess its quality.
In clinical practice
Evidence from five peer-reviewed, published studies contradict the FDA advisory on increased risk for congenital cardiac malformations with paroxetine use during pregnancy. So, when prescribing antidepressants for depressed pregnant women, do we rely on the five negative studies or practice defensive medicine and choose SSRIs other than paroxetine?
We have good reasons to question the FDA advisory’s scientific validity, but our patients—and our lawyers—will be more comfortable if we avoid paroxetine in women of child-bearing potential for now. Excluding paroxetine and relying on other SSRIs when necessary to treat major depression during pregnancy is hardly evidence-based medicine, but it’s a legitimate practice of legally-defensive medicine.
This answers our question about how to respond to Mrs. J’s concerns:
- First, she and I would decide if she needs an antidepressant during pregnancy.
- Then, after reviewing with her the FDA warning on paroxetine and discussing its questionable scientific validity, I would recommend that she switch to another SSRI.
- If she chooses to continue paroxetine, I would ask her to sign the note that documents our discussion of the pros and cons of choosing paroxetine instead of alternatives.
- GlaxoSmithKline letter to clinicians about paroxetine during pregnancy, September 2005. www.fda.gov/medwatch/safety/2005/Paxil_dearhcp_letter.pdf
- Food and Drug Administration advisory on paroxetine during pregnancy, December 2005 (full text). www.fda.gov/cder/drug/advisory/paroxetine200512.htm.
- Citalopram • Celexa
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Paroxetine • Paxil, Paxil CR, Pexeva
- Sertraline • Zoloft
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Fleming N. Depression drugs ‘can raise birth defect risk.’ News.telegraph. Available at: www.telegraph.co.uk/news/main.jhtml?xml=/news/2005/09/01/wdep01.xml&sSheet=/news/2005/09/01/ixworld.html. Accessed February 22, 2006.
2. Lamberg L. Risks and benefits key to psychotropic drug use during pregnancy and postpartum period. JAMA 2005;294(13):1604-8.
3. Pastuszak A, Zuber C, et al. Pregnancy outcome following first trimester exposure to fluoxetine. JAMA 1993;269:2246-8.
4. Chambers CD, Johnson KA, Dick LM, et al. Birth outcomes in pregnant women taking fluoxetine. N Engl J Med 1996;335:1010-5.
5. Goldstein DJ, Corbin LA, Sundell KL. Effects of first-trimester fluoxetine exposure on the newborn. Obstet Gynecol 1997;89(5):713-8.
6. Kulin NA, Pastuszak A, Sage SR, et al. Pregnancy outcome following maternal use of the new selective serotonin reuptake inhibitors; a prospective controlled multicenter study. JAMA 1998;279:609-10.
7. Ericson A, Kallen B, Wiholm B. Delivery outcome after the use of antidepressants in early pregnancy. Eur J Clin Pharmacol 1999;55:503-8.
8. Hallberg P, Sjoblom V. The use of selective serotonin reuptake inhibitors during pregnancy and breast-feeding: a review and clinical aspects [review]. J Clin Psychopharmacol 2005;25:59-73.
9. Simon GE, Cunningham ML, Davis RL. Outcomes of prenatal antidepressant exposure. Am J Psychiatry 2002;159:2055-61.
10. Hendrick V, Smith LM, Suri R, et al. Birth outcomes after prenatal exposure to antidepressant medication. Am J Obstet Gynecol 2003;188:812-5.
11. Einarson TR, Einarson A. Newer antidepressants in pregnancy and rates of major malformations: a meta-analysis of prospective comparative studies. Pharmacoepidemiol Drug Saf 2005;14(12):823-7.
12. GlaxoSmithKline study EPIP083. GSK medicine: bupropion and paroxetine. Epidemiology study: preliminary report on bupropion in pregnancy and the occurrence of cardiovascular and major congenital malformation. Available at: http://ctr.gsk.co.uk/summary/paroxetine/epip083.pdf. Accessed February 22, 2006.
13. Honein MA, Paulozzi LJ, Cragan JD, Correa A. Evaluation of selected characteristics of pregnancy drug registries. Teratology 1999;60:356-64.
14. Alwan S, Reefhuis J, Rasmussen S, et al. Maternal use of selective serotonin re-uptake inhibitors and risk for birth defects. Birth Defects Research (Part A). Clin Molecular Teratology 2005;731:291-S143.
15. Wogelius P, Norgaard M, Muff Munk E, et al. Maternal use of selective serotonin reuptake inhibitors and risk of adverse pregnancy outcomes. Pharmacoepidemiol Drug Saf 2005;14:S143.-Available at: www.laegemiddelstyrelsen.dk/db/filarkiv/5611/Pharmacoepidemiology_2005.pdf. Accessed February 22, 2006.
1. Fleming N. Depression drugs ‘can raise birth defect risk.’ News.telegraph. Available at: www.telegraph.co.uk/news/main.jhtml?xml=/news/2005/09/01/wdep01.xml&sSheet=/news/2005/09/01/ixworld.html. Accessed February 22, 2006.
2. Lamberg L. Risks and benefits key to psychotropic drug use during pregnancy and postpartum period. JAMA 2005;294(13):1604-8.
3. Pastuszak A, Zuber C, et al. Pregnancy outcome following first trimester exposure to fluoxetine. JAMA 1993;269:2246-8.
4. Chambers CD, Johnson KA, Dick LM, et al. Birth outcomes in pregnant women taking fluoxetine. N Engl J Med 1996;335:1010-5.
5. Goldstein DJ, Corbin LA, Sundell KL. Effects of first-trimester fluoxetine exposure on the newborn. Obstet Gynecol 1997;89(5):713-8.
6. Kulin NA, Pastuszak A, Sage SR, et al. Pregnancy outcome following maternal use of the new selective serotonin reuptake inhibitors; a prospective controlled multicenter study. JAMA 1998;279:609-10.
7. Ericson A, Kallen B, Wiholm B. Delivery outcome after the use of antidepressants in early pregnancy. Eur J Clin Pharmacol 1999;55:503-8.
8. Hallberg P, Sjoblom V. The use of selective serotonin reuptake inhibitors during pregnancy and breast-feeding: a review and clinical aspects [review]. J Clin Psychopharmacol 2005;25:59-73.
9. Simon GE, Cunningham ML, Davis RL. Outcomes of prenatal antidepressant exposure. Am J Psychiatry 2002;159:2055-61.
10. Hendrick V, Smith LM, Suri R, et al. Birth outcomes after prenatal exposure to antidepressant medication. Am J Obstet Gynecol 2003;188:812-5.
11. Einarson TR, Einarson A. Newer antidepressants in pregnancy and rates of major malformations: a meta-analysis of prospective comparative studies. Pharmacoepidemiol Drug Saf 2005;14(12):823-7.
12. GlaxoSmithKline study EPIP083. GSK medicine: bupropion and paroxetine. Epidemiology study: preliminary report on bupropion in pregnancy and the occurrence of cardiovascular and major congenital malformation. Available at: http://ctr.gsk.co.uk/summary/paroxetine/epip083.pdf. Accessed February 22, 2006.
13. Honein MA, Paulozzi LJ, Cragan JD, Correa A. Evaluation of selected characteristics of pregnancy drug registries. Teratology 1999;60:356-64.
14. Alwan S, Reefhuis J, Rasmussen S, et al. Maternal use of selective serotonin re-uptake inhibitors and risk for birth defects. Birth Defects Research (Part A). Clin Molecular Teratology 2005;731:291-S143.
15. Wogelius P, Norgaard M, Muff Munk E, et al. Maternal use of selective serotonin reuptake inhibitors and risk of adverse pregnancy outcomes. Pharmacoepidemiol Drug Saf 2005;14:S143.-Available at: www.laegemiddelstyrelsen.dk/db/filarkiv/5611/Pharmacoepidemiology_2005.pdf. Accessed February 22, 2006.
The boy who longed for a ‘dry spell’
History: ‘I can’t face myself’
Jimmy, age 12, is referred to us by his pediatrician, who is concerned about his “frequent nighttime accidents.” His parents report that he wets his bed 5 to 6 times weekly and has never stayed consistently dry for more than a few days.
The accidents occur only at night, his parents say. Numerous interventions have failed, including restricting fluids after dinner and awakening the boy overnight to make him go to the bathroom.
Jimmy, a sixth-grader, wonders if he will ever stop wetting his bed. He refuses to go to summer camp or stay overnight at a friend’s house, fearful that other kids will make fun of him after an accident. Asked how “wet nights” are affecting his life, he says, “I can’t face myself in the mirror.”
The authors’ observations
Primary nocturnal enuresis is diagnosed in children age ≥5 who have never gone 6 consecutive months without an overnight accident. Pediatricians generally discover enuresis incidentally during regular checkups and refer to a psychiatrist only if the child has an emotional problem secondary to enuresis or a comorbid psychiatric disorder.
Once identified, enuresis requires a thorough assessment—including its emotional consequences, which for Jimmy are significant. In its practice parameter for treating enuresis, the American Academy of Child and Adolescent Psychiatry (AACAP)1 suggests that you:
Take an extensive developmental and family history. Find out if the child was toilet trained and started walking, talking, or running at an appropriate age. Delays in reaching developmental milestones can predict enuresis.1
Also find out if either parent had enuresis during childhood. Enuresis is heritable,2 and children often outgrow the problem at the same age as did the parent(s).
Focus on the bedwetting and the child’s reaction to it. Treat enuresis aggressively if it is hurting the child’s performance at school, social or emotional development, or self-esteem, or if the youth appears emotionally withdrawn or distressed.
Interview the child and parents separately, as each often reacts differently to the problem. In some cases, for example, the child’s bedwetting upsets the parents but the child hardly seems to care. Also, children often feel more at ease talking to a doctor alone, and parents can vent frustration without upsetting their child.
While interviewing the child, listen for psychosocial stressors that can lead to enuresis, such as parents’ marital problems, problems at school, recent hospitalization, physical or sexual abuse, or the recent birth of a sibling.
We spend about one half-hour with the child and another half-hour with the parents to thoroughly gauge enuresis’ emotional impact. To engage the child and hold his attention during that half-hour, we offer toys or play a game.
Check for physical causes. According to the AACAP practice parameter for enuresis treatment, you should:
- assess nare patency and voice quality to rule out enlarged adenoids
- check the nasal pharynx for enlarged tonsils
- palpate the abdomen to check for bladder distention or fecal impaction
- examine genitalia for abnormalities
- view the back for a sacral dimple or other sign of a vertebral or spinal cord anomaly.
Perform a urinalysis and urine culture to rule out urinary tract infection (UTI).
Order urodynamic studies or renal ultrasound if enuresis persists after two unsuccessful treatment trials, the physical examination uncovers positive findings, or the child has had a UTI.
Psychotherapy has a limited role in treating primary enuresis unless you suspect a psychological cause.1 We offered Jimmy supportive counseling to help alleviate emotional problems caused by bedwetting. He and his parents declined but agreed to reconsider later.
Further history: Toilet trained At 2
Jimmy was toilet trained at age 2 and reached all other age-appropriate developmental milestones, his mother says. Results of urine culture, repeated urinalyses, and neurologic and physical examinations are normal. Neither Jimmy nor his family have a history of UTI, dysuria, urgency, or increased urination frequency.
When Jimmy was age 9, his pediatrician prescribed imipramine, 25 mg/d, to try to stop his bedwetting. He did not respond after 6 months, so his parents stopped giving the drug to him.
A few months later, Jimmy’s parents heard about a “bedwetting alarm” designed to condition children not to urinate while asleep, but the boy and his parents viewed this treatment as “humiliating” and refused to try it. They have not attempted another intervention for 2 years.
poll here
The authors’ observations
Having found no medical or psychological basis for Jimmy’s enuresis (Box), we pondered our next clinical move.
Genetics. In more than one-half of children with enuresis, one or both parents had the disorder during childhood.
Developmental delay. Delayed functional CNS maturation can decrease arousal. Enuresis is common in children with developmental disorders, including autism, Rett’s syndrome, or pervasive developmental disorder NOS.
Irregular sleep pattern associated with specific sleep disorders, such as narcolepsy and sleep apnea. Also, children with enuresis sleep more soundly than do youths without the disorder.
Psychological problem. Considered a reaction to primary enuresis rather than its cause.
Medical condition. Enlarged adenoids, tonsils, constipation with fecal impaction, vertebral and spinal cord anomaly, and diabetes mellitus may cause enuresis.
Source: Reference 1
Among behavioral treatments, only the bedwetting alarm has shown effectiveness in clinical trials,1,3 and it carries the lowest risk of post-treatment relapse.3 Urine moistens a sensor in the bed pad or inside cloth, triggering an alarm that awakens the child when wetting starts. The child gradually awakens earlier in an enuretic episode until the sensation of bladder fullness awakens him.
Many parents/guardians and their children—particularly older youths—consider alarm systems demeaning. We again suggested this treatment to Jimmy and his parents, but they refused.
Medication. Six months of low-dose imipramine, a tricyclic antidepressant often prescribed for enuresis, produced no response. We did not restart imipramine at a higher dosage because of its association with increased arrhythmia risk.
We instead considered desmopressin acetate, a synthetic analog of ADH vasopressin that regulates diurnal variation, which is usually abnormal in children with enuresis. Desmopressin, often used to treat clozapine-induced enuresis in adults, has been associated with successful outcomes in as many as 65% of children in clinical trials.1,4
Desmopressin, however, can reduce urine production. Water intoxication or hyponatremia is rare but can lead to seizures or coma, and the risk increases with the dosage. Obtain informed consent from the parents before starting this drug. Check electrolytes 2 or 3 days after the first dose, 1 month later, then again every 2 to 3 months. Discontinue at once if serum sodium decreases significantly from baseline or is
Treatment: Meaningless response
We start Jimmy on oral desmopressin, 0.2 mg at bedtime, after discussing its benefits and risks with his parents. We increase the dosage to 0.4 mg after 3 days and to 0.6 mg the following week, as the lower dosages have not worked. Serum electrolytes, gauged before starting desmopressin and again 2 weeks later, are normal. We see Jimmy every 2 weeks to check progress and monitor for side effects.
Soon after the second dosage increase, Jimmy’s accidents gradually decrease to 2 to 3 per week, but no improvement is seen after that.
Two months later, Jimmy is still avoiding sleepovers and has trouble making friends. His parents worry about his increasing frustration, hopelessness, and low self-esteem. We again offer supportive counseling, but the boy refuses.
poll here
The authors’ observations
We were running out of treatment options. Two medication trials failed, and the family still would not try a bedwetting alarm.
Urodynamic testing usually is not ordered unless the child has a history of urge incontinence or UTI. For some treatment-resistant patients, the test can reveal detruser muscle or bladder capacity deficits that might be causing enuresis.
Testing: Below the norm
We refer Jimmy to a urologist for a urodynamic test. Results showed mild detruser muscle instability and slightly low maximum bladder capacity compared with age-predicted norms.
The authors’ observations
Based on this finding, we considered oxybutynin, an anticholinergic agent that increases bladder control by relaxing the smooth muscles. Patients with detruser instability and inadequate bladder capacity have responded well to oxybutynin in clinical trials,5,6 and combination oxybutynin/desmopressin therapy has been shown effective in treatment-resistant patients.7-9
Oxybutynin and desmopressin complement each other; reduced urinary output and bladder filling associated with desmopressin can reduce uninhibited bladder contractions, thus enhancing oxybutynin’s action.
Treatment: Happy summer
We continue desmopressin, 0.6 mg nightly, and add extended-release oxybutynin, 2.5 mg/d. We increase oxybutynin to 5 mg/d after 3 days and to 10 mg/d the following week, as Jimmy reported no side effects from the lower dosages.
We see Jimmy 1 week after adding oxybutynin, then again 3 weeks later. He reports no wet nights after 1 month of combination therapy, then wets his bed once over the next 2 months. We continue to see him every 3 to 4 weeks and check his electrolytes every 2 to 3 months. He reports no side effects
Five months after starting combination therapy, Jimmy seems much more confident. He has gone 2 months without a bedwetting accident, and his face lights up while discussing the fun he had last week in summer camp. He remains free of side effects, and his parents are thrilled with his progress.
We see Jimmy three more times, once every 2 months. He is staying “dry” but says he wishes to stop his medication because he wants to control his bladder without it.
poll here
The authors’ observations
Medications and behavioral treatments can preserve the child’s self-esteem until he or she outgrows enuresis (Table).
No guidelines address drug regimen duration. Tapering Jimmy’s medications after 7 to 8 months seemed reasonable, but children with enuresis often relapse after stopping treatment. Researchers have recorded relapse rates as high as 60% after stopping imipramine and 80% after stopping desmopressin.1,4
Taper medications slowly to avoid withdrawal, immediate relapse, and anticholinergic effects. If the child relapses, restart medication at the previous therapeutic dosage(s), then start tapering after the child has been accident-free for 3 months.
Table
Medication strategies for treating enuresis
| Medication | Dosage | Risks |
|---|---|---|
| Desmopressin acetate (first-line) | Start with 0.2-mg tablet or 1 to 2 10-μg puffs of nasal spray (half in each nostril) in children age >6; increase to 0.6 mg/d or 4 puffs daily after 1 week if necessary Stop after approximately 6 months without an accident | High relapse rate Reduced urine production Water intoxication, hyponatremia are rare but can result in seizures, coma |
| Oxybutynin (second-line) | 2.5 to 5 mg tid (immediate-release) or 15 mg/d (extended-release) Start at 5 mg at bedtime for children age >5; increase to 15 mg/d after 1 to 2 weeks if needed Stop after approximately 6 months without an accident | High relapse rate Anticholinergic effects (dry mouth, facial flushing, drowsiness, decreased GI motility) Few efficacy studies done Mostly used with other medication |
| Desmopressin with oxybutynin or imipramine; medication plus alarm method (third-line) | Dosages of individual medications as listed | Limited data available Positive results seen in resistant cases, particularly in older children |
| Imipramine (last option) | 1 to 2.5 mg/kg/d Start with 25 mg/d at bedtime; if no response, increase in weekly 25-mg increments to 50 mg/d for children ages 7 to 12 or up to 75 mg/d for children age >12 Stop after approximately 6 months without an accident | High relapse rate after stopping medication Risk of arrhythmias (order ECG when starting medication, 1 month later, then every 6 months) Fatal in overdose (do not prescribe >75 mg/d in enuresis) Associated with suicidal behavior in youths (carries FDA “black box” warning) |
Follow-up: Still dry
After discussing the relapse risk with Jimmy’s parents, we withdraw both oxybutynin and desmopressin over 2 months, reducing each dosage 25% every 2 weeks. We see Jimmy every 4 to 6 weeks during the taper period, then for two bimonthly follow-up visits. He reports no adverse effects and has been accident-free for 8 months.
After consulting with his pediatrician and family, we refer Jimmy, now age 13, back to the pediatrician. We have not seen him for more than 1 year.
Related resources
- National Association For Continence. www.nafc.org.
- Mayo ME, Burns MW. Urodynamic studies in children who wet. Br J Urol 1990 65;641-5.
- Desmopressin • DDAVP
- Imipramine • Tofranil
- Oxybutynin • Ditropan
Dr. Williams is a speaker for Wyeth.
Dr. Singh reports no financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
1. Practice parameter for the assessment and treatment of children and adolescents with enuresis. J Am Acad Child Adolesc Psychiatry 2004;43:1540-50.
2. Bakwin H. The genetics of enuresis. In: Kolvin I, MacKeith RC, Meadow SR (eds). Bladder control and enuresis. London: Heinemann Medical; 1973.
3. Jensen IN, Kristensen G. Alarm treatment: analysis of response and relapse. Scand J Urol Nephrol Suppl 1999;202:73-5.
4. Thompson S, Rey JM. Functional enuresis: is desmopressin the answer? J Am Acad Child Adolesc Psychiatry 1995;34:266-71.
5. Kosar A, Arikan N, Dincel C. Effectiveness of oxybutynin hydrochloride in the treatment of enuresis nocturna—a clinical and urodynamic study. Scand J Urol Nephrol 1999;33:115-8.
6. Persson-Junemann C, Seemann O, Kohrmann KU, et al. Comparison of urodynamic findings and response to oxybutynin in nocturnal enuresis. Eur Urol 1993;24:92-6.
7. Martin-Crespo R, Luque R. [In which patients is the oxybutynin-desmopressin combination therapy indicated?] Cir Pediatr 2003;16:181-5. Spanish.
8. Caione P, Arena F, Biraghi M, et al. Nocturnal enuresis and daytime wetting: a multicentric trial with oxybutynin and desmopressin. Eur Urol 1997;31:459-63.
9. Lee T, Suh HJ, Lee HJ, Lee JE. Comparison of effects of treatment of primary nocturnal enuresis with oxybutynin plus desmopressin, desmopressin alone or imipramine alone: a randomized controlled clinical trial. J Urol 2005;174:1084-7.
History: ‘I can’t face myself’
Jimmy, age 12, is referred to us by his pediatrician, who is concerned about his “frequent nighttime accidents.” His parents report that he wets his bed 5 to 6 times weekly and has never stayed consistently dry for more than a few days.
The accidents occur only at night, his parents say. Numerous interventions have failed, including restricting fluids after dinner and awakening the boy overnight to make him go to the bathroom.
Jimmy, a sixth-grader, wonders if he will ever stop wetting his bed. He refuses to go to summer camp or stay overnight at a friend’s house, fearful that other kids will make fun of him after an accident. Asked how “wet nights” are affecting his life, he says, “I can’t face myself in the mirror.”
The authors’ observations
Primary nocturnal enuresis is diagnosed in children age ≥5 who have never gone 6 consecutive months without an overnight accident. Pediatricians generally discover enuresis incidentally during regular checkups and refer to a psychiatrist only if the child has an emotional problem secondary to enuresis or a comorbid psychiatric disorder.
Once identified, enuresis requires a thorough assessment—including its emotional consequences, which for Jimmy are significant. In its practice parameter for treating enuresis, the American Academy of Child and Adolescent Psychiatry (AACAP)1 suggests that you:
Take an extensive developmental and family history. Find out if the child was toilet trained and started walking, talking, or running at an appropriate age. Delays in reaching developmental milestones can predict enuresis.1
Also find out if either parent had enuresis during childhood. Enuresis is heritable,2 and children often outgrow the problem at the same age as did the parent(s).
Focus on the bedwetting and the child’s reaction to it. Treat enuresis aggressively if it is hurting the child’s performance at school, social or emotional development, or self-esteem, or if the youth appears emotionally withdrawn or distressed.
Interview the child and parents separately, as each often reacts differently to the problem. In some cases, for example, the child’s bedwetting upsets the parents but the child hardly seems to care. Also, children often feel more at ease talking to a doctor alone, and parents can vent frustration without upsetting their child.
While interviewing the child, listen for psychosocial stressors that can lead to enuresis, such as parents’ marital problems, problems at school, recent hospitalization, physical or sexual abuse, or the recent birth of a sibling.
We spend about one half-hour with the child and another half-hour with the parents to thoroughly gauge enuresis’ emotional impact. To engage the child and hold his attention during that half-hour, we offer toys or play a game.
Check for physical causes. According to the AACAP practice parameter for enuresis treatment, you should:
- assess nare patency and voice quality to rule out enlarged adenoids
- check the nasal pharynx for enlarged tonsils
- palpate the abdomen to check for bladder distention or fecal impaction
- examine genitalia for abnormalities
- view the back for a sacral dimple or other sign of a vertebral or spinal cord anomaly.
Perform a urinalysis and urine culture to rule out urinary tract infection (UTI).
Order urodynamic studies or renal ultrasound if enuresis persists after two unsuccessful treatment trials, the physical examination uncovers positive findings, or the child has had a UTI.
Psychotherapy has a limited role in treating primary enuresis unless you suspect a psychological cause.1 We offered Jimmy supportive counseling to help alleviate emotional problems caused by bedwetting. He and his parents declined but agreed to reconsider later.
Further history: Toilet trained At 2
Jimmy was toilet trained at age 2 and reached all other age-appropriate developmental milestones, his mother says. Results of urine culture, repeated urinalyses, and neurologic and physical examinations are normal. Neither Jimmy nor his family have a history of UTI, dysuria, urgency, or increased urination frequency.
When Jimmy was age 9, his pediatrician prescribed imipramine, 25 mg/d, to try to stop his bedwetting. He did not respond after 6 months, so his parents stopped giving the drug to him.
A few months later, Jimmy’s parents heard about a “bedwetting alarm” designed to condition children not to urinate while asleep, but the boy and his parents viewed this treatment as “humiliating” and refused to try it. They have not attempted another intervention for 2 years.
poll here
The authors’ observations
Having found no medical or psychological basis for Jimmy’s enuresis (Box), we pondered our next clinical move.
Genetics. In more than one-half of children with enuresis, one or both parents had the disorder during childhood.
Developmental delay. Delayed functional CNS maturation can decrease arousal. Enuresis is common in children with developmental disorders, including autism, Rett’s syndrome, or pervasive developmental disorder NOS.
Irregular sleep pattern associated with specific sleep disorders, such as narcolepsy and sleep apnea. Also, children with enuresis sleep more soundly than do youths without the disorder.
Psychological problem. Considered a reaction to primary enuresis rather than its cause.
Medical condition. Enlarged adenoids, tonsils, constipation with fecal impaction, vertebral and spinal cord anomaly, and diabetes mellitus may cause enuresis.
Source: Reference 1
Among behavioral treatments, only the bedwetting alarm has shown effectiveness in clinical trials,1,3 and it carries the lowest risk of post-treatment relapse.3 Urine moistens a sensor in the bed pad or inside cloth, triggering an alarm that awakens the child when wetting starts. The child gradually awakens earlier in an enuretic episode until the sensation of bladder fullness awakens him.
Many parents/guardians and their children—particularly older youths—consider alarm systems demeaning. We again suggested this treatment to Jimmy and his parents, but they refused.
Medication. Six months of low-dose imipramine, a tricyclic antidepressant often prescribed for enuresis, produced no response. We did not restart imipramine at a higher dosage because of its association with increased arrhythmia risk.
We instead considered desmopressin acetate, a synthetic analog of ADH vasopressin that regulates diurnal variation, which is usually abnormal in children with enuresis. Desmopressin, often used to treat clozapine-induced enuresis in adults, has been associated with successful outcomes in as many as 65% of children in clinical trials.1,4
Desmopressin, however, can reduce urine production. Water intoxication or hyponatremia is rare but can lead to seizures or coma, and the risk increases with the dosage. Obtain informed consent from the parents before starting this drug. Check electrolytes 2 or 3 days after the first dose, 1 month later, then again every 2 to 3 months. Discontinue at once if serum sodium decreases significantly from baseline or is
Treatment: Meaningless response
We start Jimmy on oral desmopressin, 0.2 mg at bedtime, after discussing its benefits and risks with his parents. We increase the dosage to 0.4 mg after 3 days and to 0.6 mg the following week, as the lower dosages have not worked. Serum electrolytes, gauged before starting desmopressin and again 2 weeks later, are normal. We see Jimmy every 2 weeks to check progress and monitor for side effects.
Soon after the second dosage increase, Jimmy’s accidents gradually decrease to 2 to 3 per week, but no improvement is seen after that.
Two months later, Jimmy is still avoiding sleepovers and has trouble making friends. His parents worry about his increasing frustration, hopelessness, and low self-esteem. We again offer supportive counseling, but the boy refuses.
poll here
The authors’ observations
We were running out of treatment options. Two medication trials failed, and the family still would not try a bedwetting alarm.
Urodynamic testing usually is not ordered unless the child has a history of urge incontinence or UTI. For some treatment-resistant patients, the test can reveal detruser muscle or bladder capacity deficits that might be causing enuresis.
Testing: Below the norm
We refer Jimmy to a urologist for a urodynamic test. Results showed mild detruser muscle instability and slightly low maximum bladder capacity compared with age-predicted norms.
The authors’ observations
Based on this finding, we considered oxybutynin, an anticholinergic agent that increases bladder control by relaxing the smooth muscles. Patients with detruser instability and inadequate bladder capacity have responded well to oxybutynin in clinical trials,5,6 and combination oxybutynin/desmopressin therapy has been shown effective in treatment-resistant patients.7-9
Oxybutynin and desmopressin complement each other; reduced urinary output and bladder filling associated with desmopressin can reduce uninhibited bladder contractions, thus enhancing oxybutynin’s action.
Treatment: Happy summer
We continue desmopressin, 0.6 mg nightly, and add extended-release oxybutynin, 2.5 mg/d. We increase oxybutynin to 5 mg/d after 3 days and to 10 mg/d the following week, as Jimmy reported no side effects from the lower dosages.
We see Jimmy 1 week after adding oxybutynin, then again 3 weeks later. He reports no wet nights after 1 month of combination therapy, then wets his bed once over the next 2 months. We continue to see him every 3 to 4 weeks and check his electrolytes every 2 to 3 months. He reports no side effects
Five months after starting combination therapy, Jimmy seems much more confident. He has gone 2 months without a bedwetting accident, and his face lights up while discussing the fun he had last week in summer camp. He remains free of side effects, and his parents are thrilled with his progress.
We see Jimmy three more times, once every 2 months. He is staying “dry” but says he wishes to stop his medication because he wants to control his bladder without it.
poll here
The authors’ observations
Medications and behavioral treatments can preserve the child’s self-esteem until he or she outgrows enuresis (Table).
No guidelines address drug regimen duration. Tapering Jimmy’s medications after 7 to 8 months seemed reasonable, but children with enuresis often relapse after stopping treatment. Researchers have recorded relapse rates as high as 60% after stopping imipramine and 80% after stopping desmopressin.1,4
Taper medications slowly to avoid withdrawal, immediate relapse, and anticholinergic effects. If the child relapses, restart medication at the previous therapeutic dosage(s), then start tapering after the child has been accident-free for 3 months.
Table
Medication strategies for treating enuresis
| Medication | Dosage | Risks |
|---|---|---|
| Desmopressin acetate (first-line) | Start with 0.2-mg tablet or 1 to 2 10-μg puffs of nasal spray (half in each nostril) in children age >6; increase to 0.6 mg/d or 4 puffs daily after 1 week if necessary Stop after approximately 6 months without an accident | High relapse rate Reduced urine production Water intoxication, hyponatremia are rare but can result in seizures, coma |
| Oxybutynin (second-line) | 2.5 to 5 mg tid (immediate-release) or 15 mg/d (extended-release) Start at 5 mg at bedtime for children age >5; increase to 15 mg/d after 1 to 2 weeks if needed Stop after approximately 6 months without an accident | High relapse rate Anticholinergic effects (dry mouth, facial flushing, drowsiness, decreased GI motility) Few efficacy studies done Mostly used with other medication |
| Desmopressin with oxybutynin or imipramine; medication plus alarm method (third-line) | Dosages of individual medications as listed | Limited data available Positive results seen in resistant cases, particularly in older children |
| Imipramine (last option) | 1 to 2.5 mg/kg/d Start with 25 mg/d at bedtime; if no response, increase in weekly 25-mg increments to 50 mg/d for children ages 7 to 12 or up to 75 mg/d for children age >12 Stop after approximately 6 months without an accident | High relapse rate after stopping medication Risk of arrhythmias (order ECG when starting medication, 1 month later, then every 6 months) Fatal in overdose (do not prescribe >75 mg/d in enuresis) Associated with suicidal behavior in youths (carries FDA “black box” warning) |
Follow-up: Still dry
After discussing the relapse risk with Jimmy’s parents, we withdraw both oxybutynin and desmopressin over 2 months, reducing each dosage 25% every 2 weeks. We see Jimmy every 4 to 6 weeks during the taper period, then for two bimonthly follow-up visits. He reports no adverse effects and has been accident-free for 8 months.
After consulting with his pediatrician and family, we refer Jimmy, now age 13, back to the pediatrician. We have not seen him for more than 1 year.
Related resources
- National Association For Continence. www.nafc.org.
- Mayo ME, Burns MW. Urodynamic studies in children who wet. Br J Urol 1990 65;641-5.
- Desmopressin • DDAVP
- Imipramine • Tofranil
- Oxybutynin • Ditropan
Dr. Williams is a speaker for Wyeth.
Dr. Singh reports no financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
History: ‘I can’t face myself’
Jimmy, age 12, is referred to us by his pediatrician, who is concerned about his “frequent nighttime accidents.” His parents report that he wets his bed 5 to 6 times weekly and has never stayed consistently dry for more than a few days.
The accidents occur only at night, his parents say. Numerous interventions have failed, including restricting fluids after dinner and awakening the boy overnight to make him go to the bathroom.
Jimmy, a sixth-grader, wonders if he will ever stop wetting his bed. He refuses to go to summer camp or stay overnight at a friend’s house, fearful that other kids will make fun of him after an accident. Asked how “wet nights” are affecting his life, he says, “I can’t face myself in the mirror.”
The authors’ observations
Primary nocturnal enuresis is diagnosed in children age ≥5 who have never gone 6 consecutive months without an overnight accident. Pediatricians generally discover enuresis incidentally during regular checkups and refer to a psychiatrist only if the child has an emotional problem secondary to enuresis or a comorbid psychiatric disorder.
Once identified, enuresis requires a thorough assessment—including its emotional consequences, which for Jimmy are significant. In its practice parameter for treating enuresis, the American Academy of Child and Adolescent Psychiatry (AACAP)1 suggests that you:
Take an extensive developmental and family history. Find out if the child was toilet trained and started walking, talking, or running at an appropriate age. Delays in reaching developmental milestones can predict enuresis.1
Also find out if either parent had enuresis during childhood. Enuresis is heritable,2 and children often outgrow the problem at the same age as did the parent(s).
Focus on the bedwetting and the child’s reaction to it. Treat enuresis aggressively if it is hurting the child’s performance at school, social or emotional development, or self-esteem, or if the youth appears emotionally withdrawn or distressed.
Interview the child and parents separately, as each often reacts differently to the problem. In some cases, for example, the child’s bedwetting upsets the parents but the child hardly seems to care. Also, children often feel more at ease talking to a doctor alone, and parents can vent frustration without upsetting their child.
While interviewing the child, listen for psychosocial stressors that can lead to enuresis, such as parents’ marital problems, problems at school, recent hospitalization, physical or sexual abuse, or the recent birth of a sibling.
We spend about one half-hour with the child and another half-hour with the parents to thoroughly gauge enuresis’ emotional impact. To engage the child and hold his attention during that half-hour, we offer toys or play a game.
Check for physical causes. According to the AACAP practice parameter for enuresis treatment, you should:
- assess nare patency and voice quality to rule out enlarged adenoids
- check the nasal pharynx for enlarged tonsils
- palpate the abdomen to check for bladder distention or fecal impaction
- examine genitalia for abnormalities
- view the back for a sacral dimple or other sign of a vertebral or spinal cord anomaly.
Perform a urinalysis and urine culture to rule out urinary tract infection (UTI).
Order urodynamic studies or renal ultrasound if enuresis persists after two unsuccessful treatment trials, the physical examination uncovers positive findings, or the child has had a UTI.
Psychotherapy has a limited role in treating primary enuresis unless you suspect a psychological cause.1 We offered Jimmy supportive counseling to help alleviate emotional problems caused by bedwetting. He and his parents declined but agreed to reconsider later.
Further history: Toilet trained At 2
Jimmy was toilet trained at age 2 and reached all other age-appropriate developmental milestones, his mother says. Results of urine culture, repeated urinalyses, and neurologic and physical examinations are normal. Neither Jimmy nor his family have a history of UTI, dysuria, urgency, or increased urination frequency.
When Jimmy was age 9, his pediatrician prescribed imipramine, 25 mg/d, to try to stop his bedwetting. He did not respond after 6 months, so his parents stopped giving the drug to him.
A few months later, Jimmy’s parents heard about a “bedwetting alarm” designed to condition children not to urinate while asleep, but the boy and his parents viewed this treatment as “humiliating” and refused to try it. They have not attempted another intervention for 2 years.
poll here
The authors’ observations
Having found no medical or psychological basis for Jimmy’s enuresis (Box), we pondered our next clinical move.
Genetics. In more than one-half of children with enuresis, one or both parents had the disorder during childhood.
Developmental delay. Delayed functional CNS maturation can decrease arousal. Enuresis is common in children with developmental disorders, including autism, Rett’s syndrome, or pervasive developmental disorder NOS.
Irregular sleep pattern associated with specific sleep disorders, such as narcolepsy and sleep apnea. Also, children with enuresis sleep more soundly than do youths without the disorder.
Psychological problem. Considered a reaction to primary enuresis rather than its cause.
Medical condition. Enlarged adenoids, tonsils, constipation with fecal impaction, vertebral and spinal cord anomaly, and diabetes mellitus may cause enuresis.
Source: Reference 1
Among behavioral treatments, only the bedwetting alarm has shown effectiveness in clinical trials,1,3 and it carries the lowest risk of post-treatment relapse.3 Urine moistens a sensor in the bed pad or inside cloth, triggering an alarm that awakens the child when wetting starts. The child gradually awakens earlier in an enuretic episode until the sensation of bladder fullness awakens him.
Many parents/guardians and their children—particularly older youths—consider alarm systems demeaning. We again suggested this treatment to Jimmy and his parents, but they refused.
Medication. Six months of low-dose imipramine, a tricyclic antidepressant often prescribed for enuresis, produced no response. We did not restart imipramine at a higher dosage because of its association with increased arrhythmia risk.
We instead considered desmopressin acetate, a synthetic analog of ADH vasopressin that regulates diurnal variation, which is usually abnormal in children with enuresis. Desmopressin, often used to treat clozapine-induced enuresis in adults, has been associated with successful outcomes in as many as 65% of children in clinical trials.1,4
Desmopressin, however, can reduce urine production. Water intoxication or hyponatremia is rare but can lead to seizures or coma, and the risk increases with the dosage. Obtain informed consent from the parents before starting this drug. Check electrolytes 2 or 3 days after the first dose, 1 month later, then again every 2 to 3 months. Discontinue at once if serum sodium decreases significantly from baseline or is
Treatment: Meaningless response
We start Jimmy on oral desmopressin, 0.2 mg at bedtime, after discussing its benefits and risks with his parents. We increase the dosage to 0.4 mg after 3 days and to 0.6 mg the following week, as the lower dosages have not worked. Serum electrolytes, gauged before starting desmopressin and again 2 weeks later, are normal. We see Jimmy every 2 weeks to check progress and monitor for side effects.
Soon after the second dosage increase, Jimmy’s accidents gradually decrease to 2 to 3 per week, but no improvement is seen after that.
Two months later, Jimmy is still avoiding sleepovers and has trouble making friends. His parents worry about his increasing frustration, hopelessness, and low self-esteem. We again offer supportive counseling, but the boy refuses.
poll here
The authors’ observations
We were running out of treatment options. Two medication trials failed, and the family still would not try a bedwetting alarm.
Urodynamic testing usually is not ordered unless the child has a history of urge incontinence or UTI. For some treatment-resistant patients, the test can reveal detruser muscle or bladder capacity deficits that might be causing enuresis.
Testing: Below the norm
We refer Jimmy to a urologist for a urodynamic test. Results showed mild detruser muscle instability and slightly low maximum bladder capacity compared with age-predicted norms.
The authors’ observations
Based on this finding, we considered oxybutynin, an anticholinergic agent that increases bladder control by relaxing the smooth muscles. Patients with detruser instability and inadequate bladder capacity have responded well to oxybutynin in clinical trials,5,6 and combination oxybutynin/desmopressin therapy has been shown effective in treatment-resistant patients.7-9
Oxybutynin and desmopressin complement each other; reduced urinary output and bladder filling associated with desmopressin can reduce uninhibited bladder contractions, thus enhancing oxybutynin’s action.
Treatment: Happy summer
We continue desmopressin, 0.6 mg nightly, and add extended-release oxybutynin, 2.5 mg/d. We increase oxybutynin to 5 mg/d after 3 days and to 10 mg/d the following week, as Jimmy reported no side effects from the lower dosages.
We see Jimmy 1 week after adding oxybutynin, then again 3 weeks later. He reports no wet nights after 1 month of combination therapy, then wets his bed once over the next 2 months. We continue to see him every 3 to 4 weeks and check his electrolytes every 2 to 3 months. He reports no side effects
Five months after starting combination therapy, Jimmy seems much more confident. He has gone 2 months without a bedwetting accident, and his face lights up while discussing the fun he had last week in summer camp. He remains free of side effects, and his parents are thrilled with his progress.
We see Jimmy three more times, once every 2 months. He is staying “dry” but says he wishes to stop his medication because he wants to control his bladder without it.
poll here
The authors’ observations
Medications and behavioral treatments can preserve the child’s self-esteem until he or she outgrows enuresis (Table).
No guidelines address drug regimen duration. Tapering Jimmy’s medications after 7 to 8 months seemed reasonable, but children with enuresis often relapse after stopping treatment. Researchers have recorded relapse rates as high as 60% after stopping imipramine and 80% after stopping desmopressin.1,4
Taper medications slowly to avoid withdrawal, immediate relapse, and anticholinergic effects. If the child relapses, restart medication at the previous therapeutic dosage(s), then start tapering after the child has been accident-free for 3 months.
Table
Medication strategies for treating enuresis
| Medication | Dosage | Risks |
|---|---|---|
| Desmopressin acetate (first-line) | Start with 0.2-mg tablet or 1 to 2 10-μg puffs of nasal spray (half in each nostril) in children age >6; increase to 0.6 mg/d or 4 puffs daily after 1 week if necessary Stop after approximately 6 months without an accident | High relapse rate Reduced urine production Water intoxication, hyponatremia are rare but can result in seizures, coma |
| Oxybutynin (second-line) | 2.5 to 5 mg tid (immediate-release) or 15 mg/d (extended-release) Start at 5 mg at bedtime for children age >5; increase to 15 mg/d after 1 to 2 weeks if needed Stop after approximately 6 months without an accident | High relapse rate Anticholinergic effects (dry mouth, facial flushing, drowsiness, decreased GI motility) Few efficacy studies done Mostly used with other medication |
| Desmopressin with oxybutynin or imipramine; medication plus alarm method (third-line) | Dosages of individual medications as listed | Limited data available Positive results seen in resistant cases, particularly in older children |
| Imipramine (last option) | 1 to 2.5 mg/kg/d Start with 25 mg/d at bedtime; if no response, increase in weekly 25-mg increments to 50 mg/d for children ages 7 to 12 or up to 75 mg/d for children age >12 Stop after approximately 6 months without an accident | High relapse rate after stopping medication Risk of arrhythmias (order ECG when starting medication, 1 month later, then every 6 months) Fatal in overdose (do not prescribe >75 mg/d in enuresis) Associated with suicidal behavior in youths (carries FDA “black box” warning) |
Follow-up: Still dry
After discussing the relapse risk with Jimmy’s parents, we withdraw both oxybutynin and desmopressin over 2 months, reducing each dosage 25% every 2 weeks. We see Jimmy every 4 to 6 weeks during the taper period, then for two bimonthly follow-up visits. He reports no adverse effects and has been accident-free for 8 months.
After consulting with his pediatrician and family, we refer Jimmy, now age 13, back to the pediatrician. We have not seen him for more than 1 year.
Related resources
- National Association For Continence. www.nafc.org.
- Mayo ME, Burns MW. Urodynamic studies in children who wet. Br J Urol 1990 65;641-5.
- Desmopressin • DDAVP
- Imipramine • Tofranil
- Oxybutynin • Ditropan
Dr. Williams is a speaker for Wyeth.
Dr. Singh reports no financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
1. Practice parameter for the assessment and treatment of children and adolescents with enuresis. J Am Acad Child Adolesc Psychiatry 2004;43:1540-50.
2. Bakwin H. The genetics of enuresis. In: Kolvin I, MacKeith RC, Meadow SR (eds). Bladder control and enuresis. London: Heinemann Medical; 1973.
3. Jensen IN, Kristensen G. Alarm treatment: analysis of response and relapse. Scand J Urol Nephrol Suppl 1999;202:73-5.
4. Thompson S, Rey JM. Functional enuresis: is desmopressin the answer? J Am Acad Child Adolesc Psychiatry 1995;34:266-71.
5. Kosar A, Arikan N, Dincel C. Effectiveness of oxybutynin hydrochloride in the treatment of enuresis nocturna—a clinical and urodynamic study. Scand J Urol Nephrol 1999;33:115-8.
6. Persson-Junemann C, Seemann O, Kohrmann KU, et al. Comparison of urodynamic findings and response to oxybutynin in nocturnal enuresis. Eur Urol 1993;24:92-6.
7. Martin-Crespo R, Luque R. [In which patients is the oxybutynin-desmopressin combination therapy indicated?] Cir Pediatr 2003;16:181-5. Spanish.
8. Caione P, Arena F, Biraghi M, et al. Nocturnal enuresis and daytime wetting: a multicentric trial with oxybutynin and desmopressin. Eur Urol 1997;31:459-63.
9. Lee T, Suh HJ, Lee HJ, Lee JE. Comparison of effects of treatment of primary nocturnal enuresis with oxybutynin plus desmopressin, desmopressin alone or imipramine alone: a randomized controlled clinical trial. J Urol 2005;174:1084-7.
1. Practice parameter for the assessment and treatment of children and adolescents with enuresis. J Am Acad Child Adolesc Psychiatry 2004;43:1540-50.
2. Bakwin H. The genetics of enuresis. In: Kolvin I, MacKeith RC, Meadow SR (eds). Bladder control and enuresis. London: Heinemann Medical; 1973.
3. Jensen IN, Kristensen G. Alarm treatment: analysis of response and relapse. Scand J Urol Nephrol Suppl 1999;202:73-5.
4. Thompson S, Rey JM. Functional enuresis: is desmopressin the answer? J Am Acad Child Adolesc Psychiatry 1995;34:266-71.
5. Kosar A, Arikan N, Dincel C. Effectiveness of oxybutynin hydrochloride in the treatment of enuresis nocturna—a clinical and urodynamic study. Scand J Urol Nephrol 1999;33:115-8.
6. Persson-Junemann C, Seemann O, Kohrmann KU, et al. Comparison of urodynamic findings and response to oxybutynin in nocturnal enuresis. Eur Urol 1993;24:92-6.
7. Martin-Crespo R, Luque R. [In which patients is the oxybutynin-desmopressin combination therapy indicated?] Cir Pediatr 2003;16:181-5. Spanish.
8. Caione P, Arena F, Biraghi M, et al. Nocturnal enuresis and daytime wetting: a multicentric trial with oxybutynin and desmopressin. Eur Urol 1997;31:459-63.
9. Lee T, Suh HJ, Lee HJ, Lee JE. Comparison of effects of treatment of primary nocturnal enuresis with oxybutynin plus desmopressin, desmopressin alone or imipramine alone: a randomized controlled clinical trial. J Urol 2005;174:1084-7.
4 drugs can improve autism’s repetitive behaviors
Autism’s repetitive behaviors and restricted interests interfere with adaptive functioning, social interactions, and learning. No medications are FDA-approved for autistic disorder, but some selective serotonin reuptake inhibitors (SSRIs) and atypical antipsychotics and an anticonvulsant have reduced repetitive behaviors in controlled trials. We discuss how that evidence shapes our approach to patients with or without a comorbid family history of bipolar disorder.
Evidence for SSRIs
Repetitive behaviors and restricted interests are autism’s third core domain, as defined by DSM-IVTR criteria.1 For an autistic disorder diagnosis, a patient must show at least one of these behaviors:
- encompassing preoccupations with stereotyped or restricted patterns of interest
- inflexible routines or rituals
- stereotyped, repetitive motor mannerisms
- or persistent preoccupation with parts of objects.
As in obsessive-compulsive disorder (OCD), rituals and restricted interests are thought to decrease anxiety in autism, whereas self-stimulatory behaviors and stereotypy may regulate arousal. The behaviors persist2,3 but may change across the lifespan.
Because SSRIs improve OCD’s repetitive behaviors, clinicians have also used them to treat autism’s repetitive behaviors, though without supporting data. Recently, however, fluoxetine and fluvoxamine have shown efficacy for autism’s repetitive behaviors in randomized, controlled trials. Results indicate:
- In children, fluoxetine is probably better-tolerated than available dosing forms of fluvoxamine.
- In adults, fluvoxamine is well-tolerated and can improve repetitive behavior.
SSRIs and suicidal ideation in autism. The increased risk of suicidality reported with SSRIs when treating depression and OCD has not been seen in children with autism. But because fewer children with autism have been treated with SSRIs, we recommend that you try to assess suicidal ideation during SSRI treatment in those able to express such concerns (Box). Starting with low SSRI dosages (Table 1) and increasing slowly may help prevent behavioral activation, a possible risk factor for suicidality.
Suicidal ideation has not been reported in studies of selective serotonin reuptake inhibitors (SSRIs) in autism. Even so, children and adolescents with autistic disorder are not excluded from the FDA black-box warning of increased risk of suicidality with SSRIs.
Children with obsessive-compulsive disorder (OCD) treated with SSRIs have shown evidence of suicidal thoughts. Thus, higher-functioning children and adults with autism might think about suicide when they become aware of their deficits.
For lower-functioning patients (generally, those who receive medication), we need markers of possible suicidal ideation other than their reports of symptoms. In clinical trials, investigators measure behavioral activation symptoms as risk factors for suicidality.
Thus, when you start an SSRI in a patient with autistic disorder, educate the caregivers to watch for agitation, increased energy, poor sleep, disinhibition, or new hyperactivity. Encourage them to contact you immediately if these signs of activation occur.
Ask higher-functioning patients taking SSRIs about suicidal thinking in a step-wise fashion: thoughts of death, thoughts of their own death, intent, plan, and finally possible attempts.
Table 1
4 drugs with evidence of benefit for autism’s repetitive behaviors*
| Medication | Suggested target daily dosage |
|---|---|
| Fluoxetine7 | Children: Start at 2.5 mg/d; maximum 20 mg/d |
| Adults: Start at 10 to 20 mg/d; maximum 60 mg/d | |
| Fluvoxamine10 | Children: Not first-line; start at 12.5 mg/d; maximum 150 to200 mg/d |
| Adults: Start at 25 mg/d; maximum 300 mg/d | |
| Risperidone13,16 | Children: Start at 0.25 mg/d; maximum 3 mg/d |
| Adults: Start at 2 mg/d; maximum 4 mg/d | |
| Valproate20 | Children: Start at 125 mg (sprinkles); titrate to clinical effect and blood levels of 50 of 120 mcg/mL |
| Adults: Start at 250 mg; increase by 250mg/week to clinical effect and blood levels of 50 to 120 mcg/mL | |
| *Data from randomized, placebo-controlled trials | |
Fluoxetine. In the first open-label study of fluoxetine in children and adults with autistic disorder, global functioning improved significantly in 15 of 23 patients, as measured by the Clinical Global Impressions (CGI) scale.4 Autism symptoms also improved in follow-up, open-label trials, but these did not target repetitive behaviors specifically.5,6
Our group conducted the first randomized, placebo-controlled study of fluoxetine’s effect on repetitive behaviors in children with autism.7 We measured obsessions and compulsions in 45 children, ages 5 to 16, with the Children’s Yale-Brown Obsessive Compulsive Scale (CY-BOCS). This 10-item, clinician-rated questionnaire uses a 5-point scale to rate repetitive behaviors by time spent, distress, interference, resistance, and control.
Using a crossover design—two 8-week phases of active or placebo treatment separated by a 4-week washout—we started liquid fluoxetine at 2.5 mg/d and slowly increased the dosage to clinical effect or a maximum of 0.8 mg/kg/day. Mean final dosage was 9.9 (±4.35) mg/d.
Repetitive behaviors improved, even though we used relatively low dosages to avoid side effects. The mean baseline CY-BOCS compulsion score of 13.15 dropped to 11.6 with fluoxetine and to 12.9 with placebo. Fluoxetine’s effect size was moderate to large, and we found no suicidal ideation with this SSRI.
Fluvoxamine. Repetitive behaviors did not change—as measured with the CY-BOCS—when 18 children with autism received fluvoxamine, 1.5 mg/kg/day, in a 10-week prospective, open-label trial.8 Most patients (72%) reported at least one side effect, and 3 discontinued the SSRI because of behavioral activation. Ten completed the trial. Likewise in a randomized trial, children who received fluvoxamine experienced troublesome side effects and limited benefit.9
Compared with outcomes in children, fluvoxamine has shown greater efficacy in adults. In a 12-week, double-blind, placebo-controlled trial of 30 adults with autism, 8 of 15 treated with fluvoxamine (50 mg/d initially and titrated to 300 mg/d) were rated as responders, compared with none of 15 receiving placebo.
Repetitive behaviors and adaptive functioning improved significantly with fluvoxamine, as measured with the Yale-Brown Obsessive Compulsive Scale (YBOCS) and Vineland Adaptive Behavior Scale, respectively.10 The SSRI was well-tolerated, with only mild nausea and sedation reported.
Thus, fluvoxamine may be useful for treating repetitive behaviors but probably is not a first choice for children with autism. Results might be more favorable in children if fluvoxamine were available in doses <12.5 mg.
Citalopram. Open-label data support using citalopram in autism.11 In a retrospective chart review, 10 of 15 children (73%) were reported “much improved” with citalopram (mean dosage 16.9 mg/d [+/-12.1]), but the review did not specifically address repetitive behaviors. Two patients stopped taking the SSRI because of side effects; agitation, aggressiveness, sedation, and lip dyskinesia were reported.
The National Institutes of Health is sponsoring a multicenter trial of citalopram (starting dosage 2.5 mg/d, up to 20 mg/d) for repetitive behaviors in 144 children with autism, Results are expected in 2007 (see Related resources).
Escitalopram. Some early, open-label evidence suggests that escitalopram may be well-tolerated in autism, but its efficacy for treating repetitive behaviors has not been studied.
Sertraline. One of three reported open-label studies of sertraline in patients with autism measured repetitive behaviors.12 In this study, 42 adults with autism spectrum disorder were treated for 12 weeks with sertraline, 50 to 200 mg/d. One-half were rated “much improved”—mostly in aggressive and repetitive behaviors—with the CGI improvement scale. Sertraline was well-tolerated, although 3 patients dropped out because of persistent agitation.
No randomized trials have examined sertraline in autism.
Clinical recommendations
Family history of bipolar disorder guides our treatment of patients with an autism spectrum disorder and disabling repetitive behaviors (Algorithm).
Algorithm Suggested medications to treat repetitive behaviors in autism
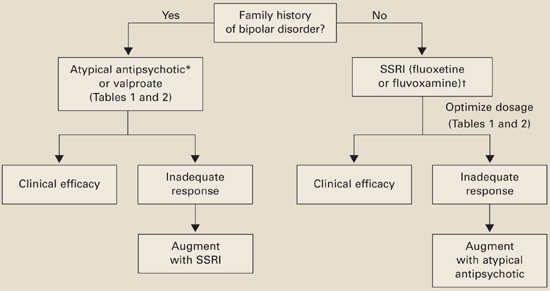
*Risperidone has the most evidence of efficacy, but aripiprazole may be useful for patients with weight-gain problems.
†For children, controlled data support using liquid fluoxetine, starting at 2.5 mg/d.Without bipolar history. SSRIs are usually first-line therapy (although patients with significant irritability and aggression may be an exception and require an atypical antipsychotic first).
If you reach the maximal SSRI dosage without a desired effect, consider adding an atypical (risperidone has the strongest supporting data) or valproate. If behavioral activation symptoms emerge and a lower dosage does not ameliorate them or reduces the clinical effect, consider switching to an atypical or valproate.
With bipolar history. Consider starting with an atypical or valproate; augment with an SSRI if needed.
Monitor for side effects with each medication (Table 2).
Table 2
Medication side effects and recommended monitoring
| Medication | Side effects | Recommended monitoring |
|---|---|---|
| Fluoxetine | Anxiety, insomnia, GI disturbance, appetite and weight changes, mania/hypomania activation, suicidal ideation | Observe closely when starting treatment and while increasing dosage |
| Fluvoxamine | Somnolence, nervousness, insomnia, agitation, GI disturbance, suicidal ideation | Observe closely when starting treatment and while increasing dosage |
| Risperidone | Drowsiness, weight gain, hyperglycemia, GI disturbance, extrapyramidal symptoms, neuroleptic malignant syndrome | Obtain metabolic profile, including serum glucose and lipids |
| Monitor for weight gain and clinical signs of extrapyramidal symptoms | ||
| Valproate | Rash, headaches, weight gain, ataxia, alopecia, GI disturbance, hyperammonemic encephalopathy, sedation, thrombocytopenia, polycystic ovary syndrome, pancreatitis, liver failure, teratogenic effects | CBC with platelets, liver function tests, valproate levels |
| Therapeutic blood levels: 50 to 120 mcg/mL |
Evidence for atypical antipsychotics
Atypicals have been used in autistic disorder to treat irritability and impulsive aggression. Risperidone also has been shown to reduce repetitive behaviors in a controlled trial.13 No evidence or only open-label trials support the use of other atypicals in patients with autism.
Risperidone’s side effects may include metabolic syndrome. No studies have examined lipid profiles and insulin resistance after risperidone treatment in patients with autism, but weight gain has been reported in the Research Units on Pediatric Psychopharmacology (RUPP) trial andothers.13-15 Carefully assess the risk-benefit ratio when you consider using risperidone to treat repetitive behaviors in patients with autism.
Atypical antipsychotics also may increase dyskinesia risk, although extrapyramidal symptoms (EPS) have not been reported in studies of patients with autism and repetitive behaviors. Because EPS could develop after clinical trials are completed, long-term naturalistic studies are needed to address this concern.
Risperidone. The double-blind, placebo-controlled RUPP trial examined risperidone’s efficacy in treating autism’s core symptoms (primarily irritability) in 101 children.13 Mean dosages after 8 weeks and during a 16-week open-label extension for 63 children were 2 and 2.1 mg/d, respectively.
Repetitive behavior—as measured with the CY-BOCS, using RUPP trial data16—improved significantly with risperidone compared with placebo. During the 8-week controlled trial, CY-BOCS scores improved from 15.51 (SD±2.73) to 11.65 (SD±4.02) in the risperidone group, compared with 15.18 (SD±3.88) to 14.21 (SD±4.81) in the placebo group. This response was maintained through the open-label trial.
Side effects included weight gain, fatigue, drowsiness, and drooling. No children receiving risperidone dropped out because of side effects. No EPS were reported, based on weekly Abnormal Involuntary Movement scale and Simpson-Angus scale scores.
Olanzapine. Only open-label studies have examined olanzapine in autism, and one systematically measured repetitive behaviors.17 Eight children with autism or other pervasive developmental disorders were given olanzapine, mean dosage 7.8 (±4.7) mg/d at the end of the 12-week trial.
Repetitive behaviors did not change significantly, as measured with YBOCS. Seven of eight patients completed the trial. Mean weight for the group after 12 weeks was 156±55 lbs, compared with 137±56 lbs at baseline.
Quetiapine. No data support using quetiapine for autism’s repetitive behaviors. Quetiapine, 100 to 350 mg/d (1.6 to 5.2 mg/kg/day) was poorly tolerated in a 16-week open-label safety and efficacy trial among 6 mentally retarded boys with autistic disorder. Side effects included a possible seizure, behavioral activation, increased appetite, and weight gain (0.9 to 8.2 kg). Two patients completed the trial.18
Ziprasidone. Small open-label studies and anecdotal reports of ziprasidone in autism have not examined this drug’s effect on repetitive behaviors.
Aripiprazole. Anecdotal information suggests that clinicians are using this medication to treat patients with autism, but no supporting data exist.
Evidence for valproate
Preliminary trials by our group suggest that valproate may reduce repetitive behaviors in autism. In a retrospective, open-label study, 14 patients (mean age 17) with autism spectrum disorder received divalproex sodium (mean 768±582 mg/d) for a mean 11 months. Ten patients (71%) showed sustained improvement in function, as measured by the CGI-improvement scale, and valproate was generally well-tolerated.19
We then measured valproate’s effect on repetitive behaviors in an 8-week, double-blind, placebo-controlled study of 13 patients (mean age 9) with autism spectrum disorder. Repetitive behaviors improved significantly compared with placebo, as measured by the CY-BOCS, in those who received divalproex (mean 833.93±326.21 mg/d).20
Further studies are needed to replicate this finding. Although it is too early to make general recommendations, valproate may be a reasonable choice for children with autism and epilepsy or affective instability.
- Hollander E, Phillips AT, Yeh CC. Targeted treatments for symptom domains in child and adolescent autism. Lancet 2003;362:732-4.
- Hollander E (ed). Autism spectrum disorders. New York: Marcel Decker; 2003.
- National Institute of Health multicenter study of citalopram for repetitive behaviors in autism. http://www.clinicaltrials.gov/ct/show/nct00086645?order=2
Drug brand names
- Aripiprazole • Abilify
- Citalopram • Celexa
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Sertraline • Zoloft
- Valproic acid • Depakote, Depakene
- Ziprasidone • Geodon
Disclosures
Dr. Anagnostou reports no financial interest with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Hollander receives research/grant support from and is a consultant to Abbott Laboratories. He also receives support from the National Institutes of Health (STAART Center) to investigate orphan drug status for fluoxetine in treating autism symptoms.
1. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC; American Psychiatric Association; 2000.
2. Seltzer MM, Shattuck P, Abbeduto L, Greenberg JS. Trajectory of development in adolescents and adults with autism. MRDD Research Reviews 2004;10:234-47.
3. Howlin P, Goode S, Hutton J, Rutter M. Adult outcome for children with autism. J Child Psychol Psychiatry 2004;45:212-29.
4. Cook EH, Rowlett R, Jaselinkis C, Leventhal BL. Fluoxetine treatment of children and adults with autistic disorder and mental retardation. J Am Acad Child Adolesc Psychiatry 1992;31:739-45.
5. DeLong GR, Teague LA, Kamran MM. Effects of fluoxetine treatment in young children with idiopathic autism. Dev Med Child Neurol 1998;40:551-62.
6. DeLong GR, Ritch CR, Burch S. Fluoxetine response in children with autistic spectrum disorders: correlation with familial major affective disorder and intellectual achievement. Dev Med Child Neurol 2002;44:652-9.
7. Hollander E, Phillips A, Chaplin W, et al. A placebo-controlled cross over trial of liquid fluoxetine on repetitive behaviors in childhood and adolescent autism. Neuropsychopharmacology 2005;30:582-9.
8. Martin A, Koenig K, Anderson GM, Scahill L. Low-dose fluvoxamine treatment of children and adolescents with pervasive developmental disorders: a prospective, open-label study. J Autism Dev Disord 2003;33:77-85.
9. McDougle CJ, Kresch LE, Posey DJ. Repetitive thoughts and behavior in pervasive developmental disorders: treatment with serotonin reuptake inhibitors. J Autism Dev Disord 2000;30:427-35.
10. McDougle CJ, Naylor ST, Cohen DJ, et al. A double-blind, placebo-controlled study of fluvoxamine in adults with autistic disorder. Arch Gen Psychiatry 1996;53:1001-8.
11. Namerow LB, Thomas P, Bostic JQ, et al. Use of citalopram in pervasive developmental disorders. J Dev Behav Pediatr. 2003;24(2):104-8.
12. McDougle CJ, Brodkin ES, Naylor ST, et al. Sertraline in adults with pervasive developmental disorders: a prospective open-label investigation. J Clin Psychopharmacol 1998;18:62-6.
13. McCracken JT, McGough J, Shah B, et al. Research Units on Pediatric Psychopharmacology Autism Network. Risperidone in children with autism and serious behavioral problems. N Engl J Med 2002;347:314-21.
14. Troost PW, Lahuis BE, Steenhuis MP, et al. Long-term effects of risperidone in children with autism spectrum disorders: A placebo discontinuation study. J Am Acad Child Adolesc Psychiatry 2005;44:1137-44.
15. Shea S, Turgay A, Carroll A, et al. Risperidone in the treatment of disruptive behavioral symptoms in children with autistic and other pervasive developmental disorders. Pediatrics 2004;114:e634-e641.
16. McDougle CJ, Scahill L, Aman MG, et al. Risperidone for the core symptom domains of autism: results from the study by the autism network of the research units on pediatric psychopharmacology. Am J Psychiatry 2005;162:1142-8.
17. Potenza MN, Holmes JP, Kanes SJ, McDougle CJ. Olanzapine treatment of children, adolescents and adults with pervasive developmental disorders: an open-label pilot study. J Clin Psychopharmacol 1999;19:37-44.
18. Martin A, Koenig K, Scahill L, Bregman J. Open-label quetiapine in the treatment of children and adolescents with autistic disorder. J Child Adolesc Psychopharmacol 1999;9:99-107.
19. Hollander E, Dolgoff-Kaspar R, Cartwright C, et al. An open trial of divalproex sodium in autism spectrum disorders. J Clin Psychiatry 2001;62:530-4.
20. Hollander E, Soorya LV, Wasserman S, et al. Divalproex sodium vs. placebo in the treatment of repetitive behaviours in autism spectrum disorder. Int J Neuropsychopharmacol 2006;9:209-13.
Autism’s repetitive behaviors and restricted interests interfere with adaptive functioning, social interactions, and learning. No medications are FDA-approved for autistic disorder, but some selective serotonin reuptake inhibitors (SSRIs) and atypical antipsychotics and an anticonvulsant have reduced repetitive behaviors in controlled trials. We discuss how that evidence shapes our approach to patients with or without a comorbid family history of bipolar disorder.
Evidence for SSRIs
Repetitive behaviors and restricted interests are autism’s third core domain, as defined by DSM-IVTR criteria.1 For an autistic disorder diagnosis, a patient must show at least one of these behaviors:
- encompassing preoccupations with stereotyped or restricted patterns of interest
- inflexible routines or rituals
- stereotyped, repetitive motor mannerisms
- or persistent preoccupation with parts of objects.
As in obsessive-compulsive disorder (OCD), rituals and restricted interests are thought to decrease anxiety in autism, whereas self-stimulatory behaviors and stereotypy may regulate arousal. The behaviors persist2,3 but may change across the lifespan.
Because SSRIs improve OCD’s repetitive behaviors, clinicians have also used them to treat autism’s repetitive behaviors, though without supporting data. Recently, however, fluoxetine and fluvoxamine have shown efficacy for autism’s repetitive behaviors in randomized, controlled trials. Results indicate:
- In children, fluoxetine is probably better-tolerated than available dosing forms of fluvoxamine.
- In adults, fluvoxamine is well-tolerated and can improve repetitive behavior.
SSRIs and suicidal ideation in autism. The increased risk of suicidality reported with SSRIs when treating depression and OCD has not been seen in children with autism. But because fewer children with autism have been treated with SSRIs, we recommend that you try to assess suicidal ideation during SSRI treatment in those able to express such concerns (Box). Starting with low SSRI dosages (Table 1) and increasing slowly may help prevent behavioral activation, a possible risk factor for suicidality.
Suicidal ideation has not been reported in studies of selective serotonin reuptake inhibitors (SSRIs) in autism. Even so, children and adolescents with autistic disorder are not excluded from the FDA black-box warning of increased risk of suicidality with SSRIs.
Children with obsessive-compulsive disorder (OCD) treated with SSRIs have shown evidence of suicidal thoughts. Thus, higher-functioning children and adults with autism might think about suicide when they become aware of their deficits.
For lower-functioning patients (generally, those who receive medication), we need markers of possible suicidal ideation other than their reports of symptoms. In clinical trials, investigators measure behavioral activation symptoms as risk factors for suicidality.
Thus, when you start an SSRI in a patient with autistic disorder, educate the caregivers to watch for agitation, increased energy, poor sleep, disinhibition, or new hyperactivity. Encourage them to contact you immediately if these signs of activation occur.
Ask higher-functioning patients taking SSRIs about suicidal thinking in a step-wise fashion: thoughts of death, thoughts of their own death, intent, plan, and finally possible attempts.
Table 1
4 drugs with evidence of benefit for autism’s repetitive behaviors*
| Medication | Suggested target daily dosage |
|---|---|
| Fluoxetine7 | Children: Start at 2.5 mg/d; maximum 20 mg/d |
| Adults: Start at 10 to 20 mg/d; maximum 60 mg/d | |
| Fluvoxamine10 | Children: Not first-line; start at 12.5 mg/d; maximum 150 to200 mg/d |
| Adults: Start at 25 mg/d; maximum 300 mg/d | |
| Risperidone13,16 | Children: Start at 0.25 mg/d; maximum 3 mg/d |
| Adults: Start at 2 mg/d; maximum 4 mg/d | |
| Valproate20 | Children: Start at 125 mg (sprinkles); titrate to clinical effect and blood levels of 50 of 120 mcg/mL |
| Adults: Start at 250 mg; increase by 250mg/week to clinical effect and blood levels of 50 to 120 mcg/mL | |
| *Data from randomized, placebo-controlled trials | |
Fluoxetine. In the first open-label study of fluoxetine in children and adults with autistic disorder, global functioning improved significantly in 15 of 23 patients, as measured by the Clinical Global Impressions (CGI) scale.4 Autism symptoms also improved in follow-up, open-label trials, but these did not target repetitive behaviors specifically.5,6
Our group conducted the first randomized, placebo-controlled study of fluoxetine’s effect on repetitive behaviors in children with autism.7 We measured obsessions and compulsions in 45 children, ages 5 to 16, with the Children’s Yale-Brown Obsessive Compulsive Scale (CY-BOCS). This 10-item, clinician-rated questionnaire uses a 5-point scale to rate repetitive behaviors by time spent, distress, interference, resistance, and control.
Using a crossover design—two 8-week phases of active or placebo treatment separated by a 4-week washout—we started liquid fluoxetine at 2.5 mg/d and slowly increased the dosage to clinical effect or a maximum of 0.8 mg/kg/day. Mean final dosage was 9.9 (±4.35) mg/d.
Repetitive behaviors improved, even though we used relatively low dosages to avoid side effects. The mean baseline CY-BOCS compulsion score of 13.15 dropped to 11.6 with fluoxetine and to 12.9 with placebo. Fluoxetine’s effect size was moderate to large, and we found no suicidal ideation with this SSRI.
Fluvoxamine. Repetitive behaviors did not change—as measured with the CY-BOCS—when 18 children with autism received fluvoxamine, 1.5 mg/kg/day, in a 10-week prospective, open-label trial.8 Most patients (72%) reported at least one side effect, and 3 discontinued the SSRI because of behavioral activation. Ten completed the trial. Likewise in a randomized trial, children who received fluvoxamine experienced troublesome side effects and limited benefit.9
Compared with outcomes in children, fluvoxamine has shown greater efficacy in adults. In a 12-week, double-blind, placebo-controlled trial of 30 adults with autism, 8 of 15 treated with fluvoxamine (50 mg/d initially and titrated to 300 mg/d) were rated as responders, compared with none of 15 receiving placebo.
Repetitive behaviors and adaptive functioning improved significantly with fluvoxamine, as measured with the Yale-Brown Obsessive Compulsive Scale (YBOCS) and Vineland Adaptive Behavior Scale, respectively.10 The SSRI was well-tolerated, with only mild nausea and sedation reported.
Thus, fluvoxamine may be useful for treating repetitive behaviors but probably is not a first choice for children with autism. Results might be more favorable in children if fluvoxamine were available in doses <12.5 mg.
Citalopram. Open-label data support using citalopram in autism.11 In a retrospective chart review, 10 of 15 children (73%) were reported “much improved” with citalopram (mean dosage 16.9 mg/d [+/-12.1]), but the review did not specifically address repetitive behaviors. Two patients stopped taking the SSRI because of side effects; agitation, aggressiveness, sedation, and lip dyskinesia were reported.
The National Institutes of Health is sponsoring a multicenter trial of citalopram (starting dosage 2.5 mg/d, up to 20 mg/d) for repetitive behaviors in 144 children with autism, Results are expected in 2007 (see Related resources).
Escitalopram. Some early, open-label evidence suggests that escitalopram may be well-tolerated in autism, but its efficacy for treating repetitive behaviors has not been studied.
Sertraline. One of three reported open-label studies of sertraline in patients with autism measured repetitive behaviors.12 In this study, 42 adults with autism spectrum disorder were treated for 12 weeks with sertraline, 50 to 200 mg/d. One-half were rated “much improved”—mostly in aggressive and repetitive behaviors—with the CGI improvement scale. Sertraline was well-tolerated, although 3 patients dropped out because of persistent agitation.
No randomized trials have examined sertraline in autism.
Clinical recommendations
Family history of bipolar disorder guides our treatment of patients with an autism spectrum disorder and disabling repetitive behaviors (Algorithm).
Algorithm Suggested medications to treat repetitive behaviors in autism
*Risperidone has the most evidence of efficacy, but aripiprazole may be useful for patients with weight-gain problems.
†For children, controlled data support using liquid fluoxetine, starting at 2.5 mg/d.Without bipolar history. SSRIs are usually first-line therapy (although patients with significant irritability and aggression may be an exception and require an atypical antipsychotic first).
If you reach the maximal SSRI dosage without a desired effect, consider adding an atypical (risperidone has the strongest supporting data) or valproate. If behavioral activation symptoms emerge and a lower dosage does not ameliorate them or reduces the clinical effect, consider switching to an atypical or valproate.
With bipolar history. Consider starting with an atypical or valproate; augment with an SSRI if needed.
Monitor for side effects with each medication (Table 2).
Table 2
Medication side effects and recommended monitoring
| Medication | Side effects | Recommended monitoring |
|---|---|---|
| Fluoxetine | Anxiety, insomnia, GI disturbance, appetite and weight changes, mania/hypomania activation, suicidal ideation | Observe closely when starting treatment and while increasing dosage |
| Fluvoxamine | Somnolence, nervousness, insomnia, agitation, GI disturbance, suicidal ideation | Observe closely when starting treatment and while increasing dosage |
| Risperidone | Drowsiness, weight gain, hyperglycemia, GI disturbance, extrapyramidal symptoms, neuroleptic malignant syndrome | Obtain metabolic profile, including serum glucose and lipids |
| Monitor for weight gain and clinical signs of extrapyramidal symptoms | ||
| Valproate | Rash, headaches, weight gain, ataxia, alopecia, GI disturbance, hyperammonemic encephalopathy, sedation, thrombocytopenia, polycystic ovary syndrome, pancreatitis, liver failure, teratogenic effects | CBC with platelets, liver function tests, valproate levels |
| Therapeutic blood levels: 50 to 120 mcg/mL |
Evidence for atypical antipsychotics
Atypicals have been used in autistic disorder to treat irritability and impulsive aggression. Risperidone also has been shown to reduce repetitive behaviors in a controlled trial.13 No evidence or only open-label trials support the use of other atypicals in patients with autism.
Risperidone’s side effects may include metabolic syndrome. No studies have examined lipid profiles and insulin resistance after risperidone treatment in patients with autism, but weight gain has been reported in the Research Units on Pediatric Psychopharmacology (RUPP) trial andothers.13-15 Carefully assess the risk-benefit ratio when you consider using risperidone to treat repetitive behaviors in patients with autism.
Atypical antipsychotics also may increase dyskinesia risk, although extrapyramidal symptoms (EPS) have not been reported in studies of patients with autism and repetitive behaviors. Because EPS could develop after clinical trials are completed, long-term naturalistic studies are needed to address this concern.
Risperidone. The double-blind, placebo-controlled RUPP trial examined risperidone’s efficacy in treating autism’s core symptoms (primarily irritability) in 101 children.13 Mean dosages after 8 weeks and during a 16-week open-label extension for 63 children were 2 and 2.1 mg/d, respectively.
Repetitive behavior—as measured with the CY-BOCS, using RUPP trial data16—improved significantly with risperidone compared with placebo. During the 8-week controlled trial, CY-BOCS scores improved from 15.51 (SD±2.73) to 11.65 (SD±4.02) in the risperidone group, compared with 15.18 (SD±3.88) to 14.21 (SD±4.81) in the placebo group. This response was maintained through the open-label trial.
Side effects included weight gain, fatigue, drowsiness, and drooling. No children receiving risperidone dropped out because of side effects. No EPS were reported, based on weekly Abnormal Involuntary Movement scale and Simpson-Angus scale scores.
Olanzapine. Only open-label studies have examined olanzapine in autism, and one systematically measured repetitive behaviors.17 Eight children with autism or other pervasive developmental disorders were given olanzapine, mean dosage 7.8 (±4.7) mg/d at the end of the 12-week trial.
Repetitive behaviors did not change significantly, as measured with YBOCS. Seven of eight patients completed the trial. Mean weight for the group after 12 weeks was 156±55 lbs, compared with 137±56 lbs at baseline.
Quetiapine. No data support using quetiapine for autism’s repetitive behaviors. Quetiapine, 100 to 350 mg/d (1.6 to 5.2 mg/kg/day) was poorly tolerated in a 16-week open-label safety and efficacy trial among 6 mentally retarded boys with autistic disorder. Side effects included a possible seizure, behavioral activation, increased appetite, and weight gain (0.9 to 8.2 kg). Two patients completed the trial.18
Ziprasidone. Small open-label studies and anecdotal reports of ziprasidone in autism have not examined this drug’s effect on repetitive behaviors.
Aripiprazole. Anecdotal information suggests that clinicians are using this medication to treat patients with autism, but no supporting data exist.
Evidence for valproate
Preliminary trials by our group suggest that valproate may reduce repetitive behaviors in autism. In a retrospective, open-label study, 14 patients (mean age 17) with autism spectrum disorder received divalproex sodium (mean 768±582 mg/d) for a mean 11 months. Ten patients (71%) showed sustained improvement in function, as measured by the CGI-improvement scale, and valproate was generally well-tolerated.19
We then measured valproate’s effect on repetitive behaviors in an 8-week, double-blind, placebo-controlled study of 13 patients (mean age 9) with autism spectrum disorder. Repetitive behaviors improved significantly compared with placebo, as measured by the CY-BOCS, in those who received divalproex (mean 833.93±326.21 mg/d).20
Further studies are needed to replicate this finding. Although it is too early to make general recommendations, valproate may be a reasonable choice for children with autism and epilepsy or affective instability.
- Hollander E, Phillips AT, Yeh CC. Targeted treatments for symptom domains in child and adolescent autism. Lancet 2003;362:732-4.
- Hollander E (ed). Autism spectrum disorders. New York: Marcel Decker; 2003.
- National Institute of Health multicenter study of citalopram for repetitive behaviors in autism. http://www.clinicaltrials.gov/ct/show/nct00086645?order=2
Drug brand names
- Aripiprazole • Abilify
- Citalopram • Celexa
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Sertraline • Zoloft
- Valproic acid • Depakote, Depakene
- Ziprasidone • Geodon
Disclosures
Dr. Anagnostou reports no financial interest with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Hollander receives research/grant support from and is a consultant to Abbott Laboratories. He also receives support from the National Institutes of Health (STAART Center) to investigate orphan drug status for fluoxetine in treating autism symptoms.
Autism’s repetitive behaviors and restricted interests interfere with adaptive functioning, social interactions, and learning. No medications are FDA-approved for autistic disorder, but some selective serotonin reuptake inhibitors (SSRIs) and atypical antipsychotics and an anticonvulsant have reduced repetitive behaviors in controlled trials. We discuss how that evidence shapes our approach to patients with or without a comorbid family history of bipolar disorder.
Evidence for SSRIs
Repetitive behaviors and restricted interests are autism’s third core domain, as defined by DSM-IVTR criteria.1 For an autistic disorder diagnosis, a patient must show at least one of these behaviors:
- encompassing preoccupations with stereotyped or restricted patterns of interest
- inflexible routines or rituals
- stereotyped, repetitive motor mannerisms
- or persistent preoccupation with parts of objects.
As in obsessive-compulsive disorder (OCD), rituals and restricted interests are thought to decrease anxiety in autism, whereas self-stimulatory behaviors and stereotypy may regulate arousal. The behaviors persist2,3 but may change across the lifespan.
Because SSRIs improve OCD’s repetitive behaviors, clinicians have also used them to treat autism’s repetitive behaviors, though without supporting data. Recently, however, fluoxetine and fluvoxamine have shown efficacy for autism’s repetitive behaviors in randomized, controlled trials. Results indicate:
- In children, fluoxetine is probably better-tolerated than available dosing forms of fluvoxamine.
- In adults, fluvoxamine is well-tolerated and can improve repetitive behavior.
SSRIs and suicidal ideation in autism. The increased risk of suicidality reported with SSRIs when treating depression and OCD has not been seen in children with autism. But because fewer children with autism have been treated with SSRIs, we recommend that you try to assess suicidal ideation during SSRI treatment in those able to express such concerns (Box). Starting with low SSRI dosages (Table 1) and increasing slowly may help prevent behavioral activation, a possible risk factor for suicidality.
Suicidal ideation has not been reported in studies of selective serotonin reuptake inhibitors (SSRIs) in autism. Even so, children and adolescents with autistic disorder are not excluded from the FDA black-box warning of increased risk of suicidality with SSRIs.
Children with obsessive-compulsive disorder (OCD) treated with SSRIs have shown evidence of suicidal thoughts. Thus, higher-functioning children and adults with autism might think about suicide when they become aware of their deficits.
For lower-functioning patients (generally, those who receive medication), we need markers of possible suicidal ideation other than their reports of symptoms. In clinical trials, investigators measure behavioral activation symptoms as risk factors for suicidality.
Thus, when you start an SSRI in a patient with autistic disorder, educate the caregivers to watch for agitation, increased energy, poor sleep, disinhibition, or new hyperactivity. Encourage them to contact you immediately if these signs of activation occur.
Ask higher-functioning patients taking SSRIs about suicidal thinking in a step-wise fashion: thoughts of death, thoughts of their own death, intent, plan, and finally possible attempts.
Table 1
4 drugs with evidence of benefit for autism’s repetitive behaviors*
| Medication | Suggested target daily dosage |
|---|---|
| Fluoxetine7 | Children: Start at 2.5 mg/d; maximum 20 mg/d |
| Adults: Start at 10 to 20 mg/d; maximum 60 mg/d | |
| Fluvoxamine10 | Children: Not first-line; start at 12.5 mg/d; maximum 150 to200 mg/d |
| Adults: Start at 25 mg/d; maximum 300 mg/d | |
| Risperidone13,16 | Children: Start at 0.25 mg/d; maximum 3 mg/d |
| Adults: Start at 2 mg/d; maximum 4 mg/d | |
| Valproate20 | Children: Start at 125 mg (sprinkles); titrate to clinical effect and blood levels of 50 of 120 mcg/mL |
| Adults: Start at 250 mg; increase by 250mg/week to clinical effect and blood levels of 50 to 120 mcg/mL | |
| *Data from randomized, placebo-controlled trials | |
Fluoxetine. In the first open-label study of fluoxetine in children and adults with autistic disorder, global functioning improved significantly in 15 of 23 patients, as measured by the Clinical Global Impressions (CGI) scale.4 Autism symptoms also improved in follow-up, open-label trials, but these did not target repetitive behaviors specifically.5,6
Our group conducted the first randomized, placebo-controlled study of fluoxetine’s effect on repetitive behaviors in children with autism.7 We measured obsessions and compulsions in 45 children, ages 5 to 16, with the Children’s Yale-Brown Obsessive Compulsive Scale (CY-BOCS). This 10-item, clinician-rated questionnaire uses a 5-point scale to rate repetitive behaviors by time spent, distress, interference, resistance, and control.
Using a crossover design—two 8-week phases of active or placebo treatment separated by a 4-week washout—we started liquid fluoxetine at 2.5 mg/d and slowly increased the dosage to clinical effect or a maximum of 0.8 mg/kg/day. Mean final dosage was 9.9 (±4.35) mg/d.
Repetitive behaviors improved, even though we used relatively low dosages to avoid side effects. The mean baseline CY-BOCS compulsion score of 13.15 dropped to 11.6 with fluoxetine and to 12.9 with placebo. Fluoxetine’s effect size was moderate to large, and we found no suicidal ideation with this SSRI.
Fluvoxamine. Repetitive behaviors did not change—as measured with the CY-BOCS—when 18 children with autism received fluvoxamine, 1.5 mg/kg/day, in a 10-week prospective, open-label trial.8 Most patients (72%) reported at least one side effect, and 3 discontinued the SSRI because of behavioral activation. Ten completed the trial. Likewise in a randomized trial, children who received fluvoxamine experienced troublesome side effects and limited benefit.9
Compared with outcomes in children, fluvoxamine has shown greater efficacy in adults. In a 12-week, double-blind, placebo-controlled trial of 30 adults with autism, 8 of 15 treated with fluvoxamine (50 mg/d initially and titrated to 300 mg/d) were rated as responders, compared with none of 15 receiving placebo.
Repetitive behaviors and adaptive functioning improved significantly with fluvoxamine, as measured with the Yale-Brown Obsessive Compulsive Scale (YBOCS) and Vineland Adaptive Behavior Scale, respectively.10 The SSRI was well-tolerated, with only mild nausea and sedation reported.
Thus, fluvoxamine may be useful for treating repetitive behaviors but probably is not a first choice for children with autism. Results might be more favorable in children if fluvoxamine were available in doses <12.5 mg.
Citalopram. Open-label data support using citalopram in autism.11 In a retrospective chart review, 10 of 15 children (73%) were reported “much improved” with citalopram (mean dosage 16.9 mg/d [+/-12.1]), but the review did not specifically address repetitive behaviors. Two patients stopped taking the SSRI because of side effects; agitation, aggressiveness, sedation, and lip dyskinesia were reported.
The National Institutes of Health is sponsoring a multicenter trial of citalopram (starting dosage 2.5 mg/d, up to 20 mg/d) for repetitive behaviors in 144 children with autism, Results are expected in 2007 (see Related resources).
Escitalopram. Some early, open-label evidence suggests that escitalopram may be well-tolerated in autism, but its efficacy for treating repetitive behaviors has not been studied.
Sertraline. One of three reported open-label studies of sertraline in patients with autism measured repetitive behaviors.12 In this study, 42 adults with autism spectrum disorder were treated for 12 weeks with sertraline, 50 to 200 mg/d. One-half were rated “much improved”—mostly in aggressive and repetitive behaviors—with the CGI improvement scale. Sertraline was well-tolerated, although 3 patients dropped out because of persistent agitation.
No randomized trials have examined sertraline in autism.
Clinical recommendations
Family history of bipolar disorder guides our treatment of patients with an autism spectrum disorder and disabling repetitive behaviors (Algorithm).
Algorithm Suggested medications to treat repetitive behaviors in autism
*Risperidone has the most evidence of efficacy, but aripiprazole may be useful for patients with weight-gain problems.
†For children, controlled data support using liquid fluoxetine, starting at 2.5 mg/d.Without bipolar history. SSRIs are usually first-line therapy (although patients with significant irritability and aggression may be an exception and require an atypical antipsychotic first).
If you reach the maximal SSRI dosage without a desired effect, consider adding an atypical (risperidone has the strongest supporting data) or valproate. If behavioral activation symptoms emerge and a lower dosage does not ameliorate them or reduces the clinical effect, consider switching to an atypical or valproate.
With bipolar history. Consider starting with an atypical or valproate; augment with an SSRI if needed.
Monitor for side effects with each medication (Table 2).
Table 2
Medication side effects and recommended monitoring
| Medication | Side effects | Recommended monitoring |
|---|---|---|
| Fluoxetine | Anxiety, insomnia, GI disturbance, appetite and weight changes, mania/hypomania activation, suicidal ideation | Observe closely when starting treatment and while increasing dosage |
| Fluvoxamine | Somnolence, nervousness, insomnia, agitation, GI disturbance, suicidal ideation | Observe closely when starting treatment and while increasing dosage |
| Risperidone | Drowsiness, weight gain, hyperglycemia, GI disturbance, extrapyramidal symptoms, neuroleptic malignant syndrome | Obtain metabolic profile, including serum glucose and lipids |
| Monitor for weight gain and clinical signs of extrapyramidal symptoms | ||
| Valproate | Rash, headaches, weight gain, ataxia, alopecia, GI disturbance, hyperammonemic encephalopathy, sedation, thrombocytopenia, polycystic ovary syndrome, pancreatitis, liver failure, teratogenic effects | CBC with platelets, liver function tests, valproate levels |
| Therapeutic blood levels: 50 to 120 mcg/mL |
Evidence for atypical antipsychotics
Atypicals have been used in autistic disorder to treat irritability and impulsive aggression. Risperidone also has been shown to reduce repetitive behaviors in a controlled trial.13 No evidence or only open-label trials support the use of other atypicals in patients with autism.
Risperidone’s side effects may include metabolic syndrome. No studies have examined lipid profiles and insulin resistance after risperidone treatment in patients with autism, but weight gain has been reported in the Research Units on Pediatric Psychopharmacology (RUPP) trial andothers.13-15 Carefully assess the risk-benefit ratio when you consider using risperidone to treat repetitive behaviors in patients with autism.
Atypical antipsychotics also may increase dyskinesia risk, although extrapyramidal symptoms (EPS) have not been reported in studies of patients with autism and repetitive behaviors. Because EPS could develop after clinical trials are completed, long-term naturalistic studies are needed to address this concern.
Risperidone. The double-blind, placebo-controlled RUPP trial examined risperidone’s efficacy in treating autism’s core symptoms (primarily irritability) in 101 children.13 Mean dosages after 8 weeks and during a 16-week open-label extension for 63 children were 2 and 2.1 mg/d, respectively.
Repetitive behavior—as measured with the CY-BOCS, using RUPP trial data16—improved significantly with risperidone compared with placebo. During the 8-week controlled trial, CY-BOCS scores improved from 15.51 (SD±2.73) to 11.65 (SD±4.02) in the risperidone group, compared with 15.18 (SD±3.88) to 14.21 (SD±4.81) in the placebo group. This response was maintained through the open-label trial.
Side effects included weight gain, fatigue, drowsiness, and drooling. No children receiving risperidone dropped out because of side effects. No EPS were reported, based on weekly Abnormal Involuntary Movement scale and Simpson-Angus scale scores.
Olanzapine. Only open-label studies have examined olanzapine in autism, and one systematically measured repetitive behaviors.17 Eight children with autism or other pervasive developmental disorders were given olanzapine, mean dosage 7.8 (±4.7) mg/d at the end of the 12-week trial.
Repetitive behaviors did not change significantly, as measured with YBOCS. Seven of eight patients completed the trial. Mean weight for the group after 12 weeks was 156±55 lbs, compared with 137±56 lbs at baseline.
Quetiapine. No data support using quetiapine for autism’s repetitive behaviors. Quetiapine, 100 to 350 mg/d (1.6 to 5.2 mg/kg/day) was poorly tolerated in a 16-week open-label safety and efficacy trial among 6 mentally retarded boys with autistic disorder. Side effects included a possible seizure, behavioral activation, increased appetite, and weight gain (0.9 to 8.2 kg). Two patients completed the trial.18
Ziprasidone. Small open-label studies and anecdotal reports of ziprasidone in autism have not examined this drug’s effect on repetitive behaviors.
Aripiprazole. Anecdotal information suggests that clinicians are using this medication to treat patients with autism, but no supporting data exist.
Evidence for valproate
Preliminary trials by our group suggest that valproate may reduce repetitive behaviors in autism. In a retrospective, open-label study, 14 patients (mean age 17) with autism spectrum disorder received divalproex sodium (mean 768±582 mg/d) for a mean 11 months. Ten patients (71%) showed sustained improvement in function, as measured by the CGI-improvement scale, and valproate was generally well-tolerated.19
We then measured valproate’s effect on repetitive behaviors in an 8-week, double-blind, placebo-controlled study of 13 patients (mean age 9) with autism spectrum disorder. Repetitive behaviors improved significantly compared with placebo, as measured by the CY-BOCS, in those who received divalproex (mean 833.93±326.21 mg/d).20
Further studies are needed to replicate this finding. Although it is too early to make general recommendations, valproate may be a reasonable choice for children with autism and epilepsy or affective instability.
- Hollander E, Phillips AT, Yeh CC. Targeted treatments for symptom domains in child and adolescent autism. Lancet 2003;362:732-4.
- Hollander E (ed). Autism spectrum disorders. New York: Marcel Decker; 2003.
- National Institute of Health multicenter study of citalopram for repetitive behaviors in autism. http://www.clinicaltrials.gov/ct/show/nct00086645?order=2
Drug brand names
- Aripiprazole • Abilify
- Citalopram • Celexa
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Sertraline • Zoloft
- Valproic acid • Depakote, Depakene
- Ziprasidone • Geodon
Disclosures
Dr. Anagnostou reports no financial interest with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Hollander receives research/grant support from and is a consultant to Abbott Laboratories. He also receives support from the National Institutes of Health (STAART Center) to investigate orphan drug status for fluoxetine in treating autism symptoms.
1. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC; American Psychiatric Association; 2000.
2. Seltzer MM, Shattuck P, Abbeduto L, Greenberg JS. Trajectory of development in adolescents and adults with autism. MRDD Research Reviews 2004;10:234-47.
3. Howlin P, Goode S, Hutton J, Rutter M. Adult outcome for children with autism. J Child Psychol Psychiatry 2004;45:212-29.
4. Cook EH, Rowlett R, Jaselinkis C, Leventhal BL. Fluoxetine treatment of children and adults with autistic disorder and mental retardation. J Am Acad Child Adolesc Psychiatry 1992;31:739-45.
5. DeLong GR, Teague LA, Kamran MM. Effects of fluoxetine treatment in young children with idiopathic autism. Dev Med Child Neurol 1998;40:551-62.
6. DeLong GR, Ritch CR, Burch S. Fluoxetine response in children with autistic spectrum disorders: correlation with familial major affective disorder and intellectual achievement. Dev Med Child Neurol 2002;44:652-9.
7. Hollander E, Phillips A, Chaplin W, et al. A placebo-controlled cross over trial of liquid fluoxetine on repetitive behaviors in childhood and adolescent autism. Neuropsychopharmacology 2005;30:582-9.
8. Martin A, Koenig K, Anderson GM, Scahill L. Low-dose fluvoxamine treatment of children and adolescents with pervasive developmental disorders: a prospective, open-label study. J Autism Dev Disord 2003;33:77-85.
9. McDougle CJ, Kresch LE, Posey DJ. Repetitive thoughts and behavior in pervasive developmental disorders: treatment with serotonin reuptake inhibitors. J Autism Dev Disord 2000;30:427-35.
10. McDougle CJ, Naylor ST, Cohen DJ, et al. A double-blind, placebo-controlled study of fluvoxamine in adults with autistic disorder. Arch Gen Psychiatry 1996;53:1001-8.
11. Namerow LB, Thomas P, Bostic JQ, et al. Use of citalopram in pervasive developmental disorders. J Dev Behav Pediatr. 2003;24(2):104-8.
12. McDougle CJ, Brodkin ES, Naylor ST, et al. Sertraline in adults with pervasive developmental disorders: a prospective open-label investigation. J Clin Psychopharmacol 1998;18:62-6.
13. McCracken JT, McGough J, Shah B, et al. Research Units on Pediatric Psychopharmacology Autism Network. Risperidone in children with autism and serious behavioral problems. N Engl J Med 2002;347:314-21.
14. Troost PW, Lahuis BE, Steenhuis MP, et al. Long-term effects of risperidone in children with autism spectrum disorders: A placebo discontinuation study. J Am Acad Child Adolesc Psychiatry 2005;44:1137-44.
15. Shea S, Turgay A, Carroll A, et al. Risperidone in the treatment of disruptive behavioral symptoms in children with autistic and other pervasive developmental disorders. Pediatrics 2004;114:e634-e641.
16. McDougle CJ, Scahill L, Aman MG, et al. Risperidone for the core symptom domains of autism: results from the study by the autism network of the research units on pediatric psychopharmacology. Am J Psychiatry 2005;162:1142-8.
17. Potenza MN, Holmes JP, Kanes SJ, McDougle CJ. Olanzapine treatment of children, adolescents and adults with pervasive developmental disorders: an open-label pilot study. J Clin Psychopharmacol 1999;19:37-44.
18. Martin A, Koenig K, Scahill L, Bregman J. Open-label quetiapine in the treatment of children and adolescents with autistic disorder. J Child Adolesc Psychopharmacol 1999;9:99-107.
19. Hollander E, Dolgoff-Kaspar R, Cartwright C, et al. An open trial of divalproex sodium in autism spectrum disorders. J Clin Psychiatry 2001;62:530-4.
20. Hollander E, Soorya LV, Wasserman S, et al. Divalproex sodium vs. placebo in the treatment of repetitive behaviours in autism spectrum disorder. Int J Neuropsychopharmacol 2006;9:209-13.
1. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC; American Psychiatric Association; 2000.
2. Seltzer MM, Shattuck P, Abbeduto L, Greenberg JS. Trajectory of development in adolescents and adults with autism. MRDD Research Reviews 2004;10:234-47.
3. Howlin P, Goode S, Hutton J, Rutter M. Adult outcome for children with autism. J Child Psychol Psychiatry 2004;45:212-29.
4. Cook EH, Rowlett R, Jaselinkis C, Leventhal BL. Fluoxetine treatment of children and adults with autistic disorder and mental retardation. J Am Acad Child Adolesc Psychiatry 1992;31:739-45.
5. DeLong GR, Teague LA, Kamran MM. Effects of fluoxetine treatment in young children with idiopathic autism. Dev Med Child Neurol 1998;40:551-62.
6. DeLong GR, Ritch CR, Burch S. Fluoxetine response in children with autistic spectrum disorders: correlation with familial major affective disorder and intellectual achievement. Dev Med Child Neurol 2002;44:652-9.
7. Hollander E, Phillips A, Chaplin W, et al. A placebo-controlled cross over trial of liquid fluoxetine on repetitive behaviors in childhood and adolescent autism. Neuropsychopharmacology 2005;30:582-9.
8. Martin A, Koenig K, Anderson GM, Scahill L. Low-dose fluvoxamine treatment of children and adolescents with pervasive developmental disorders: a prospective, open-label study. J Autism Dev Disord 2003;33:77-85.
9. McDougle CJ, Kresch LE, Posey DJ. Repetitive thoughts and behavior in pervasive developmental disorders: treatment with serotonin reuptake inhibitors. J Autism Dev Disord 2000;30:427-35.
10. McDougle CJ, Naylor ST, Cohen DJ, et al. A double-blind, placebo-controlled study of fluvoxamine in adults with autistic disorder. Arch Gen Psychiatry 1996;53:1001-8.
11. Namerow LB, Thomas P, Bostic JQ, et al. Use of citalopram in pervasive developmental disorders. J Dev Behav Pediatr. 2003;24(2):104-8.
12. McDougle CJ, Brodkin ES, Naylor ST, et al. Sertraline in adults with pervasive developmental disorders: a prospective open-label investigation. J Clin Psychopharmacol 1998;18:62-6.
13. McCracken JT, McGough J, Shah B, et al. Research Units on Pediatric Psychopharmacology Autism Network. Risperidone in children with autism and serious behavioral problems. N Engl J Med 2002;347:314-21.
14. Troost PW, Lahuis BE, Steenhuis MP, et al. Long-term effects of risperidone in children with autism spectrum disorders: A placebo discontinuation study. J Am Acad Child Adolesc Psychiatry 2005;44:1137-44.
15. Shea S, Turgay A, Carroll A, et al. Risperidone in the treatment of disruptive behavioral symptoms in children with autistic and other pervasive developmental disorders. Pediatrics 2004;114:e634-e641.
16. McDougle CJ, Scahill L, Aman MG, et al. Risperidone for the core symptom domains of autism: results from the study by the autism network of the research units on pediatric psychopharmacology. Am J Psychiatry 2005;162:1142-8.
17. Potenza MN, Holmes JP, Kanes SJ, McDougle CJ. Olanzapine treatment of children, adolescents and adults with pervasive developmental disorders: an open-label pilot study. J Clin Psychopharmacol 1999;19:37-44.
18. Martin A, Koenig K, Scahill L, Bregman J. Open-label quetiapine in the treatment of children and adolescents with autistic disorder. J Child Adolesc Psychopharmacol 1999;9:99-107.
19. Hollander E, Dolgoff-Kaspar R, Cartwright C, et al. An open trial of divalproex sodium in autism spectrum disorders. J Clin Psychiatry 2001;62:530-4.
20. Hollander E, Soorya LV, Wasserman S, et al. Divalproex sodium vs. placebo in the treatment of repetitive behaviours in autism spectrum disorder. Int J Neuropsychopharmacol 2006;9:209-13.
SSRI use during pregnancy
Untreated depression can have serious consequences, but many pregnant women resist taking antidepressants because they overestimate the risk of birth defects.Paroxetine in pregnancy”). Further study is needed to define the risks of teratogenesis with paroxetine compared with other antidepressants.
Third-trimester exposure
In a recent meta-analysis, infants exposed to SSRIs in utero showed an increased risk for prematurity (OR; 2.03) and low birth weight (OR; 2.37).15 Other studies, however, showed no differences in these risks in SSRI-exposed infants or attributed the results to untreated maternal depression or smoking.16
A Medline search across the last 20 years17 found 26 case reports, three prospective controlled cohort studies, and other records of >400 women who received fluoxetine, sertraline, or paroxetine in the third trimester. The authors found the evidence “ambiguous” as to the cause of adverse events and concluded that the risk of not treating major depression with adequate SSRI therapy at that stage of pregnancy “most likely” outweighs the risk of harm to infants.
Transient neonatal complications. Thirty percent of neonates exposed to SSRIs in the third trimester experience transient adaptation problems, which peak 48 hours after birth18 (Table 3). Symptoms may include initial lack of crying, increased muscle tonus, flush, irritability, jitteriness, hypothermia, abnormal breathing, and disrupted sleep and motor activity.2,19,20
Transient neonatal symptoms from SSRI exposure are thought to be a serotonin withdrawal syndrome or serotonin overstimulation.21 The syndrome is usually mild, self-limited, and requires only supportive treatments. All antidepressants’ labels warn of these effects.
Table 3
Neonatal SSRI withdrawal: Symptoms, causes, and treatment
| Symptoms | Initial lack of crying |
| Increased muscle tonus | |
| Irritability, jitteriness | |
| Abnormal breathing pattern | |
| Disrupted sleep and motor activity | |
| Hypotheses of cause | Serotonin overstimulation or withdrawal |
| Treatment | Close observation |
| Supportive measures |
Recommendation. Some authors have recommended tapering antidepressants in the third trimester, but the risk of postpartum depression appears to outweigh any potential benefit from discontinuation. Because birth timing is unpredictable, some women whose antidepressants are tapered off could be without medication for a long time.
Thus, we recommend:
- continuing SSRIs during late pregnancy
- monitoring the newborn for 48 hours for transient neonatal adaptation symptoms or PPHN.2,17,18
Long-term effects of SSRI exposure
Do SSRIs during pregnancy have long-term effects on infants’ neurodevelopment? Study results are mixed. For example:
- A prospective, controlled, cohort trial found no adverse effects on IQ, language, or behavioral development in children ages 15 months to 6 years whose mothers took tricyclic antidepressants (N=46) or fluoxetine (N=40) during pregnancy, compared with 36 unexposed controls.23
- Another prospective study showed lower Bayley Psychomotor Developmental Index scores in 31 SSRI-exposed infants compared with 13 infants born to depressed mothers not on antidepressants. Reduced body control, coordination, and fine motor skills might suggest possible subtle effects of SSRIs on motor development in exposed infants, the authors concluded.24
Case continued: A healthy delivery
Ms. P’s depression improves a few weeks after she restarts an SSRI. She delivers a healthy term baby with Apgar score of 7. The baby initially does not cry, awakens easily, and shows mild irritability. His mother’s SSRI use, her severe depression during part of the pregnancy, or some other factor may have caused his mild neonatal complications.
Nursing staff carefully observe the infant for 2 days in the newborn nursery, and his irritability fades away. Ms. P decides to continue taking antidepressants to care for herself and the baby.
Weighing treatment options
For each woman with a history of depression who is pregnant or intends to conceive, we recommend a risk-benefit analysis of her depression severity and need for an antidepressant:
Moderate to severe depression (history of recurrent depressive episodes, hospitalization, or suicidality). Strongly consider medication. If your patient is taking an SSRI, counsel her about:
- the 70% risk of depression relapse if she stops the medication, even for the first trimester
- risks of untreated depression during pregnancy (poor self-care, preterm labor, birth complications, and increased risk for poor stress adaptations in children).
Choosing an SSRI. No one SSRI is the safest choice for all women, especially when data on breast-feeding come into play.
- Fluoxetine has been studied more than other SSRIs during pregnancy; most evidence is reassuring, except for transient neonatal complications. With its long half-life, fluoxetine is not recommended during breastfeeding because it may accumulate in infant sera.
- Sertraline has shown low umbilical cord to maternal serum ratios in small samples and has reassuring breast-feeding data.
- Citalopram, compared with sertraline, has been studied more in pregnancy but has a higher fetal-to-maternal serum ratio (as does escitalopram). These SSRIs are usually second-line for starting a new antidepressant during pregnancy but could be first-line if they have worked well for a patient or she has had adverse effects with fluoxetine or sertraline.
You may need to increase SSRI dosages as pregnancy progresses. Increased metabolism and weight gain during pregnancy can lower SSRI serum levels, allowing depressive symptoms to re-emerge in the third trimester. Counsel the patient to continue taking the antidepressant for at least 12 months postpartum, then re-evaluate the need for medication based on her history.
Paroxetine precautions. If your patient is taking paroxetine and wishes to become pregnant, consider switching to another SSRI (using a slow cross-taper) unless paroxetine has been the only effective medication (Table 4). When discussing risks of any SSRI, explain that the baseline risk for congenital malformations is 3%. Paroxetine might increase this risk by 1% and other SSRIs by less.
If a woman becomes pregnant while taking paroxetine, often the time when cardiac defects occur is passed or will be before you slowly taper the medication to avoid withdrawal. If the patient’s depression has been severe, the risk of shifting her to an untested SSRI is probably higher than the possible 1% increased risk of fetal malformation. If she has taken paroxetine during the first-trimester, refer for ultrasound to monitor for cardiac anomalies.
Table 4
Recommendations for managing paroxetine risk during pregnancy
| Patient status | Recommendation |
|---|---|
| Taking paroxetine and planning pregnancy | Advise of possible 1% increase in risk of fetal malformation |
| Switch to another SSRI unless paroxetine has been the only successful therapy for depression | |
| If stopping paroxetine, slowly taper to avoid withdrawal symptoms | |
| Taking paroxetine and is pregnant | Advise of possible 1% increase in risk of fetal malformation |
| Continue paroxetine; a slow taper probably could not be completed before the first-trimester period associated with increased risk of fetal cardiac defects | |
| If any paroxetine exposure in first trimester, order ultrasound to monitor for fetal malformations |
- California Teratogen Information Service (CTIS). Pediatric department, University of California San Diego Medical Center. www.otispregnancy.org/ctis.html
- MGH Center for Women’s Mental Health, Massachusetts General Hospital. Psychiatric disorders during pregnancy and postpartum. www.womensmentalhealth.com
- MOTHERISK Web site. Teratogen information and updates on reproductive risk research. The Hospital for Sick Children, University of Toronto. www.motherisk.org
- Citalopram • Celexa
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Paroxetine • Paxil
- Sertraline • Zoloft
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Bonari L, Koren G, Einarson TR, et al. Use of antidepressants by pregnant women: evaluation of perception of risk, efficacy of evidence-based counseling, and determinants of decision making. Arch Women Ment Health 2005;8:214-20.
2. Hallberg P, Joblom V. The use of selective serotonin reuptake inhibitors during pregnancy and breast-feeding: a review and clinical aspects. J Clin Psychopharmacol 2005;25:59-73.
3. Larsson C, Sydsjo G, Josefsson A. Health, sociodemographic data, and pregnancy outcome in women with antepartum depressive symptoms. Obstet Gynecol 2004;104(3):469-66.
4. Bonari L, Pinto N, Ahn E, et al. Perinatal risks of untreated depression during pregnancy. Can J Psychiatry 2004;49(11):726-35.
5. Cohen L, Altshuler LL, Harlow BL, et al. Relapse of major depression during pregnancy in women who maintain or discontinue antidepressant treatment. JAMA 2006;295(5):499-507.
6. Hendrick V, Altshuler L. Management of major depression during pregnancy. Am J Psychiatry 2002;159:1667-73.
7. Sandman CA, Glynn L, Wadhwa PD, et al. Maternal hypothalamic-pituitary-adrenal dysregulation during the third trimester influences human fetal responses. Dev Neurosci 2003;25(1):41-9.
8. Huot RL, Brennan PA, Stowe ZN, et al. Negative affect in offspring of depressed mothers is predicted by infant cortisol levels at 6 months and maternal depression during pregnancy, but not postpartum. Ann NY Acad Sci 2004;1032:234-6.
9. Gutteling BM, de Weerth C, Buitelaar JK. Prenatal stress and children’s cortisol reaction to the first day of school. Psychoneuroendocrinology 2005;20:541-9.
10. Hendrick V, Stowe ZN, Altshuler LL, et al. Placental passage of antidepressant medications. Am J Psychiatry 2003;5:993-6.
11. GlaxoSmithKline study EPIP083. GSK medicine: buproprion and paroxetine. Epidemiology study: preliminary report on bupropion in pregnancy and the occurrence of cardiovascular and major congenital malformation. Available at: http://ctr.gsk.co.uk/summary/paroxetine/epip083.pdf. Accessed March 13, 2006.
12. Ericson A, Kallen B, Wiholm BE. Delivery outcome after the use of antidepressants in early pregnancy. Eur J Clin Pharmacol 1999;55:503-8.
13. Kulin NA, Pastuszak A, Sage S, et al. Pregnancy outcome following maternal use of the new selective serotonin reuptake inhibitors. A prospective controlled multicenter study. JAMA 1998;279:609-10.
14. Kallen BA, Otterblad Olausson P. Maternal drug use in early pregnancy and infant cardiovascular defect. Reprod Toxicol 2003;17:255-61.
15. Lattimore K, Donn S, Kaciroti N, et al. Selective serotonin reuptake inhibitor use during pregnancy and effects on the fetus and newborn: a meta-analysis. J Perinatol 2005;25:595-604.
16. Levy L, Ragan K, Hower-Hartley A, et al. Psychiatric disorders in pregnancy. Neurol Clin 2004;22:863-93.
17. Nordeng H, Spigset O. Treatment with selective serotonin reuptake inhibitors in the third trimester of pregnancy: effects on the infant. Drug Saf 2005;28(7):565-81.
18. Levinson-Castiel R, Merlob P, Linder N, et al. Neonatal abstinence syndrome after in utero exposure to selective serotonin reuptake inhibitors to term infants. Arch Pediatr Adolesc Med 2006;160:173-6.
19. Oberlander TF, Misri S, Fitzgerald CE, et al. Pharmacologic factors associated with transient neonatal symptoms following prenatal psychotropic medication exposure. J Clin Psychiatry 2004;65(2):230-7.
20. Zeskind PS, Stephens L. Maternal selective serotonin reuptake inhibitor use during pregnancy and newborn neurobehavior. Pediatrics 2004;113:368-75.
21. Moses-Kolko E, Bogen D, Perel J, et al. Neonatal signs after late in utero exposure to serotonin reuptake inhibitors. JAMA 2005;293:2372-83.
22. Chambers CD, Hernandez-Diaz S, Van Marter LJ, et al. Selective serotonin-reuptake inhibitors and risk of persistent pulmonary hypertension of the newborn. N Engl J Med 2006;354(6):579-87.
23. Nulman I, Rovet J, Stewart DE, et al. Child development following exposure to tricyclic antidepressants or fluoxetine throughout fetal life: a prospective, controlled study. Am J Psychiatry 2002;159:1889-95.
24. Casper RC, Fleisher BE, Lee-Ancajas JC, et al. Follow-up of children of depressed mothers exposed or not exposed to antidepressant drugs during pregnancy. J Pediatr 2003;4(142):402-8.
25. Spinelli M, Endicott J. Controlled clinical trial of interpersonal psychotherapy versus parenting education program for depressed pregnant women. Am J Psychiatry 2003;160:555-62.
26. Oren D, Wisner K, Spinelli M, et al. An open trial of morning light therapy for treatment of antepartum depression. Am J Psychiatry 2002;159:666-9.
Untreated depression can have serious consequences, but many pregnant women resist taking antidepressants because they overestimate the risk of birth defects.Paroxetine in pregnancy”). Further study is needed to define the risks of teratogenesis with paroxetine compared with other antidepressants.
Third-trimester exposure
In a recent meta-analysis, infants exposed to SSRIs in utero showed an increased risk for prematurity (OR; 2.03) and low birth weight (OR; 2.37).15 Other studies, however, showed no differences in these risks in SSRI-exposed infants or attributed the results to untreated maternal depression or smoking.16
A Medline search across the last 20 years17 found 26 case reports, three prospective controlled cohort studies, and other records of >400 women who received fluoxetine, sertraline, or paroxetine in the third trimester. The authors found the evidence “ambiguous” as to the cause of adverse events and concluded that the risk of not treating major depression with adequate SSRI therapy at that stage of pregnancy “most likely” outweighs the risk of harm to infants.
Transient neonatal complications. Thirty percent of neonates exposed to SSRIs in the third trimester experience transient adaptation problems, which peak 48 hours after birth18 (Table 3). Symptoms may include initial lack of crying, increased muscle tonus, flush, irritability, jitteriness, hypothermia, abnormal breathing, and disrupted sleep and motor activity.2,19,20
Transient neonatal symptoms from SSRI exposure are thought to be a serotonin withdrawal syndrome or serotonin overstimulation.21 The syndrome is usually mild, self-limited, and requires only supportive treatments. All antidepressants’ labels warn of these effects.
Table 3
Neonatal SSRI withdrawal: Symptoms, causes, and treatment
| Symptoms | Initial lack of crying |
| Increased muscle tonus | |
| Irritability, jitteriness | |
| Abnormal breathing pattern | |
| Disrupted sleep and motor activity | |
| Hypotheses of cause | Serotonin overstimulation or withdrawal |
| Treatment | Close observation |
| Supportive measures |
Recommendation. Some authors have recommended tapering antidepressants in the third trimester, but the risk of postpartum depression appears to outweigh any potential benefit from discontinuation. Because birth timing is unpredictable, some women whose antidepressants are tapered off could be without medication for a long time.
Thus, we recommend:
- continuing SSRIs during late pregnancy
- monitoring the newborn for 48 hours for transient neonatal adaptation symptoms or PPHN.2,17,18
Long-term effects of SSRI exposure
Do SSRIs during pregnancy have long-term effects on infants’ neurodevelopment? Study results are mixed. For example:
- A prospective, controlled, cohort trial found no adverse effects on IQ, language, or behavioral development in children ages 15 months to 6 years whose mothers took tricyclic antidepressants (N=46) or fluoxetine (N=40) during pregnancy, compared with 36 unexposed controls.23
- Another prospective study showed lower Bayley Psychomotor Developmental Index scores in 31 SSRI-exposed infants compared with 13 infants born to depressed mothers not on antidepressants. Reduced body control, coordination, and fine motor skills might suggest possible subtle effects of SSRIs on motor development in exposed infants, the authors concluded.24
Case continued: A healthy delivery
Ms. P’s depression improves a few weeks after she restarts an SSRI. She delivers a healthy term baby with Apgar score of 7. The baby initially does not cry, awakens easily, and shows mild irritability. His mother’s SSRI use, her severe depression during part of the pregnancy, or some other factor may have caused his mild neonatal complications.
Nursing staff carefully observe the infant for 2 days in the newborn nursery, and his irritability fades away. Ms. P decides to continue taking antidepressants to care for herself and the baby.
Weighing treatment options
For each woman with a history of depression who is pregnant or intends to conceive, we recommend a risk-benefit analysis of her depression severity and need for an antidepressant:
Moderate to severe depression (history of recurrent depressive episodes, hospitalization, or suicidality). Strongly consider medication. If your patient is taking an SSRI, counsel her about:
- the 70% risk of depression relapse if she stops the medication, even for the first trimester
- risks of untreated depression during pregnancy (poor self-care, preterm labor, birth complications, and increased risk for poor stress adaptations in children).
Choosing an SSRI. No one SSRI is the safest choice for all women, especially when data on breast-feeding come into play.
- Fluoxetine has been studied more than other SSRIs during pregnancy; most evidence is reassuring, except for transient neonatal complications. With its long half-life, fluoxetine is not recommended during breastfeeding because it may accumulate in infant sera.
- Sertraline has shown low umbilical cord to maternal serum ratios in small samples and has reassuring breast-feeding data.
- Citalopram, compared with sertraline, has been studied more in pregnancy but has a higher fetal-to-maternal serum ratio (as does escitalopram). These SSRIs are usually second-line for starting a new antidepressant during pregnancy but could be first-line if they have worked well for a patient or she has had adverse effects with fluoxetine or sertraline.
You may need to increase SSRI dosages as pregnancy progresses. Increased metabolism and weight gain during pregnancy can lower SSRI serum levels, allowing depressive symptoms to re-emerge in the third trimester. Counsel the patient to continue taking the antidepressant for at least 12 months postpartum, then re-evaluate the need for medication based on her history.
Paroxetine precautions. If your patient is taking paroxetine and wishes to become pregnant, consider switching to another SSRI (using a slow cross-taper) unless paroxetine has been the only effective medication (Table 4). When discussing risks of any SSRI, explain that the baseline risk for congenital malformations is 3%. Paroxetine might increase this risk by 1% and other SSRIs by less.
If a woman becomes pregnant while taking paroxetine, often the time when cardiac defects occur is passed or will be before you slowly taper the medication to avoid withdrawal. If the patient’s depression has been severe, the risk of shifting her to an untested SSRI is probably higher than the possible 1% increased risk of fetal malformation. If she has taken paroxetine during the first-trimester, refer for ultrasound to monitor for cardiac anomalies.
Table 4
Recommendations for managing paroxetine risk during pregnancy
| Patient status | Recommendation |
|---|---|
| Taking paroxetine and planning pregnancy | Advise of possible 1% increase in risk of fetal malformation |
| Switch to another SSRI unless paroxetine has been the only successful therapy for depression | |
| If stopping paroxetine, slowly taper to avoid withdrawal symptoms | |
| Taking paroxetine and is pregnant | Advise of possible 1% increase in risk of fetal malformation |
| Continue paroxetine; a slow taper probably could not be completed before the first-trimester period associated with increased risk of fetal cardiac defects | |
| If any paroxetine exposure in first trimester, order ultrasound to monitor for fetal malformations |
- California Teratogen Information Service (CTIS). Pediatric department, University of California San Diego Medical Center. www.otispregnancy.org/ctis.html
- MGH Center for Women’s Mental Health, Massachusetts General Hospital. Psychiatric disorders during pregnancy and postpartum. www.womensmentalhealth.com
- MOTHERISK Web site. Teratogen information and updates on reproductive risk research. The Hospital for Sick Children, University of Toronto. www.motherisk.org
- Citalopram • Celexa
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Paroxetine • Paxil
- Sertraline • Zoloft
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Untreated depression can have serious consequences, but many pregnant women resist taking antidepressants because they overestimate the risk of birth defects.Paroxetine in pregnancy”). Further study is needed to define the risks of teratogenesis with paroxetine compared with other antidepressants.
Third-trimester exposure
In a recent meta-analysis, infants exposed to SSRIs in utero showed an increased risk for prematurity (OR; 2.03) and low birth weight (OR; 2.37).15 Other studies, however, showed no differences in these risks in SSRI-exposed infants or attributed the results to untreated maternal depression or smoking.16
A Medline search across the last 20 years17 found 26 case reports, three prospective controlled cohort studies, and other records of >400 women who received fluoxetine, sertraline, or paroxetine in the third trimester. The authors found the evidence “ambiguous” as to the cause of adverse events and concluded that the risk of not treating major depression with adequate SSRI therapy at that stage of pregnancy “most likely” outweighs the risk of harm to infants.
Transient neonatal complications. Thirty percent of neonates exposed to SSRIs in the third trimester experience transient adaptation problems, which peak 48 hours after birth18 (Table 3). Symptoms may include initial lack of crying, increased muscle tonus, flush, irritability, jitteriness, hypothermia, abnormal breathing, and disrupted sleep and motor activity.2,19,20
Transient neonatal symptoms from SSRI exposure are thought to be a serotonin withdrawal syndrome or serotonin overstimulation.21 The syndrome is usually mild, self-limited, and requires only supportive treatments. All antidepressants’ labels warn of these effects.
Table 3
Neonatal SSRI withdrawal: Symptoms, causes, and treatment
| Symptoms | Initial lack of crying |
| Increased muscle tonus | |
| Irritability, jitteriness | |
| Abnormal breathing pattern | |
| Disrupted sleep and motor activity | |
| Hypotheses of cause | Serotonin overstimulation or withdrawal |
| Treatment | Close observation |
| Supportive measures |
Recommendation. Some authors have recommended tapering antidepressants in the third trimester, but the risk of postpartum depression appears to outweigh any potential benefit from discontinuation. Because birth timing is unpredictable, some women whose antidepressants are tapered off could be without medication for a long time.
Thus, we recommend:
- continuing SSRIs during late pregnancy
- monitoring the newborn for 48 hours for transient neonatal adaptation symptoms or PPHN.2,17,18
Long-term effects of SSRI exposure
Do SSRIs during pregnancy have long-term effects on infants’ neurodevelopment? Study results are mixed. For example:
- A prospective, controlled, cohort trial found no adverse effects on IQ, language, or behavioral development in children ages 15 months to 6 years whose mothers took tricyclic antidepressants (N=46) or fluoxetine (N=40) during pregnancy, compared with 36 unexposed controls.23
- Another prospective study showed lower Bayley Psychomotor Developmental Index scores in 31 SSRI-exposed infants compared with 13 infants born to depressed mothers not on antidepressants. Reduced body control, coordination, and fine motor skills might suggest possible subtle effects of SSRIs on motor development in exposed infants, the authors concluded.24
Case continued: A healthy delivery
Ms. P’s depression improves a few weeks after she restarts an SSRI. She delivers a healthy term baby with Apgar score of 7. The baby initially does not cry, awakens easily, and shows mild irritability. His mother’s SSRI use, her severe depression during part of the pregnancy, or some other factor may have caused his mild neonatal complications.
Nursing staff carefully observe the infant for 2 days in the newborn nursery, and his irritability fades away. Ms. P decides to continue taking antidepressants to care for herself and the baby.
Weighing treatment options
For each woman with a history of depression who is pregnant or intends to conceive, we recommend a risk-benefit analysis of her depression severity and need for an antidepressant:
Moderate to severe depression (history of recurrent depressive episodes, hospitalization, or suicidality). Strongly consider medication. If your patient is taking an SSRI, counsel her about:
- the 70% risk of depression relapse if she stops the medication, even for the first trimester
- risks of untreated depression during pregnancy (poor self-care, preterm labor, birth complications, and increased risk for poor stress adaptations in children).
Choosing an SSRI. No one SSRI is the safest choice for all women, especially when data on breast-feeding come into play.
- Fluoxetine has been studied more than other SSRIs during pregnancy; most evidence is reassuring, except for transient neonatal complications. With its long half-life, fluoxetine is not recommended during breastfeeding because it may accumulate in infant sera.
- Sertraline has shown low umbilical cord to maternal serum ratios in small samples and has reassuring breast-feeding data.
- Citalopram, compared with sertraline, has been studied more in pregnancy but has a higher fetal-to-maternal serum ratio (as does escitalopram). These SSRIs are usually second-line for starting a new antidepressant during pregnancy but could be first-line if they have worked well for a patient or she has had adverse effects with fluoxetine or sertraline.
You may need to increase SSRI dosages as pregnancy progresses. Increased metabolism and weight gain during pregnancy can lower SSRI serum levels, allowing depressive symptoms to re-emerge in the third trimester. Counsel the patient to continue taking the antidepressant for at least 12 months postpartum, then re-evaluate the need for medication based on her history.
Paroxetine precautions. If your patient is taking paroxetine and wishes to become pregnant, consider switching to another SSRI (using a slow cross-taper) unless paroxetine has been the only effective medication (Table 4). When discussing risks of any SSRI, explain that the baseline risk for congenital malformations is 3%. Paroxetine might increase this risk by 1% and other SSRIs by less.
If a woman becomes pregnant while taking paroxetine, often the time when cardiac defects occur is passed or will be before you slowly taper the medication to avoid withdrawal. If the patient’s depression has been severe, the risk of shifting her to an untested SSRI is probably higher than the possible 1% increased risk of fetal malformation. If she has taken paroxetine during the first-trimester, refer for ultrasound to monitor for cardiac anomalies.
Table 4
Recommendations for managing paroxetine risk during pregnancy
| Patient status | Recommendation |
|---|---|
| Taking paroxetine and planning pregnancy | Advise of possible 1% increase in risk of fetal malformation |
| Switch to another SSRI unless paroxetine has been the only successful therapy for depression | |
| If stopping paroxetine, slowly taper to avoid withdrawal symptoms | |
| Taking paroxetine and is pregnant | Advise of possible 1% increase in risk of fetal malformation |
| Continue paroxetine; a slow taper probably could not be completed before the first-trimester period associated with increased risk of fetal cardiac defects | |
| If any paroxetine exposure in first trimester, order ultrasound to monitor for fetal malformations |
- California Teratogen Information Service (CTIS). Pediatric department, University of California San Diego Medical Center. www.otispregnancy.org/ctis.html
- MGH Center for Women’s Mental Health, Massachusetts General Hospital. Psychiatric disorders during pregnancy and postpartum. www.womensmentalhealth.com
- MOTHERISK Web site. Teratogen information and updates on reproductive risk research. The Hospital for Sick Children, University of Toronto. www.motherisk.org
- Citalopram • Celexa
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Paroxetine • Paxil
- Sertraline • Zoloft
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Bonari L, Koren G, Einarson TR, et al. Use of antidepressants by pregnant women: evaluation of perception of risk, efficacy of evidence-based counseling, and determinants of decision making. Arch Women Ment Health 2005;8:214-20.
2. Hallberg P, Joblom V. The use of selective serotonin reuptake inhibitors during pregnancy and breast-feeding: a review and clinical aspects. J Clin Psychopharmacol 2005;25:59-73.
3. Larsson C, Sydsjo G, Josefsson A. Health, sociodemographic data, and pregnancy outcome in women with antepartum depressive symptoms. Obstet Gynecol 2004;104(3):469-66.
4. Bonari L, Pinto N, Ahn E, et al. Perinatal risks of untreated depression during pregnancy. Can J Psychiatry 2004;49(11):726-35.
5. Cohen L, Altshuler LL, Harlow BL, et al. Relapse of major depression during pregnancy in women who maintain or discontinue antidepressant treatment. JAMA 2006;295(5):499-507.
6. Hendrick V, Altshuler L. Management of major depression during pregnancy. Am J Psychiatry 2002;159:1667-73.
7. Sandman CA, Glynn L, Wadhwa PD, et al. Maternal hypothalamic-pituitary-adrenal dysregulation during the third trimester influences human fetal responses. Dev Neurosci 2003;25(1):41-9.
8. Huot RL, Brennan PA, Stowe ZN, et al. Negative affect in offspring of depressed mothers is predicted by infant cortisol levels at 6 months and maternal depression during pregnancy, but not postpartum. Ann NY Acad Sci 2004;1032:234-6.
9. Gutteling BM, de Weerth C, Buitelaar JK. Prenatal stress and children’s cortisol reaction to the first day of school. Psychoneuroendocrinology 2005;20:541-9.
10. Hendrick V, Stowe ZN, Altshuler LL, et al. Placental passage of antidepressant medications. Am J Psychiatry 2003;5:993-6.
11. GlaxoSmithKline study EPIP083. GSK medicine: buproprion and paroxetine. Epidemiology study: preliminary report on bupropion in pregnancy and the occurrence of cardiovascular and major congenital malformation. Available at: http://ctr.gsk.co.uk/summary/paroxetine/epip083.pdf. Accessed March 13, 2006.
12. Ericson A, Kallen B, Wiholm BE. Delivery outcome after the use of antidepressants in early pregnancy. Eur J Clin Pharmacol 1999;55:503-8.
13. Kulin NA, Pastuszak A, Sage S, et al. Pregnancy outcome following maternal use of the new selective serotonin reuptake inhibitors. A prospective controlled multicenter study. JAMA 1998;279:609-10.
14. Kallen BA, Otterblad Olausson P. Maternal drug use in early pregnancy and infant cardiovascular defect. Reprod Toxicol 2003;17:255-61.
15. Lattimore K, Donn S, Kaciroti N, et al. Selective serotonin reuptake inhibitor use during pregnancy and effects on the fetus and newborn: a meta-analysis. J Perinatol 2005;25:595-604.
16. Levy L, Ragan K, Hower-Hartley A, et al. Psychiatric disorders in pregnancy. Neurol Clin 2004;22:863-93.
17. Nordeng H, Spigset O. Treatment with selective serotonin reuptake inhibitors in the third trimester of pregnancy: effects on the infant. Drug Saf 2005;28(7):565-81.
18. Levinson-Castiel R, Merlob P, Linder N, et al. Neonatal abstinence syndrome after in utero exposure to selective serotonin reuptake inhibitors to term infants. Arch Pediatr Adolesc Med 2006;160:173-6.
19. Oberlander TF, Misri S, Fitzgerald CE, et al. Pharmacologic factors associated with transient neonatal symptoms following prenatal psychotropic medication exposure. J Clin Psychiatry 2004;65(2):230-7.
20. Zeskind PS, Stephens L. Maternal selective serotonin reuptake inhibitor use during pregnancy and newborn neurobehavior. Pediatrics 2004;113:368-75.
21. Moses-Kolko E, Bogen D, Perel J, et al. Neonatal signs after late in utero exposure to serotonin reuptake inhibitors. JAMA 2005;293:2372-83.
22. Chambers CD, Hernandez-Diaz S, Van Marter LJ, et al. Selective serotonin-reuptake inhibitors and risk of persistent pulmonary hypertension of the newborn. N Engl J Med 2006;354(6):579-87.
23. Nulman I, Rovet J, Stewart DE, et al. Child development following exposure to tricyclic antidepressants or fluoxetine throughout fetal life: a prospective, controlled study. Am J Psychiatry 2002;159:1889-95.
24. Casper RC, Fleisher BE, Lee-Ancajas JC, et al. Follow-up of children of depressed mothers exposed or not exposed to antidepressant drugs during pregnancy. J Pediatr 2003;4(142):402-8.
25. Spinelli M, Endicott J. Controlled clinical trial of interpersonal psychotherapy versus parenting education program for depressed pregnant women. Am J Psychiatry 2003;160:555-62.
26. Oren D, Wisner K, Spinelli M, et al. An open trial of morning light therapy for treatment of antepartum depression. Am J Psychiatry 2002;159:666-9.
1. Bonari L, Koren G, Einarson TR, et al. Use of antidepressants by pregnant women: evaluation of perception of risk, efficacy of evidence-based counseling, and determinants of decision making. Arch Women Ment Health 2005;8:214-20.
2. Hallberg P, Joblom V. The use of selective serotonin reuptake inhibitors during pregnancy and breast-feeding: a review and clinical aspects. J Clin Psychopharmacol 2005;25:59-73.
3. Larsson C, Sydsjo G, Josefsson A. Health, sociodemographic data, and pregnancy outcome in women with antepartum depressive symptoms. Obstet Gynecol 2004;104(3):469-66.
4. Bonari L, Pinto N, Ahn E, et al. Perinatal risks of untreated depression during pregnancy. Can J Psychiatry 2004;49(11):726-35.
5. Cohen L, Altshuler LL, Harlow BL, et al. Relapse of major depression during pregnancy in women who maintain or discontinue antidepressant treatment. JAMA 2006;295(5):499-507.
6. Hendrick V, Altshuler L. Management of major depression during pregnancy. Am J Psychiatry 2002;159:1667-73.
7. Sandman CA, Glynn L, Wadhwa PD, et al. Maternal hypothalamic-pituitary-adrenal dysregulation during the third trimester influences human fetal responses. Dev Neurosci 2003;25(1):41-9.
8. Huot RL, Brennan PA, Stowe ZN, et al. Negative affect in offspring of depressed mothers is predicted by infant cortisol levels at 6 months and maternal depression during pregnancy, but not postpartum. Ann NY Acad Sci 2004;1032:234-6.
9. Gutteling BM, de Weerth C, Buitelaar JK. Prenatal stress and children’s cortisol reaction to the first day of school. Psychoneuroendocrinology 2005;20:541-9.
10. Hendrick V, Stowe ZN, Altshuler LL, et al. Placental passage of antidepressant medications. Am J Psychiatry 2003;5:993-6.
11. GlaxoSmithKline study EPIP083. GSK medicine: buproprion and paroxetine. Epidemiology study: preliminary report on bupropion in pregnancy and the occurrence of cardiovascular and major congenital malformation. Available at: http://ctr.gsk.co.uk/summary/paroxetine/epip083.pdf. Accessed March 13, 2006.
12. Ericson A, Kallen B, Wiholm BE. Delivery outcome after the use of antidepressants in early pregnancy. Eur J Clin Pharmacol 1999;55:503-8.
13. Kulin NA, Pastuszak A, Sage S, et al. Pregnancy outcome following maternal use of the new selective serotonin reuptake inhibitors. A prospective controlled multicenter study. JAMA 1998;279:609-10.
14. Kallen BA, Otterblad Olausson P. Maternal drug use in early pregnancy and infant cardiovascular defect. Reprod Toxicol 2003;17:255-61.
15. Lattimore K, Donn S, Kaciroti N, et al. Selective serotonin reuptake inhibitor use during pregnancy and effects on the fetus and newborn: a meta-analysis. J Perinatol 2005;25:595-604.
16. Levy L, Ragan K, Hower-Hartley A, et al. Psychiatric disorders in pregnancy. Neurol Clin 2004;22:863-93.
17. Nordeng H, Spigset O. Treatment with selective serotonin reuptake inhibitors in the third trimester of pregnancy: effects on the infant. Drug Saf 2005;28(7):565-81.
18. Levinson-Castiel R, Merlob P, Linder N, et al. Neonatal abstinence syndrome after in utero exposure to selective serotonin reuptake inhibitors to term infants. Arch Pediatr Adolesc Med 2006;160:173-6.
19. Oberlander TF, Misri S, Fitzgerald CE, et al. Pharmacologic factors associated with transient neonatal symptoms following prenatal psychotropic medication exposure. J Clin Psychiatry 2004;65(2):230-7.
20. Zeskind PS, Stephens L. Maternal selective serotonin reuptake inhibitor use during pregnancy and newborn neurobehavior. Pediatrics 2004;113:368-75.
21. Moses-Kolko E, Bogen D, Perel J, et al. Neonatal signs after late in utero exposure to serotonin reuptake inhibitors. JAMA 2005;293:2372-83.
22. Chambers CD, Hernandez-Diaz S, Van Marter LJ, et al. Selective serotonin-reuptake inhibitors and risk of persistent pulmonary hypertension of the newborn. N Engl J Med 2006;354(6):579-87.
23. Nulman I, Rovet J, Stewart DE, et al. Child development following exposure to tricyclic antidepressants or fluoxetine throughout fetal life: a prospective, controlled study. Am J Psychiatry 2002;159:1889-95.
24. Casper RC, Fleisher BE, Lee-Ancajas JC, et al. Follow-up of children of depressed mothers exposed or not exposed to antidepressant drugs during pregnancy. J Pediatr 2003;4(142):402-8.
25. Spinelli M, Endicott J. Controlled clinical trial of interpersonal psychotherapy versus parenting education program for depressed pregnant women. Am J Psychiatry 2003;160:555-62.
26. Oren D, Wisner K, Spinelli M, et al. An open trial of morning light therapy for treatment of antepartum depression. Am J Psychiatry 2002;159:666-9.