User login
Venetoclax plus ibrutinib appears to suit elderly and high-risk patients with CLL
A combination of venetoclax and ibrutinib may be a safe and effective treatment option for previously untreated elderly and high-risk patients with chronic lymphocytic leukemia (CLL), according to investigators of a phase 2 trial of the combination.
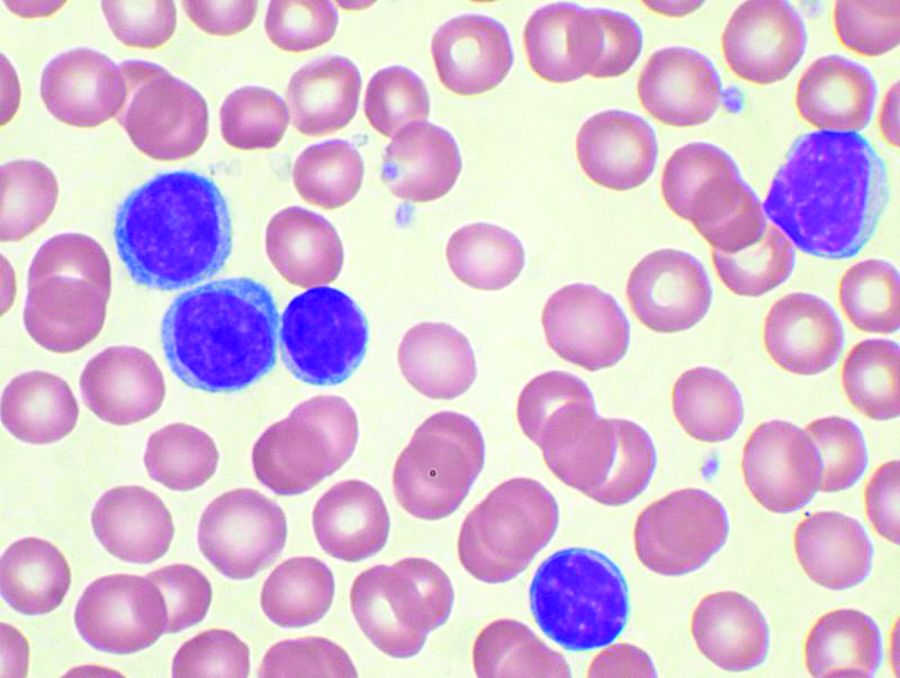
About 88% of patients achieved complete remission or complete remission with incomplete count recovery after 12 cycles of treatment, reported lead author Nitin Jain, MD, of the University of Texas MD Anderson Cancer Center, Houston, and colleagues.
There were no new safety signals for the combination of ibrutinib, an irreversible inhibitor of Bruton’s tyrosine kinase, and venetoclax, a B-cell lymphoma 2 protein inhibitor, the investigators noted.
“This combination was reported to be safe and active in patients with mantle cell lymphoma,” they wrote in the New England Journal of Medicine. “Given the clinically complementary activity, preclinical synergism, and nonoverlapping toxic effects, we examined the safety and efficacy of combined ibrutinib and venetoclax treatment in previously untreated patients with CLL.”
In particular, the investigators recruited older patients, as this is a common population that can be challenging to treat. “Because CLL typically occurs in older adults, the majority of patients who need treatment are older than 65 years of age,” the investigators wrote. “This group of patients often has unacceptable side effects and has a lower rate of complete remission and undetectable minimal residual disease with chemoimmunotherapy than younger patients.”
The open-label, phase 2 trial enrolled 80 elderly and high-risk patients with previously untreated CLL. Eligibility required an age of at least 65 years or presence of at least one high-risk genetic feature; namely, mutated TP53, unmutated IgVH, or chromosome 11q deletion.
In order to reduce the risk of tumor lysis syndrome, ibrutinib (420 mg once daily) was given as monotherapy for three 28-day cycles. From the fourth cycle onward, venetoclax was also given, with weekly dose escalations to a target dose of 400 mg once daily. The combination was given for 24 cycles, with treatment continuation offered to patients who were still positive for minimal residual disease.
The median patient age was 65 years, with 30% of the population aged 70 years or older. A large majority (92%) had at least one high-risk genetic feature.
Following initiation with three cycles of ibrutinib, most patients had partial responses, the investigators wrote; however, with the addition of venetoclax, responses improved over time. Of all 80 patients, 59 (74%) had a best response of complete remission or complete remission with incomplete count recovery.
After six cycles, 51 out of 70 patients (73%) achieved this marker. After 12 cycles, 29 of 33 patients (88%) had this response, with 61% of the same group demonstrating undetectable minimal residual disease in bone marrow.
After 18 cycles, 25 of 26 patients (96%) had complete remission or complete remission with incomplete count recovery, 18 of which (69%) were negative for minimal residual disease. Three patients completed 24 cycles of combined therapy, all of whom achieved complete remission or complete remission with incomplete count recovery and undetectable minimal residual disease.
Focusing on patients aged 65 years or older, 74% had complete remission or complete remission with incomplete count recovery after six cycles of therapy and nearly half (44%) had undetectable minimal residual disease. After 12 cycles, these rates increased to 94% and 76%, respectively. Responses were also seen across genetically high-risk subgroups.
One patient died from a cryptococcal infection of the central nervous system; this was deemed unrelated to treatment, as symptoms began prior to initiation of treatment and only one dose of ibrutinib was given.
The estimated 1-year progression-free survival rate was 98% and the estimated overall survival rate was 99%. At the time of publication, no patients had disease progression.
Among all patients, 60% experienced grade 3 or higher adverse events, the most common being neutropenia (48%).
Almost half of the patient population (44%) required dose reductions of ibrutinib, most commonly because of atrial fibrillation, and 24% required dose reductions of venetoclax, most often because of neutropenia.
“Our data showed that combination therapy with ibrutinib and venetoclax was effective in patients with CLL, with no new toxic effects from the combination that were not reported previously for the individual agents,” the investigators wrote, adding that the efficacy findings were also “substantially better” than what has been reported with monotherapy for each of the agents in patients with CLL.
The study was funded by AbbVie, the University of Texas MD Anderson Cancer Center Chronic Lymphocytic Leukemia Moon Shot program, the Andrew Sabin Family Foundation, and the CLL Global Research Foundation. The investigators reported relationships with AbbVie, Incyte, Celgene, and other companies.
SOURCE: Jain N et al. N Engl J Med. 2019;380:2095-103.
In addition to noting the “impressive” results from combining venetoclax and ibrutinib as frontline CLL therapy, Adrian Wiestner, MD, PhD, highlighted the lack of a Kaplan-Meier curve in the paper published by Jain et al. in the New England Journal of Medicine.
“Here, assessment of minimal residual disease has replaced the progression-free survival curve of old, indicating a possible shift in focus away from traditional clinical trial endpoints and toward even more stringent measures of clinical efficacy that may be central to regulatory decisions,” Dr. Wiestner wrote.
Dr. Wiestner of the National Institutes of Health made his remarks in an accompanying editorial (N Engl J Med. 2019 May 29. doi: 10.1056/NEJMe1904362). He reported grants from with Merck, Pharmacyclics (an AbbVie company), and Acerta Pharma.
In addition to noting the “impressive” results from combining venetoclax and ibrutinib as frontline CLL therapy, Adrian Wiestner, MD, PhD, highlighted the lack of a Kaplan-Meier curve in the paper published by Jain et al. in the New England Journal of Medicine.
“Here, assessment of minimal residual disease has replaced the progression-free survival curve of old, indicating a possible shift in focus away from traditional clinical trial endpoints and toward even more stringent measures of clinical efficacy that may be central to regulatory decisions,” Dr. Wiestner wrote.
Dr. Wiestner of the National Institutes of Health made his remarks in an accompanying editorial (N Engl J Med. 2019 May 29. doi: 10.1056/NEJMe1904362). He reported grants from with Merck, Pharmacyclics (an AbbVie company), and Acerta Pharma.
In addition to noting the “impressive” results from combining venetoclax and ibrutinib as frontline CLL therapy, Adrian Wiestner, MD, PhD, highlighted the lack of a Kaplan-Meier curve in the paper published by Jain et al. in the New England Journal of Medicine.
“Here, assessment of minimal residual disease has replaced the progression-free survival curve of old, indicating a possible shift in focus away from traditional clinical trial endpoints and toward even more stringent measures of clinical efficacy that may be central to regulatory decisions,” Dr. Wiestner wrote.
Dr. Wiestner of the National Institutes of Health made his remarks in an accompanying editorial (N Engl J Med. 2019 May 29. doi: 10.1056/NEJMe1904362). He reported grants from with Merck, Pharmacyclics (an AbbVie company), and Acerta Pharma.
A combination of venetoclax and ibrutinib may be a safe and effective treatment option for previously untreated elderly and high-risk patients with chronic lymphocytic leukemia (CLL), according to investigators of a phase 2 trial of the combination.
About 88% of patients achieved complete remission or complete remission with incomplete count recovery after 12 cycles of treatment, reported lead author Nitin Jain, MD, of the University of Texas MD Anderson Cancer Center, Houston, and colleagues.
There were no new safety signals for the combination of ibrutinib, an irreversible inhibitor of Bruton’s tyrosine kinase, and venetoclax, a B-cell lymphoma 2 protein inhibitor, the investigators noted.
“This combination was reported to be safe and active in patients with mantle cell lymphoma,” they wrote in the New England Journal of Medicine. “Given the clinically complementary activity, preclinical synergism, and nonoverlapping toxic effects, we examined the safety and efficacy of combined ibrutinib and venetoclax treatment in previously untreated patients with CLL.”
In particular, the investigators recruited older patients, as this is a common population that can be challenging to treat. “Because CLL typically occurs in older adults, the majority of patients who need treatment are older than 65 years of age,” the investigators wrote. “This group of patients often has unacceptable side effects and has a lower rate of complete remission and undetectable minimal residual disease with chemoimmunotherapy than younger patients.”
The open-label, phase 2 trial enrolled 80 elderly and high-risk patients with previously untreated CLL. Eligibility required an age of at least 65 years or presence of at least one high-risk genetic feature; namely, mutated TP53, unmutated IgVH, or chromosome 11q deletion.
In order to reduce the risk of tumor lysis syndrome, ibrutinib (420 mg once daily) was given as monotherapy for three 28-day cycles. From the fourth cycle onward, venetoclax was also given, with weekly dose escalations to a target dose of 400 mg once daily. The combination was given for 24 cycles, with treatment continuation offered to patients who were still positive for minimal residual disease.
The median patient age was 65 years, with 30% of the population aged 70 years or older. A large majority (92%) had at least one high-risk genetic feature.
Following initiation with three cycles of ibrutinib, most patients had partial responses, the investigators wrote; however, with the addition of venetoclax, responses improved over time. Of all 80 patients, 59 (74%) had a best response of complete remission or complete remission with incomplete count recovery.
After six cycles, 51 out of 70 patients (73%) achieved this marker. After 12 cycles, 29 of 33 patients (88%) had this response, with 61% of the same group demonstrating undetectable minimal residual disease in bone marrow.
After 18 cycles, 25 of 26 patients (96%) had complete remission or complete remission with incomplete count recovery, 18 of which (69%) were negative for minimal residual disease. Three patients completed 24 cycles of combined therapy, all of whom achieved complete remission or complete remission with incomplete count recovery and undetectable minimal residual disease.
Focusing on patients aged 65 years or older, 74% had complete remission or complete remission with incomplete count recovery after six cycles of therapy and nearly half (44%) had undetectable minimal residual disease. After 12 cycles, these rates increased to 94% and 76%, respectively. Responses were also seen across genetically high-risk subgroups.
One patient died from a cryptococcal infection of the central nervous system; this was deemed unrelated to treatment, as symptoms began prior to initiation of treatment and only one dose of ibrutinib was given.
The estimated 1-year progression-free survival rate was 98% and the estimated overall survival rate was 99%. At the time of publication, no patients had disease progression.
Among all patients, 60% experienced grade 3 or higher adverse events, the most common being neutropenia (48%).
Almost half of the patient population (44%) required dose reductions of ibrutinib, most commonly because of atrial fibrillation, and 24% required dose reductions of venetoclax, most often because of neutropenia.
“Our data showed that combination therapy with ibrutinib and venetoclax was effective in patients with CLL, with no new toxic effects from the combination that were not reported previously for the individual agents,” the investigators wrote, adding that the efficacy findings were also “substantially better” than what has been reported with monotherapy for each of the agents in patients with CLL.
The study was funded by AbbVie, the University of Texas MD Anderson Cancer Center Chronic Lymphocytic Leukemia Moon Shot program, the Andrew Sabin Family Foundation, and the CLL Global Research Foundation. The investigators reported relationships with AbbVie, Incyte, Celgene, and other companies.
SOURCE: Jain N et al. N Engl J Med. 2019;380:2095-103.
A combination of venetoclax and ibrutinib may be a safe and effective treatment option for previously untreated elderly and high-risk patients with chronic lymphocytic leukemia (CLL), according to investigators of a phase 2 trial of the combination.
About 88% of patients achieved complete remission or complete remission with incomplete count recovery after 12 cycles of treatment, reported lead author Nitin Jain, MD, of the University of Texas MD Anderson Cancer Center, Houston, and colleagues.
There were no new safety signals for the combination of ibrutinib, an irreversible inhibitor of Bruton’s tyrosine kinase, and venetoclax, a B-cell lymphoma 2 protein inhibitor, the investigators noted.
“This combination was reported to be safe and active in patients with mantle cell lymphoma,” they wrote in the New England Journal of Medicine. “Given the clinically complementary activity, preclinical synergism, and nonoverlapping toxic effects, we examined the safety and efficacy of combined ibrutinib and venetoclax treatment in previously untreated patients with CLL.”
In particular, the investigators recruited older patients, as this is a common population that can be challenging to treat. “Because CLL typically occurs in older adults, the majority of patients who need treatment are older than 65 years of age,” the investigators wrote. “This group of patients often has unacceptable side effects and has a lower rate of complete remission and undetectable minimal residual disease with chemoimmunotherapy than younger patients.”
The open-label, phase 2 trial enrolled 80 elderly and high-risk patients with previously untreated CLL. Eligibility required an age of at least 65 years or presence of at least one high-risk genetic feature; namely, mutated TP53, unmutated IgVH, or chromosome 11q deletion.
In order to reduce the risk of tumor lysis syndrome, ibrutinib (420 mg once daily) was given as monotherapy for three 28-day cycles. From the fourth cycle onward, venetoclax was also given, with weekly dose escalations to a target dose of 400 mg once daily. The combination was given for 24 cycles, with treatment continuation offered to patients who were still positive for minimal residual disease.
The median patient age was 65 years, with 30% of the population aged 70 years or older. A large majority (92%) had at least one high-risk genetic feature.
Following initiation with three cycles of ibrutinib, most patients had partial responses, the investigators wrote; however, with the addition of venetoclax, responses improved over time. Of all 80 patients, 59 (74%) had a best response of complete remission or complete remission with incomplete count recovery.
After six cycles, 51 out of 70 patients (73%) achieved this marker. After 12 cycles, 29 of 33 patients (88%) had this response, with 61% of the same group demonstrating undetectable minimal residual disease in bone marrow.
After 18 cycles, 25 of 26 patients (96%) had complete remission or complete remission with incomplete count recovery, 18 of which (69%) were negative for minimal residual disease. Three patients completed 24 cycles of combined therapy, all of whom achieved complete remission or complete remission with incomplete count recovery and undetectable minimal residual disease.
Focusing on patients aged 65 years or older, 74% had complete remission or complete remission with incomplete count recovery after six cycles of therapy and nearly half (44%) had undetectable minimal residual disease. After 12 cycles, these rates increased to 94% and 76%, respectively. Responses were also seen across genetically high-risk subgroups.
One patient died from a cryptococcal infection of the central nervous system; this was deemed unrelated to treatment, as symptoms began prior to initiation of treatment and only one dose of ibrutinib was given.
The estimated 1-year progression-free survival rate was 98% and the estimated overall survival rate was 99%. At the time of publication, no patients had disease progression.
Among all patients, 60% experienced grade 3 or higher adverse events, the most common being neutropenia (48%).
Almost half of the patient population (44%) required dose reductions of ibrutinib, most commonly because of atrial fibrillation, and 24% required dose reductions of venetoclax, most often because of neutropenia.
“Our data showed that combination therapy with ibrutinib and venetoclax was effective in patients with CLL, with no new toxic effects from the combination that were not reported previously for the individual agents,” the investigators wrote, adding that the efficacy findings were also “substantially better” than what has been reported with monotherapy for each of the agents in patients with CLL.
The study was funded by AbbVie, the University of Texas MD Anderson Cancer Center Chronic Lymphocytic Leukemia Moon Shot program, the Andrew Sabin Family Foundation, and the CLL Global Research Foundation. The investigators reported relationships with AbbVie, Incyte, Celgene, and other companies.
SOURCE: Jain N et al. N Engl J Med. 2019;380:2095-103.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point:
Major finding: After 12 cycles of treatment with venetoclax and ibrutinib, 88% of patients had complete remission or complete remission with incomplete count recovery.
Study details: A randomized, open-label, phase 2 study involving 80 elderly and high-risk patients with chronic lymphocytic leukemia.
Disclosures: The study was funded by AbbVie, the University of Texas MD Anderson Cancer Center Chronic Lymphocytic Leukemia Moon Shot program, the Andrew Sabin Family Foundation, and the CLL Global Research Foundation. The investigators reported relationships with AbbVie, Incyte, Celgene, and other companies.
Source: Jain N et al. N Engl J Med. 2019;380:2095-103.
Daratumumab regimen shows benefit in transplant-ineligible myeloma
For patients with newly diagnosed multiple myeloma who are ineligible for autologous stem cell transplantation (ASCT), adding daratumumab to lenalidomide and dexamethasone provides better outcomes than standard therapy alone, based on an interim analysis from the phase 3 MAIA trial.
A greater proportion of patients in the daratumumab group had complete responses and were alive without disease progression after a median follow-up of 28 months, reported lead author Thierry Facon, MD, of the University of Lille (France) and colleagues, who also noted that daratumumab was associated with higher rates of grade 3 or 4 pneumonia, neutropenia, and lymphopenia.
“For patients who are ineligible for stem-cell transplantation, multiagent regimens, including alkylating agents, glucocorticoids, immunomodulatory drugs, proteasome inhibitors, and new agents, are the standard of care,” the investigators wrote in the New England Journal of Medicine.
The findings from MAIA add clarity to the efficacy and safety of daratumumab in this setting, building on previous phase 3 myeloma trials in the same area, such as ALCYONE, CASTOR, and POLLUX, the investigators noted.
MAIA was an open-label, international trial involving 737 patients with newly diagnosed multiple myeloma who were ineligible for ASCT. Patients were randomized in a 1:1 ratio to receive either daratumumab, lenalidomide, and dexamethasone (daratumumab group; n = 368) or lenalidomide and dexamethasone alone (control group; n = 369).
On a 28-day cycle, all patients received oral lenalidomide 25 mg on days 1-21 and oral dexamethasone 40 mg on days 1, 8, 15, and 22. Patients in the daratumumab group received intravenous daratumumab dosed at 16 mg/kg once a week for cycles 1 and 2, every 2 weeks for cycles 3-6, and then every 4 weeks thereafter. Treatment was continued until unacceptable toxic effects or disease progression occurred.
The primary end point was progression-free survival (PFS). Various secondary end points were also evaluated, including time to progression, complete responses, overall survival, and others.
Among the 737 randomized patients, 729 ultimately underwent treatment. The median patient age was 73 years.
Generally, efficacy measures favored adding daratumumab. After a median follow-up of 28.0 months, disease progression or death had occurred in 26.4% of patients in the daratumumab group, compared with 38.8% in the control group.
The median PFS was not reached in the daratumumab group, compared with 31.9 months in the control group. There was a 44% lower risk of disease progression or death among patients who received daratumumab, compared with the control group (hazard ratio, 0.56, P less than .001).
This PFS trend was consistent across most subgroups, including those for sex, age, and race, with the exception of patients with baseline hepatic impairment.
Additional efficacy measures added weight to the apparent benefit of adding daratumumab. For instance, more patients in the daratumumab group achieved a complete response or better (47.6% vs. 24.9%) and were negative for minimum residual disease (24.2% vs. 7.3%).
In terms of safety, more patients in the daratumumab group than the control group developed grade 3 or higher neutropenia (50% vs. 35.3%), lymphopenia (15.1% vs. 10.7%), infections (32.1% vs. 23.3%) or pneumonia (13.7% vs. 7.9%).
In contrast, grade 3 or 4 anemia was less common in the daratumumab group than the control group (11.8% vs. 19.7%). Overall, the rate of serious adverse events was similar for both groups (approximately 63%), as was the rate of adverse events resulting in death (approximately 6%-7%).
“In this trial involving patients with newly diagnosed multiple myeloma who were ineligible for stem-cell transplantation, the addition of daratumumab to lenalidomide and dexamethasone resulted in significantly longer progression-free survival, a higher response rate, an increased depth of response, and a longer duration of response than lenalidomide and dexamethasone alone,” the investigators concluded.
The study was funded by Janssen Research and Development. The investigators reported relationships with Janssen, Celgene, Takeda, Sanofi, and other companies.
SOURCE: Facon T et al. N Engl J Med. 2019;380:2104-15.
The findings from the phase 3 MAIA trial highlight the “superior efficacy” of adding daratumumab to lenalidomide and dexamethasone for patients with newly diagnosed multiple myeloma who are ineligible for stem cell transplantation, Jacob Laubach, MD, commented in an accompanying editorial.
Dr. Laubach noted several important clinical implications of the study findings, including that the use of CD38-targeting monoclonal antibody therapy was associated with a significant improvement in the number of patients who had a complete response to therapy and who were negative for minimal residual disease.
However, with daratumumab as a component of induction and maintenance therapy for patients with multiple myeloma who are ineligible for transplantation, it is important to consider the feasibility of retreatment with CD38-targeting therapy in patients who become resistant to daratumumab-containing regimens.
Jacob Laubach, MD, is at the Dana-Farber Cancer Institute in Boston. He reported having no financial disclosures. He made his remarks in an editorial in the New England Journal of Medicine (2019;380:2172-3).
The findings from the phase 3 MAIA trial highlight the “superior efficacy” of adding daratumumab to lenalidomide and dexamethasone for patients with newly diagnosed multiple myeloma who are ineligible for stem cell transplantation, Jacob Laubach, MD, commented in an accompanying editorial.
Dr. Laubach noted several important clinical implications of the study findings, including that the use of CD38-targeting monoclonal antibody therapy was associated with a significant improvement in the number of patients who had a complete response to therapy and who were negative for minimal residual disease.
However, with daratumumab as a component of induction and maintenance therapy for patients with multiple myeloma who are ineligible for transplantation, it is important to consider the feasibility of retreatment with CD38-targeting therapy in patients who become resistant to daratumumab-containing regimens.
Jacob Laubach, MD, is at the Dana-Farber Cancer Institute in Boston. He reported having no financial disclosures. He made his remarks in an editorial in the New England Journal of Medicine (2019;380:2172-3).
The findings from the phase 3 MAIA trial highlight the “superior efficacy” of adding daratumumab to lenalidomide and dexamethasone for patients with newly diagnosed multiple myeloma who are ineligible for stem cell transplantation, Jacob Laubach, MD, commented in an accompanying editorial.
Dr. Laubach noted several important clinical implications of the study findings, including that the use of CD38-targeting monoclonal antibody therapy was associated with a significant improvement in the number of patients who had a complete response to therapy and who were negative for minimal residual disease.
However, with daratumumab as a component of induction and maintenance therapy for patients with multiple myeloma who are ineligible for transplantation, it is important to consider the feasibility of retreatment with CD38-targeting therapy in patients who become resistant to daratumumab-containing regimens.
Jacob Laubach, MD, is at the Dana-Farber Cancer Institute in Boston. He reported having no financial disclosures. He made his remarks in an editorial in the New England Journal of Medicine (2019;380:2172-3).
For patients with newly diagnosed multiple myeloma who are ineligible for autologous stem cell transplantation (ASCT), adding daratumumab to lenalidomide and dexamethasone provides better outcomes than standard therapy alone, based on an interim analysis from the phase 3 MAIA trial.
A greater proportion of patients in the daratumumab group had complete responses and were alive without disease progression after a median follow-up of 28 months, reported lead author Thierry Facon, MD, of the University of Lille (France) and colleagues, who also noted that daratumumab was associated with higher rates of grade 3 or 4 pneumonia, neutropenia, and lymphopenia.
“For patients who are ineligible for stem-cell transplantation, multiagent regimens, including alkylating agents, glucocorticoids, immunomodulatory drugs, proteasome inhibitors, and new agents, are the standard of care,” the investigators wrote in the New England Journal of Medicine.
The findings from MAIA add clarity to the efficacy and safety of daratumumab in this setting, building on previous phase 3 myeloma trials in the same area, such as ALCYONE, CASTOR, and POLLUX, the investigators noted.
MAIA was an open-label, international trial involving 737 patients with newly diagnosed multiple myeloma who were ineligible for ASCT. Patients were randomized in a 1:1 ratio to receive either daratumumab, lenalidomide, and dexamethasone (daratumumab group; n = 368) or lenalidomide and dexamethasone alone (control group; n = 369).
On a 28-day cycle, all patients received oral lenalidomide 25 mg on days 1-21 and oral dexamethasone 40 mg on days 1, 8, 15, and 22. Patients in the daratumumab group received intravenous daratumumab dosed at 16 mg/kg once a week for cycles 1 and 2, every 2 weeks for cycles 3-6, and then every 4 weeks thereafter. Treatment was continued until unacceptable toxic effects or disease progression occurred.
The primary end point was progression-free survival (PFS). Various secondary end points were also evaluated, including time to progression, complete responses, overall survival, and others.
Among the 737 randomized patients, 729 ultimately underwent treatment. The median patient age was 73 years.
Generally, efficacy measures favored adding daratumumab. After a median follow-up of 28.0 months, disease progression or death had occurred in 26.4% of patients in the daratumumab group, compared with 38.8% in the control group.
The median PFS was not reached in the daratumumab group, compared with 31.9 months in the control group. There was a 44% lower risk of disease progression or death among patients who received daratumumab, compared with the control group (hazard ratio, 0.56, P less than .001).
This PFS trend was consistent across most subgroups, including those for sex, age, and race, with the exception of patients with baseline hepatic impairment.
Additional efficacy measures added weight to the apparent benefit of adding daratumumab. For instance, more patients in the daratumumab group achieved a complete response or better (47.6% vs. 24.9%) and were negative for minimum residual disease (24.2% vs. 7.3%).
In terms of safety, more patients in the daratumumab group than the control group developed grade 3 or higher neutropenia (50% vs. 35.3%), lymphopenia (15.1% vs. 10.7%), infections (32.1% vs. 23.3%) or pneumonia (13.7% vs. 7.9%).
In contrast, grade 3 or 4 anemia was less common in the daratumumab group than the control group (11.8% vs. 19.7%). Overall, the rate of serious adverse events was similar for both groups (approximately 63%), as was the rate of adverse events resulting in death (approximately 6%-7%).
“In this trial involving patients with newly diagnosed multiple myeloma who were ineligible for stem-cell transplantation, the addition of daratumumab to lenalidomide and dexamethasone resulted in significantly longer progression-free survival, a higher response rate, an increased depth of response, and a longer duration of response than lenalidomide and dexamethasone alone,” the investigators concluded.
The study was funded by Janssen Research and Development. The investigators reported relationships with Janssen, Celgene, Takeda, Sanofi, and other companies.
SOURCE: Facon T et al. N Engl J Med. 2019;380:2104-15.
For patients with newly diagnosed multiple myeloma who are ineligible for autologous stem cell transplantation (ASCT), adding daratumumab to lenalidomide and dexamethasone provides better outcomes than standard therapy alone, based on an interim analysis from the phase 3 MAIA trial.
A greater proportion of patients in the daratumumab group had complete responses and were alive without disease progression after a median follow-up of 28 months, reported lead author Thierry Facon, MD, of the University of Lille (France) and colleagues, who also noted that daratumumab was associated with higher rates of grade 3 or 4 pneumonia, neutropenia, and lymphopenia.
“For patients who are ineligible for stem-cell transplantation, multiagent regimens, including alkylating agents, glucocorticoids, immunomodulatory drugs, proteasome inhibitors, and new agents, are the standard of care,” the investigators wrote in the New England Journal of Medicine.
The findings from MAIA add clarity to the efficacy and safety of daratumumab in this setting, building on previous phase 3 myeloma trials in the same area, such as ALCYONE, CASTOR, and POLLUX, the investigators noted.
MAIA was an open-label, international trial involving 737 patients with newly diagnosed multiple myeloma who were ineligible for ASCT. Patients were randomized in a 1:1 ratio to receive either daratumumab, lenalidomide, and dexamethasone (daratumumab group; n = 368) or lenalidomide and dexamethasone alone (control group; n = 369).
On a 28-day cycle, all patients received oral lenalidomide 25 mg on days 1-21 and oral dexamethasone 40 mg on days 1, 8, 15, and 22. Patients in the daratumumab group received intravenous daratumumab dosed at 16 mg/kg once a week for cycles 1 and 2, every 2 weeks for cycles 3-6, and then every 4 weeks thereafter. Treatment was continued until unacceptable toxic effects or disease progression occurred.
The primary end point was progression-free survival (PFS). Various secondary end points were also evaluated, including time to progression, complete responses, overall survival, and others.
Among the 737 randomized patients, 729 ultimately underwent treatment. The median patient age was 73 years.
Generally, efficacy measures favored adding daratumumab. After a median follow-up of 28.0 months, disease progression or death had occurred in 26.4% of patients in the daratumumab group, compared with 38.8% in the control group.
The median PFS was not reached in the daratumumab group, compared with 31.9 months in the control group. There was a 44% lower risk of disease progression or death among patients who received daratumumab, compared with the control group (hazard ratio, 0.56, P less than .001).
This PFS trend was consistent across most subgroups, including those for sex, age, and race, with the exception of patients with baseline hepatic impairment.
Additional efficacy measures added weight to the apparent benefit of adding daratumumab. For instance, more patients in the daratumumab group achieved a complete response or better (47.6% vs. 24.9%) and were negative for minimum residual disease (24.2% vs. 7.3%).
In terms of safety, more patients in the daratumumab group than the control group developed grade 3 or higher neutropenia (50% vs. 35.3%), lymphopenia (15.1% vs. 10.7%), infections (32.1% vs. 23.3%) or pneumonia (13.7% vs. 7.9%).
In contrast, grade 3 or 4 anemia was less common in the daratumumab group than the control group (11.8% vs. 19.7%). Overall, the rate of serious adverse events was similar for both groups (approximately 63%), as was the rate of adverse events resulting in death (approximately 6%-7%).
“In this trial involving patients with newly diagnosed multiple myeloma who were ineligible for stem-cell transplantation, the addition of daratumumab to lenalidomide and dexamethasone resulted in significantly longer progression-free survival, a higher response rate, an increased depth of response, and a longer duration of response than lenalidomide and dexamethasone alone,” the investigators concluded.
The study was funded by Janssen Research and Development. The investigators reported relationships with Janssen, Celgene, Takeda, Sanofi, and other companies.
SOURCE: Facon T et al. N Engl J Med. 2019;380:2104-15.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: For patients with newly diagnosed multiple myeloma who are ineligible for autologous stem cell transplantation, adding daratumumab to lenalidomide and dexamethasone standard therapy provides better outcomes than standard therapy alone.
Major finding: After 28-month follow-up, 26.4% of patients in the daratumumab group had disease progression or died, compared with 38.8% in the control group.
Study details: A randomized, open-label, phase 3 trial involving 737 patients with newly diagnosed multiple myeloma.
Disclosures: The study was funded by Janssen Research and Development. The investigators reported relationships with Janssen, Celgene, Takeda, Sanofi, and other companies.
Source: Facon T et al. N Engl J Med. 2019;380:2104-15.
FDA approves lenalidomide/rituximab for previously treated FL, MZL
The Food and Drug Administration has approved lenalidomide (Revlimid), in combination with rituximab, for the treatment of adult patients with previously treated follicular or marginal zone lymphoma.

FDA approval is based on results from the randomized, double-blind, phase 3 AUGMENT trial, which evaluated lenalidomide/rituximab versus rituximab and placebo in patients with previously treated follicular or marginal zone lymphoma. The median progression-free survival in those receiving lenalidomide/rituximab was 39.4 months, compared with 14.1 months for those receiving rituximab/placebo (odds ratio, 0.46; 95% confidence interval, 0.34-0.62; P less than .0001).
A numeric trend was seen in overall survival over the follow-up period of 28.3 months (16 vs. 26 deaths; hazard ratio, 0.61; 95% CI, 0.33-1.13).
The most common adverse events associated with lenalidomide/rituximab are neutropenia, diarrhea, constipation, cough, fatigue, rash, pyrexia, leukopenia, pruritus, upper respiratory tract infections, abdominal pain, anemia, headache, and thrombocytopenia. Lenalidomide also contains a boxed warning for embryo-fetal toxicity, hematologic toxicity, and venous and arterial thromboembolism.
“Chemotherapy continues to be a standard of care for indolent forms of NHL, but most patients will relapse or become refractory to their current treatment. This approval represents a new therapeutic option for previously treated patients with follicular and marginal zone lymphomas, including those who relapse or no longer respond to initial treatment,” Meghan Gutierrez, CEO of the Lymphoma Research Foundation, said in a statement.
The Food and Drug Administration has approved lenalidomide (Revlimid), in combination with rituximab, for the treatment of adult patients with previously treated follicular or marginal zone lymphoma.
FDA approval is based on results from the randomized, double-blind, phase 3 AUGMENT trial, which evaluated lenalidomide/rituximab versus rituximab and placebo in patients with previously treated follicular or marginal zone lymphoma. The median progression-free survival in those receiving lenalidomide/rituximab was 39.4 months, compared with 14.1 months for those receiving rituximab/placebo (odds ratio, 0.46; 95% confidence interval, 0.34-0.62; P less than .0001).
A numeric trend was seen in overall survival over the follow-up period of 28.3 months (16 vs. 26 deaths; hazard ratio, 0.61; 95% CI, 0.33-1.13).
The most common adverse events associated with lenalidomide/rituximab are neutropenia, diarrhea, constipation, cough, fatigue, rash, pyrexia, leukopenia, pruritus, upper respiratory tract infections, abdominal pain, anemia, headache, and thrombocytopenia. Lenalidomide also contains a boxed warning for embryo-fetal toxicity, hematologic toxicity, and venous and arterial thromboembolism.
“Chemotherapy continues to be a standard of care for indolent forms of NHL, but most patients will relapse or become refractory to their current treatment. This approval represents a new therapeutic option for previously treated patients with follicular and marginal zone lymphomas, including those who relapse or no longer respond to initial treatment,” Meghan Gutierrez, CEO of the Lymphoma Research Foundation, said in a statement.
The Food and Drug Administration has approved lenalidomide (Revlimid), in combination with rituximab, for the treatment of adult patients with previously treated follicular or marginal zone lymphoma.
FDA approval is based on results from the randomized, double-blind, phase 3 AUGMENT trial, which evaluated lenalidomide/rituximab versus rituximab and placebo in patients with previously treated follicular or marginal zone lymphoma. The median progression-free survival in those receiving lenalidomide/rituximab was 39.4 months, compared with 14.1 months for those receiving rituximab/placebo (odds ratio, 0.46; 95% confidence interval, 0.34-0.62; P less than .0001).
A numeric trend was seen in overall survival over the follow-up period of 28.3 months (16 vs. 26 deaths; hazard ratio, 0.61; 95% CI, 0.33-1.13).
The most common adverse events associated with lenalidomide/rituximab are neutropenia, diarrhea, constipation, cough, fatigue, rash, pyrexia, leukopenia, pruritus, upper respiratory tract infections, abdominal pain, anemia, headache, and thrombocytopenia. Lenalidomide also contains a boxed warning for embryo-fetal toxicity, hematologic toxicity, and venous and arterial thromboembolism.
“Chemotherapy continues to be a standard of care for indolent forms of NHL, but most patients will relapse or become refractory to their current treatment. This approval represents a new therapeutic option for previously treated patients with follicular and marginal zone lymphomas, including those who relapse or no longer respond to initial treatment,” Meghan Gutierrez, CEO of the Lymphoma Research Foundation, said in a statement.
Genetic analysis identifies prognostic markers in CLL
A genetic analysis of patients with chronic lymphocytic leukemia treated with frontline, rituximab-based regimens found that deletion 11q22 and unmutated IgVH status may predict worse prognosis.
Michaela Spunarova, MD, of Masaryk University, Brno, Czech Republic, and colleagues conducted a genetic analysis of 177 patients with chronic lymphocytic leukemia (CLL). The results of the analysis were published in Leukemia Research.
The study focused on patients with CLL with an intact TP53 gene, looking at recurrently muted genes in CLL, genomic aberrations by fluorescence in situ hybridization, and IgVH status, according to the researchers.
The team analyzed the effects of these mutations on progression-free survival (PFS) following frontline treatment with bendamustine and rituximab (BR) or fludarabine, cyclophosphamide, and rituximab (FCR) therapeutic regimens.
Dr. Spunarova and colleagues used next-generation sequencing to analyze DNA from the patient samples. Data on 11q22, 13q14, trisomy 12, and IgVH mutation status were also considered in the analyses of PFS.
After analysis, the researchers validated that unmutated IgVH status is an indicator of poor prognosis in CLL patients with wild-type TP53 treated with frontline FCR.
When looking at both BR and FCR regimens, a single 11q22 deletion, lacking an ATM mutation on the other allele, resulted in the shortest PFS, at a median of just 16 months.
“Based on our data, special attention should be given to CLL patients harboring a sole 11q22 deletion, with no ATM mutation on the other allele, who manifest particularly short PFS,” they noted.
The researchers acknowledged a key limitation of the study was the small sample size. As a result, the results should be interpreted in a careful manner.
The study was funded by the Ministry of Health of the Czech Republic. The authors reported having no conflicts of interest.
SOURCE: Spunarova M et al. Leuk Res. 2019 Jun;81:75-81.
A genetic analysis of patients with chronic lymphocytic leukemia treated with frontline, rituximab-based regimens found that deletion 11q22 and unmutated IgVH status may predict worse prognosis.
Michaela Spunarova, MD, of Masaryk University, Brno, Czech Republic, and colleagues conducted a genetic analysis of 177 patients with chronic lymphocytic leukemia (CLL). The results of the analysis were published in Leukemia Research.
The study focused on patients with CLL with an intact TP53 gene, looking at recurrently muted genes in CLL, genomic aberrations by fluorescence in situ hybridization, and IgVH status, according to the researchers.
The team analyzed the effects of these mutations on progression-free survival (PFS) following frontline treatment with bendamustine and rituximab (BR) or fludarabine, cyclophosphamide, and rituximab (FCR) therapeutic regimens.
Dr. Spunarova and colleagues used next-generation sequencing to analyze DNA from the patient samples. Data on 11q22, 13q14, trisomy 12, and IgVH mutation status were also considered in the analyses of PFS.
After analysis, the researchers validated that unmutated IgVH status is an indicator of poor prognosis in CLL patients with wild-type TP53 treated with frontline FCR.
When looking at both BR and FCR regimens, a single 11q22 deletion, lacking an ATM mutation on the other allele, resulted in the shortest PFS, at a median of just 16 months.
“Based on our data, special attention should be given to CLL patients harboring a sole 11q22 deletion, with no ATM mutation on the other allele, who manifest particularly short PFS,” they noted.
The researchers acknowledged a key limitation of the study was the small sample size. As a result, the results should be interpreted in a careful manner.
The study was funded by the Ministry of Health of the Czech Republic. The authors reported having no conflicts of interest.
SOURCE: Spunarova M et al. Leuk Res. 2019 Jun;81:75-81.
A genetic analysis of patients with chronic lymphocytic leukemia treated with frontline, rituximab-based regimens found that deletion 11q22 and unmutated IgVH status may predict worse prognosis.
Michaela Spunarova, MD, of Masaryk University, Brno, Czech Republic, and colleagues conducted a genetic analysis of 177 patients with chronic lymphocytic leukemia (CLL). The results of the analysis were published in Leukemia Research.
The study focused on patients with CLL with an intact TP53 gene, looking at recurrently muted genes in CLL, genomic aberrations by fluorescence in situ hybridization, and IgVH status, according to the researchers.
The team analyzed the effects of these mutations on progression-free survival (PFS) following frontline treatment with bendamustine and rituximab (BR) or fludarabine, cyclophosphamide, and rituximab (FCR) therapeutic regimens.
Dr. Spunarova and colleagues used next-generation sequencing to analyze DNA from the patient samples. Data on 11q22, 13q14, trisomy 12, and IgVH mutation status were also considered in the analyses of PFS.
After analysis, the researchers validated that unmutated IgVH status is an indicator of poor prognosis in CLL patients with wild-type TP53 treated with frontline FCR.
When looking at both BR and FCR regimens, a single 11q22 deletion, lacking an ATM mutation on the other allele, resulted in the shortest PFS, at a median of just 16 months.
“Based on our data, special attention should be given to CLL patients harboring a sole 11q22 deletion, with no ATM mutation on the other allele, who manifest particularly short PFS,” they noted.
The researchers acknowledged a key limitation of the study was the small sample size. As a result, the results should be interpreted in a careful manner.
The study was funded by the Ministry of Health of the Czech Republic. The authors reported having no conflicts of interest.
SOURCE: Spunarova M et al. Leuk Res. 2019 Jun;81:75-81.
FROM LEUKEMIA RESEARCH
FDA: Faulty hematology analyzers face class I recall
The Food and Drug Administration is alerting laboratories and providers to a class I recall on Beckman Coulter hematology analyzers because of the potential for inaccurate platelet count results.
A class I recall indicates reasonable probability of serious adverse health consequences or death associated with use, according to the FDA.
The recall is related to the devices’ platelet analyzing function; among other uses, these devices help assess patients fitness for surgery, so a faulty reading on platelet counts could result in increased risk for life-threatening bleeding during a procedure in patients who have unidentified severe thrombocytopenia, according to a statement from the agency.
“Because this may cause serious injury, or even death, to a patient, we are urging health care professionals to be aware of the potential for inaccurate diagnostic results with these analyzers and to take appropriate actions including the use of alternative diagnostic testing or confirming analyzer results with manual scanning or estimate of platelets,” Tim Stenzel, MD, PhD, director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health, said in the statement.
The recall applies to the UniCel DxH 800 Coulter Cellular Analysis System, UniCel DxH 600 Coulter Cellular Analysis System, and UniCel DxH 900 Coulter Cellular Analysis System. The faulty devices were first identified in 2018, and the manufacturer released an urgent medical device correction letter at that time. The company has more recently released a software patch for the devices, but the FDA has not yet assessed whether it resolves the problem. The agency has released detailed actions and recommendations related to these devices.
At this time, the FDA is unaware of any serious adverse events that have been directly linked to these devices, but the agency recommends that any events be reported through its MedWatch reporting system.
The Food and Drug Administration is alerting laboratories and providers to a class I recall on Beckman Coulter hematology analyzers because of the potential for inaccurate platelet count results.
A class I recall indicates reasonable probability of serious adverse health consequences or death associated with use, according to the FDA.
The recall is related to the devices’ platelet analyzing function; among other uses, these devices help assess patients fitness for surgery, so a faulty reading on platelet counts could result in increased risk for life-threatening bleeding during a procedure in patients who have unidentified severe thrombocytopenia, according to a statement from the agency.
“Because this may cause serious injury, or even death, to a patient, we are urging health care professionals to be aware of the potential for inaccurate diagnostic results with these analyzers and to take appropriate actions including the use of alternative diagnostic testing or confirming analyzer results with manual scanning or estimate of platelets,” Tim Stenzel, MD, PhD, director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health, said in the statement.
The recall applies to the UniCel DxH 800 Coulter Cellular Analysis System, UniCel DxH 600 Coulter Cellular Analysis System, and UniCel DxH 900 Coulter Cellular Analysis System. The faulty devices were first identified in 2018, and the manufacturer released an urgent medical device correction letter at that time. The company has more recently released a software patch for the devices, but the FDA has not yet assessed whether it resolves the problem. The agency has released detailed actions and recommendations related to these devices.
At this time, the FDA is unaware of any serious adverse events that have been directly linked to these devices, but the agency recommends that any events be reported through its MedWatch reporting system.
The Food and Drug Administration is alerting laboratories and providers to a class I recall on Beckman Coulter hematology analyzers because of the potential for inaccurate platelet count results.
A class I recall indicates reasonable probability of serious adverse health consequences or death associated with use, according to the FDA.
The recall is related to the devices’ platelet analyzing function; among other uses, these devices help assess patients fitness for surgery, so a faulty reading on platelet counts could result in increased risk for life-threatening bleeding during a procedure in patients who have unidentified severe thrombocytopenia, according to a statement from the agency.
“Because this may cause serious injury, or even death, to a patient, we are urging health care professionals to be aware of the potential for inaccurate diagnostic results with these analyzers and to take appropriate actions including the use of alternative diagnostic testing or confirming analyzer results with manual scanning or estimate of platelets,” Tim Stenzel, MD, PhD, director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health, said in the statement.
The recall applies to the UniCel DxH 800 Coulter Cellular Analysis System, UniCel DxH 600 Coulter Cellular Analysis System, and UniCel DxH 900 Coulter Cellular Analysis System. The faulty devices were first identified in 2018, and the manufacturer released an urgent medical device correction letter at that time. The company has more recently released a software patch for the devices, but the FDA has not yet assessed whether it resolves the problem. The agency has released detailed actions and recommendations related to these devices.
At this time, the FDA is unaware of any serious adverse events that have been directly linked to these devices, but the agency recommends that any events be reported through its MedWatch reporting system.
FVIII/W ratio may help predict relapse in hemophilia A
, according to a retrospective analysis.
Marc Trossaert, MD, PhD, of the CHU de Nantes, France, and colleagues conducted a retrospective analysis of 64 consecutive patients diagnosed with acquired hemophilia A over a period of 15 years (2000-2015). Data were obtained from institutional databases at the Toulouse and Nantes university hospitals in France.
Data collected included patient demographics, comorbidities, biological factors, and information related to immunosuppressive therapy. The findings of the study were published in Haemophilia.
To ascertain normal parameters and uses of the FVIII/W ratio, the team assessed FVIII:C and VWF:Ag levels of 40 healthy individuals and normal parameters of the ratio were defined using these levels.
Among the 64 patients with acquired hemophilia A who were enrolled in the study, 55 patients achieved complete remission. Of that group, 44 patients did not relapse. Researchers had follow-up data of at least 1 year for 22 of these patients. They found that the FVIII/W ratio remained within normal parameters for all 22 patients.
Researchers had follow-up data on 5 of the 11 patients who relapsed during the study period. For 4 of the patients, a decrease of FVIII/W ratio was the first indicator of relapse. In the fifth patient, an abnormal activated partial thromboplastin time (aPTT) displayed before the changes were observed in the FVIII/W ratio.
Dr. Trossaert and his colleagues acknowledged that a key limitation of the study was the retrospective design.
“We cannot eliminate the fact that in these patients less frequent testing may have influenced the chance of seeing a low FVIII/W ratio,” they wrote.
The biomarker now needs to be studied in larger cohorts, the researchers suggested.
No funding sources were reported. The authors reported having no conflicts of interest.
SOURCE: Trossaert M et al. Haemophilia. 2019 May 2. doi: 10.1111/hae.13752.
, according to a retrospective analysis.
Marc Trossaert, MD, PhD, of the CHU de Nantes, France, and colleagues conducted a retrospective analysis of 64 consecutive patients diagnosed with acquired hemophilia A over a period of 15 years (2000-2015). Data were obtained from institutional databases at the Toulouse and Nantes university hospitals in France.
Data collected included patient demographics, comorbidities, biological factors, and information related to immunosuppressive therapy. The findings of the study were published in Haemophilia.
To ascertain normal parameters and uses of the FVIII/W ratio, the team assessed FVIII:C and VWF:Ag levels of 40 healthy individuals and normal parameters of the ratio were defined using these levels.
Among the 64 patients with acquired hemophilia A who were enrolled in the study, 55 patients achieved complete remission. Of that group, 44 patients did not relapse. Researchers had follow-up data of at least 1 year for 22 of these patients. They found that the FVIII/W ratio remained within normal parameters for all 22 patients.
Researchers had follow-up data on 5 of the 11 patients who relapsed during the study period. For 4 of the patients, a decrease of FVIII/W ratio was the first indicator of relapse. In the fifth patient, an abnormal activated partial thromboplastin time (aPTT) displayed before the changes were observed in the FVIII/W ratio.
Dr. Trossaert and his colleagues acknowledged that a key limitation of the study was the retrospective design.
“We cannot eliminate the fact that in these patients less frequent testing may have influenced the chance of seeing a low FVIII/W ratio,” they wrote.
The biomarker now needs to be studied in larger cohorts, the researchers suggested.
No funding sources were reported. The authors reported having no conflicts of interest.
SOURCE: Trossaert M et al. Haemophilia. 2019 May 2. doi: 10.1111/hae.13752.
, according to a retrospective analysis.
Marc Trossaert, MD, PhD, of the CHU de Nantes, France, and colleagues conducted a retrospective analysis of 64 consecutive patients diagnosed with acquired hemophilia A over a period of 15 years (2000-2015). Data were obtained from institutional databases at the Toulouse and Nantes university hospitals in France.
Data collected included patient demographics, comorbidities, biological factors, and information related to immunosuppressive therapy. The findings of the study were published in Haemophilia.
To ascertain normal parameters and uses of the FVIII/W ratio, the team assessed FVIII:C and VWF:Ag levels of 40 healthy individuals and normal parameters of the ratio were defined using these levels.
Among the 64 patients with acquired hemophilia A who were enrolled in the study, 55 patients achieved complete remission. Of that group, 44 patients did not relapse. Researchers had follow-up data of at least 1 year for 22 of these patients. They found that the FVIII/W ratio remained within normal parameters for all 22 patients.
Researchers had follow-up data on 5 of the 11 patients who relapsed during the study period. For 4 of the patients, a decrease of FVIII/W ratio was the first indicator of relapse. In the fifth patient, an abnormal activated partial thromboplastin time (aPTT) displayed before the changes were observed in the FVIII/W ratio.
Dr. Trossaert and his colleagues acknowledged that a key limitation of the study was the retrospective design.
“We cannot eliminate the fact that in these patients less frequent testing may have influenced the chance of seeing a low FVIII/W ratio,” they wrote.
The biomarker now needs to be studied in larger cohorts, the researchers suggested.
No funding sources were reported. The authors reported having no conflicts of interest.
SOURCE: Trossaert M et al. Haemophilia. 2019 May 2. doi: 10.1111/hae.13752.
FROM HAEMOPHILIA
Novel chromogenic assay looks accurate in hemophilia A diagnosis
, according to recent study findings.
“The original one‐stage clotting assay is still the most widely used method for measuring FVIII activity in these patients, although the chromogenic assay is recognized to be less prone to the variability related to the use of different reagents and to the presence of interferences,” Cristina Novembrino, MD, of the Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, in Milan, and colleagues wrote in Haemophilia. “The choice of the proper assay is a crucial point in the frame of diagnosis, particularly in moderate or mild [hemophilia A] patients.”
The BIOPHEN FVIII:C assay, used on the Sysmex CS‐2400 analyzer, is a novel chromogenic diagnostic tool used to analyze FVIII clotting activity in patients with hemophilia A of all severity levels. The researchers evaluated the diagnostic and clinical capabilities of the assay in 60 patients with hemophilia A and 120 healthy controls.
Dr. Novembrino and colleagues used samples of FVIII deficient plasma and Actin FS to compare the novel tool to a one-stage assay and another chromogenic assay.
After analysis, the researchers found that the inter‐assay and intra‐assay coefficient of variation were less than 6%. The mean recovery and limit of detection were 91.7% (range, 79.8%-98.6%) and 0.2%, respectively.
The linearity test revealed positive results of up to 1/128 dilution (r = 0.99).
“The K coefficient was 0.91 when BIOPHEN FVIII:C was compared with the historical classification of the patients, demonstrating an optimal diagnostic accuracy in hemophilia A,” the researchers wrote.
The novel assay may be an appropriate laboratory tool for the diagnosis and therapeutic monitoring of patients with hemophilia A, they added.
Sysmex Corporation and Hyphen BioMed provided the instrument and reagents for the study. One of the authors is an employee of Sysmex Corporation. The authors reported having no other conflicts of interest.
SOURCE: Novembrino C et al. Haemophilia. 2019 May 2. doi: 10.1111/hae.13746.
, according to recent study findings.
“The original one‐stage clotting assay is still the most widely used method for measuring FVIII activity in these patients, although the chromogenic assay is recognized to be less prone to the variability related to the use of different reagents and to the presence of interferences,” Cristina Novembrino, MD, of the Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, in Milan, and colleagues wrote in Haemophilia. “The choice of the proper assay is a crucial point in the frame of diagnosis, particularly in moderate or mild [hemophilia A] patients.”
The BIOPHEN FVIII:C assay, used on the Sysmex CS‐2400 analyzer, is a novel chromogenic diagnostic tool used to analyze FVIII clotting activity in patients with hemophilia A of all severity levels. The researchers evaluated the diagnostic and clinical capabilities of the assay in 60 patients with hemophilia A and 120 healthy controls.
Dr. Novembrino and colleagues used samples of FVIII deficient plasma and Actin FS to compare the novel tool to a one-stage assay and another chromogenic assay.
After analysis, the researchers found that the inter‐assay and intra‐assay coefficient of variation were less than 6%. The mean recovery and limit of detection were 91.7% (range, 79.8%-98.6%) and 0.2%, respectively.
The linearity test revealed positive results of up to 1/128 dilution (r = 0.99).
“The K coefficient was 0.91 when BIOPHEN FVIII:C was compared with the historical classification of the patients, demonstrating an optimal diagnostic accuracy in hemophilia A,” the researchers wrote.
The novel assay may be an appropriate laboratory tool for the diagnosis and therapeutic monitoring of patients with hemophilia A, they added.
Sysmex Corporation and Hyphen BioMed provided the instrument and reagents for the study. One of the authors is an employee of Sysmex Corporation. The authors reported having no other conflicts of interest.
SOURCE: Novembrino C et al. Haemophilia. 2019 May 2. doi: 10.1111/hae.13746.
, according to recent study findings.
“The original one‐stage clotting assay is still the most widely used method for measuring FVIII activity in these patients, although the chromogenic assay is recognized to be less prone to the variability related to the use of different reagents and to the presence of interferences,” Cristina Novembrino, MD, of the Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, in Milan, and colleagues wrote in Haemophilia. “The choice of the proper assay is a crucial point in the frame of diagnosis, particularly in moderate or mild [hemophilia A] patients.”
The BIOPHEN FVIII:C assay, used on the Sysmex CS‐2400 analyzer, is a novel chromogenic diagnostic tool used to analyze FVIII clotting activity in patients with hemophilia A of all severity levels. The researchers evaluated the diagnostic and clinical capabilities of the assay in 60 patients with hemophilia A and 120 healthy controls.
Dr. Novembrino and colleagues used samples of FVIII deficient plasma and Actin FS to compare the novel tool to a one-stage assay and another chromogenic assay.
After analysis, the researchers found that the inter‐assay and intra‐assay coefficient of variation were less than 6%. The mean recovery and limit of detection were 91.7% (range, 79.8%-98.6%) and 0.2%, respectively.
The linearity test revealed positive results of up to 1/128 dilution (r = 0.99).
“The K coefficient was 0.91 when BIOPHEN FVIII:C was compared with the historical classification of the patients, demonstrating an optimal diagnostic accuracy in hemophilia A,” the researchers wrote.
The novel assay may be an appropriate laboratory tool for the diagnosis and therapeutic monitoring of patients with hemophilia A, they added.
Sysmex Corporation and Hyphen BioMed provided the instrument and reagents for the study. One of the authors is an employee of Sysmex Corporation. The authors reported having no other conflicts of interest.
SOURCE: Novembrino C et al. Haemophilia. 2019 May 2. doi: 10.1111/hae.13746.
FROM HAEMOPHILIA
Low-dose FVIII prophylaxis reduced bleeding in hemophilia A
The findings have implications for low resource countries, the researchers reported.
Novie A. Chozie, MD, of Universitas Indonesia in Jakarta, along with colleagues, conducted a parallel-group, randomized controlled study of 50 children with severe hemophilia A. Study participants were randomized to receive either low‐dose FVIII prophylaxis (n = 25) or on‐demand treatment (n = 25) for a total of 12 months. The findings were published in Haemophilia.
Participants in the prophylaxis arm received FVIII at a dose of 10 IU/kg, infused twice weekly. If patients experienced an episode of acute bleeding, prophylaxis was delayed until the episode resolved.
The primary outcome was the number of total and joint bleeding events from the start of therapy to 12 months. Secondary outcomes included evidence of FVIII inhibitor, Hemophilia Joint Health Score (HJHS), and Hemophilia Early Arthropathy Detection Ultrasound (HEAD‐US) score.
After analysis, the team found that the number of joint and total bleeding events was significantly lower in the prophylaxis group. For total bleeding events, there was a median of 8 events with the prophylaxis group, compared with 25 in the on-demand treatment group (P less than .001). There was a median of three joint bleeding events in the prophylaxis group versus nine in the on-demand group (P less than .001).
Patients in the prophylaxis arm also showed improved joint function (P = .004), while those in the on‐demand arm showed evidence of deterioration (P = .001).
Two key limitations of the study were short duration of follow-up and single‐center design, which could have limited the generalizability of the results.
“In countries with limited resources, low‐dose prophylaxis is strongly recommended as a therapeutic option for severe haemophilia A [in] children,” the researchers wrote.
The study was funded by Grifols. The authors reported having no conflicts of interest.
SOURCE: Chozie NA et al. Haemophilia. 2019 May 2. doi: 10.1111/hae.13770.
The findings have implications for low resource countries, the researchers reported.
Novie A. Chozie, MD, of Universitas Indonesia in Jakarta, along with colleagues, conducted a parallel-group, randomized controlled study of 50 children with severe hemophilia A. Study participants were randomized to receive either low‐dose FVIII prophylaxis (n = 25) or on‐demand treatment (n = 25) for a total of 12 months. The findings were published in Haemophilia.
Participants in the prophylaxis arm received FVIII at a dose of 10 IU/kg, infused twice weekly. If patients experienced an episode of acute bleeding, prophylaxis was delayed until the episode resolved.
The primary outcome was the number of total and joint bleeding events from the start of therapy to 12 months. Secondary outcomes included evidence of FVIII inhibitor, Hemophilia Joint Health Score (HJHS), and Hemophilia Early Arthropathy Detection Ultrasound (HEAD‐US) score.
After analysis, the team found that the number of joint and total bleeding events was significantly lower in the prophylaxis group. For total bleeding events, there was a median of 8 events with the prophylaxis group, compared with 25 in the on-demand treatment group (P less than .001). There was a median of three joint bleeding events in the prophylaxis group versus nine in the on-demand group (P less than .001).
Patients in the prophylaxis arm also showed improved joint function (P = .004), while those in the on‐demand arm showed evidence of deterioration (P = .001).
Two key limitations of the study were short duration of follow-up and single‐center design, which could have limited the generalizability of the results.
“In countries with limited resources, low‐dose prophylaxis is strongly recommended as a therapeutic option for severe haemophilia A [in] children,” the researchers wrote.
The study was funded by Grifols. The authors reported having no conflicts of interest.
SOURCE: Chozie NA et al. Haemophilia. 2019 May 2. doi: 10.1111/hae.13770.
The findings have implications for low resource countries, the researchers reported.
Novie A. Chozie, MD, of Universitas Indonesia in Jakarta, along with colleagues, conducted a parallel-group, randomized controlled study of 50 children with severe hemophilia A. Study participants were randomized to receive either low‐dose FVIII prophylaxis (n = 25) or on‐demand treatment (n = 25) for a total of 12 months. The findings were published in Haemophilia.
Participants in the prophylaxis arm received FVIII at a dose of 10 IU/kg, infused twice weekly. If patients experienced an episode of acute bleeding, prophylaxis was delayed until the episode resolved.
The primary outcome was the number of total and joint bleeding events from the start of therapy to 12 months. Secondary outcomes included evidence of FVIII inhibitor, Hemophilia Joint Health Score (HJHS), and Hemophilia Early Arthropathy Detection Ultrasound (HEAD‐US) score.
After analysis, the team found that the number of joint and total bleeding events was significantly lower in the prophylaxis group. For total bleeding events, there was a median of 8 events with the prophylaxis group, compared with 25 in the on-demand treatment group (P less than .001). There was a median of three joint bleeding events in the prophylaxis group versus nine in the on-demand group (P less than .001).
Patients in the prophylaxis arm also showed improved joint function (P = .004), while those in the on‐demand arm showed evidence of deterioration (P = .001).
Two key limitations of the study were short duration of follow-up and single‐center design, which could have limited the generalizability of the results.
“In countries with limited resources, low‐dose prophylaxis is strongly recommended as a therapeutic option for severe haemophilia A [in] children,” the researchers wrote.
The study was funded by Grifols. The authors reported having no conflicts of interest.
SOURCE: Chozie NA et al. Haemophilia. 2019 May 2. doi: 10.1111/hae.13770.
FROM HAEMOPHILIA
Family history plays a large role in bleeding disorder diagnosis in women
Disease severity and family history appear to play a significant role in the age of diagnosis for women with congenital bleeding disorders, according to recent survey findings.
A European multinational survey has identified delays in diagnosis and other challenges faced by girls and women with congenital bleeding disorders.
“The aim of this survey, carried out by the European Haemophilia Consortium (EHC), was to provide the patient voice of their lived experiences with congenital bleeding disorders,” wrote Declan Noone of the EHC in Brussels, and colleagues. The findings were published in Haemophilia.
The researchers conducted a survey of 709 girls and women with various congenital bleeding disorders from 32 countries, primarily located in Western Europe. Most respondents were adults, with just 3.8% under age 18 years.
The questionnaire was administered to eligible patients at various hemophilia treatment centers. More than half of respondents were hemophilia carriers and nearly 28% had von Willebrand disease.
The survey explored the effects of bleeding disorders on several activities of daily life, including symptoms, physical activity, and reproductive ability.
After analysis, the researchers found that overall the median age at diagnosis of a bleeding disorder was 16 years (range, 2-28 years) among respondents. Having a family history of a bleeding disorder resulted in a significantly younger median age at diagnosis (6 years; range, 0-26 years) versus those without a family history (17 years; range 5-28 years; P less than .01).
Disease severity also appears to play a role. Women with type 3 von Willebrand disease had a median age of diagnosis of 1 year old, compared with 19.3 years old for type 2 disease (P less than .01).
Respondents reported a substantial disease burden on activities of daily life, especially for women with platelet function disorders and other factor deficiency.
Women without a known family history of a bleeding disorders reported a significantly greater impact on their physical life, social life, and romantic life (P less than .01 for all domains), compared with women with a family history of bleeding disorders.
There were no statistically significant differences across types of bleeding disorders on questions related to reproductive life. However, the researchers reported that “surprisingly,” 25% of women reported that having a bleeding disorder “has had a severe impact on their decision or has prevented them from having children.
“The bleeding symptom of biggest impact on daily life is [heavy menstrual bleeding], reported by 55% of women,” the researchers wrote.
The researchers acknowledged that a key limitation of the survey was the composition of the sample: predominantly of patients from Western Europe. As a result, the findings may not be generalizable to all patient populations.
No funding sources were reported. The authors reported having no conflicts of interest.
SOURCE: Noone D et al. Haemophilia. 2019 Apr 29. doi: 10.1111/hae.13722.
Disease severity and family history appear to play a significant role in the age of diagnosis for women with congenital bleeding disorders, according to recent survey findings.
A European multinational survey has identified delays in diagnosis and other challenges faced by girls and women with congenital bleeding disorders.
“The aim of this survey, carried out by the European Haemophilia Consortium (EHC), was to provide the patient voice of their lived experiences with congenital bleeding disorders,” wrote Declan Noone of the EHC in Brussels, and colleagues. The findings were published in Haemophilia.
The researchers conducted a survey of 709 girls and women with various congenital bleeding disorders from 32 countries, primarily located in Western Europe. Most respondents were adults, with just 3.8% under age 18 years.
The questionnaire was administered to eligible patients at various hemophilia treatment centers. More than half of respondents were hemophilia carriers and nearly 28% had von Willebrand disease.
The survey explored the effects of bleeding disorders on several activities of daily life, including symptoms, physical activity, and reproductive ability.
After analysis, the researchers found that overall the median age at diagnosis of a bleeding disorder was 16 years (range, 2-28 years) among respondents. Having a family history of a bleeding disorder resulted in a significantly younger median age at diagnosis (6 years; range, 0-26 years) versus those without a family history (17 years; range 5-28 years; P less than .01).
Disease severity also appears to play a role. Women with type 3 von Willebrand disease had a median age of diagnosis of 1 year old, compared with 19.3 years old for type 2 disease (P less than .01).
Respondents reported a substantial disease burden on activities of daily life, especially for women with platelet function disorders and other factor deficiency.
Women without a known family history of a bleeding disorders reported a significantly greater impact on their physical life, social life, and romantic life (P less than .01 for all domains), compared with women with a family history of bleeding disorders.
There were no statistically significant differences across types of bleeding disorders on questions related to reproductive life. However, the researchers reported that “surprisingly,” 25% of women reported that having a bleeding disorder “has had a severe impact on their decision or has prevented them from having children.
“The bleeding symptom of biggest impact on daily life is [heavy menstrual bleeding], reported by 55% of women,” the researchers wrote.
The researchers acknowledged that a key limitation of the survey was the composition of the sample: predominantly of patients from Western Europe. As a result, the findings may not be generalizable to all patient populations.
No funding sources were reported. The authors reported having no conflicts of interest.
SOURCE: Noone D et al. Haemophilia. 2019 Apr 29. doi: 10.1111/hae.13722.
Disease severity and family history appear to play a significant role in the age of diagnosis for women with congenital bleeding disorders, according to recent survey findings.
A European multinational survey has identified delays in diagnosis and other challenges faced by girls and women with congenital bleeding disorders.
“The aim of this survey, carried out by the European Haemophilia Consortium (EHC), was to provide the patient voice of their lived experiences with congenital bleeding disorders,” wrote Declan Noone of the EHC in Brussels, and colleagues. The findings were published in Haemophilia.
The researchers conducted a survey of 709 girls and women with various congenital bleeding disorders from 32 countries, primarily located in Western Europe. Most respondents were adults, with just 3.8% under age 18 years.
The questionnaire was administered to eligible patients at various hemophilia treatment centers. More than half of respondents were hemophilia carriers and nearly 28% had von Willebrand disease.
The survey explored the effects of bleeding disorders on several activities of daily life, including symptoms, physical activity, and reproductive ability.
After analysis, the researchers found that overall the median age at diagnosis of a bleeding disorder was 16 years (range, 2-28 years) among respondents. Having a family history of a bleeding disorder resulted in a significantly younger median age at diagnosis (6 years; range, 0-26 years) versus those without a family history (17 years; range 5-28 years; P less than .01).
Disease severity also appears to play a role. Women with type 3 von Willebrand disease had a median age of diagnosis of 1 year old, compared with 19.3 years old for type 2 disease (P less than .01).
Respondents reported a substantial disease burden on activities of daily life, especially for women with platelet function disorders and other factor deficiency.
Women without a known family history of a bleeding disorders reported a significantly greater impact on their physical life, social life, and romantic life (P less than .01 for all domains), compared with women with a family history of bleeding disorders.
There were no statistically significant differences across types of bleeding disorders on questions related to reproductive life. However, the researchers reported that “surprisingly,” 25% of women reported that having a bleeding disorder “has had a severe impact on their decision or has prevented them from having children.
“The bleeding symptom of biggest impact on daily life is [heavy menstrual bleeding], reported by 55% of women,” the researchers wrote.
The researchers acknowledged that a key limitation of the survey was the composition of the sample: predominantly of patients from Western Europe. As a result, the findings may not be generalizable to all patient populations.
No funding sources were reported. The authors reported having no conflicts of interest.
SOURCE: Noone D et al. Haemophilia. 2019 Apr 29. doi: 10.1111/hae.13722.
FROM HAEMOPHILIA
NGS comparable to FC for minimal residual disease assessment
NEW ORLEANS – Next-generation sequencing of peripheral blood is at least as effective as flow cytometry of bone marrow for assessing minimal residual disease, according to a new study.
Researchers compared bone marrow flow cytometry (FC) and peripheral blood next-generation sequencing (NGS) for minimal residual disease (MRD) assessment in pediatric and young adult patients with B-cell acute lymphoblastic leukemia (B-ALL) who received treatment with tisagenlecleucel. There was a high level of concordance between the assays, but the NGS assay detected more MRD-positive samples and NGS results provided a longer lead time to relapse.
Michael A. Pulsipher, MD, of the Children’s Hospital Los Angeles, presented these results at the annual meeting of the American Society of Pediatric Hematology/Oncology.
The researchers analyzed samples from pediatric and young adult patients aged 2-25 years who had relapsed or refractory B-ALL and received treatment with tisagenlecleucel on the ELIANA or ENSIGN trials.
The patients had received at least two prior lines of therapy and were ineligible for allogeneic transplant. They received a single dose of tisagenlecleucel. MRD was assessed before tisagenlecleucel infusion, at various time points after infusion, and at relapse.
Dr. Pulsipher and his colleagues compared MRD results from an NGS assay – Adaptive Biotechnologies’ clonoSEQ – using peripheral blood and results from FC of bone marrow. NGS and FC results were available for 237 samples from 83 patients.
After treatment, NGS detected more MRD-positive samples at each sensitivity level tested (10-4, 10-5, and 10-6). At 10-6, NGS detected 18% more MRD-positive samples than did FC – 50% and 32%, respectively.
Detection of MRD positivity prior to relapse was faster with NGS than with FC. In 17 of 34 patients with morphological relapse, NGS provided a median lead time of 67 days. FC provided a median lead time of 39 days in 11 of the 34 patients.
About 80% of patients who had an MRD status of zero by NGS at day 28 remained relapse-free for up to 3 years.
Among complete responders (n = 50), the duration of response was significantly longer in patients who had an MRD status of zero at day 28 by NGS than in patients who had an MRD status greater than zero (P = .0003). Overall survival was significantly better among patients with an MRD status of zero as well (P = .0004).
Dr. Pulsipher said additional studies are needed to confirm these findings and determine the best way to know if a patient has been cured or needs additional therapy after tisagenlecleucel.
Dr. Pulsipher reported relationships with Adaptive Biotech, Novartis, Incyte, Amgen, Bellicum Pharmaceuticals, Medac Pharma, and Miltenyi Biotec. ELIANA and ENSIGN were funded by Novartis, which markets tisagenlecleucel as Kymriah.
SOURCE: Pulsipher MA et al. ASPHO 2019, Abstract 2001.
NEW ORLEANS – Next-generation sequencing of peripheral blood is at least as effective as flow cytometry of bone marrow for assessing minimal residual disease, according to a new study.
Researchers compared bone marrow flow cytometry (FC) and peripheral blood next-generation sequencing (NGS) for minimal residual disease (MRD) assessment in pediatric and young adult patients with B-cell acute lymphoblastic leukemia (B-ALL) who received treatment with tisagenlecleucel. There was a high level of concordance between the assays, but the NGS assay detected more MRD-positive samples and NGS results provided a longer lead time to relapse.
Michael A. Pulsipher, MD, of the Children’s Hospital Los Angeles, presented these results at the annual meeting of the American Society of Pediatric Hematology/Oncology.
The researchers analyzed samples from pediatric and young adult patients aged 2-25 years who had relapsed or refractory B-ALL and received treatment with tisagenlecleucel on the ELIANA or ENSIGN trials.
The patients had received at least two prior lines of therapy and were ineligible for allogeneic transplant. They received a single dose of tisagenlecleucel. MRD was assessed before tisagenlecleucel infusion, at various time points after infusion, and at relapse.
Dr. Pulsipher and his colleagues compared MRD results from an NGS assay – Adaptive Biotechnologies’ clonoSEQ – using peripheral blood and results from FC of bone marrow. NGS and FC results were available for 237 samples from 83 patients.
After treatment, NGS detected more MRD-positive samples at each sensitivity level tested (10-4, 10-5, and 10-6). At 10-6, NGS detected 18% more MRD-positive samples than did FC – 50% and 32%, respectively.
Detection of MRD positivity prior to relapse was faster with NGS than with FC. In 17 of 34 patients with morphological relapse, NGS provided a median lead time of 67 days. FC provided a median lead time of 39 days in 11 of the 34 patients.
About 80% of patients who had an MRD status of zero by NGS at day 28 remained relapse-free for up to 3 years.
Among complete responders (n = 50), the duration of response was significantly longer in patients who had an MRD status of zero at day 28 by NGS than in patients who had an MRD status greater than zero (P = .0003). Overall survival was significantly better among patients with an MRD status of zero as well (P = .0004).
Dr. Pulsipher said additional studies are needed to confirm these findings and determine the best way to know if a patient has been cured or needs additional therapy after tisagenlecleucel.
Dr. Pulsipher reported relationships with Adaptive Biotech, Novartis, Incyte, Amgen, Bellicum Pharmaceuticals, Medac Pharma, and Miltenyi Biotec. ELIANA and ENSIGN were funded by Novartis, which markets tisagenlecleucel as Kymriah.
SOURCE: Pulsipher MA et al. ASPHO 2019, Abstract 2001.
NEW ORLEANS – Next-generation sequencing of peripheral blood is at least as effective as flow cytometry of bone marrow for assessing minimal residual disease, according to a new study.
Researchers compared bone marrow flow cytometry (FC) and peripheral blood next-generation sequencing (NGS) for minimal residual disease (MRD) assessment in pediatric and young adult patients with B-cell acute lymphoblastic leukemia (B-ALL) who received treatment with tisagenlecleucel. There was a high level of concordance between the assays, but the NGS assay detected more MRD-positive samples and NGS results provided a longer lead time to relapse.
Michael A. Pulsipher, MD, of the Children’s Hospital Los Angeles, presented these results at the annual meeting of the American Society of Pediatric Hematology/Oncology.
The researchers analyzed samples from pediatric and young adult patients aged 2-25 years who had relapsed or refractory B-ALL and received treatment with tisagenlecleucel on the ELIANA or ENSIGN trials.
The patients had received at least two prior lines of therapy and were ineligible for allogeneic transplant. They received a single dose of tisagenlecleucel. MRD was assessed before tisagenlecleucel infusion, at various time points after infusion, and at relapse.
Dr. Pulsipher and his colleagues compared MRD results from an NGS assay – Adaptive Biotechnologies’ clonoSEQ – using peripheral blood and results from FC of bone marrow. NGS and FC results were available for 237 samples from 83 patients.
After treatment, NGS detected more MRD-positive samples at each sensitivity level tested (10-4, 10-5, and 10-6). At 10-6, NGS detected 18% more MRD-positive samples than did FC – 50% and 32%, respectively.
Detection of MRD positivity prior to relapse was faster with NGS than with FC. In 17 of 34 patients with morphological relapse, NGS provided a median lead time of 67 days. FC provided a median lead time of 39 days in 11 of the 34 patients.
About 80% of patients who had an MRD status of zero by NGS at day 28 remained relapse-free for up to 3 years.
Among complete responders (n = 50), the duration of response was significantly longer in patients who had an MRD status of zero at day 28 by NGS than in patients who had an MRD status greater than zero (P = .0003). Overall survival was significantly better among patients with an MRD status of zero as well (P = .0004).
Dr. Pulsipher said additional studies are needed to confirm these findings and determine the best way to know if a patient has been cured or needs additional therapy after tisagenlecleucel.
Dr. Pulsipher reported relationships with Adaptive Biotech, Novartis, Incyte, Amgen, Bellicum Pharmaceuticals, Medac Pharma, and Miltenyi Biotec. ELIANA and ENSIGN were funded by Novartis, which markets tisagenlecleucel as Kymriah.
SOURCE: Pulsipher MA et al. ASPHO 2019, Abstract 2001.
REPORTING FROM 2019 ASPHO CONFERENCE
Key clinical point: Major finding: At the highest sensitivity level tested, next-generation sequencing detected 18% more minimal residual disease–positive samples than did flow cytometry – 50% and 32%, respectively.
Study details: An analysis of samples from pediatric and young adult patients with B-cell acute lymphoblastic leukemia who received treatment with tisagenlecleucel on the ELIANA and ENSIGN trials.
Disclosures: The speaker reported relationships with Adaptive Biotech, Novartis, Incyte, Amgen, Bellicum Pharmaceuticals, Medac Pharma, and Miltenyi Biotec. The ELIANA and ENSIGN trials were funded by Novartis, which markets tisagenlecleucel as Kymriah.
Source: Pulsipher MA et al. ASPHO 2019, Abstract 2001.