User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
Combo treatment may improve quality of life in CTCL
LA JOLLA, CALIF. — Treatment with brentuximab vedotin (BV) and lenalidomide (len) may improve quality of life (QOL) for patients with cutaneous T-cell lymphoma (CTCL), according to the principal investigator of a phase 2 trial.

In this small trial, most CTCL patients experienced relief from pruritus after one cycle of treatment with BV-len.
Investigators also observed durable responses to the combination, although two patients experienced tumor flare prior to response.
“Because of the tumor flare, we decreased the dose of lenalidomide ... and, since then, it has not been a major problem,” said Basem M. William, MD, principal investigator of the trial and a professor at Ohio State University in Columbus.
“We’re trying to be more reassuring to patients that, if they experience a little bit of tumor flare, as long as it’s not dangerous or life-threatening, if they can hold on with the treatment, this might translate to a later durable response.”
Dr. William and his colleagues presented results from this ongoing, phase 2 trial (NCT03409432) at the annual T-cell Lymphoma Forum.
Thus far, the investigators have treated 12 patients with relapsed or refractory CTCL or peripheral T-cell lymphoma (PTCL). The CTCL patients had received at least two lines of skin-directed therapy or one line of systemic therapy, and the PTCL patients had received at least one line of systemic therapy.
Dr. William and his colleagues reported results for 10 patients. Six patients had mycosis fungoides (MF), two had Sézary syndrome (SS), one had CD30+ lymphoproliferative disorder, and one had systemic anaplastic large-cell lymphoma (ALCL).
The patients’ median age was 59 (range, 49-74), there were nine males, and patients had received a median of 2 (range, 1-10) prior therapies.
The first seven patients received BV at 1.2 mg/kg and len at 20 mg daily every 3 weeks. However, after the investigators observed tumor flare in two patients, the dose of len was lowered to 10 mg.
Safety
The investigators said all adverse events (AEs) were reversible by stopping therapy, there were no grade 4 AEs, and none of the patients had grade 3 or higher neuropathy.
“We have not seen an excess of neuropathy, which is very important because both brentuximab and lenalidomide are known to cause neuropathy,” Dr. William said. “So we were fairly concerned that there would be a synergistic neurotoxic effect, which we don’t want, but we haven’t seen that.”
The most common treatment-related AE was neutropenia. Grade 3 neutropenia occurred in four patients.
Other grade 3 AEs, which occurred in patients on the 20 mg dose of len, were thrombocytopenia (n = 1), dyspnea (n = 1), vertigo (n = 1), drug rash with eosinophilia and systemic symptoms (DRESS) syndrome (n = 1), and tumor flare (n = 1).
Three patients discontinued treatment because of AEs — thrombocytopenia, tumor flare, and DRESS syndrome.
Tumor flare and response
“We did see tumor flare in two initial patients treated with the higher dose of lenalidomide, and we had to remove them from the study for their safety,” Dr. William said. “One of them had a full-blown DRESS syndrome. For their safety, we did have to remove them, but both did experience durable remissions after.”
One of the patients with tumor flare, who had MF, didn’t require treatment for 6 months after going off study. The other patient, who had SS, cleared the clone from his blood but developed DRESS syndrome.
In all, three patients achieved a response to treatment. The ALCL patient had a complete response, and two MF patients achieved a partial response.
Two MF patients and one SS patient had stable disease. The remaining four patients — two with MF, one with SS, and one with lymphoproliferative disorder — progressed.
QOL
The investigators used the Skindex-16 to assess the effect of treatment on QOL.
Five of six evaluable patients with CTCL had a 50% or greater reduction in their Skindex-16 scores after two cycles of treatment. In fact, most patients had relief from pruritus after one cycle, Dr. William said.
“Patients with cutaneous T-cell lymphoma, their biggest problem is with the symptom burden, with pruritus,” he said. “They’re really miserable from all the itching they have. They cannot sleep at night. So we’re fairly excited that most of the patients we’ve treated so far had relief from pruritus just after one cycle.”
Dr. William said he and his colleagues are excited about the overall results they have observed with BV-len, although it’s “still pretty early” in the trial. The investigators are planning to enroll a total of 42 patients and may open the trial at a second center.
The study is sponsored by Ohio State University and the lenalidomide is provided by Celgene. Dr. William reported relationships with miRagen Therapeutics, GuidePoint, Kyowa Kirin, and Celgene.
The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
LA JOLLA, CALIF. — Treatment with brentuximab vedotin (BV) and lenalidomide (len) may improve quality of life (QOL) for patients with cutaneous T-cell lymphoma (CTCL), according to the principal investigator of a phase 2 trial.
In this small trial, most CTCL patients experienced relief from pruritus after one cycle of treatment with BV-len.
Investigators also observed durable responses to the combination, although two patients experienced tumor flare prior to response.
“Because of the tumor flare, we decreased the dose of lenalidomide ... and, since then, it has not been a major problem,” said Basem M. William, MD, principal investigator of the trial and a professor at Ohio State University in Columbus.
“We’re trying to be more reassuring to patients that, if they experience a little bit of tumor flare, as long as it’s not dangerous or life-threatening, if they can hold on with the treatment, this might translate to a later durable response.”
Dr. William and his colleagues presented results from this ongoing, phase 2 trial (NCT03409432) at the annual T-cell Lymphoma Forum.
Thus far, the investigators have treated 12 patients with relapsed or refractory CTCL or peripheral T-cell lymphoma (PTCL). The CTCL patients had received at least two lines of skin-directed therapy or one line of systemic therapy, and the PTCL patients had received at least one line of systemic therapy.
Dr. William and his colleagues reported results for 10 patients. Six patients had mycosis fungoides (MF), two had Sézary syndrome (SS), one had CD30+ lymphoproliferative disorder, and one had systemic anaplastic large-cell lymphoma (ALCL).
The patients’ median age was 59 (range, 49-74), there were nine males, and patients had received a median of 2 (range, 1-10) prior therapies.
The first seven patients received BV at 1.2 mg/kg and len at 20 mg daily every 3 weeks. However, after the investigators observed tumor flare in two patients, the dose of len was lowered to 10 mg.
Safety
The investigators said all adverse events (AEs) were reversible by stopping therapy, there were no grade 4 AEs, and none of the patients had grade 3 or higher neuropathy.
“We have not seen an excess of neuropathy, which is very important because both brentuximab and lenalidomide are known to cause neuropathy,” Dr. William said. “So we were fairly concerned that there would be a synergistic neurotoxic effect, which we don’t want, but we haven’t seen that.”
The most common treatment-related AE was neutropenia. Grade 3 neutropenia occurred in four patients.
Other grade 3 AEs, which occurred in patients on the 20 mg dose of len, were thrombocytopenia (n = 1), dyspnea (n = 1), vertigo (n = 1), drug rash with eosinophilia and systemic symptoms (DRESS) syndrome (n = 1), and tumor flare (n = 1).
Three patients discontinued treatment because of AEs — thrombocytopenia, tumor flare, and DRESS syndrome.
Tumor flare and response
“We did see tumor flare in two initial patients treated with the higher dose of lenalidomide, and we had to remove them from the study for their safety,” Dr. William said. “One of them had a full-blown DRESS syndrome. For their safety, we did have to remove them, but both did experience durable remissions after.”
One of the patients with tumor flare, who had MF, didn’t require treatment for 6 months after going off study. The other patient, who had SS, cleared the clone from his blood but developed DRESS syndrome.
In all, three patients achieved a response to treatment. The ALCL patient had a complete response, and two MF patients achieved a partial response.
Two MF patients and one SS patient had stable disease. The remaining four patients — two with MF, one with SS, and one with lymphoproliferative disorder — progressed.
QOL
The investigators used the Skindex-16 to assess the effect of treatment on QOL.
Five of six evaluable patients with CTCL had a 50% or greater reduction in their Skindex-16 scores after two cycles of treatment. In fact, most patients had relief from pruritus after one cycle, Dr. William said.
“Patients with cutaneous T-cell lymphoma, their biggest problem is with the symptom burden, with pruritus,” he said. “They’re really miserable from all the itching they have. They cannot sleep at night. So we’re fairly excited that most of the patients we’ve treated so far had relief from pruritus just after one cycle.”
Dr. William said he and his colleagues are excited about the overall results they have observed with BV-len, although it’s “still pretty early” in the trial. The investigators are planning to enroll a total of 42 patients and may open the trial at a second center.
The study is sponsored by Ohio State University and the lenalidomide is provided by Celgene. Dr. William reported relationships with miRagen Therapeutics, GuidePoint, Kyowa Kirin, and Celgene.
The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
LA JOLLA, CALIF. — Treatment with brentuximab vedotin (BV) and lenalidomide (len) may improve quality of life (QOL) for patients with cutaneous T-cell lymphoma (CTCL), according to the principal investigator of a phase 2 trial.
In this small trial, most CTCL patients experienced relief from pruritus after one cycle of treatment with BV-len.
Investigators also observed durable responses to the combination, although two patients experienced tumor flare prior to response.
“Because of the tumor flare, we decreased the dose of lenalidomide ... and, since then, it has not been a major problem,” said Basem M. William, MD, principal investigator of the trial and a professor at Ohio State University in Columbus.
“We’re trying to be more reassuring to patients that, if they experience a little bit of tumor flare, as long as it’s not dangerous or life-threatening, if they can hold on with the treatment, this might translate to a later durable response.”
Dr. William and his colleagues presented results from this ongoing, phase 2 trial (NCT03409432) at the annual T-cell Lymphoma Forum.
Thus far, the investigators have treated 12 patients with relapsed or refractory CTCL or peripheral T-cell lymphoma (PTCL). The CTCL patients had received at least two lines of skin-directed therapy or one line of systemic therapy, and the PTCL patients had received at least one line of systemic therapy.
Dr. William and his colleagues reported results for 10 patients. Six patients had mycosis fungoides (MF), two had Sézary syndrome (SS), one had CD30+ lymphoproliferative disorder, and one had systemic anaplastic large-cell lymphoma (ALCL).
The patients’ median age was 59 (range, 49-74), there were nine males, and patients had received a median of 2 (range, 1-10) prior therapies.
The first seven patients received BV at 1.2 mg/kg and len at 20 mg daily every 3 weeks. However, after the investigators observed tumor flare in two patients, the dose of len was lowered to 10 mg.
Safety
The investigators said all adverse events (AEs) were reversible by stopping therapy, there were no grade 4 AEs, and none of the patients had grade 3 or higher neuropathy.
“We have not seen an excess of neuropathy, which is very important because both brentuximab and lenalidomide are known to cause neuropathy,” Dr. William said. “So we were fairly concerned that there would be a synergistic neurotoxic effect, which we don’t want, but we haven’t seen that.”
The most common treatment-related AE was neutropenia. Grade 3 neutropenia occurred in four patients.
Other grade 3 AEs, which occurred in patients on the 20 mg dose of len, were thrombocytopenia (n = 1), dyspnea (n = 1), vertigo (n = 1), drug rash with eosinophilia and systemic symptoms (DRESS) syndrome (n = 1), and tumor flare (n = 1).
Three patients discontinued treatment because of AEs — thrombocytopenia, tumor flare, and DRESS syndrome.
Tumor flare and response
“We did see tumor flare in two initial patients treated with the higher dose of lenalidomide, and we had to remove them from the study for their safety,” Dr. William said. “One of them had a full-blown DRESS syndrome. For their safety, we did have to remove them, but both did experience durable remissions after.”
One of the patients with tumor flare, who had MF, didn’t require treatment for 6 months after going off study. The other patient, who had SS, cleared the clone from his blood but developed DRESS syndrome.
In all, three patients achieved a response to treatment. The ALCL patient had a complete response, and two MF patients achieved a partial response.
Two MF patients and one SS patient had stable disease. The remaining four patients — two with MF, one with SS, and one with lymphoproliferative disorder — progressed.
QOL
The investigators used the Skindex-16 to assess the effect of treatment on QOL.
Five of six evaluable patients with CTCL had a 50% or greater reduction in their Skindex-16 scores after two cycles of treatment. In fact, most patients had relief from pruritus after one cycle, Dr. William said.
“Patients with cutaneous T-cell lymphoma, their biggest problem is with the symptom burden, with pruritus,” he said. “They’re really miserable from all the itching they have. They cannot sleep at night. So we’re fairly excited that most of the patients we’ve treated so far had relief from pruritus just after one cycle.”
Dr. William said he and his colleagues are excited about the overall results they have observed with BV-len, although it’s “still pretty early” in the trial. The investigators are planning to enroll a total of 42 patients and may open the trial at a second center.
The study is sponsored by Ohio State University and the lenalidomide is provided by Celgene. Dr. William reported relationships with miRagen Therapeutics, GuidePoint, Kyowa Kirin, and Celgene.
The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
REPORTING FROM TCLF 2019
Key clinical point:
Major finding: Five of six evaluable CTCL patients had a 50% or greater reduction in their Skindex-16 scores after two cycles of treatment.
Study details: A phase 2 study with results reported for 10 patients.
Disclosures: The study is sponsored by Ohio State University and the lenalidomide is provided by Celgene. The principal investigator reported relationships with miRagen Therapeutics, GuidePoint, Kyowa Kirin, and Celgene.

Zanubrutinib receives breakthrough designation for MCL
The (MCL) who have received at least one prior therapy.

Zanubrutinib (BGB-3111) is a Bruton’s tyrosine kinase inhibitor being developed by BeiGene as a potential treatment for B-cell malignancies.
Researchers have evaluated zanubrutinib in a phase 2 trial (NCT03206970) of patients with relapsed/refractory MCL. Results from this trial were presented at the 2018 annual meeting of the American Society of Hematology (Abstract 148).
As of March 27, 2018, 86 patients had been enrolled in the trial and received treatment. They had a median of two prior lines of therapy and they received zanubrutinib at 160 mg twice daily.
Eighty-five patients were evaluable for efficacy. The overall response rate was 83.5% (71/85), and the complete response rate was 58.8% (50/85). At a median follow-up of 24.1 weeks, the median duration of response and median progression-free survival had not been reached. The estimated 24-week progression-free survival rate was 82%. The most common adverse events (AEs) in this trial were decrease in neutrophil count (31.4%), rash (29.1%), upper respiratory tract infection (29.1%), and decrease in platelet count (22.1%). Common grade 3 or higher AEs included neutrophil count decrease (11.6%) and lung infection (5.8%).
Four patients had fatal treatment-emergent AEs. One death was caused by a traffic accident, one was due to cerebral hemorrhage, and one resulted from pneumonia. The fourth death occurred in a patient with infection, but the cause of death was unknown.
Breakthrough therapy designation is designed to expedite the development and review of a therapy for a serious or life-threatening disease, following preliminary clinical evidence indicating it demonstrates substantial improvement over existing therapies.
The (MCL) who have received at least one prior therapy.
Zanubrutinib (BGB-3111) is a Bruton’s tyrosine kinase inhibitor being developed by BeiGene as a potential treatment for B-cell malignancies.
Researchers have evaluated zanubrutinib in a phase 2 trial (NCT03206970) of patients with relapsed/refractory MCL. Results from this trial were presented at the 2018 annual meeting of the American Society of Hematology (Abstract 148).
As of March 27, 2018, 86 patients had been enrolled in the trial and received treatment. They had a median of two prior lines of therapy and they received zanubrutinib at 160 mg twice daily.
Eighty-five patients were evaluable for efficacy. The overall response rate was 83.5% (71/85), and the complete response rate was 58.8% (50/85). At a median follow-up of 24.1 weeks, the median duration of response and median progression-free survival had not been reached. The estimated 24-week progression-free survival rate was 82%. The most common adverse events (AEs) in this trial were decrease in neutrophil count (31.4%), rash (29.1%), upper respiratory tract infection (29.1%), and decrease in platelet count (22.1%). Common grade 3 or higher AEs included neutrophil count decrease (11.6%) and lung infection (5.8%).
Four patients had fatal treatment-emergent AEs. One death was caused by a traffic accident, one was due to cerebral hemorrhage, and one resulted from pneumonia. The fourth death occurred in a patient with infection, but the cause of death was unknown.
Breakthrough therapy designation is designed to expedite the development and review of a therapy for a serious or life-threatening disease, following preliminary clinical evidence indicating it demonstrates substantial improvement over existing therapies.
The (MCL) who have received at least one prior therapy.
Zanubrutinib (BGB-3111) is a Bruton’s tyrosine kinase inhibitor being developed by BeiGene as a potential treatment for B-cell malignancies.
Researchers have evaluated zanubrutinib in a phase 2 trial (NCT03206970) of patients with relapsed/refractory MCL. Results from this trial were presented at the 2018 annual meeting of the American Society of Hematology (Abstract 148).
As of March 27, 2018, 86 patients had been enrolled in the trial and received treatment. They had a median of two prior lines of therapy and they received zanubrutinib at 160 mg twice daily.
Eighty-five patients were evaluable for efficacy. The overall response rate was 83.5% (71/85), and the complete response rate was 58.8% (50/85). At a median follow-up of 24.1 weeks, the median duration of response and median progression-free survival had not been reached. The estimated 24-week progression-free survival rate was 82%. The most common adverse events (AEs) in this trial were decrease in neutrophil count (31.4%), rash (29.1%), upper respiratory tract infection (29.1%), and decrease in platelet count (22.1%). Common grade 3 or higher AEs included neutrophil count decrease (11.6%) and lung infection (5.8%).
Four patients had fatal treatment-emergent AEs. One death was caused by a traffic accident, one was due to cerebral hemorrhage, and one resulted from pneumonia. The fourth death occurred in a patient with infection, but the cause of death was unknown.
Breakthrough therapy designation is designed to expedite the development and review of a therapy for a serious or life-threatening disease, following preliminary clinical evidence indicating it demonstrates substantial improvement over existing therapies.
Chidamide may be more effective in PTCL than previously thought
LA JOLLA, CALIF. – Real-world data suggest chidamide may be more effective against relapsed or refractory peripheral T-cell lymphoma (PTCL) than a pivotal study indicated.

Single-agent chidamide produced an overall response rate of 47.0% in a real-world study of more than 1,000 patients, compared with the 28.0% overall response rate that was observed in the phase 2 study of chidamide (Ann Oncol. 2015 Aug;26[8]:1766-71).
Yuqin Song, MD, PhD, of Peking University Cancer Hospital and Institute in Beijing, China, presented data from the real-world study at the annual T-cell Lymphoma Forum.
Dr. Song said this study is the largest cohort of real-world patients with relapsed or refractory PTCL. She and her colleagues analyzed data on 1,064 patients treated at 216 sites across China between February 2015 and December 2017.
The patients had a median age of 54 years, 63.9% were male, and 88.1% had stage III-IV disease.
Disease subtypes included PTCL not otherwise specified (NOS, 38.0%), angioimmunoblastic T-cell lymphoma (AITL, 29.1%), extranodal natural killer T-cell lymphoma (ENKTL, 13.4%), anaplastic large-cell lymphoma (ALCL, 9.1%), and others (10.3%), including cutaneous T-cell lymphoma (CTCL).
Fifty-two percent of patients (n = 553) received chidamide as a single agent, and 48% (n = 511) received the drug with other agents. The most common treatment regimens combined with chidamide were the following
- Cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP, 20.7%).
- Gemcitabine, dexamethasone, and cisplatin (GDP, 11.8%).
- Etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin (EPOCH, 9.8%).
- Patients with ENKTL received chidamide with L-asparaginase (35.4%) or without it (64.5%).
The median follow-up was 4.9 months (range, 0-36.2 months). Across disease subtypes, the overall response rate was 47.0% with single-agent chidamide and 65.4% when chidamide was given in combination with other agents (P less than .01).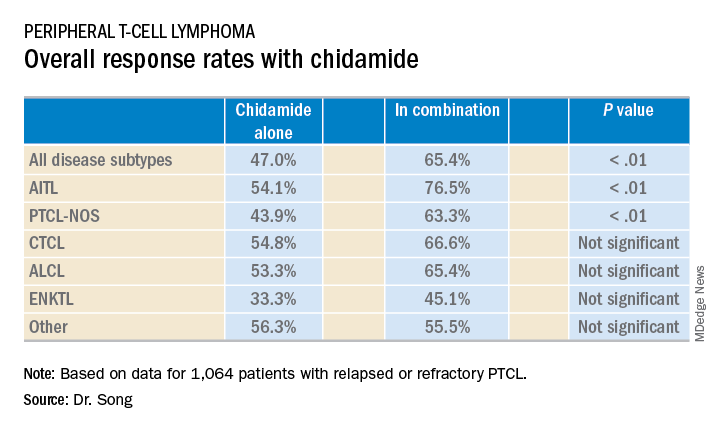
The median overall survival was 400 days for all patients, 342 days for patients treated with chidamide alone, and 457 days for patients who received combination therapy. The 1-year overall survival rates were 52%, 48%, and 56%, respectively.
Dr. Song said these data verify the efficacy of chidamide as a single agent and suggest chidamide might lead to improved survival in refractory or relapsed PTCLs.
Chidamide was generally well tolerated in this study, Dr. Song said. There were no unexpected adverse events (AEs) and most were grade 1 or 2.
The most common AEs (of any grade) observed with single-agent chidamide were neutropenia (42.9%), thrombocytopenia (40.5%), fatigue (38.3%), anemia (31.6%), and nausea/vomiting (21.0%).
The most common AEs observed with chidamide in combination were neutropenia (61.4%), thrombocytopenia (58.5%), fatigue (56.2%), anemia (54.2%), nausea/vomiting (30.7%), and fever (22.1%).
This study was supported by the Union for China Lymphoma Investigators and the Chinese Society of Clinical Oncology. Dr. Song did not disclose any conflicts of interest.
The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
LA JOLLA, CALIF. – Real-world data suggest chidamide may be more effective against relapsed or refractory peripheral T-cell lymphoma (PTCL) than a pivotal study indicated.
Single-agent chidamide produced an overall response rate of 47.0% in a real-world study of more than 1,000 patients, compared with the 28.0% overall response rate that was observed in the phase 2 study of chidamide (Ann Oncol. 2015 Aug;26[8]:1766-71).
Yuqin Song, MD, PhD, of Peking University Cancer Hospital and Institute in Beijing, China, presented data from the real-world study at the annual T-cell Lymphoma Forum.
Dr. Song said this study is the largest cohort of real-world patients with relapsed or refractory PTCL. She and her colleagues analyzed data on 1,064 patients treated at 216 sites across China between February 2015 and December 2017.
The patients had a median age of 54 years, 63.9% were male, and 88.1% had stage III-IV disease.
Disease subtypes included PTCL not otherwise specified (NOS, 38.0%), angioimmunoblastic T-cell lymphoma (AITL, 29.1%), extranodal natural killer T-cell lymphoma (ENKTL, 13.4%), anaplastic large-cell lymphoma (ALCL, 9.1%), and others (10.3%), including cutaneous T-cell lymphoma (CTCL).
Fifty-two percent of patients (n = 553) received chidamide as a single agent, and 48% (n = 511) received the drug with other agents. The most common treatment regimens combined with chidamide were the following
- Cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP, 20.7%).
- Gemcitabine, dexamethasone, and cisplatin (GDP, 11.8%).
- Etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin (EPOCH, 9.8%).
- Patients with ENKTL received chidamide with L-asparaginase (35.4%) or without it (64.5%).
The median follow-up was 4.9 months (range, 0-36.2 months). Across disease subtypes, the overall response rate was 47.0% with single-agent chidamide and 65.4% when chidamide was given in combination with other agents (P less than .01).
The median overall survival was 400 days for all patients, 342 days for patients treated with chidamide alone, and 457 days for patients who received combination therapy. The 1-year overall survival rates were 52%, 48%, and 56%, respectively.
Dr. Song said these data verify the efficacy of chidamide as a single agent and suggest chidamide might lead to improved survival in refractory or relapsed PTCLs.
Chidamide was generally well tolerated in this study, Dr. Song said. There were no unexpected adverse events (AEs) and most were grade 1 or 2.
The most common AEs (of any grade) observed with single-agent chidamide were neutropenia (42.9%), thrombocytopenia (40.5%), fatigue (38.3%), anemia (31.6%), and nausea/vomiting (21.0%).
The most common AEs observed with chidamide in combination were neutropenia (61.4%), thrombocytopenia (58.5%), fatigue (56.2%), anemia (54.2%), nausea/vomiting (30.7%), and fever (22.1%).
This study was supported by the Union for China Lymphoma Investigators and the Chinese Society of Clinical Oncology. Dr. Song did not disclose any conflicts of interest.
The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
LA JOLLA, CALIF. – Real-world data suggest chidamide may be more effective against relapsed or refractory peripheral T-cell lymphoma (PTCL) than a pivotal study indicated.
Single-agent chidamide produced an overall response rate of 47.0% in a real-world study of more than 1,000 patients, compared with the 28.0% overall response rate that was observed in the phase 2 study of chidamide (Ann Oncol. 2015 Aug;26[8]:1766-71).
Yuqin Song, MD, PhD, of Peking University Cancer Hospital and Institute in Beijing, China, presented data from the real-world study at the annual T-cell Lymphoma Forum.
Dr. Song said this study is the largest cohort of real-world patients with relapsed or refractory PTCL. She and her colleagues analyzed data on 1,064 patients treated at 216 sites across China between February 2015 and December 2017.
The patients had a median age of 54 years, 63.9% were male, and 88.1% had stage III-IV disease.
Disease subtypes included PTCL not otherwise specified (NOS, 38.0%), angioimmunoblastic T-cell lymphoma (AITL, 29.1%), extranodal natural killer T-cell lymphoma (ENKTL, 13.4%), anaplastic large-cell lymphoma (ALCL, 9.1%), and others (10.3%), including cutaneous T-cell lymphoma (CTCL).
Fifty-two percent of patients (n = 553) received chidamide as a single agent, and 48% (n = 511) received the drug with other agents. The most common treatment regimens combined with chidamide were the following
- Cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP, 20.7%).
- Gemcitabine, dexamethasone, and cisplatin (GDP, 11.8%).
- Etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin (EPOCH, 9.8%).
- Patients with ENKTL received chidamide with L-asparaginase (35.4%) or without it (64.5%).
The median follow-up was 4.9 months (range, 0-36.2 months). Across disease subtypes, the overall response rate was 47.0% with single-agent chidamide and 65.4% when chidamide was given in combination with other agents (P less than .01).
The median overall survival was 400 days for all patients, 342 days for patients treated with chidamide alone, and 457 days for patients who received combination therapy. The 1-year overall survival rates were 52%, 48%, and 56%, respectively.
Dr. Song said these data verify the efficacy of chidamide as a single agent and suggest chidamide might lead to improved survival in refractory or relapsed PTCLs.
Chidamide was generally well tolerated in this study, Dr. Song said. There were no unexpected adverse events (AEs) and most were grade 1 or 2.
The most common AEs (of any grade) observed with single-agent chidamide were neutropenia (42.9%), thrombocytopenia (40.5%), fatigue (38.3%), anemia (31.6%), and nausea/vomiting (21.0%).
The most common AEs observed with chidamide in combination were neutropenia (61.4%), thrombocytopenia (58.5%), fatigue (56.2%), anemia (54.2%), nausea/vomiting (30.7%), and fever (22.1%).
This study was supported by the Union for China Lymphoma Investigators and the Chinese Society of Clinical Oncology. Dr. Song did not disclose any conflicts of interest.
The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
REPORTING FROM TCLF 2019
Key clinical point:
Major finding: Single-agent chidamide had an overall response rate of 47.0% among relapsed/refractory PTCL patients, compared with 65.4% when used in combination with other agents (P less than .01).
Study details: A real-world cohort of 1,064 relapsed/refractory PTCL patients treated at 216 sites across China between February 2015 and December 2017.
Disclosures: The study was supported by the Union for China Lymphoma Investigators and the Chinese Society of Clinical Oncology. Dr. Song did not disclose any conflicts of interest.
Checkmate 436: Two-drug combo is ‘promising’ for PMBCL
SAN DIEGO – Nivolumab plus brentuximab vedotin may be a new treatment option for patients with relapsed/refractory primary mediastinal large B-cell lymphoma (PMBCL), according to investigators from the CheckMate 436 trial.
Interim results from this phase 1/2 trial revealed an overall response rate of 70%, including a complete response rate of 27%.
“It’s very promising ... to see this level of activity in this advanced, relapsed/refractory population,” said Joseph E. Eid, MD, senior vice president of Bristol-Myers Squibb, which is sponsoring CheckMate 436 in collaboration with Seattle Genetics.
Dr. Eid noted that adverse events (AEs) observed with this regimen were consistent with the safety profiles of nivolumab and brentuximab vedotin alone.
These results were presented as a poster at the annual meeting of the American Society of Hematology.
Dr. Eid noted that patients with relapsed or refractory PMBCL have limited treatment options.
“The initial therapy works well in 70% to 80% of patients but the patients who fail don’t have good options,” he said.
Prior research has shown that PMBCL is often characterized by overexpression of the PD-1 ligands PD-L1 and PD-L2, and most PMBCL expresses CD30.
Dr. Eid said CheckMate 436 (NCT02581631) was designed to “take advantage” of these characteristics by employing the anti-PD-1 checkpoint inhibitor nivolumab and the anti-CD30 antibody-drug conjugate brentuximab vedotin.
The interim analysis of this trial included 30 patients with relapsed/refractory PMBCL. Their median age at enrollment was 35.5 and 57% of patients were female. More than half of the patients (60%) had refractory disease, 23% had relapsed disease, and 17% had both.
The median number of prior therapies was two and 13% of patients had prior autologous stem cell transplant.
The patients received nivolumab at 240 mg and brentuximab vedotin at 1.8 mg/kg every 3 weeks until progression or unacceptable toxicity.
At a median follow-up of 6.1 months, 10 patients were still on treatment. Reasons for discontinuation included maximum clinical benefit, disease progression, AEs unrelated to treatment, patient request, and other concerns.
The rate of treatment-related AEs was 83%. The most common of these were neutropenia (27%), peripheral neuropathy (20%), hyperthyroidism (13%), rash (10%), and thrombocytopenia (10%).
Grade 3-4 treatment-related AEs included neutropenia (27%), thrombocytopenia (7%), decreased neutrophil count (7%), hypersensitivity (3%), diarrhea (3%), and maculopapular rash (3%).
The rate of serious treatment-related AEs was 10%. This included grade 3-4 diarrhea and maculopapular rash and grade 5 acute kidney injury.
The acute kidney injury was the only fatal AE considered treatment related. There were three other deaths in the trial, but they were considered unrelated to treatment.
The complete response rate was 27% (n = 8), and the partial response rate was 43% (n = 13), for an overall response rate of 70% (n = 21).
“The early indication is that 70% response is a pretty good outcome in a relapsed/refractory population that, otherwise, their outcome is pretty dismal,” Dr. Eid said.
Ten percent of patients (n = 3) had stable disease, 13% (n = 4) progressed, and investigators were unable to determine the status for 7% of patients (n = 2).
The median time to response was 1.3 months, and the median time to complete response was 3.0 months. The median duration of response and complete response were not reached.
Overall and progression-free survival data are not yet mature.
Still, the investigators concluded that nivolumab and brentuximab vedotin “may provide a new treatment option” for patients with relapsed/refractory PMBCL.
This trial is supported by Bristol-Myers Squibb in collaboration with Seattle Genetics. Investigators reported relationships with Bristol-Myers Squibb, Seattle Genetics, and a range of other companies.
SOURCE: Moskowitz AJ et al. ASH 2018. Abstract 1691.
SAN DIEGO – Nivolumab plus brentuximab vedotin may be a new treatment option for patients with relapsed/refractory primary mediastinal large B-cell lymphoma (PMBCL), according to investigators from the CheckMate 436 trial.
Interim results from this phase 1/2 trial revealed an overall response rate of 70%, including a complete response rate of 27%.
“It’s very promising ... to see this level of activity in this advanced, relapsed/refractory population,” said Joseph E. Eid, MD, senior vice president of Bristol-Myers Squibb, which is sponsoring CheckMate 436 in collaboration with Seattle Genetics.
Dr. Eid noted that adverse events (AEs) observed with this regimen were consistent with the safety profiles of nivolumab and brentuximab vedotin alone.
These results were presented as a poster at the annual meeting of the American Society of Hematology.
Dr. Eid noted that patients with relapsed or refractory PMBCL have limited treatment options.
“The initial therapy works well in 70% to 80% of patients but the patients who fail don’t have good options,” he said.
Prior research has shown that PMBCL is often characterized by overexpression of the PD-1 ligands PD-L1 and PD-L2, and most PMBCL expresses CD30.
Dr. Eid said CheckMate 436 (NCT02581631) was designed to “take advantage” of these characteristics by employing the anti-PD-1 checkpoint inhibitor nivolumab and the anti-CD30 antibody-drug conjugate brentuximab vedotin.
The interim analysis of this trial included 30 patients with relapsed/refractory PMBCL. Their median age at enrollment was 35.5 and 57% of patients were female. More than half of the patients (60%) had refractory disease, 23% had relapsed disease, and 17% had both.
The median number of prior therapies was two and 13% of patients had prior autologous stem cell transplant.
The patients received nivolumab at 240 mg and brentuximab vedotin at 1.8 mg/kg every 3 weeks until progression or unacceptable toxicity.
At a median follow-up of 6.1 months, 10 patients were still on treatment. Reasons for discontinuation included maximum clinical benefit, disease progression, AEs unrelated to treatment, patient request, and other concerns.
The rate of treatment-related AEs was 83%. The most common of these were neutropenia (27%), peripheral neuropathy (20%), hyperthyroidism (13%), rash (10%), and thrombocytopenia (10%).
Grade 3-4 treatment-related AEs included neutropenia (27%), thrombocytopenia (7%), decreased neutrophil count (7%), hypersensitivity (3%), diarrhea (3%), and maculopapular rash (3%).
The rate of serious treatment-related AEs was 10%. This included grade 3-4 diarrhea and maculopapular rash and grade 5 acute kidney injury.
The acute kidney injury was the only fatal AE considered treatment related. There were three other deaths in the trial, but they were considered unrelated to treatment.
The complete response rate was 27% (n = 8), and the partial response rate was 43% (n = 13), for an overall response rate of 70% (n = 21).
“The early indication is that 70% response is a pretty good outcome in a relapsed/refractory population that, otherwise, their outcome is pretty dismal,” Dr. Eid said.
Ten percent of patients (n = 3) had stable disease, 13% (n = 4) progressed, and investigators were unable to determine the status for 7% of patients (n = 2).
The median time to response was 1.3 months, and the median time to complete response was 3.0 months. The median duration of response and complete response were not reached.
Overall and progression-free survival data are not yet mature.
Still, the investigators concluded that nivolumab and brentuximab vedotin “may provide a new treatment option” for patients with relapsed/refractory PMBCL.
This trial is supported by Bristol-Myers Squibb in collaboration with Seattle Genetics. Investigators reported relationships with Bristol-Myers Squibb, Seattle Genetics, and a range of other companies.
SOURCE: Moskowitz AJ et al. ASH 2018. Abstract 1691.
SAN DIEGO – Nivolumab plus brentuximab vedotin may be a new treatment option for patients with relapsed/refractory primary mediastinal large B-cell lymphoma (PMBCL), according to investigators from the CheckMate 436 trial.
Interim results from this phase 1/2 trial revealed an overall response rate of 70%, including a complete response rate of 27%.
“It’s very promising ... to see this level of activity in this advanced, relapsed/refractory population,” said Joseph E. Eid, MD, senior vice president of Bristol-Myers Squibb, which is sponsoring CheckMate 436 in collaboration with Seattle Genetics.
Dr. Eid noted that adverse events (AEs) observed with this regimen were consistent with the safety profiles of nivolumab and brentuximab vedotin alone.
These results were presented as a poster at the annual meeting of the American Society of Hematology.
Dr. Eid noted that patients with relapsed or refractory PMBCL have limited treatment options.
“The initial therapy works well in 70% to 80% of patients but the patients who fail don’t have good options,” he said.
Prior research has shown that PMBCL is often characterized by overexpression of the PD-1 ligands PD-L1 and PD-L2, and most PMBCL expresses CD30.
Dr. Eid said CheckMate 436 (NCT02581631) was designed to “take advantage” of these characteristics by employing the anti-PD-1 checkpoint inhibitor nivolumab and the anti-CD30 antibody-drug conjugate brentuximab vedotin.
The interim analysis of this trial included 30 patients with relapsed/refractory PMBCL. Their median age at enrollment was 35.5 and 57% of patients were female. More than half of the patients (60%) had refractory disease, 23% had relapsed disease, and 17% had both.
The median number of prior therapies was two and 13% of patients had prior autologous stem cell transplant.
The patients received nivolumab at 240 mg and brentuximab vedotin at 1.8 mg/kg every 3 weeks until progression or unacceptable toxicity.
At a median follow-up of 6.1 months, 10 patients were still on treatment. Reasons for discontinuation included maximum clinical benefit, disease progression, AEs unrelated to treatment, patient request, and other concerns.
The rate of treatment-related AEs was 83%. The most common of these were neutropenia (27%), peripheral neuropathy (20%), hyperthyroidism (13%), rash (10%), and thrombocytopenia (10%).
Grade 3-4 treatment-related AEs included neutropenia (27%), thrombocytopenia (7%), decreased neutrophil count (7%), hypersensitivity (3%), diarrhea (3%), and maculopapular rash (3%).
The rate of serious treatment-related AEs was 10%. This included grade 3-4 diarrhea and maculopapular rash and grade 5 acute kidney injury.
The acute kidney injury was the only fatal AE considered treatment related. There were three other deaths in the trial, but they were considered unrelated to treatment.
The complete response rate was 27% (n = 8), and the partial response rate was 43% (n = 13), for an overall response rate of 70% (n = 21).
“The early indication is that 70% response is a pretty good outcome in a relapsed/refractory population that, otherwise, their outcome is pretty dismal,” Dr. Eid said.
Ten percent of patients (n = 3) had stable disease, 13% (n = 4) progressed, and investigators were unable to determine the status for 7% of patients (n = 2).
The median time to response was 1.3 months, and the median time to complete response was 3.0 months. The median duration of response and complete response were not reached.
Overall and progression-free survival data are not yet mature.
Still, the investigators concluded that nivolumab and brentuximab vedotin “may provide a new treatment option” for patients with relapsed/refractory PMBCL.
This trial is supported by Bristol-Myers Squibb in collaboration with Seattle Genetics. Investigators reported relationships with Bristol-Myers Squibb, Seattle Genetics, and a range of other companies.
SOURCE: Moskowitz AJ et al. ASH 2018. Abstract 1691.
REPORTING FROM ASH 2018
Key clinical point:
Major finding: The overall response rate was 70%, including a complete response rate of 27%.
Study details: A phase 1/2 study of 30 patients.
Disclosures: This trial is supported by Bristol-Myers Squibb in collaboration with Seattle Genetics, and investigators reported relationships with a range of other companies.
Source: Moskowitz AJ et al. ASH 2018, Abstract 1691.
ECHELON-2: BV-CHP boosts survival in PTCL
SAN DIEGO – A newly approved treatment regimen provides a survival benefit over standard therapy for patients with CD30-positive peripheral T-cell lymphomas (PTCLs), according to new research presented at the annual meeting of the American Society of Hematology.
In the ECHELON-2 trial, patients who received brentuximab vedotin (BV) plus cyclophosphamide, doxorubicin, and prednisone (CHP) had superior progression-free survival (PFS) and overall survival (OS), compared with patients who received standard treatment with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP).
These results supported the recent U.S. approval of BV in combination with CHP for adults with previously untreated, systemic anaplastic large cell lymphoma or other CD30-expressing PTCLs.
“ECHELON-2 is the first prospective trial in peripheral T-cell lymphoma to show an overall survival benefit over CHOP,” said Steven M. Horwitz, MD, of Memorial Sloan Kettering Cancer Center, with locations in New York and New Jersey.
Dr. Horwitz presented data from this trial at the ASH meeting. Results were simultaneously published in the Lancet (2018 Dec 3. doi: 10.1016/S0140-6736[18]32984-2).
ECHELON-2 (NCT01777152) enrolled 452 patients with previously untreated, CD30-positive PTCL. Subtypes included ALK-positive or ALK-negative systemic anaplastic large-cell lymphoma, PTCL not otherwise specified, angioimmunoblastic T-cell lymphoma, enteropathy-associated T-cell lymphoma, and adult T-cell leukemia/lymphoma.
Patients were randomized to receive BV-CHP plus placebo (n = 226) or CHOP plus placebo (n = 226) every 3 weeks for six to eight cycles.
At baseline, the median age was 58 in the BV-CHP arm and the CHOP arm. The majority of patients were male – 59% in the BV-CHP arm and 67% in the CHOP arm – and most patients had stage III/IV disease, 81% and 80%, respectively.
In all, 89% of patients in the BV-CHP arm and 81% in the CHOP arm completed six or more cycles of their assigned treatment.
The overall response rate was 83% in the BV-CHP arm and 72% in the CHOP arm (P = .0032). The complete response rates were 68% and 56%, respectively (P = .0066).
At a median follow-up of 36.2 months, the median PFS was 48.2 months in the BV-CHP arm and 20.8 months in the CHOP arm. The rate of death or progression was 42% in the BV-CHP arm and 55% in the CHOP arm (hazard ratio = 0.71, P = .011).
At a median follow-up of 42.1 months, the median OS was not reached in either treatment arm. The rate of death was 23% in the BV-CHP arm and 32% in the CHOP arm (HR = 0.66, P = .0244).
Dr. Horwitz noted that this study was not powered to determine differences in PFS or OS by PTCL subtypes.
BV-CHP had a safety profile comparable with that of CHOP, Dr. Horwitz said.
The rate of adverse events (AEs) was 99% in the BV-CHP arm and 98% in the CHOP arm. Grade 3 or higher AEs occurred in 66% and 65% of patients, respectively. Serious AEs occurred in 39% and 38%, respectively.
Three percent of patients in the BV-CHP arm and 4% of those in the CHOP arm had fatal AEs.
The study was funded by Seattle Genetics, Millennium Pharmaceuticals, and the National Institutes of Health. Dr. Horwitz reported relationships with Seattle Genetics, Millennium Pharmaceuticals, and other companies.
SOURCE: Horwitz S et al. ASH 2018, Abstract 997.
SAN DIEGO – A newly approved treatment regimen provides a survival benefit over standard therapy for patients with CD30-positive peripheral T-cell lymphomas (PTCLs), according to new research presented at the annual meeting of the American Society of Hematology.
In the ECHELON-2 trial, patients who received brentuximab vedotin (BV) plus cyclophosphamide, doxorubicin, and prednisone (CHP) had superior progression-free survival (PFS) and overall survival (OS), compared with patients who received standard treatment with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP).
These results supported the recent U.S. approval of BV in combination with CHP for adults with previously untreated, systemic anaplastic large cell lymphoma or other CD30-expressing PTCLs.
“ECHELON-2 is the first prospective trial in peripheral T-cell lymphoma to show an overall survival benefit over CHOP,” said Steven M. Horwitz, MD, of Memorial Sloan Kettering Cancer Center, with locations in New York and New Jersey.
Dr. Horwitz presented data from this trial at the ASH meeting. Results were simultaneously published in the Lancet (2018 Dec 3. doi: 10.1016/S0140-6736[18]32984-2).
ECHELON-2 (NCT01777152) enrolled 452 patients with previously untreated, CD30-positive PTCL. Subtypes included ALK-positive or ALK-negative systemic anaplastic large-cell lymphoma, PTCL not otherwise specified, angioimmunoblastic T-cell lymphoma, enteropathy-associated T-cell lymphoma, and adult T-cell leukemia/lymphoma.
Patients were randomized to receive BV-CHP plus placebo (n = 226) or CHOP plus placebo (n = 226) every 3 weeks for six to eight cycles.
At baseline, the median age was 58 in the BV-CHP arm and the CHOP arm. The majority of patients were male – 59% in the BV-CHP arm and 67% in the CHOP arm – and most patients had stage III/IV disease, 81% and 80%, respectively.
In all, 89% of patients in the BV-CHP arm and 81% in the CHOP arm completed six or more cycles of their assigned treatment.
The overall response rate was 83% in the BV-CHP arm and 72% in the CHOP arm (P = .0032). The complete response rates were 68% and 56%, respectively (P = .0066).
At a median follow-up of 36.2 months, the median PFS was 48.2 months in the BV-CHP arm and 20.8 months in the CHOP arm. The rate of death or progression was 42% in the BV-CHP arm and 55% in the CHOP arm (hazard ratio = 0.71, P = .011).
At a median follow-up of 42.1 months, the median OS was not reached in either treatment arm. The rate of death was 23% in the BV-CHP arm and 32% in the CHOP arm (HR = 0.66, P = .0244).
Dr. Horwitz noted that this study was not powered to determine differences in PFS or OS by PTCL subtypes.
BV-CHP had a safety profile comparable with that of CHOP, Dr. Horwitz said.
The rate of adverse events (AEs) was 99% in the BV-CHP arm and 98% in the CHOP arm. Grade 3 or higher AEs occurred in 66% and 65% of patients, respectively. Serious AEs occurred in 39% and 38%, respectively.
Three percent of patients in the BV-CHP arm and 4% of those in the CHOP arm had fatal AEs.
The study was funded by Seattle Genetics, Millennium Pharmaceuticals, and the National Institutes of Health. Dr. Horwitz reported relationships with Seattle Genetics, Millennium Pharmaceuticals, and other companies.
SOURCE: Horwitz S et al. ASH 2018, Abstract 997.
SAN DIEGO – A newly approved treatment regimen provides a survival benefit over standard therapy for patients with CD30-positive peripheral T-cell lymphomas (PTCLs), according to new research presented at the annual meeting of the American Society of Hematology.
In the ECHELON-2 trial, patients who received brentuximab vedotin (BV) plus cyclophosphamide, doxorubicin, and prednisone (CHP) had superior progression-free survival (PFS) and overall survival (OS), compared with patients who received standard treatment with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP).
These results supported the recent U.S. approval of BV in combination with CHP for adults with previously untreated, systemic anaplastic large cell lymphoma or other CD30-expressing PTCLs.
“ECHELON-2 is the first prospective trial in peripheral T-cell lymphoma to show an overall survival benefit over CHOP,” said Steven M. Horwitz, MD, of Memorial Sloan Kettering Cancer Center, with locations in New York and New Jersey.
Dr. Horwitz presented data from this trial at the ASH meeting. Results were simultaneously published in the Lancet (2018 Dec 3. doi: 10.1016/S0140-6736[18]32984-2).
ECHELON-2 (NCT01777152) enrolled 452 patients with previously untreated, CD30-positive PTCL. Subtypes included ALK-positive or ALK-negative systemic anaplastic large-cell lymphoma, PTCL not otherwise specified, angioimmunoblastic T-cell lymphoma, enteropathy-associated T-cell lymphoma, and adult T-cell leukemia/lymphoma.
Patients were randomized to receive BV-CHP plus placebo (n = 226) or CHOP plus placebo (n = 226) every 3 weeks for six to eight cycles.
At baseline, the median age was 58 in the BV-CHP arm and the CHOP arm. The majority of patients were male – 59% in the BV-CHP arm and 67% in the CHOP arm – and most patients had stage III/IV disease, 81% and 80%, respectively.
In all, 89% of patients in the BV-CHP arm and 81% in the CHOP arm completed six or more cycles of their assigned treatment.
The overall response rate was 83% in the BV-CHP arm and 72% in the CHOP arm (P = .0032). The complete response rates were 68% and 56%, respectively (P = .0066).
At a median follow-up of 36.2 months, the median PFS was 48.2 months in the BV-CHP arm and 20.8 months in the CHOP arm. The rate of death or progression was 42% in the BV-CHP arm and 55% in the CHOP arm (hazard ratio = 0.71, P = .011).
At a median follow-up of 42.1 months, the median OS was not reached in either treatment arm. The rate of death was 23% in the BV-CHP arm and 32% in the CHOP arm (HR = 0.66, P = .0244).
Dr. Horwitz noted that this study was not powered to determine differences in PFS or OS by PTCL subtypes.
BV-CHP had a safety profile comparable with that of CHOP, Dr. Horwitz said.
The rate of adverse events (AEs) was 99% in the BV-CHP arm and 98% in the CHOP arm. Grade 3 or higher AEs occurred in 66% and 65% of patients, respectively. Serious AEs occurred in 39% and 38%, respectively.
Three percent of patients in the BV-CHP arm and 4% of those in the CHOP arm had fatal AEs.
The study was funded by Seattle Genetics, Millennium Pharmaceuticals, and the National Institutes of Health. Dr. Horwitz reported relationships with Seattle Genetics, Millennium Pharmaceuticals, and other companies.
SOURCE: Horwitz S et al. ASH 2018, Abstract 997.
REPORTING FROM ASH 2018
Key clinical point:
Major finding: The rate of death or progression was 42% in the BV-CHP arm and 55% in the CHOP arm (hazard ratio = 0.71, P = .011), while the rate of death alone was 23% and 32%, respectively (HR = 0.66, P = .0244).
Study details: A phase 3 trial of 452 patients with peripheral T-cell lymphoma.
Disclosures: The study was funded by Seattle Genetics, Millennium Pharmaceuticals, and the National Institutes of Health. Dr. Horwitz reported relationships with Seattle Genetics, Millennium Pharmaceuticals, and other companies.
Source: Horwitz S et al. ASH 2018, Abstract 997.

Ibrutinib-rituximab ‘new standard of care’ in younger CLL patients
SAN DIEGO – The combination of ibrutinib and rituximab was associated with a two-thirds reduction in chronic lymphocytic leukemia (CLL) progression, compared with standard chemoimmunotherapy in patients younger than 70 years old, interim results of a phase 3 randomized trial showed.
Among 529 patients with previously untreated, symptomatic CLL randomly assigned to ibrutinib-rituximab (IR) or to chemoimmunotherapy with fludarabine, cyclophosphamide, and rituximab (FCR), the IR regimen was associated with a 65% reduction in risk for disease progression, which was the trial’s primary endpoint.
The IR regimen was also associated with better overall survival out to 4 years of follow-up, reported Tait D. Shanafelt, MD, of Stanford (Calif.) University.
“This establishes ibrutinib-based therapy as the most effective treatment tested to date in this disease for untreated patients,” he said at a media briefing prior at the annual meeting of the American Society of Hematology.
The study results are likely to dethrone FCR as the most active chemoimmunotherapy regimen against CLL, Dr. Shanafelt said.
In the ECOG-ACRIN Cancer Research Group E1912 trial, 529 patients aged 70 or younger with previously untreated CLL were enrolled and randomly assigned on a 2:1 basis to either standard therapy with six cycles of FCR according to standard protocols (175 patients), or IR, with ibrutinib 420 mg daily for each cycle, and rituximab delivered 50 mg/m2 intravenously on day 1 of cycle 2, and 325 mg/m2 on day 2 of the same cycle, and 500 mg/m2 on day 1 for all remaining cycles (354 patients).
From cycle 8 until progression, patients in the IR arm received daily ibrutinib 420 mg.
Dr. Shanafelt presented results from both an intention-to-treat analysis and a per-protocol analysis excluding 22 patients in the IR arm and 9 patients in the FCR arm who were randomized but later found not to meet eligibility criteria.
After a mean follow-up of 34 months, there were 37 PFS events in the IR arm, compared with 40 events in the FCR arm in an intention-to-treat analysis. The difference translated into a hazard ratio for progression of 0.35 with IR (P less than .00001).
The results were similar in the per-protocol analysis, with an HR of 0.32 favoring IR (P less than .00001).*
There were four deaths in the IR arm, compared with 10 in the FCR arm at the time of the data lock, translating into a hazard ratio (HR) for overall survival of 0.17 (P less than .0003) in the intention-to-treat analysis, and 0.13 in the per-protocol analysis (P less than .0001).
Dr. Shanafelt noted that although the overall number of deaths was relatively small, there were twice as many patients enrolled in the IR arm as in the FCR arm, meaning that the rate of deaths in the FCR arm was fivefold higher than in the IR arm.
In a subgroup analysis of PFS, IR was superior to FCR regardless of patient age, sex, performance status, disease stage, or the presence or absence of the 11q23.3 deletion.
PFS was also significantly better with IR in patients with unmutated immunoglobulin heavy chain variable (IGHV) regions (HR 0.26, P less than .00001), but not in patients with mutated IGHV.*
Grade 3 or greater treatment-related adverse events occurred in 58.5% of patients in the IR arm, compared with 72.1% of patients in the FCR arm. Specific events that occurred significantly less often with IR included neutropenia (22.7% vs. 43.7%), anemia (2.6% vs. 12.0%), thrombocytopenia (2.9% vs. 13.9%), any infection (7.1% vs. 19.0%), and neutropenic fever (2.3% vs. 15.8%; P less than .001 for all comparisons).
Events that occurred more frequently with IR than FCR included atrial fibrillation (2.9% vs. 0%, P = .04), and hypertension (7.4% vs. 1.9%, P = .01).
Dr. Shanafelt acknowledged that one possible barrier to the IR regimen is cost; the monthly cost of ibrutinib maintenance is about $10,000, he said, although he noted that cost considerations were not studied in the trial.
“Future trials testing novel agent combinations to see if we can eliminate the need for chronic therapy should be pursued,” he said.
The trial was sponsored by the National Cancer Institute with additional support from Pharmacyclics. Dr. Shanafelt reported patents and royalties from the Mayo Clinic, and research funding from Celgene, GlaxoSmithKline, Genentech, Abbvie, Pharmacyclics, and Janssen.
SOURCE: Shanafelt TD et al. ASH 2018, Abstract LBA-4.
*Correction, 12/12/2018: An earlier version of this story misstated the P value in two comparisons.
SAN DIEGO – The combination of ibrutinib and rituximab was associated with a two-thirds reduction in chronic lymphocytic leukemia (CLL) progression, compared with standard chemoimmunotherapy in patients younger than 70 years old, interim results of a phase 3 randomized trial showed.
Among 529 patients with previously untreated, symptomatic CLL randomly assigned to ibrutinib-rituximab (IR) or to chemoimmunotherapy with fludarabine, cyclophosphamide, and rituximab (FCR), the IR regimen was associated with a 65% reduction in risk for disease progression, which was the trial’s primary endpoint.
The IR regimen was also associated with better overall survival out to 4 years of follow-up, reported Tait D. Shanafelt, MD, of Stanford (Calif.) University.
“This establishes ibrutinib-based therapy as the most effective treatment tested to date in this disease for untreated patients,” he said at a media briefing prior at the annual meeting of the American Society of Hematology.
The study results are likely to dethrone FCR as the most active chemoimmunotherapy regimen against CLL, Dr. Shanafelt said.
In the ECOG-ACRIN Cancer Research Group E1912 trial, 529 patients aged 70 or younger with previously untreated CLL were enrolled and randomly assigned on a 2:1 basis to either standard therapy with six cycles of FCR according to standard protocols (175 patients), or IR, with ibrutinib 420 mg daily for each cycle, and rituximab delivered 50 mg/m2 intravenously on day 1 of cycle 2, and 325 mg/m2 on day 2 of the same cycle, and 500 mg/m2 on day 1 for all remaining cycles (354 patients).
From cycle 8 until progression, patients in the IR arm received daily ibrutinib 420 mg.
Dr. Shanafelt presented results from both an intention-to-treat analysis and a per-protocol analysis excluding 22 patients in the IR arm and 9 patients in the FCR arm who were randomized but later found not to meet eligibility criteria.
After a mean follow-up of 34 months, there were 37 PFS events in the IR arm, compared with 40 events in the FCR arm in an intention-to-treat analysis. The difference translated into a hazard ratio for progression of 0.35 with IR (P less than .00001).
The results were similar in the per-protocol analysis, with an HR of 0.32 favoring IR (P less than .00001).*
There were four deaths in the IR arm, compared with 10 in the FCR arm at the time of the data lock, translating into a hazard ratio (HR) for overall survival of 0.17 (P less than .0003) in the intention-to-treat analysis, and 0.13 in the per-protocol analysis (P less than .0001).
Dr. Shanafelt noted that although the overall number of deaths was relatively small, there were twice as many patients enrolled in the IR arm as in the FCR arm, meaning that the rate of deaths in the FCR arm was fivefold higher than in the IR arm.
In a subgroup analysis of PFS, IR was superior to FCR regardless of patient age, sex, performance status, disease stage, or the presence or absence of the 11q23.3 deletion.
PFS was also significantly better with IR in patients with unmutated immunoglobulin heavy chain variable (IGHV) regions (HR 0.26, P less than .00001), but not in patients with mutated IGHV.*
Grade 3 or greater treatment-related adverse events occurred in 58.5% of patients in the IR arm, compared with 72.1% of patients in the FCR arm. Specific events that occurred significantly less often with IR included neutropenia (22.7% vs. 43.7%), anemia (2.6% vs. 12.0%), thrombocytopenia (2.9% vs. 13.9%), any infection (7.1% vs. 19.0%), and neutropenic fever (2.3% vs. 15.8%; P less than .001 for all comparisons).
Events that occurred more frequently with IR than FCR included atrial fibrillation (2.9% vs. 0%, P = .04), and hypertension (7.4% vs. 1.9%, P = .01).
Dr. Shanafelt acknowledged that one possible barrier to the IR regimen is cost; the monthly cost of ibrutinib maintenance is about $10,000, he said, although he noted that cost considerations were not studied in the trial.
“Future trials testing novel agent combinations to see if we can eliminate the need for chronic therapy should be pursued,” he said.
The trial was sponsored by the National Cancer Institute with additional support from Pharmacyclics. Dr. Shanafelt reported patents and royalties from the Mayo Clinic, and research funding from Celgene, GlaxoSmithKline, Genentech, Abbvie, Pharmacyclics, and Janssen.
SOURCE: Shanafelt TD et al. ASH 2018, Abstract LBA-4.
*Correction, 12/12/2018: An earlier version of this story misstated the P value in two comparisons.
SAN DIEGO – The combination of ibrutinib and rituximab was associated with a two-thirds reduction in chronic lymphocytic leukemia (CLL) progression, compared with standard chemoimmunotherapy in patients younger than 70 years old, interim results of a phase 3 randomized trial showed.
Among 529 patients with previously untreated, symptomatic CLL randomly assigned to ibrutinib-rituximab (IR) or to chemoimmunotherapy with fludarabine, cyclophosphamide, and rituximab (FCR), the IR regimen was associated with a 65% reduction in risk for disease progression, which was the trial’s primary endpoint.
The IR regimen was also associated with better overall survival out to 4 years of follow-up, reported Tait D. Shanafelt, MD, of Stanford (Calif.) University.
“This establishes ibrutinib-based therapy as the most effective treatment tested to date in this disease for untreated patients,” he said at a media briefing prior at the annual meeting of the American Society of Hematology.
The study results are likely to dethrone FCR as the most active chemoimmunotherapy regimen against CLL, Dr. Shanafelt said.
In the ECOG-ACRIN Cancer Research Group E1912 trial, 529 patients aged 70 or younger with previously untreated CLL were enrolled and randomly assigned on a 2:1 basis to either standard therapy with six cycles of FCR according to standard protocols (175 patients), or IR, with ibrutinib 420 mg daily for each cycle, and rituximab delivered 50 mg/m2 intravenously on day 1 of cycle 2, and 325 mg/m2 on day 2 of the same cycle, and 500 mg/m2 on day 1 for all remaining cycles (354 patients).
From cycle 8 until progression, patients in the IR arm received daily ibrutinib 420 mg.
Dr. Shanafelt presented results from both an intention-to-treat analysis and a per-protocol analysis excluding 22 patients in the IR arm and 9 patients in the FCR arm who were randomized but later found not to meet eligibility criteria.
After a mean follow-up of 34 months, there were 37 PFS events in the IR arm, compared with 40 events in the FCR arm in an intention-to-treat analysis. The difference translated into a hazard ratio for progression of 0.35 with IR (P less than .00001).
The results were similar in the per-protocol analysis, with an HR of 0.32 favoring IR (P less than .00001).*
There were four deaths in the IR arm, compared with 10 in the FCR arm at the time of the data lock, translating into a hazard ratio (HR) for overall survival of 0.17 (P less than .0003) in the intention-to-treat analysis, and 0.13 in the per-protocol analysis (P less than .0001).
Dr. Shanafelt noted that although the overall number of deaths was relatively small, there were twice as many patients enrolled in the IR arm as in the FCR arm, meaning that the rate of deaths in the FCR arm was fivefold higher than in the IR arm.
In a subgroup analysis of PFS, IR was superior to FCR regardless of patient age, sex, performance status, disease stage, or the presence or absence of the 11q23.3 deletion.
PFS was also significantly better with IR in patients with unmutated immunoglobulin heavy chain variable (IGHV) regions (HR 0.26, P less than .00001), but not in patients with mutated IGHV.*
Grade 3 or greater treatment-related adverse events occurred in 58.5% of patients in the IR arm, compared with 72.1% of patients in the FCR arm. Specific events that occurred significantly less often with IR included neutropenia (22.7% vs. 43.7%), anemia (2.6% vs. 12.0%), thrombocytopenia (2.9% vs. 13.9%), any infection (7.1% vs. 19.0%), and neutropenic fever (2.3% vs. 15.8%; P less than .001 for all comparisons).
Events that occurred more frequently with IR than FCR included atrial fibrillation (2.9% vs. 0%, P = .04), and hypertension (7.4% vs. 1.9%, P = .01).
Dr. Shanafelt acknowledged that one possible barrier to the IR regimen is cost; the monthly cost of ibrutinib maintenance is about $10,000, he said, although he noted that cost considerations were not studied in the trial.
“Future trials testing novel agent combinations to see if we can eliminate the need for chronic therapy should be pursued,” he said.
The trial was sponsored by the National Cancer Institute with additional support from Pharmacyclics. Dr. Shanafelt reported patents and royalties from the Mayo Clinic, and research funding from Celgene, GlaxoSmithKline, Genentech, Abbvie, Pharmacyclics, and Janssen.
SOURCE: Shanafelt TD et al. ASH 2018, Abstract LBA-4.
*Correction, 12/12/2018: An earlier version of this story misstated the P value in two comparisons.
REPORTING FROM ASH 2018
Key clinical point:
Major finding: The hazard ratio for disease progression with IR versus FCR was 0.35 (P less than .00001).
Study details: Interim analysis of a phase 3 trial in 529 patients aged 70 or younger with newly diagnosed CLL.
Disclosures: The trial was sponsored by the National Cancer Institute with additional support from Pharmacyclics. Dr. Shanafelt reported patents and royalties from the Mayo Clinic, and research funding from Celgene, GlaxoSmithKline, Genentech, Abbvie, Pharmacyclics, and Janssen.
Source: Shanafelt TD et al. ASH 2018, Abstract LBA-4.

2018: A banner year for hematology drug approvals
SAN DIEGO – It was banner year for new hematology drug approvals, according to R. Angelo de Claro, MD, of the Food and Drug Administration.
, including 12 first-time approvals, 5 new biosimilars, and 15 new indications for previously approved drugs, Dr. de Claro, clinical team leader in the FDA’s division of hematology products in Silver Spring, Md., said during an overview of the approvals at the annual meeting of the American Society of Hematology.
These include six new approvals for first-line treatment, and eight for pediatric indications, he said.
Highlights were discussed at two ASH-FDA joint symposia at the meeting, including one focused on the malignant hematology approvals, and another on the nonmalignant hematology approvals. In a video interview, Dr. de Claro provides some additional insight into their importance and about what might lie ahead.
“I think what’s exciting is that you have drug development occurring in more common conditions such as chronic lymphocytic leukemia, as well as in rare conditions, including hairy cell leukemia – and the first-ever approval in hemophagocytic lymphohistiocytosis,” he said. “It’s been very busy at the FDA; stay tuned ... the year’s not done yet. There could be more coming and we certainly anticipate more applications in the future.”
Dr. de Claro is an FDA employee. He reported having no other relevant disclosures.
SAN DIEGO – It was banner year for new hematology drug approvals, according to R. Angelo de Claro, MD, of the Food and Drug Administration.
, including 12 first-time approvals, 5 new biosimilars, and 15 new indications for previously approved drugs, Dr. de Claro, clinical team leader in the FDA’s division of hematology products in Silver Spring, Md., said during an overview of the approvals at the annual meeting of the American Society of Hematology.
These include six new approvals for first-line treatment, and eight for pediatric indications, he said.
Highlights were discussed at two ASH-FDA joint symposia at the meeting, including one focused on the malignant hematology approvals, and another on the nonmalignant hematology approvals. In a video interview, Dr. de Claro provides some additional insight into their importance and about what might lie ahead.
“I think what’s exciting is that you have drug development occurring in more common conditions such as chronic lymphocytic leukemia, as well as in rare conditions, including hairy cell leukemia – and the first-ever approval in hemophagocytic lymphohistiocytosis,” he said. “It’s been very busy at the FDA; stay tuned ... the year’s not done yet. There could be more coming and we certainly anticipate more applications in the future.”
Dr. de Claro is an FDA employee. He reported having no other relevant disclosures.
SAN DIEGO – It was banner year for new hematology drug approvals, according to R. Angelo de Claro, MD, of the Food and Drug Administration.
, including 12 first-time approvals, 5 new biosimilars, and 15 new indications for previously approved drugs, Dr. de Claro, clinical team leader in the FDA’s division of hematology products in Silver Spring, Md., said during an overview of the approvals at the annual meeting of the American Society of Hematology.
These include six new approvals for first-line treatment, and eight for pediatric indications, he said.
Highlights were discussed at two ASH-FDA joint symposia at the meeting, including one focused on the malignant hematology approvals, and another on the nonmalignant hematology approvals. In a video interview, Dr. de Claro provides some additional insight into their importance and about what might lie ahead.
“I think what’s exciting is that you have drug development occurring in more common conditions such as chronic lymphocytic leukemia, as well as in rare conditions, including hairy cell leukemia – and the first-ever approval in hemophagocytic lymphohistiocytosis,” he said. “It’s been very busy at the FDA; stay tuned ... the year’s not done yet. There could be more coming and we certainly anticipate more applications in the future.”
Dr. de Claro is an FDA employee. He reported having no other relevant disclosures.
REPORTING FROM ASH 2018

Phase 3 study confirms biosimilarity of PF-05280586 with rituximab
SAN DIEGO – The potential rituximab biosimilar drug PF-05280586 showed efficacy, safety, immunogenicity, pharmacokinetics, and pharmacodynamics similar to those of rituximab at up to 26 weeks in a randomized phase 3 study of treatment-naive patients with CD20-positive low tumor burden follicular lymphoma (LTB-FL).

The primary endpoint of overall response rate at 26 weeks was 75.5% in 196 patients randomized to receive PF-05280586, and 70.7% in 198 patients who received a rituximab reference product sourced from the European Union (MabThera; rituximab‑EU), Jeff Sharman, MD, reported at the annual meeting of the American Society of Hematology.
“This resulted in a difference between the two arms of 4.66%,” said Dr. Sharman of Willamette Valley Cancer Institute and Research Center, Springfield, Ore.
The 95% confidence interval for this difference ... was entirely contained within the prespecified equivalence margin, he said.
“Depth of response was a key secondary endpoint, and rates of complete response were 29.3% and 30.4%, respectively,” he said, noting that rates of partial response, stable response, and progressive disease were also similar between the two study arms.
Estimated 1-year progression-free survival (PFS) rates were also highly similar at 76.4% and 81.2% in the PF-05280586 and rituximab-EU arms.
Rapid depletion in CD19-positive B-cell counts was observed in both groups after initial dosing, with recovery by week 39 and a sustained increase until the end of week 52.
Treatment-emergent adverse events (TEAEs) occurred in 78.6% vs. 72.1% of patients in the PF‑05280586 vs. rituximab‑EU arms, respectively, and the rates of serious adverse events and grade 3 events were similar in the groups, as were rates of infusion interruptions or infusion-related reactions (IRRs), Dr. Sharman said.
IRRs occurred in about 25% of patients in each arm, and most were grade 1 or 2. Grade 3 IRRs occurred in 2.6% vs. 0.5% of patients in the groups, respectively, and no grade 4 IRRs occurred.
Rates of anti-drug antibodies were also similar in the two groups, as were serum drug concentrations – regardless of anti-drug antibody status, he noted.
Study subjects were adults with a mean age of 60 years and histologically confirmed CD20-positive grade 1-3a follicular lymphoma with no prior rituximab or system therapy for B-cell non-Hodgkin lymphoma (NHL). They had Ann Arbor disease stages II (26.9%), III (44.2%) or IV (28.9%), ECOG performance status of 0-1, and at least 1 measurable disease lesion identifiable on imaging.
Risk level as assessed by the Follicular Lymphoma International Prognostic Index–2 was low in 28.4%, medium in 66%, and high in 5.6% of patients.
Treatment with each agent was given at intravenous doses of 375 mg/m2 weekly for 4 weeks at days 1, 8, 15, and 22.
PF-05280586 is being developed by Pfizer, and in this 52-week double-blind study – the largest study to date of the early use of the potential rituximab biosimilar in patients with previously untreated CD20-positive LTB-FL – the primary endpoint was met, demonstrating its therapeutic equivalence with rituximab-EU for overall response rate at week 26, Dr. Sharman said.
“These results therefore confirm the biosimilarity of PF-05280586 with rituximab-EU,” he concluded.
Of note, the reporting of these findings comes on the heels of the first Food and Drug Administration approval of a biosimilar rituximab product for the treatment of NHL; Celltrion’s product Truxima (formerly CT-P10), a biosimilar of Genentech’s Rituxan (rituximab), was approved Nov. 28 to treat adults with CD20-positive, B-cell NHL, either as a single agent or in combination with chemotherapy.
The PF-0528056 study was sponsored by Pfizer. Dr. Sharman has been a consultant for, and/or received research funding and honoraria from Acerta, Pharmacyclics (an AbbVie Company), Pfizer, TG Therapeutics, Abbvie, and Genentech.
SOURCE: Sharman J et al. ASH 2018: Abstract 394.
SAN DIEGO – The potential rituximab biosimilar drug PF-05280586 showed efficacy, safety, immunogenicity, pharmacokinetics, and pharmacodynamics similar to those of rituximab at up to 26 weeks in a randomized phase 3 study of treatment-naive patients with CD20-positive low tumor burden follicular lymphoma (LTB-FL).
The primary endpoint of overall response rate at 26 weeks was 75.5% in 196 patients randomized to receive PF-05280586, and 70.7% in 198 patients who received a rituximab reference product sourced from the European Union (MabThera; rituximab‑EU), Jeff Sharman, MD, reported at the annual meeting of the American Society of Hematology.
“This resulted in a difference between the two arms of 4.66%,” said Dr. Sharman of Willamette Valley Cancer Institute and Research Center, Springfield, Ore.
The 95% confidence interval for this difference ... was entirely contained within the prespecified equivalence margin, he said.
“Depth of response was a key secondary endpoint, and rates of complete response were 29.3% and 30.4%, respectively,” he said, noting that rates of partial response, stable response, and progressive disease were also similar between the two study arms.
Estimated 1-year progression-free survival (PFS) rates were also highly similar at 76.4% and 81.2% in the PF-05280586 and rituximab-EU arms.
Rapid depletion in CD19-positive B-cell counts was observed in both groups after initial dosing, with recovery by week 39 and a sustained increase until the end of week 52.
Treatment-emergent adverse events (TEAEs) occurred in 78.6% vs. 72.1% of patients in the PF‑05280586 vs. rituximab‑EU arms, respectively, and the rates of serious adverse events and grade 3 events were similar in the groups, as were rates of infusion interruptions or infusion-related reactions (IRRs), Dr. Sharman said.
IRRs occurred in about 25% of patients in each arm, and most were grade 1 or 2. Grade 3 IRRs occurred in 2.6% vs. 0.5% of patients in the groups, respectively, and no grade 4 IRRs occurred.
Rates of anti-drug antibodies were also similar in the two groups, as were serum drug concentrations – regardless of anti-drug antibody status, he noted.
Study subjects were adults with a mean age of 60 years and histologically confirmed CD20-positive grade 1-3a follicular lymphoma with no prior rituximab or system therapy for B-cell non-Hodgkin lymphoma (NHL). They had Ann Arbor disease stages II (26.9%), III (44.2%) or IV (28.9%), ECOG performance status of 0-1, and at least 1 measurable disease lesion identifiable on imaging.
Risk level as assessed by the Follicular Lymphoma International Prognostic Index–2 was low in 28.4%, medium in 66%, and high in 5.6% of patients.
Treatment with each agent was given at intravenous doses of 375 mg/m2 weekly for 4 weeks at days 1, 8, 15, and 22.
PF-05280586 is being developed by Pfizer, and in this 52-week double-blind study – the largest study to date of the early use of the potential rituximab biosimilar in patients with previously untreated CD20-positive LTB-FL – the primary endpoint was met, demonstrating its therapeutic equivalence with rituximab-EU for overall response rate at week 26, Dr. Sharman said.
“These results therefore confirm the biosimilarity of PF-05280586 with rituximab-EU,” he concluded.
Of note, the reporting of these findings comes on the heels of the first Food and Drug Administration approval of a biosimilar rituximab product for the treatment of NHL; Celltrion’s product Truxima (formerly CT-P10), a biosimilar of Genentech’s Rituxan (rituximab), was approved Nov. 28 to treat adults with CD20-positive, B-cell NHL, either as a single agent or in combination with chemotherapy.
The PF-0528056 study was sponsored by Pfizer. Dr. Sharman has been a consultant for, and/or received research funding and honoraria from Acerta, Pharmacyclics (an AbbVie Company), Pfizer, TG Therapeutics, Abbvie, and Genentech.
SOURCE: Sharman J et al. ASH 2018: Abstract 394.
SAN DIEGO – The potential rituximab biosimilar drug PF-05280586 showed efficacy, safety, immunogenicity, pharmacokinetics, and pharmacodynamics similar to those of rituximab at up to 26 weeks in a randomized phase 3 study of treatment-naive patients with CD20-positive low tumor burden follicular lymphoma (LTB-FL).
The primary endpoint of overall response rate at 26 weeks was 75.5% in 196 patients randomized to receive PF-05280586, and 70.7% in 198 patients who received a rituximab reference product sourced from the European Union (MabThera; rituximab‑EU), Jeff Sharman, MD, reported at the annual meeting of the American Society of Hematology.
“This resulted in a difference between the two arms of 4.66%,” said Dr. Sharman of Willamette Valley Cancer Institute and Research Center, Springfield, Ore.
The 95% confidence interval for this difference ... was entirely contained within the prespecified equivalence margin, he said.
“Depth of response was a key secondary endpoint, and rates of complete response were 29.3% and 30.4%, respectively,” he said, noting that rates of partial response, stable response, and progressive disease were also similar between the two study arms.
Estimated 1-year progression-free survival (PFS) rates were also highly similar at 76.4% and 81.2% in the PF-05280586 and rituximab-EU arms.
Rapid depletion in CD19-positive B-cell counts was observed in both groups after initial dosing, with recovery by week 39 and a sustained increase until the end of week 52.
Treatment-emergent adverse events (TEAEs) occurred in 78.6% vs. 72.1% of patients in the PF‑05280586 vs. rituximab‑EU arms, respectively, and the rates of serious adverse events and grade 3 events were similar in the groups, as were rates of infusion interruptions or infusion-related reactions (IRRs), Dr. Sharman said.
IRRs occurred in about 25% of patients in each arm, and most were grade 1 or 2. Grade 3 IRRs occurred in 2.6% vs. 0.5% of patients in the groups, respectively, and no grade 4 IRRs occurred.
Rates of anti-drug antibodies were also similar in the two groups, as were serum drug concentrations – regardless of anti-drug antibody status, he noted.
Study subjects were adults with a mean age of 60 years and histologically confirmed CD20-positive grade 1-3a follicular lymphoma with no prior rituximab or system therapy for B-cell non-Hodgkin lymphoma (NHL). They had Ann Arbor disease stages II (26.9%), III (44.2%) or IV (28.9%), ECOG performance status of 0-1, and at least 1 measurable disease lesion identifiable on imaging.
Risk level as assessed by the Follicular Lymphoma International Prognostic Index–2 was low in 28.4%, medium in 66%, and high in 5.6% of patients.
Treatment with each agent was given at intravenous doses of 375 mg/m2 weekly for 4 weeks at days 1, 8, 15, and 22.
PF-05280586 is being developed by Pfizer, and in this 52-week double-blind study – the largest study to date of the early use of the potential rituximab biosimilar in patients with previously untreated CD20-positive LTB-FL – the primary endpoint was met, demonstrating its therapeutic equivalence with rituximab-EU for overall response rate at week 26, Dr. Sharman said.
“These results therefore confirm the biosimilarity of PF-05280586 with rituximab-EU,” he concluded.
Of note, the reporting of these findings comes on the heels of the first Food and Drug Administration approval of a biosimilar rituximab product for the treatment of NHL; Celltrion’s product Truxima (formerly CT-P10), a biosimilar of Genentech’s Rituxan (rituximab), was approved Nov. 28 to treat adults with CD20-positive, B-cell NHL, either as a single agent or in combination with chemotherapy.
The PF-0528056 study was sponsored by Pfizer. Dr. Sharman has been a consultant for, and/or received research funding and honoraria from Acerta, Pharmacyclics (an AbbVie Company), Pfizer, TG Therapeutics, Abbvie, and Genentech.
SOURCE: Sharman J et al. ASH 2018: Abstract 394.
REPORTING FROM ASH 2018
Key clinical point: PF-05280586 shows biosimilarity to rituximab at up to 26 weeks.
Major finding: ORR at 26 weeks was 75.5% vs. 70.7% with PF-05280586 vs. rituximab, respectively.
Study details: A phase 3 study of 394 patients.
Disclosures: This study was sponsored by Pfizer. Dr. Sharman has been a consultant for, and/or received research funding and honoraria from Acerta, Pharmacyclics (an AbbVie Company), Pfizer, TG Therapeutics, Abbvie, and Genentech.
Source: Sharman J et al. ASH 2018: Abstract 394.
CLL resistance mechanism to venetoclax identified
SAN DIEGO – A recurrent mutation in BCL2, the therapeutic target of venetoclax (Venclexta), appears to be a major contributor to drug resistance in patients with chronic lymphocytic leukemia (CLL), investigators reported.
The mutation has been detected in some patients with CLL up to 2 years before resistance to venetoclax actually develops, said lead author Piers Blombery, MBBS, from the Peter MacCallum Cancer Center in Melbourne.
“We have identified the first acquired BCL2 mutation developed in patients clinically treated with venetoclax,” he said in a late-breaking oral abstract session at the annual meeting of the American Society of Hematology.
The mutation, which the investigators have labeled BCL2 Gly101Val, “is a recurrent and frequent mediator of resistance and may be detected years before clinical relapse occurs,” he added.
The paper was published online in Cancer Discovery (2018 Dec 4. doi: 10.1158/2159-8290.CD-18-1119) to coincide with the presentation at ASH.
Despite the demonstrated efficacy of venetoclax as continuous therapy in patients with relapsed or refractory CLL, the majority of patients experience disease progression, prompting the investigators to explore molecular mechanisms of secondary resistance.
To do this, they analyzed paired samples from 15 patients with CLL, enrolled in clinical trials of venetoclax, collected both before the start of venetoclax therapy and at the time of disease progression.
In seven of the patients, they identified a novel mutation that showed up at the time of progression, but was absent from the pre-venetoclax samples. The mutation first became detectable from about 19 to 42 months after the start of therapy and preceded clinical progression by as much as 25 months, the investigators found.
They pinned the mutation down to the BH3-binding groove on BCL2, the same molecular site targeted by venetoclax. They found that the mutation was not present in samples from 96 patients with venetoclax-naive CLL nor in any other B-cell malignancies. Searches for references to the mutation in both a cancer database (COSMIC) and a population database (gnomAD) came up empty.
In other experiments, they determined that cell lines overexpressing BCL2 Gly101Val are resistant to venetoclax, and that in the presence of venetoclax in vitro, BCL2 Gly101Val-expressing cells have a growth advantage, compared with wild type cells.
Additionally, they showed that the mutation results in impaired venetoclax binding in vitro.
“BCL2 Gly101Val is observed subclonally, implicating multiple mechanisms of venetoclax resistance in the same patient,” Dr. Blombery said.
In an interview, Dr. Blombery said that the identification of the resistance mutation is a strong rationale for using combination therapy to treat patients with relapsed or refractory CLL to help prevent or attenuate selection pressures that lead to resistance.
The investigators were supported by the Wilson Center for Lymphoma Genomics, Snowdome Foundation, National Health Medical Research Council, Leukemia and Lymphoma Society, Leukemia Foundation, Cancer Council of Victoria, and Australian Cancer Research Foundation. Dr. Blombery reported having no relevant disclosures.
SOURCE: Blombery P et al. ASH 2018, Abstract LBA-7.
SAN DIEGO – A recurrent mutation in BCL2, the therapeutic target of venetoclax (Venclexta), appears to be a major contributor to drug resistance in patients with chronic lymphocytic leukemia (CLL), investigators reported.
The mutation has been detected in some patients with CLL up to 2 years before resistance to venetoclax actually develops, said lead author Piers Blombery, MBBS, from the Peter MacCallum Cancer Center in Melbourne.
“We have identified the first acquired BCL2 mutation developed in patients clinically treated with venetoclax,” he said in a late-breaking oral abstract session at the annual meeting of the American Society of Hematology.
The mutation, which the investigators have labeled BCL2 Gly101Val, “is a recurrent and frequent mediator of resistance and may be detected years before clinical relapse occurs,” he added.
The paper was published online in Cancer Discovery (2018 Dec 4. doi: 10.1158/2159-8290.CD-18-1119) to coincide with the presentation at ASH.
Despite the demonstrated efficacy of venetoclax as continuous therapy in patients with relapsed or refractory CLL, the majority of patients experience disease progression, prompting the investigators to explore molecular mechanisms of secondary resistance.
To do this, they analyzed paired samples from 15 patients with CLL, enrolled in clinical trials of venetoclax, collected both before the start of venetoclax therapy and at the time of disease progression.
In seven of the patients, they identified a novel mutation that showed up at the time of progression, but was absent from the pre-venetoclax samples. The mutation first became detectable from about 19 to 42 months after the start of therapy and preceded clinical progression by as much as 25 months, the investigators found.
They pinned the mutation down to the BH3-binding groove on BCL2, the same molecular site targeted by venetoclax. They found that the mutation was not present in samples from 96 patients with venetoclax-naive CLL nor in any other B-cell malignancies. Searches for references to the mutation in both a cancer database (COSMIC) and a population database (gnomAD) came up empty.
In other experiments, they determined that cell lines overexpressing BCL2 Gly101Val are resistant to venetoclax, and that in the presence of venetoclax in vitro, BCL2 Gly101Val-expressing cells have a growth advantage, compared with wild type cells.
Additionally, they showed that the mutation results in impaired venetoclax binding in vitro.
“BCL2 Gly101Val is observed subclonally, implicating multiple mechanisms of venetoclax resistance in the same patient,” Dr. Blombery said.
In an interview, Dr. Blombery said that the identification of the resistance mutation is a strong rationale for using combination therapy to treat patients with relapsed or refractory CLL to help prevent or attenuate selection pressures that lead to resistance.
The investigators were supported by the Wilson Center for Lymphoma Genomics, Snowdome Foundation, National Health Medical Research Council, Leukemia and Lymphoma Society, Leukemia Foundation, Cancer Council of Victoria, and Australian Cancer Research Foundation. Dr. Blombery reported having no relevant disclosures.
SOURCE: Blombery P et al. ASH 2018, Abstract LBA-7.
SAN DIEGO – A recurrent mutation in BCL2, the therapeutic target of venetoclax (Venclexta), appears to be a major contributor to drug resistance in patients with chronic lymphocytic leukemia (CLL), investigators reported.
The mutation has been detected in some patients with CLL up to 2 years before resistance to venetoclax actually develops, said lead author Piers Blombery, MBBS, from the Peter MacCallum Cancer Center in Melbourne.
“We have identified the first acquired BCL2 mutation developed in patients clinically treated with venetoclax,” he said in a late-breaking oral abstract session at the annual meeting of the American Society of Hematology.
The mutation, which the investigators have labeled BCL2 Gly101Val, “is a recurrent and frequent mediator of resistance and may be detected years before clinical relapse occurs,” he added.
The paper was published online in Cancer Discovery (2018 Dec 4. doi: 10.1158/2159-8290.CD-18-1119) to coincide with the presentation at ASH.
Despite the demonstrated efficacy of venetoclax as continuous therapy in patients with relapsed or refractory CLL, the majority of patients experience disease progression, prompting the investigators to explore molecular mechanisms of secondary resistance.
To do this, they analyzed paired samples from 15 patients with CLL, enrolled in clinical trials of venetoclax, collected both before the start of venetoclax therapy and at the time of disease progression.
In seven of the patients, they identified a novel mutation that showed up at the time of progression, but was absent from the pre-venetoclax samples. The mutation first became detectable from about 19 to 42 months after the start of therapy and preceded clinical progression by as much as 25 months, the investigators found.
They pinned the mutation down to the BH3-binding groove on BCL2, the same molecular site targeted by venetoclax. They found that the mutation was not present in samples from 96 patients with venetoclax-naive CLL nor in any other B-cell malignancies. Searches for references to the mutation in both a cancer database (COSMIC) and a population database (gnomAD) came up empty.
In other experiments, they determined that cell lines overexpressing BCL2 Gly101Val are resistant to venetoclax, and that in the presence of venetoclax in vitro, BCL2 Gly101Val-expressing cells have a growth advantage, compared with wild type cells.
Additionally, they showed that the mutation results in impaired venetoclax binding in vitro.
“BCL2 Gly101Val is observed subclonally, implicating multiple mechanisms of venetoclax resistance in the same patient,” Dr. Blombery said.
In an interview, Dr. Blombery said that the identification of the resistance mutation is a strong rationale for using combination therapy to treat patients with relapsed or refractory CLL to help prevent or attenuate selection pressures that lead to resistance.
The investigators were supported by the Wilson Center for Lymphoma Genomics, Snowdome Foundation, National Health Medical Research Council, Leukemia and Lymphoma Society, Leukemia Foundation, Cancer Council of Victoria, and Australian Cancer Research Foundation. Dr. Blombery reported having no relevant disclosures.
SOURCE: Blombery P et al. ASH 2018, Abstract LBA-7.
REPORTING FROM ASH 2018
Key clinical point:
Major finding: The mutation was identified in samples from seven patients after venetoclax therapy, but not in any of the pretherapy samples.
Study details: Genetic analysis of CLL mutations in 15 patients enrolled in clinical trials of venetoclax.
Disclosures: The investigators were supported by the Wilson Center for Lymphoma Genomics, Snowdome Foundation, National Health Medical Research Council, Leukemia and Lymphoma Society, Leukemia Foundation, Cancer Council of Victoria, and Australian Cancer Research Foundation. Dr. Blombery reported having no relevant disclosures.
Source: Blombery P et al. ASH 2018, Abstract LBA-7.

JULIET: CAR T cells go the distance in r/r DLBCL
SAN DIEGO – Two-thirds of adults with relapsed or refractory diffuse large B-cell lymphoma who had early responses to chimeric antigen receptor T-cell (CAR T) therapy with tisagenlecleucel (Kymriah) remain in remission with no evidence of minimal residual disease, according to an updated analysis of the JULIET trial.
In the single-arm, open-label trial, the overall response rate after 19 months of follow-up was 54%, including 40% complete remissions and 14% partial remissions. The median duration of response had not been reached at the time of data cutoff, and the median overall survival had not been reached for patients with a complete remission. Overall survival in this heavily pretreated population as a whole (all patients who received CAR T-cell infusions) was 11.1 months.
Adverse events were similar to those previously reported and were manageable, according to investigator Richard Thomas Maziarz, MD, from the Oregon Health & Science Knight Cancer Institute in Portland.
In this video interview at the annual meeting of the American Society of Hematology, Dr. Maziarz discusses the promising results using CAR T cells in this difficult to treat population.
SAN DIEGO – Two-thirds of adults with relapsed or refractory diffuse large B-cell lymphoma who had early responses to chimeric antigen receptor T-cell (CAR T) therapy with tisagenlecleucel (Kymriah) remain in remission with no evidence of minimal residual disease, according to an updated analysis of the JULIET trial.
In the single-arm, open-label trial, the overall response rate after 19 months of follow-up was 54%, including 40% complete remissions and 14% partial remissions. The median duration of response had not been reached at the time of data cutoff, and the median overall survival had not been reached for patients with a complete remission. Overall survival in this heavily pretreated population as a whole (all patients who received CAR T-cell infusions) was 11.1 months.
Adverse events were similar to those previously reported and were manageable, according to investigator Richard Thomas Maziarz, MD, from the Oregon Health & Science Knight Cancer Institute in Portland.
In this video interview at the annual meeting of the American Society of Hematology, Dr. Maziarz discusses the promising results using CAR T cells in this difficult to treat population.
SAN DIEGO – Two-thirds of adults with relapsed or refractory diffuse large B-cell lymphoma who had early responses to chimeric antigen receptor T-cell (CAR T) therapy with tisagenlecleucel (Kymriah) remain in remission with no evidence of minimal residual disease, according to an updated analysis of the JULIET trial.
In the single-arm, open-label trial, the overall response rate after 19 months of follow-up was 54%, including 40% complete remissions and 14% partial remissions. The median duration of response had not been reached at the time of data cutoff, and the median overall survival had not been reached for patients with a complete remission. Overall survival in this heavily pretreated population as a whole (all patients who received CAR T-cell infusions) was 11.1 months.
Adverse events were similar to those previously reported and were manageable, according to investigator Richard Thomas Maziarz, MD, from the Oregon Health & Science Knight Cancer Institute in Portland.
In this video interview at the annual meeting of the American Society of Hematology, Dr. Maziarz discusses the promising results using CAR T cells in this difficult to treat population.
REPORTING FROM ASH 2018
